

ABSTRACT
Fibrodysplasia ossificans progressiva (FOP) is a devastating disease of progressive heterotopic bone formation for which effective treatments are currently unavailable. FOP is caused by dominant gain‐of‐function mutations in the receptor ACVR1 (also known as ALK2), which render the receptor inappropriately responsive to activin ligands. In previous studies, we developed a genetic mouse model of FOP that recapitulates most clinical aspects of the disease. In this model, genetic loss of the wild‐type Acvr1 allele profoundly exacerbated heterotopic ossification, suggesting the hypothesis that the stoichiometry of wild‐type and mutant receptors dictates disease severity. Here, we tested this model by producing FOP mice that conditionally overexpress human wild‐type ACVR1. Injury‐induced heterotopic ossification (HO) was completely blocked in FOP mice when expression of both the mutant and wild‐type receptor were targeted to Tie2‐positive cells, which includes fibro/adipogenic progenitors (FAPs). Perinatal lethality of Acvr1 R206H/+ mice was rescued by constitutive ACVR1 overexpression, and these mice survived to adulthood at predicted Mendelian frequencies. Constitutive overexpression of ACVR1 also provided protection from spontaneous abnormal skeletogenesis, and the incidence and severity of injury‐induced HO in these mice was dramatically reduced. Analysis of pSMAD1/5/8 signaling both in cultured cells and in vivo indicates that ACVR1 overexpression functions cell‐autonomously by reducing osteogenic signaling in response to activin A. We propose that ACVR1 overexpression inhibits HO by decreasing the abundance of ACVR1(R206H)‐containing signaling complexes at the cell surface while increasing the representation of activin‐A‐bound non‐signaling complexes comprised of wild‐type ACVR1. © 2022 The Authors. Journal of Bone and Mineral Research published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research (ASBMR).
Keywords: FIBRODYSPLASIA OSSIFICANS PROGRESSIVA (FOP), GENETIC ANIMAL MODELS, HETEROTOPIC OSSIFICATION, ACVR1, ACTIVIN A, BMP SIGNALING, FOP THERAPEUTICS, FIBRO/ADIPOGENIC PROGENITORS
Introduction
Heterotopic ossification (HO), the formation of bone at extraskeletal anatomical locations, can result from traumatic injury or disease. The most extreme manifestation of HO is represented by the rare, autosomal‐dominant genetic disorder fibrodysplasia ossificans progressiva (FOP), in which heterotopic bone forms progressively throughout the life of the individual, resulting in devastating effects on health, life expectancy, and quality of life.( 1 , 2 ) Abnormal skeletal outgrowths extending from the surface of existing bone (referred to here as exostosis) is also a common pathological feature of FOP.( 3 ) The great majority of FOP cases are caused by a single arginine to histidine amino acid substitution (R206H) in the intracellular glycine/serine‐rich domain of the type 1 bone morphogenetic protein (BMP) receptor ACVR1.( 4 , 5 ) This amino acid substitution not only renders cells mildly hypersensitive to BMP ligands( 6 , 7 ) but also alters signaling specificity such that SMAD 1/5/8 phosphorylation and osteogenic differentiation is elicited by activin ligands,( 8 , 9 , 10 ) which normally activate SMAD 2/3 and function as inhibitors of BMP signaling.( 8 , 11 , 12 ) In fact, recent data have shown that activin A is both necessary and sufficient for HO flares, and its inhibition suppresses growth of nascent lesions in mouse models of FOP.( 8 , 10 , 13 , 14 ) The safety and efficacy of systemic delivery of an anti‐activin A neutralizing antibody are currently being evaluated in individuals with FOP (Clinical Trial # NCT03188666).
The relationship between physiological triggers of HO, such as soft tissue injury, inflammation, and immune cell activation, and inappropriate activation of ACVR1(R206H) by osteogenic ligands in cells responsible for heterotopic skeletogenesis remains obscure. Also unexplained is the common occurrence of so‐called spontaneous HO, in which episodes of HO lesion formation occur in the absence of known triggers.( 15 ) To interrogate these and other disease pathophysiological mechanisms and to test potential therapeutic agents and approaches, we developed a knockin mouse model of FOP (Acvr1 tnR206H ) in which the FOP‐causing gene is expressed under the control of the endogenous Acvr1 promoter upon Cre‐dependent removal of the floxed stop cassette upstream of the mutant exon.( 10 ) Lineage‐tracing studies in FOP mice support the conclusion that fibro/adipogenic progenitors (FAPs)—originally defined as muscle‐resident bipotent mesenchymal progenitors( 16 , 17 )—are a key offending cell type for both injury‐induced and spontaneous disease.( 10 , 13 , 18 , 19 ) Targeting Acvr1 R206H expression to Pdgfra‐positive cell populations, which in the postnatal musculoskeletal system are highly enriched for FAPs( 16 , 20 , 21 ) and also include periosteal cells,( 10 ) recapitulates HO at essentially all anatomical sites and tissues that affect individuals with FOP, including back and appendicular musculature, tendons and ligaments, major joints, and the jaw.( 13 ) Identification of skeletogenic progenitors responsible for HO now allows consideration of cell‐specific therapies for FOP, such as gene therapies or other therapeutic interventions that alter the developmental capacity specifically of offending cell types.
In mice, as in humans, FOP mutations in Acvr1 result in gain‐of‐function receptor activity that is manifested in the heterozygous state. Notably, in FOP mice, genetic loss of the remaining wild‐type Acvr1 allele in Tie2+ skeletogenic progenitors (predominantly FAPs) causes a profound increase in HO volume after muscle injury.( 10 ) These data suggest that the relative stoichiometry of wild‐type and mutant ACVR1 receptors dictates disease severity in FOP mice. To test this hypothesis, we developed knockin mice that conditionally overexpress a wild‐type human ACVR1 cDNA (R26 nACVR1 ) and assessed whether ACVR1 overexpression mitigates injury‐induced and spontaneous HO in FOP mice. We also tested the capacity of global overexpression of ACVR1 during prenatal development to rescue perinatal lethality of mice that express Acvr1 R206H under endogenous transcriptional control.( 22 ) Collectively, the data demonstrate that overexpression of wild‐type ACVR1 is well tolerated and effectively dampens the pathological effects of the mutant FOP receptor.
Materials and Methods
Generation of R26 nACVR1 and R26 ACVR1 knockin mice
R26 NACVR1 knockin line was designed to express HA‐tagged human ACVR1 (hACVR1‐HA) upon Cre‐dependent excision of the floxed PGKNeo‐stop cassette located between CAG promoter and hACVR1‐HA. hACVR1‐HA sequence was generated by insertion of a HA linker annealed by the oligomers 5′‐ACCCATACGACGTACCAGATTACGCTAGTCTCTAGCTCGA‐3′ and 5′‐GCTAGAGACTAGCGTAATCTGGTACGTCGTATGGGT‐3′ between the HincII, which is located at the 3′ end of the ACVR1 coding sequence, and the XhoI sites of pCMV‐SPORT6‐hACVR1 plasmid (Invitrogen, Carlsbad, CA, USA). The R26NALK2 knockin plasmid, pR26‐CNhAHA, was constructed by replacing the nlslacZ‐3xSV40pA/FRT/EGFP‐bGHpA sequence of pR26‐CLNFZG( 23 ) with the HA‐tagged human ACVR1 followed by a SV40pA. The detail cloning strategy and complete sequence of the plasmids are available on request.
ES cell electroporation and production of chimeric mice were performed by the Center for Mouse Genome Modification (CMGM) at UConn Health. The pR26‐CNhAHA plasmid was linearized with Acc65I and electroporated into 129S6/C57BL/6 hybrid ES cells (D1: established by CMGM). Nested PCR was used to screen for homologous recombination on both 5′ and 3′ ends with the following primers: 5′ end: 1st PCR, 5′‐TTCCTCTCAATATGCTGCACACAAA‐3′ and 5′‐GGAACTCCATATATGGGCTATGAACT‐3′, 2nd PCR, 5′‐AGGCAGGGAAAACGACAAAATCTGG‐3′ and 5′‐GGAACTCCATATATGGGCTATGAACT‐3′; 3′ end: 1st PCR, 5′‐TGTGGTTTGTCCAAACTCATCAATGT‐3′ and 5′‐GATGCCCAATTCCAACTGTGAAGAC‐3′, 2nd PCR, 5′‐TGTGGTTTGTCCAAACTCATCAATGT‐3′ and 5′‐TGGTTCACACCACAAATGAACAGTGC‐3′. The targeted allele generated 2.4 kb and 5.8 kb diagnostic products with the 5′ and 3′ nested PCR reactions, respectively. Chimeric mice were produced from three targeted ES clones by aggregation with CD1 embryos. Germline transmission of the targeted allele was assessed by PCR for the floxed PGKNeo‐stop cassette/hACVR1 boundary (5′‐GATCAGCAGCCTCTGTTCCACA‐3′ and 5′‐CACGTCTCGGGGATTGAGGC‐3′; 653 bp) and the nested PCR for the 5′ and 3′ recombination regions. One R26 nACVR1 knockin line was established.
Mice, crosses, genotyping, and husbandry
Tie2‐Cre transgenic mice were kindly provided by Dr Tom Sato (UT Southwestern). Pdgfra‐Cre transgenic mice (Tg(Pdgfra‐cre)1Clc, #013148), Hprt1 Cre (Hprt tm1(CAG−cre)Mnn , #004302), Hprt1 iCreER (Hprt tm345(icre/ERT2)Ems /Mmjax, MMRRC stock #037075), and R26 FLPo (Gt(ROSA)26Sor tm2(FLP*)Sor , #012930) knockin mice were obtained from Jackson Laboratory, (Bar Harbor, ME, USA). R26 NG( 23 ), Alk2 Flox( 24 , 25 ) (referred to here as Acvr1 flox ), and Acvr1 tnR206H( 10 ) were described previously. Hprt1 iCre was generated by a cross between Hprt1 iCreER/+ and R26 FLPo/+ mice. Acvr1 loxP was generated by a cross between Acvr1 flox/+ and Hprt1 iCre/+ mice. Acvr1 R206H and R26 ACVR1 were generated by a cross between Acvr1 tnR206H/+ ;R26 nACVR1/+ and Hprt1 iCre/+ mice. The Acvr1 R206H allele can be maintained in the presence of the R26 ACVR1 allele. Transmission of the constitutively recombined Acvr1 R206H allele was verified by the absence of tdTomato red fluorescence and the presence of a 203 bp PCR product using the following primers: 5′‐CAACAGGGTTATCTGATGG‐3′ and 5′‐TCACATGTCCAGAGTTGCT‐3′. The following primers were used for genotyping of the other alleles: Acvr1 tnR206H : 5′‐GCTAACCATGTTCATGCCTTC‐3′ and 5′‐AGCGCATGAACTCCTTGATGAC‐3′ (144 bp); Acvr1 flox : 5′‐TGCTGTCTTTTAACTCCTGGGATC‐3′ and 5′‐TCTCACCTTCCCTATGACTCTTAG‐3′ (0.51 kbp); Acvr1 loxP : 5′‐TGCTGTCTTTTAACTCCTGGGATC‐3′ and 5′‐TCTAAGAGCCATGACAGAGGTTG‐3′ (0.6 kbp); Hprt iCre : 5′‐GCTAAAGAGTTGAACGCAAAGGTG‐3′ and 5′‐GGGCTATGAACTAATGACCCCGTA‐3′ (0.6 kbp); Tie2‐Cre, Pdgfra‐Cre and Hprt1 Cre : 5′‐GCGGTCTGGCAGTAAAAACTATC‐3′ and 5′‐GTTCGAACGCTAGAGCCTGTTT‐3′ (174 bp); R26 ACVR1 : 5′‐GCTAACCATGTTCATGCCTTC‐3′ and 5′‐CACGTCTCGGGGATTGAGGC‐3′ (688 bp) and R26 NG : 5′‐GATCAGCAGCCTCTGTTCCACA‐3′ and 5′‐CGCTGAACTTGTGGCCGTTTAC‐3′ (264 bp). Experimental mice carrying Cre were either hemizygous (Tie2‐Cre and Pdgfra‐Cre transgenes) or heterozygous (Hprt iCre and Hprt Cre knockin alleles) for the Cre deleter allele.
Experimental mice were maintained on a mixed background predominantly composed of FVB, CD1, and C57BL/6 strains. Mice were housed singly (males) or at a maximum of 4 mice per cage (females) in a negative pressure vented micro‐isolation container system (Animal Care Systems Optimice IVC Rack System) with enrichment structures. Mice were maintained on 2918 Irradiated Teklad Global 18% Rodent Diet (Envigo, Indianapolis, IN, USA) and food was available ad libitum.
Muscle injury by cardiotoxin injection
Muscle injury was performed by injecting 100 μL of 10 μM Naja pallida cardiotoxin (Lotoxan, #L8102) in PBS into the tibialis anterior muscle. HO formation was analyzed at 3, 8, and 20 to 24 days post‐injury.
Histology, immunohistochemistry, and μCT imaging
Samples were either directly embedded in Tissue Plus O.C.T. compound (Thermo Fisher Scientific, Waltham, MA, USA) immediately after dissection or fixed in 4% paraformaldehyde/PBS, washed in PBS, and incubated in 30% sucrose in PBS before embedding. For phospho‐SMAD1/5/8 immunohistochemical detection, cryosections of paraformaldehyde‐fixed samples were subjected to antigen‐retrieval as described.( 26 ) For HRP/DAB‐based signal detection, sections were incubated in 0.1% H2O2/PBS for 30 minutes. Sections were blocked in 2% BSA/PBS/0.2% Triton X‐100 (PBST) supplemented with 50 mM glycine with primary antibodies in PBST supplemented with 10 mM glycine. A mixture of rabbit anti‐pSmad1/5/9 (D5B10) and anti‐pSMAD1/5 (41D10) monoclonal antibodies (Cell Signaling Technology, Danvers, MA, USA; #13820 and #9516, respectively), diluted 1:100 each, was used for pSMAD1/5/8 detection. Rabbit anti‐SOX9 antibody (Millipore, Burlington, MA, USA; #AB5535) and rabbit anti‐HA antibody (Zymed, Thermo Fisher Scientific, #71‐5500) were used at 1:1000 and 1:50 dilutions, respectively. Biotin‐XX‐goat anti‐rabbit IgG (Thermo Fisher Scientific, #B2770) was used as a secondary antibody at 1:2000 dilution in PBS/5% goat serum/0.2% Triton X‐100. Alexa Fluor 647‐streptavidin (Thermo Fisher Scientific, #S21374) was used for fluorescent detection of the target proteins.
Alcian blue/alizarin red whole mount skeletal staining( 27 ) and μCT imaging were performed as described.( 10 )
Cell isolation and culturing for Acvr1 mRNA and protein quantification
Total hindlimb muscle tissue was dissected from adult mice, physically dissociated into small pieces, and incubated in 0.2% collagenase type 1/DMEM/1X penicillin–streptomycin (Pen‐Strep, Caisson, Smithfield, UT, USA; #PSL01) for 90 minutes at 37°C with gentle shaking. Digestion was terminated by addition of FBS to a final concentration of 10%. After filtering through a polyester fiber filter, the lysate was centrifuged at 800g for 2 minutes. The cells were subjected to a red‐blood cell lysis, filtered through a 100 μm cell strainer (Falcon, #352360) and centrifuged at 300g for 5 minutes. After counting with a hemocytometer, cells were centrifuged at 800g for 2 minutes and resuspended in DMEM/10% FBS/1X Pen‐Strep.
Cells (2.5 × 106) were plated in a 6 cm tissue culture dish and cultured for 90 minutes in DMEM/10% FBS/1X Pen‐Strep. After removing nonadherent cells by flushing with PBS, adherent cells were further cultured overnight in DMEM/10% DMEM/1X Pen‐Strep. After rinsing with PBS, cells were processed for Acvr1 mRNA or protein quantification.
Acvr1 mRNA quantification
RNA from the adherent cells was extracted using QIAGEN (Valencia, CA, USA) RNeasy Micro Kit according to the manufacturer's manual. RNA from 14.5 dpc whole fetuses was prepared using QIAGEN RNeasy Midi Kit. Complementary DNA (cDNA) was synthesized using Bio‐Rad (Hercules, CA, USA) iScript Advanced cDNA Synthesis Kit (#1725038) according to the manufacturer's instructions. Absolute quantification of target transcripts was determined by Bio‐Rad QX200 Droplet Digital PCR system using QX200 ddPCR EvaGreen Supermix (#1864033) according to the manufacturer's instructions. The following PCR primer sets were used for each gene transcripts: Acvr1 wild type: 5′‐CAGCACTCTAGCGGAACTAC‐3′ and 5′‐CTCCAACAGGGTTATCTGGC‐3′ (106‐bp), Acvr1 R206H : 5′‐CAGCACTCTAGCGGAACTAC‐3′ and 5′‐CAACAGGGTTATCTGATGG‐3′ (103‐bp), β Actin: 5′‐TAGGCACCAGGGTGTGATGG‐3′ and 5′‐GTACATGGCTGGGGTGTTGAA‐3′ (286‐bp), Pcna: 5′‐TGGAGAGCTTGGCAATGGGAA‐3′ and 5′‐GCAAACGTTAGGTGAACAGGCT‐3′ (108‐bp), R26 ACVR1 : 5′‐AATCCATCCGCAAGACTCAC‐3′ and 5′‐CTCGAGCTAGAGACTAGCGT‐3′ (129‐bp). After droplet generation with the QX200 Droplet Generator (Bio‐Rad), thermal cycling was performed with a C1000 Thermal Cycler (Bio‐Rad) as follows: 95°C for 5 minutes, 40 cycles of 95°C for 30 seconds and 60°C for 60 seconds, followed by one each of 4°C for 5 minutes and 98°C for 10 minutes, all at a ramp rate of 2°C per second. The droplets were subsequently read by a QX200 Droplet Reader and the data were analyzed with QuantaSoft v.1.7.4 software (Bio‐Rad).
Western blotting for detection of ACVR1
Adherent cells were collected in 100 μL 2X RIPA buffer supplemented with 2 mM PMSF. Total volume of cell lysate was adjusted to 200 μL with PBS. The 14.5 dpc fetus was homogenized in 4 ml of 1X RIPA buffer supplemented with 1 mM PMSF, rocked for 2 hours at 4°C, centrifuged at 8,160 x g for 20 minutes at 4°C, and the supernatant collected. After protein quantification, an equal volume of 2X Laemmli SDS‐sample buffer was added to the protein lysate. A Bio‐Rad Mini Trans‐Blot system was used to perform SDS‐PAGE and protein transfer onto PVDF membrane according to the manufacturer's instructions. Immunoblotting was performed using SuperSignal West Pico PLUS (Thermo Fisher Scientific, #34577) according to the manufacturer's instructions. For sequential immunodetection of multiple epitopes, stripping was performed by incubating used membrane in 62.5 mM Tris–HCl pH 6.8/1% SDS/100 mM β‐mercaptoethanol for 40 minutes at 50°C followed by rinsing in water. The following antibodies and dilutions were used: rabbit monoclonal anti‐ACVR1 antibody (Abcam, Cambridge, MA, USA; #155981) diluted 1:1000, rabbit anti‐PCNA antibody (Santa Cruz Biotechnology, Dallas, TX, USA; #FL‐261) diluted 1:1000, mouse monoclonal anti‐HA antibody (2‐2.2.14, Thermo Fisher Scientific, #26183) diluted 1:104, rabbit anti‐mouse IgG (Alexa Fluor 647‐conjugated, Thermo Fisher Scientific, #A21239) diluted 1:104 and HRP‐conjugated goat anti‐rabbit IgG (Cell Signaling Technology, #7074) diluted 1:104. Chemiluminescence was detected using FluorChem HD2 (ProteinSimple, San Jose, CA, USA) or IVIS SpectrumCT (PerkinElmer, Waltham, MA, USA) instruments.
Cell isolation, culturing, and Western blotting for pSMAD1/5/8
Total hindlimb skeletal muscle was harvested from mice of the indicated genotypes, minced, enzymatically digested, and filtered as previously described.( 10 ) The resulting crude mixture of cells from each mouse was seeded into a 100 mm cell culture dish (NEST, Woodbridge, NJ, USA; #704004) and grown to 60% to 80% confluence, over 3 to 4 days, in growth media containing 20% fetal bovine serum (FBS) (R&D Systems, Minneapolis, MN, USA; #S11150) and 1% Pen/Strep (Sigma, St. Louis, MO, USA; P4333) in Dulbecco's Modified Eagle Medium (DMEM) (Life Technologies, Carlsbad, CA, USA; #11995‐073). Crude plastic‐adherent cells were then dissociated from the culture dishes using Accumax (Innovative Cell Technologies, San Diego, CA, USA; AM105), blocked with 10% FBS in PBS, and stained for 30 minutes on ice with the following fluorescently conjugated antibodies at indicated dilutions: anti‐PDGFRα‐APC (1:100; eBioscience, San Diego, CA, USA; #17‐1401‐81), anti‐SCA‐1‐v450 (1:400; BD Bioscience, San Jose, CA, USA; #560653), anti‐CD31‐bv711 (1:500; BD Biosciences, #740690), and anti‐CD45‐bv711 (1:500; BD Biosciences, #563709). 7‐AAD was also added to cell suspensions at 0.5 μg/mL 10 minutes before to identify and exclude dead/dying cells. Using a FACSAria II flow cytometer (BD Biosciences), control FAPs, with and without the R26 ACVR1 allele, were isolated as live single cells expressing the cell surface profile, CD31‐CD45‐SCA‐1 + PDGFRα+. R206H‐FAPs, with and without the R26 ACVR1 allele, were identified with the additional selection criteria tdTomato‐GFP+, which selects for only Acvr1 tnR206H ‐recombined FAPs.
FACS‐isolated FAPs were seeded in wells of 6‐well plates at 2000 cells/cm2 in growth media. After reaching near confluence, FAPs were serum starved in DMEM for 2 hours before a 1‐hour incubation with either activin A or BMP6 at the indicated concentrations. FAPs were washed with cold PBS, scraped off the dish in cold RIPA lysis buffer, and vortexed to complete lysis. Total protein concentration was measured using the DC protein assay (Bio‐Rad), and equal amounts of total protein were loaded per lane of the same gel for SDS‐PAGE. After the transfer step, PVDF membranes were blocked in 5% non‐fat milk in PBS with Tween‐20, and incubated with rabbit anti‐pSMAD1/5/8 (1:1,000; Cell Signaling Technology, #13820) and mouse anti‐β‐actin (1:2,500; Cell Signaling Technology, #3700) overnight at 4°C. Goat anti‐rabbit IgG HRP‐linked antibody (Cell Signaling Technology, #7074) was used either as a secondary antibody to detect pSMAD1/5/8 or as a tertiary antibody after incubation with rabbit anti‐mouse IgG Alexa Fluor 647 to detect β‐actin. Membranes were incubated in SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific) and imaged using the IVIS SpectrumCT (PerkinElmer). pSMAD1/5/8 and β‐actin bands were imaged separately, as separate secondary antibody incubations of the same membrane were required. Blots were quantified using ImageJ, wherein the integrated density values (arbitrary units) of pSMAD1/5/8 bands were divided by those of β‐actin bands in the same sample to generate normalized integrated density values. The normalized integrated density values from individual, representative blots are displayed as bar graphs, which were generated in GraphPad Prism v7.05.
Results
Characterization of mice that conditionally overexpress human ACVR1
To produce mice that conditionally overexpress the wild‐type ACVR1 receptor, the human ACVR1 cDNA was knocked into the constitutively expressed Rosa26 locus,( 28 ) using a strategy and design similar to that used to produce the Cre‐dependent eGFP reporter R26 NG .( 23 ) A floxed Neo cassette was positioned upstream of ACVR1, and the constitutively active CAG promoter/enhancer sequence( 29 ) was provided to ensure high‐level expression. Finally, an in‐frame hemagglutinin sequence (HA) was added at the 3′ end of ACVR1 to allow discrimination between human and mouse proteins, which are >98% identical. The unrecombined, Neo‐containing allele is designated R26 nACVR1 , whereas R26 ACVR1 denotes the Cre‐recombined allele (Supplemental Fig. S1 A).
One R26 nACVR1 mouse line was established and maintained by routine breeding schemes. To validate expression of human ACVR1 from the R26 nACVR1 allele, R26 nACVR1 mice were crossed with Tie2‐Cre mice,( 30 ) and the HA peptide was detected by immunofluorescence on skeletal muscle sections derived from adult Acvr1 tnR206H/+ ;R26 nACVR1/+ ;Tie2‐Cre mice. As expected, based on the known expression of Tie2 ( 21 , 30 , 31 ), the HA tag was immunodetected in the vasculature and in a fraction of interstitial mononuclear cells (referred to as Tie2+ progenitors, (21 ) or FAPs( 10 )), but not in muscle fibers, which strongly expressed tdTomato from the unrecombined Acvr1 tnR206H allele (Supplemental Fig. S1 B, C). Quantification of ACVR1 overexpression is provided below.
As BMP signaling regulates development of many tissues, we first determined whether widespread overexpression of ACVR1 is compatible with viability. Males carrying the R26 nACVR1 allele were crossed with Hprt iCre/+( 32 ) females to generate embryos in which the R26 nACVR1 allele was globally recombined (R26 ACVR1 ). Although most Hprt iCre hemizygous male mice died before weaning irrespective of inheritance of R26 nACVR1 (data not shown), R26 ACVR1/+ ;Hprt iCre/+ females developed to adulthood and were fertile. For subsequent analyses, both male and female R26 ACVR1/+ mice (germline‐recombined at the R26 nACVR1 locus and lacking Hprt iCre ) were generated using standard breeding schemes. The R26 ACVR1 allele was transmitted to offspring of weaning age at Mendelian frequencies (50.8%; 62 of 122 pups). R26 ACVR1/+ mice were fertile and did not show any gross physical or behavioral abnormalities.
Acvr1‐null embryos exhibit multiple developmental defects and die during gastrulation.( 33 , 34 , 35 ) To test whether R26 ACVR1 can functionally replace the endogenous Acvr1 gene, animals were produced that are homozygous null for Acvr1 and heterozygous for R26 ACVR1 . Two null alleles were utilized: the unrecombined Acvr1 tnR206H allele,( 10 ) and a second, independently‐derived null allele (referred to here as Acvr1 loxP ) produced by crossing a floxed conditional Acvr1 allele( 24 ) (referred to here as Acvr1 flox ) with Hprt iCre . Notably, Acvr1 loxP/loxP ;R26 ACVR1/+ , Acvr1 tnR206H/tnR206H ;R26 ACVR1/+ , and Acvr1 tnR2006H/loxP ;R26 ACVR1/+ mice were viable and fertile and recovered at Mendelian frequencies at weaning (Table 1). These data demonstrate that the R26 ACVR1 allele can functionally replace the endogenous Acvr1 gene.
Table 1.
The R26 ACVR1 Allele Can Functionally Substitute for the Endogenous Acvr1 Gene
| R26 genotype | R26 +/+ | R26 ACVR1/+ | ||||
|---|---|---|---|---|---|---|
| Acvr1 genotype | Acvr1 +/+ | Acvr1 +/− | Acvr1 −/− | Acvr1 +/+ | Acvr1 +/− | Acvr1 −/− |
| No. mice at weaning | 3 | 17 | 0 | 5 | 10 | 7 |
| Expected (%) | 12.5 | 25 | 12.5 | 12.5 | 25 | 12.5 |
| Adjusted expected (%) | 14.3 | 28.6 | 0 | 14.3 | 28.6 | 14.3 |
| Actual (%) | 7.1 | 40.5 | 0 | 11.9 | 23.8 | 16.7 |
Mice were generated by crossing Acvr1 +/−;R26 ACVR1/+ and Acvr1 +/− parents. Two Acvr1 null alleles were used (Acvr1 tnR206H and Acvr1 loxP ) and are collectively designated as Acvr1 − . Expected percentages are based strictly on predicted Mendelian frequencies, whereas adjusted expected percentages account for the known embryonic lethality of Acvr1 −/− mice and assume complete rescue by R26 ACVR1 .
Overexpression of ACVR1 rescues perinatal lethality of FOP mice
Progeny that are heterozygous for the R206H‐encoding germline mutation in Acvr1 die perinatally.( 19 , 22 ) To further characterize the effects of Acvr1 R206H expression, the Acvr1 tnR206H allele was globally recombined in the early embryo so that the pattern and timing of Acvr1 R206H expression were dictated exclusively by Acvr1 promoter activity. Acvr1 tnR206H/+ males were crossed with females that were heterozygous for the ubiquitously expressed Hprt Cre allele, and skeletal development and survival were assessed. Unlike the Hprt iCre allele used above, recombination of the paternally inherited Acvr1 tnR206H allele after fertilization was not dependent on inheritance of the Hprt Cre allele, presumably because of accumulation of Cre in the egg.( 36 ) Thus, globally recombined offspring had the genotype of Acvr1 R206H/+ , Acvr1 R206H/+ ; Hprt Cre/+ (females), or Acvr1 R206H/+ ;Hprt Cre /Y (males). Mice were born at the expected Mendelian frequency, but all pups carrying the recombined Acvr1 R206H allele (17 of 34 neonates) died within 24 hours of birth and exhibited grossly visible malformation of the hindlimbs (Fig. 1F ). Although the precise cause of death was not determined, affected pups lacked a milk spot in their bellies, raising the likelihood that death resulted from dehydration and nutritional deficits.
Fig. 1.
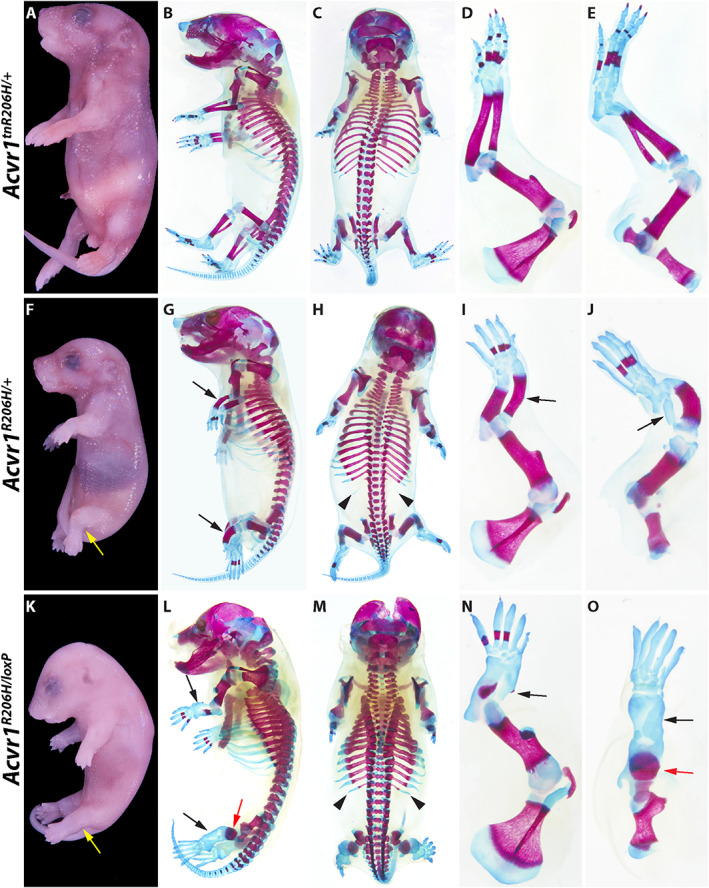
Representative examples of severe skeletal malformations caused by prenatal expression of Acvr1 R206H . (A–E) Acvr1 tnR206H/+ control P0 neonate. (F–J) Acvr1 R206H/+ neonate. (K–O) Acvr1 R206H/loxP neonate. Developmental defects are aggravated by loss of the wild‐type Acvr1 allele. (A, F, K) Whole‐mount views after skin removal. Yellow arrows indicate abnormal positioning of the hindlimbs. Left side (B, G, L) and dorsal (C, H, M) views of ABAR‐stained whole‐mount neonates. ABAR‐stained left forelimbs (D, I, N) and hindlimbs (E, J, O). Black and red arrows indicate severely shortened zeugopodal bones and femur, respectively; black arrowheads indicate L1 supernumerary ribs. Also note missing or abnormal ossification centers and thickened ribs in Acvr1 tnR206H/+ and Acvr1 R206H/loxP neonates. Additional defects are described in the text.
Newborn Acvr1 R206H/+ mice did not exhibit HO, but alcian blue/alizarin red (ABAR) staining of P0 pups revealed pronounced skeletal defects, particularly of the forelimbs and hindlimbs (Fig. 1G–J ; Table 2) (n = 20). Bones of the hindlimb zeugopod (tibia/fibula) were grossly misshapen, the diaphyses were shortened and thickened, and the fibula lacked an ossification center (Fig. 1G–J ). The digits of both the forelimb and hindlimb lacked distal ossification centers, the proximal ossification centers of digits 1 and 5 (forelimb) or digit 5 (hindlimb) were absent (Fig. 1I, J ), most digital joints were absent (Fig. 1F–J ), and cutaneous syndactyly was evident (Supplemental Fig. S2 A–D). Shortening of the fibula and defects in the formation of the metacarpophalangeal joints were already observed—although differences were often subtle—in embryos by 13.5 dpc (Supplemental Fig. S2 G, H). The axial skeleton also showed abnormalities, including rib thickenings in all Acvr1 R206H/+ P0 mice (Fig. 1 G, H and Supplemental Fig. S3 C), the presence of supernumerary ribs associated with the first lumbar vertebrae (L1) in the majority of these mice, and spina bifida (Fig. 1H and Supplemental Fig. S3 C; n = 20 mice). Mice lacking the remaining wild‐type Acvr1 allele (Acvr1 R206H/loxP ) survived to birth, demonstrating that Acvr1 R206H can fulfill the requirement of Acvr1 in gastrulation.( 33 , 34 , 35 ) Importantly, however, loss of the wild‐type Acvr1 allele produced even more pronounced skeletal abnormalities and live newborns were not recovered. Skeletal defects included more severely truncated and malformed stylopods and zeugopods, further disruption of ossification including complete loss of autopod ossification in the hindlimbs, and thicker ribs (Fig. 1L–O ). The exacerbated phenotype of Acvr1 R206H/loxP mice suggests that wild‐type ACVR1 partially suppresses the pathogenic developmental effects of ACVR1(R206H) in Acvr1 R206H/+ cells, a conclusion consistent with our previous findings.( 10 ) Perinatal lethality and most aforementioned skeletal malformations were fully penetrant, providing robust phenotypic criteria by which to evaluate the ability of ACVR1 overexpression to mitigate the deleterious effects of prenatal Acvr1 R206H expression.
Table 2.
Summary of Phenotypes of Different Genotypic Classes, as Assessed by ABAR Staining of P0 Mice
| Phenotype | Wild type | R26 ACVR1/+ | Acvr1 R206H/+ | Acvr1 R206H/+ ; R26 ACVR1/+ | Acvr1 R206H/− | Acvr1 R206H/− ; R26 ACVR1/+ |
|---|---|---|---|---|---|---|
| Found dead | 1/18 (5.6%) | 1/14 (7.1%) | 3/20 (6.0%) | 0/27 (0%) | 10/10 (100%) | 1/15 (6.7%) |
| Spina bifida | 0/16 (0%) | 0/13 (0%) | 20/20 (100%) | 0/27 (0%) | 8/8 (100%) | 0/15 (0%) |
| Costovertebral fusion | 0/16 (0%) | 0/14 (0%) | 20/20 (100%) | 0/27 (0%) | 8/8 (100%) | 0/15 (0%) |
| Intervertebral fusion | 0/16 (0%) | 0/14 (0%) | 0/20 (0%) | 0/27 (0%) | 8/8 (100%) | 0/15 (0%) |
| L1 supernumerary rib | 2/16 (12.5%) | 5/14 (35.7%) | 16/20 (80%) | 13/27 (48.1%) | 8/8 (100%) | 9/15 (60.0%) |
| Zeugopod truncation | 0/18 (0%) | 0/14 (0%) | 20/20 (100%) | 0/27 (0%) | 9/9 (100%) | 0/15 (0%) |
| Metacarpal/metatarsal‐phalangeal fusion | 0/18 (0%) | 0/13 (0%) | 20/20 (100%) | 0/27 (0%) | 9/9 (100%) | 0/14 (0%) |
Differences in numbers of mice scored within a group reflect damage during processing and inability to score a particular phenotype. For Acvr1 R206H/− genotypes, the null allele was either the unrecombined Acvr1 tnR206H allele or the germline‐recombined Acvr1 loxP allele.
To determine whether ACVR1 overexpression can rescue lethality of FOP neonates, Acvr1 tnR206H/+ ;R26 nACVR1/+ males were crossed to Hprt iCre/+ females, and offspring were genotyped at weaning. As expected, no Acvr1 R206H/+ mice lacking R26 ACVR1 were recovered. Notably, however, mice carrying both the globally recombined Acvr1 R206H and R26 ACVR1 alleles were viable and exhibited no externally visible abnormalities at weaning. Subsequent breeding of Acvr1 R206H/+ ;R26 ACVR1/+ and wild‐type mice confirmed R26 ACVR1 ‐dependent survival of Acvr1 R206H/+ mice, which were recovered at expected Mendelian frequencies at weaning (Table 3). Using several crossing schemes, a total of 90 adult mice were produced that were heterozygous for both the Acvr1 R206H and R26 ACVR1 alleles. Except for one mouse that died prematurely (see below), mice showed no reduction in life span up to the latest experimental endpoint of 17 months of age, and mice were fertile. Skeletal development showed a concomitant normalization, as assessed by ABAR staining of 16.5 dpc fetuses (Fig. 2A–C ) and P0 pups (Table 2). Although 13 of 27 mice exhibited supernumerary ribs at L1 on at least one side (approximately 40% of all left and right sides), ACVR1 overexpression rescued the rib thickening malformations and zeugopodal bone defects of the appendicular skeleton in all mice (Fig. 2C ; Supplemental Fig. S3 D; Table 2). Global expression of R26 ACVR1 also rescued the perinatal‐lethal phenotype of the more severely affected Acvr1 R206H/− mice (either Acvr1 R206H/loxP or Acvr1 R206H/tnR206H ) (n = 21 at weaning age or older) (Fig. 3E ; Table 2).
Table 3.
The R26 ACVR1 Allele Can Rescue Perinatal Lethality of Acvr1 R206H/+ Mice
| Genotype | Wild type | Acvr1 R206H/+ | R26 ACVR1/+ | Acvr1 R206H/+ ;R26 ACVR1/+ |
|---|---|---|---|---|
| No. of mice at weaning | 22 | 0 | 19 | 19 |
| Expected (%) | 25.0 | 25.0 | 25.0 | 25.0 |
| Adjusted expected (%) | 33.3 | 0.0 | 33.3 | 33.3 |
| Actual (%) | 36.7 | 0.0 | 31.7 | 31.7 |
Mice were generated by crossing Acvr1 R206H/+ ;R26 ACVR1/+ and wild‐type parental mice. Expected percentages are based strictly on predicted Mendelian frequencies, whereas adjusted expected percentages account for perinatal lethality of Acvr1 R206H/+ mice and assume complete rescue by R26 ACVR1 .
Fig. 2.
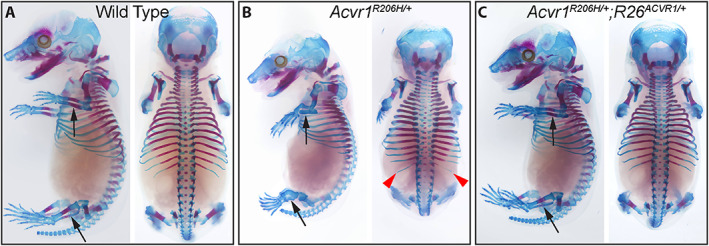
Suppression of Acvr1 R206H ‐induced skeletal defects by overexpression of wild‐type ACVR1. (A–C) Left and dorsal views of ABAR‐stained 16.5 dpc fetuses. (A) Wild‐type, (B) Acvr1 R206H/+ , and (C) Acvr1 R206H/+ ;R26 ACVR1/+ fetuses. Most of the skeletal abnormalities exhibited by Acvr1 R206H/+ mice at P0 are evident by 16.5 dpc. Black arrows = zeugopod; red arrowheads = L1 supernumerary ribs.
Fig. 3.
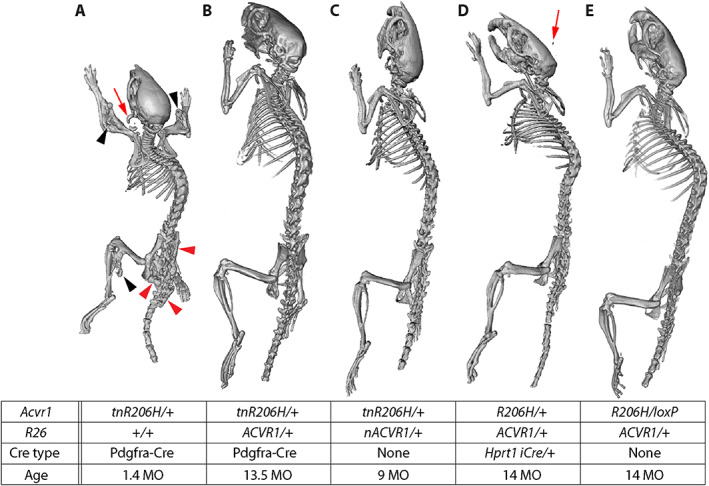
Suppression of spontaneous heterotopic ossification (HO) by overexpression of wild‐type ACVR1 in two models of FOP. The table shows the genotype and age (months) of each mouse. (A–E) Representative μCT images of juvenile (A) and adult (B–E) mice. Note that a juvenile Acvr1 R206H/+ ;Pdgfra‐Cre mouse (A) was used because of the poor survival and health of adult mice of this genotype. A control mouse that lacked Cre is shown in (C). The mouse in (E) inherited the recombined alleles shown through the germline, and their expression was therefore independent of Cre. Black and red arrowheads = spontaneous exostosis associated with the appendicular and axial skeleton, respectively; red arrows = HO at the ear‐tag site.
Inhibition of juvenile‐onset spontaneous HO in FOP mice
Patient histories indicate that most new bone growth in FOP patients occurs spontaneously, without a known injury or inflammatory trigger.( 37 ) In previous analyses in which Acvr1 tnR206H recombination was targeted to Pdgfra‐positive progenitors, FOP mice exhibited aggressive, juvenile‐onset spontaneous HO that was fully penetrant, characterized by HO at multiple anatomical locations, and survival to a median age of 39 days.( 13 ) Here, we addressed whether ACVR1 overexpression reduces the occurrence and severity of spontaneous disease. In the present study, 20 of 23 Acvr1 tnR206H/+ ;Pdgfra‐Cre mice imaged by micro‐computed tomography (μCT) between 33 and 90 days of age showed heterotopic bone growth (Fig. 3A ). Affected mice typically exhibited widespread HO, with most mice having lesions at both axial and appendicular anatomical locations, as previously described.( 13 ) Strikingly, all Acvr1 tnR206H/+ ;Pdgfra‐Cre mice carrying the R26 nACVR1 allele survived to adulthood and appeared healthy, with no cases of premature death (89 mice; up to 15 months old at the time of death). Twelve mice between 5 weeks and 15 months of age were imaged by μCT. No skeletal malformations were detected in 8 mice (Fig. 3B ), whereas 3 mice exhibited a small aberrant skeletal overgrowth (exostosis) of the distal tibia (imaged at 13, 44, and 64 weeks of age) (Supplemental Fig. S4 F; Table 4). In Acvr1 tnR206H/+ ;Pdgfra‐Cre mice, ACVR1 overexpression reduced the incidence and severity of exostosis (Table 4), which is a common skeletal overgrowth phenotype in patients with FOP.( 3 ) Importantly, however, we have not observed exostosis among non‐FOP mice overexpressing ACVR1.
Table 4.
Effect of ACVR1 Overexpression on Spontaneous HO in the Pdgfra‐Cre FOP Model
| Phenotype | Control | Acvr1 tnR206H/+ ; Pdgfra‐Cre | Acvr1 tnR206H/+ ; R26 (n)ACVR1/+ ; Pdgfra‐Cre |
|---|---|---|---|
| HO in the tagged ear | 0/8 (0%) | 17/18 (94.4%) | 0/7 (0%) |
| HO in trunk or limbs | 0/11 (0%) | 6/25 (24%) | 0/12 (0%) |
| Exostosis | 0/11 (0%) | 22/25 (88.0%) | 3/12 (25.0%) |
Heterotopic ossification (HO) was defined as ectopic bone that was not attached to endogenous bone. The genotype, R26 (n)ACVR1 , refers to either R26 nACVR1 or germline‐recombined R26 ACVR1 . Control mice lacked Acvr1 tnR206H , the Cre‐deleter allele, or both.
ACVR1 overexpression confers long‐term protection from spontaneous HO in Acvr1 R206H germline mice
We next assessed the degree to which overexpression of ACVR1 mitigates spontaneous HO in mice carrying the germline‐recombined alleles, Acvr1 R206H and R26 ACVR1 . Among the 90 adult mice produced that were heterozygous for both the Acvr1 R206H and R26 ACVR1 alleles (7 weeks old or older), one mouse was found dead at 52 days of age, and imaging revealed fusion between zygomatic and mandibular bones but no other sites of spontaneous bone growth. Twenty‐five Acvr1 R206H/+ ;R26 ACVR1/+ mice were scanned by μCT (Table 5). Strikingly, of 8 mice imaged between 7 and 9 months of age, none exhibited spontaneous HO or other skeletal abnormalities. Three of these mice showed a small degree of heterotopic bone growth associated with the ear tag site, presumably resulting from minor injury caused by ear tagging. Seventeen additional mice in this cohort were imaged at 12 months of age or older. Among this older group, 5 mice showed exostosis of the distal tibia (Supplemental Fig. S4 A–E), and 6 mice had minor HO at the ear tag site (Supplemental Fig. S4 C, D; Table 5). Importantly, these overgrowths were dependent on the presence of the Acvr1 R206H allele, although it is unclear whether these exostoses occurred because of greater expression of Acvr1 R206H or reduced expression of the R26 ACVR1 allele in periosteal cells or other responsible cell types. Comparisons could not be made to Acvr1 R206H/+ mice, as these mice die at P0 (see above), although severe spontaneous HO at least as pronounced as that exhibited by Acvr1 tnR206H/+ ;Pdgfra‐Cre mice (Fig. 3A )( 13 ) would be expected. Collectively, these data demonstrate that ACVR1 overexpression confers long‐term protection from spontaneous HO at all anatomical sites that typify disease manifestations in FOP mouse models and human patients.
Table 5.
Incidence of Abnormal Skeletogenesis in Aged Acvr1 R206H/+ ;R26 ACVR1/+ Mice
| Phenotype | Control | R26 ACVR1/+ | Acvr1 R206H/+ ;R26 ACVR1/+ |
|---|---|---|---|
| HO in the tagged ear | 0/7 (0%) | 0/6 (0%) | 9/25 (36.0%) |
| HO in trunk or limbs | 0/7 (0%) | 0/6 (0%) | 0/25 (0%) |
| Exostosis | 0/7 (0%) | 0/6 (0%) | 5/25 (20.0%) |
Animals were analyzed between 7 and 17 months of age. All 5 Acvr1 R206H/+ ;R26 ACVR1/+ mice that developed exostosis were 13 months of age or older. Heterotopic ossification (HO) was defined as ectopic bone that was not attached to endogenous bone. Note that the comparison group, Acvr1 R206H , was not available, as these mice die perinatally. Control mice lacked both the Acvr1 R206H and R26 ACVR1 alleles.
Overexpression of ACVR1 in mesenchymal progenitors protects FOP mice from injury‐induced heterotopic ossification
Using a Tie2‐Cre transgenic driver( 30 ) to direct recombination of Acvr1 tnR206H to Tie2+ mesenchymal cells, the majority of which have the marker profile of FAPs,( 10 , 21 , 31 ) consistently results in robust HO after pinch‐ or cardiotoxin‐induced muscle injury.( 10 ) To test the consequence of ACVR1 overexpression, adult Acvr1 tnR206H/+ ;Tie2‐Cre mice that either carry or lack the R26 nACVR1 overexpression allele were subjected to cardiotoxin‐induced injury of the tibialis anterior (TA) muscle, and HO and exostosis evaluated by μCT. Seventeen of 20 Acvr1 tnR206H/+ ;Tie2‐Cre FOP mice assessed at approximately 3 weeks post‐injury (20 to 24 days) developed extraskeletal bone, and many of them (n = 11) exhibited exostosis originating from the tibia, often manifested as localized thickening of the tibia near the middle of the tibial shaft (Fig. 4A ; Supplemental Fig. S5 A; Table 6). Notably, HO was undetectable among Acvr1 tnR206H/+ ;Tie2‐Cre mice that carried either the R26 nACVR1 or R26 ACVR1 alleles, and exostoses were absent (n = 18) (Fig. 4B ; Table 6). Whole‐mount ABAR staining of 14 of these mice revealed that both heterotopic cartilage and bone were absent (data not shown). We previously showed that genetic loss of the remaining wild‐type Acvr1 allele in Acvr1 tnR206H/flox ;Tie2‐Cre mice causes profound exacerbation of injury‐induced HO.( 10 ) ACVR1 overexpression also effectively blocked this extreme manifestation of trauma‐induced HO (Supplemental Fig. S5 B, C).
Fig. 4.
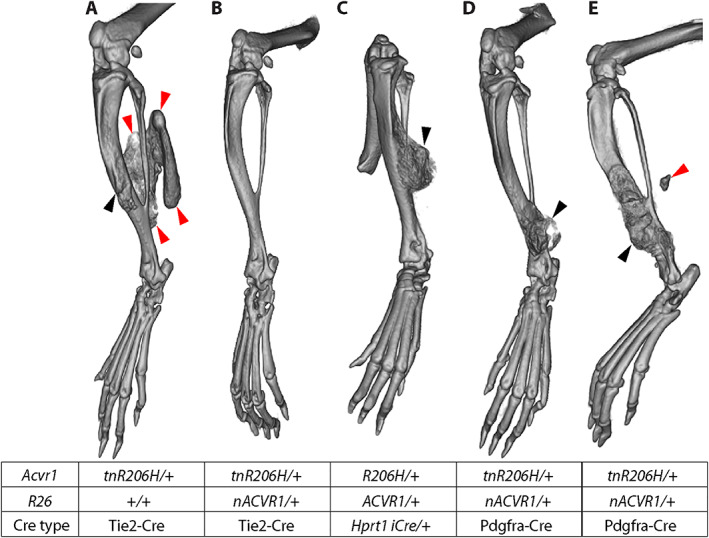
Inhibition of injury‐induced heterotopic ossification (HO) by ACVR1 overexpression. The table shows the genotype of each mouse. (A–E) μCT images captured 21 days after cardiotoxin injection of the transverse abdominal (TA) muscle. Bone growth in (C) represents the most pronounced HO observed in mice of this genotype. In one Acvr1 tnR206H/+ ;R26 nACVR1/+ ;Pdgfra‐Cre mouse (E), a small volume of heterotopic bone was observed in the soft tissue near, but well‐separated from, the tibia. Red arrowheads = HO that likely originated in soft tissue; black arrowheads = exostosis originating from the tibial surface.
Table 6.
ACVR1 Overexpression Protects From Injury‐Induced Heterotopic Ossification (HO)
| Phenotype | Control | Acvr1 tnR206H/+ ; Tie2‐Cre | Acvr1 tnR206H/+ ; R26(n)ACVR1/+ ; Tie2‐Cre | Acvr1 R206H/+ ; R26 ACVR1/+ | Acvr1 tnR206H/+ ; R26 (n)ACVR1/+ ; Pdgfra‐Cre |
|---|---|---|---|---|---|
| HO | 0/17 (0%) | 16/20 (80.0%) | 0/18 (0%) | 0/10 (0%) | 1/6 (16.7%) |
| Exostosis | 0/17 (0%) | 11/20 (55.5%) | 0/18 (0%) | 7/10 (70.0%) | 6/6 (100%) |
The tibialis anterior (TA) muscle was injured with cardiotoxin and assessed at 21 days post‐injury. HO was defined as ectopic bone that was not attached to endogenous bone. The genotype, R26 (n)ACVR1 , refers to either R26 nACVR1 or germline‐recombined R26 ACVR1 . The Acvr1 tnR206H/+ ;Pdgfra‐Cre comparison group was not included because of their poor health and propensity to develop spontaneous skeletal lesions, which complicates assessment of injury‐induced exostosis. The Acvr1 R206H/+ comparison group was not available, as these mice die perinatally. Control mice lacked Acvr1 tnR206H , the Cre‐deleter allele, or both.
We typically do not use adult Acvr1 tnR206H/+ ;Pdgfra‐Cre mice as a model of injury‐induced HO, both because aggressive spontaneous HO that begins by 4 weeks of age( 13 ) complicates analyses and because obtaining healthy adult mice in sufficient numbers is difficult because of their poor fitness and survival(12) (present study). Of note, however, only 1 of 6 adult Acvr1 tnR206H/+ ;Pdgfra‐Cre mice carrying the R26 nACVR1 allele developed extraskeletal bone after cardiotoxin‐induced injury of the transverse abdominal (TA) muscle (Fig. 4E ; Table 6), and this heterotopic bone was minor compared with the typical HO response of juvenile Acvr1 tnR206H/+ ;Pdgfra‐Cre mice (data not shown) and adult Acvr1 tnR206H/+ ;Tie2‐Cre mice (Fig. 4A ). However, exostoses at the mid to distal tibia were observed in all Acvr1 tnR206H/+ ;Pdgfra‐Cre;R26 nACVR1/+ mice after injury of the TA muscle (n = 6) (Fig. 4D, E ; Table 6). The degree to which the R26 nACVR1 allele reduced the severity of the exostosis phenotype could not be assessed because of poor survival of the Acvr1 tnR206H/+ ;Pdgfra‐Cre comparison group and their propensity to develop spontaneous skeletal lesions, which complicates assessment of injury‐induced exostosis. Remarkably, even germline‐recombined Acvr1 R206H/+ ;R26 ACVR1/+ mice did not develop extraskeletal bone after cardiotoxin‐induced injury of the TA muscle (n = 10). The only evident abnormality, which ranged in severity but was often pronounced, was localized outgrowth of endogenous bone in most mice (7 of 10 mice), sometimes extending from the tibia and invading soft tissues (Fig. 4C ).
Quantification of mRNA from the R26 ACVR1 and endogenous Acvr1 alleles
The functionality of the R26 ACVR1 allele was shown by its ability to substitute for the endogenous Acvr1 gene and to rescue perinatal lethality of Acvr1 R206H/+ mice. To quantify expression from the R26 ACVR1 allele, mRNA was isolated from adherent unfractionated cells derived from hindlimb muscles and associated soft tissues of adult Acvr1 R206H/+ ;R26 ACVR1/+ mice (these include FAPs, as well as other uncharacterized mesenchymal cell types; unpublished observations) and mRNA quantified by RT‐droplet digital PCR (ddPCR) with primers that distinguish transcripts from the Acvr1 R206H , wild‐type Acvr1, and R26 ACVR1 alleles. Human ACVR1 mRNA was 40‐ to 50‐fold more abundant than endogenous Acvr1 R206H and Acvr1 mRNAs, which were equivalently expressed (Fig. 5A ), and abundance of the endogenous alleles was not affected by the presence of R26 ACVR1 (Fig. 5 A, B). A similar ratio of exogenous to endogenous transcripts was observed in 14.5 dpc Acvr1 R206H/+ ;R26 ACVR1/+ mouse fetuses (Fig. 5C ).
Fig. 5.
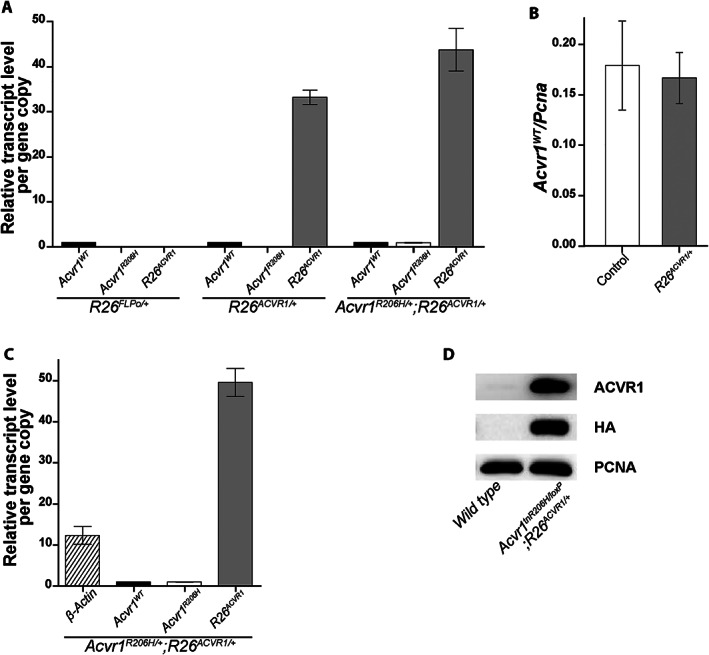
Quantification of exogenous ACVR1 mRNA and protein. (A, C) RT‐ddPCR quantification of ACVR1 mRNA from cultured adherent cells derived from total adult leg muscles and surrounding soft tissues (A) or whole 14.5 dpc fetuses (C). In (A), R26 FLPo/+ mice served as a control for possible nonspecific effects of a gene knockin at the Rosa locus. The transcript level of each gene allele was normalized to that of Acvr1 WT . (B) Effect of R26 ACVR1 on transcriptional activity of the endogenous Acvr1 gene. The transcript level of Acvr1 WT was normalized to Pcna, which was used as an internal control. (D) Western blot of HA‐tagged ACVR1 protein from lysates of cultured adherent cells derived from total adult leg muscles and surrounding soft tissues. Antibody specificity is shown in the right column. Note that Acvr1 tnR206H/loxP cells cannot produce functional full‐length ACVR1 protein; the signal detected with the anti‐ACVR1 antibody represents exogenous ACVR1 only. Endogenous ACVR1 in wild‐type cells is barely detectable under these conditions.
ACVR1 protein showed a strong signal on Western blots of extracts from R26 ACVR1 ‐expressing adherent cells using an anti‐ACVR1 antibody (Fig. 5D ). As human and mouse ACVR1 proteins could not be distinguished with anti‐ACVR1 antibodies, analysis of exogenous ACVR1 expression utilized cells lacking a functional Acvr1 allele (derived from Acvr1 tnR206H/loxP ;R26 ACVR1/+ mice). Whereas the signal with an anti‐ACVR1 antibody reflected a marked increase in protein abundance in R26 ACVR1 ‐expressing cells compared with wild‐type cells, the fold increase (calculated to range from 17‐ to 57‐fold between biological replicates) could not be accurately quantified because of the barely detectable signal in wild‐type cells (Fig. 5D ). Expression of exogenous ACVR1 was confirmed using an anti‐HA antibody (Fig. 5D ), which detects the ACVR1‐HA fusion protein.
Effects of ACVR1 overexpression on activin A‐induced osteogenic signaling
We next tested whether overexpression of wild‐type ACVR1 affects pSMAD1/5/8 osteogenic signaling of Acvr1 R206H ‐expressing cells in response to activin A and BMP6 ligands. Whereas BMP6 can signal through ACVR1 and activate osteogenic signaling of both wild‐type and FOP cells, most studies have shown that osteogenic signaling in response to activin A is dependent on ACVR1(R206H).( 8 , 9 , 10 , 11 , 12 ) Fluorescence‐activated cell sorting was used to isolate FAPs from total hindlimb muscles of control mice (R26 NG/+ ), ACVR1‐overexpressing mice (R26 ACVR1/NG ), and FOP mice that either carried (Acvr1 tnR206H/+ ;R26 ACVR1/NG ;Tie2‐Cre) or lacked (Acvr1 tnR206H/+ ;R26 NG/+ ;Tie2‐Cre) the germline‐transmitted R26 ACVR1 allele, as previously described.( 10 , 21 , 31 ) Cultured FAPs were serum‐starved for 2 hours, treated with different concentrations of activin A or BMP6 for 1 hour, and pSMAD1/5/8 quantified by Western blotting. Consistent with previous results, R206H‐FAPs, but not control FAPs lacking Acvr1 tnR206H , induced SMAD1/5/8 phosphorylation in response to activin A,( 10 ) and the response was dose‐dependent (Fig. 6 A, C). Importantly, ACVR1 overexpression dampened the response to activin A, although the reduction in pSMAD1/5/8 levels was modest at the highest concentration of activin A (4 nM; 100 ng/mL) (Fig. 6 A, C). As expected, BMP6 stimulated concentration‐dependent SMAD1/5/8 phosphorylation of control FAPs (Fig. 6 B, D). Interestingly, however, overexpression of ACVR1 did not sensitize cells to BMP6 at any ligand input concentration; in fact, pSMAD1/5/8 levels were modestly reduced in ACVR1‐overexpressing cells (Fig. 6 B, D). The finding that ACVR1 overexpression does not confer hyperresponsiveness to BMP6 suggests the limited availability of one or more positively acting signaling components, such as obligatory type II receptors, or positively acting effectors downstream of receptor engagement.
Fig. 6.
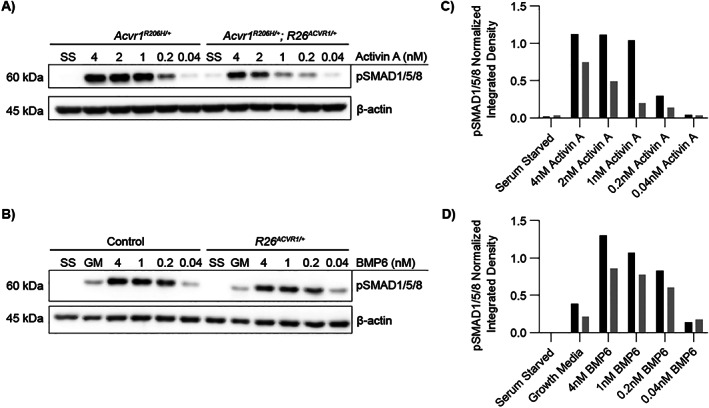
Effects of ACVR1 overexpression on activin A‐ and BMP6‐induced SMAD1/5/8 phosphorylation. (A–D) Representative Western blots and corresponding quantification of pSMAD1/5/8 from lysates of fibro/adipogenic progenitors (FAPs) of the indicated genotypes. After serum starvation for 2 hours, cells were incubated with the indicated concentrations of activin A (A) or BMP6 (B) for 1 hour before cell lysis. β‐actin was used as an internal control to normalize pSMAD1/5/8 signal intensity. (C) pSMAD1/5/8 quantification of the blot in (A). (D) pSMAD1/5/8 quantification of the blot in (B). In (C) and (D), the integrated densities of pSMAD1/5/8 and β‐actin bands were quantified using ImageJ, and normalized pSMAD1/5/8 values are shown.
ACVR1 overexpression reduces fibroproliferative cell expansion and BMP signaling after muscle injury
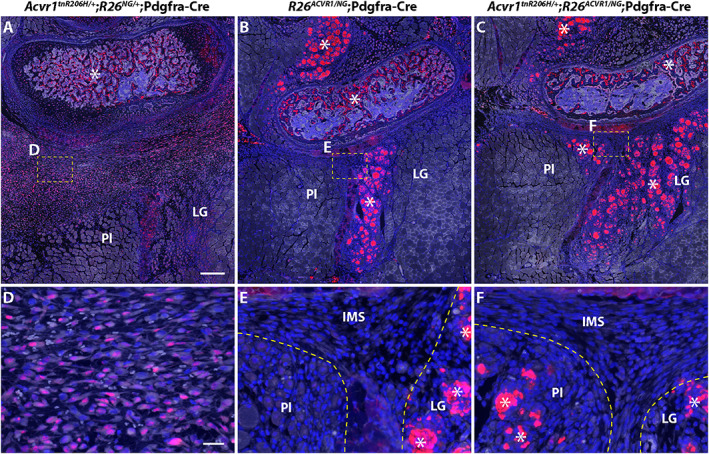
We next addressed whether inhibition of injury‐induced HO by ACVR1 overexpression was associated with reduced osteogenic signaling. For this analysis, the FOP mouse model Acvr1 tnR206H/+ ;R26 NG/+ ;Pdgfra‐Cre was used, as the osteogenic response is typically greater than when recombination is driven by Tie2‐Cre.( 10 , 13 , 21 ) Juvenile mice were used for this analysis to avoid the aforementioned complications of using adult Acvr1 tnR206H/+ ;R26 NG/+ ;Pdgfra‐Cre mice for injury studies. First, the distribution of pSMAD1/5/8‐positive cells was determined 3 days after injection of cardiotoxin into the TA muscle of FOP mice. With the relatively large volume of cardiotoxin used (100 μL), surrounding muscles, including the EDL, peroneus longus, plantaris, and gastrocnemius, often showed varying extents of damage (unpublished observations). Interestingly, unlike induction of HO within the TA by intramuscular injection of BMP2,( 21 ) HO typically developed posterior to, but separated from, the fibula, and on the surface of the tibia, rather than within the TA itself. This was also observed when the Tie2‐Cre driver was used (Fig. 4A and Supplemental Fig. S5 A, B) and in a TA pinch‐injury model (data not shown). As the ultimate anatomical location of HO varied between mice and likely emanated from connective tissues between muscles rather than intramuscularly, surveys of cryosections of the entire lower hindlimb at day 3 post‐injury were undertaken to identify areas of mesenchymal hypercellularity. Irrespective of genotype (control, FOP, and ACVR1‐overexpressing mice), muscle injury resulted in increased mesenchymal cellularity (fibroproliferative response) of intermuscular fascia (typically, the posterior and transverse intermuscular septa) and loss or poor definition of septal boundaries (Fig, 7A, B, D, E and Supplemental Fig. S6). Importantly, however, FOP mice exhibited a much more pronounced fibroproliferative response, which is characteristic of early, preskeletal FOP lesions in mice and humans( 22 , 38 ) (Figs. 7A and 8A and Supplemental Fig. S6 B). Cells that stained intensely for both pSMAD1/5/8 and the cartilage/pre‐cartilage marker SOX9, which is first detected before histologically identifiable cartilage (day 5 to 6 in this model), were abundant in the fibroproliferative regions of FOP mice (Figs. 7 A, D and 8 B, C). The majority of SOX9‐positive cells expressed the R26 NG lineage tracer and were negative for tdTomato (recombined at the Acvr1 tnR206H locus), although some tdTomato‐positive cells also stained for SOX9 (Fig. 8B–E ). Whether these unrecombined SOX9‐positive cells would have contributed to definitive HO at endpoint could not be determined, although our previous data indicated that the great majority of definitive HO cells express Acvr1 R206H .( 10 ) Some mesenchymal cells within regenerating muscles located near fibroproliferative areas (typically, gastrocnemius and plantaris muscles) also expressed pSMAD1/5/8 and SOX9, although apparent protein levels were substantially lower than in mesenchymal cells of presumptive HO regions (Fig. 8 F, G and Supplemental Fig. S7 G–I; data not shown). In both control and FOP mice, centrally localized nuclei within nascent regenerating fibers stained weakly for pSMAD1/5/8 (Fig. S7 A–C) and SOX9 (data not shown). SOX9 expression in wild‐type muscle at this stage is consistent with a previous report of transient SOX9 expression during early myogenic differentiation.( 39 )
Fig. 7.
ACVR1 overexpression greatly reduces the number of pSMAD1/5/8‐positive cells in the hindlimbs of juvenile (33‐day‐old) mice 3 days after cardiotoxin‐induced injury of the transverse abdominal (TA) muscle. Fibroproliferation was observed in the intramuscular septa posterior to the fibula in all genotypes, but was most pronounced in Acvr1 tnR206H/+ ;R26 NG/+ ;Pdgfra‐Cre mice. (A–C) Representative low‐magnification images of cross sections of the lower leg showing the presumptive heterotopic ossification (HO)‐forming region located posterior to the fibula of a Acvr1 tnR206H/+ ;R26 NG/+ ;Pdgfra‐Cre mouse (A), and the corresponding regions of R26 ACVR1/NG ;Pdgfra‐Cre (B) and Acvr1 tnR206H/+ ;R26 ACVR1/NG ;Pdgfra‐Cre (C) mice. (D–F) Higher magnification of fibroproliferative regions in the intermuscular septa corresponding to the boxed areas in (A–C). Note that GFP signal from the recombined R26 NG reporter was destroyed during antigen retrieval for pSMAD1/5/8 detection. Cells with nuclear‐localized pSMAD1/5/8 signal (red) were abundant in the intermuscular septum of Acvr1 tnR206H/+ ; R26 NG/+ ;Pdgfra‐Cre mice (A, D) but not in R26 ACVR1/NG ;Pdgfra‐Cre (B, E) or Acvr1 tnR206H/+ ;R26 ACVR1/NG ;Pdgfra‐Cre (C, F) mice. Autofluorescence through the green channel is shown in white to better reveal tissue structure. Damaged muscles are outlined by hatched lines (E, F). The strong non‐nuclear fluorescent signal, which was present in all genotypes and considered non‐specific staining, was observed in, but not limited to, damaged myofibers and the trabecular bone (examples indicated by asterisks). Sections were counterstained with DAPI (blue).
Fig. 8.
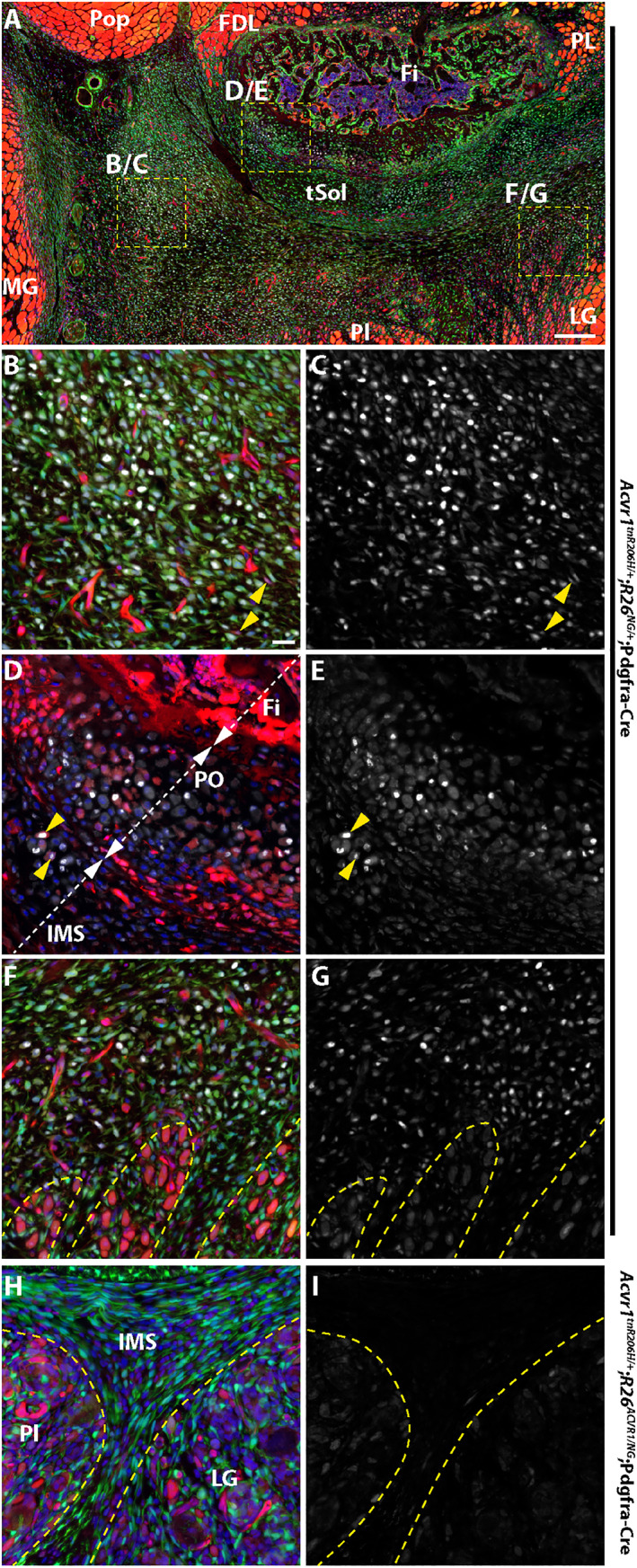
ACVR1 overexpression inhibits induction of SOX9 in fibroproliferative cells in the hindlimbs of juvenile (33‐day‐old) mice after cardiotoxin‐induced injury of the transverse abdominal (TA) muscle. Tissue was collected 3 days post‐injury. Markers are as follows: SOX9 (white, A–I); tdTomato from the unrecombined Acvr1 tnR206H allele (red, A, B, D, F, H); GFP from the recombined R26 NG reporter (green, A, B, F, H); DAPI (blue, A, B, D, F, H). (A–G) Representative images from an Acvr1 tnR206H/+ ;R26 NG/+ ;Pdgfra‐Cre mouse. (A) Low‐magnification image of a cross section of the lower leg showing the presumptive heterotopic ossification (HO)‐forming region located posterior to the fibula. Boxed areas correspond to regions shown at higher magnification in panels B/C, D/E, and F/G. (B, C) Portion of the fibroproliferative region of the transverse intermuscular septum. SOX9‐positive cells are abundant. The great majority of SOX9+ cells are recombined at the Acvr1 tnR206H locus, although a few unrecombined, tdTomato‐positive cells are weakly positive for SOX9 (yellow arrowheads). Most of the red fluorescence in (B) represents unrecombined vascular elements. (D, E) High‐magnification view of a portion of the fibular periosteum (PO) and juxtaposed intermuscular septum (IMS). A small portion of the posterior fibula (Fi) is shown. Some cells of the periosteum are SOX9+. A small number of Acvr1 tnR206H ‐unrecombined cells are positive for SOX9 (examples indicated by yellow arrowheads). (F, G) Image representing the boundary between the SOX9+ fibroproliferative region and the regenerating lateral gastrocnemius. (H, I) Representative image from an Acvr1 tnR206H/+ ;R26 ACVR1/NG ;Pdgfra‐Cre mouse. The intermuscular septum is largely devoid of SOX9+ cells. Damaged muscles are outlined by hatched lines in (F–I). FDL = flexor digitorum longus; Fi = fibula; LG = lateral head of gastrocnemius; MG = medial head of gastrocnemius; Pl = plantaris; Pop = popliteus; tSol = soleus tendon; IMS = intermuscular septum. Images in (B–I) are at the same magnification.
FOP mice that overexpress ACVR1 also exhibited increased cellularity of intermuscular septa after injury of the TA muscle (Figs. 7 C, F and 8H ). Importantly, however, they lacked the pronounced fibroproliferative cell accumulations and strong pSMAD1/5/8 and SOX9 staining that typify early lesions in FOP mice (Figs. 7 C, F and 8H ). Like FOP and control mice, FOP mice carrying the R26 ACVR1 allele exhibited weak pSMAD1/5/8 and SOX9 staining of nascent myonuclei and intramuscular interstitial cells (Fig. S7 C, F, I; data not shown). Collectively, these data show that ACVR1 overexpression greatly diminishes osteogenic signaling and mesenchyme expansion in anatomical locations typically associated with HO but does not affect BMP signaling in nascent muscle fibers in regenerating muscle.
Discussion
The impetus to explore the possible therapeutic applicability of manipulating the stoichiometric balance of wild‐type and mutant ACVR1 receptors was based on our previous finding that loss of the wild‐type ACVR1 receptor in FOP mice profoundly exacerbated trauma‐induced HO, which remained activin A dependent.( 10 ) Here, we developed knockin mice that conditionally overexpress wild‐type ACVR1 and assessed whether overproduction of the wild‐type receptor in FOP mice has the opposite effect, namely, to mitigate the pathogenic effects of ACVR1(R206H). We showed that global overexpression of human ACVR1 is well tolerated and has no obvious negative effects on development, longevity, or reproduction. This strategy was highly effective at mitigating or abrogating most of the deleterious effects of Acvr1 R206H expression, including injury‐induced and spontaneous HO, and the major developmental skeletal abnormalities and perinatal lethality of Acvr1 R206H/+ mice. Additionally, the protective effects of the R26 ACVR1 allele allowed us to generate the first adult mice that carry a germline Acvr1 R206H mutation,( 22 ) providing the most rigorous and physiologically relevant demonstration of the therapeutic efficacy of ACVR1 overexpression to date.
We note that although HO of apparent soft tissue origin was effectively inhibited in all FOP models, not all manifestations of abnormal bone growth were eliminated by ACVR1 overexpression. In particular, exostosis associated with the tibial surface, which typically resulted in localized thickening of the tibia, was common after cardiotoxin‐induced injury of the TA muscle of Acvr1 tnR206H/+ ;R26 nACVR1/+ ;Pdgfra‐Cre and Acvr1 R206H/+ ;R26 ACVR1/+ mice. In some Acvr1 R206H/+ ;R26 ACVR1/+ mice, substantial ectopic bone growth apparently emanating from the tibial surface invaded the surrounding soft tissues. We cannot evaluate the degree to which ACVR1 overexpression mitigated HO in these models because of the lack of comparison groups due to inviability (Acvr1 R206H/+ ) or the complicating effect of aggressive, juvenile‐onset spontaneous HO (Lees‐Shepard and colleagues(13 ) and present study). We note, however, that the R26 ACVR1 allele did effectively suppress exostosis in Acvr1 tnR206H/+ ;Tie‐Cre mice, even when this phenotype was greatly exacerbated by loss of the wild‐type Acvr1 allele. These data demonstrate that the tibial overgrowth phenotype can be blocked by ACVR1 overexpression in certain experimental settings. As the specificity of Tie2‐Cre and Pdgfra‐Cre expression is overlapping but distinct,( 10 , 13 , 21 , 30 , 40 ) differences in ACVR1 and/or Acvr1 R206H expression levels in distinct cell types targeted by these Cre drivers is one possible explanation for apparent differences in efficacy of ACVR1 overexpression in these models.
ACVR1 overexpression probably dampens activin‐dependent BMP signaling by reducing the absolute number or density of ligand‐engaged ACVR1(R206H)‐containing signaling complexes at the surface of skeletogenic progenitors. Activin A and other members of the TGFβ ligand superfamily signal through heterotetrameric complexes that are typically composed of two type I and two type II receptor serine‐threonine kinases. Members of the activin/inhibin and BMP/GDF subgroups share type II receptors (ACVR2A, ACVR2B, and BMPR2 for BMP/GDF ligands and predominately ACVR2A and ACVR2B for activin/inhibin ligands), and association with these type II receptors is required for type I receptor signaling activity,( 41 , 42 ) including that of both ACVR1 and ACVR1(R206H).( 43 , 44 , 45 ) In this context, competition between overexpressed wild‐type ACVR1 and ACVR1(R206H) for limiting quantities of one or more obligate type II receptors is an attractive possibility, as it would directly result in fewer ACVR1(R206H)‐containing tetrameric signaling complexes at the cell surface. Receptor complexes composed of wild‐type ACVR1 should serve as an activin A sink, since activin A treatment of wild‐type cells stimulates very weak( 45 , 46 ) or no detectable( 8 , 9 , 10 , 11 , 12 ) SMAD1/5/8 phosphorylation. Indeed, an elegant recent study provided a link between activin A‐receptor complexes, signaling, and HO; an activin A mutein that cannot form a nonsignaling complex with wild‐type ACVR1 was a more potent inducer of HO than wild‐type activin A in FOP mice.( 12 ) Thus, if activin A is present at subsaturating levels, the increased representation of wild‐type ACVR1 should further reduce pathogenic signaling. Although the molecular details differ, a conceptually related phenomenon was recently described for the type I TGFβ receptor TGFβRI, which competes with the type I receptor ACVRL1 (ALK1) for ACTRIIB (ACVR2B) binding, thereby constraining BMP9 signaling in the growth plate through highly responsive ACVRL1/ACTRIIB complexes.( 47 ) More relatedly, competition for the shared type II receptor BMPRII probably explains the inhibitory effects of transfected wild‐type ACVR1 on BMP2‐ and BMP4‐dependent osteogenic signaling through BMPR1A and BMPR1B in cell culture models.( 7 )
The composition of ACVR1(R206H)‐containing receptor complexes that activate BMP signaling pathways in response to activin A remains uncertain. Given the promiscuity that characterizes interactions within and between the type I and type II receptor families,( 42 , 48 , 49 ) many distinct ACVR1(R206H)‐containing complexes probably exist on the surface of heterozygous FOP cells. In terms of the type I receptor component, these include ACVR1(R206H) homodimers and heteromers in which the type I partner is either wild‐type ACVR1 or other type I receptors. Although definitive data are lacking, several observations point to receptor complexes containing ACVR1(R206H) homodimers as key drivers of HO in FOP. First, the greatly exacerbated HO phenotype of FOP mice lacking the wild‐type Acvr1 allele in skeletogenic progenitors( 10 ) suggests that complexes composed of ACVR1(R206H) homodimers exhibit increased or dysregulated BMP signaling activity compared with their ACVR1/ACVR1(R206H) heteromeric counterparts. Second, ACVR1(R206H) does not require typical type I receptor partners to activate pSMAD1/5/8. In zebrafish embryos, for example, Acvr1/Bmpr1 heteromeric complexes are required to transduce BMP signals in dorsoventral patterning,( 50 , 51 ) whereas Bmpr1 is not required for signaling via exogenously provided ACVR1(R206H) in this model.( 52 ) Similarly, ACVR1B/C is required to induce SMAD1/5 phosphorylation by activin A in wild‐type HEK293T cells, but these type I receptors are dispensable in Acvr1 R206H/R206H cells.( 45 ) Third, although not demonstrated to be solely attributable to ACVR1(R206H) homomeric complexes, Acvr1 R206H/R206H HEK293T cells exhibited a modestly extended duration of receptor activation in response to activin A.( 45 ) Finally, in contradistinction to wild‐type cells, pSMAD1/5 activation driven by activin A‐dependent receptor clustering did not require ACVR2A/B kinase activity in Acvr1 R206H/R206H cells.( 45 ) Regarding the present study, probabilistic estimates predict that overexpression of wild‐type ACVR1 in FOP mice would most dramatically reduce representation of ACVR1(R206H) homodimeric complexes, under the reasonable assumption that wild‐type and mutant ACVR1 receptors have similar binding affinities for receptor partners. From these observations, we hypothesize that tetrameric complexes containing ACVR1(R206H) homodimers represent the main drivers of pathological bone formation in FOP. By extension, it is reasonable to propose that ACVR1 overexpression inhibits HO in FOP mice both by competing for essential signaling components and by titrating ACVR1(R206H) into inactive or less active heteromeric receptor complexes.
Given the essential roles of the TGFβ superfamily in mesoderm formation, skeletogenesis, and many other developmental and physiological processes,( 53 , 54 ) one might predict that overexpression of ACVR1 at levels reported here would have deleterious effects, which could result from any of several gain‐of‐function or dominant‐negative mechanisms. Examples include hypersensitizing cells to BMP ligands that signal through ACVR1,( 41 , 55 ) reducing SMAD2/3 signaling by sequestration of activins into inactive complexes,( 8 , 12 ) or dampening signaling by other TGFβ family ligands and type I receptors by reducing availability of shared type I or type II receptors.( 7 , 41 , 42 , 50 , 51 ) Interestingly, however, overexpression of the Acvr1 orthologs in the zebrafish early embryo( 56 , 57 ) or chicken limb bud( 58 ) did not perturb development or limb skeletogenesis, respectively. Consistent with these observations, we showed that global overexpression of human ACVR1 ( 24 , 34 , 35 ) was well tolerated, having no obvious negative developmental effects. In addition, adult mice that globally overexpress ACVR1 did not develop HO in response to muscle injury. This lack of responsiveness was not due to competition between activin A and BMPs for binding to ACVR1‐containing receptor complexes,( 8 , 11 , 12 ) as globally recombined R26 ACVR1/+ mice were refractile to injury‐induced HO even under conditions of antibody‐mediated inhibition of activin A (unpublished observations). These data could indicate that BMPs that signal through ACVR1, such as BMP5, BMP6, and BMP7,( 41 , 59 , 60 ) are not available at sufficient levels in skeletal muscles and associated connective tissues to elicit an osteogenic response from presumptively hyperresponsive cells. Alternatively, ACVR1 overexpression may not hypersensitize skeletogenic progenitors to BMP ligands because of insufficient quantities of one or more signal transduction components required for ACVR1 to signal, including obligatory type II receptors. The latter possibility is consistent with Western blot analyses, which showed that ACVR1‐overexpressing FAPs did not exhibit enhanced SMAD1/5/8 phosphorylation at any BMP6 input concentration tested. To the contrary, pSMAD1/5/8 levels were modestly reduced in ACVR1‐overexpressing cells.
To date, three pharmacological treatment modalities have shown promise in preclinical models of FOP, all of which involve systemic inhibition of signaling pathways involved in diverse cellular processes and tissue homeostasis.( 61 ) These include anti‐activin A antibodies to block productive ligand‐receptor interactions, small molecule inhibitors of ACVR1 kinase activity, and inhibition of cartilage differentiation by retinoic acid receptor gamma (RARγ) agonists.( 41 , 61 ) Although the RARγ agonist palovarotene was recently approved by Canada Health for adults and children 8 years old (females) or 10 years old (males) and older, a palovarotene clinical trial (Ipsen: NCT03312634) was earlier placed on hold for children under the age of 14 because of incidents of premature growth plate closure, a consequence consistent with the known toxic effects of palovarotene in FOP mice( 13 , 62 ) (but see Chakkalakal and colleagues( 63 )). Antibody‐mediated inhibition of activin A was highly effective at reducing the incidence of HO in a phase II clinical trial for adult patients with FOP (Regeneron: NCT03188666), although this trial was also placed on hold because of possible adverse effects. Finally, we and others recently showed in preclinical studies that bivalent anti‐ACVR1 blocking antibodies have the surprising effect of severely exacerbating HO in Acvr1 R206H FOP mouse models.( 64 , 65 ) Given this challenging therapeutic landscape, assessing potential new treatments for FOP deserves consideration.
Although findings thus far demonstrate that ACVR1 overexpression in FOP and control mice is well tolerated, additional preclinical safety and efficacy studies are required to evaluate whether ACVR1 overexpression should be considered a possible therapeutic modality. One primary challenge will be development of efficient delivery systems to increase ACVR1 levels in relevant cell types. We note, however, that specific targeting of FAPs and other skeletal progenitors may not be necessary, considering the lack of observed deleterious effects of widespread overexpression of ACVR1 shown here. An additional essential requirement will be to establish delivery methods that minimally activate the host immune system, given the well‐known connection between tissue inflammation as both a trigger and manifestation of flare‐ups in patients with FOP.( 66 , 67 , 68 )
Disclosures
All authors state that they have no conflicts of interest.
Author contributions
Masakazu Yamamoto: Conceptualization; formal analysis; investigation; methodology; validation; visualization; writing – review and editing. Sean J Stoessel: Conceptualization; formal analysis; investigation; methodology; validation; visualization; writing – review and editing. Shoko Yamamoto: Investigation; visualization. David J Goldhamer: Conceptualization; formal analysis; funding acquisition; investigation; methodology; project administration; supervision; writing – original draft; writing – review and editing.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/jbmr.4617.
Supporting information
Appendix S1. Supplemental Information
Acknowledgments
We thank Dr Vesa Kaartinen for Acvr1 knockout mice; Acceleron Pharma for the anti‐activin A antibody; Dr Wu He, director of the Flow Cytometry Facility, for technical assistance with FACS; and members of the Goldhamer Lab for helpful discussions throughout the course of this work. This work was funded by NIH grants R01AR072052 and R21AR074584 to DJG.
One Sentence Summary: Overexpression of wild‐type ACVR1 inhibits heterotopic ossification in FOP mice.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Bossche LV, Vanderstraeten G. Heterotopic ossification: a review. J Rehabil Med. 2005;37(3):129‐136. [DOI] [PubMed] [Google Scholar]
- 2. Kaplan FS, Glaser DL, Hebela N, Shore EM. Heterotopic ossification. J Am Acad Orthop Surg. 2004;12(2):116‐125. [DOI] [PubMed] [Google Scholar]
- 3. Morales‐Piga A, Bachiller‐Corral J, González‐Herranz P, et al. Osteochondromas in fibrodysplasia ossificans progressiva: a widespread trait with a streaking but overlooked appearance when arising at femoral bone end. Rheumatol Int. 2015;35(10):1759–67. [DOI] [PubMed] [Google Scholar]
- 4. Shore EM, Xu M, Feldman GJ, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38(5):525–7. [DOI] [PubMed] [Google Scholar]
- 5. Fukuda T, Kohda M, Kanomata K, et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J Biol Chem. 2009;284(11):7149–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Culbert AL, Chakkalakal SA, Theosmy EG, Brennan TA, Kaplan FS, Shore EM. Alk2 regulates early chondrogenic fate in fibrodysplasia ossificans progressiva heterotopic endochondral ossification. Stem Cells. 2014;32(5):1289‐1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hildebrand L, Stange K, Deichsel A, Gossen M, Seemann P. The Fibrodysplasia Ossificans Progressiva (FOP) mutation p.R206H in ACVR1 confers an altered ligand response. Cell Signal. 2017;29:23‐30. [DOI] [PubMed] [Google Scholar]
- 8. Hatsell SJ, Idone V, Wolken DMA, et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med. 2015; 7(303):303ra137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hino K, Ikeya M, Horigome K, et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc Natl Acad Sci USA. 2015;112(50):15438–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lees‐Shepard JB, Yamamoto M, Biswas AA, et al. Activin‐dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat Commun. 2018;9(1):471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olsen OE, Wader KF, Hella H, et al. Activin A inhibits BMP‐signaling by binding ACVR2A and ACVR2B. Cell Commun Signaling. 2015;13(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aykul S, Corpina RA, Goebel EJ, et al. Activin a forms a non‐signaling complex with ACVR1 and type II Activin/BMP receptors via its finger 2 tip loop. eLife. 2020;9:e54582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lees‐Shepard JB, Nicholas S‐AE, Stoessel SJ, et al. Palovarotene reduces heterotopic ossification in juvenile FOP mice but exhibits pronounced skeletal toxicity. eLife. 2018;18;7:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Upadhyay J, Xie L, Huang L, et al. The expansion of heterotopic bone in Fibrodysplasia Ossificans Progressiva is Activin A‐dependent. J Bone Miner Res. 2017;38(5):525. [DOI] [PubMed] [Google Scholar]
- 15. Kaplan FS, Pignolo RJ, Shore EM. Granting immunity to FOP and catching heterotopic ossification in the act. Semin Cell Dev Biol. 2016;49:30‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Joe AWB, Yi L, Natarajan A, et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol. 2010;12(2):153–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol. 2010;12(2):143‐152. [DOI] [PubMed] [Google Scholar]
- 18. Dey D, Bagarova J, Hatsell SJ, et al. Two tissue‐resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci Transl Med. 2016;8(366):366ra163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lees‐Shepard JB, Goldhamer DJ. Stem cells and heterotopic ossification: lessons from animal models. Bone. 2018;109:178‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Uezumi A, Fukada S, Yamamoto N, et al. Identification and characterization of PDGFR|[alpha]||[plus]| mesenchymal progenitors in human skeletal muscle. Cell Death Dis. 2014;5(4):e1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wosczyna MN, Biswas AA, Cogswell CA, Goldhamer DJ. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP‐dependent osteogenic activity and mediate heterotopic ossification. J Bone Miner Res. 2012;27(5):1004‐1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chakkalakal SA, Zhang D, Culbert AL, et al. An Acvr1 R206H knock‐in mouse has fibrodysplasia ossificans progressiva. J Bone Miner Res. 2012;27(8):1746–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamamoto M, Shook NA, Kanisicak O, et al. A multifunctional reporter mouse line for Cre‐ and FLP‐dependent lineage analysis. Genesis. 2009;47(2):107–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kaartinen V, Nagy A. Removal of the floxed neo gene from a conditional knockout allele by the adenoviral Cre recombinase in vivo. Genesis. 2001;31(3):126‐129. [DOI] [PubMed] [Google Scholar]
- 25. Kaartinen V, Dudas M, Nagy A, Sridurongrit S, Lu MM, Epstein JA. Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development. 2004;131(14):3481‐3490. [DOI] [PubMed] [Google Scholar]
- 26. Yamamoto M, Legendre NP, Biswas AA, et al. Loss of MyoD and Myf5 in skeletal muscle stem cells results in altered myogenic programming and failed regeneration. Stem Cell Rep. 2018;10(3):956–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Inouye M. Differential staining of cartilage and bone in fetal mouse skeleton by alcian blue and alizarin red S. Off J Congenit Anom Res Assoc Jpn. 1976;16(3):171‐173. [Google Scholar]
- 28. Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70‐71. [DOI] [PubMed] [Google Scholar]
- 29. Niwa H, Yamamura K, Miyazaki J. Efficient selection for high‐expression transfectants with a novel eukaryotic vector. Gene. 1991;108(2):193‐199. [DOI] [PubMed] [Google Scholar]
- 30. Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2‐Cre transgenic mice: a new model for endothelial cell‐lineage analysis in vivo. Dev Biol. 2001;230(2):230‐242. [DOI] [PubMed] [Google Scholar]
- 31. Biswas AA, Goldhamer DJ. FACS fractionation and differentiation of skeletal‐muscle resident multipotent Tie2+ progenitors. In: Kyba M, ed. Skeletal Muscle Regeneration in the Mouse. Methods in Molecular Biology, Vol 1460. New York, NY: Springer; 2016: pp. 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Korecki AJ, Hickmott JW, Lam SL, et al. Twenty‐seven tamoxifen‐inducible iCre‐driver mouse strains for eye and brain; including seventeen carrying a new inducible‐first constitutive‐ready allele. Genetics. 2019;211(4):1155–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Komatsu Y, Scott G, Nagy A, Kaartinen V, Mishina Y. BMP type I receptor ALK2 is essential for proper patterning at late gastrulation during mouse embryogenesis. Dev Dyn. 2007;236(2):512‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gu Z, Reynolds EM, Song J, et al. The type I serine/threonine kinase receptor ActRIA (ALK2) is required for gastrulation of the mouse embryo. Development. 1999;126(11):2551–61. [DOI] [PubMed] [Google Scholar]
- 35. Mishina Y, Crombie R, Bradley A, Behringer RR. Multiple roles for Activin‐like Kinase‐2 signaling during mouse embryogenesis. Dev Biol. 1999;213(2):314‐326. [DOI] [PubMed] [Google Scholar]
- 36. Tang S‐HE, Silva FJ, Tsark WMK, Mann JR. A Cre/loxP‐deleter transgenic line in mouse strain 129S1/SvImJ. Genesis. 2002;32(3):199‐202. [DOI] [PubMed] [Google Scholar]
- 37. Pignolo RJ, Bedford‐Gay C, Liljesthröm M, et al. The natural history of flare‐ups in Fibrodysplasia Ossificans Progressiva (FOP): a comprehensive global assessment. J Bone Miner Res. 2016;31(3):650–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Suda RK, Billings PC, Egan KP, et al. Circulating osteogenic precursor cells in heterotopic bone formation. Stem Cells. 2009;27(9):2209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hernández‐Hernández JM, Delgado‐Olguín P, Aguillón‐Huerta V, Furlan‐Magaril M, Recillas‐Targa F, Coral‐Vázquez RM. Sox9 represses alpha‐sarcoglycan gene expression in early myogenic differentiation. J Mol Biol. 2009;394(1):1‐14. [DOI] [PubMed] [Google Scholar]
- 40. Roesch K, Jadhav AP, Trimarchi JM, et al. The transcriptome of retinal Müller glial cells. J Comp Neurol. 2008. 509(2):225–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sanchez‐Duffhues G, Williams E, Goumans M‐J, Heldin C‐H, Ten Dijke P. Bone morphogenetic protein receptors: structure, function and targeting by selective small molecule kinase inhibitors. Bone. 2020;138:115472. [DOI] [PubMed] [Google Scholar]
- 42. Yadin D, Knaus P, Mueller TD. Structural insights into BMP receptors: specificity, activation and inhibition. Cytokine Growth Factor Rev. 2016;27:13‐34. [DOI] [PubMed] [Google Scholar]
- 43. Bagarova J, Vonner AJ, Armstrong KA, et al. Constitutively active ALK2 receptor mutants require type II receptor cooperation. Mol Cell Biol. 2013;33(12):2413–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Le VQ, Wharton KA. Hyperactive BMP signaling induced by ALK2 R206H requires type II receptor function in a drosophila model for classic fibrodysplasia ossificans progressiva. Dev Dyn. 2011;241(1):200‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ramachandran A, Mehić M, Wasim L, et al. Pathogenic ACVR1R206H activation by Activin A‐induced receptor clustering and autophosphorylation. EMBO J. 2021;40(14):e106317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haupt J, Xu M, Shore EM. Variable signaling activity by FOP ACVR1 mutations. Bone. 2018;109:232‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang W, Chun H, Baek J, et al. The TGFβ type I receptor TGFβRI functions as an inhibitor of BMP signaling in cartilage. Proc National Acad Sci. 2019;116(31):15570–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mueller TD, Nickel J. Promiscuity and specificity in BMP receptor activation. FEBS Lett. 2012;586(14):1846‐1859. [DOI] [PubMed] [Google Scholar]
- 49. Nickel J, Mueller TD. Specification of BMP signaling. Cells. 2019;8(12):1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Little SC, Mullins MC. BMP heterodimers assemble hetero‐type I receptor complexes that pattern the DV axis. Nat Cell Biol. 2009;11(5):637‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tajer B, Dutko JA, Little SC, Mullins MC. BMP heterodimers signal via distinct type I receptor class functions. Proc Natl Acad Sci. 2021;118(15):e2017952118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Allen RS, Tajer B, Shore EM, Mullins MC. Fibrodysplasia ossificans progressiva mutant ACVR1 signals by multiple modalities in the developing zebrafish. Elife. 2020;9:e53761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brazil DP, Church RH, Surae S, Godson C, Martin F. BMP signalling: agony and antagony in the family. Trends Cell Biol. 2015;25(5):249‐264. [DOI] [PubMed] [Google Scholar]
- 54. Zinski J, Tajer B, Mullins MC. TGF‐β family signaling in early vertebrate development. Cold Spring Harbor Perspect Biol. 2018;10(6):a033274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Scharpfenecker M, van Dinther M, Liu Z, et al. BMP‐9 signals via ALK1 and inhibits bFGF‐induced endothelial cell proliferation and VEGF‐stimulated angiogenesis. J Cell Sci. 2007;120(6):964–72. [DOI] [PubMed] [Google Scholar]
- 56. Mintzer KA, Lee MA, Runke G, Trout J, Whitman M, Mullins MC. Lost‐a‐fin encodes a type I BMP receptor, Alk8, acting maternally and zygotically in dorsoventral pattern formation. Development. 2001;128(6):859‐869. [DOI] [PubMed] [Google Scholar]
- 57. Bauer H, Lele Z, Rauch GJ, Geisler R, Hammerschmidt M. The type I serine/threonine kinase receptor Alk8/Lost‐a‐fin is required for Bmp2b/7 signal transduction during dorsoventral patterning of the zebrafish embryo. Development. 2001;128(6):849‐858. [DOI] [PubMed] [Google Scholar]
- 58. Haupt J, Deichsel A, Stange K, et al. ACVR1 p.Q207E causes classic fibrodysplasia ossificans progressiva and is functionally distinct from the engineered constitutively active ACVR1 p.Q207D variant. Hum Mol Genet. 2014;23(20):5364–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Saremba S, Nickel J, Seher A, Kotzsch A, Sebald W, Mueller TD. Type I receptor binding of bone morphogenetic protein 6 is dependent on N‐glycosylation of the ligand. FEBS J. 2008;275(1):172‐183. [DOI] [PubMed] [Google Scholar]
- 60. Heinecke K, Seher A, Schmitz W, Mueller TD, Sebald W, Nickel J. Receptor oligomerization and beyond: a case study in bone morphogenetic proteins. BMC Biol. 2009;7(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ventura F, Williams E, Ikeya M, et al. Challenges and opportunities for drug repositioning in fibrodysplasia ossificans progressiva. Biomedicines. 2021;9(2):213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Goldhamer DJ, Lees‐Shepard JB. Response to comment on “Palovarotene reduces heterotopic ossification in juvenile FOP mice but exhibits pronounced skeletal toxicity”. eLife. 2019;30:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chakkalakal SA, Uchibe K, Convente MR, et al. Palovarotene inhibits heterotopic ossification and maintains limb mobility and growth in mice with the human ACVR1(R206H) fibrodysplasia ossificans progressiva (FOP) mutation. J Bone Miner Res. 2016;31(9):1666–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lees‐Shepard JB, Stoessel SJ, Chandler J, et al. An anti‐ACVR1 antibody exacerbates heterotopic ossification by fibro‐adipogenic progenitors in fibrodysplasia ossificans progressiva mice. J Clin Invest. 2022;132(12):e153795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aykul S, Huang L, Wang L, et al. Anti‐ACVR1 antibodies exacerbate heterotopic ossification in fibrodysplasia ossificans progressiva (FOP) by activating FOP‐mutant ACVR1. J Clin Invest. 2022;132(12):e153792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kaplan FS, Shore EM, Gupta R, et al. Immunological features of fibrodysplasia ossificans progressiva and the dysregulated BMP4 pathway. Clin Rev Bone Miner Metab. 2005;3(3–4):189–93. [Google Scholar]
- 67. Convente MR, Wang H, Pignolo RJ, Kaplan FS, Shore EM. The immunological contribution to heterotopic ossification disorders. Curr Osteoporos Rep. 2015;13(2):116‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Barruet E, Morales BM, Cain CJ, et al. NF‐κB/MAPK activation underlies ACVR1‐mediated inflammation in human heterotopic ossification. JCI Insight. 2018;3(22):e122958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supplemental Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.