

ABSTRACT
Synaptic connections between neurons are essential for every facet of human cognition and are thus regulated with extreme precision. Rho-family GTPases, molecular switches that cycle between an active GTP-bound state and an inactive GDP-bound state, comprise a critical feature of synaptic regulation. Rho-GTPases are exquisitely controlled by an extensive suite of activators (GEFs) and inhibitors (GAPs and GDIs) and interact with many different signalling pathways to fulfill their roles in orchestrating the development, maintenance, and plasticity of excitatory synapses of the central nervous system. Among the mechanisms that control Rho-GTPase activity and signalling are cell surface receptors, GEF/GAP complexes that tightly regulate single Rho-GTPase dynamics, GEF/GAP and GEF/GEF functional complexes that coordinate multiple Rho-family GTPase activities, effector positive feedback loops, and mutual antagonism of opposing Rho-GTPase pathways. These complex regulatory mechanisms are employed by the cells of the nervous system in almost every step of development, and prominently figure into the processes of synaptic plasticity that underlie learning and memory. Finally, misregulation of Rho-GTPases plays critical roles in responses to neuronal injury, such as traumatic brain injury and neuropathic pain, and in neurodevelopmental and neurodegenerative disorders, including intellectual disability, autism spectrum disorder, schizophrenia, and Alzheimer’s Disease. Thus, decoding the mechanisms of Rho-GTPase regulation and function at excitatory synapses has great potential for combatting many of the biggest current challenges in mental health.
KEYWORDS: Rac1, RhoA, Cdc42, LTP, LTD, NMDA receptor, AMPA receptor, dendritic spine
0. Introduction
No less a transformative connector of the world than Sir Timothy Berners-Lee (inventor of the World Wide Web) noted, ‘There are billions of neurons in our brains, but … the brain has no knowledge until connections are made between neurons. All that we know, all that we are, comes from the way our neurons are connected.’ Interneuronal connections are called synapses, and the adult human brain has ~1015 thereof [1]. Synapses mediate information flow and storage in the brain and are essential for all behaviours in humans and most other metazoans. Accordingly, synaptic pathologies underlie many pathophysiological conditions, so decoding their dynamic molecular nature is critical for human health.
Synapses form where specialized neuronal regions come into apposition, separated by a synaptic cleft of ~20 nm [2]. In canonical synapses, information flows from the axon of one neuron to the dendrite or cell body of another. The axonal (presynaptic) side contains neurotransmitter within synaptic vesicles that fuse with the synaptic membrane in response to Ca2+ signals arising from an all-or-nothing electrochemical event, the action potential. Neurotransmitter is released into the synaptic cleft, diffuses to the dendritic (postsynaptic) side, and interacts with receptors, eliciting electrochemical responses in the downstream neuron. Synapses can be broadly divided into excitatory, inhibitory, and neuromodulatory classes. In adults, excitatory synapses of the central nervous system (CNS) typically utilize the neurotransmitter glutamate, which triggers electrochemical depolarization, or activation, of the downstream neuron [3,4]. CNS inhibitory synapses primarily utilize the neurotransmitters γ-aminobutyric acid (GABA) and/or glycine, which cause hyperpolarization, or inactivation, of downstream neurons [5]. Finally, neuromodulatory synapses utilize a wide variety of neurotransmitters, including dopamine and serotonin, that generally bind to metabotropic G-protein coupled receptors (GPCRs) and can profoundly alter the biochemistry of the postsynaptic neuron [6,7]. Here, we focus on excitatory glutamatergic synapses in the CNS, though we also include discussion of inhibitory and neuromodulatory synapses to illustrate the broad effects of Rho-GTPases, the primary theme of this review.
Excitatory postsynapses typically form on small (1–2 µm), actin-rich projections from the dendrite called dendritic spines [8] (in this review, spines) (Figure 1A). Spines emerge as long, thin filopodia early in development, but those that make stable axonal contacts mature into thin spines, which have a roughly spherical ‘head’ at the contact site [8]. Further maturation or synaptic potentiation leads to shorter spines with wider heads, e.g. the classical mature ‘mushroom’ spine [8] (Figure 1A). The primary functional component of the postsynapse is the postsynaptic density (PSD), an electron-dense macromolecular structure apposed to the synaptic cleft containing neurotransmitter receptors, scaffolds, and regulatory proteins [9]. In glutamatergic synapses, there are two primary types of ionotropic glutamate receptors. α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptors (AMPARs) mediate fast depolarization of the postsynaptic membrane in response to glutamate and are the key receptors for excitatory neurotransmission [10] (Figure 1B). Functional AMPARs are composed of a pair of dimers, each containing 1 GluA2 subunit and 1 GluA1, GluA3, or GluA4 subunit [10]. Activating N-methyl-d-aspartate receptors (NMDARs) requires both glutamate binding and membrane depolarization [11,12]. NMDARs carry Ca2+ currents that control synaptic plasticity, and are comprised of 4 subunits: 2 GluN1 subunits and 2 GluN2A-D or GluN3A-B subunits [11]. Downstream of NMDARs, excitatory synapses undergo two primary forms of functional plasticity, long-term potentiation (LTP) and long-term depression (LTD) [13]. The ultimate effect of LTP is an increase in cell-surface synaptic AMPARs, yielding a stronger synapse, while LTD results in the opposite [14]. LTP and LTD are essential for cognition, and both are generally accompanied by the parallel process of structural plasticity, in which spine heads grow or shrink in accord with the functional properties of the resident synapses. Thus, spine morphology can be used as a proxy for synaptic strength.
Figure 1.
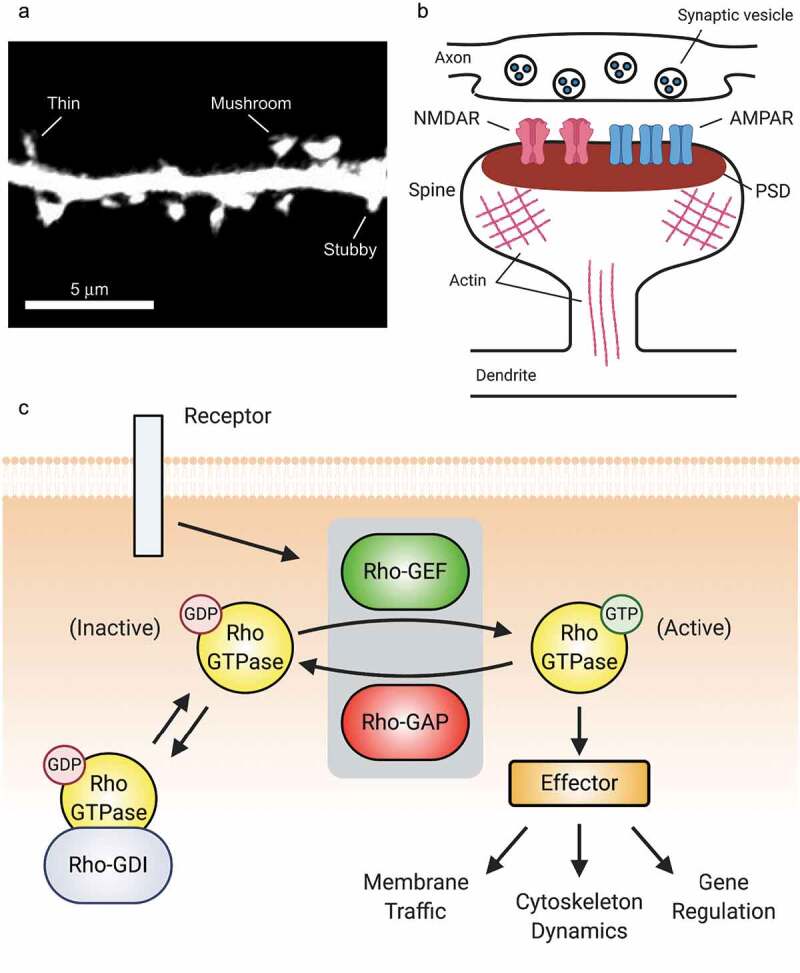
Rho-GTPases are master regulators of dendritic spines. (A) Confocal image of a dendritic segment showing spines from a mature rat hippocampal neuron expressing green fluorescent protein. Spine morphology is diverse, ranging from filopodia-like protrusions (spine precursors) to more mature thin, stubby, or mushroom-shaped structures (shown). The shape of a spine is highly correlated with the strength of its associated synapse, with the strongest synapses located on mushroom-shaped spines. Image by C. A. Cronkite. (B). Schematic of a dendritic spine and associated glutamatergic excitatory synapse. (C) Overview of Rho-GTPase signalling. Rho-GTPase activity is tightly regulated in space and time by GEFs, GAPs, and GDIs. GEFs activate Rho-GTPases by facilitating GDP/GTP exchange, whereas GAPs inactivate Rho-GTPases by enhancing GTP hydrolysis. GDIs also inhibit Rho-GTPases by sequestering them in an inactive state in the cytosol.
Doubtlessly due to the critical importance of precise synaptic regulation, myriad mechanisms do so. Rho-GTPases are an essential component of this machinery. Rho-GTPases comprise a subfamily of the Ras superfamily of small (20–25 kDa) GTPases. Generally speaking, they function as molecular switches: when bound to GTP, they are active and associate with and activate downstream proteins, called effectors (Figure 1C). Through their inherent GTPase activity, they ultimately become GDP-bound and inactive. Rho-GTPases regulate actin and microtubule cytoskeletal dynamics, membrane traffic, and gene regulation [15]. The most studied (the so-called canonical) family members are Ras homolog family member A (RhoA), Ras-related C3 botulinum toxin substrate 1 (Rac1), and cell division control protein 42 homolog (Cdc42) [15]. In the broadest terms, Rac1 and Cdc42 promote growth and synaptic strength, while RhoA opposes these functions [15,16]. Rho-GTPases are activated by guanine nucleotide exchange factors (GEFs) that stabilize the nucleotide-free conformation of the GTPase [17,18], and inactivated by GTPase-activating proteins (GAPs) that stimulate the GTPases’ enzymatic activity [19] (Figure 1C). While inactive, Rho-GTPases can be sequestered by guanine nucleotide dissociation inhibitors (GDIs), which prevent GTPase activation by GEFs [20]. While to date there are 22 known Rho-GTPases in the mammalian genome [21], there are nearly 90 GEFs, 60 GAPs, and 3 GDIs that combine with Rho-GTPase effectors to form an immensely complex regulatory network that crafts exquisitely precise spatiotemporal activation patterns that are essential for proper synaptic function. In this review, the term Rho-GEF or Rho-GAP designates some protein that so regulates any Rho-GTPase, while naming a Rho-GTPase within the term, e.g. Rac1-GEF, signifies specificity towards that particular Rho-GTPase; this specificity may not be absolute.
This review highlights Rho-GTPase synaptic signalling. In the first section, we consider GTPase regulation through (i) upstream signals, (ii) GEF/GAP coordination of single Rho-GTPases, (iii) coordination of multiple Rho-GTPases by GEF/GAP and GEF/GEF complexes, and (iv) Rho-GTPase effectors. In the second section, we look at the consequences of these elaborate control mechanisms in (i) neurodevelopment, (ii) synaptic plasticity, (iii) neuronal injury, and (iv) neurodevelopmental and neurodegenerative disease. Each topic could fill a review, so our objective is to provide a useful synthesis of current trends and topics, not an encyclopaedic compendium, on this fascinating and underappreciated topic.
1. Mechanisms of Rho-GTPase regulation
A textbook view of cellular signalling can be summarized thusly: ligand engages receptor, receptor activates secondary messenger(s), secondary messenger propagates signal, feedback inactivates receptor. This simple model is correct in its essentials but an overly simple description of most cellular signals. Signals are often tightly regulated in space and time, yielding highly specific activation patterns. Among those for which these features are known are such ubiquitous and critical signals as Ca2+ [22–25], cAMP [26–28], and a wide variety of kinases, including PKA [29,30], PKC [30,31], PI3K/Akt [32], FAK [33], GSK-3β [34] and receptor tyrosine kinases [29,35]. The analogous textbook model of Rho-GTPase function is: signal activates GEF, GEF activates Rho-GTPase, Rho-GTPase activates effectors, effectors mediate effects, and GAP inactivates Rho-GTPase (Figure 1C). This model likewise oversimplifies and trivializes the myriad of connections, interactions, and complexities that craft exquisitely precise spatiotemporal Rho-GTPase signals that regulate the development and essential functions of neurons and other cells in the nervous system.
Signalling complexity and spatiotemporal dynamics are not merely diverting esoterica. The exquisite features of these messages greatly increase the encoding power of any given signal. For example, synaptic Ca2+ gives rise to both LTP and LTD, depending on the context and nature of the Ca2+ signal [36,37]. This phenomenon is specific to neither Ca2+ nor excitatory synapses: Ca2+ elicits manifold responses in other cell types [25,38], and cAMP/PKA signalling occurs within an network of A-kinase anchoring proteins (AKAPs) that plays a critical role in determining the outcome of cAMP signals [39,40]. The large number of dedicated regulators and effectors alone suggests that these features apply to Rho-GTPase signalling. Such an extensive, multifunctional signalling apparatus could have multiple effects on any given phenomenon. Indeed, we describe several cases in which a Rho-GTPase both positively and negatively regulates some phenomenon. Besides reflecting the aforementioned complexity and importance of the spatiotemporal features of Rho-GTPase signals, they make the critical point that cellular outcomes are not linear phenomena of the ‘if some is good, more is better’ ilk. Rather, proper cell signalling delivers optimal levels of Rho-GTPase signals to specific loci at specific times to yield functional outcomes. In this section, we describe phenomena that sculpt these features of Rho-GTPase signalling.
1A. Receptors modulate Rho-GTPase signalling
Transmembrane receptors on the plasma membrane guide Rho-GTPases in regulating excitatory synapses [16]. These receptors promote dendritic spine growth and synapse formation by controlling the balance between Rho-GTPase pathways mediating synaptogenic signals and those that antagonize these actions, tightly regulating synapse formation and refinement in development [41,42]. After neuronal circuit establishment, receptors continue to exert precise control over Rho-GTPases to regulate spine and synapse maturation, maintenance, and plasticity [43]. For instance, the receptor tyrosine kinase EphB2, the adhesion-GPCR brain-specific angiogenesis inhibitor 1 (BAI1/ADGRB1), NMDARs, and the neurotrophin receptor TrkB are critical mediators of these processes. Here, we explore the ability of these receptors to regulate Rho-GTPases and the crosstalk between receptors that guides synapse formation and function.
Individual receptors coordinate multiple Rho-GTPase signalling pathways. Ephs are membrane-associated receptor tyrosine kinases that signal in response to cell-cell interactions [41]. Mammalian Ephs are divided into A and B subclasses, including 8 EphAs (EphA1-8) and 5 EphBs (EphB1-4 and 6) [44]. EphB2 and its ephrin-B ligands are essential for regulating excitatory synapse formation during early development and synaptic plasticity at mature synapses [45–47]. A major mechanism by which EphB2 regulates these processes is through Rho-GTPases. EphB2 orchestrates Rho-GTPase signalling in neurons by recruiting and phosphorylating Rho-GTPase regulatory proteins, altering their enzymatic activity, subcellular localization, and/or molecular interactions [44,48–50] (Figure 2A).
Figure 2.
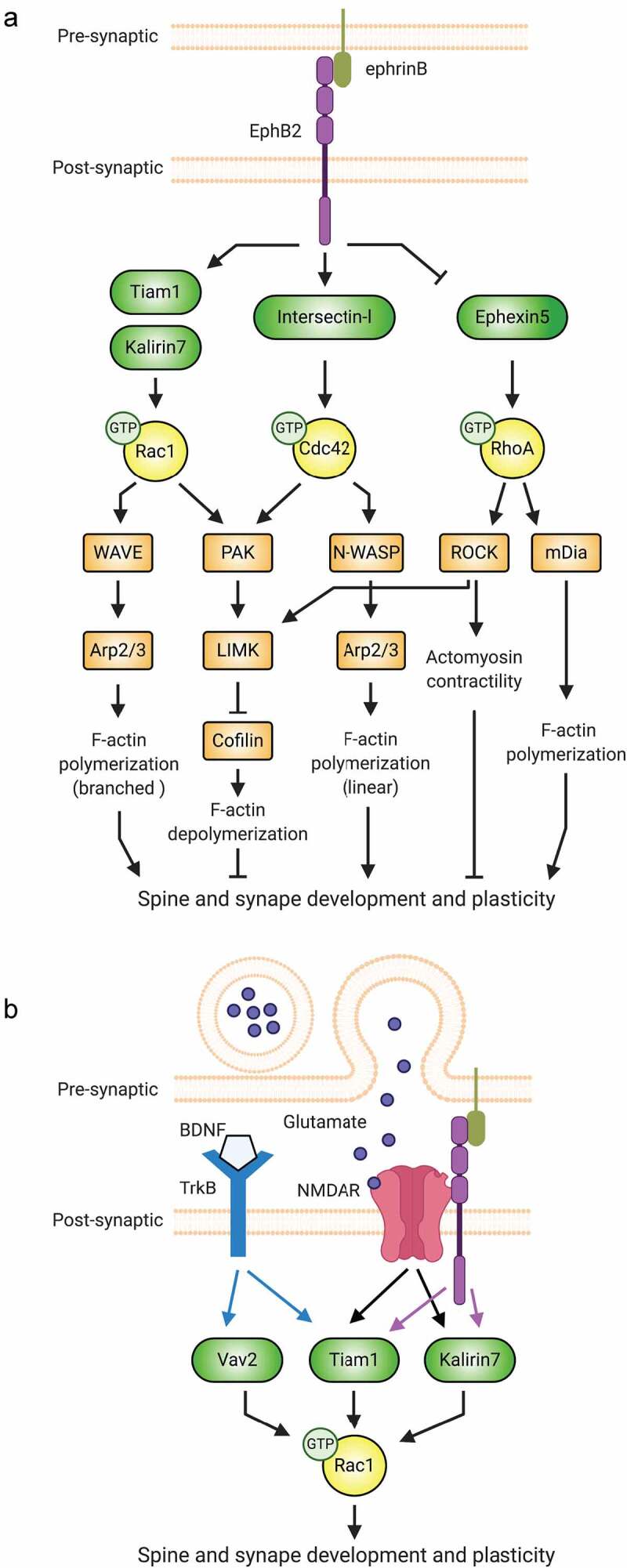
Synaptic receptors signal to Rho-GTPases via multiple pathways. (A) By regulating the function of different Rho-family GEFs, the EphB2 receptor tyrosine kinase controls Rac1, Cdc42, and RhoA signalling important for the actin cytoskeletal remodelling that drives spine and synapse development. (B) At synapses, the actions of individual Rho-GTPases are coordinated by multiple receptors.
To promote spinogenesis, EphB2 coordinates the activities of Rac1, Cdc42, and RhoA (Figure 2A). In developing neurons, the RhoA-GEF Ephexin5 associates with EphB2, restricting spine and synapse formation [51]. Ephrin-B stimulation induces EphB2-mediated phosphorylation of Ephexin5, driving the latter’s association with the E3 ubiquitin ligase Ube3A. Proteasomal degradation of Ephexin5 ensues and RhoA activity decreases, enabling spine and synapse development to proceed [51]. EphB2 also promotes Rac1 and Cdc42 activation, prompting filamentous actin (F-actin) polymerization, which is crucial for spine formation and growth [47] (Figure 2A). For example, activated EphB2 phosphorylates the Rac1-GEF Tiam1 (T-lymphocyte invasion and metastasis 1), increasing its GEF activity and EphB2-association [52]. The recruitment of Tiam1 to activated EphB2 receptors induces localized Rac1-dependent actin remodelling and spine formation [52]. EphB2 also physically interacts with and activates the Cdc42-GEF Intersectin-l, which together with neural Wiskott-Aldrich syndrome protein (N-WASP) promotes Cdc42-dependent actin polymerization and spine morphogenesis [53,54]. Thus, EphB2 drives global RhoA inhibition and targeted Rac1 and Cdc42 activation to promote spinogenesis (Figure 2A). Intriguingly, in addition to inhibiting overall spine outgrowth in a RhoA-GEF-dependent manner, Ephexin5 has also been shown to accumulate at sites of future spines and to be required for activity-dependent new spine growth [55]. Moreover, other studies have provided evidence suggesting that RhoA may cooperate with Rac1 and Cdc42 to prime specific locations of spine formation [43,56,57], in addition to its canonical role in globally suppressing spine formation.
EphB2 can also signal to individual Rho-GTPases via multiple GEFs: for instance, it activates Rac1 by the Rac1-GEF Kalirin-7 in addition to Tiam1 [58]. Loss of either GEF spurs synapse loss and aberrant spine morphology, suggesting that these GEFs function at different times or places and/or induce distinct Rac1 signalling. Thus, individual receptors can direct distinct cellular functions via different GEFs for a particular Rho-GTPase. In another striking example, the adhesion-GPCR BAI1 promotes Rac1 activation resulting in either phagocytosis or excitatory synaptogenesis, depending on the Rac1-GEF engaged: BAI1 signalling via the Rac1-GEF dedicator of cytokinesis 180 (DOCK180) mediates phagocytosis, while signalling through Tiam1 drives synaptogenesis [59–61].
Multiple receptors cooperate to ensure precise regulation of Rho-GTPases. In spine remodelling, several synaptic receptors impinge upon Rho-GTPases [42,57], and growing evidence indicates that crosstalk between NMDARs, TrkB, and EphB2 is required (Figure 2B). Like EphB2, NMDARs regulate activity-dependent spine remodelling by modulating Rho-GTPase activity via multiple GEFs. In response to NMDAR activation, Tiam1 is phosphorylated in a calcium-dependent manner, resulting in Rac1-mediated actin dynamics and spine morphogenesis. Inhibiting Tiam1 blocks these actions in primary hippocampal neurons [62]. Similarly, in primary cortical neurons, NMDARs activate Rac1 via Kalirin-7 to induce enlargement of mature spines [63]. In organotypic hippocampal slices, RNAi knockdown (KD) of either Tiam1 or Kalirin-7 reduces the long-lasting structural remodelling induced by single spine glutamate stimulation, suggesting both are involved in activity-dependent spine remodelling, as in EphB2 signalling [64]. Critically, NMDAR-dependent Rac1 regulation and spine morphogenesis may be modulated by EphB2, since EphB receptors interact directly with NMDARs and enhance their function through tyrosine phosphorylation [65,66] and Tiam1 recruitment to EphB2-NMDAR complexes [62].
Brain-derived neurotrophic factor (BDNF) and its receptor TrkB also regulate spine formation and function [67]. Similar to EphB2 and NMDARs, TrkB activates Tiam1 [68,69] and Rac1-dependent spine remodelling [70]. Disruption of TrkB-Tiam1-Rac1 signalling with TrkB mutants in primary neurons abolishes BDNF-induced spine formation and enlargement [70]. TrkB also signals to Kalirin-7 in neurons [71], and while the TrkB/Kalirin-7 pathway is unexplored in spine morphogenesis, we expect that it plays some role therein. In addition to Tiam1 and Kalirin-7, BDNF-TrkB signalling activates Vav family Rac1-GEFs in neurons [72]. While CA1 pyramidal neurons from Vav2/Vav3 double-knockout (dKO) mouse hippocampal slice cultures display normal spine density and cumulative spine length under basal conditions, they fail to undergo rapid spine head growth in response to BDNF, suggesting a role for Vav GEFs in BDNF/TrkB-induced synaptic remodelling [72]. Interestingly, these TrkB-mediated pathways may function downstream of NMDARs to modulate changes in spine morphology. While exogenous BDNF stimulation of hippocampal slices is sufficient to drive rapid spine head growth, the same stimulation in the absence of glutamatergic transmission yields no spine growth or synapse potentiation [73]. More recently, a Förster resonance energy transfer (FRET)-based probe to monitor TrkB activity in organotypic hippocampal slices revealed that glutamate stimulation of single spines increased TrkB activity [74]. These examples demonstrate the importance of receptor regulation of Rho-GTPase signalling in spines and highlight the need to further elucidate the mechanisms of these complex signalling networks.
1B. GEF/GAP complexes that target single Rho-GTPases
Live cell measurements reveal that Rho-GTPase signalling dynamics occur at very short distances (µm scale) and on very fast (subminute) time scales during cell migration, axon guidance, and spine plasticity [43,75,76]. How is this precise spatiotemporal control of Rho-GTPases accomplished? One mechanism for achieving tightly controlled on/off cycling of a specific Rho-GTPase is the targeting of specific subcellular pools of that GTPase by GEF/GAP complexes. At first blush, such modules may seem pointless because the components counter the actions of one another. However, the effects of cycling rate-altering mutations, such as Cdc42 F28L [77] or Rac1 P29S [78], suggest that Rho-GTPase turnover often needs to be very tightly regulated. Fine tuning by GEF/GAP complexes may thus guide Rho-GTPase activity to highly specific and dynamic spatiotemporal optima.
The Rac1-GEF Tiam1 and Rac1-GAP Breakpoint cluster region (Bcr) protein form a complex that balances Rac1 activity during synaptogenesis [50] (Figure 3A). Individually, Tiam1 and Bcr play opposing roles in neurons: Tiam1 loss causes dendrite arbour simplification and lowers spine and synapse numbers, while Bcr loss yields spine and arbour overgrowth [50,52,59,62,79–81]. However, the colocalization and physical interaction of Tiam1/Bcr at excitatory synapses unveils an intriguing mechanism for precisely and dynamically regulating Rac1. Tiam1/Bcr complex disruption results in overactive Rac1, increased spine density and size, and converts a spinogenic ephrin-B/EphB2 signal into spine loss [50]. Functional Tiam1 inhibition with peptides or chemical inhibitors reverses these phenotypes [50]. In another interesting twist, the Tiam1/Bcr complex appears to be dynamically regulated, allowing Rac1 signalling to toggle between tightly regulated and more global patterns of Rac1 activation in response to signals [50] (Figure 3A). Another Rac1-GEF/GAP interaction is found in the synaptic CNK2 complex that includes the Rac1-GAP ARHGAP39/Vilse and the Rac1-GEFs α- and β-PIX, in addition to Rac1 effectors, PIX modulators and Rac1 itself [82]. Disruption of the CNK2/Vilse association by mutating CNK2 results in excessive Rac1 activity and spine abnormalities [82], suggesting a function analogous to Tiam1/Bcr.
Figure 3.
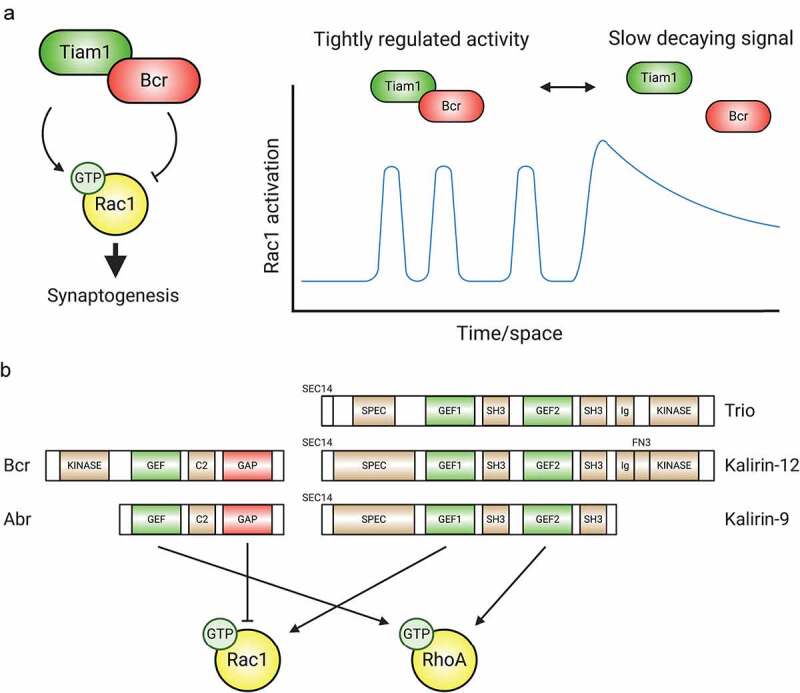
GEFs, GAPs, and multifunctional regulatory proteins tightly control Rho-GTPase activity. (A) The Tiam1/Bcr GEF/GAP complex regulates Rac1 activity during synaptogenesis. By forming a complex whose association can be modulated, the Rac1-GEF Tiam1 and the Rac1-GAP Bcr provide tight spatiotemporal regulation of Rac1 activation. (B) Tandem GEF/GAPs and GEF/GEF proteins contribute additional precise regulation to coordinated Rho-GTPase signalling. C2: protein kinase C conserved region 2, SEC14: domain in phosphatidylinositol transfer protein Sec14, SPEC: spectrin-like repeats, SH3: Src homology 3 domain, CC: coiled coil, Ig/FN3: Ig/fibronectin III.
GEF/GAP complexes are not restricted to spines. Tiam1 and Bcr associate with members of the partition-defective (Par) polarity complex in cortical astrocytes [83]. Tiam1 is recruited by the Par complex (Par3, Par6, and atypical protein kinase C zeta (PKCζ)), where it activates Rac1 [84]. Bcr also interacts with the Par complex, inhibiting Rac1 and PKCζ activity [83]. Bcr knockout (KO) results in faster migration, and defective directionality and cytoskeletal organization that are rescued by wild-type Bcr [83]. The interaction between the RhoA-GEF Ect2 and RhoA-GAP MgcRacGAP/CYK-4 in cytokinesis highlights yet another aspect of Rho-GTPase regulation [85,86]. Metazoan cytokinesis is regulated by the centralspindlin complex, consisting of the kinesin MKLP-1 and MgcRacGAP. In Xenopus laevis embryos, MgcRacGAP recruits Ect2 to the centralspindlin complex at the cell equator where they create a RhoA activity zone essential for cytokinetic ring formation [85]. GAP-dead mutants of MgcRacGAP lead to unrestrained and unstable RhoA activity. It is hypothesized that MgcRacGAP binds to Ect2-activated RhoA and transiently anchors it. RhoA-GTP can then bind to a nearby effector to initiate signalling or be inactivated by MgcRacGAP [85], maintaining a constant and demarcated RhoA flux at the cell equator. Both proteins are expressed in the brain [87,88], though their roles there require further investigation.
A variation on the GEF/GAP model is GEF/GAP compartmentalization, leading to defined zones of Rho-GTPase activity [89]. This GEF/GAP relationship is found during posterior spiracle formation in the Drosophila embryo. Spiracles are organs formed by tissue invagination due to apical constriction and basolateral membrane elongation. Activated Rho1, a Drosophila RhoA homologue, is apically restricted due to the apical distribution of two GEFs, RhoGEF64C and RhoGEF2 and the basaolateral distribution of the Rho1-GAP Crossveinless-c (Cv-c) [90]. Basolateral Rho1 suppression is crucial to cell polarity during morphogenesis, as unrestricted Rho1 inhibits invagination. Interestingly, Cv-c is also involved in directional elongation of dendrites in Drosophila dorsal da neurons by suppressing Rho1 activity [91]. The identity of a GEF partner of Cv-c in dendrites is not yet known.
Many more such GEF/GAP pairs that tightly regulate specific Rho-GTPases may exist. An important advantage of these associations is that multiple pools of a Rho-GTPase could be simultaneously regulated at distinct subcellular locations. Furthermore, a combination of different GEFs, GAPs, effectors and variable interacting proteins could offer Rho-GTPases more flexibility to control distinct downstream pathways.
1C. Coordination of multiple Rho-GTPases by multi-functional Rho-GTPase regulatory proteins and complexes
Signals can also engage GEF/GAP or GEF/GEF composites that target multiple Rho-GTPases (Figure 3B). For example, Bcr contains a RhoA-GEF domain [92] in addition to its Rac1-GAP domain [93]. The synaptic role of Bcr’s RhoA-GEF function is currently unknown, but both Bcr’s RhoA-GEF and Rac1-GAP activities are essential for dendritic growth arrest in hippocampal neurons [80]. In addition to directly regulating neuronal development, Bcr loss also causes astrocytic hyperexcitabilty, hypertrophy, and Rac1 hyperactivation [83,94]. As many excitatory synapses in the forebrain contain astrocytic processes (the so-called ‘tripartite synapse’) [95], Bcr’s multifunctional nature may also affect synapses through its regulation of astrocytes. Bcr is not unique. Activated Bcr-like (Abr) is a partial Bcr duplication, including the GEF and GAP domains [96]. Abr plays roles similar to Bcr [50,81], and Bcr and Abr partially compensate for each other at synapses [50,81], though Abr is less enriched at synapses and cannot replace Bcr in dendrite growth arrest or polarized cell migration [80,81,83]. The Rac1/Cdc42-GEF β-PIX binds to the RhoA-GAP slit-robo GAP1 (srGAP1) [97], coordinating Cdc42 and RhoA signalling in collagen-stimulated fibroblast membrane protrusion and migration [97]. While it is not yet known whether this GEF/GAP complex functions at synapses, both proteins are widely expressed in the brain and regulate neuron development individually [98,99].
What is the purpose of multi-GTPase GEF/GAP complexes? In all examples above, the GTPases inhibited by GEF/GAP complexes tend to oppose the activity of those that are activated. This suggests that, to reach some threshold, the signalling machinery requires both events to occur within a well-defined space. This requirement may be due to the magnitude of the signalling change required, and complexes may also integrate signals if the GEF and GAP activities are regulated independently. For example, Bcr’s GAP activity is activated by phosphorylation at Y177 by Fyn [100], but its GEF activity is activated by BAI1 [80].
Tandem GEFs also exist: Trio, whose GEF1 domain targets Rac1 and RhoG [101] and whose GEF2 domain targets RhoA [102], is an example (Figure 3B). Trio functions in neuronal migration [103], neurite extension [104–106], and cerebellar parallel fibre formation [103], but also in Slit2-mediated axonal growth cone collapse [107,108]. Two splice variants of the KALRN gene, Kalirin-9 and Kalirin-12, possess a Trio-like GEF doublet [109]. Both proteins are expressed in neurons during early postnatal development, regulate dendritogenesis, and have overlapping function [110,111]. Kalirin-9 also plays a role in cortical neurite extension [109]. Interestingly, Kalirin-9 is synaptic, where it colocalizes with the postsynaptic scaffold PSD95 and promotes spinogenesis [110]. Besides RhoG/Rac1 and RhoA GEF tandems, endothelia possess a DOCK4/DOCK9 complex [112], whose members possess Rac1- [112] and Cdc42- [113] GEF domains, respectively. This complex is critical for sprouting and tubule formation [112], but DOCK4 and DOCK9 expression strongly overlap in the forebrain [114,115] and DOCK4 functions in dendrite and spine development [115,116]. Finally, Ras-GRF2 has GEF domains for both Rac1 and H-Ras, a Ras-GTPase, and is located at hippocampal synapses where it regulates NMDAR-mediated synaptic plasticity [117–119].
The logic of GEF tandems is less clear than that of the GEF/GAP duets. What purpose do they serve? First, GEF tandems, by recruiting signalling molecules and creating molecular microenvironments for Rho-GTPase signals and bringing antagonistic Rho-GTPase signals into these milieus, create a localized and dynamic signal via mutual antagonism, akin to that of the Tiam1/Bcr complex, but downstream of the GTPases (see section 1D), creating distinct inhibitory dynamics better suited for some cellular processes. Second, activating one GEF could occlude the other, forcing pathways to choose between mutually exclusive outcomes. This scenario has been proposed in the telencephalon, with netrin-1 activating Trio’s Rac1-GEF domain and Slit2 activating its RhoA-GEF domain [107]. Third, when GEF tandems activate non-opposing pathways, signal amplification may ensue. A potential example of this is the Pol II CTD phosphorylation code, a ‘shortcut’ between Rho-GTPases and transcription. Herein, DOCK4 and DOCK9 activate Rac1 and Cdc42, which each target a specific phosphatase for degradation. This action prevents the dephosphorylation and inhibition of a major subunit of RNA polymerase, resulting in enhanced transcription [120]. Here, DOCK4 and DOCK9 are not known to be complexed, but since they do interact, it is a possibility. Finally, GEF tandems may determine the temporal sequences of signalling processes. For instance, DOCK9 is a Rac1 effector [112], so complexing it to the Rac1 activator DOCK4 would facilitate signalling through Cdc42, DOCK9’s target. Since complex formation is regulated [112], this pathway could operate in different sequential modes. Further, the Ras-GEF domain of Ras-GRF2 functions after its Rac1-GEF domain [117,119]. To wit, Rac1 is required for the initial steps of LTP, while H-Ras signalling mediates the transition to stably potentiated synapses and spines. It is likely that all of these scenarios or some combination thereof play out in synapses and elsewhere.
1D. Downstream effectors that regulate Rho-GTPase activity
Effectors and Rho-GDI dissociation. p21-activated kinases (PAKs) are highly conserved serine/threonine kinases that are well-described, multifunctional effectors of Rac1/Cdc42. In addition to mediating the effects of Rac1/Cdc42 on neuronal morphology, migration, and synapse development and function [121,122], PAKs provide feedback to modulate Rho-GTPase activity (Figure 4A). In fact, one of the most compelling regulatory mechanisms of Rho-GTPase/GDI complexes is through PAK [123]. PAK1 phosphorylates Rho-GDI on Ser101 and Ser174, promoting dissociation from Rac1 but not RhoA [124]; Cdc42 stimulates Rac1 release from Rho-GDI in this manner [124]. On the other hand, RhoA dissociation is promoted by protein kinase C (PKC) phosphorylation of Rho-GDI at Ser96 [125] or Ser34 [126], though this may not be regulated by Rho-GTPases. GEFs may also play a role in dissociation of Rho-GTPases from GDIs [127], but this remains to be determined.
Figure 4.
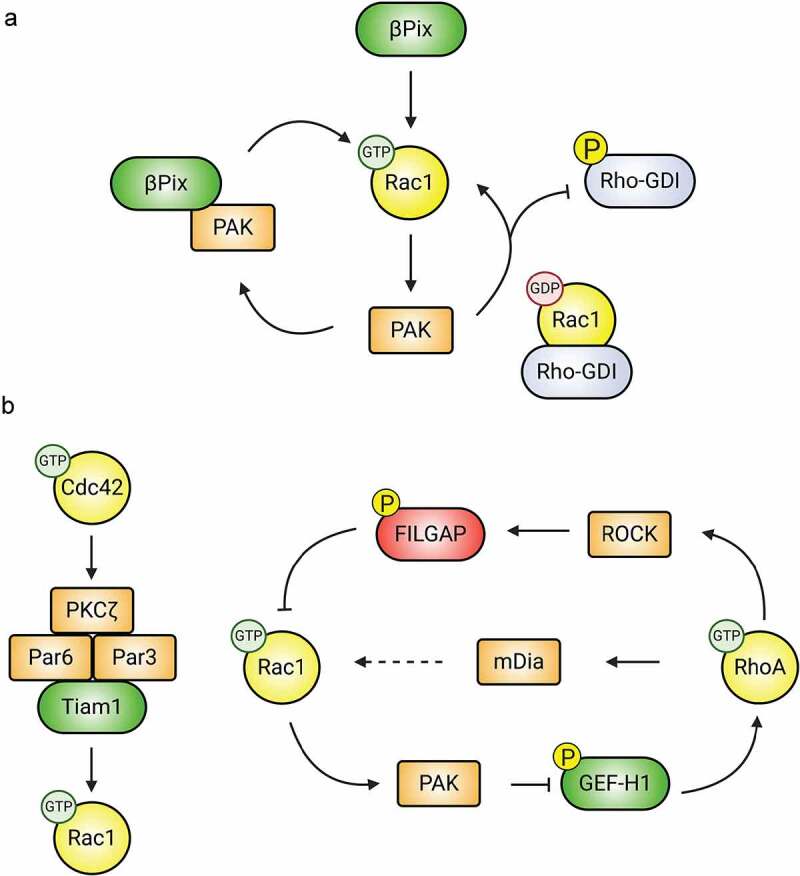
Downstream effectors modulate Rho-GTPase activity. (A) PAK regulates the activity of Rac1. Activated PAK associates with the Rac-GEF βPIX, increasing its GEF activity. Additionally, active PAK promotes Rac1 activation by phosphorylating and inactivating Rho-GDI, resulting in the release of sequestered Rac1. (B) Downstream effectors mediate crosstalk between Rac1, Cdc42, and RhoA. Following activation, Cdc42 recruits members of the Par polarity complex (Par3, Par6, and PKCζ), which in turn associate with the Rac1-GEF Tiam1, promoting local Rac1 activation. Similarly, PAK, ROCK, and mDia mediate crosstalk between Rac1 and RhoA.
Effectors balancing upstream Rho-GTPase signals. GEFs activate Rho-GTPases, but also assemble specific effectors and other signalling components (Figure 4A). For example, PIX proteins are Rac1- and Cdc42-GEFs that bind to group I PAKs and activate them through Rac1/Cdc42 and an independent T1 domain [128]. PAKs, in turn, promote PIX’s GEF activity, creating a positive feedback loop [129]. Both proteins form complexes with GIT proteins, and one such complex, consisting of GIT1, PIX, Rac1, and PAK, regulates spine and synapse formation, cell adhesion, migration, neurite extension, and synaptic plasticity [130,131]. PAK also cooperates with the CNS-specific β-Pix-d isoform [132] to phosphorylate Stathmin1, promoting tubulin acetylation and neurite morphogenesis during development [133]. This is not the only GEF/effector positive Rho-GTPase feedback loop. Tiam1’s association with the actin-related protein (Arp)2/3 complex promotes its localization at key sites and enhances its own Rac1-GEF activity; moreover, it promotes Arp2/3 actin nucleation activity through Rac1 [134]. The recently proposed reciprocally activating kinase effector complex (RAKEC) involves Tiam1 and Ca2+/calmodulin kinase II α (CaMKIIα) activating Rac1 and downstream actin regulators [64]. Here, Tiam1 binds to CaMKIIα, interrupting the autoinhibition of CaMKIIα and prolonging its activity. This, in turn, leads to prolonged phosphorylation of Tiam1, stimulating its Rac1-GEF activity. Downstream effectors also confer specificity to Rho-GTPase signalling. Insulin receptor substrate protein of 53 kDa (IRSp53) is a scaffold and an effector of Rac1 and Cdc42. Tiam1/IRSp53 interactions promote the association of IRSp53 with both the scaffold WASP-family verprolin homologous protein 2 (WAVE2) and activated Rac1 itself. This enhances Rac1 effects on actin, presumably at the expense of Cdc42-mediated effects [135].
Downstream effectors allowing crosstalk between Rho-GTPases. Collaborations between GEFs and GAPs with downstream effectors also mediates Rho-GTPase crosstalk. For example, activated Cdc42 binds to the PAR complex (PAR3/6-PKCζ) [136] and mediates its effects on the cytoskeleton through Rac1 activation by Tiam1, which is also recruited to the Par complex (Figure 4B). These proteins do not always work together, as Par6 acts independently of Par3 in spines to negatively regulate RhoA through p190RhoGAP [137], possibly through GAP-stimulating phosphorylation of p190RhoGAP by PKCζ. The aforementioned DOCK4/DOCK9 complex may regulate crosstalk between Rac1 and Cdc42. Rac1 inhibits RhoA via PAK phosphorylation of RhoA-specific GEFs (Figure 4B). To wit, PAK-mediated phosphorylation of the RhoA-GEF GEF-H1 sequesters it to microtubules and renders it inactive towards RhoA [138]. Other PAK-regulated RhoA-GEFs include ArhGEF1 [139], PDZ-GEF [140], and Net1 [141]. Another contributor to the antagonistic relationship between RhoA and Rac1 signalling is the RhoA effector Rho-associated protein kinase (ROCK), which suppresses Rac1 through phosphorylation and activation of the Rac1-GAP FilGAP [142] (Figure 4B). The RhoA effector myosin II triggers dissociation of β-PIX from adhesion proteomes, locally inhibiting Rac1 [143]. Interestingly, RhoA also can stimulate the activity of Rac1 through mDia, but the mechanism behind this is still unknown [144].
2. Outputs and consequences of Rho-GTPase activity
There would be little point in charting the intricate Rho-GTPase regulatory mechanisms if the resultant activation patterns didn’t matter. Fortunately, these patterns are deeply consequential for cognition and all other known functions of the nervous system. In this section, we look at both physiological and pathophysiological consequences of Rho-GTPase signalling in the CNS. It is impossible to summarize this huge literature here, but we will highlight key conceptual points and, where possible, avoid restating phenomena necessarily mentioned above.
2A. Rho-GTPases in neuronal development
By orchestrating actin and microtubule cytoskeletal dynamics in response to external stimuli, Rho-GTPases regulate key aspects of neuronal development, including migration, axonal/dendritic outgrowth and guidance, and synapse development and remodelling [42,145]. This section looks at some of the effects of Rho-GTPases on neurodevelopment, touching on some additional regulatory mechanisms. To illustrate the breadth of Rho-GTPase function, we consider many processes within the general heading of neurodevelopment, extending beyond synapses themselves.
RhoA. Unsurprisingly, RhoA plays important roles in many neurodevelopmental processes. For example, RhoA KO from neuroprogenitor cells (NPCs) impedes cerebellar morphogenesis, affecting foliation, lamination and neuronal migration [146]. Likewise, in the developing cerebral cortex, RhoA KO disrupts neural progenitor adherens junctions and migration due to alterations in radial glial morphology [147,148]. Prior to migration, nascent neurons undergo a critical multipolar-bipolar transition. KD of the mammalian Ste20-like kinase 3 (Mst3), which normally inhibits RhoA through phosphorylation at Ser26, perturbs this multipolar-to-bipolar transition and retards radial migration, and RhoA KD rescues these defects [149]. Additionally, RhoA inhibits neuronal process formation. KD of the RhoA-GEF ARHGEF1 or pharmacological blockade of RhoA signalling markedly enhances neurite outgrowth [150], whereas overexpression of ARHGEF1 restricts neurite formation [151]. RhoA also plays a key role in axon repulsion during development [152,153] and later on inhibits axon regrowth after CNS injury [154,155]. Extracellular β-amyloid (Aβ) promotes RhoA activation, leading to growth cone collapse and neurite contraction reminiscent of development, but with implications for Alzheimer’s disease [156]. These findings suggest a general role for RhoA as a brake on neuronal migration and development even prior to synaptogenesis.
As mentioned in Sec. 1A, RhoA limits dendritic spine and excitatory synapse formation during development. Many lines of evidence support this canonical RhoA function; in addition those already mentioned, increases in RhoA function caused by KO or KD of negative regulators, such as hnRNP-Q1 [157], Par6C [158], Rnd3 [158], LGI1 (which inhibits Nogo receptor 1) [159], PKCε [160], and the RhoA-GAPs oligophrenin-1 [161] and ARHGAP10 [162], all associate with decreases in spine and excitatory synapse density, most of which are reversed by inhibition of RhoA signalling [157,158,160,161]. Elimination of hippocampal spines as a result of exposure to the stress-related corticotropin-releasing hormone requires RhoA [163]. Conversely, KD or inhibition of the RhoA-GEF GEF-H1/Lfc increases spine density in hippocampal neurons [164]. Besides these postnatal RhoA signalling events, recent studies have looked at prenatal development and observed a similar inhibitory role for RhoA at these earliest stages of synaptogenesis in both rabbits [165] and organoids derived from human induced pluripotent stem cells [166]. However, it is possible to overgeneralize from these results. To wit, loss of the RhoA-GAP oligophrenin-1 has no effect on spine density, though it does cause spine shortening due to RhoA overactivation [161]. Further, loss of or mutations in the kinase TAOK2 leads to decreases in cortical and hippocampal spine densities that are rescued by pharmacologically activating RhoA [167]. Thus, RhoA plays various roles in spine and synapse development that may vary depending on the context provided by specific signalling pathways.
Rac1. Rac1 also plays a fundamental role in many neurodevelopmental processes. During development, the transition of neurons from a multipolar to a bipolar morphology requires precisely regulated actin and microtubule dynamics. Both constitutively active and dominant-negative (DN) Rac1 inhibit radial migration of cortical neurons and cause ectopic accumulation of multipolar neurons, suggesting that the multipolar-to-bipolar transition requires Rac1 turnover [168], in addition to RhoA inhibition [149]. Overexpression of the Rac1-GEF P-Rex1 inhibits this transition, leading to abnormal neuronal migration [169]. Rac1 KD also disrupts F-actin assembly and perturbs neuronal migration [170]. Moreover, Rac1 is a critical regulator of cytoskeletal dynamics in multiple neuronal types. Rac1 KO causes axon growth defects in sensory and motor neurons of the central and peripheral nervous systems, and these cell-autonomous defects are related to neuron loss in motor neurons and retinal ganglion cells [171], consistent with neuron survival-dependent axon targeting. Rac1 also regulates axon guidance. In Caenorhabditis elegans, a single mutation in the switch 1 region (G30E) in Rac1/CED-10 resulted in an axonal growth defect [172,173]. In contrast, other mutations of Rac1/CED-10 in the switch 2 region (G60R) or membrane targeting region (V190G) showed defects in axon guidance, but not growth, indicating that different Rac1 regions endow context-sensitive functions, and specific molecules interact with these domains to drive distinct developmental processes [172,173]. KO of Rac1/CED-10 also prevents growth cone formation, which in turn causes circuit defects [174]. Rac1 is also involved in dendritic arbour formation. For example, the Rac1-GEF DOCK4 regulates axon-dendrite polarity and dendrite arborization through Rac1 and actin dynamics [175]. Importantly, DOCK4 plays a more prominent role in dendritic branching than dendrite elongation, which may explain its association with autism and dyslexia [175].
Section 1A illustrates several examples of Rac1 playing a positive role in spine and synapse development. These are only part of a relatively large literature pointing to such a role for Rac1. Rac1 expression in neurons increases throughout synaptogenesis, and its early ectopic expression drives spine formation and AMPAR recruitment thereto [176]. Increasing Rac1 activation, either through the activation of the Rac1-GEFs Kalirin-7 [58,177], β-PIX [131,178] or Tiam1 [52,62] or the KO or KD of the Rac1-GAPs Bcr/Abr [50], srGAP2 [179], RICH2 [180,181], or p250GAP [182] drives spino- and synaptogenesis during postnatal development. On the other hand, suppressing Rac1 activation using dominant negative Rac1 [183], the small molecule inhibitor EHT 1864 [184], Rac1-GAP overexpression [50,184], or Rac1 KO (see below) decreases spine density and synaptic function. Specific Rac1 inhibition also serves as a developmental brake. For instance, the spine-localized Rac1-GAP ArhGAP12 in developing CA1 pyramidal neurons inhibits Rac1 early in development, maintaining silent synapses, i.e. excitatory synapses lacking AMPARs [185]. ArhGAP12 KD prompts premature synaptic un-silencing, high levels of immature spines and synapses, increased AMPAR currents, and increased frequency and amplitude baseline electrical events without affecting NMDAR currents [185]. Interestingly, G-actin occludes Rac1 binding to ArhGAP12 [186], suggesting another feedback mechanism between Rho-GTPases and the actin cytoskeleton.
Despite all of these results, the relationship between Rac1 activation and spine formation is not a simple linear one. Rac1 overactivation due to constitutively active Rac1 expression or mutation of the Rac1-GAP α2-chimaerin does not lead to proportionally higher spine densities, but to abnormal spine morphologies [183,187]. Even more surprisingly, spine density is increased by KO of the Rac1-GEF α-PIX, which lowers Rac1 activation [188]. Moreover, KO of the arginine methyltransferase Prmt8 leads to increased Rac1 activation in neurons, but blocks spine maturation [189]. Thus, like RhoA, Rac1 function in spine and synapse formation and maturation is complex and cannot be reduced to one simple rule.
In addition to its role in excitatory synapse development, Rac1 mediates the formation of inhibitory synapses, as evidenced by increased numbers of inhibitory synapses in L2/3 cortical pyramidal neurons when the Rac1-GAP srGAP2A is knocked down [190]. Interestingly, srGAP2A KD also leads to increased dendritic spines and excitatory synapses in the same neurons [179,190], and the effects on both synapse types require srGAP2A’s Rac1-GAP activity [190]. This raises the interesting possibility that the same pool of Rac1 regulates the formation of both synapse types, especially as srGAP2A interacts with the excitatory scaffold Homer1 and the inhibitory scaffold gephyrin by different domains [190], suggesting that this pool of Rac1 is involved in setting up the excitatory/inhibitory (E/I) balance required for proper brain function. It is also intriguing that srGAP2A is inhibited by the product of the human-specific gene duplication srGAP2C [179,190,191], which binds to the former and causes its degradation [191], suggesting that tight regulation of the srGAP2A-regulated Rac1 pool plays a role in specifically human intellect.
Rac3. Rac3 is usually coexpressed with Rac1 in the brain; compared to Rac1 single knockout (KO), dKO of Rac1 and Rac3 causes stronger reductions of spines [192], cortical GABAergic interneuron migration and microtubule dynamics [193], and cortical–hippocampal GABAergic interneuron motility [194], indicating a functional overlap between Rac1 and Rac3. Interestingly, re-expression of Rac3 or Rac1 in dKO hippocampal neurons causes distinct effects: Rac1 restores spine density, whereas Rac3 restores spine size [195]. Similarly, in the cortical–hippocampal GABAergic interneuronal network, loss of either Rac1 or Rac3 leads to a moderate loss of parvalbumin-positive interneurons, but has different effects on the development of hippocampal circuits [196]. These differences may underlie the obvious behavioural and neurological differences observed in Rac1 DN and Rac3 KO mice. Compared to the Rac3 KO, the Rac1 mutants show higher excitability and reduced spontaneous inhibitory currents in hippocampal pyramidal neurons [196]. Interestingly, cannabinoid receptor 1-positive terminals are increased in the hippocampal CA1 region of the Rac1 mutants, and incubation with cannabinoid receptor antagonists partially normalized spontaneous currents in pyramidal cells [196]. Thus, though one Rho-GTPase may compensate for another, both may retain specific functions.
Cdc42. In many ways, the developmental functions of Cdc42 resemble those of Rac1. Conditional KO of Cdc42 in cerebellar granule cell precursor (GCPs) leads to abnormalities in the cerebellar lobe, including foliation defects, loss of columnar tissue in the external germinal layer, and disordered parallel fibre organization at the molecular layer [197]. Notably, GCPs lacking Cdc42 have a multipolar morphology and fail to form migration junctions with glial fibres. Altered phosphorylation of Cdc42 regulators and effectors, including PAK1/2/4, cytoskeletal proteins Pxn, Fmn2, Dbn1 and Map2, and polar regulators Numbl and Scrib, suggest that changes in cytoskeletal structure may be the basis of the change in GCP polarity, while the change in cell-cell adhesion may lead to defects in the axon fasciculation and migration of GCPs lacking Cdc42 [197].
Likewise, Cdc42 is generally regarded as playing a role similar to that of Rac1 in spine and synapse development. For example, Cdc42 activity is required for spino- and synaptogenesis in hippocampal neurons [198], including in adult born neurons of the dentate gyrus [199]. However, as described below (Sec. 2B), one reason for this dual GTPase requirement is that the two proteins affect actin polymerization in different ways, both of which are required for spinogenesis. Accordingly, Cdc42 function cannot be replaced by Rac1 in these processes [199–201]. In a mouse with a chromosomal deletion similar to that observed in schizophrenia, it was reported that the neuron-specific palmitoylated splice variant of Cdc42 is required for the stabilization of spines already formed [202]. Determining the precise role of Cdc42 vis-à-vis Rac1 signalling and the roles that its splice variants play will be an interesting challenge in the future.
Cdc42 is also thought to play a role in inhibitory synaptogenesis. Much of this story revolves around a Cdc42-GEF encoded by ARHGEF9 known as collybistin (CB) in rodents [203] and hPEM-2 in humans [204]. CB/hPEM-2 localizes to a subset of inhibitory synapses that vary in prevalence depending on brain region, ranging from 40–80% [205]. While it is clear that CB/hPEM-2 can help to drive gephyrin and inhibitory neurotransmitter receptor clustering [203,206,207], conflicting results as to whether [207] or not [206] its Cdc42-GEF activity is required have been reported. Moreover, depending on alternative splicing, CB may or may not possess an N-terminal SH3 domain that contributes to Cdc42 regulation [207]. Both dominant negative and constitutively active mutants of Cdc42 drive increasing size of gephyrin clusters in hippocampal neurons, suggesting that Cdc42 turnover stipulates inhibitory synapse size [207]. Uncovering the precise role of Cdc42 in inhibitory thus presents yet another interesting challenge.
Other Rho-GTPases. Besides the canonical Rho-GTPases, atypical Rho-GTPases also contribute to axon guidance. For instance, CHW-1 and CRP-1, related to Cdc42, work redundantly with Cdc42 in axon pathfinding and neuronal migration in Caenorhabditis elegans [208]. Overexpression of CHW-1 or CHW-1 GTPase mutants alters axon guidance, indicating that appropriate levels of CHW-1 expression and activities are critical for this process [208]. KD of Rho-BTB, another Rho-GTPase, reduced dendrite numbers in Drosophila dendritic arborization neurons, suggesting a role in dendritic development [209]. Rnd3 KD in the embryonic cerebral cortex interferes with interactive nuclear migration of radial glial stem cells, disrupting their apical attachment and changing the orientations of their cleavage planes. These defects were rescued by co-expression of the active form of cofilin (see below) [210]. RhoG promotes neuronal migration [211] and neurite outgrowth [104,211,212], opposes dendritic branching [213], and is required for spinogenesis in hippocampal neurons [214]. The roles of the non-canonical Rho-GTPases are likely underappreciated and much work remains in this area.
2B. Spine and synapse remodelling
Spine structure is supported chiefly by actin filaments, and Rho-GTPases play an essential role in regulating the actin dynamics that underlie spine and synapse formation, maintenance, plasticity, and elimination [215]. Rho-GTPases regulate actin assembly and disassembly through effectors, many of which are mentioned above (Figure 2A). In this section, we will treat additional aspects of Rho-GTPase signalling at spines and synapses.
Direct effects on the F-actin regulatory machinery in spines. The critical regulator cofilin severs F-actin, producing new barbed ends for polymerization or causing actin disassembly [216,217]. Actin control by cofilin is necessary for proper mature spine density and morphology [218], changes in spine morphology related to LTD of hippocampal synapses in mature (but not juvenile) mice [219], and spine loss in CA1 pyramidal neurons in response to sleep deprivation [220]. Moreover, cofilin is a nexus downstream of multiple signals [220,221], including Rho-GTPases. RhoA inhibits cofilin function by stimulating phosphorylation of Ser3 on LIM kinase (LIMK) by ROCK; LIMK then phosphorylates and inhibits cofilin [222] (Figure 2A). Activation of the serotonin receptor 5-HT4R locally activates RhoA and stimulates cofilin phosphorylation, ultimately driving spine and synapse maturation [223]. RhoA/ROCK may also mediate synaptoxicity in primary cortical neurons via cofilin phosphorylation downstream of Aβ [224]. Interestingly, Rac1 and Cdc42 also activate LIMK via PAK1, inhibiting cofilin activity [225] (Figure 2A). However, Rac1 counteracts PAK1/LIMK-mediated cofilin inhibition by activating the cofilin phosphatase slingshot 1 (SSH1) [226], suggesting a modulation of cofilin turnover. Disruption of Rac1/cofilin signalling leads to spine and synapse loss in Fragile X Syndrome, the most common genetic cause of intellectual disability [227], and in aluminium toxicity [228].
The Arp2/3 complex is a seven-protein complex and one of the most important cellular actin nucleators, creating branched actin networks and capping pointed ends of actin filaments [229–231]. Arp2/3 is required for the formation and remodelling of the dense, highly branched actin cytoskeleton that controls spine morphology and function [215,229]. In all contexts, Arp2/3 requires activation through nucleation promoting factors (NPFs) such as Wiskott-Aldrich Syndrome protein (WASP) or the WAVE1/2 complex [232,233] (Figure 2A). Cdc42 activates Arp2/3 through WASP and the closely related N-WASP to drive actin polymerization and spine and synapse development in hippocampal neurons [53,198,234]. Likewise, Rac1 regulates Arp2/3-mediated branched actin polymerization in spines through WAVE activation via Cdk5 or IRSp53 [234–237]. Thus, though both Rac1 and Cdc42 relay positive synaptogenic signals through F-actin, they do so via distinct mechanisms with differing effects.
Diaphanous formins (mDia1 and 2) are Rho-GTPase effectors that contain formin homology 1 and 2 domains (FH1/2), the latter binding to the barbed end of F-actin and driving unbranched actin polymerization [238] (Figure 2A). mDia’s FH1 domain interacts with profilin-bound actin monomer and other species, facilitating actin polymerization [238]. mDias are well-positioned to affect the spine cytoskeleton, though the data remain incomplete. mDia2-mediated elongation of F-actin is essential for filopodial formation and elongation in the initial phase of spinogenesis and ultimately for normal mature spine density; this is regulated by the Rho-GTPase RhoF/Rif [218]. mDia1/2 are also RhoA effectors, though the implications of this axis in spines are not well understood. Protein kinase A (PKA)-mediated phosphorylation of RhoA at Ser188 occludes its activation of ROCK, shunting RhoA activation elsewhere, including mDia1 [239]. Further afield, Aβ-mediated RhoA activation leads to mDia1-mediated ectopic stabilization of neuronal microtubules and precipitates spine loss [240].
In addition to actin dynamics, Rho-GTPases regulate actin function in spines. For example, myosin IIB is a cytoskeletal motor protein essential for actomyosin contractility that is necessary for spine maintenance. In primary neurons, myosin IIb KD or inhibition caused spine heads to elongate and become more filopodia-like and mediated synapse loss [241]. Myosin is downstream of RhoA in at least two ways: ROCK phosphorylates the myosin light chain, increasing its ATPase activity [242] and phosphorylates and inactivates myosin light chain phosphatase [243]; both may play a role in CA1 spine loss caused by chronic restraint stress [244]. Interestingly, the two ROCK isoforms phosphorylate myosin light chains differently, leading to differing outcomes: ROCK1 targets Thr18 and drives the formation of actomyosin bundles that confer spine polarity, while ROCK2 targets Ser19 and regulates contractile force in the spine head by attenuating Rac1 activity at this site [245]. Furthermore, Rac1 activation is required for proper spine localization of myosin II and normal retrograde actin flow through spines [246], and can cause protein kinase C (PKC)-dependent phosphorylation of the heavy chain of myosin IIa, regulating its subcellular localization in tissue culture cells [247].
Rho-GTPases in glutamatergic receptor trafficking. As noted above, the ultimate consequences of LTP and LTD are the insertion and removal, respectively, of surface AMPARs from excitatory synapses. AMPAR traffic requires actin and its regulators [248–250], so it is not surprising that Rho-GTPases, too, play a critical role (Figure 5). Rac1 is required for synaptic AMPAR insertion [176] during development and in NMDAR-mediated LTP in the hippocampus and nucleus accumbens [63,251–256]. Depending on the context and neuron type, this occurs downstream of several Rac1-GEFs, including DOCK4 [251], Trio [254], and Kalirin-7 [63]. Paradoxically, Rac1 activation is also required for hippocampal AMPAR endocytosis and resultant LTD, also downstream of NMDARs [253,257–261]. This Rac1 activation requires the Rac1-GEFs P-Rex1 [259] and Tiam1 [257], and is mediated by both Rac1’s canonical actin regulatory proteins [261] and Jun N-terminal kinase 1 (JNK-1) [258]. Less is known about the role of Cdc42 in AMPAR traffic, but it functions to increase synaptic AMPAR levels downstream of EphB2 [262] and NMDARs [263]. As in the case of Rac1, the role of RhoA in AMPAR traffic is complex. RhoA overactivation through KD of negative regulators produces the expected decreases in surface AMPARs [260,264] and, accordingly, the RhoA-GAP oligophrenin-1 is required for LTP formation in CA1 neurons [265]. However, oligophrenin-1 KO inhibits LTD formation and AMPAR endocytosis, effects reversed by pharmacologic RhoA signalling inhibition [266]. Furthermore, RhoA activity affects AMPAR subunit composition, increasing GluA3 at the expense of GluA1 [267].
Figure 5.
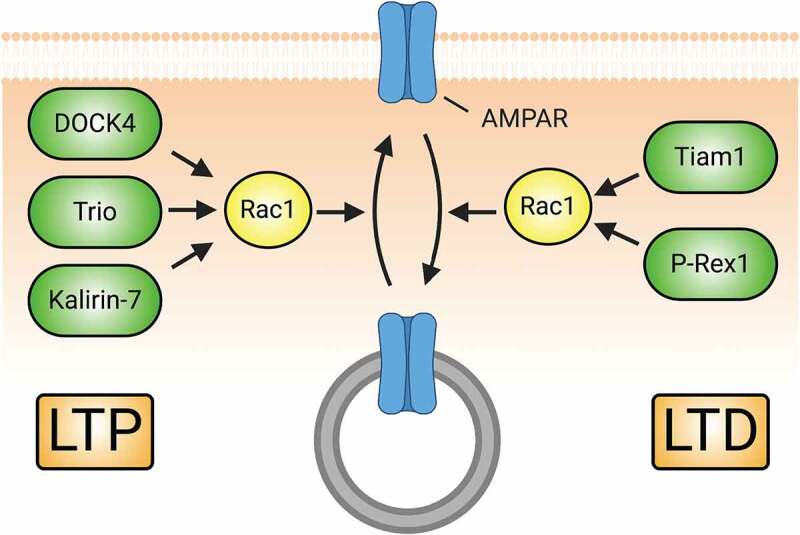
Rac1-GEFs differentially coordinate synaptic AMPAR insertion (LTP) and endocytosis (LTD) through Rac1-mediated actin cytoskeletal modifications. The surface expression of AMPARs during development and in response to LTP relies on Rac1 activity. Rac1-GEFs like DOCK4, Trio, and Kalirin-7 can activate Rac1 following LTP-inducing NMDAR activity. Subsequent Rac1-mediated actin cytoskeletal remodelling promotes the synaptic insertion of AMPARs. Conversely, Rac1 activity can also drive the downregulation of AMPAR surface expression in conditions promoting LTD. NMDAR-mediated signalling of Tiam1 and P-Rex1 activate Rac1 to promote synaptic AMPAR endocytosis.
Despite the role that Rho-GTPases play in transducing NMDAR-mediated signalling, less is known about the role they play in regulating NMDAR traffic. Rac1 overexpression rescues lowered surface NMDAR expression in DOCK4 KO mouse neurons [251], but perturbation of Bcr localization in hippocampal neurons causes hyperactivation of both Rac1 and RhoA and leads to a loss of NMDARs from synapses [260]. More work is required to understand the role that Rho-GTPases play in this process.
Rho-GTPases in heterosynaptic plasticity. Heterosynaptic plasticity refers to phenomena in which plasticity at one synapse affects that of nearby synapses. Excitatory synaptic activity leads to a spread of activated RhoA and Rac1 into the dendritic shaft and neighbouring spines, while active Cdc42 remains in the stimulated spine [43,56]. Diffusion of RhoA and Rac1 alone to the nearby spines does not induce plasticity [56], but it is necessary for synaptic crosstalk in which spines near an activated spine undergo expansion in response to subthreshold inputs [56,268], discussed in [269]. Although the precise mechanism of RhoA- and Rac1-mediated potentiation of nearby spines needs further investigation, it is possible that these GTPases exert their effects by increasing tonic levels of their target signalling cascades, decreasing the activation energy for LTP in response to further stimuli. It is easy to imagine how this pattern of diffusing Rho-GTPases might also play a role in heterosynaptic depression, in which activated spines causes shrinkage and/or retraction of nearby unstimulated spines [270], but this is not yet known.
Rho-GTPases in memory. Memory comprises a critical cognitive domain, and, as key synaptic regulators, Rho-GTPases play prominent roles therein. In addition to its role in spine and synapse formation, Rac1 plays a key role in LTP, a cellular process thought to underlie learning and memory [271]. KO of Rac1 from forebrain neurons impedes LTP formation [252], as does interfering with Rac1 signalling downstream of BDNF by preventing Tiam1 recruitment to TrkB receptors with a TrkB mutant [70]. Other manipulations that alter Rac1 signalling, including disrupting its spine localization [272], suppressing its activation through iodine deficiency [273], or inducing exaggerated signalling through KO of Rac1-GAPs [81] also strongly inhibit the formation of LTP. Critically, these and other disruptions of Rac1 signalling greatly impair spatial learning, working memory, object recognition memory, and fear memory in mice [70,81,252,274]. In addition, Rac1 KO in the dentate gyrus inhibits adult neurogenesis, which also impairs working memory [275]. Moreover, Rac1 controls the association between Cdk5 and p35 in the hippocampus and functions to prevent memory extinction therein, though this extinction can be stimulated by direct action of Cdk5 on the Rac1-effector PAK1 [276]. In addition to its role in the hippocampus in regulating LTP and memory, Rac1 is also required for auditory fear memory formation in the basolateral amygdala (BLA) [277].
As is often the case with Rho-GTPases, it is not that simple: Rac1 also opposes memory formation and plays a critical role in active forgetting. In mice, selective Rac1 activation in spines recently potentiated by motor learning erases the motor memory [278]. Similarly, retrograde interference introduced 22 hr post-training increases Rac1 levels and induced forgetting [279] and dominant negative and constitutively active Rac1 in the CA1 enhanced and diminished fear memory, respectively, at 24 h, but not 1 h, after training [280]. Rac1 inhibition through pharmacology or targeted expression of DN mutants in excitatory hippocampal neurons extends object recognition memory and contextual fear memory, whereas Rac1 activation via drugs or stimulation of photoactivatable Rac1 shows the opposite effect [279,281]. Likewise, in CA1, Rac1 is regulated by the Rac1-GAP α2-chimaerin, whose expression is triggered by learning tasks. KD of α2-chimaerin increases Rac1 activation and impedes LTP and memory formation, while overexpression of α2-chimaerin suppresses Rac1 activity, promoting LTP and memory formation [280]. Further, loss of the Rac1-GEF Tiam1 in dentate granule cells facilitates contextual fear learning [79]. Social isolation accelerates forgetting in mice through Rac1 activity [282]. Rac1 also mediates active forgetting in the nucleus accumbens [283] and in Drosophila [284]. Interestingly, Rac1 is hyperactive in humans with Alzheimer’s Disease and animal models thereof [285], and memory in both mouse and Drosophila models can be improved through pharmacological Rac1 inhibition [285]. Similarly, pharmacological inhibition of Rac1 signalling also improves contextual memory in a mouse model of Fragile X Syndrome [286]. Thus, Rac1 promotes both formation and dissolution of memories, though perhaps on different time scales.
Intriguingly, Cdc42, despite its generally similar functions and effectors, exhibits the opposite effects. To wit, Cdc42 conditional KO mice have impaired remote memory recall after fear conditioning and Morris water maze training [287]. This striking difference in function might arise from the differing methods of KO, local vs. global, of Rac1 and Cdc42 in the models used. Among Rho-GTPase effectors, cofilin and WAVE complex-formin activity in Rac1-mediated forgetting and WASP-Arp2/3 activity in Cdc42-mediated forgetting is reported in Drosophila [288,289]. In mice, Arp2/3 and vasodilator-stimulated phosphoprotein (VASP) function in the lateral amygdala is important for long-term memory maintenance [290].
Despite its role in opposing spine and synapse formation in development (Sec. 2A), RhoA can also play a role in the formation of memories. RhoA is involved in the formation of conditioned aversive memories in the hippocampus in mice [291], and also of spatial working memory in the dorsal striatum of rats [292]. BDNF stimulates RhoA synthesis at spines in the CA1 and CA3 regions of the hippocampus via mTOR, and blocking this synthesis impairs LTP consolidation [293]. Interestingly, this memory-initiated RhoA signal is transient, as it is degraded by calpain-1 [293]. RhoA is also implicated in the formation of addiction, due to its role in the formation of conditioned place preference for morphine in rats [294]. Clearly, the precise role of Rho-GTPases, including downstream pathways in different brain regions, and their mutual interactions in both memory and active forgetting needs further clarification.
2C. Rho-GTPase signalling associated with neuronal injury
Traumatic brain injury. Traumatic brain injury (TBI) induces pathological changes, including inflammation, neural circuit disruption, and neuronal and glial death, that lead to long-term cognitive, motor and emotional disabilities [295,296]. TBI causes immediate necrotic damage to the brain, including neurons, but also pathological signalling. Strikingly, RhoA activation increases for hours to months after TBI [297–299]. This activates a ROCK and phosphatase and tensin homolog (PTEN)-pathway that inactivates the pro-survival protein Akt, further decreasing neuronal survival [300]. Moreover, RhoA activation can provoke apoptosis via p38 or JNK by activating pro-apoptotic members of the Bcl-2 family [301]. These injuries are unlikely to be reversible, and thus necessitate immediate action, so the connection between TBI and RhoA is potentially very important. One approach to exploiting this connection is by inhibiting RhoA/ROCK signalling with Fasudil or Y-27632. Indeed, fasudil treatment rescues TBI-induced motor, cognitive, and synaptic deficits in the cortical contusion model of rodent TBI, similar to the effects seen with neuronal RhoA ablation [302]. Since TBI induces synapse loss [303] and Rac1 promotes process growth and synapse formation and maintenance, Rac1 is positioned to potentially enhance regeneration and/or recovery following TBI [145,304]. Moreover, by signalling through PAK, Rac1 is poised to oppose RhoA’s effects on neuronal survival through activation of mitogen activated protein kinase (MAPK) pathways that inhibit pro-apoptotic Bad and Bax and increase the expression of pro-survival Bcl-xL members of the Bcl-2 family [305–308]. Rac1 also stimulates the pro-survival PI3K/Akt pathway, enhancing neuronal survival [145].
Neuropathic pain. Emerging evidence suggests that Rho-GTPase-regulated spine remodelling in the spinal cord plays important roles in the development of chronic pain, which may explain why pain can persist for months or years after injury [309,310]. After spinal cord or peripheral nerve injury, animals exhibit symptoms of thermal hyperalgesia and tactile allodynia, accompanied by increased spine density, rearrangement of dendritic spines, and enhanced mEPSCs in spinal cord dorsal horn neurons located in lamina IV–V [311,312]. Meanwhile, NSC23766, an inhibitor of Rac1 activation, can normalize the morphology of spines after injury, reduce traumatic hyperexcitability, and increase pain thresholds [311,312]. Similar phenomena were observed in the STZ-induced diabetic neuropathic pain and burn injury models [313,314]. Further studies have shown that superficial dorsal horn neurons located in lamina II are also involved in pain memory after spinal cord injury. Although the total density of dendritic spines on lamina II neurons after spinal cord injury does not change, thin spine density decreases and mushroom spine density increases; NSC23766 reduces these changes and attenuates neuropathic pain [315,316]. KD of the Rac1-GAP srGAP3 during the maintenance phase enhances Rac1 activity, promotes maturation of spines, and increases the persistence of neuropathic pain [317]. These observations suggest that Rac1 signalling pathways regulate dendritic spine remodelling and may explain pain analgesia after spinal cord injury or peripheral injury.
RhoA signalling may also contribute to the development of neuropathic pain. Activation of RhoA/ROCK2 and increased plasma membrane levels of RhoA are found in the spinal cord of neuropathologic pain model animals, and the ROCK2 inhibitor Fasudil abrogated pain hypersensitivity and increased levels of phosphorylated RhoA and ROCK2 [318,319]. Injury-induced overactivation of the RhoA/LIMK/cofilin pathway has been suggested to yield a cytoskeletal scaffold for the increased trafficking of nociceptive signalling factors, resulting in chronic neuropathic pain [320]. Indeed, treatments that reduce RhoA signalling, such as the Rho kinase inhibitor Y-27632, prevent actin filament disruption in the dorsal root ganglion and attenuate chronic constriction injury-induced neuropathic pain [320].
Cdc42 could also have a key role in proposed therapies for neuropathic pain. Neural stem cells (NSCs) have considerable ability to self-renew and generate neurons in mammalian brains, and NSC transplantation may promote peripheral nerve regeneration and provide a new method for the treatment of peripheral nerve injury [321]. It has been reported that mRNA and protein levels of Cdc42 and the number of myelinated axon fibres per nerve in animals that underwent sciatic nerve injury are significantly less than in uninjured animals [322]. Cdc42 mRNA and protein levels and the myelinated fibres/nerve ratio increased in animals that received NSC transplants, suggesting that NSCs promoted myelination in regenerated nerves [322]. Furthermore, overexpression of Cdc42 promoted myelination of the regenerated nerve and neural stem cells migration and proliferation, whereas suppression of Cdc42 by miR-7 had the opposite effects, suggesting that Cdc42 expression influences peripheral nerve injury repair through the proliferation and migration of NSCs [322]. Together, these observations suggest that Rho-GTPases, including Rac1, RhoA and Cdc42, play key roles in the induction of and in strategies for the treatment of neuropathic pain.
2D. Diseases associated with Rho-GTPase dysfunction
As would be expected from the critical roles enumerated above, dysfunctions of Rho-GTPase signalling pathways underlie a number of neurodevelopmental and neurodegenerative diseases. Here, we highlight recent developments in Rho-GTPase signalling impacting diseases that exact high clinical burdens.
Schizophrenia. Schizophrenia is a psychiatric disease characterized by defects in cortical network circuitry. Cellularly, schizophrenia consistently associates with decreases in spine density [323–326]. Rho-GTPase abnormalities are implicated in the neuropathology of schizophrenia, such as reduced pro-spinogenic Cdc42 signalling [327]. Genetic analyses of schizophrenia cases revealed mutational burdens on cytoskeleton regulating genes, including that which encodes IRSp53, BAIAP2 [328,329]. Hypomethylation of BAIAP2 is associated with reduced spine density in the superior temporal gyrus observed in schizophrenia patients [330]. IRSp53 regulates Rac1 and Cdc42 in dendrites during synaptogenesis by anchoring GEFs, GAPs, and other regulators on its SH3 domain (Figure 6A), including Tiam1, BAI1, WAVE2, Kalirin-7, and Bcr/Abr [331,332], and its deletion in mice recapitulates decreased spine densities and abnormal behaviours observed in schizophrenic humans, including hyperactivity and cognitive dysfunction [333,334]. Additionally, a 50% reduction in IRSp53 levels upregulates NMDAR function, an effect partially ameliorated by the NMDAR antagonist memantine to some therapeutic benefit [333,335,336]. Restricted KO of IRSp53 in dorsal telencephalic neurons increases the ratio of evoked excitatory and inhibitory synaptic transmission in male, but not female, mice [337]. The combination of altered GEF/GAP and NMDAR function upstream of Rac/Cdc42 activity may account for the defects observed in some schizophrenia patients. Further support for this idea is provided by the evidence that Kalirin mRNA, which encodes several Rho-GEFs, is decreased in schizophrenia post-mortem analyses, along with increased Kalirin-9 and decreased Kalirin-7 protein levels [338]. Notably, Kalirin-7 has also been shown to interact with the schizophrenia risk factor Disrupted-in-Schizophrenia 1 (DISC1), which controls spine size by restricting the duration and intensity of Rac1 activation by Kalirin-7 in response to NMDAR activation [339].
Figure 6.
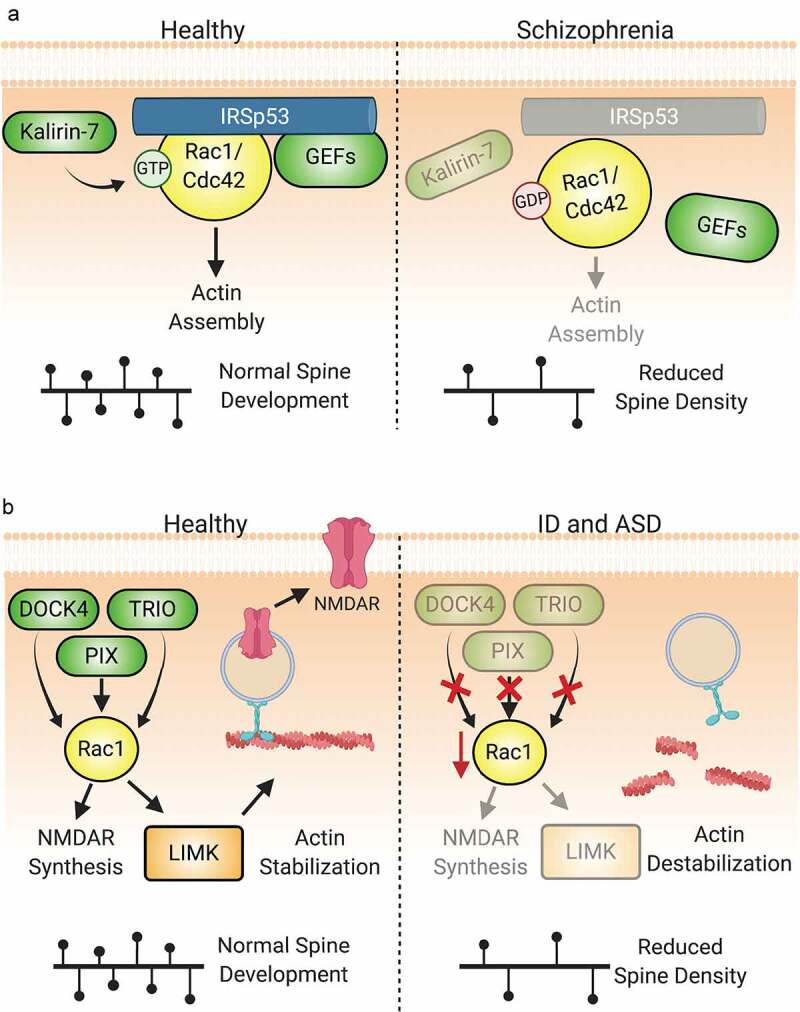
Dysregulation of Rho-GTPase signalling is involved in the pathologies of numerous neuropsychiatric disorders. (A) IRSp53 serves as a scaffold to mediate the interactions of Rac1 and Cdc42 with regulatory proteins. Under normal conditions, IRSp53 promotes actin stabilization and synaptic maturation by promoting Rac1 and Cdc42 activity. Loss of IRSp53 activity through mutation or hypomethylation of BAIAP2, the gene encoding IRSp53, is associated with the development of schizophrenia. The reduction in IRSp53 levels limits the activity of GEFs to activate Rac1 and Cdc42, thereby diminishing pro-spinogenic Rho-GTPase-mediated actin cytoskeletal changes and reducing dendritic spine density. Additionally, diminished levels of Kalirin-7 at the protein and mRNA levels have been observed in schizophrenia patients. This reduction can directly lead to decreased size and density of dendiritic spines. (B) Numerous Rho-GEFs and -GAPs are implicated in the pathophysiology of intellectual disability (ID). For example, multiple mutations in the Rac-GEFs TRIO and α-PIX have been found in individuals with ID. Loss-of-function of these proteins diminish Rac1 activation and the downstream signals associated with its activity. Likewise, DOCK4 is one of many Rho-GEFs that may be linked to the pathogenesis of autism spectrum disorder. The dysregulation of DOCK4 protein levels or activity reduces Rac1-mediated actin stabilization in addition to NMDAR subunit translation. This results in the reduction of both excitatory synapses through spine loss and LTP by the reduction in available NMDARs.
Intellectual Disability and Autism Spectrum Disorder. Perhaps the most significant link between Rho GTPase dysfunction and neurological pathology is in intellectual disability (ID) [340–342]. ID is a heterogeneous neurodevelopmental disorder that adversely affects intellectual, adaptive, and social functioning. While some aetiologies like lissencephaly and microcephaly cause gross structural brain alterations, a significant number of cases present without these changes. Given the cognitive deficits associated with ID, synaptic dysfunction appears to be the most likely cause [343]. Genetic studies have supported this by linking a majority of the hundreds of mutations associated with ID to the pre- and post-synaptic compartments, including Rho-GTPase family members such as oligophrenin-1, PAK3, ARHGEF9, TRIO, and β-PIX among many others [344–346]. The confluence of Rho-GEF and -GAP mutations and aberrations of synaptic development and/or plasticity leading to deficits in information processing in ID has placed Rho-GTPase dysfunction at the molecular centre of this disorder [347].
There has been much attention given to the evident relationship between Rho-GTPases and ID and it has been extensively reviewed [340,346]. The relationship between Rho-GTPase function and autism spectrum disorder (ASD), while often coincident with ID, has received less attention and can provide further insight into the role of Rho-GTPases in synaptic dysfunction [348]. ASD arises from disruptions in neurodevelopment, leading to impaired communication and social interaction and repetitive behaviours and interests, often with significant comorbidities. The genetic causes of ASD are very complex due to its polygenism: nearly 2,000 genes are linked to ASD aetiology [349]; many of these genes are involved in synapse development and maturation [350,351]. Between 8–12% of Rho-GEFs, Rho-GAPs, and Rho effectors are included in ASD susceptibility gene databases [349]. By focusing on the Rac1-GEF DOCK4 (Figure 6B) [352], it is possible to see how Rho-GTPase dysfunction may be involved in ASD pathogenesis. There are many documented SNPs, microdeletions, and duplications of the DOCK4 gene in autism patients [353,354]. In fact, both homozygous and heterozygous KOs of this gene in mice revealed altered social, anxiety, and memory functioning [355]. In addition to decreased dendritic spine density, these mice exhibited decreased CA1 mEPSCs, primarily NMDAR-mediated, and impaired LTP. Both the behavioural and cellular dysfunctions are Rac1-dependent, as Rac1 overexpression increased NMDAR subunit protein synthesis, restored dendritic spine density, and corrected behavioural symptoms [355].
Alzheimer’s Disease. Alzheimer’s disease (AD) presents with progressive decline in cognition and is associated with the formation of amyloid plaques (aggregates of Aβ) and neurofibrillary tangles (aggregates of tau). Cognitive decline is more strongly associated with the dysfunction and loss of excitatory synapses associated with learning and memory than aggregate build-up and neuronal atrophy [356–358]. Zhao et al. first reported that PAK1 and PAK3 were significantly lost in neurons of AD patients and in animal models [359]. Downregulation of either PAK1 or PAK3 does not dramatically affect neuronal morphology or function, but loss of both leads to significant decreases in mature spine and synapse density and synaptic plasticity (Figure 7A) [360]. This effect is related to Aβ by an abnormal activation and subsequent loss of cytoplasmic PAK1 and PAK3, leading to the loss of F-actin and spines themselves [361–363]. Greater reductions in both cytoplasmic and synaptic PAK have indeed been associated with more severe presentations of AD [361,363].
Figure 7.
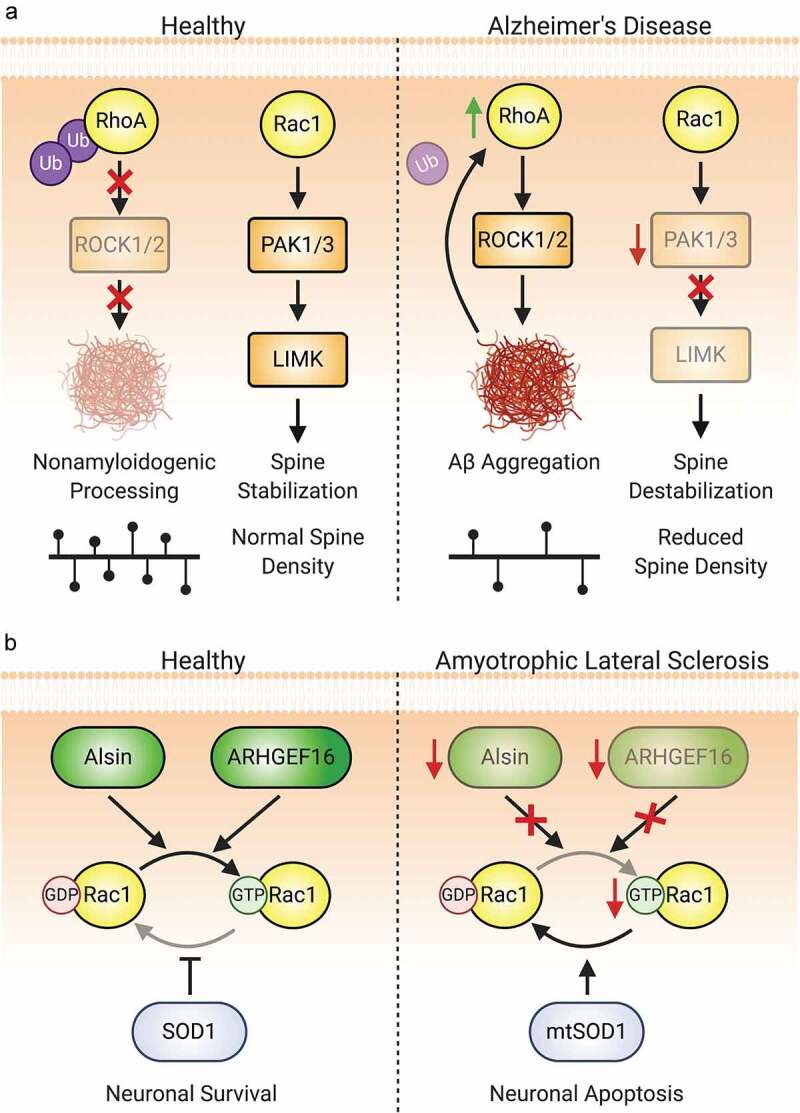
Aberrant Rho-GTPase signalling is also implicated in neurodegenerative diseases. (A) Both Rac1 and RhoA dysregulation are implicated in the pathogenesis of Alzheimer’s disease. A reduction in the ubiquitination and degradation of RhoA leading to an increase in ROCK1/2 activity promotes the aggregation of Aβ. This in turn further downregulates Ube3A-mediated ubiquitination of RhoA, propagating the aberrant signalling and increased amyloid plaque development. Rac1 signalling is also dysregulated due to reduced levels of the Rac1 effectors PAK1 and PAK3. The decrease in LIMK activation disinhibits cofilin, promoting actin cleavage and spine loss. (B) Rac1 activity is also implicated in several aetiologies of amyotrophic lateral sclerosis (ALS). Loss-of-function or downregulation of Rac1-GEFs such as Alsin and ARHGEF16 are associated with both juvenile- and adult-onset ALS. Mutations in SOD1, strongly associated with ALS development, also affect Rac1. Mutant SOD1, as well as an increased oxidative environment found in ALS, inhibit Rac1 by promoting GTP hydrolysis.
Aberrant RhoA signalling is also implicated in AD-associated synaptic loss (Figure 7A). It has long been appreciated that RhoA plays a role in AD pathology via the therapeutic use of NSAIDs that inhibit RhoA signalling [364], and to the reduction of Aβ aggregates in the brain by the ROCK1/2 inhibitor Y-27632 [364–366]. Excessive RhoA activation drives ROCK hyperactivation, leading to pathologic processing of amyloidogenic APP into toxic Aβ [367]. Additionally, as ROCK phosphorylates LIMK, aberrant RhoA activation leads to F-actin dysregulation. The E3 ligase Ube3A regulates RhoA protein levels and is downregulated in AD [368]. Ube3A also regulates the RhoA-GEF Ephexin-5 [51]. Aβ-triggered depletion of Ube3A leads to RhoA accumulation and overactivation and its deleterious downstream signals [368,369]. Thus, Rac1, Cdc42, and RhoA dysfunction all contribute to the synaptic loss and progressive cognitive decline observed in AD.
Amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that presents with progressive and severe muscle atrophy secondary to loss of neurons along the corticospinal tract, usually leading to respiratory failure and death. Of the 10% of cases with genetic aetiologies, ~20% are caused by mutations in the gene encoding superoxide dismutase 1 (SOD1), an enzyme that detoxifies superoxide free radicals. The roles of Rho-GTPases in ALS have emerged, with decreased Rac1 and increased RhoA activity being implicated in neuronal death (Figure 7B). In neurons with mutant SOD1, constitutively active Rac1 prevents, whereas dominant negative Rac1 induces, neuronal apoptosis [370]. Moreover, loss-of-function mutations in alsin, a Rac1-GEF, cause juvenile-onset ALS [371,372], and hypermethylation and downregulation of the Rac1-GEF-encoding ARHGEF16 are seen in patients with sporadic ALS (Figure 7B) [373]. Conversely, Rho/ROCK signalling is increased in diseased spinal cord neurons [374–376]. In mice expressing mutant SOD1, active GTP-bound Rac1 is significantly decreased in motor neurons at disease onset but not prior [377]. While the amount of active RhoA in ALS mouse spinal cord was not different from healthy controls, RhoB was redistributed to neuronal processes of the diseased neurons [377]. Pharmacological inhibition of RhoB signalling through ROCK inactivation significantly increased neuronal survival [378,379].
3. Conclusions
Despite decades of progress, much remains unknown about both the exquisite regulatory mechanisms and expansive and spatiotemporally precise downstream signals and outputs of Rho-GTPases. The extent to which this complicated and daunting puzzle can be solved will determine much about our knowledge of neurons, glia, and other cell types. Since Rho-GTPases play such pivotal roles in the cognitive and emotional processes that distinguish our species, this quest has much to teach about ourselves but will also provide tools for improving human health. To this end, it will be useful to have more tools with clinical potential that target specific Rho-GTPase pathways, rather than globally targeting the multifunctional GTPases themselves. GEFs and GAPs have more inherent specificity in terms of expression patterns than Rho-GTPases and are potential targets for this enterprise. Additionally, while we have mentioned a few Rho-GTPases besides RhoA, Rac1, and Cdc42, relatively little is understood about the at least 19 remaining Rho-GTPases. Improving technologies for monitoring the spatiotemporal dynamics of Rho-GTPases, their regulators, and their downstream signalling pathways make this challenge a thrilling one, with the promise of many more exciting discoveries.
Acknowledgments
We apologize to colleagues we did not cite due to space limitations. J.G.D. is supported by NIH R01 CA219667; F.A.B. by NIH T32 GM008231; and K.F.T. by NIH R01 NS062829, R01 MH109511, and R01 MH103108. All figures were created with BioRender.com. The authors declare that they have no conflict of interest and that there are no competing financial interests.
Funding Statement
This work was supported by the National Cancer Institute [CA219667]; National Institute of General Medical Sciences [GM008231]; National Institute of Mental Health [MH103108]; National Institute of Mental Health [MH109511]; National Institute of Neurological Disorders and Stroke [NS062829].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Pakkenberg B, et al. Aging and the human neocortex. Exp Gerontol. 2003;38(1–2):95–99. DOI: 10.1016/S0531-5565(02)00151-1. [DOI] [PubMed] [Google Scholar]
- [2].Markus EJ, Petit TL, LeBoutillier JC.. Synaptic structural changes during development and aging. Developmental Brain Research. 1987;35(2):239–248. [DOI] [PubMed] [Google Scholar]
- [3].Freeman AR. Polyfunctional role of glutamic acid in excitatory synaptic transmission. Prog Neurobiol. 1976;6(2):137–153. [DOI] [PubMed] [Google Scholar]
- [4].Jahr CE, Lester RA. Synaptic excitation mediated by glutamate-gated ion channels. Curr Opin Neurobiol. 1992;2(3):270–274. [DOI] [PubMed] [Google Scholar]
- [5].Mody I, De Koninck Y, Otis TS, et al. Bridging the cleft at GABA synapses in the brain. Trends Neurosci. 1994;17(12):517–525. [DOI] [PubMed] [Google Scholar]
- [6].Harvey JD, Heinbockel T. Neuromodulation of synaptic transmission in the main olfactory bulb. Int J Environ Res Public Health. 2018;15(10):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Palacios-Filardo J, Mellor JR. Neuromodulation of hippocampal long-term synaptic plasticity. Curr Opin Neurobiol. 2019;54:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci. 2008;31(1):47–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu Y-T, Tao C-L, Lau P-M, et al. Postsynaptic protein organization revealed by electron microscopy. Curr Opin Struct Biol. 2019;54:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Henley JM, Wilkinson KA. Synaptic AMPA receptor composition in development, plasticity and disease. Nat Rev Neurosci. 2016;17(6):337–350. [DOI] [PubMed] [Google Scholar]
- [11].Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nature Reviews Neuroscience. 2013;14(6):383–400. [DOI] [PubMed] [Google Scholar]
- [12].Vyklicky V, Korinek M, Smejkalova T, et al. Structure, function, and pharmacology of NMDA receptor channels. Physiol Res/Acad Scient Bohemoslo. 2014;63(1):S191–203. Suppl. [DOI] [PubMed] [Google Scholar]
- [13].Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55(7):1081–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Diering GH, Huganir RL. The AMPA receptor code of synaptic plasticity. Neuron. 2018;100(2):314–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Govek -E-E, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19(1):1–49. [DOI] [PubMed] [Google Scholar]
- [16].Duman JG, Mulherkar S, Tu Y-K, et al. Mechanisms for spatiotemporal regulation of Rho-GTPase signaling at synapses. Neurosci Lett. 2015;601:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gadea G, Blangy A. Dock-family exchange factors in cell migration and disease. Eur J Cell Biol. 2014;93(10–12):466–477. [DOI] [PubMed] [Google Scholar]
- [18].Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nature Reviews Molecular Cell Biology. 2005;6(2):167–180. [DOI] [PubMed] [Google Scholar]
- [19].Huang G-H, Sun Z-L, Li H-J, et al. Rho GTPase-activating proteins: regulators of Rho GTPase activity in neuronal development and CNS diseases. Mol Cell Neurosci. 2017;80:18–31. [DOI] [PubMed] [Google Scholar]
- [20].DerMardirossian C, Bokoch GM. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005;15(7):356–363. [DOI] [PubMed] [Google Scholar]
- [21].Wennerberg K. Rho-family GTPases: it’s not only Rac and Rho (and I like it). J Cell Sci. 2004;117(8):1301–1312. doi: 10.1242/jcs.01118. [DOI] [PubMed] [Google Scholar]
- [22].Agarwal A, Wu P-H, Hughes EG, et al. Transient opening of the mitochondrial permeability transition pore induces microdomain calcium transients in astrocyte processes. Neuron. 2017;93(3):587–605.e7. DOI: 10.1016/j.neuron.2016.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Goldberg JH, Tamas G, Aronov D, et al. Calcium microdomains in aspiny dendrites. Neuron. 2003;40(4):807–821. [DOI] [PubMed] [Google Scholar]
- [24].Oheim M, Kirchhoff F, Stühmer W. Calcium microdomains in regulated exocytosis. Cell Calcium. 2006;40(5–6):423–439. [DOI] [PubMed] [Google Scholar]
- [25].Willoughby D, Wachten S, Masada N, et al. Direct demonstration of discrete Ca2+ microdomains associated with different isoforms of adenylyl cyclase. J Cell Sci. 2010;123(1):107–117. doi: 10.1242/jcs.062067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Averaimo S, Assali A, Ros O, et al. A plasma membrane microdomain compartmentalizes ephrin-generated cAMP signals to prune developing retinal axon arbors. Nat Commun. 2016;7(1):12896. DOI: 10.1038/ncomms12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bhogal NK, Hasan A, Gorelik J. The development of compartmentation of cAMP signaling in Cardiomyocytes: the role of T-Tubules and Caveolae Microdomains. J Cardiovasc Dev Dis. 2018;5(2):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Burdyga A, Surdo NC, Monterisi S, et al. Phosphatases control PKA-dependent functional microdomains at the outer mitochondrial membrane. Proc Natl Acad Sci U S A. 2018;115(28):E6497–E6506. DOI: 10.1073/pnas.1806318115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Abrahamsen H, Vang T, Taskén K. Protein kinase A intersects SRC signaling in membrane microdomains. J Biol Chem. 2003;278(19):17170–17177. [DOI] [PubMed] [Google Scholar]
- [30].Sim AT, Scott JD. Targeting of PKA, PKC and protein phosphatases to cellular microdomains. Cell Calcium. 1999;26(5):209–217. [DOI] [PubMed] [Google Scholar]
- [31].Monastyrskaya K, Hostettler A, Buergi S, et al. The NK1 receptor localizes to the plasma membrane microdomains, and its activation is dependent on lipid raft integrity. J Biol Chem. 2005;280(8):7135–7146. [DOI] [PubMed] [Google Scholar]
- [32].Gao X, Lowry PR, Zhou X, et al. PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc Natl Acad Sci U S A. 2011;108(35):14509–14514. DOI: 10.1073/pnas.1019386108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Seong J, Ouyang M, Kim T, et al. Detection of focal adhesion kinase activation at membrane microdomains by fluorescence resonance energy transfer. Nat Commun. 2011;2(1):406. DOI: 10.1038/ncomms1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sui Z, Kovács AD, Maggirwar SB. Recruitment of active glycogen synthase kinase-3 into neuronal lipid rafts. Biochem Biophys Res Commun. 2006;345(4):1643–1648. [DOI] [PubMed] [Google Scholar]
- [35].Delos Santos RC, Garay C, Antonescu CN. Charming neighborhoods on the cell surface: plasma membrane microdomains regulate receptor tyrosine kinase signaling. Cell Signal. 2015;27(10):1963–1976. [DOI] [PubMed] [Google Scholar]
- [36].Mateos-Aparicio P, Rodriguez-Moreno A. Calcium signaling. advances in experimental medicine and biology. In: Islam M, editor. Calcium dynamics and synaptic plasticity. Cham: Springer; 2020. p. 965–984. [DOI] [PubMed] [Google Scholar]
- [37].Stanton PK. LTD, LTP, and the sliding threshold for long-term synaptic plasticity. Hippocampus. 1996;6(1):35–42. [DOI] [PubMed] [Google Scholar]
- [38].Ng S-W, Nelson C, Parekh AB. Coupling of Ca2+ microdomains to spatially and temporally distinct cellular responses by the tyrosine kinase syk. J Biol Chem. 2009;284(37):24767–24772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dema A, Perets E, Schulz MS, et al. Pharmacological targeting of AKAP-directed compartmentalized cAMP signalling. Cell Signal. 2015;27(12):2474–2487. [DOI] [PubMed] [Google Scholar]
- [40].Wild AR, Dell’Acqua ML. Potential for therapeutic targeting of AKAP signaling complexes in nervous system disorders. Pharmacol Ther. 2018;185:99–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sheffler-Collins SI, Dalva MB. EphBs: an integral link between synaptic function and synaptopathies. Trends Neurosci. 2012;35(5):293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tolias KF, Duman JG, Um K. Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog Neurobiol. 2011;94(2):133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Murakoshi H, Wang H, Yasuda R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature. 2011;472(7341):100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Henderson NT, Dalva MB. EphBs and ephrin-Bs: trans-synaptic organizers of synapse development and function. Mol Cell Neurosci. 2018;91:108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Henkemeyer M, Itkis OS, Ngo M, et al. Multiple EphB receptor tyrosine kinases shape dendritic spines in the hippocampus. J Cell Biol. 2003;163(6):1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kania A, Klein R. Mechanisms of ephrin–Eph signalling in development, physiology and disease. Nature Reviews Molecular Cell Biology. 2016;17(4):240–256. [DOI] [PubMed] [Google Scholar]
- [47].Sloniowski S, Ethell IM. Looking forward to EphB signaling in synapses. Semin Cell Dev Biol. 2012;23(1):75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hruska M, Dalva MB. Ephrin regulation of synapse formation, function and plasticity. Mol Cell Neurosci. 2012;50(1):35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Klein R. Bidirectional modulation of synaptic functions by Eph/ephrin signaling. Nat Neurosci. 2009;12(1):15–20. [DOI] [PubMed] [Google Scholar]
- [50].Um K, Niu S, Duman JG, et al. Dynamic control of excitatory synapse development by a Rac1 GEF/GAP regulatory complex. Dev Cell. 2014;29(6):701–715. DOI: 10.1016/j.devcel.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Margolis SS, Salogiannis J, Lipton DM, et al. EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell. 2010;143(3):442–455. DOI: 10.1016/j.cell.2010.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tolias KF, Bikoff JB, Kane CG, et al. The Rac1 guanine nucleotide exchange factor Tiam1 mediates EphB receptor-dependent dendritic spine development. Proc Natl Acad Sci U S A. 2007;104(17):7265–7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Irie F, Yamaguchi Y. EphB receptors regulate dendritic spine development via intersectin, Cdc42 and N-WASP. Nat Neurosci. 2002;5(11):1117–1118. [DOI] [PubMed] [Google Scholar]
- [54].Thomas S, Ritter B, Verbich D, et al. Intersectin regulates dendritic spine development and somatodendritic endocytosis but not synaptic vesicle recycling in hippocampal neurons. J Biol Chem. 2009;284(18):12410–12419. DOI: 10.1074/jbc.M809746200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hamilton AM, Lambert JT, Parajuli LK, et al. A dual role for the RhoGEF Ephexin5 in regulation of dendritic spine outgrowth. Mol Cell Neurosci. 2017;80:66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hedrick NG, Harward SC, Hall CE, et al. Rho GTPase complementation underlies BDNF-dependent homo- and heterosynaptic plasticity. Nature. 2016;538(7623):104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hedrick NG, Yasuda R. Regulation of Rho GTPase proteins during spine structural plasticity for the control of local dendritic plasticity. Curr Opin Neurobiol. 2017;45:193–201. [DOI] [PubMed] [Google Scholar]
- [58].Penzes P, Beeser A, Chernoff J, et al. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron. 2003;37(2):263–274. DOI: 10.1016/S0896-6273(02)01168-6. [DOI] [PubMed] [Google Scholar]
- [59].Duman JG, Tzeng CP, Tu Y-K, et al. The adhesion-GPCR BAI1 regulates synaptogenesis by controlling the recruitment of the Par3/Tiam1 polarity complex to synaptic sites. J Neurosci. 2013;33(16):6964–6978. DOI: 10.1523/JNEUROSCI.3978-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Park D, Tosello-Trampont A-C, Elliott MR, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450(7168):430–434. DOI: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- [61].Tu Y-K, Duman JG, Tolias KF. The adhesion-GPCR BAI1 promotes excitatory synaptogenesis by coordinating bidirectional trans-synaptic signaling. J Neurosci. 2018;38(39):8388–8406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tolias KF, Bikoff JB, Burette A, et al. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45(4):525–538. DOI: 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- [63].Xie Z, Srivastava DP, Photowala H, et al. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. 2007;56(4):640–656. DOI: 10.1016/j.neuron.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Saneyoshi T, Matsuno H, Suzuki A, et al. Reciprocal activation within a kinase-effector complex underlying persistence of structural LTP. Neuron. 2019;102(6):1199–1210. doi: 10.1016/j.neuron.2019.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Dalva MB, Takasu MA, Lin MZ, et al. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell. 2000;103(6):945–956. DOI: 10.1016/S0092-8674(00)00197-5. [DOI] [PubMed] [Google Scholar]
- [66].Takasu MA. Modulation of NMDA Receptor- dependent calcium influx and gene expression through EphB receptors. Science. 2002;295(5554):491–495. [DOI] [PubMed] [Google Scholar]
- [67].Yoshii A, Constantine-Paton M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol. 2010;70(5):304–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Miyamoto Y, Yamauchi J, Tanoue A, et al. TrkB binds and tyrosine-phosphorylates Tiam1, leading to activation of Rac1 and induction of changes in cellular morphology. Proc Natl Acad Sci U S A. 2006;103(27):10444–10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhou P, Porcionatto M, Pilapil M, et al. Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron. 2007;55(1):53–68. DOI: 10.1016/j.neuron.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lai K-O, Wong ASL, Cheung M-C, et al. TrkB phosphorylation by Cdk5 is required for activity-dependent structural plasticity and spatial memory. Nat Neurosci. 2012;15(11):1506–1515. DOI: 10.1038/nn.3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Yan Y, Eipper BA, Mains RE. Kalirin is required for BDNF-TrkB stimulated neurite outgrowth and branching. Neuropharmacology. 2016;107:227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Hale CF, Dietz KC, Varela JA, et al. Essential role for vav Guanine nucleotide exchange factors in brain-derived neurotrophic factor-induced dendritic spine growth and synapse plasticity. J Neurosci. 2011;31(35):12426–12436. DOI: 10.1523/JNEUROSCI.0685-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Tanaka J-I, Horiike Y, Matsuzaki M, et al. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 2008;319(5870):1683–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Harward SC, Hedrick NG, Hall CE, et al. Autocrine BDNF-TrkB signalling within a single dendritic spine. Nature. 2016;538(7623):99–103. DOI: 10.1038/nature19766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Iseppon F, Napolitano LM, Torre V, et al. Combining FRET and optical tweezers to study RhoGTPases spatio-temporal dynamics upon local stimulation. J Biol Meth. 2017;4(1):e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Pertz O. Spatio-temporal Rho GTPase signaling - where are we now?. J Cell Sci. 2010;123(11):1841–1850. doi: 10.1242/jcs.064345. [DOI] [PubMed] [Google Scholar]
- [77].Vanni C, Ottaviano C, Guo F, et al. Constitutively active Cdc42 mutant confers growth disadvantage in cell transformation. Cell Cycle. 2005;4(11):1675–1682. DOI: 10.4161/cc.4.11.2170. [DOI] [PubMed] [Google Scholar]
- [78].Davis MJ, Ha BH, Holman EC, et al. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc Natl Acad Sci U S A. 2013;110(3):912–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Cheng JX, Scala F, Blanco FA, et al. The Rac-GEF Tiam1 promotes dendrite and synapse stabilization of dentate granule cells and restricts hippocampal-dependent memory functions. J Neurosci. 2020. DOI: 10.1523/JNEUROSCI.3271-17.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Duman JG, Mulherkar S, Tu Y-K, et al. The adhesion-GPCR BAI1 shapes dendritic arbors via Bcr-mediated RhoA activation causing late growth arrest. eLife. 2019;8:e47566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Oh D, Han S, Seo J, et al. Regulation of synaptic Rac1 activity, long-term potentiation maintenance, and learning and memory by BCR and ABR Rac GTPase-activating proteins. J Neurosci. 2010;30(42):14134–14144. DOI: 10.1523/JNEUROSCI.1711-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Lim J, Ritt DA, Zhou M, et al. The CNK2 scaffold interacts with vilse and modulates Rac cycling during spine morphogenesis in hippocampal neurons. Curr Biol. 2014;24(7):786–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Narayanan AS, Reyes SB, Um K, et al. The Rac-GAP Bcr is a novel regulator of the Par complex that controls cell polarity. Mol Biol Cell. 2013;24(24):3857–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Mertens AEE, Pegtel DM, Collard JG. Tiam1 takes PARt in cell polarity. Trends Cell Biol. 2006;16(6):308–316. [DOI] [PubMed] [Google Scholar]
- [85].Miller AL, Bement WM. Regulation of cytokinesis by Rho GTPase flux. Nat Cell Biol. 2009;11(1):71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Somers WG, Saint R. A RhoGEF and Rho family GTPase-activating protein complex links the contractile ring to cortical microtubules at the onset of cytokinesis. Dev Cell. 2003;4(1):29–39. [DOI] [PubMed] [Google Scholar]
- [87].Reiter LT, Seagroves TN, Bowers M, et al. Expression of the Rho-GEF Pbl/ECT2 is regulated by the UBE3A E3 ubiquitin ligase. Hum Mol Genet. 2006;15(18):2825–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Van De Putte T, Zwijsen A, Lonnoy O, et al. Mice with a homozygous gene trap vector insertion in mgcRacGAP die during pre-implantation development. Mech Dev. 2001;102(1–2):33–44. DOI: 10.1016/S0925-4773(01)00279-9. [DOI] [PubMed] [Google Scholar]
- [89].Bement WM, Miller AL, Von Dassow G. Rho GTPase activity zones and transient contractile arrays. BioEssays. 2006;28(10):983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Simões S, Denholm B, Azevedo D, et al. Compartmentalisation of Rho regulators directs cell invagination during tissue morphogenesis. Development. 2006;133(21):4257–4267. DOI: 10.1242/dev.02588. [DOI] [PubMed] [Google Scholar]
- [91].Sato D, Sugimura K, Satoh D, et al. Crossveinless-c, the Drosophila homolog of tumor suppressor DLC1, regulates directional elongation of dendritic branches via down-regulating Rho1 activity. Genes to Cells : Devoted to Molecular & Cellular Mechanisms. 2010;15(5):485–500. [DOI] [PubMed] [Google Scholar]
- [92].Daubon T, Chasseriau J, El Ali A, et al. Differential motility of p190bcr-abl- and p210bcr-abl-expressing cells: respective roles of Vav and Bcr-Abl GEFs. Oncogene. 2008;27(19):2673–2685. DOI: 10.1038/sj.onc.1210933. [DOI] [PubMed] [Google Scholar]
- [93].Ridley AJ, Self AJ, Kasmi F, et al. rho family GTPase activating proteins p190, bcr and rhoGAP show distinct specificities in vitro and in vivo. Embo J. 1993;12(13):5151–5160. DOI: 10.1002/j.1460-2075.1993.tb06210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Kaartinen V, Gonzalez-Gomez I, Voncken JW, et al. Abnormal function of astroglia lacking Abr and Bcr RacGAPs. Development. 2001;128(21):4217–4227. [DOI] [PubMed] [Google Scholar]
- [95].Cresto N, Pillet L-E, Billuart P, et al. Do astrocytes play a role in intellectual disabilities?. Trends Neurosci. 2019;42(8):518–527. [DOI] [PubMed] [Google Scholar]
- [96].Heisterkamp N, Kaartinen V, Van Soest S, et al. Human ABR encodes a protein with GAPrac activity and homology to the DBL nucleotide exchange factor domain. J Biol Chem. 1993;268(23):16903–16906. [PubMed] [Google Scholar]
- [97].Kutys ML, Yamada KM. An extracellular-matrix-specific GEF–GAP interaction regulates Rho GTPase crosstalk for 3D collagen migration. Nat Cell Biol. 2014;16(9):909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Saneyoshi T, Wayman G, Fortin D, et al. Activity-Dependent Synaptogenesis: regulation by a CaM-Kinase Kinase/CaM-Kinase I/βPIX Signaling Complex. Neuron. 2008;57(1):94–107. DOI: 10.1016/j.neuron.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Wong K, Ren X-R, Huang Y-Z, et al. Signal transduction in neuronal migration: roles of GTPase activating proteins and the small GTPase Cdc42 in the Slit-Robo pathway. Cell. 2001;107(2):209–221. DOI: 10.1016/S0092-8674(01)00530-X. [DOI] [PubMed] [Google Scholar]
- [100].Park A-R, Oh D, Lim S-H, et al. Regulation of dendritic arborization by BCR Rac1 GTPase-activating protein, a substrate of PTPRT. J Cell Sci. 2012;125(19):4518–4531. doi: 10.1242/jcs.105502. [DOI] [PubMed] [Google Scholar]
- [101].May V, Schiller MR, Eipper BA, et al. Kalirin Dbl-homology guanine nucleotide exchange factor 1 domain initiates new axon outgrowths via RhoG-mediated mechanisms. J Neurosci. 2002;22(16):6980–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Bellanger JM, Estrach S, Schmidt S, et al. Different regulation of the Trio Dbl-Homology domains by their associated PH domains. Biol Cell. 2003;95(9):625–634. DOI: 10.1016/j.biolcel.2003.10.002. [DOI] [PubMed] [Google Scholar]
- [103].Peng Y-J, He W-Q, Tang J, et al. Trio is a key guanine nucleotide exchange factor coordinating regulation of the migration and morphogenesis of granule cells in the developing cerebellum. J Biol Chem. 2010;285(32):24834–24844. DOI: 10.1074/jbc.M109.096537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Estrach S, Schmidt S, Diriong S, et al. The human Rho-GEF trio and its target GTPase RhoG are involved in the NGF pathway, leading to neurite outgrowth. Curr Biol. 2002;12(4):307–312. DOI: 10.1016/S0960-9822(02)00658-9. [DOI] [PubMed] [Google Scholar]
- [105].Van Haren J, Boudeau J, Schmidt S, et al. Dynamic microtubules catalyze formation of navigator-TRIO complexes to regulate neurite extension. Curr Biol. 2014;24(15):1778–1785. DOI: 10.1016/j.cub.2014.06.037. [DOI] [PubMed] [Google Scholar]
- [106].Neubrand VE, Thomas C, Schmidt S, et al. Kidins220/ARMS regulates Rac1-dependent neurite outgrowth by direct interaction with the RhoGEF Trio. J Cell Sci. 2010;123(12):2111–2123. doi: 10.1242/jcs.064055. [DOI] [PubMed] [Google Scholar]
- [107].Backer S, Lokmane L, Landragin C, et al. Trio GEF mediates RhoA activation downstream of Slit2 and coordinates telencephalic wiring. Development. 2018;145(19):19. [DOI] [PubMed] [Google Scholar]
- [108].Tao T, Sun J, Peng Y, et al. Distinct functions of Trio GEF domains in axon outgrowth of cerebellar granule neurons. J Genet Genomics. 2019;46(2):87–96. DOI: 10.1016/j.jgg.2019.02.003. [DOI] [PubMed] [Google Scholar]
- [109].Penzes P, Johnson RC, Kambampati V, et al. Distinct roles for the two Rho GDP/GTP exchange factor domains of kalirin in regulation of neurite growth and neuronal morphology. J Neurosci. 2001;21(21):8426–8434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Deo AJ, Cahill ME, Li S, et al. Increased expression of Kalirin-9 in the auditory cortex of schizophrenia subjects: its role in dendritic pathology. Neurobiol Dis. 2012;45(2):796–803. DOI: 10.1016/j.nbd.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Yan Y, Eipper BA, Mains RE. Kalirin-9 and Kalirin-12 play essential roles in dendritic outgrowth and branching. Cereb Cortex. 2015;25(10):3487–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Abraham S, Scarcia M, Bagshaw RD, et al. A Rac/Cdc42 exchange factor complex promotes formation of lateral filopodia and blood vessel lumen morphogenesis. Nat Commun. 2015;6(1):7286. DOI: 10.1038/ncomms8286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Kwofie MA, Skowronski J. Specific recognition of Rac2 and Cdc42 by DOCK2 and DOCK9 guanine nucleotide exchange factors. J Biol Chem. 2008;283(6):3088–3096. [DOI] [PubMed] [Google Scholar]
- [114].Kuramoto K, Negishi M, Katoh H. Regulation of dendrite growth by the Cdc42 activator Zizimin1/Dock9 in hippocampal neurons. J Neurosci Res. 2009;87(8):1794–1805. [DOI] [PubMed] [Google Scholar]
- [115].Ueda S, Fujimoto S, Hiramoto K, et al. Dock4 regulates dendritic development in hippocampal neurons. J Neurosci Res. 2008;86(14):3052–3061. [DOI] [PubMed] [Google Scholar]
- [116].Huang M, Liang C, Li S, et al. Two autism/dyslexia linked variations of DOCK4 disrupt the gene function on rac1/rap1 activation, neurite outgrowth, and synapse development. Front Cell Neurosci. 2019;13:577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Brambilla R, Gnesutta N, Minichiello L, et al. A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nature. 1997;390(6657):281–286. DOI: 10.1038/36849. [DOI] [PubMed] [Google Scholar]
- [118].Li S. Distinct roles for Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and Ras-GRF2 in the induction of long-term potentiation and long-term depression. J Neurosci. 2006;26(6):1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Schwechter B, Rosenmund C, Tolias KF. RasGRF2 Rac-GEF activity couples NMDA receptor calcium flux to enhanced synaptic transmission. Proc Natl Acad Sci U S A. 2013;110(35):14462–14467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Zhang B, Zhong X, Sauane M, et al. Modulation of the Pol II CTD phosphorylation code by Rac1 and Cdc42 small GTPases in cultured human cancer cells and its implication for developing a synthetic-lethal cancer therapy. Cells. 2020;9(3):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Boda B, Nikonenko I, Alberi S, et al. Central nervous system functions of PAK protein family: from spine morphogenesis to mental retardation. Mol Neurobiol. 2006;34(1–2):67–80. [DOI] [PubMed] [Google Scholar]
- [122].Nikolić M. The Pak1 kinase: an important regulator of neuronal morphology and function in the developing forebrain. Mol Neurobiol. 2008;37(2–3):187–202. [DOI] [PubMed] [Google Scholar]
- [123].Garcia-Mata R, Boulter E, Burridge K. The “invisible hand”: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011;12(8):493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].DerMardirossian C, Schnelzer A, Bokoch GM. Phosphorylation of RhoGDI by Pak1 mediates dissociation of Rac GTPase. Mol Cell. 2004;15(1):117–127. [DOI] [PubMed] [Google Scholar]
- [125].Knezevic N, Roy A, Timblin B, et al. GDI-1 phosphorylation switch at serine 96 induces RhoA activation and increased endothelial permeability. Mol Cell Biol. 2007;27(18):6323–6333. DOI: 10.1128/MCB.00523-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Dovas A, Choi Y, Yoneda A, et al. Serine 34 phosphorylation of rho guanine dissociation inhibitor (RhoGDIalpha) links signaling from conventional protein kinase C to RhoGTPase in cell adhesion. J Biol Chem. 2010;285(30):23296–23308. DOI: 10.1074/jbc.M109.098129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Robbe K, Otto-Bruc A, Chardin P, et al. Dissociation of GDP dissociation inhibitor and membrane translocation are required for efficient activation of Rac by the Dbl homology-pleckstrin homology region of Tiam. J Biol Chem. 2003;278(7):4756–4762. [DOI] [PubMed] [Google Scholar]
- [128].Feng Q, Albeck JG, Cerione RA, et al. Regulation of the Cool/Pix proteins: key binding partners of the Cdc42/Rac targets, the p21-activated kinases. J Biol Chem. 2002;277(7):5644–5650. [DOI] [PubMed] [Google Scholar]
- [129].Obermeier A. PAK promotes morphological changes by acting upstream of Rac. Embo J. 1998;17(15):4328–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Za L. betaPIX controls cell motility and neurite extension by regulating the distribution of GIT1. J Cell Sci. 2006;119(13):2654–2666. doi: 10.1242/jcs.02996. [DOI] [PubMed] [Google Scholar]
- [131].Zhang H. A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J Neurosci. 2005;25(13):3379–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Kim T, Park D. Molecular cloning and characterization of a novel mouse betaPix isoform. Mol Cells. 2001;11(1):89–94. [PubMed] [Google Scholar]
- [133].Kwon Y, Jeon YW, Kwon M, et al. βPix-d promotes tubulin acetylation and neurite outgrowth through a PAK/Stathmin1 signaling pathway. Plos One. 2020;15(4):e0230814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Ten Klooster JP, Evers EE, Janssen L, et al. Interaction between Tiam1 and the Arp2/3 complex links activation of Rac to actin polymerization. Biochem J. 2006;397(1):39–45. DOI: 10.1042/BJ20051957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Connolly BA, Rice J, Feig LA, et al. Tiam1-IRSp53 complex formation directs specificity of rac-mediated actin cytoskeleton regulation. Mol Cell Biol. 2005;25(11):4602–4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Nishimura T, Yamaguchi T, Kato K, et al. PAR-6–PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat Cell Biol. 2005;7(3):270–277. DOI: 10.1038/ncb1227. [DOI] [PubMed] [Google Scholar]
- [137].Zhang H, Macara IG. The PAR-6 polarity protein regulates dendritic spine morphogenesis through p190 RhoGAP and the Rho GTPase. Dev Cell. 2008;14(2):216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Zenke FT, Krendel M, DerMardirossian C, et al. p21-activated kinase 1 phosphorylates and regulates 14-3-3 binding to GEF-H1, a microtubule-localized Rho exchange factor. J Biol Chem. 2004;279(18):18392–18400. [DOI] [PubMed] [Google Scholar]
- [139].Rosenfeldt H, Castellone MD, Randazzo PA, et al. Rac inhibits thrombin-induced Rho activation: evidence of a Pak-dependent GTPase crosstalk. J Mol Signal. 2006;1:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Barac A, Basile J, Vázquez-Prado J, et al. Direct interaction of p21-activated kinase 4 with PDZ-RhoGEF, a G protein-linked Rho guanine exchange factor. J Biol Chem. 2004;279(7):6182–6189. [DOI] [PubMed] [Google Scholar]
- [141].Alberts AS, Qin H, Carr HS, et al. PAK1 negatively regulates the activity of the Rho exchange factor NET1. J Biol Chem. 2005;280(13):12152–12161. [DOI] [PubMed] [Google Scholar]
- [142].Ohta Y, Hartwig JH, Stossel TP. FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nat Cell Biol. 2006;8(8):803–814. [DOI] [PubMed] [Google Scholar]
- [143].Kuo J-C, Han X, Hsiao C-T, et al. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat Cell Biol. 2011;13(4):383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Tsuji T, Ishizaki T, Okamoto M, et al. ROCK and mDia1 antagonize in Rho-dependent Rac activation in Swiss 3T3 fibroblasts. J Cell Biol. 2002;157(5):819–830. DOI: 10.1083/jcb.200112107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Stankiewicz TR, Linseman DA. Rho family GTPases: key players in neuronal development, neuronal survival, and neurodegeneration. Front Cell Neurosci. 2014;8:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Mulherkar S, Uddin MD, Couvillon AD, et al. The small GTPases RhoA and Rac1 regulate cerebellar development by controlling cell morphogenesis, migration and foliation. Dev Biol. 2014;394(1):39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Cappello S, Böhringer CRJ, Bergami M, et al. A radial glia-specific role of RhoA in double cortex formation. Neuron. 2012;73(5):911–924. DOI: 10.1016/j.neuron.2011.12.030. [DOI] [PubMed] [Google Scholar]
- [148].Katayama K, Melendez J, Baumann JM, et al. Loss of RhoA in neural progenitor cells causes the disruption of adherens junctions and hyperproliferation. Proc Natl Acad Sci USA. 2011;108(18):7607–7612. DOI: 10.1073/pnas.1101347108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Tang J, Ip JPK, Ye T, et al. Cdk5-dependent Mst3 phosphorylation and activity regulate neuronal migration through RhoA inhibition. J Neurosci. 2014;34(22):7425–7436. DOI: 10.1523/JNEUROSCI.5449-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Xiang X, Zhuang X, Li S, et al. Arhgef1 is expressed in cortical neural progenitor cells and regulates neurite outgrowth of newly differentiated neurons. Neurosci Lett. 2017;638:27–34. [DOI] [PubMed] [Google Scholar]
- [151].Xiang X, Li S, Zhuang X, et al. Arhgef1 negatively regulates neurite outgrowth through activation of RhoA signaling pathways. FEBS Lett. 2016;590(17):2940–2955. [DOI] [PubMed] [Google Scholar]
- [152].Katayama K, Leslie JR, Lang RA, et al. Left-right locomotor circuitry depends on RhoA-driven organization of the neuroepithelium in the developing spinal cord. J Neurosci. 2012;32(30):10396–10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Mulherkar S, Liu F, Chen Q, et al. The small GTPase RhoA is required for proper locomotor circuit assembly. Plos One. 2013;8(6):e67015. DOI: 10.1371/journal.pone.0067015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Fujita Y, Yamashita T. Axon growth inhibition by RhoA/ROCK in the central nervous system. Front Neurosci. 2014;8:338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Hu J, Selzer ME. RhoA as a target to promote neuronal survival and axon regeneration. Neural Regen Res. 2017;12(4):525–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Tsushima H, Emanuele M, Polenghi A, et al. HDAC6 and RhoA are novel players in Abeta-driven disruption of neuronal polarity. Nat Commun. 2015;6(1):7781. DOI: 10.1038/ncomms8781. [DOI] [PubMed] [Google Scholar]
- [157].Xing L, Yao X, Williams KR, et al. Negative regulation of RhoA translation and signaling by hnRNP-Q1 affects cellular morphogenesis. Mol Biol Cell. 2012;23(8):1500–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Lesiak A, Pelz C, Ando H, et al. A genome-wide screen of CREB occupancy identifies the RhoA inhibitors Par6C and Rnd3 as regulators of BDNF-induced synaptogenesis. Plos One. 2013;8(6):e64658. DOI: 10.1371/journal.pone.0064658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Thomas RA, Gibon J, Chen CXQ, et al. The nogo receptor ligand LGI1 regulates synapse number and synaptic activity in hippocampal and cortical neurons. eNeuro. 2018;5(4):4. DOI: 10.1523/ENEURO.0185-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Schaffer TB, Smith JE, Cook EK, et al. PKCε inhibits neuronal dendritic spine development through dual phosphorylation of Ephexin5. Cell Rep. 2018;25(9):2470–2483. doi: 10.1016/j.celrep.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Govek -E-E, Newey SE, Akerman CJ, et al. The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat Neurosci. 2004;7(4):364–372. [DOI] [PubMed] [Google Scholar]
- [162].Sekiguchi M, Sobue A, Kushima I, et al. ARHGAP10, which encodes Rho GTPase-activating protein 10, is a novel gene for schizophrenia risk. Transl Psychiatry. 2020;10(1):247. DOI: 10.1038/s41398-020-00917-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Chen Y, Kramár EA, Chen LY, et al. Impairment of synaptic plasticity by the stress mediator CRH involves selective destruction of thin dendritic spines via RhoA signaling. Mol Psychiatry. 2013;18(4):485–496. DOI: 10.1038/mp.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Kang M-G, Guo Y, Huganir RL. AMPA receptor and GEF-H1/Lfc complex regulates dendritic spine development through RhoA signaling cascade. Proc Natl Acad Sci U S A. 2009;106(9):3549–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Klebe D, Tibrewal M, Sharma DR, et al. Reduced hippocampal dendrite branching, spine density and neurocognitive function in premature rabbits, and reversal with estrogen or TrkB agonist treatment. Cereb Cortex. 2019;29(12):4932–4947. DOI: 10.1093/cercor/bhz033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Wilson E, Rudisill T, Kirk B, et al. Cytoskeletal regulation of synaptogenesis in a model of human fetal brain development. J Neurosci Res. 2020;98(11):2148–2165. [DOI] [PubMed] [Google Scholar]
- [167].Richter M, Murtaza N, Scharrenberg R, et al. Altered TAOK2 activity causes autism-related neurodevelopmental and cognitive abnormalities through RhoA signaling. Mol Psychiatry. 2019;24(9):1329–1350. DOI: 10.1038/s41380-018-0025-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Konno D, Yoshimura S, Hori K, et al. Involvement of the phosphatidylinositol 3-kinase/rac1 and cdc42 pathways in radial migration of cortical neurons. J Biol Chem. 2005;280(6):5082–5088. [DOI] [PubMed] [Google Scholar]
- [169].Li Q, Wang L, Ma Y, et al. P-Rex1 overexpression results in aberrant neuronal polarity and psychosis-related behaviors. Neurosci Bull. 2019;35(6):1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Yang T, Sun Y, Zhang F, et al. POSH localizes activated Rac1 to control the formation of cytoplasmic dilation of the leading process and neuronal migration. Cell Rep. 2012;2(3):640–651. DOI: 10.1016/j.celrep.2012.08.007. [DOI] [PubMed] [Google Scholar]
- [171].Hua ZL, Emiliani FE, Nathans J. Rac1 plays an essential role in axon growth and guidance and in neuronal survival in the central and peripheral nervous systems. Neural Dev. 2015;10(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Nørgaard S, Deng S, Cao W, et al. Distinct CED-10/Rac1 domains confer context-specific functions in development. PLoS Genet. 2018;14(9):e1007670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Norgaard S, Pocock R. Rac GTPases: domain-specific functions in neuronal development. Neural Regen Res. 2019;14(8):1367–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Guan L, Ma X, Zhang J, et al. The calponin family member CHDP-1 Interacts with Rac/CED-10 to promote cell protrusions. PLoS Genet. 2016;12(7):e1006163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Xiao Y, Peng Y, Wan J, et al. The atypical guanine nucleotide exchange factor Dock4 regulates neurite differentiation through modulation of Rac1 GTPase and actin dynamics. J Biol Chem. 2013;288(27):20034–20045. DOI: 10.1074/jbc.M113.458612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Wiens KM. Rac1 induces the clustering of AMPA receptors during spinogenesis. J Neurosci. 2005;25(46):10627–10636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Risher WC, Kim N, Koh S, et al. Thrombospondin receptor α2δ-1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1. J Cell Biol. 2018;217(10):3747–3765. DOI: 10.1083/jcb.201802057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Dhar M, Wayman GA, Zhu M, et al. Leptin-induced spine formation requires TrpC channels and the CaM kinase cascade in the hippocampus. J Neurosci. 2014;34(30):10022–10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Charrier C, Joshi K, Coutinho-Budd J, et al. Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation. Cell. 2012;149(4):923–935. DOI: 10.1016/j.cell.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Sarowar T, Grabrucker S, Boeckers TM, et al. Object phobia and altered rhoa signaling in amygdala of mice lacking RICH2. Front Mol Neurosci. 2017;10:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Sarowar T, Grabrucker S, Föhr K, et al. Enlarged dendritic spines and pronounced neophobia in mice lacking the PSD protein RICH2. Mol Brain. 2016;9(1):28. DOI: 10.1186/s13041-016-0206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Impey S, Davare M, Lesiak A, et al. An activity-induced microRNA controls dendritic spine formation by regulating Rac1-PAK signaling. Mol Cell Neurosci. 2010;43(1):146–156. DOI: 10.1016/j.mcn.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20(14):5329–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Raynaud F, Moutin E, Schmidt S, et al. Rho-GTPase-activating protein interacting with Cdc-42-interacting protein 4 homolog 2 (Rich2): a new Ras-related C3 botulinum toxin substrate 1 (Rac1) GTPase-activating protein that controls dendritic spine morphogenesis. J Biol Chem. 2014;289(5):2600–2609. DOI: 10.1074/jbc.M113.534636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Ba W, Selten MM, Van Der Raadt J, et al. ARHGAP12 functions as a developmental brake on excitatory synapse function. Cell Rep. 2016;14(6):1355–1368. DOI: 10.1016/j.celrep.2016.01.037. [DOI] [PubMed] [Google Scholar]
- [186].Diring J, Mouilleron S, McDonald NQ, et al. RPEL-family rhoGAPs link Rac/Cdc42 GTP loading to G-actin availability. Nat Cell Biol. 2019;21(7):845–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Valdez CM, Murphy GG, Beg AA. The Rac-GAP alpha2-chimaerin regulates hippocampal dendrite and spine morphogenesis. Mol Cell Neurosci. 2016;75:14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Ramakers GJA, Wolfer D, Rosenberger G, et al. Dysregulation of Rho GTPases in the αPix/Arhgef6 mouse model of X-linked intellectual disability is paralleled by impaired structural and synaptic plasticity and cognitive deficits. Hum Mol Genet. 2012;21(2):268–286. DOI: 10.1093/hmg/ddr457. [DOI] [PubMed] [Google Scholar]
- [189].Lo LH-Y, Dong R, Lyu Q, et al. The protein arginine methyltransferase PRMT8 and substrate G3BP1 control Rac1-PAK1 signaling and actin cytoskeleton for dendritic spine maturation. Cell Rep. 2020;31(10):107744. [DOI] [PubMed] [Google Scholar]
- [190].Fossati M, Pizzarelli R, Schmidt ER, et al. SRGAP2 and its human-specific paralog co-regulate the development of excitatory and inhibitory synapses. Neuron. 2016;91(2):356–369. DOI: 10.1016/j.neuron.2016.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Schmidt ERE, Kupferman JV, Stackmann M, et al. The human-specific paralogs SRGAP2B and SRGAP2C differentially modulate SRGAP2A-dependent synaptic development. Sci Rep. 2019;9(1):18692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Corbetta S, Gualdoni S, Ciceri G, et al. Essential role of Rac1 and Rac3 GTPases in neuronal development. Faseb J. 2009;23(5):1347–1357. DOI: 10.1096/fj.08-121574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Tivodar S, Kalemaki K, Kounoupa Z, et al. Rac-GTPases regulate microtubule stability and axon growth of cortical GABAergic interneurons. Cereb Cortex. 2015;25(9):2370–2382. DOI: 10.1093/cercor/bhu037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Vaghi V, Pennucci R, Talpo F, et al. Rac1 and rac3 GTPases control synergistically the development of cortical and hippocampal GABAergic interneurons. Cereb Cortex. 2014;24(5):1247–1258. DOI: 10.1093/cercor/bhs402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Pennucci R, Gucciardi I, De Curtis I. Rac1 and Rac3 GTPases differently influence the morphological maturation of dendritic spines in hippocampal neurons. Plos One. 2019;14(8):e0220496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Pennucci R, Talpo F, Astro V, et al. Loss of either Rac1 or Rac3 GTPase differentially affects the behavior of mutant mice and the development of functional GABAergic networks. Cerebral Cortex (New York, N.Y. : 1991). 2016;26(2):873–890. DOI: 10.1093/cercor/bhv274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Govek -E-E, Wu Z, Acehan D, et al. Cdc42 regulates neuronal polarity during cerebellar axon formation and glial-guided migration. iScience. 2018;1:35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Wegner AM, Nebhan CA, Hu L, et al. N-wasp and the arp2/3 complex are critical regulators of actin in the development of dendritic spines and synapses. J Biol Chem. 2008;283(23):15912–15920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Vadodaria KC, Brakebusch C, Suter U, et al. Stage-specific functions of the small Rho GTPases Cdc42 and Rac1 for adult hippocampal neurogenesis. J Neurosci. 2013;33(3):1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Chen Y, Liang Z, Fei E, et al. Axin regulates dendritic spine morphogenesis through Cdc42-dependent signaling. Plos One. 2015;10(7):e0133115. DOI: 10.1371/journal.pone.0133115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Kim Y, Ha CM, Chang S. SNX26, a GTPase-activating protein for Cdc42, interacts with PSD-95 protein and is involved in activity-dependent dendritic spine formation in mature neurons. J Biol Chem. 2013;288(41):29453–29466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Moutin E, Nikonenko I, Stefanelli T, et al. Palmitoylation of cdc42 promotes spine stabilization and rescues spine density deficit in a mouse model of 22q11.2 deletion syndrome. Cerebral Cortex (New York, N.Y. : 1991). 2017;27(7):3618–3629. DOI: 10.1093/cercor/bhw183. [DOI] [PubMed] [Google Scholar]
- [203].Kins S, Betz H, Kirsch J. Collybistin, a newly identified brain-specific GEF, induces submembrane clustering of gephyrin. Nat Neurosci. 2000;3(1):22–29. [DOI] [PubMed] [Google Scholar]
- [204].Reid T, Bathoorn A, Ahmadian MR, et al. Identification and characterization of hPEM-2, a guanine nucleotide exchange factor specific for Cdc42. J Biol Chem. 1999;274(47):33587–33593. [DOI] [PubMed] [Google Scholar]
- [205].Patrizi A, Viltono L, Frola E, et al. Selective localization of collybistin at a subset of inhibitory synapses in brain circuits. J Comp Neurol. 2012;520(1):130–141. [DOI] [PubMed] [Google Scholar]
- [206].Reddy-Alla S, Schmitt B, Birkenfeld J, et al. PH-domain-driven targeting of collybistin but not Cdc42 activation is required for synaptic gephyrin clustering. Eur J Neurosci. 2010;31(7):1173–1184. DOI: 10.1111/j.1460-9568.2010.07149.x. [DOI] [PubMed] [Google Scholar]
- [207].Tyagarajan SK, Ghosh H, Harvey K, et al. Collybistin splice variants differentially interact with gephyrin and Cdc42 to regulate gephyrin clustering at GABAergic synapses. J Cell Sci. 2011;124(16):2786–2796. doi: 10.1242/jcs.086199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Alan JK, Robinson SK, Magsig KL, et al. The Atypical Rho GTPase CHW-1 works with SAX-3/Robo to mediate axon guidance in caenorhabditis elegans. G3 (Bethesda). 2018;8(6):1885–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Straub J, Konrad EDH, Grüner J, et al. Missense variants in RHOBTB2 cause a developmental and epileptic encephalopathy in humans, and altered levels cause neurological defects in drosophila. The American Journal of Human Genetics. 2018;102(1):44–57. DOI: 10.1016/j.ajhg.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Pacary E, Azzarelli R, Guillemot F. Rnd3 coordinates early steps of cortical neurogenesis through actin-dependent and -independent mechanisms. Nat Commun. 2013;4(1):1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Tian D, Diao M, Jiang Y, et al. Anillin regulates neuronal migration and neurite growth by linking rhog to the actin cytoskeleton. Curr Biol. 2015;25(9):1135–1145. DOI: 10.1016/j.cub.2015.02.072. [DOI] [PubMed] [Google Scholar]
- [212].Namekata K, Watanabe H, Guo X, et al. Dock3 regulates BDNF-TrkB signaling for neurite outgrowth by forming a ternary complex with Elmo and RhoG. Genes Cells. 2012;17(8):688–697. DOI: 10.1111/j.1365-2443.2012.01616.x. [DOI] [PubMed] [Google Scholar]
- [213].Schulz J, Franke K, Frick M, et al. Different roles of the small GTPases Rac1, Cdc42, and RhoG in CALEB/NGC-induced dendritic tree complexity. J Neurochem. 2016;139(1):26–39. [DOI] [PubMed] [Google Scholar]
- [214].Kim J-Y, Oh MH, Bernard LP, et al. The RhoG/ELMO1/Dock180 signaling module is required for spine morphogenesis in hippocampal neurons. J Biol Chem. 2011;286(43):37615–37624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Spence EF, Soderling SH. Actin out: regulation of the synaptic cytoskeleton. J Biol Chem. 2015;290(48):28613–28622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Carlier MF, Laurent V, Santolini J, et al. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136(6):1307–1322. DOI: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].dos Remedios, C.G., Chhabra, D., Kekic, M., et al . Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol Rev. 2003;83(2):433–473. DOI: 10.1152/physrev.00026.2002. [DOI] [PubMed] [Google Scholar]
- [218].Hotulainen P, Llano O, Smirnov S, et al. Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol. 2009;185(2):323–339. DOI: 10.1083/jcb.200809046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Cao F, Zhou Z, Pan X, et al. Developmental regulation of hippocampal long-term depression by cofilin-mediated actin reorganization. Neuropharmacology. Pt A 2017;112: 66–75.doi: 10.1016/j.neuropharm.2016.08.017 [DOI] [PubMed] [Google Scholar]
- [220].Havekes R, Park AJ, Tudor JC, et al. Sleep deprivation causes memory deficits by negatively impacting neuronal connectivity in hippocampal area CA1. eLife. 2016;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Zhang Z, Ye M, Li Q, et al. The schizophrenia susceptibility gene OPCML regulates spine maturation and cognitive behaviors through Eph-Cofilin signaling. Cell Rep. 2019;29(1):49–61. doi: 10.1016/j.celrep.2019.08.091. [DOI] [PubMed] [Google Scholar]
- [222].Maekawa M, et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285(5429):895–898. DOI: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- [223].Schill Y, Bijata M, Kopach O, et al. Serotonin 5-HT4 receptor boosts functional maturation of dendritic spines via RhoA-dependent control of F-actin. Commun Biol. 2020;3(1):76. DOI: 10.1038/s42003-020-0791-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Rush T, Martinez-Hernandez J, Dollmeyer M, et al. Synaptotoxicity in Alzheimer’s disease involved a dysregulation of actin cytoskeleton dynamics through cofilin 1 phosphorylation. J Neurosci. 2018;38(48):10349–10361. DOI: 10.1523/JNEUROSCI.1409-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Edwards DC, Sanders LC, Bokoch GM, et al. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1(5):253–259. [DOI] [PubMed] [Google Scholar]
- [226].Kligys K, Claiborne JN, DeBiase PJ, et al. The slingshot family of phosphatases mediates Rac1 regulation of cofilin phosphorylation, laminin-332 organization, and motility behavior of keratinocytes. J Biol Chem. 2007;282(44):32520–32528. DOI: 10.1074/jbc.M707041200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Pyronneau A, He Q, Hwang J-Y, et al. Aberrant Rac1-cofilin signaling mediates defects in dendritic spines, synaptic function, and sensory perception in fragile X syndrome. Sci Signal. 2017;10(504):504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Yang X, Cao Z, Zhang J, et al. Dendritic spine loss caused by AlCl3 is associated with inhibition of the Rac 1/cofilin signaling pathway. Environ Pollut. 2018;243(B):1689–1695. doi: 10.1016/j.envpol.2018.09.145. [DOI] [PubMed] [Google Scholar]
- [229].Chou F-S, Wang P-S. The Arp2/3 complex is essential at multiple stages of neural development. Neurogenesis. 2016;3(1):e1261653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Mullins RD, Heuser JA, Pollard TD. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci U S A. 1998;95(11):6181–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Welch MD, Iwamatsu A, Mitchison TJ. Actin polymerization is induced by Arp 2/3 protein complex at the surface of Listeria monocytogenes. Nature. 1997;385(6613):265–269. [DOI] [PubMed] [Google Scholar]
- [232].Stradal TEB, Scita G. Protein complexes regulating Arp2/3-mediated actin assembly. Curr Opin Cell Biol. 2006;18(1):4–10. [DOI] [PubMed] [Google Scholar]
- [233].Yarar D, To W, Abo A, et al. The Wiskott–Aldrich syndrome protein directs actin-based motility by stimulating actin nucleation with the Arp2/3 complex. Curr Biol. 1999;9(10):555–558. [DOI] [PubMed] [Google Scholar]
- [234].Rotty JD, Wu C, Bear JE. New insights into the regulation and cellular functions of the ARP2/3 complex. Nature Reviews Molecular Cell Biology. 2013;14(1):7–12. [DOI] [PubMed] [Google Scholar]
- [235].Abou-Kheir W, Isaac B, Yamaguchi H, et al. Membrane targeting of WAVE2 is not sufficient for WAVE2-dependent actin polymerization: a role for IRSp53 in mediating the interaction between Rac and WAVE2. J Cell Sci. 2008;121(3pt):379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Miki H, Yamaguchi H, Suetsugu S, et al. IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature. 2000;408(6813):732–735. [DOI] [PubMed] [Google Scholar]
- [237].Sanchez AM, Flamini MI, Fu X-D, et al. Rapid signaling of estrogen to WAVE1 and moesin controls neuronal spine formation via the actin cytoskeleton. Mol Endocrinol. 2009;23(8):1193–1202. DOI: 10.1210/me.2008-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Zigmond SH. Formin-induced nucleation of actin filaments. Curr Opin Cell Biol. 2004;16(1):99–105. [DOI] [PubMed] [Google Scholar]
- [239].Nusser N, Gosmanova E, Makarova N, et al. Serine phosphorylation differentially affects RhoA binding to effectors: implications to NGF-induced neurite outgrowth. Cell Signal. 2006;18(5):704–714. DOI: 10.1016/j.cellsig.2005.06.010. [DOI] [PubMed] [Google Scholar]
- [240].Qu X, Yuan FN, Corona C, et al. Stabilization of dynamic microtubules by mDia1 drives Tau-dependent Aβ1–42 synaptotoxicity. J Cell Biol. 2017;216(10):3161–3178. DOI: 10.1083/jcb.201701045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Ryu J, Liu L, Wong TP, et al. A critical role for myosin IIb in dendritic spine morphology and synaptic function. Neuron. 2006;49(2):175–182. DOI: 10.1016/j.neuron.2005.12.017. [DOI] [PubMed] [Google Scholar]
- [242].Hirose M, Ishizaki T, Watanabe N, et al. Molecular dissection of the Rho-associated protein kinase (p160ROCK)-regulated neurite remodeling in neuroblastoma N1E-115 cells. J Cell Biol. 1998;141(7):1625–1636. DOI: 10.1083/jcb.141.7.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Amano M, Ito M, Kimura K, et al. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J Biol Chem. 1996;271(34):20246–20249. DOI: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- [244].Castañeda P, Muñoz M, García-Rojo G, et al. Association of N-cadherin levels and downstream effectors of Rho GTPases with dendritic spine loss induced by chronic stress in rat hippocampal neurons. J Neurosci Res. 2015;93(10):1476–1491. DOI: 10.1002/jnr.23602. [DOI] [PubMed] [Google Scholar]
- [245].Newell-Litwa KA, Badoual M, Asmussen H, et al. ROCK1 and 2 differentially regulate actomyosin organization to drive cell and synaptic polarity. J Cell Biol. 2015;210(2):225–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Tatavarty V, Das S, Yu J. Polarization of actin cytoskeleton is reduced in dendritic protrusions during early spine development in hippocampal neuron. Mol Biol Cell. 2012;23(16):3167–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Pasapera AM, Plotnikov SV, Fischer RS, et al. Rac1-dependent phosphorylation and focal adhesion recruitment of myosin IIA regulates migration and mechanosensing. Curr Biol. 2015;25(2):175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Allison DW, Gelfand VI, Spector I, et al. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci. 1998;18(7):2423–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Kerr JM, Blanpied TA. Subsynaptic AMPA receptor distribution is acutely regulated by actin-driven reorganization of the postsynaptic density. J Neurosci. 2012;32(2):658–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Spence EF, Kanak DJ, Carlson BR, et al. The arp2/3 complex is essential for distinct stages of spine synapse maturation, including synapse unsilencing. J Neurosci. 2016;36(37):9696–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Guo D, Peng Y, Wang L, et al. Autism-like social deficit generated by Dock4 deficiency is rescued by restoration of Rac1 activity and NMDA receptor function. Mol Psychiatry. 2019. DOI: 10.1038/s41380-019-0472-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Haditsch U, Leone DP, Farinelli M, et al. A central role for the small GTPase Rac1 in hippocampal plasticity and spatial learning and memory. Mol Cell Neurosci. 2009;41(4):409–419. DOI: 10.1016/j.mcn.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Martinez LA, Tejada-Simon MV. Pharmacological inactivation of the small GTPase Rac1 impairs long-term plasticity in the mouse hippocampus. Neuropharmacology. 2011;61(1–2):305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Sadybekov A, Tian C, Arnesano C, et al. An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat Commun. 2017;8(1):601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Tian C, Kay Y, Sadybekov A, et al. An intellectual disability-related missense mutation in Rac1 prevents LTP induction. Front Mol Neurosci. 2018;11:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Wang X, Cahill ME, Werner CT, et al. Kalirin-7 mediates cocaine-induced AMPA receptor and spine plasticity, enabling incentive sensitization. J Neurosci. 2013;33(27):11012–11022. DOI: 10.1523/JNEUROSCI.1097-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Benoist M, Palenzuela R, Rozas C, et al. MAP1B-dependent Rac activation is required for AMPA receptor endocytosis during long-term depression. Embo J. 2013;32(16):2287–2299. DOI: 10.1038/emboj.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Kim MJ, Futai K, Jo J, et al. Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron. 2007;56(3):488–502. [DOI] [PubMed] [Google Scholar]
- [259].Li J, Chai A, Wang L, et al. Synaptic P-Rex1 signaling regulates hippocampal long-term depression and autism-like social behavior. Proc Natl Acad Sci USA. 2015;112(50):E6964–72. DOI: 10.1073/pnas.1512913112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Yang XY, Stanley RE, Ross AP, et al. Sestd1 encodes a developmentally dynamic synapse protein that complexes with BCR Rac1-GAP to regulate forebrain dendrite, spine and synapse formation. Cereb Cortex. 2019;29(2):505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Zhou Z, Hu J, Passafaro M, et al. GluA2 (GluR2) regulates metabotropic glutamate receptor-dependent long-term depression through N-cadherin-dependent and cofilin-mediated actin reorganization. J Neurosci. 2011;31(3):819–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Hussain NK, Thomas GM, Luo J, et al. Regulation of AMPA receptor subunit GluA1 surface expression by PAK3 phosphorylation. Proc Natl Acad Sci USA. 2015;112(43):E5883–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Brachet A, Norwood S, Brouwers JF, et al. LTP-triggered cholesterol redistribution activates Cdc42 and drives AMPA receptor synaptic delivery. J Cell Biol. 2015;208(6):791–806. DOI: 10.1083/jcb.201407122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Shen W, Kilander MBC, Bridi MS, et al. Tomosyn regulates the small RhoA GTPase to control the dendritic stability of neurons and the surface expression of AMPA receptors. J Neurosci Res. 2020;98(6):1213–1231. DOI: 10.1002/jnr.24608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Nadif Kasri N, Nakano-Kobayashi A, Malinow R, et al. The Rho-linked mental retardation protein oligophrenin-1 controls synapse maturation and plasticity by stabilizing AMPA receptors. Genes Dev. 2009;23(11):1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Khelfaoui M, Pavlowsky A, Powell AD, et al. Inhibition of RhoA pathway rescues the endocytosis defects in Oligophrenin1 mouse model of mental retardation. Hum Mol Genet. 2009;18(14):2575–2583. DOI: 10.1093/hmg/ddp189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Hayashi T, Yoshida T, Ra M, et al. IL1RAPL1 associated with mental retardation and autism regulates the formation and stabilization of glutamatergic synapses of cortical neurons through RhoA signaling pathway. Plos One. 2013;8(6):e66254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450(7173):1195–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Duman JG, Tolias KF. Rhogtpases spread the word for synaptic crosstalk. Dev Cell. 2016;39(2):136–138. [DOI] [PubMed] [Google Scholar]
- [270].Oh WC, Parajuli LK, Zito K. Heterosynaptic structural plasticity on local dendritic segments of hippocampal CA1 neurons. Cell Rep. 2015;10(2):162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Whitlock JR. Learning induces long-term potentiation in the hippocampus. Science. 2006;313(5790):1093–1097. [DOI] [PubMed] [Google Scholar]
- [272].Reinhard JR, Kriz A, Galic M, et al. The calcium sensor Copine-6 regulates spine structural plasticity and learning and memory. Nat Commun. 2016;7(1):11613. DOI: 10.1038/ncomms11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Min H, Dong J, Wang Y, et al. Marginal iodine deficiency affects dendritic spine development by disturbing the function of rac1 signaling pathway on cytoskeleton. Mol Neurobiol. 2017;54(1):437–449. DOI: 10.1007/s12035-015-9657-5. [DOI] [PubMed] [Google Scholar]
- [274].Sun W, Yang J, Hong Y, et al. Lanthanum chloride impairs learning and memory and induces dendritic spine abnormality by down-regulating Rac1/PAK signaling pathway in hippocampus of offspring rats. Cell Mol Neurobiol. 2020;40(3):459–475. DOI: 10.1007/s10571-019-00748-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Haditsch U, Anderson MP, Freewoman J, et al. Neuronal Rac1 is required for learning-evoked neurogenesis. J Neurosci. 2013;33(30):12229–12241. DOI: 10.1523/JNEUROSCI.2939-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Sananbenesi F, Fischer A, Wang X, et al. A hippocampal Cdk5 pathway regulates extinction of contextual fear. Nat Neurosci. 2007;10(8):1012–1019. DOI: 10.1038/nn1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Gao Q, Yao W, Wang J, et al. Post-training activation of Rac1 in the basolateral amygdala is required for the formation of both short-term and long-term auditory fear memory. Front Mol Neurosci. 2015;8:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Hayashi-Takagi A, Yagishita S, Nakamura M, et al. Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature. 2015;525(7569):333–338. DOI: 10.1038/nature15257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Liu Y, Du S, Lv L, et al. Hippocampal activation of rac1 regulates the forgetting of object recognition memory. Curr Biol. 2016;26(17):2351–2357. DOI: 10.1016/j.cub.2016.06.056. [DOI] [PubMed] [Google Scholar]
- [280].Lv L, Liu Y, Xie J, et al. Interplay between α2-chimaerin and Rac1 activity determines dynamic maintenance of long-term memory. Nat Commun. 2019;10(1):5313. DOI: 10.1038/s41467-019-13236-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Jiang L, Mao R, Zhou Q, et al. Inhibition of rac1 activity in the hippocampus impairs the forgetting of contextual fear memory. Mol Neurobiol. 2016;53(2):1247–1253. DOI: 10.1007/s12035-015-9093-6. [DOI] [PubMed] [Google Scholar]
- [282].Liu Y, Lv L, Wang L, et al. Social isolation induces Rac1-dependent forgetting of social memory. Cell Rep. 2018;25(2):288–295. doi: 10.1016/j.celrep.2018.09.033. [DOI] [PubMed] [Google Scholar]
- [283].Zhao J, Ying L, Liu Y, et al. Different roles of Rac1 in the acquisition and extinction of methamphetamine-associated contextual memory in the nucleus accumbens. Theranostics. 2019;9(23):7051–7071. DOI: 10.7150/thno.34655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Zhang X, Li Q, Wang L, et al. Active protection: learning-activated Raf/MAPK activity protects labile memory from Rac1-independent forgetting. Neuron. 2018;98(1):142–155. doi: 10.1016/j.neuron.2018.02.025. [DOI] [PubMed] [Google Scholar]
- [285].Wu W, Du S, Shi W, et al. Inhibition of Rac1-dependent forgetting alleviates memory deficits in animal models of Alzheimer’s disease. Protein Cell. 2019;10(10):745–759. DOI: 10.1007/s13238-019-0641-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Martinez LA, Tejada-Simon MV. Pharmacological rescue of hippocampal fear learning deficits in fragile X syndrome. Mol Neurobiol. 2018;55(7):5951–5961. [DOI] [PubMed] [Google Scholar]
- [287].Kim IH, Wang H, Soderling SH, et al. Loss of Cdc42 leads to defects in synaptic plasticity and remote memory recall. eLife. 2014;3(3). DOI: 10.7554/eLife.02839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Gao Y, Shuai Y, Zhang X, et al. Genetic dissection of active forgetting in labile and consolidated memories in Drosophila. Proc Natl Acad Sci U S A. 2019;116(42):21191–21197. DOI: 10.1073/pnas.1903763116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Shuai Y, Lu B, Hu Y, et al. Forgetting is regulated through Rac activity in Drosophila. Cell. 2010;140(4):579–589. [DOI] [PubMed] [Google Scholar]
- [290].Basu S, Kustanovich I, Lamprecht R. Arp2/3 and VASP are essential for fear memory formation in lateral amygdala. eNeuro. 2016;3(6):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Wang J, Wang Y, Hou Y, et al. The small GTPase RhoA, but not Rac1, is essential for conditioned aversive memory formation through regulation of actin rearrangements in rat dorsal hippocampus. Acta Pharmacol Sin. 2013;34(6):811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [292].Kang S, Ling Q, Liu W, et al. Down-regulation of dorsal striatal RhoA activity and impairment of working memory in middle-aged rats. Neurobiol Learn Mem. 2013;103:3–10. [DOI] [PubMed] [Google Scholar]
- [293].Briz V, Zhu G, Wang Y, et al. Activity-dependent rapid local RhoA synthesis is required for hippocampal synaptic plasticity. J Neurosci. 2015;35(5):2269–2282. DOI: 10.1523/JNEUROSCI.2302-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Fakira AK, Massaly N, Cohensedgh O, et al. Morphine-associated contextual cues induce structural plasticity in hippocampal CA1 pyramidal neurons. Neuropsychopharmacology. 2016;41(11):2668–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].Barker-Collo S, Theadom A, Jones K, et al. Depression and anxiety across the first 4 years after mild traumatic brain injury: findings from a community-based study. Brain Inj. 2018;32(13–14):1651–1658. [DOI] [PubMed] [Google Scholar]
- [296].Whelan-Goodinson R, Ponsford J, Johnston L, et al. Psychiatric disorders following traumatic brain injury: their nature and frequency. J Head Trauma Rehabil. 2009;24(5):324–332. [DOI] [PubMed] [Google Scholar]
- [297].Dubreuil CI, Marklund N, Deschamps K, et al. Activation of Rho after traumatic brain injury and seizure in rats. Exp Neurol. 2006;198(2):361–369. [DOI] [PubMed] [Google Scholar]
- [298].Dubreuil CI, Winton MJ, McKerracher L. Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. J Cell Biol. 2003;162(2):233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [299].Erschbamer MK, Hofstetter CP, Olson L. RhoA, RhoB, RhoC, Rac1, Cdc42, and Tc10 mRNA levels in spinal cord, sensory ganglia, and corticospinal tract neurons and long-lasting specific changes following spinal cord injury. J Comp Neurol. 2005;484(2):224–233. [DOI] [PubMed] [Google Scholar]
- [300].Li Z, Dong X, Wang Z, et al. Regulation of PTEN by Rho small GTPases. Nat Cell Biol. 2005;7(4):399–404. DOI: 10.1038/ncb1236. [DOI] [PubMed] [Google Scholar]
- [301].Zhou H, Sun Y, Zhang L, et al. The RhoA/ROCK pathway mediates high glucose-induced cardiomyocyte apoptosis via oxidative stress, JNK, and p38MAPK pathways. Diabetes Metab Res Rev. 2018;34(6):e3022. [DOI] [PubMed] [Google Scholar]
- [302].Mulherkar S, Firozi K, Huang W, et al. RhoA-ROCK inhibition reverses synaptic remodeling and motor and cognitive deficits caused by traumatic brain injury. Sci Rep. 2017;7(1):10689. DOI: 10.1038/s41598-017-11113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [303].Mulherkar S, Tolias KF. RhoA-ROCK signaling as a therapeutic target in traumatic brain injury. Cells. 2020;9(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [304].Campbell JN, Low B, Kurz JE, et al. Mechanisms of dendritic spine remodeling in a rat model of traumatic brain injury. J Neurotrauma. 2012;29(2):218–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Loucks FA, Le SS, Zimmermann AK, et al. Rho family GTPase inhibition reveals opposing effects of mitogen-activated protein kinase kinase/extracellular signal-regulated kinase and Janus kinase/signal transducer and activator of transcription signaling cascades on neuronal survival. J Neurochem. 2006;97(4):957–967. DOI: 10.1111/j.1471-4159.2006.03802.x. [DOI] [PubMed] [Google Scholar]
- [306].Schürmann A, Mooney AF, Sanders LC, et al. p21-activated kinase 1 phosphorylates the death agonist bad and protects cells from apoptosis. Mol Cell Biol. 2000;20(2):453–461. DOI: 10.1128/MCB.20.2.453-461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Stankiewicz TR, Loucks FA, Schroeder EK, et al. Signal transducer and activator of transcription-5 mediates neuronal apoptosis induced by inhibition of Rac GTPase activity. J Biol Chem. 2012;287(20):16835–16848. DOI: 10.1074/jbc.M111.302166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [308].Stankiewicz TR, Ramaswami SA, Bouchard RJ, et al. Neuronal apoptosis induced by selective inhibition of Rac GTPase versus global suppression of Rho family GTPases is mediated by alterations in distinct mitogen-activated protein kinase signaling cascades. J Biol Chem. 2015;290(15):9363–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Kalpachidou T, Spiecker L, Kress M, et al. Rho gtpases in the physiology and pathophysiology of peripheral sensory neurons. Cells. 2019;8(6):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Tan AM, Waxman SG. Spinal cord injury, dendritic spine remodeling, and spinal memory mechanisms. Exp Neurol. 2012;235(1):142–151. [DOI] [PubMed] [Google Scholar]
- [311].Tan AM, Chang Y-W, Zhao P, et al. Rac1-regulated dendritic spine remodeling contributes to neuropathic pain after peripheral nerve injury. Exp Neurol. 2011;232(2):222–233. [DOI] [PubMed] [Google Scholar]
- [312].Tan AM, Stamboulian S, Chang Y-W, et al. Neuropathic pain memory is maintained by Rac1-regulated dendritic spine remodeling after spinal cord injury. J Neurosci. 2008;28(49):13173–13183. DOI: 10.1523/JNEUROSCI.3142-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Tan AM, Samad OA, Fischer TZ, et al. Maladaptive dendritic spine remodeling contributes to diabetic neuropathic pain. J Neurosci. 2012;32(20):6795–6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [314].Tan AM, Samad OA, Liu S, et al. Burn injury-induced mechanical allodynia is maintained by Rac1-regulated dendritic spine dysgenesis. Exp Neurol. 2013;248:509–519. [DOI] [PubMed] [Google Scholar]
- [315].Cao XC, Pappalardo LW, Waxman SG, et al. Dendritic spine dysgenesis in superficial dorsal horn sensory neurons after spinal cord injury. Mol Pain. 2017;13:1744806916688016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Zhao P, Hill M, Liu S, et al. Dendritic spine remodeling following early and late Rac1 inhibition after spinal cord injury: evidence for a pain biomarker. J Neurophysiol. 2016;115(6):2893–2910. DOI: 10.1152/jn.01057.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Chen Z, Zhang S, Nie B, et al. Distinct roles of srGAP3-Rac1 in the initiation and maintenance phases of neuropathic pain induced by paclitaxel. J Physiol. 2020;598(12):2415–2430. DOI: 10.1113/JP279525. [DOI] [PubMed] [Google Scholar]
- [318].Ohsawa M, Ishikura K-I, Mutoh J, et al. Involvement of inhibition of RhoA/Rho kinase signaling in simvastatin-induced amelioration of neuropathic pain. Neuroscience. 2016;333:204–213. [DOI] [PubMed] [Google Scholar]
- [319].Xu H, Peng C, Chen X-T, et al. Chemokine receptor CXCR4 activates the RhoA/ROCK2 pathway in spinal neurons that induces bone cancer pain. Mol Pain. 2020;16:1744806920919568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Qiu Y, Chen WY, Wang ZY, et al. Simvastatin attenuates neuropathic pain by inhibiting the rhoa/limk/cofilin pathway. Neurochem Res. 2016;41(9):2457–2469. DOI: 10.1007/s11064-016-1958-1. [DOI] [PubMed] [Google Scholar]
- [321].Stenudd M, Sabelström H, Frisén J. Role of endogenous neural stem cells in spinal cord injury and repair. JAMA Neurol. 2015;72(2):235–237. [DOI] [PubMed] [Google Scholar]
- [322].Zhou N, Hao S, Huang Z, et al. MiR-7 inhibited peripheral nerve injury repair by affecting neural stem cells migration and proliferation through cdc42. Mol Pain. 2018;14:1744806918766793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [323].Konopaske GT, Lange N, Coyle JT, et al. Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry. 2014;71(12):1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [324].Moyer CE, Shelton MA, Sweet RA. Dendritic spine alterations in schizophrenia. Neurosci Lett. 2015;601:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [325].Shelton MA, Newman JT, Gu H, et al. Loss of microtubule-associated protein 2 immunoreactivity linked to dendritic spine loss in schizophrenia. Biol Psychiatry. 2015;78(6):374–385. DOI: 10.1016/j.biopsych.2014.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [326].Sweet RA, Henteleff RA, Zhang W, et al. Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology. 2009;34(2):374–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].Datta D, Arion D, Corradi JP, et al. Altered expression of CDC42 signaling pathway components in cortical layer 3 pyramidal cells in schizophrenia. Biol Psychiatry. 2015;78(11):775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [328].Fromer M, Pocklington AJ, Kavanagh DH, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506(7487):179–184. DOI: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [329].Purcell SM, Moran JL, Fromer M, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506(7487):185–190. DOI: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [330].McKinney B, Ding Y, Lewis DA, et al. DNA methylation as a putative mechanism for reduced dendritic spine density in the superior temporal gyrus of subjects with schizophrenia. Transl Psychiatry. 2017;7(2):e1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [331].Kang J, Park H, Kim E. IRSp53/BAIAP2 in dendritic spine development, NMDA receptor regulation, and psychiatric disorders. Neuropharmacology. 2016;100:27–39. [DOI] [PubMed] [Google Scholar]
- [332].Krugmann S, Jordens I, Gevaert K, et al. Cdc42 induces filopodia by promoting the formation of an IRSp53:Mena complex. Curr Biol. 2001;11(21):1645–1655. [DOI] [PubMed] [Google Scholar]
- [333].Chung W, Choi SY, Lee E, et al. Social deficits in IRSp53 mutant mice improved by NMDAR and mGluR5 suppression. Nat Neurosci. 2015;18(3):435–443. DOI: 10.1038/nn.3927. [DOI] [PubMed] [Google Scholar]
- [334].Sawallisch C, Berhörster K, Disanza A, et al. The insulin receptor substrate of 53 kDa (IRSp53) limits hippocampal synaptic plasticity. J Biol Chem. 2009;284(14):9225–9236. DOI: 10.1074/jbc.M808425200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [335].Bobsin K, Kreienkamp H-J. Severe learning deficits of IRSp53 mutant mice are caused by altered NMDA receptor-dependent signal transduction. J Neurochem. 2016;136(4):752–763. [DOI] [PubMed] [Google Scholar]
- [336].Kim M-H, Choi J, Yang J, et al. Enhanced NMDA receptor-mediated synaptic transmission, enhanced long-term potentiation, and impaired learning and memory in mice lacking IRSp53. J Neurosci. 2009;29(5):1586–1595. DOI: 10.1523/JNEUROSCI.4306-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [337].Kim Y, Noh YW, Kim K, et al. Irsp53 deletion in glutamatergic and gabaergic neurons and in male and female mice leads to distinct electrophysiological and behavioral phenotypes. Front Cell Neurosci. 2020;14:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [338].Remmers C, Sweet RA, Penzes P. Abnormal kalirin signaling in neuropsychiatric disorders. Brain Res Bull. 2014;103:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [339].Hayashi-Takagi A, Takaki M, Graziane N, et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 2010;13(3):327–332. DOI: 10.1038/nn.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [340].Ba W, Van Der Raadt J, Nadif Kasri N. Rho GTPase signaling at the synapse: implications for intellectual disability. Exp Cell Res. 2013;319(15):2368–2374. [DOI] [PubMed] [Google Scholar]
- [341].Newey SE, Velamoor V, Govek -E-E, et al. Rho GTPases, dendritic structure, and mental retardation. J Neurobiol. 2005;64(1):58–74. [DOI] [PubMed] [Google Scholar]
- [342].Ramakers GJA. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002;25(4):191–199. [DOI] [PubMed] [Google Scholar]
- [343].Humeau Y, Gambino F, Chelly J, et al. X-linked mental retardation: focus on synaptic function and plasticity. J Neurochem. 2009;109(1):1–14. [DOI] [PubMed] [Google Scholar]
- [344].Van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annu Rev Genet. 2011;45(1):81–104. [DOI] [PubMed] [Google Scholar]
- [345].Lee S, Rudd S, Gratten J, et al. Gene networks associated with non-syndromic intellectual disability. J Neurogenet. 2018;32(1):6–14. [DOI] [PubMed] [Google Scholar]
- [346].Zamboni V, Jones R, Umbach A, et al. Rho gtpases in intellectual disability: from genetics to therapeutic opportunities. Int J Mol Sci. 2018;19(6):6. DOI: 10.3390/ijms19061821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [347].Li J, Zhang W, Yang H, et al. Spatiotemporal profile of postsynaptic interactomes integrates components of complex brain disorders. Nat Neurosci. 2017;20(8):1150–1161. DOI: 10.1038/nn.4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [348].Srivastava AK, Schwartz CE. Intellectual disability and autism spectrum disorders: causal genes and molecular mechanisms. Neurosci Biobehav Rev. 2014;46(2):161–174. doi: 10.1016/j.neubiorev.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [349].Guo D, Yang X, Shi L. Rho gtpase regulators and effectors in autism spectrum disorders: animal models and insights for therapeutics. Cells. 2020;9(4):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [350].Bourgeron T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. 2015;16(9):551–563. [DOI] [PubMed] [Google Scholar]
- [351].Joensuu M, Lanoue V, Hotulainen P. Dendritic spine actin cytoskeleton in autism spectrum disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2018;84: 362–381. Pt B. [DOI] [PubMed] [Google Scholar]
- [352].Maestrini E, Pagnamenta AT, Lamb JA, et al. High-density SNP association study and copy number variation analysis of the AUTS1 and AUTS5 loci implicate the IMMP2L–DOCK4 gene region in autism susceptibility. Mol Psychiatry. 2010;15(9):954–968. DOI: 10.1038/mp.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [353].Liang S, Wang X, Zou M, et al. Family-based association study of ZNF533, DOCK4 and IMMP2L gene polymorphisms linked to autism in a northeastern Chinese Han population. Journal of Zhejiang University SCIENCE B. 2014;15(3):264–271. DOI: 10.1631/jzus.B1300133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [354].Pagnamenta AT, Bacchelli E, De Jonge MV, et al. Characterization of a family with rare deletions in CNTNAP5 and DOCK4 suggests novel risk loci for autism and dyslexia. Biol Psychiatry. 2010;68(4):320–328. DOI: 10.1016/j.biopsych.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [355].Guo H, Duyzend MH, Coe BP, et al. Genome sequencing identifies multiple deleterious variants in autism patients with more severe phenotypes. Genet Med. 2019;21(7):1611–1620. DOI: 10.1038/s41436-018-0380-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [356].Jack CR, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119–128. DOI: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [357].Scheff SW, Price DA, Schmitt FA, et al. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2006;27(10):1372–1384. [DOI] [PubMed] [Google Scholar]
- [358].Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298(5594):789–791. [DOI] [PubMed] [Google Scholar]
- [359].Zhao L, Ma Q-L, Calon F, et al. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci. 2006;9(2):234–242. DOI: 10.1038/nn1630. [DOI] [PubMed] [Google Scholar]
- [360].Huang W, Zhou Z, Asrar S, et al. p21-activated kinases 1 and 3 control brain size through coordinating neuronal complexity and synaptic properties. Mol Cell Biol. 2011;31(3):388–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [361].Lauterborn JC, Cox CD, Chan SW, et al. Synaptic actin stabilization protein loss in down syndrome and Alzheimer disease. Brain Pathol. 2020;30(2):319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [362].Ma Q-L, Yang F, Calon F, et al. p21-activated kinase-aberrant activation and translocation in Alzheimer disease pathogenesis. J Biol Chem. 2008;283(20):14132–14143. DOI: 10.1074/jbc.M708034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [363].Nguyen T-V-V, Galvan V, Huang W, et al. Signal transduction in Alzheimer disease: p21-activated kinase signaling requires C-terminal cleavage of APP at Asp664. J Neurochem. 2008;104(4):1065–1080. DOI: 10.1111/j.1471-4159.2007.05031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [364].Zhou Y, et al. Nonsteroidal anti-inflammatory drugs can lower amyloidogenic Abeta42 by inhibiting rho. Science. 2003;302(5648):1215–1217. DOI: 10.1126/science.1090154. [DOI] [PubMed] [Google Scholar]
- [365].Uehata M, Ishizaki T, Satoh H, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389(6654):990–994. DOI: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- [366].Wang JY, Wigston DJ, Rees HD, et al. LIM kinase 1 accumulates in presynaptic terminals during synapse maturation. J Comp Neurol. 2000;416(3):319–334. [DOI] [PubMed] [Google Scholar]
- [367].Tang BL, Liou YC. Novel modulators of amyloid-? Precursor protein processing. J Neurochem. 2007;100(2):314–323. [DOI] [PubMed] [Google Scholar]
- [368].Olabarria M, Pasini S, Corona C, et al. Dysfunction of the ubiquitin ligase E3A Ube3A/E6-AP contributes to synaptic pathology in Alzheimer’s disease. Commun Biol. 2019;2(1):111. DOI: 10.1038/s42003-019-0350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [369].Singh BK, Vatsa N, Kumar V, et al. Ube3a deficiency inhibits amyloid plaque formation in APPswe/PS1δE9 mouse model of Alzheimer’s disease. Hum Mol Genet. 2017;26(20):4042–4054. [DOI] [PubMed] [Google Scholar]
- [370].Pesaresi MG, Amori I, Giorgi C, et al. Mitochondrial redox signalling by p66Shc mediates ALS-like disease through Rac1 inactivation. Hum Mol Genet. 2011;20(21):4196–4208. DOI: 10.1093/hmg/ddr347. [DOI] [PubMed] [Google Scholar]
- [371].Castellanos-Montiel MJ, Chaineau M, Durcan TM. The neglected genes of ALS: cytoskeletal dynamics impact synaptic degeneration in ALS. Front Cell Neurosci. 2020;14:594975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [372].Hadano S, Kunita R, Otomo A, et al. Molecular and cellular function of ALS2/alsin: implication of membrane dynamics in neuronal development and degeneration. Neurochem Int. 2007;51(2–4):74–84. [DOI] [PubMed] [Google Scholar]
- [373].Figueroa-Romero C, Hur J, Bender DE, et al. Identification of epigenetically altered genes in sporadic amyotrophic lateral sclerosis. Plos One. 2012;7(12):e52672. DOI: 10.1371/journal.pone.0052672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [374].Conti A, Riva N, Pesca M, et al. Increased expression of Myosin binding protein H in the skeletal muscle of amyotrophic lateral sclerosis patients. Biochim Biophys Acta. 2014;1842(1):99–106. DOI: 10.1016/j.bbadis.2013.10.013. [DOI] [PubMed] [Google Scholar]
- [375].Hu JH, Chernoff K, Pelech S, et al. Protein kinase and protein phosphatase expression in the central nervous system of G93A mSOD over-expressing mice. J Neurochem. 2003;85(2):422–431. [DOI] [PubMed] [Google Scholar]
- [376].Tönges L, Günther R, Suhr M, et al. Rho kinase inhibition modulates microglia activation and improves survival in a model of amyotrophic lateral sclerosis. Glia. 2014;62(2):217–232. DOI: 10.1002/glia.22601. [DOI] [PubMed] [Google Scholar]
- [377].Stankiewicz TR, Pena C, Bouchard RJ, et al. Dysregulation of Rac or Rho elicits death of motor neurons and activation of these GTPases is altered in the G93A mutant hSOD1 mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2020;136:104743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [378].Lingor P, Weber M, Camu W, et al. ROCK-ALS: protocol for a randomized, placebo-controlled, double-blind phase iia trial of safety, tolerability and efficacy of the Rho Kinase (ROCK) inhibitor fasudil in amyotrophic lateral sclerosis. Front Neurol. 2019;10:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [379].Takata M, Tanaka H, Kimura M, et al. Fasudil, a rho kinase inhibitor, limits motor neuron loss in experimental models of amyotrophic lateral sclerosis. Br J Pharmacol. 2013;170(2):341–351. DOI: 10.1111/bph.12277. [DOI] [PMC free article] [PubMed] [Google Scholar]