

Abstract
Chromatin immunoprecipitation followed by sequencing (ChIP-seq) is widely used to identify factor binding to genomic DNA and chromatin modifications. ChIP-seq data analysis is affected by genomic regions that generate ultra-high artifactual signals. To remove these signals from ChIP-seq data, the Encyclopedia of DNA Elements (ENCODE) project developed comprehensive sets of regions defined by low mappability and ultra-high signals called blacklists for human, mouse (Mus musculus), nematode (Caenorhabditis elegans), and fruit fly (Drosophila melanogaster). However, blacklists are not currently available for many model and nonmodel species. Here, we describe an alternative approach for removing false-positive peaks called greenscreen. Greenscreen is easy to implement, requires few input samples, and uses analysis tools frequently employed for ChIP-seq. Greenscreen removes artifactual signals as effectively as blacklists in Arabidopsis thaliana and human ChIP-seq dataset while covering less of the genome and dramatically improves ChIP-seq peak calling and downstream analyses. Greenscreen filtering reveals true factor binding overlap and occupancy changes in different genetic backgrounds or tissues. Because it is effective with as few as two inputs, greenscreen is readily adaptable for use in any species or genome build. Although developed for ChIP-seq, greenscreen also identifies artifactual signals from other genomic datasets including Cleavage Under Targets and Release Using Nuclease. We present an improved ChIP-seq pipeline incorporating greenscreen that detects more true peaks than other methods.
Greenscreen is a user-friendly method to identify and remove artifactual signals from ChIP-seq data from any species.
IN A NUTSHELL.
Background: Chromatin immunoprecipitation followed by sequencing (ChIP-seq) and other genomic approaches reveal transcription factor occupancy at target loci, providing insight into gene activation and repression. These methods rely on amplification of factor-associated and control DNA and amplification artifacts have been identified that obscure detection of biologically meaningful binding events.
Question: We asked whether we can develop a simple, versatile method to remove these artifacts. We then asked how incorporation of this tool into an improved ChIP-seq analysis pipeline affects insight into factor occupancy.
Findings: We were able to remove artifactual signals using a combination of common ChIP-seq analysis tools and control samples in a method we call greenscreen. A greenscreen filter can be generated in any new organism with a single ChIP experiment that has at least two controls (input samples). We developed and tested greenscreen filters for Arabidopsis, rice, and human and found that filtering out peaks that overlap with greenscreen regions is required to detect similarities and differences between biological ChIP replicates, to test for overlap in genome occupancy by different transcription factors and to quantify changes in factor binding in different conditions. When linked to an optimized ChIP-seq pipeline we present, greenscreen furthermore leads to identification of more true peaks. The greenscreen tool thus improves ability to answer biological questions with ChIP-seq and related approaches.
Next steps: We would want to develop, test, and optimize greenscreen filters for other plant species. We would like to know whether greenscreen or an adapted version thereof can filter artifactual signals from other types of genomic datasets that measure facture binding, chromatin accessibility or genome architecture.
Introduction
Chromatin immunoprecipitation followed by sequencing (ChIP-seq) probes the association of a factor or modification with chromatin (Johnson et al., 2007). After factor crosslinking to chromatin and shearing of the genomic DNA, DNA fragments associated with the factor of interest are enriched by immunoprecipitation and sequenced after crosslink reversal (Johnson et al., 2007). ChIP-seq produces both true-positive signals and artifactual signal during the process of sequence enrichment (Kharchenko et al., 2008; Park, 2009; Kidder et al., 2011; Chen et al., 2012; Bailey et al., 2013).
Guidelines for accurate analyses of ChIP-seq data suggest using experimental controls, including input DNA or mock ChIP, to account for areas of the genome with sequencing efficiency biases (Kharchenko et al., 2008; Park, 2009, p. 200; Kidder et al., 2011; Chen et al., 2012; Landt et al., 2012; Bailey et al., 2013). Input control samples are identical to a given experimental sample except that they are not subjected to immunoprecipitation (Johnson et al., 2007). Mock samples are ChIP reactions where either the genetic background lacks the antigen or antiserum is employed that does not bind to the antigen (Kharchenko et al., 2008; Park, 2009; Kidder et al., 2011; Chen et al., 2012; Landt et al., 2012; Bailey et al., 2013). In the absence of sequencing biases, input DNA should appear uniformly distributed across the genome, whereas no peaks are expected for the mock ChIP experiment (Kharchenko et al., 2008; Park, 2009).
However, some regions in the genome give rise to amplified artifactual signals that are not efficiently removed through normalization with experimental controls, which affect experimental analysis (Kharchenko et al., 2008; Kundaje, 2013; Carroll et al., 2014; Amemiya et al., 2019). These ultrahigh signals are present in ChIP, mock, and input samples at various levels. The failure to remove these artifactual signals prevents accurate estimates of sample quality, replicate concordance, and identification of factor binding sites (Kundaje, 2013; Carroll et al., 2014; Amemiya et al., 2019). To identify and mask out these regions from downstream analysis, the Encyclopedia of DNA Elements (ENCODE) project curated a filter called “blacklist” for mouse (Mus musculus), human (Homo sapiens), fruit fly (Drosophila melanogaster), and nematode (Caenorhabditis elegans) (Kundaje, 2013; Carroll et al., 2014; Amemiya et al., 2019).
Ultra-high signals often occur near assembly gaps and in other genomic regions with low copy repeat elements and have high ratios of multi-mapped to unique reads (Carroll et al., 2014; Amemiya et al., 2019). The genomic regions that give rise to artifactual signals are invariant for a given species with regards to developmental stage/tissue sampled, yet the signal strength in these regions can vary between experiments (Kundaje, 2013; Carroll et al., 2014; Amemiya et al., 2019; Wimberley and Heber, 2020). Where these signals arise is also sensitive to the genome build, as new genomic regions prone to artifactual signal may be added and others lost due to the addition or resolution of genome gaps or the inclusion of centromeric regions or additional satellite sequences. Therefore, new blacklists have been generated for successive genome assemblies (Amemiya et al., 2019).
The blacklist filter identifies areas of the genome that have low mappability rates or contain high artifactual signals. UMap software (Karimzadeh et al., 2018) uses genome assembly files to measure a region’s mappability, a metric for how uniquely all predicted read-length fragments map to the genome. In addition, blacklists identify high signal regions in inputs (top 0.1% given quantile-normalization of read depth) (Amemiya et al., 2019). Next, these artifactual signal regions are merged within a certain distance (20 kb for human and 5 kb for Drosophila blacklists) only if the merged region maintains an overall average signal intensity in the top 1% (Amemiya et al., 2019). ENCODE blacklists were generated using several hundred inputs, and it was recommended that users employ these curated blacklist regions to mask out reads that overlap with them before applying ChIP-seq peak-calling software such as MACS2 (Zhang et al., 2008; Amemiya et al., 2019).
However, blacklists are not available for most species. In addition, the blacklist generation pipeline requires tools not frequently used in ChIP-seq analysis and that require considerable amounts of RAM and disc storage (minimum requirements are RAM: 64+ GB; CPU: 24+ cores, 3.4+ GHz/core; https://github.com/Boyle-Lab/Blacklist) (Amemiya et al., 2019). Finally, existing blacklists for H. sapiens, M. musculus, D. melanogaster, and C. elegans employed hundreds of inputs (Carroll et al., 2014; Amemiya et al., 2019). Given that such large input numbers are not available for most other model or nonmodel species, there is a need for a facile tool that enables the identification of artifactual signal regions with few inputs.
To address this need, we developed an alternative approach for removing ultra-high signal peaks from ChIP-seq datasets called greenscreen. We hypothesized that we could identify regions of ultra-high noise from a small number of inputs with a common peak-calling tool, MACS2 (2.2.7.1) (Zhang et al., 2008). We show here that our method is robust with as few as two inputs and performs as well as the blacklist in masking artifactual signals from Arabidopsis thaliana (Arabidopsis) and human ChIP-seq datasets. Because of these attributes, and because it utilizes software that is frequently used for ChIP-seq peak calling, greenscreen can readily be applied to any model and nonmodel species, as we show here with rice (Oryza sativa). In addition to increased versatility and ease of implementation, greenscreen masks less of the genome and fewer genes than blacklists. Greenscreen filtering uncovers true ChIP-seq replicate concordance, true factor binding overlaps, and factor occupancy changes under different conditions. We present a ChIP-seq pipeline that incorporates greenscreen and identifies a larger number of true peaks than other methods.
Results
Development of the greenscreen mask
To design artifactual signal masks, we first focused on Arabidopsis, a model plant for which there is currently no blacklists available. We collected inputs and identified 20 that passed our quality control (see “Materials and methods”). These inputs were derived from different tissues and were generated in different laboratories (Supplemental Table S1). Ultrahigh signal peaks were present in these inputs (Supplemental Figure S1A), as previously observed for human (Kundaje, 2013; Carroll et al., 2014; Amemiya et al., 2019).
First, we generated a blacklist for Arabidopsis as a positive control using UMap (version 1.1.0) (Karimzadeh et al., 2018) based on the TAIR10 A. thaliana genome assembly (Berardini et al., 2015). Employing the 20 inputs in Supplemental Table S1 and the UMap output, we applied the recently published blacklist tool (Supplemental Figure S2; Amemiya et al., 2019). We manually adjusted the merge parameter for regions with artifactual signal to 5 kb as for the Drosophila blacklist, since Arabidopsis and Drosophila have similar genome sizes. The resulting Arabidopsis blacklist masked 2.39% of the genome, including 173 protein-coding genes (Table 1 and Supplemental Tables S2 and S3). As previously described (Amemiya et al., 2019), we removed reads that overlapped with blacklist regions prior to ChIP-seq peak calling with MACS2 (Figure 1A and Supplemental Figure S2).
Table 1.
Comparison of blacklist and greenscreen filter regions
| Arabidopsis greenscreen | Arabidopsis blacklist | Human greenscreen | Human blacklist | |
|---|---|---|---|---|
| Percentage of the genome masked | 0.41 | 2.49 | 0.01 | 7.36 |
| Protein-coding genes masked | 26 | 172 | 20 | 3,390 |
All filters for artifactual signal removal were generated in this study, except for the human blacklist, which was published previously (Amemiya et al., 2019).
Figure 1.

Generating ultrahigh signal blacklist and greenscreen masks for Arabidopsis. A, ChIP-seq analysis workflow (top). Generation of the greenscreen filter (bottom). The steps, from raw fastq collection to peak calling in MACS2 and peak filtering (q-value < 10−10), are identical for ChIP-seq and greenscreen, except that the latter employs inputs. To generate the greenscreen filter, input peaks (MACS2 q-value < 10−10) are concatenated (three red arrows), and those within a set distance (5 kb for Arabidopsis) are merged into one region. Greenscreen regions that contained peaks in fewer than half of the 20 inputs (Supplemental Table S1) were removed from the filter. To apply the greenscreen filter, ChIP-seq peaks are called in MACS2 (q-value < 10−10) using the appropriate controls. Subsequently, peaks that overlap with the greenscreen regions are removed. In contrast, blacklist removes reads from ChIP-seq and control samples before peak calling in MACS2. B, Box-and-whisker/swarmplot of the length of artifactual signal regions [log10(base pairs)] in the blacklist masks (n = 83, mean = 4.1) and the greenscreen mask (n = 36, mean = 3.8). Two sample one-sided t test **P = 1.6e−3. C, Grouped box-and-whisker plot of average normalized read signal in input or ChIP-seq data (Moyroud et al., 2011; Sayou et al., 2016; Collani et al., 2019; Goretti et al., 2020; Romera-Branchat et al., 2020; Zhu et al., 2020; Jin et al., 2021). Peaks that do not overlap with greenscreen or blacklist regions are shown in blue, peaks that overlap with blacklist regions are shown in black, and peaks that overlap with greenscreen regions are shown in green. Bootstrapping (n = 100) of 500 random nonoverlapping regions (500 bp in length). Nonartifactual (Input mean: 10; ChIP-seq mean: 36), blacklist (Input mean: 28; ChIP-seq mean: 39), or greenscreen (Input mean: 139; ChIP-seq mean: 178.3). Kruskal–Wallis H test was performed to identify differences among the three input groups (P = 0.0) or ChIP groups (P = 0.0). Gray bars above the boxplots show one-sided Mann–Whitney U rank test comparisons with Holms multiple test correction. Inputs: nonartifactual signal regions and blacklist (***P = 0.0) or greenscreen (***P = 0.0), greenscreen relative to blacklist (***P = 0.0). ChIP-seq samples: nonartifactual signal regions and blacklist (P = 0.48) or greenscreen (***P = 9.9e−258) and greenscreen relative to blacklist (***P = 4.3e−307). D, Box-and-whisker plot of the frequency of 100 randomly sampled 500-bp regions from nonartifact, blacklist, or greenscreen sites residing within 1 kb of an assembly gap. Nonartifactual (mean = 0.5), blacklist regions (mean = 11.5), and greenscreen regions (mean = 9.4). n = 1,000 trials with replacement were conducted. ANOVA was performed to test for differences among the three groups (P = 0). Welch’s one-sided t test with Holm’s correction relative to nonartifactual regions: blacklist ***P = 0, greenscreen, ***P = 0. Welch’s two-sided t test comparing blacklist and greenscreen ***P = 1.8e−52. B–D Legend: Types of filters applied.
To design the Arabidopsis greenscreen mask, we employed MACS2 (Zhang et al., 2008), a tool routinely used to identify ChIP-seq peaks. Following the same steps commonly used to identify peaks in ChIP-seq experiments, we trimmed and mapped each of the 20 inputs and called input “peaks” with MACS2 using the entire genome as background (Figure 1A). The broad peak setting was applied to call peaks in each of the input samples (Figure 1A). We optimized the threshold of significance for the mean q-value for each base pair in the input peaks such that the resulting greenscreen filter maximizes overlap between a ChIP-seq and an orthogonal ChIP-chip dataset for the same transcription factor (Figure 1A and Supplemental Table S3). As ChIP-chip is not based on next-generation sequencing, it is not subject to ultrahigh artifactual signal peaks (Supplemental Figure S3A). At the same time, we strove to minimize the number of genomic regions and genes masked out by the greenscreen filter (Figure 1A and Supplemental Table S3). Using these criteria, we chose MACS2 q < 10−10 as the cutoff to identify input peaks. MACS2-called regions in each input were concatenated into a single list and, to maximize artifactual signal removal, regions within a certain distance were merged (three red arrows in Figure 1A). Using the same criteria as described above for the q-value cutoff, we selected the merge distance. Merging regions within 5 kb was optimal; it resulted in slightly higher overlap with the orthogonal ChIP-chip datasets than the 2.5 kb merge, which also performed well (Supplemental Table S3). Finally, to minimize over masking, we restricted the greenscreen mask to regions where significant input peaks were called in at least half (i.e. 10) of the input samples (see “Materials and methods” for details; Figure 1A). Our final greenscreen filter masks ∼0.41% of the genome and covers 26 protein-coding genes (Table 1 and Supplemental Table S3). Most of the greenscreen regions (99.9%) overlap with the blacklist (Supplemental Figure S3). Because greenscreen covers less of the genome than blacklist, we apply the greenscreen filter after ChIP-seq peak calling in MACS2. Removing peaks that overlap with greenscreen regions prevents the retention of artifactual ChIP peaks at the outer edges of ultrahigh signal regions (Figure 1A).
As expected, both the blacklist and greenscreen filter regions overlap with ultra-high signal in the inputs and ChIP datasets (Supplemental Figure S1A). Consistent with the higher genome coverage of the blacklist filter, blacklist regions were found to be significantly broader than greenscreen regions (Figure 1B). We then compared the read signal strength across the identified blacklist and greenscreen regions. Given their variable lengths, we iteratively bootstrapped 100 sample populations of 500 nonoverlapping 500 bp regions from the blacklist and greenscreen filters and measured the mean signal for each bootstrap. On average, the inferred read signal was significantly lower in blacklist regions than in greenscreen regions (Figure 1C). Thus, the blacklist may over mask, leading to potential false negatives (Supplemental Figure S1, B–D).
Like blacklist regions developed for human samples, Arabidopsis artifactual signals are frequently found near assembly gaps (Kundaje, 2013; Carroll et al., 2014; Amemiya et al., 2019; Supplemental Figure S1). To determine how frequently blacklist or greenscreen regions are found within 1 kb of an assembly gap, we generated 1,000 bootstrap populations of 100 nonoverlapping 500 bp regions from greenscreen, blacklist, and nonartifactual signal regions. Both blacklist and greenscreen regions were significantly more likely to be located near assembly gaps than random genomic regions that did not overlap with blacklist or greenscreen regions (Figure 1D).
Efficacy of artifactual signal removal by greenscreen
Prior studies have identified metrics suitable for assessing artifactual signal removal from ChIP-seq and control samples. One such metric is the standardized standard deviation (SSD), which measures the variation in signal across the genome normalized over read depth (Planet et al., 2012; Carroll et al., 2014). Since SSD is calculated before peak calling, we computed the SSD with ChIPQC (1.26.0) htSeqTools (Carroll et al., 2014) before and after removing reads that overlap with either greenscreen or blacklist regions. Both filters significantly reduced the input SSD scores to the expected value of ∼1 (Figure 2A). Thus, despite covering less of the genome, the greenscreen mask is as effective as the blacklist at removing regions of strong artifactual signal. We also compared the efficacy of the Arabidopsis blacklist and greenscreen in removing artifactual signals from ChIP-seq replicates by testing for a reduction in the SSD (Planet et al., 2012; Carroll et al., 2014). Applying the greenscreen filter to ChIP-seq replicates caused the SSD values to decrease, with similar efficiency as filtering out reads with the Arabidopsis blacklist (Figure 2B; Zhu et al., 2020).
Figure 2.

Efficacy of artifact removal with greenscreen. A, Box-and-whisker plot/swarmplot of SSD in 20 inputs. All reads (mean = 3.9), reads after applying blacklist (mean = 1.2), and reads after applying greenscreen (mean = 1.2). Kruskal–Wallis H test was performed to test for differences among the three groups: ***P = 2.9e−8. P-values (α = 0.001). One-sided Mann–Whitney U rank test with Holms correction relative to samples without mask: blacklist ***P = 5.8e−07, greenscreen ***P = 5.8e−07, and two-sided Mann–Whitney U rank test with Holms multiple test correction blacklist relative to greenscreen: NS (not significant, P = 0.9) (B) Bar graphs of SSD values of two ChIP-seq replicates (Zhu et al., 2020, p. 2) after blacklist or greenscreen read masking relative to the no mask control. C, SCC curves of two ChIP-seq replicates (GSE141894) (Zhu et al., 2020). Without an artifactual signal mask (blue), a phantom peak is seen at a read length of 75 bp. D, Bar plot of two ChIP-seq replicates (Zhu et al., 2020). RSC values after applying the blacklist or greenscreen read masks relative to the no mask control. Legends indicate filters appplied.
Another commonly used metric to assess the quality of a blacklist is strand cross-correlation (SCC) of all the reads in an experiment (Carroll et al., 2014). While true protein binding sites show strand-specific enrichment toward the 5-prime ends of reads, peaks found in input controls lack this pattern (Kharchenko et al., 2008; Landt et al., 2012). When plotting the SCC at given distances between reads from opposite strands, ChIP-seq experiments have a peak at the fragment length (usually between 200 and 350 bp), while input samples have a peak at the read length (Kharchenko et al., 2008). Most ChIP-seq samples have a so-called “phantom” peak at the read length in addition to the fragment length peak (Landt et al., 2012). When we measured ChIP-seq replicate SCC after masking reads using blacklist or greenscreen, the “phantom” peak at the read length position (75 bp) disappeared (Figure 2C). Again, the Arabidopsis blacklist and greenscreen were equally effective at removing this artifactual signal (Figure 2C). To quantify phantom peak removal, we computed the relative strand correlation (RSC), defined as the SCC at the fragment size divided by the SCC at the read size (Kharchenko et al., 2008; Landt et al., 2012). The lower the read length signal (characteristic of inputs), the greater the RSC of the ChIP-seq experiment. Masking ChIP-seq reads or peaks that overlap with artifactual signals using either blacklist or greenscreen regions resulted in a similar increase in RSC (Figure 2D). The combined data suggest that the greenscreen pipeline removes artifactual ultra-high signals as effectively as the Arabidopsis blacklist.
Effect of greenscreen or blacklist filters on ChIP-seq replicate concordance
Having established greenscreen as an effective tool for ultra-high signal removal based on established metrics (Carroll et al., 2014; Amemiya et al., 2019), we then investigated its effect on the assessment of ChIP-seq replicate reproducibility. Correlations between peak signals are often used to evaluate the quality of biological replicates (Schmitz et al., 2022). However, highly reproducible artifactual signal common to all replicates distorts this metric (Amemiya et al., 2019). Previous studies showed that removing artifactual signal by applying blacklist masks reveals the true correlation structure between ChIP-seq replicates (Amemiya et al., 2019). To test replicate reproducibility before and after masking by blacklist or greenscreen, we analyzed published data from ChIP-seq experiments, where ChIP-seq for the same proteins was conducted in different laboratories. We clustered ChIP-seq replicates and experiments on pairwise Pearson correlation coefficients of read signals within called peaks and assessed how well these samples clustered compared with biological expectation.
In particular, we employed ChIP-seq datasets for the transcription factors FD, TERMINAL FLOWER 1 (TFL1), and LEAFY (LFY) conducted in different laboratories (Moyroud et al., 2011; Sayou et al., 2016; Collani et al., 2019; Goretti et al., 2020; Romera-Branchat et al., 2020; Zhu et al., 2020; Jin et al., 2021). Two of the transcription factors probed are related: TFL1 is a transcriptional co-regulator that is recruited to chromatin by the bZIP transcription factor FD (Zhu et al., 2020), while LFY binds to different genomic locations (Winter et al., 2011). Our expectation is therefore that the TFL1 and FD ChIP-seq replicates will cluster together more often than either does with LFY ChIP-seq replicates (Yk = 2, Figure 3A).
Figure 3.

Clustering of ChIP-seq replicates. A, Biological expectation for clustering of 21 ChIP-seq replicates. Because TFL1 is recruited by FD, we expect all TFL1 and FD samples to cluster into one group and all LFY samples to cluster into a second group (k = 2). Color: ChIP for different factors (LFY, FD, and TFL1) (Moyroud et al., 2011; Sayou et al., 2016; Collani et al., 2019; Goretti et al., 2020; Romera-Branchat et al., 2020; Zhu et al., 2020; Jin et al., 2021); Symbol: lab that conducted the ChIP. ChIP-seq peaks were called by MACS2 using published input controls, or mock controls if input was not available. B–E, Heatmaps of pairwise Pearson correlation coefficients. Samples were sorted using unsupervised hierarchical clustering (left of heatmaps). Legend: low (left) to high (right) correlation. Below: Rand index of clustering success relative to biological expectation. Artifactual signals were either not masked (c(Yk=2, Yk′=2) = 0.51) (B), masked using greenscreen (c(Yk = 2, Yk′ = 2) = 1.00) (C), masked using the blacklist (c(Yk = 2, Yk′ = 2) = 1.00) (D), or masked using random genomic regions length matched to greenscreen regions (c(Yk = 2, Yk′ = 2) = 0.51) (E). F–I, PCA of the top two principal components. The percent of variance explained by each principal component is listed in the x- and y-axis label. PCAs were calculated using signals from the union of all ChIP-seq replicate peak sets. Artifactual signals were either not masked (F), masked using greenscreen (G), masked using a blacklist (H), or masked using random genomic regions (I).
For blacklist, we first removed mapped reads from ChIP-seq replicates that overlapped with blacklist regions. We then called significant peaks in MACS2 using q-value < 10−10 and experiment-matched input controls. When experiment-matched inputs were unavailable, we used experiment-matched mock controls for peak calling in MACS2 instead. Conversely, for greenscreen, we first called significant ChIP-seq peaks in MACS2 (using q-value < 10−10) using experiment-matched inputs (or mock if that was the only control available). Following peak calling, we removed the significant peaks that overlapped with greenscreen regions.
To evaluate the blacklist and greenscreen filter, we calculated pairwise Pearson correlation coefficients on the replicates, performed unsupervised hierarchical clustering, and generated Rand index values (Yk = 2, Yk′ = 2) to measure how similar the identified clusters were to biological expectation (Rand, 1971). Without artifactual signal removal, LFY, TFL1, and FD ChIP-seq samples did not cluster according to biological expectation, yielding a low Rand index of 0.56 (Figure 3B). A similar spurious correlation structure was observed when we computed Pearson correlation coefficients for the reads found in greenscreen regions (Supplemental Figure S4). Hence, the correlation structure observed without filtering is likely due to artifactual signal. Indeed, when we applied either the blacklist read filter or the greenscreen peak mask to the ChIP-seq replicates, the expected correlation structure emerged, with LFY and FD/TFL1 in separate clusters, yielding a Rand index of 1 (Figure 3, C and D). In contrast, random genomic regions of similar length distribution that did not overlap with artifactual signal regions did not improve the pairwise correlation coefficients or the Rand index (Figure 3E).
The relationships between replicates can also be visualized by transforming the peak signals of each experiment from a multi-dimensional to a lower dimensional space using principal component analysis (PCA) to project the two principal components that make up the most variance in each ChIP-seq replicate. PCA conducted without an artifactual signal mask or with a random mask only slightly separated LFY ChIP-seq replicates from the TFL1 and FD experiments in the second principal component (Figure 3F). However, after greenscreen or blacklist masking, the first principal component clearly distinguished the replicates or experiments based on the factor assayed, as expected (Figure 3, G and H). Random masks were indistinguishable from no mask (Figure 3, B and I). Our combined data reveal that greenscreen is as effective as blacklisting in improving analysis of ChIP-seq replicate concordance.
Although ENCODE blacklists were generated using hundreds of inputs (Carroll et al., 2014; Amemiya et al., 2019), in Arabidopsis, 20 inputs sufficed to generate effective masks for the removal of artifactual reads. This prompted us to test the performance of greenscreen filters derived from fewer inputs. We tested 1, 2, 3, 4, 6, or 10 inputs randomly chosen from our 20 inputs (Supplemental Table S1) to build greenscreen masks and applied them to the ChIP-seq replicates. We found that as few as two inputs yielded a robust increase in the Rand index for unsupervised clustering of the LFY, FD, and TFL1 ChIP-seq experiments (Figure 4A). Next, we contrasted the effect of generating a greenscreen mask using three experiment-matched inputs to that derived from three unmatched inputs from Supplemental Table S1. Inputs generated for a given experiment were as effective at removing artifactual signals as the unrelated inputs based on Rand indices (Figure 4B). We conclude that greenscreen can be applied to remove artifactual signals from ChIP-seq datasets in any new organism using a single ChIP-seq experiment with at least two experiment-matched inputs.
Figure 4.
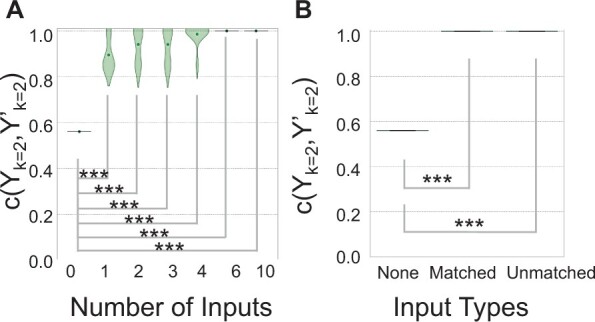
As few as three inputs effectively remove artifactual signals using greenscreen masks. A, Violin plot of Rand index values c(Yk = 2, Yk′ = 2,Figure 3A) for greenscreen filters generated using different numbers of inputs derived from subsamples (n = 0, 1, 2, 3, 4, 6, and 10) of the 20 random inputs (Supplemental Table S1). Central dot: mean. Subsampling was conducted 10 times with replacement. One-sample t test (n = 10) P-values relative to no mask: 1 input ***P = 1.4 − 07; 2 inputs ***P = 8.4e−08; 3 inputs ***P = 3.4e−08; 4 inputs: ***P = 7.3e−9; 6 inputs ***P = 0; 10 inputs ***P = 0. B, Box-and-whisker plot of Rand index values c(Yk = 2, Yk′ = 2) for greenscreen filters derived from five different combinations of three experiment-matched inputs (Zhu et al., 2020; Jin et al., 2021) or three unmatched inputs from Supplemental Table S1. One-sample t test (n = 5) P-values relative to no mask: matched inputs ***P = 0; unmatched inputs ***P = 0.
Greenscreen effectively masks artifactual signals in larger genomes
To test the efficacy of greenscreen in a large and more repetitive genome, we focused on human cell lines. We developed a greenscreen filter from 20 inputs selected randomly from the hundreds of inputs used for the human blacklist (Amemiya et al., 2019). We essentially followed the procedure employed for the Arabidopsis greenscreen filter (inputs using MACS2 [q-value < 10−10], peaks present in ≥10 inputs). However, we merged signal peaks into contiguous regions if they were less than 20 kb apart, as was done for the human blacklist (Amemiya et al., 2019), due to the larger genome size (Supplemental Table S4). We then tested our greenscreen filter versus the published human blacklist, which was generated using 636 human inputs (Amemiya et al., 2019), on 42 ChIP-seq replicates derived from 20 ChIP-seq datasets. The mapped reads from these datasets were from 9 laboratories and 13 different cell lines (Wimberley and Heber, 2020). We performed peak calling and applied the blacklist or greenscreen filters as described above for Arabidopsis.
Based on Pearson correlation analysis, the human ChIP-seq samples showed a correlation structure before masking (Figure 5A). Application of the greenscreen or blacklist filter yielded nearly identical results and revealed a different Pearson correlation structure (Figure 5, B and C). Importantly, the filtering correctly recovered known correlations between factors. For example, RNA binding protein HNRNPK and PCBP2 occupy similar states of ENCODE-annotated genome segmentation that differ from those occupied by RNA binding protein FUS (Xiao et al., 2019). This relationship was also revealed by PCA after either greenscreen or blacklist filtering (Figure 5, D–F).
Figure 5.

Greenscreen is as effective as blacklist in removing human artifactual signals. A–C, Signals within merged peak regions of ChIP-seq replicates from 20 different ChIP experiments conducted in 13 human cell lines by 9 labs (Wimberley and Heber, 2020) were used to calculate and plot Pearson correlation coefficient values. In heatmaps, pairwise correlations between HNRNKP, PCBP2, and FUS are highlighted by white boxes and arrows. D–F, Scatterplot of samples transformed using the top two principal components. The percent of variance explained by each principal component is listed in the x- and y-axis label. Arrows show HNRNKP, PCBP2, and FUS samples. A and D, No filter, (B, E) greenscreen filter, and (C, F) blacklist filter. Color: ChIP for different factors; Letter: lab that conducted the ChIP.
Given the known genome segmentation state preferences of HNRNKP, PCBP2, and FUS (Xiao et al., 2019), we calculated a Rand index for unsupervised hierarchical clustering of Pearson correlation coefficients before masking (c(Yk = 2, Yk′ = 2) = 0.56), after blacklist (c(Yk = 2, Yk′ = 2) = 1.0), or after greenscreen masking (c(Yk = 2, Yk′ = 2) = 1.0). Blacklist and greenscreen filters increased the Rand index in a similar manner using Pearson correlation analysis (Supplemental Figure S5, A–C). Likewise, samples transformed using PCA showed the biological relationship more clearly in the top principal component after blacklist or greenscreen masking than without masking (Supplemental Figure S5, D–F). Thus, greenscreen is as effective as the ENCODE blacklist in removing artifactual signal from ChIP-seq datasets derived from organisms with large, repeat-rich genomes.
We then used this Rand index metric to test the efficacy of human greenscreen filters generated with fewer inputs. Randomly sampling 10 times groups of 10, 6, 4, and 3 different input controls, we found that as few as three inputs were sufficient to generate a greenscreen filter that effectively removed artifactual signal in human ChIP-seq datasets (Rand index c(Yk = 2,Yk′ = 2) = 1.0 with no variance) while only covering 0.01% of the genome and masking 26 transcripts (Supplemental Table S3). Thus, although the published human blacklist masks used over 600 inputs and masked over 227,162 kb and 3,390 transcripts (Amemiya et al., 2019; Table 1 and Supplemental Table S4), greenscreen filters generated with 20 or 3 inputs, and covering less of the genome, were as effective as the blacklist in removing artifactual signal in ChIP-seq datasets from human cell lines.
To further test the efficacy of greenscreen on larger genomes, we developed and applied a greenscreen filter for ChIP-seq experiments from rice using 20 inputs. The rice greenscreen filter covered 0.01% of the genome and 20 protein-coding genes. Masking peaks that overlapped with the rice greenscreen filter enhanced clustering based on biological expectation (Supplemental Figure S6 and Supplemental Tables S5 and S6).
Additional methods have been developed to assess factor binding to genomic locations, including Cleavage Under Targets and Release Using Nuclease (Cut&Run), which targets micrococcal nuclease to a chromatin-bound factor to specifically liberate factor-associated genomic DNA (Skene and Henikoff, 2017; Skene et al., 2018). Cut&Run requires less tissue and is often performed without crosslinking (Skene and Henikoff, 2017; Zheng and Gehring, 2019). We found that Cut&Run experiments also harbor artifactual ultrahigh signal peaks and that these peaks overlap with greenscreen regions (Figure 6; Zheng and Gehring, 2019). The data suggest that greenscreen could also be applied to other types of genomic datasets that probe associations of a factor with chromatin or chromatin modifications.
Figure 6.

Ultrahigh signals in Arabidopsis Cut & Run datasets correlate with greenscreen regions. Top: greenscreen and blacklist regions. Below: Cut&Run bedgraph files for IgG, H3, and H3K27me3 from fixed and non-fixed Arabidopsis tissues (Zheng and Gehring, 2019). Ultrahigh artifactual peaks overlap with greenscreen regions.
A ChIP-seq analysis pipeline that incorporates greenscreen removes type I errors and identifies more true peaks
ChIP-seq peak calling by MACS2 given a q-value cutoff (q-value < 10−10) rejects the null hypothesis that the signal identified is noise because this signal is significantly higher than background. However, for significant peaks that overlap with greenscreen regions (i.e. peaks also found in input), the null hypothesis is correct, resulting in a type I error or false positive peaks. We, therefore, assessed the effects of greenscreen, blacklist, and different commonly used ChIP-seq analysis parameters on the false positive rate. For ChIP-seq analysis, we merged the LFY peak signals from replicates in MACS2 after down sampling to achieve equal genome coverage (see “Materials and methods” for details) and identified significant (MACS2 summit q-value < 10−10 cutoff) peaks using either no control, input, or mock controls. We identified false positive peaks as peaks that overlapped with the combined greenscreen and blacklist regions. ChIP-seq peak calling on an LFY ChIP-seq dataset (Jin et al., 2021) without controls yielded a large number of false positive peaks (193, Figure 7A). In contrast, ChIP-seq peak calling in MACS2 using input controls removed more than half of the false positive peaks, while calling ChIP-seq peaks in MACS2 using mock controls eliminated all false positive peaks in the LFY dataset. Similar results were obtained for an FD ChIP-seq experiment (Zhu et al., 2020) analyzed in the analogous manner (Supplemental Figure S7).
Figure 7.

Optimizing the ChIP-seq peak calling pipeline with filtering. A, Stacked bar chart to assess the impact of calling LFY ChIP-seq peaks in MACS2 using no control (-), input control (I) or mock control (M), duplicate removal (MACS2 keep dup auto [D] or no duplicates retained [-]), and masking (none [-], greenscreen [G], and blacklist [B]). Total peak number (“n” value under the x-axis). LFY ChIP-seq peaks (Jin et al., 2021) that overlap with LFY ChIP-chip peaks (Winter et al., 2011) (bottom bars), and peaks that overlap with the union of blacklist and greenscreen regions (orange bars with peak numbers; top bars). B, Stacked bar chart to assess the impact of increasing the ChIP-seq peak summit q-value threshold for calling LFY ChIP-seq peaks. Bar colors as in (A). C, Horizontal stacked bar charts for ChIP-seq peaks (MACS2 “–keep-dup=auto”) called using input controls. Top: ChIP-seq peaks that overlap with greenscreen (n = 68). Bottom: ChIP-seq peaks do not overlap greenscreen (n = 1677). Peaks were assigned to genes (legend) or could not be assigned as previously described (Jin et al., 2021) . Genes rapidly differentially expressed in response to factor binding (round 2 annotation) (Winter et al., 2011; Jin et al., 2021). D, Horizontal stacked bar charts for LFY ChIP-seq peaks that were lost when using mock instead of input control in MACS2 (n = 693; top) or when removing all duplicates instead of using MACS2 “–keep-dup=auto” (n = 456; bottom). Peak-to-gene assignment and identification of differentially expressed genes were performed as described in (C).
We then examined the effect of using no control, input control, or mock control in MACS2 for significant peak calling on the peak overlap between the above-mentioned LFY ChIP-seq dataset conducted on root explants (Jin et al., 2021) and an orthogonal LFY ChIP-chip dataset from seedlings (Winter et al., 2011), which does not overlap with greenscreen (Supplemental Figure S3A). MACS2 ChIP-seq peak calling without controls (q-value <10−10) resulted in the largest peak overlap between the two LFY binding datasets. About 99% of this peak overlap was retained when we called LFY ChIP-seq peaks in MACS2 peak using input controls. In contrast, MACS2 peak calling using mock controls only retained 77% of the overlap between the two LFY ChIP datasets. Similar results were obtained when we compared two FD ChIP-seq datasets generated in different laboratories and under different plant growth conditions (Collani et al., 2019; Zhu et al., 2020; Supplemental Figures S3B and S7). Thus, peak calling in MACS2 using mock controls likely increases the false negative rate.
Since using input controls for MACS2 peak calling retained high peak overlap between ChIP-seq datasets for the same factor while eliminating some false positive peaks, we tested the effect of duplicate removal, artifactual signal masking, and summit q-value thresholds on input normalized ChIP datasets. It is quite common to remove all duplicates from ChIP-seq data analysis even though a more nuanced approach was proposed, as duplicate reads are known to contribute to true ChIP-seq signal (Chen et al., 2012, p. 201; Bailey et al., 2013; Carroll et al., 2014; Tian et al., 2019). One such approach is the MACS2 default “–keep-dup=auto,” which removes duplicates in excess of expectation based on the effective genome length and sampling depth, and those that do not fit a binomial distribution at a given location (P ≤ 1e−5) (Zhang et al., 2008). We found that removing all duplicates reduced, but did not eliminate, LFY or FD peaks overlapping with artifactual signals (Figure 7A and Supplemental Figure S7A). In addition, removing all duplicate reads reduced the peak overlap between the two orthogonal LFY or two orthogonal FD ChIP-seq datasets (Figure 7A and Supplemental Figure S7A). We conclude that retaining some duplicate reads enhances peak detection. As expected, the greenscreen or blacklist filters removed most peaks that overlapped with universal artifacts, and neither approach adversely affected peak overlap between the two LFY or between the two FD datasets (Figure 7A and Supplemental Figure S7A). A small number of ChIP-seq peaks were in blacklist but not in greenscreen regions, and hence remained after applying the greenscreen filter. However, these regions were characterized by low input signal and are likely evidence of over masking by the blacklist (Supplemental Figures S10 and S11).
Finally, we examined the effect of increasing peak calling stringency on artifactual signal removal by decreasing the minimum summit q-value threshold. This approach did not remove artifactual peaks. This was expected, as artifactual peaks contain ultra-high signals and thus have highly significant summit q-values. Lowering the q-value also reduced the peak overlap between the LFY or the FD datasets (Figure 7B and Supplemental Figure S7B). We conclude that greenscreen or blacklist filters applied to ChIP-seq datasets effectively remove type I errors and uncover true binding peaks.
Properties of peaks that are lost when using mock ChIP reactions as MACS2 controls or when removing all duplicates
In parallel, we examined the properties of the 1,677 LFY and 5,294 FD ChIP-seq peaks called by applying the above ChIP-seq pipeline [replicate down sampling before merge, “–keep-dup=auto” in MACS2, ChIP-seq peak calling using input controls and removing peaks that overlap with the greenscreen filter], as well as the properties of the peaks removed by the greenscreen mask. We assigned peaks to genes as previously described (Zhu et al., 2020; Jin et al., 2021) (see “Materials and methods). Most of the LFY (>70%) and FD (>90%) peaks identified using our ChIP-seq pipeline that includes greenscreen were located near protein-coding or microRNA genes. Furthermore, more than half or one quarter of these genes, respectively, were rapidly differentially expressed after LFY or FD activation (Figure 7C and Supplemental Figure S7C). In contrast, most of the peaks removed by greenscreen in the LFY (>90%) and FD (>80%) ChIP-seq datasets were not located near genes (nor were they differentially expressed). Moreover, the ChIP-seq peaks that overlapped with greenscreen regions had SCC signal peaks at the read length (Supplemental Figure S8), like the input (Landt et al., 2012). The combined data support the conclusion that greenscreen removes false positives from ChIP-seq datasets.
The distinct properties of the true and artifactual ChIP-seq peaks prompted us to examine the peaks lost when using mock rather than input control in MACS2 (Figure 7A and Supplemental Figure S7A), and the peaks lost when removing all duplicates rather than using the MACS2 preset “–keep-dup=auto” (Figure 7A and Supplemental Figure S7A). We found that the peaks removed when using mock controls to call peaks in MACS2, or when excluding all duplicate reads from the analysis, mapped near genes, and showed differential expression at similar levels to true ChIP-seq peaks (Figure 7D and Supplemental Figure S7D). Our data suggest that calling peaks using MACS2 duplicate presets and input controls, followed by the greenscreen filter, increases the number of potential true positive peaks.
To further test this hypothesis, we applied the ChIP-seq pipeline (replicate down sampling before merge, “–keep-dup=auto” in MACS2, input controls, and greenscreen filter) to all ChIP-seq datasets described in this article (Moyroud et al., 2011; Sayou et al., 2016; Collani et al., 2019; Goretti et al., 2020; Romera-Branchat et al., 2020; Zhu et al., 2020; Jin et al., 2021). Relative to these published datasets, our pipeline generally identified at least twice as many peaks (n value in each heatmap row; Figure 8A and Supplemental Table S7). To assess whether these newly identified peaks are true peaks, we performed a pairwise peak overlap analysis. We computed the fraction of peaks in a given experiment (rows in Figure 8) that overlapped with ChIP-seq peaks identified in a second experiment (columns). For pairwise comparisons using the same DNA binding factor, peak overlap was consistently higher with the new ChIP-seq analysis pipeline. Hypergeometric tests revealed equal significance for the peak overlaps of the new pipeline and the published (smaller) peak sets (Supplemental Figure S9). Thus, the new ChIP-seq pipeline, which includes greenscreen masking, identified more true positive peaks than published procedures. Moreover, the newly identified target genes are differentially expressed at similar rates to those identified by the published approaches (Supplemental Figures S10–S12).
Figure 8.

Improved ChIP-seq pipeline calls many more true peaks than other methods. A, Heatmap for the peaks from experiment X (in rows) that overlap with the peaks from experiment Y (in columns) divided by the total number of peaks in experiment X. A, The number of peaks per experiment are listed on the perimeter of the heatmap. ChIP-seq samples of three factors (LFY, FD, and TFL1) conducted in four different laboratories (Moyroud et al., 2011; Sayou et al., 2016; Collani et al., 2019; Goretti et al., 2020; Romera-Branchat et al., 2020; Zhu et al., 2020; Jin et al., 2021) were analyzed. MACS2 controls were matched to the corresponding publications. B, Peaks were either identified using the optimized ChIP-seq pipeline (published previously = false) or as published (published previously = true). Note that published datasets from the Wagner lab were already greenscreen filtered (Zhu et al., 2020; Jin et al., 2021). Scale below the heatmap: percent overlap. Raw numbers for the heatmap are listed in Supplemental Table S4. B and C, Examples of new peaks identified (arrows) by applying the ChIP-seq pipeline (equal genome coverage of each replicate, MACS2 keep-dup = auto and input controls, greenscreen filter) to previously published TFL1, FD (B), and LFY (C) datasets (Moyroud et al., 2011; Sayou et al., 2016; Collani et al., 2019; Goretti et al., 2020; Romera-Branchat et al., 2020; Zhu et al., 2020; Jin et al., 2021). Browser view of ChIP-seq signals (RPK10M; all scales show range 0–220, except for LFY_W_2021 [range 0–110]). Below: significant peaks (summit q < 10−10) marked by horizontal bars, with the saturation proportional to the summit q-value (as for the narrowPeak file format in ENCODE).
In addition, the new ChIP-seq pipeline identified genes in pathways previously linked to FD/TFL1 (Zhu et al., 2020) in several of the datasets analyzed, including the chromatin regulators PICKLE RELATED 1 and BRAHMA, the strigolactone phytohormone response factor SUPPRESSOR of MAX2 LIKE 6, and the meristem identity regulators LATE MERISTEM IDENTITY 2 and FRUITFULL (Figure 8B). For LFY, several new target genes were identified in all datasets, such as the transcription factor PEAR1, which controls vascular development, and the auxin conjugating protein DWARF in LIGHT1, while in other cases, new targets were found in only some of the datasets (chromatin regulator JMJ30) (Staswick et al., 2005; Gan et al., 2014; Miyashima et al., 2019; Figure 8C). Thus, the improved ChIP-seq pipeline removes artifactual signal, calls more true peaks, and identifies additional biologically relevant target genes.
Greenscreen filtering improves the detection of factor binding site overlap and changes in factor occupancy
Artifactual signal removal also improves estimates of factor binding overlap between ChIP-seq datasets. For example, when considering binding peak overlap in the LFY, FD, and TFL1 ChIP-seq datasets described above, LFY and FD apparently occupy similar genomic regions in some experiments without masking or after applying a random mask based on Pearson correlation analysis and PCA (Figure 9, A, D, E, and H). Application of the greenscreen (or the blacklist) filter clearly separated the LFY from the FD/TFL1 bound regions by Pearson correlation and in the first principal component of the PCA (Figure 9, B, C, F, and G). In addition, application of the greenscreen filter allowed us to detect factor binding under different conditions after greenscreen filtering. For example, LFY ChIP-seq data obtained in root explants were clearly separated from data obtained in reproductive tissues (Figure 9F). Thus, removing artifactual signal by greenscreen is critical for deriving biologically relevant information from comparative ChIP-seq binding analyses.
Figure 9.

Artifactual signal masks reveal biologically relevant relationships between Arabidopsis ChIP-seq datasets. Pearson correlation coefficient values for FD, LFY, and TFL1 ChIP experiments (Moyroud et al., 2011; Sayou et al., 2016; Collani et al., 2019; Goretti et al., 2020; Romera-Branchat et al., 2020; Zhu et al., 2020; Jin et al., 2021) using our ChIP-seq pipeline and averaged replicates (see “Materials and methods” for details). For MACS2 peak calling, input controls were used; when none were available, mock controls were employed. Above: legend for color codes and symbols. A–D, Heatmaps after unsupervised hierarchical clustering of the samples based on Pearson correlation coefficients. A and D, Without an artifactual signal mask or using random genomic regions as a mask, ChIP-seq experiments do not cluster based on biological expectation. B and C, After greenscreen peak masking, samples cluster by factor, as quantified by the Rand index. The same result was obtained with the blacklist. E–H, PCA plots of the top two principal components. The percent of variance explained by each of these principal components is listed in the x- and y-axis label, respectively. Artifactual signals were either not masked (E), masked using greenscreen (F), masked using a blacklist (G), or masked using random genomic regions length matched to greenscreen regions (H).
The importance of masking for the biological conclusions derived from ChIP-seq datasets was further underscored by our analysis of the enhancer co-occupancy of different transcription factors. A database for Human and Arabidopsis ChIP-seq and DNA affinity purification sequencing (DAP-seq) datasets called ReMap 2020 includes binding information for 372 Arabidopsis transcriptional regulators (179 ChIP-seq and 330 DAP-seq datasets) (Chèneby et al., 2020). This catalog can be used to identify enhancers bound by many transcription factors, possible stretch or super enhancers (Chèneby et al., 2020). However, since artifactual signal masking was not performed, it was difficult to distinguish genomic regions to which many proteins bind from regions of high artifactual signal. Indeed, we found that 6,664 peaks and 91 “cis-regulatory modules” (CRMs) identified by Remap 2020 overlapped with greenscreen regions (Figure 10). About 17 of these CRMs contained 100 or more different transcription factor binding peaks. Hence, analysis of factor binding and the specific identification of hotspots should include artifactual signal filters.
Figure 10.

Transcription factor binding hotspots overlap with artifactual signals. Genome browser view of Arabidopsis Chr2: 3,237,883–3,511,517. Header shows blacklist or greenscreen regions (top), signals from the 20 inputs used to generate the greenscreen filter (below), and ChIP-seq factor binding hotspots called CRMs defined according to Remap 2020 as merged regions of all nonredundant peaks (Chèneby et al., 2020). Tracks: nonredundant Remap 2020 binding regions (average start and stop sites of overlapping target sites for each given transcription factor) (Chèneby et al., 2020). Different colors represent different transcription factors (Chèneby et al., 2020). Bottom: Araport 11 Gene models (Cheng et al., 2017).
Finally, masking artifactual signals allows changes in factor binding to be properly detected, for example in different genetic backgrounds. In Arabidopsis, PRC2 is recruited to target loci by class I BPC and C1-2iD Zn-finger transcription factors (Xiao et al., 2017). Without greenscreen filtering, depletion of PRC2 recruiting factors appeared to have subtle effects on PRC2 occupancy (Figure 11), perhaps due to shared ultrahigh artifactual signal between the two ChIP-seq datasets. Indeed, application of the greenscreen filter dramatically reduced background noise and revealed the true contribution of the PRC2 recruiting factors to PRC2 occupancy. In summary, employing the greenscreen filter improves the detection of ChIP-seq peak and allows accurate detection of changes in factor occupancy under different conditions.
Figure 11.
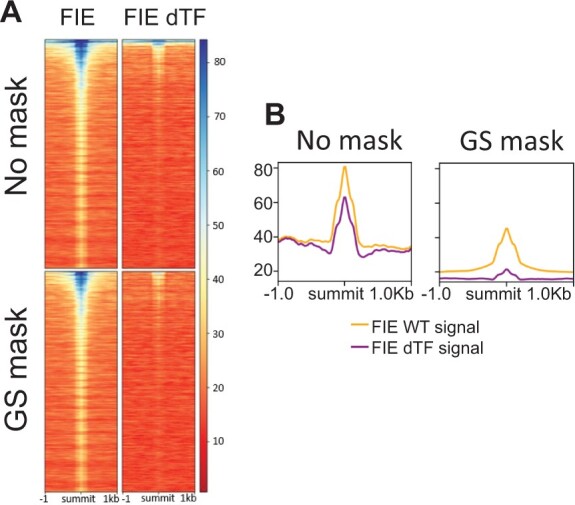
ChIP for the PRC2 component FIE in control plants and in plants that lack PRC2-recruiting transcription factors. A, ChIP-seq signal in a 2-kb region flanking significant FIE peak summits (q-value < 10−10, MACS2) ranked by increasing MACS2 summit q-values without green screening (top heatmaps) and with green screening (bottom heatmaps). ChIP was performed in the wild-type (FIE; left) or in plants that lack PRC2-recruiting transcription factors (FIE dTF; right) (Xiao et al., 2017). Note the ultra-high signal at the top of the heatmaps generated with no filter. Legend on right: signal strength (RP10M). B, Mean signal of significant FIE ChIP-seq (RP10M) ±1 kb of significant FIE peak summits without mask (right) and after applying the greenscreen mask (left). Shown are FIE binding signals in the wild-type (FIE WT) and in plants depleted of PRC2-recruiting transcription factors (FIE dTF).
Discussion
Artifactual signals obscure true correlations between ChIP-seq replicates or experiments, estimates of changes in factor binding in different genetic backgrounds or tissues, and the identification of bona fide multi-factor high-occupancy regions. The underlying mechanism by which ChIP-seq artifacts arise remains unknown; they are likely caused by multiple factors and depend on the quality of the genome annotation (Kundaje, 2013; Carroll et al., 2014; Amemiya et al., 2019). Artifactual signal in ChIP-seq dataset has previously been described in many species (Kharchenko et al., 2008; Park, 2009; Kidder et al., 2011; Chen et al., 2012; Landt et al., 2012; Bailey et al., 2013; Kundaje, 2013; Carroll et al., 2014; Amemiya et al., 2019), and reads are commonly masked from downstream analysis in human, mouse, nematodes, and fruit fly using curated blacklists (Landt et al., 2012; Kundaje, 2013; Carroll et al., 2014; Amemiya et al., 2019). However, in other model and nonmodel organisms, the identification and removal of the artifactual regions has not been standardized. Here, we developed the greenscreen method to identify and filter out the artifactual signal using a small number of inputs with tools commonly used for ChIP-seq analysis.
Like for blacklists, greenscreen parameters should be optimized based on both the size of the genome and the quality of the genome build. Of particular importance is the ultrahigh signal merge parameter. This parameter should be defined empirically by testing greenscreen performance based on Pearson correlation analysis of ChIP-seq data and Rand index values from clustering analyses, maximizing overlap with orthologous datasets while minimizing overlap with unrelated datasets and the percentage of the genome masked (Figure 3 and Supplemental Table S3). We chose a 5-kb merge parameter for Arabidopsis, as it results in a slightly higher overlap with orthogonal ChIP-chip datasets compared with the 2.5 kb merge parameter (Supplemental Table S3). Independently, ENCODE blacklist chose a 5-kb merge parameter for Drosophila, which has a similar genome size as Arabidopsis (Amemiya et al., 2019). For humans, we employed a 20-kb merge, since this merge parameter is used for the human ENCODE blacklist (Amemiya et al., 2019). Finally, for rice, we employed a 10-kb merge parameter to generate our greenscreen mask, as the rice genome is intermediate in size between that of Arabidopsis and human (Jackson, 2016). In contrast, input peak calling (MACS2 < 10−10) and the number of inputs needed to build the greenscreen filter were the same for Arabidopsis and human (Figures 3 and 9 and Supplemental Tables S3 and S4).
By identifying and masking ChIP-seq peaks with the greenscreen filter, we improved enrichment of true-positive peaks and decreased erroneous correlations, revealing true biological signals. The application of greenscreen masking to Arabidopsis ChIP-seq datasets from different laboratories was as effective at removing artifactual signal as the Arabidopsis blacklist based on metrics commonly employed to assess ultrahigh artifactual signal removal (SSD, RSC, and SCC) (Carroll et al., 2014; Wimberley and Heber, 2020). Both filters performed equally well in revealing true ChIP-seq replicate and experiment concordance using Pearson correlation analysis or PCA for both Arabidopsis and human ChIP-seq data. In both Arabidopsis and human ChIP-seq datasets, greenscreen masks a smaller percentage of the genome (0.41% and 0.01%, respectively) than blacklisting. The biggest difference between blacklist and greenscreen is that blacklist software identifies broader regions. Additional experiments are needed to determine whether blacklist software is over- masking the genome. In summary, by applying metrics that assess artifactual signal removal, such as SCC, RSC, and SSD, and by revealing correlation structures between ChIP-seq replicates and experiments that conform with prior data, we showed that greenscreen removes false positive peaks from ChIP-seq datasets as effectively as blacklists.
In nonmodel or new model genomes, few sequenced inputs are generally available. In Arabidopsis, greenscreen filters based on 20 inputs performed as well as the Arabidopsis blacklist we generated. Moreover, a greenscreen filter derived from 20 human ChIP-seq inputs generated a remarkably similar correlation structure for human ChIP-seq replicates after filtering as did the published blacklist, which is based on over 600 inputs (Amemiya et al., 2019). Indeed, based on the Rand index, as few as two to three inputs are effective at artifactual signal removal. The inputs can be from the same experiment as the ChIP-seq datasets: experiment-matched and unmatched inputs performed equally well. Hence, greenscreen can be used to improve ChIP-seq data analysis in any new species using a single ChIP experiment with as few as two matched inputs. An additional advantage of the small number of inputs required is that it is easy to generate a new greenscreen filter if a new reference genome build is released, or under conditions where massive genome re-arrangements occur, such as in cancer cell lines (Ballouz et al., 2019; Ghandi et al., 2019). This flexibility is also useful for partially assembled genomes or polypoid genomes, like those of many crops.
We present a flexible ChIP-seq analysis pipeline that incorporates greenscreen. This pipeline uses replicates with equal genome overage, input controls, and the MACS2 preset “–keep-dup=auto.” We employed down sampling of high-quality replicates to equal genome coverage to prevent the replicate with the highest sequencing depth from dominating the analysis in MACS2. Other approaches have been proposed to address this issue (Yang et al., 2014), and future efforts are needed to improve replicate handling in ChIP-seq analysis. In addition, while mock controls can remove artifactual signal (Xu et al., 2021), they also cause the loss of ChIP-seq peaks found in orthogonal datasets for the same factor. Because of this and their low overall signal and high variability, we conclude that mock ChIPs are not a suitable control for ChIP-seq peak calling in MACS2. However, mock ChIP datasets are important controls because they are very sensitive to potential contamination, another common Type I error in ChIP-seq datasets. Removing all duplicates does not effectively remove artifactual signal and leads to the loss of ChIP-seq peaks that overlap with orthogonal datasets for the same factor, which is in agreement with prior studies showing that duplicate reads contribute to ChIP-seq signal (Chen et al., 2012; Bailey et al., 2013; Carroll et al., 2014). The improved ChIP-seq pipeline, which includes the greenscreen filter, calls more significant peaks compared with other analyses. The additional peaks show strong overlap with other ChIP-seq datasets for the same factor and are linked to functionally important, differentially expressed genes and pathways.
In summary, our ChIP-seq pipeline, which incorporates greenscreen, removes false positive peaks as effectively as benchmarked approaches and displays high sensitivity for true peak detection. Moreover, greenscreen filters are generated with common ChIP-seq analysis tools and using very few inputs. Hence, greenscreen can readily be adapted to any organism or genome, as shown here with rice.
Materials and methods
Identification of artifactual signals in ChIP-seq
Single-end reads from 20 ChIP-seq input controls in A. thaliana were retrieved from different experiments (Supplemental Table S1). FASTQC (version 0.11.5) (Simon, 2012) was used to assess the quality of each sample. Inputs were not considered for downstream analysis if the average reads did not have sequencing qualities above Phred33 score 30. After passing the sequencing quality criteria, inputs were cleaned with Trimmomatic version 0.39 (LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36) (Bolger et al., 2014). If needed, the remaining adaptor sequence was removed using the Trimmomatic ILLUMINACLIP function (2:30:10). Trimmed reads were then mapped with bowtie2 version 2.4.1 (Langmead and Salzberg, 2012, p. 2) to the TAIR10 (Berardini et al., 2015) Arabidopsis genome using default parameters. Reads that did not map, did not generate a primary alignment, did not pass quality checks, did not map to a nuclear chromosome, or had MAPQ < 30 were eliminated from downstream analyses using samtools version 1.7 (htslib version 1.7) (Li et al., 2009) view (-F 772 -q 30).
To ensure that the samples were in fact ChIP-seq input controls, we generated SCC plots using the MACS2 version 2.2.7.1 predict function (Zhang et al., 2008) and ChIPQC 1.26.0 (Carroll et al., 2014) for each input and removed any sample that returned a ChIP-seq experiment signature from a Watson and Crick strand correlation test (Supplemental Figure S13). SCC metrics are typically used for quality control of ChIP-seq to quantify an experiment’s signal-to-noise ratio (Carroll et al., 2014; Wimberley and Heber, 2020). In ChIP-seq, distinct Watson and Crick strand read enrichment occurs on opposite sides of a factor binding site at a distance of at least a DNA fragment length apart. Since input does not include immunoprecipitation of factor-bound DNA sequences, the input should not show enrichment at the fragment size or above on a strand-cross correlation plot (Carroll et al., 2014).
Blacklist generation
Blacklist generation requires both uniquely mappable regions in the genome and mapped input reads (Supplemental Figure S2). Uniquely mappable sites were annotated using UMap (version 1.1.0) (Karimzadeh et al., 2018) (–kmer 8 12) on the TAIR10 A. thaliana genome assembly (Berardini et al., 2015). In parallel, high-quality mapped reads (MAPQ ≥ 30) from our 20 input samples (Supplemental Table S1) were retained for import into the blacklist tool and employed in the greenscreen pipeline (described below). The blacklist tool is hard-coded for blacklist regions in the human genome. To account for the smaller genome size of Arabidopsis, we manually modified the code (blacklist.cpp, line 469) to merge regions within 5 kb rather than 20 kb, as is done for Drosophila (Amemiya et al., 2019), which shares a comparable genome size with Arabidopsis. To apply the blacklist for ChIP-seq analyses, mapped reads overlapping with blacklist regions are removed with the samtools view function.
Greenscreen generation
Utilizing the same mapped input reads that we used to generate the blacklist, we identified peaks from each input sample individually using MACS2 version 2.2.7.1 (Zhang et al., 2008, 2) (—keepdup “auto” —no model –extsize [READ LENGTH] —broad –nolambda -g 101274395). By default, MACS2 identifies significant signals with a dynamic Poisson distribution by capturing the local backgrounds in its lambda parameter (Zhang et al., 2008, p. 2). We set “–nolambda” to ensure that we specifically capture ultra-high signals above the global background. Additionally, bypassing the default MACS2 shifting model, which extends reads based on what MACS2 estimates to be the samples fragment length, reads were extended in the 5′–3′ direction based on each sample’s read length, as determined by ChIPQC 1.26.0 (Carroll et al., 2014). The effective genome size was fixed to 85% of the full Arabidopsis genome size (Chen and Kaufmann, 2017).
To optimize the greenscreen mask, we strove to minimize Type I error (i.e. false positive) ChIP peaks called by MACS2 while also minimizing the percent of the genome and the number of genes masked (Supplemental Table S3). Improvement of ChIP-seq was measured as the enrichment of peak overlap between ChIP-seq and ChIP-chip datasets for the same transcription factor (Supplemental Table S3). In addition, we quantified the result of unsupervised clustering of pairwise Pearson correlations between ChIP-seq replicates for the same factors from different laboratories with that based on biological expectation by calculating Rand index values (see Figure 3). These combined investigations defined optimal greenscreen artifactual peaks as those with an MACS2 q-value < 10−10 and optimal input peak merging with a maximum merge distance of 5 kb. After removing peaks with q-value ≥ 10−10 (column 9 in the broadPeak output file) from each of the 20 inputs, we concatenated all input peak regions. Lastly, we removed those regions that did not have significant artifactual peaks in at least half of the 20 inputs analyzed. To apply the greenscreen for ChIP-seq analyses, ChIP peaks overlapping with greenscreen regions are removed with the bedtools intersect function.
ChIP-seq peak calling with blacklist or greenscreen filters
Raw Arabidopsis ChIP-seq read data were obtained and cleaned using Trimmomatic version 0.39 (LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 ILLUMINACLIP:TruSeq3-SE.fa:2:30:10 MINLEN:36). After trimming, the reads were mapped to the TAIR10 Arabidopsis genome using default parameters in bowtie2 version 2.4.1. Reads were then filtered for mapped primary alignments that passed quality checks and MAPQ ≥ 30 using samtools view (-F 772 -q 30). Blacklist and greenscreen are two different approaches to remove noise from ChIP-seq data. Unlike greenscreen masking, blacklist masking is performed before peak calling. To implement blacklist masks, the reads in ChIP-seq and control samples that overlapped with blacklist regions were removed from downstream analysis using samtools view (-U [MASKED_READS] -o [ARTIFACT_READS] -L [BLACKLIST]). BLACKLIST is a bed file containing the blacklist regions. The two outputs, MASKED_READS and ARTIFACT_READS, contain reads that map outside or inside of blacklist regions, respectively.
To pool controls or ChIP-seq replicates, reads were randomly down-sampled using biostar145820 (Wang et al., 2019, p. 201) (–seed 42 -n [DOWNSAMPLED READ DEPTH]) to match the read-depth of the replicate with the lowest read depth. Downsampled reads from each replicate were input to MACS2. When pooling ChIP-seq replicates, use of high-quality replicates is recommended defined as replicates that have peak signals with Pearson correlation coefficient >0.7 after applying the greenscreen filter.
Next, MACS2 version 2.2.7.1 (Zhang et al., 2008, 2) (-t [CHIP(s)] -c [CONTROL(s)] —keepdup “auto” —nomodel –extsize [fragment_length] -g 101274395) was utilized to perform peak-calling on the ChIP-seq samples. For blacklist, reads that did not overlap with the blacklist mask were used for ChIP-seq peak calling in MACS2. For greenscreen, unfiltered reads were employed. In both cases, peaks were called relative to experiment-matched normalization controls: input when available; otherwise mock control (Zhang et al., 2008, p. 2). Reads were extended within MACS2 software in the 5′–3′ direction based on each sample’s fragment size determined by ChIPQC 1.26.0 (Carroll et al., 2014). To balance removing duplicates generated from PCR amplification versus duplicate reads that originate from independent fragments, it is recommended that instead of eliminating all duplicates, a duplicate threshold per genomic location should be set based on an experiment’s sequencing depth (Chen et al., 2012; Bailey et al., 2013; Carroll et al., 2014; Tian et al., 2019). Therefore, duplicate reads were managed in MACS2 v2.2.7.1 by setting “–keepdups auto.” Peaks with a summit q-value (column 9 in MACS2 narrowPeak output) less than 10−10 were retained (awk “$9>=10”). Additional ChIP-seq quality measures were discussed previously (Schmitz et al., 2022).
To apply the greenscreen after identification of significant ChIP-seq peaks as described above, we masked peaks that overlap with greenscreen regions using bedtools (version 2.26.0) (Quinlan and Hall, 2010) intersect (-wa -v -a [UNMASKED PEAKS] -b [GREENSCREEN]). UNMASKED PEAKS refers to the q-value filtered output of MACS2 in narrowPeak format. GREENSCREEN is a bed file containing the greenscreen regions.
To assign peaks to genes, we used Araport11 gene annotation of the Arabidopsis genome (Cheng et al., 2017, 1). Peaks with intragenic summits were first assigned to the genes to which they were intrinsic. Remaining peaks with summits at least 4-kb upstream of a gene were then assigned to the closest upstream peak. If data were available for rapid (immediate early) gene expression changes after factor binding, we mapped the remaining orphan peaks within 10 kb (upstream or downstream) of significantly differentially expressed genes (round two annotation) (Zhu et al., 2020; Jin et al., 2021).
ChIP-seq Pearson correlation and PCA plots
Pearson correlation and PCA plots assess the signal within regions of interest. All of the ChIP peaks called in each replicate (Figure 3) or pooled samples (Figure 9) were concatenated and merged to a final bed file to be used as regions of interest. Signal files were generated after read extension to the respective sample’s fragment size and normalization over all mapped ChIP-seq reads or over all reads left after blacklist masking. Blacklist masking requires read removal, while greenscreen relies on peak removal.
The normalized signal within each of the selected regions was arranged into a matrix. Pairwise Pearson’s correlation was calculated across the columns, and unsupervised hierarchical clusters (k = 2) were then generated using Ward’s clustering methods (Pearson, 1896; Batagelj, 1988). Heatmaps and dendrograms were plotted to display these results. To quantify how well the unsupervised clusters match our hypothesis of how they should cluster based on the known biology, we calculated the Rand index (Rand, 1971).
Similarly, to visualize the samples using the top two principal components, the signals within each of the sample’s peak regions were also measured using deeptools (3.5.1) multiBigwigSummary (Ramírez et al., 2014). Values for each sample in the top two principal components were plotted with the deeptools (3.5.1) plotPCA function (Ramírez et al., 2014).
Generation of the human greenscreen filter
To identify inputs suitable for greenscreen analysis, we applied ChIPQC to the hg38 mapped inputs used for the human blacklist (Amemiya et al., 2019) and selected those that showed a high cross-coverage score at a shift size equal to the read length (RSC < 3.5, see Supplemental Figure S11). We optimized the MACS2 q-value to maximize overlap between EZH2_R1 and EZH2_R2, minimize the overlap between EZH2_R1/R2 and FUS_R1/R2, and minimize the percentage of the genome covered (Supplemental Table S4). The optimal MACS2 q-value was <10−10. We merged peaks with a distance of less than 20 kb, as was done for the human blacklist (Amemiya et al., 2019), and removed any regions that did not show significant artifactual peaks in at least half of the inputs analyzed.
Generation of the rice greenscreen filter
Single- and paired-end reads from 20 ChIP-seq input controls in O. sativa (cv. Nipponbare) were retrieved from different experiments (Supplemental Table S5). Trimmed reads were mapped to the RGAP version 7.0 rice genome (Kawahara et al., 2013) with bowtie2 version 2.4.1 (Langmead and Salzberg, 2012) using default parameters. Reads that did not map, did not generate a primary alignment, did not pass quality checks, did not map to a nuclear chromosome, or had MAPQ < 30 were eliminated from downstream analyses using samtools version 1.7 (htslib version 1.7) (Li et al., 2009) view (-F 772 -q 30).
As for Arabidopsis and human, ChIPQC 1.26.0 and the MACS2 version 2.2.7.1 predict function (Zhang et al., 2008; Carroll et al., 2014) were applied to the mapped inputs to confirm the input features of the samples. Peaks from each input sample were called using MACS2 version 2.2.7.1 (—keepdup “auto” —no model –extsize [READ LENGTH] —broad –nolambda -g 373128865, or, —keepdup “auto” —no model -f BAMPE —broad –nolambda -g 373128865, for single or paired-end reads, respectively). Peaks with a q-value ≥10−10 (Column 9 in the broadPeak output file) were removed. Taking into account the intermediate size of the rice genome compared with Arabidopsis and human (Jackson 2016), input “peaks” were merged that were less than 10 kb apart. Finally, we removed regions that did not have significant artifactual peaks in at least half (10) of the inputs analyzed.
Statistical analyses
Assuming the central limit theorem (Fischer, 2011; Rouaud, 2013), metrics from a sufficiently large sample size (n > 30) of independent identically distributed values follow a normal distribution. Otherwise, we applied the Shapiro–Wilk test (Shapiro and Wilk, 1965) (α ≤ 0.05) to test whether calculated metrics were normally distributed. If the sample size was sufficient or the values failed to reject the Shapiro–Wilk test, parametric statistical tests were applied. To test if more than two groups’ values originated from a statistically equal population mean, an analysis of variance (ANOVA) (Girden, 1992) (α ≤ 0.001) test was applied if all group values showed a normal distribution. Otherwise, a nonparametric Kruskal–Wallis H test (Kruskal and Wallis, 1952) was performed, and we proceeded with the analysis if the null hypothesis (no difference between the medians) was rejected. A Student’s t test (Student, 1908) was used to compare two groups from normal distributions with equal variance. A Welch t test (Welch, 1947) was applied to compare two groups from normal distributions with unequal variance. One sample t tests were applied if one group value was invariant, and two sample t tests were used for all other comparisons. The Mann–Whitney U rank (Mann and Whitney, 1947) was conducted on paired groups, such as metrics before and after masking artifactual regions, which was not assumed to show a normal distribution. Note that to correct for the fact that the Mann–Whitney U rank compares a discrete statistic against a continuous distribution, a 0.5 continuity correction was applied to the z-score. We applied one-sided statistical tests when we expected a difference in one direction only; otherwise two-sided tests were employed. To account for multiple statistical tests, P-values were adjusted using Holm’s correction (Holm, 1979; Supplemental Data Set S1).
Accession numbers
Sequence data from this article can be found in The Arabidopsis Information Resource (https://www.arabidopsis.org) under the following accession numbers: TFL1 (At5g03840), FD (At4g35900), LFY (At5g61850), and FIE (At3g20740).
Sequence data from this article can be found in the GenBank/EMBL libraries under the following accession numbers: The Arabidopsis ChIP-seq data we analyzed were from the following publication ids: PRJNA132641 (Moyroud et al., 2011), PRJNA270526 (Sayou et al., 2016), PRJNA594407(Jin et al., 2021), PRJNA560053 (Romera-Branchat et al., 2020), PRJEB24874 (Collani et al., 2019), PRJEB28959 (Goretti et al., 2020), PRJNA595112 (Zhu et al., 2020), and PRJNA377528 (Xiao et al., 2017). The Cut&Run data we analyzed were from PRJNA509360 (Zheng and Gehring, 2019). The rice ChIP-seq data we analyzed were from the following publication ids: PRJNA588458 (Ren et al., 2021), PRJNA399280 (Chung et al., 2018), and PRJNA527848 (Li et al., 2019). The following 20 ENCODE input controls were used to generate the greenscreen for the hg38 genome assembly: ENCFF448TFZ, ENCFF438KJC, ENCFF880UAU, ENCFF516YKX, ENCFF349KXI, ENCFF251JQE, ENCFF495KCW, ENCFF881SJD, ENCFF433SPB, ENCFF352RKQ, ENCFF299YGP, ENCFF522TXM, ENCFF272RAI, ENCFF019HKT, ENCFF908NWF, ENCFF383ZXS, ENCFF695MWS, ENCFF048BXG, ENCFF295VUB, and ENCFF476YAR. The source for the 20 Arabidopsis and rice inputs is listed in Supplemental Tables S1 and S2, respectively.
A github repository is available at: https://github.com/sklasfeld/GreenscreenProject and contains all scripts and files used to generate Greenscreen and analyze ChIP-Seq experiments, as well as a detailed tutorial.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. Artifactual signals are present in input and ChIP samples.
Supplemental Figure S2. Workflow for generating a blacklist.
Supplemental Figure S3. Relationship between regions of artifactual ChIP-signal and supporting experiments.
Supplemental Figure S4. ChIP-seq artifactual signal distribution.
Supplemental Figure S5. Unsupervised clustering and heatmaps of signals in FUS, HNRNPK, and PCBP2 human ChIP-seq samples.
Supplemental Figure S6. Rice greenscreen mask enhances clustering in ChIP-seq datasets based on biological expectation.
Supplemental Figure S7. Optimizing ChIP-seq peak calling by filtering.
Supplemental Figure S8. SCC plots of LFY and FD ChIP-seq reads within greenscreen regions show enrichment at a shift size equal to the read length.
Supplemental Figure S9. Improved ChIP-seq pipeline results in more true peaks.
Supplemental Figure S10. Significant peaks found in LFY ChIP-seq data that overlap with blacklist regions but are not masked by the greenscreen pipeline.
Supplemental Figure S11. Significant peaks found in FD ChIP-seq data that overlap with blacklist regions but are not masked by the greenscreen pipeline.
Supplemental Figure S12. Properties of new peaks identified by our updated ChIP-seq pipeline.
Supplemental Figure S13. SCC profiles of published inputs.
Supplemental Table S1. Inputs used for Arabidopsis greenscreen and blacklist.
Supplemental Table S2. Arabidopsis greenscreen regions.
Supplemental Table S3. Optimization of greenscreen parameters.
Supplemental Table S4. Optimization of the human greenscreen mask.
Supplemental Table S5. Inputs used for rice greenscreen.
Supplemental Table S6. Rice greenscreen regions.
Supplemental Table S7. Overlap between ChIP-seq datasets analyzed using the pipeline proposed here compared with published datasets.
Supplemental Data Set S1. Statistical tests used in figures.
Supplementary Material
Acknowledgments
We thank Tian Huang for help with the development of the improved ChIP-seq pipeline and Dr. Roberto Bonasio for comments on the manuscript.
Funding
This work was supported by the National Science Foundation Division of Integrative Organismal Systems grants 1953279 and 1905062.
Conflict of interest statement. All authors declare no conflict of interest.
Contributor Information
Samantha Klasfeld, Department of Biology, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA.
Thomas Roulé, Department of Biology, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA.
Doris Wagner, Department of Biology, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA.
S.K. and D.W. designed the research. S.K. and T.R. performed the research and S.K. developed new computational tools. S.K. and D.W. analyzed the data and wrote the manuscript with feedback from T.R.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell) is: Doris Wagner (wagnerdo@upenn.sas.edu).
References
- Amemiya HM, Kundaje A, Boyle AP (2019) The ENCODE blacklist: identification of problematic regions of the genome. Sci Rep 9: 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T, Krajewski P, Ladunga I, Lefebvre C, Li Q, Liu T, Madrigal P, Taslim C, Zhang J (2013) Practical guidelines for the comprehensive analysis of ChIP-seq data. PLoS Comput Biol 9: e1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballouz S, Dobin A, Gillis JA (2019) Is it time to change the reference genome? Genome Biol 20: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batagelj V (1988) Generalized ward and related clustering problems. In HH Bock, ed, Classification and Related Methods of Data Analysis. Springer Berlin Heidelberg, Berlin, Germany, pp 67–74 [Google Scholar]
- Berardini TZ, Reiser L, Li D, Mezheritsky Y, Muller R, Strait E, Huala E (2015) The Arabidopsis information resource: making and mining the “gold standard” annotated reference plant genome. Genesis 53: 474–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll TS, Liang Z, Salama R, Stark R, de Santiago I (2014) Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front Genet 5: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Kaufmann K (2017) Integration of genome-wide TF binding and gene expression data to characterize gene regulatory networks in plant development. Methods Mol Biol 1629: 239–269 [DOI] [PubMed] [Google Scholar]
- Chen Y, Negre N, Li Q, Mieczkowska JO, Slattery M, Liu T, Zhang Y, Kim TK, He HH, Zieba J, et al. (2012) Systematic evaluation of factors influencing ChIP-seq fidelity. Nat Methods 9: 609–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chèneby J, Ménétrier Z, Mestdagh M, Rosnet T, Douida A, Rhalloussi W, Bergon A, Lopez F, Ballester B (2020) ReMap 2020: a database of regulatory regions from an integrative analysis of human and Arabidopsis DNA-binding sequencing experiments. Nucleic Acids Res 48: D180–D188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Krishnakumar V, Chan AP, Thibaud-Nissen F, Schobel S, Town CD (2017) Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J 89: 789–804 [DOI] [PubMed] [Google Scholar]
- Chung PJ, Jung H, Choi YD, Kim JK (2018) Genome-wide analyses of direct target genes of four rice NAC-domain transcription factors involved in drought tolerance. BMC Genomics 19: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collani S, Neumann M, Yant L, Schmid M (2019) FT modulates genome-wide DNA-binding of the bZIP transcription factor FD. Plant Physiol 180: 367–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer H (2011) A History of the Central Limit Theorem: From Classical to Modern Probability Theory. Springer, Berlin, Germany [Google Scholar]
- Gan ES, Xu Y, Wong JY, Goh JG, Sun B, Wee WY, Huang J, Ito T (2014) Jumonji demethylases moderate precocious flowering at elevated temperature via regulation of FLC in Arabidopsis. Nat Commun 5: 5098. [DOI] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Andreev-Drakhlin AY, et al. (2019) Next-generation characterization of the cancer cell line encyclopedia. Nature 569: 503–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girden ER (1992) ANOVA: Repeated Measures. Sage, Thousand Oaks, California [Google Scholar]
- Goretti D, Silvestre M, Collani S, Langenecker T, Méndez C, Madueño F, Schmid M (2020) TERMINAL FLOWER1 functions as a mobile transcriptional cofactor in the shoot apical meristem. Plant Physiol 182: 2081–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm S (1979) A simple sequentially rejective multiple test procedure. Scand J Stat 6: 65–70 [Google Scholar]
- Jackson SA (2016) Rice: The first crop genome. Rice 9: 1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Klasfeld S, Zhu Y, Fernandez Garcia M, Xiao J, Han SK, Konkol A, Wagner D (2021) LEAFY is a pioneer transcription factor and licenses cell reprogramming to floral fate. Nat Commun 12: 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B (2007) Genome-wide mapping of in vivo protein–DNA interactions. Science 316: 1497–1502 [DOI] [PubMed] [Google Scholar]
- Karimzadeh M, Ernst C, Kundaje A, Hoffman MM (2018) Umap and Bismap: Quantifying genome and methylome mappability. Nucleic Acids Res 46: e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, Tanaka T, Wu J, Zhou S, et al. (2013) Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, Park PJ (2008) Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol 26: 1351–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidder BL, Hu G, Zhao K (2011) ChIP-Seq: technical considerations for obtaining high-quality data. Nat Immunol 12: 918–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruskal WH, Wallis WA (1952) Use of ranks in one-criterion variance analysis. J Am Stat Assoc 47: 583–621 [Google Scholar]
- Kundaje A (2013) A Comprehensive Collection of Signal Artifact Blacklist Regions in the Human Genome. https://personal.broadinstitute.org/anshul/projects/encode/rawdata/blacklists/hg19-blacklist-README.pdf
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. (2012) ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22: 1813–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R;. 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Chang Y, Ma S, Shen J, Hu H, Xiong L (2019) Genome-wide identification of SNAC1-targeted genes involved in drought response in rice. Front Plant Sci 10: 982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann HB, Whitney DR (1947) On a test of whether one of two random variables is stochastically larger than the other. Ann Math Stat 18: 50–60 [Google Scholar]
- Miyashima S, Roszak P, Sevilem I, Toyokura K, Blob B, Heo JO, Mellor N, Help-Rinta-Rahko H, Otero S, Smet W, et al. (2019) Mobile PEAR transcription factors integrate positional cues to prime cambial growth. Nature 565: 490–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyroud E, Minguet EG, Ott F, Yant L, Posé D, Monniaux M, Blanchet S, Bastien O, Thévenon E, Weigel D, et al. (2011) Prediction of regulatory interactions from genome sequences using a biophysical model for the Arabidopsis LEAFY transcription factor. Plant Cell 23: 1293–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PJ (2009) ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet 10: 669–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson K (1896) VII. Mathematical contributions to the theory of evolution—III. Regression, heredity, and panmixia. Phil Trans R Soc Lond A 187: 253–318 [Google Scholar]
- Planet E, Attolini CSO, Reina O, Flores O, Rossell D (2012) htSeqTools: high-throughput sequencing quality control, processing and visualization in R. Bioinformatics 28: 589–590 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T (2014) deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42: W187–W191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand WM (1971) Objective criteria for the evaluation of clustering methods. Null 66: 846–850 [Google Scholar]
- Ren Y, Huang Z, Jiang H, Wang Z, Wu F, Xiong Y, Yao J (2021) A heat stress responsive NAC transcription factor heterodimer plays key roles in rice grain filling. J Exp Bot 72: 2947–2964 [DOI] [PubMed] [Google Scholar]
- Romera-Branchat M, Severing E, Pocard C, Ohr H, Vincent C, Née G, Martinez-Gallegos R, Jang S, Andrés F, Madrigal P, et al. (2020) Functional divergence of the Arabidopsis florigen-interacting bZIP transcription factors FD and FDP. Cell Rep 31: 107717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouaud M (2013) Probability, statistics and estimation. In Propagation of Uncertainties. p 191. https://www.incertitudes.fr/book.pdf
- Sayou C, Nanao MH, Jamin M, Posé D, Thévenon E, Grégoire L, Tichtinsky G, Denay G, Ott F, Peirats-Llobet M, et al. (2016) A SAM oligomerization domain shapes the genomic binding landscape of the LEAFY transcription factor. Nat Commun 7: 11222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RJ, Marand AP, Zhang X, Mosher RA, Turck F, Chen X, Axtell MJ, Zhong X, Brady SM, Megraw M, et al. (2022) Quality control and evaluation of plant epigenomics data. Plant Cell 34: 503–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro SS, Wilk MB (1965) An analysis of variance test for normality (complete samples). Biometrika 52: 591–611 [Google Scholar]
- Simon A (2012) FastQC: A Quality Control Tool for High Throughput Sequence Data. Babraham Institute, Babraham [Google Scholar]
- Skene PJ, Henikoff JG, Henikoff S (2018) Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc 13: 1006–1019 [DOI] [PubMed] [Google Scholar]
- Skene PJ, Henikoff S (2017) An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6: e21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staswick PE, Serban B, Rowe M, Tiryaki I, Maldonado MT, Maldonado MC, Suza W (2005) Characterization of an Arabidopsis enzyme family that conjugates amino acids to indole-3-acetic acid. Plant Cell 17: 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Student (1908) The probable error of a mean. Biometrika 6: 1–25 [Google Scholar]
- Tian S, Peng S, Kalmbach M, Gaonkar KS, Bhagwate A, Ding W, Eckel-Passow J, Yan H, Slager SL (2019) Identification of factors associated with duplicate rate in ChIP-seq data. PLoS ONE 14: e0214723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Li S, Li Y, Xu Y, Wang Y, Zhang R, Sun W, Chen Q, Wang XJ, Li C, et al. (2019) MED25 connects enhancer-promoter looping and MYC2-dependent activation of jasmonate signalling. Nat Plants 5: 616–625 [DOI] [PubMed] [Google Scholar]
- Welch BL (1947) The generalization of ‘STUDENT’S’ problem when several different population variances are involved. Biometrika 34: 28–35 [DOI] [PubMed] [Google Scholar]
- Wimberley CE, Heber S (2020) PeakPass: automating ChIP-Seq blacklist creation. J Comput Biol 27: 259–268 [DOI] [PubMed] [Google Scholar]
- Winter CM, Austin RS, Blanvillain-Baufumé S, Reback MA, Monniaux M, Wu MF, Sang Y, Yamaguchi A, Yamaguchi N, Parker JE, et al. (2011) LEAFY target genes reveal floral regulatory logic, cis motifs, and a link to biotic stimulus response. Dev Cell 20: 430–443 [DOI] [PubMed] [Google Scholar]
- Xiao J, Jin R, Yu X, Shen M, Wagner JD, Pai A, Song C, Zhuang M, Klasfeld S, He C, et al. (2017) Cis and trans determinants of epigenetic silencing by Polycomb repressive complex 2 in Arabidopsis. Nat Genet 49: 1546–1552 [DOI] [PubMed] [Google Scholar]
- Xiao R, Chen JY, Liang Z, Luo D, Chen G, Lu ZJ, Chen Y, Zhou B, Li H, Du X, et al. (2019) Pervasive chromatin-RNA binding protein interactions enable RNA-based regulation of transcription. Cell 178: 107–121.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kudron MM, Victorsen A, Gao J, Ammouri HN, Navarro FCP, Gevirtzman L, Waterston RH, White KP, Reinke V, et al. (2021) To mock or not: a comprehensive comparison of mock IP and DNA input for ChIP-seq. Nucleic Acids Res 49: e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Fear J, Hu J, Haecker I, Zhou L, Renne R, Bloom D, McIntyre LM (2014) Leveraging biological replicates to improve analysis in ChIP-seq experiments. Comput Struct Biotechnol J 9: e201401002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XY, Gehring M (2019) Low-input chromatin profiling in Arabidopsis endosperm using CUT&RUN. Plant Reprod 32: 63–75 [DOI] [PubMed] [Google Scholar]
- Zhu Y, Klasfeld S, Jeong CW, Jin R, Goto K, Yamaguchi N, Wagner D (2020) TERMINAL FLOWER 1-FD complex target genes and competition with FLOWERING LOCUS T. Nat Commun 11: 5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.