

ABSTRACT
Access to DNA is a prerequisite to the execution of essential cellular processes that include transcription, replication, chromosomal segregation, and DNA repair. How the proteins that regulate these processes function in the context of chromatin and its dynamic architectures is an intensive field of study. Over the past decade, genome-wide assays and new imaging approaches have enabled a greater understanding of how access to the genome is regulated by nucleosomes and associated proteins. Additional mechanisms that may control DNA accessibility in vivo include chromatin compaction and phase separation – processes that are beginning to be understood. Here, we review the ongoing development of accessibility measurements, we summarize the different molecular and structural mechanisms that shape the accessibility landscape, and we detail the many important biological functions that are linked to chromatin accessibility.
KEYWORDS: Accessibility, ATAC-seq, chromatin, compaction, HP1, heterochromatin, linker histones, MNase-seq, phase separation, transcription
Chromatin is composed of DNA packaged into nucleosomes and decorated by associated proteins and RNAs. Its variable composition, structure, and dynamics can regulate genomic processes ranging from the molecular scale, affecting transcriptional factor binding, to functional outcomes, such as DNA repair and replication. An important aspect of chromatin-based regulation lies in the controlled ability of factors to gain and maintain physical access to DNA – often referred to as DNA or chromatin accessibility. Nucleosomes and other DNA-bound proteins are major regulators of accessibility, and their contributions will be described throughout this review, in which we detail the defining measurements, the regulatory mechanisms, and the physiological relevance of chromatin accessibility.
Defining and measuring chromatin accessibility
A continuum of chromatin states
Within a nucleus, there is a spectrum of DNA accessibility states that range from hyper-accessible, which is often referred to as ‘open’ chromatin, to more moderate states of accessibility known as ‘permissive’ chromatin, and to more inaccessible or repressive states denoted as ‘closed’ chromatin [1]. Open and permissive states are often transcriptionally active chromatin and are often interchangeably used with the term ‘euchromatin’, whereas closed states are commonly referred to as ‘heterochromatin’. However, it is important to note that these terms originated to qualitatively describe large genomic domains, and with an updated understanding of chromatin’s complexity, euchromatin and heterochromatin are not necessarily the best descriptors of accessibility. Assessing the relationships between DNA and proteins has provided a more useful quantitative framework for defining chromatin accessibility. Three possible ways to frame this relationship are through (1) the diffusion of proteins into the neighborhood of a genomic locus (Figure 1(a)), (2) the ability of a protein to bind and/or modify DNA in a non-sequence specific manner (Figure 1(b)), or (3) the ability of a protein to bind/modify DNA at a specific motif (Figure 1(c)). In the following subsections and in Table 1, we discuss the strategies, results, and caveats of broadly used assays that characterize chromatin accessibility.
Figure 1.
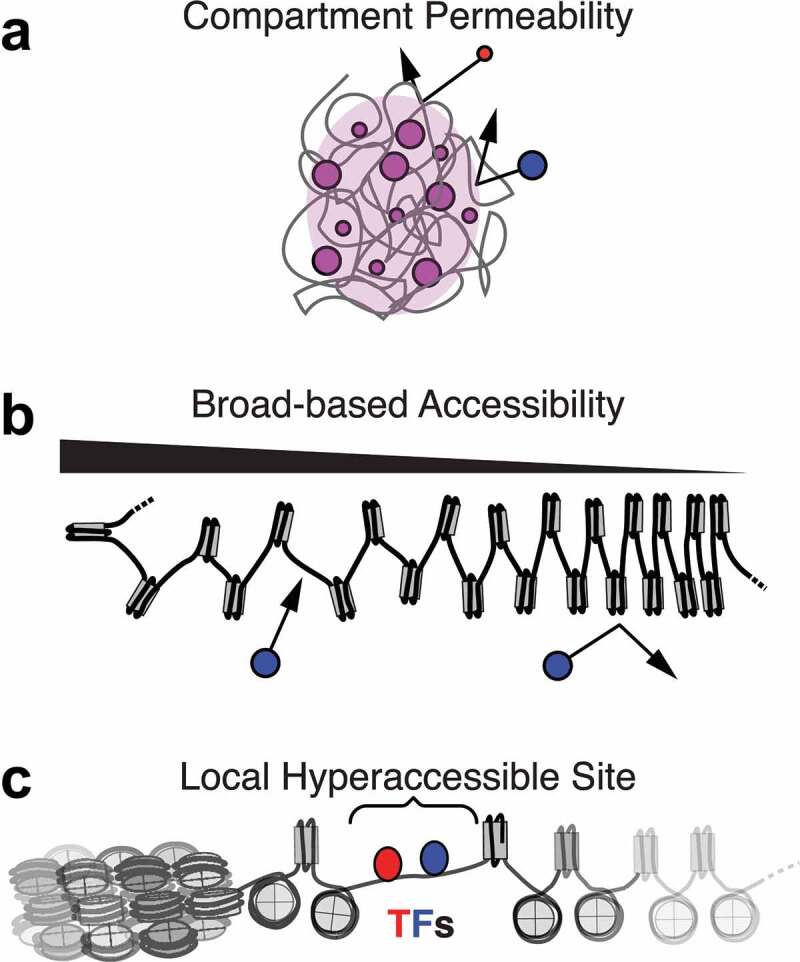
Types of accessibility. (a) Permeability of a nuclear volume (‘compartment’) determines whether proteins can diffuse into the same space as a genomic locus. (b) Broad-based accessibility of chromatin in a set of genomic loci determines the ability of proteins to nonspecifically access and scan the genomic DNA. Chromatin fiber compaction is shown as a mechanism for modulating broad-based accessibility, but many other mechanisms are possible. (c) Local hyper-accessible sites are narrow loci where proteins can access DNA with similar ease as de-chromatinized DNA. These sites are thought to have low nucleosome occupancy for several reasons, including competition from transcription factors (TFs) (illustrated, blue and red) that exclude nucleosomes.
Table 1.
DNA sequencing-based methods for mapping nucleosomes and/or chromatin accessibility.
| Assay | Description | References |
|---|---|---|
| MPE-seq | Radical-based linker DNA cleavage method that maps nucleosomal and sub-nucleosomal protected DNA fragments with minimal sequence bias [2]. | Ishii et al. 2015 |
| KAS-seq | Genome-wide mapping of ssDNA produced by transcriptionally active RNA polymerases. Compatible with low cell number [3]. | Wu et al. 2020 |
| FAIRE-seq | Non-nuclease genome-wide accessibility assay that uses formaldehyde fixation and sonication to enrich for hyper-accessible regions [4,5]. | Nagy et al. 2003 Giresi et al. 2007 |
| DNase-seq | Maps accessible regions of the genome with a bias toward hyper-accessible regions, e.g. enhancers and promoters [6]. | Boyle et al. 2008 |
| MNase-seq | Can map either hyper-accessible regions or nucleosome positions depending on enzyme dosage. MNase has strong cleavage bias based on base pair composition [7]. | Albert et al. 2007 |
| MACC | MNase accessibility metric determined by combining high MNase and low MNase measurements [8]. | Mieczkowski et al. 2016 |
| q-MNase | Similar to MACC, it uses titration of MNase, but also incorporates spike-in controls in experiment and analyses [9]. | Chereji et al. 2019 |
| MNase-SSP | Uses a single-stranded DNA library prep to map MNase-digested fragments. This method greatly lessens the base composition cleavage bias of standard double-stranded preps, and also efficiently captures sub-nucleosome-sized fragments [10]. | Ramani et al. 2019 |
| Array-seq | Long-read sequencing of partially digested chromatin by MNase. Main feature of interrogation is nucleosome phasing [11,12]. | Baldi et al. 2018 |
| ATAC-seq | Maps hyper-accessible regions using Tn5 transposase, which cuts and inserts sequencing adapters into cellular DNA in a single step. Compatible with single-cell protocols [13]. | Buenrostro et al. 2013 Buenrostro et al. 2018 |
| NA-seq | DNA accessibility measured by restriction enzyme and sequencing. Can probe hyper-accessible and other regions of the genome in the same assay. Resolution is dependent on number of restriction sites in the genome [14]. | Gargiulo et al. 2009 |
| RED-seq | Derivative of NA-seq that is performed on permeabilized cells and has an updated library prep workflow [15]. | Chen et al. 2014 |
| qDA-seq | Similar to NA-seq and RED-seq. Restriction enzymes are titrated to measure both initial cut rate and absolute accessibility [16]. | Chereji et al. 2019 |
| ORE-seq | Equivalent to qDA-seq. Cross-verified results with ODM-seq [17]. | Oberbeckmann et al. 2019 |
| ODM-seq | Methyltransferase accessibility assay. Nuclei are treated with M.SssI and M.CviPI followed by bisulfite-seq to measure cytosine methylation (5mC). Accessibility measurements were cross-verified with ORE-seq [18]. | Elisa Oberbeckmann et al. 2019 |
| DamID | Genetically encoded DNA adenine methyltransferase domain fused to an endogenous protein or by itself is expressed in living cells and modifies GATC sequences, which are detected by bisulfite-seq. Resolution of assay is limited by its cognate GATC site [19,20]. | van Steenlsel and Henikoff 2000 Sha et al. 2010 |
| NOMe-seq, MAPit-patch | Methyltransferase-based nucleosome footprinting and accessibility assays. Nuclei are treated with methyltransferase M.CviPI followed by bisulfite-seq [21,22]. | Kelly et al. 2012 Nabilsi et al. 2014 |
| dSMF | Dual-enzyme single-molecule footprinting. Treated nuclei with methyltransferases M.SssI and M.CviPI followed by bisulfite-seq of 300 bp fragments [23]. | Krebs et al. 2017 |
| Fiber-seq | Chromatin footprinting in nuclei with N6-adenine methyltransferases (Btr192IV, EcoGI, EcoGII, Hia5, or Hin1523) followed by long-read sequencing (PacBio) [24]. | Stergachis et al. 2020 |
| SAMOSA | Similar to Fiber-seq. Used M.EcoGII. Has been applied to in vitro chromatin arrays and to low nuclei samples [25]. | Abdulhay et al. 2020 |
| SMAC-Seq | Similar to Fiber-seq and SAMOSA, except that cells were treated with M.CviPI, M.EcoGII, and M.SssI. Long-read libraries were sequenced with Nanopore [26]. | Shipony et al. 2020 |
| ATAC-see, ATAC-PALM | Attached fluorophores onto the Tn5 adapters that allow for visualization of hyper-accessible regions by standard or super-resolution fluorescence microscopy. Can also sequence the samples using modified ATAC-seq protocol [27,28]. | Chen et al. 2016 Xie et al. 2020 |
| dCas9 live tracking | Accessibility measured by microscopy-based protein tracking and binding kinetics of single-molecule dCas9 particles that have been targeted to a specific locus or loci [29,30]. | Knight et al. 2015 Fu et al. 2016 |
| Micro-C, Micro-C XL | Derivative of Hi-C that uses exonuclease digestion instead of restriction enzymes to capture both short-range contacts (>1kb) and mid- to long-range contacts (kb to Mb scale) [31,32]. | Hsieh et al. 2015 Hsieh et al. 2016 |
| RICC-seq | Uses ionizing radiation with sequencing to probe short-range chromatin contacts (>1kb) [33,34]. | Rydberg et al. 1998 Risca et al. 2017 |
| Loop-seq | High throughput in vitro assay to assess inherent DNA flexibility of genomic sequence [35]. | Basu et al. 2021 |
| Gradient-seq | Derivative of FAIRE-seq with an added sucrose gradient, which allows for assessing the physical properties of chromatin. Can also be coupled with mass spectrometry [36]. | Nicetto et al. 2019 |
| CATCH-IT | Determine long-term nucleosome dynamics using a chemical-based biotin-labeling, MNase digestion, and a biotin-streptavidin purification [37]. | Deal et al. 2010 |
| Time-ChIP | Derivative of SNAP-tag using a biotin pulse-chase strategy for determining long-term histone turnover [38]. | Deaton et al. 2016 |
| dCas9-DD-BirA | Similar to CATCH-IT and Time-ChIP, but uses in vivo BirA enzyme fused to dCas9 to label nucleosomes with biotin and determine long-term turnover rates by ChIP-seq [39]. | Escobar et al. 2019 |
Measuring accessibility and related quantities using chemical probes
Although not widely employed, some chemical probes have proven informative in determining chromatin accessibility. Such approaches are based on the long-established family of chemical footprinting methods used in vitro to determine binding sites of proteins on DNA [40]. For example, methidiumpropyl-EDTA sequencing (MPE-seq) [2] uses a radical-based cleavage agent coupled to a DNA intercalator to fragment DNA at regions of low nucleosome occupancy with minimal DNA sequence bias. Chemical mapping of nucleosome by copper-generated hydroxyl radicals generated near the nucleosome dyad has also been used to measure nucleosome positioning and occupancy, which are closely related to DNA accessibility [41,42]. Another recent chemical chromatin mapping method of note, KAS-seq [3], does not probe accessibility but measures the single-stranded DNA produced at regions of active transcription and elsewhere, providing an activity measurement complementary to DNA accessibility.
Measuring accessibility with nucleases
There are a variety of techniques to quantify genome-wide accessibility, and many of them use the same basic principle of measuring how well an enzyme can access genomic template and subsequently cleave or modify the DNA. Unlike small molecules (e.g. hydroxyl radicals), enzymes do not access the DNA uniformly, as their size limits their ability to localize and interact with DNA depending on the landscape of proteins that are already bound to a given locus, and their sequence preferences, which vary for each enzyme, modulate their propensity to cut or modify a given site on DNA [43]. Early iterations of genome-wide accessibility assays include DNase-seq (DNaseI with sequencing) [6] and MNase-seq (Micrococcal Nuclease with sequencing) [7], as well as the more recently developed ATAC-seq (Assay for Transposase Accessible Chromatin) [13]. Accessibility signals from these assays are enriched in regulatory regions, such as promoters and enhancers. However, each of the probing nucleases and its respective assay have cleavage and size-selection biases that limit the interpretations of the resulting accessibility metric [1,8,9].
DNase-seq and ATAC-seq report on hyper-accessible regions, especially when analyzing sub-nucleosome sized fragments. These smaller fragments represent read inserts that are surrounded by multiple cut sites in a nearby region, whereas accessible regions that are less densely cleaved result in longer ‘orphaned’ fragments that are effectively removed by size-selection. This size-selection leads to a preferential readout of stretches of DNA that are devoid of or have higher turnover rates of nucleosomes. A clear demonstration of this bias towards hyper-accessibility can be seen in the coverage of the mitochondrial genome, which, unlike the nuclear genome, is not bound by nucleosomes and its coverage is overrepresented in the earlier iterations of ATAC-seq that were not optimized to exclude mitochondria [13,44]. Thus, DNase-seq and ATAC-seq are primarily measurements of hyper-accessibility and nucleosome depletion (Figure 1).
The enrichment of hyper-accessible sites can be an advantage when a global view of active or poised regulatory sites, such as promoters and enhancers, is desired with the least amount of sequencing. Since most of the signal is highly concentrated at nucleosome-depleted regions, most of the sequenced reads report on active regions. The efficiency of ATAC-seq has enabled its success in profiling cellular differentiation and diversity within tissues at single-cell resolution [14,45–47].
MNase-seq can measure a broader range of dynamic states, but important considerations and experimental measures must be taken to rigorously quantify accessibility. MNase has dual endo- and exo-nuclease activities – the latter of which is highly processive and preferentially digests inter-nucleosomal DNA, often referred to as linker DNA – but it can also partially digest nucleosome-bound DNA upon longer incubations. MNase also has a strong preference to digest AT-rich DNA [43,48]. Thus, MNase-seq will yield different results depending on the enzymatic dosage and the underlying sequence. At lower enzyme concentrations, nucleosome-depleted DNA will be enriched, whereas higher concentrations enrich for nucleosome-bound DNA. Using a titration-based strategy, coupled with either histone ChIP-seq (chromatin immunoprecipitation with sequencing) or mononucleosome spike-ins, recent studies have developed sophisticated quantitative frameworks, referred to as MACC or q-MNase, to address the above-mentioned biases and produce more rigorous interpretations of accessibility using MNase-seq data [8,9]. These approaches can separate position, stability, and occupancy measurements of nucleosomes. The resulting metrics of both methods corroborate higher accessibility at enhancers, promoters, and at transcription start sites in a transcription-dependent manner. However, the quantitative differences between regulatory regions and other genomic regions are more modest relative to results from other methods, like DNase-seq and ATAC-seq. Additionally, upon titrating MNase, the differences between the overall accessibility of heterochromatin and euchromatin (outside of hyper-accessible nucleosome-depleted regions) are smaller than expected [8,9].
Two additional methods have used experimental refinements of MNase-seq. Instead of the standard double-stranded library prep, MNase-SSP uses a single-stranded DNA library preparation that greatly reduces the base pair composition bias of the sequencing library [10]. Additionally, it seems to allow for more efficient capture of subnucleosome-sized fragments. Array-seq is another variant of MNase-seq; however, it uses longer chromatin fragments that have only been partially digested by MNase and then sequenced using long-read sequencing [11,12]. The data resulting from Array-seq reports on the phasing of oligonucleosomes, an aspect of chromatin organization which may play important roles of chromatin accessibility.
Relative vs. Absolute Accessibility Quantifications
Global changes in accessibility are not well-quantified using DNase-seq or ATAC-seq data, because differences in sample processing, cell permeabilization propensity, and clustering efficiency on sequencers that can affect the total amount of reads per cell obtained from different samples, necessitating normalization of libraries. Instead, these data sets generally report on relative distributions of accessibility through calling peaks of hyper-accessible chromatin and then analyzing differential read counts within each peak after normalization for the total amount of reads in each sample (for two examples of normalization strategies, see [49,50]). Broad-based changes in accessibility between samples therefore pose a challenge. For example, in a hypothetical experiment that increases accessibility at all genomic loci equally, this difference, when compared against the control sample, would be indistinguishable from technical sample-to-sample variation in reaction efficiency and would therefore be normalized out in the analysis. One approach that has been employed to handle large numbers of peaks that change accessibility is to derive the sample-specific scaling factors that account for overall sequencing depth and efficiency using a subset of low-variance peaks that remain stable across samples [49]. Spike-in-based strategies for cross-sample normalization are challenging [51] because spike-in and main sample reads likely compete for clustering on the sequencer. Alternatively, ATAC-seq data can be used to some extent as a broad-based accessibility probe by comparing the shape of the fragment size distribution histogram between genomic regions, but this approach is only possible within individual samples, where the technical variables are constant [13,52]. Sequence biases can confound these distributions and must be carefully controlled.
Given the variability in accessibility measurements yielded by DNase-seq, ATAC-seq, and MNase-seq, recent efforts sought to create more accurate accessibility maps and to determine the absolute levels of nucleosome occupancy. Two major technical reasons why many genome-wide assays lack absolute quantitative power are because (1) size-selection leads to a diminished and unrepresentative pool of accessible sites, and (2) enzymatic reactions typically do not reach saturation. To address these issues, different versions of restriction enzyme-based accessibility assays were developed (NA-seq and RED-seq) [15,16] and optimized (ORE-seq and qDA-seq) [17,18]. These assays use restriction enzyme digestion of nuclei followed by de-chromatinization, sonication, sequencing, and computational filtering of reads based on genomic coordinates of restriction sites. Restriction enzymes are strongly inhibited by nucleosomes, making it possible to rigorously relate the fraction of cleaved sites to the fraction of sites occupied by nucleosomes when the cleavage reaction is titrated up to saturation of all cleavage sites. One technical weakness is that number of recognition sites for a given enzyme limits the resolution of restriction enzyme-based assays. To address this limitation, an orthogonal technique was developed using a DNA methyltransferase and bisulfite sequencing (OME-seq), which also allows for the quantification of non-nucleosome protein occupancy [18]. Together, these absolute accessibility assays have confirmed the over-estimation of absolute accessibility by DNase-seq and ATAC-seq, validated the localized increases in accessibility at regulatory regions, and provided further support for similar levels of accessibility between broad euchromatin and heterochromatin domains. For example, in mouse liver cells absolute accessibility differs less than 2-fold between euchromatin and heterochromatin regions, ranging from 30% to 35% in promoters to 20% in H3K9me3 regions [17].
Methyltransferase-Based accessibility assays: single-molecule resolution and long-range correlations
Labeling DNA with methyltransferases as an approach for measuring accessibility developed in parallel with nuclease-based assay [53–56]. It has some disadvantages, which limited its use prior to the advent of long-read sequencing, and advantages, which are now coming to the fore as technology to directly read out base methylation in long DNA fragments has advanced. One major consideration when choosing between methods is the amount of endogenous DNA methylation in the organism being assayed, which can somewhat limit the resolution of methylation-based footprinting.
The key distinction between methylation and cleavage-based assays is that methylation is non-destructive and retains molecular contiguity. DamID [19] is an approach that takes advantage of this property and measures DNA accessibility or contacts with a protein in intact cells without the DNA damage response that would be triggered by a nuclease. DamID uses a heterologous DNA adenine methyltransferase domain fused to an endogenous protein to modify GATC sites that come in contact with the fusion protein and then identifies methylated sites through microarray or sequencing. This technique has mostly been used to profile the genomic contacts of nuclear proteins. DamID can also be used to probe chromatin accessibility more broadly. For example, expressing the Dam protein without fusion to an endogenous protein yields methylation patterns that roughly correspond with nucleosome positions from MNase-seq data and that seem to be evenly dispersed throughout the genome [20]. The GATC motif limits resolution, which prompted refinement of this assay with the use of the more permissive cytosine methyltransferases M.SssI (CpG) and M.CviPI (GpC) [18,21,23,57]. Similar to DamID, these other methyltransferases can be expressed in the cells [58] or they can be added exogenously after a nuclear permeabilization step [18,21,57]. Short-read sequencing can detect these modifications after bisulfite conversion of methylated cytosine to uracil and in silico mapping of these sites. Comparing methyltransferase sequencing and restriction enzyme sequencing results, both with the same biological sample and at enzymatic saturation, yielded similar results [18]. Thus, methyltransferase sequencing can be optimized to create high-resolution and highly accurate maps of absolute chromatin accessibility.
As the technology to directly read out methylated bases has advanced [59], long-read methyltransferase sequencing has recently been established, representing a powerful new way to measure accessibility at single-molecule resolution (dSMF, Fiber-seq, SAMOSA, SMAC-Seq) [23–26,60]. One key innovation of these protocols is the use of new enzymes. Promiscuous adenine methylases such as Btr192IV, EcoGI, EcoGII, Hia5, and Hin1523 modify bases that are not typically methylated in eukaryotes. M.CviPI which methylates GpC sites, unlike the CpG sites that are endogenously methylated. When multiple methyltransferases are combined, the assay resolution can be improved to a few base pairs.
Long-read accessibility measurements have several advantages over nuclease-based approaches. First, because they directly measure the methylation state of multi-kilobase DNA molecules extracted from cells that can span multiple regulatory regions, they offer single-molecule information about the correlation between accessibility of transcription factor (TF) binding sites within the same regulatory element or between distinct regulatory elements such as adjacent enhancers, or enhancers and promoters. Individual TF footprints can be detected, and their cooperative interactions can be studied through their correlative binding patterns and interactions with nucleosomes [24,26,61,62]. Since the signal consists of multiple methylation events on the same molecule rather than multiple cleavages of several molecules, it is possible to observe whether accessible sites are activated in a graded or all-or-none mode [24]. Interestingly, this work found that most sites appear to open in an all-or-none fashion and intermediate values found in bulk reflect the fraction of cells with hyper-accessibility at the site in question. Methylation-based long-read methods can also concurrently measure accessibility and nucleosome spacing, the latter of which is incalculable for poorly positioned nucleosomes using short reads [63]. Additionally, long reads allow for resolving features of repetitive loci that typically remain unmappable [25,60]. Initial experiments have found that some heterochromatin regions are enriched for irregular nucleosome spacing [25]. Lastly, these methods can simultaneously measure endogenous and exogenous DNA methylation, profiling how endogenous methylation correlates with the accessibility landscape [64].
How do these long-read methyltransferase sequencing experiments compare to absolute accessibility measurements? The ability to correlate accessibility between sites is a major advantage of methylation-based approaches. Another important consideration is understanding what assay conditions best capture the biological realities of accessibility (discussed in more detail in the subsection below, Assay Conditions). It would also be useful to benchmark new accessibility measurements against an orthogonal method that has been designed to measure absolute occupancy [18]. Lastly, a major limitation of the current long-read platforms is their low coverage depth to sufficiently investigate whole genomes. Upon careful optimization of workflows and the lowering of cost per read, long-read methyltransferase sequencing assays will likely play an increasingly key role in defining accessibility maps.
Visualizing accessibility
Imaging-based accessibility measurements have also been developed, but due to the technical challenges involved, they have lagged behind sequencing-based methods. There have been massive recent achievements in the imaging of chromosome conformations in sequence-space using Oligopaints and super-resolution microscopy [65–67]. Although these methods can map the underlying chromosome structures of different genomic regions, including more or less accessible regions defined by other assays, their resolution remains limited, and they cannot report on protein dynamics relative to genome sequence. Rigorous accessibility measurements in sequence space have employed tracking of a fluorescent HaloTag probe attached to a nuclease deficient CRISPR/cas protein (dCas9) that can target specific sequences [29]. Coupled with sequencing readouts, this Cas9-based technique may provide a useful orthogonal approach to verifying absolute accessibility maps.
ATAC-see is another visual accessibility measurement, based on a slightly modified ATAC protocol that includes a transposase with an attached fluorophore and fixation of the sample [27]. A subsequent iteration of this technique, ATAC-PALM, has been adapted to super resolution microscopy [28]. These assays maintain the use of sequencing barcodes that allow for multimodal results capturing genome-wide hyper-accessibility measurements, imaging of the broad nuclear organization of accessible sites, and even the incorporation of Oligopaint hybridization to visualize specific loci. Thus far, these assays have shown that the nuclear localization of accessibility is cell type-dependent and that accessibility can provide broad clues about chromatin-based processes, like the role of CTCF in organizing accessible chromatin [28], condensate formation [68], or neutrophil extracellular traps (NETosis) [27]. Overall, imaging-based techniques of chromatin accessibility are in the early stages of development with the potential to provide powerful insights into our understanding of the in vivo spatial dynamics of chromatin accessibility.
Assay conditions: intact cells vs. permeabilized, snapshot vs. dynamic
With a variety of different techniques and normalizations to measure accessibility, a critical question is how the experimental conditions of a given assay affect its readout. One important variable in assays is the cell or nucleus state. Many of the assays described above, except for DamID and live imaging, are done by permeabilizing cells, incubating them with enzymes, and then halting the reactions after a specified amount of time that can vary widely between experimenters. Measurements from permeabilized cells may keep the chromosomes and proteins largely within the nuclei, but many small molecules of the nucleoplasm are released, potentially disrupting native conformations and activities of chromatin. Assays with prolonged incubation of permeabilized cells may allow ATPase-dependent nucleosome remodelers to remain active and ultimately increase the perceived accessibility or increase background signal. Isolation of nuclei, which removes much of the ATP pool, seems to minimize changes in accessibility along the linear genome during longer incubations that are needed to reach enzymatic saturation [17]. It remains unclear, however, to what degree permeabilization or isolation of nuclei changes the internal spatial organization of chromatin. It is reasonable to hypothesize that weak three-dimensional folding interactions and biochemical effects, like polymer collapse and phase separated condensates, become altered or diminished under these conditions. It also remains unclear the degree to which these kinds of spatial features regulate accessibility in vivo (see section below, Regulation of Accessibility).
A method that is often used to maintain both linear and spatial organization of chromatin during sample handling is formaldehyde fixation [31,69]. Comparisons of fixed and non-fixed samples can offer insight into the effects of fixation. For example, when initially incorporated into the ATAC-see protocol, formaldehyde fixation produced slightly biased results [27]. However, ATAC-PALM assay conditions were subsequently updated to yield similar sequencing results with and without fixation [28]. The details of the fixation conditions are critical considerations. In one methyltransferase study, optimal sequencing results relied on fixation to match absolute occupancy scores from restriction enzyme sequencing from purified budding yeast nuclei [18]. In contrast, another methyltransferase sequencing study of mouse embryonic stem cells found minimal differences between fixed and unfixed conditions [61]. Additional studies comparing fixed and unfixed conditions will be needed to clarify this discrepancy. Downsides of formaldehyde fixation are that it is relatively slow and incomplete, it can be reversed over time (although this is also an advantage for recovering DNA), proteins can diffuse for 1 hour after fixation, its small size limits the fixation distance, and it underrepresents more transient or weaker contacts [31,70,71]. Each of these deficiencies can contribute to an incomplete understanding of the chromatin landscape or even fixation artifacts [71].
In contrast to the permeabilized, isolated, and/or fixed nuclei, accessibility can be measured under natural dynamics of intact cells. One such strategy is to genetically encode a methyltransferase and track in vivo accessibility over prolonged times. This approach has been done at low depth for Dam expression in C. elegans in a tissue-specific manner [20]. Another study in budding yeast used an M.EcoGII fusion protein coupled with long-read sequencing [58]. Adapting this long-read method with an M.EcoGII that is not fused to any other protein and that is under the control of a finely tuned inducible promoter may provide a more biologically relevant view of the in vivo kinetics of genome-wide chromatin accessibility.
In sum, careful experimental considerations must be made when designing experiments and making conclusions about accessibility assays.
Correlates of accessibility
Many genome-wide readouts correlate with accessibility measurements and are often used as shorthand for accessibility or lack thereof, including nucleosome positions, histone modifications, transcription, and chromatin compaction. Accessibility is often anti-correlated with positioning and density of nucleosomes, which often are primary determinants of accessibility due to their abundance and stability. For example, FAIRE (formaldehyde-assisted isolation of regulatory elements) [4,5], was an early sequencing-based method for profiling active regulatory elements by fragmenting crosslinked chromatin and enriching for DNA fragments that were not bound to protein (presumably, consisting largely of histones).
However, as discussed above, absolute accessibility measurements demonstrate that many of these assays often overestimate nucleosome occupancy, differences in occupancy between epigenomic states, or the extent to which nucleosomes block access to the underlying DNA. ChIP-seq or DamID peaks of histone modifications, such as lysine acetylation of H3 and H4 (H3K9ac, H3K27ac, H4K16ac) and lysine methylation of H3 (H3K4me) often occur around hyper-accessible sites, but the code of histone modifications does not simplistically correspond to accessibility. For example, some sites may be accessible before a strong histone acetylation signal appears [72], and acute perturbation of acetylation, despite dramatically impacting transcription, has little effect on the accessibility landscape [73].
Similarly, the repressive marks histone H3 lysine 9 di- or trimethylation (H3K9me3) and H3 lysine 27 trimethylation (H3K27me3) or genomic regions that tend to contact these loci, being part of the ‘B compartment’ measured by Hi-C [74] (discussed in further depth in the next section) are often referred to as low accessibility states or described as ‘compact’. Although this description is likely accurate when compared to hyper-accessible regulatory regions, absolute broad accessibility in these heterochromatin regions is rarely measured. As discussed in more mechanistic detail below, models of chromatin compaction are subject to ongoing revision and do not fit into simplistic views of open and closed. Several promising technological developments are making it possible to begin making direct measurements of chromatin compaction. First, recent advances in super-resolution imaging [65–67,75] and electron tomography [76–78] have made it possible to visualize the densities, locations, and, to some degree, relative orientations of nucleosomes in fixed or frozen cell nuclei. Live-cell imaging of chromatin with fluorescence correlation spectroscopy and single-molecule imaging with tracking [79–82] have begun to add dynamic information to those pictures, reporting on both fluctuations and material properties of chromatin as well as regulated, active motion in response to events such as DNA damage. Micro-C, a proximity ligation-based method based on Hi-C that digests chromatin with MNase [31,32,70,78,83] and RICC-seq [33], a method that analyzes clustered DNA damage patterns induced by ionizing radiation, are being investigated as probes of local chromatin fiber compaction. Finally, loop-seq and gradient-seq are two recently developed techniques that can directly probe physical properties of the genome and are starting to provide key insights into fundamental determinants of accessibility [35,36]. Loop-seq uses an elegant in vitro system to interrogate the flexibility of short DNA oligos that, when combined with high throughput oligo synthesis and sequencing, can assess the inherent mechanical code of DNA from large portions of a genome. Loop-seq applied to budding yeast has shown that underlying mechanical properties of the local genome sequences correlate with nucleosome occupancy. Gradient-seq assess the density of chromatin by crosslinking cells, sonicating, and then separating chromatin using a sucrose gradient. Chromatin that is more labile to sonication correlates well with active regions of the genome, whereas sonication-resistant chromatin correlates strongly with repression-associated features, such as H3K9me3, DNA methylation, and LADs. Intriguingly, sonication-resistant regions are better predictors of transcriptional repression and resistance to transcriptional activation under reprogramming than any single repression-associated feature alone [36,84]. Gradient-seq also permits isolation and mass spectrometry-based identification of the proteins associated with each compaction state of the chromatin, revealing molecular regulators [84,85]. Although these approaches are still relatively new, they hold promise to characterize the context-dependent relationships between chromatin compaction, accessibility, and functional outcomes like transcription.
In sum, although many genome-wide parameters often correlate with accessibility measurements, more precise language when describing experimental results and interpretations is useful, especially given the ongoing efforts to accurately quantify chromatin accessibility and compaction.
Regulation of accessibility by the chromatin fiber and nuclear compartments
In the cellular context, access to chromatinized DNA can be controlled at several levels. Indeed, the three modes of accessibility discussed in the previous section are each a prerequisite for the next, i.e. if a DNA-binding protein is to access its target locus, the protein must access the nuclear space containing the locus (also called a compartment), and be able to non-specifically bind the DNA therein, and then find its sequence-specific binding site. The mechanisms that give rise to the narrow, hyper-accessible regions, identified by DNase-seq and ATAC-seq peaks, involve a combination of competition for DNA binding between TFs and nucleosomes or the action of pioneer factors, local remodeling of nucleosomes by ATPases, and modulation of binding at specific sites by DNA methylation. These hyper-accessible regions have been well reviewed elsewhere [1,64]. Therefore, we will focus here on the mechanisms that regulate access to either chromatinized DNA in general or genomic compartments, modulating broad-based accessibility of genomic loci (not hyper-accessibility) (Figure 1(b)), or modulating which parts of the nucleus can be accessed by proteins of interest (Figure 1(a)). We will first describe factors that regulate accessibility at the chromatin fiber level and models of local chromatin compaction, and then relate them to the emerging picture of long-range chromatin compaction, phase separation of chromatin, and the generation of nuclear compartments with self-segregating compositions.
Molecular factors regulating broad chromatin accessibility
The extensive co-mapping of chromatin accessibility using sequencing-based assays and either chromatin-associated proteins or histone modifications across large numbers of cell types and organisms has revealed strong correlations between chromatin accessibility and several molecular factors, which we detail below. It is important to note, however, that these correlations do not on their own establish causality or reveal the underlying mechanism regulating accessibility. For example, chromatin accessibility also correlates with transcription, and transcriptional activity could affect local chromatin accessibility rather than the other way around. Furthermore, due to the high complexity of eukaryotic chromatin, it is likely that multiple mechanisms are in play at any given locus, and that the impact of the molecular factors below depends on the local chromatin context.
Nucleosome remodelers and chaperones
In addition to the localized removal of nucleosomes to generate hyper-accessible sites at active promoters and enhancers [1], nucleosome remodelers also play a role in broadly regulating the spacing of nucleosomes [18,86,87], which can influence chromatin fiber geometry [88–90], as well as the turnover rate of histones or nucleosomes [89], thus regulating the occupancy of underlying DNA loci. Fast histone turnover and chromatin accessibility are generally correlated [91,92]; however, they are not inextricably linked. A study of nucleosome turnover with two orthogonal methods, time-ChIP and CATCH-IT, showed that in embryonic stem cells, heterochromatin containing the histone variant H3.3 exhibits very fast nucleosome turnover with no associated increase in accessibility and that this dynamic but inaccessible state requires the H3.3 chaperone DAXX [93].
Histone variants
The H3.3 example above [93] illustrates how histone variants can interact with chaperones and remodelers to alter chromatin accessibility as part of the epigenomic context of a locus. Other histone variants are associated with high-accessibility regions, such as H2AZ, which is enriched at transcription start sites and is preferentially disrupted by RSC remodeling complexes [94–96]. MacroH2A is a notable family of variants of histone H2A that are generally, though not exclusively, associated with transcriptionally repressed chromatin and contain a positively charged linker domain [97,98], similar to the ‘tail’ domains of linker histones (see below). In line with repressive functions, macroH2A2 restricts cell reprogramming, but, another member of this family, MacroH2A1.1, promotes both accessibility and transcription at some of its target genes through histone acetylation [97]. In most cases, the mechanisms by which macroH2A family variants modulate the local chromatin context remain poorly understood.
Linker histones
Linker histones, the best-studied of which are the H1 histone family (recently reviewed in depth [99]), are globular proteins which, much like the core histones (H2A, H2B, H3 and H4), have unstructured and positively charged N- and C-terminal ‘tail’ domains. Their globular domains associate with the nucleosome at or near the ‘dyad’ position midway between the DNA entry and exit points, and their tail domains are thought to help shield the charge on linker DNA between nucleosomes. Linker histones can reversibly bind to chromatin with residence times (~3 minutes) that are shorter than core histones (hours) but are longer compared to most other chromatin factors (<1 second to 10s of seconds) [100]. Unlike other factors that must be recruited to the loci where they are found, linker histone is highly abundant and dynamic in most cells, binding nearly everywhere in the genome, with the exception of some H1 subtypes that are depleted near the promoters of active genes [101]. Despite its ability to passively dissociate from chromatin, active removal or replacement of H1 may play an important role in regulating chromatin-based processes, such as transcriptional activation. Local eviction of linker histone is in many cases, the first step toward nucleosome remodeling and generation of a hyper-accessible site [1,102]. On the other hand, higher H1 densities on chromatin are associated with transcriptionally repressed and less accessible chromatin [99,103]. Globally, the H1-to-nucleosome ratio tends to increase between stem cells and differentiated cells. Linker histone mutations have been found to associate with several cancers, particularly concentrating in lymphoma [104]. Reduction of linker histone levels generally leads to a corresponding reduction in the spacing between nucleosomes [105,106], reduction in the size of nucleosome clusters measured by super-resolution fluorescence [107], reduction in the density of heterochromatin as measured by transmission electron microscopy, a small increase in total nuclear size, and changes in accessibility that depend on the cell type and the initial H1-to-nucleosome ratio [52,108].
Histone acetylation
Histone acetylation (reviewed recently in depth [109]) is the most conserved type of histone post-translational modification [110]; therefore, it is likely to be the most fundamental ‘epigenetic’ (in the sense of relating to chromatin-based control of gene expression) mechanism of regulating DNA-based processes, with the exception of moving and removing nucleosomes (covered in more detail below, ‘Local Compaction and the 30-nm Fiber Model’). It has been observed at multiple sites concentrated on the N-terminal unstructured tail domains of histones H2 and H4 and is maintained through a balance between histone acetyltransferases (HATs) and deacetylases (HDACs) [109]. Early in vitro experiments showed that acetylation facilitates access of basal TFs to their binding sites [111]. Histone acetylation, particularly on the H4 tail, leads to de-compaction of synthetic oligonucleosome fibers through what is thought to be disruption of the docking between a basic patch on the H4 tail, which is necessary for compaction, and an acidic patch on the face of the nucleosome [112–115]. The details of this mechanism have also been explored through extensive simulations [109,115–117]. Functionally, acetylation correlates with chromatin accessibility [1], enhancer/promoter activation [109,118], histone turnover [119], and eviction of linker histones [102]. Recruitment of acetyltransferases is a key step in the eukaryotic transcriptional activation process [120,121], and cell states with high plasticity or high overall accessibility, such as embryonic stem cells [122] or activated B cells [123], also have high levels of acetylated chromatin. Upon HDAC inhibition in B cells, increased chromatin acetylation contributed to spreading of chromatin from the nuclear lamina toward the nuclear interior, but surprisingly, the increase in acetylation did not significantly change the size of nucleosome clusters or the search time of TFs diffusing to their binding site [123]. Conversely, massive deacetylation is associated with genome- or chromosome-wide reduction of transcriptional activity and loss of at least a part of hyper-accessible sites (as measured by ATAC-seq) in specialized states, such as mitosis [124], quiescence [78,83], and X chromosome inactivation [125–127].
HP1 family proteins
H3K9me2/3-marked chromatin generally has the highest DNA density among epigenomic states [128] and is associated with constitutive heterochromatin. H3K9me2/3 covers much of the repressed repetitive genome, and more recently it was found to also participate in the silencing of some developmentally regulated loci [36,129,130]. Loss of H3K9me2/3 marks leads to genomic instability caused by inappropriate and widespread transcription of repetitive regions [131,132]. H3K9me3 is recognized and bound by the chromodomains of HP1 family proteins, which can then dimerize through their chromoshadow domains, bridging nearby or distal non-adjacent nucleosomes along chromatin [133,134]. HP1 family proteins serve as interaction hubs, helping H3K9 methyltransferases and demethylases remain localized to chromatin, enhancing their stability [135], and introducing feedback in the maintenance of heterochromatin [136]. HP1α has been shown to contribute to the mechanical elasticity of chromosomes in both interphase and mitotic mammalian cells, consistent with function as a crosslinking factor [137]. However, in rapid degron-based HP1α depletion, little transcriptional dysregulation has been observed [137]. A potential link between the transcriptionally repressive function of H3K9me2/3 and its role in chromatin compaction was found using an assay for chromatin density or condensation in which formaldehyde-crosslinked chromatin is lightly sonicated and fractionated on density gradients [84]. Chromatin’s propensity to resist sonication, taken as a measure of compaction, correlated strongly with H3K9me2/3 and also predicted repressed states of genes and resistance to activation during ectopic trans-differentiation better than histone modifications alone [84,138,139]. H3K9me2/3-marked chromatin may also be compacted because it is also enriched in linker histone binding [52,99,108], and in Drosophila, linker histone H1 interacts with the H3K9me3 methyltransferase Su(var)3–9 [140].
Polycomb family proteins
H3K27me3 chromatin is dynamically regulated during development by polycomb repressive complexes PRC1 and PRC2, and this modification is often referred to as facultative heterochromatin [141,142]. Compact clusters or ‘hubs’ of transcriptionally repressed and H3K27me3-marked chromatin have been detected by Hi-C and microscopy [143–146]. The core canonical PRC1 can compact chromatin in vitro, even in the absence of histone tails [147–149]. PRC2 can also bind H3K27me3 through its EED subunit, and like HP1α, PCR2 can bridge non-adjacent nucleosomes in vitro [150]. H3K27me3 is also found over most of the transcriptionally repressed inactive X chromosome, called the Barr body in female mammalian cells [151]. Although it is not as compact as H3K9me3-decorated constitutive heterochromatin, the Barr body excludes RNAPII and hyper-accessible chromatin [152]. Genes on the inactive X chromosome that escape silencing are consistently found on the outer periphery of the silent chromosome [153,154]. A large number of recent studies into the mechanism of transcriptional silencing on the inactivated X chromosome (reviewed by Heard and colleagues [126,151]) together show that PRC1 and PRC2 are involved in helping to compact the chromosome, and thus, they facilitate spreading of the long noncoding RNA that directs and coordinates silencing by Xist. However, transcriptional silencing is mediated by the SPEN protein, which further recruits the NuRD remodeling and deacetylase complex and NCoR corepressor as effectors to deacetylate and silence chromatin [126]. PRC2 complexes also silence many developmentally regulated loci and generally, their binding and the chromatin marks they deposit and bind, H3K27me3 and H2AK119ub (in vertebrates), are anti-correlated with gene expression, but surprisingly not correlated with very low accessibility as measured by ATAC-seq and other similar assays [155]. Several models for the mechanism of polycomb-based repression have been proposed, including blocking transcription elongation rather than initiation or compaction of the chromatin fiber [156,157].
Physical factors regulating broad chromatin accessibility
Compaction of DNA is necessary to fit genomic DNA into cell nuclei. The radius of gyration of a piece of DNA the length of the diploid genome would be about 187 µm, and that of an average-sized chromosome about 27 µm (or slightly less, because these numbers are based on low-salt measurements), while the nucleus is typically about 10 µm across [158,159]. Nucleosomes compact the length of DNA about 6- to 7-fold, but the radius of gyration of this more compacted chromatin fiber becomes more difficult to estimate because its effective stiffness depends strongly on the arrangement, the dynamic geometry, and the interactions of nucleosomes [160]. Careful estimates of chromatin stiffness from yeast suggest that it would have an average compaction of 53–65 bp/nm and a persistence length of 52–85 nm [161], properties which would yield a minimum radius of gyration for a contiguous diploid human genome of about 40 µm. Additional compaction of the genome, beyond simple winding of nucleosomes, must therefore be occurring.
Chromatin ‘condensation’ has largely been the term invoked when discussing the dramatic micron-scale changes in density of mitotic chromosomes, while ‘compaction’ is usually applied to the denser regions of heterochromatin in interphase nuclei, as well as to shortening of DNA through the formation of nucleosomes (the units of chromatin) and through their local interactions along the fiber to fold it further still. Chromatin condensation and compaction on microscopic length scales have been studied for over a century. Condensed mitotic chromosomes were observed by Walther Flemming in the 1880s [162] and heterochromatin was coined as a term to describe the dense regions of chromatin in the nuclei of some cells observed by Emil Heitz in the 1920s [163,164]. Despite this long history, our understanding of the molecular details and functions of dense chromatin has remained surprisingly limited, and many major questions remain unanswered. How does heterochromatin repress transcription? What is the relationship between chromatin compaction and DNA accessibility to DNA-binding proteins? Does chromatin compaction depend on different factors at different length scales?
One emerging theme is that compaction may look different depending on the length scale over which it is probed. Chromosome condensation can occur over long length scales (hundreds of nanometers to microns) through looping by cohesin or condensin complexes, as has been documented for mitotic chromosomes [165], while still potentially leaving the chromatin fiber extended when measured at the length scale of tens of nanometers. For example, condensins can still generate remarkably normal, albeit more fragile, condensed mitotic chromosomes under conditions of extreme nucleosome depletion [166]. Furthermore, different epigenomic states can have different fractal scaling of compaction as a function of genomic distance. In interphase nuclei, inactive chromatin scales by a simple power law at all length scales probed, but polycomb repressed chromatin exhibits different behavior below and above the length scale of ~30–50 kb [65], with chromatin fibers apparently having low compaction below that length scale, but then probably looped into more compact conformations above that length scale in a manner that depends on the polyhomeotic protein. Such findings suggest that the different length scales of chromatin organization may be more independently regulated than previously thought. Connections between length scales do exist, of course. For example, a recent study of mitotic chromosome condensation [167] explored the interaction between linker histone (regulator of short-range chromatin fiber behavior that binds individual nucleosomes) and condensins (large ring-like complexes with ATPase activity that can extrude loops of DNA) [168]. In vitro, linker histone inhibits condensin binding to nucleosome arrays, which has been proposed to play a role in tuning the compaction and topology of mitotic chromosomes [167]. Thus, it is important to consider both the distinctions and the connections in compaction at different length scales.
Local chromatin compaction, the 30-nm fiber, and steric exclusion models
In the classical model that emerged soon after the discovery of nucleosomes [169–171], the proposed relationship between chromatin compaction and DNA accessibility is straightforward. A highly compact, 30 nm-diameter fiber was thought to be formed through close stacking of nearby nucleosomes into a helix, though the detailed structure of that helix was debated [172]. This state was thought to be either the repressed or the default ‘ground state’ of chromatin, and because the fiber was tightly and stably packed with linker DNA buried in its center, it would be easy to envision how both access to binding sites on DNA and nucleosome dynamics would be suppressed. Accessible or transcriptionally active chromatin, generally acetylated, would be unfolded from this helix into an unstructured fiber with nucleosomes connected by accessible DNA linkers resembling ‘beads on a string’ [173]. The 30-nm fiber would then be folded into higher order loops anchored to protein scaffolds to form heterochromatin or condensed mitotic chromosomes. This hierarchical model, in which compaction and accessibility are mutually exclusive and in which compaction starts at the nanoscale interactions between nucleosomes, has proven inadequate to explain the much more complex picture of chromatin organization that has emerged over the last decade.
Multiple methods have shown that chromatin is much more disordered and dynamic than the classical 30-nm fiber model proposes. Several electron tomography studies have found that chromatin fiber diameters, nucleosome clusters, inter-nucleosome spacers, and DNA entry-exit angles are highly heterogeneous in most cells [76,77,107,174,175], in contrast to the regular 30-nm fiber models based on in vitro reconstitution of chromatin on regular arrays of nucleosome positioning sites [175–177]. Proximity ligation and X-ray scattering experiments have also failed to find long-range ordered 30-nm fibers in most cell types [32,70,174,178,179]. One possible exception is the transcriptionally inert chromatin of chicken red blood cells, in which very high linker histone-to-nucleosome ratios appear to package chromatin into something resembling 30-nm fibers [78,176]. These results suggest that the long-range order of 30-nm fibers formed in vitro on regularly spaced arrays of strong nucleosome positioning sites is difficult to find in most cell types, with the exception of avian erythrocytes and similar cell types with mostly inactive chromatin and very high levels of linker histone.
Despite a relatively disordered landscape, some observations point to local inter-nucleosome interactions as a potential regulatory point for chromatin structure (Figure 2(a)). Clusters of nucleosomes identified by super-resolution microscopy of histones, termed ‘clutches’, change their size upon cell differentiation, changes in histone modifications, and changes in the global linker histone-to-nucleosome ratio [75,89,107,123]. The molecular regulators of this change were dissected in B cells, and interestingly, the size of the clutches was altered by cellular ATP levels, thought to drive the activity of chromatin remodeling complexes already associated with their target loci, but not by histone acetylation levels [123]. However, it is not clear to what extent the decompaction of nucleosome clutches is driven by changes in nucleosome spacing and occupancy or by the eviction of linker histone or by other factors. Extensive simulation, experimental and analytical work has shown that the chromatin fiber’s folding into either clutches or regular fibers and its stiffness depend on inter-nucleosome spacing [88–90,160,180–184]. In vitro, linker histone promotes compaction of nucleosome arrays into 30-nm fibers, though new results show that even these fibers can adopt heterogeneous conformations, which may explain how the 30-nm fiber can be reconciled with the nucleosome clutches model [185]. In vivo linker histone levels increase from pluripotent stem cells to differentiated cells and are necessary for proper differentiation and organismal development [99,106]. Simulations suggest that higher linker histone-to-nucleosome ratios increase the self-stacking and the effective stiffness of chromatin fibers, even when they are not forming true 30-nm fibers [186,187]. Proximity ligation and sequencing experiments in yeast also show that local contacts between nearby nucleosomes along the chromatin fiber, perhaps in tetranucleosome units, are mediated by interactions between the unstructured tail of histone H4 and the acidic patch on the nucleosome face composed of regions of histones H2A and H2B [32,188]. More recent sequencing-based experiments in mammalian cells with both Micro-C and RICC-seq also show DNA–DNA contacts consistent with zig-zag-like tetranucleosome stacking [33,70,179], which recapitulates the kind of tetranucleosome units observed by more recent cryo-EM and X-ray scattering [177,185]. Dynamic measurements of the fluctuations of reconstituted chromatin fibers in vitro show that nucleosomes can stack with each other transiently on scales of hundreds of milliseconds, forming tetranucleosome units within the chromatin fiber [189,190]. All together, this evidence supports the idea that partially disordered segments of the 30-nm fiber or its basic architectural motif, a tetranucleosome unit with zig-zag stacking of nucleosomes and the nucleosome–nucleosome contacts therein, occur commonly in chromatin in a dynamic mode that depends on local regulation by irregularities in nucleosome spacing, histone modifications, and chromatin-binding proteins.
Figure 2.
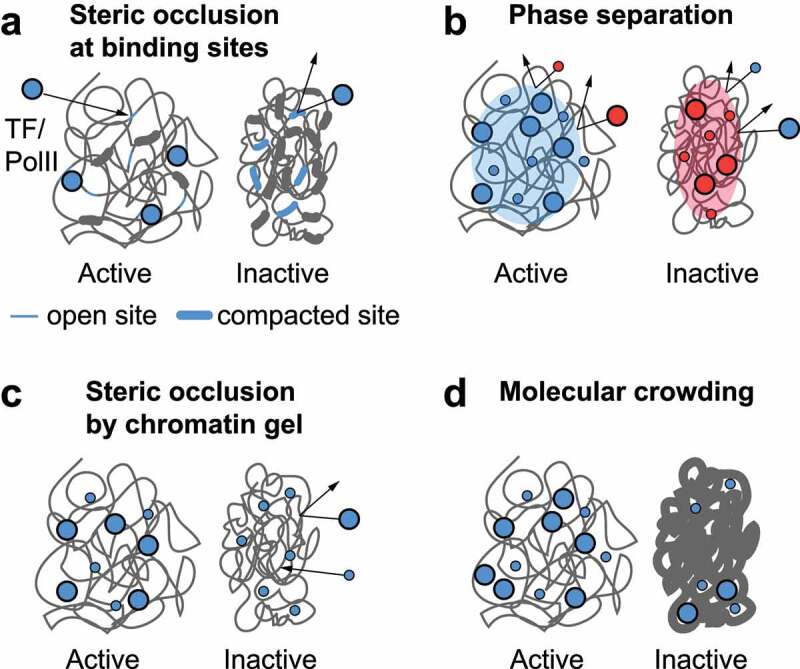
Models of accessibility control mechanisms by chromatin. (a) Steric occlusion at binding sites by nucleosomes or oligo-nucleosome contacts can prevent productive binding interactions and reduce the effective concentration of a TF or polymerase (blue circles) in a genomic region. If all binding sites are obscured, the protein is not concentrated in the genomic region, even though diffusion may be unaffected. (b) Liquid-liquid phase separation of chromatin and associated proteins can prevent proteins from entering three-dimensional regions of the nucleus (compartments) based on the proteins’ chemical properties such as charge. (c) If chromatin is crosslinked into a gel, it would exclude proteins larger than the pore size of the gel regardless of their chemical properties. (d) Volume exclusion due to crowding can reduce the concentration of soluble protein in a manner that depends more weakly on size than a gel (c).
How does the local compaction of chromatin into larger nucleosome clutches or more stable or more abundant tetranucleosome units affect chromatin accessibility? The classical model would predict that DNA-binding proteins are sterically excluded from deacetylated heterochromatin through local fiber compaction. DNA editing with Cas9 is less efficient in heterochromatin than in euchromatin [191,192], but a systematic study suggests that this effect is due largely to slower kinetics in heterochromatin, and can be overcome with longer exposure to Cas9 or higher expression levels [193]. Similar effects are seen in explicit assays of chromatin accessibility. DNase-seq [194] and ATAC-seq [13,44] are performed with limiting amounts of enzymes that have strong preferences for nucleosome-depleted DNA and any amount of cleavage in heterochromatin is generally considered noise in the assay as it was originally designed [195]. However, ATAC-seq has been used to estimate accessibility changes due to linker histone depletion in heterochromatin and has found measurable changes [52]. Studies that characterized accessibility by titrating MNase (MACC and q-MNase) [8,9], found that presumably dynamic nucleosomes can occupy sites of high overall accessibility in a bulk population measurement [8] and that overall accessibility was similar between heterochromatin and euchromatin in Drosophila cells, although nucleosome fragility at specific sites differed significantly. More recently, other enzymes have been employed to get around the problematic processivity of MNase. qDA-seq [17] and ORE-seq [18] are similar methods that use titration of a restriction endonuclease until saturation followed by sequencing of all fragments to obtain absolute cleavage rates at each restriction enzyme site. Only qDA-seq was applied to mammalian cells, which have appreciable compact heterochromatin, and the absolute accessibility measured was surprisingly similar between euchromatin and heterochromatin. H3K9me3-marked chromatin was only about 30% less accessible than the body of a transcribed gene, while H3K27me3-marked chromatin was about 30% more accessible [17]. The most dramatic changes in accessibility, 2–3 fold in sub-saturating conditions, were at the well-defined hyperaccessible sites found by assays like ATAC-seq – promoters, enhancers and insulators – though, interestingly, some promoter accessibility was observed in some inactive genes [17]. This observation is concordant with a recent perturbation study in preprint that found precise stimulus-induced changes in transcription did not necessarily correlate with changes in accessibility [196]. Together, these results suggest that although nucleosome volume density is certainly higher in heterochromatin [77], small enzymes can still access their binding sites on DNA.
Several systems in which the short-range structure of chromatin is perturbed have yielded further insights. In yeast, local contacts between nearby nucleosomes in the chromatin fiber increase between log phase and quiescence, coinciding with large-scale transcriptional repression [78]. This repressive folding of the chromatin fiber is consistent with disordered local contacts rather than an ordered 30-nm fiber and depends on acetylation of the basic patch of the histone H4 N-terminal tail domain. Furthermore, unfolding of the chromatin fiber induced upon global chromatin acetylation in log-phase cells was found to be a prerequisite for and not a consequence of transcriptional activation, though importantly, it was not sufficient for transcriptional activation. Because chromatin accessibility was not directly probed, RNA polymerase II (RNAPII) occupancy and transcriptional activity must be used as a proxy to interpret this data. Recent experiments currently in preprint [125] investigating a different deacetylation event at the early stages of X chromosome inactivation in female mammalian cells, found that RNAPII has shorter residence times on chromatin in the inactivated X chromosome than elsewhere in the nucleus, but that its diffusion in the unbound state is unaffected, despite the overall higher compaction level of the inactivated X chromosome. In earlier work, it was found that histone deacetylation is the event that temporally best correlates with the reduction of RNAPII density within the X chromosome territory [152], but additional experiments found that the concerted action of both the deacetylase HDAC3 and the corepressor complexes NCoR/SMRT and NuRD, organized by the transcriptional repressor SPEN, are necessary to effect transcriptional repression and the associated loss of accessibility at promoters and enhancers found in the inactive X chromosomes [151,152,197]. These complexes couple deacetylation with nucleosome remodeling, and it is likely that both factors contribute to the observed changes in accessibility.
In B cells, TFs were tracked at the single-molecule level using fluorescence microscopy and their residence time on chromatin as well as the number of random collisions before stable binding events were reduced in activated cells [123]. This effect required ATP and did not occur upon inhibition of acetylation with trichostatin A, indicating that the dynamics of TFs searching for binding sites are sped up by ATP-dependent nucleosome remodeling and not by chromatin acetylation. The control of nucleosome clutch size, imaged by super-resolution fluorescence microscopy under the same conditions, was similarly dependent on ATP and not on acetylation, indicating that the sizes of local nucleosome clusters is correlated with TF search dynamics.
Lastly, mitotic chromosomes have also been investigated as a natural perturbation of chromatin structure. In mitosis, transcription is globally downregulated, and chromatin is both broadly deacetylated and phosphorylated, which blocks the recognition of the heterochromatin marks H3K9me2/3 [198]. The 30-nm fibers predicted by the classical model were not detected by X-ray scattering experiments [174,178]. Based on electron tomography measurements, chromatin fibers are highly irregular [76,77], and the volume density of chromatin in condensed mitotic chromosomes is approximately as high as it is in heterochromatin (~40–50%) [77]. Despite this high density, multiple studies using ATAC-seq have found that mitotic chromosomes retain accessibility at promoters and at some, but not all, enhancers [71,199–201]. Live-cell fluorescence measurements indicated that a previous model that invoked the eviction of most TFs from mitotic chromosomes was based on a fixation artifact. In contrast, Sox2 retains access to mitotic chromosomes but has a more dynamic interaction with chromatin with a lower residence time attributed to the loss of stabilization by global inactivation of transcription [71]. ESRRB was shown by ChIP-seq to bind specifically at many, but not all, of its sites in mitotic mouse embryonic stem cells [202]. At promoter-distal regulatory regions where ESRRB binding was lost in mitosis, the ordered nucleosome arrays found in interphase and in some, but not all, of DNA accessible sites were also lost, whereas both were maintained at bound (‘bookmarked’) sites. Similar mitotic bookmarking behavior was also observed at CTCF binding sites during mitosis in mouse embryonic stem cells, with the strongest motifs being more strongly bound [201]. Surprisingly, this bookmarking effect appears to be cell type-dependent for reasons that are not yet understood [201,203]. Other TFs exhibit reduced residence time on mitotic chromosomes [29,202]. Overall, although the chromatin environment of condensed mitotic chromosomes with a high nucleosome density and transcription is globally inhibited [204,205], TFs can still access chromatin and where they bind is regulated by factors more complex than simple steric exclusion or loss of accessibility.
Above the level of short stretches of the chromatin fiber, it has been suggested that RNAPII may be excluded from compact chromatin through formation of a gel with a defined pore size that excludes molecules beyond a certain size (Figure 2(c)). This has been tested by measuring the ability of exogenous factors, protein and otherwise, to penetrate dense heterochromatin domains using either microscopy or DNA cleavage or modification as assays. Fluorescent dextrans of varying sizes were excluded from heterochromatin in mammalian cells to an extent that depended on molecular size – by a factor of up to 50% for 90-nm diameter 500 kDa dextrans – and on the local DNA density, which can vary six-fold between euchromatin and heterochromatin [206]. In mitotic cells, 40-nm nanoparticles were partially excluded from condensed mitotic chromosomes [206]. These data are consistent with chromatin acting as a molecular crowding agent. In vitro, crowding can serve to enhance the effective binding affinity of protein–protein interactions, and this was observed for fluorescently tagged chromatin-binding proteins – two linker histones and RCC1 [206]. However, the detailed dependence of the anomalous diffusion on chromatin density could not be explained by a random crowding effect. More consistent results were produced from modeling of a fractal organization of chromatin on length scales below 100 nm that restrict protein motion through volume exclusion more than simple reaction-diffusion would and does not depend on molecular size [206] (Figure 2(d)). Experiments tracking the diffusion of Cas9 similarly found heterochromatin to be permeable, albeit at lower efficiency [29]. The density of fluorescent molecules in heterochromatin was reduced by 30% compared to euchromatin and diffusion was found to be slower.
Chromatin compaction and regulation of accessibility by phase separation
Liquid–liquid phase separation (LLPS) of intracellular components has emerged as an organizing principle of biological systems. In the case of macromolecules, it is a phenomenon in which molecular species with weak, multi-valent interactions, such as nucleic acids and proteins, spontaneously form two liquid ‘phases’ with different concentrations of the key molecules as a more energetically favorable state than the corresponding homogeneous mixture. The physics of the process and examples in many biological contexts have been extensively reviewed elsewhere [207–211].
LLPS, as it relates to chromatin accessibility and transcriptional regulation, has been found to occur both for chromatin-associated proteins, including HP1α [212–214], linker histone [215], RNA-protein complexes such as those found in the nucleolus [216], other nuclear bodies [217], a diversity of other factors [218–223], and for chromatin itself [215]. LLPS, as formally defined, applies to systems at equilibrium, and therefore, it can be difficult to characterize and define rigorously in vivo, where most processes that regulate chromatin involve molecular activities that expend energy and push the system out of equilibrium. Dynamic evolution from liquid states to more solid-like gel states can also occur, and the material properties of chromatin may depend on when they are observed [214,224]. Lastly, some local clusters of molecules like RNA polymerase are both transient and consisting of small numbers of molecules, making it difficult to determine whether they fit the definition of a macroscopic phenomenon like LLPS. Instead, such phenomena have been termed ‘condensates’ [222,223,225].
Phase-separated droplets appear when unmodified reconstituted chromatin is at concentrations beyond a threshold that depends on the mono- and di-valent salt concentration, the length of nucleosome arrays, and the presence of linker histone [215,226–228]. Because this state is consistent with physiological salt and concentrations of chromatin present in eukaryotic nuclei [229], this phase-separated state may represent the ‘ground state’ of chromatin in cells in the absence of histone modifications and enzymatic activity. Inside the droplets, chromatin can be concentrated up to 10,000-fold relative to bulk solution, showing that LLPS itself can act as a long-range chromatin compaction mechanism. The structure of the chromatin fiber within these phase-separated droplets is still under investigation, but early results show significant inter-digitation of nucleosome fibers, as well as much more heterogeneity than is present in 30-nm fibers [226,227]. Interdigitation is also consistent with the sensitivity of LLPS to the spacing of nucleosomes, i.e. the 10 n +5 bp inter-nucleosome linker lengths, which are also favored in vivo, promote phase separation and are generally less favorable to 30-nm fiber formation than 10 n bp linkers [215]. Multi-scale simulations have identified nucleosome ‘breathing’ (partial unwrapping of DNA) at physiological salt as a factor that drives multi-valent interactions between nucleosomes and causes chromatin to form heterogeneous condensates rather than ordered 30-nm fibers [230]. The extent of chromatin liquidity inside the droplets under certain buffer conditions is under some debate, but there are a variety of physiological-like conditions at which liquid-like behavior is robustly observed without the need for any special bundlers or crowding agents [215,228,231,232].
The phase-separating behavior of chromatin in vitro is modulated by regulatory factors and modifications, echoing the functions of these factors in vivo. Consistent with its compaction of isolated fibers and its compaction of chromatin in vivo [52,183], linker histone increases the concentration of nucleosomes, and hence the density of chromatin in phase-separated droplets [215]. Linker histone can also compact naked DNA in vitro, forming denser and more gel-like droplets with single-stranded DNA, and more liquid-like droplets with double-stranded DNA [150]. The globular domain of linker histone, which binds at the nucleosome entry/exit point, is dispensable for this activity and association of the positively charged and largely unstructured C-terminal tail domain of linker histone with nucleosomes via a viral LANA peptide has nearly the same effect [215]. There is some evidence that the unstructured C-terminal tail domain can become alpha-helical when bound to DNA [233], and the detailed mechanism by which the unstructured C- and N-terminal domains help to fold chromatin remains under investigation, as cryo-EM structures of chromatin fibers do not show a clear alpha-helix [177]. In keeping with the importance of positively charged, unstructured domains for phase separation, the positively charged basic patch in the N-terminal tail of histone H4 is also necessary for phase separation. Promiscuous acetylation of the nucleosome arrays by the acetyltransferase p300, which targets several lysine residues in the H4 basic patch, abrogates and actively dissolves phase separated droplets [215].
This observation is consistent with the unfolding and de-compaction of chromatin fibers of acetylated chromatin, and with the self-association of de-acetylated chromatin in vivo. The two self-associating chromatin compartments found by Hi-C, termed A (active) and B (inactive) largely follow the same organizing principle based on acetylation state [74,234]. Several pieces of evidence support phase separation of chromatin with similar histone modifications as the mechanism for compartment interactions [235]. First, the compartment associations are independent of active loop extrusion and are antagonized by it, as observed when compartment contacts appear stronger when cohesin is depleted from chromatin and appear weaker when cohesin’s stability on chromatin is enhanced [234,236,237]. Topological domains can also contain mixed compartment chromatin, showing that they are not the unit of compartment segregation [66]. Second, simulations compared against nuclear organization of the two compartments under normal and perturbed conditions are more consistent with the segregation of compartments being driven by inactive, deacetylated chromatin rather than active chromatin [238] and models with a combination of loop extrusion and polymer phase separation best recapitulate experimental data [239]. Third, work now in preprint using extremely high-coverage Hi-C to obtain high genomic resolution found that chromatin self-segregates into compartment interactions down to the scale of genes, and correlates very closely with the local nucleosome modification landscape [240].
Acetylated chromatin does not form phase-separated compartments or droplets on its own, but condensates of acetylated chromatin have been proposed to form through association with regulatory proteins that have the capacity to phase-separate themselves. In vitro, the acetyl-lysine ‘reader’ protein Brd4 and oligomers of the acetyl-lysine binding bromodomain both drive phase separation of acetylated chromatin into droplets with lower chromatin density [215]. Similar condensate-promoting behavior has also been observed for many other proteins, which can also phase separate on their own and are associated with transcriptional activation [109,222], transcriptional elongation [241], or with gene-body ubiquitination [242]. RNAPII itself has been found to concentrate in condensates with TFs and transcriptional coactivators [225,243–245]. Importantly, phase-separated droplets formed by acetylated chromatin (in association with acetyl-lysine binding proteins) and those formed by unmodified chromatin do not mix [215], adding further support to chromatin phase separation as a mechanism for the segregation of active and inactive compartments within the nucleus (Figure 2(b)). Additional immiscible phases may be formed by combinations of proteins that make up specialized nuclear bodies like the nucleolus [224].
Proteins with independent phase-separation behavior also associate with heterochromatin. The prime example is HP1α, which was one of the first examples of nuclear proteins found to undergo liquid–liquid phase separation at high concentrations in vitro [213,214]. HP1α forms droplets with liquid-like behavior in the early stages of heterochromatin formation in Drosophila embryos. It binds DNA, H3K9me3 on the H3 N-terminal tail, and other HP1α molecules, forming multi-valent interactions. Both chromatin and naked DNA readily partition into HP1α droplets. Compaction of DNA with significant resistance to opposing force has also been directly demonstrated by HP1α in vitro [246] and through bridging in silico.[247] Interestingly, HP1 proteins may promote phase separation of chromatin through additional mechanisms beyond bridging H3K9me3-marked nucleosomes through oligomerization. The yeast homolog Swi6 was found to increase the dynamics of the nucleosome core, exposing amino acid residues that are normally hidden [248]. Restraining this dynamic rearrangement through crosslinks impaired phase separation of chromatin by Swi6. Together, these data point to a mechanism by which HP1 is locally concentrated, possibly by binding to patches of H3K9me3, nucleating a phase-separated condensate that compacts the chromatin and reinforces deposition of more H3K9me3 through its interaction with the methyltransferase Suv39H1, which causes further compaction and concentration of HP1, H3K9me3-marked chromatin, and Suv39H1 [212,247,249].
Similar phase-separating and chromatin compaction behavior has also been observed for the canonical PRC1, the polycomb group repressive complex which binds the H3K27me3 mark associated with facultative heterochromatin [148,250,251]. However, the role of polycomb group proteins appears to be supplemental to compaction by deacetylation and linker histone, as they often associate with chromatin after transcriptional silencing has already occurred [151] and they prefer to bind to compacted chromatin fibers [252]. Nevertheless, in experiments with a switchable PRC1 phase separation system, PRC1 condensates can induce chromatin compaction in cells that is maintained after the condensates are dissolved [251]. Some evidence also exists that the PRC2 complex containing EZH1 can compact chromatin through dimerization and bridging of nucleosomes [157].
RNA, a major component of the nucleolus and many other early examples of LLPS, drives phase separation in a variety of nuclear and cytoplasmic condensates [253,254]. Broadly, RNA can act as a multi-valent scaffold to promote phase-separation of proteins that interact with it, either by concentrating those proteins at its site of transcription or through multiple repeats of their binding sites such as those present on satellite RNAs [255]. The valency of this mechanism with respect to accessibility and transcription depends on the proteins being concentrated and the local context. For example, SAF-A, an RNA-binding protein, has been implicated in both maintenance of the de-compaction of transcriptionally active chromatin and in X chromosome inactivation [151]. A microphase separation model for segregation of transcriptionally active and inactive chromatin through RNA-based recruitment of proteins associated with active chromatin and exclusion of inactive chromatin [256]. In contrast to this model, the RNA-binding factor, SAF-B, contributes to the stabilization of compacted pericentric heterochromatin [257].
LLPS presents an alternative way that the nucleus and the genome can be partitioned and access to specific loci can be controlled. Rather than physically blocking proteins from diffusing into a compartment entirely or based on size, phase separation can in principle filter proteins based on chemical features such as their surface charge or more generally, their affinity for proteins or nucleic acids that are concentrated within the condensate. This spatial filtering by LLPS is just beginning to be systematically investigated, and results so far are context and probe dependent.
The dCas9 reporter experiments discussed above showed that diffusion of exogenous proteins into heterochromatin is not blocked [29]. The inactivated X chromosome, despite being enriched in PRC complexes rather than HP1 proteins, has been proposed to form a phase-separated compartment [258]. However, work now in preprint investigating this hypothesis by tracking RNAPII inside and outside the inactive X volume found no change in the diffusion properties of the enzyme [125]. More systematic work is needed to understand to what extent these examples happen to be proteins that are not excluded or to what extent lack of exclusion is a global phenomenon.
Evidence supporting exclusion of proteins from phase separated compartments has also been gathered. Work in preprint suggests that CTCF is excluded from H3K9me3-marked and HP1-bound chromatin through a mechanism that is independent of methylation of its binding sites [259]. In mitotic chromosomes, deacetylated chromosomes with a high density of nucleosomes were found to exclude microtubules, to exclude tubulin and other negatively charged proteins, to be permeable to neutral proteins like the reporter DsRed, and to concentrate a positively charged DsRed fusion [124]. Dextrans behaved similarly, depending on their charge. Protein charge therefore appears to be an important determinant of which proteins can partition into dense chromatin condensates, and its regulation by post-translational modifications such as acetylation and phosphorylation merits further study in many contexts involving potential LLPS. Specific interactions can also lead to differences in permeability of nuclear compartments. In an extreme example, the Tn5 transposase, which normally has a strong preference for hyper-accessible, de-compacted chromatin, can be targeted to transpose into heterochromatic regions through fusion of a chromodomain that binds H3K9me3 [260].
Yet another possibility is that the snapshots of varying protein density across the nucleus we observe with static or aggregate measurements reflect not exclusion, but differential dynamics. This was found to be the case for RNAPII in the inactive X chromosome, where its bound fraction was drastically reduced while the diffusion of free molecules was unaffected [125]. Such a phenomenon has also been studied in depth in the context of viral replication domains, where proteins are concentrated not by differential diffusion but by accumulation on viral genomes [261]. In cases where this phenomenon is operative, the local structure of chromatin, local nucleosome dynamics, and their regulation of binding site accessibility for the test protein take primacy.
Compaction and accessibility regulation by looping and tethering
Condensed mitotic chromosomes are a special case of high-density chromatin. A recent tour de force of sequencing-based experiments and modeling elucidated the mechanism of mitotic chromosome condensation by condensin I and condensin II complexes through two tiers of extruded loops, arranged in a ‘spiral staircase,’ whose anchors self-organize into a chromosome axis [165,235]. Although this compaction is important for the mechanical stability of mitotic chromosomes and fidelity of chromosome segregation, local accessibility of chromatin does not appear to be globally affected by it – rather, some hyper-accessible sites lose accessibility in a way that is likely more controlled by local factors like availability of TFs, transcriptional coactivators, and polymerases to bind DNA (see discussion of mitotic bookmarking above) [202,262,263]. In yeast, condensin has been implicated in helping to mediate quiescence-associated transcriptional silencing, but this mechanism is supplemental to changes in local chromatin fiber compaction associated with deacetylation [78,83]. The molecular and physical details of how condensin contributes to global transcriptional silencing are not yet understood but have been proposed to involve restriction of promoter-enhancer interactions [83]. More direct effects on chromatin compaction and accessibility may be possible, as cohesin complexed with DNA of 3 kb or more has been shown to form condensates with liquid-like behavior [264].
Links between chromatin and the nuclear lamina also have the potential to promote compaction of associated chromatin domains or to consolidate these domains around the nuclear periphery. Theoretical modeling showed that even if chromatin is equally likely to bind the nuclear lamina regardless of epigenomic state, the more compact state will be selectively recruited to the lamina [265]. Two proteins have so far been implicated in binding both chromatin and the nuclear lamina: lamin B receptor [266–269] and PRR14 [270]. Both of these proteins bind HP1 [268,270,271], providing one mechanism for heterochromatin to be tethered specifically to the lamina. Prdm16 and ZKSCAN3 are two additional candidates for this role [272]. The Barr body is associated with the nuclear lamina via lamin B receptor, though this tethering has been shown to play only a minor role in transcriptional silencing [126,269,273]. There is abundant evidence supporting a transcriptionally repressive role for the lamina-adjacent nuclear compartment, reviewed elsewhere [272,274,275], and LADs appear to make up a major part, though not all of the B compartment identified by Hi-C. LADs are enriched in H3K9me2/3 marks, while their boundaries are enriched in H3K27me3-marked chromatin and CTCF-binding sites [275]. The other major location that houses heterochromatin in mammalian cells is the periphery of the nucleolus. Nucleolus-associated domains (NADs) partially but not completely overlap LADs – the former are enriched in telomeric and centromeric or peri-centromeric chromatin, while the latter are enriched in developmentally regulated genes [272]. Although transcriptional activation and histone acetylation have been shown to lead to disassociation of loci from the lamina, the mechanisms underlying this phenomenon and the direction of causality between association with the lamina and transcriptional repression have not yet been elucidated [272]. Transcriptional repression and compartmentalization are maintained by redundant mechanisms, and individual perturbations, including ones as dramatic as partially inverting the spatial organization of heterochromatin in the nucleus by knocking out the lamin B receptor, did not change the compartment map measurable by Hi-C [276]. Overall, the accessibility and compaction properties of lamina-associated chromatin appear to be driven mainly by their local epigenomic features, such as histone or DNA methylation and their transcriptional state. Histone turnover seems to be one mechanism that directly connects peripheral localization of chromatin to accessibility and transcription. A recent study in fission yeast found that a conserved nuclear rim protein, Amo1/NUPL2, and a conserved RNA processing complex, RIXC, are required for heterochromatin peripheral localization, which promotes a FACT/HP1-based mechanism to suppress histone turnover and maintain a transcriptional repression [91]. It will be interesting to determine if this pathway or other factors similarly couple tethering of heterochromatin to the nuclear periphery and low histone turnover in mammals. Alternative functions of the peripheral localization of heterochromatin may include stabilizing the mechanical stiffness of the nucleus [137] or protecting the genome from radiation [277–280].
The Importance of being open
Transcription factor binding
TF binding is a critical step in gene expression, and it is intimately associated with chromatin accessibility. Notably, the vast majority of TF binding sites, (94.4%) of ENCODE-mapped TF ChIP-seq peaks, fall within hyper-accessible regions [194]. Experiments that track accessibility throughout development or after TF-induction indicate that accessibility can play a casual role in promoting TF binding and vice versa [281–285]. For example, ChIP-seq of PPARγ, a core activator of adipocyte expression and differentiation, indicates that 33% of PPARγ-bound sites in terminally differentiated adipocytes were already accessible during the earliest stages of adipogenesis [281,282]. Accessibility measurements made before and after induction of the glucocorticoid receptor (GR) TF have enabled more precisely controlled experiments with clearer conclusions that point to casual relationships in both directions. Analogous to PPARγ binding, many of GR-bound sites (70–95%, depending on the depth of accessibility measurements) are in regions that were accessible prior to hormone induction [283,284]. Further genomic and genetic analyses found that Activator Protein 1 (AP1) TF binding was enriched at GR-bound sites and that AP1 is required to establish the basal chromatin accessibility that allows GR binding [284]. Systematic in vivo analyses led to a similar conclusion that hyper-accessibility is a primary determinant of TF binding in the genome, whereas cooperative effects of nearby TFs and sequences play secondary roles [281]. Lastly, the role of accessibility in promoting TF binding and activation of target genes has been demonstrated by removing the barriers to TF binding. For example, H3K9me2/3 and H3K27me3 methyltransferases are required to block the activation of target genes and differentiation upon TF overexpression [286]. This requirement is also observed throughout development of nematodes and of cultured human cells [36,130,286]. These histone methyltransferases are typically referred to as repressive factors, as their corresponding modifications correlate with lowered hyper-accessibility. Thus, TF-based phenotypes linked to chromatin accessibility can manifest at the level of organismal development.
Not all TFs preferentially bind to accessible regions. Pioneer TFs, such as FOXA1, C/EBP, and NF-YA, can initiate transcription and/or promote accessibility by binding to nucleosome-occupied sites [281,287,288]. In zebrafish zygotic genome activation, the TFs Nanog, Pou5f3, and Sox19b pioneer the creation of accessible chromatin and activation at more than half of enhancers genome-wide by recruitment of acetyltransferase activity [289]. However, pioneer TFs do not only bind to occluded sites, as they also exhibit considerable direct DNA-binding at accessible sites [290]. The repressive factor NRSF/REST [291], as well as CTCF, which binds to insulator elements [281,290], are also examples of TFs that do not preferentially bind to nucleosome free sites. Systematic analyses of TF affinities to reconstituted nucleosomes support variable binding mechanisms, some that are favored when DNA is not occluded and other binding regimes that are compatible, or even possibly enhanced, when DNA is wrapped around nucleosomes [292,293]. Thus, chromatin accessibility plays critical roles, both positively and negatively, in the regulation of TF binding.
Transcription
Beyond the binding of TFs, transcriptional activities, which include initiation, elongation, termination, and RNA processing, are also intricately linked to chromatin accessibility. Cytological studies have been key to establishing this relationship, as they have shown that transcription localizes to areas in the nucleus that are less compact and often associated with more accessible chromatin. For example, when genes on the inactivated mammalian X chromosome ‘escape’ silencing and are actively transcribed, they preferentially localize away from the nearby inactive X compartment, which remains transcriptionally silenced and is a more chromatin dense area [294,295]. Super resolution microscopy experiments have extended this observation genome-wide – RNAPII localizes within regions of less densely stained ‘nanodomains’ of DNA and are proximal to histone marks associated with hyper-accessible chromatin (e.g. H3K9ac and H3K27ac) [296]. These microscopy-based experiments show a clear correlation between accessibility and transcription.
Genetic manipulation of nucleosome supply has established strong evidence for a causal link between accessibility and transcription. To this end, the budding yeast model has been especially powerful, as it harbors only a few copies of histone genes that are readily manipulated. Decreasing histone levels by half, either by deletion or by inducible silencing of histone genes, consistently leads to a substantial increase in global mRNA abundance [297–300]. Additionally, histone depletion preferentially de-silences genes that are more proximal to large repressive domains (e.g. telomeres) [298], indicating that nucleosome density is critical, yet only one layer of repressing gene expression. In other eukaryotes, including many metazoans, histone genes are present in hundreds of copies, making them difficult to genetically manipulate. However, the maturation of histone transcripts is distinct from other genes in its requirement for specialized RNA processing factors, and thus, depleting these processing factors can selectively reduce histone levels [301]. Severe histone depletion leads to confounding DNA damage and cell cycle defects, thus only modest depletion of histones is possible in vertebrates [302]. Under these conditions, there is less gene de-repression, but transcriptional elongation rates and misreguation of splicing are significantly increased. A more targeted approach, which used an inducible Cas9 fused to the histone acetyltransferase p300, found that after induction there was increased hyper-accessibility specifically at the targeted promoters and that the corresponding genes have reduced timing between transcription events, yet total transcription levels remain the same [303]. Importantly, misregulated increases in chromatin accessibility have implications in disease states, such as cancers [50,304–306]. Thus, increasing DNA accessibility by reducing total nucleosome abundance can lead to runaway transcription, however, understanding the full consequences of accessibility requires more extensive analyses that scrutinize various aspects of transcription.
The relationship between accessibility and transcription is intertwined such that there are various counter influences that transcription exerts on chromatin organization both at large- and short-scales (10s to 100s of kbs and sub-kilobase resolution, respectively). Measuring chromosome structures at the onset or upon the inhibition of transcription has been key to addressing the directionality of effects. A classic example is the naturally occurring transcriptional induction at the onset of gene expression of the zygotic genome during early development [307]. In this developmental regime, the genome adopts large-scale chromosome organization, such as topologically associated domains (TADs) and loops on the order of 10s to 100s of kb [308]. Transcription is required for the establishment of these broad compartment contact patterns in humans, but not required in many other organisms [308]. Across organisms, the inhibition of global transcription before the onset of zygotic expression consistently limits the number of contacts made within a TAD [309,310], further supporting the role of transcription in large-scale genome organization.
Chromatin accessibility at the short-range scale is also drastically reshaped with a burst of accessibility concurrent with the onset of zygotic transcription, and that pattern becomes refined throughout development as epigenomic domains are formed [311,312]. Tracking chromatin accessibility and transcription through a single round of DNA replication in both yeast and cultured mammalian cells yields analogous conclusions. Specifically, transcription is required to re-establish accessibility after DNA replication [313,314]. Complementary experiments have also been done by inhibiting global transcription, upon which there is a decrease in intra-TAD contacts [310,315], accessibility [310], and short-range chromatin compaction [315]. These inactivation studies employed both chemical inhibition and RNAPII degrons to test whether there is a separation of function between active transcription and other possible architectural roles that the RNAPII machinery may provide. Both types of transcriptional inhibition led to similar accessibility outcomes, indicating that active transcription, not architectural scaffolding, regulates accessibility. Another feature of transcription that may play a role in increasing accessibility is torsion created from the processivity of RNAPII. Positive DNA supercoiling resulting from this torsion might increase accessibility by destabilizing nucleosomes, the effects of which have been measured within gene bodies [316,317] and at the larger-scale of genome compartments [317,318]. Overall, there is a preponderance of evidence that transcription has a major impact on chromatin accessibility in many developmental contexts, often promoting increased accessibility. Recent work shows that perturbations that alter specific transcriptional programs without affecting cell identity create a complex relationship between transcription and accessibility [72,196]. Although some genes concomitantly changed both expression and hyper-accessibility, at many genes, strong changes in gene expression came with no short-term change in hyper-accessibility. Transcription-linked accessibility may therefore be a feature that accumulates over long-term transcriptional activation at these sites, or it may only be characteristic of a subset of genes, including developmentally controlled genes.
DNA repair
Studying genome-wide chromatin accessibility in the context of DNA damage and repair can be more challenging because unlike transcription, DNA repair pathways typically act on lesions that are more randomized throughout the genome and are often not anchored to specified binding motifs or transcription start/termination sites. Anchored sites that do exist in the DNA repair context are commonly referred to as ‘hotspots’, which are correlated with a variety of genomic sequences, secondary structures, and features that may or may not interface with chromatin accessibility. For example, hairpin-forming palindromic sequences promote inverted duplications, R-loop-dependent mutations are enriched at G-rich regions that are likely to form secondary structures, and repetitive satellite sequences are especially prone to different types of mutations [319,320]. Coincidently one of the earliest genetic processes to be linked to hotspots, meiotic recombination, is strongly associated with chromatin accessibility. Although most mammals use the PRDM9 sequence motif to define sites of meiotic double-stranded breaks (DSBs), the local chromatin landscape influences which motifs are bound, and upon binding, PRDM9 modifies the chromatin to establish a nucleosome-depleted region [321]. In organisms that lack PRDM9, like dogs, plants, or yeasts, DSB preferentially target nucleosome-depleted regions, such as gene promoters [322,323]. Another example of DNA damage that is associated with accessibility is class-switch recombination during antigen development that involves the targeting of AID (Activation-induced cytidine deaminase) to accessible and actively transcribed regions in the Igh locus to initiate DSBs, and in turn, a recombinant Igh locus [324]. Thus, although many sequence-dependent features are primary determinants underlying the initiation of naturally occurring DNA damage, there are notable examples of programmed DSB systems that are tightly associated with chromatin accessibility.
Chromatin may play a broader role in shielding DNA against damage-inducing sources like ultraviolet (UV) or higher energy radiation. Some of the better characterizations of this effect are genome-wide maps of UV and alkylation damage in budding yeast [325]. Tracking the initial lesion landscape induced by either UV or the alkylating agent methyl methanesulfonate (MMS) show a strong 10-bp periodicity that results from a preference of lesions to occur at the solvent accessible regions within well-positioned nucleosome and at some TF binding sites [279,280], indicating that chromatin-bound proteins can provide protection to wrapped DNA. Analogous observations have been found in cultured human cells upon gamma irradiation. Specifically, genome-wide sequencing of radiation-dependent breaks reveals footprints of modest protection at well-positioned nucleosomes and TF binding sites, however, there is minimal overall difference in protection relative to genomic compartments that display differential levels of accessibility [33]. The tardigrade protein Dsup (damage suppressor) offers an intriguing innovation that could possibly link accessibility to DNA damage. Tardigrades are invertebrate extremophiles that naturally have robust protection against many abiotic factors, including large amounts of radiation [326], and the highly abundant chromatin factor, Dsup, was proposed as a candidate for radiation-based protection. Transgenic experiments in human cells and in vitro reconstitution confirm that Dsup can suppress hydroxyl radical-mediated DNA damage, and likely does so through its ability to bind nucleosomes [327,328]. Thus, chromatin may play a general role in safeguarding against various exogenous sources of DNA damage, especially at single nucleosomes, but more extensive experiments will be needed to rigorously address whether accessibility can be manipulated on a larger scale to protect the genome.
In contrast to the introduction of damage, DNA repair is critically dependent on chromatin landscape and accessibility. The intersection of chromatin and repair has been put into a stepwise framework known as the Access-Repair-Restore model [329], which has been reviewed extensively and updated to fit current understandings of DNA repair pathways [325,330–335]. Topics covered elsewhere include the roles of histone variant H2AX; chromosome de-condensation, re-condensation, and motility; repair pathway choice; and key histone chaperones and nucleosome remodelers that work in many DNA damage pathways from nucleotide excision repair to DSB repair.
One aspect of DNA repair and chromatin that has been summarized less within the Access-Repair-Restore framework is the initial phase of the access step, which begins with the sensing of a lesion. DNA damage comes in a variety of forms that launch distinct signaling and repair pathways, and thus there is likely to be substantial variability in the role of chromatin accessibility in the sensing of DNA lesions. For example, DSBs create a discontinuous DNA polymer, and this broken link in the DNA chain is sensed by DNA-end-binding proteins, like PARP1, MRN/MRX complex, Ku, or SIRT6 [336,337]. Sensing DSBs by these factors is not likely to be impeded by local nucleosome occupancy, as upon DNA breakage nucleosomes would unwrap and expose their DNA ends. To induce a potent damage signal and initiate repair, nearby nucleosomes need to be remodeled and removed, and this task may be facilitated by coupling the sensing of lesions with nucleosome remodeling. For example, PARP1 can directly modify histones and induce nucleosome disassembly [338]. SIRT6, another recently identified DSB sensor, can also modify chromatin through the recruitment of nucleosome remodelers [337].
In contrast to DSBs, stranded lesions that maintain the continuity of the chromosomal polymer, like single-stranded breaks or gaps, DNA base damage, DNA adducts, depurination, or crosslinks, are detected by different enzymes that scan DNA and can do so in the context of chromatin [325,336]. Nucleosome binding can inhibit the detection of single-stranded lesions that are located at the nucleosome dyad or when they are buried in solvent-inaccessible region of the nucleosome. Indirect evidence of this protection can be seen in the genome-wide maps following the fate of UV or MMS damage, as mutations or long-lasting lesions preferentially accumulate in regions of lowered accessibility [277,279,280]. More direct evidence of nucleosomes restricting access to DNA lesions comes from detailed in vitro analyses of DNA-embedded thymidine dimers and the binding affinities of these lesions by UV-DDB [339]. In contrast to their ability to hide within non-solvent accessible chromatin, stranded lesions can also facilitate their own exposure by creating a less stable nucleosome, which can become unwrapped [340] or shifted slightly in their DNA register [339]. In addition to the scanning of repair pathway proteins, transcription may also act as a sensor and facilitate the detection of lesions [341]. Given the close association of DNA accessibility with transcription, as detailed above in the Transcription subsection, this mode of sensing DNA lesion could certainly be affected by DNA accessibility. Thus, chromatin accessibility can play an important role in the sensing lesion depending on the type of DNA damage, and more work in this direction will shed mechanistic light on the potential crosstalk between damage sensors and nucleosome remodelers.
Replication
DNA replication is a key cell cycle event that is indirectly associated with chromatin accessibility for its initiation, which is also referred to as ‘origin firing’. In eukaryotes replication origins fire in a probabilistic manner [342]. One of the best studied replication programs is that of budding yeast, which have sequence-defined origins referred to as autonomously replicating sequences (ARSs). These sequence-encoded origins have a consensus sequence that is enriched for asymmetric dA:dT tracts, which disfavor nucleosome occupancy [343,344]. Origins that are identical in sequence, like the hundreds of origins in the ribosomal DNA array, preferentially initiate downstream of active transcription [345,346], indirectly indicating that accessibility influences origin firing efficiency. At origins throughout the rest of the genome there are thousands of possible consensus sites, yet only a few hundred are bound by the Origin Replication Complex (ORC). At these ORC-bound sites there is a strong depletion of nucleosomes and well-positioned flanking nucleosomes, reminiscent of promoters [344,347]. The maintenance of low nucleosome occupancy at replication origins seems to be independent of ORC binding, as temperature-sensitive ORC mutants maintain a nucleosome-depleted region at origins to a similar degree of WT strains [344]. However, ORC is required for coordinating the spacing of nearby nucleosomes that play key roles in origin firing [344,348,349]. A recent review has challenged the inherent ability of dA:dT tracts to establish nucleosome-depleted regions [350], suggesting that trans-factors may play important roles in helping to establish these nucleosome-depleted regions and regulating origin loading. To this end, the binding of the pioneer Forkhead family TF (Fhk1) was also found to be an important correlate of ORC loading at ~25% of origins [351,352]. Thus, the budding yeast model system has revealed that nucleosome depletion and local chromatin organization are key features and regulators of replication initiation.
Metazoan replication initiation is not controlled by defined sequences; nonetheless, it is also heavily influenced by accessibility and transcription. ORC loading in Drosophila cells is highly correlated with hyper-accessible regions [353], and origin firing in mammalian cells preferentially occurs at asymmetric dA:dT tracts [354]. More refined analyses show that mammalian origin initiation is tightly correlated with hyper-accessible sites, such as cell-specific enhancers and transcription start sites, and that the rate of firing scales with the levels of genic transcription [342,355,356]. In developing nematodes, enhancers and transcription are similarly associated with replication initiation, but the strength of this correlation does not seem to persist as development proceeds [357]. Coupling origin firing with transcription start sites has been proposed as a mechanism to help reduce replication-transcription conflicts in a cell-type specific manner [356]. Thus, chromatin accessibility is a strong indicator of replication initiation throughout eukaryotes, and it may play an important role in maintaining genome stability.
In addition to origin firing, DNA replication plays a critical role in resetting the chromatin accessibility landscape. One of the first studies to demonstrate this resetting capacity relied on the budding yeast ribosomal DNA array, which has a bimodal distribution of accessible and inaccessible chromatin states among its hundreds of repetitive units [358]. Upon cell cycle arrest in G1 or in early S phase, the bimodal accessibility distribution shifted greatly to favor accessible chromatin with nearly all repeats becoming permissive to transcription; and active transcription was required to shift the distribution towards hyper-accessibility [314]. Similar results have been reported for Drosophila and mouse cultured cells using orthogonal techniques to track genome-wide accessibility of newly replicated DNA at different timepoints after replication. Early post-replication timepoints revealed a loss of hyper-accessible sites [313,359], and these sites were gradually restored in a transcription-dependent manner [313]. DNA replication also plays an important role in resetting the epigenomic landscape, as it is required for the segregation, and thus, dilution of nucleosomes and their respective modifications upon cell division [360]. Although replication is required for segregation of all histones, the dilution of nucleosomes is especially pronounced in genes that are active and more accessible [39]. Overall, the resetting of chromatin accessibility after each round of DNA replication has wide-ranging implications from transcriptional silencing of multi-copy genes to the regulation of epigenetic inheritance.
Genome editing and invasion
The ability of exogenous proteins to access or manipulate the accessibility of host DNA can have important biomedical implications. One of the most notable examples is the differential ability of CRISPR/Cas systems to edit, repress, or activate different genomic loci. Cas nucleases use sequence-dependent recognition, but their ability to bind and cleave is inhibited by nucleosomes [29,361]. In vitro experiments indicate that unwrapping or remodeling of nucleosomes can restore DNA access to Cas enzymes [361,362], and thus the efficiency of editing roughly correlates to an average of nucleosome occupancy at a given locus. When in vivo Cas binding dynamics, cleavage patterns, or editing efficiencies are overlayed with epigenomic compartments, there are clear positive correlations between Cas cleavage efficiencies and regions that are more accessible [29,39]. This correlation is also true for off-target sites [363,364] and for the transgenic expression of traditional restriction enzymes in mammalian cells [354]. To leverage accessibility in efforts to increase editing efficiency, some histone deacetylase inhibitors, or the activation of proximal transcription, by localizing Cas activators to sites of cleavage can lead to more efficient genome editing [365–367]. Thus, chromatin accessibility provides a useful framework for understanding the functional outcomes and manipulating the design strategies of CRISPR/Cas targeting [368].
The accessibility of DNA can also have important implications during naturally occurring invasion of the genome. Many double-stranded DNA viruses and retroviruses enter the nucleus during their lifecycles, and accessibility-based mechanisms can be used by the host to repress viral gene expression. For example, herpesviruses are rapidly loaded with nucleosomes and repressive modifications upon entry into the nucleus, and successful infections require the action of viral proteins, like VP16, that can restore accessibility to the viral DNA, enabling viral gene expression [369]. Other viruses, like human papilloma virus (HPV), hepatitis B virus (HBV), human immunodeficiency virus (HIV), etc, can leverage repressive chromatin by maintaining low expression and abundance, ultimately, allowing them to escape immune surveillance and persist as chronic infections [370]. Recent structural work has highlighted one possible mechanism of immune avoidance, as nucleosome-bound DNA is able to occlude the binding sites of cGAS, a major foreign DNA sensor [371]. Viruses also employ accessibility-based mechanisms to manipulate host chromatin. For example, adenovirus Protein VII is a highly basic, histone-like protein that can bind to viral DNA and protect itself against host DNA damage response [372], possibly through its ability to limit DNA accessibility [373]. Protein VII can also suppress immune response by binding to host chromatin and stabilizing HMGB (high mobility group B) proteins, whose release from host chromatin induces a potent immune response [373]. E1A is another adenovirus protein that does not bind directly to DNA, but instead, it works in concert with host chromatin remodelers to destabilize the host chromatin accessibility landscape [374]. Influenza can similarly thwart immune response by using an exogenous histone-like protein, NS1, which competes with host chromatin remodeling complexes and affects accessibility of host genomic regions that are critical for the expression of antiviral response genes [375]. The interplay between viral and host chromatin highlights many infection-based roles of accessibility, representing an exciting and plentiful area of research with possible therapeutic applications.
Transposons are parasitic elements that correspond to an ongoing genome invasion and present a significant mutational risk. Both their expression [376] and the sites of insertion into new genomic loci [377,378] are correlated with hyper-accessibility. In early mammalian development, there is a high burst of transposon expression that occurs during large-scale genome re-organization of accessibility [376]. After this early expression, transposon activity is highly repressed, and their widespread de-repression can occur through global dysfunction in the accessibility landscape either during disease [305] or during aging [379]. Interestingly, the expression of retrotransposons during early development has also been implicated in titrating the dosage and timing of genome-wide accessibility in later developmental phases [380]. Thus, there is an intricate interplay between transposons and host chromatin accessibility.
Extrachromosomal DNAs
Renewed interest in episomes, also known as extrachromosomal circular DNA (eccDNAs), and their chromatin organization has recently highlighted their importance during tumorigensis. eccDNAs are derived from various sources, including from endogenous circularization of aberrant [381,382], or in some cases programmed [383], DNA ligation substrates. Oncogenes, such as MYC and EGFR, are often episomal and at very high copy number (up to 100s of copies), and with correspondingly high oncogene expression [384,385]. Recent studies have confirmed the presence of eccDNAs in many types of cancers, and they have also found that their abundance is a predictor of patient outcomes [386]. In addition to high copy number, increased chromatin accessibility is thought to play a role in promoting oncogene expression [386,387]. Increased accessibility can occur through enhancer hijacking [388] or through spatial clustering of eccDNAs [389]. Another possible mechanism underlying hyper-accessibility of eccDNA is their lack of a functional centromere, a feature that allows eccDNAs to resist chromatin condensation [390] and could also influence their resetting of accessibility upon DNA replication. In contrast to the growing number of reports of increased episomal accessibility, a recent preprint proposes that gene dosage alone can explain the high levels of oncogene expression [391]. Considering the high copy number potential of oncogenic eccDNAs and epigenomic dysfunction of tumor cells more generally [392], it is reasonable to question whether accessibility plays a role in episomal oncogene expression, or whether the observed accessibility has additional roles during tumorigenesis, like promoting metastases through cGAS-STING signaling [371,382]. Despite these conflicting results, eccDNAs represent an exciting new therapeutic target, as recent work has shed light into an SMC5/6-mediated silencing pathway that seems to specifically target eccDNAs [393,394]. Thus, eccDNAs are emerging as key players in many cancers, and it will be interesting to understand the degree to which accessibility influences their expression and abundance, and to understand how broadly the SMC5/6 pathway can regulate eccDNAs.
Conclusions
Unraveling the properties and consequences of chromatin accessibility has been a fruitful and rapidly developing field of study. Over the past few decades, a plethora of assays and sophisticated quantitative frameworks have fueled advances in measuring accessibility in many biological contexts. We have reviewed accessibility mapping methods, detailing their best use cases, their caveats, their limitations, and important experimental considerations, some of which include how the interpretation of accessibility data depends on the nature of the probe or on the time frame of a given assay. General conclusions from accessibility analyses implicate the importance of local nucleosome dynamics and in some cases, the permeability of nuclear compartments, the latter of which depends on phase separation, and to some extent, on electrostatic properties of chromatin. Additionally, long-lived associations with chromatin require specific chromatin binding abilities, which often require nucleosome mobilization to reveal target sites for the localization of sequence-specific proteins.
Although accessibility maps have thus far provided many important insights into fundamental DNA-based processes, more work is needed to understand both the physiologically relevant mechanisms underlying accessibility itself and its connection to functional outcomes. We have provided an overview of many chromatin factors, like HP1, linker histone, histone acetylation, etc., that directly regulate or correlate with chromatin accessibility at the level of genomic compartments. Coupled use of sophisticated genetic tools, such as CRISPR screens and inducible degrons, with powerful accessibility assays, like quantitative MNase-seq, ATAC-seq, or methyltransferase labeling with long-read sequencing, should enable a deeper understanding of the mechanisms by which these factors regulate accessibility. One mechanistic aspect that we highlight is chromatin architecture and its contributions to accessibility. Tools for measuring chromatin organization at the compartment level have been available for longer and have gone through multiple iterations, whereas assays for defining chromatin structures at the short-range scale of a few nucleosomes is still in its infancy. Important conceptual advances in the chromatin architecture field include uncoupling between the degree of compaction at long- and short-length scales, a heterogenous landscape of short-range chromatin structures, and the ability of condensates to compact and segregate chromatin. Ongoing advancements in multimodal experimental and theoretical analyses, especially at the single-cell and multi-locus level, should continue to deepen our understanding of the relationships between DNA accessibility, spatial chromatin organization, protein binding, transcription, gene expression, and downstream developmental phenotypes.
Acknowledgments
We apologize to those researchers whose work we were not able to include in this review.
Funding Statement
This work was supported by the Irma T. Hirschl/Monique Weill-Caulier Trust [Career Scientist Award] to V.I.R.; NIH Office of the Director [1DP2GM150021-01] to V.I.R.; National Institute of General Medical Sciences [F32 GM140551] to A.R.M.; NASA Space Operations Mission Directorate Human Research Program [80NSSC21K0565] to V.I.R.; Rita Allen Foundation [Scholar Award] to A.R.M.; V Foundation for Cancer Research [V Scholar Award V2019-011] to V.I.R..
Disclosure statement
The authors report no potential financial conflict of interest.
References
- 1.Klemm SL, Shipony Z, Greenleaf WJ.. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet. 2019;20(4):207–220. [DOI] [PubMed] [Google Scholar]
- 2.Ishii H, Kadonaga JT, Ren B. MPE-seq, a new method for the genome-wide analysis of chromatin structure. Proc Natl Acad Sci USA. 2015;112(27):E3457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu T, Ruitu L, Qiancheng Y . Kethoxal-assisted single-stranded DNA sequencing captures global transcription dynamics and enhancer activity in situ. Nat Methods. 2020;17(5):515–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagy PL, Cleary ML, Brown PO . Genomewide demarcation of RNA polymerase II transcription units revealed by physical fractionation of chromatin. Proc Natl Acad Sci USA. 2003;100(11): 6364–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giresi PG, Kim J, McDaniell RM, et al. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007;17(6):877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyle AP, Davis S, Shulha HP, et al. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132(2):311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Albert I, Mavrich TN, Tomsho LP, et al. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446(7135):572–576. [DOI] [PubMed] [Google Scholar]
- 8.Mieczkowski J, Cook A, Bowman SK, et al. MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat Commun. 2016;7(1):11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chereji RV, Bryson TD, Henikoff S. Quantitative MNase-seq accurately maps nucleosome occupancy levels. Genome Biol. 2019;20(1):198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramani V, Qiu R, Shendure J. High sensitivity profiling of chromatin structure by MNase-SSP. Cell Rep. 2019;26(9):2465–2476 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baldi S, Krebs S, Blum H, et al. Genome-wide measurement of local nucleosome array regularity and spacing by nanopore sequencing. Nat Struct Mol Biol. 2018;25(9):894–901. [DOI] [PubMed] [Google Scholar]
- 12.Baldi S, Jain DS, Harpprecht L, et al. Genome-wide rules of nucleosome phasing in Drosophila. Mol Cell. 2018;72(4):661–672 e4. [DOI] [PubMed] [Google Scholar]
- 13.Buenrostro JD, Giresi PG, Zaba LC, et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buenrostro JD, Corces MR, Lareau CA, et al. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell. 2018;173(6):1535–1548 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gargiulo G, Levy S, Bucci G, et al. NA-Seq: a discovery tool for the analysis of chromatin structure and dynamics during differentiation. Dev Cell. 2009;16(3):466–481. [DOI] [PubMed] [Google Scholar]
- 16.Chen PB, Zhu LJ, Hainer SJ, et al. Unbiased chromatin accessibility profiling by RED-seq uncovers unique features of nucleosome variants in vivo. BMC Genomics. 2014;15:1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chereji RV, Eriksson PR, Ocampo J, et al. Accessibility of promoter DNA is not the primary determinant of chromatin-mediated gene regulation. Genome Res. 2019;29(12):1985–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oberbeckmann E, Wolff M, Krietenstein N, et al. Absolute nucleosome occupancy map for the Saccharomyces cerevisiae genome. Genome Res. 2019;29(12):1996–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Steensel B, Henikoff S. Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat Biotechnol. 2000;18(4):424–428. [DOI] [PubMed] [Google Scholar]
- 20.Sha K. Distributed probing of chromatin structure in vivo reveals pervasive chromatin accessibility for expressed and non-expressed genes during tissue differentiation in C. elegans. BMC Genomics. 2010;11:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelly TK, Liu Y, Lay FD, et al. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012;22(12):2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nabilsi NH, Deleyrolle LP, Darst RP, et al. Multiplex mapping of chromatin accessibility and DNA methylation within targeted single molecules identifies epigenetic heterogeneity in neural stem cells and glioblastoma. Genome Res. 2014;24(2):329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krebs AR, Imanci D, Hoerner L, et al. Genome-wide single-molecule footprinting reveals high RNA Polymerase II turnover at paused promoters. Mol Cell. 2017;67(3):411–422 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stergachis AB, Debo BM, Haugen E, et al. Single-molecule regulatory architectures captured by chromatin fiber sequencing. Science. 2020;368(6498):1449–+. [DOI] [PubMed] [Google Scholar]
- 25.Abdulhay NJ, McNally CP, Hsieh LJ, et al. Massively multiplex single-molecule oligonucleosome footprinting. Elife. 20209:e59404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shipony Z, Marinov GK, Swaffer MP, et al. Long-range single-molecule mapping of chromatin accessibility in eukaryotes. Nat Methods. 2020;17(3):319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Shen Y, Draper W, et al. ATAC-see reveals the accessible genome by transposase-mediated imaging and sequencing. Nat Methods. 2016;13(12):1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie LQ, Dong P, Chen X, et al. 3D ATAC-PALM: super-resolution imaging of the accessible genome. Nat Methods. 2020;17(4):430–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knight SC, Xie L, Deng W, et al. Dynamics of CRISPR-Cas9 genome interrogation in living cells. Science. 2015;350(6262):823–826. [DOI] [PubMed] [Google Scholar]
- 30.Fu Y, Rocha PP, Luo VM, et al. CRISPR-dCas9 and sgRNA scaffolds enable dual-colour live imaging of satellite sequences and repeat-enriched individual loci. Nat Commun. 2016;7:11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsieh TS, Fudenberg G, Goloborodko A, et al. Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nat Methods. 2016;13(12):1009–1011. [DOI] [PubMed] [Google Scholar]
- 32.Hsieh THS, Weiner A, Lajoie B, et al. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 2015;162(1):108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Risca VI, Denny SK, Straight AF, et al. Variable chromatin structure revealed by in situ spatially correlated DNA cleavage mapping. Nature. 2017;541(7636):237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rydberg B, Holley WR, Mian IS, et al. Chromatin conformation in living cells: support for a zig-zag model of the 30 nm chromatin fiber. J Mol Biol. 1998;284(1):71–84. [DOI] [PubMed] [Google Scholar]
- 35.Basu A, Bobrovnikov DG, Qureshi Z, et al. Measuring DNA mechanics on the genome scale. Nature. 2021;589(7842):462–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicetto D, Donahue G, Jain T, et al. H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification. Science. 2019;363(6424):294–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deal RB, Henikoff JG, Henikoff S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science. 2010;328(5982):1161–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deaton AM, Gomez-Rodriguez M, Mieczkowski J, et al., Enhancer regions show high histone H3.3 turnover that changes during differentiation. Elife, 2016. 5:e15316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Escobar TM, Oksuz O, Saldaña-Meyer R, et al. Active and repressed chromatin domains exhibit distinct nucleosome segregation during DNA replication. Cell. 2019;179(4):953–963 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tullius TD. Physical studies of protein-DNA complexes by footprinting. Ann Rev Biophys Biophys Chem. 1989;18:213–237. [DOI] [PubMed] [Google Scholar]
- 41.Brogaard K, Xi L, Wang JP, et al. A map of nucleosome positions in yeast at base-pair resolution. Nature. 2012;486(7404):496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voong LN, Xi L, Sebeson AC, et al. Insights into nucleosome organization in mouse embryonic stem cells through chemical mapping. Cell. 2016;167(6):1555–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer CA, Liu XS. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat Rev Genet. 2014;15(11):709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Corces MR, Trevino AE, Hamilton EG, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14(10):959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shema E, Bernstein BE, Buenrostro JD. Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat Genet. 2019;51(1):19–25. [DOI] [PubMed] [Google Scholar]
- 46.Cao JY, O’Day DR, Pliner HA, et al. A human cell atlas of fetal gene expression. Science. 2020;370(6518):808–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma S, Zhang B, LaFave LM, et al. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell. 2020;183(4):1103–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chereji RV, Ocampo J, Clark DJ. MNase-Sensitive complexes in yeast: nucleosomes and non-histone barriers. Mol Cell. 2017;65(3):565–577 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Denny SK, Yang D, Chuang CH, et al. Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell. 2016;166(2):328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Corces MR, Granja JM, Shams S, et al. The chromatin accessibility landscape of primary human cancers. Science. 2018;362:6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cusack M, King HW, Spingardi P, et al. Distinct contributions of DNA methylation and histone acetylation to the genomic occupancy of transcription factors. Genome Res. 2020;30(10):1393–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Willcockson MA, Healton SE, Weiss CN, et al. H1 histones control the epigenetic landscape by local chromatin compaction. Nature. 2021;589(7841):293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bell O, Tiwari VK, Thomä NH, et al. Determinants and dynamics of genome accessibility. Nat Rev Genet. 2011;12(8):554–564. [DOI] [PubMed] [Google Scholar]
- 54.Gottschling DE, Sauria ME, Guitart X. Telomere-Proximal DNA in Saccharomyces-Cerevisiae is refractory to methyltransferase activity invivo. Proc Natl Acad Sci USA. 1992;89(9):4062–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fatemi M, Pao MM, Jeong S, et al. Footprinting of mammalian promoters: use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res. 2005;33(20):e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh J, Klar AJ. Active genes in budding yeast display enhanced in vivo accessibility to foreign DNA methylases: a novel in vivo probe for chromatin structure of yeast. Genes Dev. 1992;6(2):186–196. [DOI] [PubMed] [Google Scholar]
- 57.Pott S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. Elife. 2017;6:e23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brothers M, Rine J. Distinguishing between recruitment and spread of silent chromatin structures in Saccharomyces cerevisiae. Elife. 2022. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gouil Q, Keniry A. Latest techniques to study DNA methylation. Essays Biochem. 2019;63(6):639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gershman A, Sauria ME, Guitart X, et al. Epigenetic patterns in a complete human genome. Science. 2022;376(6588):eabj5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sonmezer C, Kleinendorst R, Imanci D, et al. Molecular co-occupancy identifies transcription factor binding cooperativity in vivo. Mol Cell. 2021;81(2):255–267 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rao S, Ahmad K, Ramachandran S. Cooperative binding between distant transcription factors is a hallmark of active enhancers. Mol Cell. 2021;81(8):1651–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baldi S, Korber P, Becker PB. Beads on a string-nucleosome array arrangements and folding of the chromatin fiber. Nat Struct Mol Biol. 2020;27(2):109–118. [DOI] [PubMed] [Google Scholar]
- 64.Isbel L, Grand RS, Schubeler D. Generating specificity in genome regulation through transcription factor sensitivity to chromatin. Nat Rev Genet. 2022. doi: 10.1038/s41576-022-00512-6. [DOI] [PubMed] [Google Scholar]
- 65.Boettiger AN, Bintu B, Moffitt JR, et al. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature. 2016;529(7586):418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Su JH, Zheng P, Kinrot SS, et al. Genome-Scale imaging of the 3D organization and transcriptional activity of chromatin. Cell. 2020;182(6):1641–1659 e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boettiger A, Murphy S. Advances in Chromatin Imaging at Kilobase-Scale Resolution. Trends Genet. 2020;36(4):273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang YT, Ye X, Dai R, et al. Phase separation of Epstein-Barr virus EBNA2 protein reorganizes chromatin topology for epigenetic regulation. Commun Biol. 2021;4(1):967. doi: 10.1038/s42003-021-02501-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oksuz BA, Yang L, Abraham S, et al. Systematic evaluation of chromosome conformation capture assays. Nat Methods. 2021;18(9):1046–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hsieh THS, Cattoglio C, Slobodyanyuk E, et al. Resolving the 3D landscape of transcription-linked mammalian chromatin folding. Mol Cell. 2020;78(3):539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Teves SS, An L, Hansen AS, et al. A dynamic mode of mitotic bookmarking by transcription factors. Elife. 20165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim DS, Risca V.I, Reynolds D. L, et al. The dynamic, combinatorial cis-regulatory lexicon of epidermal differentiation. Nat Genet. 2021;53(11):1564–1576. doi: 10.1038/s41588-021-00947-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hogg SJ, Motorna O, Cluse L. A, et al. Targeting histone acetylation dynamics and oncogenic transcription by catalytic P300/CBP inhibition. Mol Cell. 2021;81(10):2183–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lieberman-Aiden E, Van Berkum N. L, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lakadamyali M, Cosma MP. Visualizing the genome in high resolution challenges our textbook understanding. Nat Methods. 2020;17(4):371–379. [DOI] [PubMed] [Google Scholar]
- 76.Beel AJ, Azubel M, Matteï P. J, et al. Structure of mitotic chromosomes. Mol Cell. 2021;81(21):4369–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ou HD, Phan S, Deerinck T. J, et al. ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science. 2017;357:6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Swygert SG, Lin D, Portillo-Ledesma S, et al., Local chromatin fiber folding represses transcription and loop extrusion in quiescent cells. Elife. 2021;10:e72062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lerner J, Gomez-Garcia P. A, McCarthy R. L, et al. Two-Parameter mobility assessments discriminate diverse regulatory factor behaviors in chromatin. Mol Cell. 2020;79(4):677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ide S, Tamura S, Maeshima K. Chromatin behavior in living cells: lessons from single-nucleosome imaging and tracking. Bioessays. 2022;44(7):e2200043. [DOI] [PubMed] [Google Scholar]
- 81.Iida S, Shinkai S, Itoh Y, et al. Single-nucleosome imaging reveals steady-state motion of interphase chromatin in living human cells. Sci Adv. 2022;8(22):eabn5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hihara S, Pack C. G, Kaizu K, et al. Local nucleosome dynamics facilitate chromatin accessibility in living mammalian cells. Cell Rep. 2012;2(6):1645–1656. [DOI] [PubMed] [Google Scholar]
- 83.Swygert SG, Kim S, Wu X, et al. Condensin-Dependent chromatin compaction represses transcription globally during quiescence. Mol Cell. 2019;73(3):533–546 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Becker JS, Kim S, Wu X, et al. Genomic and proteomic resolution of heterochromatin and its restriction of alternate fate genes. Mol Cell. 2017;68(6):1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McCarthy RL, Kaeding K. E, Keller S. H, et al. Diverse heterochromatin-associated proteins repress distinct classes of genes and repetitive elements. Nat Cell Biol. 2021;23(8):905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moyle-Heyrman G, Zaichuk T, Xi L, et al. Chemical map of Schizosaccharomyces pombe reveals species-specific features in nucleosome positioning. Proc Natl Acad Sci USA. 2013;110(50):20158–20163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Krietenstein N, Wal M, Watanabe S, et al. Genomic nucleosome organization reconstituted with pure proteins. Cell. 2016;167(3):709–721 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Riposo J, Mozziconacci J. Nucleosome positioning and nucleosome stacking: two faces of the same coin. Mol Biosyst. 2012;8(4):1172–1178. [DOI] [PubMed] [Google Scholar]
- 89.Gomez-Garcia PA, Portillo-Ledesma S., Neguembor M. V, et al. Mesoscale modeling and single-nucleosome tracking reveal remodeling of clutch folding and dynamics in stem cell differentiation. Cell Rep. 2021;34(2):108614. doi: 10.1016/j.celrep.2020.108614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Muller O, Kepper N, Schöpflin R, et al. Changing chromatin fiber conformation by nucleosome repositioning. Biophys J. 2014;107(9):2141–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Holla S, Dhakshnamoorthy J, Folco H. D, et al. Positioning heterochromatin at the nuclear periphery suppresses histone turnover to promote epigenetic inheritance. Cell. 2020;180(1):150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Henikoff S. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat Rev Genet. 2008;9(1):15–26. [DOI] [PubMed] [Google Scholar]
- 93.Navarro C, Lyu J, Katsori A. M, et al. An embryonic stem cell-specific heterochromatin state promotes core histone exchange in the absence of DNA accessibility. Nat Commun. 2020;11(1):5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mizuguchi G, Shen X, Landry J, et al. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science. 2004;303(5656):343–348. [DOI] [PubMed] [Google Scholar]
- 95.Redon C, Pilch D, Rogakou E, et al. Histone H2A variants H2AX and H2AZ. Curr Opin Genet Dev. 2002;12(2):162–169. [DOI] [PubMed] [Google Scholar]
- 96.Cakiroglu A, Clapier C. R, Ehrensberger A. H, et al. Genome-wide reconstitution of chromatin transactions reveals that RSC preferentially disrupts H2AZ-containing nucleosomes. Genome Res. 2019;29(6):988–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hsu CJ, Meers O, Buschbeck M, et al. The role of MacroH2A histone variants in cancer. Cancers (Basel). 2021;13(12):3003. doi: 10.3390/cancers13123003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Buschbeck M, Hake SB. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat Rev Mol Cell Biol. 2017;18(5):299–314. [DOI] [PubMed] [Google Scholar]
- 99.Fyodorov DV, Zhou B. R, Skoultchi A. I, et al. Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol. 2018;19(3):192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Catez F, Ueda T, Bustin M. Determinants of histone H1 mobility and chromatin binding in living cells. Nat Struct Mol Biol. 2006;13(4):305–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Izzo A, Kamieniarz K, Schneider R. The histone H1 family: specific members, specific functions? Biol Chem. 2008;389(4):333–343. [DOI] [PubMed] [Google Scholar]
- 102.Shimada M, Chen W-Y, Nakadai T, et al. Gene-Specific H1 eviction through a transcriptional activator -> p300 -> NAP1 -> H1 pathway. Mol Cell. 2019;74(2):268–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hergeth SP, Schneider R. The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 2015;16(11):1439–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yusufova N, Kloetgen A, Teater M, et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature. 2021;589(7841):299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fan Y, Nikitina T, Morin-Kensicki EM, et al. H1 linker histones are essential for mouse development and affect nucleosome spacing in vivo. Mol Cell Biol. 2003;23(13):4559–4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fan Y, Nikitina T, Zhao J, et al. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell. 2005;123(7):1199–1212. [DOI] [PubMed] [Google Scholar]
- 107.Ricci MA, Manzo C, García-Parajo MF, et al. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell. 2015;160(6):1145–1158. [DOI] [PubMed] [Google Scholar]
- 108.Healton SE, Pinto H. D, Mishra L. N, et al. H1 linker histones silence repetitive elements by promoting both histone H3K9 methylation and chromatin compaction. Proc Natl Acad Sci USA. 2020;117(25):14251–14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shvedunova M, Akhtar A. Modulation of cellular processes by histone and non-histone protein acetylation. Nature reviews. Molecular cell biology. 2022;23(5):329–349. [DOI] [PubMed] [Google Scholar]
- 110.Gregoretti IV, Lee Y-M, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338(1):17–31. [DOI] [PubMed] [Google Scholar]
- 111.Lee DY, Hayes JJ, Pruss D, et al. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72(1):73–84. [DOI] [PubMed] [Google Scholar]
- 112.Robinson PJJ, An W, Routh A, et al. 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J Mol Biol. 2008;381(4):816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shogren-Knaak M, Ishii H, Sun J-M, et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311(5762):844–847. [DOI] [PubMed] [Google Scholar]
- 114.Liu Y, Lu C, Yang Y, et al. Influence of histone tails and H4 tail acetylations on nucleosome–nucleosome interactions. J Mol Biol. 2011;414(5):749–764 [DOI] [PubMed] [Google Scholar]
- 115.Zhang RT, Erler J, Langowski J. Histone acetylation regulates chromatin accessibility: role of H4K16 in inter-nucleosome interaction. Biophys J. 2017;112(3):450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Arya G, Schlick T. Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model. Proc Natl Acad Sci USA. 2006;103(44):16236–16241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Collepardo-Guevara R, Portella G, Vendruscolo M, et al. Chromatin unfolding by epigenetic modifications explained by dramatic impairment of internucleosome interactions: a multiscale computational study. J Am Chem Soc. 2015;137(32):10205–10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brownell JE, Zhou J, Ranalli T, et al. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84(6):843–851. [DOI] [PubMed] [Google Scholar]
- 119.Aygun O, Mehta S, Grewal SIS. HDAC-mediated suppression of histone turnover promotes epigenetic stability of heterochromatin. Nat Struct Mol Biol. 2013;20(5):547–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hassan AH, Neely KE, Workman JL. Histone acetyltransferase complexes stabilize SWI/SNF binding to promoter nucleosomes. Cell. 2001;104(6):817–827. [DOI] [PubMed] [Google Scholar]
- 121.Ortega E, Rengachari S, Ibrahim Z, et al. Transcription factor dimerization activates the p300 acetyltransferase. Nature. 2018;562(7728):538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Meshorer E, Misteli T. Chromatin in pluripotent embryonic stem cells and differentiation. Nat Rev Mol Cell Biol. 2006;7(7):540–546. [DOI] [PubMed] [Google Scholar]
- 123.Kieffer-Kwon KR, Nimura K, Rao SSP, et al. Myc Regulates Chromatin Decompaction and Nuclear Architecture during B Cell Activation. Mol Cell. 2017;67(4):566–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schneider MWG, Gibson B. A, Otsuka S, et al. A mitotic chromatin phase transition prevents perforation by microtubules. Nature. 2022;609:7925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Collombet S, Rall I, Dugast-Darzacq C, et al. RNA polymerase II depletion from the inactive X chromosome territory is not mediated by physical compartmentalization. bioRxiv. 2021. [DOI] [PMC free article] [PubMed]
- 126.Dossin F, Heard E. The molecular and nuclear dynamics of X-Chromosome inactivation. Cold Spring Harb Perspect Biol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Loda A, Collombet S, Heard E. Gene regulation in time and space during X-chromosome inactivation. Nat Rev Mol Cell Biol. 2022;23(4):231–249. [DOI] [PubMed] [Google Scholar]
- 128.Ugarte F, Sousae R, Cinquin B, et al. Progressive chromatin condensation and H3K9 methylation regulate the differentiation of embryonic and hematopoietic stem cells. Stem Cell Reports. 2015;5(5):728–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nicetto D, Zaret KS. Role of H3K9me3 heterochromatin in cell identity establishment and maintenance. Curr Opin Genet Dev. 2019;55:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Methot SP, Padeken J, Brancati G, et al. H3K9me selectively blocks transcription factor activity and ensures differentiated tissue integrity. Nat Cell Biol. 2021;23(11):1163–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zeller P, Padeken J, van Schendel R, et al. Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat Genet. 2016;48(11):1385–1395. [DOI] [PubMed] [Google Scholar]
- 132.Shen JZ, Qiu Z, Wu Q, et al. FBXO44 promotes DNA replication-coupled repetitive element silencing in cancer cells. Cell. 2021;184(2):352–369 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Canzio D, Liao M, Naber N, et al. A conformational switch in HP1 releases auto-inhibition to drive heterochromatin assembly. Nature. 2013;496(7445):377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Machida S, Takizawa Y, Ishimaru M, et al. Structural basis of heterochromatin formation by Human HP1. Mol Cell. 2018;69(3):385–+. [DOI] [PubMed] [Google Scholar]
- 135.Maeda R, Tachibana M. HP1 maintains protein stability of H3K9 methyltransferases and demethylases. Embo Rep. 2022;23:e53581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Al-Sady B, Madhani HD, Narlikar GJ. Division of labor between the chromodomains of HP1 and Suv39 methylase enables coordination of heterochromatin spread. Mol Cell. 2013;51(1):80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Strom AR, Biggs R. J, Banigan E. J, et al. HP1 alpha is a chromatin crosslinker that controls nuclear and mitotic chromosome mechanics. Elife, 2021. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yang SM, Kim B. J, Norwood Toro L, et al. H1 linker histone promotes epigenetic silencing by regulating both DNA methylation and histone H3 methylation. Proc Natl Acad Sci USA, 2013;110(5):1708–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lu XW, Wontakal SN, Emelyanov AV, et al. Linker histone H1 is essential for Drosophila development, the establishment of pericentric heterochromatin, and a normal polytene chromosome structure. Genes Dev. 2009;23(4):452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lu X, Wontakal SN, Kavi H, et al. Drosophila H1 regulates the genetic activity of heterochromatin by recruitment of Su(var)3-9. Science. 2013;340(6128):78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Li GH, Reinberg D. Chromatin higher-order structures and gene regulation. Curr Opin Genet Dev. 2011;21(2):175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Simon JA, Kingston RE. Occupying chromatin: polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol Cell. 2013;49(5):808–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Boyle S, Flyamer IM, Williamson I, et al. A central role for canonical PRC1 in shaping the 3D nuclear landscape. Genes Dev. 2020;34(13–14):931–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kundu S, Ji F, Sunwoo H, et al. Polycomb repressive complex 1 generates discrete compacted domains that change during differentiation. Mol Cell. 2017;65(3):432–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vieux-Rochas M, Fabre P. J, Leleu M, et al. Clustering of mammalian Hox genes with other H3K27me3 targets within an active nuclear domain. Proc Natl Acad Sci USA. 2015;112(15):4672–4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Williamson I, Eskeland R, Lettice LA, et al. Anterior-posterior differences in HoxD chromatin topology in limb development. Development. 2012;139(17):3157–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Francis NJ, Kingston RE, Woodcock CL. Chromatin compaction by a polycomb group protein complex. Science. 2004;306(5701):1574–1577. [DOI] [PubMed] [Google Scholar]
- 148.Grau DJ, Chapman BA, Garlick JD, et al. Compaction of chromatin by diverse polycomb group proteins requires localized regions of high charge. Genes Dev. 2011;25(20):2210–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kim J, Kingston RE. The CBX family of proteins in transcriptional repression and memory. J Biosci. 2020;45(1). [PubMed] [Google Scholar]
- 150.Leicher R, Ge E. J, Lin X, et al. Single-molecule and in silico dissection of the interaction between Polycomb repressive complex 2 and chromatin. Proc Natl Acad Sci USA. 2020. 117(48): 30465–30475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zylicz JJ, Heard E. Molecular mechanisms of facultative heterochromatin formation: an X-Chromosome perspective. Annu Rev Biochem. 2020;89(1):255–282. [DOI] [PubMed] [Google Scholar]
- 152.Zylicz JJ, Bousard A, Žumer K, et al. The implication of early chromatin changes in X Chromosome inactivation. Cell. 2019;176(1–2):182–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bonora G, Disteche CM. Structural aspects of the inactive X chromosome. Philos Trans Royal Soc B. 2017;372(1733):1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Galupa R, Heard E. X-Chromosome inactivation: a crossroads between chromosome architecture and gene regulation. Annu Rev Genet. 2018;52(1):535–566. [DOI] [PubMed] [Google Scholar]
- 155.Illingworth RS. Chromatin folding and nuclear architecture: PRC1 function in 3D. Curr Opin Genet Dev. 2019;55:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Aranda S, Mas G, Di Croce L. Regulation of gene transcription by polycomb proteins. Sci Adv. 2015;1(11):e1500737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Grau D, Zhang Y, Lee C-H, et al. Structures of monomeric and dimeric PRC2:EZH1 reveal flexible modules involved in chromatin compaction. Nat Commun. 2021;12(1):714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sun HB, Shen J, Yokota H. Size-dependent positioning of human chromosomes in interphase nuclei. Biophys J. 2000;79(1):184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Shore D, Langowski J, Baldwin RL. DNA flexibility studied by covalent closure of short fragments into circles. Proc Natl Acad Sci USA, 1981. 78(8): 4833–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Beltran B, Kannan D, MacPherson Q, et al. Geometrical heterogeneity dominates thermal fluctuations in facilitating chromatin contacts. Phys Rev Lett. 2019;123(20):208103. [DOI] [PubMed] [Google Scholar]
- 161.Arbona J-M, Herbert S, Fabre E, et al. Inferring the physical properties of yeast chromatin through Bayesian analysis of whole nucleus simulations. Genome Biol. 2017;18(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Flemming W. Zellsubstanz, Kern und Zelltheilung. Leipzig, Germany: Vogel; 1882. [Google Scholar]
- 163.Berger F. Emil Heitz, a true epigenetics pioneer. Nat Rev Mol Cell Biol. 2019;20(10):572. [DOI] [PubMed] [Google Scholar]
- 164.Heitz E. Das heterochromatin der moose. Stuttgart, Germany: Bornträger; 1928. [Google Scholar]
- 165.Gibcus JH, Samejima K, Goloborodko A, et al. A pathway for mitotic chromosome formation. Sci. 2018;359, 6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shintomi K, Inoue F, Watanabe H, et al. Mitotic chromosome assembly despite nucleosome depletion in Xenopus egg extracts. Science. 2017;356(6344):1284–1287. [DOI] [PubMed] [Google Scholar]
- 167.Choppakatla P, Dekker B, Cutts E. E, et al. Linker histone H1.8 inhibits chromatin binding of condensins and DNA topoisomerase II to tune chromosome length and individualization. Elife. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ganji M, Shaltiel IA, Bisht S, et al. Real-time imaging of DNA loop extrusion by condensin. Science. 2018;360(6384):102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Finch JT, Klug A. Solenoidal model for superstructure in chromatin. Proc Natl Acad Sci USA. 1976;73(6):1897–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhu P, Li G. Structural insights of nucleosome and the 30-nm chromatin fiber. Curr Opin Struct Biol. 2016;36:106–115. [DOI] [PubMed] [Google Scholar]
- 171.Robinson PJJ, Rhodes D. Structure of the ‘30 nm’ chromatin fibre: a key role for the linker histone. Curr Opin Struct Biol. 2006;16(3):336–343. [DOI] [PubMed] [Google Scholar]
- 172.Luger K, Dechassa ML, Tremethick DJ. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Biol. 2012;13(7):436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Roberts K, Alberts B, Johnson A, et al. Molecular biology of the cell. Garland Sci. 2002. [Google Scholar]
- 174.Eltsov M, MacLellan K. M, Maeshima K, Maeshima K., Frangakis A. S., & Dubochet J. Analysis of cryo-electron microscopy images does not support the existence of 30-nm chromatin fibers in mitotic chromosomes in situ. Proc Natl Acad Sci USA. 2008. 105(50): 19732–19737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fussner E, Strauss M, Djuric U, et al. Open and closed domains in the mouse genome are configured as 10-nm chromatin fibres. EMBO Rep. 2012;13(11):992–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Scheffer MP, Eltsov M, Frangakis AS. Evidence for short-range helical order in the 30-nm chromatin fibers of erythrocyte nuclei. Proc Natl Acad Sci USA. 2011;108(41): 16992–16997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Song F, Chen P, Sun D, et al. Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science. 2014;344(6182):376–380. [DOI] [PubMed] [Google Scholar]
- 178.Nishino Y, Eltsov M, Joti Y, et al. Human mitotic chromosomes consist predominantly of irregularly folded nucleosome fibres without a 30-nm chromatin structure. EMBO J. 2012;31(7):1644–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Krietenstein N, Abraham S, Venev SV, et al. Ultrastructural details of mammalian chromosome architecture. Mol Cell. 2020;78(3):554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Koslover EF, Fuller CJ, Straight AF, et al. Local geometry and elasticity in compact chromatin structure. Biophys J. 2010;99(12):3941–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Portillo-Ledesma S, Tsao LH, Wagley M, et al. Nucleosome clutches are regulated by chromatin internal parameters. J Mol Biol. 2021;433(6):166701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhurkin VB, Norouzi D. Topological polymorphism of nucleosome fibers and folding of chromatin. Biophys J. 2021;120(4):577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Routh A, Sandin S, Rhodes D. Nucleosome repeat length and linker histone stoichiometry determine chromatin fiber structure. Proc Natl Acad Sci USA. 2008;105(26):8872–8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kenzaki H, Takada S. Linker DNA length is a key to tri-nucleosome folding. J Mol Biol. 2021;433(6):166792. [DOI] [PubMed] [Google Scholar]
- 185.Zhou BR, Jiang J, Ghirlando R, et al. Revisit of reconstituted 30-nm nucleosome arrays reveals an ensemble of dynamic structures. J Mol Biol. 2018;430(18 Pt B):3093–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Luque A, Ozer G, Schlick T. Correlation among DNA linker length, linker histone concentration, and histone tails in chromatin. Biophys J. 2016;110(11):2309–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Perisic O, Portillo-Ledesma S, Schlick T. Sensitive effect of linker histone binding mode and subtype on chromatin condensation. Nucleic Acids Res. 2019;47(10):4948–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chodaparambil JV, Barbera A. J, Lu X, et al. A charged and contoured surface on the nucleosome regulates chromatin compaction. Nat Struct Mol Biol. 2007;14(11):1105–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Fierz B, Poirier MG. Biophysics of chromatin dynamics. Annu Rev Biophys. 2019;48:321–345. [DOI] [PubMed] [Google Scholar]
- 190.Kilic S, Felekyan S, Doroshenko O, et al. Single-molecule FRET reveals multiscale chromatin dynamics modulated by HP1alpha. Nat Commun. 2018;9(1):235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Jensen KT, Fløe L, Petersen T. S, et al. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Lett. 2017;591(13):1892–1901. [DOI] [PubMed] [Google Scholar]
- 192.Verkuijl SA, Rots MG. The influence of eukaryotic chromatin state on CRISPR-Cas9 editing efficiencies. Curr Opin Biotechnol. 2019;55: 68–73. [DOI] [PubMed] [Google Scholar]
- 193.Kallimasioti-Pazi EM, Thelakkad Chathoth K, Taylor GC, et al. Heterochromatin delays CRISPR-Cas9 mutagenesis but does not influence the outcome of mutagenic DNA repair. PLoS Biol. 2018;16(12):e2005595. doi: 10.1371/journal.pbio.2005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Thurman RE, Rynes E, Humbert R, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Buenrostro JD, Wu B, Chang H. Y, et al. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 2015;109:21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kiani K, Sanford E. M, Goyal Y, et al. Changes in chromatin accessibility are not concordant with transcriptional changes for single-factor perturbations. Mol Syst Biol. 2022;18(9):e10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Dossin F, Pinheiro I, Żylicz J. J, et al. SPEN integrates transcriptional and epigenetic control of X-inactivation. Nature. 2020;578(7795):455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Mateescu B, England P, Halgand F, et al. Tethering of HP1 proteins to chromatin is relieved by phosphoacetylation of histone H3. EMBO Rep. 2004;5(5):490–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Coux RX, Owens NDL, Navarro P. Chromatin accessibility and transcription factor binding through the perspective of mitosis. Transcription. 2020;11(5):236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hsiung CCS, Morrissey C. S, Udugama M, et al. Genome accessibility is widely preserved and locally modulated during mitosis. Genome Res. 2015;25(2):213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Owens N, Papadopoulou T, Festuccia N, et al. CTCF confers local nucleosome resiliency after DNA replication and during mitosis. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Festuccia N, Owens N, Papadopoulou T, et al. Transcription factor activity and nucleosome organization in mitosis. Genome Res. 2019;29(2):250–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Oomen ME, Hansen A. S, Liu Y, et al. CTCF sites display cell cycle-dependent dynamics in factor binding and nucleosome positioning. Genome Res. 2019;29(2):236–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Akoulitchev S, Reinberg D. The molecular mechanism of mitotic inhibition of TFIIH is mediated by phosphorylation of CDK7. Genes Dev. 1998;12(22):3541–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Segil N, Guermah M, Hoffmann A, et al. Mitotic regulation of TFIID: inhibition of activator-dependent transcription and changes in subcellular localization. Genes Dev. 1996;10(19):2389–2400. [DOI] [PubMed] [Google Scholar]
- 206.Bancaud A, Huet S, Daigle N, et al. Molecular crowding affects diffusion and binding of nuclear proteins in heterochromatin and reveals the fractal organization of chromatin. EMBO J. 2009;28(24):3785–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Falahati H, Haji-Akbari A. Thermodynamically driven assemblies and liquid-liquid phase separations in biology. Soft Matter. 2019;15(6):1135–1154. [DOI] [PubMed] [Google Scholar]
- 208.Lee DSW, Strom AR, Brangwynne CP. The mechanobiology of nuclear phase separation. APL Bioeng. 2022;6(2):021503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mittag T, Pappu RV. A conceptual framework for understanding phase separation and addressing open questions and challenges. Mol Cell. 2022;82(12):2201–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Choi JM, Holehouse AS, Pappu RV. Physical principles underlying the complex biology of intracellular phase transitions. Annu Rev Biophys. 2020;49:107–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Zhu L, Brangwynne CP. Nuclear bodies: the emerging biophysics of nucleoplasmic phases. Curr Opin Cell Biol. 2015;34:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Larson AG, Narlikar GJ. The role of phase separation in heterochromatin formation, function, and regulation. Biochemistry. 2018;57(17):2540–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Larson AG, Elnatan D, Keenen M. M, et al. Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature. 2017;547(7662):236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Strom AR, Emelyanov A. V, Mir M., et al. Phase separation drives heterochromatin domain formation. Nature. 2017;547(7662):241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Gibson BA, Schneider M. W., Jensen L. E., Gamarra N., Henry L., & Rosen M. K. Organization of chromatin by intrinsic and regulated phase separation. Cell. 2019;179(2):470–484 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lafontaine DLJ, Riback J. A., Bascetin R., & Brangwynne C. P. The nucleolus as a multiphase liquid condensate. Nat Rev Mol Cell Biol. 2021;22(3):165–182. [DOI] [PubMed] [Google Scholar]
- 217.Zhang HY, Zhao R., Tones J., Liu M., Dilley R. L., Chenoweth D. M., & Lampson M. A. Nuclear body phase separation drives telomere clustering in ALT cancer cells. Mol Biol Cell. 2020;31(18):2048–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Rippe K. Liquid-Liquid phase separation in chromatin. Cold Spring Harb Perspect Biol. 2022;14(2):a040683. doi: 10.1101/cshperspect.a040683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Erdel F, Rippe K. Formation of chromatin subcompartments by phase separation. Biophys J. 2018;114(10):2262–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.A P, Weber SC. Evidence for and against liquid-liquid phase separation in the nucleus. Noncoding RNA. 2019;5(4):50. doi: 10.3390/ncrna5040050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Shin Y, Brangwynne CP. Liquid phase condensation in cell physiology and disease. Science. 2017;357:6357. [DOI] [PubMed] [Google Scholar]
- 222.Sabari BR, Dall’Agnese A, Young RA. Biomolecular condensates in the nucleus. Trends Biochem Sci. 2020;45(11):961–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sabari BR. Biomolecular condensates and gene activation in development and disease. Dev Cell. 2020;55(1):84–96. [DOI] [PubMed] [Google Scholar]
- 224.Feric M, Vaidya N., Harmon T. S., Mitrea D. M., Zhu L., Richardson T. M., & Brangwynne C. P. Coexisting liquid phases underlie nucleolar subcompartments. Cell. 2016;165(7):1686–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Cho WK, Spille J. H., Hecht M., Lee C., Li C., Grube V., & Cisse I. I. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science. 2018;361(6400):412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Maeshima K, Rogge R., Tamura S., Joti Y., Hikima T., Szerlong H., & Hansen J. C. Nucleosomal arrays self-assemble into supramolecular globular structures lacking 30-nm fibers. EMBO J. 2016;35(10):1115–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Zhang M, Díaz-Celis C., Onoa B., Cañari-Chumpitaz C., Requejo K. I., Liu J., & Bustamante C. Molecular organization of the early stages of nucleosome phase separation visualized by cryo-electron tomography. Mol Cell. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Strickfaden H, Tolsma T. O., Sharma A., Underhill D. A., Hansen J. C., & Hendzel M. J. Condensed chromatin behaves like a solid on the mesoscale in vitro and in living cells. Cell. 2020;183(7):1772–1784 e13. [DOI] [PubMed] [Google Scholar]
- 229.Weidemann T, Wachsmuth M., Knoch T. A., Müller G., Waldeck W., & Langowski J. Counting nucleosomes in living cells with a combination of fluorescence correlation spectroscopy and confocal imaging. J Mol Biol. 2003;334(2):229–240. [DOI] [PubMed] [Google Scholar]
- 230.Farr SE, Woods E. J., Joseph J. A., Garaizar A., & Collepardo-Guevara R. Nucleosome plasticity is a critical element of chromatin liquid-liquid phase separation and multivalent nucleosome interactions. Nat Commun. 2021;12(1):2883. doi: 10.1038/s41467-021-23090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Hansen JC, Maeshima K, Hendzel MJ. The solid and liquid states of chromatin. Epigenetics Chromatin. 2021;14(1):50. doi: 10.1186/s13072-021-00424-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Gibson BA, Blaukopf C., Lou T., Doolittle L. K., Finkelstein I. J., Narlikar G., & Rosen M. K. In diverse conditions intrinsic chromatin condensates have liquid-like material properties. bioRxiv. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Roque A, Iloro I., Ponte I., Arrondo J. L. R., & Suau P. DNA-induced secondary structure of the carboxyl-terminal domain of histone H1. J Biol Chem. 2005;280(37):32141–32147. [DOI] [PubMed] [Google Scholar]
- 234.Rao SSP, Huang S. C., St Hilaire B. G., Engreitz J. M., Perez E. M., Kieffer-Kwon K. R., & Aiden E. L. Cohesin loss eliminates all loop domains. Cell. 2017;171(2):305–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Mirny L, Dekker J. Mechanisms of chromosome folding and nuclear organization: their interplay and open questions. Cold Spring Harb Perspect Biol; 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Haarhuis JHI, van der Weide R. H., Blomen V. A., Flach K. D., Teunissen H., Willems L., & de Wit E. A Mediator-cohesin axis controls heterochromatin domain formation. Nat Commun. 2022;13(1):754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Wutz G, Várnai C., Nagasaka K., Cisneros D. A., Stocsits R. R., Tang W., & Peters J. M. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017;36(24):3573–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Falk M, Feodorova Y., Naumova N., Imakaev M., Lajoie B. R., Leonhardt H., & Mirny L. A. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature. 2019;572(7771):E22. [DOI] [PubMed] [Google Scholar]
- 239.Conte M, Irani E., Chiariello A. M., Abraham A., Bianco S., Esposito A., & Nicodemi M. Loop-extrusion and polymer phase-separation can co-exist at the single-molecule level to shape chromatin folding. Nat Commun. 2022;13(1):4070. doi: 10.1038/s41467-022-31856-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Gu H, Harris H., Olshansky M., Eliaz Y., Krishna A., Kalluchi A., & Rowley M. J. Fine-mapping of nuclear compartments using ultra-deep Hi-C shows that active promoter and enhancer elements localize in the active A compartment even when adjacent sequences do not. bioRxiv. 2021.
- 241.Guo CH, Luo ZJ, Lin CQ. Phase separation, transcriptional elongation control, and human diseases. J Mol Cell Biol. 2021;13(4):314–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Gallego LD, Schneider M., Mittal C., Romanauska A., Gudino Carrillo R. M., Schubert T., & Köhler A. Phase separation directs ubiquitination of gene-body nucleosomes. Nature. 2020;579(7800):592–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Han X, Yu D., Gu R., Jia Y., Wang Q., Jaganathan A., & Zeng L. Roles of the BRD4 short isoform in phase separation and active gene transcription. Nat Struct Mol Biol. 2020;27(4):333–341. [DOI] [PubMed] [Google Scholar]
- 244.Sabari BR, Dall’Agnese A., Boija A., Klein I. A., Coffey E. L., Shrinivas K., & Young R. A. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018;361:6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Chong SS, Dugast-Darzacq C., Liu Z., Dong, P., Dailey G. M., Cattoglio C., & Tjian R. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 2018;361:6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Keenen MM, Brown D., Brennan L. D., Renger R., Khoo, H., Carlson C. R., & Redding S. HP1 proteins compact DNA into mechanically and positionally stable phase separated domains. Elife, 2021;10: e64563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.MacPherson Q, Beltran B, Spakowitz AJ. Bottom-up modeling of chromatin segregation due to epigenetic modifications. Proc Natl Acad Sci U S A. 2018 Dec 11;115(50):12739–12744. doi: 10.1073/pnas.1812268115. Epub 2018 Nov 26. PMID: 30478042; PMCID: PMC6294944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Sanulli S, et al. HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature. 2019;575(7782):390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Sandholtz SH, MacPherson Q, Spakowitz AJ. Physical modeling of the heritability and maintenance of epigenetic modifications. Proc Natl Acad Sci U S A. 2020 Aug 25;117(34):20423–20429. doi: 10.1073/pnas.1920499117. Epub 2020 Aug 10. PMID: 32778583; PMCID: PMC7456148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Guo YR, Zhao S, Wang GG. Polycomb gene silencing mechanisms: PRC2 chromatin targeting, H3K27me3 ‘readout’, and phase separation-based compaction. Trends Genet. 2021;37(6):547–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Eeftens JM, Kapoor M., Michieletto D., & Brangwynne C. P. Polycomb condensates can promote epigenetic marks but are not required for sustained chromatin compaction. Nat Commun. 2021;12(1):5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Yuan W, Wu T., Fu H., Dai C., Wu H., Liu N., & Zhu B. Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science. 2012;337(6097):971–975. [DOI] [PubMed] [Google Scholar]
- 253.Polymenidou M. The RNA face of phase separation. Science. 2018;360(6391):859–860. [DOI] [PubMed] [Google Scholar]
- 254.Guo Q, Shi X, Wang X. RNA and liquid-liquid phase separation. Noncoding RNA Res. 2021;6(2):92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Novo CL, et al. Satellite repeat transcripts modulate heterochromatin condensates and safeguard chromosome stability in mouse embryonic stem cells. Nat Commun. 2022;13(1):3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Hilbert L, Armaos A., Neumayer C., et al. Transcription organizes euchromatin via microphase separation. Nat Commun. 2021;12(1):1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Huo XR, Ji L., Zhang Y., et al. The nuclear matrix protein SAFB cooperates with major satellite RNAs to stabilize heterochromatin architecture partially through phase separation. Mol Cell. 2020;77(2):368–+. [DOI] [PubMed] [Google Scholar]
- 258.Cerase A, Armaos A., Neumayer C., et al. Phase separation drives X-chromosome inactivation: a hypothesis. Nat Struct Mol Biol. 2019;26(5):331–334. [DOI] [PubMed] [Google Scholar]
- 259.Spracklin G, Abdennur N., Imakaev M., et al. Heterochromatin diversity modulates genome compartmentalization and loop extrusion barriers. 2021. [DOI] [PMC free article] [PubMed]
- 260.Tedesco M, Udugama M. I., Pawlicki J. M., et al. Chromatin velocity reveals epigenetic dynamics by single-cell profiling of heterochromatin and euchromatin. Nat Biotechnol. 2022;40(2):235–244. [DOI] [PubMed] [Google Scholar]
- 261.McSwiggen DT, Hansen A. S., Teves S. S., et al. Evidence for DNA-mediated nuclear compartmentalization distinct from phase separation. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kadauke S, Udugama M. I., Pawlicki J. M., et al. Tissue-Specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell. 2012;150(4):725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Gonzalez I, Molliexa A, Navarro P. Mitotic memories of gene activity. Curr Opin Cell Biol. 2021;69:41–47. [DOI] [PubMed] [Google Scholar]
- 264.Ryu JK, Beltran B, Spakowitz AJ, et al. Bridging-induced phase separation induced by cohesin SMC protein complexes. Sci Adv. 2021;7(7):eabe5905. doi: 10.1126/sciadv.abe5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.MacPherson Q, Beltran B, Spakowitz AJ. Chromatin compaction leads to a preference for peripheral heterochromatin. Biophys J. 2020;118(6):1479–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Chen CK, Blanco M., Jackson C., et al. Xist recruits the X chromosome to the nuclear lamina to enable chromosome-wide silencing. Science. 2016;354(6311):468–472. [DOI] [PubMed] [Google Scholar]
- 267.McHugh CA, Perlas E., Ruiz-Blanes N., et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature. 2015;521(7551):232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Solovei I, Wang A. S., Thanisch K., et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell. 2013;152(3):584–598. [DOI] [PubMed] [Google Scholar]
- 269.Young AN, Perlas E., Ruiz-Blanes N., et al. Deletion of LBR N-terminal domains recapitulates Pelger-Huet anomaly phenotypes in mouse without disrupting X chromosome inactivation. Commun Biol. 2021;4(1):478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Dunlevy KL, Medvedeva V., Wilson J. E., et al. The PRR14 heterochromatin tether encodes modular domains that mediate and regulate nuclear lamina targeting. J Cell Sci. 2020;133(10):jcs240416. doi: 10.1242/jcs.240416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Ye Q, Worman HJ. Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. J Biol Chem. 1996;271(25):14653–14656. [DOI] [PubMed] [Google Scholar]
- 272.Manzo SG, Dauban L, van Steensel B. Lamina-associated domains: tethers and looseners. Curr Opin Cell Biol. 2022;74:80–87. [DOI] [PubMed] [Google Scholar]
- 273.Nesterova TB, Wei G., Coker H., et al. Systematic allelic analysis defines the interplay of key pathways in X chromosome inactivation. Nat Commun. 2019;10(1):3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Hoskins VE, Smith K, Reddy KL. The shifting shape of genomes: dynamics of heterochromatin interactions at the nuclear lamina. Curr Opin Genet Dev. 2021;67:163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Wong XR, Melendez-Perez AJ, Reddy KL. The nuclear lamina. Cold Spring Harb Perspect Biol. 2022;14(2):a040113. doi: 10.1101/cshperspect.a040113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Falk M, Feodorova Y., Naumova N., et al. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature. 2019;570(7761):395–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Perez BS, Wong K. M., Schwartz E. K., et al. Genome-wide profiles of UV lesion susceptibility, repair, and mutagenic potential in melanoma. Mutat Res. 2021;823:111758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Garcia-Nieto PE, Schwartz E. K., King D. A., et al. Carcinogen susceptibility is regulated by genome architecture and predicts cancer mutagenesis. EMBO J. 2017;36(19):2829–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Mao P, Brown A. J., Malc E. P., et al. Genome-wide maps of alkylation damage, repair, and mutagenesis in yeast reveal mechanisms of mutational heterogeneity. Genome Res. 2017;27(10):1674–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Mao P, Smerdon M. J., Roberts S. A., et al. Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc Natl Acad Sci USA. 2016;113(32):9057–9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Grossman SR, Zhang X., Wang L., et al. Systematic dissection of genomic features determining transcription factor binding and enhancer function. Proc Natl Acad Sci USA. 2017;114(7):E1291–E1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Siersbaek R, Nielsen R., John S., et al. Extensive chromatin remodelling and establishment of transcription factor ‘hotspots’ during early adipogenesis. EMBO J. 2011;30(8):1459–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.John S, Sabo P. J., Thurman R. E., et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43(3):264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Biddie SC, John S., Sabo P. J., et al. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell. 2011;43(1):145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Mazzoni EO, Mahony S., Peljto M., et al. Saltatory remodeling of Hox chromatin in response to rostrocaudal patterning signals. Nat Neurosci. 2013;16(9):1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Patel T, Hobert O. Coordinated control of terminal differentiation and restriction of cellular plasticity. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Lupien M, Eeckhoute J., Meyer C. A., et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132(6):958–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Sherwood RI, Hashimoto T., O’donnell C. W., et al. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat Biotechnol. 2014;32(2):171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Miao L, Tang Y., Bonneau A. R., et al. The landscape of pioneer factor activity reveals the mechanisms of chromatin reprogramming and genome activation. Mol Cell. 2022;82(5):986–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Meers MP, Janssens DH, Henikoff S. Pioneer factor-nucleosome binding events during differentiation are motif encoded. Mol Cell. 2019;75(3):562–575 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Ballas N, et al. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31(3):353–365. [DOI] [PubMed] [Google Scholar]
- 292.Michael AK, Thoma NH. Reading the chromatinized genome. Cell. 2021;184(14):3599–3611. [DOI] [PubMed] [Google Scholar]
- 293.Zhu FJ, Farnung L., Kaasinen E., et al. The interaction landscape between transcription factors and the nucleosome. Nature. 2018;562(7725):76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Chaumeil J, Le Baccon P., Wutz A., et al. A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev. 2006;20(16):2223–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Fang H, Disteche CM, Berletch JB. X inactivation and escape: epigenetic and structural features. Front Cell Dev Biol. 2019;7:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Xu J, Ma H., Jin J., et al. Super-Resolution imaging of higher-order chromatin structures at different epigenomic states in single mammalian cells. Cell Rep. 2018;24(4):873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Kim UJ, Han M., Kayne P., et al. Effects of histone H4 depletion on the cell cycle and transcription of Saccharomyces cerevisiae. EMBO J. 1988;7(7):2211–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Wyrick JJ, Holstege F. C., Jennings E. G., et al. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature. 1999;402(6760):418–421. [DOI] [PubMed] [Google Scholar]
- 299.Celona B, Weiner A., Di Felice F., et al. Substantial histone reduction modulates genomewide nucleosomal occupancy and global transcriptional output. PLoS Biol. 2011;9(6):e1001086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Gossett AJ, Lieb JD. In vivo effects of histone H3 depletion on nucleosome occupancy and position in Saccharomyces cerevisiae. PLoS Genet. 2012;8(6):e1002771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Prado F, Jimeno-Gonzalez S, Reyes JC. Histone availability as a strategy to control gene expression. RNA Biol. 2017;14(3):281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Jimeno-Gonzalez S, Payán-Bravo L., Muñoz-Cabello A. M., et al. Defective histone supply causes changes in RNA polymerase II elongation rate and cotranscriptional pre-mRNA splicing. Proc Natil Acad Sci USA, 2015. 112(48): 14840–14845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Fraser LCR, Dikdan R. J., Dey S., et al. Reduction in gene expression noise by targeted increase in accessibility at gene loci. Proc Natl Acad Sci USA, 2021;118(42). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Xu J, Ma H., Ma H., et al. Super-resolution imaging reveals the evolution of higher-order chromatin folding in early carcinogenesis. Nat Commun. 2020;11(1):1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Gomez NC, Hepperla A. J., Dumitru R., et al. Widespread chromatin accessibility at repetitive elements links stem cells with human cancer. Cell Rep. 2016;17(6):1607–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Sanghi A, Gruber J. J., Metwally A., et al. Chromatin accessibility associates with protein-RNA correlation in human cancer. Nat Commun. 2021;12(1):5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Lee MT, Bonneau AR, Giraldez AJ. Zygotic genome activation during the Maternal-to-Zygotic transition. Annu Rev Cell Dev Biol. 2014;30:581–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Ing-Simmons E, Rigau M, Vaquerizas JM. Emerging mechanisms and dynamics of three-dimensional genome organisation at zygotic genome activation. Curr Opin Cell Biol. 2022;74:37–46. [DOI] [PubMed] [Google Scholar]
- 309.Hug CB, Grimaldi A. G., Kruse K., et al. Chromatin architecture emerges during zygotic genome activation independent of transcription. Cell. 2017;169(2):216–228 e19. [DOI] [PubMed] [Google Scholar]
- 310.Jiang Y, Lun K., Li B, et al. Genome-wide analyses of chromatin interactions after the loss of Pol I, Pol II, and Pol III. Genome Biol. 2020;21(1):158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Wu J, Xu J., Liu B., et al. Chromatin analysis in human early development reveals epigenetic transition during ZGA. Nature. 2018;557(7704):256–260. [DOI] [PubMed] [Google Scholar]
- 312.Wu J, Huang B. O., Chen H. E., et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature. 2016;534(7609):652–657. [DOI] [PubMed] [Google Scholar]
- 313.Stewart-Morgan KR, Reveron-Gomez N, Groth A. Transcription restart establishes chromatin accessibility after DNA replication. Mol Cell. 2019;75(2):408–414. [DOI] [PubMed] [Google Scholar]
- 314.Wittner M, Hamper S., Stöck U., et al. Establishment and maintenance of alternative chromatin states at a multicopy gene locus. Cell. 2011;145(4):543–554. [DOI] [PubMed] [Google Scholar]
- 315.Neguembor MV, Martin L., Castells-García Á., et al. Transcription-mediated supercoiling regulates genome folding and loop formation. Mol Cell. 2021;81(15):3065–3081 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Teves SS, Henikoff S. Transcription-generated torsional stress destabilizes nucleosomes. Nat Struct Mol Biol. 2014;21(1):88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Corless S, Gilbert N. Effects of DNA supercoiling on chromatin architecture. Biophys Rev. 2016;8(Suppl 1):51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Naughton C, Avlonitis N., Corless S., et al. Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures. Nat Struct Mol Biol. 2013;20(3):387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Nesta AV, Tafur D, Beck CR. Hotspots of human mutation. Trends Genet. 2021;37(8):717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Deng SK, Yin Y., Petes T. D., et al. Mre11-Sae2 and RPA collaborate to prevent palindromic gene amplification. Mol Cell. 2015;60(3):500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Paigen K, Petkov PM. PRDM9 and its role in genetic recombination. Trends Genet. 2018;34(4):291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Cooper TJ, Garcia V, Neale MJ. Meiotic DSB patterning: a multifaceted process. Cell Cycle. 2016;15(1):13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Tock AJ, Henderson IR. Hotspots for initiation of meiotic recombination. Front Genet . 2018;9:521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Daniel JA, Nussenzweig A. The AID-induced DNA damage response in chromatin. Mol Cell. 2013;50(3):309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Meas R, Wyrick JJ, Smerdon MJ. Nucleosomes regulate base excision repair in chromatin. Mutat Res Rev Mutat Res. 2019;780:29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Hashimoto T, Kunieda T. DNA protection protein, a novel mechanism of radiation tolerance: lessons from tardigrades. Life (Basel). 2017;7(2):26. doi: 10.3390/life7020026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Hashimoto T, Horikawa D. D., Saito Y., et al. Extremotolerant tardigrade genome and improved radiotolerance of human cultured cells by tardigrade-unique protein. Nat Commun. 2016;7:12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Chavez C, Cruz-Becerra G., Fei J., et al. The tardigrade damage suppressor protein binds to nucleosomes and protects DNA from hydroxyl radicals. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Smerdon MJ. DNA repair and the role of chromatin structure. Curr Opin Cell Biol. 1991;3(3):422–428. [DOI] [PubMed] [Google Scholar]
- 330.Soria G, Polo SE, Almouzni G. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell. 2012;46(6):722–734. [DOI] [PubMed] [Google Scholar]
- 331.Polo SE, Almouzni G. Chromatin dynamics after DNA damage: The legacy of the access-repair-restore model. DNA Repair (Amst). 2015;36:114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Mohan C, Das C, Tyler J. Histone and chromatin dynamics facilitating DNA repair. DNA Repair (Amst). 2021;107:103183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Chakraborty U, Shen ZJ, Tyler J. Chaperoning histones at the DNA repair dance. DNA Repair. 2021;108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Hauer MH, Gasser SM. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017;31(22):2204–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Gursoy-Yuzugullu O, House N, Price BD. Patching broken DNA: nucleosome dynamics and the repair of DNA breaks. J Mol Biol. 2016;428(9):1846–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Andres SN, Schellenberg M. J., Wallace B. D., et al. Recognition and repair of chemically heterogeneous structures at DNA ends. Environ Mol Mutagen. 2015;56(1):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Onn L, Portillo M., Ilic S., et al. SIRT6 is a DNA double-strand break sensor. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol. 2017;18(10):610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Matsumoto S, Cavadini S., Bunker R. D., et al. DNA damage detection in nucleosomes involves DNA register shifting. Nature. 2019;571(7763):79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Cannan WJ, Rashid I., Tomkinson A. E. The human ligase IIIalpha-XRCC1 protein complex performs DNA nick repair after transient unwrapping of nucleosomal DNA. J Biol Chem. 2017;292(13):5227–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Ljungman M, Lane DP. Transcription - guarding the genome by sensing DNA damage. Nat Rev Cancer. 2004;4(9):727–737. [DOI] [PubMed] [Google Scholar]
- 342.Wang W, Klein K. N., Proesmans K., et al. Genome-wide mapping of human DNA replication by optical replication mapping supports a stochastic model of eukaryotic replication. Mol Cell. 2021;81(14):2975–2988 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Struhl K, Segal E. Determinants of nucleosome positioning. Nat Struct Mol Biol. 2013;20(3):267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Eaton ML, Galani K., Kang S., et al. Conserved nucleosome positioning defines replication origins. Genes Dev. 2010;24(8):748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Muller M, Lucchini R, Sogo JM. Replication of yeast rDNA initiates downstream of transcriptionally active genes. Mol Cell. 2000;5(5):767–777. [DOI] [PubMed] [Google Scholar]
- 346.Foss EJ, Gatbonton-Schwager T., Thiesen A. H., et al. Sir2 suppresses transcription-mediated displacement of Mcm2-7 replicative helicases at the ribosomal DNA repeats. PLoS Genet. 2019;15(5):e1008138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Donaldson AD. Shaping time: chromatin structure and the DNA replication programme. Trends Genet. 2005;21(8):444–449. [DOI] [PubMed] [Google Scholar]
- 348.Azmi IF, et al. Nucleosomes influence multiple steps during replication initiation. Elife. 2017;6:e22512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Li S, Wasserman M. R., Yurieva O., Bai L., O’Donnell M. E., & Liu S. Nucleosome-directed replication origin licensing independent of a consensus DNA sequence. Nat Commun. 2022;13(1):4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Barnes T, Korber P. The active mechanism of nucleosome depletion by poly(dA:dT) tracts in vivo. Int J Mol Sci. 2021;22(15):8233. doi: 10.3390/ijms22158233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Knott SR, Hollatz A. J., Cherney R. E., Seman M. R., & Fox C. A. Forkhead transcription factors establish origin timing and long-range clustering in S. cerevisiae. Cell. 2012;148(1–2):99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Hoggard T, Hollatz A. J., Cherney R. E., Seman M. R., & Fox C. A. The Fkh1 Forkhead associated domain promotes ORC binding to a subset of DNA replication origins in budding yeast. Nucleic Acids Res. 2021;49(18):10207–10220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.MacAlpine HK, Gordân R., Powell S. K., Hartemink A. J., & MacAlpine D. M. Drosophila ORC localizes to open chromatin and marks sites of cohesin complex loading. Genome Res. 2010;20(2):201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Canela A, Sridharan S., Sciascia N., Tubbs A., Meltzer P., Sleckman, B. P., & Nussenzweig A. DNA breaks and end resection measured genome-wide by end sequencing. Mol Cell. 2016;63(5):898–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Hansen RS, Thomas S., Sandstrom R., Canfield T. K., Thurman R. E., Weaver M., & Stamatoyannopoulos J. A. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci USA. 2010;107(1):139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Chen YH, Keegan S., Kahli M., Tonzi P., Fenyö D., Huang T. T., & Smith D. J. Transcription shapes DNA replication initiation and termination in human cells. Nat Struct Mol Biol. 2019;26(1):67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Pourkarimi E, Bellush JM, Whitehouse I. Spatiotemporal coupling and decoupling of gene transcription with DNA replication origins during embryogenesis in C. elegans. Elife. 2016;5:e21728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Dammann R, Lucchini R., Koller T., & Sogo J. M. Chromatin structures and transcription of rDNA in yeast Saccharomyces cerevisiae. Nucleic Acids Res. 1993;21(10):2331–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Ramachandran S, Henikoff S. Transcriptional regulators compete with nucleosomes post-replication. Cell. 2016;165(3):580–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Stewart-Morgan KR, Petryk N, Groth A. Chromatin replication and epigenetic cell memory. Nat Cell Biol. 2020;22(4):361–371. [DOI] [PubMed] [Google Scholar]
- 361.Horlbeck MA, Witkowsky L. B., Guglielmi B., Replogle J. M., Gilbert L. A., Villalta J. E., & Weissman J. S. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. Elife. 2016;5:12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Strohkendl I, Saifuddin FA, Gibson BA, Rosen MK, Russell R, Finkelstein IJ. Inhibition of CRISPR-Cas12a DNA targeting by nucleosomes and chromatin. Sci Adv. 2021;7(11):eabd6030. doi: 10.1126/sciadv.abd6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Wu X, Scott D. A., Kriz A. J., Chiu A. C., Hsu P. D., Dadon D. B., & Sharp P. A. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol. 2014;32(7):670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.O’Geen H, Henry I. M., Bhakta M. S., Meckler J. F., & Segal D. J. A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res. 2015;43(6):3389–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Liu B, Chen S., Rose A. L., Chen D., Cao F., Zwinderman M., & Haisma H. J. Inhibition of histone deacetylase 1 (HDAC1) and HDAC2 enhances CRISPR/Cas9 genome editing. Nucleic Acids Res. 2020;48(2):517–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Bashir S, Dang T., Rossius J., Wolf J., & Kühn R. Enhancement of CRISPR-Cas9 induced precise gene editing by targeting histone H2A-K15 ubiquitination. BMC Biotechnol. 2020;20(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Daer R, Hamna F., Barrett C. M., & Haynes K. A. Site-directed targeting of transcriptional activation-associated proteins to repressed chromatin restores CRISPR activity. APL Bioeng. 2020;4(1):016102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Horlbeck MA, Gilbert L. A., Villalta J. E., Adamson B., Pak R. A., Chen Y., & Weissman J. S. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Tsai K, Cullen BR. Epigenetic and epitranscriptomic regulation of viral replication. Nat Rev Microbiol. 2020;18(10):559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Lucic B, de Castro IJ, Lusic M. Viruses in the nucleus. Cold Spring Harb Perspect Biol. 2021;13(8):a039446. doi: 10.1101/cshperspect.a039446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Kujirai T, Zierhut C., Takizawa Y., Kim R., Negishi L., Uruma N., & Kurumizaka H. Structural basis for the inhibition of cGAS by nucleosomes. Science. 2020;370(6515):455–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Karen KA, Hearing P. Adenovirus core protein VII protects the viral genome from a DNA damage response at early times after infection. J Virol. 2011;85(9):4135–4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Avgousti DC, Herrmann C., Kulej K., Pancholi N. J., Sekulic N., Petrescu J., & Weitzman M. D. A core viral protein binds host nucleosomes to sequester immune danger signals. Nature. 2016;535(7610):173–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Lynch KL, Gooding L. R., Garnett‐Benson C., Ornelles D. A., & Avgousti D. C. Epigenetics and the dynamics of chromatin during adenovirus infections. FEBS Lett. 2019;593(24):3551–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Marazzi I, Ho J. S., Kim J., Manicassamy B., Dewell S., Albrecht R. A., & Tarakhovsky A. Suppression of the antiviral response by an influenza histone mimic. Nature. 2012;483(7390):428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Gao L, Wu K., Liu Z., Yao X., Yuan S., Tao W., & Liu J. Chromatin accessibility landscape in human early embryos and its association with evolution. Cell. 2018;173(1):248–259 e15. [DOI] [PubMed] [Google Scholar]
- 377.Perrat PN, DasGupta S., Wang J., Theurkauf W., Weng Z., Rosbash M., & Waddell S. Transposition-driven genomic heterogeneity in the Drosophila brain. Science. 2013;340(6128):91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Cost GJ, Golding A., Schlissel M. S., & Boeke J. D. Target DNA chromatinization modulates nicking by L1 endonuclease. Nucleic Acids Res. 2001;29(2):573–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Gorbunova V, Seluanov A., Mita P., McKerrow W., Fenyö D., Boeke J. D., & Sedivy J. M. The role of retrotransposable elements in ageing and age-associated diseases. Nature. 2021;596(7870):43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Jachowicz JW, Bing X., Pontabry J., Bošković A., Rando O. J., & Torres-Padilla M. E. LINE-1 activation after fertilization regulates global chromatin accessibility in the early mouse embryo. Nat Genet. 2017;49(10):1502–1510. [DOI] [PubMed] [Google Scholar]
- 381.Paulsen T, Kumar P., Koseoglu M. M., & Dutta A. Discoveries of extrachromosomal circles of DNA in normal and tumor cells. Trends Genet. 2018;34(4):270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Wang Y, Wang M., Djekidel M. N., Chen H., Liu D., Alt F. W., & Zhang Y. eccDNAs are apoptotic products with high innate immunostimulatory activity. Nature. 2021;599(7884):308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Mansisidor A, Molinar JrT., Srivastava P., Dartis D. D., Delgado A. P., Blitzblau H. G., & Hochwagen A. Genomic copy-number loss is rescued by self-limiting production of DNA circles. Mol Cell. 2018;72(3):583–593 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Vogt N, Lefèvre S. H., Apiou F., Dutrillaux A. M., Cör A., Leuraud P., & Malfoy B. Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc Natl Acad Sci USA. 2004;101(31):11368–11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Schwab M, Alitalo K., Klempnauer K. H., Varmus H. E., Bishop J. M., Gilbert F., & Trent J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature. 1983;305(5931):245–248. [DOI] [PubMed] [Google Scholar]
- 386.Kim H, Nguyen N. P., Turner K., Wu S., Gujar A. D., Luebeck J., & Verhaak R. G. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet. 2020;52(9):891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Wu S, Turner K. M., Nguyen N., Raviram R., Erb M., Santini J., & Mischel P. S. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature. 2019;575(7784):699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Morton AR, Dogan-Artun N, Faber Z. J, et al. Functional enhancers shape extrachromosomal oncogene amplifications. Cell. 2019;179(6):1330–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Hung KL, Yost K. E, Xie L, et al. ecDNA hubs drive cooperative intermolecular oncogene expression. Nature. 2021;600(7890):731–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Kruitwagen T, Chymkowitch P, Denoth-Lippuner A, et al. Centromeres license the mitotic condensation of yeast chromosome arms. Cell. 2018;175(3):780–795 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Karin Purshouse ETF, Boyle S, Singh Dewari P, et al., Oncogene expression from extrachromosomal DNA is driven by copy number amplification and does not require spatial clustering. bioRxiv. 2022. [DOI] [PMC free article] [PubMed]
- 392.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153(1):38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Decorsiere A, Mueller H, Van Breugel P. C, et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature. 2016;531(7594):386–389. [DOI] [PubMed] [Google Scholar]
- 394.Dupont L, Bloor S, Williamson J. C, et al. The SMC5/6 complex compacts and silences unintegrated HIV-1 DNA and is antagonized by Vpr. Cell Host Microbe. 2021;29(5):792–805 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]