

Abstract
Diabetic retinopathy (DR) and other diabetic vascular complications are the leading cause of death and disability in patients with suboptimum glycemic control. In the pathogenesis of diabetic vascular diseases, hyperglycemia-induced oxidative stress, DNA damage, and poly-ADP-ribose-polymerase (PARP) hyperactivation play important roles in endothelial cell impairment. Adipose differentiation-related protein FBXW7 was reported to regulate PGC-1α stability and mitochondrial homeostasis. Here, we investigated the role and mechanism of FBXW7 in repairing endothelial oxidative stress injuries under hyperglycemic conditions. FBXW7 promoted the hampered activity of homologous recombination and non-homologues end joining pathway for repairing DNA double-strand breaks damage, an initiating factor for PARP hyperactivation and diabetic vascular complications. The abundant mobilization of DNA damage repair mediated by FBXW7 suppressed PARP activation, leading to downregulation of PARP expression and activity in both human endothelial cells and diabetic rat retinas. This provided a new method for PARP inhibition, superior to PARP inhibitors for treating diabetic vascular complication. Furthermore, FBXW7 rescued downregulated NAD+ levels and ameliorated mitochondrial dysfunction, thereby reducing superoxide production under hyperglycemic conditions. These effects reversed oxidative injury and vascular leakage in diabetic rat retina, providing a potential future treatment strategy.
keywords: Diabetic retinopathy, DNA damage, Endothelial cell, Mitochondria, Oxidative stress
Graphical abstract
Abbreviations
- DR
diabetic retinopathy
- PARP
poly-ADP-ribose-polymerase
- PGC-1α
peroxisome proliferator-activated receptor γ coactivator
- NAD+
Nicotinamide adenine dinucleotide
- CVD
cardiovascular disease
- DM
diabetes mellitus
- ETC
electron transfer chain
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- PAR
poly-ADP-ribose
- T1DM
type 1 diabetes
- T2DM
type 2 diabetes
- 3AB
3-Aminobenzamide
- OXPHOS
oxidative phosphorylation
- PFVM
proliferative fibrovascular membrane
- ERM
epiretinal membrane
- DDR
DNA damage response
- DSBs
DNA double-strand breaks
- ATM
ataxia telangiectasia mutated
- ATR
ATM- and Rad3-related
- HR
homologous recombination
- NHEJ
non-homologues end-joining
- TFAM
mitochondrial transcription factor A
- Rot/Aa
rotenone and antimycin A
- H2O2
hydrogen peroxide
- OCR
oxygen consumption rate
- ECAR
extracellular acidification rate
- EB
Evans blue
- SOD
superoxide dismutase
- NOX
NADPH oxidase
- FBXW7
F-box and WD repeat domain containing 7
- ROS
reactive oxygen species
1. Introduction
Diabetic cardiovascular disease (CVD) and micro-vascular complications, such as diabetic retinopathy (DR), are the leading causes of death and disability in people with high blood glucose [1]. The prevalence of diabetic vascular complications has increased rapidly as the number of individuals with diabetes mellitus (DM) has grown [2,3]. Unfortunately, no effective intervention is currently available for these complications due to their complex pathogenesis.
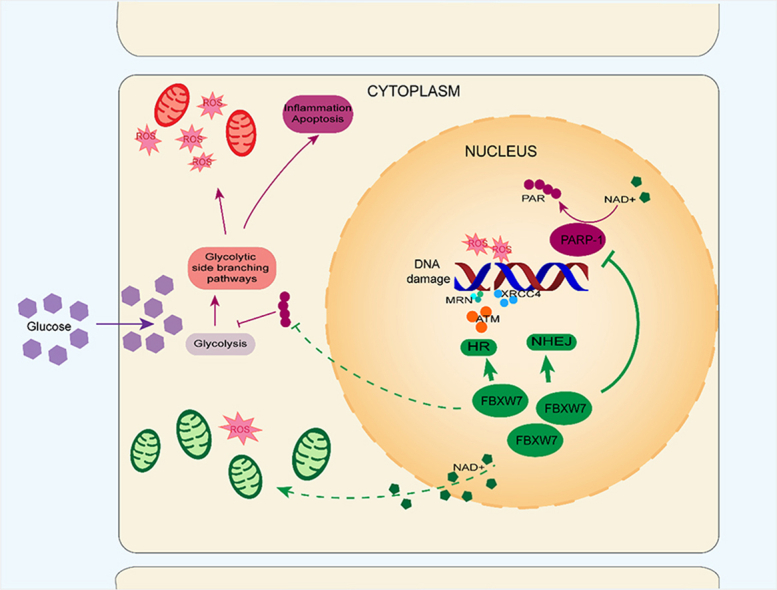
Oxidative stress plays a significant role in the pathogenesis of diabetic vascular complications. As illustrated by the widely recognized “unified mechanism theory” [4], hyperglycemia blocks the mitochondrial electron transfer chain (ETC) and produces massive amount of reactive oxygen species (ROS), that can cause DNA damage such as double-strand breaks (DSBs), and subsequent overactivation of poly-ADP-ribose-polymerase (PARP). Then, the hyper-activated PARP inhibits the activity of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by consuming substrate NAD+ and attaching poly-ADP-ribose (PAR) to it. When GAPDH is inhibited, the physiological glycolysis is interfered. Meanwhile, the glycolytic side branching pathways including the polyol, protein kinase C, and hexosamine biosynthesis pathways, are activated and advanced glycation end products are formed, which triggers oxidative stress injury in target cells, including macro- and micro-vascular cells [5]. Therefore, ROS overproduction and PARP overactivation are two crucial upstream pathogenic factors, that must be recognized [6,7].
Recent studies showed that intensive glycemic control improved, but did not prevent the progression of diabetic vascular complications in both type 1 diabetes(T1DM) and type 2 diabetes(T2DM). Besides blood glucose, a series of factors including obesity, hypertension, dyslipidemia, inflammation, and insulin resistance, underlie the onset and development of diabetic vascular complications [8]. Dyslipidemia in patients with T2DM confers a significant additional risk of adverse outcomes to patients with CVD, despite optimal management with conventional therapy [9,10]. And analysis of blood lipid profiles established a close association between the development of diabetic vasular disease and dyslipidemia [[11], [12], [13]], indicating that lipid metabolism regulatory proteins may be an available target for the treatment of diabetic vascular complications.
The F-box and WD repeat domain containing 7 (FBXW7) is an E3 ligase of the SCF (complex of SKP1, CUL1 and F-box protein) family, a well-known tumor suppressor regulating cell division, growth and differentiation through substrate proto-oncogenes degradation [14]. Besides tumor suppression, FBXW7's role in glucose and lipid metabolism gradually came to be known in recent years. FBXW7 is reported to be a critical regulator for adipocyte differentiation and lipid metabolism and is involved in the progress of metabolic syndrome including obesity, hyperglycemia, glucose intolerance, and insulin resistance [[15], [16], [17]]. Park et al. reported that FBXW7 influenced mitochondrial homeostasis through regulating the stability of the peroxisome proliferator-activated receptor γ coactivator (PGC-1α), a central regulator of mitochondrial biogenesis, in skeletal muscle and brown adipocytes, two high energy-consuming and insulin-sensitive tissues that play important roles in blood glucose control [18]. As the metabolic powerhouse where many crucial biological processes including Krebs cycle, fatty acid oxidation, and oxidative phosphorylation (OXPHOS) occur, mitochondrial dysfunction contributes to the pathogenesis of metabolic disorders such as DM. We therefore designed this research in part to explore whether and how this protein could play a role in impaired mitochondrial function under hyperglycemic conditions.
Our findings demonstrate that FBXW7 can relieve vascular endothelium oxidative damage under hyperglycemic conditions. We found that FBXW7 inhibited PARP overactivation via enhancing impaired classic DSBs damage repair potential, the effect of this protein was superior to traditional PARP inhibitors. We therefore propose that strengthening the hampered DNA repair pathway is a better way to downregulate PARP than its direct inhibition. This novel strategy also provides new insights into the prevention and treatment of diabetic vascular complications. Additionally, FBXW7 undoubtedly increased the mitochondrial DNA (mtDNA) copy number, leading to the improvement of impaired mitochondrial OXPHOS, which then dramatically decreased the level of ROS production against oxidative damage under hyperglycemic conditions. Therefore, FBXW7 shows promise as a potential therapeutic target for diabetic vascular complications.
2. Results
2.1. Expression of FBXW7 was downregulated and adenovirus-mediated FBXW7 overexpression inhibited PARP hyperactivation in DR
To investigate the change of endogenous FBXW7 in the progress of DR, pre-retinal proliferative fibrovascular membranes (PFVM) from patients with proliferative diabetic retinopathy (PDR) were collected and then tested by q-PCR, which revealed the significant downregulation of FBXW7 mRNA levels compared to macular epiretinal membranes (ERM) from nondiabetic patients (Fig. 1A). Furthermore, a diabetic rat model was established to identify the protein level of FBXW7 in the retina. Similarly, although FBXW7 was slightly increased in the diabetic retina at 4 w post-diabetic model establishment, it significantly decreased at 8 w post-diabetic model establishment (Fig. 1B).
Fig. 1.

Altered expression of FBXW7 in diabetic retina and adenovirus-mediated FBXW7 overexpression inhibited PARP hyperactivation in diabetic rat retina and endothelial cells. A: q-PCR analysis of FBXW7 mRNA in macular ERM from nondiabetic patients (CTRL) and pre-retinal PFVM from patients with proliferative DR (n = 7–9; 2-tailed unpaired t-test). B: Western blot of FBXW7 in retinas of normal rats (CTRL) and diabetic rats (DM) 4 weeks and 8 weeks post-diabetic model establishment. C: Western blot of PARP in HRECs and HUVECs cultured in normal glucose (NG) or high glucose (HG) with or without FBXW7 overexpression (FOE) for 48 h. D: PARP activity was assessed by a HT Colorimetric PARP/Apoptosis Assay in HRECs and HUVECs. E: Measurement of PAR in HRECs and HUVECs by HT PARP in vivo Pharmacodynamic Assay II. F:Measurement of PAR in retinas of the normal rats (CTRL) or diabetic rats (DM) with or without intravitreal injection of adenovirus-mediated FBXW7 overexpression (DM + FOE) after 12 weeks of treatment (n = 6). For all charts, values were presented as the mean ± SD. All the results were evaluated by ANOVA with Tukey's multiple comparison test. *P < 0.05, **P < 0.01, ***P < 0.001.
Then, we investigated whether FBXW7 could have any impact on PARP, which initiates downstream oxidative stress injuries resulting diabetic vascular complications. Adenovirus-mediated FBXW7 overexpression significantly decreased the elevated PARP expression and PARP activity in both HUVECs and HRECs under hyperglycemic conditions (Fig. 1C and D).
PARP overactivation can produce elevated levels of PAR products, which inhibits the downstream activity of GAPDH. Therefore, we next measured PAR levels. In HUVECs, high glucose-induced PAR products were significantly inhibited by FBXW7 overexpression, while a similar effect was not observed in HRECs with extremely low levels of PAR. This may be due to a brief period of high glucose incubation (Fig. 1E). To observe the effect of long-term hyperglycemia exposure, PAR levels in rat retinal tissues were measured at 12 w post-diabetic model establishment, demonstrating that increased PAR products were reversed by adenovirus-mediated FBXW7 overexpression via vitreous cavity injection in diabetic rats (Fig. 1F).
2.2. FBXW7 improved the damaged DNA repair activities in HUVECs under hyperglycemic conditions and was superior to traditional PARP inhibitors
Large amounts of ROS induced by hyperglycemia aggravated DNA damage in endothelial cells, which leads to overactivation of PARP with comprehensive activity for DNA damage repair. We explored whether FBXW7 affected the DNA damage response (DDR). Additionally, as PARP inhibitors are well-known classical chemical reagents for the treatment of diabetic vascular complications, we compared FBXW7 with PARP inhibitors.
Homologous recombination (HR) and non-homologues end-joining (NHEJ) are the most common ways to repair DSBs, the most deleterious form of DNA damage, which destroy genomic integrity and threaten cell survival [19]. We first determined the protein level of γH2AX (a sensitive marker of DSBs) and the phosphorylation level of ataxia telangiectasia mutated (ATM) and ATM- and Rad3-related (ATR) kinases (two critical PI3Ks initiating DSB repair (DSBR)). As shown in Fig. 2A, increased γH2AX, p-ATM and p-ATR under high-glucose conditions were reversed by FBXW7 overexpression, while no perceptible change was detected after the treatment of PARP inhibitors 3-Aminobenzamide (3AB) and PJ34, indicating no necessity for DSBR mobilization due to the obviously decreased DSB lesions after FBXW7 overexpression. To further confirm this result, we treated HUVECs with H2O2 as moderate ROS for DNA damage inducement and conducted alkaline comet assays to evaluate whether there was any difference in recovery capability among these groups. The average tail moment in the high-glucose condition, which was higher than that of the control, declined after FBXW7 overexpression, demonstrating that DNA damage repair is severely impaired following high glucose treatment, but was rescued by FBXW7 overexpression. On the contrary, PJ34, rather than 3AB, further weakened DNA repair capability, indicating the different DNA damage repair potential of FBXW7 and PARP inhibitors (Fig. 2B).
Fig. 2.

FBXW7 improved the damaged DNA repair activity in HUVECs under hyperglycemic condition and was superior to traditional PARP inhibitors. A: Western blot showing the expression of DNA damage marker γH2AX and DDR protein p-ATM and p-ATR in HUVECs cultured in NG, HG, HG + FOE, HG+3AB (10 μM), and HG + PJ34 (10 μM) for 48 h. B: Comet assay for DNA damage assessment in HUVECs after H2O2 treatment. Representative cells and quantification of DNA tail moments were shown. Scale bar:100 μm. C: Relative HR (mCherry) and NHEJ (GFP) activity measured by DR-GFP assay were shown. D: q-PCR indicated the expression of genes involved in DNA damage repair in HUVECs cultured for 48 h.
E: Western blot of the MRN complex in HUVECs post H2O2 treatment. F: Immunofluorescence of ATM (green) and FBXW7(red) in HUVECs post H2O2 treatment. Scale bar: 25 μm. G: The cell cycle assay of HUVECs was measured by flow cytometry and quantification of cells in S phase was analyzed. H: Western blot of γH2AX and PARP in HUVECs cultured in NG, HG or high glucose plus endogenous FBXW7 knockdown (HG + shFB) for 48 h. I: Comet assay for DNA damage assessment was conducted in HUVECs post H2O2 treatment. Representative cells and quantification of DNA tail moments were shown. Scale bar: 100 μm. J: Relative HR (mCherry) and NHEJ (GFP) activity were measured by DR-GFP reporter assay in HUVECs cultured in NG, HG, and HG + shFB for 48 h. All the results were analyzed by ANOVA with Tukey's multiple comparison test and presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Then, the capacity for DNA repair was tested with the DR-GFP assay [20,21], a canonical technique for detecting DSBR potential. The activity of HR and NHEJ, both of which play a major role in DSBR, decreased significantly in the high-glucose condition, and was improved by FBXW7 overexpression. PARP inhibitors did not show similar effects; PJ34 even worsened NHEJ repair activity (Fig. 2C). Meanwhile, the mRNA levels of ATM, ATR, XRCC4, and MSH2, which are widely recognized as important elements of DSBR pathways, were also provoked significantly (Fig. 2D).
As DSBR activity relies on the MRE11-RAD50-NBS1 (MRN) complex [22,23], we hypothesized that FBXW7 overexpression mobilized its expression. Confirmed by Western blot, high glucose levels decreased expression of the MRN complex, which was improved by FBXW7 overexpression as expected (Fig. 2E). The nuclear foci representing the recruitment of DSBR kinases to DNA damage sites were also assessed. Compared to the control group, high glucose decreased the foci number of ATM during the recovery from H2O2 treatment, which was improved by FBXW7 overexpression; no perceptible change was detected after treatment with PARP inhibitors (Fig. 2F). Generally, DSBR is accompanied by cell cycle changes. Under hyperglycemic conditions, FBXW7 overexpression arrested the cell cycle at G1-S boundaries and shortened the S phase, which provided sufficient time for DNA damage repair (Fig. 2G).
Conversely, we also confirmed the role of FBXW7 in DSBR via adenovirus-mediated endogenous FBXW7 knockdown. In HUVECs, increased γH2AX and the average tail moment under high-glucose conditions were further upregulated after FBXW7 knockdown, as demonstrated by Western blot and alkaline comet assay (Fig. 2H and I). The aggravated DNA damage mediated by FBXW7 knockdown indicated poorer DNA damage repair potential, which was afterwards verified with the DR-GFP assay (Fig. 2J). Moreover, we noticed that the effect of FBXW7 on PARP inhibition vanished after FBXW7 knockdown, suggesting a certain relationship between the two proteins (Fig. 2H).
In sum, endothelial cells have obvious DSBs and impaired DNA repair ability under hyperglycemic conditions. FBXW7 enhanced DNA repair potential and had advantages over traditional PARP inhibitors.
2.3. The function of FBXW7 in promoting DNA damage repair was related to ATM
To explore the potential mechanism for DNA damage repair ability, inhibitors of ATM (KU55933), DNA-PKcs (NU7441), and ATR (VE821) were administrated, as ATM, ATR and DNA-PKcs activation are the pivotal steps for major DSBR. The improvement of HR and NHEJ activity by FBXW7 overexpression was inhibited by all these inhibitors (Fig. 3A). Meanwhile, the effect of FBXW7 overexpression on declining DNA damage disappeared (Fig. 3B), demonstrating that its efficiency on strengthening DSBR activity is comprehensive.
Fig. 3.

The effect of FBXW7 on promoting DNA damage was related to ATM. A: DR-GFP assay measured the HR and NHEJ activity in HUVECs cultured in HG + FOE with or without ATM inhibitor (KU55933), DNA-PKcs inhibitor (NU7441) or ATR inhibitor (VE821). B: Comet assay measured DNA damage in HUVECs cultured in NG, HG, HG + FOE with or without KU55933, NU7441 or VE821 after H2O2 treatment. Scale bar: 100 μm. C: Western blot of the MRN complex in HUVECs cultured in the same conditions as fig.B. D: Western blot of p-S/TQ representing p-FBXW7 in HUVECs cultured in the same conditions as fig.B. E: Co-IP was conducted to detect the interaction between FBXW7 and ATM or XRCC4. Anti-FLAG antibody was used for immunoprecipitation and anti-ATM antibody and anti-XRCC4 antibody were used for subsequent Western blot. F: Ubiquitination assay was conducted by Co-IP. Anti-HA antibody was used for immunoprecipitation and anti-XRCC4 antibody was used for subsequent Western blot. G: Immunofluorescence of γH2AX (green) and XRCC4 (red) in HUVECs cultured in NG, HG or HG with FBXW7 overexpression or knockdown after H2O2 treatment. Scale bar: 10 μm. All the results were analyzed by ANOVA with Tukey's multiple comparison test and presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Next, we observed that the elevated expression levels of the MRN complex after FBXW7 overexpression were downregulated (Fig. 3C) when ATM and ATR were inhibited, suggesting a close relationship between FBXW7 and ATM or ATR.
Zhang et al. reported that FBXW7 containing two evolutionarily conserved serine/glutamine (SQ) motifs, can serve as a substrate of ATM for phosphorylation modifications when exposed to ionizing radiation. In this way, it can play an important role in DNA damage repair in MiaPaCa-2, a pancreatic ductal carcinoma cell [24]. In this study, we explored whether this effect exists in endothelial cells when exposed to hyperglycemia and oxidative stress. The decreased expression of p-S/TQ under high-glucose conditions, which represented the phosphorylation of FBXW7, was elevated significantly after FBXW7 overexpression. This rescue was abrogated upon ATM inhibition, indicating FBXW7 overexpression improved DNA repair, probably depending on ATM (Fig. 3D).
Co-IP was next conducted to confirm the direct interaction between ATM, DSBR downstream effector protein and FBXW7. The results showed that FBXW7 interacted with not only ATM but also XRCC4, a critical DDR protein stabilizing DNA ligase IV to help tether the broken DSB ends together in NHEJ during the recovery from H2O2 treatment (Fig. 3E).
As FBXW7 is an E3 ligase, the ubiquitin assay was further employed to illustrate that FBXW7 increased the ubiquitination modification level of XRCC4 (Fig. 3F). Immunofluorescence staining was conducted afterwards to demonstrate that decreased expression of XRCC4 in the nucleus under hyperglycemic conditions, accompanying an increased foci number of γH2AX, can be reversed significantly by FBXW7 overexpression, while this effect was abrogated after endogenous FBXW7 knockdown (Fig. 3G). These results verified that the ubiquitination of XRCC4, mediated by FBXW7, increased its stability rather than degradation to improve DSBR activity. While activated DNA-PKcs phosphorylates XRCC4 at serines 325/326 which promotes binding of XRCC4 to FBXW7 [24], NHEJ activity mediated by FBXW7 overexpression was inhibited by NU7441 correspondingly (Fig. 3A and B).
In sum, we have obtained solid evidence that ATM phosphorylated FBXW7 and recruited it to DSBs sites, which stabilized XRCC4 by polyubiquitylation to facilitate NHEJ in diabetic vascular endothelial cells.
2.4. FBXW7 decreased ROS levels by alleviating mitochondrial dysfunction in hyperglycemic states
Massive ROS production induced by hyperglycemia contributes to endothelial damage and the progression of DR. The elevated ROS level in both HUVECs and HRECs under high-glucose conditions, was decreased dramatically after FBXW7 overexpression (Fig. 4A). The same effect was also observed in retinal tissues at 12 w post-diabetic model establishment. However, adenovirus-mediated FBXW7 knockdown significantly increased the ROS level in the retinal ganglion cell layer, inner nuclear layer, and outer nuclear layer, even as early as 8 w post-diabetic model establishment (Fig. 4B).
Fig. 4.

FBXW7 decreased ROS level through alleviating mitochondria dysfunction in hyperglycemic state. A: Total intracellular ROS was measured by DCF probes in HUVECs and HRECs by flow cytometry. B: In situ superoxide was detected by DHE probes in frozen eye sections in normal rats, diabetic rats or diabetic rats with FBXW7 overexpression or knockdown. Representative images and quantitated superoxide level relative to normal rats were presented (n = 6). Scale bar:100 μm. C: mtROS was measured by MitoSOX probes in HRECs and HUVECs by flow cytometry. D: Mitochondria stress test was measured to detect mitochondrial respiration function in HUVECs cultured in NG, HG or HG + FOE. OCR was measured and data was presented normalized to NG. E: As did mitochondria stress test in HUVECs cultured in NG, HG, HG+3AB, and HG + PJ34.
F: Mitochondrial respiration function measured in HUVECs with or without H2O2 acute injection induced oxidative stress. G: q-PCR illustrated the mRNA levels of COX5B, ERRα, HIF-1α, PGC-1α, S6K1, and ATP5O in HUVECs. H: Mitochondria DNA copy number (relative to nuclear DNA) was detected in HUVECs. I: Total intracellular ROS was measured in HUVECs cultured in NG, HG and HG + FBXW7 with or without ATM inhibitor KU55933 for 48 h. J: Total intracellular ROS was measured in HUVECs cultured in NG, HG and HG + FBXW7 with or without rotenone and antimycin A for 48 h. All the results were analyzed by ANOVA with Tukey's multiple comparison test and presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
As known, hyperglycemia activates NADPH oxidase (NOX) and damages mitochondrial function, two important sources for ROS production. Hyperglycemia stimulates the expression of NOX1, NOX2 and NOX4, three abundant isoforms in endothelium, while no obvious expression changes are observed after FBXW7 overexpression or knockdown (Figs. S1A and S1B). Furthermore, the effect of FBXW7 overexpression on decreasing ROS level remained even after the administration of apocynin, a selective NOX inhibitor, indicating that this effect may be related to mitochondria, but not NOX (Fig. S1C). Therefore, we confirmed that FBXW7 eliminated mitochondrial ROS (mtROS) in both HUVECs and HRECs, as demonstrated by MitoSOX red mitochondrial superoxide indicator (Fig. 4C).
A close relationship exists between endothelial mitochondrial dysfunction and ROS generation in hyperglycemic states. To confirm the impact of FBXW7 overexpression on mitochondrial respiration, HUVECs were subjected to the Mito Stress Test. High glucose impaired maximal respiration, while FBXW7 overexpression significantly elevated both maximal respiration and spare respiratory capacity, illustrating that FBXW7 can restore hampered mitochondrial function (Fig. 4D). However, on the contrary, PARP inhibitors did not show a beneficial effect, and PJ34 even further damaged the entire mitochondrial respiration process (Fig. 4E). Furthermore, this effect still existed even after employing H2O2 acute injection, with the overall decline of mitochondrial function rescued by FBXW7 overexpression (Fig. 4F).
Next, to elucidate how FBXW7 overexpression contributes to its enhancement of mitochondria function, the mRNA level of important regulatory genes involved in mitochondrial metabolism was measured to determine whether these genes were fully mobilized. FBXW7 overexpression increased the mRNA levels of COX5B, ATP5O, ERRα, S6K1, HIF-1α, and PGC-1α, while PJ34 increased S6K1, and HIF-1α mRNA, and 3AB merely increased the PGC-1α mRNA level (Fig. 4G), indicating that mitochondrial mass may have increased. We subsequently evaluated the mitochondrial abundance by employing a q-PCR assay. As expected, high glucose levels incurred the decline of mitochondria DNA (mtDNA) copy number, which was recovered by FBXW7 overexpression, while no evident change was observed following PARP inhibitor administration (Fig. 4H). These results confirmed that FBXW7 overexpression improved mitochondrial function in diabetic vascular endothelial cells by strengthening mitochondrial biogenesis.
DNA damage and mitochondrial dysfunction both contribute to ROS production. We wondered which could be responsible for FBXW7's ROS elimination effect. After ATM inhibitor KU55933 was administrated to restrain FBXW7's promotion of DNA damage repair, there was no change in the decline in ROS level resulting from FBXW7 overexpression compared to the control group (Fig. 4I). However, while mitochondrial ETC was entirely interrupted by rotenone and antimycin A (Rot/Aa), no significant difference was observed among these groups (Fig. 4J), demonstrating that the degree to which FBXW7 reduced intracellular ROS levels depended on its improvement of mitochondrial function.
2.5. FBXW7 reserved intracellular NAD + levels and SIRT3 expression to improve mitochondrial function in HUVECs in hyperglycemic states
In addition to the above results, we also detected a possible mechanism for mitochondrial respiration improvement and mtDNA copy number maintenance. Mitochondrial transcription factor A (TFAM), is considered essential for mtDNA maintenance [25]. The expression and function of TFAM are regulated by SIRT3, a deacetylase that is critical for recovering the normal activity of respiratory enzymes in mitochondria [26]. Therefore, the expression of TFAM and SIRT3 were detected, and their expression declined in high-glucose conditions was markedly increased by FBXW7 overexpression, while no significant improvement was observed with either of the two PARP inhibitors (Fig. 5A).
Fig. 5.

FBXW7 reserved intracellular NAD + level and SIRT3 expression to improve mitochondria function in HUVECs in hyperglycemic state. A: Western blot illustrated the expression of SIRT3 and TFAM in HUVECs. B: Western blot analyzed the expression of PGC-1α and TFAM in HUVECs cultured in NG, HG, HG + FOE, and HG + FOE+3-TYP. C: Mitochondria DNA copy number was detected by q-PCR in HUVECs cultured in the same conditions as fig.B. D: Mitochondria stress test measured respiration function in HUVECs cultured in corresponding conditions as fig.B. E:Intracellular ROS was measured in HUVECs cultured in NG, HG, and HG + FBXW7 with or without 3-TYP for 48 h. F: Intracellular NAD + levels were determined by colorimetric measurement. Data was presented as relative NAD + level in HUVECs cultured in NG, HG and HG + FOE. Intracellular total SOD activity (G), catalase activity (H) and GSH content (I) were measured in the same condition as fig.F. All the results were analyzed by ANOVA with Tukey's multiple comparison test and presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
We further evaluated whether FBXW7 ameliorated impaired mitochondrial respiration under high-glucose conditions depending on SIRT3 activity. The administration of 3-TYP, a SIRT3 inhibitor, not only decreased the expression of TFAM and PGC-1α in the FBXW7 overexpression group (Fig. 5B), but also abolished its protective effect on mtDNA copy number maintenance (Fig. 5C). Next, the beneficial impact of FBXW7 overexpression on mitochondrial respiration was further crippled after SIRT3 inhibition, with obviously compromised maximal respiration and spare respiratory capacity (Fig. 5D). Furthermore, the effect of FBXW7 on intracellular ROS alleviation disappeared after SIRT3 inhibition, which further confirmed that FBXW7 relied on SIRT3 activity to improve mitochondrial function (Fig. 5E).
SIRT3 is a member of the sirtuin family in mitochondria and regulates mitochondrial function depending on intracellular NAD + level [27]. In mammals, sirtuins and PARPs are the two major NAD + responsive proteins [28], which means they compete with each other in utilizing NAD + to participate in many important intracellular activities including energy metabolism and DNA damage repair. As shown above, FBXW7 overexpression decreased PARP hyperactivation under high-glucose conditions; therefore an increase of intracellular NAD + levels was detected (Fig. 5F). This suggests that the effect of FBXW7 on improving SIRT3 activity is due to the change in intracellular NAD + level, which accounts for the improvement of mitochondrial function.
In addition, we also evaluated whether FBXW7 affected the antioxidant capacity in HUVECs. Superoxide dismutase (SOD), catalase (CAT) and glutathione (GSH), the main ways to eliminate intracellular ROS, were detected. The increased activities of SOD and CAT under high glucose, accompanying significant decrease of antioxidant GSH level, demonstrated an abnormal state of antioxidation, which were rescued by FBXW7 overexpression (Fig. 5G, H and I).
These results showed that FBXW7 reduced mtROS production mainly by improving the mitochondrial function, thus normalizing ROS scavenging enzyme activity and reducing potential.
2.6. FBXW7 downregulated PARP through DNA damage repair promotion
As displayed in Fig. 1, Fig. 2, FBXW7 significantly inhibited the overactivation of PARP, showing an obvious advantage over classical PARP inhibitors due to its enhancement of DSBR capability. Therefore, determining the molecular mechanism behind FBXW7 inspired us to develop a new therapeutic strategy for PARP inhibition, which may be a potential target for treatment of diabetic vascular complications [6,7].
We found that FBXW7 interacted with PARP as Co-IP demonstrated (Fig. 6A). Since FBXW7 is an E3 ligase, we at first explored whether FBXW7 promoted the polyubiquitination modification of PARP and degraded it by the proteasome system. However, employing a ubiquitination assay, we observed no increase of PARP ubiquitination modification in the FBXW7 overexpressing group (Fig. 6B). Consistently, the decline in PARP remained even after inhibition of the degradation activity of both lysosome and proteasome (Fig. 6C), verifying that ubiquitination degradation was not responsible for the decline of PARP by FBXW7 overexpression.
Fig. 6.

FBXW7 downregulated PARP through DNA damage repair promotion. A: Co-IP illustrated the interaction between FBXW7 and PARP. Anti-FLAG antibody was used for immunoprecipitation and anti-PARP antibody was used for subsequent Western blot. B: For ubiquitination assay, HA-Ub adenovirus was pretreated overnight and H2O2 treatment and MG132 administration subsequently followed. Anti-HA was used for immunoprecipitation and PARP was further detected by Western blot(left). Anti-PARP was used for immunoprecipitation and HA was further detected by Western blot(right). C: Western blot of PARP expression. HUVECs were cultured in NG, HG, and HG + FBXW7 with or without subsequent administration of proteasome inhibitor MG132 and lysosome inhibitor leupeptin hemisulfate (leu) for 2 h or 4 h. Western blot of PARP and γH2AX expression in HUVECs cultured in NG, HG, and HG + FBXW7 with or without the administration of 3-TYP (D), Rot/Aa (E), APO(F) and KU55933 (G). Figure C was analyzed by ANOVA with Tukey's multiple comparison test and figure D, E, F and G were analyzed by Sidak's multiple comparison. All the data were presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Next, considering that FBXW7 strengthened mitochondrial function to decrease ROS production, we employed 3-TYP and Rot/Aa to completely block mitochondria. Although the effect of FBXW7 on ROS elimination was abrogated by 3-TYP and Rot/Aa (Fig. 4, Fig. 5J), the expression of PARP and γH2AX remained decreased (Fig. 6D and E). The same effect was observed when apocynin was administrated to inhibit NOX activity (Fig. 6F). These results indicated that decrease in ROS level as a result of FBXW7 overexpression did not account for FBXW7's effect on PARP inhibition.
Subsequently, we noticed another possibility that both FBXW7 and PARP were involved in DSBR. As shown in Fig. 2, silencing FBXW7 aggravated DSB lesions and worsen DSBR ability, while overactivated PARP. These results suggest that FBXW7 and PARP interact with each other in one way or another. Therefore, we inhibited DNA repair activity with KU55933, an ATM inhibitor, and then evaluated the expression levels of γH2AX and PARP. The effect of PARP inhibition exerted by FBXW7 was eliminated after ATM inhibition (Fig. 6G), demonstrating that FBXW7 declined PARP through DNA damage repair promotion. In addition, we also noticed that FBXW7 still reduced the expression of γH2AX even after ATM inhibition, perhaps owing to its protective effect against ROS production as shown in Fig. 4I.
2.7. FBXW7 relieved oxidative stress damage in diabetic rat retinas
The breakdown of the blood barrier in the retina mediated by hyperglycemia-induced oxidative stress incurs vascular leakage, which contributes to diabetic macular edema and vision loss [29]. To analyze the degree of vascular dysfunction, an Evans blue (EB) assay was conducted to detect retinal vascular leakage, and periodic acid-Schiff staining was conducted after retinal trypsin digestion to detect the structural changes in retinal vessels. Retinal vascular leakage increased in diabetic rats at 12 w post-diabetic model established, while adenovirus-mediated endogenous FBXW7 silence distinctly further impaired vascular permeability as early as 8 w post-diabetic model establishment, when compared with the control (Fig. 7A). The protective effect of FBXW7 overexpression on vascular leakage was observed in mouse retinas at 5 months post-diabetic model established (Fig. 7B). Moreover, the increased number of acellular capillaries in the retina of diabetic mice was significantly reduced by FBXW7 overexpression compared to those of control mice (Fig. 7C).
Fig. 7.

FBXW7 relieved oxidative stress damage in diabetic rat retina. A: Quantitative retina vascular leakage was measured by EB dye in normal rats, diabetic rats with or without FBXW7 silence at 8 or 12 weeks post diabetic model established (n = 6). B: Representative fluorescence images indicating vascular leakage in normal mice, diabetic mice with or without FBXW7 overexpression at 5 months post diabetes induced were presented (n = 6). Scale bar: 500 μm. C: Retinal trypsin digestion and PAS staining were conducted for counting the formation of acellular capillary among the same mice as fig.B (n = 6–10). Representative images were shown. Scale bar: 50 μm. D: The protein oxidation levels in normal rats, diabetic rats with or without FBXW7 overexpression were measured (n = 6–10). E: Inflammation related protein NF-κB and apoptosis related proteins BAX and BCL-2 were measured by Western blot in rats retinal tissues as in fig.D. All the results were analyzed by ANOVA with Tukey's multiple comparison test and presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
The protein carbonylation level in rat retinal tissues, a widely recognized marker of oxidative damage, was also measured [30]. FBXW7 overexpression notably reduced protein carbonylation in diabetic rat retinas (Fig. 7D), indicating its protective effect against oxidative injury. Meanwhile, FBXW7 overexpression increased BCL-2 and reduced BAX and NF-κB expression in diabetic rat retinas (Fig. 7E), exerting a positive effect on the alleviation of apoptosis and inflammation, reducing the formation of acellular capillaries.
In summary, FBXW7 overexpression reduced oxidative damage, inflammation and apoptosis in retinal tissues and alleviated the pathological changes of DR.
3. Discussion
Vascular endothelium dysfunction is a central factor in the pathogenesis of diabetic vascular complications. As “unified mechanism theory” illustrates, ROS production, DNA damage and subsequent PARP activation are three important upstream pathogenic factors in sequence, that initiate downstream oxidative stress injury for diabetic vascular complications. Here, we first reported that FBXW7 relieved the three detrimental processes, showing the effect of antioxidative injuries. FBXW7 suppressed the hyperactivation of PARP by a completely different pathway compared to classical PARP inhibitors and showed marked superiority, which opens up possibilities for new therapeutic approaches and treatments. PARP hyperactivation modifies GAPDH with PAR, thereby reducing its activity and activating glycolytic side branching pathways. Apart from glycolysis inhibition and downstream pathway activation, PARP activation exhausts intracellular NAD+ and ATP, leading to energy failure and cell death [31]. PAR, the products of PARP hyperactivation, induce the nuclear translocation of mitochondrial-associated apoptosis-inducing factors and leads to parthanatos [32]. Therefore, PARP inhibition has been recognized as a potential target for vascular diseases. It has been reported that the PARP-inhibitor PJ34 not only normalized vascular rings exhibiting impaired endothelium-dependent relaxation dysfunction, but also reversed these effects in diabetic mice [33]. Another PARP inhibitor, 4-AB, downregulated inflammation and oxidative stress markers and alleviated structural changes and collagen deposits in the hearts of diabetic rats [34]. However, the long-term use of PARP inhibitors has exposed serious side effects such as loss of genome stability and premature aging which gradually came to be known in recent years [35].
Endothelial cell cultures exposed to high levels of glucose exhibit increased DNA strand breaks, and the efficiency of DNA lesion repair represents a mechanism for different individual susceptibilities to diabetic vascular complications [36]. Our research confirmed that massive DNA damage and significant DNA damage repair defects exist in endothelial cells under hyperglycemic conditions. DSBs are the most detrimental forms of DNA damage and contribute to vascular diseases [37,38]. It is worth noting that inefficient DNA damage repair induced cell growth arrest, senescence, apoptosis, and inflammation response [38]. Vascular smooth muscle cells in human atherosclerotic plaques exhibited increased levels of DSBs, which closely correlated with disease severity [39]. Additionally, exposure to high concentration of carbohydrates impaired cellular DNA repair potential and persistent DNA damage leads to diabetes related pulmonary and renal fibrosis. These effects can be reversed by restoring DNA repair, indicating the relationship between metabolite-driven impairment of DNA repair and diabetic complications [40].
Obvious DNA damage and deficiency in HR and NHEJ activity in the hyperglycemic state may be a possible interpretation of PARP hyperactivity. HR and NHEJ are the predominant pathways for DSBs [41]. As a responder in DDR, PARP not only engaged in base excision repair and single strand break repair but also participated in an alternative NHEJ pathway when Ku was limited or absent for DSBs repair. PARP-1, the most abundant isoform of the PARPs superfamily, binds to DNA ends in direct competition with Ku and works as a backup of the classical NHEJ pathway [42,43]. Therefore, the administration of PARP inhibitors prevents DNA damage repair, which may lead to genome instability. Moreover, due to the different mechanisms to inhibit PARP activity, certain PARP inhibitors lead to a phenomenon termed ‘PARP trapping’ in which PARP inhibitors induced PARP dissociation from damaged DNA, and trapped PARP-DNA complexes blocked DNA repair [44]. Thus, inhibiting PARP activity by chemical inhibitors has a greater influence than the lack of enzyme itself regarding DNA damage repair [45], which limits the treatment for diabetic vascular complications. Previous study confirmed that PJ34 augmented injury in mouse retinal cells suffering from oxidative stress, including oxidative damage and apoptosis [46]. At the same time, our research showed that neither of these inhibitors alleviated DNA damage under hyperglycemic and oxidative stress conditions, while PJ34 administration further decreased DNA repair potential in the hyperglycemia state. Therefore, limitations and deficiencies remain in the administration of PARP inhibitors because of the disadvantageous effects in DNA damage repair.
Unlike traditional PARP inhibitors, our research first reported that FBXW7 provided a new method for downregulating PARP expression and activity, which demonstrates advantages in regard to diabetic vascular complications. FBXW7 improved HR and NHEJ activity and therefore decreased DNA damage, indicated by γH2AX and alkaline comet assays. Under hyperglycemic and oxidative stress conditions, FBXW7 promoted the ubiquitination modification of XRCC4 and retained it for DNA damage repair in endothelial cells, which is consistent with irradiation-induced DNA damage [24]. Upon DNA damage, FBXW7 also upregulated the MRN complex for DDR, which was regulated by ATM and ATR, two DNA-damage checkpoint kinases [47,48].
Therefore, the downregulation of PARP activity relies on highly efficient DNA damage repair by FBXW7 via enhancing the major DSBs repair pathway HR and NHEJ, which was superior to PARP inhibitors. Knocking down endogenous FBXW7 or inhibiting repair activity by KU55933 both reversed the downregulation of PARP. Mitochondria inhibitors Rot/Aa were administrated to exclude the interference of ROS production, while intracellular ROS level did not influence the role of FBXW7 on PARP downregulation. Although Co-IP revealed interaction between FBXW7 and PARP, no significant increase in ubiquitin modification or afterwards proteasomal degradation were observed. These results all confirmed that FBXW7 downregulated PARP hyperactivation via strengthening efficient DNA damage repair. Therefore, timely repairing of DNA damage prevented the progression of diabetic complications, which was confirmed by an in vivo assay [40].
Regarding treatment for diabetes and diabetic complications, a recent consensus put great emphasis on the prevention of CVD in diabetes treatment and recommended medication with proven cardiovascular benefits. The coexistence of diabetes and dyslipidemia increases cardiovascular disease risk [49]. Optimum control of blood glucose, lipids and blood pressure reduced the risk for diabetic retinopathy [50]. We first reported that FBXW7, as an adipose differentiation-related protein, had a beneficial effect on endothelial cells, the central factor in the pathogenesis of diabetic vascular complications.
Our study demonstrated that FBXW7 decreased oxidative injury in diabetic rat retinal tissues. FBXW7 downregulated ROS levels in both endothelial cells and retina tissues, exerting an anti-oxidative injury effect. Similarly, FBXW7 decreased PARP expression and activity in both endothelial cells and retinal tissues, as well as reducing PAR products, leading to less protein oxidation and inflammation. Vascular leakage and the formation of acellular capillaries, two pathological processes in DR, were reduced after FBXW7 overexpression. There is evidence that inhibition of upstream ROS by uncoupling protein-1, manganese superoxide dismutase, or PARP inhibition can block hyperglycemia-induced activation of multiple downstream pathways of vascular damage [4,5,51,]. Therefore, FBXW7 may be a preferred target for treating diabetic vascular complications due to its coexisting beneficial effects, anti-oxidative stress, and PARP downregulation.
Several studies have observed the downregulation of FBXW7 in the progress of neovascularization in DR. In high-glucose states, FBXW7 downregulated VEGF expression and accelerated ubiquitination degradation of c‐Myc, which binds to the HDAC2 promoter to promote angiogenesis [53]. Additionally, FBXW7 inhibited the proliferation of retinal endothelial cells by negatively regulating the Notch1 pathway [54]. In contrast to the above literature, we first focused on the anti-oxidative effect of FBXW7 in endothelial cells under hyperglycemic conditions. FBXW7 was markedly downregulated in the liver of obese mouse and human subjects, while mice with liver-specific knockout of FBXW7 developed hyperglycemia, glucose intolerance, and insulin resistance even on a normal diet [15]. Our results show that although the expression of FBXW7 slightly increased at the early stage of DR progress, it gradually decreased with the progress of DR. Therefore, FBXW7 upregulation may exert beneficial effects on DR and metabolism regulation, such as blood glucose control and insulin resistance prevention.
In addition, we further illuminated the association between ROS alleviation and PARP inhibition by FBXW7. The blocked ETC is the source of electron leakage and superoxide production because of accelerated the tricarboxylic acid cycle and overloaded electron donors (NADH and FADH2) under hyperglycemic conditions [4]. ROS, the by-product of oxidative respiration, was significantly increased when mitochondria were damaged in hyperglycemic states. In turn, sustained exposure to ROS damages mitochondria function and downregulates mtDNA biogenesis, leading to a vicious cycle between ROS production and mitochondrial dysfunction [55]. As a result, the release of cytochrome c from mitochondria and translocation of BAX-induced apoptosis and contributed to the loss of capillary cells [56].
An observational study demonstrated that mtDNA copy number might serve as a marker for mitochondrial dysfunction and be negatively related to the risk of metabolic syndrome and T2DM [57]. Exposure to hyperglycemia significantly decreased mtDNA copy number, while FBXW7 increased mtDNA copy number, indicating mitochondrial abundance elevation. Hyperglycemia also damaged maximal oxygen consumption and spare oxygen consumption for HUVECs, but this kind of respiration damage can be normalized by FBXW7. A similar beneficial effect of FBXW7 was observed when H2O2 induced oxidative stress in HUVECs. Consequently, the blocked ETC was alleviated, and ROS production was decreased under hyperglycemia conditions. The mechanism for BAX downregulation by FBXW7 overexpression can partially be attributed to mitochondrial function improvement.
In hyperglycemic states, FBXW7 promoted DNA damage repair, leading to the reverse of PARP activity, and therefore maintained intracellular NAD+, the substrate of PARP and SIRT3. SIRT3 mostly localizes to the mitochondrial matrix and regulates substrate deacetylase and mitochondrial metabolism such as ETC/OXPHOS, ROS detoxification, and mitochondrial dynamics [58]. In the hyperglycemia state, FBXW7 elevated intracellular NAD+ and downregulated the expression of SIRT3 and TFAM, which is essential for mitochondrial gene transcription and mtDNA replication, contributing to the alleviation of mitochondria function. Consistent with our research, Dikalova et al. reported that SIRT3 depletion promoted endothelial dysfunction, vascular hypertrophy, vascular inflammation, and end-organ damage in mice with hypertension [59]. Zhang et al. reported that mitochondrial dysfunction caused by TFAM inactivation induced ROS production in cardiomyocytes [60]. The effect of FBXW7 on mtDNA copy number maintenance and respiration promotion was reversed by the SIRT3 inhibitor 3-TYP. Furthermore, 3-TYP eliminated the anti-oxidative effect of FBXW7.
As an essential organelle for many aspects of cellular homeostasis, mitochondria not only regulates intracellular energy production, biosynthesis, and cellular stress responses, but also influence an organism's physiology by regulating communication between cells and tissues [61]. Therefore, mitochondria dysfunction drives a series of diseases, including metabolic, cardiovascular, neurodegenerative, and neuromuscular diseases, leading to an intensive search for new therapeutic and preventive strategies aimed at invigorating mitochondrial function [62]. Our research is the first to report that FBXW7 improved damaged mitochondrial function in hyperglycemia states, which could be a target for disease with malfunctional mitochondria and tissues with high mitochondria content and high oxygen consumption.
However, previous research has reported that FBXW7 downregulated PGC-1α by ubiquitin-mediated proteolysis, leading to the deterioration of mitochondria biogenesis and function [18,63,64], which seems contrary to our conclusion. PGC-1α, an inducible coactivator, is preferentially expressed in tissues with high oxygen consumption, and its expression level can be regulated by FBXW7 isoforms. Of the three isoforms FBXW7α, FBXW7β and FBXW7γ generated by alternative splicing, nuclear FBXW7α promotes the stability of PGC-1α while cytoplasmic FBXW7β leads to ubiquitin-mediated degradation of PGC-1α [65]. FBXW7α, the most highly expressed and stable isoform, is more abundant than the other isoforms [66]. As Zhao et al. reported in liver tissues, we observed no significant change of PGC-1α expression in HUVECs after FBXW7 overexpression in hyperglycemic states, which may partially due to the differences between cells and tissues [15]. In line with the low abundance of mitochondria in vascular endothelium, endothelium prefers glycolysis for ATP production and glycolytic flux is more than 200-folds higher than glucose oxidation flux in HUVECs [67], which may account for the difference between endothelial cells and cells with high oxygen consumption.
We conducted in vivo and in vitro assays to demonstrate that FBXW7 promoted DNA damage repair, eliminated ROS, and inhibited PARP activity, which are three key links in the unified mechanism theory for diabetic vascular complications. However, apart from models for DR, we did not examine other diabetic complications such as CVD, atherosclerosis, and diabetic nephropathy in this study. Further researches will be conducted to verify the role of FBXW7 in other diabetic complications.
In summary, we first reported and clarified the anti-oxidative stress effect of FBXW7 in endothelial cells. FBXW7 improved detrimental processes contributing to the pathogenesis of DR, including DNA damage, mitochondrial dysfunction, ROS elimination, and PARP overactivation, which could be a potential target for diabetic vascular complication treatment and superior to existing PARP inhibitors.
4. Methods
4.1. Adenoviruses
Adenoviruses ad-shFB (expressing a short hairpin RNA (shRNA) targeting human FBXW7 (GenBank NM_018315)), ad-FOE, ad-Ub (overexpressing human FBXW7 mRNA and ubiquitin mRNA), and vehicle control sh-ctrl (Ad-U6-CMV-MCS), ad-ctrl (Ad-CMV-MCS-3FLAG) were purchased from OBiO Technology (Shanghai, China). The shRNAs were mixed before transfection. The knockdown target sequences are shown in Supplementary Table 1.
4.2. Cell culture and adenovirus infection
Human primary umbilical vein endothelial cells (HUVECs, five to six passages) (ScienCell, CA, USA) and human primary retinal micro-vascular endothelial cells (HREC, three to four passages) (Cell Systems, WA, USA) were cultured in ECM (ScienCell, CA, USA) containing 5% FBS, 1% ECGS, 100 U/mL penicillin and 100 μg/ml streptomycin at 5.5 mM d-glucose concentration for normal glucose and 30 mM for high glucose. Endothelial cells were infected with sh-FBXW7, ad-FBXW7, and ad-Ubiquitin at a multiplicity of infection of 100, or corresponding vehicle control at approximately 70% confluence. Inhibitors for PARP1(3AB and PJ34), ATM (KU55933), DNA-PKcs (NU7441), ATR (VE821), NOX (apocynin), SIRT3 (3-TYP), lysosome (leupeptin hemisulfate) and proteasome (MG132), as well as mitochondrial electron transport chain complex Ⅰ (rotenone), complex Ⅲ (antimycin A), shown in Supplementary Table 2, were used individually or in combination with the adenoviruses. To induce severe oxidative stress and cause DNA damage, 300 μM hydrogen peroxide (H2O2) treatment was used to incubate HUVECs for 30 min in a 37 °C incubator. After H2O2 treatment, cells were returned to the previous medium for another 2 h for recovery. Each experiment was repeated independently at least three times.
4.3. Human fundus pathological membrane tissues
Human macular epiretinal membrane (ERM) tissues harvested from nondiabetic patients and pre-retinal PFVM from patients with DR were used for quantitative real-time PCR. DR was evaluated according to the diagnostic criteria of the American Academy of Ophthalmology set at the 2001 Annual Meeting. Patient details including age, gender, blood pressure and blood glucose are included in Supplementary Table 3. Tissues were collected with informed consent following the guidelines of the Helsinki Declaration. This study was approved by the ethics committee of Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
4.4. Western blot analysis
HUVECs, HRECs, and rat retinal tissues were lysed in cell lysis buffer (Beyotime Biotechnology, Shanghai, China) containing 1 mM PMSF, protease/phosphatase inhibitor cocktail (Roche, IN, USA). 30–50 μg protein samples were used for Western blot using 4–20% SDS PAGE gels and transferred to a 0.22 μm PVDF membrane (Roche). Then, membranes were incubated in primary antibody at 4 °C overnight. The dilution for the primary antibody was shown in Supplementary Table 4. The secondary antibody (Jackson, PA, USA) was incubated at room temperature for 1 h. Immunoreactive bands were visualized using ECL Plus HRP substrate (Millipore, MA, USA) with an Amersham Imager 600 (GE, CT, USA).
4.5. Quantitative real-time PCR
Total RNA from cells and tissues was isolated using TRIzol reagent (Invitrogen, CA, USA). cDNAs were prepared using a PrimeScript™ RT Reagent Kit (TaKaRa, Japan) and analyzed by real-time PCR using TB Green Premix Ex Taq™(TaKaRa) by ViiA 7 (Applied Biosystems, DE, USA). Relative mRNA levels were quantified by normalizing with β-actin. The primer sequences are available in Supplementary Table 5.
4.6. Alkaline comet assay
DNA damage levels were evaluated using an alkaline comet assay (Trevigen, MD, USA) and a specialized electrophoresis system (Trevigen). HUVECs seeded in six-well plates were treated with 300 μM H2O2 for 30 min, washed in PBS and left to recover in fresh media for 2 h. After being digested with trypsin and washed once in PBS, the HUVECs suspension (1 × 105 cells/mL) in ice-cold PBS was harvested. 20 μl of this cell suspension was mixed gently with 180 μL of pre-warmed low melting point agarose; 50 μl of this mixture was then placed on each well of comet slides. After solidification of the gels, slides were immersed in a lysis solution at 4 °C overnight, protecting them from light. Slides were incubated in alkaline solution (1 mM EDTA, 200 mM NaOH, pH > 13) for 40 min in the dark for DNA unwinding and then placed in a horizontal electrophoresis chamber filled with alkaline solution for electrophoresis (21 V, 30 min). For dehydration of the gels, slides were placed in 70% ethanol for 5 min. After air-drying, all samples were stained with SYBR Gold(Invitrogen). Five fields of view per slide were taken using confocal microscopy (Leica Microsystems, Wetzlar, Germany). The DNA tail moment representing DNA damage severity was scored using comet analysis software (R&D, MN, USA).
4.7. Immunofluorescence microscopy
HUVECs seeded in eight-well Millicell EZ slides (Merck, Darmstadt, Germany) were fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature (RT). They were then permeabilized with 0.5%Triton X-100 (Sigma Aldrich) for 10 min at RT followed by 30 min of blocking in a blocking buffer (Beyotime Biotechnology). Cells were subsequently incubated overnight with anti-FBXW7(1:1000) (Abcam, ab109617), anti-ATM (1:400) (Abcam, ab98), anti-XRCC4(1:1000) (Genetex,109632) or anti-γH2AX (1:400) (Millipore,05–636) at 4 °C, and subsequently for 1 h in diluted secondary antibody (1:500) (Invitrogen, A11029 and A11037). DNA was stained with DAPI at 25 °C for 10 min. Images were captured with a confocal microscopy (Leica, Wetzlar, Germany) using a 20 × or 63 × oil-immersion lens.
4.8. Cell cycle assay
HUVECs digested with trypsin were fixed (absolute ethanol/PBS = 7:3 vol/vol) overnight. FxCycle™ PI/RNase Staining Solution (Life Technologies, OR, USA) was used to stain cells at RT for 30min in the dark. The cell cycle was analyzed using flow cytometry (Beckman, CA, USA).
4.9. DR-GFP assay
The HR/NHEJ assay was performed as previously described [20,21]. Briefly, HUVECs were seeded in six-well plates and incubated in either normal glucose or high glucose, as detailed above. When cells reached approximately 70% confluence, adenovirus was infected overnight, followed by administration of an inhibitor. Cells were transfected 24 h later, with 1.6 μg of pLCN-double-strand break (DSB) Repair Reporter DNA damage response (DDR) (Addgene Cat.No.98895, MA, USA), 1.6 μg of pCAGGS DRR mCherry Donor EF1 BFP plasmid (Addgene Cat.No.98896), and 1.6 μg of pCBAsceI (Addgene Cat.No.26477) plasmid with lipofectamine 2000 (Invitrogen). The pCBAsceI or mCherry Donor EF1 BFP plasmid alone was used alone as a control. GFP and mCherry signal analyses were measured using flow cytometry (Beckman) 48 h after transfection.
4.10. Measurement of intracellular ROS
HUVECs and HRECs were incubated with diluted DCFH-DA probes (10 μM) (Molecular Probes, OR, USA) in serum-free medium for 30 min protecting from light. After washing with cold PBS to remove uncombined probes, the cells were harvested and analyzed via flow cytometry (Beckman). Tempol, a SOD mimetic, was administrated for the specificity of the fluorescence signal.
4.11. Measurement of mitochondria ROS (mtROS)
Harvested cells were washed and then resuspended in pre-warmed Hanks’ balanced salt solution (HBSS) buffer containing 5 μM MitoSOX (Invitrogen) at 37 °C for 25 min. After washing with HBSS buffer, the cells were measured and analyzed via flow cytometry (Beckman).
4.12. Antioxidant capacity evaluation
According to the manufacturer's instructions, total SOD activity, total GSH (Beyotime Biotechnology) and catalase activity (Cayman Chemical, MI, USA) were measured using colorimetric assay.
4.13. Coimmunoprecipitation (Co-IP)
Co-IP was performed using a Pierce Co-IP kit (Invitrogen) according to the manufacturer's instructions. After exposure to 300 μM H2O2, HUVECs were incubated with MG132 (10 μM) for 2 h and then lysed in IP lysis buffer containing a protease/phosphatase inhibitor cocktail and 1 mM PMSF. The anti-FLAG antibody (Thermo Fisher Scientific, 91878) was immobilized with AminoLink Plus coupling resin for 2 h at RT. Cell lysates were incubated overnight with anti-FLAG-conjugated agarose resin at 4 °C. After washing, the eluted immunoprecipitant samples were analyzed via Western blot.
4.14. Ubiquitination assay
HUVECs were infected with ad-Ubiquitin-HA, with or without ad-FBXW7 overnight. After exposure to 300 μM H2O2, cells were incubated with MG132 (10 μM) for 2 h and then lysed for Co-IP assay. Anti-HA antibody (Thermo Fisher Scientific, 26183) or anti-PARP1 antibody (Proteintech Group, 66520-1-Ig) was immobilized with coupling resin for 2 h. Afterwards, cell lysis was proceeded in the same manner as the procedures described for Co-IP.
4.15. Mitochondrial respiration assay
HUVECs were seeded in XFe 24-well microplates (Agilent Technologies, CA, USA) at a density of 20000 cells per well with corresponding treatment. Extracellular acidification rate (ECAR) indicating the glycolytic levels, and oxygen consumption rate (OCR) indicating the capacity of mitochondrial respiration, were measured with a Mito Stress Test Kit using the Seahorse XFe24 Analyzer (Agilent Technologies) following the manufacturer's instructions. Briefly, RPMI 1640 media (Seahorse) was supplemented with 2 mM l-glutamine, 10 mM glucose and 1 mM sodium pyruvate. The sensor cartridge was incubated overnight with 1 mL XF Calibrant (Agilent Technologies, pH7.4) per well at 37 °C without CO2. OCR and ECAR were detected by sequential injections of compounds including oligomycin (1.5 μM), FCCP (1 μM) and Rot/Aa (0.5 μM) at the specified time points according to the instructions of the manufacturer. For acute injection, H2O2 (200 μM) was acutely injected into microplates during the measurement of basal OCR, which oxidatively damaged the HUVECs. Basal respiration, maximal respiration, spare respiratory capacity, and ATP production were measured and normalized with cell numbers and analyzed using Wave software (Agilent Technologies).
4.16. Mitochondria DNA copy number determination
Total DNA from HUVECs was extracted using the TaKaRa MiniBEST Universal Genomic DNA Extraction Kit (TaKaRa) and quantified using NanoDrop 2000 (Thermo Fisher Scientific). Mitochondrial DNA level was detected using the Human Mitochondrial DNA Monitoring Primer Set (TaKaRa) with q-PCR. The procedures for DNA extraction and mtDNA quantification were conducted according to the manufacturer's manual.
4.17. Intracellular NAD + level measurement
NAD + levels were measured from whole-cells extracts using an NAD+/NADH quantification kit with the colorimetric method (Abcam, MA, USA). HUVECs were scraped and washed twice with ice-cold PBS and then centrifuged for 5 min at 375×g at 4 °C. The supernatant was discarded, and metabolites were extracted from the HUVECs pellet with the buffers supplied with the commercial assay kits according to the manufacturer's instructions. Colorimetric measurements were made at 450 nm absorbance and at 25 °C. NAD + levels were determined from a standard calibration curve. The value was normalized according to protein concentrations.
4.18. Animals
Eight-week-old male Sprague-Dawley rats (200–250 g body weight) and C57BL/6J mice (20–25 g body weight) were purchased from Shanghai Laboratory Animal Center (Shanghai, China). All animals were bred in an air-conditioned room with a 12-h light-dark cycle and had free access to food and water. DR was induced by intraperitoneal injection of STZ (Sigma Aldrich); rats were injected with 65 mg/kg in 10 mM of a sodium citrate buffer (pH 4.5) once, and mice were injected with 55 mg/kg for six consecutive days. The animals were fasted for 12 h before STZ injection. Control animals were administered with the sodium citrate buffer only. Animals were categorized as diabetic when their blood glucose level exceeded 16.7 mM, and then randomly assigned to different experimental groups, n = 6–12 for each group.
4.19. Intravitreal injection
Animals were anesthetized with a 10:50 mix of ketamine (100 mg/mL) and xylazine (20 mg/mL). The pupils were dilated with tropicamide (1%). For rats, a sclerotomy incision was created ∼0.5 mm posterior to the limbus with a blade. Then, 1 × 108 pfu of adenovirus that would cause either the overexpressing or knocking down of FBXW7 was injected into the vitreous cavity with a 33-gauge glass injector connected to syringe. The first injection was administrated immediately after the diabetic model was established, and the second injection followed 4 w later. Rats were sacrificed at 8 or 12 w post-diabetic model establishment.
For mice, a sclerotomy incision was created through the sclera posterior to the limbus with a 33-gauge injector and 1 × 108 pfu of adenovirus that would cause overexpression of FBXW7 was injected into the vitreous cavity. The first injection was administered 3 m after the diabetic model was established; the second injection followed a month later. Mice were sacrificed 5 months post-diabetes induction.
4.20. In situ superoxide detection
As previously described, 2 μM of dihydroethidium (DHE) (Life Technologies) was used to detect superoxide in fresh frozen eye sections [68]. The fluorescence intensity of retinal sections was determined under a fluorescence microscope (Olympus, Tokyo, Japan) and measured using ImageJ software (National Institutes of Health). Data were normalized using values from non-diabetic rats.
4.21. Protein oxidation assay
Retinal tissues were sonicated and homogenized in a pre-chilled lysis buffer containing a protease inhibitor cocktail (Roche) and PMSF on ice. Protein oxidation levels were detected using an OxiSelect Protein Carbonyl ELISA Kit (Cell Biolabs, CA, USA) according to the manufacturer's instructions. The protein oxidation amount was normalized against the total protein concentrations.
4.22. PARP activity and PAR quantitative assay
The PARP activity in HUVECs and HRECs was determined using the HT Colorimetric PARP/Apoptosis Assay (Trevigen) as instructed in the manual. For the PAR quantitative assay, HUVECs, HRECs, and retinal tissues were sonicated in a lysis buffer on ice. The extracts were incubated with 20% (w/v) SDS and the PAR levels were measured via chemiluminescent reading (Biotek Synergy2, VT, USA) using the HT PARP in vivo Pharmacodynamic Assay II (Trevigen) according to the manufacturer's instructions and were normalized to the total cellular protein concentrations.
4.23. Evans blue assay
Retinal vascular permeability was determined by Evans blue (EB) staining, which was covalently linked to albumin as an indicator of albumin leakage into the retina from the capillary. EB dye (100 mg/mL, Sigma Aldrich) was dissolved in PBS, sonicated for 5 min, and filtered through a 0.45 μm filter (Millipore).
Quantitative vascular permeability measurements were taken according to procedures previously documented in literature [69]. EB dye (30 mg/kg) was injected via the femoral vein. After the dye was circulated for 1 h with the animals on a warm pad, the rats were perfused with citrate buffer (0.05 M pH = 3.5) via the left ventricle. The retinas were then dissected from the eyeballs and dried for 5 h. After measuring retinal dry weight, EB dye was extracted by solubilization overnight in formamide (0.2 mL per retina) at 78 °C. The resulting suspensions were centrifuged at 120,000×g at 4 °C for 30 min. EB dye in the supernatant was spectrophotometrically detected by absorbance at 620 nm (blue signal) and 740 nm (background subtracted). Blood samples were centrifuged at 3600×g at 25 °C for 15 min. The concentration of EB dye was calculated from a standard curve of EB dye in formamide and normalized to dry retina weight. The leakage of EB dye was calculated as: retinal EB content (μg)/retina dry weight (g)/averaged plasma EB concentration (μg/μl) × circulation time (h).
For immunofluorescence observation, procedures were referred to previous literatures [70,71]. Mice were injected with EB dye (45 mg/kg) via the femoral vein and blood samples were collected. After the dye circulated for 1 h, mice were sacrificed and their eyes were enucleated and immersed in 4% PFA for 1 h at 25 °C. After removing the cornea, lens, and vitreous body, the retina was carefully removed from the choroid and sclera. The retinal cups were cut radically to lie flat. Z-stack images were captured through a × 10 objective.
4.24. Retinal trypsin digestion assay
The formation of acellular capillaries was detected by a retinal trypsin digestion assay. Eyes were enucleated from mice and fixed in 4% PFA for 24 h at 4 °C. Then retina was dissected under a microscope and washed with PBS for 6 h. Retinas were incubated overnight in a solution of 0.25% trypsin in a 0.1 M Tris buffer (pH 7.8) at 37 °C. After trypsin digestion, vasculature networks were isolated by gentle but thorough percussion and then mounted on glass slides. When the vessels turned white, retinal vasculature was stained with periodic acid-Schiff hematoxylin and observed under light microscopy (Olympus). The number of acellular capillaries was counted in 10 random fields ( × 400 magnification) in a masked manner and was standardized to capillary area (per mm2 capillary area).
4.25. Statistics
All experimental data are expressed as the means ± standard deviations of at least three independent experiments. Statistical analyses were performed using a two-tailed Student's t-test or one-way ANOVA followed by Tukey's test or Sidak's test in GraphPad Prism 8 (GraphPad Software) or SPSS22.0 (IBM), and P < 0.05 was considered significant.
4.26. Study approval
The in vivo animal experiments complied with the requirements of the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. Animal research was approved by the Laboratory Animal Ethics Committee of Shanghai General Hospital (No. SC049).
The study using human retinal samples was approved by the ethics committee of Shanghai General Hospital. The experiment was carried out in accordance with the Declaration of Helsinki. All patients or their families provided written informed consent.
Conflict-of-interest statement
Shanghai General Hospital holds a patent (ZL 2018 1 1020687. X) in regard to the application of FBXW7 and its up-regulator in the prevention or treatment of diabetic retinopathy, licensed by China National Intellectual Property Administration. FW, XX, JD, XY and QG are inventors of this patent, which may be considered as potential conflict of interests.
Author contributions
SL, FW and JD performed most of the experimental work; DS, QG, SC, SZ, HL and XY prepared and incubated human endothelial cells and established diabetic animal models; SC, NW and JZ performed in vivo animal experiments; SL, JW and SZ are involved in vitro cell experiments. FW, XX along with SL and JD participated in conception and design, analysis and interpretation of the data; SL and XY drafted the article with contributions from FW.
Acknowledgements
The authors thank all the members in Shanghai Key Laboratory of Ocular Fundus Diseases for expert technical assistance. The work was financially supported by Natural Science Foundation of China (Grant No.81970812, 81700842), Project of National Key R&D Program of China (Grant No.2016YFC0904800, 2019YFC0840607), National Science and Technology Major Project of China (Grant No.2017ZX09304010), Excellent medical young talent projects of Shanghai General Hospital (Grant No.06N1702019), Science and Technology Research Project of Songjiang District (No. 2020SJ300).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102530.
Contributor Information
Xun Xu, Email: drxuxun@sjtu.edu.cn.
Fang Wei, Email: weifang73@hotmail.com.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Harding J.L., Pavkov M.E., Magliano D.J., Shaw J.E., Gregg E.W. Global trends in diabetes complications: a review of current evidence. Diabetologia. 2018;62(1):3–16. doi: 10.1007/s00125-018-4711-2. [DOI] [PubMed] [Google Scholar]
- 2.Ogurtsova K., da Rocha Fernandes J.D., Huang Y., et al. IDF Diabetes Atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017;128:40–50. doi: 10.1016/j.diabres.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 3.Vos T., Allen C., Arora M., et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1545–1602. doi: 10.1016/S0140-6736(16)31678-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 5.Du X., Matsumura T., Edelstein D., et al. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Invest. 2003;112(7):1049–1057. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szabo C., Biser A., Benko R., Bottinger E., Susztak K. Poly(ADP-ribose) polymerase inhibitors ameliorate nephropathy of type 2 diabetic Leprdb/db mice. Diabetes. 2006;55(11):3004–3012. doi: 10.2337/db06-0147. [DOI] [PubMed] [Google Scholar]
- 7.Giacco F., Brownlee M. Oxidative stress and diabetic complications. Circ. Res. 2010;107(9):1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eid S., Sas K.M., Abcouwer S.F., et al. New insights into the mechanisms of diabetic complications: role of lipids and lipid metabolism. Diabetologia. 2019;62(9):1539–1549. doi: 10.1007/s00125-019-4959-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganda O.P., Bhatt D.L., Mason R.P., Miller M., Boden W.E. Unmet need for adjunctive dyslipidemia therapy in hypertriglyceridemia management. J. Am. Coll. Cardiol. 2018;72(3):330–343. doi: 10.1016/j.jacc.2018.04.061. [DOI] [PubMed] [Google Scholar]
- 10.Chamberlain J.J., Johnson E.L., Leal S., Rhinehart A.S., Shubrook J.H., Peterson L. Cardiovascular disease and risk management: review of the American diabetes association standards of medical care in diabetes 2018. Ann. Intern. Med. 2018;168(9):640–650. doi: 10.7326/M18-0222. [DOI] [PubMed] [Google Scholar]
- 11.Sibley S.D., Hokanson J.E., Steffes M.W., et al. Increased small dense LDL and intermediate-density lipoprotein with albuminuria in type 1 diabetes. Diabetes Care. 1999;22(7):1165–1170. doi: 10.2337/diacare.22.7.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Group A.S., Group A.E.S., Chew E.Y., et al. Effects of medical therapies on retinopathy progression in type 2 diabetes. N. Engl. J. Med. 2010;363(3):233–244. doi: 10.1056/NEJMoa1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keech A.C., Mitchell P., Summanen P.A., et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007;370(9600):1687–1697. doi: 10.1016/S0140-6736(07)61607-9. [DOI] [PubMed] [Google Scholar]
- 14.Welcker M., Clurman B.E. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer. 2008;8(2):83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 15.Zhao J., Xiong X., Li Y., et al. Hepatic F-box protein FBXW7 maintains glucose homeostasis through degradation of fetuin-A. Diabetes. 2018;67(5):818–830. doi: 10.2337/db17-1348. [DOI] [PubMed] [Google Scholar]
- 16.Bengoechea-Alonso M.T., Ericsson J. The ubiquitin ligase Fbxw7 controls adipocyte differentiation by targeting C/EBPalpha for degradation. Proc. Natl. Acad. Sci. U. S. A. 2010;107(26):11817–11822. doi: 10.1073/pnas.0913367107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Onoyama I., Suzuki A., Matsumoto A., et al. Fbxw7 regulates lipid metabolism and cell fate decisions in the mouse liver. J. Clin. Invest. 2011;121(1):342–354. doi: 10.1172/JCI40725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park J.H., Kang H.J., Lee Y.K., et al. Inactivation of EWS reduces PGC-1alpha protein stability and mitochondrial homeostasis. Proc. Natl. Acad. Sci. U. S. A. 2015;112(19):6074–6079. doi: 10.1073/pnas.1504391112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ceccaldi R., Rondinelli B., D'Andrea A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26(1):52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arnoult N., Correia A., Ma J., et al. Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature. 2017;549(7673):548–552. doi: 10.1038/nature24023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fukumoto T., Zhu H., Nacarelli T., et al. N(6)-Methylation of adenosine of FZD10 mRNA contributes to PARP inhibitor resistance. Cancer Res. 2019;79(11):2812–2820. doi: 10.1158/0008-5472.CAN-18-3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uziel T., Lerenthal Y., Moyal L., Andegeko Y., Mittelman L., Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22(20):5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lavin M.F., Kozlov S., Gatei M., Kijas A.W. ATM-dependent phosphorylation of all three members of the MRN complex: from sensor to adaptor. Biomolecules. 2015;5(4):2877–2902. doi: 10.3390/biom5042877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Q., Karnak D., Tan M., Lawrence T.S., Morgan M.A., Sun Y. FBXW7 facilitates nonhomologous end-joining via K63-linked polyubiquitylation of XRCC4. Mol. Cell. 2016;61(3):419–433. doi: 10.1016/j.molcel.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang D., Kim S.H., Hamasaki N. Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion. 2007;7(1–2):39–44. doi: 10.1016/j.mito.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 26.Bagul P.K., Katare P.B., Bugga P., Dinda A.K., Banerjee S.K. SIRT-3 modulation by resveratrol improves mitochondrial oxidative phosphorylation in diabetic heart through deacetylation of TFAM. Cells. 2018;7(12) doi: 10.3390/cells7120235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Canto C., Sauve A.A., Bai P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Aspect. Med. 2013;34(6):1168–1201. doi: 10.1016/j.mam.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rajman L., Chwalek K., Sinclair D.A. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metabol. 2018;27(3):529–547. doi: 10.1016/j.cmet.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang Q., Yang C. Oxidative stress and diabetic retinopathy: molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020;37 doi: 10.1016/j.redox.2020.101799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fedorova M., Bollineni R.C., Hoffmann R. Protein carbonylation as a major hallmark of oxidative damage: update of analytical strategies. Mass Spectrom. Rev. 2014;33(2):79–97. doi: 10.1002/mas.21381. [DOI] [PubMed] [Google Scholar]
- 31.Cipriani G., Rapizzi E., Vannacci A., Rizzuto R., Moroni F., Chiarugi A. Nuclear poly(ADP-ribose) polymerase-1 rapidly triggers mitochondrial dysfunction. J. Biol. Chem. 2005;280(17):17227–17234. doi: 10.1074/jbc.M414526200. [DOI] [PubMed] [Google Scholar]
- 32.Fatokun A.A., Dawson V.L., Dawson T.M. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014;171(8):2000–2016. doi: 10.1111/bph.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soriano F.G., Pacher P., Mabley J., Liaudet L., Szabo C. Rapid reversal of the diabetic endothelial dysfunction by pharmacological inhibition of poly(ADP-ribose) polymerase. Circ. Res. 2001;89(8):684–691. doi: 10.1161/hh2001.097797. [DOI] [PubMed] [Google Scholar]
- 34.Zakaria E.M., El-Bassossy H.M., El-Maraghy N.N., Ahmed A.F., Ali A.A. PARP-1 inhibition alleviates diabetic cardiac complications in experimental animals. Eur. J. Pharmacol. 2016;791:444–454. doi: 10.1016/j.ejphar.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Li F., Drel V.R., Szabo C., Stevens M.J., Obrosova I.G. Low-dose poly(ADP-ribose) polymerase inhibitor-containing combination therapies reverse early peripheral diabetic neuropathy. Diabetes. 2005;54(5):1514–1522. doi: 10.2337/diabetes.54.5.1514. [DOI] [PubMed] [Google Scholar]
- 36.Lorenzi M., Montisano D.F., Toledo S., Barrieux A. High glucose induces DNA damage in cultured human endothelial cells. J. Clin. Invest. 1986;77(1):322–325. doi: 10.1172/JCI112295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Polo S.E., Jackson S.P. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25(5):409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uryga A., Gray K., Bennett M. DNA damage and repair in vascular disease. Annu. Rev. Physiol. 2016;78:45–66. doi: 10.1146/annurev-physiol-021115-105127. [DOI] [PubMed] [Google Scholar]
- 39.Mahmoudi M., Gorenne I., Mercer J., Figg N., Littlewood T., Bennett M. Statins use a novel Nijmegen breakage syndrome-1-dependent pathway to accelerate DNA repair in vascular smooth muscle cells. Circ. Res. 2008;103(7):717–725. doi: 10.1161/CIRCRESAHA.108.182899. [DOI] [PubMed] [Google Scholar]
- 40.Kumar V., Agrawal R., Pandey A., et al. Compromised DNA repair is responsible for diabetes-associated fibrosis. EMBO J. 2020;39(11) doi: 10.15252/embj.2019103477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scully R., Panday A., Elango R., Willis N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019;20(11):698–714. doi: 10.1038/s41580-019-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang M., Wu W., Wu W., et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34(21):6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pandey N., Black B.E. Rapid detection and signaling of DNA damage by PARP-1. Trends Biochem. Sci. 2021;46(9):744–757. doi: 10.1016/j.tibs.2021.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murai J., Huang S.Y., Das B.B., et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curtin N.J., Szabo C. Poly(ADP-ribose) polymerase inhibition: past, present and future. Nat. Rev. Drug Discov. 2020;19(10):711–736. doi: 10.1038/s41573-020-0076-6. [DOI] [PubMed] [Google Scholar]
- 46.Martin-Guerrero S.M., Casado P., Munoz-Gamez J.A., et al. Poly(ADP-Ribose) Polymerase-1 inhibition potentiates cell death and phosphorylation of DNA damage response proteins in oxidative stressed retinal cells. Exp. Eye Res. 2019;188 doi: 10.1016/j.exer.2019.107790. [DOI] [PubMed] [Google Scholar]
- 47.Trenz K., Smith E., Smith S., Costanzo V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006;25(8):1764–1774. doi: 10.1038/sj.emboj.7601045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lavin M.F. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene. 2007;26(56):7749–7758. doi: 10.1038/sj.onc.1210880. [DOI] [PubMed] [Google Scholar]
- 49.Jaiswal M., Schinske A., Pop-Busui R. Lipids and lipid management in diabetes. Best Pract. Res. Clin. Endocrinol. Metabol. 2014;28(3):325–338. doi: 10.1016/j.beem.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 50.Cheung N., Mitchell P., Wong T.Y. Diabetic retinopathy. Lancet. 2010;376(9735):124–136. doi: 10.1016/S0140-6736(09)62124-3. [DOI] [PubMed] [Google Scholar]
- 51.Garcia Soriano F., Virag L., Jagtap P., et al. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat. Med. 2001;7(1):108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- 53.Hu L., Lv X., Li D., et al. The anti-angiogenesis role of FBXW7 in diabetic retinopathy by facilitating the ubiquitination degradation of c-Myc to orchestrate the HDAC2. J. Cell Mol. Med. 2021;25(4):2190–2202. doi: 10.1111/jcmm.16204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shao J., Fan G., Yin X., et al. A novel transthyretin/STAT4/miR-223-3p/FBXW7 signaling pathway affects neovascularization in diabetic retinopathy. Mol. Cell. Endocrinol. 2019;498 doi: 10.1016/j.mce.2019.110541. [DOI] [PubMed] [Google Scholar]
- 55.Santos J.M., Tewari S., Kowluru R.A. A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Radic. Biol. Med. 2012;53(9):1729–1737. doi: 10.1016/j.freeradbiomed.2012.08.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kowluru R.A., Abbas S.N. Diabetes-induced mitochondrial dysfunction in the retina. Invest. Ophthalmol. Vis. Sci. 2003;44(12):5327–5334. doi: 10.1167/iovs.03-0353. [DOI] [PubMed] [Google Scholar]
- 57.Fazzini F., Lamina C., Raftopoulou A., et al. Association of mitochondrial DNA copy number with metabolic syndrome and type 2 diabetes in 14 176 individuals. J. Intern. Med. 2021;290(1):190–202. doi: 10.1111/joim.13242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J., Xiang H., Liu J., Chen Y., He R.R., Liu B. Mitochondrial Sirtuin 3: new emerging biological function and therapeutic target. Theranostics. 2020;10(18):8315–8342. doi: 10.7150/thno.45922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dikalova A.E., Pandey A., Xiao L., et al. Mitochondrial deacetylase Sirt3 reduces vascular dysfunction and hypertension while Sirt3 depletion in essential hypertension is linked to vascular inflammation and oxidative stress. Circ. Res. 2020;126(4):439–452. doi: 10.1161/CIRCRESAHA.119.315767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang D., Li Y., Heims-Waldron D., et al. Mitochondrial cardiomyopathy caused by elevated reactive oxygen species and impaired cardiomyocyte proliferation. Circ. Res. 2018;122(1):74–87. doi: 10.1161/CIRCRESAHA.117.311349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nunnari J., Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148(6):1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sorrentino V., Menzies K.J., Auwerx J. Repairing mitochondrial dysfunction in disease. Annu. Rev. Pharmacol. Toxicol. 2018;58:353–389. doi: 10.1146/annurev-pharmtox-010716-104908. [DOI] [PubMed] [Google Scholar]
- 63.Abbate F., Badal B., Mendelson K., et al. FBXW7 regulates a mitochondrial transcription program by modulating MITF. Pigment Cell Melanoma Res. 2018;31(5):636–640. doi: 10.1111/pcmr.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Olson B.L., Hock M.B., Ekholm-Reed S., et al. SCFCdc4 acts antagonistically to the PGC-1alpha transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes Dev. 2008;22(2):252–264. doi: 10.1101/gad.1624208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trausch-Azar J.S., Abed M., Orian A., Schwartz A.L. Isoform-specific SCF(Fbw7) ubiquitination mediates differential regulation of PGC-1alpha. J. Cell. Physiol. 2015;230(4):842–852. doi: 10.1002/jcp.24812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grim J.E., Gustafson M.P., Hirata R.K., et al. Isoform- and cell cycle-dependent substrate degradation by the Fbw7 ubiquitin ligase. J. Cell Biol. 2008;181(6):913–920. doi: 10.1083/jcb.200802076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eelen G., de Zeeuw P., Treps L., Harjes U., Wong B.W., Carmeliet P. Endothelial cell metabolism. Physiol. Rev. 2018;98(1):3–58. doi: 10.1152/physrev.00001.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chan E.C., van Wijngaarden P., Liu G.S., Jiang F., Peshavariya H., Dusting G.J. Involvement of Nox2 NADPH oxidase in retinal neovascularization. Invest. Ophthalmol. Vis. Sci. 2013;54(10):7061–7067. doi: 10.1167/iovs.13-12883. [DOI] [PubMed] [Google Scholar]
- 69.Xu Q., Qaum T., Adamis A.P. Sensitive blood-retinal barrier breakdown quantitation using Evans blue. Invest. Ophthalmol. Vis. Sci. 2001;42(3):789–794. [PubMed] [Google Scholar]
- 70.Fan L., Yan H. FTY720 attenuates retinal inflammation and protects blood-retinal barrier in diabetic rats. Invest. Ophthalmol. Vis. Sci. 2016;57(3):1254–1263. doi: 10.1167/iovs.15-18658. [DOI] [PubMed] [Google Scholar]
- 71.Hernandez C., Bogdanov P., Sola-Adell C., et al. Topical administration of DPP-IV inhibitors prevents retinal neurodegeneration in experimental diabetes. Diabetologia. 2017;60(11):2285–2298. doi: 10.1007/s00125-017-4388-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.