

Abstract
Antigen heterogeneity that results in tumor antigenic escape is one of the major obstacles to successful chimeric antigen receptor (CAR) T cell therapies in solid tumors including glioblastoma multiforme (GBM). To address this issue and improve the efficacy of CAR T cell therapy for GBM, we developed an approach that combines CAR T cells with inhibitor of apoptosis protein (IAP) antagonists, a new class of small molecules that mediate the degradation of IAPs, to treat GBM. Here, we demonstrated that the IAP antagonist birinapant could sensitize GBM cell lines and patient-derived primary GBM organoids to apoptosis induced by CAR T cell-derived cytokines, such as tumor necrosis factor. Therefore, birinapant could enhance CAR T cell-mediated bystander death of antigen-negative GBM cells, thus preventing tumor antigenic escape in antigen-heterogeneous tumor models in vitro and in vivo. In addition, birinapant could promote the activation of NF-κB signaling pathways in antigen-stimulated CAR T cells, and with a birinapant-resistant tumor model we showed that birinapant had no deleterious effect on CAR T cell functions in vitro and in vivo. Overall, we demonstrated the potential of combining the IAP antagonist birinapant with CAR T cells as a novel and feasible approach to overcoming tumor antigen heterogeneity and enhancing CAR T cell therapy for GBM.
Keywords: IAP antagonist, birinapant, CAR T cell, glioblastoma, antigen heterogeneity
Graphical abstract
Antigen heterogeneity is a major obstacle to successful CAR T cell therapies in solid tumors. Using an antigen-heterogeneous glioblastoma model, Milone and colleagues showed that combining the IAP antagonist birinapant with CAR T cells could enhance bystander tumor death, thus limiting antigenic escape and improving the efficacy of CAR T cell therapy.
Introduction
Despite the success of chimeric antigen receptor (CAR) T cell therapies in treating hematologic malignancies,1,2 CAR T cell therapies have not been effective in treating solid tumors, including glioblastoma multiforme (GBM).3,4 GBM is the most common and aggressive brain cancer, and it remains incurable with a median survival time of 16.9 months under the current standard of care.5 CAR T cell therapies targeting GBM have shown evidence of anti-tumor activity in early-phase studies.6,7,8 However, there remain numerous challenges. A phase I clinical trial of CAR T cells targeting the highly tumor-specific antigen, epidermal growth factor receptor variant III (EGFRvIII), in patients with recurrent GBM showed CAR T cell infiltration into tumors, with evidence of CAR T cell activation and EGFRvIII reduction.7 Nevertheless, the tumors continued to progress, suggesting antigenic escape as an important mechanism of resistance, likely related to the heterogeneous expression of EGFRvIII within GBM tumors.9 To address the issue of antigen heterogeneity and improve the efficacy of CAR T cell therapy, various approaches are under investigation. Here, we hypothesize that combining CAR T cells with small molecules that antagonize inhibitor of apoptosis proteins (IAPs) can limit antigenic escape in GBM by sensitizing antigen-negative tumor cells to apoptosis induced by CAR T cell-derived cytokines.
IAPs are frequently upregulated in many types of tumors including GBM,10,11 in which they inhibit apoptotic pathways.10 Family members such as cellular IAP1 (c-IAP1) and c-IAP2 can ubiquitinate receptor-interacting protein 1 (RIP1) to block the extrinsic apoptotic pathway mediated by the death receptors, such as tumor necrosis factor (TNF) receptor 1;12 X chromosome-linked IAP (XIAP) can directly bind to and inhibit caspase 3, 7, and 9 to prevent the downstream execution of apoptosis.13,14 Endogenously, IAPs are antagonized by second mitochondria-derived activator of caspase (SMAC).15 To mimic the function of endogenous SMAC peptide, IAP antagonists (also known as SMAC mimetics or IAP inhibitors) have been developed as small molecules that can bind to and induce the conformational change of IAPs, which subsequently results in their auto-ubiquitination and proteasomal degradation.16 By downregulating IAPs, IAP antagonists could unblock apoptotic pathways in GBM cells, thus sensitizing them to apoptosis induced by TNF-α through TNF receptor 1.16,17 Since TNF-α is a central cytokine produced by activated CAR T cells while they kill antigen-positive tumor cells,18 combining IAP antagonists with CAR T cells may limit tumor antigenic escape by enhancing bystander cell death of antigen-negative tumor via TNF-α-triggered apoptosis.
In addition to the anti-tumor activity, IAP antagonists have been demonstrated to increase immune cell functions by promoting nuclear factor κB (NF-κB)-inducing kinase (NIK)-mediated NF-κB pathways.19,20 NIK is constitutively repressed by c-IAP1/2 proteins, which ubiquitinate NIK for proteasomal degradation via the adaptor proteins TNFR-associated factor 2 (TRAF2) and TRAF3.16,21 By inducing the degradation of c-IAP1/2 proteins, IAP antagonists would release NIK to activate the non-canonical as well as the canonical NF-κB pathways,16,22,23,24 and both pathways could promote T cell survival, proliferation, and cytokine production such as TNF-α.25
IAP antagonists have shown promising anti-tumor effects when combined with other immunostimulatory agents in preclinical settings, including in a GBM model, in which IAP antagonists synergized with oncolytic virus and immune checkpoint inhibitors to treat GBM in mice.26 More recently, IAP antagonists were demonstrated to be the top candidates that sensitize B cell acute lymphoblastic leukemia and diffuse large B cell lymphoma cells to anti-CD19 CAR T cell-mediated killing in a compound screening assay.27 In addition, IAP antagonists were shown to enhance mouse CAR T cell anti-tumor efficacy in a colorectal cancer model.28 Here, we investigated whether IAP antagonists could enhance CAR T cell therapy in an antigen-heterogeneous GBM setting by limiting tumor antigenic escape through increased bystander tumor cytotoxicity. Taking advantage of an IAP antagonist-resistant tumor model, we also explored the effects of IAP antagonist treatment on human CAR T cell functions. In our study, we showed that the IAP antagonist birinapant promotes CAR T cell-mediated bystander death of the antigen-negative GBM cells in vitro and in vivo by sensitizing them to TNF-α-induced apoptosis, and birinapant does not negatively impact CAR T cell functions in vitro and in vivo, while it may enhance CAR T cell persistence in a donor-dependent manner. Overall, our data support the potential for combining IAP antagonists with CAR T cells as a novel approach to addressing the issue of antigen heterogeneity and improving the efficacy of CAR T cell therapy for GBM.
Results
The IAP antagonist birinapant sensitizes GBM cell lines and primary GBM organoids to cytokine-induced apoptosis
Several IAP antagonists have reached clinical phase development, although their anti-tumor efficacy appears very limited despite evidence of on-target pharmacodynamic activity.29 We chose to focus on birinapant30 in combination with CAR T cells, as birinapant is one of the most clinically advanced among the IAP antagonists with a well-established safety profile.29 As expected, birinapant efficiently induced the degradation of c-IAP1 in GBM cell lines within 2 h (Figure 1A). c-IAP2 was not detectable among the cell lines tested. We also observed that birinapant sensitized GBM cells to TNF-α-induced cell death (Figure 1B). While TNF-α alone did not affect tumor cell viability and birinapant alone only had a modest or limited effect, TNF-α combined with birinapant exhibited synergistic killing of the GBM cell lines M059K and U118. To confirm that the cell death induced by birinapant and TNF-α was due to apoptosis, we used imaging-based assays and demonstrated an increased percentage of cells with active caspase 3/7 after treatment with birinapant and TNF-α for 24 h compared with the vehicle control, indicating increased apoptotic cell death (Figure 1C). However, we also found that GBM cell lines such as U87 and U251 were resistant to the combination of birinapant with TNF-α (Figure 1B), despite the induction of c-IAP1 downregulation by birinapant (Figure 1A) and detectable cell-surface expression of TNF receptor 1 (Figure S1A). Given that cell lines may not retain the same behaviors as primary tumors, we further investigated the sensitivity of patient-derived GBM organoids (GBOs) to birinapant and TNF-α. This GBO tumor model has been shown to maintain the transcriptome and mutations of the original tumors, including the heterogeneous expression of antigens.31 Upon screening eight GBO cultures from different patients with birinapant and either TNF-α or CAR T cell-derived conditioned medium, we observed that half of the GBOs (#7790, #9469, #9468, and #9446) showed increased tumor cell death when birinapant was present compared with TNF-α or the conditioned medium alone (Figure 1D). Based on these results, we decided to explore whether combining birinapant with CAR T cells could enhance bystander killing of tumor cells lacking the CAR target antigen, a situation found in natural tumors with intratumor heterogeneous antigen expression.
Figure 1.

The IAP antagonist birinapant sensitizes GBM cell lines and primary GBM organoids to cytokine-induced apoptosis
(A) Relative abundance of c-IAP1 protein normalized to β-actin loading control by western blotting assay from GBM cell lines treated with vehicle (0.1% DMSO) or 1 μM birinapant for 2 h. (B) Relative cell viability of GBM cell lines after treatment with vehicle (0.1% DMSO), 1 μM birinapant, 100 pg/mL TNF-α, or 100 pg/mL TNF-α with 1 μM birinapant for 48 h. (C) Percentage of M059K cells with active caspase 3/7 by imaging after treatment with vehicle (0.1% DMSO) or 1 μM birinapant with 100 pg/mL TNF-α for 24 h. (D) Relative cell viability of primary GBOs after treatment with vehicle (0.1% DMSO), 1 μM birinapant, 1 ng/mL TNF-α, 1 ng/mL TNF-α with 1 μM birinapant, 1:2 diluted CAR T cell-derived conditioned medium (CM), or 1:2 diluted CM with 1 μM birinapant for 5 days. Cell viability was normalized to the vehicle control in (B) and (D). p values were determined by paired Student’s t test (A), one-way ANOVA with Dunnett’s multiple comparisons test (B), unpaired Student’s t test (C), or one-way ANOVA with Šídák’s multiple comparisons test (D) using data from three independent experiments. ∗p < 0.05.
Birinapant promotes CAR T cell-mediated bystander death of GBM cells and enhances apoptosis in CAR T cell-treated antigen-heterogeneous primary GBOs
EGFRvIII is a tumor-specific antigen found in approximately 30% of GBM cases.32,33 However, established GBM cell lines do not express this antigen, so we generated GBM cell lines that express EGFRvIII by lentiviral transduction followed by cell sorting, and our human CAR T cells were generated to specifically target EGFRvIII (2173-28ζ). To verify the specificity of our CAR T cells, we co-cultured the CAR T cells or non-transduced (NTD) T cells of the same donor with M059K-parental or M059K-EGFRvIII cells, and we showed that our CAR T cells only directly killed M059K-EGFRvIII cells but spared antigen-negative M059K-parental cells (Figure 2A). Next, to test whether combining birinapant with CAR T cells could enhance the bystander killing of antigen-negative GBM cells that are expected to contribute to tumor antigenic escape, we established a co-cultured system in which luciferase-labeled M059K-parental cells were mixed with unlabeled M059K-EGFRvIII cells to permit monitoring of the bystander killing. In this antigen-heterogeneous tumor setting, the bystander cell death of M059K-parental cells after 24 h was significantly enhanced by birinapant in combination with the CAR T cells compared with the CAR T cells alone (Figure 2B), and the combined treatment of birinapant and the CAR T cells eliminated the bystander M059K-parental cells after 48 h, which was not achieved by either single agent (Figure S1C). The CAR T cell specificity (Figure S1B) and the enhanced bystander killing with birinapant (Figures 2B and S1C) was also demonstrated with another sensitive cell line, U118.
Figure 2.

Birinapant promotes CAR T cell-mediated bystander death of GBM cells and enhances apoptosis in CAR T cell-treated antigen-heterogeneous primary GBOs
(A) Relative cell viability of M059K-parental or M059K-EGFRvIII cells after co-culture with NTD T or CAR T cells at the indicated E/T ratios for 24 h. (B) Relative cell viability of the bystander GBM parental cells in a co-culture with EGFRvIII-expressing GBM cells and NTD T or CAR T cells (at E/T ratio of 1:10) in the presence of vehicle (0.1% DMSO) or 1 μM birinapant for 24 h. (C) Relative cell viability of GBM cells after treatment with vehicle (0.1% DMSO), 1 μM birinapant, 1:100 diluted CAR T cell-derived conditioned medium (CM), or 1:100 diluted CM with 1 μM birinapant for 24 h. (D) Relative cell viability of M059K cells after treatment with 1 μM birinapant with 1:100 diluted CM in the presence of the indicated concentrations of infliximab for 24 h. (E) Quantification of cleaved caspase 3 intensity by confocal microscopy in primary GBOs after co-culture with CAR T cells (at E/T ratio of approximately 1:30) in the presence of vehicle (0.01% DMSO) or 1 μM birinapant for 48 h. Cell viability was normalized to the vehicle control without T cells in (A), (B), (C), and (D). p values were determined by two-way ANOVA with Dunnett’s multiple comparisons test (A), one-way ANOVA with Šídák’s multiple comparisons test (B) or with Dunnett’s multiple comparisons test (C and D) using data from three independent experiments, or Welch’s t test (E). Significance was compared with CAR T versus M059K-parental at the indicated E/T ratio in (A), and with 0 μg/mL infliximab in (D). Results are presented as means ± SD in (A), (D) and (E). ∗p < 0.05.
To investigate whether this enhanced bystander cell death was mediated by soluble components released from the CAR T cells, we prepared CAR T cell-derived conditioned medium by stimulating the CAR T cells with EGFRvIII-expressing tumor cells and harvesting the supernatant from the culture after 24 h. We treated luciferase-labeled GBM-parental cells with birinapant and the conditioned medium and quantified the cell viability. We showed that birinapant with the conditioned medium significantly killed M059K and U118-parental cells (Figure 2C), indicating that the soluble components produced by the activated CAR T cells were enough to induce the bystander cell death of sensitive tumor cells. To further ask whether this bystander cell death depended on TNF-α, we applied varying concentrations of the TNF-α-neutralizing infliximab to the cell culture when treating M059K cells with birinapant and the conditioned medium or 10 pg/mL TNF-α (positive control), which was equivalent to the concentration of TNF-α in the diluted conditioned medium (Figure S1D). As shown in Figure 2D, infliximab could partially rescue the viability of M059K cells treated with birinapant and the conditioned medium while the rescuing effect plateaued at higher infliximab concentrations, which indicated that TNF-α played an important role in mediating the bystander cell death, although other secretory products from the CAR T cells were likely involved. To explore whether other death receptor ligands could also induce tumor cell death when combined with birinapant, we treated M059K and U118 cells with TNF-related apoptosis-inducing ligand (TRAIL), which is expressed by CAR T cells. Similar to TNF-α, TRAIL treatment alone did not affect tumor cell viability, but combined with birinapant TRAIL induced significant M059K and U118 cytotoxicity (Figure S1E). However, the presence of TRAIL-neutralizing antibody failed to significantly rescue the viability of M059K cells treated with birinapant and the conditioned medium (Figure S1F), indicating that TNF-α was the dominant cytokine in the conditioned medium inducing M059K cell death with birinapant.
Next, we combined birinapant with CAR T cells to treat the sensitive GBOs and quantified the apoptosis marker, cleaved caspase 3, by confocal imaging. Because the sensitive GBOs that we screened did not have EGFRvIII mutation, we used another CAR construct, 806-28ζ, which recognizes overexpression of wild-type EGFR on tumor cells in addition to EGFRvIII.34 Our data showed that cleaved caspase 3 was significantly upregulated in the presence of birinapant for three of the GBOs compared with the CAR T cells alone (Figure 2E). Although there was no significant increase in apoptosis with GBO #9446 despite its sensitivity to the combination of birinapant and TNF-α, this may have been due to high baseline cytotoxicity by CAR T cells alone, as evidenced by the high cleaved caspase 3 signals, precluding the ability to see further enhancement by birinapant. Overall, these results indicate that combining birinapant with CAR T cells is a promising approach to enhance the CAR T cell therapies for GBM patients, and this primary tumor model can be capitalized to screen patient tumor biopsies to determine their sensitivity to birinapant. Furthermore, these GBOs can be utilized for uncovering biomarkers that predict the sensitivity of patients’ tumors to birinapant as well as for developing methods to overcome the resistance.
Birinapant promotes the activation of NF-κB pathways in CAR T cells and has no negative impact on CAR T cell functions in vitro
Since we are combining CAR T cells with birinapant to treat cancer, it is important to understand birinapant’s effects on CAR T cells. We hypothesized that birinapant would enhance CAR T cell functions by promoting NF-κB pathways. NIK is constitutively repressed by c-IAP1/2, so eliminating c-IAP1/2 in CAR T cells by birinapant should relieve the repression of NIK, which activates NF-κB pathways, leading to increased cell survival, proliferation, and cytokine production. By western blotting assays, we first confirmed that birinapant significantly induced the degradation of c-IAP1/2 in primary T cells of multiple donors (Figure S2A). We then evaluated the abundance of NF-κB subunit p52 relative to its precursor, p100, after birinapant treatment, and we detected significantly increased p52/p100 ratios, which was reflective of the activation of the non-canonical NF-κB pathway (Figure S2B). In addition, we detected birinapant-mediated activation of the canonical NF-κB pathway in the treated primary T cells, which was indicated by the upregulation of phosphorylated p65 (Figure S2C). To further investigate whether birinapant promotes the activation of both the non-canonical and canonical NF-κB pathways in antigen-stimulated CAR T cells, we stimulated our EGFRvIII-specific 2173-28ζ CAR T cells of multiple donors with EGFRvIII-conjugated m450 Dynabeads in the presence of birinapant. A significantly increased p52/p100 ratio was observed with birinapant compared with the vehicle control, indicating the activation of the non-canonical NF-κB pathway (Figure 3A). Although the canonical NF-κB pathway was activated by the CAR signaling alone, birinapant further promoted the activation of the canonical NF-κB pathway (phosphorylated p65 upregulation) in the stimulated CAR T cells (Figure 3B).
Figure 3.

Birinapant promotes the activation of NF-κB pathways in CAR T cells and has no negative impact on CAR T cell functions in vitro
(A) Relative abundance of NF-κB subunit p52 normalized to its precursor, p100, by western blotting assay from non-stimulated CAR T cells or CAR T cells stimulated by EGFRvIII-conjugated m450 Dynabeads (at bead-to-cell ratio of 3:1) in the presence of vehicle (0.1% DMSO), 1 μM birinapant, or 10 μM birinapant for 24 h. (B) Relative abundance of phosphorylated p65 normalized to total p65 by western blotting assay from samples as in (A). (C) Schematic of in vitro repeated stimulation assay. (D) Percentage of EdU + CAR T cells by flow cytometry after 1-hour incubation with 10 μM EdU at 37°C on day 12 and day 19. (E) CAR T cell viability on day 12 and day 19, calculated as (live CAR T cell count)/(live + dead CAR T cell count) × 100%. (F) CAR T cell population doubling curve for each donor. p values were determined by randomized block (matching the data for each donor) one-way ANOVA with Dunnett’s multiple comparisons test (A and B) using data from three independent experiments with different T cell donors, or randomized block (matching the data for each donor) two-way ANOVA with Dunnett’s multiple comparisons test (D and E) using data from three independent experiments, each performed with all four donors side by side. Results are presented as means ± SD in (F). ∗p < 0.05; ns, not significant.
To determine how birinapant affects CAR T cell viability, proliferation, and TNF-α production in vitro, we performed repeated stimulation assays with our 2173-28ζ CAR T cells of multiple donors in the presence or absence of birinapant. As illustrated in Figure 3C, we used the birinapant-resistant U87-EGFRvIII cell line to repeatedly restimulate 2173-28ζ CAR T cells every 7 days. The supernatant was collected from the cell culture after 24 h of each restimulation, followed by ELISA assays to detect TNF-α concentration. We enumerated the CAR T cells by imaging-based cell counting with live and dead cell staining, and the birinapant concentrations were kept constant when feeding cells. To compare proliferation rates, we performed 5-ethynyl-2′-deoxyuridine (EdU) incorporation assays with flow cytometry on day 12 and day 19. We found that birinapant promoted CAR T cell proliferation as shown by the increased fraction of EdU+ cells in birinapant-treated CAR T cells compared with the vehicle control on day 12 and day 19 (Figure 3D). Birinapant also had a minimal impact on CAR T cell viability, and a significant increase in CAR T cell viability was observed on day 12 with 1 μM birinapant treatment (representative data from day 12 and day 19 are shown in Figure 3E). Although the overall CAR T cell expansion after 21 days was not significantly different between birinapant and the vehicle-treated CAR T cells when the data of all the donors were pooled (Figure S2D), birinapant did enhance the proliferation and survival of CAR T cells of two donors (ND520 and ND535) at late time points after repetitive stimulations (Figure 3F), and it was noticeable that these donors appeared to have the poorest expansion with the vehicle. When we compared the cell expansion after the third antigen restimulation, the effect of birinapant was significant with the pooled data (Figure S2E), indicating increased persistence of CAR T cells by birinapant. Our data also showed that a higher dose of birinapant could mildly promote TNF-α production by CAR T cells after the repetitive stimulations (Figure S2F), and the effect was not significant with a lower dose of birinapant. Additionally, we did not observe a significant difference in CAR T cell effector function such as degranulation (Figure S2G) or direct killing of the birinapant-resistant U87-EGFRvIII cells (Figure S2H) in the presence of birinapant. Overall, the positive effects of birinapant on CAR T cells were not prominent, but importantly there was no evidence of birinapant having a negative impact on CAR T cell function in vitro, supporting the potential for combining CAR T cells with this small molecule.
Birinapant does not significantly affect CAR T cell functions in vivo
Given the promising in vitro results of combining birinapant with CAR T cell therapy, we investigated the effect of birinapant on CAR T cells in vivo. To isolate the effect of birinapant on CAR T cells from its anti-tumor effect, we first evaluated the combination using the birinapant-resistant tumor U87-EGFRvIII, which was resistant to the cytokine and birinapant-mediated cytotoxicity (Figures 1B and S3A). NSG mice with pre-established subcutaneous U87-EGFRvIII tumors were intravenously (i.v.) administered 5 × 106 2173-28ζ CAR T cells, followed by intraperitoneal (i.p.) injections with either birinapant (10 mg/kg) or the vehicle (12.5% Captisol) every 3 days for a total of 10 doses beginning on the day of CAR T cell administration. Tumor regression was observed in both groups, with similar kinetics and complete regression by day 12 (Figure 4A). Higher concentrations of TNF-α and interferon-γ (IFN-γ) were detected in the plasma of mice treated with birinapant compared with the vehicle control on day 7 (Figure 4B). To study whether birinapant may enhance CAR T cell persistence in vivo, we rechallenged the mice with new U87-EGFRvIII tumors on day 22. Mice treated with birinapant or the vehicle control during the primary tumor challenge were able to efficiently defend against the secondary tumor challenge (Figure 4A). CAR T cell concentrations (Figure 4C) and immunophenotype (Figure 4D) in peripheral blood were similar between birinapant and the vehicle-treated mice. A similar study performed with a lower CAR T cell dose demonstrated very similar results albeit, with an expected less efficient tumor clearance during primary and secondary tumor challenges (Figures S3B–S3E). Overall, these data indicate that CAR T cell engraftment and functions remain intact during birinapant treatment, which is important when combining this small molecule with CAR T cell therapy to enhance anti-tumor activity.
Figure 4.

Birinapant does not significantly affect CAR T cell functions in vivo
NSG mice were subcutaneously implanted with 0.5 × 106 birinapant-resistant U87-EGFRvIII-CBG cells on the right flank. After 6 days (day 0), 5 × 106 2173-28ζ CAR T cells were intravenously injected. The vehicle (12.5% Captisol) or birinapant at 10 mg/kg were given by intraperitoneal injections every 3 days for a total of 10 doses since day 0. The mice were rechallenged with 3 × 106 U87-EGFRvIII-CBG cells subcutaneously implanted in the left flank on day 22. (A) Geometric mean of bioluminescent total flux by imaging from U87-EGFRvIII-CBG tumors in each treatment group at the indicated time points. (B) Concentration of TNF-α and IFN-γ in mouse plasma on day 7 by ELISA. (C) Geometric mean of human CAR T cell (CD45+/CD3+) count in mouse blood in each treatment group at the indicated time points. (D) Percentage of CD4 (CD4+/CD8−), CD8 (CD4−/CD8+), naïve-like (CCR7+/CD45RO−), central memory (CCR7+/CD45RO+), effector memory (CCR7−/CD45RO+), and effector (CCR7−/CD45RO−) T cell subset among total human CAR T cells (CD45+/CD3+) in mouse blood on day 21 by flow cytometry. BLI, bioluminescent imaging. p values were determined by unpaired t tests in (B). Results are presented as geometric means ± geometric SD in (A) and (C), and as means ± SD in (B). ∗p < 0.05.
Birinapant enhances CAR T cell treatment in vivo for GBM by preventing tumor antigenic escape
To determine whether combining birinapant with CAR T cells could enhance the bystander death of birinapant-sensitive tumors and limit tumor antigenic escape in vivo for GBM, we used the GBM cell line U118, which was subject to TNF-α-mediated killing when co-treated with birinapant (Figure 1B). Since U118 lacks EGFRvIII, we generated U118-EGFRvIII cell line expressing click beetle green (CBG) luciferase, while the U118-parental cell line was labeled with click beetle red (CBR) luciferase. By imaging the bioluminescence with a set of emission filters for mice bearing pure U118-parental-CBR tumors or pure U118-EGFRvIII-CBG tumors, we established a spectral unmixing library that allowed us to differentiate the CBR and CBG bioluminescence signal from the two U118 tumor lines in vivo. NSG mice were then subcutaneously implanted with a mixture of U118-parental-CBR cells with U118-EGFRvIII-CBG cells (4:1 ratio) to mimic the heterogeneous expression of tumor antigen EGFRvIII, followed by i.v. treatment with either 2173-28ζ CAR T cells (0.5 × 106) or NTD T cells of the same donor, combined with birinapant (10 mg/kg) or the vehicle (12.5% Captisol) i.p. every 3 days for a total of 10 doses. Although tumor size as assessed by caliper measurement showed complete regression (Figures 5A and 5B) and the EGFRvIII-specific CAR T cells efficiently eliminated U118-EGFRvIII-CBG cells regardless of the presence of birinapant (Figure 5C), the CAR T cells with the vehicle incompletely eliminated antigen-negative U118-parental-CBR tumor cells, as all mice had a bioluminescent signal that was 1–2 orders of magnitude above the background up to day 33 (Figures 5D and 5E). In contrast, the combined treatment of the EGFRvIII-specific CAR T cells with birinapant led to a greater reduction in U118-parental-CBR tumor signal to background levels by day 33 for most mice (Figures 5D and 5E). Similar results were observed in a repeat experiment (Figure S4). In addition, in both experiments, birinapant did not significantly affect the tumor growth compared with the vehicle among mice given the NTD T cells (Figures 5A, 5B, and S4D). Western blotting assay with the tumor tissue lysates from these mice demonstrated c-IAP1 downregulation by birinapant (Figure S4E), confirming the pharmacologic effect of birinapant, but also illustrating a requirement for CAR T cells for tumor regression.
Figure 5.

Birinapant enhances CAR T cell treatment in vivo for GBM by preventing tumor antigenic escape
NSG mice were subcutaneously implanted with a mixture of 4 × 106 U118-parental-CBR and 1 × 106 U118-EGFRvIII-CBG cells on the right flank. After 14 days (day 0), 0.5 × 106 2173-28ζ CAR T cells or the equivalent number of NTD T cells were intravenously injected. The vehicle (12.5% Captisol) or birinapant at 10 mg/kg were given by intraperitoneal injections every 3 days for 10 total doses since day 0. (A) Tumor size of each mouse by caliper measurement at the indicated time points. (B) Tumor size by caliper measurement on day 21. (C) Geometric mean of spectrally unmixed bioluminescent total flux from U118-EGFRvIII-CBG cells in each treatment group at the indicated time points. (D) Geometric mean of spectrally unmixed bioluminescent total flux from U118-parental-CBR cells in each treatment group at the indicated time points. (E) Log10 of spectrally unmixed bioluminescent total flux from U118-parental-CBR cells on day 33. (F) Mean of mouse net weight change from day 0. BLI, bioluminescent imaging. Solid and dotted black lines in (C), (D), and (E) indicate median and range of background total flux from non-tumor area of each mouse at the indicated time points. p values were determined by unpaired t tests (B and E). Results are presented as means ± SD in (B), (E), and (F), and as geometric means ± geometric SD in (C) and (D). ∗p < 0.05; ns, not significant.
Lastly, we evaluated our approach of combining birinapant with CAR T cells in an orthotopic mouse model of GBM, in which 4:1 mixed U118-parental-CBR and U118-EGFRvIII (without luciferase) cells were implanted into the intracranial space of NSG mice. In an initial experiment, similar to what was observed in the subcutaneous model, the bioluminescent signal from antigen-negative U118-parental-CBR cells was reduced to background levels in all mice treated with the EGFRvIII-specific CAR T cells (1 × 106) and birinapant, whereas antigenic escape was observed in two out of six mice treated with the CAR T cells and the vehicle (Figures S5A and S5B). A repeat experiment with a suboptimal CAR T cell dose (0.3 × 106), at which the CAR T cells and the vehicle failed to eliminate antigen-negative U118-parental-CBR tumor cells (Figure 6A), demonstrated enhanced bystander tumor death by the CAR T cells and birinapant in this model, with half of the mice achieving a bioluminescent signal at background levels by day 33 (Figure 6B). In aggregate, these results demonstrate that combining birinapant with CAR T cells enhances bystander death of antigen-negative tumor cells and prevents antigenic escape in antigen-heterogeneous tumors.
Figure 6.

Birinapant enhances CAR T cell treatment for intracranial GBM by promoting bystander tumor death
NSG mice were intracranially implanted with a mixture of 200 × 103 U118-parental-CBR and 50 × 103 U118-EGFRvIII (without luciferase) cells. After 7 days (day 0), 0.3 × 106 2173-28ζ CAR T cells were intravenously injected. The vehicle (12.5% Captisol) or birinapant at 20 mg/kg were given by intraperitoneal injections every 3 days for 10 total doses since day 0. (A) Bioluminescent total flux from U118-parental-CBR cells in each mouse at the indicated time points. (B) Log10 of bioluminescent total flux from U118-parental-CBR cells in each mouse on day 33. (C) Mean of mouse net weight change from day 0. Solid and dotted black lines in (A) and (B) indicate median and range of background total flux from non-tumor area of each mouse at the indicated time points. BLI, bioluminescent imaging. p values were determined by unpaired t tests in (B). Results are presented as geometric means ± geometric SD in (A) and as means ± SD in (B) and (C). ∗p < 0.05.
Since there was some evidence of increased serum cytokines in the birinapant-resistant U87-EGFRvIII model (Figure 4B), we also explored whether birinapant promotes the cytokine release in the orthotopic U118 model. We evaluated 32 human cytokines in mouse plasma on day 7 using a validated multiplex assay.35 However, owing to the low dose of CAR T cells (0.3 × 106) administered in this experiment (in contrast to 5 × 106 used in Figure 4), we did not detect meaningful levels of cytokines for comparison between the two treatment groups (Table S1).
Although the ability to assess toxicity is very limited in xenograft studies, there was no weight loss (Figure 5, Figure 6F, 6C, S4F, and S5C) or sign of illness such as lethargy, hunching posture at rest, or hair loss exhibited by the mice treated with birinapant in combination with CAR T cells. In addition, histopathologic analysis of major organs (lungs, livers, and kidneys) from the mouse experiment as in Figure 6 showed infiltration by atypical mononuclear cells, which is consistent with the frequently observed graft-versus-host disease lesions in mouse xenograft models with human T cells. These infiltrates were mild and observed almost entirely in the CAR T cells with vehicle group. There were other mild pathologic changes noted in these same tissues; however, there was no discernible difference in the incidence of these other lesions when comparing the birinapant-treated and vehicle control groups, as shown in Table S2. Human TNF-α is biologically active in mice,36 and birinapant has been shown to have activity in mouse cell lines.28 The absence of any appreciable toxicity is therefore noteworthy and suggests that combining birinapant with CAR T cells may not increase off-target toxicity in normal tissues; however, the safety of this combination therapy can only really be tested in human clinical trials.
Discussion
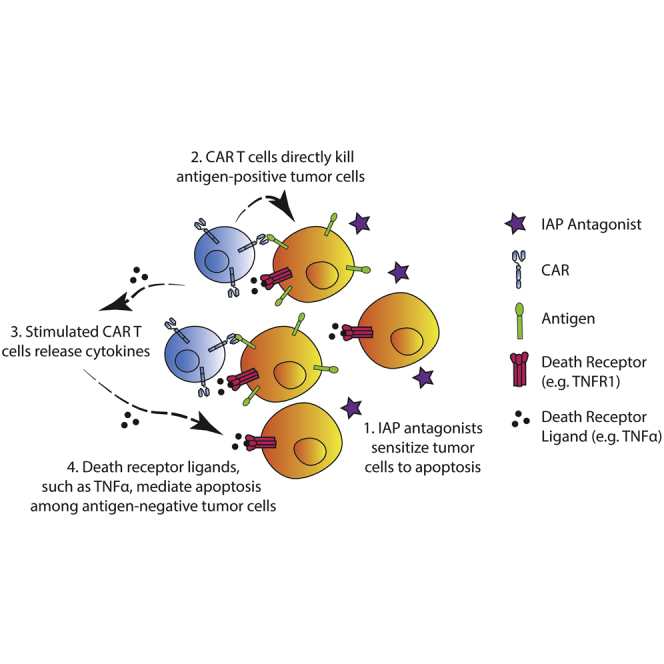
Antigen heterogeneity that results in tumor antigenic escape is one of the major obstacles to successful CAR T cell therapies, especially in solid tumors such as GBM.3,4 Approaches to overcoming resistance due to antigen loss have largely focused on CAR T cells with multiple antigen specificities. Our results suggest that approaches that enhance bystander cytotoxicity of CAR T cells might be a viable alternative strategy. Using a GBM model with heterogeneously expressed antigen, we showed that combining the IAP antagonist birinapant with CAR T cells could sensitize antigen-negative tumor cells to cytokine-induced apoptosis, therefore preventing antigenic escape and improving the efficacy of CAR T cell therapy (Figure 7).
Figure 7.

Schematic of the proposed working model for combining the IAP antagonist birinapant with CAR T cell therapy to treat antigen-heterogeneous GBM
(1) Birinapant sensitizes tumor cells to death receptor-mediated cell death. (2) Antigen-specific CAR T cells are stimulated by antigen-positive cells and mediate on-target cytotoxicity. (3) Stimulated CAR T cells release cytokines, such as TNF-α. (4) TNF-α, together with other death receptor ligands, induces apoptosis among the rest of antigen-negative tumor cells.
Nevertheless, our data with glioblastoma cell lines and primary GBOs suggest that not all tumors are subject to birinapant and cytokine-mediated apoptosis (Figure 1). A published report37 also described varied sensitivity of cancer cell lines to cytotoxicity induced by an IAP antagonist combined with TNF-α or TRAIL, and most of the resistant cell lines became sensitive after knockdown of cellular FLICE-like inhibitory protein (c-FLIP), an alternative anti-apoptotic protein that stops the extrinsic apoptotic pathway independently of c-IAP1/2. Indeed, we observed that knockout of c-FLIP in U87 cells significantly increased their sensitivity to apoptotic cell death upon treatment with birinapant and TNF-α (Figure S6A), although knockout of c-FLIP in U251 cells only mildly reduced their resistance to this combined treatment. These results indicate that c-FLIP plays an important role in regulating the resistance to birinapant and death receptor ligand-mediated cytotoxicity, though it may not be the sole mechanism. Although c-FLIP would be a reasonable target to modify resistance to apoptosis in tumor cells, the design of c-FLIP-specific inhibitors has proven challenging, in part due to the fact that c-FLIP functions by structurally resembling caspase 8,38 a crucial initiator caspase along the extrinsic apoptotic pathway. Drugs such as paclitaxel and bortezomib may indirectly downregulate c-FLIP, which could render U87 and U251 cells more susceptible to birinapant with TNF-α, although both drugs were potent cytotoxic agents by themselves, eclipsing the effect of birinapant and TNF-α (Figures S6B and S6C). Anti-sense oligonucleotide approaches may be another method to inhibit c-FLIP39 and to provide a potential solution for resistant tumors, although targeting c-FLIP may also impact CAR T cell viability, as discussed below, and requires further assessment of its safety. There are also likely other mechanisms that confer tumors resistance to birinapant, such as loss or mutations in genes that are critical to apoptotic pathways. Our primary GBO model may provide insight into these mechanisms in GBM as well as provide a platform for uncovering biomarkers that predict tumor sensitivity and developing methods to overcome the resistance. Meanwhile, the patient-derived GBOs can be directly applied for sensitivity screening to stratify the patients who are most likely to benefit from our approach.
Multiple IAP antagonists have been developed in the past two decades, with a handful of them being investigated in clinical studies for cancer, either as a single agent or as combination therapy. IAP antagonists are categorized into monovalent or bivalent compounds based on their structure, with the bivalent structure being more potent to antagonize IAPs.40 Birinapant is a bivalent compound with tolerable toxicity and higher anti-tumor activity than monovalent IAP antagonists in preclinical studies.10,30 Our studies focused on birinapant, as this IAP antagonist is one of the most advanced in the clinical setting with a reasonable safety profile, while other IAP antagonist combinations with CAR T cells may also be worth exploring, since differences in pharmacokinetics and pharmacodynamics exist. Although birinapant has been well tolerated in phase I/II clinical trials29 and there was a lack of evidence for toxicity with mice treated with birinapant and CAR T cells in our studies, the safety of our combined treatment in humans is yet to be studied. Although highly speculative, this combination therapy may even reduce the risk of cytokine-mediated toxicity by lowering the CAR T cell dose necessary to achieve anti-tumor efficacy; because birinapant could sensitize tumor cells to cytokine-induced apoptosis, less cytokine would be required to result in an anti-tumor effect, which should translate to a lower CAR T cell dose needed to achieve efficacy with less risk of inflammation and systemic cytokine-mediated toxicity such as cytokine release syndrome (CRS). Although neurotoxicity by CAR T cell therapy for brain tumor always remains a concern, we note that GBM patients treated with EGFRvIII-targeting CAR T cells did not show appreciable CRS, neurotoxicity, or any other significant adverse effects in an early-phase clinical trial, despite evidence of CAR T cell trafficking to the GBM tumor with associated reduction of the EGFRvIII antigen.7
Drug penetration into tumors is an important limitation for small-molecule therapeutics, and the blood-brain barrier (BBB) represents one of the formidable challenges. It has been shown that IAP antagonists can effectively reach intracranial tumors in preclinical murine GBM models,26 consistent with the enhanced anti-tumor efficacy with birinapant observed in our study. However, we cannot rule out the possibility that intracranial implantations may cause disruption of the BBB in the mouse models. Even though a compromised BBB is seen in nearly all GBM patients, tumor regions with an intact BBB are prevalent among patients.41 Therefore, whether birinapant can efficiently cross the human BBB remains a relevant question in these cases and requires further investigation.
IAP antagonists have been shown to modulate innate and adaptive immune cell functions by inhibiting c-IAP1/2.19 In the context of combining with CAR T cells, our data suggest that birinapant does not negatively affect CAR T cell functions in our in vitro and in vivo models. CAR T cells could resist death receptor-mediated apoptosis in the absence of c-IAPs most likely thanks to the upregulation of c-FLIP short (c-FLIPS) upon T cell activation,42 which is mediated by the co-stimulatory signals from CD28 and 4-1BB of T cells.43,44 c-FLIPS prevents the formation of the death-inducing signaling complex, thus stopping the activation of the extrinsic apoptotic pathway.42,43 In addition, we observed significantly enhanced activation of NF-κB pathways by birinapant in antigen-stimulated CAR T cells (Figures 3A and 3B), particularly the non-canonical NF-κB pathway, whose activation is independent of 28ζ-CAR signaling.45 Although the enhanced NF-κB signaling induced by birinapant only resulted in increased CAR T cell persistence for selected donors in our in vitro repeated stimulation assay (Figure 3F), it is noticeable that birinapant particularly benefited CAR T cells that did not expand well (donors ND520 and ND535), in contrast to CAR T cells that demonstrated robust, exponential expansion (donors ND410 and ND539). T cell expansion is influenced by the composition of the T cell subsets in the donor. The ability of birinapant treatment to rescue the poorest expanding donor T cells may reflect differences in its effects on T cells at various stages of differentiation and deserves further exploration.
Overall, we demonstrated that co-administration of IAP antagonists such as birinapant can enhance CAR T cell therapy for GBM by leveraging T cell cytokines for bystander tumor cell killing, thereby limiting antigenic escape in the setting of antigen-heterogeneous tumor. Although the specific combination of birinapant administered with EGFRvIII-specific CAR T cells as described in our study might address only a portion of GBMs, given the short life expectancy of GBM patients under the current standard of care this approach would still represent an important advance for patients with this disease. In addition, other CAR targets with higher frequency of expression such as IL13Rα2 or Her2 might increase the addressable population. Although beyond the scope of our current study, the primary GBO model also provides a useful tool for exploring the mechanisms of resistance to this combination therapy as well as for identification of potential biomarkers that predict tumor sensitivity to IAP antagonist-enhanced apoptosis. Moreover, our approach of combining CAR T cells with IAP antagonists might also address antigenic escape issues faced by CAR T cell therapies in other tumor contexts beyond GBM and should be further investigated.
Materials and methods
Human GBM cell lines and modifications
The human GBM cell lines M059K (American Type Culture Collection [ATCC]), U118 (ATCC), U87 (ATCC), and U251 (Millipore Sigma) were cultured in Eagle’s minimum essential medium (ATCC) supplemented with 10% fetal bovine serum (Millipore Sigma), 10 mM HEPES (Gibco), 100 U/mL penicillin/streptomycin (Gibco), and 1× GlutaMax (Gibco) at 37°C in a CO2 incubator. Cell lines were modified by lentiviral transduction followed by cell sorting to steadily express CBG luciferase, CBR luciferase, EGFRvIII, or a combination of the modifications. Each cell line was expanded after receipt or cell sorting, then aliquoted to be cryopreserved in fetal bovine serum (Millipore Sigma) with 5% dimethyl sulfoxide (DMSO; Millipore Sigma). After resuscitation, cells were subcultured every 3 days and used for experiments within 1 month. Cells in culture were regularly tested to be mycoplasma negative by a MycoAlert PLUS Mycoplasma Detection Kit (Lonza). M059K, U118, and U87 cell lines were authenticated by short tandem repeat (STR) profiling at ATCC prior to purchase. U251 cell line was authenticated by STR profiling at the Genomic Analysis Core at University of Pennsylvania.
Patient-derived GBM organoids
Patient GBM tissues were collected at the Hospital of the University of Pennsylvania under an approved protocol by the University of Pennsylvania’s Institutional Review Board (IRB). Patient samples were de-identified before tissue processing. A total of eight patient cases (including recurrent cases) were used in this study to generate the data shown. Informed patient consent was obtained before the surgery.
Primary GBOs were generated and cultured as previously described.31,46 In brief, fresh GBM tissue was microdissected into 0.5–1 mm3 pieces in a sterile dissection hood. The tumor pieces were washed by PBS (Thermo Fisher Scientific) three times and treated with RBC lysis buffer (Thermo Fisher Scientific) for 5 min, followed by washing with Dulbecco’s modified Eagle’s medium (DMEM):F12 medium (Thermo Fisher Scientific) three times. The tumor pieces were then transferred to a 6-well culture plate (Corning) with 4 mL of GBO medium per well that contained 50% DMEM:F12 (Thermo Fisher Scientific), 50% Neurobasal medium (Thermo Fisher Scientific), 1× GlutaMax (Thermo Fisher Scientific), 1× non-essential amino acids (Thermo Fisher Scientific), 1× penicillin/streptomycin (Thermo Fisher Scientific), 1× N2 supplement (Thermo Fisher Scientific), 1× B27 supplement without vitamin A (Thermo Fisher Scientific), 1× 2-mercaptoethanol (Thermo Fisher Scientific), and 2.5 mg/mL human insulin (Millipore Sigma) and placed on an orbital shaker at 120 rpm in a 37°C, 5% CO2, and 90% humidity sterile incubator. The GBO medium was sterilized by filtration through a 0.2-μm polyethersulfone membrane and was changed every 2 days. The GBOs were passaged by cutting into half without dissociation.
Generation of human CAR T cells
Primary human T cells were acquired from the Human Immunology Core at the University of Pennsylvania, where peripheral blood mononuclear cells were collected from anonymous healthy donors by apheresis, followed by T cell isolation with Lymphoprep and RosetteSep Human T cell Enrichment Cocktail kits per the manufacturer’s instructions (STEMCELL Technologies). All specimens were collected under a protocol approved by the IRB at the University of Pennsylvania, and written informed consent was obtained from each donor. Primary human T cells were cultured in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum (Millipore Sigma), 10 mM HEPES (Gibco), and 100 U/mL penicillin/streptomycin (Gibco) at 37°C in a CO2 incubator. CAR constructs composed of 2173 or 806 scFv were previously described.18,34 2173 scFv specifically targets EGFRvIII, and 806 scFv is tuned to target overexpressed EGFR wild type found on tumor cells in addition to EGFRvIII. We engineered 2173-28ζ and 806-28ζ CAR constructs that consist of CD28 and CD3ζ signaling domains. The process to generate human CAR T cells was previously described.47 In brief, primary human T cells were first activated with anti-CD3 and anti-CD28 monoclonal antibodies-coated beads (Invitrogen) in the medium at 1 × 106 cells/mL for 24 h, followed by transduction with lentiviral vectors at multiplicity of infection of 3, after which the transduced T cells were enumerated by a Multisizer 4e Coulter Counter (Beckman Coulter) every other day and fed with the medium to 0.8 × 106 cells/mL density, and the CAR expression rate was determined by flow cytometry on day 7. When the median cell size fell to around 300 fL, as measured by Coulter Counter (Beckman Coulter), the CAR T cells were considered rested and were either immediately used in assays or cryopreserved in fetal bovine serum (Millipore Sigma) with 5% DMSO (Millipore Sigma). For cryopreserved CAR T cells, the CAR T cells were quickly thawed with pre-warmed medium and pelleted by centrifugation, then cultured in the medium at 37°C overnight to recover, and the viable cell number was decided by hemocytometer with trypan blue staining (Corning) before being used for functional assays.
Lentiviral vector preparation
Lentiviral vectors were produced by transfecting 293-T cells. 293-T cells were seeded in T150 flasks and cultured in RPMI 1640 medium (Gibco) supplemented 10% fetal bovine serum (Millipore Sigma), 10 mM HEPES (Gibco), and 100 U/mL penicillin/streptomycin (Gibco) at 37°C in a CO2 incubator to grow to 80% confluence, after which the cells were incubated briefly with a cocktail of 3 mL of Opti-MEM (Invitrogen) with 90 μL Lipofectamine 2000 transfection reagent (Invitrogen) that was gently mixed with 15 μg of the vector plasmid of interest and the packaging plasmids of 7 μg of pCL-VSVG, 18 μg of pRSV-REV, and 18 μg of pGAG-POL (Nature Technology). Additional culture medium was added thereafter, and the transfected 293-T cells were cultured at 37°C in a CO2 incubator. After 24 h, supernatants that contained lentiviral vectors were collected and filtered through 0.45-mm syringe filters (Argos Technologies), then centrifuged at 18,000 × g for 20 h at 4°C. The pellets of lentiviral vectors were resuspended in 2 mL of culture medium, aliquoted, and stored at −80°C.
Drugs and reagents
Birinapant was purchased from MedChemExpress and reconstituted either with DMSO (Millipore Sigma) for in vitro assays or with 12.5% Captisol (CyDex Pharmaceuticals) in sterile water (adjusted to pH 4.6 as the vehicle) for in vivo experiments, and aliquoted for storage in −80°C. Human TNF-α (Miltenyi Biotec), recombinant human TRAIL (R&D Systems), anti-TRAIL antibody (clone RIK-2; Invitrogen), and paclitaxel and bortezomib (Millipore Sigma) were acquired from the vendors and stored as instructed. Infliximab, the chimeric monoclonal anti-TNF antibody, was acquired from the pharmacy at the Hospital of the University of Pennsylvania. CAR T cell-derived conditioned medium was produced by stimulating 2173-28ζ CAR T cells with EGFRvIII-expressing GBM cells for 24 h, and the supernatant was harvested, aliquoted, and stored at −80°C. Recombinant human EGFRvIII protein with hFc-tag (SinoBiological) and M-450 Tosylactivated Dynabeads (Thermo Fisher Scientific) were used to prepare EGFRvIII-conjugated beads, as instructed by the manufacturer. The conjugated beads were counted by a Multisizer 4e Coulter Counter (Beckman Coulter) and stored at 4°C. To stimulate CAR T cells, every three beads were used for one CAR T cell after PBS (Corning) washes on a magnet rack.
Flow cytometry and cell sorting
For flow cytometry with cultured cells, the antibodies used included goat-anti-human immunoglobulin G (IgG) F(ab′)₂-AF647 (Jackson) to detect 2173-28ζ CAR expression, goat-anti-mouse IgG F(ab′)₂-AF647 (Jackson) to detect 806-28ζ CAR expression, mouse-anti-human EGFRvIII (Millipore Sigma; clone DH8.3) followed by goat-anti-mouse IgG (H + L)-AF647 secondary antibody to detect EGFRvIII expression and for cell sorting, and mouse-anti-human TNFR1-PE (R&D Systems; clone 16803) to detect TNF receptor 1. For flow cytometry analysis, cells were sampled and washed with PBS (Corning), incubated with antibodies in PBS (Corning) at room temperature in darkness for 30 min, washed again with PBS (Corning) twice, and stained with secondary antibodies if necessary (followed by additional wash steps), then evaluated on a BD LSR Fortessa flow cytometer. The cell-surface expression of the proteins of interest was analyzed by FlowJo (Tree Star). For cell sorting, cells were washed with PBS (Corning), incubated with antibodies in PBS (Corning) on ice in darkness followed by wash steps as described above, then kept in cold PBS (Corning) with 1% BSA on ice before and after sorting on a BD Influx cell sorter. Sorted cells were pelleted by centrifugation and returned to culture with warm medium. Because CBG and CBR luciferase were co-transduced with GFP (separated by a T2A self-cleaving peptide) to the cells, GFP signal was used as a surrogate to sort the luciferase-expressing cells.
For flow cytometry with mouse blood samples to evaluate human CAR T cell count and phenotypes in mouse studies, the staining panel included mouse-anti-human CD45-APC (BD Biosciences; clone HI30), mouse-anti-human CD3-BV605 (BioLegend; clone OKT3), mouse-anti-human CD4-BV510 (BioLegend; clone OKT4), mouse-anti-human CD8-APC-H7 (BD Biosciences; clone SK1), mouse-anti-human CCR7-FITC (BD Biosciences; clone 150503), and mouse-anti-human CD45RO-PE (BioLegend; clone UCHL1), and a separate panel was composed of mouse-anti-human PD1-BV421 (BioLegend; clone EH12.2H7), mouse-anti-human LAG3-PerCP/Cyanine5.5 (BioLegend; clone11C3C65), rabbit-anti-human TIM3-AF700 (R&D Systems; clone 344823), and mouse-anti-human CD45-FITC (BD Biosciences; clone HI30). After incubating mouse blood with the antibodies in PBS (Corning), 1× FACS Lysing Solution (BD Biosciences) with CountBright Absolute Counting Beads (Thermo Fisher Scientific) was added to the mouse blood, and the samples were collected on a BD LSR Fortessa flow cytometer with proper compensation setup and fluorescence-minus-one controls, followed by analysis with FlowJo (Tree Star) to calculate CD45+/CD3+ human CAR T cell concentration in mouse blood and to characterize T cell phenotypes.
Western blotting assay
Cell lysates were prepared with radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, and 50 mM Tris-HCl [pH 8.0]) supplemented with cOmplete proteasome inhibitor (Roche) and PhosSTOP phosphatase inhibitor (Roche), as previously described.45 In brief, cells were harvested from the culture, washed with cold PBS (Corning), and resuspended in the working stock of cold RIPA buffer. The lysates were incubated on ice for 30 min while being vortexed at maximum speed for 10 s at 10-min intervals, followed by centrifugation at 16,000 × g for 10 min in a refrigerated centrifuge. For tumor tissues harvested from mice, repeated sonication (QSonica Q55 Sonicator) was applied during the incubation on ice. The supernatants were collected, aliquoted, and immediately frozen at −80°C. On the day of performing western blotting assay, cell lysates were thawed on ice, and protein concentrations were measured by DC Protein Assay (Bio-Rad) per the manufacturer’s instructions. Equal quantities of protein were added to Laemmli buffer (Bio-Rad) containing 2-mercaptoethanol (Bio-Rad) and boiled at 95°C for 5 min, then loaded into NuPAGE gels (Invitrogen) for electrophoresis in SureLock Mini-Cell (Invitrogen). After separation, the proteins were transferred from the gels to nitrocellulose membranes (Bio-Rad) in Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad) immersed in 10% methanol NuPAGE transfer buffer (Thermo Fisher Scientific). After transfer, the membranes were rinsed in Tris-buffered saline (TBS), then blocked for 1 h in Intercept (TBS) Blocking Buffer (LI-COR). The membranes were then incubated with primary antibodies for the proteins of interest in Intercept (TBS) Blocking Buffer (LI-COR) with 0.5% Tween 20 (Bio-Rad) at 4°C overnight. The primary antibodies used included β-actin (Cell Signaling Technology; clone 8H10D10), c-IAP1 (CST; clone D5G9), c-IAP2 (CST; clone 58C7), XIAP (CST; clone D2Z8W), phospho-p65 (CST; clone 93H1), total p65 (CST; clone L8F6), and p52 (Millipore Sigma; catalog #05-361). Primary antibodies from the same host species (rabbit or mouse) were avoided for incubation at the same time. The membranes were washed with TBS containing 0.1% Tween 20 (TBS-T) the next day and incubated with donkey-anti-rabbit-IRDye 800CW (LI-COR), donkey-anti-mouse-IRDye 680RD (LI-COR), or both secondary antibodies in Intercept (TBS) Blocking Buffer (LI-COR) with 0.5% Tween 20 for 40 min at room temperature. The membranes were then washed again with TBS-T, rinsed once in TBS, and kept in TBS at 4°C before imaging with Odyssey CLx (LI-COR). The fluorescent signals from the proteins of interest were quantified by Image Studio (LI-COR) with median background correction setup for western blotting analysis. To detect additional proteins on membranes, the membranes were first reblocked in Intercept (TBS) Blocking Buffer (LI-COR) for 1 h at room temperature, then processed as described earlier.
In vitro viability assays
The human GBM cell lines used for in vitro viability assays were modified to express CBG luciferase. D-Luciferin (PerkinElmer) substrate solution was added to the cell culture at the indicated time points for each assay and incubated at 37°C for 10 min before quantifying the bioluminescent signal from each well with a Synergy H4 Hybrid Reader (BioTek). The in vitro viability assays with patient-derived GBOs were performed with a CellTiter-Glo 3D Cell Viability Assay Kit (Promega) per the manufacturer’s instructions. All the in vitro viability assays were performed at least three times with triplicate wells for each condition, and the bioluminescent signals were normalized to the vehicle-treated control wells as 100% viability.
Imaging-based detection of cleaved caspase activity
For adherent GBM cell lines, cells were seeded and treated in CellCarrier Ultra microplates (PerkinElmer) for the indicated time and stained with Live Caspase 3/7 ViaStain with Hoechst (Nexcelom Bioscience) per the manufacturer’s instructions. The plates were then imaged by Celigo Image Cytometer (Nexcelom Bioscience), followed by data processing on the same machine. Nucleus masks by Hoechst staining were used to define the total cell population in each well, and the percentage of tumor cells with positive fluorescent signal due to cleaved caspase 3/7 activity was analyzed. The assay was performed three times with triplicate wells for each condition.
For primary GBOs, one GBO spheroid was seeded with 806-28ζ CAR T cells in GBO medium at CAR T cell/GBO cell ratio of approximately 1:30 in each well of the U-bottomed 96-well plates. After co-culturing for 24 h, GBOs were transferred to 6-well culture plates and cultured with GBO medium containing 0.01% DMSO or 1 μM birinapant under normal culture conditions. After desired amount of time, GBOs were fixed with 4% formaldehyde solution (Polysciences) for 30 min at room temperature, followed by PBS (Thermo Fisher Scientific) wash and 30% sucrose (Millipore Sigma) incubation at 4°C overnight. Fixed GBOs were cut into 25-μm sections and stained based on a published protocol. In brief, sectioned tissue was attached onto charged slides followed by permeabilization and blocking for 1 h at room temperature. After buffer aspiration, samples were incubated in diluted primary antibodies at 4°C overnight. The primary antibodies used included cleaved caspase 3 (CST; clone Asp175), human CD3 (BioLegend; clone SK7), and Ki67 (BD Biosciences; clone B56). The next day, samples were washed three times for 5 min with 1× TBS-T (Thermo Fisher Scientific) and incubated with diluted secondary antibodies for 2 h at room temperature. The secondary antibodies used included donkey-anti-goat-AF647 (Thermo Fisher Scientific), donkey-anti-mouse-AF488 (Thermo Fisher Scientific), donkey-anti-rabbit-AF555, donkey-anti-rabbit-AF647 (Thermo Fisher Scientific), and donkey-anti-human-AF-488 (Jackson). Samples were washed three times for 5 min with 1× TBS-T (Thermo Fisher Scientific) and mounted with antifade mounting medium (Vector Laboratories) on glass coverslips. Sample images were taken using Zeiss confocal microscopy and converted to .tif files using Zen 2 Blue software. The images were further analyzed using ImageJ software. Background was removed using default parameters. Desired quantification area was defined by DAPI staining (Sigma-Aldrich). The mean intensity of cleaved caspase 3 signal was quantified in the same area. The mean intensity was normalized to the control condition and further plotted.
c-FLIP knockout by CRISPR
GBM cells were transduced with the lentiCRISPR-v2 system (Addgene) as described in Sanjana et al.48 with gRNA targeting c-FLIP. The cells were selected and maintained in 10 μg/mL puromycin in the culture medium. To confirm c-FLIP knockout, genomic DNA from puromycin-selected cells was extracted with a Blood & Cell Culture DNA kit (Qiagen) for AMPLICON-EZ Sequencing (Genewiz).
In vitro repeated stimulation assay
Cryopreserved CAR T cell aliquots of four different healthy donors were thawed and cultured overnight to recover as described earlier. The next day (day 0), CAR T cells were washed with PBS (Corning) and counted for viable cells on a hemocytometer with trypan blue staining (Corning). To restimulate CAR T cells with stimulator cells, birinapant-resistant U87-EGFRvIII cells were washed and added to the CAR T cell cultures in regular T cell medium in the presence or absence of birinapant. For every three CAR T cells, one U87-EGFRvIII cell was added to stimulate the CAR T cells. After 24 h of restimulation, supernatants were sampled from the cultures, aliquoted, and stored at −80°C. Later, ELISA assays were performed to detect TNF-α and IFN-γ concentrations in the collected supernatants with Quantikine ELISA kits (R&D Systems) per the manufacturer’s instructions. To count live and dead CAR T cells on day 3, day 5, and day 7 after the restimulation, CAR T cells were sampled and stained with Calcein Violet-AM (BioLegend) for live cells and propidium iodide (Thermo Fisher Scientific) for dead cells in PBS (Corning) in darkness for 20 min at room temperature, then enumerated by a Celigo Image Cytometer (Nexcelom Bioscience). Each experimental group was sampled in triplicate wells for counting. After excluding GFP + U87-EGFRvIII cells, the CAR T cell viability was calculated as (live cell count)/(live cell count + dead cell count) × 100%. Following cell counting on day 3 and day 5, CAR T cells were fed with the medium containing DMSO (vehicle) or birinapant to 0.8 × 106 live cells/mL density, keeping the birinapant concentration constant. On day 7, 1 × 106 live CAR T cells were pulled from each group to be restimulated by U87-EGFRvIII cells again with or without birinapant, starting another 7-day cycle. A total of three cycles were conducted for each experiment, lasting 21 days. Accumulated CAR T cell expansions were calculated based on the portions of live CAR T cells when pulled for restimulations. To compare proliferation rates, CAR T cells were sampled for EdU incorporation assays with Click-iT Plus EdU Flow Cytometry Assay Kit (Thermo Fisher Scientific) on day 12 and day 19 of each experiment. Sampled CAR T cells were incubated with 10 μM EdU in the medium at 37°C for 1 h, followed by PBS (Corning) wash and intracellular staining as instructed by the manufacturer, and flow cytometry was used to examine the EdU incorporations by cells entering the S phase of the cell cycle, which indicates relative proliferation rates. This experiment was performed with CAR T cells of four different healthy donors side by side and repeated three times with the same donors.
Degranulation assay
CAR T cells were stimulated with birinapant-resistant U87-EGFRvIII cells at E/T ratio of 1:1 in the presence or absence of birinapant. Immediately after the co-culture started, mouse-anti-human CD107a-BV650 antibody (BioLegend; clone H4A3) was added to the cell culture. After 1 h of incubation, GolgiStop and GolgiPlug (BD Biosciences) was added to the cell culture at 1:1,000 dilution. After a total of 5 h of co-culture, cells were collected for PBS (Corning) wash, followed by staining with LIVE/DEAD Fixable Violet Dead Cell Stain (Invitrogen) and mouse-anti-human CD3-AF488 antibody (BD Biosciences; clone UCHT1). Samples were collected on a BD LSR Fortessa flow cytometer with proper compensation setup, followed by analysis with FlowJo (Tree Star) to evaluate the expression of CD107a among live CD3+ cells. Unstimulated CAR T cells were used as negative control when analyzing the data.
Mouse experiments
All mouse experiments were conducted in accordance with IACUC-approved protocols. Gender-balanced NSG mice were provided by the Stem Cell and Xenograft Core facility at the University of Pennsylvania and housed under pathogen-free conditions. Mice in each treatment group were randomized among the cages and balanced for genders. Mouse weight was recorded on a weekly basis, and mice were monitored for sign of illness such as weight loss, hair loss, lethargy, and hunching posture at rest.
For the subcutaneous U87-EGFRvIII mouse model, 0.5 × 106 U87-EGFRvIII cells with CBG luciferase were washed in PBS (Corning) and prepared in 50% Matrigel (Corning) in PBS (Corning) on ice, then subcutaneously injected in the right flank of each NSG mouse. Six days after tumor implantation, 5 × 106 or 0.5 × 106 2173-28ζ CAR T cells in PBS (Corning) were injected i.v. to each mouse, and the date was marked as day 0 of the experiment. The vehicle (12.5% Captisol in sterile water) or birinapant at dosage of 10 mg/kg mouse weight was given i.p. every 3 days for 10 total doses since day 0. To evaluate tumor growth or reduction, mice were injected i.p. with D-luciferin (PerkinElmer) substrate solution and anesthetized by isoflurane after 30 min for bioluminescent imaging by the IVIS Lumina S5 Imaging System (PerkinElmer) at 3-day intervals. The total flux of bioluminescent signal was quantified using Living Image Software (PerkinElmer). Retro-orbital bleeding for each mouse was performed weekly to evaluate human CAR T cell count and phenotypes in mouse blood by flow cytometry (see “flow cytometry and cell sorting” for staining panels and methods). Mouse plasma from leftover blood on day 7 for the study with 5 × 106 CAR T cells was used for ELISA assays for TNF-α and IFN-γ with Quantikine ELISA kits (R&D Systems) per the manufacturer’s instructions. To rechallenge mice with new U87-EGFRvIII-CBG tumor, 3 × 106 U87-EGFRvIII-CBG cells were washed in PBS (Corning) and prepared in 50% Matrigel (Corning) in PBS (Corning) on ice, then subcutaneously injected in the left flank of mice on the given date.
For the subcutaneous antigen-heterogeneous U118 mouse model, 4 × 106 U118-parental-CBR cells mixed with 1 × 106 U118-EGFRvIII-CBG cells were washed in PBS (Corning) and prepared in 50% Matrigel (Corning) in PBS (Corning) on ice, then subcutaneously injected in the right flank of each NSG mouse. Six or 14 days after tumor implantation, 0.5 × 106 2173-28ζ CAR T cells or the equivalent number of non-transduced (NTD) T cells in PBS (Corning) were injected i.v. to each mouse, and the date was marked as day 0 of the experiment. The vehicle (12.5% Captisol in sterile water) or birinapant at dosage of 10 mg/kg mouse weight was given i.p. every 3 days for 10 total doses since day 0. To monitor specific bioluminescent signals from U118-parental-CBR cells and U118-EGFRvIII-CBG cells, mice were injected i.p. with D-luciferin (PerkinElmer) substrate solution and anesthetized after 30 min for bioluminescent imaging by the IVIS Lumina S5 Imaging System (PerkinElmer) at 3-day intervals. The imaging was done in sequence with a set of emission filters of 520 nm, 570 nm, 620 nm, 670 nm, and 710 nm, and the sequence images were analyzed by Living Image Software (PerkinElmer) using a spectral unmixing library pre-established with pure subcutaneous U118-CBR and U118-CBG cells. The total fluxes of the differentiated CBG and CBR bioluminescence were then quantified. Tumor volume was measured by caliper and calculated as volume = ½ × length × width2. Non-palpable tumors were designated as 1 mm3 and palpable but non-measurable tumors were designated as 50 mm3. Tumors in mice given NTD T cells in the repeat study were harvested on day 28 and rinsed in cold PBS (Corning) before dissection and cell lysis for western blotting assay as described earlier.
For intracranial antigen-heterogeneous U118 mouse model, 200 × 103 U118-parental-CBR cells mixed with 50 × 103 U118-EGFRvIII (without luciferase) cells were washed in PBS (Corning) and resuspended in PBS (Corning) on ice, then implanted intracranially in each NSG mouse. The surgeries were performed using a stereotactic surgical setup, and tumor cells were implanted in the right parietal lobe with the following coordinates: 2 mm to the sagittal suture, 2 mm to the lambdoid suture, and 2 mm deep. During and 24 h after the surgery, mice were injected with painkillers consisting of meloxicam (5 mg/kg; Butler Animal Health Holding) and bupivacaine (2 mg/kg for during the surgery and 0.7 mg/kg for 24 h post surgery; Butler Animal Health Holding). Seven days after tumor implantation, 1 × 106 or 0.3 × 106 2173-28ζ CAR T cells in PBS (Corning) were injected i.v. to each mouse, and the date was marked as day 0 of the experiment. The vehicle (12.5% Captisol in sterile water) or birinapant at dosage of 20 mg/kg mouse weight was given i.p. every 3 days for 10 total doses since day 0. To monitor the bioluminescent signals from U118-parental-CBR cells, mice were injected i.p. with D-luciferin (PerkinElmer) substrate solution and anesthetized after 30 min for bioluminescent imaging by the IVIS Lumina S5 Imaging System (PerkinElmer) at 3-day intervals. Retro-orbital bleeding was done on day 7 for the repeat experiment with 0.3 × 106 CAR T cells, and the mouse plasma was evaluated for 32-plex human cytokines via Luminex performed by the Translational and Correlative Studies Laboratory of the University of Pennsylvania; heart, liver, kidney, and lung from each mouse were harvested at the endpoint of this experiment and fixed in 10% formalin (Lab Alley). These tissues were then processed, sectioned, stained with hematoxylin and eosin (H&E), and assessed by a veterinary pathologist at the Comparative Pathology Core of the University of Pennsylvania School of Veterinary Medicine in a blinded fashion without prior knowledge of the experimental intervention and group distribution. Lesions were scored on a scale of 0–4 ranging from unremarkable (score of 0) to severe/marked change (score of 4).
Statistical analysis
The statistical analyses were performed as noted in the figure legends using Prism9 (GraphPad) to determine the p values. Significance is reported if p < 0.05 (marked by an asterisk).
Acknowledgments
We thank A. Wang, J. Leferovich, J. Glover, and F. Chen for technical assistance, and S. Nunez-Cruz, V. Bhoj, S. Richman, R. O’Connor, S. Ghassemi, F. Miao, D. Powell Jr., X. Yang, Y. Fan, and C. June for helpful discussions. We also thank the Human Immunology Core, Cytomics and Cell Sorting Resource Laboratory, Stem Cell and Xenograft Core, Translational and Correlative Studies Laboratory, and Comparative Pathology Core at the University of Pennsylvania for their technical services. This research was supported by institutional funds as well as grants from the Glioblastoma Translational Center of Excellence at Abramson Cancer Center (to M.C.M. and D.M.O.), National Institutes of Health (R35NS116843 to H.S., R35NS097370 to G.-l.M.), and the Sheldon G. Adelson Medical Research Foundation (to G.-l.M.).
Author contributions
Conceptualization, E.Z.S., B.I.P., and M.C.M.; data curation, E.Z.S. and X.W.; formal analysis, E.Z.S. and C.-A.A.; funding acquisition, M.C.M., D.M.O., H.S., and G.-l.M.; investigation, E.Z.S. and X.W.; methodology, E.Z.S., X.W., and B.I.P.; project administration, E.Z.S. and M.C.M.; resources, M.C.M., D.M.O., Z.A.B., H.S., G.-l.M., R.T., L.Z., and C.A.; supervision, M.C.M., Q.Z., Z.A.B., and H.S.; validation, M.C.M.; visualization, E.Z.S.; writing – original draft, E.Z.S.; writing – review & editing, E.Z.S., M.C.M., Z.A.B., H.S., X.W., Q.Z., and B.I.P.
Declaration of interests
R.T., Z.A.B., D.M.O., and M.C.M. are inventors on multiple patents relating to CAR T cell therapy for GBM. M.C.M. is also a founder and scientific advisor for Verismo Therapeutics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.11.004.
Supplemental information
Table S1. Concentration of human cytokines in mouse plasma on day 7 by Luminex from the experiment of Figure 6
Heart, liver, kidney, and lung from each mouse were harvested at the endpoint of this experiment, followed by fixation in 10% formalin, processing, sectioning, H&E staining, and histopathologic assessment by a veterinary pathologist in a blinded fashion without prior knowledge of the experimental intervention and group.
Document S2. Article plus supplemental information
Data availability
The data generated in this study are available upon request from the corresponding author.
References
- 1.June C.H., Sadelain M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018;379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song E.Z., Milone M.C. Pharmacology of chimeric antigen receptor-modified T cells. Annu. Rev. Pharmacol. Toxicol. 2021;61:805–829. doi: 10.1146/annurev-pharmtox-031720-102211. [DOI] [PubMed] [Google Scholar]
- 3.Bagley S.J., Desai A.S., Linette G.P., June C.H., O'Rourke D.M. CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neuro Oncol. 2018;20:1429–1438. doi: 10.1093/neuonc/noy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maggs L., Cattaneo G., Dal A.E., Moghaddam A.S., Ferrone S. CAR T cell-based immunotherapy for the treatment of glioblastoma. Front Neurosci. 2021;15:662064. doi: 10.3389/fnins.2021.662064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lara-Velazquez M., Shireman J.M., Lehrer E.J., Bowman K.M., Ruiz-Garcia H., Paukner M.J., Chappell R.J., Dey M. A comparison between chemo-radiotherapy combined with immunotherapy and chemo-radiotherapy alone for the treatment of newly diagnosed glioblastoma: a systematic review and meta-analysis. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.662302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown C.E., Alizadeh D., Starr R., Weng L., Wagner J.R., Naranjo A., Ostberg J.R., Blanchard M.S., Kilpatrick J., Simpson J., et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Rourke D.M., Nasrallah M.P., Desai A., Melenhorst J.J., Mansfield K., Morrissette J.J.D., Martinez-Lage M., Brem S., Maloney E., Shen A., et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl Med. 2017;9 doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmed N., Brawley V., Hegde M., Bielamowicz K., Kalra M., Landi D., Robertson C., Gray T.L., Diouf O., Wakefield A., et al. HER2-Specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3:1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biernat W., Huang H., Yokoo H., Kleihues P., Ohgaki H. Predominant expression of mutant EGFR (EGFRvIII) is rare in primary glioblastomas. Brain Pathol. 2004;14:131–136. doi: 10.1111/j.1750-3639.2004.tb00045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fulda S., Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012;11:109–124. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- 11.Weber R.G., Sommer C., Albert F.K., Kiessling M., Cremer T. Clinically distinct subgroups of glioblastoma multiforme studied by comparative genomic hybridization. Lab Invest. 1996;74:108–119. [PubMed] [Google Scholar]
- 12.Bertrand M.J., Milutinovic S., Dickson K.M., Ho W.C., Boudreault A., Durkin J., Gillard J.W., Jaquith J.B., Morris S.J., Barker P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 13.Deveraux Q.L., Takahashi R., Salvesen G.S., Reed J.C. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 14.Deveraux Q.L., Leo E., Stennicke H.R., Welsh K., Salvesen G.S., Reed J.C. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. Embo j. 1999;18:5242–5251. doi: 10.1093/emboj/18.19.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chai J., Du C., Wu J.W., Kyin S., Wang X., Shi Y. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature. 2000;406:855–862. doi: 10.1038/35022514. [DOI] [PubMed] [Google Scholar]
- 16.Varfolomeev E., Blankenship J.W., Wayson S.M., Fedorova A.V., Kayagaki N., Garg P., Zobel K., Dynek J.N., Elliott L.O., Wallweber H.J., et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 17.Vince J.E., Wong W.W., Khan N., Feltham R., Chau D., Ahmed A.U., Benetatos C.A., Chunduru S.K., Condon S.M., McKinlay M., et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131:682–693. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 18.Johnson L.A., Scholler J., Ohkuri T., Kosaka A., Patel P.R., McGettigan S.E., Nace A.K., Dentchev T., Thekkat P., Loew A., et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl Med. 2015;7:275ra222. doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dougan S.K., Dougan M. Regulation of innate and adaptive antitumor immunity by IAP antagonists. Immunotherapy. 2018;10:787–796. doi: 10.2217/imt-2017-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dougan M., Dougan S., Slisz J., Firestone B., Vanneman M., Draganov D., Goyal G., Li W., Neuberg D., Blumberg R., et al. IAP inhibitors enhance co-stimulation to promote tumor immunity. J. Exp. Med. 2010;207:2195–2206. doi: 10.1084/jem.20101123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao G., Zhang M., Harhaj E.W., Sun S.C. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J. Biol. Chem. 2004;279:26243–26250. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 22.Thu Y.M., Richmond A. NF-kappaB inducing kinase: a key regulator in the immune system and in cancer. Cytokine Growth Factor Rev. 2010;21:213–226. doi: 10.1016/j.cytogfr.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao G., Harhaj E.W., Sun S.C. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 24.Zarnegar B., Yamazaki S., He J.Q., Cheng G. Control of canonical NF-kappaB activation through the NIK-IKK complex pathway. Proc. Natl. Acad. Sci. USA. 2008;105:3503–3508. doi: 10.1073/pnas.0707959105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerondakis S., Siebenlist U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb Perspect. Biol. 2010;2:a000182. doi: 10.1101/cshperspect.a000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beug S.T., Beauregard C.E., Healy C., Sanda T., St-Jean M., Chabot J., Walker D.E., Mohan A., Earl N., Lun X., et al. Smac mimetics synergize with immune checkpoint inhibitors to promote tumour immunity against glioblastoma. Nat. Commun. 2017;8 doi: 10.1038/ncomms14278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dufva O., Koski J., Maliniemi P., Ianevski A., Klievink J., Leitner J., Pölönen P., Hohtari H., Saeed K., Hannunen T., et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood. 2020;135:597–609. doi: 10.1182/blood.2019002121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michie J., Beavis P.A., Freeman A.J., Vervoort S.J., Ramsbottom K.M., Narasimhan V., Lelliott E.J., Lalaoui N., Ramsay R.G., Johnstone R.W., et al. Antagonism of IAPs enhances CAR T-cell efficacy. Cancer Immunol. Res. 2019;7:183–192. doi: 10.1158/2326-6066.cir-18-0428. [DOI] [PubMed] [Google Scholar]
- 29.Chang Y.-C., Cheung C.H.A. An updated review of smac mimetics, LCL161, birinapant, and GDC-0152 in cancer treatment. Appl. Sci. 2021;11:335. [Google Scholar]
- 30.Condon S.M., Mitsuuchi Y., Deng Y., LaPorte M.G., Rippin S.R., Haimowitz T., Alexander M.D., Kumar P.T., Hendi M.S., Lee Y.H., et al. Birinapant, a smac-mimetic with improved tolerability for the treatment of solid tumors and hematological malignancies. J. Med. Chem. 2014;57:3666–3677. doi: 10.1021/jm500176w. [DOI] [PubMed] [Google Scholar]
- 31.Jacob F., Salinas R.D., Zhang D.Y., Nguyen P.T.T., Schnoll J.G., Wong S.Z.H., Thokala R., Sheikh S., Saxena D., Prokop S., et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell. 2020;180:188–204.e122. doi: 10.1016/j.cell.2019.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Humphrey P.A., Wong A.J., Vogelstein B., Zalutsky M.R., Fuller G.N., Archer G.E., Friedman H.S., Kwatra M.M., Bigner S.H., Bigner D.D. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc. Natl. Acad. Sci. USA. 1990;87:4207–4211. doi: 10.1073/pnas.87.11.4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Padfield E., Ellis H.P., Kurian K.M. Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front Oncol. 2015;5:5. doi: 10.3389/fonc.2015.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thokala R., Binder Z.A., Yin Y., Zhang L., Zhang J.V., Zhang D.Y., Milone M.C., Ming G.L., Song H., O'Rourke D.M. High-affinity chimeric antigen receptor with cross-reactive scFv to clinically relevant EGFR oncogenic isoforms. Front Oncol. 2021;11:664236. doi: 10.3389/fonc.2021.664236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grupp S.A., Kalos M., Barrett D., Aplenc R., Porter D.L., Rheingold S.R., Teachey D.T., Chew A., Hauck B., Wright J.F., et al. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. New Engl. J. Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis M., Tartaglia L.A., Lee A., Bennett G.L., Rice G.C., Wong G.H., Chen E.Y., Goeddel D.V. Cloning and expression of cDNAs for two distinct murine tumor necrosis factor receptors demonstrate one receptor is species specific. Proc. Natl. Acad. Sci. USA. 1991;88:2830–2834. doi: 10.1073/pnas.88.7.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheung H.H., Mahoney D.J., Lacasse E.C., Korneluk R.G. Down-regulation of c-FLIP Enhances death of cancer cells by smac mimetic compound. Cancer Res. 2009;69:7729–7738. doi: 10.1158/0008-5472.CAN-09-1794. [DOI] [PubMed] [Google Scholar]
- 38.Safa A.R., Pollok K.E. Targeting the anti-apoptotic protein c-FLIP for cancer therapy. Cancers (Basel) 2011;3:1639–1671. doi: 10.3390/cancers3021639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Logan A.E., Wilson T.R., Fenning C., Cummins R., Kay E., Johnston P.G., Longley D.B. In vitro and in vivo characterisation of a novel c-FLIP-targeted antisense phosphorothioate oligonucleotide. Apoptosis. 2010;15:1435–1443. doi: 10.1007/s10495-010-0533-5. [DOI] [PubMed] [Google Scholar]
- 40.Cong H., Xu L., Wu Y., Qu Z., Bian T., Zhang W., Xing C., Zhuang C. Inhibitor of apoptosis protein (IAP) antagonists in anticancer agent discovery: current status and perspectives. J. Med. Chem. 2019;62:5750–5772. doi: 10.1021/acs.jmedchem.8b01668. [DOI] [PubMed] [Google Scholar]
- 41.Sarkaria J.N., Hu L.S., Parney I.F., Pafundi D.H., Brinkmann D.H., Laack N.N., Giannini C., Burns T.C., Kizilbash S.H., Laramy J.K., et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2018;20:184–191. doi: 10.1093/neuonc/nox175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kirchhoff S., Muller W.W., Krueger A., Schmitz I., Krammer P.H. TCR-mediated up-regulation of c-FLIPshort correlates with resistance toward CD95-mediated apoptosis by blocking death-inducing signaling complex activity. J. Immunol. 2000;165:6293–6300. doi: 10.4049/jimmunol.165.11.6293. [DOI] [PubMed] [Google Scholar]
- 43.Kirchhoff S., Muller W.W., Li-Weber M., Krammer P.H. Up-regulation of c-FLIPshort and reduction of activation-induced cell death in CD28-costimulated human T cells. Eur. J. Immunol. 2000;30:2765–2774. doi: 10.1002/1521-4141(200010)30:10<2765::aid-immu2765>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 44.Starck L., Scholz C., Dorken B., Daniel P.T. Costimulation by CD137/4-1BB inhibits T cell apoptosis and induces Bcl-xL and c-FLIP(short) via phosphatidylinositol 3-kinase and AKT/protein kinase B. Eur. J. Immunol. 2005;35:1257–1266. doi: 10.1002/eji.200425686. [DOI] [PubMed] [Google Scholar]
- 45.Philipson B.I., O'Connor R.S., May M.J., June C.H., Albelda S.M., Milone M.C. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-kappaB signaling. Sci. Signal. 2020;13 doi: 10.1126/scisignal.aay8248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jacob F., Ming G.L., Song H. Generation and biobanking of patient-derived glioblastoma organoids and their application in CAR T cell testing. Nat. Protoc. 2020;15:4000–4033. doi: 10.1038/s41596-020-0402-9. [DOI] [PubMed] [Google Scholar]
- 47.Milone M.C., Fish J.D., Carpenito C., Carroll R.G., Binder G.K., Teachey D., Samanta M., Lakhal M., Gloss B., Danet-Desnoyers G., et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Concentration of human cytokines in mouse plasma on day 7 by Luminex from the experiment of Figure 6
Heart, liver, kidney, and lung from each mouse were harvested at the endpoint of this experiment, followed by fixation in 10% formalin, processing, sectioning, H&E staining, and histopathologic assessment by a veterinary pathologist in a blinded fashion without prior knowledge of the experimental intervention and group.
Document S2. Article plus supplemental information
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.