

Abstract
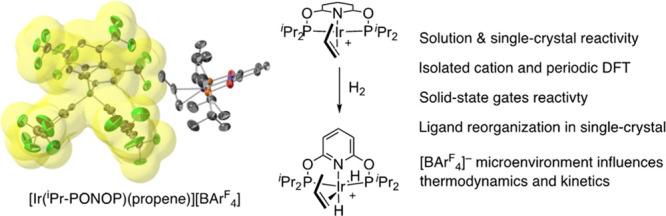
The reactivity of the Ir(I) PONOP pincer complex [Ir(iPr-PONOP)(η2-propene)][BArF4], 6, [iPr-PONOP = 2,6-(iPr2PO)2C6H3N, ArF = 3,5-(CF3)2C6H3] was studied in solution and the solid state, both experimentally, using molecular density functional theory (DFT) and periodic-DFT computational methods, as well as in situ single-crystal to single-crystal (SC-SC) techniques. Complex 6 is synthesized in solution from sequential addition of H2 and propene, and then the application of vacuum, to [Ir(iPr-PONOP)(η2-COD)][BArF4], 1, a reaction manifold that proceeds via the Ir(III) dihydrogen/dihydride complex [Ir(iPr-PONOP)(H2)H2][BArF4], 2, and the Ir(III) dihydride propene complex [Ir(iPr-PONOP)(η2-propene)H2][BArF4], 7, respectively. In solution (CD2Cl2) 6 undergoes rapid reaction with H2 to form dihydride 7 and then a slow (3 d) onward reaction to give dihydrogen/dihydride 2 and propane. DFT calculations on the molecular cation in solution support this slow, but productive, reaction, with a calculated barrier to rate-limiting propene migratory insertion of 24.8 kcal/mol. In the solid state single-crystals of 6 also form complex 7 on addition of H2 in an SC-SC reaction, but unlike in solution the onward reaction (i.e., insertion) does not occur, as confirmed by labeling studies using D2. The solid-state structure of 7 reveals that, on addition of H2 to 6, the PONOP ligand moves by 90° within a cavity of [BArF4]− anions rather than the alkene moving. Periodic DFT calculations support the higher barrier to insertion in the solid state (ΔG‡ = 26.0 kcal/mol), demonstrating that the single-crystal environment gates onward reactivity compared to solution. H2 addition to 6 to form 7 is reversible in both solution and the solid state, but in the latter crystallinity is lost. A rare example of a sigma amine-borane pincer complex, [Ir(iPr-PONOP)H2(η1-H3B·NMe3)][BArF4], 5, is also reported as part of these studies.
1. Introduction
The addition of dihydrogen and an alkene to metal centers, followed by migratory insertion and reductive elimination of an alkane, are elementary transformations in organometallic synthesis and catalysis.1 As well as being the key steps in catalytic alkene hydrogenation, the microscopic reverse of hydrogenation, alkane dehydrogenation, allows for the generation of simple alkenes from their corresponding alkanes,2 by C–H oxidative addition and β-elimination, Scheme 1A. Iridium complexes based upon the Ir(pincer)H2 motif (pincer = R-PCP, R-POCOP, or R-PONOP, R = alkyl or aryl, Scheme 1B) are often the systems of choice for alkane dehydrogenation reactions,3,4 operating by key, but yet to be experimentally observed, highly reactive Ir(pincer) intermediates such as 14-electron5 (a) or alkane σ-complexes6,7 (e). Under transfer dehydrogenation conditions intermediate a is generated from dihydride b using a sacrificial alkene in a complementary hydrogenation cycle.8,9
Scheme 1. (A) Simplified Alkene Hydrogenation and DeHydrogenation Catalytic Cycles. (B) Exemplar Ir(pincer) Complexes R = Alkyl or Aryl.

Anions not shown.
We have an interest in the generation and reactivity of σ-alkane complexes (i.e., e) using single-crystal to single-crystal (SC-SC10,11) solid-state molecular organometallic chemistry techniques (SMOM).12 For example, addition of H2 to crystalline [Rh(chelating-phosphine)(alkene)][BArF4] precursors [ArF = 3,5-(CF3)2C6H3] results in the formation of the corresponding cationic alkane σ-complexes in a solid/gas SC-SC transformation by hydrogenation of the alkene to form an alkane, which remains bound to the metal center.13−15 In solution σ-alkane complexes are transient (lifetimes of μs to hrs), being observed using in situ spectroscopic methods, often at low temperatures.16−18 The stability of the σ-alkane complexes in the solid state comes from the [BArF4]− anions that provide a microenvironment of secondary noncovalent interactions organized in a tertiary periodic anionic motif.15 This stability also allows these well-defined σ-alkane complexes to be directly connected with the products of C–H activation, for example, in stoichiometric acceptorless alkane dehydrogenation at room temperature.14,15
We recently showed that this synthetic methodology can be extended to allow for the isolation of a cobalt σ-alkane complex.19 In contrast, the iridium analogue cannot be isolated, and a hydride-bridged dimer results.20 Inspired by the work of Brookhart and co-workers in exploring the solid/gas SC-SC reactivity of aryl-fluorinated (ArF) pincer Ir(ArF-POCOP)H2 complexes with alkenes and hydrogen in which hydride-bridging dimers do not form,21 and their reports of [M(tBu-PONOP)(CH4)][BArF4] complexes that are σ-alkane complexes (M = Rh22) or alkyl hydrides (M = Ir23), respectively, in solution at low temperature, we were interested in the SC-SC reactivity of cationic [Ir(PONOP)(propene)][BArF4] complexes with H2 to explore if a propane15 (or propyl hydride) complex was accessible. A further motivation for this was the study of the elementary steps and intermediates of alkene hydrogenation/alkane dehydrogenation using Ir-pincer complexes by SC-SC reactivity. Propane to propene dehydrogenation is particularly interesting, as it is industrially relevant,24 and solid-phase supported Ir-pincer catalysts have used propene as a sacrificial acceptor in the gas-phase dehydrogenation of light alkanes25 as well as catalyzing direct propene hydrogenation.26
In this contribution we report the synthesis of the new complex [Ir(iPr-PONOP)(propene)][BArF4] and compare its reactivity with H2 in solution and the solid state, Scheme 2. While this Ir(I) complex reversibly adds H2 in a solid-state SC-SC reaction that results in significant and unexpected reorientation of the ligand framework, onward reaction to form propane does not occur. By comparison, in solution the formation of the Ir(III) dihydrogen/dihydride complex [Ir(iPr-PONOP)(H2)H2][BArF4] results alongside the formation of propane, but with more mechanistic complexity than in the solid state. While, ultimately, a σ-propane complex was not accessible in the solid state, these observations reveal the role of the solid-state environment in mechanically gating organometallic reactivity. A rare example of a σ-bound amine-borane pincer complex is also reported as part of this study.
Scheme 2. This Work.

[BArF4]− anions not shown.
2. Results and Discussion
2.1. Solution Synthesis of [Ir(iPr-PONOP)(H2)H2][BArF4], [Ir(iPr-PONOP)H2][BArF4], and [Ir(iPr-PONOP)H2(η1-H3B·NMe3)][BArF4]
A suitable entry point into the synthesis of the desired Ir(I) propene complex is an [Ir[(PONOP)H2]+ precursor, in the anticipation that reaction with excess propene would generate an Ir(I) propene-bound complex alongside free propane from hydrogenation.27,28 The precursor to this, [Ir(iPr-PONOP)(η2-COD)][BArF4]291, was synthesized from addition of free ligand, iPr-PONOP, to [Ir(COD)2][BArF4] (COD = cyclooctadiene, see the Supporting Information). In the 1H NMR spectrum of complex 1 signals due to bound [δ 4.45] and unbound [δ 5.68] alkene are observed, showing the COD does not reversibly dissociate or undergo intramolecular exchange on the NMR time scale. Resonances assigned to the iPr-PONOP ligand indicate overall C2v symmetry for the cation, and consistent with this in the 31P{1H} NMR spectrum a single environment is observed.30 A simple rotation of the COD ligand would account for these data, as also suggested to occur for the Rh analogue [Rh(iPr-PONOP)(η2-COD)][BArF4].31 Addition of excess H2 (4 bar) to complex 1 in CD2Cl2 solution resulted in the rapid formation (5 min) of the dihydrogen/dihydride complex [Ir(iPr-PONOP)(H2)H2][BArF4], 2 (∼85%), alongside free COD and a complex identified as [Ir(iPr-PONOP)H2(η2-COE)][BArF4], 3 (∼15%), Scheme 3, (COE = cyclooctene). No free COE was observed. Complex 3 was characterized in the 1H NMR spectrum by the observation of two bound alkene [δ 3.00 and 2.93] and two inequivalent hydride environments [δ −10.41 and −17.77], while an AB doublet is observed in the 31P{1H} NMR spectrum that shows trans J(PP) coupling [280 Hz]. Given that COD does not dissociate rapidly in complex 1, these observations are best explained by addition of H2 to form an unobserved dihydride complex [Ir(iPr-PONOP)H2(η2-COD)][BArF4] from which H2 and COE (the latter formed from slow hydrogenation of COD) must bind competitively compared with COD. Further hydrogenation of COE or COD to cyclooctane (COA) takes much longer (3 d, 4 bar H2), after which time only the pale yellow complex 2 and COA are observed.
Scheme 3. Synthesis of Complex 2.

[BArF4]− anions are not shown.
Attempts to isolate complex 2 at the end of reaction by recrystallization led to the formation of intractable oils, while removal of the H2 atmosphere resulted in the reversible loss of bound H2 (vide infra). Characterization was thus performed in situ under a H2 atmosphere. Complex 2 has a highly fluxional manifold of hydride ligands. At 298 K a single slightly broadened resonance is observed in the 1H NMR spectrum, δ −8.79, that integrates to 4 H. This chemical shift is very similar to that reported for [Ir(tBu-PONOP)H4][BArF4], δ −8.9, which is also highly fluxional.32 Dissolved H2 is also observed as a broadened signal, suggesting a slow exchange occurs with 2 at room temperature. In the 31P{1σH} NMR spectrum a single environment is observed at δ 181.2. Spin-lattice relaxation time (T1) measurements33 (500 MHz) on the hydride signal showed that, at 295 K T1 = 119 ± 8 ms, while at 253 K T1 = 38 ± 1 ms. At the low temperature the hydride signal is now observed as a sharper triplet [δ −8.81, t, J(PH) = 7.5 Hz] and still relative integral 4 H. The fluxional behavior and the short T1 time at 253 K suggest rapidly interconverting isomers that sit on a rather flat potential energy surface, for which there is a significant contribution to the time-averaged structure from complexes with a dihydrogen ligand rather than an Ir(V) tetrahydride. Similar behavior has been studied in detail for Ir(tBu-POCOP)H4 (T1 = 110 ms) and Ir(tBu-PCP)H4 (T1 = 130 ms) but where a significant contribution from a Ir(V) tetrahydride is suggested.34
Replacing the H2 atmosphere over complex 2 with D2 resulted in the disappearance of the hydride signal at δ −8.79 in the 1H NMR spectrum and the observation of HD(dissolved) at δ 4.55 [triplet, J(HD) = 43 Hz]. This confirms intermolecular exchange with D2 and also that subsequent intramolecular H/D exchange between the hydrides is occurring, either by a σ-CAM (σ-complex assisted metathesis) process or a Ir(V)-tetrahydride.35−37 In the 2H NMR spectrum a broad signal is observed at δ −8.8.
In the absence of an H2 atmosphere complex 2 slowly (3 h) loses H2 in CD2Cl2 solution to form 16-electron [Ir(iPr-PONOP)H2][BArF4], 4, Figure 1. This process can be accelerated by freeze/pump/thawing. In the resulting 1H NMR spectrum a single broad hydride environment is observed at δ −19.82, for which T1 measured at 295 K is fully consistent with a classical dihydride structure (1870 ± 10 ms).33 These data can be compared with those for the closely related dihydride [Ir(tBu-PONOP)H2][BArF4] (δ −25.07, T1(min) = 873 ms).37
Figure 1.

Synthesis of and solid-state structure of complex 5. Displacement ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angles (deg): Ir–P1, 2.2621(7); Ir–P2, 2.2660(6); Ir–N1, 2.103(2); Ir–B 2.771(4); Ir–H1C–B, 134(1). [BArF4]− anions are not shown.
The behavior of the [Ir(iPr-PONOP)H4]+ cation was also studied with density functional theory (DFT) calculations (see the Supporting Information for details) that identified [Ir(iPr-PONOP)(η2-H2)(H)2]+ with cis-hydrides (i.e., Figure 1) as the most stable form. The alternative trans isomer (with η2-H2trans to N) corresponds to a transition state for intramolecular H/H(D) exchange that proceeds with a barrier of 11.1 kcal/mol. Dissociation of H2 from 2 entails a barrier of 9.6 kcal/mol and forms square-pyramidal 4 with an axial hydride, at +1.4 kcal/mol, consistent with facile intermolecular H2/D2 exchange but endergonic H2 loss that should still be possible upon application of a vacuum. Hydride exchange in 4 is computed to be extremely facile and proceeds via a C2v-symmetric dihydride isomer, 4′, that lies only 2.9 kcal/mol above 4, consistent with the symmetric structure suggested by 1H NMR spectroscopy.38
While we were unable to structurally characterize complex 4, it reacts rapidly with H3B·NMe3 to quantitatively form an 18-electron σ-amine-borane complex, [Ir(iPr-PONOP)H2(η1-H3B·NMe3)][BArF4], 5, Figure 1, for which small number of colorless cuboid crystals were obtained from a CD2Cl2/pentane recrystallization, allowing for a single-crystal X-ray diffraction study. The resulting solid-state structure shows a pseudo-octahedral Ir(III) complex, with two cis hydride ligands (located, but not freely refined), a PONOP ligand, and an η1-coordinated H3B·NMe3 ligand. The latter coordination mode is signaled by a long Ir···B distance [2.771(4) Å] and a rather open Ir–H–B angle [134(1)°].13 Structurally characterized complexes with η1-coordination modes of amine-boranes39−42 have been previously reported. The solution NMR spectroscopic data (CD2Cl2) are fully consistent with the solid-state structure. Two Ir–hydride environments are observed in the 1H NMR spectrum, at δ −20.01 (td) and −15.91 (td), and a broad, relative integral 3 H, signal at δ −2.20 is assigned to the Ir··H3B group that is undergoing rapid site exchange between terminal and bridging B–H groups.42 The 11B{1H} and 31P{1H} NMR spectra of complex 5 are unremarkable.
2.2. Synthesis of [Ir(iPr-PONOP)(propene)][BArF4] in Solution
Addition of excess propene to in situ generated, pale yellow, complex 2 resulted in the immediate formation of the alkene dihydride complex [Ir(iPr-PONOP)H2(η2-propene)][BArF4], 7, Scheme 4. While the onward reaction of 7 is slow (see Section 2.3 for characterization and reactivity), simply pumping a solution to dryness results in H2 loss and the formation of [Ir(iPr-PONOP)(η2-propene)][BArF4], 6. Complex 6 thus formed can be isolated in crystalline form in 94% yield from a subsequent 1,2-difluorobenzene/pentane recrystallization. Analogous Ir(tBu-PCP)(propene) and Ir(tBu-POCOP) complexes are known.43,44
Scheme 4. Solution Synthesis of Complex 6.

[BArF4]− anions are not shown.
In CD2Cl2 solution a single set of resonances for the propene ligand are observed in the 1H and 13C{1H} NMR spectra of complex 6, alongside four closely overlapping iPr methyl, two methine, and two pyridyl environments, the last in a 1:2 ratio. A single environment is observed in the 31P{1H} NMR spectrum, δ 188.6. These solution data point to a fluxional process for the propene that gives time-averaged C2 symmetry on the NMR time scale. This process is not frozen out at 183 K; that is, a single 31P environment is still observed. A simple rotation around the (C=C)–Ir–N vector accounts for this,45 as also suggested for complex 1.
2.3. Addition of H2 to [Ir(iPr-PONOP)(propene)][BArF4] in Solution, and Characterization of [Ir(iPr-PONOP)H2(propene)][BArF4]
With the synthesis of complex 6 in hand its reactivity with H2 in CD2Cl2 solution was studied to determine baseline solution reactivity for comparison with the solid state. Addition of H2 (4 bar) to orange complex 6 rapidly (on time of mixing) resulted in the formation of a pale yellow solution that contains dihydride propene complex 7, alongside dihydrogen/dihydride 2 and dissolved propene, all in approximately equal amounts, Scheme 5. Over the course of 3 d under H2 this mixture slowly turns to give complex 2 and propane as the only components. As for the COD complex 1 these observations are best accounted for by competitive binding of propene (i.e., complex 7) and H2 (complex 2). Evidence for an equilibrium between 2 and 7 is provided by the addition of H2 to a solution of complex 6, shaking the NMR tube to ensure reaction with H2 to form 7, and then removal of noncondensable gases (H2) from a frozen (77 K) solution under vacuum. Under these conditions of low H2 concentration but persistent propene, complex 7 is the sole organometallic product observed on thawing under Ar. This also allows for its definitive characterization free from other products.
Scheme 5. Reactivity of Complex 5 in CD2Cl2 Solution.

[BArF4]− anions not shown.
In CD2Cl2 solution (Ar atmosphere, 298 K) the 13C{1H} NMR spectrum of complex 7 displays two alkene environments (δ 82.5, 62.0). In the 1H NMR spectrum sharp signals for two inequivalent Ir–H groups, at δ −11.64 and −16.50, that also show coupling to two 31P nuclei are observed alongside a single set of resonances due the bound propene. The iPr methyl groups resolve as at least five broad signals, and the methine signals are also complex. These data report on cis-hydride environments, with the propene in an axial coordination site. Two tightly coupled AB doublets are observed in the 31P{1H} NMR spectrum at δ 168.2 and 170.0 that show trans PP coupling [J(PP) = 320 Hz]. Inequivalent phosphine environments are expected for propene coordinated via a π-face in the axial position that is not undergoing reversible dissociation that is fast on the NMR time scale. At low temperature (185 K) the 31P{1H} NMR spectrum becomes significantly more complex, suggesting the resolution of two different isomeric species associated with the orientation of the propene ligand, which is undergoing fast rotation at room temperature. Two sets of hydride resonances are now observed at chemical shifts consistent with their frequency averaging at room temperature.
Under an Ar atmosphere in a sealed NMR tube complex 7 does not lose H2 in solution. However, as stated, pumping to dryness results in the reformation of 6, showing that H2 addition is reversible, but its loss is likely an endergonic process. Over a 24 h period decomposition occurs in solution (∼25%) to unidentified products, and attempts to recrystallize 7 led to poorly diffracting crystalline material from which no structural solution was possible.
DFT calculations confirm the facile addition of H2 to 6 to form 7 (ΔG‡ = 15.9 kcal/mol; ΔG° = −4.4 kcal/mol, Figure 2) and that this reaction, with a return barrier of 20.3 kcal/mol, should be reversible if H2 is removed from the system. In contrast, the onward hydrogenation of propene from 7 is much less accessible due to the significant barriers associated with the propene migratory insertion step. The lowest-energy process is shown in Figure 2 and has an overall barrier of 24.8 kcal/mol via TS(7–8iso)M, consistent with a slow process at room temperature. TS(7–8iso)M leads to an isopropyl intermediate, 8iso, and geometrically this step is best described as a hydride migration onto the propene ligand (labeled “M”) that is coupled to movement of the spectator hydride such that the isopropyl ligand occupies the axial site in 8iso as the Talkyl isomer. A second pathway where propene inserts (labeled “I”) into the adjacent Ir–H bond proceeds via TS(7–8iso)I at +26.0 kcal/mol and leads to the more stable TH isomer of 8iso at −0.2 kcal/mol. Hydride migration and propene insertion transition states to form the n-propyl species (8n) were also located (TS(7–8n)M: +21.9 kcal/mol; TS(7–8n)I: 24.7 kcal/mol) and gave the Talkyl and TH isomers of 8n at +4.3 kcal/mol and −3.1 kcal/mol, respectively (see the Supporting Information). All these migratory insertion transition states exhibit “late” geometries; for example, in TS(7–8iso)M the forming C–H and Ir–Calkyl bonds (1.21 Å/2.19 Å) are already close to their computed values in Talkyl8iso (1.10 Å/2.15 Å). The high barriers to insertion are likely linked to the formation of a strongly donating alkyl ligand trans to the spectator hydride, although movement of the spectator hydride ligands does serve to mitigate this effect (the H–Ir–Calkyl angle is ca. 140° in all cases).
Figure 2.
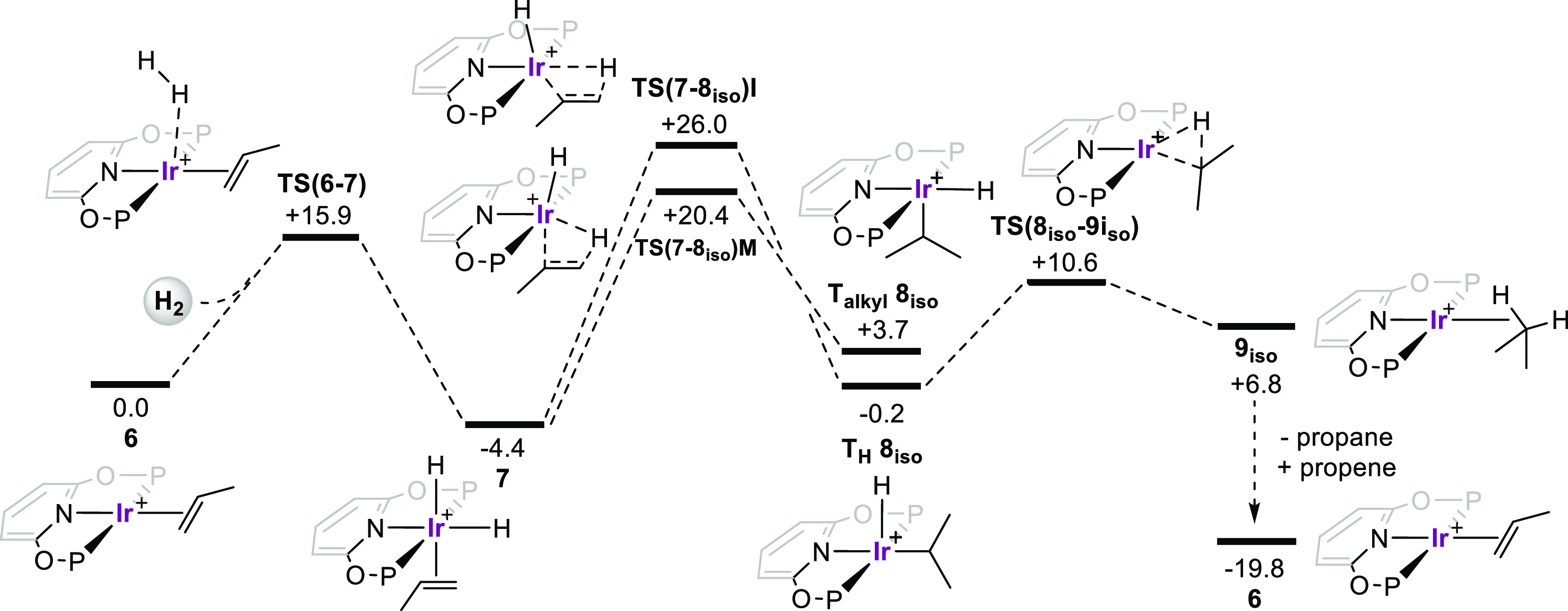
Computed free energy profile (kcal/mol) for propene hydrogenation from 6 modeled in dichloromethane solvent (P = PiPr2; level of theory: BP86[D3BJ,CH2Cl2]/Def2TZVP//BP86/SDD (Rh, P, with polarization on P); 6-31G** on other atoms.
Once formed, Talkyl8iso must first isomerize to TH8iso in order to access C–H reductive coupling via TS(8iso-9iso) (+10.6 kcal/mol) to give the propane complex 9iso (+6.8 kcal/mol). The reaction is then driven to completion by displacement of propane by either propene (ΔG° = −19.8 kcal/mol relative to 6) or (if present) H2 to give 2 (ΔG° = −14.7 kcal/mol). The calculations thus indicate the equilibrium between 2 (+ propene) and 7 (+ H2) will favor the latter (ΔG = −5.1 kcal/mol) while 2 is still accessible under H2 pressure.
2.4. SC-SC Reactivity in the Solid State
In contrast to the solution behavior, addition of H2 to finely crushed crystalline samples of complex 6 (∼50 mg, 15 min, 4 bar) did not result in the formation of tetrahydride complex 2 or the hydrogenation of propene to propane, even after 5 d under H2. Instead room-temperature 31P{1H}/13C{1H} solid-state NMR (SSNMR) and solution (CD2Cl2) spectroscopy of dissolved crystalline material showed the formation of the dihydride alkene complex [Ir(iPr-PONOP)H2(η2-propene)][BArF4], 7, with data identical to those obtained by solution methods. Under a system open to Ar, in the solid-state complex 7 loses H2 to reform 6, a process that is speeded up by application of a dynamic vacuum (2 h). No decomposition is observed, unlike in solution. Because of the experimental challenges of keeping samples under a H2 atmosphere when manipulating in the solid state, these samples show 90% complex 7 with the remainder being complex 6. Analysis of single crystals of complex 7 hydrogenated ex situ in the bulk, by rapid transfer to the diffractometer, leads to a structural refinement in which the electron density due to the alkene ligand could not be satisfactorily modeled, a consequence of partial H2 loss to reform complex 6. Analysis of material that had completely reformed complex 6 after H2 loss showed only weak Bragg peaks and showed evidence of significant crystal cracking (scanning electron microscopy (SEM), Supporting Information). As the resulting 31P{1H} SSNMR spectrum of this material was indistinguishable from initially synthesized complex 6, this suggests retention of short-range order but loss of bulk crystallinity.46 The formation of microcrystalline, or amorphous, products as a response to solid-state reactivity in single crystals is well-documented.47−50
To circumvent this problem of H2 loss in complex 7 we used in situ SC-SC techniques on the I19 Beamline at the Diamond Light Source for its synthesis from 6. This allowed for the collection of data under an atmosphere of H2 and thus the formation of complex 7 without reforming 6. The solid-state structure of complex 6 is shown in Figure 2. Data were collected at 273 K allowing for the subsequent reaction with H2 to be followed crystallographically in situ. The structural solution is good [R(2σ) = 4.0%, Rint = 4.3%, P1̅ space group] and shows a pseudo-square-planar cation, as expected for an Ir(I) complex. The propene is disordered essentially equally between two sites, related to one another by a noncrystallographic mirror plane (inset left Figure 2 and Figure 3), accounted for by coordination of a different π-face of propene in each. The C–C distances in the propene are consistent with a double and a single bond [1.271(8) and 1.56(1) Å, respectively]. The Ir···C distances show alkene coordination with a non-interacting methyl group: Ir–C1, 2.138(5); Ir–C2; 2.209(5); Ir–C3, 3.15(5) Å. Data collection at 150 K on a laboratory-based diffractometer (Cu source), while more precise [R(2σ) = 1.9%, Rint = 2.0%], showed the same disorder indicating that there are no dynamic processes operating in the solid state that involve reorientation of the propene.51
Figure 3.

Solid-state structures of the cations in complexes 6 and 7 (from an SC-SC reaction) as determined by single-crystal X-ray diffraction (273 K). [BArF4]− anions and hydrogen atoms are not shown; displacement ellipsoids are shown at the 30% probability level. Hydrides were not located in 7. Only one disordered component of the propene is shown in each case. Selected bond distances (Å) and angles (deg): Complex 6 Ir–C1, 2.138(5); Ir–C2, 2.209(5); Ir–P1, 2.2603(7); Ir–P2, 2.2635(7); Ir–N, 2.044(2); C1–C2, 1.271(8); C2–C3 1.56(1); ∠Ir–C1–C2/P1–P2–N–Ir, 66.7(4). Complex 7 Ir–C1A, 2.36(6); Ir–C2A, 2.25(5); Ir–P1, 2.26(1); Ir–P2, 2.26(1); Ir–N, 2.12(2); C1A–C2A, 1.31(8); C2–C3, 1.46(6); ∠C1–C2/N–Ir, 48(3)°.
The supporting [BArF4]− anions present a skewed-bicapped square prism motif, in which 10 anions surround two crystallographically identical cations.52 This is discussed in more detail in Section 2.6.
Addition of H2 (2 bar) to the same single crystal used for the structural analysis of complex 6 at 273 K led to the formation of complex 7, for which a satisfactory structural solution could be determined, Figure 3 [R(2σ) = 9.3%, Rint = 8.7%, P1̅ space group]. However, there was a significant deterioration in data quality on hydrogenation resulting in a drop in resolution from 0.65 Å in 6 to 1.2 Å for 7. Complex 7 has a pseudo-octahedral coordination geometry, in which the propene has moved from the equatorial position found in 6 to an axial position. The remaining coordination sites are occupied by the hydrides, which were not located, but their presence is confirmed by solution NMR spectroscopy on dissolved crystals. In this SC-SC transformation the disorder observed in the propene ligand in 6 is retained in complex 7, with two orientations observed, in an ∼50:50 ratio, Figures 3 and 4. Because of the lower quality of data the C–C and C=C distances in the propene were restrained to sensible distances [1.46(6), 1.31(8) Å, respectively], and detailed discussion of the metrics of propene binding are not appropriate. Confidence in the assignment of C=C and C–C bonds comes from the orientation of the disordered propene fragments that map onto those observed in starting 6, the longer Ir···C distance to the third propene carbon atom [Ir–C3A, 3.29(4) Å], and solid-state NMR data that support the formation of two isomers, as discussed next.
Figure 4.
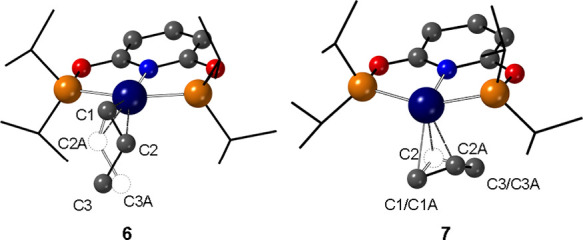
Comparison of structures of complexes 6 and 7 orientated to compare the propene coordination and disorder. [BArF4]− anions are not shown.
2.5. Solid-State NMR Analysis of Complexes 6 and 7
In the 31P{1H} SSNMR spectrum of complex 6 two 31P environments are observed [δ 189.5, 194.1], as an AB doublet showing large 31P–31P coupling, consistent with trans-disposed, crystallographically distinct, phosphines, J(PP) = 310 Hz.53 While the two, noncrystallographically generated, disordered components observed in the solid-state structure suggest there should be four 31P environments, we suggest a coincidence of signals results in only two being observed. In contrast, in the 13C{1H} NMR spectrum four signals are observed in the bound alkene region [δ 57.4, 56.6, 44.8, 42.9], fully consistent with the solid-state structure. In the 31P{1H} SSNMR spectrum of complex 7 a complex set of overlapping resonances is observed, centered around δ 174,54 while in the 13C{1H} SSNMR spectrum two sets of alkene signals are observed. These data are consistent with two isomers being formed for complex 7 in the solid state, leading on from the two isomers observed for propene binding in complex 6 that are related by the relative orientation of the methyl group.
2.6. Movement of the PONOP-Ligand on the SC-SC Transformation
Oxidative addition of H2 to complex 6 results in the alkene in 7 now being orientated orthogonal to its starting position, Figures 3 and 4. We initially interpreted this as coming from movement of the alkene ligand around the coordination sphere of the iridium in the SC-SC transformation. However, inspection of the wider crystalline environment of [BArF4]− anions surrounding the cationic metal center shows that, surprisingly, it is the iPr-PONOP ligand that moves on oxidative addition of H2, not the alkene. Figure 5A shows that the [BArF4]− anions in 6 form a skewed bicapped square prism (BCSP) that surrounds two crystallographically related [Ir(iPr-PONOP)H2(η2-propene)]+ cations. BCSP motifs have been reported previously in SMOM chemistry.55,56 The cations are orientated so that the propene is directed into a pocket formed by three of the [BArF4]− aryl groups of a single anion, with the pyridyl group directly trans to the propene. On addition of H2 in the SC-SC transformation the skewed BCSP motif is retained (difference in unit cell volumes = 1.9%), but there is a small contra-translation of the [BArF4]− anions in upper and lower basal planes. The orientation of the propene is also retained in the same pocket of aryl groups. What has changed significantly is the orientation of the PONOP ligand, which has pivoted ∼90° around the P–Ir–P vector so that it now sits orthogonal to the propene. Figure 5B,C shows this schematically and by an overlay of the crystallographically determined structures, respectively. While such large movements of metal–ligand fragments on SC-SC transformations are precedented, they are rare.57−59 In the case here, this reorientation may explain why the crystal quality degrades significantly on cycling H2 addition/removal, that is, 6-7-6.
Figure 5.

(A) Arrangement of anions and cations for complexes 6 and 7. The anion motif is described by B atoms of the [BArF4]− (with selected anions shown, without F atoms), and enclosed cations are depicted in ball-stick format. The propene ligand in each is highlighted as a hatched circle, and only one disordered component is shown (see Figure 3). Arrows indicate the PONOP pyridyl ligand that moves on H2 addition. (B) Diagram of 6 and 7 orientated with regard to propene, showing the movement of the PONOP ligand. (C) Overlaid 6 (blue) and 7 (red) stick representations in the same orientation as shown in (B).
The addition of H2 to 6 to form 7 also results in a change in the oxidation state of the metal, accompanied by a move from pseudo square planar to octahedral. SC-SC transformations that involve an overall change in oxidation state have been reported previously,10 for example, Mn(II)/Mn(IV);60 Co(II)/Co(III);61 Rh(I)/Rh(III);13,62,63 Ir(I)/Ir(III);21 Ir(III)/Ir(V);64 and Au(I)/Au(III).65
2.7. Differences between Solution and Solid-State Reactivity
That the dihydrogen/dihydride complex 2 is formed in solution on hydrogenation of complex 6 while only complex 7 formed in the solid-state could arise from two restraining conditions imposed by the crystalline microenvironment of [BArF4]− anions. First, the reversible dissociation of propene from complex 7 that occurs in solution on addition of H2 is clearly disfavored in the solid state. Second, onward reactivity of the propene hydride to ultimately form free propane, by migratory insertion and reductive elimination, and 2 could be disfavored due to local steric effects of the proximal [BArF4]− anion that hinder either of these transformations. In both solid state and solution complex 7 undergoes H2 loss to reform 6. We thus turned to computational studies in the extended solid state to understand these fundamental steps in greater detail and compare with solution DFT studies on the molecular cations reported above.
2.8. Computational Studies in the Solid State
The structures of 6 and 7 in the extended solid state were computed with periodic DFT calculations using the PBE-D3 functional. Geometries for both species were based on the experimental crystal structures, and subsequent reactivity studies considered changes at one of the two Ir centers present in each unit cell (see the Supporting Information for full details). Key stationary points are presented in Figure 6, where the profile in red is computed using the unit cell of 6, while that in blue uses the unit cell of 7.
Figure 6.

Computed free energy profiles (kcal/mol) for reversible H2 addition to 6 and propene migratory insertion in 7 (P = PiPr2). Data are computed either in the unit cell of 6 (red) or 7 (blue). The heavy open V indicates the relative position of the adjacent BArF4– anion. Level of theory: periodic-DFT: PBE-D3/DZVP-MOLOPT-SR-GTH/GTH-PBE (plane wave cutoff = 500 Ry).
H2 addition to 6 to form 7 has a computed free energy change of −4.8 kcal/mol, indicating that, while 7 is favored thermodynamically, 6 may still be accessible from 7 under conditions of H2 loss. Kinetically, however, H2 loss directly from 7 is inaccessible with a barrier of 44.1 kcal/mol; the extended crystal environment within 7 therefore blocks this process. In contrast, H2 oxidative addition to 6 is feasible (ΔG‡ = +14.4 kcal/mol) and forms a propene dihydride complex within the crystal environment of 6. This species, 7*, is 10.9 kcal/mol less stable than 7, and we propose that one reason for this arises from the movement of the propene ligand to outside the pocket defined by the adjacent [BArF4]− anion (represented by the open “V” in Figure 6) that occurs upon H2 addition. On the basis of the experimentally observed, reversible H2 addition to 6 to give 7, we speculate that the reorganization of the crystal lattice (that occurs with the net movement of the pincer ligand relative to the anionic framework) must occur at 7* to move between the “red” (7*) and “blue” (7) crystal lattices.66 Viewed in reverse, and assuming facile rearrangement between the two crystal lattices, the loss of H2 from 7 can occur with an overall barrier of 19.2 kcal/mol via TS(6–7*).
The positioning of the propene relative to the [BArF4]− anion pocket appears to be a key factor in determining the energy of other stationary points in these systems. Thus, in 6* the propene sits outside the pocket, and this structure is 16.3 kcal/mol higher than that of 6. Similarly, H2 reductive elimination from 7 is also strongly disfavored, as this requires the propene to move out of the pocket in order to restore square-planar coordination at the Ir(I) center in 6*. The preferred pathways for propene migratory insertion are also impacted by the crystal environment. Thus, for 7 the hydride migration pathway is favored via TS(7–8iso)M at +21.2 kcal/mol, as this allows the propene to remain in the anion pocket. For 7* alkene insertion into the hydride bond via TS(7*-8iso*)I at +21.9 kcal/mol is preferred, as this permits the propene to remain within the pocket. In contrast, propene insertion in 7 and hydride migration in 7* have barriers in excess of 40 kcal/mol as they necessitate movement of propene out of the anion pocket (see the Supporting Information). This demonstrates a significant impact of the solid-state environment: in solution the barriers to the four different migratory insertion steps spanned only 5.6 kcal/mol, whereas in the solid state the range is over 30 kcal/mol.
The barrier to propene migratory insertion in 7 in the solid state is 26.0 kcal/mol, slightly higher than that of 24.8 kcal/mol computed in solution. Thus, the slow hydrogenation of propene that does occur in solution is somewhat more disfavored in the solid state. Experimentally, formation of propane is not observed in the solid state.67 The possibility of reversible migratory insertion in the solid state was probed by addition of D2 (15 min) to finely crushed 6. This resulted in the formation of [Ir(iPr-PONOP)D2(propene)][BArF4], d2-7, as indicted by a featureless high-field region of the 1H NMR spectrum of dissolved material, and two Ir–D signals observed in the 2H NMR spectrum at chemical shifts essentially the same as for 7. Longer reactions times (3 d) did not result in H/D exchange into the bound propene as measured by 1H and 2H NMR spectroscopy, consistent with the significant computed barrier to migratory insertion. Reversible alkene insertion in Ir(pincer)(alkene)D2 complexes is known, as determined by H/D exchange experiments into the bound alkene.28 Interestingly there is an example of insertion being reversible, but no H/D exchange is observed.27 DFT calculations suggest that this is due to a high barrier for ring opening of the insertion product, which has an agostic alkyl group. Such a scenario is not happening in our systems.
3. Conclusions
While still relatively uncommon, organometallic synthesis and reactivity in the single-crystalline environment is becoming an increasingly appreciated methodology.10,11 However, examples where solution and crystalline-phase reactivity can be directly compared experimentally68 and computationally are rare, in part a consequence of the stabilizing role that the solid state has on reactive complexes that are challenging to access in solution (e.g., σ-alkane complexes6). With [Ir(iPr-PONOP)(η2-propene)][BArF4] we have such a platform through reactivity with H2 to initially form [Ir(iPr-PONOP)(η2-propene)H2][BArF4]. Subsequent migratory insertion and reductive elimination of propane occurs in solution but in the solid state does not. While the solution-phase chemistry is more complex than that observed in the SC-SC transformation, with equilibria operating through reversible alkene dissociations that are not available in the solid state, the solid-state crystalline microenvironment, and, in particular, the positioning of the propene ligand relative to the [BArF4]− aryl groups, is a key factor in determining both the relative (higher) barrier to migratory insertion and the observed structures. The significantly more stable arrangement when propene sits in the pocket formed by the aryl groups in the crystalline lattice leads to both iPr-PONOP ligand movement and plasticity in the anion lattice to accommodate this motif on reaction of 6 with H2 to form 7. While this reorganization provides access to the most stable structures in the solid state, a consequence is that the barriers to onward reactivity (e.g., migratory insertion) are significant, meaning that the targeted complex that has propane interacting with the Ir center is not accessible via this route. What will be interesting to explore is if engineering both the anion-motif and metal–ligand fragment in related SMOM systems allows for a reduction in these barriers through ground-state destabilization and/or transition-state stabilization.69 These are concepts that are familiar in enzyme catalysis, where the primary, secondary, and tertiary structures also work in concert to lower barriers to elementary reaction steps.70
Acknowledgments
The EPSRC (EP/M024210/2, EP/T019867/1), SCG Chemicals, The Clarendon Trust, The Leverhulme Trust (RPG-2020-184), Diamond Light Source for funding (PhD studentship to AM).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.2c00274.
The authors declare no competing financial interest.
Dedication
Dedicated to Professor Maurice Brookhart (Brook) for his immense contributions to organometallic synthesis and catalysis, and his generosity and support of others in the field.
Supplementary Material
References
- Hartwig J. F.Organotransition Metal Chemistry. From Bonding to Catalysis ;Hartwig J. F., Ed.; University Science Books: Sausalito, CA, 2010. [Google Scholar]
- Goldberg K. I.; Goldman A. S. Large-Scale Selective Functionalization of Alkanes. Acc. Chem. Res. 2017, 50, 620–626. 10.1021/acs.accounts.6b00621. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Bhatti T. M.; Goldman A. S. Dehydrogenation of Alkanes and Aliphatic Groups by Pincer-Ligated Metal Complexes. Chem. Rev. 2017, 117, 12357–12384. 10.1021/acs.chemrev.7b00247. [DOI] [PubMed] [Google Scholar]
- Choi J.; MacArthur A. H. R.; Brookhart M.; Goldman A. S. Dehydrogenation and Related Reactions Catalyzed by Iridium Pincer Complexes. Chem. Rev. 2011, 111, 1761–1779. 10.1021/cr1003503. [DOI] [PubMed] [Google Scholar]
- More generally 14-electron group 9 ML3 complexes are known. See, for example,; Chaplin A. B. Rhodium(I) Complexes of the Conformationally Rigid IBioxMe4 Ligand: Isolation of a Stable Low-Coordinate T-Shaped Complex. Organometallics 2014, 33, 624–626. 10.1021/om401223w. [DOI] [Google Scholar]; and references therein.
- Weller A. S.; Chadwick F. M.; McKay A. I. Transition Metal Alkane-Sigma Complexes: Synthesis, Characterization, and Reactivity. Adv. Organomet. Chem. 2016, 66, 223–276. 10.1016/bs.adomc.2016.09.001. [DOI] [Google Scholar]
- Crimmin M. R.; Kong R. Y.; Phillips N. In Comprehensive Coordination Chemistry III ;Constable E. C., Parkin G., Que L. Jr, Eds.; Elsevier: Oxford, England, 2021. [Google Scholar]
- Renkema K. B.; Kissin Y. V.; Goldman A. S. Mechanism of Alkane Transfer-Dehydrogenation Catalyzed by a Pincer-Ligated Iridium Complex. J. Am. Chem. Soc. 2003, 125, 7770–7771. 10.1021/ja0289200. [DOI] [PubMed] [Google Scholar]
- Göttker-Schnetmann I.; Brookhart M. Mechanistic Studies of the Transfer Dehydrogenation of Cyclooctane Catalyzed by Iridium Bis(Phosphinite) p-XPCP Pincer Complexes. J. Am. Chem. Soc. 2004, 126, 9330–9338. 10.1021/ja048393f. [DOI] [PubMed] [Google Scholar]
- Reid K. A.; Powers D. C. In Crystallo Organometallic Chemistry. Chem. Commun. 2021, 57, 4993–5003. 10.1039/D1CC01684A. [DOI] [PubMed] [Google Scholar]
- Pike S. D.; Weller A. S. Organometallic Synthesis, Reactivity and Catalysis in the Solid State Using Well-Defined Single-Site Species. Philos. Trans. R. Soc. A 2015, 373, 20140187. 10.1098/rsta.2014.0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick F. M.; McKay A. I.; Martinez-Martinez A. J.; Rees N. H.; Krämer T.; Macgregor S. A.; Weller A. S. Solid-State Molecular Organometallic Chemistry. Single-Crystal to Single-Crystal Reactivity and Catalysis with Light Hydrocarbon Substrates. Chem. Sci. 2017, 8, 6014–6029. 10.1039/C7SC01491K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Martínez A. J.; Tegner B. E.; McKay A. I.; Bukvic A. J.; Rees N. H.; Tizzard G. J.; Coles S. J.; Warren M. R.; Macgregor S. A.; Weller A. S. Modulation of σ-Alkane Interactions in [Rh(L2)(Alkane)]+ Solid-State Molecular Organometallic (SMOM) Systems by Variation of the Chelating Phosphine and Alkane: Access to η2,η2-σ-Alkane Rh(I), η1-σ-Alkane Rh(III) Complexes, and Alkane Encapsulation. J. Am. Chem. Soc. 2018, 140, 14958–14970. 10.1021/jacs.8b09364. [DOI] [PubMed] [Google Scholar]
- McKay A. I.; Bukvic A. J.; Tegner B. E.; Burnage A. L.; Martínez-Martínez A. J.; Rees N. H.; Macgregor S. A.; Weller A. S. Room Temperature Acceptorless Alkane Dehydrogenation from Molecular σ-Alkane Complexes. J. Am. Chem. Soc. 2019, 141, 11700–11712. 10.1021/jacs.9b05577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukvic A. J.; Burnage A. L.; Tizzard G. J.; Martínez-Martínez A. J.; McKay A. I.; Rees N. H.; Tegner B. E.; Krämer T.; Fish H.; Warren M. R.; et al. A Series of Crystallographically Characterized Linear and Branched σ-Alkane Complexes of Rhodium: From Propane to 3-Methylpentane. J. Am. Chem. Soc. 2021, 143, 5106–5120. 10.1021/jacs.1c00738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calladine J. A.; Duckett S. B.; George M. W.; Matthews S. L.; Perutz R. N.; Torres O.; Vuong K. Q. Manganese Alkane Complexes: An IR and NMR Spectroscopic Investigation. J. Am. Chem. Soc. 2011, 133, 2303–2310. 10.1021/ja110451k. [DOI] [PubMed] [Google Scholar]
- Yau H. M.; McKay A. I.; Hesse H.; Xu R.; He M.; Holt C. E.; Ball G. E. Observation of Cationic Transition Metal–Alkane Complexes with Moderate Stability in Hydrofluorocarbon Solution. J. Am. Chem. Soc. 2016, 138, 281–288. 10.1021/jacs.5b10583. [DOI] [PubMed] [Google Scholar]
- Watson J. D.; Field L. D.; Ball G. E. Binding Methane to a Metal Centre. Nat. Chem. 2022, 14, 801–804. 10.1038/s41557-022-00929-w. [DOI] [PubMed] [Google Scholar]
- Boyd T. M.; Tegner B. E.; Tizzard G. J.; Martínez-Martínez A. J.; Neale S. E.; Hayward M. A.; Coles S. J.; Macgregor S. A.; Weller A. S. A Structurally Characterized Cobalt(I) σ-Alkane Complex. Angew. Chem., Int. Ed. 2020, 59, 6177–6181. 10.1002/anie.201914940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick F. M.; Olliff N.; Weller A. S. A Convenient Route to a Norbornadiene Adduct of Iridium with Chelating Phosphines, [Ir(R2PCH2CH2PR2)(NBD)][BAr4F] and a Comparison of Reactivity with H2 in Solution and the Solid–State. J. Organomet. Chem. 2016, 812, 268–271. 10.1016/j.jorganchem.2015.12.001. [DOI] [Google Scholar]
- Huang Z.; White P. S.; Brookhart M. Ligand Exchanges and Selective Catalytic Hydrogenation in Molecular Single Crystals. Nature 2010, 465, 598–601. 10.1038/nature09085. [DOI] [PubMed] [Google Scholar]
- Bernskoetter W. H.; Hanson S. K.; Buzak S. K.; Davis Z.; White P. S.; Swartz R.; Goldberg K. I.; Brookhart M. Investigations of Iridium-Mediated Reversible C–H Bond Cleavage: Characterization of a 16-Electron Iridium(III) Methyl Hydride Complex. J. Am. Chem. Soc. 2009, 131, 8603–8613. 10.1021/ja901706b. [DOI] [PubMed] [Google Scholar]
- Bernskoetter W. H.; Schauer C. K.; Goldberg K. I.; Brookhart M. Characterization of a Rhodium(I) σ-Methane Complex in Solution. Science 2009, 326, 553–556. 10.1126/science.1177485. [DOI] [PubMed] [Google Scholar]
- Docherty S. R.; Rochlitz L.; Payard P.-A.; Copéret C. Heterogeneous Alkane Dehydrogenation Catalysts Investigated Via a Surface Organometallic Chemistry Approach. Chem. Soc. Rev. 2021, 50, 5806–5822. 10.1039/D0CS01424A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Zhou T.; Emge T. J.; Mironov O.; Saxton R. J.; Krogh-Jespersen K.; Goldman A. S. Dehydrogenation of N-Alkanes by Solid-Phase Molecular Pincer-Iridium Catalysts. High Yields of α–Olefin Product. J. Am. Chem. Soc. 2015, 137, 9894–9911. 10.1021/jacs.5b05313. [DOI] [PubMed] [Google Scholar]
- Syed Z. H.; Kaphan D. M.; Perras F. A.; Pruski M.; Ferrandon M. S.; Wegener E. C.; Celik G.; Wen J.; Liu C.; Dogan F.; et al. Electrophilic Organoiridium(III) Pincer Complexes on Sulfated Zirconia for Hydrocarbon Activation and Functionalization. J. Am. Chem. Soc. 2019, 141, 6325–6337. 10.1021/jacs.9b00896. [DOI] [PubMed] [Google Scholar]
- Bézier D.; Guan C.; Krogh-Jespersen K.; Goldman A. S.; Brookhart M. Experimental and Computational Study of Alkane Dehydrogenation Catalyzed by a Carbazolide-Based Rhodium Pnp Pincer Complex. Chem. Sci. 2016, 7, 2579–2586. 10.1039/C5SC04794C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C.; Kim B. G.; Guironnet D.; Brookhart M.; Guan C.; Wang D. Y.; Krogh-Jespersen K.; Goldman A. S. Synthesis and Characterization of Carbazolide-Based Iridium PNP Pincer Complexes. Mechanistic and Computational Investigation of Alkene Hydrogenation: Evidence for an Ir(III)/Ir(V)/Ir(III) Catalytic Cycle. J. Am. Chem. Soc. 2014, 136, 6672–6683. 10.1021/ja501572g. [DOI] [PubMed] [Google Scholar]
- The analogous complex [Ir(iPr-PNP)(η2-COD)][BArF4] has recently been reported.; Chapp S. M.; Schley N. D. Group-Transfer Reactions of a Cationic Iridium Alkoxycarbene Generated by Ether Dehydrogenation. Inorg. Chem. 2020, 59, 7143–7149. 10.1021/acs.inorgchem.0c00609. [DOI] [PubMed] [Google Scholar]
- In the 31P{1H} SSNMR spectrum of complex 1 an AB doublet is obserbed with trans 31P-31P coupling, J(PP) = 330 Hz.
- Johnson A.; Royle C. G.; Brodie C. N.; Martínez-Martínez A. J.; Duckett S. B.; Weller A. S. η2-Alkene Complexes of [Rh(PONOP-iPr)(L)]+ Cations (L = COD, NBD, Ethene). Intramolecular Alkene-Assisted Hydrogenation and Dihydrogen Complex [Rh(PONOP-iPr)(η-H2)]+. Inorg. Chem. 2021, 60, 13903–13912. 10.1021/acs.inorgchem.0c03687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos J.; Kundu S.; Pahls D. R.; Brookhart M.; Carmona E.; Cundari T. R. Mechanism of Hydrogenolysis of an Iridium–Methyl Bond: Evidence for a Methane Complex Intermediate. J. Am. Chem. Soc. 2013, 135, 1217–1220. 10.1021/ja310982v. [DOI] [PubMed] [Google Scholar]
- Hamilton D. G.; Crabtree R. H. An NMR Method for Distinguishing Classical from Nonclassical Structures in Transition Metal Polyhydrides. J. Am. Chem. Soc. 1988, 110, 4126–4133. 10.1021/ja00221a005. [DOI] [Google Scholar]
- Hebden T. J.; Goldberg K. I.; Heinekey D. M.; Zhang X.; Emge T. J.; Goldman A. S.; Krogh-Jespersen K. Dihydrogen/Dihydride or Tetrahydride? An Experimental and Computational Investigation of Pincer Iridium Polyhydrides. Inorg. Chem. 2010, 49, 1733–1742. 10.1021/ic902163w. [DOI] [PubMed] [Google Scholar]
- Perutz R. N.; Sabo-Etienne S. The σ-CAM Mechanism: σ-Complexes as the Basis of σ-Bond Metathesis at Late-Transition-Metal Centers. Angew. Chem., Int. Ed. 2007, 46, 2578–2592. 10.1002/anie.200603224. [DOI] [PubMed] [Google Scholar]
- Perutz R. N.; Sabo-Etienne S.; Weller A. S. Metathesis by Partner Interchange in σ-Bond Ligands: Expanding Applications of the σ-CAM Mechanism. Angew. Chem., Int. Ed. 2022, 61, e202111462. 10.1002/anie.202111462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlater M.; Schultz K. M.; Bernskoetter W. H.; Cartwright-Sykes A.; Heinekey D. M.; Brookhart M. Dihydrogen Complexes of Iridium and Rhodium. Inorg. Chem. 2012, 51, 4672–4678. 10.1021/ic202630x. [DOI] [PubMed] [Google Scholar]
- Both 2′ (the tetrahydride form of 2) and 4′ were located as minima on the electronic surface along with transition states linking then to 2 and 4, respectively. However, the inclusion of zero-point energy and thermochemical and entropy corrections suggested these would not be minima on the free energy surface. No local minimum corresponding to a Ir(I) bis-dihydrogen tautomer could be located. See the Supporting Information
- Johnson H. C.; McMullin C. L.; Pike S. D.; Macgregor S. A.; Weller A. S. Dehydrogenative Boron Homocoupling of an Amine-Borane. Angew. Chem., Int. Ed. 2013, 52, 9776–9780. 10.1002/anie.201304382. [DOI] [PubMed] [Google Scholar]
- Ledger A. E. W.; Ellul C. E.; Mahon M. F.; Williams J. M. J.; Whittlesey M. K. Ruthenium Bidentate Phosphine Complexes for the Coordination and Catalytic Dehydrogenation of Amine– and Phosphine–Boranes. Chem.-Eur. J. 2011, 17, 8704–8713. 10.1002/chem.201100101. [DOI] [PubMed] [Google Scholar]
- Adams G. M.; Colebatch A. L.; Skornia J. T.; McKay A. I.; Johnson H. C.; Lloyd-Jones G. C.; Macgregor S. A.; Beattie N. A.; Weller A. S. Dehydropolymerization of H3B·NMeH2 to Form Polyaminoboranes Using [Rh(Xantphos-Alkyl)] Catalysts. J. Am. Chem. Soc. 2018, 140, 1481–1495. 10.1021/jacs.7b11975. [DOI] [PubMed] [Google Scholar]
- Shimoi M.; Nagai S.-I.; Ichikawa M.; Kawano Y.; Katoh K.; Uruichi M.; Ogino H. Coordination Compounds of Monoborane–Lewis Base Adducts: Syntheses and Structures of [M(CO)5(η1-BH3·L)] (M = Cr, Mo, W; L = NMe3, PMe3, PPh3). J. Am. Chem. Soc. 1999, 121, 11704–11712. 10.1021/ja990828p. [DOI] [Google Scholar]
- Biswas S.; Huang Z.; Choliy Y.; Wang D. Y.; Brookhart M.; Krogh-Jespersen K.; Goldman A. S. Olefin Isomerization by Iridium Pincer Catalysts. Experimental Evidence for an η3-Allyl Pathway and an Unconventional Mechanism Predicted by DFT Calculations. J. Am. Chem. Soc. 2012, 134, 13276–13295. 10.1021/ja301464c. [DOI] [PubMed] [Google Scholar]
- Rimoldi M.; Fodor D.; van Bokhoven J. A.; Mezzetti A. Catalytic Hydrogenation of Liquid Alkenes with a Silica-Grafted Hydride Pincer Iridium(III) Complex: Support for a Heterogeneous Mechanism. Catal. Sci. Technol. 2015, 5, 4575–4586. 10.1039/C5CY00837A. [DOI] [Google Scholar]
- Rubio M.; Suárez A.; del Río D.; Galindo A.; Álvarez E.; Pizzano A. Rhodium Complexes with Pincer Diphosphite Ligands. Unusual Olefin in-Plane Coordination in Square-Planar Compounds. Organometallics 2009, 28, 547–560. 10.1021/om800830w. [DOI] [Google Scholar]
- Harris R. K.; Wasylishen R. E.; Duer M. J.. NMR Crystallography ;John Wiley & Sons: Chichester, England, 2009. [Google Scholar]
- Albrecht M.; Lutz M.; Schreurs A. M. M.; Lutz E. T. H.; Spek A. L.; van Koten G. Self-Assembled Organoplatinum(II) Supermolecules as Crystalline, SO2 Gas-Triggered Switches. J. Chem. Soc., Dalton Trans. 2000, 3797–3804. 10.1039/b006419j. [DOI] [Google Scholar]
- Braga D.; Cojazzi G.; Emiliani D.; Maini L.; Grepioni F. Reversible Gas–Solid Reactions between the Organometallic Zwitterion [(η5-C5H4COOH)(η5-C5H4COO)CoIII] and Vapors of Trifluoroacetic and Tetrafluoroboric Acids. Organometallics 2002, 21, 1315–1318. 10.1021/om010936w. [DOI] [Google Scholar]
- Albrecht M.; Lutz M.; Spek A. L.; van Koten G. Organoplatinum Crystals for Gas-Triggered Switches. Nature 2000, 406, 970–974. 10.1038/35023107. [DOI] [PubMed] [Google Scholar]
- Oliván M.; Marchenko A. V.; Coalter J. N.; Caulton K. G. Gas/Solid Reactivity of Unsaturated Ruthenium-Containing Molecular Solids. J. Am. Chem. Soc. 1997, 119, 8389–8390. 10.1021/ja971608j. [DOI] [Google Scholar]
- Braga D. Dynamical Processes in Crystalline Organometallic Complexes. Chem. Rev. 1992, 92, 633–665. 10.1021/cr00012a007. [DOI] [Google Scholar]
- The asymmetric unit contains one [BArF4]− anion and one [Ir(iPr-PONOP)(propene)]+ cation.
- Pregosin P. S.NMR in Organometallic Chemistry ;Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- This complexity can be intepreted as two sets of overlapping AB doublets with trans PP coupling, for which one is a very tightly coupled doublet that is a virtual singlet. See the Suppporting Information
- Martínez-Martínez A. J.; Royle C. G.; Furfari S. K.; Suriye K.; Weller A. S. Solid–State Molecular Organometallic Catalysis in Gas/Solid Flow (Flow-SMOM) as Demonstrated by Efficient Room Temperature and Pressure 1-Butene Isomerization. ACS Catal. 2020, 10, 1984–1992. 10.1021/acscatal.9b03727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick F. M.; Rees N. H.; Weller A. S.; Krämer T.; Iannuzzi M.; Macgregor S. A. A Rhodium–Pentane Sigma-Alkane Complex: Characterization in the Solid State by Experimental and Computational Techniques. Angew. Chem., Int. Ed. 2016, 55, 3677–3681. 10.1002/anie.201511269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay A. I.; Krämer T.; Rees N. H.; Thompson A. L.; Christensen K. E.; Macgregor S. A.; Weller A. S. Formation of a σ-Alkane Complex and a Molecular Rearrangement in the Solid-State: [Rh(Cyp2PCH2CH2PCyp2)(η2:η2-C7H12)][BArF4]. Organometallics 2017, 36, 22–25. 10.1021/acs.organomet.6b00645. [DOI] [Google Scholar]
- Bogadi R. S.; Levendis D. C.; Coville N. J. Solid-State Reaction Study of the Trans-to-Cis Isomerization of (η5-C5H4Me)Re(CO)[P(OPh)3]Br2: A New Mechanism for the Isomerization Reaction. J. Am. Chem. Soc. 2002, 124, 1104–1110. 10.1021/ja010695j. [DOI] [PubMed] [Google Scholar]
- Edwards A. J.; Burke N. J.; Dobson C. M.; Prout K.; Heyes S. J. Solid State NMR and X-Ray Diffraction Studies of Structure and Molecular Motion in Ansa-Titanocenes. J. Am. Chem. Soc. 1995, 117, 4637–4653. 10.1021/ja00121a021. [DOI] [Google Scholar]
- Gallagher A. T.; Lee J. Y.; Kathiresan V.; Anderson J. S.; Hoffman B. M.; Harris T. D. A Structurally-Characterized Peroxomanganese(IV) Porphyrin from Reversible O2 Binding within a Metal–Organic Framework. Chem. Sci. 2018, 9, 1596–1603. 10.1039/C7SC03739B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller M. S.; Haag A.; McKee V.; McKenzie C. J. No Sorption, in-Crystal Nitrite and Nitrate Production with Arylamine Oxidation in Gas–Solid Single Crystal to Single Crystal Reactions. Chem. Commun. 2019, 55, 10551–10554. 10.1039/C9CC05315H. [DOI] [PubMed] [Google Scholar]
- Ozerov O. V.; Guo C.; Papkov V. A.; Foxman B. M. Facile Oxidative Addition of N–C and N–H Bonds to Monovalent Rhodium and Iridium. J. Am. Chem. Soc. 2004, 126, 4792–4793. 10.1021/ja049659l. [DOI] [PubMed] [Google Scholar]
- Burgun A.; Coghlan C. J.; Huang D. M.; Chen W.; Horike S.; Kitagawa S.; Alvino J. F.; Metha G. F.; Sumby C. J.; Doonan C. J. Mapping-out Catalytic Processes in a Metal–Organic Framework with Single-Crystal X-Ray Crystallography. Angew. Chem., Int. Ed. 2017, 56, 8412–8416. 10.1002/anie.201611254. [DOI] [PubMed] [Google Scholar]
- Chaplin A. B.; Green J. C.; Weller A. S. C–C Activation in the Solid State in an Organometallic σ-Complex. J. Am. Chem. Soc. 2011, 133, 13162–13168. 10.1021/ja2047599. [DOI] [PubMed] [Google Scholar]
- Sethi N. K.; Whitwood A. C.; Bruce D. W. Sequential X-Ray-Induced Single-Crystal to Single-Crystal Transformation Followed by Topotactic Reduction in a Potassium Crown Ether Complex of Tetrachloroaurate(III). Inorg. Chem. 2018, 57, 13524–13532. 10.1021/acs.inorgchem.8b02130. [DOI] [PubMed] [Google Scholar]
- Attempts to model the change in the crystal lattice starting from 7, for example, by moving the pincer ligand in combination with adjusting the unit cell parameters, were unsuccessful.
- The lowest-energy transition states for propene insertion to form the n-propyl isomers 8n and 8n* were computed to be marginally higher than the isopropyl analogues (25.1 and 23.8 kcal/mol, respectively).
- Wang C.-H.; Das A.; Gao W.-Y.; Powers D. C. Probing Substrate Diffusion in Interstitial MOF Chemistry with Kinetic Isotope Effects. Angew. Chem., Int. Ed. 2018, 57, 3676–3681. 10.1002/anie.201713244. [DOI] [PubMed] [Google Scholar]
- Gordon B. M.; Lease N.; Emge T. J.; Hasanayn F.; Goldman A. S. Reactivity of Iridium Complexes of a Triphosphorus-Pincer Ligand Based on a Secondary Phosphine. Catalytic Alkane Dehydrogenation and the Origin of Extremely High Activity. J. Am. Chem. Soc. 2022, 144, 4133–4146. 10.1021/jacs.1c13309. [DOI] [PubMed] [Google Scholar]
- Benkovic S. J.; Hammes-Schiffer S. A Perspective on Enzyme Catalysis. Science 2003, 301, 1196–1202. 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.