

Keywords: DNA methylation, environmental epigenetics, DMRs
Abstract
Evidence from human epidemiological studies suggests that exertional heat stroke (EHS) results in an elevated risk of long-term cardiovascular and systemic disease. Previous results using a preclinical mouse model of EHS demonstrated severe metabolic imbalances in ventricular myocardium developing at 9–14 days of recovery. Whether this resolves over time is unknown. We hypothesized that the long-term effects of EHS on the heart reflect retained maladaptive epigenetic responses. In this study, we evaluated genome-wide DNA methylation, RNA-Seq, and metabolomic profiles of the left ventricular myocardium in female C57BL/6 mice, 30 days after EHS (exercise in 37.5°C; n = 7–8), compared with exercise controls. EHS mice ran to loss of consciousness, reaching core temperatures of 42.4 ± 0.2°C. All mice recovered quickly. After 30 days, the left ventricles were rapidly frozen for DNA methyl sequencing, RNA-Seq, and untargeted metabolomics. Ventricular DNA from EHS mice revealed >13,000 differentially methylated cytosines (DMCs) and >900 differentially methylated regions (DMRs; ≥5 DMCs with ≤300 bp between each CpG). Pathway analysis using DMRs revealed alterations in genes regulating basic cell functions, DNA binding, transcription, and metabolism. Metabolomics and mRNA expression revealed modest changes that are consistent with a return to homeostasis. Methylation status did not predict RNA expression or metabolic state at 30 days. We conclude that EHS induces a sustained DNA methylation memory lasting over 30 days of recovery, but ventricular gene expression and metabolism return to a relative homeostasis at rest. Such long-lasting alterations to the DNA methylation landscape could alter responsiveness to environmental or clinical challenges later in life.
INTRODUCTION
Exertional heat stroke (EHS) is an acute and potentially lethal environmental stress that is characterized by elevated core temperature, neurological dysfunction, multiorgan damage, and systemic inflammatory responses (1–3). As such, EHS is the most severe form of exertional heat injury. The incidence of EHS in the general population is not well known. However, exertional heat injury, of which EHS is the most severe form, was reported in 75,000 individuals across the US emergency rooms in 2006, and the numbers have been steadily rising (4). In the active military, across the globe, the incidence of EHS is 0.2–10 per 1,000 person-years of exposure (5), and in the US military active service, there were between 475 and 600 cases per year between 2017 and 2021 (6). Mortality is generally low in the US Military with modern recovery and monitoring methods, estimated to be ∼2% of cases (7); however, mortality estimates as a result of EHS are much higher in the general, worldwide population (8).
Mounting clinical evidence has shown that some individuals surviving an EHS exposure have a greater risk of suffering long-term health complications. For example, epidemiological studies have described increased incidence of delayed-onset cardiovascular disease (9, 10), chronic kidney disease (9–11), and neurological syndromes (12), as well as earlier onset of all-cause mortality compared with suitably matched control groups (9, 10). However, in these retrospective epidemiological studies, it is not possible to separate whether individuals with propensities toward medical disorders are more likely to suffer heat stroke, or whether the exposure to heat stroke leads to decrements in long-term health outcomes.
Previously, we used a preclinical mouse model of EHS to investigate possible metabolic underpinnings of delayed heart pathology by quantifying the metabolomic profile of the myocardium from 30 min to 14 days of recovery following a single EHS exposure (13). The ventricular myocardium of female mice began to accumulate free fatty acids, ceramides, and diacylglycerols beginning at 9 days, a response characteristic of a number of myocardial disorders, including heart failure (14, 15). In addition, widespread abnormalities were seen in tricarboxylic acid cycle (TCA) intermediates, components of glycolysis, and purine metabolism as well as indications of increased oxidative stress and inflammation (13). Whether this slowly evolving pathological profile at 14 days persists even longer or eventually resolves is unknown. Therefore, in this study, we extended the recovery from EHS to 30 days to evaluate the progression of this metabolomic profile over time. We hypothesized that the mice would continue to show signs of metabolic dysfunction at this extended time point.
Recently, we investigated possible underlying molecular mechanisms for such long-term health consequences of EHS exposure. We previously discovered that EHS exposure in mice leads to significant long-term reprogramming of DNA methylation in immune cells at 4 and 30 days of recovery, which predicted an attenuated inflammatory response in whole blood, an outcome reminiscent of clinical observations (16, 17). Although epigenetic responses to stress stimuli can often lead to adaptations that favor health and survival (18–20), severe or persistent stressors can alternatively lead to cumulative maladaptive epigenetic responses (21, 22). We hypothesized that exposure to EHS in mice would create such a maladaptive epigenetic profile in the heart, and that this profile would contribute to, or predict the developing stress-induced long-term cardiovascular complications (13). To test these hypotheses, we quantified the reprogramming of the DNA methylome of the ventricular myocardium along with resting mRNA expression and resting metabolomics after 30 days of recovery from EHS in mice compared with sham exercise controls (EXC).
METHODS
Ethical Approval
All animal protocols were approved by the University of Florida Institutional Animal Care and Use Committee under protocol No. 201910745. We adhered to the “Guide for the Care and Use of Laboratory Animals” as prepared by the Committee for the Update of the Guide for the Care and Use of Laboratory Animals of the Institute for Laboratory Animal Research, National Research Council. Reported information about the procedures conformed to the “Animal Research Reporting of in vivo Experiments (ARRIVE) guidelines”(23). Throughout and following the EHS procedure, we carefully monitored animals for signs of dehydration, weight loss, impaired mobility, or physiological signs of underlying disorders such as labored breathing or respiratory distress. No animals had to be removed because of reaching these humane endpoints.
Animal Treatment and Surgical Procedures
In all, 16 C57BL6/J female mice were used (Jackson Laboratories, Bar Harbor, ME). Only female mice were used, as males exhibit a much lower heat tolerance and exercise duration during the identical EHS protocol (24), with little or no metabolomic crisis at 14 days (13). All mice were 8–10 wk of age upon arrival, making them ∼4 mo old at sample collection. A standard chow diet and water were provided ad libitum. Mice were housed in groups until they were implanted with telemetry devices, after which they were individually housed and maintained on a 12-h dark/12-h light cycle at 19°C–22°C and 30%–60% humidity.
Implant surgery for telemetry devices (G-2 E-mitter, Star Life Sciences, Oakmont, PA) to measure core temperature (Tc) was performed under isoflurane anesthesia (5.0% for induction; 1.5% for maintenance), as previously described (16, 25). Subcutaneous buprenorphine injections were given every 12 h for 48 h following surgery to minimize pain. The mice then recovered for 2 wks. Following recovery, in-cage running wheels (Model 0297-0521, Columbus Instruments, Columbus, OH), modified to fit our cages, were installed for voluntary exercise training. During the third week of wheel running, the mice were taken to the laboratory to undergo four familiarization/exercise trials using forced running wheels (Lafayette Instruments, #80840B, Lafayette, IN) on four separate days and then allowed to rest for 2 days before being subjected to the EHS or EXC protocol (16, 25).
Exertional Heat Stroke and Recovery
The EHS protocol began at ∼0800. A complete description of the protocol has been previously published (25). Briefly, the mouse cage was placed in an environmental chamber (Thermo Forma, 3940, Thermo Fisher, Waltham, MA) set to 37.5°C and ≤ 40% relative humidity. After equilibration (∼30 min), mice were quickly placed in an enclosed forced running wheel. Once the Tc of the mice stabilized at 36–37.5°C (∼5 min), the forced running wheel protocol was initiated and the EHS trial begun. EXC mice were run simultaneously and matched for both intensity and duration in a twin incubator at 22°C. The sessions were terminated at “symptom limitation” when the EHS mice exhibited apparent loss of consciousness and motor control. After EHS, the mice were quickly weighed and remained in their respective chamber, in their own housing for 2 h, while being kept at 37.5° (16). They continued to be monitored for 12 h before being returned to the vivarium. Both EHS and EXC mice were then allowed to recover in the vivarium for 30 days with standard care.
At the time of tissue sample collection, mice were anesthetized with isoflurane and euthanized by thoracotomy and removal of the heart. The heart was quickly washed in sterile, ice-cold phosphate-buffered saline (PBS) and kept on ice while the left ventricle was isolated. The left ventricles were divided into two components, and then each component was frozen immediately in liquid nitrogen and stored at −80°C.
Metabolomic Analyses of Ventricular Cardiac Muscle
Frozen heart ventricles (n = 7/group) were sent for metabolomics analysis to Metabolon, Inc. (Durham, NC). One tissue sample in each group was excluded due to technical difficulties during tissue digestion. The samples were prepared and analyzed using Metabolon’s standard protocols for their HD4 platform using gas chromatography/mass spectrometry. Briefly, samples were extracted by rapid cell disruption in methanol under vigorous shaking for 2 min (Glen Mills GenoGrinder 2000, Clifton, NJ) to precipitate protein and dissociate metabolites. The samples were centrifuged and the extract was divided into five fractions: two for analysis by two separate reverse phase (RP)/UPLC-MS/MS (ultrahigh performance liquid chromatography-tandem mass spectrometry) methods using positive ion mode electrospray ionization (ESI), one for analysis by RP/UPLC-MS/MS using negative ion mode ESI, one for analysis by HILIC/UPLC-MS/MS (Hydrophilic UPLC-MS/MS) using negative ion mode ESI, and one reserved for backup. Samples were placed briefly on a TurboVap solvent evaporator (Zymark, Clackamas, OR) to concentrate the metabolites. A number of quality control standards were analyzed in concert with the experimental samples.
Proprietary software (Metabolon Inc.) was used to match ions to an in-house library of standards for metabolite identification and quantification. Peak areas were first normalized to eliminate batch effects and then each group of metabolites was tested for normality using the Shapiro–Wilk test. When one or more groups were nonparametric, comparisons were performed using the Mann–Whitney U test. For parametric data, equality of variances was tested using the F test and groups with unequal variances were tested using Welch’s modified T test, whereas groups with equal variances were tested using the Student’s t test. The raw P values were reported for each significant metabolite of interest, and Benjamini–Hochberg correction for multiple testing was also performed. We accepted a β < 20% for multiple testing based on recommendations for biological samples (26) compared to a more stringent cutoff of <0.05 that was used for genomics data. Power and effect size estimates were performed using G*Power (27). A total of 631 metabolites were identified from the available library. The total list of metabolites analyzed is reported in the supplement (see https://doi.org/10.6084/m9.figshare.20483664.v1).
Enzymatic Methyl Sequencing and Analysis for DNA Methylation
DNA was isolated from left ventricles (n = 4/group for sequencing) using the AllPrep DNA/RNA Mini Kit (Qiagen, #80204, Germantown, MD). DNA quality and concentration were assessed using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham MA), then flash-frozen in liquid nitrogen and stored at −80°C for whole genome methylation sequencing at a later time point. The individual DNA samples were sent to the University of Florida Interdisciplinary Center for Biotechnology Research (ICBR) NextGen DNA Sequencing core. Libraries were constructed according to the NEBNext Enzymatic Methyl-seq protocol (Cat. No. E7120S), using ∼200 ng genomic DNA. Samples were fragmented in a Covaris S220 ultrasonic disruptor to obtain DNA fragments of 300 bp average size. AMPure magnetic beads (Omega Biotek #M1386-01) with a 1:1 bead-to-sample ratio were used to purify the samples. Purified DNA fragments were end-repaired, adaptor-ligated, unmethylated cytosine bases converted to uracil (NEBNext EM-seq kit), and cleaned again with AMPure beads. Cleaned products for each sample were amplified and barcoded using the NEBNext Q5U Master Mix for four cycles, followed by purification with AMPure magnetic beads (0.8:1.0 beads/sample). Libraries were then examined for size (Agilent TapeStation; DNA5000 Screen Tape; #5067–5592) and quantified by Qubit fluorometry (Thermo Fisher). Typically, library yield was ∼0.6–1.0 µg (in 20 µL Tris-HCl, pH 8.0), with fragments ranging from 250 to 800 bp (mean ∼400 bp).
For sequencing, equimolar amounts of barcoded methyl-Seq libraries were pooled, followed by treatment with “Free Adapter Blocking Reagent” (Illumina Cat. No. 20024145) to minimize index-hopping rates. The library pool was diluted to 2.5 nM and sequenced (one S4 lane, 2 × 150 cycles) according to the XP (lane splitting) Illumina NovaSeq6000 sequencing protocol using 180 pM loading concentration. PhiX (20%) was spiked-in to increase sequence diversity. A total of 2.5–3.0 billion paired-end reads were obtained from a single S4 lane (Q30 ≥ 84%; clusters passing filter = 80.5%).
Resulting FASTQ files were trimmed for low-quality bases and adaptor sequences were removed using Trim-Galore! (28). Filtered reads were then aligned to the Mus musculus genome reference, build mm9, using “Bismark” software (29). Methylation levels were determined using Bismark’s methylation extractor helper function for every CpG site with at least 10× coverage, counting regions of mate-pair overlap only once.
Differentially methylated CpGs (DMCs) for each individual sample in each respective group (n = 4/group) were calculated using the R (30) package methylKit (31), which uses a logistic regression test to compare the fraction of methylated cytosines across the experimental and control groups. DMCs were determined to be significant if they had a read depth of at least 10× per sample, a difference in methylation of ≥ |10%| with a false discovery rate (FDR)-corrected P value (32), i.e., Q < 0.05. Differentially methylated regions (DMRs) were calculated using “metilene” software in de novo mode (33). A DMR was considered significant if it had ≥ 5 CpGs with a maximum distance of 300 bp between consecutive sites, a read depth of at least 10× per sample, a change in methylation of at least ±10%, a 2D-KS (Kolmogorov–Smirnov test) P ≤ 0.05, and an FDR-corrected Mann–Whitney U test, Q ≤ 0.05. Greater than 600,000 regions of the genome were tested for differential methylation and >74,000 individual CpGs exhibited significant (uncorrected) methylation changes.
All presented DMCs and DMRs were additionally confirmed visually using “big wig” files on Integrative Genomics Viewer v. 2.4.8. These “big wig” files were produced for each individual sample (n = 4/group); the files visualize the read coverage of each CpG and the fraction at which a given CpG was methylated over the total number of reads at a given CpG for each respective sample in each respective group.
RNA Sequencing Library Preparation
For mRNA isolation, parts of the remaining frozen ventricular samples from the same hearts used for DNA methylation analysis (n = 4/group) were separately homogenized using a Powergen 125 homogenizer (Cole-Parmer, Vernon Hills, IL). The RNA was isolated using the RNeasy Fibrous Tissue Mini Kit (Qiagen, Germantown MD) according to the manufacturer’s instructions. The RNA quality was tested initially using a Nanodrop spectrophotometry (Cole-Parmer, Vernon Hill, IL) at a wavelength ratio of 260/280 (all samples met a value ≥ 2.0) and later tested for integrity using an RNA integrity number (RIN) (all samples exceeded 7.0). The RNA samples were used to construct mRNA libraries by the University of Florida Interdisciplinary Center for Biotechnology Research, Gene Expression and Genotyping Core Lab using the NEBNext Ultra II Directional Prep (New England Biolabs). Each library was barcoded, then enriched by 13 cycles of amplification, and purified with Agencourt AMPure beads (Beckman Coulter). Equimolar amounts of each library were pooled and sequenced in paired-end mode (2 × 150 nt) on a NovaSeq 6000 sequencer.
FASTQ reads were filtered based on sequencing quality, and contaminating adaptor sequences were trimmed using Trim-Galore! with default settings (34). Trimmed and filtered FASTQ reads were aligned using the STAR aligner (35) to the Mus musculus genome, build GRCm38, and counts were obtained using RSEM (36). Differential gene expression analysis was performed using custom scripts based on the R packages edgeR (37), along with limma (38) and voom (39). Genes with zero counts in all samples were removed and genes with very low expression were removed using the filterByExprs function implemented by edgeR (37). Mean-variance modeling was performed on normalized count data, followed by linear modeling using limma; P values are from a “Moderated” t test implemented in limma (38). The Moderated t test uses information from all selected genes to calculate variance. The resulting P value is adjusted for multiple hypothesis testing using the FDR method (32). Genes were considered to be significantly differentially expressed if they had an FDR corrected P value ≤ 0.05 and a fold change of at ≥ ± 1.5.
Evaluation of DMRs Using Gene Ontology Overrepresentation Analysis
The raw DMC, DMR, RNA-Seq, and metabolomics files can be found in EXCEL format at the permanent link within the University of Florida Digital Collections repository https://original-ufdc.uflib.ufl.edu//IR00011706/00001. Gene set enrichment analysis (over-representation analysis) was performed for those genes that were associated with at least one DMR using the Consensus PathDB (CPDB) from the Max Planck Institute for Molecular Genetics, using predefined Gene Ontology (GO)-based sets (40). Genes were considered associated with a DMR if the DMR fell within the gene body or within 15 kb of the transcription start site (TSS), a region with a high probability of affecting gene regulation. GO analysis is the international standard for gene function classification, which fully describes the properties of genes and gene products in organisms. In brief, all the identified genes are mapped to each term in the GO database, gene numbers for each term were calculated, and then a hypergeometric test was used to find significantly enriched GO terms. P values derived from a hypergeometric test of the number of significant genes and gene-set genes were FDR-corrected using the Benjamini–Hochberg method (32).
RESULTS
The mean core temperatures and performance measurements in this specific group of animals during the EHS protocol are summarized in Table 1. Of note, this is a relatively severe model in which the mice exhibit a blunted post-EHS hypothermia response as the mice remain in the environmental chamber at 37.5°C during 2 h of recovery. This change in protocol minimizes or eliminates the typical rodent post-heat stroke hypothermia response.
Table 1.
Average exercise and temperature responses to EHS
| Exercise Duration, min | Exercise Time (>41°C <Tc,max), min | Loss Body Mass, % | Tc,min | Tc,max | Time from Tc,max to Tc,min, min | Ascending Thermal Area (Time•Tc > 39.5°C) | |
|---|---|---|---|---|---|---|---|
| EHS | 260 ± 30 | 29.3 ± 14.5 | 12.2 ± 1.3 | 35.4 ± 2.0 °C | 42.4 ± 0.2 °C | 50.5 ± 30.9 | 197.1 ± 39.2 °C (min•°C) |
| EXC | 260 ± 30 | N/A | 6.4 ± 1.9 | N/A | N/A | N/A | N/A |
Values are means ± SD. EHS, exertional heat stroke; EXC, matched exercise control; Tc,max, maximum core temperature at symptom limitation; Tc,min, minimum temperature reached during recovery.
Reprogramming of the DNA Methylome 30 Days after EHS
Following 30 days of recovery from EHS, DNA obtained from the left ventricles exhibited profound DNA methylation reprogramming compared with that from sham EXC mice (Fig. 1A). In total, there were 13,894 DMCs that reached an inclusion threshold of ≥±10% change in methylation and Q < 0.05. Of those, 6,912 were hypomethylated and 6,982 were hypermethylated in EHS myocardiums compared with sham EXC. DMCs were found to be located across all genomic regions (Fig. 1B), with the largest fractions located in intergenic regions and introns. In addition, a significant number of DMCs were found within promoters as well as 5′-UTRs (untranslated regions) and 3′-UTRs.
Figure 1.

Genome-wide DNA methylation analyses of individual differentially methylated cytosines (DMCs). A: number of hypomethylated (blue) vs. hypermethylated (red) DMCs. Threshold inclusion was > 10% change in methylation and Q < 0.05. B: fraction of DMCs within each genomic region.
To get a better understanding of the predominant location of significant changes in cytosine methylation, we plotted % methylation differences of individual DMCs, across the entire genome on a continuous scale of distance, originating from the translation start site, TSS (+1 centered at 0 bp), for each respective gene (Fig. 2A). Both significantly hypomethylated (black symbols) and hypermethylated CpGs (deep auburn symbols) were most concentrated proximal to the TSS, but a few were distributed over hundreds of thousands of base pairs from the TSS. Focusing on a range of distances ±5 kb from the TSS in Fig. 2B (expanded x scale), a frequency histogram of the number of significantly hyper- and hypomethylated DMCs (auburn open circles) within bin sizes of 200 bp was found to be in very high abundance at and just downstream of the TSS.
Figure 2.
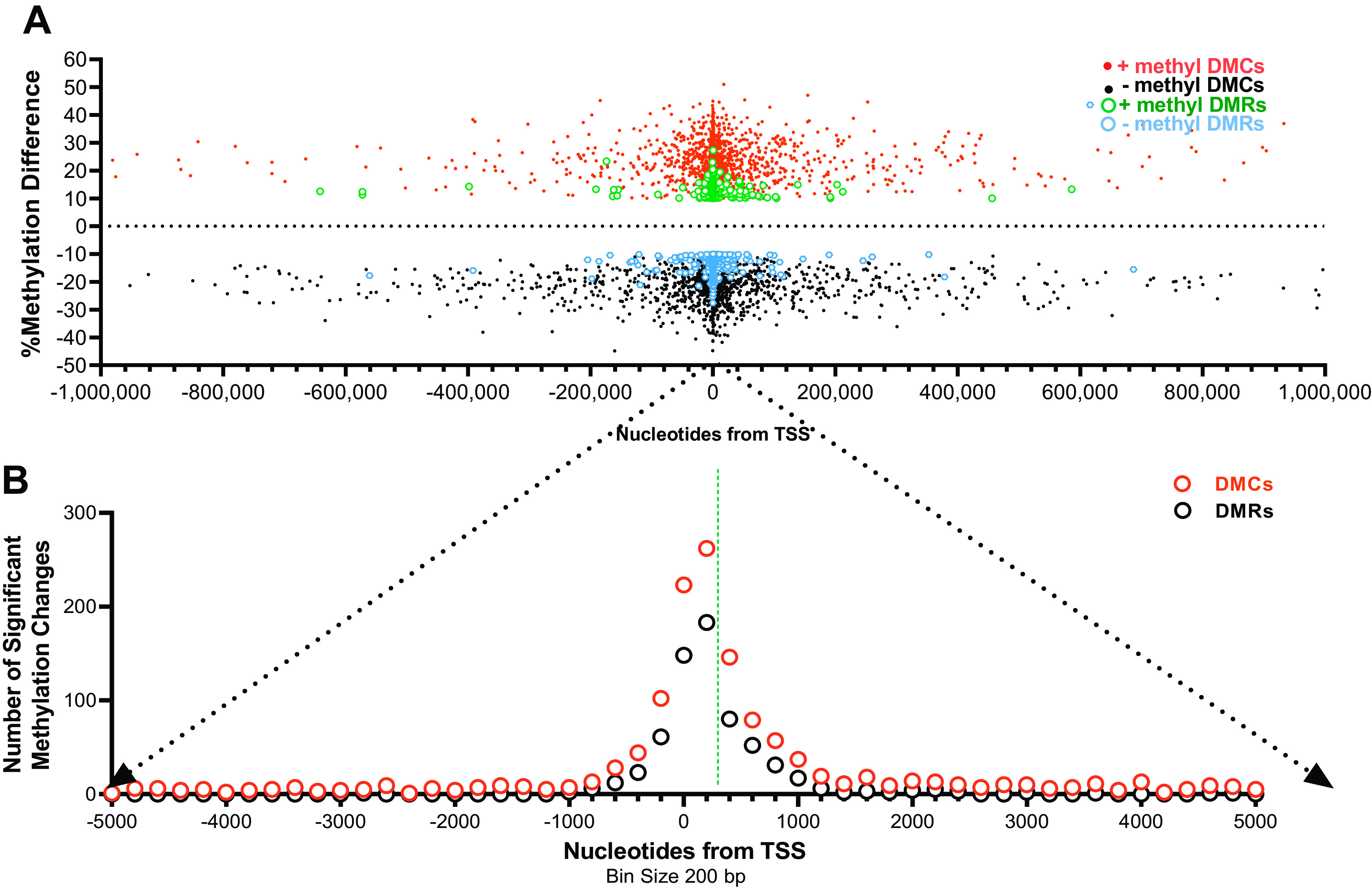
Gene location of individual differentially methylated cytosines (DMCs) and regions (DMRs). A: scatterplot of individual DMCs, hypermethylation (red) or hypomethylation (black), and DMRs, hypermethylation (green) or hypomethylation (blue) as a function of distance from the transcription start site (TSS). B: histogram of the frequency of DMCs and DMRs as a function of distance from the TSS. Note this is an expansion of the axis from “A” as indicated by the black dotted arrows.
Supplemental Tables S1 and S2 provide a list of the topmost 25 hypermethylated and hypomethylated DMCs, respectively, restricted to regions of the promoter, the first intron and first exon (41, 42). These top 25 DMCs had increases in methylation from 31% to 45% (Supplemental Table S1) and decreases in methylation from 36% to 45% (Supplemental Table S2).
The DMC data were further refined to identify differentially methylated regions (DMRs), thought to have more biological significance than DMCs in affecting gene transcription and histone behavior (43). In this analysis, a DMR was defined as a region with ≥ 5 CpGs that are all hypo- or hypermethylated and located within 300 bp of each other. As shown in Fig. 3A, a total of 947 significant DMRs within 897 genes were detected between the EHS and EXC groups, with 57% acquiring significant hypomethylation. These DMRs exhibited a minimum of ±10% average change in methylation and Q < 0.05. The majority of the DMRs were found within 1 kb of the TSS (Fig. 2, A and B). As shown in Fig. 3B, 69% were confined to the promoter region, the 5′-UTR, or the first exon and first intron, locations all considered to have a strong capacity for impacting gene regulation (41, 42). The distribution of fractional methylation levels (i.e., the average underlying total fraction of methylated cytosines within a given DMR, averaged within bins representing specific gene locations) for each group was plotted across all affected genomic features, which revealed significant changes only in the promoter regions, with an overall reduction in methylation in EHS samples (Fig. 3C). Of note, the box plot in Fig. 3C illustrates that the predominant state of most DMRs in the resting state is methylation, as ∼70% of CpGs outside of CpG islands are expected to normally be in a methylated state (44, 45). This means that when considering percent methylation changes seen in specific DMRs and DMCs, it is with respect to the inherent background methylation state of a given CpG in the EXC control samples.
Figure 3.

Genome-wide DNA methylation analyses of differentially methylated regions (DMRs). A: frequency of hypomethylation (blue) vs. hypermethylation (red). B: genomic location of a given DMR. Threshold inclusion was > ± 10% average change in methylation and Q < 0.05. C: box plot of actual fractional methylation levels for each group of samples across genomic features affected by DMRs. White dots represent mean; the black horizontal lines represent medians; the boxes are interquartile ranges. **P ≤ 0.005 by Mann–Whitney U test. DMC, differentially methylated cytosine; EXC, exercise control; EHS, exertional heat stroke.
Hierarchical clustering of significant DMRs clearly distinguished between the EHS and EXC populations in terms of both relatively hypomethylated (cyan) and hypermethylated (red) DMRs (Fig. 4A). The top 25 hypermethylated DMRs based on the mean methylated differences between EHS and EXC are listed in Table 2, and the top 25 hypermethylated DMRs are listed in Table 3. Figure 4B is an unbiased PCA plot of the top 600,000 DMRs identified, based on overall variance among all eight measurements (both EXC and ESH) for a given gene, independent of tests of significance. The plot shows that DMR analysis can clearly separate between EHS and EXC groupings of subjects within two principal components.
Figure 4.

Multivariate analysis of differentially methylated regions (DMRs). A: heat map/hierarchical clustering of all significant DMRs identified in the left ventricle. All DMRs in each row represent those with Q < 0.05 and ± 10% change in methylation. The change in methylation is scaled from −2 to +2-fold across all samples (columns, 4/group) with the mean level depicted in black, hypermethylated DMRs (403) in red, and hypomethylated DMRs (544) in cyan. B: unbiased PCA plot of individual samples demonstrating clustering of exercise control (EXC) and exertional heat stroke (EHS) treatments. PCA, principal component analysis.
Table 2.
Top 25 genes with the highest average changes in hypermethylation within DMRs after EHS
| Gene Name | %Mean Meth Diff EHS-EXC | Number CpGs | Q Value | Location | Distance to TSS, bp | Gene Description |
|---|---|---|---|---|---|---|
| Mok | 27 | 5 | 0.00109 | Intron 1 | 333 | MOK protein kinase |
| Kcnma1 | 23 | 5 | 0.000243 | Promoter-TSS | −670 | K+ large conductance Ca+2-activated channel |
| Ndufa8 | 20 | 5 | 0.002019 | Promoter-TSS | −10 | NADH dehydrogenase (ubiquinone) subcomplex |
| Trim62 | 19 | 5 | 0.004109 | Exon 1 | 182 | Tripartite motif-containing 62 |
| Rps15a | 19 | 5 | 0.00525 | Promoter-TSS | 45 | Ribosomal protein S15A |
| Dmr | 18 | 5 | 0.002525 | Promoter-TSS | −43 | Dmrt1 related/ncRNA |
| Mgst3 | 18 | 5 | 0.003195 | Promoter-TSS | −144 | Microsomal glutathione S-transferase 3 |
| Hsd17b7 | 18 | 5 | 0.001276 | Promoter-TSS | −50 | Hydroxysteroid (17-beta) dehydrogenase 7 |
| Shox2 | 18 | 5 | 0.000202 | Intron 1 | 767 | Short stature homeobox 2 |
| Plcd1 | 18 | 5 | 0.00525 | Intron 1 | 4,813 | Phospholipase C, delta 1 |
| Tmem42 | 18 | 5 | 0.008 | Promoter-TSS | −173 | Transmembrane protein 42 |
| Mecom | 18 | 5 | 0.00525 | Intron 1 | 821 | MDS1 and EVI1 complex locus |
| Zkscan16 | 17 | 7 | 0.00183 | Intron 1 | 268 | Zinc finger with KRAB and SCAN domains 16 |
| Map3k5 | 17 | 5 | 0.002647 | Promoter-TSS | −480 | Mitogen-activated protein kinase kinase kinase 5 |
| Gpr62 | 17 | 5 | 0.004961 | Exon 1 | 236 | G protein-coupled receptor 62 |
| Sass6 | 17 | 5 | 0.00597 | Promoter-TSS | −253 | Spindle assembly 6 homolog (C. elegans) |
| Ahcy | 17 | 12 | 3.72E-05 | CpG island | 204 | S-adenosylhomocysteine hydrolase |
| Sidt2 | 17 | 8 | 5.47E-05 | 5′-UTR | 329 | SID1 transmembrane family, member 2 |
| Sh3bgrl2 | 17 | 5 | 0.001639 | Intron 1 | 188 | SH3 domain binding glutamic acid-rich protein like 2 |
| Triobp | 17 | 5 | 0.009435 | Exon 1 | 132 | TRIO and F-actin binding protein |
| Egr4 | 16 | 5 | 0.002129 | Promoter-TSS | −269 | Early growth response 4 |
| Camk2n1 | 16 | 5 | 0.00525 | Intron 1 | 383 | Ca+2/calm.-dependent protein kinase II inhibitor |
| R3hcc1l | 16 | 6 | 0.00183 | Promoter-TSS | 26 | R3H domain and coiled-coil containing 1 like |
| Gnal | 16 | 6 | 0.005178 | Exon 1 | 476 | Guanine nucleotide binding protein |
| Hspd1 | 16 | 5 | 0.008452 | Intron 1 | 821 | Heat shock protein 1 (chaperonin) |
EHS, exertional heat stroke; Meth diff, methylation difference; #CpGs, number of methylation sites within each DMR (differentially methylated region); TSS, transcription start site; Q value = P value adjusted from multiple comparisons (Benjamini–Hochberg).
Table 3.
Top 25 genes with the highest average changes in hypomethylation within DMRs after EHS
| Gene Name | %Mean Meth Diff EHS-EXC | Number CpGs | Q Value | Location | Distance to TSS, bp | Gene Description |
|---|---|---|---|---|---|---|
| LOC100861615 | −28 | 5 | 0.004109 | Promoter-TSS | −102 | Alpha takusan-like |
| Rasgef1b | −25 | 11 | 0.000101 | Promoter-TSS | −211 | RasGEF domain family, member 1B |
| Hars | −23 | 8 | 0.001685 | Promoter-TSS | 189 | Histidyl-tRNA synthetase |
| Uggt2 | −22 | 8 | 0.000295 | Promoter-TSS | −10 | UDP-glucose glycoprotein glucosyltransferase 2 |
| Iqcc | −22 | 5 | 0.008452 | Promoter-TSS | 18 | IQ motif containing C |
| Gramd3 | −21 | 5 | 0.003863 | Intron 1 | 675 | GRAM domain containing 3 |
| Runx1 | −20 | 5 | 0.001069 | Promoter-TSS | −192 | Runt-related transcription factor 1 |
| Myb | −20 | 5 | 0.000665 | Intron 1 | 1,876 | Myeloblastosis oncogene |
| Efna3 | −20 | 8 | 0.001069 | Intron 1 | 3,561 | Ephrin A3 |
| Cep85 | −20 | 5 | 0.001276 | Promoter-TSS | −73 | Centrosomal protein 85 |
| Dynll1 | −20 | 10 | 0.001685 | Promoter-TSS | −21 | Dynein light chain LC8-type 1 |
| Gins2 | −20 | 7 | 0.001533 | Promoter-TSS | −400 | GINS complex subunit 2 (Psf2 homolog) |
| Gmps | −20 | 5 | 0.009094 | Intron 1 | 603 | Guanine monophosphate synthetase |
| D330050I16Rik | −20 | 16 | 0.000665 | Promoter-TSS | 7 | RIKEN cDNA D330050I16 gene |
| Magi1 | −19 | 5 | 0.003863 | Exon 1 | 816 | Membrane-associated guanylate kinase, |
| Slc4a11 | −19 | 7 | 0.007141 | Promoter-TSS | −201 | Solute carrier family 4, NaHCO3 transporter-like |
| Trafd1 | −19 | 5 | 0.006348 | Intron 1 | 624 | TRAF type zinc finger domain containing 1 |
| Gpam | −19 | 5 | 0.00283 | Intron 1 | 331 | Glycerol-3-phosphate acyltransferase, mitochondrial |
| Ube4b | −19 | 7 | 0.001779 | 5′-UTR | 420 | Ubiquitination factor E4B |
| Pxn | −19 | 7 | 0.001131 | Intron 1 | 9,764 | Paxillin |
| Rab40c | −19 | 6 | 0.002937 | Promoter-TSS | −342 | Rab40C, member RAS oncogene family |
| Gmcl1 | −18 | 10 | 0.001031 | Promoter-TSS | −209 | Germ cell-less homolog 1 (Drosophila) |
| Numb | −18 | 5 | 0.00525 | Intron 1 | 240 | Numb gene homolog (Drosophila) |
| Rhob | −18 | 5 | 0.001359 | Exon 1 | 750 | Ras homolog gene family, member B |
| Tbc1d4 | −18 | 10 | 0.001685 | Promoter-TSS | 15 | TBC1 domain family, member 4 |
EHS, exertional heat stroke; Meth diff, methylation difference; #CpGs, number of methylation sites within each DMR (differentially methylated region); TSS, transcription start site; Q value = P value adjusted from multiple comparisons (Benjamini–Hochberg).
To explore pathways of genes enriched in DMRs, we performed Gene Ontology analysis, using those genes associated with at least one significant DMR. For GO analysis, genes were only considered whether they had an associated DMR within the gene body or within 15 kb of the TSS, regions that would likely affect gene regulation. A total of 864 genes met these criteria. GO analysis was performed using ShinyGO (46). The resulting significant GO (FDR-corrected Fisher’s exact test P ≤ 0.05) terms were plotted in Fig. 5, with the most significant represented in the dark blue dots at the top. These overrepresented pathways were related to many basic cell functions, including DNA and RNA metabolism, DNA transcription, protein modifications, and protein degradation.
Figure 5.

Gene Ontology (GO) enrichment analysis. Significantly enriched GO terms from DMR results using ShinyGO (y-axis) are plotted with the fold enrichment along the x-axis. The color of the dots represents the −log10(Q value) and the size of the dots are based on the number of genes with DMRs assigned to the enriched GO term on the left. DMR, differentially methylated region; FDR, false discovery rate.
The percent methylation calculated for DMRs is performed by averaging across all CpGs within the DMR, and therefore large changes in methylation in a few DMCs can, in some cases, be averaged out and appear proportionally small or below the threshold (33). We tested further whether the DMR definition used, as described in the section Enzymatic Methyl Sequencing and Analysis for DNA Methylation, is sensitive enough to detect such individual genes that have high methylation changes but with < 5 CpGs. More specifically, a secondary DMR analysis was executed on genes that contained a differentially methylated region with ≥ 2 and <5 CpGs that included CpGs among the highest hypo- or hypermethylated DMCs and were also located within 250 bp of each. This subanalysis of DMRs is based on the observation that in some genes, even one DMC can influence transcription factor binding and DNA structure, but the effects can be greatly amplified if there is a similar change in methylation on neighboring CpGs within ∼200 bp from each other (47). Our results are provided in Supplemental Table S3 (hypermethylation) and Supplemental Table S4 (hypomethylation). Note that the average distance between DMCs within these groups was only 7.9 ± 42 bp (means ± SD) apart. The data were further restricted for specificity with the purpose of trying to identify the most reliable methylation events by using DMCs with Q values < 0.01, which were located within the promoter, 5′-UTR, first exon, or first intron. As denoted in Supplemental Tables S3 and S4, the majority of these genes were also found within previously defined DMRs (designated by asterisks), demonstrating that in general, the definition we used for a DMR was relatively inclusive of this secondary category of DMRs. However, note that the most highly hypomethylated cytosine pair (Agfg2, encodes “ArfGAP with FG repeats 2” protein) and the most highly hypermethylated pair (Gdf6, encodes “Growth Differentiation Factor 6” protein), each of which exhibited 30%–45% changes in methylation, were not identified by our original definition of a DMR.
Representative Genes that Exhibit Strong Differentially Methylated Regions
It is instructive to highlight a few of the most highly altered DMRs to provide an indication of their potential significance. For example, the gene Ndufa8 (encodes for the protein, “Tripartite Motif Containing 62”) in Table 2 and Fig. 6A codes for an essential component of mitochondrial NADH dehydrogenase in complex I (NADH:Ubiquinone Oxidoreductase Subunit A8). It had five hypermethylated DMCs located very close to the TSS, modifications that should be associated with gene suppression (48). However, the mRNA expression measurements for Ndufa8 exhibited almost identical outcomes in both groups. Other examples of hypermethylated DMRs include Trim62 that encodes a ubiquitin E3 ligase involved with autophagy (49) and Kcnma1 that encodes a critically important BKc channel (Ca+2-dependent K+ channel, “potassium calcium-activated channel subfamily M alpha 1”) in mitochondria (50). Many of these strongly hypermethylated DMRs point to genes that relate to mitochondrial function or remodeling. The gene Ahcy is also interesting. It had a DMR with 12 DMCs and an average increase of ∼17% cytosine methylation in a CpG island. The enzyme encoded by Ahcy is S-adenosylhomocysteine hydrolase (SAHH) that is part of the methionine degradation pathway. Its activity in degrading S-adenosylhomocysteine (SAH) to homocysteine is important in regulating the level of intracellular SAH, which serves as a potent end-product inhibitor of many different types of methyltransferases (51, 52). Disorders of homocysteine regulation are considered an independent risk factor for cardiovascular disease (52).
Figure 6.

Integrative Genomics Viewer (IGV) tracks of Ndufa8 (A) and Uggt2 (B) after 30 days of recovery from EHS or EXC. The sequence coordinates of the promoters of Ndufa8 (chromosome 2; divergently transcribed relative to Mom5) and Uggt2 (chromosome 14) are indicated at the top of each panel. The first eight tracks indicate the sequencing coverage, i.e., the number of times that each CpG was sequenced (combined top and bottom strands) in each sample (EXC, blue bars; EHS, red bars). Only CpGs sequenced ≥10× are shown, with all coverage scales set from 0 to 60 reads. The % methylation tracks are all scaled from 0 to 100%. EXC, exercise control; EHS, exertional heat stroke.
An example of a DMR that was strongly hypomethylated was on the gene Uggt2 (Fig. 6B and Table 3), which encodes the protein UDP-glucose glycoprotein glucosyltransferase 2, an important pathway in the detection and repair of damaged proteins within the endoplasmic reticulum (53). It is involved with ongoing tissue repair or resolution of protein damage. The DMR on the Uggt2 gene had eight DMCs within the transcriptional regulating regions, with an average loss of 22% methylation, whereas another DMR on the same gene had 15 DMCs with an average loss of 10% methylation. A loss of methylation in the promoter region is generally associated with release from transcriptional suppression (48).
Changes in RNA-Seq 30 Days after EHS
RNA-Seq was performed on the same ventricular samples (n = 4/group) in which the DNA methylation measurements were made. These were performed to assign possible physiological meaning to the observed changes to the DNA methylome in a resting state. In general, the differences in expression for all genes measured between the EHS and EXC mice were relatively modest, as shown in the volcano plot in Fig. 7A. The mRNA from 23 genes was suppressed >2-fold (uncorrected P < 0.05) and mRNA from 21 genes was increased >2-fold (uncorrected P < 0.05). All mRNAs that exhibited >1.5-fold increase or decrease in expression were illustrated in a hierarchical heat map in Fig. 7B, which clearly sort into distinct EHS and EXC groups. The significance of mRNA from only six genes reached FDR-corrected Q < 0.05 and were all downregulated. Their names are listed in Fig. 7A. Two of these genes, Lgals4 (encodes Galectin 4) and Mdm4 (encodes the Mdm4 protein), are associated with disorders of cardiac function in animal models (54, 55). In Fig. 7C, the RNA-Seq outcomes are evaluated by principal component analysis (PCA), which again distinguish the experimental subjects into treatment-defined groups.
Figure 7.
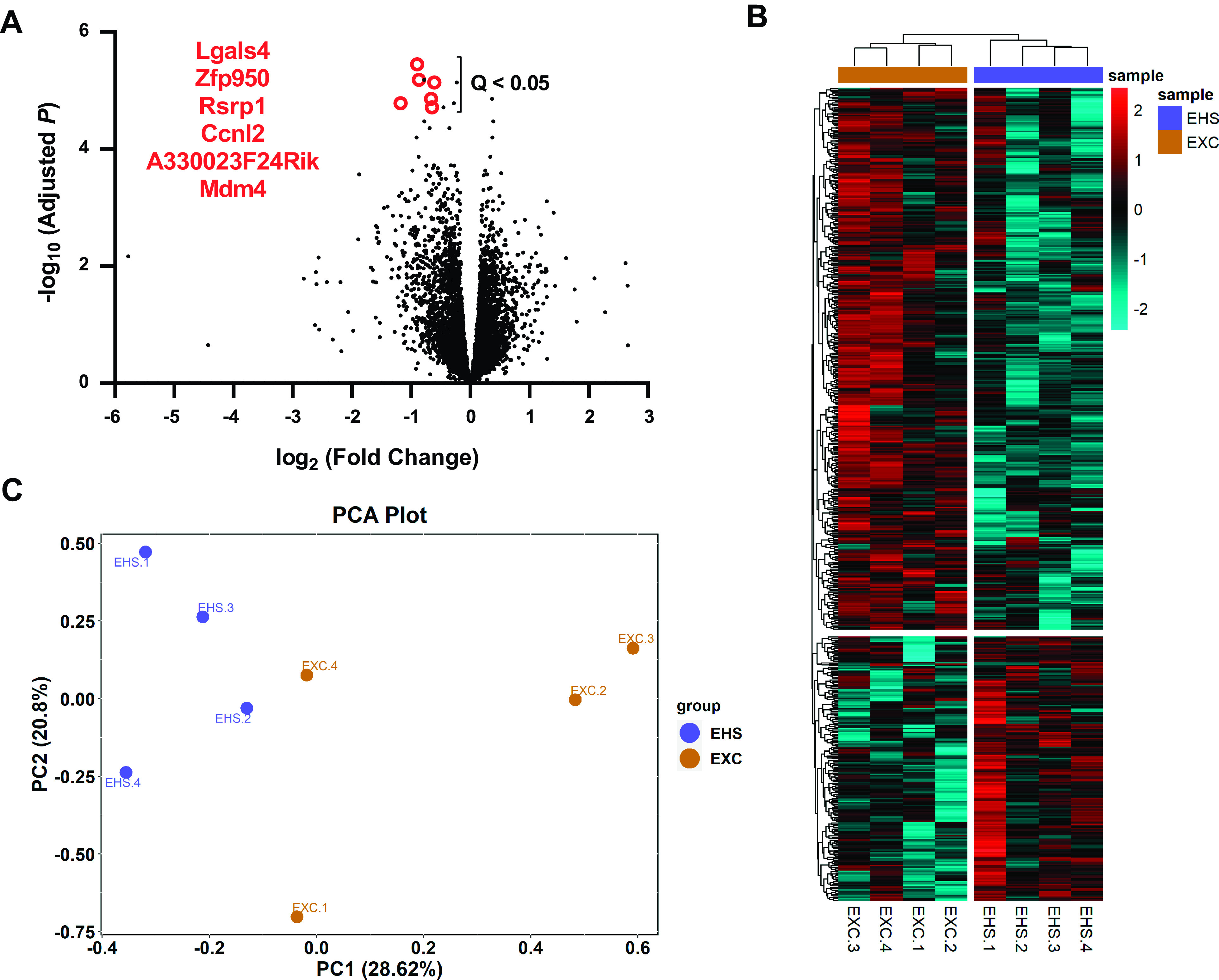
RNA-Seq results of EHS vs. EXC (n = 4/group). A: volcano plot of the log of fold changes in expression to all 14,696 RNA detected. Red outlined points and gene names are the six mRNAs that reached an FDR Q < 0.05. All were significantly downregulated. The post hoc power analysis revealed levels of β > 0.98 for all six identified mRNAs. B: heat map of the hierarchical expression of mRNA of all genes that exhibited ±1.5-fold differences between EHS and EXC. The color code is normalized to bright red (+2) representing the highest fold changes and cyan representing the lowest fold changes (−2). C: unbiased PCA plot of mRNA results using the genes that exhibited the highest overall variance compared to the mean (coefficient of determination) of all 8 samples (EXC + EHS) for a given gene. EXC, exercise control; EHS, exertional heat stroke; PCA, principal component analysis.
Finally, direct correlations were performed between DMR methylation results and RNA-Seq results (Supplemental Fig. S1), hypothesizing that mRNA expression would reflect underlying changes in DNA methylation. No clear relationship could be identified between genes that had significant alterations in DNA methylation with genes that had differences in expression, suggesting that changes in gene transcription in this basal state of recovery in mice previously exposed to EHS were not primarily related to the methylation status of a given gene.
Alterations in the Metabolomic Profile 30 Days after EHS
Metabolomic analyses were performed, in part, to determine if any measurements of DNA methylation were useful in predicting the metabolic state of the heart. In addition, we aimed to determine if residual metabolic abnormalities that we previously reported after 9–14 days of recovery (13) persisted at 30 days. In general, after 30 days of recovery from EHS, the ventricular myocardium exhibited an apparent general resolution of the majority of extensive metabolic disorders we previously observed after 2 wk (13). Only a few residual abnormalities appeared to persist, as shown in Fig. 8. Of the 807 detected metabolites, 11 reached an uncorrected P value of <0.05, and only one metabolite, 16-hydroxypalmitate, reached an FDR threshold of Q < 0.2. These modest metabolomic findings are consistent with a return to a relative metabolic homeostasis in the ventricular myocardium over this longer recovery period.
Figure 8.

Significantly different left ventricular metabolites. All data are expressed as a fraction of the mean exercise control value for that metabolite. Red bars: exertional heat stroke (EHS) vs. white bars: matched sham exercise controls (EXC); n = 7/group, means ± SD. Only 16-hydroxypalmitate was within a Benjamini–Hochberg FDR threshold of Q < 0.2. Uncorrected P values ***P < 0.001, **< 0.01, *< 0.05. The average post hoc power calculations for these 11 significant comparisons was 0.84, ranging from 0.57 to 0.99. FDR, false discovery rate.
DISCUSSION
By 30 days of recovery from EHS, left ventricles from female mice developed substantial reprogramming of the DNA methylome compared with matched EXC mice. A wide variety of single DMCs and multiple, contiguous CpG targets (DMRs) were affected near the TSS on genes involved with basic cell functions of cell signaling, stress adaptation, remodeling, and metabolism. The physiological significance of these changes for the heart, both in the short and long term, remains unclear. However, it is likely that they result in alterations in gene responsiveness at a very fundamental level of cell organization that could contribute to loss or gain of resilience upon subsequent environmental-, nutritional-, or stress-related challenges. However, at baseline, only modest changes in mRNA expression could be identified. Although these did not relate well to methylation records of the same genes, there were unique transcriptional phenotypes between the EHS and EXC groups of mice. The results of metabolomic analyses of the same left ventricles provide evidence for considerable metabolic recovery at 30 days compared with the 14 days responses we previously reported (13), with some very modest residual effects. Therefore, the myocardium appears to have largely returned to a new level of metabolic and transcriptional homeostasis while holding on to a considerably altered DNA methylation landscape.
DNA Methylation Responses of the Ventricular Myocardium
In designing this study, we thought it possible to link the changes in DNA methylation with both the metabolomic and RNA-Seq results obtained from the same hearts. However, the metabolomic differences at 30 days were weak in EHS compared with EXC and only mildly consistent with the severe disturbances previously identified at 14 days (16). Furthermore, mRNA expression differences between EHS and EXC did not correlate with the methylation measurements. We also looked for DNA methylation and RNA-Seq changes in genes that reflect expected physiological responses to heat stress such as heat shock protein (HSP) expression, antioxidant defense systems, and inflammatory response pathways. The altered responsiveness of these three categories of genes was previously identified in this model in different kinds of in vitro settings (13, 16). However, we observed little or no statistically significant methylation changes for any related genes with the exception of a single hypermethylated CpG on Hspd1 (encodes mitochondrial HSP60 protein, Table 1). Alterations in common antioxidant genes, such as those responsible for catalase, SODs, glutathione peroxidase and peroxiredoxin, were also not significant. In terms of inflammatory signaling, one gene was hypermethylated, Nfkb2 (Supplemental Table S2, encodes “nuclear factor kappa B subunit 2”) that was previously shown to acquire hypermethylated DMCs within immune cells at 30 days of recovery (16). Other genes associated with inflammatory processes were hypomethylated, including Csf1 (encodes “colony stimulating factor 1”), Mllt3 (encodes “myeloid/lymphoid or mixed-lineage leukemia; translocated to 3”) (Supplemental Table S1), Nfatc2ip (encodes “nuclear factor of activated T cells 2 interacting protein”; Supplemental Table S3), and Agfg2 (Supplemental Table S1), which may suggest altered inflammatory responsiveness in recovering EHS mice. Again, with these specific examples of strong methylation changes, there was no correlation with resting expression levels of the same genes from the same tissues.
Metabolomics of the Ventricular Myocardium at 30 Days of Recovery
We found only a few metabolite differences between EHS and EXC mice at this 30-day time point in the ventricular myocardium. There were some limitations to the measurements and comparisons with our previous work (13) as the library of metabolites that were evaluated for the current study had shifted from the analysis performed after only 14 days of recovery. In addition, we did not have sufficient samples to run additional lipidomics platforms that were performed at the 14-day time point. Despite these limitations, it is clear that the extensive metabolomic disturbances we observed previously were largely absent at 30 days. There were several notable exceptions that provided some evidence showing persistence at 30 days. For example, the reduced α-ketoglutarate, a product of isocitrate metabolism in the TCA cycle, appeared to be reduced (uncorrected P < 0.05) both at 30 days and 14 days of recovery (13). Concurrently, the gene that codes for the enzyme that mediates isocitrate to α-ketoglutarate (Idh2) exhibited two DMCs near the TSS that were strongly methylated. However, there were no differences in the expression level for this gene using RNA-Seq. A reduction in α-ketoglutarate has been observed in early-stage heart failure (56). Furthermore, the responsible enzyme, isocitrate dehydrogenase, is a target for a variety of stress-induced posttranslational modifications, including oxidation (57), that can affect the progression of heart disease and responses to oxidative stress. α-Ketoglutarate is also a key metabolite regulating ten-eleven translocase (TET)-dependent DNA demethylation (58), and therefore could have implications for epigenetic regulation in this model. The most significantly elevated ventricular metabolite at 30 days in the EHS group was 16-hydroxypalmitate (P < 0.001, Fig. 6). This species was not available in the library that was used for the 14-day assessment. However, 16-hydroxyplamitate is an alternative omega oxidation product of palmitic acid, considered the most common saturated fatty acid in humans, and one that was highly elevated in ventricular samples at 14 days (13). It is elevated when normal β-oxidation in the mitochondria is defective, as reported in acute kidney disease (59). Therefore, these somewhat limited results that parallel the previous 14-day study indicate that some metabolic disturbances could linger for as long as 30 days.
Changes in RNA Expression
Based on a nonbiased comparison of the mRNA samples, the EXC and EHS groups were identifiably different in the basal state as indicated by several comparisons. However, these differences in expression were modest, with only a few mRNAs reaching statistical significance with > ± 2-fold difference. Furthermore, these changes did not seem to be directly related to the DNA methylation status of the corresponding gene, even when restricted to methylation changes within the promoter. Although this outcome is based on a limited sample size, it suggests that the hearts of mice recovering from EHS for 30 days have reached a new state of transcriptional homeostasis. This is not unexpected, because in fully differentiated cells, genome-wide changes in methylation of most genes do not seem to correlate well with the expression of that gene in the nonstressed state (60, 61). In homeostasis, genes in differentiated cells are likely to be kept at steady-state expression by upstream transcriptional regulators that are under normal feedback regulation. This is in contrast to actively differentiating or developing cells in which methylation status within CpG islands or in the promoter regions has predictable influences on gene expression and phenotype (62). The importance of DNA methylation status in differentiated cells may well be more evident following exposure to a secondary stress when cells must undergo rapid acceleration or de-acceleration of their transcriptional machinery. This effect was evident in our previous work on in vitro bone marrow-derived monocytes, obtained 30 days after EHS exposure, where significant transcriptional changes in the expression of heat shock-related proteins were only observed after a second heat shock exposure. Of the three heat shock-related proteins found significantly different between EHS and EXC following this second heat shock exposure, the transcriptional changes of two heat shock-related proteins were predicted by EHS-mediated changes in DNA methylation (e.g., HSP90 and HSP72 isoforms) (16). As such, we speculate that the functional significance of long-term DNA methylation changes in the heart in this setting may impose their greatest influence following exposure to secondary effectors of heart disease, such as atherosclerosis, aging, poor diet, subsequent heat exposure, or ischemia.
Limitations
We evaluated the methylation profile of genomic DNA harvested from the composites of left ventricles that contain many different cell types. This approach has at least one advantage in that it scans all cell types within the myocardium for epigenetic changes, before focusing on any one phenotype, and without extensive stress induced by cell preparation or cell isolation. A disadvantage is that the specific cellular phenotypes where DNA methylation was affected could not be separated out. Different estimates have been made of the number of different types of cells within the ventricular myocardium. Depending on the method used, several groups have estimated, in both humans and mice, that the cardiomyocyte population in the left ventricle is 25%–49% of the total cells (63–65). The endothelium, leukocytes, and mesenchymal cells make up the balance. Therefore, in stoichiometric terms, the DNA methylation patterns of the gross specimen we used represent a wide spectrum of phenotypes, with less than half representing cardiac myocytes. If we had used a method that isolated only myonuclear DNA, e.g., using fluorescence-activated cell sorting as recently described for DNA methylation of myocytes (63), we may have missed the overall signal. For example, in failing versus nonfailing adult human hearts, the CpG methylation pattern in myonuclei is not significantly different from controls (63). Nevertheless, future work will need to further refine these observations to specific cell subtypes and over a variety of time points during recovery. A second limitation was the relatively small sample size, particularly for RNA-Seq, which likely could have resulted in missing subtle differences in expression between groups.
A third limitation is that this study only evaluated methylation changes to DNA, but other epigenetic mechanisms such as posttranslational modifications of histones, long noncoding RNAs, microRNAs, etc., are also important to the epigenetic responses of these cells. Changes to the nucleosome can be directly or indirectly linked to DNA methylation (66), but they can also provide unique contributions. Our goal here was to look for changes that might represent epigenetic signatures of long-term myocardial vulnerability to health challenges over the life of the animal. Since DNA methylation is generally regarded as a relatively stable epigenetic change, at least relative to other mechanisms (67), it seemed to be an appropriate first choice to evaluate.
A fourth limitation of this study was that we limited our analyses to only female mice. This was due to the fact that we only saw the development of cardiac abnormalities at this time point in female mice. Our laboratory has been pursuing the development of an altered model that is specifically targeted to males in the hopes of pursuing the effects in this sex in future experiments.
Conclusions
For the first time, we have established that there is a persistent and highly significant altered DNA methylation pattern within the left ventricle in response to an intense and brief environmental heat and exercise stimulus resulting in heat stroke. How long this persists, its significance with respect to the functional stability of the myocardium over time, the specific phenotypes that are reflected in this pattern, the signaling mechanisms responsible for initiating these changes in methylation, and its relationship to gene transcription and other epigenetic mechanisms, such as posttranscriptional modification of histones, will require additional study. In addition, we established that the remarkable disorders to the ventricular metabolome that we previously observed between 9 and 14 days after EHS exposure (13) have largely resolved to a new metabolic homeostasis. This does not mean that the metabolic resiliency of the heart would be prepared for further stress exposures or perturbations, but rather that the heart has compensated to bring the system back to relative equilibrium. This outcome is likely to also apply to the DNA methylation results, where the biological effects of alterations in the methylome would most likely emerge following a second stress (e.g., ischemia, circulatory collapse, or additional environmental exposures), when transcriptional responses need to be recruited rapidly for repair or survival.
DATA AVAILABILITY
The raw FASTQ and GEO data sets for DNA methylation analysis and RNAseq can be found under accession number PRJNA884710 at https://www.ncbi.nlm.nih.gov/bioproject/884710.
The raw DMC, DMR, RNA-Seq, and metabolomics files can be found in EXCEL format at the permanent link within the University of Florida Digital Collections repository at https://original-ufdc.uflib.ufl.edu//IR00011706/00001.
SUPPLEMENTAL DATA
Supplemental Tables S1–S4: https://doi.org/10.6084/m9.figshare.16933240.v2.
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.21235437.v2.
Supplemental list of metabolites analyzed (Metabolomics Library of Detected Metabolites Metabolon Inc): https://doi.org/10.6084/m9.figshare.20483664.v1.
GRANTS
The research was supported by contracts from the US Department of Defense, BA180078 (to T.L. Clanton), with supplemental support from the B.K. and Betty Stevens Endowment at the University of Florida (to T.L. Clanton).
DISCLAIMERS
The opinions or assertions herein are the private views of the authors and are not construed as official or as reflecting the views of the Army or the Department of Defense. Citations of commercial organizations and trade names in this report do not constitute an official Department of the Army endorsement or approval of the products or services of these organizations.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
K.O.M. and T.L.C. conceived and designed research; K.O.M. performed experiments; K.O.M., J.O.B., M.P.K., and T.L.C. analyzed data; K.O.M., J.O.B., M.P.K., and T.L.C. interpreted results of experiments; J.O.B. and T.L.C. prepared figures; K.O.M. and T.L.C. drafted manuscript; K.O.M., J.O.B., M.P.K., and T.L.C. edited and revised manuscript; K.O.M., J.O.B., M.P.K., and T.L.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the help and expertise of the Interdisciplinary Center for Biotechnology Research Gene Expression and Genotyping Core at the University of Florida. Graphical abstract image created with Biorender.com and published with permission.
REFERENCES
- 1. King MA, Leon LR, Mustico DL, Haines JM, Clanton TL. Biomarkers of multi-organ injury in a pre-clinical model of exertional heat stroke. J Appl Physiol (1985) 118: 1207–1220, 2015. doi: 10.1152/japplphysiol.01051.2014. [DOI] [PubMed] [Google Scholar]
- 2. O'Connor FG, Casa DJ, Bergeron MF, Carter R, Deuster P, Heled Y, Kark J, Leon L, McDermott B, O'Brien K, Roberts WO, Sawka M. American College of Sports Medicine Roundtable on exertional heat stroke–return to duty/return to play: conference proceedings. Curr Sports Med Rep 9: 314–321, 2010. doi: 10.1249/JSR.0b013e3181f1d183. [DOI] [PubMed] [Google Scholar]
- 3. Leon LR, Bouchama A. Heat stroke. Compr Physiol 5: 611–647, 2015. doi: 10.1002/cphy.c140017. [DOI] [PubMed] [Google Scholar]
- 4. Nelson NG, Collins CL, Comstock RD, McKenzie LB. Exertional heat-related injuries treated in emergency departments in the U.S., 1997-2006. Am J Prev Med 40: 54–60, 2011. doi: 10.1016/j.amepre.2010.09.031. [DOI] [PubMed] [Google Scholar]
- 5. Alele FO, Malau-Aduli BS, Malau-Aduli AEO, J. Crowe M. Epidemiology of exertional heat illness in the military: a systematic review of observational studies. Int J Environ Res Public Health 17: 7037, 2020. doi: 10.3390/ijerph17197037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Update: Heat illness, active component, U.S. Armed Forces. MSMR 28: 10–15, 2021. [PubMed] [Google Scholar]
- 7. Donham BP, Frankfurt SB, Cartier RA, O'Hara SM, Sieg VC. Low incidence of death and renal failure in United States Military Service members hospitalized with exertional heat stroke: a retrospective cohort study. Mil Med 185: 362–367, 2020. doi: 10.1093/milmed/usz214. [DOI] [PubMed] [Google Scholar]
- 8. Bouchama A, Abuyassin B, Lehe C, Laitano O, Jay O, O’Connor FG, Leon LR. Classic and exertional heatstroke. Nat Rev Dis Primers 8: 8, 2022. doi: 10.1038/s41572-021-00334-6. [DOI] [PubMed] [Google Scholar]
- 9. Wallace RF, Kriebel D, Punnett L, Wegman DH, Amoroso PJ. Prior heat illness hospitalization and risk of early death. Environ Res 104: 290–295, 2007. doi: 10.1016/j.envres.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 10. Wang J-C, Chien W-C, Chu P, Chung C-H, Lin C-Y, Tsai S-H. The association between heat stroke and subsequent cardiovascular diseases. PLoS ONE 14: e0211386, 2019. doi: 10.1371/journal.pone.0211386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tseng M-F, Chou C-L, Chung C-H, Chen Y-K, Chien W-C, Feng C-H, Chu P. Risk of chronic kidney disease in patients with heat injury: a nationwide longitudinal cohort study in Taiwan. PLoS ONE 15: e0235607, 2020. [Erratum in PLoS ONE 15: e0238826, 2020]. doi: 10.1371/journal.pone.0235607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lawton EM, Pearce H, Gabb GM. Review article: Environmental heatstroke and long-term clinical neurological outcomes: a literature review of case reports and case series 2000-2016. Emerg Med Australas 31: 163–173, 2019. doi: 10.1111/1742-6723.12990. [DOI] [PubMed] [Google Scholar]
- 13. Laitano O, Garcia CK, Mattingly AJ, Robinson GP, Murray KO, King MA, Ingram B, Ramamoorthy S, Leon LR, Clanton TL. Delayed metabolic dysfunction in myocardium following exertional heat stroke in mice. J Physiol 598: 967–985, 2020. doi: 10.1113/JP279310. [DOI] [PubMed] [Google Scholar]
- 14. Kurisu S, Inoue I, Kawagoe T, Ishihara M, Shimatani Y, Nishioka K, Umemura T, Nakamura S, Yoshida M, Sato H. Myocardial perfusion and fatty acid metabolism in patients with tako-tsubo-like left ventricular dysfunction. J Am Coll Cardiol 41: 743–748, 2003. doi: 10.1016/s0735-1097(02)02924-8. [DOI] [PubMed] [Google Scholar]
- 15. Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 18: 1692–1700, 2004. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 16. Murray KO, Brant JO, Iwaniec JD, Sheikh LH, Carvalho L. D, Garcia CK, Robinson GP, Alzahrani JM, Riva A, Laitano O, Kladde MP, Clanton TL. Exertional heat stroke leads to concurrent long-term epigenetic memory, immunosuppression and altered heat shock response in female mice. J Physiol 599: 119–141, 2021. doi: 10.1113/JP280518. [DOI] [PubMed] [Google Scholar]
- 17. Murray KO, Clanton TL, Horowitz M. Epigenetic responses to heat: from adaptation to maladaptation. Exp Physiol 107: 1144–1158, 2022. doi: 10.1113/EP090143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Horowitz M. Epigenetics and cytoprotection with heat acclimation. J Appl Physiol (1985) 120: 702–710, 2016. doi: 10.1152/japplphysiol.00552.2015. [DOI] [PubMed] [Google Scholar]
- 19. Tetievsky A, Horowitz M. Posttranslational modifications in histones underlie heat acclimation-mediated cytoprotective memory. J Appl Physiol (1985) 109: 1552–1561, 2010. doi: 10.1152/japplphysiol.00469.2010. [DOI] [PubMed] [Google Scholar]
- 20. Tetievsky A, Cohen O, Eli-Berchoer L, Gerstenblith G, Stern MD, Wapinski I, Friedman N, Horowitz M. Physiological and molecular evidence of heat acclimation memory: a lesson from thermal responses and ischemic cross-tolerance in the heart. Physiol Genomics 34: 78–87, 2008. doi: 10.1152/physiolgenomics.00215.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zannas AS, West AE. Epigenetics and the regulation of stress vulnerability and resilience. Neuroscience 264: 157–170, 2014. doi: 10.1016/j.neuroscience.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zannas AS, Chrousos GP. Epigenetic programming by stress and glucocorticoids along the human lifespan. Mol Psychiatry 22: 640–646, 2017. doi: 10.1038/mp.2017.35. [DOI] [PubMed] [Google Scholar]
- 23. Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLOS Biol 8: e1000412, 2010. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Garcia CK, Mattingly AJ, Robinson GP, Laitano O, King MA, Dineen SM, Leon LR, Clanton TL. Sex-dependent responses to exertional heat stroke in mice. J Appl Physiol (1985) 125: 841–849, 2018. doi: 10.1152/japplphysiol.00220.2018. [DOI] [PubMed] [Google Scholar]
- 25. King MA, Alzahrani JM, Clanton TL, Laitano O. A preclinical model of exertional heat stroke in mice. J Vis Exp July 1; (173), 2021. doi: 10.3791/62738. [DOI] [PubMed] [Google Scholar]
- 26. McDonald JH. Handbook of Biological Statistics (3rd ed.). Baltimore, MD: Sparky House Publishing, 2019. http://www.biostathandbook.com/ [Google Scholar]
- 27. Faul F, Erdfelder E, Lang A-G, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39: 175–191, 2007. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
- 28. Krueger F, James F, Ewels P, Afyounian E, Schuster-Boecklar B. Babraham Bioinformatics - Trim Galore! [Online]. https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ [14 Sep. 2021].
- 29. Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27: 1571–1572, 2011. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2020. https://www.r-project.org. [Google Scholar]
- 31. Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol 13: R87, 2012. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B 57: 289–300, 1995. doi: 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 33. Jühling F, Kretzmer H, Bernhart SH, Otto C, Stadler PF, Hoffmann S. metilene: fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Res 26: 256–262, 2016. doi: 10.1101/gr.196394.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17: 10–12, 2011. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 35. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21, 2013. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323, 2011. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140, 2010. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47, 2015. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15: R29, 2014. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB–a database for integrating human functional interaction networks. Nucleic Acids Res 37: D623–D628, 2009. doi: 10.1093/nar/gkn698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Anastasiadi D, Esteve-Codina A, Piferrer F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin 11: 37, 2018. doi: 10.1186/s13072-018-0205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brenet F, Moh M, Funk P, Feierstein E, Viale AJ, Socci ND, Scandura JM. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One 6: e14524, 2011. doi: 10.1371/journal.pone.0014524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Choudhury SR, Ashby C, Tytarenko R, Bauer M, Wang Y, Deshpande S, Den J, Schinke C, Zangari M, Thanendrarajan S, Davies FE, van Rhee F, Morgan GJ, Walker BA. The functional epigenetic landscape of aberrant gene expression in molecular subgroups of newly diagnosed multiple myeloma. J Hematol Oncol 13: 108, 2020. doi: 10.1186/s13045-020-00933-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jang HS, Shin WJ, Lee JE, Do JT. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes (Basel) 8: 148, 2017. doi: 10.3390/genes8060148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jeziorska DM, Murray RJS, De Gobbi M, Gaentzsch R, Garrick D, Ayyub H, Chen T, Li E, Telenius J, Lynch M, Graham B, Smith AJH, Lund JN, Hughes JR, Higgs DR, Tufarelli C. DNA methylation of intragenic CpG islands depends on their transcriptional activity during differentiation and disease. Proc Natl Acad Sci USA 114: E7526–E7535, 2017. doi: 10.1073/pnas.1703087114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.ShinyGO: a graphical gene-set enrichment tool for animals and plants | Bioinformatics | Oxford Academic [Online]. https://academic.oup.com/bioinformatics/article/36/8/2628/5688742 [15 Aug. 2022]. [DOI] [PMC free article] [PubMed]
- 47. Tenayuca J, Cousins K, Yang S, Zhang L. Computational modeling approach in probing the effects of cytosine methylation on the transcription factor binding to DNA. Curr Top Med Chem 17: 1778–1787, 2017. doi: 10.2174/1568026617666161116142031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ehrlich M, Lacey M. DNA methylation and differentiation: silencing, upregulation and modulation of gene expression. Epigenomics 5: 553–568, 2013. doi: 10.2217/epi.13.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schmidt F, Kny M, Zhu X, Wollersheim T, Persicke K, Langhans C, Lodka D, Kleber C, Weber-Carstens S, Fielitz J. The E3 ubiquitin ligase TRIM62 and inflammation-induced skeletal muscle atrophy. Crit Care 18: 545, 2014. doi: 10.1186/s13054-014-0545-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cordeiro B, Terentyev D, Clements RT. BKCa channel activation increases cardiac contractile recovery following hypothermic ischemia/reperfusion. Am J Physiol Heart Circ Physiol 309: H625–H633, 2015. doi: 10.1152/ajpheart.00818.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ponnaluri VKC, Estève P-O, Ruse CI, Pradhan S. S-adenosylhomocysteine hydrolase participates in DNA methylation inheritance. J Mol Biol 430: 2051–2065, 2018. doi: 10.1016/j.jmb.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 52. Barroso M, Handy DE, Castro R. The link between hyperhomocysteinemia and hypomethylation: implications for cardiovascular disease. J Inborn Errors Metab Screen 5: 1–15, 2017. doi: 10.1177/2326409817698994. [DOI] [Google Scholar]
- 53. Adams BM, Canniff NP, Guay KP, Larsen ISB, Hebert DN. Quantitative glycoproteomics reveals cellular substrate selectivity of the ER protein quality control sensors UGGT1 and UGGT2. eLife 9: e63997, 2020. doi: 10.7554/eLife.63997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Eijssen LMT, van den Bosch BJC, Vignier N, Lindsey PJ, van den Burg CMM, Carrier L, Doevendans PA, van der Vusse GJ, Smeets HJM. Altered myocardial gene expression reveals possible maladaptive processes in heterozygous and homozygous cardiac myosin-binding protein C knockout mice. Genomics 91: 52–60, 2008. doi: 10.1016/j.ygeno.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 55. Xiong S, Van Pelt CS, Elizondo-Fraire AC, Fernandez-Garcia B, Lozano G. Loss of Mdm4 results in p53-dependent dilated cardiomyopathy. Circulation 115: 2925–2930, 2007. doi: 10.1161/CIRCULATIONAHA.107.689901. [DOI] [PubMed] [Google Scholar]
- 56. Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, Vega RB, Attie AD, Muoio DM, Kelly DP. Energy metabolic re-programming in the hypertrophied and early stage failing heart: a multi-systems approach. Circ Heart Fail 7: 1022–1031, 2014. doi: 10.1161/CIRCHEARTFAILURE.114.001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Benderdour M, Charron G, DeBlois D, Comte B, Des Rosiers C. Cardiac mitochondrial NADP+-isocitrate dehydrogenase is inactivated through 4-hydroxynonenal adduct formation: an event that precedes hypertrophy development. J Biol Chem 278: 45154–45159, 2003. doi: 10.1074/jbc.M306285200. [DOI] [PubMed] [Google Scholar]
- 58. Carey BW, Finley LWS, Cross JR, Allis CD, Thompson CB. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518: 413–416, 2015. doi: 10.1038/nature13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Grams ME, Tin A, Rebholz CM, Shafi T, Köttgen A, Perrone RD, Sarnak MJ, Inker LA, Levey AS, Coresh J. Metabolomic alterations associated with cause of CKD. Clin J Am Soc Nephrol 12: 1787–1794, 2017. doi: 10.2215/CJN.02560317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liu A, Dai Y, Mendez EF, Hu R, Fries GR, Najera KE, Jiang S, Meyer TD, Stertz L, Jia P, Walss-Bass C, Zhao Z. Genome-wide correlation of DNA methylation and gene expression in postmortem brain tissues of opioid use disorder patients. Int J Neuropsychopharmacol 24: 879–891, 2021. doi: 10.1093/ijnp/pyab043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Weyrich A, Benz S, Karl S, Jeschek M, Jewgenow K, Fickel J. Paternal heat exposure causes DNA methylation and gene expression changes of Stat3 in Wild guinea pig sons. Ecol Evol 6: 2657–2666, 2016. doi: 10.1002/ece3.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lim YC, Li J, Ni Y, Liang Q, Zhang J, Yeo GSH, Lyu J, Jin S, Ding C. A complex association between DNA methylation and gene expression in human placenta at first and third trimesters. PLoS One 12: e0181155, 2017. doi: 10.1371/journal.pone.0181155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gilsbach R, Schwaderer M, Preissl S, Grüning BA, Kranzhöfer D, Schneider P, Nührenberg TG, Mulero-Navarro S, Weichenhan D, Braun C, Dreßen M, Jacobs AR, Lahm H, Doenst T, Backofen R, Krane M, Gelb BD, Hein L. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat Commun 9: 391, 2018. doi: 10.1038/s41467-017-02762-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D'Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD. Revisiting cardiac cellular composition. Circ Res 118: 400–409, 2016.doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Litviňuková M, Talavera-López C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, Nadelmann ER, Roberts K, Tuck L, Fasouli ES, DeLaughter DM, McDonough B, Wakimoto H, Gorham JM, Samari S, Mahbubani KT, Saeb-Parsy K, Patone G, Boyle JJ, Zhang H, Zhang H, Viveiros A, Oudit GY, Bayraktar OA, Seidman JG, Seidman CE, Noseda M, Hubner N, Teichmann SA. Cells of the adult human heart. Nature 588: 466–472, 2020. doi: 10.1038/s41586-020-2797-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li E, Zhang Y. DNA Methylation in Mammals. Cold Spring Harb Perspect Biol 6: a019133, 2014. doi: 10.1101/cshperspect.a019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schär P, Fritsch O. DNA repair and the control of DNA methylation. Prog Drug Res 67: 51–68, 2011. doi: 10.1007/978-3-7643-8989-5_3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Tables S1–S4: https://doi.org/10.6084/m9.figshare.16933240.v2.
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.21235437.v2.
Supplemental list of metabolites analyzed (Metabolomics Library of Detected Metabolites Metabolon Inc): https://doi.org/10.6084/m9.figshare.20483664.v1.
Data Availability Statement
The raw FASTQ and GEO data sets for DNA methylation analysis and RNAseq can be found under accession number PRJNA884710 at https://www.ncbi.nlm.nih.gov/bioproject/884710.
The raw DMC, DMR, RNA-Seq, and metabolomics files can be found in EXCEL format at the permanent link within the University of Florida Digital Collections repository at https://original-ufdc.uflib.ufl.edu//IR00011706/00001.