

SUMMARY
The analogy of mitochondria as powerhouses has expired. Mitochondria are living, dynamic, maternally inherited, energy-transforming, biosynthetic, and signaling organelles that actively transduce biological information. We argue that mitochondria are the processor of the cell, and together with the nucleus and other organelles they constitute the mitochondrial information processing system (MIPS). In a three-step process, mitochondria (1) sense and respond to both endogenous and environmental inputs through morphological and functional remodeling; (2) integrate information through dynamic, network-based physical interactions and diffusion mechanisms; and (3) produce output signals that tune the functions of other organelles and systemically regulate physiology. This input-to-output transformation allows mitochondria to transduce metabolic, biochemical, neuroendocrine, and other local or systemic signals that enhance organismal adaptation. An explicit focus on mitochondrial signal transduction emphasizes the role of communication in mitochondrial biology. This framework also opens new avenues to understand how mitochondria mediate inter-organ processes underlying human health.
INTRODUCTION
Our collective view of mitochondria evolved from that of dynamic cytoplasmic granules or “bioblasts”1 to bean-shaped ATP-synthesizing chemiosmotic machines,2,3 motivating the popular “powerhouse of the cell” analogy.4 Subsequently, mitochondria became known as maternally inherited organelles5 with their own genome, in which mutations can cause disease,6,7 setting off the field of mitochondrial medicine. The imaging of living mitochondria dynamically exchanging proteins and DNA8 and triggering death via propagating apoptotic waves9,10 later sparked an era of mitochondria as dynamic organelles undergoing constant fusion/fission events, enabling functional complementation11,12 and mitochondrial quality control.13 Most recently, the omics era and a new quantitative handle on intermediate metabolism have cast mitochondria as biosynthetic and signaling organelles14,15 that produce signals influencing cell and organism behaviors via metabokine/mitokine signaling,16–18 mitonuclear crosstalk,19–22 and remodeling of the epigenomic machinery across species.23–26 Through these theoretical transitions, mitochondria have become the most studied organelle in the biomedical sciences (Figure 1).
Figure 1. Modern historical landmarks in mitochondrial research illustrate the need for an integrative view of this multifaceted organelle.

Proportion of biomedical publications by organelle, corrected for total published articles across biomedicine. Selected discoveries that challenged prior views about mitochondria are noted, as well as some historical landmarks for context. Figure adapted from Picard et al.390 with data retrieved from https://pubmed.ncbi.nlm.nih.gov/ on February 12, 2022.
Outside of their intracellular roles as organelles, mitochondria also operate beyond the confines of the cell.27 They undergo physical transfer from cell to cell,28–30 influence neurotransmitter metabolism and inter-cellular communication at the neural synapse,31,32 synthesize all circulating steroid hormones that ensure sexual reproduction and species survival in mammals,33 and as discussed below, even contain receptors for systemic hormones.34 These discoveries are not only blurring cellular boundaries, but also revealing mitochondria in a light that emphasizes communication—i.e., the bidirectional transfer of information from organelle to organism—as a natural aspect of their biology.
It is a particularly exciting time for mitochondrial biology. Community resources like MitoCarta35 and MitoCoP,36 together with spectacular theoretical advances in mitochondrial research, have brought insights to diverse fields across the biological sciences and medicine.37 This includes, but is not limited to, immunometabolism, behavioral neuroscience, and psychobiology. As examples, energy metabolism in general and mitochondria in particular play permissive and instructive roles in stem cell differentiation and in the acquisition of immunometabolic phenotypes,38 influence whether animals are socially dominant or submissive,39 and influence how multiple organ systems in mice respond to evoked stress.40 In humans, mitochondrial energy production capacity also appears to dynamically respond to subjective psychosocial experiences,41,42 providing a foundation to begin understanding the mind-mitochondria connection. These discoveries are contributing to mechanistically linking sub-cellular bioenergetic processes to physiological, health-related organismal phenotypes. Thus, scientific progress not only among mitochondrial biology but also more broadly in the biomedical sciences43 has been and likely will continue to be catalyzed by increasingly accurate and integrative models of mitochondrial behavior.
In this perspective, we argue that as we move toward increasingly accurate mechanistic models of the role of mitochondria in human health, we need an understanding of mitochondrial behavior extending far beyond energetics. As echoed by others,44,45 the “powerhouse” analogy promotes an overly simplistic picture of this beautifully complex organelle. The outdated mechanical analogy is too unidimensional to guide integrative scientific thinking. The challenge ahead is to integrate current prevailing perspectives of mitochondria as inherited, dynamic, energy-transforming, signaling organelles whose influence extends to all cellular compartments, and to the whole organism. Here we propose that our existing knowledge of mitochondrial biology can be integrated under the common framework of mitochondrial signal transduction. Consequently, a more integrative and accurate analogy portrays mitochondria as the processor of the cell—or more precisely as the mitochondrial information processing system (MIPS).
WHY MITOCHONDRIAL SIGNAL TRANSDUCTION?
Signal transduction involves input-to-output transformation. It is a generalizable process in biology, taking place across all living, complex adaptive systems. Signal transduction allows single cells to sense, migrate toward, and respond to stimuli,46 and enables organelle networks to interact and accomplish complex cellular operations that isolated organelles could not accomplish.47 At the organ level, signal transduction also allows the brain to receive, integrate, and process multiple streams of sensory information to generate a coherent internal representation of the outside world.48 This process is analogous to the way in which an antenna or sensor coupled to information processing systems—as in a cell phone, for example—receives and converts simple signals (e.g., radio waves) into intelligible outputs of different kinds (e.g., sounds, images, etc.). In the same way, cellular signal transduction systems such as the MIPS convert complex combinations of ions, proteins, nutrients, and energetic states into goal-driven genetic programs, which guide the reorganization of metabolic pathways and drive adaptive behaviors (to grow and divide, contract, secrete, die, etc.). Signal transduction allows cells and organisms to respond and adapt to environmental demands.
Through evolution, the endosymbiotic incorporation of mitochondria marked the transition from a selfish unicellular world to a multicellular reality.49 In multicellular organisms, a vital priority became the metabolic coordination and cell-to-cell cooperation toward a shared common goal—to sustain the organism. Cellular cooperation produced a sort of “social contract” among increasingly specialized cells.50 Thus unified as an organism, cells make decisions not based solely on their individual states hardwired in the genome, but based on the collective state of the organism established through information exchange and communication between organs, cells, and organelles.51 This collective principle implies that to ensure survival, cellular and organismal decisions must be matched to the local and systemic energetic constraints, which are reflected in the bioenergetic state of mitochondria.52,53 For an organism, achieving faster responses to changing bioenergetic conditions means faster transitions to new optimal states. This, in turn, maximizes energetic efficiency and minimizes the risk of damage.54 Therefore, the role of mitochondria in optimizing cellular and organismal behavior toward health—defined as optimal responsiveness to challenges55—requires mechanisms that transduce information, from organelle to organism.
THE PILLARS OF MITOCHONDRIAL SIGNAL TRANSDUCTION
Before reviewing the specific molecular components and mechanisms that support signal transduction within the MIPS, we describe some general features of signal transduction. Signal transduction within the MIPS is an extension of the traditional process of receptor-mediated detection of (extra)cellular signals, signal amplification, and transduction into downstream secondary messengers (Figure 2). Mitochondrial signal transduction consists of three main processes:
Figure 2. Three-step model of mitochondrial signal transduction.

As the mitochondrial information processing system (MIPS), mitochondria are input integrators and output generators. Within the cytoplasm, mitochondria are topologically positioned at the interface between incoming signals from the outside extracellular space and the inside compartment of the nucleus where the (epi) genome is stored. In a three-step process, mitochondria receive, integrate, and generate signals that contribute to cellular and organismal adaptation. All mitochondria have the potential to perform sensing, integration, and signaling steps. Here the contributions to signal transduction are color-coded and matched to specific topologies for illustrative purposes. ER, endoplasmic reticulum; LD, lipid droplet; Per, peroxisome; mtDNA, mitochondrial DNA; NO, nitric oxide; ΔGp, phosphorylation potential.
Sensing: The ability of mitochondria to detect metabolic and hormonal inputs, and to transform these inputs into morphological, biochemical, and functional mitochondrial states.
Integration: The pooling of multiple inputs into common effectors driven by the exchange of information among mitochondria and other organelles, and influenced by the current state of the mitochondrial network and of the cell.
Signaling: The production of mitochondrial outputs, or signals, that transmit information locally to direct metabolic pathway fluxes and influence other organelles, including nuclear gene expression, and systemically to regulate the physiology and organismal behavior.
The flow of information through the MIPS proceeds sequentially as follows: incoming signals are sensed by molecular receptors and biological structures on/within mitochondria (sensing), which exchange with each other molecular signals and labile states such as membrane potential via fusion/fission processes and other (non)physical interactions (integration), and which simultaneously release signaling factors such as metabolites, cofactors, proteins, nucleic acids, and heat that propagate information beyond the mitochondrial membranes (signaling). Figure 3 illustrates the repertoire of known mitochondrial substrates and mechanisms available for signal transduction. This broad repertoire emphasizes communication at different levels of biological organization, including protein-protein interactions (molecular), inter-organelle communication (sub-cellular organelles), autocrine or inter-cellular paracrine transfer (cells), and endocrine information transfer (organs and systems).
Figure 3. The hallmarks of mitochondrial signal transduction.

Depicted is the mitochondrial repertoire of mechanisms and substrates through which mitochondria receive, integrate, and transmit intracellular and systemic signals.
In the following three sections, we first review the known molecular machinery responsible for mitochondrial input sensing (step 1), the processes enabling information integration within mitochondrial networks (step 2), and the resulting signals that communicate mitochondrial states intracellularly and systemically (step 3). To avoid the natural tendency to emphasize only specific well-known examples that would naturally narrow the spectrum of physiological processes to which mitochondrial signal transduction may apply, we discuss the mechanisms involved in the sensing, integration, and signaling stages sequentially. Recognizing that mitochondria are not all created equal, we then discuss tissue-specific mitochondrial features relevant to signal transduction. We close by considering outstanding questions and opportunities that an integrated mitochondrial signal transduction perspective raises for cell biology and clinical/translational research.
MITOCHONDRIAL SENSING
The main molecular features that enable mitochondrial sensing include traditional ligand-activated receptors, transporters, and biochemical reactions such as the oxidative phosphorylation (OxPhos) system within the outer and inner mitochondrial membranes (OMM and IMM) and the mitochondrial genome (Figure 4). These components enable mitochondria to rapidly and selectively sense changes in specific biochemical inputs. In the same way that a capsaicin receptor allows a sensory cell on the taste bud to depolarize and convert the spicy molecule into an action potential (recognizable as information by the brain), the mitochondrial sensing machinery converts simple inputs into biochemical, functional, and/or morphological changes (eventually converted to outputs that cells recognize). Mitochondrial inputs range in their nature from atoms, gases, and ions to small molecules and metabolites, proteins, lipids, DNA, and temperature, as well as physical interactions with surrounding organelles (Figure 3). In this section, we cover the molecular machinery that allows the MIPS to selectively sense and respond to extrinsic and intrinsic inputs (MIPS step 1 of 3).
Figure 4. MIPS step 1: Sensing.

As in excitable cells where a broad variety of chemical inputs (e.g., neurotransmitters) converge onto membrane potential variations, extrinsic and intrinsic MIPS inputs trigger molecular changes that converge into morpho-functional mitochondrial states. Mitochondria sense extrinsic and intrinsic information through four main classes of mechanisms.
(A) Canonical DNA-binding “nuclear” receptors for steroid hormones including glucocorticoids (GC), estrogen (ER), and androgen (AR) exist in mitochondria or can translocate upon ligand binding.
(B) G protein-coupled receptors (GPCRs) embedded within mitochondrial membranes including the angiotensin (AT1R and AT2R), the cannabinoid (mtCB1), melatonin (MT1), and purine (Py2Rs) receptors, and possibly others (e.g., GPR35).
(C) Metabolite and ion carriers/transporters such as the ADP/ATP carrier protein (AAC, also adenine nucleotide translocator [ANT]) and the SLC25 family of transporters. Also shown are some gases and ions that either freely diffuse through the IMM or whose import/export is mediated by other carriers/transporters.
(D) Acquired sequence variation in the mtDNA sequence, including mutations and deletions that cause functional changes within the OxPhos system. The top path shows nucleotide availability/imbalance, and the bottom path shows exogenous toxins that can interfere with electron transport chain function and secondarily cause mtDNA instability.
AAC/ANT, ADP/ATP carrier or adenine nucleotide translocator; AT2Rs, angiotensin receptors; AR, androgen receptor; ERβ, estrogen receptor beta; GR, glucocorticoid receptor; GRE, glucocorticoid response element (used here as an example for other gene regulatory elements); IMS, intermembrane space; MCU, mitochondrial calcium uniporter; mtCB1, mitochondrial cannabinoid receptor; MT1, melatonin 1 receptor; mtDNA, mitochondrial DNA; NO, nitric oxide; O2, molecular oxygen; OxPhos, oxidative phosphorylation; P2YRs, purine receptors; ROS, reactive oxygen species; SLC25s, solute carriers family 25; T3, triiodothyronine; ΔpH+Δψm, mitochondrial proton motive force.
Canonical “nuclear” receptors
Mitochondria contain ligand-activated transcription factors traditionally known as “nuclear” receptors. These receptors are expressed and generally reside in the cytoplasm or directly in mitochondria, homo- or hetero-dimerize upon ligand binding, and then translocate either to the nucleus or to the mitochondrial matrix where they interact with target DNA sequences. Limitations exist around the experimental evidence underlying the localization of these receptors within mitochondria, calling for further research using rigorous designs and sensitive molecular approaches.56 The most well-documented examples include receptors for thyroid hormones, sex hormones (estrogen and androgen), and stress-related glucocorticoids (Figure 4A).
Thyroid hormones (T3, T4, and related metabolites) have potent effects on tissue oxidative capacity and systemic energy expenditure through their dual action on nuclear gene expression and directly on mitochondria. In isolated mitochondria, respiratory chain activity is modulated by triiodothyronine (T3) without changes in protein synthesis—taking place in vitro or “in organello”—substantiating the direct sensitivity of mitochondria to circulating thyroid hormones.57 Two putative resident mitochondrial thyroid hormone receptors have been described. The p28 receptor likely resides in the IMM, lacks a DNA-binding domain, and binds T3 with high affinity.58 It appears responsible for the rapidity (within 2 min in vitro, <30 min in vivo) with which mitochondrial respiration responds to thyroid hormone stimulation.59,60 The other p43 receptor is a 43 kDa member of the c-Erb α1 DNA-binding family residing in the mitochondrial matrix.61 Mitochondria respond to T3 by increasing mitochondrial DNA (mtDNA) transcription and changing the ration of mRNA and rRNA.62,63 Most studies on mitochondrial T3 response have been performed on liver mitochondria, but mitochondrial thyroid sensing may exhibit high tissue specificity.64 In mice, manipulating the expression of the mitochondrial T3 receptor p43 in different tissues triggers selective transcriptional and enzymatic effects on mtDNA-encoded OxPhos components (e.g., not on complex II proteins or activity),65 as well as broad cellular and physiological effects,66 highlighting the likely physiological significance of mitochondrial thyroid hormone sensing.
There are two isoforms of the estrogen receptor related to mitochondria: ERα and ERβ. They have relatively low homology for their ligand-binding domains and trans-activational domains, and therefore have strongly divergent functions.67 ERα primarily influences nuclear gene expression, including activation of mitochondrial biogenesis and other aspects of mitochondrial biology,68 whereas ERβ translocates to mitochondria and is found at high levels in murine and human mitochondria from neurons and cardiomyocytes.69 Under baseline conditions in cancer cell lines, the majority of ERβ is primarily located in mitochondria.70 Activation of mitochondrial ERβ results in anti-apoptotic effects by disrupting Bad-Bcl-X(L) and Bad-Bcl-2 interactions.71 Estrogen receptor signaling may also increase mitochondrial OxPhos capacity in genetically compromised cells from patients with Leber’s hereditary optic neuropathy (LHON), either directly by acting on the mitochondrial OxPhos system or indirectly by reducing oxidative stress.72
Mitochondria sense androgenic hormones via the canonical androgen receptor (AR). In cultured prostate cells, a substantial fraction of total AR may localize to mitochondria.73 The mitochondrial AR influences mtDNA levels, mtDNA transcription and translation, and RC protein abundance and complex activity.73 This enables both genetic and functional mitochondrial responses to circulating androgen levels. In the human sperm mid-piece, both the AR and ERβ localize to mitochondria.74 Mitochondrial responses to estrogens and androgens via these DNA-binding receptors may in part account for sexually dimorphic mitochondrial features and functions.75,76
Mitochondria also contain the glucocorticoid receptor (GR) and thus can respond to glucocorticoid hormones, including the psychological stress mediator cortisol and corticosterone. Two major GR isoforms have been defined, GRα (predominant, ~90% of transcripts) and GRβ (minor, ~10%), which differ only in their distal domain from exon 9 alternative splicing.77 Another isoform, GRγ, is produced through alternative splicing (includes an intronic codon between exon 3 and 4) and differs from GRα and GRβ isoforms by a single amino acid.78 Within mitochondria, the active GR receptors interact with mtDNA glucocorticoid response element (GRE) sequence motifs to influence mitochondrial rRNA synthesis79 and gene expression. The mtDNA contains 8 putative GREs: 2 within the D loop,80 1 in the 12S rRNA, 1 in tRNALeu(UUR), 3 in COX I, and 1 in COX III.81 Compared to the nuclear genome, which contains approximately 680 GREs (1 GRE for every ~37 protein coding gene), the abundance of GREs in mtDNA is substantially higher, at 1 mtDNA GRE for every ~1.6 protein coding gene.82 Mitochondrial glucocorticoid sensing is likely primarily mediated by GRγ,83 acting on D loop GREs to promote expression of all polycistronic rRNA and mRNA genes.84 It is worth noting that reduced mitochondrial GR localization may occur under certain pathological states, potentially hindering the mitochondrial sensing of glucocorticoid levels.85
Thus, conserved DNA-binding receptors allow mitochondria to sense the broad class of metabolism-regulating, sex- and stress-related hormones conveying systemic information about the state of the organism to core biochemical and genetic elements within the MIPS.
G protein-coupled receptors
The mitochondrial sensory system also includes one of the evolutionarily more recent innovations, the G protein-coupled receptors (GPCRs). Mitochondrial GPCRs sit in the OMM and IMM and are specific to hormones such as angiotensin II,86 melatonin,87 endocannabinoids,88 and purines.89 Mitochondria-localized GPCRs influence core mitochondrial functions including ion uptake, OxPhos, nitric oxide synthesis, apoptotic signaling, and reactive oxygen species (ROS) production,90 illustrating their potentially broad action spectrum (Figure 4B).
Angiotensin type 1 and 2 receptors (AT1R and AT2R) are present on both the nuclear membrane and on the IMM.91 Despite lacking a canonical mitochondrial targeting sequence, transfection of full-length GFP-tagged AT2R naturally results in their mitochondrial localization.86 Mitochondrial AT2R appears to colocalize with its endogenous ligand Angiotensin II (thus forming an endogenous renin-angiotensin system) in several cell types including (from most to least abundant) mouse hepatocytes, cardiomyocytes, and renal tubule cells, and in human monocytes.91 Functionally, activation of AT2R on isolated mitochondria was found to increase nitric oxide production by ~25% with a proportional decrease in complex I-driven O2 consumption capacity,86 supporting the physiological effects of the angiotensin GPCR on mitochondrial oxidative capacity.
Mitochondria also contain the functional melatonin GPCR MT1. In mouse brain neuron mitochondria, MT1 is located on the OMM with its signal transduction apparatus coupled to cyclic AMP in the intermembrane space.87 In isolated mitochondria, activation of MT1 by melatonin could partially inhibit permeability transition and subsequent cytochrome c (Cyt c) release, and expectedly conferred downstream protection from ischemic injury, permeability transition pore (PTP) opening, and subsequent cell death.87 Other potential functions of mitochondrial melatonin signaling involve redox modulation as melatonin is a potent antioxidant,92 and may also include stimulation of mitochondrial biogenesis.93 However, further research in neurons and other cell types is needed to disentangle the effects of MT1 signaling on mitochondria versus on the plasma membrane.
Another functional GPCR localized to mitochondria is the type I cannabinoid receptor (mtCB1). mtCB1 is expressed in neurons,88 astrocytes,94 and skeletal muscle myofibers.95 Similar to CB1 localized at the plasma membrane of neuronal synapses where their signaling inhibits neurotransmitter release, mtCB1 receptors on the OMM signal through intra-mitochondrial Gαi protein activation and inhibition of soluble-adenylyl cyclase (sAC),34 which inhibits protein kinase A (PKA)-dependent phosphorylation of target OxPhos subunits.34 Functionally, mitochondrial endocannabinoid sensing through CB1 reduces complex I activity and mitochondrial respiration.88 In mice, the effects of mtCB1 signaling affect neuronal function and memory formation,34 suggesting that mitochondrial endocannabinoid sensing also has downstream effects on the behavior of the organism.
Mitochondria sense and respond to cytoplasmic purine (ATP, ADP, and AMP) levels via the purine GPCRs P2Y1 and P2Y2. These receptors, whose precise sub-organellar location remains unclear, have been suggested to be coupled to phospholipase C (PLC) and downstream regulation of the mitochondrial calcium uniporter (MCU).89 In hepatocytes, activation of mitochondrial P2Y1 stimulates Ca2+ uptake whereas activation of P2Y2 inhibits uptake.96 A receptor of the same family, the P2X7 ionotropic purinoceptor (P2Y7R), also appears to be present in both the plasma membrane and in the OMM of cultured mouse and human cells.97 Deletion of P2X7 reduces transmembrane potential and respiratory capacity and leads to the accumulation of NADH (i.e., reductive stress) likely secondary to complex I inhibition.97 The presence of surface ADP-sending GPCRs in mitochondria, along with the ADP/ATP carrier and F1/F0 ATP synthase system (discussed below), illustrates the potential value of redundant mechanisms allowing the MIPS to sense particularly critical cytoplasmic signals such as the cytoplasmic ADP:ATP ratio.
Other receptors may also localize to the MIPS. For example, GPCRs internalized from the plasma membrane, such as the kynurenic acid-activated GPR35, may translocate to the OMM and modulate the OxPhos system under stress conditions.98 The ligand-gated ion channel α7 nicotinic acetylcholine receptor (nAChR) on the OMM may also enable mitochondria to sense acetylcholine (and nicotine) to modulate Ca2+ transients, permeability transition, and mtDNA release.99,100 Additional work is required to discover and validate additional mitochondrial receptors, and to determine the ligand specificity and functional significance of mitochondrial GPCRs as components of the mitochondrial sensing machinery.
Metabolite signaling
In a process highly integrated with cytoplasmic micronutrient sensors such as mTORC1 and AMPK, mitochondria sense and respond to metabolite levels and availability through specific carriers and transporters embedded within the IMM (Figure 4C). One of the classic MIPS inputs that triggers responses among the OxPhos system and multiple downstream mitochondrial processes is the phosphorylation potential (ΔGp), reflected simplistically in ADP levels.101 An increase in cytoplasmic ADP concentration (or more accurately a decrease in ΔGp, or ATP:ADP ratio) is rapidly transmitted to the mitochondrial matrix via monomers of the ADP/ATP carrier spanning the IMM.102 This sets the rotary FoF1 ATP synthase into motion, transiently dissipating the protonmotive force (ΔpH+Δψm: pH gradient + mitochondrial membrane potential).103 In this single step, the shift in ATP/ADP levels causes a consequential thermodynamic shift that sends biochemical ripples sequentially accelerating (1) proton pumping and electron flow by respiratory chain complexes I, III, and IV; (2) the reduction of O2 to H2O by Cyt c oxidase (complex IV); and (3) the oxidation of reducing equivalents NADH and FADH2 at complexes I and II, respectively, thus (4) driving the oxidation of metabolic intermediates in the TCA/Krebs cycle, which in turn (5) increases CO2 production and (6) increases the diffusion gradient for the uptake of cytoplasmic substrates/metabolites into the matrix.101 In isolated mitochondria, this ADP-induced change in state from low flux to actively respiring mitochondria is associated with profound conformation changes of the mitochondrial cristae membranes—from the orthodox to condensed state.104 This simple example illustrates the breadth and complexity of ultrastructural and biochemical responses that a single input, such as ADP, can elicit within the MIPS.
Mitochondrial metabolite uptake and sensing are broadly enabled by >50 proteins from the SLC25 family of transporters, known as the mitochondrial carrier system (MCS).105 These transmembrane IMM proteins are essential to both MIPS sensing and also to fulfill the biosynthetic functions of mitochondria, allowing the export of the ingredients for growth and repair, i.e., several amino acids and one-carbon carrying folate forms, carboxylic acids, fatty acids, cofactors, inorganic ions, and nucleotides to the whole cell.106 In relation to mitochondrial sensing, the MCS provides sensory capacity to small molecule metabolite carriers including pyruvate (MPC1/2),107,108 dicarboxylate (malate-phosphate, DIC) and tricarboxylate (citratemalate, CTP) carriers, aspartate-glutamate (AGC1/2), the phosphate carrier105 and NAD carrier,109 and many others. Some mitochondrial transporters are often alternatively expressed variants of genes encoding plasma membrane carriers, as for the mitochondrial glutamine transporter that allow mitochondria to respond directly to cytoplasmic glutamine levels.110 In cancer cells, the diverse MCS enables mitochondria to sense cytoplasmic levels of key amino acids via the TCA cycle, and to undergo secondary morphological changes.111 Thus, metabolite carriers and transporters allow the MIPS to sense and respond to inputs from numerous metabolic pathways.
We also note that the bioenergetic state of the cell exerts regulatory control on dozens of mitochondrial enzymatic reactions. As a result, high ATP:ADP and NADH:NAD+ ratios favor anabolism and promote export of metabolites, shifting metabolism toward non-oxygen-dependent glycolysis and biosynthesis.112 In cases of OxPhos defects, NADH accumulates and reduce the availability of NAD+, causing reductive stress,113,114 driving the flux of metabolic pathways toward mitochondrial anabolic pathways contributing to disease.115,116 The collective action of metabolic inputs on mitochondrial OxPhos and the NADH:NAD+ ratio also translates into the availability of downstream substrates, including coenzyme A (CoA) and NADP(H). Changes in CoA availability affect the conversion of acetyl to acyl-CoA used to post-translationally acetylate and functionally modulate not only cytoplasmic enzymes and nuclear chromatin,117 but also dozens of mitochondrial proteins.118 Together, these biochemical dial systems provide a set of molecular cascades that dynamically integrate and convert biochemical inputs into functional mitochondrial recalibrations,119 thereby allowing the MIPS to sense a broad array of biochemical inputs about the dynamic bioenergetic state of the cell.
Electrophysiology of mitochondria: Ion signaling
Mitochondria sense and respond to ions, which is a logical consequence of the relatively high membrane potential across the IMM, generating a large diffusion potential for charged atoms and molecules (Figure 4C). One of the most studied examples of ionic mitochondrial sensing is calcium (Ca2+). The MCU enables rapid Ca2+ uptake within seconds.120,121 Mitochondrial Ca2+ uptake from the cytoplasm and ER triggers rapid changes in mitochondrial physiology through post-translational modifications (PTMs) of dehydrogenases that increase TCA cycle activity122 and results in membrane potential changes.123 Although genetic ablation of the MCU is not lethal in most mouse strains, its loss prevents mitochondria from sensing surrounding Ca2+ levels123 and may impair mitochondrial fusion during cell-cycle division,124 highlighting the significance of mitochondrial Ca2+ sensing on the MIPS.
Mitochondrial Ca2+ is also linked with sodium (Na+) signaling. The organellar Ca2+ levels are affected by the activity of the mitochondrial Na+/Ca2+ exchanger (NCLX). NCLX sits in the IMM and extrudes matrix Ca2+ in exchange for cytoplasmic Na+.125 Entry of sodium into the cytosol during an action potential in neurons, or in response to glucose stimulation in the pancreatic beta cell, triggers the extrusion of calcium from the mitochondria, preventing mitochondrial calcium overload and subsequent cell death.126 By lowering the intra-mitochondrial Ca2+ levels ([Ca2+]mito) in cardiomyocytes and brown adipocytes, NCLX activity thereby decreases intra-mitochondrial calcium levels and directly regulates mitochondrial PTP (mPTP) dynamics and downstream signaling.127,128 But in addition to decreasing [Ca2+]mito levels, NCLX activity rapidly elevates matrix [Na+] levels,129 likely preventing collapse of the MIPS through the flux of Ca2+ and Na+ ions.
Mitochondria sense and also respond to surrounding concentrations of various atoms and ions including magnesium (Mn), inorganic phosphate (Pi), chloride (Cl), iron (Fe), and possibly others including lithium (Li),130 although the underlying mechanisms for the most part are not resolved. Mitochondria are also sensitive to divalent gases such as nitric oxide (NO), which acts directly on complexes I and IV by chemically modifying sensitive residues, thereby modulating respiration.131 One fairly well-studied input is molecular oxygen (O2), which is sensed directly by a combination of complex I-132,133 and complex III-derived134 bursts of ROS during hypoxia. In low-oxygen conditions (hypoxia), acidification of the mitochondrial matrix in a complex I-dependent manner solubilizes intra-mitochondrial calcium-phosphate deposits, which increases soluble [Ca2+]mito (even in the absence of MCU). In mouse embryonic fibroblasts, hypoxia activates NCLX and causes a 2- to 3-fold increase in mitochondrial matrix Na+, which interacts with IMM phospholipids to decrease membrane fluidity and promote superoxide formation by semiquinone,129 transforming information about oxygen availability into molecular information within the MIPS. Mitochondrial O2 sensing undoubtedly complements cytoplasmic sensors such the hypoxia-inducible factor 1 alpha (HIF-1α) pathway that primarily act on the nucleus.135 Overall, several evolutionarily ancient ion channels, transporters, and mechanisms based on chemical modifications thus ensure that mitochondria can sense and rapidly respond to their surrounding intracellular ionic environment.
Intrinsic mtDNA defects
In addition to cytoplasmic signals sensed through canonical receptors and carriers, mitochondria also dynamically recalibrate their structure and internal processes in response to intrinsic signals, such as those arising from the mitochondrial genome (Figure 4D). The mtDNA codes for 37 canonical genes, including 13 protein-coding mRNA sequences,136 plus small mitochondria-derived peptides (MDPs).137 As the mtDNA can be affected by external factors (e.g., mutagens, nucleotide availability) and produce outputs (RNA, proteins) that influence and shape the OxPhos system and downstream mitochondrial behaviors, the mtDNA is a component of the mitochondrial sensing system.
Defects in the mtDNA sequence, which are either inherited138 or acquired,139 alter the synthesis of OxPhos subunits, which consequently impair respiratory capacity, OxPhos function, and Δψm, culminating in disease.140,141 Because most biochemical reactions taking place within mitochondria are directly or indirectly linked to OxPhos and Δψm, including substrate and ion uptake, mtDNA perturbations have widespread consequences for several metabolic pathways.113,142 Within skeletal muscle, for example, acquired somatic mtDNA deletions can disrupt OxPhos enzyme activities and trigger local mitochondrial proliferation when in proximity to a nucleus143 (see section on mitochondrial signaling). Milder mtDNA variants are also sensed, are translated in metabolite levels,144 and result in variation in cellular and organismal phenotypes that influence lifespan and disease risk.145,146 Moreover, because some mutagens and toxins may preferentially affect the mtDNA relative to the nuclear DNA—in part owing to the negatively charged matrix compartment (which attracts positively charged molecules), rapid replication, poor repair, or other factors—the mitochondrial genome maintenance and expression systems may act as a cellular “sentinel” of genotoxic stress.147
Certain intrinsic mitochondrial inputs such as nucleotide availability and genotoxic molecules may not influence any of the mitochondrial biochemical processes directly, and may in fact only be sensed through the (replicating) mtDNA. For example, low nucleotide availability impairs OxPhos function specifically through the decline in mtDNA copy number,148 which may or may not induce nucleotide salvage or cytoplasmic nucleotide synthesis pathways. We propose that the mtDNA replisome is an actively communicating structure, which tunes its function based on cellular nucleotide pools and the local mtDNA expression machinery. The actual signals contributing to mtDNA communication within the MIPS are still poorly understood.
Summary of mitochondrial sensing
Mitochondria are equipped with a surprisingly wide variety of receptors and molecular features that give them the ability to sense hormonal, metabolic, ionic, genetic, and other inputs. With such sensitivity to a broad spectrum of inputs, the MIPS senses both the local biochemical conditions surrounding each organelle and systemic neuroendocrine signals produced in distant anatomical locations of the organism: by other cells, within other organs. Mitochondrial behavior is therefore not only driven by changes in nuclear gene expression—which produce the sensing components—but also more acutely and reversibly by biochemical and endocrine inputs that dynamically modulate their biochemical, genetic, ultrastructural, and physiological properties.
The evolutionary co-opting of a variety of DNA-binding receptors, GPCRs, and transporters suggests that increasing the range of inputs that mitochondria were capable of sensing must have positively contributed to the organism’s adaptive capacity. As a result, the diverse mitochondrial sensing machinery has been evolutionarily selected and likely also enriched in mitochondrial membranes relative to other organelles. Defining the full spectrum of inputs directly sensed by the MIPS across different cell types is an outstanding research challenge. Expanding our understanding of the inputs that directly shape mitochondrial biology could illuminate new disease pathways, independent or upstream of OxPhos or other well-defined disease-causing mechanisms.
Next, we turn our attention to dynamic factors that physically and functionally connect sensing mitochondria as interactive networks capable of signal processing and integration.
MITOCHONDRIAL SIGNAL INTEGRATION
In its simplest form, signal integration is the process by which inputs are converted into common second messengers containing transformed information about the inputs. For example, within cells, multiple cell surface receptors converge on the production of common chemical secondary messenger molecules such as cyclic AMP or Ca2+, which in turn trigger broad-acting downstream response(s).149 Because secondary messengers are shared products for multiple receptors, multiple stimuli converge on the same signaling hubs. Another good example of this concept takes place in neurons: dozens of neurotransmitters and modulators signal via ionotropic and metabotropic receptors to converge on a single cellular property—the plasma membrane potential. The temporal combination of inputs determines whether or not an action potential is generated.150 As a result, in neural networks (as in mitochondrial networks) membrane potential serves as an integrating hub for signal transduction. The convergence of inputs onto chemical second messengers and membrane potential thus allows cells to produce coherent, integrated, and robust responses simultaneously shaped by multiple inputs.
Another core concept for signal integration is that large-scale functional networks bind small competent units into larger scale computational agents.51 Cells and organs integrate and compute information as cell collectives.151 For instance, in the brain no single neuron (unit) can accomplish the sophisticated brain computations required to coordinate and sustain the rest of the body. Glial cells and neurons accomplish remarkable feats of integration through cell-to-cell communication, creating a functional collective (the brain) that naturally integrates or computes information.48 Similarly, mitochondria are functionally linked and operate as “social” collectives within the cell cytoplasm.152 For our purposes, integration refers to the functional computations (i.e., the transformation of inputs into outputs) that take place within the MIPS between the sensing and signaling steps.
A third and final relevant concept to signal integration states that computational processes are influenced by the structural properties of the network itself—i.e., how individual units are arranged and connected relative to other ones.153,154 The interactions between mitochondria, defined as the probability of direct information exchange between individual organelles, is termed “connectivity.” Across physical, biological, and social networks, the extent and nature of the connectivity between units largely define the network properties155,156 (Figure 5). Thus, the information processing capacity of the MIPS, created by networks of co-existing mitochondria, must be determined jointly by both the intrinsic properties of mitochondria (their sensing machinery, mtDNA, OxPhos system, etc.) and their functional connectivity with one another.
Figure 5. MIPS step 2: Signal integration.

The physical and functional binding of multiple energized units (mitochondria) into sparsely connected networks naturally gives rise to signal integration.
(A) Mechanisms of mitochondrial network remodeling and inter-organellar communication (mito-mito, mito-other organelles) among the MIPS.
(B) Conceptual representation of the organism’s organ network and of the brain, where information from one group of units (e.g., neurons) is transmitted to other units, giving rise to computational agents. Information processing is not a private property of brains; it is a generalizable property of all life forms.
(C) Four examples of network properties that may be used to define the organization of mitochondrial collectives processing biochemical, metabolic, endocrine, and other inputs into coherent outputs.
Below we discuss how physical inter-organellar interactions, diffusible signals, dynamic morphological transitions, motility, and sub-cellular positioning dynamically define the architecture and connectivity of mitochondrial networks, which are the basis for mitochondrial signal integration (MIPS step 2 of 3).
Mechanisms of homologous mitochondrial communication
Several types of physical interactions enable transient information exchanges among mitochondria. Mitochondrial “kiss-and-run” involves the partial fusion of mitochondrial membranes among motile mitochondria in plants157 and cultured mammalian cells.13,158 These rapid interactions occur in the span of seconds to minutes and require the OMM mitofusins (MFN1/2) and IMM optic atrophy 1 (OPA1). Kiss-and-run events enable the exchange of proteins and membrane potential,158 and possibly mtDNA nucleoids, although likely only in some cell types.159
Inter-mitochondrial junctions (IMJs) are close OMM-OMM contact sites anatomically similar to cell-cell gap junctions. The juxtaposition of highly electron-dense mitochondrial membranes, originally visualized in cardiomyocytes160 between electrically connected mitochondria,161 increases in frequency with cellular energy demand (e.g., exercise162) and with mitochondrial volume density.163 At IMJs, which are evolutionarily conserved from mollusks to mammals, internal cristae membranes exhibit a remarkable degree of coordination (i.e., cristae alignment) across the two juxtaposed mitochondria, revealing the exchange of information between the two linked organelles.163 Artificially linking energized mitochondria in vitro via synthetic linkers was sufficient to recapitulate IMJs and trigger cristae remodeling,163 and the iron-sulfur cluster containing OMM protein MitoNEET may be one of the IMJ tethering proteins.164 Functionally, IMJs may provide the physical basis for the propagation of membrane potential and other physicochemical signals even in the absence of protein exchanges and complete mitochondrial fusion.165,166
Mitochondrial nanotunnels are thin ~100-nm-wide double-membrane protrusions that arise from donor mitochondria, extend over distances up to several microns, and can interact and fuse with a receiver mitochondrion.167 Nanotunnels transport matrix proteins and therefore represent a mechanism of protein sharing and communication even between non-adjacent mitochondria.168 In cultured cells, nanotunnels can be induced by the pulling action of the kinesin motor protein Kif5b.169 In vivo, the existence of nanotunnels has been limited to tissues where mitochondrial motility is restricted such as in the densely packed cytoplasm of human skeletal muscles170 and rat cardiomyocytes,171 suggesting that physically constrained mitochondria that cannot encounter diverse fusion partners reach out to other functional mitochondria via nanotunnels. In patients with mitochondrial disease, mitochondria with compromised OxPhos function due to mtDNA mutations were found to have ~6-fold more nanotunnels than in healthy controls.170 This suggests that mitochondrial nanotunnels may preferentially arise or stabilize between mitochondria with impaired OxPhos capacity as a mean of functional complementation,167 or as a mean of increasing the effective functional connectivity among the mitochondrial network of the MIPS. Among other biological networks, enhancing the structural connectivity between individual units alters global network properties and can enhance robustness and computational/cognitive properties.172,173
Mitochondria also communicate via diffusible signals. One well-described example of diffusion-based mito-mito communication is ROS-induced ROS release (RIRR). Among the relatively uncluttered cytoplasm of cultured cells, mitochondria can generate and propagate waves of ROS production progressing through sequential PTP opening at rates of ~5 μm/min.174 This soluble form of signaling relies mostly on the physical proximity of mitochondria. In cardiomyocytes, proximity-based propagation depends on the production and diffusion of superoxide anions (O2.—) and H2O2.175 Similarly, mitochondria can propagate waves of apoptotic signaling by sequentially undergoing permeability transition: waves are propagated by groups of mitochondria that sequentially uptake and release Ca2+, which neighboring mitochondria then uptake and release, and so on.10 The mito-mito transmission of information via diffusible signals within the MIPS may also be facilitated by some of the physical structures described above, particularly inter-organellar tethers.
Mitochondrial dynamics: Fusion and fission
Mitochondrial fusion is a well-described process whereby two adjacent and generally motile mitochondria encounter each other and interact via the outwardly protruding domains of mitofusins (MFN1/2) and accessory proteins, leading to the sequential merging of the OMM and IMM of both organelles.176 After fusion, the two original mitochondria form a unified organelle with a continuous matrix and membrane system. Organelle fusion allows the exchange of all matrix, IMM, IMS, and OMM components, including mtDNA, proteins and RC complexes, lipids, metabolites, ions, and membrane potential.
Experiments tracking the diffusion of photo-activable green fluorescent protein (mtPAGFP) in cultured mammalian cells177 and in vivo178 show that fusing mitochondria readily exchange molecular material.179 In cardiomyocytes cultured ex vivo, mtPAGFP is exchanged through kiss-and-run fusion and nanotunnels and becomes distributed to the entire mitochondrial network within ~10 h.168 In immortalized cell lines, the rate of mitochondrial fusion for each organelle is significantly faster, at one fusion event every ~5–20 min.13 Mitochondrial membrane fusion therefore leads to the mixture and homogenization of mitochondrial protein distribution (i.e., mitochondria are more similar to each other). On the other hand, ablation of mitochondrial fusion by double Mfn1/2 silencing in mouse embryonic fibroblasts drastically increases mitochondrial heterogeneity (some mitochondria have a lot of protein x, others have little of it) within the MIPS.12 Ex vivo studies of post-mitotic tissues and cells have made it clear that mitochondria in post-mitotic cells have lower fusion rates than cancer cells and immortalized cell lines (e.g., Eisner et al.180). Moreover, the cytoplasm of certain tissues can inhibit ex vivo mitochondrial fusion rates.181 But in post-mitotic cells in which mitochondrial movement is restricted by cytoskeletal elements, fusion and fission can take place without displacement of mitochondria. This can be viewed as fire-doors in a long corridor—rapidly and reversibly modulating the network connectivity.
The functional relevance of dynamics to mitochondrial signal transduction is that larger mitochondria with larger matrix volume and lower surface-area-to-volume ratio respond differently to incoming signals. One example is the ability of mitochondria to handle histamine-induced rises in cytoplasmic [Ca2+]. Relative to small fragmented mitochondria, larger tubular mitochondria in the same cell uptake Ca2+ at a similar rate but recover more quickly (within 30 s).182 In response to hyperglycemia, mitochondrial fragmentation precedes hyperglycemia-induced ROS production.183,184 Hyperglycemia increases ROS production within ~30 min, and fragmented mitochondria produce ~50% more ROS than filamentous mitochondria in the same cell.182 Again, the sequential events of sensing and responses illustrate how the functional responses of mitochondria to environmental inputs and stimuli are not rigidly set by genetically encoded states, but rather dynamically regulated by shape changes that remodel the network properties of the MIPS. Distinct fission signatures (i.e., where the fission event occurs along the mitochondrial tubule) are associated with the fate—degradation or biogenesis—of the resulting mitochondrial fragments,185 possibly influencing long-term network properties.
The mitochondrial network also responds to metabolic signals. Mitochondrial fusion and fission are modulated by the cellular metabolic state,186 and in turn regulate mtDNA stability in vitro and in vivo.187 For example, the MIPS responds to substrate deprivation by undergoing MFN-dependent fusion,188,189 whereas metabolic oversupply can inhibit mitochondrial fusion and lead to higher DRP1-dependent fragmentation in cultured cells184,190,191 and in skeletal muscle in vivo.192 Morphological changes underlie intra-mitochondrial functional changes that optimize coupling efficiency (i.e., the coupling of oxygen consumption to ATP synthesis) to best match the dynamic metabolic state.186,193 In brown adipocytes, mitochondrial fission decreases coupling efficiency in a DRP-1 and free fatty acid-dependent mechanism,194 reflecting an intra-mitochondrial morpho-functional response that increases fatty acid utilization and heat production. Like neural connections that come and go through activity-dependent sprouting and pruning,195 mitochondrial interactions and connections also persist and vanish over variable time periods, modulating information flow within the MIPS network.
Motility
Mitochondrial motility refers to the ability of mitochondria to travel to and from different parts of the cell. Motility influences MIPS structure as mitochondria stretch into their common tubular structure by adhering to cytoskeletal elements such as microtubules and actin filaments. When mitochondria fall off the cytoskeleton, they lose their tubular shape. Motility of an individual mitochondrion is also the strongest predictor of mitochondrial fusion.196 Remarkably, the highest probability for a successful meeting between two mitochondria to develop into a fusion event is when one mitochondrion is moving while the other is stationary. The lowest probability is when both mitochondria have been stationary, even if they are juxtaposed.177 On the other hand, fission is commonly followed with movement of the two daughter mitochondria so that they are not juxtaposed anymore (e.g., Kleele et al.185). The two fission products can therefore subsequently interact, possibly fuse, and thus share their content and more labile states with other units within the network. Both microtubules and actin filaments play a role in mitochondrial fission; for example, forcing the depolymerization of microtubules prevents the cytoplasmic redistribution of mitochondria in response to stress.197 Directional motility is facilitated by cytoskeletal elements, but non-directional Brownian movement also appears to be a contributor to mitochondrial motility.198
Motility is influenced by the sensing of environmental signals.199 The molecular sensors responsible for transducing metabolic and biochemical signals into motility implicate a complex of proteins that connect mitochondria to the motor machinery, the dynein and kinesin. Dynein and kinesin-1 walk the mitochondria on microtubules and thus any movement requires their attachment to the mitochondrial surface.200 The molecular complex connecting mitochondria to these motor proteins includes Miro and Milton, whose regulations have been well defined in neurons.201 When mitochondria enter an area with high calcium concentrations, Miro detaches from the motor proteins, resulting in the mitochondria falling off the cytoskeleton and becoming stationary; as a result, mitochondria stop their movement and accumulate in areas with increased calcium, where they can contribute to calcium buffering.202 Similarly, Milton (Trak1) is inactivated by GlucNAC when glucose concentrations increase, leading to a similar arrest of mitochondrial movement in neurons in response to hyperglycemia.203 In cultured cells, inter-mitochondrial tethering events similar to IMJs (without fusion) regulated by lysosomes204 occur ~10× more frequently than fusion/fission events, limiting mitochondrial motility and therefore regulating mitochondrial distribution within the cytoplasm.205 Overall, motility is a mechanism that dynamically redistributes mitochondria and together with fusion and fission determines the network structure of the intracellular mitochondrial collective.
Communication with other organelles
The MIPS engages in functional interactions with the ER lysosomes, peroxisomes, lipid droplets, and likely other organelles. This topic has been elegantly reviewed elsewhere.206,207 Mitochondrial metabolism is directly supported by surrounding organelles that provide various substrates, lipid intermediates, and ionic signals that not only supply substrates, but also communicate information about the overall state of the cell. In particular, input from the nucleus provides hundreds of proteins that sustain and confer mitochondria with both their molecular sensory machinery and the machinery for fusion/fission dynamics and motility that influence their propensity to adopt certain network configurations.
Mitochondrial cortisol synthesis is exemplary of this inter-organelle inter-dependence, requiring the transfer of cholesterol from lipid droplets to mitochondria and its import across mitochondrial membranes, followed by shuttling of steroidogenic intermediates from mitochondria to the ER, and from the ER back to the mitochondrial matrix, where cortisol is finally synthesized.208 The synthesis of the mitochondrial IMM phospholipid cardiolipin similarly involves the shuttling of lipid intermediates between mitochondria and ER at mitochondria-associated membranes (MAMs) through the ERMES209 and ER-mitochondria complex (EMC).210 Punctual, localized, and pulsatile redox-based communication between mitochondria and the ER can also propagate signals from single mitochondria to the ER and other mitochondria.211 These examples illustrate the functional inter-dependence of mitochondria and other organelles, and the existence of conserved mechanisms for information exchange, propagating the state of the MIPS to other organelles, and vice versa.
Summary of mitochondrial signal integration
After describing the molecular machinery allowing mitochondria to sense and dynamically respond to intracellular and systemic inputs, here we have discussed the mechanisms allowing mitochondria to communicate and exchange information among each other and with other organelles. As the MIPS physically and functionally interacts as a mitochondrial collective with other organelles, it generates distributed representations of the biochemical and energetic conditions of the cells and organism. In turn, these capacities to sense and integrate information are adaptive, allowing mitochondria to tune and optimize their morpho-functional states to changing intracellular and environmental conditions.
Note that soluble communication mechanisms undoubtedly complement more complete forms of mitochondrial communication involving the merging and more-or-less complete union of mitochondria through membrane fusion. If diffusible signaling and transient protein exchange are analogous to “kiss-and-run,” more stable physical mitochondrial contacts such as IMJs and nanotunnels may reflect “engage-and-hold,” whereas complete mitochondrial fusion is analogous to “marry-and-mix.” Thus, mitochondrial interactions can be relatively transient (ion efflux lasts a few milliseconds), selective (nanotunnels connect with only one acceptor mito), and reversible (inter-organellar tethers can dissociate). The nature of these interactions is consistent with other plasticity mechanisms in biology, such as those modulating synaptic function within neural networks, which similarly integrate inputs and compute information.212
However, the ultimate unit of evolution and adaptation is not the mitochondrial network or the individual cell. It is the cell collective that constitutes the organism. Therefore, the goal of mitochondrial sensing and integrating information must be to optimize adaptation and health of the organism itself. Biologically, this becomes possible if the information sensed and integrated by the MIPS is then communicated to the cell and to the rest of the organism. This logic brings us to consider how mitochondrial inputs are converted and transmitted into meaningful cellular and organismal outputs or signals, through mitochondrial signaling (MIPS step 3 of 3).
MITOCHONDRIAL SIGNALING
Several well-established and emerging signaling pathways link mitochondrial behavior to gene expression within the cell nucleus. Moreover, beyond the cell, the MIPS releases signals in the systemic circulation, influencing metabolic processes in neighboring cells and distant target organs. Several elements of mitochondrial signaling have been extensively covered elsewhere, such as apoptotic signaling,213 ROS-mediated signaling,214,215 and metabolic intermediates.216 Here we only briefly cover these areas and expand the discussion of mitochondrial signaling to a broader spectrum of mitochondrial outputs that serve as intracellular and/or systemic outputs, including small metabolites, proteins, DNA, steroid hormones, and non-molecular signals including heat (Figure 6). We also discuss how the potency and specificity of signal transduction may be influenced by the sub-cellular localization of signaling mitochondria.
Figure 6. MIPS step 3: Signaling.
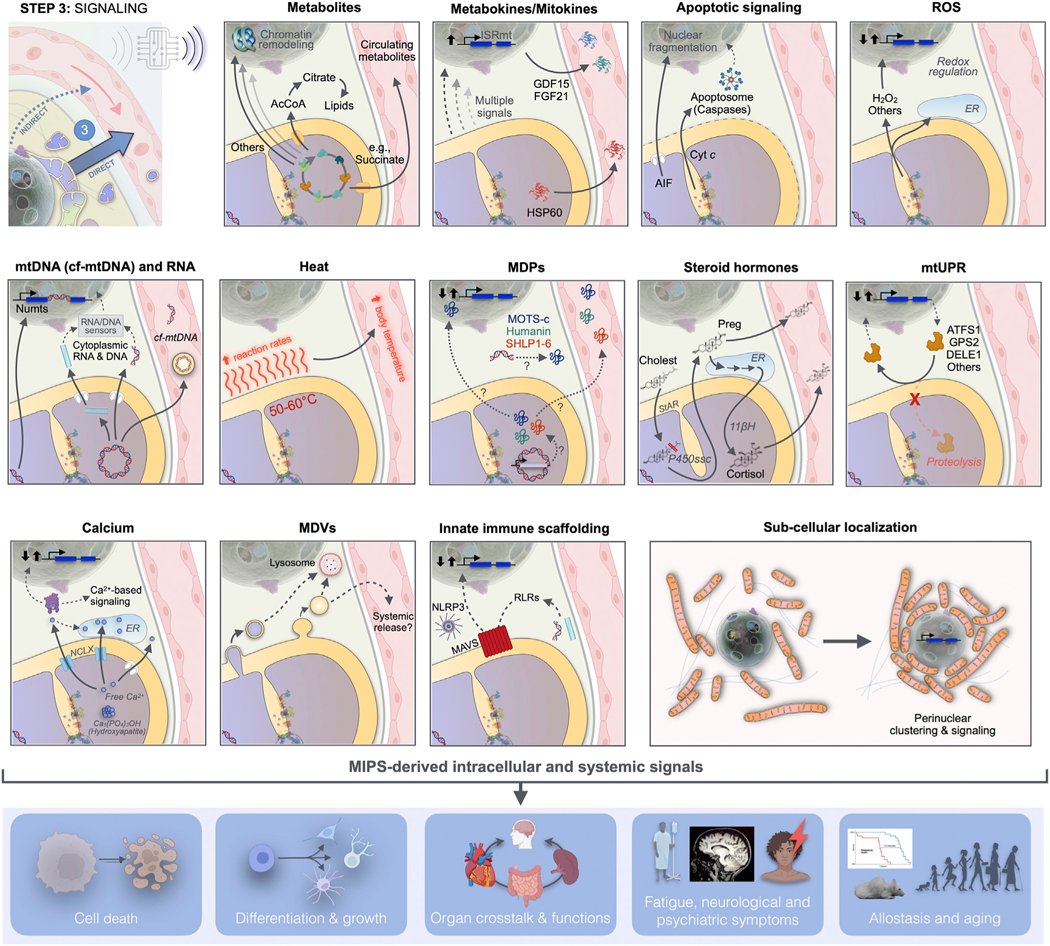
Mitochondria synthesize and release signals evolved to influence cellular and organismal functions. Mitochondrial signals arise from various mitochondrial compartments and reach the cytoplasm, nucleus, and other organelles, where they induce cell-autonomous responses. These responses are transmitted to the systemic circulation either directly as mitochondria-derived metabolites and mitokines, or indirectly through transcriptional regulation of nuclear genes encoding metabokines or other hormone-like mediators. Mito-nuclear signaling is a form of signal amplification and integration. The MIPS converts metabolic signals into extracellular proteinaceous, secreted factors, allowing mitochondria to signal their state well beyond the confine of the cell in which they reside. AcCoA, acetyl coenzyme A; AIF, apoptosis inducible factor; ATFS1, activating transcription factor associated with stress-1; cf-mtDNA, cell-free mitochondrial DNA; Cholest, cholesterol; Cyt c, cytochrome c; DELE1, DAP3-binding cell death enhancer 1; ER, endoplasmic reticulum; FGF21, fibroblast growth factor 21; GDF15, growth differentiation factor 15; GPS2, G-protein pathway suppressor 2; HSP60, heat shock protein 60; ISRmt, integrated stress response; MAVS, mitochondrial antiviral signaling; MDVs, mitochondria-derived vesicles; MPDs, mitochondria-derived peptides; NLRP3, NLR family pyrin domain containing 3; Numts, nuclear mitochondrial DNA segments; Preg, pregnenolone; P450ssc, side chain cleavage enzyme cytochrome P450; RLRs, RIG-I-like receptors; StAR, steroidogenic acute regulatory protein; UPRmito, mitochondrial unfolded protein response; 11βH, 11β-hydroxylase (mitochondrial cytochrome P450 11B1).
Apoptotic signaling
The first use of the term “mitochondrial signaling” appeared in 1999 in relation to the release of the pro-apoptotic mitochondrial output Cyt c.217 Cyt c is a small heme protein normally residing in the IMS where it shuttles electrons between OxPhos complexes III and IV. However, in response to the convergence of specific inputs such as ROS, high [Ca2+], and low [ATP], especially among a fragmented and poorly connected mitochondrial network,218 mitochondria undergo permeability transition through PTP opening.219 PTP opening triggers the cytoplasmic release of Cyt c where it interacts with and activates pro-caspases,9 as well as other mediators of the intrinsic apoptotic pathway, including the apoptosis inducing factor (AIF) and the endonuclease EndoG that translocates to the nucleus and fragments the nuclear genome, and Smac/Diablo (reviewed in Wang and Youle220). In cancer cells, Cyt c released during non-lethal permeability transition (i.e., “flickering mode”) can also play non-apoptotic signaling roles involving the activation of the nuclear ATF4-dependent integrated stress responses (ISRs; see below).221 To prevent the assembly of pro-apoptotic molecular complexes at the OMM, mitochondria can also recruit anti-apoptotic proteins from the Bcl2 family. Functionally, PTP opening is closely linked to mitochondrial Ca2+ release and signaling, which is under the control of increasingly well-defined cristae-regulating mechanisms.222 Thus, the MIPS contains a number of powerful cellular life-or-death signals coordinately released based on their integrated representation of biochemical conditions both within mitochondria and the cytoplasm.223
Mitochondrial metabolite signaling
Mitochondria speak the language of the epigenome. It is likely that the endosymbiosis of mitochondria and the MIPS preceded the development of the histone code, such that current epigenetic nuclear mechanisms have developed to couple gene expression to the metabolic state of the cell in the context of mito-nuclear communication.224 As a result, the nuclear genome is densely wrapped with abundant histone proteins (mainly H2A, H2B, H3, and H4) that contain hydrophilic tails, which are heavily post-translationally modified by the metabo-chemical perinuclear and nuclear environment.225
Most substrates or cofactors required by histone-modifying enzymes to alter histone structure and downstream gene expression are direct products of mitochondrial metabolism.226,227 These include metabolites from the TCA cycle216 and from one-carbon metabolism.228 For example, the methylation of histones and DNA by histone methyltransferases (HMTs) and DNA methyltransferases (DMTs), respectively, requires S-adenosylmethionine (SAM) derived from serine metabolism as part of the folate cycle and one-carbon metabolism pathway.116,229 On the other hand, the reverse demethylation reaction requires the cofactor α-ketoglutarate (αKG), a TCA cycle metabolite. Several mitochondrial-derived metabolites are involved in PTMs of histones (and other proteins). These include lactate (i.e., lactylation),230 a metabolite derived from glycolysis that increases in concentration when mitochondrial OxPhos is impaired; dopamine (i.e., dopaminylation),231 whose catabolism via the OMM-bound monoamine oxidase involves the respiratory chain;232 β-hydroxybutyrate (i.e., β-hydroxybutyration),233 a ketone body synthesized in the mitochondrial matrix under low carbohydrate conditions; and many others.
Mitochondrial metabolites are epigenome-modifying MIPS outputs. mtDNA-depleted Rho0 cells were initially used to demonstrate that mitochondrial outputs alter nuclear DNA methylation.26 In a similar model comparing a series of human cell lines with varying mutation load (i.e., heteroplasmy) of the pathogenic mtDNA 3243A>G mutation, heteroplasmy ranging from 0% to 100% influenced in a dose-response manner DMT gene expression and global transcriptional signatures.23 In the same model, mtDNA heteroplasmy altered acetyl-CoA and αKG levels and yielded downstream changes in H4K16ac and H3K9me3 status.234 Acute mtDNA depletion in immortalized cells also triggered a physiologically meaningful decrease in the mitochondrial acetyl-CoA pool and downstream histone acetylation,235 illustrating the range of epigenomic effects of the MIPS. Finally, a longitudinal study in primary human fibroblasts tracking DNA methylation changes over several months showed that both genetic and pharmacological OxPhos defects caused conserved, age-related hyper- and hypomethylation at thousands of genomic locations encoding developmental programs and cell-cell signaling components.236 A publicly available multi-omic, longitudinal dataset is available to explore the influence of bioenergetic perturbations on the epigenome and transcriptome of aging human fibroblasts.237 Together, these findings illustrate some mechanisms whereby intrinsic mtDNA-related and OxPhos inputs are transduced into epigenome-remodeling outputs.
However, one point that remains largely unclear is how MIPS-induced molecular and epigenetic modifications are temporally, spatially, and molecularly targeted, as well as their functional consequences on gene expression and cellular phenotypes.226 Furthermore, this scientific challenge is compounded by the existence of multiple active TCA cycle enzymes directly in the nucleus.238 The presence of mitochondrial enzymes in the nucleus, mostly documented to date in cancer cell lines, suggests that at least in some cell types, mitochondria may not be the only source of chromatin-modifying metabolites.
In recent years, other mitochondrial metabolites and molecular features have emerged as broad-acting intracellular signals. For example, the levels of TCA cycle metabolites succinate and fumarate are regulated by electron flux through the OxPhos system and more directly by the enzymes fumarate hydratase and succinate dehydrogenase (comprehensively reviewed in Martinez-Reyes and Chandel216). These metabolites are released in the cytoplasm, where they regulate signaling pathways involved in hypoxia sensing, immune activation, inflammation, and oncogenic transformation. TCA cycle metabolites also are enzymatically converted to metabolic derivatives such as itaconate and 2-hydroxyglutarate, among others. Itaconate is produced from the TCA cycle metabolite aconitate by aconitate decarboxylase and then acts either on intra-mitochondrial enzymes, for example by inhibiting succinate dehydrogenase,239 or on transcription factors in the cytoplasm/nucleus, for example by inhibiting NF-κB signaling.240 Two isomers of 2-hydroxyglutarate (2-HG) are produced from αKG by the mitochondrial or cytoplasmic malate dehydrogenases (MDH2, MDH1, respectively) in an NADH-dependent manner, and also promoted by acidic pH.241 In the nucleus, 2-HG then inhibits the demethylation of histone tails and DNA by the teneleven translocation hydroxylases (TET1–3) and plays important roles in cell fate transitions that affect oncogenesis242 and immune activation.243 Besides soluble metabolites, larger mitochondrial lipids also play important signaling roles.244 For example, the IMM lipid cardiolipin participates in a variety of cell signaling events, translocating to the OMM during stress and serving as a signaling platform relevant to mitophagy, apoptotic signaling, and other functions.245
In addition to TCA cycle flux, NADH/NAD+ ratio, and pH, the presence of carrier proteins on the IMM can influence MIPS metabolite signaling. For example, in mesenchymal stem cells age-related changes in the citrate carrier expression regulate the cytoplasmic export of acetyl-CoA to drive histone acetylation levels, increase chromatin accessibility, and influence stem cell differentiation.246 Thus, the nature and strength of mitochondrial outputs are likely regulated not only by rapidly changing fluxes through specific intra-mitochondrial metabolic pathways, but also by the relatively stable, albeit malleable, composition and abundance of IMM carriers and transporters.
Beyond the cell, metabolites also act in a cell-non-autonomous manner. A well-studied example is succinate, an obligatory mitochondrial TCA cycle intermediate that accumulates in equilibrium with the coenzyme Q redox state influenced by oxygen tension, ΔpH+Δψm, and ATP demand.247 Succinate has been reported to signal extracellularly and perhaps systemically through at least one cell surface GPCR, the succinate receptor 1 (SUCNR1), on immune and other cell types to regulate inflammatory processes.248 On target immune (and possibly other) cell types, succinate may also be imported via the monocarboxylate transporter 1 (MCT1), where it acts intracellularly to inhibit TCA cycle activity and signal transduction pathways inhibiting interferon secretion.249 Thus, metabolite outputs from the MIPS collectively have broad-acting cell-autonomous and cell-non-autonomous effects on the epigenome, nuclear gene expression, and cell behavior.
One other mitochondrial metabolite is worth special mention for its well-known role in circadian biology: melatonin (N-acetyl-t-methoxytryptamine). Melatonin is an evolutionarily ancient bacterial molecule preceding endosymbiosis that has strong antioxidant properties (reviewed in Reiter et al.92). Mitochondria not only contain the MT1 melatonin GPCR, but also synthesize melatonin from the amino acid L-tryptophan (with serotonin as an intermediate) via two enzymatic reactions catalyzed by enzymes in the mitochondrial matrix (arylalkylamine N-acetyltransferase [AANAT] and acetyl serotonin methyltransferase [ASMT]).87 Like other mitochondrial metabolites, systemic melatonin concentration exhibits strong diurnal variation (almost undetectable during the day, peaking at night; e.g., Paul et al.250). It modulates sleep/wake cycles in some animals,251 and its oral consumption in humans may modulate sleep onset.252 Thus, mitochondria-derived melatonin potentially acts locally in an “automitocrine” and cell-autonomous manner, in a paracrine manner between cells/neurons, as well as systemically via the bloodstream,253 illustrating the broad reach of MIPS-derived metabolites/hormones in mammalian physiology.
Together, mitochondria-derived metabolic outputs represent complementary signals that integrate and transduce the bioenergetic state of the MIPS into signals intelligible to core cellular signal transduction machinery that orchestrate a broad array of cellular and organismal behaviors.
Mitochondrial ROS signaling
ROS are diffusible molecules, particularly hydrogen peroxide (H2O2) produced from the dismutation of superoxide anion (O2• −) by the matrix and IMS antioxidant systems. Mitochondrial ROS originate predominately from OxPhos complexes I and III254 and travel to the cytoplasm and nucleus where they trigger redox-sensitive gene-regulatory processes.197,255 Mitochondrial ROS signaling215 and guidelines for their measurements256 have previously been reviewed in detail, so here we mainly focus on recent developments in this area.
Mitochondrial ROS regulate various internal mitochondrial states and systemic signaling. For example, in brown adipose tissue mitochondrial ROS production post-translationally modifies UCP1 at Cys253 to increase uncoupling and enable thermoregulation, whereas pharmacological depletion of mitochondrial ROS with MitoQ prevented IMM uncoupling and heat production.257 In the mitochondrial matrix of heme-synthesizing mitochondria in adipocytes, H2O2 oxidizes bilirubin to form biliverdin, which is exported from mitochondria by the ATP binding cassette (ABC) transporter ABCB10.258 And in secretory pancreatic beta cells, glucose-stimulated insulin secretion is similarly driven by H2O2 accumulation,259 illustrating how mitochondrial ROS signals within the mitochondrion and intracellularly to trigger the release of systemic endocrine signals such as insulin.
Likely owing to the central role of oxygen in the evolution of aerobic creatures, mitochondria-derived ROS have broad effects on nuclear transcriptional regulation. In cultured cells, elevated ROS production secondary to respiratory chain dysfunction, or mimicked with the addition of the mitochondria-targeted redox cycling agent paraquat (MitoPQ), was sufficient to activate proteins of the mitogen-activated protein kinase (MAPK), including JNK signaling, which induces a secondary signal, namely nuclear chromatin release into the cytoplasm.260 Similarly, eliciting high levels of temporally controlled ROS specifically in mitochondria using a chemoptogenetic tool elevated nuclear hydrogen peroxide levels and induced telomere damage.261 In mice, silencing the mitochondrial matrix antioxidant enzyme manganese superoxide dismutase (MnSOD) during development showed that mitochondria-derived ROS activated the cytoplasmic/nuclear Nrf2 and PPARγ/PGC-1α pathway, leading to lasting adaptive hormetic responses that persist in adult animals.262 Similar results were obtained in mice treated with low-dose rotenone (a complex I inhibitor that increases mitochondrial ROS emission) during embryonic and post-natal development, which altered nuclear DNA methylation and modified coat color.263 In aging human fibroblasts, mitochondrial signaling via ROS is also necessary and sufficient to activate the Nf-kB pathway and senescence features, including the senescence-associated secretory profile (SASP).264 In fact, experimentally depleting mitochondria from human fibroblasts by using a Parkin-overexpression/FCCP treatment prevented the acquisition of senescence characteristics,265 providing compelling evidence that MIPS signaling—including but likely not limited to ROS—is required to trigger complex cellular states like senescence. Moreover, the SASP can propagate senescence phenotypes to neighboring bystander cells both in vitro264 and in vivo,266 illustrating one of many pathways whereby mitochondrial signaling propagates systemically in a cell-non-autonomous manner to influence organismal behavior and lifespan.17,267
Besides mito-nuclear signaling, ROS production by individual mitochondria also locally contributes to communication with the ER.211 Even in distant neural arborizations, far from the nucleated cell body, mitochondrial ROS contribute to local synaptic activity.268 In response to plasma membrane photodamage, mitochondria at the site of injury were also shown to respond in a DRP1-dependent manner by increasing repair-promoting ROS production.269 Thus, the site-specific roles of mitochondrial ROS across sub-cellular locations illustrate the significance and potential specificity of localized ROS outputs from the MIPS as drivers of gene regulation and cellular functions.
Mitochondria synthesize sex and stress hormones
One of the most powerful types of mammalian hormones are steroid molecules, broadly categorized into three major classes: (1) the sex-defining testosterone, estrogens, and progestins produced in the gonads; (2) the stress hormones that promote stress adaptation via metabolic and salt balance regulation including glucocorticoids and mineralocorticoids produced in the adrenal glands; and (3) neurosteroids produced in the nervous system.270,271 Their release is regulated by trophic pituitary hormones from the brain (adrenocorticotropic hormone, ACTH; follicular stimulatory hormone, FSH; and luteineizing, LH) mediated by GPCR-coupled cyclic AMP-protein kinase A (cAMP-PKA) or Ca2+-PKC signaling in steroid-producing cells.208 In steroidogenic tissues, the rate-limiting step to synthesize all steroid hormones takes place within mitochondria.
Mitochondria produce steroid hormones from cholesterol, the initial substrate to all steroids. The import of cholesterol through the OMM and IMM requires microtubule and microfilament dynamics as well as protein synthesis272 and is accomplished by the steroidogenic acute regulatory protein (StAR, from the STARD1 gene) in the OMM.273 Whereas mitochondrial import of StAR through the TIM/TOMM translocator complex leads to its proteolytic degradation, stabilization of StAR at the OMM,274 in association with Tom22 and VDAC2,275,276 delivers cholesterol to the matrix-facing IMM side chain cleavage enzyme cytochrome P450 (CYP450scc) protein. CYP450scc then catalyzes the rate-limiting reaction for steroidogenesis, which convert cholesterol into pregnanolone, the common precursor to all steroid hormones.33 Pregnanolone synthesized in the matrix is then exported to the ER where other enzymes sequentially catalyze its transformation into progesterone and other steroid intermediates.208 In steroidogenic mitochondria from the adrenal cortex zona fasciculata cells, the downstream steroid intermediate returns to the mitochondrial matrix, possibly through the MAM, where the terminal reaction is catalyzed by the mitochondrial matrix enzyme 11β-hydroxylase (11βH, also “mitochondrial cytochrome P450 11B1” encoded by CYP11B1 in humans) that produces cortisol.277 Mitochondrial synthesis of systemically acting steroids occurs rapidly within minutes, and its synthesis arrest is equally rapid.208 The rapid, redox-sensitive, protein import-dependent regulation of this process illustrates how multiple intrinsic factors can influence mitochondrial steroidogenic outputs.
The evolutionary basis for positioning steroidogenesis in mitochondria remains uncertain but may have involved the uniquely reducing conditions of the mitochondrial matrix.278 The conversion of cholesterol to pregnanolone by P450scc requires the reductive action of 3 high-energy NADPH molecules. As a result, the loss of the matrix-facing NADPH-generating enzyme nicotinamide nucleotide transhydrogenase (NNT) inhibits steroidogenesis, causing hypocortisolemia in mice and humans.279 Developmentally, steroid hormones drive energetically expensive transcriptional and physiological programs that must incur substantial cellular energetic costs in target tissues. As a result, it is possible that to optimize fitness, these hormones should only be produced in proportion with the energetic capacity of target tissues. Assuming that the function of mitochondria is partially harmonized across both source steroidogenic and target catabolic energy-consuming tissues, we postulate that the mitochondrial localization of steroidogenesis enzymes may reflect the product of system-level adaptation aiming to couple mitochondrial bioenergetic capacity and steroid hormone signaling across the organism.
Mitochondrial genome signaling: Intracellularly
The circular mtDNA is typically contained in the mitochondrial matrix, insulated from the cytoplasm by two membranes. However, the enclosure of mtDNA within the mitochondrial IMM and OMM is naturally disrupted under certain physiological conditions. This includes mtDNA instability caused by the partial loss of the mtDNA-associated protein TFAM, which triggers mtDNA-dependent antiviral gene expression programs in the nucleus.22 mtDNA release is a relevant signaling mechanism because both the cytoplasm and the extracellular surface of immune and non-immune cells harbor DNA sensors that recognize mtDNA fragments as a damage-associated molecular pattern (DAMP).280 DNA (viral, bacterial, and mtDNA) is sensed by multiple innate immune receptors including cGAS (cyclic GMP-AMP synthase), TLR9 (toll-like receptor 9), and the NLRP3 (NOD-, LRR-, and pyrin domain-containing protein 3) and AIM2 (absent in melanoma) inflammasomes. Sensing of mtDNA triggers signaling cascades that either converge on cytokine- and interferon-producing transcription factors including IRF3/7, MAPK, and NF-κB or engage Caspase-1 for processing and secretion of IL-1b and IL-18.281 The cytosolic release of mitochondrial double-stranded RNA (mt-dsRNA) can also act as a DAMP and is detected by the RIG-I-like (RLR) receptors RIG-I and MDA5. Once engaged, these sensors translocate to mitochondria and activate the mitochondrial antiviral signaling protein (MAVS), which assembles as filaments on the mitochondrial surface282 in a membrane potential-dependent manner to act as an antiviral signaling platform.283,284
Current thinking around innate immune signaling suggests that the mitochondrial network is both a source of stimulatory ligands and acts as the major signaling hub for the four major pattern recognition receptor families (TLRs, NOD, RLRs, and cytosolic DNA sensors [CDSs]).285 Most of the effects of both exogenous (bacterial, viral) nucleic acids and endogenous mtDNA/mtRNA signaling are likely mediated via these pathways. As an example of mitochondrial signal transduction, when mitochondria in human fibroblasts and cancer cells detect the genotoxic agent doxorubicin, mtDNA damage (sensing) eventually leads to the cytoplasmic release of mtDNA fragments (signaling), which trigger nuclear DNA repair mechanisms in the nucleus and cGAS-STING-dependent activation of interferon-stimulated gene (ISG) expression.147
Regarding the mechanism(s) responsible for facilitating mtDNA extrusion into the cytoplasm, two molecular pathways have been described. One mechanism tested in mouse embryonic fibroblasts and mice with lupus-like disease involves VDAC oligomerization in the OMM, stabilized by short mtDNA fragments, forming a large-scale pore that enables the cytoplasmic extrusion of 100- to 400-bp-long mtDNA fragments.286 Similarly, in bone marrow-derived macrophages 500- to 650-bp-long mtDNA fragments are cleaved from the circular genome by the mitochondrial protein flap-structure-specific endonuclease 1 (FEN1) and released in a VDAC-dependent manner into the cytoplasm where it activates the inflammasome and cGAS-STING signaling.287 The other described mechanism of cytoplasmic mtDNA extrusion consists of BAX/BAK-mediated pore formation in the OMM, followed by herniation of the IMM at the surface of mitochondria during apoptosis.288 Under conditions of genotoxic stress, BAX/BAK-mediated herniation also appears to release ds-mtRNAs to activate ISGs in the nucleus.289,290 A third non-specific mechanism may involve the rupture of mitochondrial membranes, possibly secondary to swelling, which, for example, may occur in skeletal muscle of patients with primary mtDNA mutations.291
Another, more permanent way in which the mitochondrial genome can carry information to the nucleus is via the translocation of mtDNA segments to the nucleus followed by their insertion within the coding sequence as nuclear mtDNA insertions (NUMTs, pronounced “nu-mites”).292 This process, termed “numptogenesis,” has traditionally been understood as horizontal gene transfer, having occurred multiple times during the evolution of single-celled and multicellular organisms.293 As a result, multiple germline NUMTs are shared across individuals. In the case of mitochondria, mtDNA gene transfer is also likely to explain how the majority of the genes from the original proteobacterium’s genome have migrated to the nucleus such that >98% of the mitochondrial proteome is now encoded in the nucleus.294
But the transfer of mtDNA sequences to the nucleus may also occur over a cell’s lifespan. In yeast, mitochondria lacking the mitochondrial protease Yme1 (yeast mtDNA escape 1), a member of the AAA family of ATPase that degrades IMS/IMM proteins to regulate mitochondrial cristae dynamics,295 produce 77-fold more mito-nuclear transfer of NUMTs along with a 50% reduction in lifespan.296 Similarly, in cancerous mammalian cells, mitochondria without the human homolog YME1L1 generate ~4-fold more NUMTs, recapitulating the abnormally elevated number of NUMTs in ovarian tumors297 and other cancer types.298 Numptogenesis may also occur at a steady rate over days to weeks in healthy replicating primary human fibroblasts in vitro, and over a person’s lifespan in brain tissue (unpublished data). Whether the effects of NUMTs on nuclear genome instability and cellular aging is a bona fide, regulated form of MIPS signaling remains to be established.
Mitochondrial genome signaling as extracellular, cell-free mtDNA
mtDNA copies are often well in excess of the number of copies required to transcriptionally sustain OxPhos.299 An emerging notion suggested by Shadel et al. is that the mtDNA molecules do not only supply RNA and OxPhos subunits but in fact exist as sentinels of genotoxic stress and other insults.147 This suggests that the hundreds of mtDNA genomes in each cell may represent a pool of signaling molecules—in the same way the neurotransmitters are produced and stored in presynaptic boutons, awaiting release.
Beyond mtDNA detected in the cytoplasm and nucleus, a substantial amount of circulating cell-free mtDNA (cf-mtDNA; as well as nuclear DNA, cf-nDNA) is released extracellularly, detectable in various biofluids from healthy individuals (reviewed in Trumpff et al.300). In blood (serum or plasma, which contain different cf-mtDNA levels301) and cerebrospinal fluid, cf-mtDNA is elevated in some although not all individuals with primary OxPhos defects,302–304 during pregnancy,305 after physiological stress such as exercise,306 minutes after acute psychological stress,306,307 and hours to days after intensely stressful life events,308 highlighting the dynamic release of cf-mtDNA. In saliva, cf-mtDNA also increases several-fold during the morning sleep-wake transition.309 Notably, in critically ill individuals cf-mtDNA levels are also dramatically elevated, and cf-mtDNA abundance (copies per mL) is a strong predictor of mortality,310 highlighting the potential physiological significance of cf-mtDNA in humans.
Because the majority of the initial work on cf-mtDNA was conducted in the context of sepsis and inflammatory disorders, and given that the molecular features of the mitochondrial genome and associated proteins are bacterial in origin, the pro-inflammatory aspects of cf-mtDNA signaling have been emphasized.311 The role of mitochondrial signaling in the control of inflammation has been expertly reviewed elsewhere285 and leaves little doubt that intracellularly, cf-mtDNA is immunogenic.
However, in relation to extracellular cf-mtDNA in biofluids, emerging evidence suggests that (1) the majority of blood and saliva circulating cf-mtDNA in human plasma may not be naked (required to be accessible to DNA receptors) but rather contained within sedimentable cargo, possibly as circulating whole mitochondria,309,312,313 and (2) cf-mtDNA is abundant in healthy individuals who do not show signs of systemic inflammation. Therefore, a critical re-appraisal of the evidence reveals that by itself, circulating cf-mtDNA in its physiological forms in human blood is unlikely pro-inflammatory.300 Cf-mtDNA levels in blood, cerebrospinal fluid,314 and saliva309 also do not consistently correlate with inflammatory markers. The physiological role of cf-mtDNA in general remains unclear. Technically, how mtDNA fragments are physiologically released from the matrix to the cytoplasm, into the extracellular space, and into biofluids also remains to be established.
Together, these findings highlight the influence of mtDNA signaling beyond autocrine/cytoplasmic mito-nuclear signaling. Emerging work in multiple biofluids implicates paracrine (cell-to-cell) and potential endocrine (systemic) roles of cf-mtDNA signaling among the repertoire of signaling mechanisms available to transduce information from the MIPS to the organism.
Non-apoptotic nuclear-encoded proteins sequestered in mitochondria
A different class of mitochondrial outputs includes a group of nuclear-encoded proteins that are normally imported and degraded by functional energized mitochondria but fail to be imported when mitochondria are de-energized (i.e., depolarized). In non-mammalian systems and cultured cells, stress conditions that induce mitochondrial depolarization inhibit protein uptake and cause their accumulation in the cytoplasm where they act as transcription factors.27 Based on initial studies using unfolded protein stress in the mitochondrial matrix,315 this response was coined the mitochondrial unfolded protein response (mtUPR). The mtUPR involves close physical contact and functional interactions between mitochondria and the ER.316
Known pathways involving nuclear-encoded proteins include ATFS-1 (activating transcription factor associated with stress-1, in C. elegans),19 GPS2 (G-protein pathway suppressor 2, in mammalian cells),20 and DELE1 (DAP3-binding cell death enhancer 1, in mammalian cells).317,318 In an OMA1 protease-dependent manner, the mitochondrial network acts as an active sink that normally shunts these proteins and prevents their interactions with nuclear transcription factors including EIF2α.317,318 Via these proteins, mitochondrial depolarization promotes ATF4 and ATF5 expression, the master regulators of the mtUPR.319,320 Interestingly, inhibiting mitochondrial translation interferes with cytoplasmic translation and triggers ATF4/ATF5-dependent signaling, marking the interconnectedness of intra-mitochondrial and cytoplasmic protein homeostasis.321 The ATF4/ATF5 transcription factors overlap with those of the mitochondrial ISR (ISRmt) well defined in mammals. However, in cultured human cells at least, different mitochondrial perturbations selectively induce the mtUPR and ISRmt in a relatively mutually exclusive manner.322 This result suggests that the mtUPR and ISRmt stress response pathways have either evolved separately or diverged in their specificity, highlighting the existence of at least two well-defined nuclear transcriptional programs induced by MIPS signaling in mammals.
Systemic mitochondrial signaling via the nucleus
MIPS signaling induces nuclear programs that remodel catabolic and anabolic biosynthetic pathways within the cell, and also shapes metabolism systemically. In mammalian models, intrinsic mtDNA transcriptional and translational defects,16,115,323 inhibition of autophagy,324 IMM uncoupling,325 and both pharmacological and genetic OxPhos defects236,326 are transduced to the nucleus via mechanisms that at least in part involve mitochondria-derived signals inducing the ATF4- and ATF5-regulated ISRmt. In cultured human cells, loss of hundreds of nuclear-encoded mitochondrial genes, although not all, selectively triggers the ISRmt.322
In animals, the ISRmt produces two main nuclear-encoded systemic signals: fibroblast growth factor 21 (FGF21) and growth differentiation factor 15 (GDF15)—two proteins with overlapping but distinct systemic metabolic effects.327 In mouse models with molecular alterations in skeletal muscle mitochondria, the muscle-derived FGF21 protein travels to distant tissues where it is necessary for some tissue-specific effects such as white adipose tissue browning328 and glucose uptake and mitochondrial biogenesis in the dorsal hippocampus.18 However, FGF21 is dispensable for other systemic effects such as glucose tolerance, insulin resistance, anorexia, and weight regulation.328–330 The other well-studied metabokine GDF15 is most highly expressed in secretory tissues (syncytiotrophoblasts, epithelial cells, and glandular cells; https://www.proteinatlas.org/) and has wide-ranging systemic effects linking mitochondrial OxPhos dysfunction, metabolism, and inflammation.331 Mitochondrial translation defects (Crif1KO) in skeletal muscle329 or in adipocytes,332 or chronic skeletal muscle mitochondrial uncoupling (uncoupling protein 1 [UCP1] overexpression),333 induce both FGF21 and GDF15 secretion from the affected tissues, where GDF15 is required to increase systemic energy expenditure and other behavioral and physiological recalibrations.
Consistent with the functional interplay of the MIPS and the organism, ISRmt signaling occurs at least in part via a periphery-to-brain signaling axis. Mice and humans express the receptor for GDF15 GFRAL most highly in brain tissue (e.g., Mullican et al.334), but GFRAL may be expressed in many tissues at low levels (https://www.proteinatlas.org/) and is stress inducible in other cell types including macrophages.335 Therefore, GDF15-GFRAL signaling provides a mechanism whereby mitochondrial outputs from peripheral tissues use the brain—an organismal integration center—to transduce the functional state of a tissue’s mitochondria to regulate systemic metabolism.336,337 Adding to this systemic signaling picture, in mice GDF15 signaling upregulates hypothalamic corticotropin-releasing hormone (CRH)338 and activates the downstream HPA axis and secretion of corticosterone, a mitochondria-derived hormone released in response to psychosocial stress.339 Consistent with the production of metabokines in response to intra-mitochondrial defects converging on OxPhos capacity, FGF21 and GDF15 are circulating biomarkers of subgroups of mitochondrial diseases in adults and children.113,304,340–342
Thus, the secreted nuclear-encoded metabokines FGF21 and GDF15 convey information about the state of the MIPS in one tissue/organ to the whole organism. However, the contributions of these stress-induced, nuclear-encoded mitochondrial signaling outputs to the maintenance of human health or to disease progression remain only partially explored.
Mitochondria-derived peptides
The discovery of alternative open reading frames (ORFs) within the mtDNA sequence led to the identification of MDPs released within the cell systemic circulation (for a comprehensive review, see Reynolds et al.343). Eight MDPs have been reported: Humanin, a 24-amino-acid peptide encoded within the 16S rRNA gene initially discovered to have neuroprotective effects in neuronal cultures344 and subsequently linked to longevity across invertebrates, small mammals, and humans;345 6 small humanin-like peptides (SHLP1–6) that functionally overlap with humanin;346 and MOTS-c (mitochondrial ORF within the twelve S rRNA type-c), a 16-amino-acid peptide initially identified to promote insulin sensitivity and prevent age-related insulin resistance in mice.347 When the mtDNA is selectively depleted with chronic ethidium bromide treatment, or mtDNA transcription is selectively inhibited with actinonin, the expression of MDPs is lost, confirming their origin in the mitochondrial genome.347 However, where MPDs are transcribed and translated (mitochondrial matrix or cytoplasm) remains uncertain.
Functionally, once in the cytoplasm, MOTS-c translocates to the nucleus in an AMP-activated protein kinase (AMPK)-dependent manner, where it regulates stress-induced gene expression and promotes cell survival.21 In mice, MOTS-c and Humanin are also found in blood and act in a cell-non-autonomous manner to apparently regulate systemic metabolism.348 In humans, like other mitochondrial outputs, MOTS-c349 and Humanin350 levels increase dynamically in skeletal muscle and in circulation upon exercise. Thus, in addition to nDNA-encoded metabokine proteins FGF21 and GDF15, mitochondria release mtDNA-encoded peptides that act as both intracellular and systemic signaling mediators. The range of physiological functions for MDPs is only beginning to be uncovered, but MOTS-c may increase running capacity (i.e., endurance) in mice and improve resilience to metabolic starvation in cultured myotubes.349 Other mitochondrial resident proteins, including heat shock protein 60 (HSP60)351 and TFAM,352,353 have been identified in blood, suggesting that several canonical mitochondrial proteins are released systemically by the MIPS where they have metabolic, inflammatory, or other systemic signaling roles.
Mitochondrial heat signaling and thermodynamic gradients
Temperature is a powerful effector of biological change. Without heat, biological processes do not proceed. For instance, growth and degradation are greatly reduced at 4◦C, whereas optimal temperatures accelerate enzyme kinetics, membrane fluidity, and organismal development.354 Therefore, the diffusion of heat and the ensuing changes in biochemical activities represent a form of signaling, where information about the state of an organelle is transferred from one sub-cellular compartment to another. Because—according to the second law of thermodynamics—the flow of heat always proceeds from warmer to colder locations, the flow of information also must preferentially (although not exclusively) occur from warmer to colder structures.
Among the cell and the organism, mitochondria are the warmest compartment and the major heat source. Body temperature in endotherms is primarily derived from respiratory chain activity.355 Exemplary of this phenomenon, mitochondria in brown adipocytes express high levels of UCP1 that increases proton leak across the IMM, accelerating upstream heat-producing biochemical reactions in a Ca2+-dependent manner.356 Using temperature-sensitive fluorescent probes, initial studies found that the warmest cellular compartments in cultured cells were the nucleus (which is surrounded by perinuclear mitochondria) and mitochondria.357 Uncoupling of OxPhos with the uncoupler FCCP increases mitochondrial matrix temperature by 6◦C–9◦C, as would be expected from relieving the electrochemical energy gradient across the IMM and subsequent cascading acceleration of biochemical reactions upstream from the OxPhos system.358 Accordingly, the biochemical activity of mammalian mitochondrial respiratory chain enzymes was found to be maximal around 50◦C.359 Consistent with this finding, refined live-cell imaging with mitochondria-targeted temperature-sensitive probe showed that mitochondria function at internal temperatures around 50◦C, well above the core body temperature of 37◦C,359 suggesting that the MIPS radiates heat-based signals into the cell. Thus, the temperature gradient between mitochondria (warmest compartment) and other sub-cellular structures likely provides mitochondria with a thermodynamically privileged position in signal transduction.
Sub-cellular mitochondrial localization
The sub-cellular positioning of cellular structures influences their functions and ability to signal to other organelles. Within the cytoplasm, the MIPS is topologically positioned at the interface between the naturally inert nuclear genome and the dynamic extracellular environment. In many cell types, mitochondria directly contact or hover only hundreds of nanometers away from the nucleus. At the nuclear surface, diffusible mitochondrial signals can travel through nuclear pores to reach the nucleoprotein complex of the chromatin.360 To travel 1 μm—from the mitochondrial IMS to the chromatin—the theoretical isotropic diffusion time for a small 40 kDa protein is 0.02 s,361 whereas small metabolites like ATP362 or amino acids363 travel ~20–100 times faster, closing the 1 μm gap in less than 1 ms. Physical proximity, particularly at high temperature, favors rapid communication.
The position of the mitochondrial network within the cytoplasm can influence signaling behavior. In response to stressors such as hypoxia in cultured endothelial cells, mitochondria redistribute and cluster around the nucleus within <3 h where they promote a pro-oxidant intranuclear state.197 Inhibiting mitochondrial motility with the microtubule depolymerizing agent nocodazole or dynein knockdown effectively prevents perinuclear clustering.197 In this case, the reduced physical proximity appeared to decrease the potency of mito-nuclear signaling, hindering HIF-1α binding to the nDNA hypoxia response element nucleotide sequence. The formation of physical contact sites between mitochondria and the nuclear envelope by the OMM-based translocator protein (TSPO) also enables cholesterol redistribution to the nucleus and initiates pro-survival nuclear transcriptional programs that are blunted without mito-nuclear proximity,255 highlighting the influence of proximity and physical interactions in mito-nuclear signaling.
Mitochondrial positioning also shapes cell behavior away from the nucleus. In developing neurons, mitochondrial positioning at specific locations along axons determines the location of branch points.364 In ganglion cell dendrites of the retina, mitochondria positioning at terminal branch points and presynaptic sites also stabilizes mature dendritic structures.365 In neurons, both within presynaptic boutons31 and at specific locations near dendritic synapses, mitochondria contribute both to local ATP synthesis and Ca2+ handling, locally influencing neurotransmitter release and, as a direct result, cell-cell communication.366 Thus, the regulated topological positioning of mitochondria within the cell can minimize diffusion distances and likely optimize the potency and/or nature of MIPS outputs, illustrating how mitochondrial positioning guides signaling and various cellular functions.
Summary of mitochondrial signaling
In addition to their elaborate sensing (step 1) and networking (step 2) capabilities, mitochondria also possess a wide array of signaling (step 3) mechanisms that transduce mitochondrial states to the cell and organism. The nature of these signals includes ions, metabolites, chemical species (e.g., ROS), DNA, and proteins. These outputs carry—possibly with a fairly high degree of specificity based on their circumstantial combinations—information about various aspects of mitochondrial biology to the cell nucleus and other organelles. At specific sub-cellular locations and in specific cell types, adaptive mitochondrial signals are transformed into transcriptional, cellular, and humoral physiological responses that inform and influence organismal functions. Less specific factors, including temperature and physical distances, may modulate the potency of these signals. Emerging evidence also suggest that systemic MIPS outputs may include mitochondria-derived vesicles (MDVs),367 extracellular vesicles with mitochondrial cargo,368,369 and even whole mitochondria313 that travel from source cells/tissues to functionally impact distant target organ systems. Finally, the description of overlapping molecular connections between mitochondria and target nuclear genetic programs, such as the mtUPR and ISRmt, emphasizes the evolved sensitivity of the mammalian genome to MIPS outputs. Notably, several mitochondrial outputs reach the bloodstream, the biofluid that metabolically connects all cells and organs, giving systemic organismal access to MIPS signaling.
TISSUE SPECIFICITY IN MITOCHONDRIAL FUNCTIONS AND BEHAVIORS
Although we have so far considered mitochondria as a more-or-less uniform family of organelles, mitochondria are not all created equal. From their shared origin in the oocyte,370 mitochondria undergo profound specialization as different cell types and tissues mature during embryonic development. This gives rise to somatic mitochondria that differ in their protein composition371,372 and functions (respiratory properties, ROS production, PTP sensitivity, β-oxidation capacity).373 These develop-mentally acquired characteristics represent tissue-specific mitochondrial phenotypes. Analogous to functionally and molecularly distinct cell types, there are functionally and molecularly distinct mitochondrial types, or “mitotypes.”374
Different tissues and cell types contain markedly different mitotypes that likely influence MIPS signal transduction. For example, cardiomyocyte mitotypes in the heart are optimized for ATP synthesis, adrenal cortex mitotypes specialize for steroidogenesis, and liver mitotypes specialize for ketogenesis, serine metabolism, and anaplerosis. Even within a given organ, neighboring cell types can acquire distinct mitotypes. For example, mitochondria from adjacent oxidative versus glycolytic skeletal muscle fiber types acquire vastly different proteomes that match their functional specialization.375 Likewise, circulating human immune cell subtypes (B cells, naive or activated T cells, monocytes, neutrophils, etc.) exhibit markedly different OxPhos profiles and mtDNA copies per cell.374 And in the mouse brain, mitochondria exhibit regional376 as well cell-type-specific functional and molecular diversity.372 These divergent cell-type-specific mitochondrial features, along with cell-specific metabolic requirements, may explain why in mice spongiform neurodegeneration is caused by the loss of mtDNA specifically in astrocytes but not in neurons, for example.377 In mitochondrial disease, large intracellular and cell-to-cell heterogeneity in mitochondrial phenotypes also develops as mutant mitochondria proliferate,378 such that adjacent muscle fibers can show different stages or even opposite responses to OxPhos defects.379
Diverse mitotypes also populate different sub-cellular compartments within individual cells.201 In neurons, the cell body (i.e., soma) and synaptic boutons have remarkably different mitochondrial proteomes.380 Similarly, muscle fibers contain two mitotype sub-populations (subsarcolemmal [SS] and inter-myofibfillar [IMF]) with quantitatively distinct proteomes.381 Adipocytes also are populated by at least two main mitotypes: mitochondria proximal to lipid droplets (i.e., peridroplet, or PDMs) and mitochondria not in immediate contact with lipid droplets.382 These two adipocyte-specific mitotypes exhibit distinct bioenergetic, proteomic, and fusion dynamics. Thus, the organism is composed of a wide spectrum of molecularly and functionally specialized mitotypes.
Relevance of mitochondrial functional specialization to sensing, integration, and signaling
Mitochondrial sensing (MIPS step 1): in multicellular organisms, individual cell types express a narrow set of cell-type-specific receptors and are therefore sensitive to a narrow set of inputs. For example, the sensory organs in animals exhibit specific sensitivities to a narrow set of inputs: the eyes sense light but do not perceive sound nor taste, and neither the inner ear nor the tongue respond to light. Each set of neurons within sense organs specifically responds to select inputs. Similarly, the expression levels of dozens of mitochondrial genes and proteins35,383 including mitochondrial sensory components—transporters, receptors, enzymes—are relatively specialized across both mouse and human tissues (A.S. Monzel, personal communication). The functional specialization of mitotypes across tissues and cell types may thus produce unique mitochondrial sensory systems tailored for specific inputs in different cell types.
Mitochondrial signal integration (MIPS step 2): several aspects of mitochondrial morphology, dynamics, and motility vary between tissues and cell types.201 In human and mouse skeletal muscle, SS/perinuclear mitotypes are spheroids whereas IMF mitotypes that intersperse sarcomeres have a branched, elongated morphology.170 In neurons, despite sharing a continuous cytoplasm, somatic mitochondria form a partially connected network surround the cell nucleus, elongated branched dendritic mitochondria extend for tens of microns, whereas axonal and synaptic mitochondria are mostly punctate.384 In relation to motility, skeletal muscle mitochondria are remarkably stationary180 compared to neuron axonal and dendritic mitochondria that exhibit greater motility.385 From first principles, these profound differences in the morphology, topology, and dynamic properties of the MIPS predict that mitochondrial information transfer, integration, and computation differ between tissues and cell types. Although currently technically challenging, improving imaging (e.g., Wolf et al.386) and experimental (e.g., Berry et al.387) technologies combining spatial and temporal resolution should eventually allow us to map and empirically manipulate information flow through mitochondrial networks.
Mitochondrial signaling (MIPS step 3): tissue-specific mitotypes influence both the nature and magnitude of MIPS outputs and cellular responses. For example, susceptibility to PTP opening, Ca2+ buffering capacity, and ROS emission characteristics differ remarkably between glycolytic and oxidative skeletal muscle mitotypes.373 This has implications for inter-organelle crosstalk. The ISRmt response also is induced cell-specifically; proliferating myoblasts and myotubes exhibit different ISRmt responses to the same mitochondrial perturbations.326 In the mouse brain, progressive mtDNA depletion in Twinkle-KO astrocytes but not neurons induces the ISRmt.377,388 Mitochondrial activation of the ISRmt and its interaction with other pathways, such as the mTORC1, is also tissue-specific.115,379,388,389 Thus, MIPS signaling as well as the cellular responses to MIPS signals both differ between cell types and tissues. Our current understanding of how mitotypes from different tissues differentially signal intracellularly and systemically is in its infancy.
OUTSTANDING QUESTIONS
To understand how mitochondria contribute to organismal health and disease, several important challenges remain. As reviewed above, the MIPS performs several functions that ensure rapid cellular and systemic responses commensurate with the energetic and biochemical state of the organism. As a result, mitochondria contribute to healthy cellular and physiological adaptation. Clinically, it is clear that mitochondrial diseases involve primary genetic defects affecting molecular processes other than the OxPhos machinery (i.e., not all mitochondrial diseases are disorders of ATP deficiency).390 Certain tissues and organs also specifically become affected, whereas others are relatively spared.391 How do non-energetic mitochondrial functions influence health and disease?
The three-step mitochondrial signal transduction framework described here raises several questions, some of which are listed below. Providing answers to these and other emerging questions would advance our understanding of the instructive role that the mitochondria play in human health. Because tissue-specific mitochondrial phenotypes (i.e., mitotypes) are the integrators of cell, tissue, and organismal metabolic inputs, and because MIPS outputs modulate not only a large fraction of the human genome but also complex animal behaviors, the following questions broadly concern the biological and biomedical sciences.
Are defects in the sensing, integration, and signaling functions of the MIPS sufficient to perturb physiological adaptation in the organism, leading to disease? Communication between cells is essential to maintaining organismal integrity, and perturbing communication alone is sufficient to cause health disorders.392,393 For example, impaired mitochondrial fusion (and/or inter-organellar interactions) causes human disease.394,395 Is impaired mitochondrial signal transduction—in the absence of OxPhos defects—a cause of metabolic and other types of health disorders?
How are specific mitochondrial functional impairments communicated to the cell? Different molecular defects converging on downstream OxPhos deficiency can cause different gene-regulatory signals,322 cellular responses,23 and disease manifestations.140 In general, critical biological processes tend to exhibit redundancy (several effectors exist to sense or communicate the same inputs), which, coupled to a diversity of downstream interacting genetic programs (e.g., innate immunity, ISRmt, mtUPR), affords a diversity of potential cellular and organismal responses. Are MIPS output signals, or combinations thereof, cell- or tissue-specific? What determines the exact transcriptional responses they elicit?
How generalizable or species-specific are mitochondrial signal transduction mechanisms? Biological mechanisms and therapeutic processes in rodents and invertebrates often align only partially with humans,396,397 and important differences also exist between mouse strains,398 for example. Furthermore, different cell types harboring distinct mitotypes may exhibit different responses to metabolic stressors.379 Systematic exploration of human health and disease states, as well as experimental models for specific molecular features that mimic as closely as possible human physiology or pathology, will increase the likelihood that our findings in model systems will be of biologic, diagnostic, or therapeutic value in humans.
Did the role of mitochondria as an information processing system contribute to the evolutionary turning point of endosymbiosis? Argument accounting for the role of mitochondria as a harbinger of multicellular, complex life includes the protection from oxygen399 and a rise in energy supply,400 although these possibilities have been challenged.401 Communication and information exchange via optimized biological structures—epitomized at the scale of the organism by the nervous system360—afforded an unprecedented acceleration and complexification of social behaviors among animals. This raises the possibility that the acquired ability of cells to sense their environment, efficiently transduce information, and communicate with each other via the MIPS may have been a decisive factor in the evolution and diversification of multicellular life.
SUMMARY
The past decades of mitochondrial research have produced remarkable advances in our knowledge of how mitochondria function and behave within the cell. More recently, accumulating evidence revealed how mitochondria communicate extensively with other organelles, between cells, and even across organ systems. Integrating these notions under a common framework suggests that a central role of mitochondria is to transduce information, functioning as a distributed information processing system. The most advanced known form of signal transduction occurs in the brain, which efficiently integrates sensory inputs to develop precise internal representations of the outside world, and secondarily deploys optimal organismal responses and behaviors that promote adaptation and survival. We propose that mitochondria perform a similar, albeit more primitive, form of signal transduction. The MIPS integrates the constant flow of molecular and non-molecular inputs about the energetic and metabolic states of the system, and secondarily deploys in collaboration with the nucleus an array of outputs that guide cellular and organismal adaptation (Figure 7).
Figure 7. Core MIPS components for mitochondrial signal transduction.

(A) All mitochondria contain the machinery and capacity to sense, integrate, and signal information. Based on evidence reviewed above, mitochondria emerge as “the processor of the cell.”
(B) Electron micrograph of a cultured cell (human 143B) with mitochondria (blue, green, and red) and nucleus (yellow) highlighted; dotted red lines indicate cell-cell boundaries. The functions of the MIPS are determined by intrinsic properties of individual mitochondria and surrounding organelles, constantly shaped by the dynamic remodeling of organelle networks over minutes to hours. Topologically, the MIPS sits at the interface of the extracellular space harboring endocrine, metabolic, and biochemical signals, and of the (epi)genome in the nucleus.
(C) The mito-nuclear unit enhances and amplifies the effects of mitochondrial outputs, tapping into a rich variety of evolved nuclear DNA-encoded (epi)genetic stress response pathways that communicate local mitochondrial states to the organism. This includes the signaling of peripherally derived metabokines on the brain.
(D) The framework of mitochondrial signaling from organelle to organism. In multicellular animals, the MIPS is a core regulatory element acting both subcellularly and systemically to optimize adaptation and organismal health.
The mitochondrial signal transduction framework highlights how mitochondria simultaneously contribute central roles to energy transformation and biosynthesis, as well as to signal transduction. This framework also helps to situate our increasingly precise, mechanistic, and reductionistic investigations of mitochondrial features, activities, functions, and behaviors within the context of the organism and its environment. The view of mitochondria as a distributed information processing system—or as a “portal” positioned at the interface of the outside environment and the inner world of the cell’s (epi)genome—integrates all historical domains of mitochondrial biology. As a result, the mitochondrial signal transduction framework emphasizes the need for knowledge integration across sub-fields of mitochondrial science. This also highlights the many ways in which multiple domains of mitochondrial biology beyond energetics may be linked to organismal health.
Improving human health is the shared goal of the biomedical community. This collective effort involves building increasingly accurate theories, models, and testable hypotheses about the processes that not only falter in advanced stages of diseases, but also those that enable optimal adaptation so that health is achieved. Health is the ability to deploy optimal responses to challenges.55 Organismal health emerges from the functional interconnections and crosstalk between cellular and physiological systems, behaviors, and psychosocial states that regulate biology, and vice versa.402–405 As we begin to map the basis of health beyond the absence of disease,393,406 it appears crucial to mechanistically decipher two prominent forces related to mitochondria. The first is energy, which brings otherwise inert genes to life and powers the functions of cells and organs. The second is communication or signal transduction, which connects and thus turns parts into wholes. Signal transduction turns cells into cell collectives and binds organs together as an organism. The organism—not the cell—is the ultimate evolutionary unit upon which selection pressures act and where health manifests.
Therefore, articulating the role of multifaceted mitochondria in signal transduction across the organism can help us achieve our shared goal of improving human health in three main ways: (1) by broadening and prioritizing the health-relevant mitochondrial biology questions to test, (2) by selecting the ideal human study design or animal model systems in which to address them, and (3) by connecting more effectively new molecular, cellular, and physiological discoveries in mitochondrial biology to human health.
ACKNOWLEDGMENTS
The authors are grateful to Anu Suomalainen for extensive input to this manuscript; Jodi Nunnari and Vamsi Mootha for discussions and Phillip West for edits that helped shape parts of this review; Mary Elizabeth Sutherland, Eugene Mosharov, Guy Las, Daniel Dagan, Mario Ost, Alex Junker, Otto Kalliokoski, Robert Taylor, Stavros Fanourakis, and Gilles Gouspillou for comments, suggestions, or edits; and members of the Mitochondrial Psychobiology Laboratory for stimulating discussions. The authors’ work was supported by NIH grants R35GM119793, R01MH119336, R01MH122706, R01AG066828, and R21MH123927 and the Baszucki Brain Research Fund to M.P.
Footnotes
DECLARATION OF INTERESTS
O.S.S. is a co-founder of Capacity and Inspire Bio.
REFERENCES
- 1.Altman R (1890). Die Elementarorganismen und ihre Beziehungen zu den Zellen (Veit). [Google Scholar]
- 2.Mitchell P. (1961). Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191, 144–148. 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 3.Chance B, Williams GR, Holmes WF, and Higgins J. (1955). Respiratory enzymes in oxidative phosphorylation. V. A mechanism for oxidative phosphorylation. J. Biol. Chem. 217, 439–451. 10.1016/s0021-9258(19)57193-9. [DOI] [PubMed] [Google Scholar]
- 4.Siekevitz P. (1957). Powerhouse of the Cell (Scientific American). https://www.scientificamerican.com/article/powerhouse-of-the-cell/.
- 5.Giles RE, Blanc H, Cann HM, and Wallace DC (1980). Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 77, 6715–6719. 10.1073/pnas.77.11.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holt IJ, Harding AE, and Morgan-Hughes JA (1988). Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 331, 717–719. 10.1038/331717a0. [DOI] [PubMed] [Google Scholar]
- 7.Wallace DC, Zheng X, Lott MT, Shoffner JM, Hodge JA, Kelley RI, Epstein CM, and Hopkins LC (1988). Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell 55, 601–610. 10.1016/0092-8674(88)90218-8. [DOI] [PubMed] [Google Scholar]
- 8.Nunnari J, Marshall WF, Straight A, Murray A, Sedat JW, and Walter P. (1997). Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol. Biol. Cell 8, 1233–1242. 10.1091/mbc.8.7.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu X, Kim CN, Yang J, Jemmerson R, and Wang X. (1996). Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157. 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 10.Pacher P, and Hajnoczky G. (2001). Propagation of the apoptotic signal by mitochondrial waves. EMBO J. 20, 4107–4121. 10.1093/emboj/20.15.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ono T, Isobe K, Nakada K, and Hayashi JI (2001). Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat. Genet. 28, 272–275. 10.1038/90116. [DOI] [PubMed] [Google Scholar]
- 12.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, and Chan DC (2010). Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289. 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandel N. (2015). Evolution of mitochondria as signaling organelles. Cell Metab. 22, 204–206. 10.1016/j.cmet.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 15.Nunnari J, and Suomalainen A. (2012). Mitochondria: in sickness and in health. Cell 148, 1145–1159. 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tyynismaa H, Carroll CJ, Raimundo N, Ahola-Erkkila S, Wenz T, Ruhanen H, Guse K, Hemminki A, Peltola-Mjosund KE, Tulkki V, et al. (2010). Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 19, 3948–3958. 10.1093/hmg/ddq310. [DOI] [PubMed] [Google Scholar]
- 17.Durieux J, Wolff S, and Dillin A. (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91. 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forsstrom S, Jackson CB, Carroll CJ, Kuronen M, Pirinen E, Pradhan S, Marmyleva A, Auranen M, Kleine IM, Khan NA, et al. (2019). Fibroblast growth factor 21 drives dynamics of local and systemic stress responses in mitochondrial myopathy with mtDNA deletions. Cell Metab. 30, 1040–1054.e7. 10.1016/j.cmet.2019.08.019. [DOI] [PubMed] [Google Scholar]
- 19.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, and Haynes CM (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590. 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cardamone MD, Tanasa B, Cederquist CT, Huang J, Mahdaviani K, Li W, Rosenfeld MG, Liesa M, and Perissi V. (2018). Mitochondrial retrograde signaling in mammals is mediated by the transcriptional cofactor GPS2 via direct mitochondria-to-nucleus translocation. Mol. Cell 69, 757–772.e7. 10.1016/j.molcel.2018.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim KH, Son JM, Benayoun BA, and Lee C. (2018). The mitochondrial-encoded peptide MOTS-c translocates to the nucleus to regulate nuclear gene expression in response to metabolic stress. Cell Metab. 28, 516–524.e7. 10.1016/j.cmet.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, O’Hearn S, Levy S, Potluri P, Lvova M, et al. (2014). Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. USA 111, E4033–E4042. 10.1073/pnas.1414028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian Y, Garcia G, Bian Q, Steffen K, Joe L, Wolff S, Meyer B, and Dillin A. (2016). Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell 165, 1197–1208. 10.1016/j.cell.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lozoya OA, Martinez-Reyes I, Wang T, Grenet D, Bushel P, Li J, Chandel N, Woychik RP, and Santos JH (2018). Mitochondrial nicotinamide adenine dinucleotide reduced (NADH) oxidation links the tricarboxylic acid (TCA) cycle with methionine metabolism and nuclear DNA methylation. PLoS Biol. 16, e2005707. 10.1371/journal.pbio.2005707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smiraglia D, Kulawiec M, Bistulfi GL, Ghoshal S, and Singh KK (2008). A novel role for mitochondria in regulating epigenetic modifications in the nucleus. Cancer Biol. Ther. 7, 1182–1190. 10.4161/cbt.7.8.6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bar-Ziv R, Bolas T, and Dillin A. (2020). Systemic effects of mitochondrial stress. EMBO Rep. 21, e50094. 10.15252/embr.202050094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berridge MV, Herst PM, Rowe MR, Schneider R, and McConnell MJ (2018). Mitochondrial transfer between cells: methodological constraints in cell culture and animal models. Anal. Biochem. 552, 75–80. 10.1016/j.ab.2017.11.008. [DOI] [PubMed] [Google Scholar]
- 29.Levoux J, Prola A, Lafuste P, Gervais M, Chevallier N, Koumaiha Z, Kefi K, Braud L, Schmitt A, Yacia A, et al. (2021). Platelets facilitate the wound-healing capability of mesenchymal stem cells by mitochondrial transfer and metabolic reprogramming. Cell Metab. 33, 688–690. 10.1016/j.cmet.2021.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Nicolas-Avila JA, Lechuga-Vieco AV, Esteban-Martinez L, Sanchez-Diaz M, Diaz-Garcia E, Santiago DJ, Rubio-Ponce A, Li JL, Balachander A, Quintana JA, et al. (2020). A network of macrophages supports mitochondrial homeostasis in the heart. Cell 183, 94–109.e23. 10.1016/j.cell.2020.08.031. [DOI] [PubMed] [Google Scholar]
- 31.Sun T, Qiao H, Pan PY, Chen Y, and Sheng ZH (2013). Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep. 4, 413–419. 10.1016/j.celrep.2013.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanellopoulos AK, Mariano V, Spinazzi M, Woo YJ, McLean C, Pech U, Li KW, Armstrong JD, Giangrande A, Callaerts P, et al. (2020). Aralar sequesters GABA into hyperactive mitochondria, causing social behavior deficits. Cell 180, 1178–1197.e20. 10.1016/j.cell.2020.02.044. [DOI] [PubMed] [Google Scholar]
- 33.Miller WL (2013). Steroid hormone synthesis in mitochondria. Mol. Cell. Endocrinol. 379, 62–73. 10.1016/j.mce.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 34.Hebert-Chatelain E, Desprez T, Serrat R, Bellocchio L, Soria-Gomez E, Busquets-Garcia A, Pagano Zottola AC, Delamarre A, Cannich A, Vincent P, et al. (2016). A cannabinoid link between mitochondria and memory. Nature 539, 555–559. 10.1038/nature20127. [DOI] [PubMed] [Google Scholar]
- 35.Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, Goodman RP, Grabarek Z, Haas ME, Hung WHW, et al. (2021). MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547. 10.1093/nar/gkaa1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morgenstern M, Peikert CD, Lubbert P, Suppanz I, Klemm C, Alka O, Steiert C, Naumenko N, Schendzielorz A, Melchionda L, et al. (2021). Quantitative high-confidence human mitochondrial proteome and its dynamics in cellular context. Cell Metab. 33, 2464–2483.e18. 10.1016/j.cmet.2021.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Picard M, Trumpff C, and Burelle Y. (2019). Mitochondrial psychobiology: foundations and applications. Curr. Opin. Behav. Sci. 28, 142–151. 10.1016/j.cobeha.2019.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buck MD, Sowell RT, Kaech SM, and Pearce EL (2017). Metabolic instruction of immunity. Cell 169, 570–586. 10.1016/j.cell.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hollis F, van der Kooij MA, Zanoletti O, Lozano L, Canto C, and Sandi C. (2015). Mitochondrial function in the brain links anxiety with social subordination. Proc. Natl. Acad. Sci. USA 112, 15486–15491. 10.1073/pnas.1512653112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Picard M, McManus MJ, Gray JD, Nasca C, Moffat C, Kopinski PK, Seifert EL, McEwen BS, and Wallace DC (2015). Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc. Natl. Acad. Sci. USA 112, E6614–E6623. 10.1073/pnas.1515733112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gumpp AM, Boeck C, Behnke A, Bach AM, Ramo-Fernandez L, Welz T, Gundel H, Kolassa IT, and Karabatsiakis A. (2020). Childhood maltreatment is associated with changes in mitochondrial bioenergetics in maternal, but not in neonatal immune cells. Proc. Natl. Acad. Sci. USA 117, 24778–24784. 10.1073/pnas.2005885117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Picard M, Prather AA, Puterman E, Cuillerier A, Coccia M, Aschbacher K, Burelle Y, and Epel ES (2018). A mitochondrial health index sensitive to mood and caregiving stress. Biol. Psychiatry 84, 9–17. 10.1016/j.biopsych.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hill BG, Shiva S, Ballinger S, Zhang J, and Darley-Usmar VM (2019). Bioenergetics and translational metabolism: implications for genetics, physiology and precision medicine. Biol. Chem. 401, 3–29. 10.1515/hsz-2019-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McBride HM, Neuspiel M, and Wasiak S. (2006). Mitochondria: more than just a powerhouse. Curr. Biol. 16, R551–R560. 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 45.Chakrabarty RP, and Chandel NS (2022). Beyond ATP, new roles of mitochondria. Biochemist 44, 2–8. 10.1042/bio_2022_119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kellogg RA, Tian C, Lipniacki T, Quake SR, and Tay S. (2015). Digital signaling decouples activation probability and population heterogeneity. eLife 4, e08931. 10.7554/eLife.08931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gottschling DE, and Nystrom T. (2017). The upsides and downsides of organelle interconnectivity. Cell 169, 24–34. 10.1016/j.cell.2017.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lynn CW, and Bassett DS (2019). The physics of brain network structure, function and control. Nat. Rev. Phys. 1, 318–332. 10.1038/s42254-019-0040-8. [DOI] [Google Scholar]
- 49.Sagan L. (1967). On the origin of mitosing cells. J. Theor. Biol. 14, 225–IN6. 10.1016/0022-5193(67)90079-3. [DOI] [PubMed] [Google Scholar]
- 50.Birch J. (2017). The multicellular organism as a social phenomenon. In The Philosophy of Social Evolution (Oxford University Press; ), pp. 165–188. 10.1093/oso/9780198733058.003.0007. [DOI] [Google Scholar]
- 51.Levin M. (2021). Bioelectric signaling: reprogrammable circuits underlying embryogenesis, regeneration, and cancer. Cell 184, 1971–1989. 10.1016/j.cell.2021.02.034. [DOI] [PubMed] [Google Scholar]
- 52.Kerr R, Jabbari S, and Johnston IG (2019). Intracellular energy variability modulates cellular decision-making capacity. Sci. Rep. 9, 20196. 10.1038/s41598-019-56587-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Katajisto P, Dohla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, Zoncu R, Chen W, Weinberg RA, and Sabatini DM (2015). Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 348, 340–343. 10.1126/science.1260384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nijhout HF, Sadre-Marandi F, Best J, and Reed MC (2017). Systems biology of phenotypic robustness and plasticity. Integr. Comp. Biol. 57, 171–184. 10.1093/icb/icx076. [DOI] [PubMed] [Google Scholar]
- 55.Sterling P. (2020). What Is Health? Allostasis and the Evolution of Human Design (MIT Press; ). [DOI] [PubMed] [Google Scholar]
- 56.Rubalcava-Gracia D, Garcia-Villegas R, and Larsson NG (2022). No role for nuclear transcription regulators in mammalian mitochondria? Mol. Cell. 10.1016/j.molcel.2022.09.010. [DOI] [PubMed] [Google Scholar]
- 57.Wrutniak-Cabello C, Casas F, and Cabello G. (2001). Thyroid hormone action in mitochondria. J. Mol. Endocrinol. 26, 67–77. 10.1677/jme.0.0260067. [DOI] [PubMed] [Google Scholar]
- 58.Sterling K, Campbell GA, and Brenner MA (1984). Purification of the mitochondrial triiodothyronine (T3) receptor from rat liver. Acta Endocrinol. 105, 391–397. 10.1530/acta.0.1050391. [DOI] [PubMed] [Google Scholar]
- 59.Sterling K, Lazarus JH, Milch PO, Sakurada T, and Brenner MA (1978). Mitochondrial thyroid hormone receptor: localization and physiological significance. Science 201, 1126–1129. 10.1126/science.210507. [DOI] [PubMed] [Google Scholar]
- 60.Sterling K, Milch PO, Brenner MA, and Lazarus JH (1977). Thyroid hormone action: the mitochondrial pathway. Science 197, 996–999. 10.1126/science.196334. [DOI] [PubMed] [Google Scholar]
- 61.Wrutniak C, Cassar-Malek I, Marchal S, Rascle A, Heusser S, Keller JM, Flechon J, Daua M, Samarut J, Ghysdael J, and Cabello G. (1995). A 43-kDa protein related to c-Erb A alpha 1 is located in the mitochondrial matrix of rat liver. J. Biol. Chem. 270, 16347–16354. 10.1074/jbc.270.27.16347. [DOI] [PubMed] [Google Scholar]
- 62.Casas F, Rochard P, Rodier A, Cassar-Malek I, Marchal-Victorion S, Wiesner RJ, Cabello G, and Wrutniak C. (1999). A variant form of the nuclear triiodothyronine receptor c-ErbAα1 plays a direct role in regulation of mitochondrial RNA synthesis. Mol. Cell Biol. 19, 7913–7924. 10.1128/MCB.19.12.7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Enriquez JA, Fernandez-Silva P, Garrido-Perez N, Lopez-Perez MJ, Perez-Martos A, and Montoya J. (1999). Direct regulation of mitochondrial RNA synthesis by thyroid hormone. Mol. Cell Biol. 19, 657–670. 10.1128/MCB.19.1.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fernandez-Vizarra E, Enriquez JA, Perez-Martos A, Montoya J, and Fernandez-Silva P. (2008). Mitochondrial gene expression is regulated at multiple levels and differentially in the heart and liver by thyroid hormones. Curr. Genet. 54, 13–22. 10.1007/s00294-008-0194-x. [DOI] [PubMed] [Google Scholar]
- 65.Casas F, Pessemesse L, Grandemange S, Seyer P, Gueguen N, Baris O, Lepourry L, Cabello G, and Wrutniak-Cabello C. (2008). Overexpression of the mitochondrial T3 receptor p43 induces a shift in skeletal muscle fiber types. PLoS One 3, e2501. 10.1371/journal.pone.0002501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wrutniak-Cabello C, Casas F, and Cabello G. (2018). Thyroid hormone action: the p43 mitochondrial pathway. Methods Mol. Biol. 1801, 163–181. 10.1007/978-1-4939-7902-8_14. [DOI] [PubMed] [Google Scholar]
- 67.Mosselman S, Polman J, and Dijkema R. (1996). ERb: identification and characterization of a novel human estrogen receptor. FEBS Lett. 392, 49–53. 10.1016/0014-5793(96)00782-x. [DOI] [PubMed] [Google Scholar]
- 68.Klinge CM (2017). Estrogens regulate life and death in mitochondria. J. Bioenerg. Biomembr. 49, 307–324. 10.1007/s10863-017-9704-1. [DOI] [PubMed] [Google Scholar]
- 69.Yang SH, Liu R, Perez EJ, Wen Y, Stevens SM Jr., Valencia T, Brun-Zinkernagel AM, Prokai L, Will Y, Dykens J, et al. (2004). Mitochondrial localization of estrogen receptor β. Proc. Natl. Acad. Sci. USA 101, 4130–4135. 10.1073/pnas.0306948101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Solakidi S, Psarra AM, and Sekeris CE (2005). Differential subcellular distribution of estrogen receptor isoforms: localization of ERα in the nucleoli and ERβ in the mitochondria of human osteosarcoma SaOS-2 and hepatocarcinoma HepG2 cell lines. Biochim. Biophys. Acta 1745, 382–392. 10.1016/j.bbamcr.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 71.Zhang G, Yanamala N, Lathrop KL, Zhang L, Klein-Seetharaman J, and Srinivas H. (2010). Ligand-independent antiapoptotic function of estrogen receptor-beta in lung cancer cells. Mol. Endocrinol. 24, 1737–1747. 10.1210/me.2010-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Giordano C, Montopoli M, Perli E, Orlandi M, Fantin M, Ross-Cisneros FN, Caparrotta L, Martinuzzi A, Ragazzi E, Ghelli A, et al. (2011). Oestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathy. Brain 134, 220–234. 10.1093/brain/awq276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bajpai P, Koc E, Sonpavde G, Singh R, and Singh KK (2019). Mitochondrial localization, import, and mitochondrial function of the androgen receptor. J. Biol. Chem. 294, 6621–6634. 10.1074/jbc.RA118.006727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Solakidi S, Psarra AM, Nikolaropoulos S, and Sekeris CE (2005). Estrogen receptors α and β (ERα and ERβ) and androgen receptor (AR) in human sperm: localization of ERβ and AR in mitochondria of the midpiece. Hum. Reprod. 20, 3481–3487. 10.1093/humrep/dei267. [DOI] [PubMed] [Google Scholar]
- 75.Ventura-Clapier R, Moulin M, Piquereau J, Lemaire C, Mericskay M, Veksler V, and Garnier A. (2017). Mitochondria: a central target for sex differences in pathologies. Clin. Sci. (Lond.) 131, 803–822. 10.1042/CS20160485. [DOI] [PubMed] [Google Scholar]
- 76.Junker A, Wang J, Gouspillou G, Ehinger JK, Elmer E, Sjovall F, Fisher-Wellman KH, Neufer PD, Molina AJA, Ferrucci L, and Picard M. (2022). Human studies of mitochondrial biology demonstrate an overall lack of binary sex differences: a multivariate meta-analysis. FASEB J. 36, e22146. 10.1096/fj.202101628R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, Brad Thompson E, Rosenfeld MG, and Evans RM (1985). Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 318, 635–641. 10.1038/318635a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rivers C, Levy A, Hancock J, Lightman S, and Norman M. (1999). Insertion of an amino acid in the DNA-binding domain of the glucocorticoid receptor as a result of alternative splicing. J. Clin. Endocrinol. Metab. 84, 4283–4286. 10.1210/jcem.84.11.6235. [DOI] [PubMed] [Google Scholar]
- 79.Yu FL, and Feigelson P. (1970). A comparative study of RNA synthesis in rat hepatic nuclei and mitochondria under the influence of cortisone. Biochim. Biophys. Acta 213, 134–141. 10.1016/0005-2787(70)90014-6. [DOI] [PubMed] [Google Scholar]
- 80.Demonacos C, Djordjevic-Markovic R, Tsawdaroglou N, and Sekeris CE (1995). The mitochondrion as a primary site of action of glucocorticoids: the interaction of the glucocorticoid receptor with mitochondrial DNA sequences showing partial similarity to the nuclear glucocorticoid responsive elements. J. Steroid Biochem. Mol. Biol. 55, 43–55. 10.1016/0960-0760(95)00159-w. [DOI] [PubMed] [Google Scholar]
- 81.Tsiriyotis C, Spandidos DA, and Sekeris CE (1997). The mitochondrion as a primary site of action of glucocorticoids: mitochondrial nucleotide sequences, showing similarity to hormone response elements, confer dexamethasone inducibility to chimaeric genes transfected in LATK-cells. Biochem. Biophys. Res. Commun. 235, 349–354. 10.1006/bbrc.1997.6787. [DOI] [PubMed] [Google Scholar]
- 82.Polman JAE, Welten JE, Bosch DS, de Jonge RT, Balog J, van der Maarel SM, de Kloet ER, and Datson NA (2012). A genome-wide signature of glucocorticoid receptor binding in neuronal PC12 cells. BMC Neurosci. 13, 118. 10.1186/1471-2202-13-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morgan DJ, Poolman TM, Williamson AJK, Wang Z, Clark NR, Ma’ayan A, Whetton AD, Brass A, Matthews LC, and Ray DW (2016). Glucocorticoid receptor isoforms direct distinct mitochondrial programs to regulate ATP production. Sci. Rep. 6, 26419. 10.1038/srep26419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Psarra AMG, and Sekeris CE (2011). Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: role of the mitochondrial glucocorticoid receptor. Biochim. Biophys. Acta 1813, 1814–1821. 10.1016/j.bbamcr.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 85.Simoes DCM, Psarra AMG, Mauad T, Pantou I, Roussos C, Sekeris CE, and Gratziou C. (2012). Glucocorticoid and estrogen receptors are reduced in mitochondria of lung epithelial cells in asthma. PLoS One 7, e39183. 10.1371/journal.pone.0039183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, Smith BJ, Burks TN, Cohn RD, Fedarko NS, et al. (2011). Identification and characterization of a functional mitochondrial angiotensin system. Proc. Natl. Acad. Sci. USA 108, 14849–14854. 10.1073/pnas.1101507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suofu Y, Li W, Jean-Alphonse FG, Jia J, Khattar NK, Li J, Baranov SV, Leronni D, Mihalik AC, He Y, et al. (2017). Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc. Natl. Acad. Sci. USA 114, E7997–E8006. 10.1073/pnas.1705768114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Benard G, Massa F, Puente N, Lourenco J, Bellocchio L, Soria-Gomez E, Matias I, Delamarre A, Metna-Laurent M, Cannich A, et al. (2012). Mitochondrial CB(1) receptors regulate neuronal energy metabolism. Nat. Neurosci. 15, 558–564. 10.1038/nn.3053. [DOI] [PubMed] [Google Scholar]
- 89.Belous AE, Jones CM, Wakata A, Knox CD, Nicoud IB, Pierce J, and Chari RS (2006). Mitochondrial calcium transport is regulated by P2Y1- and P2Y2-like mitochondrial receptors. J. Cell. Biochem. 99, 1165–1174. 10.1002/jcb.20985. [DOI] [PubMed] [Google Scholar]
- 90.Jong YJI, Harmon SK, and O’Malley KL (2018). Intracellular GPCRs play key roles in synaptic plasticity. ACS Chem. Neurosci. 9, 2162–2172. 10.1021/acschemneuro.7b00516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Abadir PM, Walston JD, and Carey RM (2012). Subcellular characteristics of functional intracellular renin-angiotensin systems. Peptides 38, 437–445. 10.1016/j.peptides.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reiter RJ, Sharma R, Pires de Campos Zuccari DA, de Almeida Chuffa LG, Manucha W, and Rodriguez C. (2021). Melatonin synthesis in and uptake by mitochondria: implications for diseased cells with dysfunctional mitochondria. Future Med. Chem. 13, 335–339. 10.4155/fmc-2020-0326. [DOI] [PubMed] [Google Scholar]
- 93.Guo P, Pi H, Xu S, Zhang L, Li Y, Li M, Cao Z, Tian L, Xie J, Li R, et al. (2014). Melatonin improves mitochondrial function by promoting MT1/SIRT1/PGC-1 alpha-dependent mitochondrial biogenesis in cadmium-induced hepatotoxicity in vitro. Toxicol. Sci. 142, 182–195. 10.1093/toxsci/kfu164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gutierrez-Rodriguez A, Bonilla-Del Rio I, Puente N, Gomez-Urquijo SM, Fontaine CJ, Egana-Huguet J, Elezgarai I, Ruehle S, Lutz B, Robin LM, et al. (2018). Localization of the cannabinoid type-1 receptor in subcellular astrocyte compartments of mutant mouse hippocampus. Glia 66, 1417–1431. 10.1002/glia.23314. [DOI] [PubMed] [Google Scholar]
- 95.Mendizabal-Zubiaga J, Melser S, Benard G, Ramos A, Reguero L, Arrabal S, Elezgarai I, Gerrikagoitia I, Suarez J, Rodriguez De Fonseca F, et al. (2016). Cannabinoid CB1 receptors are localized in striated muscle mitochondria and regulate mitochondrial respiration. Front. Physiol. 7, 476. 10.3389/fphys.2016.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Belous A, Wakata A, Knox CD, Nicoud IB, Pierce J, Anderson CD, Pinson CW, and Chari RS (2004). Mitochondrial P2Y-like receptors link cytosolic adenosine nucleotides to mitochondrial calcium up-take. J. Cell. Biochem. 92, 1062–1073. 10.1002/jcb.20144. [DOI] [PubMed] [Google Scholar]
- 97.Sarti AC, Vultaggio-Poma V, Falzoni S, Missiroli S, Giuliani AL, Boldrini P, Bonora M, Faita F, Di Lascio N, Kusmic C, et al. (2021). Mitochondrial P2X7 receptor localization modulates energy metabolism enhancing physical performance. Function (Oxf). 2, zqab005. 10.1093/function/zqab005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wyant GA, Yu W, Doulamis IP, Nomoto RS, Saeed MY, Duignan T, McCully JD, and Kaelin WG Jr. (2022). Mitochondrial remodeling and ischemic protection by G protein-coupled receptor 35 agonists. Science 377, 621–629. 10.1126/science.abm1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lykhmus O, Gergalova G, Koval L, Zhmak M, Komisarenko S, and Skok M. (2014). Mitochondria express several nicotinic acetylcholine receptor subtypes to control various pathways of apoptosis induction. Int. J. Biochem. Cell Biol. 53, 246–252. 10.1016/j.biocel.2014.05.030. [DOI] [PubMed] [Google Scholar]
- 100.Lu B, Kwan K, Levine YA, Olofsson PS, Yang H, Li J, Joshi S, Wang H, Andersson U, Chavan SS, and Tracey KJ (2014). α7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol. Med. 20, 350–358. 10.2119/molmed.2013.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nicholls DG, and Fergusson SJ (2013). Bioenergetics, 4 Edition (Academic Press; ). [Google Scholar]
- 102.Ruprecht JJ, King MS, Zogg T, Aleksandrova AA, Pardon E, Crichton PG, Steyaert J, and Kunji ER (2019). The molecular mechanism of transport by the mitochondrial ADP/ATP carrier. Cell 176, 435–447.e15. 10.1016/j.cell.2018.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nelson MA, McLaughlin KL, Hagen JT, Coalson HS, Schmidt C, Kassai M, Kew KA, McClung JM, Neufer PD, Brophy P, et al. (2021). Intrinsic OXPHOS limitations underlie cellular bioenergetics in leukemia. eLife 10, e63104. 10.7554/eLife.63104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hackenbrock CR (1968). Ultrastructural bases for metabolically linked mechanical activity in mitochondria. II. Electron transport-linked ultrastructural transformations in mitochondria. J. Cell Biol. 37, 345–369. 10.1083/jcb.37.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Taylor EB (2017). Functional properties of the mitochondrial carrier system. Trends Cell Biol. 27, 633–644. 10.1016/j.tcb.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ruprecht JJ, and Kunji ER (2020). The SLC25 mitochondrial carrier family: structure and mechanism. Trends Biochem. Sci. 45, 244–258. 10.1016/j.tibs.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, et al. (2012). A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100. 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ERS, and Martinou JC (2012). Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93–96. 10.1126/science.1218530. [DOI] [PubMed] [Google Scholar]
- 109.Kory N, Uit de Bos J, van der Rijt S, Jankovic N, Gura M, Arp N, Pena IA, Prakash G, Chan SH, Kunchok T, et al. (2020). MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci. Adv. 6, eabe5310. 10.1126/sciadv.abe5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yoo HC, Park SJ, Nam M, Kang J, Kim K, Yeo JH, Kim JK, Heo Y, Lee HS, Lee MY, et al. (2020). A variant of SLC1A5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab. 31, 267–283.e12. 10.1016/j.cmet.2019.11.020. [DOI] [PubMed] [Google Scholar]
- 111.Abdullah MO, Zeng RX, Margerum CL, Papadopoli D, Monnin C, Punter KB, Chu C, Al-Rofaidi M, Al-Tannak NF, Berardi D, et al. (2022). Mitochondrial hyperfusion via metabolic sensing of regulatory amino acids. Cell Rep. 40, 111198. 10.1016/j.celrep.2022.111198. [DOI] [PubMed] [Google Scholar]
- 112.Lane N. (2022). Transformer: The Deep Chemistry of Life and Death (WW Norton). [Google Scholar]
- 113.Sharma R, Reinstadler B, Engelstad K, Skinner OS, Stackowitz E, Haller RG, Clish CB, Pierce K, Walker MA, Fryer R, et al. (2021). Circulating markers of NADH-reductive stress correlate with mitochondrial disease severity. J. Clin. Invest. 131, 136055. 10.1172/JCI136055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Goodman RP, Markhard AL, Shah H, Sharma R, Skinner OS, Clish CB, Deik A, Patgiri A, Hsu YHH, Masia R, et al. (2020). Hepatic NADH reductive stress underlies common variation in metabolic traits. Nature 583, 122–126. 10.1038/s41586-020-2337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Khan NA, Nikkanen J, Yatsuga S, Jackson C, Wang L, Pradhan S, Kivela R, Pessia A, Velagapudi V, and Suomalainen A. (2017). mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab. 26, 419–428.e5. 10.1016/j.cmet.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 116.Nikkanen J, Forsstrom S, Euro L, Paetau I, Kohnz R, Wang L, Chilov D, Viinamaki J, Roivainen A, Marjamaki P, et al. (2016). Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metab. 23, 635–648. 10.1016/j.cmet.2016.01.019. [DOI] [PubMed] [Google Scholar]
- 117.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Carrico C, Meyer JG, He W, Gibson BW, and Verdin E. (2018). The mitochondrial acylome emerges: proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metab. 27, 497–512. 10.1016/j.cmet.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Spinelli JB, and Haigis MC (2018). The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 20, 745–754. 10.1038/s41556-018-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.De Stefani D, Raffaello A, Teardo E, Szabo I, and Rizzuto R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Glancy B, and Balaban RS (2012). Role of mitochondrial ca(2+) in the regulation of cellular energetics. Biochemistry 51, 2959–2973. 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wescott AP, Kao JPY, Lederer WJ, and Boyman L. (2019). Voltage-energized calcium-sensitive ATP production by mitochondria. Nat. Metab. 1, 975–984. 10.1038/s42255-019-0126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Koval OM, Nguyen EK, Santhana V, Fidler TP, Sebag SC, Rasmussen TP, Mittauer DJ, Strack S, Goswami PC, Abel ED, and Grumbach IM (2019). Loss of MCU prevents mitochondrial fusion in G1-S phase and blocks cell cycle progression and proliferation. Sci. Signal. 12, eaav1439. 10.1126/scisignal.aav1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Palty R, Ohana E, Hershfinkel M, Volokita M, Elgazar V, Beharier O, Silverman WF, Argaman M, and Sekler I. (2004). Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J. Biol. Chem. 279, 25234–25240. 10.1074/jbc.M401229200. [DOI] [PubMed] [Google Scholar]
- 126.Nita II, Hershfinkel M, Kantor C, Rutter GA, Lewis EC, and Sekler I. (2014). Pancreatic beta-cell Na+ channels control global Ca2+ signaling and oxidative metabolism by inducing Na+ and Ca2+ responses that are propagated into mitochondria. FASEB J. 28, 3301–3312. 10.1096/fj.13-248161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, et al. (2017). The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 545, 93–97. 10.1038/nature22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Assali EA, Jones AE, Veliova M, Acin-Perez R, Taha M, Miller N, Shum M, Oliveira MF, Las G, Liesa M, et al. (2020). NCLX prevents cell death during adrenergic activation of the brown adipose tissue. Nat. Commun. 11, 3347. 10.1038/s41467-020-16572-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hernansanz-Agustin P, Choya-Foces C, Carregal-Romero S, Ramos E, Oliva T, Villa-Pina T, Moreno L, Izquierdo-Alvarez A, Cabrera-Garcia JD, Cortes A, et al. (2020). Na(+) controls hypoxic signalling by the mitochondrial respiratory chain. Nature 586, 287–291. 10.1038/s41586-020-2551-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.de Sousa RT, Streck EL, Zanetti MV, Ferreira GK, Diniz BS, Brunoni AR, Busatto GF, Gattaz WF, and Machado-Vieira R. (2015). Lithium increases leukocyte mitochondrial complex I activity in bipolar disorder during depressive episodes. Psychopharmacology (Berl) 232, 245–250. 10.1007/s00213-014-3655-6. [DOI] [PubMed] [Google Scholar]
- 131.Lundberg JO, and Weitzberg E. (2022). Nitric oxide signaling in health and disease. Cell 185, 2853–2878. 10.1016/j.cell.2022.06.010. [DOI] [PubMed] [Google Scholar]
- 132.Arias-Mayenco I, Gonzalez-Rodriguez P, Torres-Torrelo H, Gao L, Fernandez-Aguera MC, Bonilla-Henao V, Ortega-Saenz P, and Lopez-Barneo J. (2018). Acute O2 sensing: role of coenzyme QH2/Q ratio and mitochondrial ROS compartmentalization. Cell Metab. 28, 145–158.e4. 10.1016/j.cmet.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 133.Fernández-Agüera M, Gao L, Gonzalez-Rodriguez P, Pintado C, Arias-Mayenco I, Garcia-Flores P, Garcia-Perganeda A, Pascual A, Ortega-Saenz P, and Lopez-Barneo J. (2015). Oxygen sensing by arterial chemoreceptors depends on mitochondrial complex I signaling. Cell Metab. 22, 825–837. 10.1016/j.cmet.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 134.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, and Schumacker PT (2005). Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 1, 401–408. 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 135.Semenza GL (2009). Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology 24, 97–106. 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- 136.Gustafsson CM, Falkenberg M, and Larsson NG (2016). Maintenance and expression of mammalian mitochondrial DNA. Annu. Rev. Biochem. 85, 133–160. 10.1146/annurev-biochem-060815-014402. [DOI] [PubMed] [Google Scholar]
- 137.Merry TL, Chan A, Woodhead JST, Reynolds JC, Kumagai H, Kim SJ, and Lee C. (2020). Mitochondrial-derived peptides in energy metabolism. Am. J. Physiol. Endocrinol. Metab. 319, E659–E666. 10.1152/ajpendo.00249.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chomyn A, Martinuzzi A, Yoneda M, Daga A, Hurko O, Johns D, Lai ST, Nonaka I, Angelini C, and Attardi G. (1992). MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc. Natl. Acad. Sci. USA 89, 4221–4225. 10.1073/pnas.89.10.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Payne BAI, Wilson IJ, Hateley CA, Horvath R, Santibanez-Koref M, Samuels DC, Price DA, and Chinnery PF (2011). Mitochondrial aging is accelerated by anti-retroviral therapy through the clonal expansion of mtDNA mutations. Nat. Genet. 43, 806–810. 10.1038/ng.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, Suomalainen A, Thorburn DR, Zeviani M, and Turnbull DM (2016). Mitochondrial diseases. Nat. Rev. Dis. Primers 2, 16080. 10.1038/nrdp.2016.80. [DOI] [PubMed] [Google Scholar]
- 141.Taylor RW, and Turnbull DM (2005). Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 6, 389–402. 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shaham O, Slate NG, Goldberger O, Xu Q, Ramanathan A, Souza AL, Clish CB, Sims KB, and Mootha VK (2010). A plasma signature of human mitochondrial disease revealed through metabolic profiling of spent media from cultured muscle cells. Proc. Natl. Acad. Sci. USA 107, 1571–1575. 10.1073/pnas.0906039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Vincent AE, Rosa HS, Pabis K, Lawless C, Chen C, Grunewald A, Rygiel KA, Rocha MC, Reeve AK, Falkous G, et al. (2018). Sub-cellular origin of mitochondrial DNA deletions in human skeletal muscle. Ann. Neurol. 84, 289–301. 10.1002/ana.25288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cai N, Gomez-Duran A, Yonova-Doing E, Kundu K, Burgess AI, Golder ZJ, Calabrese C, Bonder MJ, Camacho M, Lawson RA, et al. (2021). Mitochondrial DNA variants modulate N-formylmethionine, proteostasis and risk of late-onset human diseases. Nat. Med. 27, 1564–1575. 10.1038/s41591-021-01441-3. [DOI] [PubMed] [Google Scholar]
- 145.Latorre-Pellicer A, Moreno-Loshuertos R, Lechuga-Vieco AV, Sanchez-Cabo F, Torroja C, Acin-Perez R, Calvo E, Aix E, Gonzalez-Guerra A, Logan A, et al. (2016). Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535, 561–565. 10.1038/nature18618. [DOI] [PubMed] [Google Scholar]
- 146.Yonova-Doing E, Calabrese C, Gomez-Duran A, Schon K, Wei W, Karthikeyan S, Chinnery PF, and Howson JMM (2021). An atlas of mitochondrial DNA genotype-phenotype associations in the UK Biobank. Nat. Genet. 53, 982–993. 10.1038/s41588-021-00868-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wu Z, Oeck S, West AP, Mangalhara KC, Sainz AG, Newman LE, Zhang XO, Wu L, Yan Q, Bosenberg M, et al. (2019). Mitochondrial DNA stress signalling protects the nuclear genome. Nat. Metab. 1, 1209–1218. 10.1038/s42255-019-0150-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chanprasert S, Wang J, Weng SW, Enns GM, Boue DR, Wong BL, Mendell JR, Perry DA, Sahenk Z, Craigen WJ, et al. (2013). Molecular and clinical characterization of the myopathic form of mitochondrial DNA depletion syndrome caused by mutations in the thymidine kinase (TK2) gene. Mol. Genet. Metab. 110, 153–161. 10.1016/j.ymgme.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 149.Sassone-Corsi P. (2012). The cyclic AMP pathway. Cold Spring Harbor Perspect. Biol. 4, a011148. 10.1101/cshperspect.a011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kandel ER, Koester JD, Mack SH, and Siegelbaum SA (2021). Principles of Neural Science, Sixth edition (Appleton & Lange; ). [Google Scholar]
- 151.Keidar Haran T, and Keren L. (2022). From genes to modules, from cells to ecosystems. Mol. Syst. Biol. 18, e10726. 10.15252/msb.202110726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Picard M, and Sandi C. (2021). The social nature of mitochondria: implications for human health. Neurosci. Biobehav. Rev. 120, 595–610. 10.1016/j.neubiorev.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hein AM, Rosenthal SB, Hagstrom GI, Berdahl A, Torney CJ, and Couzin ID (2015). The evolution of distributed sensing and collective computation in animal populations. eLife 4, e10955. 10.7554/eLife.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sosna MMG, Twomey CR, Bak-Coleman J, Poel W, Daniels BC, Romanczuk P, and Couzin ID (2019). Individual and collective encoding of risk in animal groups. Proc. Natl. Acad. Sci. USA 116, 20556–20561. 10.1073/pnas.1905585116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Barabasi AL, and Oltvai ZN (2004). Network biology: understanding the cell’s functional organization. Nat. Rev. Genet. 5, 101–113. 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 156.Kadushin C. (2012). Understanding Social Networks (Oxford University Press; ). [Google Scholar]
- 157.Arimura S.i., Yamamoto J, Aida GP, Nakazono M, and Tsutsumi N. (2004). Frequent fusion and fission of plant mitochondria with unequal nucleoid distribution. Proc. Natl. Acad. Sci. USA 101, 7805–7808. 10.1073/pnas.0401077101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Liu X, Weaver D, Shirihai O, and Hajnoczky G. (2009). Mitochondrial ‘kiss-and-run’: interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 28, 3074–3089. 10.1038/emboj.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yang L, Long Q, Liu J, Tang H, Li Y, Bao F, Qin D, Pei D, and Liu X. (2015). Mitochondrial fusion provides an ‘initial metabolic complementation’ controlled by mtDNA. Cell. Mol. Life Sci. 72, 2585–2598. 10.1007/s00018-015-1863-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bakeeva L, Chentsov Yu S, and Skulachev VP (1983). Intermitochondrial contacts in myocardiocytes. J. Mol. Cell. Cardiol. 15, 413–420. 10.1016/0022-2828(83)90261-4. [DOI] [PubMed] [Google Scholar]
- 161.Amchenkova AA, Bakeeva LE, Chentsov YS, Skulachev VP, and Zorov DB (1988). Coupling membranes as energy-transmitting cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. J. Cell Biol. 107, 481–495. 10.1083/jcb.107.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Picard M, Gentil BJ, McManus MJ, White K, St Louis K, Gartside SE, Wallace DC, and Turnbull DM (2013). Acute exercise remodels mitochondrial membrane interactions in mouse skeletal muscle. J. Appl. Physiol. 115, 1562–1571. 10.1152/japplphysiol.00819.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Picard M, McManus MJ, Csordas G, Varnai P, Dorn II GW, Williams D, Hajnoczky G, and Wallace DC (2015). Trans-mitochondrial coordination of cristae at regulated membrane junctions. Nat. Commun. 6, 6259. 10.1038/ncomms7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Vernay A, Marchetti A, Sabra A, Jauslin TN, Rosselin M, Scherer PE, Demaurex N, Orci L, and Cosson P. (2017). MitoNEET-dependent formation of intermitochondrial junctions. Proc. Natl. Acad. Sci. USA 114, 8277–8282. 10.1073/pnas.1706643114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Santo-Domingo J, Giacomello M, Poburko D, Scorrano L, and Demaurex N. (2013). OPA1 promotes pH flashes that spread between contiguous mitochondria without matrix protein exchange. EMBO J. 32, 1927–1940. 10.1038/emboj.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Glancy B, Hartnell LM, Combs CA, Femnou A, Sun J, Murphy E, Subramaniam S, and Balaban RS (2017). Power grid protection of the muscle mitochondrial reticulum. Cell Rep. 19, 487–496. 10.1016/j.celrep.2017.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Vincent AE, Turnbull DM, Eisner V, Hajnoczky G, and Picard M. (2017). Mitochondrial nanotunnels. Trends Cell Biol. 27, 787–799. 10.1016/j.tcb.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Huang X, Sun L, Ji S, Zhao T, Zhang W, Xu J, Zhang J, Wang Y, Wang X, Franzini-Armstrong C, et al. (2013). Kissing and nanotunneling mediate intermitochondrial communication in the heart. Proc. Natl. Acad. Sci. USA 110, 2846–2851. 10.1073/pnas.1300741110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wang C, Du W, Su QP, Zhu M, Feng P, Li Y, Zhou Y, Mi N, Zhu Y, Jiang D, et al. (2015). Dynamic tubulation of mitochondria drives mitochondrial network formation. Cell Res. 25, 1108–1120. 10.1038/cr.2015.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Vincent AE, White K, Davey T, Philips J, Ogden RT, Lawless C, Warren C, Hall MG, Ng YS, Falkous G, et al. (2019). Quantitative 3D mapping of the human skeletal muscle mitochondrial network. Cell Rep. 26, 996–1009.e4. 10.1016/j.celrep.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lavorato M, Iyer VR, Dewight W, Cupo RR, Debattisti V, Gomez L, De la Fuente S, Zhao YT, Valdivia HH, Hajnoczky G, and Franzini-Armstrong C. (2017). Increased mitochondrial nanotunneling activity, induced by calcium imbalance, affects intermitochondrial matrix exchanges. Proc. Natl. Acad. Sci. USA 114, E849–E858. 10.1073/pnas.1617788113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ji X, Ferreira T, Friedman B, Liu R, Liechty H, Bas E, Chandrashekar J, and Kleinfeld D. (2021). Brain microvasculature has a common topology with local differences in geometry that match metabolic load. Neuron 109, 1168–1187.e13. 10.1016/j.neuron.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Han JDJ, Bertin N, Hao T, Goldberg DS, Berriz GF, Zhang LV, Dupuy D, Walhout AJM, Cusick ME, Roth FP, and Vidal M. (2004). Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 430, 88–93. 10.1038/nature02555. [DOI] [PubMed] [Google Scholar]
- 174.Brady NR, Elmore SP, van Beek JJ, Krab K, Courtoy PJ, Hue L, and Westerhoff HV (2004). Coordinated behavior of mitochondria in both space and time: a reactive oxygen species-activated wave of mitochondrial depolarization. Biophys. J. 87, 2022–2034. 10.1529/biophysj.103.035097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhou L, Aon MA, Almas T, Cortassa S, Winslow RL, and O’Rourke B. (2010). A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput. Biol. 6, e1000657. 10.1371/journal.pcbi.1000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Friedman JR, and Nunnari J. (2014). Mitochondrial form and function. Nature 505, 335–343. 10.1038/nature12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Twig G, Graf SA, Wikstrom JD, Mohamed H, Haigh SE, Elorza A, Deutsch M, Zurgil N, Reynolds N, and Shirihai OS (2006). Tagging and tracking individual networks within a complex mitochondrial web with photoactivatable GFP. Am. J. Physiol. Cell Physiol. 291, C176–C184. 10.1152/ajpcell.00348.2005. [DOI] [PubMed] [Google Scholar]
- 178.Eisner V, Cupo RR, Gao E, Csordas G, Slovinsky WS, Paillard M, Cheng L, Ibetti J, Chen SRW, Chuprun JK, et al. (2017). Mitochondrial fusion dynamics is robust in the heart and depends on calcium oscillations and contractile activity. Proc. Natl. Acad. Sci. USA 114, E859–E868. 10.1073/pnas.1617288114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Molina AJA, and Shirihai OS (2009). Monitoring mitochondrial dynamics with photoactivatable [corrected] green fluorescent protein. Methods Enzymol. 457, 289–304. 10.1016/S0076-6879(09)05016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Eisner V, Lenaers G, and Hajnoczky G. (2014). Mitochondrial fusion is frequent in skeletal muscle and supports excitation-contraction coupling. J. Cell Biol. 205, 179–195. 10.1083/jcb.201312066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Schauss AC, Huang H, Choi SY, Xu L, Soubeyrand S, Bilodeau P, Zunino R, Rippstein P, Frohman MA, and McBride HM (2010). A novel cell-free mitochondrial fusion assay amenable for high-throughput screenings of fusion modulators. BMC Biol. 8, 100. 10.1186/1741-7007-8-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Paltauf-Doburzynska J, Malli R, and Graier WF (2004). Hyperglycemic conditions affect shape and Ca2+ homeostasis of mitochondria in endothelial cells. J. Cardiovasc. Pharmacol. 44, 423–436. 10.1097/01.fjc.0000139449.64337.1b. [DOI] [PubMed] [Google Scholar]
- 183.Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, et al. (2011). Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 124, 444–453. 10.1161/CIRCULATIONAHA.110.014506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yu T, Robotham JL, and Yoon Y. (2006). Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 103, 2653–2658. 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kleele T, Rey T, Winter J, Zaganelli S, Mahecic D, Perreten Lambert H, Ruberto FP, Nemir M, Wai T, Pedrazzini T, and Manley S. (2021). Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 593, 435–439. 10.1038/s41586-021-03510-6. [DOI] [PubMed] [Google Scholar]
- 186.Liesa M, and Shirihai O. (2013). Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 17, 491–506. 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Picard M, and Turnbull DM (2013). Linking the metabolic state and mitochondrial DNA in chronic disease, health, and aging. Diabetes 62, 672–678. 10.2337/db12-1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gomes LC, Benedetto GD, and Scorrano L. (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598. 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rambold AS, Kostelecky B, Elia N, and Lippincott-Schwartz J. (2011). Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 108, 10190–10195. 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wang W, Wang Y, Long J, Wang J, Haudek S, Overbeek P, Chang B, Schumacker P, and Danesh F. (2012). Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 15, 186–200. 10.1016/j.cmet.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Molina AJ, Wikstrom JD, Stiles L, Las G, Mohamed H, Elorza A, Walzer G, Twig G, Katz S, Corkey BE, and Shirihai OS (2009). Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes 58, 2303–2315. 10.2337/db07-1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Picard M, Azuelos I, Jung B, Giordano C, Matecki S, Hussain S, White K, Li T, Liang F, Benedetti A, et al. (2015). Mechanical ventilation triggers abnormal mitochondrial dynamics and morphology in the diaphragm. J. Appl. Physiol. 118, 1161–1171. 10.1152/japplphysiol.00873.2014. [DOI] [PubMed] [Google Scholar]
- 193.Cogliati S, Frezza C, Soriano M, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes L, et al. (2013). Mitochondrial cristae shape determines respiratory chain super-complexes assembly and respiratory efficiency. Cell 155, 160–171. 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wikstrom JD, Mahdaviani K, Liesa M, Sereda SB, Si Y, Las G, Twig G, Petrovic N, Zingaretti C, Graham A, et al. (2014). Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J. 33, 418–436. 10.1002/embj.201385014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Liston C, Cichon JM, Jeanneteau F, Jia Z, Chao MV, and Gan WB (2013). Circadian glucocorticoid oscillations promote learning-dependent synapse formation and maintenance. Nat. Neurosci. 16, 698–705. 10.1038/nn.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Twig G, Liu X, Liesa M, Wikstrom JD, Molina AJA, Las G, Yaniv G, Hajnoczky G, and Shirihai OS (2010). Biophysical properties of mitochondrial fusion events in pancreatic beta-cells and cardiac cells unravel potential control mechanisms of its selectivity. Am. J. Physiol. Cell Physiol. 299, C477–C487. 10.1152/ajpcell.00427.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF, and Gillespie MN (2012). Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 5, ra47. 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Oikawa K, Imai T, Thagun C, Toyooka K, Yoshizumi T, Ishikawa K, Kodama Y, and Numata K. (2021). Mitochondrial movement during its association with chloroplasts in Arabidopsis thaliana. Commun. Biol. 4, 292. 10.1038/s42003-021-01833-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Eisner V, Picard M, and Hajnoczky G. (2018). Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 20, 755–765. 10.1038/s41556-018-0133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kruppa AJ, and Buss F. (2021). Motor proteins at the mitochondria-cytoskeleton interface. J. Cell Sci. 134, jcs226084. 10.1242/jcs.226084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Pekkurnaz G, and Wang X. (2022). Mitochondrial heterogeneity and homeostasis through the lens of a neuron. Nat. Metab. 4, 802–812. 10.1038/s42255-022-00594-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Yi M, Weaver D, and Hajnoczky G. (2004). Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J. Cell Biol. 167, 661–672. 10.1083/jcb.200406038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Pekkurnaz G, Trinidad J, Wang X, Kong D, and Schwarz T. (2014). Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell 158, 54–68. 10.1016/j.cell.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wong YC, Peng W, and Krainc D. (2019). Lysosomal regulation of inter-mitochondrial contact fate and motility in Charcot-Marie-Tooth type 2. Dev. Cell 50, 339–354.e4. 10.1016/j.devcel.2019.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.English AM, and Hughes AL (2019). Knowing when to let go: lysosomes regulate inter-mitochondrial tethering. Dev. Cell 50, 259–260. 10.1016/j.devcel.2019.07.019. [DOI] [PubMed] [Google Scholar]
- 206.Murley A, and Nunnari J. (2016). The emerging network of mitochondria-organelle contacts. Mol. Cell 61, 648–653. 10.1016/j.molcel.2016.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Csordas G, Weaver D, and Hajnoczky G. (2018). Endoplasmic reticulum-mitochondrial contactology: structure and signaling functions. Trends Cell Biol. 28, 523–540. 10.1016/j.tcb.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Selvaraj V, Stocco DM, and Clark BJ (2018). Current knowledge on the acute regulation of steroidogenesis. Biol. Reprod. 99, 13–26. 10.1093/biolre/ioy102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, and Walter P. (2009). An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325, 477–481. 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lahiri S, Chao JT, Tavassoli S, Wong AKO, Choudhary V, Young BP, Loewen CJR, and Prinz WA (2014). A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol. 12, e1001969. 10.1371/journal.pbio.1001969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Booth DM, Varnai P, Joseph SK, and Hajnoczky G. (2021). Oxidative bursts of single mitochondria mediate retrograde signaling toward the ER. Mol. Cell 81, 3866–3876.e2. 10.1016/j.molcel.2021.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kasai H, Ziv NE, Okazaki H, Yagishita S, and Toyoizumi T. (2021). Spine dynamics in the brain, mental disorders and artificial neural networks. Nat. Rev. Neurosci. 22, 407–422. 10.1038/s41583-021-00467-3. [DOI] [PubMed] [Google Scholar]
- 213.Kasahara A, and Scorrano L. (2014). Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol. 24, 761–770. 10.1016/j.tcb.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 214.Balaban RS, Nemoto S, and Finkel T. (2005). Mitochondria, oxidants, and aging. Cell 120, 483–495. 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 215.Shadel G, and Horvath T. (2015). Mitochondrial ROS signaling in organismal homeostasis. Cell 163, 560–569. 10.1016/j.cell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Martinez-Reyes I, and Chandel NS (2020). Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102. 10.1038/s41467-019-13668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Jiang S, Cai J, Wallace DC, and Jones DP (1999). Cytochrome c-mediated apoptosis in cells lacking mitochondrial DNA. J. Biol. Chem. 274, 29905–29911. 10.1074/jbc.274.42.29905. [DOI] [PubMed] [Google Scholar]
- 218.Picard M, Shirihai OS, Gentil BJ, and Burelle Y. (2013). Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am. J. Physiol. Regul. Integr. Comp. Physiol. 304, R393–R406. 10.1152/ajpregu.00584.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Bernardi P, Rasola A, Forte M, and Lippe G. (2015). The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 95, 1111–1155. 10.1152/physrev.00001.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wang C, and Youle RJ (2009). The role of mitochondria in apoptosis. Annu. Rev. Genet. 43, 95–118. 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kalkavan H, Chen MJ, Crawford JC, Quarato G, Fitzgerald P, Tait SW, Goding CR, and Green DR (2022). Sublethal cytochrome c release generates drug-tolerant persister cells. Cell 185, 3356–3374.e22. 10.1016/j.cell.2022.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Gottschalk B, Madreiter-Sokolowski CT, and Graier WF (2022). Cristae junction as a fundamental switchboard for mitochondrial ion signaling and bioenergetics. Cell Calcium 101, 102517. 10.1016/j.ceca.2021.102517. [DOI] [PubMed] [Google Scholar]
- 223.Brookes PS, Yoon Y, Robotham JL, Anders MW, and Sheu SS (2004). Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 287, C817–C833. 10.1152/ajp-cell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 224.Sebastian C, and Mostoslavsky R. (2017). The various metabolic sources of histone acetylation. Trends Endocrinol. Metab. 28, 85–87. 10.1016/j.tem.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 225.Diehl KL, and Muir TW (2020). Chromatin as a key consumer in the metabolite economy. Nat. Chem. Biol. 16, 620–629. 10.1038/s41589-020-0517-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Dai Z, Ramesh V, and Locasale JW (2020). The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 21, 737–753. 10.1038/s41576-020-0270-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Santos JH (2021). Mitochondria signaling to the epigenome: a novel role for an old organelle. Free Radic. Biol. Med. 170, 59–69. 10.1016/j.freeradbiomed.2020.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Mentch SJ, and Locasale JW (2016). One-carbon metabolism and epigenetics: understanding the specificity. Ann. N. Y. Acad. Sci. 1363, 91–98. 10.1111/nyas.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bao XR, Ong SE, Goldberger O, Peng J, Sharma R, Thompson DA, Vafai SB, Cox AG, Marutani E, Ichinose F, et al. (2016). Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife 5, e10575. 10.7554/eLife.10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al. (2019). Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580. 10.1038/s41586-019-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Lepack AE, Werner CT, Stewart AF, Fulton SL, Zhong P, Farrelly LA, Smith ACW, Ramakrishnan A, Lyu Y, Bastle RM, et al. (2020). Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science 368, 197–201. 10.1126/science.aaw8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Graves SM, Xie Z, Stout KA, Zampese E, Burbulla LF, Shih JC, Kondapalli J, Patriarchi T, Tian L, Brichta L, et al. (2020). Dopamine metabolism by a monoamine oxidase mitochondrial shuttle activates the electron transport chain. Nat. Neurosci. 23, 15–20. 10.1038/s41593-019-0556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Huang H, Zhang D, Weng Y, Delaney K, Tang Z, Yan C, Qi S, Peng C, Cole PA, Roeder RG, and Zhao Y. (2021). The regulatory enzymes and protein substrates for the lysine beta-hydroxybutyrylation pathway. Sci. Adv. 7, eabe2771. 10.1126/sciadv.abe2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kopinski PK, Janssen KA, Schaefer PM, Trefely S, Perry CE, Potluri P, Tintos-Hernandez JA, Singh LN, Karch KR, Campbell SL, et al. (2019). Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy. Proc. Natl. Acad. Sci. USA 116, 16028–16035. 10.1073/pnas.1906896116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Lozoya OA, Wang T, Grenet D, Wolfgang TC, Sobhany M, Ganini da Silva D, Riadi G, Chandel N, Woychik RP, and Santos JH (2019). Mitochondrial acetyl-CoA reversibly regulates locus-specific histone acetylation and gene expression. Life Sci. Alliance 2, e201800228. 10.26508/lsa.201800228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sturm G, Karan KR, Monzel A, Santhanam BS, Taivassalo T, Bris C, Ware SA, Cross M, Towheed A, Higgins-Chen A, et al. (2021). OxPhos dysfunction causes hypermetabolism and reduces lifespan in cells and in patients with mitochondrial diseases. Preprint at bioRxiv. 10.1101/2021.11.29.470428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Sturm G, Monzel AS, Karan KR, Michelson J, Ware SA, Cardenas A, Lin J, Bris C, Santhanam B, Murphy MP, et al. (2022). A multi-omics and bioenergetics longitudinal aging dataset in primary human fibroblasts with mitochondrial perturbations. Preprint at bioRxiv. 10.1101/2021.11.12.468448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Kafkia E, Andres-Pons A, Ganter K, Seiler M, Smith TS, Andrejeva A, Jouhten P, Pereira F, Franco C, Kuroshchenkova A, et al. (2022). Operation of a TCA cycle subnetwork in the mammalian nucleus. Sci. Adv. 8, eabq5206. 10.1126/sciadv.abq5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent E, Loginicheva E, Cervantes-Barragan L, Ma X, Huang SC, Griss T, et al. (2016). Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 24, 158–166. 10.1016/j.cmet.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, Korenfeld D, Mathyer ME, Kim H, Huang LH, et al. (2018). Electrophilic properties of itaconate and derivatives regulate the IκBζ–ATF3 inflammatory axis. Nature 556, 501–504. 10.1038/s41586-018-0052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Intlekofer AM, Wang B, Liu H, Shah H, Carmona-Fontaine C, Rustenburg AS, Salah S, Gunner MR, Chodera JD, Cross JR, and Thompson CB (2017). L-2-hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat. Chem. Biol. 13, 494–500. 10.1038/nchembio.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Colvin H, Nishida N, Konno M, Haraguchi N, Takahashi H, Nishimura J, Hata T, Kawamoto K, Asai A, Tsunekuni K, et al. (2016). Oncometabolite D-2-hydroxyglurate directly induces epithelial-mesenchymal transition and is associated with distant metastasis in colorectal cancer. Sci. Rep. 6, 36289. 10.1038/srep36289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, You J, Chia GS, Sim J, Doedens A, et al. (2016). S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature 540, 236–241. 10.1038/nature20165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Funai K, Summers SA, and Rutter J. (2020). Reign in the membrane: how common lipids govern mitochondrial function. Curr. Opin. Cell Biol. 63, 162–173. 10.1016/j.ceb.2020.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Dudek J. (2017). Role of cardiolipin in mitochondrial signaling pathways. Front. Cell Dev. Biol. 5, 90. 10.3389/fcell.2017.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Pouikli A, and Tessarz P. (2021). Metabolism and chromatin: a dynamic duo that regulates development and ageing. Bioessays 43, e2000273. 10.1002/bies.202000273. [DOI] [PubMed] [Google Scholar]
- 247.Murphy MP, and Chouchani ET (2022). Why succinate? Physiological regulation by a mitochondrial coenzyme Q sentinel. Nat. Chem. Biol. 18, 461–469. 10.1038/s41589-022-01004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Mills EL, Harmon C, Jedrychowski MP, Xiao H, Garrity R, Tran NV, Bradshaw GA, Fu A, Szpyt J, Reddy A, et al. (2021). UCP1 governs liver extracellular succinate and inflammatory pathogenesis. Nat. Metab. 3, 604–617. 10.1038/s42255-021-00389-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Gudgeon N, Munford H, Bishop EL, Hill J, Fulton-Ward T, Bending D, Roberts J, Tennant DA, and Dimeloe S. (2022). Succinate uptake by T cells suppresses their effector function via inhibition of mitochondrial glucose oxidation. Cell Rep. 40, 111193. 10.1016/j.celrep.2022.111193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Paul MA, Love RJ, Jetly R, Richardson JD, Lanius RA, Miller JC, MacDonald M, and Rhind SG (2019). Blunted nocturnal salivary melatonin secretion profiles in military-related posttraumatic stress disorder. Front. Psychiatry 10, 882. 10.3389/fpsyt.2019.00882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Gandhi A, Mosser EA, Oikonomou G, and Prober D. (2015). Melatonin is required for the circadian regulation of sleep. Neuron 85, 1193–1199. 10.1016/j.neuron.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.van Geijlswijk IM, Korzilius HPLM, and Smits MG (2010). The use of exogenous melatonin in delayed sleep phase disorder: a meta-analysis. Sleep 33, 1605–1614. 10.1093/sleep/33.12.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Cipolla-Neto J, and Amaral F.G.d. (2018). Melatonin as a hormone: new physiological and clinical insights. Endocr. Rev. 39, 990–1028. 10.1210/er.2018-00084. [DOI] [PubMed] [Google Scholar]
- 254.Lambert AJ, and Brand MD (2009). Reactive oxygen species production by mitochondria. Methods Mol. Biol. 554, 165–181. 10.1007/978-1-59745-521-3_11. [DOI] [PubMed] [Google Scholar]
- 255.Desai R, East DA, Hardy L, Faccenda D, Rigon M, Crosby J, Alvarez MS, Singh A, Mainenti M, Hussey LK, et al. (2020). Mitochondria form contact sites with the nucleus to couple prosurvival retrograde response. Sci. Adv. 6, eabc9955. 10.1126/sciadv.abc9955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Murphy MP, Bayir H, Belousov V, Chang CJ, Davies KJA, Davies MJ, Dick TP, Finkel T, Forman HJ, Janssen-Heininger Y, et al. (2022). Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 4, 651–662. 10.1038/s42255-022-00591-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Chouchani ET, Kazak L, Jedrychowski MP, Lu GZ, Erickson BK, Szpyt J, Pierce KA, Laznik-Bogoslavski D, Vetrivelan R, Clish CB, et al. (2016). Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 532, 112–116. 10.1038/nature17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Shum M, Shintre CA, Althoff T, Gutierrez V, Segawa M, Saxberg AD, Martinez M, Adamson R, Young MR, Faust B, et al. (2021). ABCB10 exports mitochondrial biliverdin, driving metabolic maladaptation in obesity. Sci. Transl. Med. 13, eabd1869. 10.1126/scitranslmed.abd1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, Reece JM, Deeney JT, Andersen ME, Corkey BE, and Collins S. (2007). Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 56, 1783–1791. 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- 260.Vizioli MG, Liu T, Miller KN, Robertson NA, Gilroy K, Lagnado AB, Perez-Garcia A, Kiourtis C, Dasgupta N, Lei X, et al. (2020). Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 34, 428–445. 10.1101/gad.331272.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Qian W, Kumar N, Roginskaya V, Fouquerel E, Opresko PL, Shiva S, Watkins SC, Kolodieznyi D, Bruchez MP, and Van Houten B. (2019). Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction. Proc. Natl. Acad. Sci. USA 116, 18435–18444. 10.1073/pnas.1910574116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Cox CS, McKay SE, Holmbeck MA, Christian BE, Scortea AC, Tsay AJ, Newman LE, and Shadel GS (2018). Mitohormesis in mice via sustained basal activation of mitochondrial and antioxidant signaling. Cell Metab. 28, 776–786.e5. 10.1016/j.cmet.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Lozoya OA, Xu F, Grenet D, Wang T, Grimm SA, Godfrey V, Waidyanatha S, Woychik RP, and Santos JH (2020). Single nucleotide resolution analysis reveals pervasive, long-lasting DNA methylation changes by developmental exposure to a mitochondrial toxicant. Cell Rep. 32, 108131. 10.1016/j.celrep.2020.108131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Nelson G, Kucheryavenko O, Wordsworth J, and von Zglinicki T. (2018). The senescent bystander effect is caused by ROS-activated NF-kB signalling. Mech. Ageing Dev. 170, 30–36. 10.1016/j.mad.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, et al. (2016). Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 35, 724–742. 10.15252/embj.201592862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.da Silva PFL, Ogrodnik M, Kucheryavenko O, Glibert J, Miwa S, Cameron K, Ishaq A, Saretzki G, Nagaraja-Grellscheid S, Nelson G, and von Zglinicki T. (2019). The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell 18, e12848. 10.1111/acel.12848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Zhang Q, Wu X, Chen P, Liu L, Xin N, Tian Y, and Dillin A. (2018). The mitochondrial unfolded protein response is mediated cell-non-autonomously by retromer-dependent Wnt signaling. Cell 174, 870–883.e17. 10.1016/j.cell.2018.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Fu ZX, Tan X, Fang H, Lau PM, Wang X, Cheng H, and Bi GQ (2017). Dendritic mitoflash as a putative signal for stabilizing long-term synaptic plasticity. Nat. Commun. 8, 31. 10.1038/s41467-017-00043-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Horn A, Raavicharla S, Shah S, Cox D, and Jaiswal JK (2020). Mitochondrial fragmentation enables localized signaling required for cell repair. J. Cell Biol. 219, e201909154. 10.1083/jcb.201909154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Miller WL (1988). Molecular biology of steroid hormone synthesis. Endocr. Rev. 9, 295–318. 10.1210/edrv-9-3-295. [DOI] [PubMed] [Google Scholar]
- 271.Reddy DS (2010). Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog. Brain Res. 186, 113–137. 10.1016/B978-0-444-53630-3.00008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Crivello JF, and Jefcoate CR (1980). Intracellular movement of cholesterol in rat adrenal cells. Kinetics and effects of inhibitors. J. Biol. Chem. 255, 8144–8151. 10.1016/s0021-9258(19)70620-6. [DOI] [PubMed] [Google Scholar]
- 273.Bose HS, Lingappa VR, and Miller WL (2002). The steroidogenic acute regulatory protein, StAR, works only at the outer mitochondrial membrane. Endocr. Res. 28, 295–308. 10.1081/erc-120016800. [DOI] [PubMed] [Google Scholar]
- 274.Bose HS, Lingappa VR, and Miller WL (2002). Rapid regulation of steroidogenesis by mitochondrial protein import. Nature 417, 87–91. 10.1038/417087a. [DOI] [PubMed] [Google Scholar]
- 275.Prasad M, Kaur J, Pawlak KJ, Bose M, Whittal RM, and Bose HS (2015). Mitochondria-associated endoplasmic reticulum membrane (MAM) regulates steroidogenic activity via steroidogenic acute regulatory protein (StAR)-voltage-dependent anion channel 2 (VDAC2) interaction. J. Biol. Chem. 290, 2604–2616. 10.1074/jbc.M114.605808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Rajapaksha M, Kaur J, Prasad M, Pawlak KJ, Marshall B, Perry EW, Whittal RM, and Bose HS (2016). An outer mitochondrial translocase, Tom22, is crucial for inner mitochondrial steroidogenic regulation in adrenal and gonadal tissues. Mol. Cell Biol. 36, 1032–1047. 10.1128/MCB.01107-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Papadopoulos V, and Miller WL (2012). Role of mitochondria in steroidogenesis. Best Pract. Res. Clin. Endocrinol. Metab. 26, 771–790. 10.1016/j.beem.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 278.Black SM, Harikrishna JA, Szklarz GD, and Miller WL (1994). The mitochondrial environment is required for activity of the cholesterol side-chain cleavage enzyme, cytochrome P450scc. Proc. Natl. Acad. Sci. USA 91, 7247–7251. 10.1073/pnas.91.15.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Meimaridou E, Kowalczyk J, Guasti L, Hughes CR, Wagner F, Frommolt P, Nurnberg P, Mann NP, Banerjee R, Saka HN, et al. (2012). Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat. Genet. 44, 740–742. 10.1038/ng.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Riley JS, and Tait SW (2020). Mitochondrial DNA in inflammation and immunity. EMBO Rep. 21, e49799. 10.15252/embr.201949799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.West AP, and Shadel GS (2017). Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 17, 363–375. 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Xu H, He X, Zheng H, Huang LJ, Hou F, Yu Z, de la Cruz MJ, Borkowski B, Zhang X, Chen ZJ, and Jiang QX (2014). Structural basis for the prion-like MAVS filaments in antiviral innate immunity. eLife 3, e01489. 10.7554/eLife.01489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Seth RB, Sun L, Ea CK, and Chen ZJ (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 122, 669–682. 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 284.Koshiba T, Yasukawa K, Yanagi Y, and Kawabata S.i. (2011). Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci. Signal. 4, ra7. 10.1126/scisignal.2001147. [DOI] [PubMed] [Google Scholar]
- 285.Marchi S, Guilbaud E, Tait SWG, Yamazaki T, and Galluzzi L. (2022). Mitochondrial control of inflammation. Nat. Rev. Immunol. 10.1038/s41577-022-00760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, Zhu J, Yoon HE, Wang X, Kerkhofs M, et al. (2019). VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 366, 1531–1536. 10.1126/science.aav4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Xian H, Watari K, Sanchez-Lopez E, Offenberger J, Onyuru J, Sampath H, Ying W, Hoffman HM, Shadel GS, and Karin M. (2022). Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 55, 1370–1385.e8. 10.1016/j.immuni.2022.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, Geoghegan ND, Chappaz S, Davidson S, San Chin H, et al. (2018). BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359, eaao6047. 10.1126/science.aao6047. [DOI] [PubMed] [Google Scholar]
- 289.Tigano M, Vargas DC, Tremblay-Belzile S, Fu Y, and Sfeir A. (2021). Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 591, 477–481. 10.1038/s41586-021-03269-w. [DOI] [PubMed] [Google Scholar]
- 290.Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rotig A, Crow YJ, Rice GI, Duffy D, Tamby C, et al. (2018). Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242. 10.1038/s41586-018-0363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Vincent AE, Ng YS, White K, Davey T, Mannella C, Falkous G, Feeney C, Schaefer AM, McFarland R, Gorman GS, et al. (2016). The spectrum of mitochondrial ultrastructural defects in mitochondrial myopathy. Sci. Rep. 6, 30610. 10.1038/srep30610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Gaziev AI, and Shaikhaev GO (2010). [Nuclear mitochondrial pseudo-genes]. Mol. Biol. (Mosk). 44, 358–368. 10.1134/s0026893310030027. [DOI] [PubMed] [Google Scholar]
- 293.Quammen D. (2018). The Tangled Tree: A Radical New History of Life (Simon & Schuster; ). [DOI] [PubMed] [Google Scholar]
- 294.Bock R. (2017). Witnessing genome evolution: experimental reconstruction of endosymbiotic and horizontal gene transfer. Annu. Rev. Genet. 51, 1–22. 10.1146/annurev-genet-120215-035329. [DOI] [PubMed] [Google Scholar]
- 295.Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, and Langer T. (2014). The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 204, 919–929. 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Cheng X, and Ivessa AS (2010). The migration of mitochondrial DNA fragments to the nucleus affects the chronological aging process of Saccharomyces cerevisiae. Aging Cell 9, 919–923. 10.1111/j.1474-9726.2010.00607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Srinivasainagendra V, Sandel MW, Singh B, Sundaresan A, Mooga VP, Bajpai P, Tiwari HK, and Singh KK (2017). Migration of mitochondrial DNA in the nuclear genome of colorectal adenocarcinoma. Genome Med. 9, 31. 10.1186/s13073-017-0420-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Ju YS, Tubio JM, Mifsud W, Fu B, Davies HR, Ramakrishna M, Li Y, Yates L, Gundem G, Tarpey PS, et al. (2015). Frequent somatic transfer of mitochondrial DNA into the nuclear genome of human cancer cells. Genome Res. 25, 814–824. 10.1101/gr.190470.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, and Letellier T. (2003). Mitochondrial threshold effects. Biochem. J. 370, 751–762. 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Trumpff C, Michelson J, Lagranha CJ, Taleon V, Karan KR, Sturm G, Lindqvist D, Fernstrom J, Moser D, Kaufman BA, and Picard M. (2021). Stress and circulating cell-free mitochondrial DNA: a systematic review of human studies, physiological considerations, and technical recommendations. Mitochondrion 59, 225–245. 10.1016/j.mito.2021.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Rosa HS, Ajaz S, Gnudi L, and Malik AN (2020). A case for measuring both cellular and cell-free mitochondrial DNA as a disease biomarker in human blood. FASEB J. 34, 12278–12288. 10.1096/fj.202000959RR. [DOI] [PubMed] [Google Scholar]
- 302.Zhao X, Yu M, Zhao Y, Zheng Y, Meng L, Du K, Xie Z, Lv H, Zhang W, Liu J, et al. (2022). Circulating cell-free mtDNA release is associated with the activation of cGAS-STING pathway and inflammation in mitochondrial diseases. J. Neurol. 269, 4985–4996. 10.1007/s00415-022-11146-3. [DOI] [PubMed] [Google Scholar]
- 303.Trifunov S, Paredes-Fuentes AJ, Badosa C, Codina A, Montoya J, Ruiz-Pesini E, Jou C, Garrabou G, Grau-Junyent JM, Yubero D, et al. (2021). Circulating cell-free mitochondrial DNA in cerebrospinal fluid as a biomarker for mitochondrial diseases. Clin. Chem. 67, 1113–1121. 10.1093/clinchem/hvab091. [DOI] [PubMed] [Google Scholar]
- 304.Maresca A, Del Dotto V, Romagnoli M, La Morgia C, Di Vito L, Capristo M, Valentino ML, Carelli V, and Group E-MS (2020). Expanding and validating the biomarkers for mitochondrial diseases. J. Mol. Med. (Berl.) 98, 1467–1478. 10.1007/s00109-020-01967-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Cushen SC, Sprouse ML, Blessing A, Sun J, Jarvis SS, Okada Y, Fu Q, Romero SA, Phillips NR, and Goulopoulou S. (2020). Cell-free mitochondrial DNA increases in maternal circulation during healthy pregnancy: a prospective, longitudinal study. Am. J. Physiol. Regul. Integr. Comp. Physiol. 318, R445–R452. 10.1152/ajpregu.00324.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Hummel EM, Hessas E, Muller S, Beiter T, Fisch M, Eibl A, Wolf OT, Giebel B, Platen P, Kumsta R, and Moser DA (2018). Cell-free DNA release under psychosocial and physical stress conditions. Transl. Psychiatry 8, 236. 10.1038/s41398-018-0264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Trumpff C, Marsland AL, Basualto-Alarcon C, Martin JL, Carroll JE, Sturm G, Vincent AE, Mosharov EV, Gu Z, Kaufman BA, and Picard M. (2019). Acute psychological stress increases serum circulating cell-free mitochondrial DNA. Psychoneuroendocrinology 106, 268–276. 10.1016/j.psyneuen.2019.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Lindqvist D, Fernstrom J, Grudet C, Ljunggren L, Traskman-Bendz L, Ohlsson L, and Westrin A. (2016). Increased plasma levels of circulating cell-free mitochondrial DNA in suicide attempters: associations with HPA-axis hyperactivity. Transl. Psychiatry 6, e971. 10.1038/tp.2016.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Trumpff C, Rausser S, Haahr R, Karan KR, Gouspillou G, Puterman E, Kirschbaum C, and Picard M. (2022). Dynamic behavior of cell-free mitochondrial DNA in human saliva. Psychoneuroendocrinology 143, 105852. 10.1016/j.psyneuen.2022.105852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Nakahira K, Kyung SY, Rogers AJ, Gazourian L, Youn S, Massaro AF, Quintana C, Osorio JC, Wang Z, Zhao Y, et al. (2013). Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med. 10, e1001577. 10.1371/journal.pmed.1001577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Boyapati RK, Tamborska A, Dorward DA, and Ho GT (2017). Advances in the understanding of mitochondrial DNA as a pathogenic factor in inflammatory diseases. F1000Res. 6, 169. 10.12688/f1000research.10397.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Song X, Hu W, Yu H, Wang H, Zhao Y, Korngold R, and Zhao Y. (2020). Existence of circulating mitochondria in human and animal peripheral blood. Int. J. Mol. Sci. 21, 2122. 10.3390/ijms21062122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Al Amir Dache Z, Otandault A, Tanos R, Pastor B, Meddeb R, Sanchez C, Arena G, Lasorsa L, Bennett A, Grange T, et al. (2020). Blood contains circulating cell-free respiratory competent mitochondria. FASEB J. 34, 3616–3630. 10.1096/fj.201901917RR. [DOI] [PubMed] [Google Scholar]
- 314.Caicedo A, Zambrano K, Sanon S, and Gavilanes AW (2021). Extracellular mitochondria in the cerebrospinal fluid (CSF): potential types and key roles in central nervous system (CNS) physiology and pathogenesis. Mitochondrion 58, 255–269. 10.1016/j.mito.2021.02.006. [DOI] [PubMed] [Google Scholar]
- 315.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, and Hoogenraad NJ (2002). A mitochondrial specific stress response in mammalian cells. EMBO J. 21, 4411–4419. 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Carreras-Sureda A, Kroemer G, Cardenas JC, and Hetz C. (2022). Balancing energy and protein homeostasis at ER-mitochondria contact sites. Sci. Signal. 15, eabm7524. 10.1126/scisignal.abm7524. [DOI] [PubMed] [Google Scholar]
- 317.Fessler E, Eckl EM, Schmitt S, Mancilla IA, Meyer-Bender MF, Hanf M, Philippou-Massier J, Krebs S, Zischka H, and Jae LT (2020). A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 579, 433–437. 10.1038/s41586-020-2076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YHT, Wiita AP, Xu K, Correia MA, and Kampmann M. (2020). Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 579, 427–432. 10.1038/s41586-020-2078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Quiros PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, Gygi SP, and Auwerx J. (2017). Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 216, 2027–2045. 10.1083/jcb.201702058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Fiorese C, Schulz A, Lin YF, Rosin N, Pellegrino M, and Haynes C. (2016). The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 26, 2037–2043. 10.1016/j.cub.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Molenaars M, Janssens GE, Williams EG, Jongejan A, Lan J, Rabot S, Joly F, Moerland PD, Schomakers BV, Lezzerini M, et al. (2020). A conserved mito-cytosolic translational balance links two longevity pathways. Cell Metab. 31, 549–563.e7. 10.1016/j.cmet.2020.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Replogle JM, Saunders RA, Pogson AN, Hussmann JA, Lenail A, Guna A, Mascibroda L, Wagner EJ, Adelman K, Lithwick-Yanai G, et al. (2022). Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq. Cell 185, 2559–2575.e28. 10.1016/j.cell.2022.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Dogan S, Pujol C, Maiti P, Kukat A, Wang S, Hermans S, Senft K, Wibom R, Rugarli E, and Trifunovic A. (2014). Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 19, 458–469. 10.1016/j.cmet.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 324.Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim DH, Hur KY, Kim HK, et al. (2013). Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 19, 83–92. 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- 325.Keipert S, Ost M, Johann K, Imber F, Jastroch M, van Schothorst EM, Keijer J, and Klaus S. (2014). Skeletal muscle mitochondrial uncoupling drives endocrine cross-talk through the induction of FGF21 as a myokine. Am. J. Physiol. Endocrinol. Metab. 306, E469–E482. 10.1152/ajpendo.00330.2013. [DOI] [PubMed] [Google Scholar]
- 326.Mick E, Titov DV, Skinner OS, Sharma R, Jourdain AA, and Mootha VK (2020). Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. eLife 9, e49178. 10.7554/eLife.49178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Kang SG, Choi MJ, Jung SB, Chung HK, Chang JY, Kim JT, Kang YE, Lee JH, Hong HJ, Jun SM, et al. (2021). Differential roles of GDF15 and FGF21 in systemic metabolic adaptation to the mitochondrial integrated stress response. iScience 24, 102181. 10.1016/j.isci.2021.102181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Ost M, Coleman V, Voigt A, van Schothorst EM, Keipert S, van der Stelt I, Ringel S, Graja A, Ambrosi T, Kipp AP, et al. (2016). Muscle mitochondrial stress adaptation operates independently of endogenous FGF21 action. Mol. Metabol. 5, 79–90. 10.1016/j.molmet.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Chung HK, Ryu D, Kim KS, Chang JY, Kim YK, Yi HS, Kang SG, Choi MJ, Lee SE, Jung SB, et al. (2017). Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J. Cell Biol. 216, 149–165. 10.1083/jcb.201607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Croon M, Szczepanowska K, Popovic M, Lienkamp C, Senft K, Brandscheid CP, Bock T, Gnatzy-Feik L, Ashurov A, Acton RJ, et al. (2022). FGF21 modulates mitochondrial stress response in cardiomyocytes only under mild mitochondrial dysfunction. Sci. Adv. 8, eabn7105. 10.1126/sciadv.abn7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Luan HH, Wang A, Hilliard BK, Carvalho F, Rosen CE, Ahasic AM, Herzog EL, Kang I, Pisani MA, Yu S, et al. (2019). GDF15 is an inflammation-induced central mediator of tissue tolerance. Cell 178, 1231–1244.e11. 10.1016/j.cell.2019.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Choi MJ, Jung SB, Lee SE, Kang SG, Lee JH, Ryu MJ, Chung HK, Chang JY, Kim YK, Hong HJ, et al. (2020). An adipocyte-specific defect in oxidative phosphorylation increases systemic energy expenditure and protects against diet-induced obesity in mouse models. Diabetologia 63, 837–852. 10.1007/s00125-019-05082-7. [DOI] [PubMed] [Google Scholar]
- 333.Ost M, Igual Gil C, Coleman V, Keipert S, Efstathiou S, Vidic V, Weyers M, and Klaus S. (2020). Muscle-derived GDF15 drives diurnal anorexia and systemic metabolic remodeling during mitochondrial stress. EMBO Rep. 21, e48804. 10.15252/embr.201948804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Mullican SE, Lin-Schmidt X, Chin CN, Chavez JA, Furman JL, Armstrong AA, Beck SC, South VJ, Dinh TQ, Cash-Mason TD, et al. (2017). GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 23, 1150–1157. 10.1038/nm.4392. [DOI] [PubMed] [Google Scholar]
- 335.Moon JS, Goeminne LJE, Kim JT, Tian JW, Kim SH, Nga HT, Kang SG, Kang BE, Byun JS, Lee YS, et al. (2020). Growth differentiation factor 15 protects against the aging-mediated systemic inflammatory response in humans and mice. Aging Cell 19, e13195. 10.1111/acel.13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Klaus S, and Ost M. (2020). Mitochondrial uncoupling and longevity - A role for mitokines? Exp. Gerontol. 130, 110796. 10.1016/j.exger.2019.110796. [DOI] [PubMed] [Google Scholar]
- 337.Lockhart SM, Saudek V, and O’Rahilly S. (2020). GDF15: a hormone conveying somatic distress to the brain. Endocr. Rev. 41, bnaa007. 10.1210/endrev/bnaa007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Gil CI, Coull BM, Jonas W, Lippert R, Ost M, and Klaus S. (2021). Mitochondrial stress-induced GDF15-GFRAL axis promotes anxiety-like behavior and CRH-dependent anorexia. bioRxiv. 10.1101/2021.09.22.461199. [DOI] [Google Scholar]
- 339.Cimino I, Kim H, Tung YCL, Pedersen K, Rimmington D, Tadross JA, Kohnke SN, Neves-Costa A, Barros A, Joaquim S, et al. (2021). Activation of the hypothalamic-pituitary-adrenal axis by exogenous and endogenous GDF15. Proc. Natl. Acad. Sci. USA 118. e2106868118. 10.1073/pnas.2106868118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Yatsuga S, Fujita Y, Ishii A, Fukumoto Y, Arahata H, Kakuma T, Kojima T, Ito M, Tanaka M, Saiki R, and Koga Y. (2015). Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann. Neurol. 78, 814–823. 10.1002/ana.24506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Lehtonen JM, Forsstrom S, Bottani E, Viscomi C, Baris OR, Isoniemi H, Hockerstedt K, Osterlund P, Hurme M, Jylhava J, et al. (2016). FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology 87, 2290–2299. 10.1212/WNL.0000000000003374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Lin Y, Ji K, Ma X, Liu S, Li W, Zhao Y, and Yan C. (2020). Accuracy of FGF-21 and GDF-15 for the diagnosis of mitochondrial disorders: a meta-analysis. Ann. Clin. Transl. Neurol. 7, 1204–1213. 10.1002/acn3.51104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Reynolds JC, Bwiza CP, and Lee C. (2020). Mitonuclear genomics and aging. Hum. Genet. 139, 381–399. 10.1007/s00439-020-02119-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, et al. (2001). A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Ab. Proc. Natl. Acad. Sci. USA 98, 6336–6341. 10.1073/pnas.101133498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Yen K, Mehta HH, Kim SJ, Lue Y, Hoang J, Guerrero N, Port J, Bi Q, Navarrete G, Brandhorst S, et al. (2020). The mitochondrial derived peptide humanin is a regulator of lifespan and healthspan. Aging (Albany NY) 12, 11185–11199. 10.18632/aging.103534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Cobb LJ, Lee C, Xiao J, Yen K, Wong RG, Nakamura HK, Mehta HH, Gao Q, Ashur C, Huffman DM, et al. (2016). Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging (Albany NY) 8, 796–809. 10.18632/aging.100943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Lee C, Zeng J, Drew B, Sallam T, Martin-Montalvo A, Wan J, Kim SJ, Mehta H, Hevener A, de Cabo R, and Cohen P. (2015). The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 21, 443–454. 10.1016/j.cmet.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Kang GM, Min SH, Lee CH, Kim JY, Lim HS, Choi MJ, Jung SB, Park JW, Kim S, Park CB, et al. (2021). Mitohormesis in hypothalamic POMC neurons mediates regular exercise-induced high-turn-over metabolism. Cell Metab. 33, 334–349.e6. 10.1016/j.cmet.2021.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Reynolds JC, Lai RW, Woodhead JST, Joly JH, Mitchell CJ, Cameron-Smith D, Lu R, Cohen P, Graham NA, Benayoun BA, et al. (2021). MOTS-c is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis. Nat. Commun. 12, 470. 10.1038/s41467-020-20790-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Woodhead JST, D’Souza RF, Hedges CP, Wan J, Berridge MV, Cameron-Smith D, Cohen P, Hickey AJR, Mitchell CJ, and Merry TL (2020). High-intensity interval exercise increases humanin, a mitochondrial encoded peptide, in the plasma and muscle of men. J. Appl. Physiol. 128, 1346–1354. 10.1152/japplphysiol.00032.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Shamaei-Tousi A, Steptoe A, O’Donnell K, Palmen J, Stephens JW, Hurel SJ, Marmot M, Homer K, D’Aiuto F, Coates AR, et al. (2007). Plasma heat shock protein 60 and cardiovascular disease risk: the role of psychosocial, genetic, and biological factors. Cell Stress Chaperones 12, 384–392. 10.1379/csc-300.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Julian MW, Shao G, Bao S, Knoell DL, Papenfuss TL, VanGundy ZC, and Crouser ED (2012). Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. J. Immunol. 189, 433–443. 10.4049/jimmunol.1101375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Chaung WW, Wu R, Ji Y, Dong W, and Wang P. (2012). Mitochondrial transcription factor A is a proinflammatory mediator in hemorrhagic shock. Int. J. Mol. Med. 30, 199–203. 10.3892/ijmm.2012.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Clarke A, and Fraser KPP (2004). Why does metabolism scale with temperature? Funct. Ecol. 18, 243–251. 10.1111/j.0269-8463.2004.00841.x. [DOI] [Google Scholar]
- 355.Nicholls DG, and Locke RM (1984). Thermogenic mechanisms in brown fat. Physiol. Rev. 64, 1–64. 10.1152/physrev.1984.64.1.1. [DOI] [PubMed] [Google Scholar]
- 356.Xue K, Wu D, Wang Y, Zhao Y, Shen H, Yao J, Huang X, Li X, Zhou Z, Wang Z, and Qiu Y. (2022). The mitochondrial calcium uniporter engages UCP1 to form a thermoporter that promotes thermogenesis. Cell Metab. 34, 1325–1341.e6. e6. 10.1016/j.cmet.2022.07.011. [DOI] [PubMed] [Google Scholar]
- 357.Okabe K, Inada N, Gota C, Harada Y, Funatsu T, and Uchiyama S. (2012). Intracellular temperature mapping with a fluorescent polymeric thermometer and fluorescence lifetime imaging microscopy. Nat. Commun. 3, 705. 10.1038/ncomms1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Nakano M, Arai Y, Kotera I, Okabe K, Kamei Y, and Nagai T. (2017). Genetically encoded ratiometric fluorescent thermometer with wide range and rapid response. PLoS One 12, e0172344. 10.1371/journal.pone.0172344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Chretien D, Benit P, Ha HH, Keipert S, El-Khoury R, Chang YT, Jastroch M, Jacobs HT, Rustin P, and Rak M. (2018). Mitochondria are physiologically maintained at close to 50 ◦C. PLoS Biol. 16, e2003992. 10.1371/journal.pbio.2003992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Picard M. (2015). Mitochondrial synapses: intracellular communication and signal integration. Trends Neurosci. 38, 468–474. 10.1016/j.tins.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 361.Kumar M, Mommer MS, and Sourjik V. (2010). Mobility of cytoplasmic, membrane, and DNA-binding proteins in Escherichia coli. Biophys. J. 98, 552–559. 10.1016/j.bpj.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Kekenes-Huskey P, Liao T, Gillette A, Hake J, Zhang Y, Michailova A, McCulloch A, and McCammon J. (2013). Molecular and subcellular-scale modeling of nucleotide diffusion in the cardiac myofilament lattice. Biophys. J. 105, 2130–2140. 10.1016/j.bpj.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Cossins BP, Jacobson MP, and Guallar V. (2011). A new view of the bacterial cytosol environment. PLoS Comput. Biol. 7, e1002066. 10.1371/journal.pcbi.1002066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Courchet J, Lewis T, Lee S, Courchet V, Liou DY, Aizawa S, and Polleux F. (2013). Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell 153, 1510–1525. 10.1016/j.cell.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Faits MC, Zhang C, Soto F, and Kerschensteiner D. (2016). Dendritic mitochondria reach stable positions during circuit development. eLife 5, e11583. 10.7554/eLife.11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Lewis TL Jr., Kwon SK, Lee A, Shaw R, and Polleux F. (2018). MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat. Commun. 9, 5008. 10.1038/s41467-018-07416-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Konig T, Nolte H, Aaltonen MJ, Tatsuta T, Krols M, Stroh T, Langer T, and McBride HM (2021). MIROs and DRP1 drive mitochondrial-derived vesicle biogenesis and promote quality control. Nat. Cell Biol. 23, 1271–1286. 10.1038/s41556-021-00798-4. [DOI] [PubMed] [Google Scholar]
- 368.Brestoff JR, Wilen CB, Moley JR, Li Y, Zou W, Malvin NP, Rowen MN, Saunders BT, Ma H, Mack MR, et al. (2021). Intercellular mitochondria transfer to macrophages regulates white adipose tissue homeostasis and is impaired in obesity. Cell Metab. 33, 270–282.e8. 10.1016/j.cmet.2020.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Crewe C, Funcke JB, Li S, Joffin N, Gliniak CM, Ghaben AL, An YA, Sadek HA, Gordillo R, Akgul Y, et al. (2021). Extracellular vesicle-based interorgan transport of mitochondria from energetically stressed adipocytes. Cell Metab. 33, 1853–1868.e11. 10.1016/j.cmet.2021.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Rodriguez-Nuevo A, Torres-Sanchez A, Duran JM, De Guirior C, Martinez-Zamora MA, and Boke E. (2022). Oocytes maintain ROS-free mitochondrial metabolism by suppressing complex I. Nature 607, 756–761. 10.1038/s41586-022-04979-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. (2008). A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123. 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Fecher C, Trovo L, Muller SA, Snaidero N, Wettmarshausen J, Heink S, Ortiz O, Wagner I, Kuhn R, Hartmann J, et al. (2019). Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 22, 1731–1742. 10.1038/s41593-019-0479-z. [DOI] [PubMed] [Google Scholar]
- 373.Picard M, Hepple RT, and Burelle Y. (2012). Mitochondrial functional specialization in glycolytic and oxidative muscle fibers: tailoring the organelle for optimal function. Am. J. Physiol. Cell Physiol. 302, C629–C641. 10.1152/ajpcell.00368.2011. [DOI] [PubMed] [Google Scholar]
- 374.Rausser S, Trumpff C, McGill MA, Junker A, Wang W, Ho SH, Mitchell A, Karan KR, Monk C, Segerstrom SC, et al. (2021). Mitochondrial phenotypes in purified human immune cell subtypes and cell mixtures. eLife 10, e70899. 10.7554/eLife.70899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Glancy B, and Balaban RS (2011). Protein composition and function of red and white skeletal muscle mitochondria. Am. J. Physiol. Cell Physiol. 300, C1280–C1290. 10.1152/ajpcell.00496.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Rosenberg A, Saggar M, Rogu P, Limoges AW, Sandi C, Mosharov EV, Dumitriu D, Anacker C, and Picard P. (2021). Mouse brain-wide mitochondrial connectivity anchored in gene, brain and behavior. Preprint at bioRxiv. 10.1101/2021.06.02.446767. [DOI] [Google Scholar]
- 377.Ignatenko O, Chilov D, Paetau I, de Miguel E, Jackson CB, Capin G, Paetau A, Terzioglu M, Euro L, and Suomalainen A. (2018). Loss of mtDNA activates astrocytes and leads to spongiotic encephalopathy. Nat. Commun. 9, 70. 10.1038/s41467-017-01859-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Vincent AE, and Picard M. (2018). Multilevel heterogeneity of mitochondrial respiratory chain deficiency. J. Pathol. 246, 261–265. 10.1002/path.5146. [DOI] [PubMed] [Google Scholar]
- 379.Mito T, Vincent AE, Faitg J, Taylor RW, Khan NA, McWilliams TG, and Suomalainen A. (2022). Mosaic dysfunction of mitophagy in mitochondrial muscle disease. Cell Metab. 34, 197–208.e5. 10.1016/j.cmet.2021.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Stauch KL, Purnell PR, and Fox HS (2014). Quantitative proteomics of synaptic and nonsynaptic mitochondria: insights for synaptic mitochondrial vulnerability. J. Proteome Res. 13, 2620–2636. 10.1021/pr500295n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Ferreira R, Vitorino R, Alves RMP, Appell HJ, Powers SK, Duarte JA, and Amado F. (2010). Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics 10, 3142–3154. 10.1002/pmic.201000173. [DOI] [PubMed] [Google Scholar]
- 382.Benador IY, Veliova M, Mahdaviani K, Petcherski A, Wikstrom JD, Assali EA, Acin-Perez R, Shum M, Oliveira MF, Cinti S, et al. (2018). Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab. 27, 869–885.e6. 10.1016/j.cmet.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Sjostedt E, Zhong W, Fagerberg L, Karlsson M, Mitsios N, Adori C, Oksvold P, Edfors F, Limiszewska A, Hikmet F, et al. (2020). An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367, eaay5947. 10.1126/science.aay5947. [DOI] [PubMed] [Google Scholar]
- 384.Faitg J, Lacefield C, Davey T, White K, Laws R, Kosmidis S, Reeve AK, Kandel ER, Vincent AE, and Picard M. (2021). 3D neuronal mitochondrial morphology in axons, dendrites, and somata of the aging mouse hippocampus. Cell Rep. 36, 109509. 10.1016/j.celrep.2021.109509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Lewis T, Turi G, Kwon SK, Losonczy A, and Polleux F. (2016). Progressive decrease of mitochondrial motility during maturation of cortical axons in vitro and in vivo. Curr. Biol. 26, 2602–2608. 10.1016/j.cub.2016.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Wolf DM, Segawa M, Kondadi AK, Anand R, Bailey ST, Reichert AS, van der Bliek AM, Shackelford DB, Liesa M, and Shirihai OS (2019). Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J. 38, e101056. 10.15252/embj.2018101056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Berry BJ, Vodičková A, Müller-Eigner A, Meng C, Ludwig C, Kaeberlein M, Peleg S, and Wojtovich AP. (2022). Optogenetic rejuvenation of mitochondrial membrane potential extends C. elegans lifespan. Preprint at bioRxiv. 10.1101/2022.05.11.491574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Ignatenko O, Nikkanen J, Kononov A, Zamboni N, Ince-Dunn G, and Suomalainen A. (2020). Mitochondrial spongiotic brain disease: astrocytic stress and harmful rapamycin and ketosis effect. Life Sci. Alliance 3. e202000797. 10.26508/lsa.202000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, et al. (2013). mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342, 1524–1528. 10.1126/science.1244360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Picard M, Wallace DC, and Burelle Y. (2016). The rise of mitochondria in medicine. Mitochondrion 30, 105–116. 10.1016/j.mito.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Suomalainen A, and Battersby BJ (2018). Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 19, 77–92. 10.1038/nrm.2017.66. [DOI] [PubMed] [Google Scholar]
- 392.Chernet B, and Levin M. (2013). endogenous voltage potentials and the microenvironment: bioelectric signals that reveal, induce and normalize cancer. J. Clin. Exp. Oncol. Suppl 1, S1–S002. 10.4172/2324-9110.S1-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.López-Otín C, and Kroemer G. (2021). Hallmarks of health. Cell 184, 33–63. 10.1016/j.cell.2020.11.034. [DOI] [PubMed] [Google Scholar]
- 394.Chan DC (2020). Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. 15, 235–259. 10.1146/annurev-pathmechdis-012419-032711. [DOI] [PubMed] [Google Scholar]
- 395.Archer SL (2013). Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 369, 2236–2251. 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 396.Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, et al. (2019). Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68. 10.1038/s41586-019-1506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Drucker D. (2016). Never waste a good crisis: confronting reproducibility in translational research. Cell Metab. 24, 348–360. 10.1016/j.cmet.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 398.Enríquez JA (2019). Mind your mouse strain. Nat. Metab. 1, 5–7. 10.1038/s42255-018-0018-3. [DOI] [PubMed] [Google Scholar]
- 399.Sung HJ, Ma W, Wang PY, Hynes J, O’Riordan TC, Combs CA, McCoy JP Jr., Bunz F, Kang JG, and Hwang PM (2010). Mitochondrial respiration protects against oxygen-associated DNA damage. Nat. Commun. 1, 5. 10.1038/ncomms1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Lane N, and Martin W. (2010). The energetics of genome complexity. Nature 467, 929–934. 10.1038/nature09486. [DOI] [PubMed] [Google Scholar]
- 401.Schavemaker PE, and Munoz-Gomez SA (2022). The role of mitochondrial energetics in the origin and diversification of eukaryotes. Nat. Ecol. Evol. 6, 1307–1317. 10.1038/s41559-022-01833-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Ayres JS (2020). The biology of physiological health. Cell 181, 250–269. 10.1016/j.cell.2020.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Picard M, and McEwen BS (2018). Psychological stress and mitochondria: a conceptual framework. Psychosom. Med. 80, 126–140. 10.1097/PSY.0000000000000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Cole SW (2014). Human social genomics. PLoS Genet. 10, e1004601. 10.1371/journal.pgen.1004601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Snyder-Mackler N, Burger JR, Gaydosh L, Belsky DW, Noppert GA, Campos FA, Bartolomucci A, Yang YC, Aiello AE, O’Rand A, et al. (2020). Social determinants of health and survival in humans and other animals. Science 368, eaax9553. 10.1126/science.aax9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Picard M. (2022). Why do we care more about disease than health? Phenomics 2, 145–155. 10.1007/s43657-021-00037-8. [DOI] [PMC free article] [PubMed] [Google Scholar]