

Abstract
Using a microwave-assisted protocol, we synthesized sixteen peptoid-capped HDAC inhibitors (HDACi) with fluorinated linkers and identified two hit compounds. In biochemical and cellular assays, 10h stood out as a potent unselective HDACi with remarkable cytotoxic potential against different therapy resistant leukemia cell lines. Of note, 10h demonstrated prominent antileukemic activity with low cytotoxic activity toward healthy cells. Moreover, 10h exhibited synergistic interactions with the DNA methyltransferase inhibitor decitabine in AML cell lines. The comparison of crystal structures of HDAC6 complexes with 10h and its non-fluorinated counterpart revealed a similar occupation of the L1 loop pocket but slight differences in the zinc coordination. The substitution pattern of the acyl residue turned out to be crucial in terms of isoform selectivity. The introduction of an isopropyl group to the phenyl ring provided the highly HDAC6 selective inhibitor 10p which demonstrated moderate synergy with decitabine and outmatched the HDAC6 selectivity of tubastatin A.
Keywords: Histone deacetylase, cancer, leukemia, synergism, fluorination
Graphical Abstract
INTRODUCTION
Epigenetic events are largely induced by environmental factors and determine the post-translational alteration of a cell phenotype.1 In today’s drug discovery, controlling epigenetic mechanisms by regulating histone modifications such as histone acetylation, methylation or phosphorylation is of growing importance.1,2 One group of enzymes involved in such processes are histone deacetylases (HDACs) which have attracted attention as promising targets for cancer therapy. Consisting of eleven zinc-dependent isoforms and the NAD+-dependent sirtuins, the HDAC family is divided into classes I–IV.3 Among those, only the class I isoforms HDAC1-3 control the acetylation levels of histones, thus modifying the chromatin structure and its accessibility for gene transcription.3 Other isoforms participate in a variety of biological processes such as the formation of protein-protein complexes, the mediation of immunosuppression via Forkhead box P3 (Foxp3) deacetylation, and the regulation of transcription factors.4,5,6,7 HDAC inhibitors (HDACi) typically comprise a capping group occupying the outside of the enzyme’s active site, a zinc-binding group chelating the catalytically active zinc ion, and a linker chain connecting the two parts. Resulting from the highly conserved nature of the enzymes, this pharmacophore model generally applies to all isoforms, including the class I enzymes.3 Being particularly cytotoxic due to their interference with cell cycle progression and proliferation, selective class I HDACi as well as pan-inhibitors are considered promising tools for the therapy of both hematological and solid tumors.8 Since their discovery, four HDACi, e.g. vorinostat, belinostat, panobinostat, and the class I-selective natural compound romidepsin, have been FDA-approved to treat hematological cancers, including multiple myeloma and T cell lymphoma.6,8,9 The class I-selective tucidinostat has recently become the first HDACi to be approved for the therapy of solid tumors by the NMPA in China.10 Another field of interest concerning class I HDACi on clinical level is their presumed ability to eradicate latent HIV reservoirs.11
Despite their potential in cancer therapy, pan-HDACi are typically associated with adverse effects. To explore the scope beyond oncology, the research on isoform-specific HDACi with reduced off-target interactions and higher tolerability is quickly evolving.8 The most promising target for isoform-selective inhibition with improved safety profiles is HDAC6, which appears to play a role in cancer, neurodegenerative diseases, inflammation, rheumatoid arthritis, and neurological disorders.12,13,14,15,16,17 The first HDAC6-preferential drug candidate ricolinostat (phase II), its chlorinated analog citarinostat (phase I), and several yet undisclosed HDAC6i considered for non-oncological conditions (phase II) are currently being assessed in clinical trials.18 Unlike other isoforms, the class IIb isoform HDAC6 is located in the cytoplasm instead of the nucleus and comprises two catalytic domains (CDs).19,20 One of which, CD1, is considered inactive toward histones while its biological role remains unclear.21 Confirmed substrates of the second catalytic domain (CD2) include α-tubulin and cortactin, which are both required for cytoskeleton formation; tau, and the chaperone protein Hsp90.21,22,23,24,25,26 HDAC6 is furthermore unique in comprising a ubiquitin-binding domain which regulates the aggresome-autophagy pathway as an important protein degradation system beside the 26S proteasome.27,28 Although recent studies imply only moderate cytotoxicity induced by selective HDAC6 inhibition29, the enzyme remains a relevant drug target with high eligibility for combination therapies.30
Supposedly triggering a beneficial alteration in gene expression patterns, the combination of HDACi and DNA-hypomethylating agents is another field of interest.31 The concurrent application of HDACi and decitabine in diffuse large B-cell lymphoma cell lines appeared to induce time-dependent synergistic effects resulting in tumor growth inhibition and increased apoptosis levels that exceeded the effect of HDACi alone.31 Other preclinical experiments hinted at synergistic effects activated by vorinostat and decitabine in mantle cell lymphoma and pancreatic cancer cell lines.31,32,33 Succeeding prior clinical trials on a pan-HDACi as an additive to decitabine, current studies are focusing on the class I-selective HDACi tucidinostat in lymphoma patients.34,35,36
A feature with increasing prevalence in modern pharmaceuticals is fluorine. Although rarely occurring in natural compounds, fluorination has become an important means to improve the bioavailability and pharmacokinetic properties of drugs.37 After adding up to 20–25% of all pharmaceuticals a decade ago, fluorinated drugs now account for one half of all blockbuster drugs.37,38 Compared to non-fluorinated analogs, the low polarization of the C-F bond and the high electron-withdrawing capabilities of fluorine substituents may significantly alter the pKa, the lipophilicity, and the metabolic stability of drugs.37,39,40 Conformational changes due to the characteristic steric demands of fluorinated groups could have an additional impact on the binding mode and the target affinity.37,39,40 Indeed, another notable benefit of drug fluorination is the possibility of 18F radiolabelling. Apart from diagnostic measures, PET-tracing of 18F-labelled compounds can be useful for biodistribution studies of early-stage drug candidates, thus facilitating the drug development process.37 Despite such advantages, fluorinated drugs bear the risk of high intrinsic toxicity owing to the release of fluorine-containing metabolites.41,42 A careful investigation of the metabolic behavior of each fluorinated drug candidate is therefore inevitable.41
In 2016, our group published results on selective HDAC6 inhibition by peptoid-based compounds obtained by the Ugi 4-component reaction (U4CR).43 Follow-up studies using co-crystals of selected peptoid-capped inhibitors in complex with the CD2 of Danio rerio (zebrafish) HDAC6 (zCD2) revealed that the compounds adopt different binding modes than the highly HDAC6-selective PET-probe bavarostat which carries a fluorine substituent in m-position to the hydroxamate in the otherwise identical linker moiety.44,45 Given that bavarostat and the unselective HDACi [11C]martinostat, a cinnamoyl-based PET probe in clinical phase I, share identical cap groups, we concluded that the remarkable HDAC6-selectivity of bavarostat is likely to derive from the unique binding mode of the fluorinated linker moiety.46 Moreover, a recent study of inhibitors similarly fluorinated and difluorinated at the meta positions of phenyl linker groups found that the compounds also exhibit HDAC6 selectivity, possibly due to interactions between the fluorine atoms and His361, Gly582, and Ser531 in loop pockets L1 and L2.47 Based on the observation that selectivity profiles can be steered through substitution patterns in the cap group area, we decided to investigate the effect of linker fluorination on scaffolds that had previously turned out to favor either HDAC6- or pan-inhibition. The resulting set of highly potent, fluorinated HDACi were then assessed in respect of their antileukemic properties.
RESULTS AND DISCUSSION
Design and Synthesis.
Aiming to determine whether the bavarostat-inspired linker fluorination would improve the inhibitory qualities of our previously reported peptoids, we designed a new set of compounds that includes fluorinated analogs of the non-selective compounds DDK115 and DDK137 as well as the HDAC6-preferential inhibitors IIe and IIf (Scheme 1).45,48 In a recent study on tetrazole-capped HDACi derived from the peptoid-scaffold, we moreover identified the 2-trifluoromethyl substituent of the acyl moiety as a source of excellent selectivity.49 An additional SAR study therefore featured several analogs with differently shaped substituents in the same position.
Scheme 1.
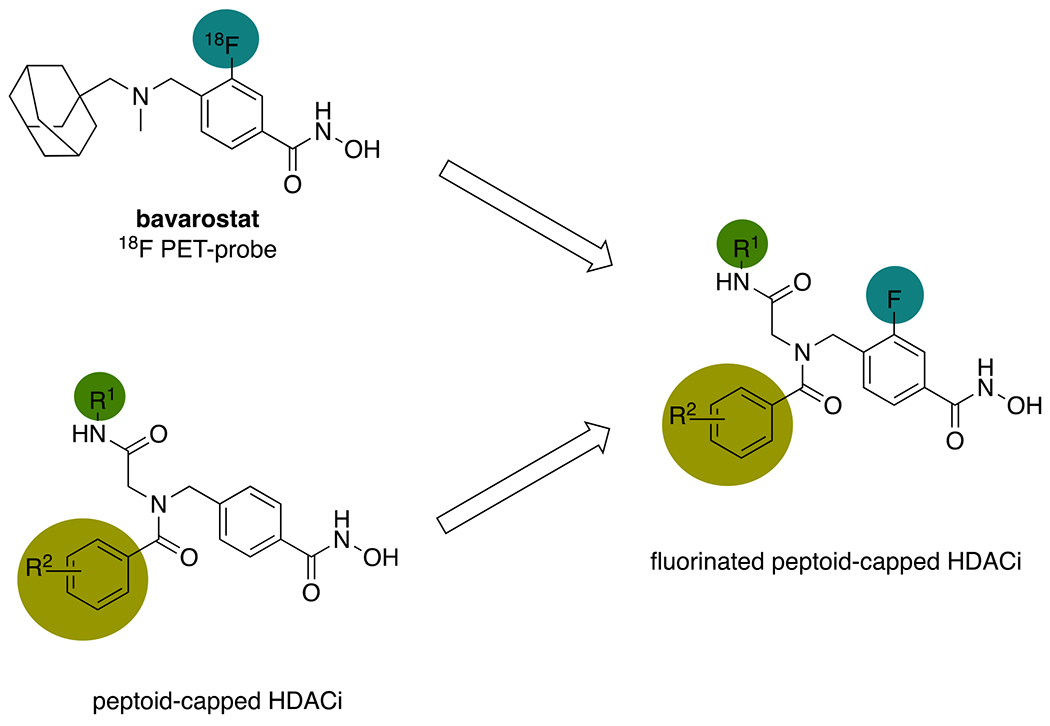
Design of fluorinated peptoid-capped HDACi inspired by the preclinical PET-probe bavarostat.
The synthesis of the 2-fluorophenyl linker was performed in accordance with a literature protocol (Scheme 2).50 Following the esterification of 1, NBS was used to introduce a bromine substituent (3) which was then substituted to give the corresponding azide 4. Next, the Staudinger reaction using triphenylphosphine was carried out to afford the desired amine, which was easily isolated as the hydrochloride salt 5. The U4CR of 5, the respective isonitriles 6, the respective carboxylic acids 7, and paraformaldehyde 8 built the peptoid scaffolds 9 within less than 4 hours of microwave irradiation (Scheme 3). After aqueous workup and recrystallization of the crude products, the resulting esters 9 were treated with a mixture of sodium hydroxide and aqueous hydroxylamine to yield the hydroxamates 10 after 15 min of stirring at 0 °C. All final compounds were isolated by precipitation from the aqueous crude mixture and exceeded 95% purity without further purification.
Scheme 2.
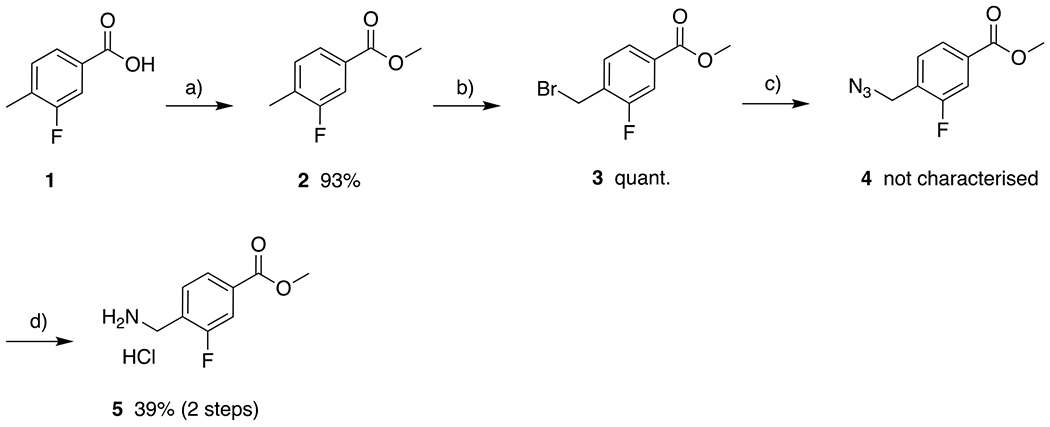
Synthesis of the fluorinated linker 5. a) SOCl2, MeOH, 0 °C to rt, 16 h; b) NBS, AIBN, DCM, reflux, 16 h; c) NaN3, DMF, 80 °C, 16 h; d) PPh3, HCl, THF, rt, 24 h.
Scheme 3.
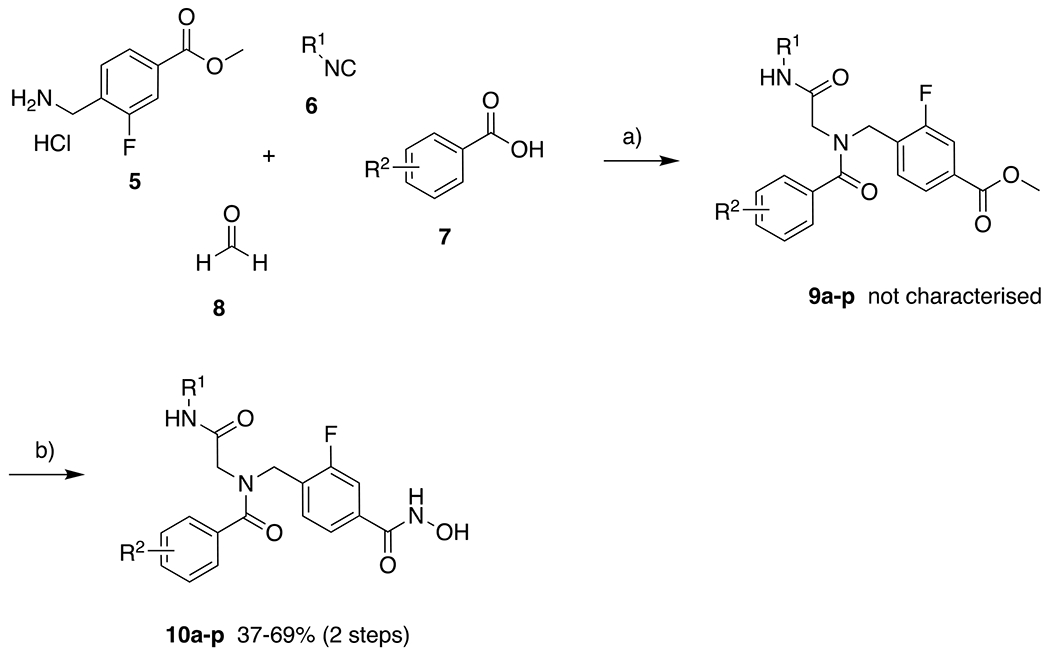
Synthesis of the fluorinated peptoid-capped HDACi 10a-p. a) Et3N, MS 4Å, MeOH, microwave, 45 °C, 150 W, 60–210 min; b) H2NOH, NaOH, MeOH, DCM, 0 °C, 15 min.
HDAC inhibition.
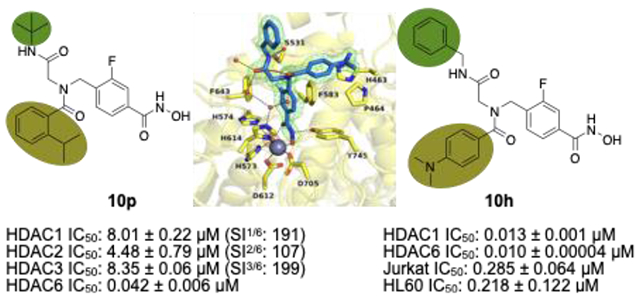
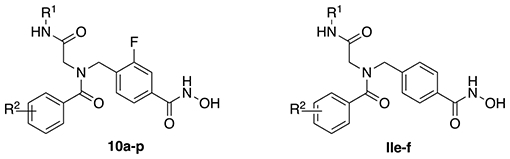
Biochemical assays were performed to evaluate the inhibitory activities of all final compounds against HDAC1 and HDAC6. The resulting IC50 values (Table 1) indicate that minor structural modifications suffice to steer between unselective inhibition and HDAC6 selectivity without affecting the overall high inhibitory activities against HDAC6 in the low nanomolar concentration range. In comparison to compounds featuring a cyclohexyl group in position R1 (10c-f; IC50 HDAC6: 0.018–0.020 μM, selectivity indices (SIHDAC1/6): 43–75), it is apparent that the tert-butyl derivatives 10k-n with identical substitution patterns at the R2 moiety display more promising selectivity profiles (SIHDAC1/6: 45–103) and similar potencies (IC50 HDAC6: 0.017–0.027 μM). The selectivity profile of the voluminous and branched iso-propyl derivative 10p (SIHDAC1/6: 191) exceeded all other analogs as well as the control compound tubastatin A (SIHDAC1/6: 178), in spite of the slightly impaired inhibition of HDAC6 (0.042 μM). Hence, 10p was singled out as the most selective inhibitor of this set. The equally bulky 2-chloro analog 10o was found to be twice as potent (IC50 HDAC6: 0.021 μM) but less selective (SIHDAC1/6: 165). Additional assays using HDAC2 and HDAC3 confirmed the low activity of 10p against other class I isoforms (IC50 HDAC2: 4.48 μM, SIHDAC2/6: 107; HDAC3: 8.35 μM, SIHDAC3/6: 199). Furthermore, we screened 10p against HDAC4 as a representative class IIa isoform. As expected, the compound turned out to be inactive (IC50 HDAC4: > 10 μM, SIHDAC4/6: > 238; Figure 1).
Table 1.
IC50 of the fluorinated HDACi 10a-p and the non-fluorinated analogs DDK115, DDK137, and IIe-f in comparison to vorinostat, ricolinostat, HPOB and Tubastatin A (Tub A) against HDACs 1 and 6.
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | HDAC1 IC50 [μM] | HDAC6 IC50 [μM] | SIa |
| 10a | c-Hexyl | 3,5-Me | 0.310 ± 0.004 | 0.020 ± 0.002 | 16 |
| 10b | c -Hexyl | 4-NMe2 | 0.066 ± 0.008 | 0.016 ± 0.004 | 4 |
| 10c | c -Hexyl | 2-F | 0.771 ± 0.006 | 0.018 ± 0.002 | 43 |
| 10d | c -Hexyl | 2-Me | 0.938 ± 0.013 | 0.018 ± 0.002 | 52 |
| 10e | c -Hexyl | 2-OMe | 1.50 ± 0.10 | 0.020 ± 0.004 | 75 |
| 10f | c -Hexyl | 2-CF3 | 1.45 ± 0.05 | 0.027 ± 0.002 | 54 |
| 10g | Bn | 3,5-Me | 0.077 ± 0.003 | 0.013 ± 0.004 | 6 |
| 10h | Bn | 4-NMe2 | 0.013 ± 0.001 | 0.010 ± 0.00004 | 1.3 |
| 10i | t-Bu | 3,5-Me | 0.573 ± 0.011 | 0.024 ± 0.002 | 24 |
| 10j | t-Bu | 4-NMe2 | 0.158 ± 0.056 | 0.020 ± 0.006 | 8 |
| 10k | t-Bu | 2-F | 1.18 ± 0.15 | 0.026 ± 0.002 | 45 |
| 10l | t-Bu | 2-Me | 1.75 ± 0.13 | 0.027 ± 0.005 | 65 |
| 10m | t-Bu | 2-OMe | 1.75 ± 0.05 | 0.017 ± 0.0001 | 103 |
| 10n | t-Bu | 2-CF3 | 2.33 ± 0.43 | 0.027 ± 0.005 | 86 |
| 10o | t-Bu | 2-Cl | 3.47 ± 0.14 | 0.021 ± 0.001 | 165 |
| 10p | t-Bu | 2-iPr | 8.01 ± 0.22 | 0.042 ± 0.006 | 191 |
| DDK115 | c-Hexyl | 3,5-Me | 0.249 ± 0.028 | 0.040 ± 0.008 | 6 |
| DDK137 | Bn | 4-NMe2 | 0.005 ± 0.0006 | 0.011 ± 0.003 | 0.5 |
| IIe b | t-Bu | 2-Me | 2.41 ± 0.086 | 0.051 ± 0.004 | 47 |
| IIf b | t-Bu | 2-OMe | 3.11 ± 0.308 | 0.038 ± 0.004 | 82 |
| Vorinostat | - | - | 0.099 ± 0.005 | 0.042 ± 0.005 | 2 |
| Givinostat | - | - | 0.035 ± 0.002 | 0.014 ± 0.0002 | 2.5 |
| Givinostatc | - | - | 0.070 ± 0.012 | 0.023 ± 0.004 | 3 |
| Ricolinostatd | - | - | 0.19 ± 0.022 | 0.018 ± 0.003 | 11 |
| Tub Ae | - | - | 2.49 ± 0.14 | 0.014 ± 0.0006 | 178 |
| HPOBe | - | - | 2.10 ± 0.23 | 0.085 ± 0.009 | 25 |
Figure 1.
Inhibition of HDAC1-3 and HDAC6 by the selected hit compounds 10h and 10p. Selectivity index (SI = IC50 (HDAC1, 2, 3, or 4)/IC50 (HDAC6)).
In accordance with previous SAR data, all inhibitors featuring 3,5-dimethyl residues in the acyl ring (10a, 10g, 10i) exhibited only low to moderate HDAC6 selectivity (SIHDAC1/6: 6–24) but high inhibitory qualities ranging from 0.013 μM to 0.024 μM against HDAC6.48,51 The two 4-dimethylamino derivatives 10b and 10h met the expectations by turning out to be strong unselective inhibitors (SI: 1.4–8) of which 10h proved to be the most potent ligand against both isoforms (IC50 HDAC1: 0.013 μM; HDAC6: 0.010 μM) and other class I enzymes (IC50 HDAC2: 0.014 μM, SIHDAC2/6: 1.4; HDAC3: 0.021 μM, SIHDAC3/6: 2.1; Figure 1). Compound 10h also displayed some inhibitory activity against HDAC4 (IC50 HDAC4 1.88 μM, SIHDAC4/6: 188; Figure 1). The tert-butyl-capped analog 10j (IC50 HDAC1: 0.158 μM; HDAC6: 0.020 μM, SI: 7.9) exhibited similar inhibition qualities as ricolinostat. In summary, those results indicate that the substitution pattern of the acyl ring is crucial in terms of isoform selectivity. Depending on the size of the residue, the sole occupation of position 2 of the aromatic ring promises a strong preference for HDAC6. The introduction of small residues in positions 3, 4, and 5, in turn, effectuates equal inhibition of HDAC1 and HDAC6 or a marginal preference for the latter. The HDAC6 selectivity achieved by appropriate R2 residues may moreover be boosted by choosing the tert-butyl motif over a cyclohexyl group in position R1. Overall, all new ligands, except for the most selective inhibitor 10p, outmatched the HDAC6 inhibition of the control compound vorinostat and were equally potent as tubastatin A and ricolinostat.
The comparison of the unselective inhibitors 10a and 10h with their non-fluorinated analogs DDK115 and DDK137 implies that linker fluorination results in decreased HDAC1 inhibition (DDK115 vs. 10a: 0.249 μM/0.310 μM; DDK137 vs. 10h: 0.005 μM/0.013 μM) accompanied by higher or similar inhibitory activity against HDAC6 (DDK115 vs. 10a: 0.040 μM/0.020 μM; DDK137 vs. 10h: 0.011 μM/0.010 μM), thus almost tripling the selectivity for HDAC6 in both cases.
In respect of the HDAC6-selective compounds IIe and IIf, linker fluorination increased the inhibitory activity toward HDAC1 (IIe vs. 10l: 2.41 μM/1.75 μM; IIf vs. 10m: 3.11 μM/1.75 μM) and nearly doubled inhibitory potency against HDAC6 (IIe vs. 10l: 0.051 μM/0.027 μM; IIf vs. 10m: 0.038 μM/0.017 μM), thereby improving the selectivity profiles.
Those results are generally in line with the observations reported by Sandrone et al. who concluded that the increased HDAC6 selectivity of HDACi with mono- or difluorinated linkers in meta-position to the hydroxamic acid results from increased HDAC6 inhibition and/or decreased inhibitory activity toward HDAC1.47 Differences in the assay setups (in particular different substrates) prohibit a direct comparison of the results published by Sandrone et al. and our results summarized in Table 1 so that we chose to include givinostat as an additional control compound. Although we (IC50 HDAC1: 0.035 μM; HDAC6: 0.014 μM, SI: 2.5) measured different IC50 values for givinostat compared to Sandrone et al. (IC50 HDAC1: 0.070 μM; HDAC6: 0.023 μM, SI: 3)47, the resulting selectivity indices were comparable (Table 1). In general, Sandrone et al. observed different reasons for the increased HDAC6 selectivity depending on whether small cap-less inhibitors or more complex larger HDACi with (hetero)aromatic caps were investigated. In the case of small cap-less HDACi, the increased HDAC6 selectivity is mainly driven by improved HDAC6 inhibition. This was exemplified by the comparison of N-hydroxybenzamide (IC50 HDAC1: 25.4 μM; HDAC6: 1.34 μM, SI: 19) and 3-fluoro-N-hydroxybenzamide (IC50 HDAC1: 29.4 μM; HDAC6: 0.453 μM, SI: 65).47 The improvement of the HDAC6 selectivity of larger HDACi with complex (hetero)aromatic caps, in contrast, originates mainly from a decreased HDAC1 inhibition.47 This can be explained by differences in the L1 loops of HDAC1 and HDAC6: the active site of HDAC1 is more restricted and thus unable to accommodate larger cap groups, whereas HDAC6 features a wider entrance area to the catalytic tunnel.47 Moreover, unlike most other HDACs which feature an aspartate in the respective position, HDAC6 possesses a serine (Ser531) on loop L2 at the entrance of the catalytic channel.47 In the case of larger HDACi with (hetero)aromatic caps, a fluorine atom in meta-position of the linker is oriented toward loop L2 which might result in a steric repulsion in HDACs featuring an aspartate at the tunnel entrance. This could explain the results observed by Sandrone et al., indicating that fluorinated phenyl linkers with a bulky cap group are more likely to reduce the inhibitory activity toward HDAC1 rather than increasing the potency against HDAC6, thus boosting the selectivity.47 Considering our data and the results published by Sandrone et al.47, it can be concluded that linker fluorination has a distinct beneficial effect on HDAC6 inhibition and/or selectivity.
In addition to the above-mentioned effects on HDAC6, Sandrone et al.47 found that the linker fluorination restores activity against class IIa HDACs. To investigate whether this is also the case for our peptoid-based HDACi, we screened DDK137, the direct non-fluorinated analogue of compound 10h, against HDAC4. In good agreement with the results of Sandrone et al.47, DDK137 (IC50 HDAC4: 2.67 μM) demonstrated reduced HDAC4 inhibition compared to 10h (IC50 HDAC4: 1.88 μM, Figure 1), thus indicating that linker fluorination can lead to improved inhibitory properties against class IIa HDACs.
Crystal Structure of HDAC6 in Complex with 10h.
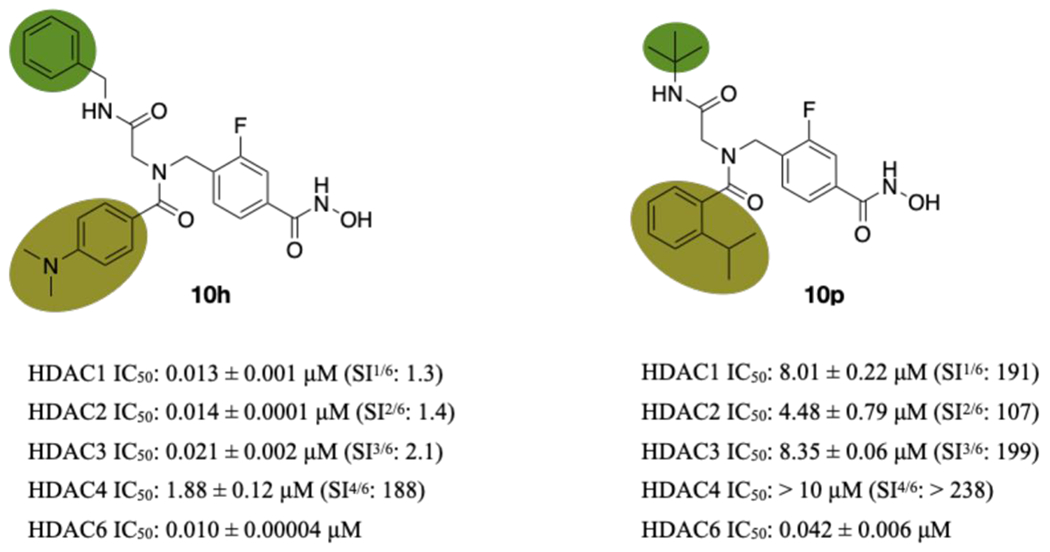
The 1.85 Å-resolution structure of the HDAC6-10h complex contains four independent copies of the enzyme-inhibitor complex in the asymmetric unit of the crystal. Hydroxamate-Zn2+ coordination differs in each of the four structures. In chain A, hydroxamate-Zn2+ coordination is modeled with bidentate geometry but the electron density for the hydroxamate group is ambiguous. In chains B and C, exclusively bidentate hydroxamate-Zn2+ coordination geometry is observed, and in chain D exclusively monodentate hydroxamate-Zn2+ coordination geometry is observed. Below, the structures of the enzyme-inhibitor complex in chain B and chain D are outlined in greater detail.
For chain B (Figure 2), the HDAC6-10h complex does not reveal any major structural rearrangements caused by inhibitor binding, and the root-mean-square deviation (rmsd) is 0.19 Å for 311 Cα atoms between the inhibitor-bound and unliganded enzyme structures. In the bidentate hydroxamate-Zn2+ chelate complex, the Zn2+–O distances are 2.2 and 2.3 Å for the N–O− and C=O groups, respectively. The side chain of Y745 additionally donates a hydrogen bond to the hydroxamate C=O group (O–O distance = 2.6 Å) and H574 accepts a hydrogen bond from the hydroxamate NH group (N–N distance = 2.8 Å).
Figure 2.
Stereoview of a Polder omit map (contoured at 4.0σ) depicting the bidentate binding mode of 10h (light gray) in the active site of HDAC6 chain B (yellow) (PDB 7U8Z). Two conformations of the aromatic ring are shown, one with the C–F group oriented into the F583-F643 aromatic crevice, and the other with the C–F group oriented toward solvent. The catalytic Zn2+ ion is shown as a grey sphere, and water molecules are shown as small red spheres. Metal coordination, hydrogen bond, and n→π* interactions are shown as solid, dashed black, and solid red lines, respectively.
The fluorophenyl linker is nestled in the aromatic crevice formed by F583 and F643; however, the aromatic ring adopts two mutually exclusive conformations (50% occupancy each). In one conformation, the C–F group is oriented into the crevice and the fluorine atom is 3.4 Å away from the hydroxyl group of S531. In the other conformation, the C–F group is oriented toward solvent. The peptoid carbonyl group is oriented toward solvent and appears to form an n→π* interaction with the adjacent benzylamide carbonyl group. The dimethylaniline group resides in the L1 loop pocket, as observed for the non-fluorinated inhibitor.45 The benzylamide carbonyl forms hydrogen bonds with bulk solvent, while the adjacent nitrogen forms a water mediated hydrogen bond with the hydroxyl side chain of S531.
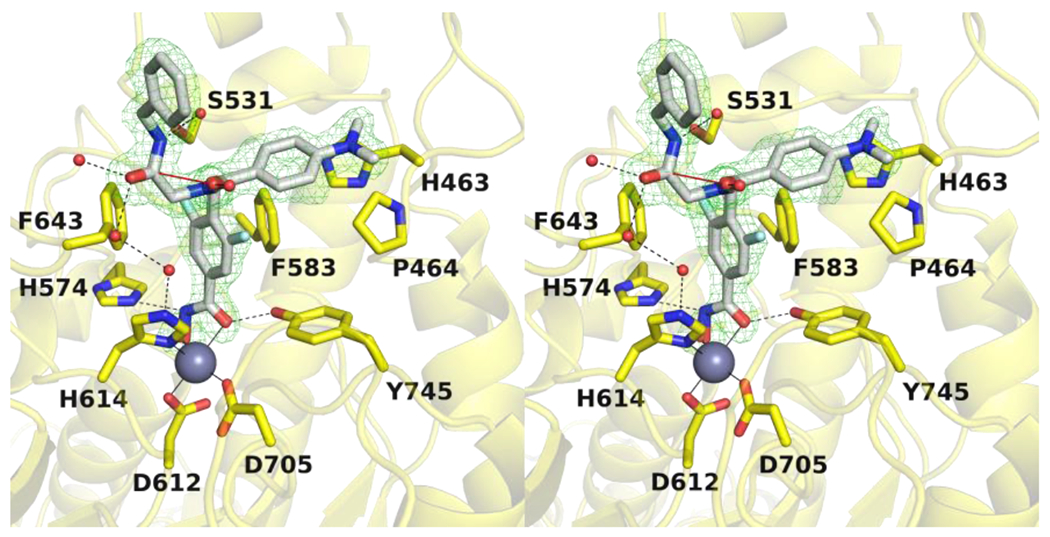
For chain D (Figure 3), the HDAC6-10h complex does not reveal any major structural rearrangements caused by inhibitor binding, and the rmsd is 0.16 Å for 302 Cα atoms between the inhibitor-bound and unliganded enzyme structures. The hydroxamate moiety adopts monodentate Zn2+ coordination geometry in which the hydroxamate N–O− group coordinates to Zn2+ at a distance of 1.8 Å. The hydroxamate C=O group accepts a short hydrogen bond from the Zn2+-coordinated water molecule (O–O distance = 2.3 Å) which in turn forms hydrogen bonds with H573 and H574. The hydroxamate NH group additionally interacts with the side chain of Y745 (O–N distance = 2.5 Å).
Figure 3.
Stereoview of a Polder omit map (contoured at 4.0σ) depicting the monodentate binding of 10h (dark gray) in the active site of HDAC6 chain D (yellow) (PDB 7U8Z). The catalytic Zn2+ ion is shown as a grey sphere, and water molecules are shown as small red spheres. Metal coordination, hydrogen bond, and n→π* interactions are shown as solid, dashed black, and solid red lines, respectively.
The fluorophenyl aromatic linker is nestled in the F583-F643 aromatic crevice with the C–F group oriented exclusively into the crevice; the fluorine atom is 3.4 Å away from the hydroxyl group of S531. Other aspects of inhibitor binding are similar to those observed in chain B described above. The peptoid carbonyl of the inhibitor capping group is oriented toward solvent and the carbonyl oxygen appears to form an n→π* interaction with the adjacent benzylamide carbonyl group. The benzylamide carbonyl and peptoid carbonyl form a hydrogen bond network with four water molecules, and this network includes Zn2+ ligand H614. The benzylamide NH group forms a water mediated hydrogen bond with the side chain of S531. Finally, the dimethylaniline group resides in the L1 loop pocket. The overall binding modes of 10h and its non-fluorinated counterpart44 are almost entirely identical, with the only major difference being variations in hydroxamate-Zn2+ coordination (Figure 4).
Figure 4.
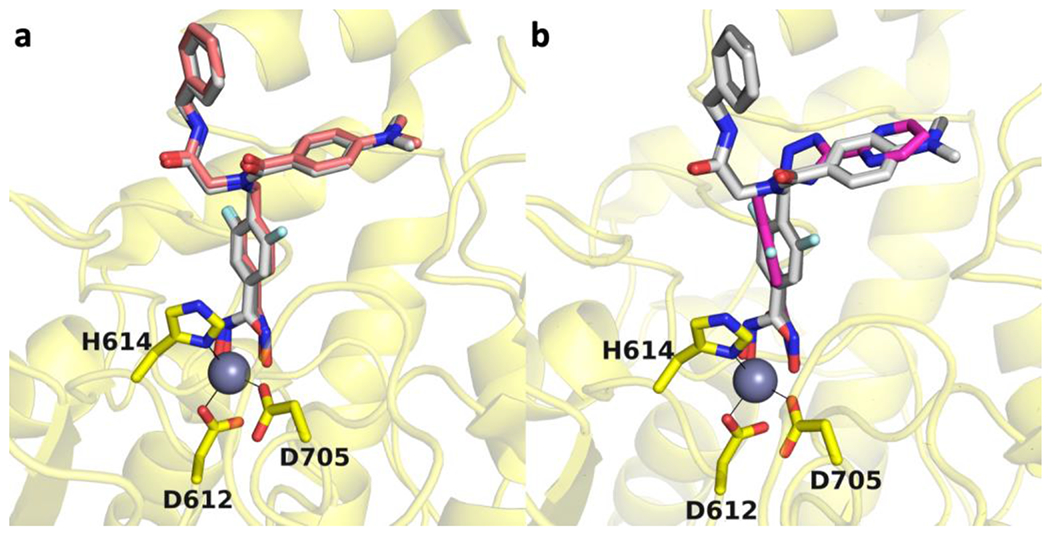
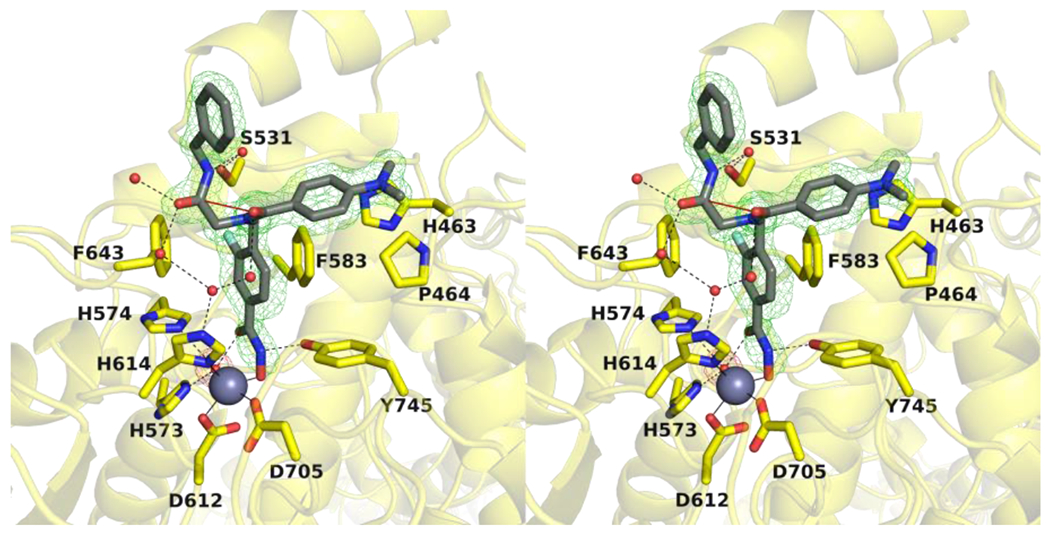
(a) Overlay of the HDAC6-10h complex (chain B, light gray; chain D, dark gray; PDB 7U8Z) with the HDAC6 complex with the non-fluorinated inhibitor (light pink; PDB 6DVN). The catalytic Zn2+ ion is shown as a grey sphere and metal coordination is indicated by solid black lines. (b) Overlay of the HDAC6-10h complex (chain B, light gray; chain D, dark gray; PDB 7U8Z) with an inhibitor containing a meta-difluorophenyl linker (PDB 7O2R).
Bavarostat is a selective inhibitor of HDAC644 and contains a meta-fluorophenyl linker that, like the meta-fluoro linker of 10h, binds in the aromatic crevice defined by F583 and F643.45 More recently, inhibitors have been developed that contain meta-fluorophenyl and meta-difluorophenyl linkers with different capping groups, and these inhibitors exhibit enhanced selectivity for inhibition of HDAC6 over HDAC1.47 The X-ray crystal structure of one of these inhibitors complexed with HDAC6 reveals that the meta-difluorophenyl linker makes favorable π-π interactions in the aromatic crevice as well as weak interaction between one of the C–F groups and S531. The fluoroaromatic ring of this inhibitor generally binds in a similar manner to the fluoroaromatic rings of bavarostat and 10h, although the inhibitor binding orientation is slightly tilted so that the rings are not coplanar (Figure 4b). Intriguingly, a key feature of isozyme selectivity derives from the reduction of inhibitory potency toward HDAC1, especially for inhibitors containing a meta-difluorophenyl linker.
Cytotoxic effects on leukemia cell lines.
The sixteen new inhibitors (10a–10p) were screened against three-selected leukemia cell lines originated from myeloid (HL60), B- (HAL01) or T- (Jurkat) lymphoid lineages (Table 2). The FDA-approved drug vorinostat, the HDAC6i HPOB, and the clinical candidate ricolinostat were used as controls. For selective HDAC6i, cytotoxicity levels are typically low and therefore, it was not surprising that neither of the IC50 values measured in presence of selective ligands lies below the micromolar concentration range.29,54 Compound 10j was found to resemble ricolinostat in terms of both HDAC inhibition and isoform selectivity. Accordingly, it induced similar levels of cytotoxicity with IC50 values ranging from 1.46 μM to 2.47 μM in all three cell lines. Two other compounds, 10c and 10d, exceeded the cytotoxic effects of HPOB throughout all screenings. As for the unselective inhibitors, the most potent derivative 10h proved to be similarly active against the B-cell acute lymphoblastic leukemia (B-ALL) cell line HAL01 and even more toxic against the T-ALL Jurkat cell line than vorinostat. Based on these results, compound 10h was picked as the most promising candidate for additional experiments.
Table 2.
Cytotoxicities of 10a-p against selected leukemia cell lines. Vorinostat, HPOB, and ricolinostat were used as controls.
| Compound | R1 | R2 | HAL01 IC50 [μM] | HL60 IC50 [μM] | Jurkat IC50 [μM] |
|---|---|---|---|---|---|
| 10a | c-Hexyl | 3,5-Me | 3.10 ± 0.91 | 2.47 ± 2.05 | 3.91 ± 0.67 |
| 10b | c -Hexyl | 4-NMe2 | 1.05±0.28 | 0.494±0.126 | 0.946±0.087 |
| 10c | c -Hexyl | 2-F | 11.8±5.14 | 6.79±3.31 | 8.97±1.73 |
| 10d | c -Hexyl | 2-Me | 9.93±1.55 | 7.72±3.45 | 9.47±3.55 |
| 10e | c -Hexyl | 2-OMe | >25 | 12.3±9.70 | 18.6±9.01 |
| 10f | c-Hexyl | 2-CF3 | 9.12±4.44 | 2.00±0.80 | >25 |
| 10g | Bn | 3,5-Me | 2.75±1.92 | 9.15±0.28 | 6.23±1.62 |
| 10h | Bn | 4-NMe2 | 0.375±0.162 | 0.218±0.122 | 0.285±0.064 |
| 10i | t-Bu | 3,5-Me | 4.93±2.60 | 4.84±2.78 | 5.47±0.86 |
| 10j | t-Bu | 4-NMe2 | 2.47±0.74 | 1.46±0.77 | 1.74±0.26 |
| 10k | t-Bu | 2-F | 22.2±3.8 | 15.7±6.87 | 19.0±4.58 |
| 10l | t-Bu | 2-Me | 23.9±1.54 | 13.6±8.19 | 23.9±1.49 |
| 10m | t-Bu | 2-OMe | >25 | 18.4±9.34 | 24.5±0.75 |
| 10n | t-Bu | 2-CF3 | >25 | 18.6±9.05 | >25 |
| 10o | t-Bu | 2-Cl | 22.9±2.64 | 12.8±8.66 | 21.6±3.37 |
| 10p | t-Bu | 2-iPr | >25 | 12.2±9.36 | >25 |
| Vorinostat | - | - | 0.299±0.045 | 0.22±0.05 | 0.470±0.023 |
| Ricolinostat | - | 2.04±0.39 | 1.54±0.16 | 2.42±0.18 | |
| HPOB | - | - | 13.9±3.89 | 11.3±7.2 | 16.1±2.52 |
Extended functional analysis of 10h in leukemia cell lines.
To study the inhibitory effects of the hit compound 10h (with highest cytotoxicity) on the growth and apoptosis induction of leukemia cells, further functional assays were carried out. First, we evaluated the effects of 10h and on the cellular viability of a broad range of leukemia cell lines originated from therapy refractory subgroups; the class I selective HDACi CI994 (tacedinaline; phase III) and the HDAC6i ricolinostat were used as controls (Figure 5A and Supplementary Table S1, Supporting Information). In order to exclude the general cytoxicity effects (i.e. therapeutic window) of 10h exposure, two healthy human fibroblast controls were also included in the assays. 10h markedly outperformed CI994 and ricolinostat in inhibiting the cellular viability of most tested leukemia cell lines at a much lower IC50 concentration range than the healthy fibroblast controls (Supplementary Table S1, Supporting Information). Next, the induction of apoptosis following the treatment of HL60 cells with 1 μM or 5 μM of 10h (for 48 h) was determined (Figure 5B). The treated cells were stained with annexin V and propidium iodide to be analyzed by flow cytometry. The significant (p < 0.0001) increase in apoptotic cells observed upon treatment with 10h at 1 μM as well as 5 μM indicates that induction of apoptosis contributes to its the antiproliferative potential. To further determine the intracellular target specificity of 10h, HL60 cells were treated with the respective IC12.5, IC25, IC50, and IC75 concentrations of 10h and subsequently immunoblotted along with the reference HDAC6i ricolinostat at its IC50 concentration (Figure 5C). After incubation with 10h, a dose-dependent increase in the acetylation of α-tubulin and histone 3 (H3) was observed. As expected, ricolinostat-treated cells showed prominent acetylation of α-tubulin and lower Ac-H3 levels than observed for 10h.
Figure 5.
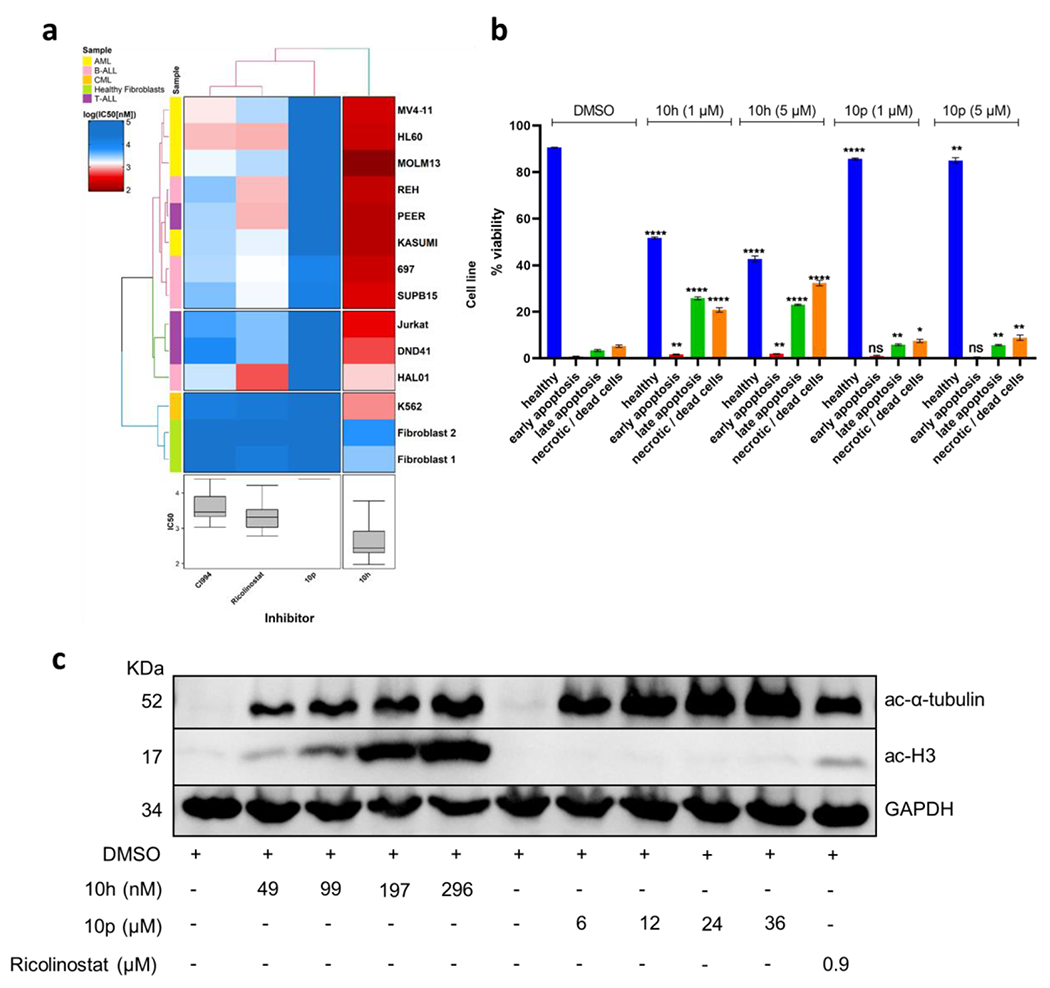
Cytotoxic and target specificity analysis of compounds 10h and 10p. (a) Comparative cellular viability (log IC50 nM) of different subgroups of leukemic cell lines and two healthy human fibroblast controls after treatment with 10h and 10p in comparison to the commercially available selective HDAC6 inhibitors ricolinostat and the HDAC class I inhibitor CI994. The IC50 data are plotted as a clustered heat map, followed by unsupervised hierarchical clustering. The horizontal axis of the dendrogram exemplifies the dissimilarity between clusters, whereas the color of the cell is related to its position along with a log IC50 (nM) gradient. (b) Annexin-PI staining of HL60 cells exposed to 10h and 10p at 1 μM and 5 μM after 48 h (n=3). (c) Immunoblot of HL60 cells after 24 h treatment with 10h, 10p, and ricolinostat at the indicated concentrations.
Resistance against single use of HDACi is often reported;55,56 however, co-administration of HDACi with either chemotherapeutic agents or targeted drugs can maximize their efficacy by minimizing the resistance development incidences and through reducing the toxicity by lowering the doses. We have previously shown the synergistic effects of HDACi in combination with proteasome inhibitors.49,57,58,59 In the present study, we focused on finding clinically relevant synergistic partners of HDACi which are frequently used in the therapy protocol for the treatment of leukemia. For instance, in previous preclinical reports, the combination of DNA hypomethylating agents along with HDACi was reported to be synergistic in different hematological malignancies, potentially by disrupting the transcriptional repressor complex involving methyl-CpG binding proteins and HDACs.31,60,61 Therefore, we evaluated the synergistic effects of 10h in combination with decitabine, a DNA methyl transferase inhibitor (DNMTi) used to treat myelodysplastic syndromes (Figure 6A). Interestingly, the combination of 10h and decitabine was found differentially synergistic in the tested myeloid lineage-derived cell lines HL60 (ZIP synergy score: 25.08) and MOLM13 (27.44) but not in the tested B- or T-lymphoid-lineage-originated leukemic cell lines HAL01 (1.18) and Jurkat (−0.87), respectively (Figure 6A). The observed synergism of 10h with decitabine later prompted us to examine the underlying molecular mechanism. The co-administration of 10h and decitabine revealed a strong induction of acetylation of H3 as compared to mono-treatment, thus indicating that class I HDAC inhibition is required for the synergistic activity with decitabine and the resulting enhancement of anticancer activity (Figure 6B).
Figure 6.
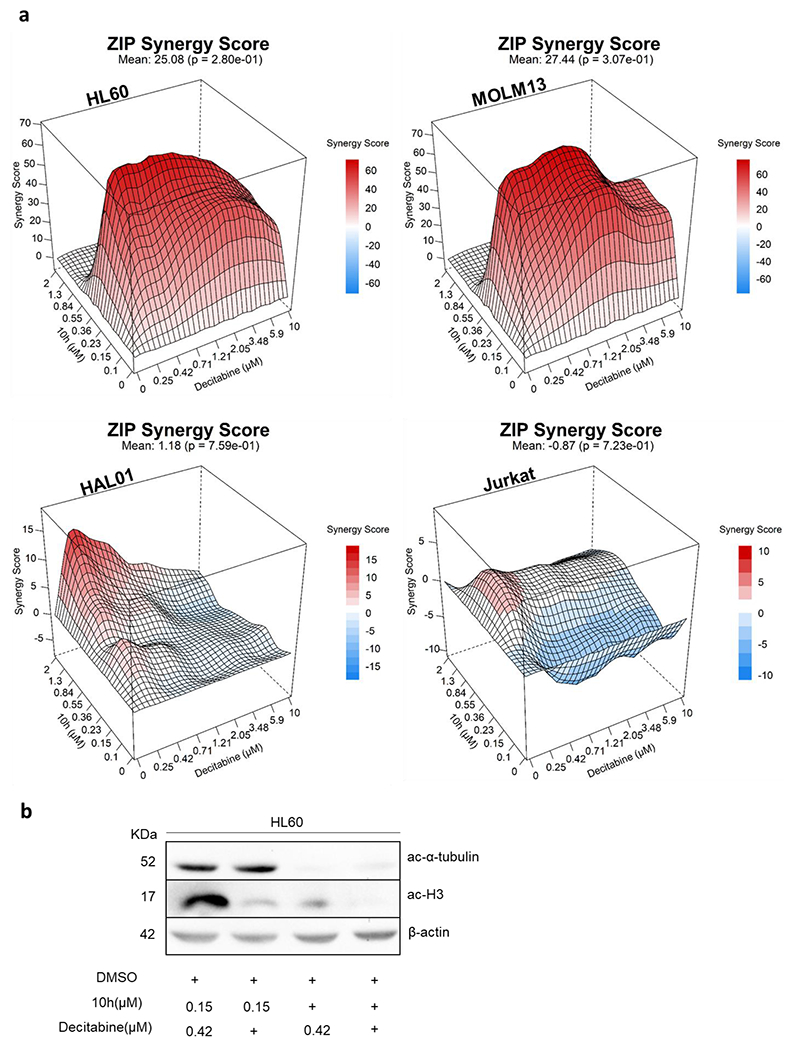
(a) Illustrative synergy plot of 10h after 72 h co-treatment of HL60, MOLM13 (AML), HAL01 (B-ALL), and Jurkat (T-ALL) leukemic cell lines with decitabine at depicted concentrations. The mean synergy score calculations were performed using ZIP model; visualizations were carried out using Synergy Finder webtool.62 (b) Immunoblotting of HL60 cells after 24 h of treatment with 10h alone and in combination with decitabine at the indicated concentrations.
Extended functional analysis of 10p in leukemia cell lines.
In parallel the inhibitory effects the most selective HDAC6i 10p on the leukemia cell growth and apoptosis induction were determined. As expected, 10p had a significantly weaker impact on the viability of tested leukemia cell lines, with IC50 values ranging in the high micromolar concentration (Figure 5A and Supplementary Table S1, Supporting Information). Similarly in the apoptosis assay, which was performed after treatment of HL60 cells with the 1 μM or 5 μM of 10p for 48h, a weaker (in comparison to 10h) although significant (p < 0.05) apoptosis induction was noticed (Figure 5B). Furthermore, in order to evaluate the intracellular specificity of 10p, HL60 cells were treated with the respective IC12.5, IC25, IC50, and IC75 concentrations of 10p and immunoblotted along with the reference HDAC6i ricolinostat at its IC50 concentration. In agreement, treatment with a selective HDAC6i 10p induced acetylation of α-tubulin, but had no effect on Ac-H3 levels even at concentrations of up to 36 μM (Figure 5C). In a similar manner, we next analyzed the combinatorial potential of 10p in combination with decitabine, which revealed a moderate (in comparison to 10h) synergism in the tested AML cell lines HL60 (7.78) and MOLM13 (7.18) (Figure 7).
Figure 7:
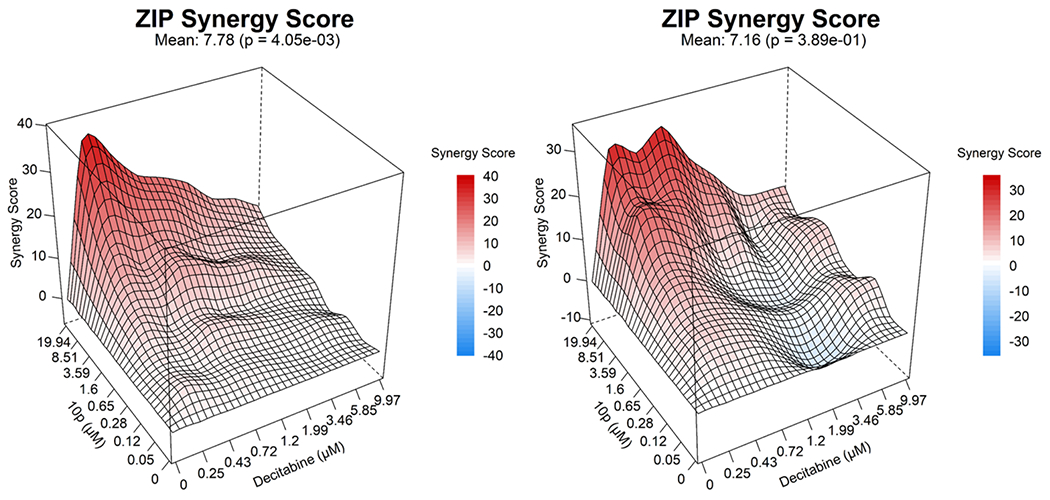
Illustrative synergy plot of 10p after 72 h co-treatment of HL60, MOLM13 (AML) leukemic cell lines with decitabine at depicted concentrations. The mean synergy score calculations were performed using ZIP model; visualizations were carried out using Synergy Finder webtool.62
Overall, these data support the therapeutic potential of 10h and 10p in leukemia, especially for AML when used in combination with decitabine.
CONCLUSIONS
Selected from a library of sixteen compounds that were synthesized using a simple, microwave-assisted 2-step protocol, 10h stood out due to its notable antileukemic effects and, importantly, with low cytotoxic activity toward healthy cells. Additional drug combinatorial drug screenings demonstrated a strong synergistic interaction of 10h with decitabine, which was especially prominent against AML cells. Designed as a pan-inhibitor, 10h proved potent inhibition of class I HDACs and HDAC6 in biochemical and cellular assays. The analysis of a crystal structure of the CD2 from zebrafish HDAC6 complexed with 10h disclosed differences in the zinc coordination but similar occupation of the L1 loop pocket in comparison to its non-fluorinated analog. The remarkable HDAC6 selectivity of our other hit compound 10p outmatched tubastatin A in biochemical enzyme inhibition assays and was confirmed in western blotting experiments. Despite moderate cytotoxicity on its own, the combination of 10p and decitabine displayed moderate synergism in AML cell lines, thus indicating the potential of 10p for combination therapies and targeted therapies involving HDAC6i. On the whole, our study provided valuable SAR data that can be used to fine-tune selectivity profiles toward HDAC6i or unselective, but highly cytotoxic pan-HDACi.
EXPERIMENTAL SECTION
Chemistry.
Dry MeOH was obtained from the MBraun MB SPS-800 solvent purification system. Except for DCM, which was purified by distillation prior to use, all reagents and solvents were purchased from commercial sources and used without further purification. Thin layer chromatography was carried out using Macherey-Nagel pre-coated aluminium foil sheets, which were visualized using UV light (254 nm). Hydroxamic acids were further stained using a 1% solution of iron(III) chloride in MeOH. 1H-NMR and 13C-NMR spectra were recorded at rt or, due to the occurrence of rotamers, at 60 °C using Bruker Avance III HD (400 MHz), and Varian/Agilent Mercury-plus (300 MHz & 400 MHz) spectrometers. Chemical shifts (δ) are quoted in parts per million (ppm). All spectra were standardized in accordance with the signals of the deuterated solvents (DMSO-d6: δH = 2.50 ppm; δC = 39.5 ppm; CDCl3: δH = 7.26 ppm; δC = 77.0 ppm). Coupling constants (J) are reported in Hertz (Hz). Mass-spectra were measured by the Leipzig University Mass Spectrometry Service via electrospray ionization (ESI) on Bruker Daltonics Impact II and Bruker Daltonics micrOTOF spectrometers. The uncorrected melting points were determined using a Barnstead Electrothermal 9100 apparatus. Analytical HPLC analysis were carried out using a Thermo Fisher Scientific UltiMate 3000 system equipped with an UltiMateTM HPG-3400SD pump, an UltiMateTM 3000 Dioden array detector, an UltiMateTM 3000 autosampler, and a TCC-3000SD standard thermostatted column compartment by Dionex. The system was operated using a Macherey-Nagel NUCLEODUR 100–5 C18 ec column (250 mm x 4.6 mm). UV absorption was detected at 254 nm with a linear gradient of 5% B to 95% B within 23 min. HPLC-grade water + 0.1% TFA (solvent A) and HPLC-grade acetonitrile + 0.1% TFA (solvent B) were used for elution at a flow rate of 1 mL/min. The purity of the final compounds was at least 95.0%.
Methyl 3-fluoro-4-methylbenzoate (2).
To a cooled (0 °C) solution of 3-fluoro-4-methylbenzoic acid (5.00 g, 32.5 mmol, 1.0 eq) in MeOH (150 mL) was added thionyl chloride (3.05 mL, 42.5 mmol, 1.3 eq) and the resulting mixture was stirred at rt for 16 h. Upon removal of the solvent under reduced pressure, the residue was dissolved in EtOAc (200 mL) and 10% HCl (50 mL) was added. The mixture was extracted with EtOAc (3 x 100 mL) and the combined organics were washed with sat. aq. NaHCO3 (2 x 20 mL) and water (1 x 20 mL). Drying over MgSO4 and evaporation of the solvent afforded 2 as a pale yellow liquid (5.10 g, 30.4 mmol, 93%). Spectroscopic data matched those reported in the literature.63
Methyl 4-(bromomethyl)-3-fluorobenzoate (3).
Synthesized according to the procedure reported in the literature.50 To a solution of 2 (4.91 g, 29.0 mmol, 1.0 eq) and NBS (7.83 g, 44.0 mmol, 1.5 eq) in DCM (150 mL) was added AIBN (476 mg, 2.90 mmol, 0.1 eq). The mixture was first refluxed for 10 h and then stirred at rt for another 16 h after which DCM (100 mL) was added. The resulting solution was washed with 1M NaOH (3 x 50 mL) and brine (2 x 50 mL) before it was dried over Na2SO4. Removal of the solvent under reduced pressure afforded the crude product 3 (quant.) as a colourless liquid that solidified upon storage at −18 °C. The crude product was used without further purification.
Methyl 4-(aminomethyl)-3-fluorobenzoate hydrochloride (5).
This compound was synthesized according to the procedure reported in the literature.50 A solution of 3 (1.62 g, 6.50 mmol, 1.0 eq) and NaN3 (510 mg, 7.80 mmol, 1.2 eq) in DMF (25 mL) was stirred at 80 °C for 12 h. After cooling to rt, brine (25 mL) was added and the resulting solution was extracted with a mixture of Et2O and cyclohexane (1:1; 3 x 100 mL). The combined organics were washed with brine (2 x 20 mL), dried over MgSO4 and concentrated under reduced pressure to afford 4 as a pale yellow liquid. The crude azide thus obtained was subsequently dissolved in a mixture of THF (40 mL) and water (4 mL) to which triphenylphosphine (3.00 g, 11.4 mmol, 1.8 eq) was added in small portions. After stirring at rt for 40 h, the solvent was evaporated. The residue was redissolved in DCM (200 mL) and extracted with 4M HCl (2 x 50 mL). The combined aqueous layers were then basified using 2M NaOH (pH 10) and extracted with DCM (3 x 100 mL). Drying of the combined organics over MgSO4 and removal of the solvent under reduced pressure afforded the crude amine which was precipitated from MeOH (2 mL), 37% HCl (0.5 mL), and Et2O (20 mL) to afford the hydrochloride salt 5 as a white solid (553 mg, 2.50 mmol, 39% over 2 steps); mp 236–241 °C; 1H NMR (400 MHz, DMSO-d6): δ 8.70 (s, 3H, NH3+), 7.87–7.62 (m, 3H, arom.), 4.12 (q, J = 5.8 Hz, 2H, CH2), 3.88 (s, 3H, OCH3) ppm; 13C NMR (101 MHz, DMSO-d6): δ 164.9, 161.2, 158.7, 132.0, 131.9, 131.7, 131.6, 126.6, 126.5, 125.2, 125.1, 115.9, 115.7, 52.6, 35.4 ppm; HRMS (m/z): MNa+ calcd for C9H10FNO2 184.0768, found 184.0771.
General procedure A.
The linker building block 5 (132 mg, 0.60 mmol, 1.2 eq), paraformaldehyde (18.0 mg, 0.60 mmol, 1.2 eq), and crushed molecular sieves 4 Å (50.0 mg) were suspended in MeOH (1.0 mL) and Et3N (0.08 mL, 0.60 mmol, 1.2 eq) was added. The mixture was subjected to microwave irradiation at 150 W and 45 °C for 20 min before the respective isonitrile (0.50 mmol, 1.0 eq) and the respective carboxylic acid (0.50 mmol, 1.0 eq) were added. The resulting mixture was again subjected to microwave irradiation at the same settings for 60 min after which the molecular sieves were removed by filtration and washed with DCM (10 mL). The filtrate was concentrated under reduced pressure and the residue was redissolved in DCM (100 mL), washed with 10% HCl (1 x 10 mL), water (1 x 10 mL), 1M NaOH (2 x 5 mL), and brine (1 x 10 mL). The organic layer was dried over MgSO4 and the solvent was removed under reduced pressure to afford the crude product 9 which was recrystallized from EtOAc (1 mL) and petrol (20 mL). The ester thus obtained was allowed to dry before it was added to a mixture of NaOH (200 mg, 5.00 mmol, 10 eq) and hydroxylamine (50% solution in water; 0.95 mL, 15.5 mmol, 31 eq) in MeOH (4 mL) and DCM (1 mL). The resulting solution was stirred at 0 °C for approx. 15 min until TLC (DCM/MeOH 9:1) indicated full conversion upon which the solvents were evaporated and the residue was dissolved in water (10 mL). Dropwise addition of 10% HCl (pH 8) induced precipitation of the hydroxamic acid 10 which was then isolated by filtration and washed with 5% HCl (2 x 3 mL) and chilled water (3 x 5 mL), successively. For compounds derived from tert-butyl isocyanide, precipitation of the product was often incomplete so that the aqueous layer was further extracted with EtOAC (3 x 30 mL). The combined organics were washed with brine (1 x 10 mL) and dried over NasSO4. Removal of the solvent under reduced pressure yielded the remaining product.
General procedure B.
The linker building block 5 (109 mg, 0.50 mmol, 1.0 eq), paraformaldehyde (15.0 mg, 0.50 mmol, 1.0 eq), and crushed molecular sieves 4 Å (50.0 mg) were suspended in MeOH (1.0 mL) and Et3N (0.08 mL, 0.60 mmol, 1.2 eq) was added. The mixture was subjected to microwave irradiation at 150 W and 45 °C for 30 min before the respective isonitrile (0.50 mmol, 1.0 eq) and 4-(dimethylamino)benzoic acid (83.0 mg, 0.50 mmol, 1.0 eq) were added. The resulting mixture was again subjected to microwave irradiation at the same settings for 3 h after which the molecular sieves were removed by filtration and washed with DCM (10 mL). The filtrate was concentrated under reduced pressure and the residue was redissolved in DCM (100 mL) and washed with 1M NaOH (2 x 5 mL), water (1 x 10 mL), and brine (1 x 10 mL). Drying over MgSO4 and evaporation of the solvent afforded the crude product 9 which was recrystallized from EtOAc (1 mL) and petrol (20 mL). The ester thus obtained was allowed to dry before it was added to a mixture of NaOH (200 mg, 5.00 mmol, 10 eq) and hydroxylamine (50% solution in water; 0.95 mL, 15.5 mmol, 31 eq) in MeOH (4 mL) and DCM (1 mL). The resulting solution was stirred at 0 °C for approx. 15 min until TLC (DCM/MeOH 9:1) indicated full conversion. The organic solvents were removed under reduced pressure and the residue was dissolved in water (10 mL). Dropwise addition of 10% HCl (pH 8) induced precipitation of the hydroxamic acid 10 which was then isolated by filtration and washed with chilled water (3 x 5 mL). For compound 10j derived from tert-butyl isocyanide, precipitation of the product was incomplete so that the aqueous layer was extracted with EtOAC (3 x 30 mL). The combined organics were washed with brine (1 x 10 mL) and dried over Na2SO4. Removal of the solvent under reduced pressure yielded the remaining product.
4-({N-[(Cyclohexylcarbamoyl)methyl]-1-(3,5-dimethylphenyl)formamido}methyl)-3-fluoro-N-hydroxybenzamide (10a).
Synthesis according to general procedure A using 3,5-dimethylbenzoic acid (75.0 mg) and cyclohexyl isocyanide (0.06 mL) afforded 10a as a white solid (86.0 mg, 0.18 mmol, 38%); mp 177 °C; tR: 8.02 min, purity: 99.7%; 1H NMR (400 MHz, DMSO-d6, 20 °C): δ 7.75 (d, J = 8.2 Hz, 1H, NH), 7.63–7.31 (m, 3H, arom.), 7.10–6.93 (m, 3H, arom.), 4.68/4.52 (2 x s, 2H, CH2), 3.91/3.72 (2 x s, 2H, CH2), 3.52 (s, 1H, CH), 2.26/1.80 (2 x s, 6H, 2 x CH3), 1.74–0.97 (m, 10H, c-Hexyl) ppm; 13C NMR (101 MHz, DMSO-d6): δ 171.6, 166.7, 162.4, 161.3, 158.9, 137.6, 135.9, 130.9, 129.8, 124.1, 122.9, 113.7, 113.4, 51.4, 47.7, 43.0, 32.4, 32.3, 25.2, 24.4, 20.8 ppm; HRMS (m/z): MNa+ calcd for C25H30FN3O4 478.2113, found 478.2128.
4-({N-[(Cyclohexylcarbamoyl)methyl]-1-[4-(dimethylamino)phenyl]formamido}methyl)-3-fluoro-N-hydroxybenzamide (10b).
Synthesis according to general procedure B using cyclohexyl isocyanide (0.06 mL) afforded 10b as a white solid (162 mg, 0.34 mmol, 69%); mp 123 °C; tR: 6.38 min, purity: 98.1%; 1H NMR (400 MHz, DMSO-d6, 20 °C): δ 11.31 (s, 1H, NH-OH), 9.17 (s, 1H, OH), 7.81 (s, 1H, NH), 7.70–7.26 (m, 5H, arom.), 6.78–6.62 (m, 2H, arom.), 4.64 (s, 2H, CH2), 3.85 (s, 2H, CH2), 3.55 (d, J = 9.8 Hz, 1H, CH), 2.93 (s, 6H, 2 x CH3), 1.76–1.45 (m, 5H, c-Hexyl), 1.31–1.02 (m, c, 5H-Hexyl) ppm; 13C NMR (101 MHz, DMSO-d6): δ 171.7, 166.9, 162.5, 161.2, 158.9, 151.3, 133.9, 129.8, 128.7, 127.5, 127.4, 123.0, 121.9, 113.7, 113.5, 111.0, 47.6, 32.3, 25.2, 24.5 ppm; HRMS (m/z): MNa+ calcd for C25H31FN4O4 493.2222, found 493.2214.
4-({N-[(Cyclohexylcarbamoyl)methyl]-1-(2-fluorophenyl)formamido}methyl)-3-fluoro-N-hydroxybenzamide (10c).
Synthesis according to general procedure A using 2-fluorobenzoic acid (70.0 mg) and cyclohexyl isocyanide (0.06 mL) afforded 10c as a pink solid (85.0 mg, 0.19 mmol, 38%); mp 179 °C; tR: 7.40 min, purity: 96.6%; 1H NMR (300 MHz, DMSO-d6, 20 °C): δ 11.30 (d, J = 4.7 Hz, 1H, NH-OH), 9.14 (s, 1H, OH), 7.77–7.16 (m, 8H, arom., NH), 4.73/4.50 (2 x s, 2H, CH2), 3.98/3.73 (2 x s, 2H, CH2), 3.47–3.40 (m, 1H, CH), 1.81–1.43 (m, 5H, c-Hexyl), 1.36–0.89 (m, 5H, c-Hexyl) ppm; 13C NMR (101 MHz, DMSO-d6): δ 166.5, 166.0, 165.9, 162.6, 161.3, 161.1, 158.9, 158.8, 158.7, 156.5, 156.3, 133.9, 131.5, 131.4, 129.8, 129.7, 129.6, 128.72, 128.68, 128.4, 127.1, 126.9, 124.8, 124.7, 124.6, 124.0, 123.8, 122.9, 115.9, 115.7, 113.9, 113.6, 50.9, 47.62, 47.55, 47.1, 47.0, 43.2, 32.4, 32.1, 25.2, 25.1, 24.5, 24.3 ppm; HRMS (m/z): M− calcd for C23H25F2N3O4 444.1740, found 444.1741.
4-({N-[(Cyclohexylcarbamoyl)methyl]-1-(2-methylphenyl)formamido}methyl)-3-fluoro-N-hydroxybenzamide (10d).
Synthesis according to general procedure A using 2-methylbenzoic acid (68.0 mg) and cyclohexyl isocyanide (0.06 mL) afforded 10d as a pink solid (84.0 mg, 0.19 mmol, 38%); mp 115 °C; tR: 7.55 min, purity: 95.1%; 1H NMR (400 MHz, DMSO-d6, 60 °C): δ 11.12 (s, 1H, NH-OH), 8.99 (s, 1H, OH), 7.71–7.37 (m, 4H, arom., NH), 7.37–7.06 (m, 4H, arom.), 4.75/4.41 (2 x s, 2H, CH2), 4.01/3.64 (2 x s, 2H, CH2), 3.48 (d, J = 9.7 Hz, 1H, CH), 2.30/2.22 (2 x s, 3H, CH3), 1.82–1.47 (m, 5H, c-Hexyl), 1.36–0.95 (m, 5H, c-Hexyl) ppm; 13C NMR (75 MHz, DMSO-d6): δ 171.1, 166.2, 135.8, 134.3, 134.1, 130.3, 128.8, 125.7, 125.6, 123.0, 113.9, 113.6, 50.6, 47.7, 47.6, 42.5, 40.1, 32.4, 32.2, 25.2, 25.1, 24.5, 24.4, 18.5 ppm; HRMS (m/z): M− calcd for C24H28FN3O4 440.1991, found 440.1988.
4-({N-[(Cyclohexylcarbamoyl)methyl]-1-(2-methoxyphenyl)formamido}methyl)-3-fluoro-N-hydroxybenzamide (10e).
Synthesis according to general procedure A using 2-methoxybenzoic acid (76.0 mg) and cyclohexyl isocyanide (0.06 mL) afforded 10e as a white solid (121 mg, 0.26 mmol, 53%); mp 130 °C; tR: 7.33 min, purity: 95.1%; 1H NMR (400 MHz, DMSO-d6, 20 °C): δ 11.31 (d, J = 6.8 Hz, 1H, NH-OH), 9.14 (s, 1H, OH), 7.69–6.92 (m, 8H, arom. NH), 4.95 (s, 1H, CH2), 4.46–3.99 (m, 2H, CH2), 3.83–3.57 (m, 4H, OCH3, CH2), 3.47–3.39 (m, 1H, CH), 1.80–1.43 (m, 5H, c-Hexyl), 1.32–0.90 (m, 5H, c-Hexyl) ppm; 13C NMR (75 MHz, DMSO-d6): δ 169.2, 169.1, 166.3, 166.2, 162.7, 161.6, 158.3, 154.9, 154.5, 133.61, 130.57, 130.4, 129.0, 127.7, 125.3, 125.1, 123.0, 120.5, 113.7, 113.4, 111.4, 55.6, 55.3, 50.7, 47.5, 42.7, 32.4, 32.2, 25.1, 24.5, 24.4 ppm; HRMS (m/z): MH+ calcd for C24H28FN3O5 458.2086, found 458.2066.
4-({N-[(Cyclohexylcarbamoyl)methyl]-1-[2-(trifluoromethyl)phenyl]formamido}methyl)-3-fluoro-N-hydroxybenzamide (10f).
Synthesis according to general procedure A using 2-(trifluoromethyl)benzoic acid (95.0 mg) and cyclohexyl isocyanide (0.06 mL) afforded 10f as a white solid (142 mg, 0.29 mmol, 57%); mp 107 °C; tR: 7.84 min, purity: 95.0%; 1H NMR (400 MHz, DMSO-d6, 20 °C): δ 11.31 (d, J = 8.4 Hz, 1H, NH-OH), 9.15 (s, 1H, OH), 7.95–7.23 (m, 8H, arom., NH), 5.27–5.07/4.60–4.24 (2 x m, 2H, CH2), 3.70–3.43 (m, 3H, CH, CH2), 1.81–1.43 (m, 5H, c-Hexyl), 1.36–0.91 (m, 5H, c-Hexyl) ppm; 13C NMR (101 MHz, DMSO-d6): δ 168.6, 168.5, 166.0, 162.5, 161.4, 159.0, 158.7, 134.2, 134.1, 134.0, 132.7, 132.0, 131.5, 131.4, 130.5, 130.0, 129.8, 129.7, 128.8, 128.7, 127.6, 126.7, 126.54, 126.49, 126.45, 125.5, 125.2, 125.0, 124.9, 122.8, 122.3, 113.8, 113.6, 50.6, 47.7, 47.5, 47.0, 42.5, 32.4, 32.1, 25.2, 25.1, 24.5, 24.3 ppm; HRMS (m/z): M− calcd for C24H25F4N3O4 494.1708, found 494.1718.
4-({N-[(Benzylcarbamoyl)methyl]-1-(3,5-dimethylphenyl)formamido}methyl)-3-fluoro-N-hydroxybenzamide (10g).
Synthesis according to general procedure A using 3,5-dimethylbenzoic acid (75.0 mg) and benzyl isocyanide (0.06 mL) afforded 10g as a dark brown solid (85.0 mg, 0.18 mmol, 37%); mp 215 °C; tR: 7.77 min, purity: 95.2%; 1H NMR (300 MHz, DMSO-d6, 20 °C): δ 11.31 (s, 1H, NH-OH), 9.14 (s, 1H, OH), 8.42 (s, 1H, NH), 7.75–6.92 (m, 11H, arom.), 4.71/4.59 (s, 2H, CH2), 4.36–4.23 (m, 2H, CH2), 4.02/3.87 (s, 2H, CH2), 2.24 (s, 6H, 2 x CH3) ppm; 13C NMR (101 MHz, DMSO-d6): δ 171.6, 167.8, 139.4, 139.3, 139.1, 139.0, 137.6, 136.1, 135.8, 131.1, 130.9, 129.9, 129.8, 129.6, 129.51, 129.45, 128.3, 127.2, 126.9, 124.1, 123.1, 123.0, 113.9, 113.7, 51.5, 43.1, 42.2, 20.8 ppm; HRMS (m/z): M− calcd for C26H26FN3O4 462.1835, found 462.1839.
4-({N-[(Benzylcarbamoyl)methyl]-1-[4-(dimethylamino)phenyl]formamido}methyl)-3-fluoro-N-hydroxybenzamide (10h).
Synthesized according to general procedure B using benzyl isocyanide (0.06 mL). The reaction time for the ester formation was reduced to 60 min (imine formation: 20 min, U4CR: 40 min). 10h was obtained as an off-white solid (162 mg, 0.34 mmol, 68%); mp 205 °C; tR: 6.35 min, purity: 97.2%; 1H NMR (400 MHz, DMSO-d6, 20 °C): δ 11.31 (s, 1H, NH-OH), 9.16 (s, 1H, OH), 8.47 (t, J = 6.0 Hz, 1H, NH), 7.68–7.43 (m, 3H, arom.), 7.39–7.16 (m, 7H, arom.), 6.74–6.58 (m, 2H, arom.), 4.69 (s, 2H, CH2), 4.30 (d, J = 5.8 Hz, 2H, CH2), 3.96 (s, 2H, CH2), 2.93 (s, 6H, NMe2) ppm; 13C NMR (101 MHz, DMSO-d6): δ 171.8, 168.2, 162.5, 158.7, 151.3, 139.2, 133.8, 128.6, 128.3, 127.2, 126.8, 123.0, 113.8, 110.9, 42.1 ppm; HRMS (m/z): MNa+ calcd for C26H27FN4O4 501.1909, found 501.1910.
4-({N-[(tert-Butylcarbamoyl)methyl]-1-(3,5-dimethylphenyl)formamido}methyl)-3-fluoro-N-hydroxybenzamide (10i).
Synthesis according to general procedure A using 3,5-dimethylbenzoic acid (75.0 mg) and tert-butyl isocyanide (0.06 mL) afforded 10i as a white solid (125 mg, 0.29 mmol, 58%); mp 162 °C; tR: 7.72 min, purity: 97.9%; 1H NMR (400 MHz, DMSO-d6): δ 11.31 (s, 1H, NH-OH), 9.14 (s, 1H, OH), 7.69–7.28 (m, 4H, arom., NH), 7.12–6.87 (m, 3H, arom.), 4.67/4.51 (2 x s, 2H, CH2), 3.88/3.69 (2 x s, 2H, CH2), 2.27 (s, 6H, 2 x CH3), 1.42/1.24/1.21 (s, 9H, t-Bu) ppm; 13C NMR (101 MHz, DMSO-d6): δ 171.6, 166.6, 162.6, 161.3, 161.1, 158.9, 137.6, 137.5, 136.0, 133.8, 131.5, 131.4, 130.8, 129.9, 128.8, 128.7, 127.4, 127.3, 124.1, 123.5, 123.0, 113.8, 113.6, 51.6, 50.2, 43.1, 28.5, 28.4, 20.8 ppm; HRMS (m/z): M− calcd for C23H28FN3O4 428.1991, found 428.1992.
4-({N-[(tert-Butylcarbamoyl)methyl]-1-[4-(dimethylamino)phenyl]formamido}methyl)-3-fluoro-N-hydroxybenzamide (10j).
Synthesis according to general procedure B using tert-butyl isocyanide (0.06 mL) afforded 10j as an off-white solid (85.0 mg, 0.19 mmol, 38%); mp 132 °C; tR: 6.10 min, purity: 95.5%; 1H NMR (400 MHz, DMSO-d6, 20 °C): δ 11.22 (s, 1H, NH-OH), 9.14 (s, 1H, OH), 7.71–7.23 (m, 6H, arom., NH), 6.78–6.62 (m, 2H, arom.), 4.63 (s, 2H, CH2), 3.82 (s, 2H, CH2), 2.93 (s, 6H, NMe2), 1.22 (s, 9H, t-Bu) ppm; 13C NMR (101 MHz, DMSO-d6): δ 171.7, 169.3, 167.3, 162.5, 161.3, 151.3, 133.7, 131.5, 131.4, 129.7, 128.8, 128.7, 127.7, 127.5, 123.0, 122.0, 113.8, 113.6, 111.0, 58.4, 50.3, 49.9, 28.43, 28.38 ppm; HRMS (m/z): MNa+ calcd for C23H29FN4O4 467.2065, found 467.2071.
4-({N-[(tert-Butylcarbamoyl)methyl]-1-(2-fluorophenyl)formamido}methyl)-3-fluoro-N-hydroxybenzamide (10k).
Synthesis according to general procedure A using 2-fluorobenzoic acid (70.0 mg) and tert-butyl isocyanide (0.06 mL) afforded 10k as an orange solid (133 mg, 0.32 mmol, 63%); mp 111 °C; tR: 7.10 min, purity: 96.8%; 1H NMR (400 MHz, DMSO-d6, 20 °C): δ 9.21 (s, 1H, OH), 7.73–7.12 (m, 8H, arom. NH), 4.72/4.47 (s, 2H, CH2), 3.95/3.75 (2 x s, 2H, CH2), 1.44/1.24/1.17/1.14 (4 x s, 9H, t-Bu) ppm; 13C NMR (101 MHz, DMSO-d6): δ 166.9, 166.4, 166.1, 162.5, 161.3, 161.2, 158.9, 158.7, 134.4, 134.0, 133.9, 131.5, 131.4, 129.9, 129.6, 128.8, 128.7, 128.5, 127.1, 126.9, 126.1, 124.8, 124.6, 124.0, 123.9, 122.9, 115.9, 115.7, 113.8, 113.6, 51.2, 50.2, 47.3, 46.9, 43.3, 28.5, 28.3, 27.9 ppm; HRMS (m/z): M− calcd for C21H23F2N3O4 418.1584, found 418.1584.
4-({N-[(tert-Butylcarbamoyl)methyl]-1-(2-methylphenyl)formamido}methyl)-3-fluoro-N-hydroxybenzamide (10l).
Synthesis according to general procedure A using 2-methylbenzoic acid (68.0 mg) and tert-butyl isocyanide (0.06 mL) afforded 10l as an off-white solid (105 mg, 0.25 mmol, 50%); mp 109 °C; tR: 7.24 min, purity: 96.4%; 1H NMR (300 MHz, DMSO-d6, 60 °C): δ 11.05 (s, 1H, NH-OH), 8.98 (s, 1H, OH), 7.70–7.12 (m, 8H, arom., NH), 4.74/4.41 (2 x s, 2H, CH2), 3.93/3.60 (2x s, 2H, CH2), 2.30/2.22 (2 x s, 3H, CH3), 1.47/1.27/1.18/1.17 (s, 9H, t-Bu) ppm; 13C NMR (101 MHz, DMSO-d6): δ 171.2, 171.0, 166.5, 162.5, 162.3, 135.9, 134.3, 134.0, 133.94, 133.86, 132.0, 131.5, 131.4, 130.3, 129.8, 128.83, 128.78, 128.7, 127.4, 127.2, 125.7, 125.6, 125.5, 123.0, 113.9, 113.6, 51.0, 50.2, 46.7, 46.5, 42.7, 28.5, 28.3, 18.5 ppm; HRMS (m/z): M− calcd for C22H26FN3O4 414.1835, found 414.1835.
4-({N-[(tert-Butylcarbamoyl)methyl]-1-(2-methoxyphenyl)formamido}methyl)-3-fluoro-N-hydroxybenzamide (10m).
Synthesis according to general procedure A using 2-methoxybenzoic acid (76.0 mg) and tert-butyl isocyanide (0.06 mL) afforded 10m as a white solid (83.0 mg, 0.19 mmol, 38%); mp 126 °C; tR: 7.54 min, purity: 95.6%; 1H NMR (300 MHz, DMSO-d6, 60 °C): δ 9.46–8.85 (br s, 1H, OH), 7.71–6.80 (m, 8H, arom., NH), 4.42 (s, 1H, CH2), 4.01–3.40 (m, 6H, OCH3, 2 x CH2), 1.47–1.09 (m, 9H, t-Bu) ppm; 13C NMR (101 MHz, DMSO-d6): δ 169.4, 169.3, 169.1, 166.7, 166.5, 162.6, 161.2, 158.7, 154.9, 154.7, 154.5, 134.0, 133.6, 133.5, 132.1, 131.5, 131.4, 130.7, 130.6, 130.4, 129.1, 129.0, 128.8, 128.7, 127.8, 127.7, 127.5, 127.4, 125.4, 125.1, 123.0, 122.8, 122.7, 120.6, 120.5, 113.7, 113.4, 111.5, 111.4, 55.6, 55.3, 51.0, 50.2, 47.7, 46.5, 42.6, 42.6, 28.5, 28.3 ppm; HRMS (m/z): M− calcd for C22H26FN3O5 430.1784, found 430.1784.
4-({N-[(tert-Butylcarbamoyl)methyl]-1-[2-(trifluoromethyl)phenyl]formamido}methyl)-3-fluoro-N-hydroxybenzamide (10n).
Synthesis according to general procedure A using 2-(trifluoromethyl)benzoic acid (95.0 mg) and tert-butyl isocyanide (0.06 mL) afforded 10n as a yellow solid (103 mg, 0.22 mmol, 44%); mp 137 °C; tR: 7.03 min, purity: 95.1%; 1H NMR (300 MHz, DMSO-d6, 60 °C): δ 10.91 (s, 1H, NH-OH), 9.00 (s, 1H, OH), 7.89–7.17 (m, 8H, arom., NH), 5.11/4.47 (2 x s, 2H, CH2), 3.90/3.57 (2 x s, 2H, CH2), 1.46/1.26/1.18 (3 x s, 9H, t-Bu) ppm; 13C NMR (75 MHz, DMSO-d6): δ 168.5, 166.4, 166.2, 162.5, 162.4, 161.8, 134.4, 134.32, 134.29, 134.1, 134.04, 133.99, 132.71, 132.68, 132.6, 132.06, 132.05, 132.0, 131.5, 131.4, 130.52, 130.45, 130.4, 129.8, 129.7, 128.8, 128.7, 127.73, 127.67, 127.6, 126.8, 126.6, 126.51, 126.49, 126.4, 125.2, 124.8, 122.9, 122.81, 122.79, 121.8, 113.91, 113.87, 113.6, 50.8, 50.3, 42.7, 42.6, 28.5, 28.3 ppm; HRMS (m/z): M− calcd for C22H23F4N3O4 468.1552, found 468.1548.
4-({N-[(tert-Butylcarbamoyl)methyl]-1-(2-chlorophenyl)formamido}methyl)-3-fluoro-N-hydroxybenzamide (10o).
Synthesis according to general procedure A using 2-chlorobenzoic acid (78.0 mg) and tert-butyl isocyanide (0.06 mL) afforded 10o as a white solid (150 mg, 0.34 mmol, 69%); mp 145 °C; tR: 7.27 min, purity: 98.6%; 1H NMR (400 MHz, DMSO-d6, 20 °C): δ 11.27 (s, 1H, NH-OH), 9.15 (s, 1H, OH), 7.71–7.24 (m, 8H, arom., NH), 5.27–5.05/4.73–4.22 (2 x m, 2H, CH2), 4.05–3.79/3.72–3.52 (2 x m, 2H, CH2), 1.24/1.14 (2 x s, 9H, t-Bu) ppm; 13C NMR (101 MHz, DMSO-d6): δ 168.3, 168.0, 166.3, 166.1, 162.5, 161.3, 161.2, 158.8, 158.7, 135.30, 135.27, 134.3, 134.2, 133.9, 133.9, 131.5, 131.4, 130.7, 130.6, 130.1, 130.04, 129.97, 129.9, 129.6, 129.4, 129.4, 128.9, 128.8, 128.7, 128.3, 128.1, 127.4, 127.3, 127.0, 126.9, 50.8, 50.3, 47.3, 46.6, 42.9, 42.9, 28.5, 28.3 ppm; HRMS (m/z): M− calcd for C21H23ClFN3O4 434.1288, found 434.1273.
4-({N-[(tert-Butylcarbamoyl)methyl]-1-[2-(propan-2-yl)phenyl]formamido}methyl)-3-fluoro-N-hydroxybenzamide (10p).
Synthesis according to general procedure A using 2-isopropylbenzoic acid (82.0 mg) and tert-butyl isocyanide (0.06 mL) afforded 10p as a white solid (132 mg, 0.30 mmol, 60%); mp 130 °C; tR: 7.96 min, purity: 96.4%; 1H NMR (400 MHz, DMSO-d6, 20 °C): δ 11.31 (s, 1H, NH-OH), 9.116 (s, 1H, OH), 7.68–7.09 (m, 8H, arom., NH), 4.98–4.82/4.64–4.08 (2 x m, 2H, CH2), 3.81–3.70/3.66–3.48 (2 x m, 2H, CH2), 3.12 (2 x p, J = 6.7/6.9 Hz, 1H, i-Pr-CH), 1.38–1.04 (m, 15H, i-Pr, t-Bu) ppm; 13C NMR (101 MHz, DMSO-d6): δ 171.1, 166.6, 166.5, 162.5, 161.4, 161.0, 159.0, 158.6, 145.1, 144.9, 135.0, 134.9, 134.0, 133.9, 131.5, 131.4, 129.6, 129.3, 129.2, 128.8, 128.7, 127.3, 127.2, 125.8, 125.7, 125.6, 125.5, 123.1, 123.0, 113.9, 113.7, 51.0, 50.3, 46.9, 42.3, 29.9, 29.8, 28.5, 28.3, 24.9, 24.2, 23.2, 22.8 ppm; HRMS (m/z): MNa+ calcd for C24H30FN3O4 466.2113, found 466.2128.
Crystallization.
Catalytic domain 2 of HDAC6 from Danio rerio (hereafter noted as simply “HDAC6”) was recombinantly expressed in E. coli BL21 (DE3) cells via inoculation with His-MBP-TEV-HDAC6-pET28a(+) vector. The protein was purified using techniques previously described.64
The HDAC6-10h complex was crystallized via sitting drop vapor diffusion at 4 °C. Briefly, a 100-nL drop of protein solution [10 mg/mL HDAC6, 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.5), 100 mM KCl, 5% glycerol (v/v), 1mM tris(2-carboxyethyl)phosphine (TCEP), 10h (2 mM), and 5% DMSO (v/v)] was added to a 100-nL drop` of precipitant solution from the PEG/ION™ screen [2% v/v Tacsimate pH 8.0, 0.1 M Tris pH 8.5, and 16% w/v PEG 3,350] and equilibrated against 80 μL of precipitant solution. Crystals appeared after 2 weeks and were soaked in a cryoprotectant solution comprised of the mother liquor solution and 20% ethylene glycol prior to looping and flash cooling in liquid nitrogen.
Data Collection and Structure Determination.
X-ray diffraction data for the HDAC6-10h complex were collected on the NSLS-II AMX beamline at Brookhaven National Laboratory. Data were indexed and integrated using iMosflm65; calculation of the Patterson map and self-rotation function indicated the presence of pseudotranslation with a peak height ~28% of that of the origin peak. Weak reflections resulting from the pseudotranslation were removed during phasing and refinement. The structure was phased by molecular replacement using the atomic coordinates of unliganded HDAC6 (PDB 5EEM)19 as a search probe in the program Phaser.66 The atomic structures were then modeled and refined using Coot67 and Phenix68 and the 10h inhibitor was built into well-defined electron density in the final stages of refinement. The quality of the final model was evaluated with MolProbity.69 Data collection and refinement statistics are recorded in Table S2 Atomic coordinates and crystallographic structure factors have been deposited in the Protein Data Bank with accession code 7U8Z.
Biological experiments
Cell Culture.
Leukemia cell lines HL60, MOLM13, MV4-11 (acute myeloid leukemia or AML), HAL01, REH, 697 (B-cell acute lymphoblastic leukemia or B-ALL), K562 (chronic myeloid leukemia), Jurkat (T-cell acute lymphoblastic leukemia) (DSMZ, Braunschweig, Germany) and two healthy fibroblast control cells (ATCC, Virginia, US) were cultured in RPMI 1640 GlutaMax mdeium supplemented with 10-20% FCS. All cell lines were grown at 37°C under humidified air supplemented with 5% CO2.
Cell Viability Assay.
The CellTiter-Glo luminescent cell viability assay (Promega, Madison) was done to determine the IC50 values of the tested compounds for every cell line. The different compounds were printed on white 384-well plates (Thermo Fisher Scientific, Waltham) with at least 5 different, increasing concentrations (5 nM – 25 μM) using a digital dispenser (D300e, Tecan, Männedorf, Switzerland). The cell viability was monitored after 72 h using the CellTiter-Glo luminescent assay using a microplate reader (Spark, Tecan). The IC50 values for the tested compounds were determined by plotting raw data (normalized to controls) using a sigmoid dose curve and nonlinear regression (GraphPad Prism Inc., San Diego, CA).
Combinatorial drug screening.
10h and decitabine were printed on white 384-well plates (Thermo Fisher Scientific, Waltham, USA) with increasing concentrations in a dose-response 8 x 8 matrices using a digital dispenser (D300e, Tecan, Maennedorf, Switzerland). 10h was printed in concentration range from 0.1 μM up to 2 μM and for decitabine from 0.25 μM up to 10 μM, in a logarithmic distribution. The viability was monitored after 72 h using CellTiter-Glo luminescent assay (as described above), using a microplate reader (Spark, Tecan). The ZIP synergy scores calculations were based on the ZIP model70 using the Synergy Finder tool (https://synergyfinder.fimm.fi/).
Annexin-V staining.
For evaluating apoptosis, cells treated with respective compounds or control at the indicated concentrations for 48 h were stained with annexin V and PI and later subjected to FACS analysis, following supplier’s guidelines (Invitrogen, Carlsbad, CA).
Immunoblotting.
Whole-cell lysates were produced after 24 h treatment with the respective compounds under standard culture conditions. Afterwards, whole-cell lysates were immunoblotted using anti-acetyl-α-tubulin (no. 5335), anti-acetyl-histone H3 (no. 9677S), and anti-β-Actin (no. 5125S) or anti-GAPDH (no. 97166), following supplier’s guidelines (Cell Signaling Technology, Danvers, MA).
Inhibition assay for HDAC1-3 and HDAC6.
In vitro inhibitory activities against human HDAC1, HDAC2, HDAC3/NcoR2, and HDAC6 were measured using a previously published protocol.71 In short, OptiPlate-96 black microplates (Perkin Elmer) were used with an assay volume of 50 μL. 5 μL test compound or control, diluted in assay buffer (50 mM Tris-HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 0.1 mg/mL BSA), were incubated with 35 μL of the fluorogenic substrate ZMAL (Z-Lys(Ac)-AMC)72 (21.43 μM in assay buffer) and 10 μL of human recombinant HDAC1 (BPS Bioscience, Catalog# 50051), HDAC2 (BPS Bioscience, Catalog# 50052), HDAC3/NcoR2 (BPS Bioscience, Catalog# 50003) or full length HDAC6 (BPS Bioscience, Catalog# 50006) at 37 °C. After an incubation time of 90 min, 50 μL of 0.4 mg/mL trypsin in trypsin buffer (50 mM Tris-HCl, pH 8.0, 100 mM NaCl) were added, followed by further incubation at 37 °C for 30 min. Fluorescence was measured with an excitation wavelength of 355 nm and an emission wavelength of 460 nm using a Fluoroskan Ascent microplate reader (Thermo Scientific). All compounds were evaluated in duplicates in at least two independent experiments
HDAC4 inhibition assay.
In vitro inhibitory activities against HDAC4 were determined using a HDAC4 assay protocol adapted from Asfaha et al.73 For compounds and controls, 3-fold serial dilutions of the respective DMSO-stock solutions were prepared in assay buffer (50 mM Tris-HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1.0 mM MgCl2·6 H2O, 0.1 mg/mL BSA). 5.0 μL of these serial dilutions were transferred into 96-well microplates (OptiPlate-96 Black, PerkinElmer). 35 μL of the fluorogenic substrate Boc-Lys(Tfa)-AMC (Bachem, Catalog# 4060676, 42.86 μM in assay buffer) and 10 μL of HDAC4 (BPS Bioscience, Catalog# 50004) enzyme solution were added. The total assay volume (50 μL, max. 1% DMSO) was incubated at 37 °C for 90 min. Subsequently, 50 μL of trypsin (1.0 mg/mL) in trypsin buffer (50 mM Tris-HCl, pH 8.0, 100 mM NaCl) were added, followed by additional 30 min of incubation at 37 °C. Fluorescence (excitation: 355 nm, emission: 460 nm) was measured using a FLUOstar OPTIMA microplate reader (BMG LABTECH). IC50 values were determined by generating normalized dose-response curves using the build-in “log(inhibitor) vs. response (three parameters)” equation provided by GraphPad Prism (GraphPad Prism 9.0, San Diego, USA). All compounds were tested in duplicates, reported mean IC50 values, including standard deviation, are calculated from at least two independent experiments. TMP26974 (Selleck Chemicals, Catalog# S7324) was used as control compound (IC50 = 0.488 ± 0.049 μM).
PAINS Analysis.
We filtered all compounds for pan-assay interference compounds (PAINS) using the online filter http://zinc15.docking.org/patterns/home/.75 No compound was flagged as PAINS.
Supplementary Material
ACKNOWLEDGMENTS
This work is based on research conducted at beamline 17-ID-1 (AMX) of the National Synchrotron Light Source II, a Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract DE-SC0012704. The Center for BioMolecular Structure (CBMS) is primarily supported by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS), through a Center Core P30 Grant (P30GM133893) and by the DOE Office of Biological and Environmental Research (KP1607011).
Funding Sources
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) (HA 7783/1-1 and 7783/1-2 to FKH) and the US National Institutes of Health (grant GM49758 to D.W.C.). S.B. acknowledges the financial support by Forschungskommission (2021-19) and DSO-Netzwerkverbundes, HHU Düsseldorf. This study was funded in part by KinderKrebsForschung e.V and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 270650915 (Research Training Group GRK 2158, TP2d to SB). A.B. is supported by the TransOnc priority program of the German Cancer Aid within grant #70112951 (ENABLE). AB additionally acknowledges the financial support from Löwenstern e.V. and from Katharina-Hardt Foundation.
ABBREVIATIONS
- AIBN
azobisisobutyronitrile
- CDCl3
chloroform-d
- DMF
dimethylformamide
- DMSO
dimethylsulfoxide
- DCM
dichloromethane
- Et2O
diethyl ether
- EtOAc
ethyl acetate
- MeOH
methanol
- min
minutes
- NBS
N-bromosuccinimide
- petrol
petroleum ether
- rt
room temperature
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- U4CR
Ugi 4-component reaction
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Supplementary tables, NMR spectra, and HPLC traces of newly synthesized compounds. (PDF) Molecular formula strings and some data. (CSV)
Accession Code
The atomic coordinates and crystallographic structure factors of the HDAC6-10h complex have been deposited in the Protein Data Bank (www.rcsb.org) with accession code 7U8Z. Authors will release the atomic coordinates upon article publication.
The authors declare no competing financial interest.
REFERENCES
- 1).Biel M; Wascholowski V; Giannis A Epigenetics – an epicenter of gene regulation: Histones and histone-modifying enzymes. Angew. Chem. Int. Ed 2005, 44, 3186–3216. [DOI] [PubMed] [Google Scholar]
- 2).Bannister AJ; Kouzarides T Regulation of chromatin by histone modifications. Cell. Res 2011, 21, 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Bertrand P Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem 2010, 45, 2095–2116. [DOI] [PubMed] [Google Scholar]
- 4).Xiao H; Jiao J; Wang L; O’Brien S; Newick K; Wang L-CS, Falkensammer E; Liu Y; Han R; Kapoor V; Hansen FK; Kurz T; Hancock WW; Beier UH HDAC5 controls the functions of Foxp3+ T-regulatory and CD8+ T cells. Int. J. Cancer 2016, 138, 2477–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Huang J; Wang L; Dahiya S; Beier UH; Han R; Samanta A; Bergman J; Sotomayor EM; Seto E; Kozikowski AP; Hancock WW Histone/protein deacetylase 11 targeting promotes Foxp3+ Treg function. Sci. Rep 2017, 7, 8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Wang P; Wang Z; Liu J Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Mielcarek M; Zielonka D; Carnemolla A; Marcinkowski JT; Guidez F HDAC4 as a potential therapeutic target in neurodegenerative diseases: a summary of recent achievements. Front. Cell. Neurosci 2015, 9, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Roche J; Bertrand P Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem 2016, 121, 451e483. [DOI] [PubMed] [Google Scholar]
- 9).Witt O; Deubzer HE; Milde T; Oehme I HDAC family: What are the cancer relevant targets? Canc. Lett 2009, 277, 8–21. [DOI] [PubMed] [Google Scholar]
- 10).Jenke R; Reßing N; Hansen FK; Aigner A; Büch T Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives. Cancers 2021, 23, 634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Stoszko M; Ne E; Abner E; Mahmoudi T A broad drug arsenal to attack a strenuous latent HIV reservoir. Curr. Opin. Virol 2019, 38, 37–53. [DOI] [PubMed] [Google Scholar]
- 12).Aldana-Masangkay GI; Sakamoto KM The role of HDAC6 in cancer. J. Biomed. Biotechnol 2011, 2011, 875824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Li T; Zhang C; Hassan S; Liu X; Song F; Chen K; Zhang W; Yang J Histone deacetylase 6 in cancer. J. Hematol. Oncol 2018, 11, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Kalin JH; Bergman JA Development and therapeutic implications of selective histone deacetylase 6 inhibitors. J. Med. Chem 2013, 56, 6297–6313. [DOI] [PubMed] [Google Scholar]
- 15).Simões-Pires C; Zwick V; Nurisso A; Schenker E; Carrupt P-A; Cuendet M HDAC6 as a target for neurodegenerative diseases: what makes it different from the other HDACs?. Mol. Neurodegener 2013, 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Sellmer A; Stangl H; Beyer M; Grünstein E; Leonhardt M; Pongratz H; Eichhorn E; Elz S; Striegl B; Jenei-Lanzl Z; Dove S; Straub RH; Krämer OH; Mahboobi S Marbostat-100 defines a new class of potent and selective antiinflammatory and antirheumatic histone deacetylase 6 inhibitors. J. Med. Chem 2018, 61, 3454–3477. [DOI] [PubMed] [Google Scholar]
- 17).Shen S; Kozikowski AP A patent review of histone deacetylase 6 inhibitors in neurodegenerative diseases (2014-2019). Expert Opin. Ther. Pat 2020, 30, 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Zhang X-H; Qin-Ma; Wu HP; Khamis MY; Li Y-H; Ma L-Y; Liu H-M A Review of progress in histone deacetylase 6 inhibitors research: Structural specificity and functional diversity. J. Med. Chem 2021, 64, 1362–1391. [DOI] [PubMed] [Google Scholar]
- 19).Hai Y; Christianson DW; Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol 2016, 12, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Zou H; Wu Y; Navre M; Sang B-C Characterization of the two catalytic domains in histone deacetylase 6. Biochem. Biophys. Res. Commun 2006, 341, 45–50. [DOI] [PubMed] [Google Scholar]
- 21).Osko JD; Christianson DW Structural basis of catalysis and inhibition of HDAC6 CD1, the enigmatic catalytic domain of histone deacetylase 6. Biochemistry 2019, 58, 4912–4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Hubbert C; Guardiola A; Shao R; Kawaguchi Y; Ito A; Nixon A; Yoshida M; Wang X-F; Yao TP HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [DOI] [PubMed] [Google Scholar]
- 23).Zhang X; Yuan Z; Zhang Y; Yong S; Salas-Burgos A; Koomen J; Olashaw N; Parsons JT; Yang X-J; Dent SR; Yao T-P; Lane WS; Seto E HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell 2007, 27, 197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Valenzuela-Fernández A; Cabrero JR; Serrador JM; Sánchez-Madrid F HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol 2008, 18, 291–297. [DOI] [PubMed] [Google Scholar]
- 25).Zhang Y; Li N; Caron C; Matthias G; Hess D; Khochbin S; Matthias P HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J 2003, 22, 1168–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Bali P; Pranpat M; Bradner J; Balasis M; Fiskus W; Guo F; Rocha K; Kumaraswamy S; Boyapalle S; Atadja P; Seto E; Bhalla K Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90. J. Biol. Chem 2005, 280, 26729–26734. [DOI] [PubMed] [Google Scholar]
- 27).Kawaguchi Y; Kovacs JJ; McLaurin A; Vance JM; Ito A; Yao T-P The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [DOI] [PubMed] [Google Scholar]
- 28).Huber EM; Groll M Inhibitors for the immuno- and constitutive proteasome: Current and future trends in drug development. Angew. Chem. Int. Ed 2012, 51, 8708–8720. [DOI] [PubMed] [Google Scholar]
- 29).Depetter Y; Geurs S; De Vreese R; Goethals S; Vandoorn E; Laevens A; Steenbrugge J; Meyer E; de Tullio P; Bracke M; D’hooghe M; De Wever O Selective pharmacological inhibitors of HDAC6 reveal biochemical activity but functional tolerance in cancer models. Int. J. Cancer 2019, 145, 735–774. [DOI] [PubMed] [Google Scholar]
- 30).Suraweera A; O’Byrne KJ; Richard DJ Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: Achieving the full therapeutic potential of HDACi. Front. Oncol 2018, 8, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Kumagai T; Wakimoto N; Yin D; Gery S; Kawamata N; Takai N; Komatsu N; Chumakov A; Imai Y; Koeffler HP Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int. J. Cancer 2007, 121, 656–665, [DOI] [PubMed] [Google Scholar]
- 32).Leshchenko VV; Kuo P-Y; Shaknovich R; Yang DT; Gellen T; Petrich A; Yu Y; Remache Y; Weniger MA; Rafiq S; Suh KS; Goy A; Wilson W; Verma A; Braunschweig I; Muthusamy N; Kahl BS; Byrd JC; Wiestner A; Melnick A; Parekh S Genomewide DNA methylation analysis reveals novel targets for drug development in mantle cell lymphoma. Blood 2010, 116, 1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Kalac M; Scotto L; Marchi E; Amengual J; Seshan VE; Bhagat G; Ulahannan N; Leshchenko VV; Temkin AM; Parekh S; Tycko B; O’Connor OA HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood 2011, 118, 5506–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).NCT04514081.
- 35).NCT04233294.
- 36).NCT04337606.
- 37).Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in medicinal chemistry. Chem. Soc. Rev 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]
- 38).Mei H; Han J; Fustero S; Medio-Simon M; Sedgwick DM; Santi C; Ruzziconi R; Soloshonok VA Fluorine-containing drugs approved by the FDA in 2018. Chem. Eur. J 2019, 25, 11797–11819. [DOI] [PubMed] [Google Scholar]
- 39).Müller K; Faeh C; Diederich F Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- 40).Meanwell NA Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- 41).Johnson BM; Shu Y-Z; Zhuo X; Meanwell NA Metabolic and pharmaceutical aspects of fluorinated compounds. J. Med. Chem 2020, 63, 6315–6386. [DOI] [PubMed] [Google Scholar]
- 42).Pan Y The dark side of fluorine. ACS Med. Chem. Lett 2019, 10, 1016–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43).Diedrich D; Hamacher A; Gertzen CGW; Alves Avelar LA; Reiss GJ; Kurz T; Gohlke H; Kassack MU; Hansen FK Rational design and diversity-oriented synthesis of peptoid-based selective HDAC6 inhibitors. Chem. Commun 2016, 52, 3219–3222. [DOI] [PubMed] [Google Scholar]
- 44).Strebl MG, Campbell AJ, Zhao W-N, Schroeder FA, Riley MA, Chindavong PS, Morin TM, Haggarty SJ, Wagner FF, Ritter T, Hooker JM HDAC6 brain mapping with [18F]Bavarostat enabled by a Ru-mediated deoxyfluorination. ACS Cent. Sci 2017, 3, 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Porter NJ; Osko JD; Diedrich D; Kurz T; Hooker JM; Hansen FK; Christianson DW Histone deacetylase 6-selective inhibitors and the influence of capping groups on hydroxamate-zinc denticity. J. Med. Chem 2018, 61, 8054–8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46).Wang C; Schroeder FA; Wey H-Y; Borra R; Wagner FF; Reis S; Kim SW; Holson EB; Haggarty SJ; Hooker JM In vivo imaging of histone deacetylases (HDACs) in the central nervous system and major peripheral organs. J. Med. Chem 2014, 57, 7999–8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47).Sandrone G; Cukier CD; Zrubek K; Marchini M; Vergani B; Caprini G; Fossati G; Steinkühler C; Stevenazzi A Role of Fluorination in the Histone Deacetylase 6 (HDAC6) Selectivity of Benzohydroxamate-Based Inhibitors. ACS Med. Chem. Lett 2021, 12, 1810–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48).Mackwitz MKW; Hesping E; Antonova-Koch Y; Diedrich D; Gebru T; Skinner-Adams T; Clarke M; Schöler A; Limbach L; Kurz T; Winzeler EA; Held J; Andrews KT; Hansen FK Structure-activity and structure-toxicity relationships of novel peptoid based histone deacetylase inhibitors with dual-stage antiplasmodial activity. ChemMedChem 2019, 14, 912–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49).Reßing N; Sönnichsen M; Osko JD; Schöler A; Schliehe-Diecks J; Skerhut A; Borkhardt A; Hauer J; Kassack MU; Christianson DW; Bhatia S; Hansen FK Multicomponent Synthesis, Binding Mode, and Structure–Activity Relationship of Selective Histone Deacetylase 6 (HDAC6) Inhibitors with Bifurcated Capping Groups. J. Med. Chem 2020, 63, 10339–10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50).Schmidt J; Rotter M; Weiser T; Wittmann S; Weizel L; Kaiser A; Heering J; Goebel T; Angioni C; Wurglics M; Paulke A; Geisslinger G; Kahnt A; Steinhilber D; Proschak E; Merk D A dual modulator of Farnesoid X receptor and soluble epoxide hydrolase to counter nonalcoholic steatohepatitis. J. Med. Chem 2017, 60, 7703–7724. [DOI] [PubMed] [Google Scholar]
- 51).Diedrich D; Stenzel K; Hesping E; Antonova-Koch Y; Gebru T; Duffy S; Fisher G; Schöler A; Meister S; Kurz T; Avery VM; Winzeler E; Held J; Andrews KT; Hansen FK One-pot, multi-component synthesis and structure-activity relation- ships of peptoid-based histone deacetylase (HDAC) inhibitors targeting malaria parasites. Eur. J. Med. Chem 2018, 158, 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52).Reßing N; Marquardt V; Gertzen CGW; Schöler A; Schramm A; Kurz T; Gohlke H; Aigner A; Remke M; Hansen FK Design, synthesis and biological evaluation of β-peptoid-capped HDAC inhibitors with anti-neuroblastoma and anti-glioblastoma activity. Med. Chem. Commun 2019, 10, 1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53).Mackwitz MKW; Hamacher A; Osko JD; Held J; Schöler A; Christianson DW; Kassack MU; Hansen FK Multicomponent synthesis and binding mode of imidazo[1,2- pubs.acs.org/jmc Article a]pyridine-capped selective HDAC6 inhibitors. Org. Lett 2018, 20, 3255–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54).Gaisina IN; Tueckmantel W; Ugolkov A; Shen S; Hoffen J; Dubrovskyi O; Mazar A; Schoon RA; Billadeau D; Kozikowski AP Identification of HDAC6-selective inhibitors of low cancer cell cytotoxicity. ChemMedChem 2016, 11, 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55).Robey RW; Chakraborty AR; Basseville A; Luchenko V; Bahr J; Zhan Z; Bates SE Histone deacetylase inhibitors: emerging mechanisms of resistance. Mol. Pharmaceutics 2011, 8, 2021–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56).Lee J-H; Choy ML; Marks PA Chapter Two - mechanisms of resistance to histone deacetylase inhibitors. Adv. Cancer Res 2012, 116, 39–86. [DOI] [PubMed] [Google Scholar]
- 57).Alves Avelar LA; Schrenk C; Sönnichsen M; Hamacher A; Hansen FK; Schliehe-Diecks J; Borkhardt A; Bhatia S; Kassack MU; Kurz T Synergistic induction of apoptosis in resistant head and neck carcinoma and leukemia by alkoxyamide-based histone deacetylase inhibitors. Eur. J. Med. Chem 2021, 211, 113095. [DOI] [PubMed] [Google Scholar]
- 58).Bhatia S; Krieger V; Groll M; Osko JD; Reßing N; Ahlert H; Borkhardt A; Kurz T; Christianson DW; Hauer J; Hansen FK Discovery of the first-in-class dual histone deacetylase- proteasome inhibitor. J. Med. Chem 2018, 61, 10299–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59).Selg C; Schöler A; Schliehe-Diecks J; Hanl M; Sinatra L; Borkhardt A; Sárosi MB; Bhatia S; Hey-Hawkins E; Hansen FK Borinostats: solid-phase synthesis of carborane-capped histone deacetylase inhibitors with a tailor-made selectivity profile. Chem. Sci 2021, 12, 11873–11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60).Yang H; Hoshino K; Sanchez-Gonzalez B; Kantarjian H; Garcia-Manero G Antileukemia activity of the combination of 5-aza-2’-deoxycytidine with valproic acid. Leuk. Res 2005, 29, 739–748. [DOI] [PubMed] [Google Scholar]
- 61).Kuendgen A; Lübbert M Current status of epigenetic treatment in myelodysplastic syndromes. Ann. Hematol 2008, 87, 601–611. [DOI] [PubMed] [Google Scholar]
- 62).Ianevski A; He L; Aittokallio T; Tang J SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics 2017, 33, 2413–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63).Jiang X; Lee GT; Prasad K; Repič O A practical synthesis of a diazepinylbenzoic acid, a retinoid X receptor antagonist. Org. Process Res. Dev 2008, 12, 1137–1141. [Google Scholar]
- 64).Osko JD; Christianson DW Methods for the expression, purification, and crystallization of histone deacetylase 6–inhibitor complexes. Methods Enzymol. 2019, 626, 447–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65).Battye TGG; Kontogiannis L; Johnson O; Powell HR; Leslie AGW iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D. Biol. Crystallogr 2011, 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69).Chen VB; Arendall WB; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70).Ianebski A; He L; Aittokallio T; Tang J SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics 2020, 36, 2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71).Erdeljac N; Bussmann K; Schöler A; Hansen FK; Gilmour R Fluorinated Analogues of the Histone Deacetylase Inhibitor Vorinostat (Zolinza): Validation of a Chiral Hybrid Bioisostere, BITE. ACS Med. Chem. Lett 2019, 10, 1336–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72).Heltweg B; Dequiedt F; Verdin E; Jung M Nonisotopic substrate for assaying both human zinc and NAD+-dependent histone deacetylases. Anal. Biochem 2003, 319, 42–48. [DOI] [PubMed] [Google Scholar]
- 73).Asfaha Y; Schrenk C; Alves Avelar LA; Lange F; Wang C; Bandolik JJ; Hamacher A; Kassack MU; Kurz T Novel alkoxyamide-based histone deacetylase inhibitors reverse cisplatin resistance in chemoresistant cancer cells. Bioorg. Med. Chem 2020, 28, 115108. [DOI] [PubMed] [Google Scholar]
- 74).Lobera M; Madauss KP; Pohlhaus DT; Wright QG; Trocha M; Schmidt DR; Baloglu E; Trump RP; Head MS; Hofmann GA; Murray-Thompson M; Schwartz B; Chakravorty S; Wu Z; Mander PK; Kruidenier L; Reid RA; Burkhart W; Turunen BJ; Rong JX; Wagner C; Moyer MB; Wells C; Hong X; Moore JT; Williams JD; Soler D; Ghosh S; Nolan MA Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol 2013, 9, 319–325. [DOI] [PubMed] [Google Scholar]
- 75).Baell JB; Holloway GA New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.