

Summary
A growing body of human literature implicates KIBRA in memory and neurodevelopmental disorders. Memory and the cellular substrates supporting adaptive cognition change across development. Using an inducible KIBRA knockout mouse, we demonstrate that adult-onset deletion of KIBRA in forebrain neurons impairs long-term spatial memory and long-term potentiation (LTP). These LTP deficits correlate with adult-selective decreases in extrasynaptic AMPA receptors under basal conditions, and we identify a role for KIBRA in LTP-induced AMPAR upregulation. In contrast, juvenile-onset deletion of KIBRA in forebrain neurons did not affect LTP and had minimal effects on basal AMPAR expression. LTP did not increase AMPAR protein expression in juvenile WT mice, providing a potential explanation for juvenile resilience to KIBRA deletion. These data suggest that KIBRA serves a unique role in adult hippocampal function through regulation of basal and activity-dependent AMPAR proteostasis that supports synaptic plasticity.
Subject areas: Molecular neuroscience, Developmental neuroscience, Cellular neuroscience
Graphical abstract
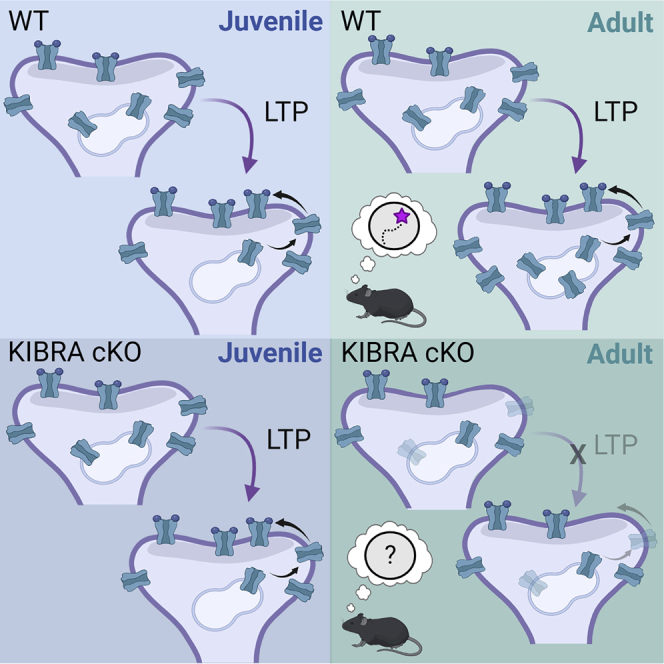
Highlights
-
•
KIBRA maintains basal expression of extrasynaptic AMPA receptors in the adult brain
-
•
Adult-onset Kibra deletion impairs memory, LTP, and LTP-induced AMPAR upregulation
-
•
Juvenile-onset Kibra deletion does not affect LTP
-
•
LTP does not induce AMPAR upregulation in juvenile WT or KIBRA cKO mice
Molecular neuroscience; Developmental neuroscience; Cellular neuroscience.
Introduction
Although it is clear that mnemonic function and information processing in the brain change across development, the mechanisms underlying these changes remain poorly understood. Several lines of evidence indicate that KIBRA (expressed in KIdney and BRAin; a.k.a. WWC1), a synaptically localized protein associated with human memory and multiple neurodevelopmental disorders, may provide insight into questions regarding differential brain function across development. The early postnatal period (1–2 years in humans, 1–3 weeks in rodents) is accompanied by rapid growth in synapse density and maturation of molecular composition of synapses.1,2 The closing of this period coincides with the late postnatal emergence of hippocampal-dependent memory3,4 and accurate representation of spatial and sequential information in the hippocampal network.5 Substantial effort has focused on understanding the synaptic changes that accompany this early period of postnatal development. However, hippocampal-dependent memory in rodents continues to mature toward adult-like performance throughout adolescence,3 and much less is known about changes in synaptic plasticity mechanisms across this critical adolescent developmental time period, which is the focus of this study. Childhood and adolescent development also coincide temporally with the onset of genetically overlapping neurodevelopmental disorders (NDDs) that have in common some form of synaptic dysfunction.6
Numerous studies demonstrate that common variants of KIBRA associate with human memory performance.7,8,9,10,11,12,13,14,15,16,17,18,19,20 Supporting a role in mnemonic function, reduction or overexpression of KIBRA impairs memory in rodents,21,22,23 and KIBRA expression is enriched in the human and rodent hippocampus,7,24 a brain structure critical for memory formation. Consistent with its effects on memory, deletion or overexpression of KIBRA impairs synaptic plasticity in rodents21,22 and blocks associative long term facilitation in Aplysia.25 However, despite robust early postnatal expression26 (Figure 1A), constitutive deletion of KIBRA impairs synaptic plasticity selectively in young adult (2–3.5-month-old) but not juvenile (3–4-week-old) mice.21 It is currently unclear if this distinction results from juvenile resilience and/or adult vulnerability to loss of KIBRA, or whether KIBRA is required for maturation of synaptic function during adolescence. Of interest, KIBRA polymorphisms associate with developmentally regulated disorders of complex cognition, including schizophrenia (SCZ)27 and Tourette,28 which also show adolescent or young adult onset. KIBRA is a postsynaptically localized scaffolding protein, and a large proportion of neuronally expressed KIBRA binding partners also associate with neurodevelopmental disorders including SCZ,29,30,31,32 bipolar disorder (BPD),33 and/or autism spectrum disorder (ASD).34,35
Figure 1.

Tamoxifen treatment rapidly reduces KIBRA expression in the hippocampus of juvenile KIBRA cKO mice
(A) Developmental expression profile of KIBRA in the hippocampus of WT mice.
(B) Experiment timeline for juvenile Kibrafloxed/floxed:CaMK2α CreERT2 mice. WT (Cre-negative, tamoxifen injected), WT’ (Cre-positive, vehicle injected), and cKO (Cre-positive, tamoxifen injected) mice received 1 injection (100mg/kg I.P.) per day for 3 days. Each mouse was given 5 days to recover from injections before experiments and tissue collection. Experiments were performed between P21-P25.
(C) Hippocampal CA1 tissue was isolated following tamoxifen or vehicle injections. Endogenous KIBRA protein levels were assessed using an anti-KIBRA antibody.
(D) Quantification normalized to VCP (one-way ANOVA, p = 0.010, post hoc comparisons shown in figure). WT, 100 ± 11%; WT′, 87 ± 17%; cKO, 40 ± 9%.
(E and F) Juvenile-onset KIBRA deletion does not affect expression of the KIBRA homolog WWC2 (unpaired t-test (total and membrane) or Mann-Whitney test (synaptic) corrected for multiple comparisons). Total, WT = 100 ± 3%, cKO = 106 ± 5%; membrane, WT = 100 ± 7%, cKO = 104 ± 14%; synaptic, WT = 100 ± 17%, cKO = 93 ± 21%. ∗p < 0.05, 0.05 <#p < 0.1, n.s. p> 0.1. Data plotted as mean ± SEM, n = number of animals, indicated on each bar.
Activity dependent changes in postsynaptic AMPAR (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor) concentration, localization, and function are highly conserved expression mechanisms of synaptic plasticity.36,37,38 In neurons, KIBRA concentrates at excitatory synapses and is enriched in membrane and postsynaptic density (PSD) fractions21,22 where it regulates trafficking of AMPA-type glutamate receptors.21,22,39 Although the specific mechanisms by which KIBRA regulates AMPAR trafficking are unknown, in non-neuronal cells KIBRA mediates recycling of endocytosed cargo through Rab11 positive recycling endosomes.40 KIBRA is a scaffolding protein with multiple protein and lipid interaction domains.41 The KIBRA interactome includes regulators of endosomal trafficking,21,40,42 actin21,43,44,45,46 and exocytic pathways,21,47,48 suggesting that KIBRA may regulate endosomal sorting of AMPARs. In addition, loss of KIBRA reduces the basal expression of multiple binding partners including PKMζ,23,25,49 Lats1/2,50 and Rab27a.47 However, it is currently unclear if KIBRA regulates AMPA proteostasis, or if KIBRA’s regulation of AMPA receptors changes with age.
Here, using inducible knockout to generate juvenile-onset or adult-onset deletion of KIBRA from CaMKIIα+ forebrain neurons, we demonstrate that loss of KIBRA causes depletion of extrasynaptic pools of AMPARs and impairs LTP selectively in the adult hippocampus. We identify a novel role for KIBRA in maintaining LTP-induced increases in AMPAR expression in the adult hippocampus, and show that loss of KIBRA expression in adult neurons impairs long-term memory.
Results
Synaptic plasticity in the juvenile brain is resilient to loss of KIBRA
Prior studies using a constitutive KIBRA KO model demonstrated that synaptic plasticity is impaired in adult (2–3.5 months old), but not juvenile (3–4 week old) mice.21 This is not because of lack of KIBRA expression at juvenile ages in WT mice. In fact, KIBRA is more robustly expressed in early postnatal development at the mRNA26 and protein (Figure 1A) levels. It is possible that developmental compensation renders the juvenile brain resilient to KIBRA loss. In support of this notion, the KIBRA homolog WWC2, whose function in neurons is unknown, is selectively upregulated in juvenile but not adult constitutive KIBRA KO mice.21 If constitutive deletion of KIBRA is compensated for by upregulation of other genes during early development, we hypothesized that rapid juvenile-onset deletion of KIBRA may allow us to observe changes in synaptic function before genetic compensation occurs. Thus, to examine whether postnatal KIBRA deletion in juvenile mice impacts synaptic function, we crossed conditional KibraFloxed/Floxed21 mice with CaMKII CreERT2 mice51 expressing tamoxifen-inducible Cre recombinase under the CaMKIIα promoter, generating mice in which genetic deletion of KIBRA is restricted primarily to excitatory forebrain neurons and is temporally controlled via tamoxifen injections, thus providing age, cell-type, and region-selective control of gene expression. Mice were injected with tamoxifen (Tam) or vehicle between P14-P16, a time at which CaMKII CreERT2 induction is fully functional.52 Synaptic transmission and plasticity were evaluated between P21-P25 (Figure 1B). Using this system, KIBRA protein expression is substantially reduced in KIBRA cKO mice (Tam-injected KibraFl/Fl:CaMKIICreERT2+) compared to WT mice (Tam-injected KibraFl/Fl:CaMKIICreERT2-) (Figures 1C and 1D). We note that although Cre-dependent KIBRA deletion is restricted to forebrain excitatory neurons, KIBRA is also expressed in a subset of inhibitory neurons and astrocytes,53,54,55 likely accounting for some residual KIBRA expression. We also find that Cre is not spontaneously active in juvenile CA1, as WT and WT’ (vehicle-injected KibraFl/Fl:CaMKIICreERT2+) mice show similar levels of KIBRA expression (Figures 1C and 1D). In contrast with the upregulation of WWC2 observed in juvenile constitutive KIBRA KO mice,21 juvenile KIBRA cKO mice express normal levels of WWC2 (Figures 1E and 1F) suggesting that long-term loss of KIBRA may produce compensatory upregulation of its homologs, but rapid postnatal deletion does not.
We next examined basal synaptic transmission in juvenile KIBRA cKO mice. Using extracellular field potential (fEPSP) recordings of Schaffer collateral synapses in acute hippocampal slices, we find that rapid deletion of KIBRA in CaMKIIα+ neurons of juvenile mice has no influence on the slope of the input-output curve across all experimental conditions (Figures 2A–2C). In addition, we observe no effect on paired-pulse facilitation at CA1 synapses (Figure 2D), indicating that presynaptic release probability is unaffected. Collectively, these data argue that reduction of KIBRA in the juvenile brain does not impact basal synaptic transmission. We then examined synaptic plasticity, finding that juvenile-onset reduction of KIBRA had no impact on LTP (Figures 2E–2G). These data establish that plasticity at juvenile synapses is independent of KIBRA function and further indicate that compensatory upregulation of WWC2, as observed in constitutive KIBRA KO mice,21 is unlikely to account for the resilience to loss of KIBRA in juvenile neurons.
Figure 2.

Juvenile-onset deletion of KIBRA does not affect basal synaptic transmission or long-term potentiation in the hippocampus
(A−C) Representative traces from input-output curves, scale bars = 2mV/5ms, (B) Summary data from juvenile input-output analysis, (C) Slopes of individual input-output curves were quantified; no differences were observed across experimental conditions (one-way ANOVA, n.s.). Mean I-O slope: WT, 4.18 ± 0.45ms−1; WT′, 3.92 ± 0.27 ms-1; cKO, 4.09 ± 0.49 ms−1.
(D) Paired pulse facilitation (fEPSP slope response 2/fEPSP slope response 1) is not altered in juvenile KIBRA cKO mice (RM two-way ANOVA, n.s. genotype X inter-stimulus interval interaction, main effect of genotype, and multiple comparisons at all inter-stimulus intervals).
(E) Hippocampal LTP induced by four trains of theta-burst stimulation is unchanged by juvenile-onset deletion of KIBRA. Scale bars= 0.25mV/5ms.
(F) STP magnitude is unaffected in juvenile KIBRA cKO mice (one-way ANOVA, n.s.). 5 min avg at gray bar, WT, 152 ± 7%; WT′, 148 ± 10%; cKO, 151 ± 7%.
(G) LTP magnitude measured at 65–70 min post LTP induction is unaffected by KIBRA deletion in the juvenile hippocampus (one-way ANOVA, n.s.). 5 min. Avg at gray bar, WT, 120 ± 3%; WT′, 125 ± 4%; cKO, 123 ± 5%. All summary data presented as mean ± SEM. B-D: WT, n = 14 slices from 4 mice; WT′, n = 7 slices from 2 mice; cKO, n = 12 slices from 4 mice. E–G: WT, n = 9 slices from 4 mice; WT′, n = 8 slices from 2 mice; cKO, n = 9 slices from 4 mice. ‘n.s.’ = p > 0.05.
Adult-onset deletion of neuronal KIBRA impairs long-term potentiation in the hippocampus
Having established a lack of impact of postnatal KIBRA deletion in juvenile mice, we next sought to determine if synaptic plasticity in the adult brain is vulnerable to adult-onset loss of KIBRA, and to disambiguate this from a role in synapse or circuit development. As older mice typically require longer and/or higher doses of tamoxifen for efficient gene excision with the CreERT2,56 we extended the tamoxifen exposure to five days to delete KIBRA in adult (2–4 month old) mice (Figure 3A). Adult KibraFl/Fl:CaMKIICreERT2+ mice that received tamoxifen injections showed approximately 70% reduction in KIBRA protein expression (Figures 3B and 3C), similar to the reduction observed in juvenile mice (% reduction in cKO mice, adult versus juvenile, unpaired t-test, p = 0.332). We also observe a modest reduction in KIBRA expression in vehicle-treated Cre-positive WT′ adult mice compared to Tam-treated Cre-negative WT mice, indicating some degree of spontaneous Cre activation in adult mice (Figures 3B and 3C).
Figure 3.

Tamoxifen treatment reduces hippocampal KIBRA expression in the adult brain
(A) Tamoxifen injection schedule for adult Kibrafloxed/floxed:CaMK2α CreERT2 mice. WT (Cre-negative, tamoxifen injected), WT’ (Cre-positive, vehicle injected), and cKO (Cre-positive, tamoxifen injected) mice received 2 injections (100mg/kg I.P.) per day for 5 days. Each mouse was given 14 days to recover from injections before experiments and tissue collection. Injections and experiments were performed after each mouse turned 2 months of age and before 4.5 months of age.
(B) Hippocampal CA1 tissue was isolated from Kibrafloxed/floxed:CaMK2α CreERT2 mice following tamoxifen or vehicle injections. Endogenous KIBRA protein levels were assessed using an anti-KIBRA antibody.
(C) Quantification normalized to VCP: One-way ANOVA with Holm-Sidak’s multiple comparisons test. (WT, 100 ± 10.58%; WT′, 60.44± 3.71%; cKO, 30.88 ± 3.39%). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. Data plotted as mean ± SEM, n = number of animals, indicated on each bar.
Similar to juvenile mice, the level of KIBRA expression in adult mice does not influence input-output relationship or paired pulse facilitation at Schaffer Collateral CA1 synapses (Figures 4A–4D), indicating that KIBRA expression is not required to maintain basal synaptic transmission in the adult brain, consistent with data from adult whole-body, constitutive KIBRA KO mice.21 To determine if KIBRA is selectively required for synaptic plasticity in the adult brain independent of potential neurodevelopmental confounds, we examined LTP in KIBRA cKO mice after adult-onset KIBRA deletion and found a significant reduction in LTP expression (Figures 4E and 4G). We also observe a trend toward decreased short-term potentiation (Figures 4E and F), a process that requires lateral diffusion of surface-localized extrasynaptic AMPARs.57 Importantly, the WT′ group shows LTP and STP similar to WT mice (Figures 4E–4G). In contrast to LTP expression, KIBRA deletion had no effect on charge transfer during the LTP induction stimulus (Theta-burst stimulation; TBS, Figures 4H and 4I), suggesting loss of KIBRA results in a deficit in LTP expression rather than NMDA receptor-dependent LTP induction. These data highlight the importance of neuronal KIBRA for expression of adult hippocampal synaptic plasticity, supporting an age-specific role for KIBRA in adaptive synaptic function.
Figure 4.

KIBRA deletion from the adult brain impairs hippocampal long-term potentiation without affecting basal synaptic transmission
(A−C) Representative traces from input-output curves, scale bars = 2mV/5ms, (B) Summary data from adult input-output analysis, (C) Slopes of individual input-output curves: WT, average I-O slope 4.98 ± 0.41 ms−1; WT′, average I-O slope 3.83 ± 0.36 ms−1; cKO, I-O slope 4.34 ± 0.39 ms−1. No difference observed across experimental conditions (one-way ANOVA, n.s.).
(D) Paired pulse facilitation (fEPSP slope response 2/fEPSP slope response1) is not altered in adult KIBRA cKO mice (repeated measures (RM) two-way ANOVA, n.s. genotype X inter-stimulus interval interaction, main effect of genotype, and multiple comparisons at all inter-stimulus intervals).
(E) Hippocampal LTP induced by four trains of theta-burst stimulation is impaired after adult-onset KIBRA deletion. Scale bars= 0.25mV/5ms.
(F) Trend toward decreased STP in adult KIBRA cKO mice (one-way ANOVA, p = 0.063). 5 min average at gray bar in E, WT, 174 ± 12%; WT′, 170 ± 13%; cKO, 141 ± 7.
(G) LTP magnitude measured at 75–80 min after LTP induction is decreased after deletion of KIBRA in the adult hippocampus (one-way ANOVA, p = 0.021). 5 min average at gray bar in E, WT, 130 ± 8%; WT′, 130 ± 7%; cKO, 110 ± 4%.
(H) LTP induction is not impaired in adult KIBRA cKO mice (one-way ANOVA, n.s. effect of genotype, genotype × train interaction, and multiple comparisons at all inter-stimulus intervals). LTP induction measured as charge transfer (Area UnderCurve, AUC) during each TBS train (40 fEPSPs per train, normalized to baseline fEPSP).
(I) Sample traces from LTP induction quantified in H. Shown is the first burst of the first train from one example recording (not averaged), Scale bar = 1.5mV/50 ms. All summary data presented as mean ± SEM. ∗p < 0.05, # 0.5 < p < 0.1, n.s. (not significant) = p > 0.1. B–D, WT, n = 14 slices from 6 mice; WT′, n = 10 slices from 3 mice; cKO, n = 16 slices from 6 mice. E–H, WT, n = 9 slices from 5 mice; WT′, n = 8 slices from 3 mice; cKO, n = 13 slices from 5 mice.
Loss of KIBRA from adult neurons reduces basal expression of extrasynaptic AMPARs in the hippocampus
We next sought to identify mechanisms which may contribute to reduction of LTP in neurons following adult-onset KIBRA deletion. AMPAR trafficking into and out of the synapse is a core mechanism driving the postsynaptic expression of synaptic plasticity.37,58 Neurons maintain distinct pools of AMPARs at excitatory synapses, including a synaptically localized pool which mediates neuronal communication, as well as extrasynaptic surface and intracellular pools that can be rapidly mobilized and inserted into the synapse to facilitate synaptic potentiation.57 KIBRA forms a complex with AMPARs21,22 and has been shown to prevent degradation of multiple binding partners,23,25,47,49,50 thus we hypothesized that loss of KIBRA disrupts AMPAR proteostasis and depletes AMPARs from extrasynaptic pools required for LTP. To test this hypothesis, we performed subcellular fractionation on hippocampal area CA1 tissue and examined total, membrane-localized, and synaptically localized (postsynaptic density-associated) AMPARs.59 As WT′ mice showed normal basal synaptic transmission and LTP, we focused further comparisons on KIBRA cKO and WT mice. Reduction of KIBRA in adult neurons decreases total expression of the AMPAR subunits GluA2 and GluA1 (Figures 5A and 5B) supporting a role for KIBRA in AMPAR proteostasis. Consistent with prior work, we also observe reduced PKMζ (Figure S1A), a brain-specific kinase that interacts with KIBRA.23,49,60 Adult-onset KIBRA deletion also decreased the basal expression of KIBRA homolog and binding partner WWC2 (Figures 5G and 5H). Total PSD95 expression was unchanged in cKO mice, indicating that loss of KIBRA does not broadly and non-selectively reduce synaptic protein expression (Figures 5A and 5B). GluA2 levels were also reduced in the membrane fraction of adult KIBRA cKO mice (Figures 5C and 5D), supporting the hypothesis that KIBRA facilitates maintenance of extrasynaptic AMPAR pools. GluA1 expression was not decreased in the KIBRA cKO membrane fraction, suggesting that under basal conditions GluA1 is depleted from intracellular stores. Synaptic expression of AMPARs was unchanged in KIBRA cKO mice (Figures 5E and 5F), consistent with our observation of normal basal synaptic transmission in these mice (Figures 4A–4D).
Figure 5.

KIBRA regulates the basal expression of extrasynaptic AMPA receptors in the adult hippocampus
(A, C, E, and G) Representative western blot images from sub-region CA1 of the adult hippocampus. Samples are normalized to loading control and quantified as % WT in B, D, F, and H (see STAR methods for details).
(B) Adult-onset KIBRA deletion from neurons decreases the total expression GluA2 and GluA1, but not the excitatory synaptic scaffold PSD-95 (unpaired t-tests, corrected for multiple comparisons). GluA2, WT = 100 ± 3%, cKO = 67 ±3; GluA1, WT = 100 ± 2%, cKO = 80 ± 4%; PSD95, WT = 100 ± 3%, cKO = 93 ± 3%.
(D) Loss of KIBRA from the adult brain decreases expression of membrane-localized GluA2 (unpaired t-tests with Welch’s correction, corrected for multiple comparisons). GluA2, WT = 100 ± 2%, cKO = 83 ± 5%; GluA1, WT = 100 ± 4%, cKO = 102 ± 2%; PSD95, WT = 100 ± 6%, cKO = 105 ± 4%.
(F) Adult-onset KIBRA deletion does not alter basal expression of synaptic AMPA receptors (unpaired Mann-Whitney tests, corrected for multiple comparisons). GluA2, WT = 100 ± 3%, cKO = 98 ± 6%; GluA1, WT = 100 ± 2%, cKO = 94 ± 8%; PSD95, WT = 100 ± 8%, cKO = 106 ± 5%.
(H) Decrease in expression of the KIBRA homolog WWC2 following adult-onset deletion of KIBRA (unpaired t-tests, corrected for multiple comparisons). Total, WT = 100 ± 4%, cKO = 70 ± 2%; membrane, WT = 100 ± 5%, cKO = 92 ± 6%; synaptic, WT = 100 ± 5%, cKO = 87 ± 7%.
(I) Representative western blot with equal protein loaded for total, cytosolic, membrane and synaptic fractions, demonstrating depletion of the postsynaptic scaffold PSD95 from the cytosolic fraction and enrichment in the membrane fraction with further enrichment in the synaptic fraction. Data shown as mean ± SEM, n on bar graphs = number of animals.
Juvenile-onset reduction of KIBRA has minimal effects on the basal expression of AMPAR complexes in the juvenile brain
To determine if normal LTP in juvenile KIBRA cKO mice corresponded with intact pools of extrasynaptic AMPARs, we investigated basal expression of AMPA receptors across subcellular fractions following juvenile-onset KIBRA deletion as described for adult mice.
We observed no changes in total, membrane-associated, or synaptic GluA1 (Figure 6). The excitatory synaptic scaffold PSD95 was similarly unaffected in juvenile KIBRA cKO mice. GluA2 expression was also unaffected in the membrane and synaptic fractions (Figures 6D–6G). Total GluA2 expression was modestly decreased in juvenile KIBRA cKO mice (Figures 6A and 6B), but this decrease was significantly smaller than was observed in adult KIBRA cKO mice (Figure 6C). Taken together, these data support the idea that dysregulation of extrasynaptic AMPAR pools contributes to impaired LTP in adult but not juvenile mice. Of interest, similar to adult-induced KIBRA cKO, we observe a decrease in PKMζ in juvenile KIBRA cKO mice (Figure S1B), suggesting that the decrease in PKMζ is unlikely to contribute to impaired LTP in adult KIBRA cKO mice.
Figure 6.

Juvenile-onset deletion of KIBRA has minimal effect on hippocampal AMPAR expression
(B, D, and F) Representative western blot images from sub-region CA1 of the juvenile hippocampus.
(A) Acute reduction of KIBRA in the juvenile hippocampus decreases total expression of AMPAR subunit GluA2 but not GluA1 or PSD95 (unpaired t-tests, corrected for multiple comparisons, Welch’s correction for GluA2). GluA2, WT = 100 ± 1%, cKO = 86 ±3; GluA1, WT = 100 ± 3%, cKO = 95 ± 4%; PSD95, WT = 100 ± 4%, cKO = 95 ± 3%.
(C) Larger decrease in total AMPAR expression in adult compared to juvenile KIBRA cKO mice (unpaired t-tests, corrected for multiple comparisons). For each group, data is shown as % decrease from respective WT (GluA2, juvenile cKO = −14 ± 4%, adult cKO = −33 ± 4%; GluA1, juvenile cKO = −3 ± 5%, adult cKO = −19 ± 4%).
(E) Juvenile-onset deletion of KIBRA does not affect expression of membrane-associated AMPARs or PSD95 in the juvenile hippocampus (unpaired t- (GluA1, GluA2) or Mann-Whitney (PSD95) tests, corrected for multiple comparisons). GluA2, WT = 100 ± 6%, cKO = 95 ± 4%; GluA1, WT = 100 ± 6%, cKO = 101 ± 11%; PSD95, WT = 100 ± 3%, cKO = 98 ± 17%.
(G) Loss of KIBRA in the juvenile brain does not alter basal expression of synaptic AMPA receptors or PSD95 (unpaired t- (GluA1, PSD95) or Mann-Whitney (GluA2) tests, corrected for multiple comparisons). GluA2, WT = 100 ± 14%, cKO = 89 ± 12%; GluA1, WT = 100 ± 7%, cKO = 85 ± 6%; PSD95, WT = 100 ± 7%, cKO = 96 ± 11%. Data shown as mean ± SEM, n on bar graphs = number of animals. ∗p < 0.05, ∗∗p < 0.01.
KIBRA is necessary for LTP-induced upregulation of AMPARs in the adult hippocampus
One mechanism by which neural activity can facilitate maintenance of synaptic potentiation is through rapid de novo synthesis of AMPARs.61,62,63 Although previous studies have examined the role of KIBRA in activity-dependent AMPAR trafficking via cell-wide pharmacological stimulation in cultured neurons with exogenously-expressed tagged AMPRs,21,22 it is unknown whether KIBRA regulates expression of endogenous AMPARs in response to physiological plasticity-inducing stimuli. Therefore, we examined whether KIBRA influences LTP-induced increases in endogenous AMPAR expression in the adult hippocampus. Acute hippocampal slices were subjected to basal stimulation (0.033Hz) or TBS LTP, which we have previously shown to be a protein synthesis-dependent form of synaptic plasticity,64 followed by microdissection of the stimulated area in CA1 at 30 or 120 min after LTP induction (Figure 7A). LTP induced an increase in GluA2 (Figures 7B and 7C) and GluA1 (Figures 7D and 7E) expression in WT but not KIBRA cKO mice. These data suggest that KIBRA not only prevents basal degradation of AMPARs, but is also necessary to maintain activity-induced increases in AMPAR expression after LTP.
Figure 7.

KIBRA is required for LTP-induced increases in AMPAR expression in the adult hippocampus
(A) Experimental design. Transverse hippocampal slices were collected from adult mice following basal stimulation (0.033 Hz) or LTP (TBS). The stimulated region of CA1 was microdissected 30 or 120 min after LTP or basal stimulation. Data from both time points was combined as no difference in AMPAR induction was observed between 30 and 120min post LTP.
(B–D) Representative GluA2 and GluA1 immunoblot images after baseline stimulation or LTP from adult WT (cre-negative tamoxifen treated) or KIBRA cKO (cre-positive tamoxifen treated) mice. LTP increases GluA2 (C) and GluA1 (E) expression in WT but not KIBRA cKO mice. Data plotted as % increase over baseline stimulation, mean ± SEM (GluA2, WT mean= 151 ± 15%, cKO = 100 ± 8%; GluA1, WT mean = 119 ± 8%, cKO = 101 ± 4%). LTP-stimulated slices were compared to baseline-stimulated slices from the same animal. Number of slices is indicated on each bar. One sample t-test, ∗p < 0.05, ∗∗p < 0.01.
LTP does not increase AMPAR expression in juvenile WT or KIBRA cKO mice
Intriguingly, when we performed similar experiments to examine the role of KIBRA in regulating activity-induced increases in AMPAR expression in the juvenile hippocampus, we found that LTP did not increase AMPAR expression in WT or KIBRA cKO mice (Figure 8). The absence of LTP-induced AMPAR upregulation in WT juvenile mice reveals a potential difference in LTP maintenance mechanisms between adult and juvenile mice and provides insight into why KIBRA function is dispensable for LTP in juvenile mice.
Figure 8.

LTP does not increase AMPAR expression in juvenile WT or KIBRA cKO mice
Transverse hippocampal slices were collected from juvenile mice following basal stimulation (0.033 Hz) or LTP (TBS). The stimulated region of CA1 was microdissected 30 min after LTP or basal stimulation.
(A and C) Representative GluA2 and GluA1 immunoblot images after baseline stimulation or LTP from juvenile WT (cre-negative tamoxifen treated) or KIBRA cKO (cre-positive tamoxifen treated) mice. LTP did not induce increases in total GluA2 (B) or GluA1 (D) expression in juvenile WT or KIBRA cKO mice. Data plotted as % increase over baseline stimulation, mean ± SEM (GluA2, WT mean = 108 ± 11%, cKO = 107 ± 7%; GluA1, WT mean = 106 ± 6%, cKO = 114 ± 7%). LTP-stimulated slices were compared to baseline-stimulated slices from the same animal. For each group, n = 18 slices from 10 mice. One sample Wilcoxon test, p > 0.05 for all comparisons shown.
Adult-onset reduction of KIBRA in forebrain neurons impairs spatial memory in adult mice
KIBRA expression is highest in excitatory neurons, but it is also present in a subset of inhibitory neurons and glial cells.53,54,55 The cell-type(s) in which KIBRA is required to support memory function are not known. Given its critical role in hippocampal LTP, we next determined the mnemonic role of KIBRA in CamKIIα+ forebrain neurons by assessing spatial memory in adult KIBRA cKO mice. Mice were trained to navigate to a rewarded escape box (target) on a modified Barnes Maze (Figure 9A). Training consisted of four trials per day over four consecutive days. We quantified the latency to reach the target on the first trial of each day as a measure of memory retention across training sessions. WT mice showed significant improvement from Day 1 to Day 2, with continued improvement thereafter (Figure 9B). However, KIBRA cKO littermates did not show significant improvement until Day 4, suggesting delayed memory retention across days during training (Figure 9B). When all trials across a day were combined, KIBRA cKO mice were not statistically different from WT littermates (Figure 9C). Thus, learning in KIBRA cKO mice was not abolished, and the impaired performance on trial 1 of Days 2 and 3 (Figure 9C) was not because of reduced levels of learning the prior day. We next tested long-term memory via a single probe trial (no target box) 7 days after the final training day (Figure 9D). Whereas WT mice showed significant preference for the target quadrant, KIBRA cKO mice had similar occupancy for the target and neighboring quadrants (Figure 9E), indicating an impairment in long-term spatial memory. KIBRA cKO mice also showed fewer cumulative entries behind the target wall (Figure 9F). Thus, KIBRA expression in CaMKIIα+ forebrain neurons appears necessary for maintenance of long-term spatial memory. Overall movement (Figures 9G and 9H), time spent in the center of the maze (Figure 9I), and weight reduction during time-restricted feeding (Figure 9J) were not different between genotypes, indicating that memory deficits in KIBRA cKO mice were not because of changes in locomotion, anxiety-like behaviors on the apparatus, or different responses to food restriction.
Figure 9.

Adult-onset KIBRA deletion from forebrain neurons impairs memory in adult mice
(A) Schematic of modified Barnes Maze arena. The target location is a covered box hidden from view by one of the identical walls positioned around the exterior of the maze. Training consisted of 4 trials per day over four days.
(B) KIBRA mice show delayed memory acquisition during training, assessed by comparing latency to reach the target box on the first trial of day 2,3, and 4 to the first trial on day 1 (Welch’s RM one-way ANOVA, cKO p = 0.0013, WT p = 0.0006, post-hoc comparisons shown in figure). Lines in violin plots = median.
(C) KIBRA cKO mice exhibit grossly normal learning as assessed by latency to reach the target box averaged across all 4 trials for each training day (RM two-way ANOVA, n.s. effect of genotype, genotype × day interaction, or WT vs KO post hoc comparison for any day). n = number of mice: WT, 6M + 5F; KIBRA cKO 8M +6F.
(D) Example maze occupancy (top) and trajectory (bottom) plots during memory retention probe test from two example mice of each genotype. ‘Max’ time for occupancy scale is indicated at the bottom left corner of each plot. Target location is as depicted in panel A.
(E) Percent occupancy per zone during memory retention (probe) test 7 days after the final training session. KIBRA cKO mice fail to show preference for target quadrant (Welch’s RM one-way ANOVA, cKO p = 0.0913, WT p = 0.0137, post-hoc comparisons shown in figure). Lines in violin plots = median.
(F) Decreased memory exhibited by KIBRA cKO mice shown by the cumulative number of entries behind the target wall over the first 5 entries during the probe trial (RM two-way ANOVA, p = 0.0484 effect of genotype).
(G and H) KIBRA cKO does not affect overall movement as shown by equivalent total distance traveled (G) and moving velocity (H, velocity when mice are moving > 3 cm/s) during the probe trial (unpaired t-test, corrected for multiple comparisons). Total velocity including pauses was also not different between genotypes (WT, 18 ±1 cm/s; cKO 16 ± 1 cm/s, ns).
(I) KIBRA cKO and WT mice spend the same amount of time in the center of the maze during the probe trial (unpaired Mann-Whitney test).
(J) No differences were observed between genotypes in weight lost because of time-restricted feeding (weight measured the last day of training/free feeding weight measured the day before maze habituation) (unpaired Mann-Whitney test). ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05, 0.05 <#p < 0.1, n.s. p> 0.1. Panels C,F,G,H,I,J, data plotted as mean ± SEM.
Discussion
KIBRA is associated with human memory7,8,9,10,11,12,13,14,15,16,17,18,19,20 as well as childhood and adolescent-emergent NDDs,27,28,29,30,31,32,33,34,35 highlighting KIBRA’s importance in cognitive function. However, the mechanisms through which KIBRA regulates adaptive cognition remain unclear. Prior work using constitutive whole-body deletion reported a role for KIBRA in plasticity in adult neurons.21 However, these studies could not dissociate whether embryonic loss of KIBRA directly impacted synaptic plasticity in mature cells or produced earlier deficits in neuronal development which later manifested as impairments in plasticity. Here, using conditional, adult- or juvenile-specific deletion of KIBRA, we demonstrate that KIBRA plays an acute role in synaptic plasticity in adult neurons. These data are consistent with a prior study that observed LTP deficits on KIBRA knockdown in organotypic hippocampal slice cultures,22 though it is difficult to evaluate temporal effects of in vivo development in cultured neurons. We find KIBRA expression in adult neurons is necessary for maintenance of extrasynaptic AMPARs and identify a novel role for KIBRA in LTP-induced upregulation of endogenous AMPAR expression. In contrast, synaptic plasticity and AMPAR expression in the juvenile brain are resilient to KIBRA deletion. Finally, our data suggest that KIBRA function in adult CaMKIIα+ (predominantly excitatory) forebrain neurons is necessary for effective memory retention.
Synaptic AMPARs are responsible for the majority of fast excitatory neurotransmission in the central nervous system. Extrasynaptic AMPARs localized to the plasma membrane and in intracellular endosomes maintain essential reservoirs for activity-dependent increases in synaptic AMPAR content.65,66 Under basal conditions, AMPARs intrinsically recycle into and out of the synaptic plasma membrane; internalized AMPARs destined for recycling back to the plasma membrane move from early to recycling endosomes, whereas receptors targeted for degradation are sorted into late endosomes for subsequent fusion with lysosomes.65,66,67,68 Thus, a shift in the balance of endosomal trafficking of AMPARs toward lysosomes can alter AMPAR proteostasis and availability of these channels for activity-induced synaptic insertion. We find that KIBRA is required to maintain extrasynaptic pools of AMPARs in adult neurons, consistent with prior studies demonstrating that KIBRA can stabilize its binding partners against degradation.23,25,47,49,50 Of interest, we also see a reduction in the KIBRA homolog WWC2 in adult KIBRA cKO neurons. The function of WWC2 in neurons is unknown, so it is not clear if decreased WWC2 expression might contribute to LTP deficits in adult neurons. However, WWC2 expression is unaffected in adult constitutive KIBRA KO mice (21, Figure S2) which show impaired LTP, arguing that reduced WWC2 expression is not necessary for LTP deficits observed in the absence of KIBRA.
One mechanism through which KIBRA may bias AMPAR trafficking away from lysosomes is through regulation of Rab family GTPases. Rab11 is enriched in recycling endosomes and disruption of Rab11 activity impairs AMPAR trafficking to the plasma membrane,65,69 consistent with our observation of decreased GluA2 in the membrane fraction following adult-onset KIBRA deletion. KIBRA regulates trafficking through recycling endosomes in non-neuronal cells40 and interacts with regulators of Rab11 activity in neurons.42 In non-neuronal cells, knockdown of KIBRA disrupts transport of transferrin receptors from early endosomes to Rab11-positive endocytic recycling compartments, resulting in lysosomal targeting and degradation of the receptors.40 In addition, KIBRA interacts with Rab27a,47 and other components of the exocyst complex21 that facilitate trafficking of newly synthesized AMPARs to synapses.70 Thus, our data support a model in which KIBRA directs endosomal AMPARs through recycling endosomes, away from late endosomes and lysosomal degradation. KIBRA also decreases ubiquitination of multiple binding partners23,47,50 and AMPAR ubiquitination directs AMPAR trafficking toward lysosomes.67 Thus, KIBRA may function through multiple pathways to maintain stable extrasynaptic pools of AMPARs. Elucidating these mechanisms is an exciting area for future study.
Phosphorylation is a well-established mechanism for regulating AMPAR trafficking and function.37,71 In addition to serving as a signaling scaffold for regulators of endosomal trafficking, KIBRA regulates a number of kinases50,60,72 including PKCγ73 which was recently reported to play an important role in regulating AMPAR phosphorylation and function73 . Identifying how different KIBRA-regulated signaling mechanisms intersect to regulate AMPARs, synaptic plasticity, and memory, and how such mechanisms change across development and celltype will be an important focus for future investigations.
Synaptic plasticity consists of distinct phases, each of which has unique mechanistic requirements. Upon LTP-inducing stimulation, extrasynaptic AMPARs residing in the plasma membrane are rapidly mobilized to synapses via lateral diffusion to support the early stages of LTP (STP).57 We observe a strong trend toward decreased STP in adult KIBRA cKO mice with no change in charge transfer during TBS induction, consistent with depleted pools of extrasynaptic surface AMPARs under basal conditions in these mice. Maintenance of later phases of LTP requires de novo protein synthesis.74,75 AMPARs are synthesized in response to LTP stimuli63 and newly synthesized AMPARs are recruited to dendritic spines in neurons activated by learning.61 We find that a protein-synthesis-dependent form of LTP64 induces rapid increases in AMPAR expression in WT, but not KIBRA cKO neurons. Considering that we observe a decrease in basal AMPAR expression and given prior work demonstrating that KIBRA prevents degradation of its binding partners, we hypothesize that KIBRA acts to prevent rapid degradation of newly synthesized AMPARs, resulting in impaired late-phase LTP in adult KIBRA cKO mice. However, our data do not rule out a role for KIBRA in activity-induced protein synthesis.43,76
We additionally confirmed that rapid postnatal deletion of KIBRA in neurons of juvenile mice has no observable impact on synaptic plasticity, consistent with the lack of effect previously reported for juvenile mice with constitutive KIBRA deletion.21 Together, these findings strongly suggest that juvenile synapses can produce and maintain synaptic plasticity via a KIBRA-independent mechanism. Juvenile mice with constitutive KIBRA deletion display elevated expression of the KIBRA homolog WWC2,21 prompting the hypothesis that WWC2 may compensate for the loss of KIBRA in juvenile mice. However, in our conditional model in which KIBRA is deleted postnatally from juvenile neurons, we do not observe a compensatory increase in WWC2. Thus, we conclude that WWC2 upregulation is not required for KIBRA-independent plasticity in juvenile neurons. Moreover, we demonstrate that, unlike in adult mice, extrasynaptic pools of AMPARs are largely intact in juvenile KIBRA cKO mice. Thus, normal plasticity in juvenile mice lacking KIBRA correlates with minimal changes in the maintenance of extrasynaptic AMPAR pools. It is unclear why KIBRA deletion has no impact on immature synaptic plasticity, although our data provide some insight as we demonstrate that LTP induces a KIBRA-dependent increase in AMPAR protein expression in the adult hippocampus but does not increase AMPAR protein expression in juvenile WT mice, suggesting an age-divergent mechanism for plasticity maintenance. Of interest, synapses from juvenile mice are also resilient to deletion of AMPAR subunits GluA1 or GluA2, as well as the AMPAR- and KIBRA-interacting protein PICK1, all of which produce impairments in plasticity when deleted from adult neurons.77,78,79 Together, these findings point to a potential broader principle that plasticity in juvenile neurons may utilize alternative or more robust mechanisms for LTP expression. In WT mice, KIBRA is strongly expressed at early developmental time points (Figure 1A, 26), arguing that KIBRA is likely playing an as-yet unidentified role in neuronal maturation. Indeed, neurons in juvenile KIBRA KO mice display morphological changes consistent with an early role for KIBRA that is distinct from its synaptic function in adult neurons.80
KIBRA protein complexes are associated with several NDDs.27,28,29,30,31,32,33,34,35 Recent genome-wide transcriptome analyses indicate that KIBRA is a developmentally regulated hub gene in ASD,34 highlighting the importance of identifying KIBRA’s function in the developing brain. Our work demonstrates that 21- to 25-day-old mice, an age roughly equivalent to a one-year-old human,1,81 display normal synaptic function and plasticity in the absence of KIBRA. Although the observed decrease in total GluA2 expression is significantly larger in adult KIBRA cKO mice, we see a modest decrease in total GluA2 expression in juvenile mice, suggesting that AMPAR proteostasis may become KIBRA-dependent during adolescence. It will be important in future work to determine the precise timeline for emergence of synaptic dysfunction in the absence of KIBRA, to identify non-synaptic functions of KIBRA during brain development, and to evaluate the function of KIBRA in other brain regions in which it is highly expressed (e.g., cortex).
In conclusion, we demonstrate an adult-selective role for KIBRA in maintaining proteostasis of extrasynaptic AMPARs under basal conditions, which correlates with impaired LTP in adult but not juvenile KIBRA cKO mice. We find that KIBRA in adult neurons is required for LTP-induced increases in endogenous AMPAR expression and for maintenance of accurate long-term memory, providing insight into the mechanisms by which this human memory-associated gene affects adaptive cognition.
Limitations of the study
A potential limitation of the current study is that KIBRA expression is not completely eliminated in our inducible conditional knockout model. It is likely that most residual KIBRA expression originates in CaMKIIα-negative inhibitory neurons and non-neuronal cells.53,54,55 However, protein quantification from bulk hippocampal tissue is not able to assess celltype-specific deletion. Thus, future studies examining the function of KIBRA in inhibitory neurons and non-neuronal brain cells will be important for a full understanding of KIBRA’s role in brain function. Importantly, the level of KIBRA reduction is similar for juvenile and adult KIBRA cKO mice. In addition, it is unlikely that intact LTP in juvenile KIBRA cKO mice is because of residual KIBRA expression given that juvenile constitutive KIBRA KO mice with no residual KIBRA expression also show intact LTP.21 Thus, regardless of this potential limitation, our data support an adult-specific role for KIBRA in synaptic plasticity. A second potential limitation of our inducible knockout model is a modest Cre leak observed in adult Cre + vehicle-injected mice. However, despite a 30–40% reduction in KIBRA in vehicle-injected WT′ mice, we observe no deficit in plasticity in this cohort. In addition, spontaneous cre activation is not observed juvenile Cre+ vehicle-injected mice (KIBRA expression is equivalent in Cre+ vehicle-treated and Cre-mice), demonstrating that Cre+ WT′ mice develop through the major period of synapse formation and maturation with normal levels of KIBRA expression. Finally, although our data provide strong support for the hypothesis that KIBRA prevents degradation of endogenous AMPARs in vivo, further analysis is required to elucidate the mechanisms by which KIBRA regulates AMPAR proteostasis, highlighting an exciting avenue for future research.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| KIBRA, mouse monoclonal | Richard Huganir | N/A |
| GluA2, mouse monoclonal | NeuroMab | Cat#L23/32 RRID:AB_2877267 |
| GluA2, rabbit polyclonal | Alomone Labs | Cat# AGC-005 RRID:AB_2039881 |
| GluA1, mouse monoclonal | NeuroMab | Cat# N355/1 RRID:AB_2877405 |
| PKC-zeta, rabbit polyclonal | Sigma Aldrich | Cat#: SAB4502380 RRID:AB_10746201 |
| WWC2, rabbit polyclonal | Sigma Aldrich | Cat# HPA044005, RRID:AB_10960552 |
| PSD-95, mouse monoclonal | NeuroMab | Cat# K28/43, RRID:AB_2877189 |
| GAPDH, rabbit monoclonal | Cell Signaling Technology | Cat# 2118, RRID:AB_561053 |
| GAPDH, chicken polyclonal | Millipore | Cat# AB2302, RRID:AB_10615768 |
| VCP, rabbit polyclonal | Cell Signaling Technology | Cat# 2648, RRID:AB_2214632 |
| Chemicals, peptides, and recombinant proteins | ||
| Tamoxifen | Sigma-Aldrich Selleck Chemicals |
Cat# T5648 Cat# S1238 |
| Odyssey Blocking Buffer (TBS) | Li-COR Biosciences | Cat#927-50000 |
| Starting Block (TBS) | Thermo Fisher Scientific | Cat# P137542 |
| Experimental models: Organisms/strains | ||
| KIBRA cKO | Makuch et al., this study | N/A |
| CaMKIIα-CreERT2 | Erdmann et al. | N/A |
| KIBRA KO | Makuch et al. | Strain# 024415 RRID:IMSR_JAX:024415 |
| Software and algorithms | ||
| GraphPad Prism | GraphPad by Dotmatics | https://www.graphpad.com |
| pCLAMP (clampex, clampfit) | Molecular Devices | https://www.moleculardevices.com/products/axon-patch-clamp-system/acquisition-and-analysis-software/pclamp-software-suite |
| Image Studio software | Li-COR | https://www.licor.com/bio/image-studio/ |
| Image Lab software | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| DeepLabCut | Mathis et al. | http://www.mackenziemathislab.org/deeplabcut-home |
| MATLAB | MathWorks | https://matlab.mathworks.com/ |
| MATLAB code for behavior analysis | This paper | https://github.com/Brad-E-Pfeiffer/KIBRABehaviorAnalysis |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lenora Volk (Lenora.Volk@utsouthwestern.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice were group housed in a climate-controlled environment on a 12 h (hr) light/dark cycle. Food and water was provided ad libitum, with the exception of food restriction prior to Barnes Maze training (detailed below). All mice were bred on the background of Charles River C57Bl/6 mice (N10+). Inducible CaMKIIa-CreERT2 hemizygous mice were bred with homozygous Kibra floxed/floxed21 mice, yielding Cre positive (Cre +) and Cre negative (Cre-) Kibra floxed/floxed experimental littermates. To induce Kibra knockout, adult mice (2–4 months old) underwent five-days of tamoxifen or vehicle (sunflower seed oil) treatment consisting of 2 daily injections (IntraPeritoneal, 100mg/kg) separated by approximately 7 h. Juvenile mice (P14 at day of first injection) underwent three-days of tamoxifen treatment consisting of 1 daily injection of tamoxifen (IP 100 mg/kg) or vehicle. Both male and female mice were used throughout all experiments. Sample size is indicated in each figure legend. All experiments were performed using protocols approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center.
Method details
Electrophysiology
Slice preparation
Mice were anesthetized with Isoflurane prior to rapid decapitation. 380μm transverse hippocampal slices were prepared using a Leica VT 1200s vibratome following dissection of the hippocampus in icecold oxygenated (95% O2/5% CO2) dissection buffer containing the following (in mM): 2.6 KCl, 1.25 NaH2PO4, 26 NaHCO3, 211 sucrose, 10 glucose, 0.75 CaCl2, 7MgCl2. Slices were recovered for at least 2h in 30°C aCSF containing the following (in mM): 125 NaCl, 3.25 KCl, 25 NaHCO3,1.25 NaH2PO4·H2O, 11 glucose, 2CaCl2, 1MgCl2.
Ex vivo slice electrophysiology
CA3 was removed prior to recording to prevent recurrent activity. Field excitatory postsynaptic potentials (fEPSPs) were digitally evoked (Cgynus Instruments, Model PG400A) at 0.033Hz with a 125μm platinum/iridium concentric bipolar electrode (FHC, Bowdoinham, ME) placed in the stratum radiatum, approximately 50 μm below the stratum pyramidale. Glass recording electrodes filled with aCSF were positioned in the stratum radiatum ∼250μm away (orthodromic) from the stimulating electrode. Signals were amplified by a differential amplifier (Model 1800; A-M Systems), digitized using an Axon Instruments Digidata 1550A (Molecular Devices), and monitored using pClamp Clampex software (Molecular Devices). Recording aCSF and temperature were identical to recovery conditions, with a flow rate of ∼3 mL/min. Input-output curves were obtained for each slice (fEPSP and fiber volley were collected at 5ms intervals from 10 to 60 μA), and responses were set to ∼45% max for LTP and paired-pulse ratio measurements. Paired-Pulse Ratios were recorded with inter-stimulus intervals of 25ms, 50ms, 100ms, 150ms, and 250ms.
Theta Burst (TBS) LTP: A stable baseline recording was obtained for a minimum of 20 min. LTP was then induced with TBS consisting of 4 trains separated by 10 s. Each train consisted of 10 bursts at 5Hz, with each burst containing 4 stimuli given at 100Hz.
AMPAR expression after TBS or basal stimulation
Slice preparation and recording conditions were as described above, however two stimulating electrodes were used in order to cover most of stratum radiuatum (see Figure 4A): one stimulating electrode was placed ∼50μm below the stratum pyramidale in proximal CA1(orthodromic to recording electrode) and the other ∼120μm below the stratum pyramidale in distal CA1 (antidromic to recording electrode). Input-out curves were obtained for each slice and responses were set at ∼45% maximum. LTP: slices received brief (3-5min.) baseline stimulation (0.033Hz) prior to and following TBS. Baseline stimulation: slices received brief baseline stimulation (0.033Hz) for approximately 10 min. Following stimulation slices were gently removed from the recording chamber and returned to the recovery chamber (oxygenated aCSF at 30°C) for 30 or 120 min. CA1 was then micro-dissected under a Lecia S6e stereomicroscope and flash frozen in liquid nitrogen. Slices were processed for western blot by homogenization in boiling (100°C) SDS protein sample buffer containing; 15% glycerol, 94mM Tris/HCl pH 6.8, 3% sodium dodecyl sulfate (SDS), 0.02% bromophenol blue, 2% beta-mercaptoethanol. Each sample was homogenized for 15 seconds using an electric pestle and boiled for 10 min. 10μL of homogenate was analyzed for AMPAR expression via western blot as described below.
Data analysis
Electrophysiological data was analyzed using Clampfit 10.7. For LTP experiments, the fEPSP slope of individual responses was normalized to the average fEPSP slope from the 20 min baseline immediately preceding TBS. LTP induction was assessed by measuring charge transfer (area under the curve) of each response from the end of the stimulation artifact + 9ms. For input-output curves, two responses were collected and averaged at each stimulation intensity. For PPR, four responses were collected and averaged at each inter-stimulus interval. Representative traces are averages of 4–8 individual responses, and stimulus artifacts have been removed for clarity.
Molecular analysis
Hippocampal dissection
Mice were briefly anaesthetized with isoflurane then rapidly decapitated. The brain was placed in icecold dissection buffer (in mM: 125 NaCl, 3.25 KCl, 25 NaHCO3, 1.25 NaH2PO4·H2O, 11 glucose, 0.75 CaCl2, 7 MgCl2) and the hippocampus was removed, followed by isolation of area CA1 from the dentate gyrus and CA3. Immediately following dissection CA1 was flash frozen in liquid nitrogen and stored at −80°C until processed as described below.
Tissue preparation for assessing KIBRA expression
Isolated dorsal CA1 tissue (both hemispheres from one mouse) was homogenized in 300 μL of homogenization buffer containing the following (in mM): 1% triton X-100, 50 Tris-HCl, pH7.5, 150 NaCl, 0.2 okadaic acid, 1 NaPPi, 5 NaF, 1 NaVO3, and Roche Complete mini protease inhibitor cocktail. Tissue was homogenized by repetitively passing the tissue through a 26-gauge needle and syringe. Protein concentration was determined by using the PierceTM Detergent Compatible Bradford Assay Kit and a BioTek Synergy H1 Microplate Reader. Samples were boiled for 10 min in SDS protein sample buffer containing the following: 10% glycerol, 62.5mM Tris/HCl pH 6.8, 2% sodium dodecyl sulfate (SDS), 0.01% bromophenol blue, 1.25% beta-mercaptoethanol.
Subcellular fractionation
Isolated CA1 tissue (both hemispheres from one mouse) was homogenized in 1 mL of homogenization buffer containing the following (in mM): 320 sucrose, 10 HEPES at pH 7.4, 1 EDTA, 0.2 okadaic acid, 1 NaPPi, 5 NaF, 1 NaVO3, Roche Complete Protease Inhibitor. 70 μL of homogenate were removed for analysis of total protein expression. The remaining homogenate was then centrifuged at 800 xg for 10 min at 4°C. The supernatant (S1) was transferred to VWR high G-force micro-centrifuge tubes and centrifuged at 15,000x g for 20 min at 4°C. The resultant supernatant (S2) was removed from the pellet (P2, membrane fraction), and the pellet was lysed in 500 μL of Milli-Q water with protease and phosphatase inhibitors containing the following (in mM): 0.2 okadaic acid, 1 NaPPi, 5 NaF, 1 NaVO3, Roche Complete Protease Inhibitor. To ensure complete lysis of P2, 2 μL of 1M HEPES at pH 7.4 was added to each sample followed by incubation with agitation for 30 min at 4°C. 70 μL were removed from each sample following incubation and stored for future analysis of the membrane fraction. The remaining lysed P2 fraction was then centrifuged at 25,000 xg for 20 min at 4°C. The lysed supernatant (LS1) was removed, and the lysed pellet (LP1) was resuspended in 250 μL of 50mM HEPES pH 7.4 with protease and phosphatase inhibitors. Each sample was then rapidly mixed with an equal volume of 250μL of 1% Triton X-100 with protease and phosphatase inhibitors and incubated with agitation at 4°C for 15 min. The resuspended LP1 was then centrifuged at 32,000 xg for 20 min at 4°C. The lysed supernatant (LS2) was discarded, thereby leaving the lysed pellet or PSD fraction. The PSD fraction was resuspended in 100μL of 50 mM HEPES pH 7.4 with proteasome and phosphatase inhibitors. Protein concentrations of each fraction were determined using the PierceTM Detergent Compatible Bradford Assay Kit. Samples were boiled for 10 min in SDS protein sample buffer containing the following: 10% glycerol, 62.5mM Tris/HCl pH 6.8, 2% sodium dodecyl sulfate (SDS), 0.01% bromophenol blue, 1.25% beta-mercaptoethanol.
Immunoblotting
To visualize the extent of the KIBRA KD, 45 μg of CA1 homogenate were loaded into 6% SDS-PAGE gels. Importantly, to verify that proteins were within a linear range for quantification, an in-gel protein concentration curve was performed by loading 50% (22.5 μg) and 150% (67.5 μg) of one sample. To ensure specificity of KIBRA Ab signal, tissue from a constitutive KIBRA knockout mouse was included, and showed no signal. Following separation, gels underwent a wet protein transfer to a PVDF membrane (Amersham Hybond P 0.45 PVDF) run in ice-cold transfer buffer at 100 V for 2 h. Membranes were blocked with a milk-based blocking solution containing the following: 1–2% milk and 0.5% TBST. The KIBRA signal was visualized using Amersham’s Highly Sensitive ECl Prime Reagent and the subsequent images were taken on a BioRad ChemiDoc. Images were taken every 30 seconds for a total exposure time of 30 min.
Hippocampal slices subjected to TBS/baseline stimulation and samples from the subcellular fractionation experiments used to examine AMPA receptor complex expression across the homogenate, plasma membrane, and synaptic fractions were loaded into either a 7% SDS-PAGE gel or 4–20% Gradient SDS- PAGE gel. Following separation, the gels underwent a wet protein transfer to a Nitrocellulose membrane (Odyssey Nitrocellulose Membrane, pore size 0.22 μm) run in ice-cold transfer buffer at 100 V for 2 h. The membranes were then incubated in Odyssey Blocking Buffer or Thermo Scientific StartingBlock (TBS) for 1 h. Following blocking, the membranes were incubated in primary antibody overnight at 4°C. Membranes were incubated with fluorescent secondary antibody for 1hat RT and imaged on a Li-Cor Odyssey Scanner (Model 9120) or Bio-Rad ChemiDoc MP.
Antibodies
Conditions for each antibody were independently optimized to determine the appropriate amount of protein needed to achieve signal within linear range for quantification, determined by in-gel concentration curves as described above. 10 μg of protein was loaded for all homogenate western blots. 5 μg of protein was loaded for both the membrane and synaptic fractions, with the exception of blots used for examining PSD95, in which 2 μg of protein was loaded for the membrane and synaptic fractions. Primary antibody dilutions: 1:500 for KIBRA and rabbit polyclonal GluA2, 1:1,000 for all other primary antibodies (see complete list in key resources table). Secondary antibodies used for detection: anti-mouse or anti-rabbit Licor IRDye 680RD or 800CWDonkey, anti-mouse IGg2a- AlexaFluor 647, HRP-conjugated anti-rabbit.
Data analysis
Western blot data was quantified using the Li-Cor image studio lite or Bio-Rad Image Studio software. Samples were normalized to a loading control (VCP, synaptophysin, or GAPDH) before being normalized to the average of all WT (Cre-negative tamoxifen-treated) samples within the same gel. Note that loading controls used for subcellular fractionation (synaptophysin and GAPDH) are present (though not enriched) in all fractions analyzed.82,83
Behavior
Apparatus
The modified Barnes Maze consisted of a 96cm diameter white circular platform with 11 equally spaced walls. Each wall was 13cm tall x 14cm wide, placed 8.5cm from the outside edge of the maze, and had a triangular back that allowed mice to explore behind the wall but prevented mice from traversing the maze by walking around the outside edge behind the walls (see Figure 5A). During training sessions, a covered target box (10.5cm long, 9cm wide, 9.5cm tall) was located behind one of the walls, and was not visible until the mouse walked behind the wall. ∼0.5cm3 pieces of peanut butter chip were placed at the back of the target box during training. Additionally, a physically inaccessible peanut butter chip was placed in the center of each wall barrier to prevent mice from using the smell of the peanut butter chip to navigate to the target box. The target box location relative to extra-maze room cues remained the same for all training sessions and probe trials. The start chamber was a 9cm cup with opaque sides. The maze was located 42cm above the floor in a brightly lit dedicated behavior room.
Food restriction and animal handling
Mice were handled 5 min per day for 5 days immediately prior to training. During the handling period, mice were introduced to peanut butter chips (one Reeses peanut butter chip per mouse each day). During training and on the day prior to the probe trial, mice underwent time-restricted feeding during which they were allowed ad lib access to food for two hours (∼6pm–8pm) per day.
Training and testing
Mice remained group housed throughout behavioral training and testing. The experimenter was blind to animal genotype and treatment condition. Behavior was conducted during the light cycle, between 9a.m. and 5p.m.
Day 0, habituation: Mice were placed in the center of the arena in an opaque start chamber. Mice were released from the start chamber 5 seconds after entering the arena, and were gently guided to the target box, and the entrance to the box was blocked. Mice remained in the target box for 2 min before being returned to their home cage.
Days 1–4, training: Mice were placed in the center of the arena in an opaque start chamber. Mice were released from the start chamber 5 seconds after entering the arena. Because mice can turn freely in the start chamber, mice began each trial facing a random direction. Mice were allowed 90 seconds to find the target box. Mice that did not enter within this time were gently guided to the target box by the experimenter. Mice remained in the closed target box for 30 seconds before being returned to the home cage. Mice underwent four training trials per day, with an inter-trial interval of 10 min. Mice were returned to holding cages between trials. The maze was cleaned with water and rotated after each trial to prevent mice from using scent cues to navigate to the target box. The target box was moved after each maze rotation such that it remained in the same location relative to the room cues.
Probe trial: The probe trial, conducted 7 days after the last training session, was identical to a training trial except that the target box was removed.
Data analysis
All training sessions and probe trials were continuously recorded on a SONY HDR-CX440 digital video camera. Videos were sampled at 30 FPS with a resolution of at least 1280 × 720. Mouse position was obtained using DeepLabCut84 to train a deep convolutional network. Briefly, a set of training frames were extracted from a representative subset of videos throughout the behavioral paradigm using a k means algorithm. Training frames were then manually labeled with the location of the animal’s body parts (head, snout, ears, and tail). After training, the model was evaluated using a train-test split to compute prediction error. Optimization was done through active refinement of poorly predicted frames and retraining to maintain high prediction accuracy across days and sessions. A final review of position estimation results was done using labeled videos to assess generalization across animals. The position data generated by Deeplabcut was then analyzed with custom MATLAB (MathWorks) programs, code available on GitHub, see key resources table. The number of entries behind each wall was scored by an experimenter blind to genotype (‘entry’ entailed the mouse’s entire body crossing the plane of the walls) and confirmed by automated MATLAB analysis of DeepLabCut-generated positional data. To calculate zone occupancy during the probe trial, the maze was divided into a 56cm diameter central zone, and the outer portion of the maze was divided into 11 equally sized sections centered each gap leading behind a wall. The target zone consisted of the section previously attached to the target box during training plus the sections on either side, the ‘right’ zone contained the three sections clockwise from the target zone, ‘opposite’ contained the three sections opposite the target zone, and ‘left’ contained the two sections counterclockwise from the target zone. Zone occupancy was normalized for the number of sections in the zone.
Sample size calculations
A priori sample size ranges were based on 1) power analysis using effect sizes estimated from our related previous experimental results and relevant published studies (G∗Power, paired and unpaired two-tailed t-tests and ANOVAs, target power 0.8, alpha 0.05) and, 2) considerable prior experience and many publications in the fields of, ex vivo electrophysiology, synaptic biochemistry, and animal behavior.
Exclusion criteria
For LTP experiments, slices that did not display a 20 min stable (less than ∼5% change) baseline were excluded. Baselines were evaluated by a separate experimenter blind to genotype/treatment type and with extensive electrophysiology experience. For behavior, one mouse (a cKO M) was removed during the experiment because the escape chamber was inadvertently included on the maze during the probe test.
Randomization and blinding
The random allocation of mice to experimental groups was driven by Mendelian Inheritance. To prevent confounders related to order or timing of electrophysiological recordings, recordings from mice of different genotypes were interleaved (e.g. WT on day 1, cKO on day 2, WT′ on day 3 and so on). For electrophysiological stimulation of slices to be used for AMPAR quantification, slices from two mice were stimulated each day (one mouse from each genotype), and the genotype sliced first alternated each day, with stimulation alternating between genotypes throughout the day to ensure similar recovery times for slices of each genotype. For behavioral experiments, the entire male cohort was trained and tested together, and subsequently the entire female cohort was trained and tested together. WT and KIBRA cKO mice were distributed throughout the running order to balance time-of-day effects, and the experimenter was blind to genotype during behavioral testing and training and analysis of maze occupancy and entry number times. The experimenter was not blind to genotype/treatment for electrophysiology and biochemistry experiments.
Quantification and statistical analysis
Statistical analysis was performed in Graph Pad Prism version 9.3.0–9.4.1. Data are plotted as mean ± SEM unless otherwise noted. The statistical test performed for each analysis is noted in the figure legend. p< 0.05 was considered significant. Data and test residuals were assessed for normality using Shapiro-wilk and D’Agostino-Pearson tests, in addition to visual inspection of QQ plots. Equality of variance between groups was assessed using an F test (t-tests) or Brown-Forsythe test (ANOVA), and examination of residual homoscedasticity plots. For data that did not meet criteria for equal variance a Welch’s correction was applied to the statistical test. Sphericity (equal variability of differences) was not assumed for repeated measures ANOVAs (Geisser-Greenhouse correction was applied). One or two-way ANOVAs with post hoc multiple comparison tests as needed or t-tests were used to evaluate significance. The Holm-Šidák correction was applied to all multiple comparisons (ANOVAs and multiple t-tests). Statistical tests were chosen based on sample size, hypotheses, and agreement with statistical assumptions. In the small number of instances where assumptions were not met for the above tests, nonparametric statistics were used (noted in figure legends).
Acknowledgments
This work was supported by National Institutes of Health Grant NIMH R01MH117149. M.L.M. was supported by the Howard Hughes Medical Institute Gilliam Fellowship for Advanced Study. L.Q. is supported by National Institutes of Health Grant F99NS120543. We thank Dr. Richard Huganir for generously sharing KIBRA cKO mice and KIBRA antibodies. We thank Dr. Brad Pfeiffer for manuscript feedback and assistance with data analysis. Graphical abstract created with BioRender.com
Author contributions
Conceptualization, M.L.M. and L.J.V.; Methodology, M.L.M. and L.J.V.; Formal analysis, M.L.M., L.D.Q., and L.J.V.; Investigation, M.L.M., T.D., and L.J.V; Writing – original draft, M.L.M. and L.J.V.; Writing – review and editing, M.L.M., L.D.Q., T.D., and L.J.V, Funding acquisition, M.L.M. and L.J.V., Supervision, L.J.V.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. One or more of the authors of this paper self-identifies as living with a disability. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. We worked to ensure sex balance in the selection of non-human subjects.
Published: December 22, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105623.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
Original code used for behavior analysis has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Semple B.D., Blomgren K., Gimlin K., Ferriero D.M., Noble-Haeusslein L.J. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013;106–107:1–16. doi: 10.1016/j.pneurobio.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lohmann C., Kessels H.W. The developmental stages of synaptic plasticity. J. Physiol. 2014;592:13–31. doi: 10.1113/jphysiol.2012.235119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alberini C.M., Travaglia A. Infantile amnesia: a critical period of learning to learn and remember. J. Neurosci. 2017;37:5783–5795. doi: 10.1523/jneurosci.0324-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dumas T.C. Late postnatal maturation of excitatory synaptic transmission permits adult-like expression of hippocampal-dependent behaviors. Hippocampus. 2005;15:562–578. doi: 10.1002/hipo.20077. [DOI] [PubMed] [Google Scholar]
- 5.Farooq U., Dragoi G. Emergence of preconfigured and plastic time-compressed sequences in early postnatal development. Science. 2019;363:168–173. doi: 10.1126/science.aav0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Volk L., Chiu S.L., Sharma K., Huganir R.L. Glutamate synapses in human cognitive disorders. Annu. Rev. Neurosci. 2015;38:127–149. doi: 10.1146/annurev-neuro-071714-033821. [DOI] [PubMed] [Google Scholar]
- 7.Papassotiropoulos A., Stephan D.A., Huentelman M.J., Hoerndli F.J., Craig D.W., Pearson J.V., Huynh K.D., Brunner F., Corneveaux J., Osborne D., et al. Common Kibra alleles are associated with human memory performance. Science. 2006;314:475–478. doi: 10.1126/science.1129837. [DOI] [PubMed] [Google Scholar]
- 8.Milnik A., Heck A., Vogler C., Heinze H.J., de Quervain D.J., Papassotiropoulos A. Association of KIBRA with episodic and working memory: a meta-analysis. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2012;159b:958–969. doi: 10.1002/ajmg.b.32101. [DOI] [PubMed] [Google Scholar]
- 9.Schaper K., Kolsch H., Popp J., Wagner M., Jessen F. KIBRA gene variants are associated with episodic memory in healthy elderly. Neurobiol. Aging. 2008;29:1123–1125. doi: 10.1016/j.neurobiolaging.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Preuschhof C., Heekeren H.R., Li S.C., Sander T., Lindenberger U., Backman L. KIBRA and CLSTN2 polymorphisms exert interactive effects on human episodic memory. Neuropsychologia. 2009;48:402–408. doi: 10.1016/j.neuropsychologia.2009.09.031. [DOI] [PubMed] [Google Scholar]
- 11.Bates T.C., Price J.F., Harris S.E., Marioni R.E., Fowkes F.G.R., Stewart M.C., Murray G.D., Whalley L.J., Starr J.M., Deary I.J. Association of KIBRA and memory. Neurosci. Lett. 2009;458:140–143. doi: 10.1016/j.neulet.2009.04.050. [DOI] [PubMed] [Google Scholar]
- 12.Almeida O.P., Schwab S.G., Lautenschlager N.T., Morar B., Greenop K.R., Flicker L., Wildenauer D. KIBRA genetic polymorphism influences episodic memory in later life, but does not increase the risk of mild cognitive impairment. J. Cell. Mol. Med. 2008;12:1672–1676. doi: 10.1111/j.1582-4934.2008.00229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rovira E., Mackie R.S., Clark N., Squire P.N., Hendricks M.D., Pulido A.M., Greenwood P.M. A role for attention during wilderness navigation: comparing effects of BDNF, KIBRA, and CHRNA4. Neuropsychology. 2016;30:709–719. doi: 10.1037/neu0000277. [DOI] [PubMed] [Google Scholar]
- 14.Vyas N.S., Ahn K., Stahl D.R., Caviston P., Simic M., Netherwood S., Puri B.K., Lee Y., Aitchison K.J. Association of KIBRA rs17070145 polymorphism with episodic memory in the early stages of a human neurodevelopmental disorder. Psychiatr. Res. 2014;220:37–43. doi: 10.1016/j.psychres.2014.07.024. [DOI] [PubMed] [Google Scholar]
- 15.Muse J., Emery M., Sambataro F., Lemaitre H., Tan H.Y., Chen Q., Kolachana B.S., Das S., Callicott J.H., Weinberger D.R., Mattay V.S. WWC1 genotype modulates age-related decline in episodic memory function across the adult life span. Biol. Psychiatr. 2014;75:693–700. doi: 10.1016/j.biopsych.2013.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duning K., Wennmann D.O., Bokemeyer A., Reissner C., Wersching H., Thomas C., Buschert J., Guske K., Franzke V., Flöel A., et al. Common exonic missense variants in the C2 domain of the human KIBRA protein modify lipid binding and cognitive performance. Transl. Psychiatry. 2013;3:e272. doi: 10.1038/tp.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kauppi K., Nilsson L.G., Adolfsson R., Eriksson E., Nyberg L. KIBRA polymorphism is related to enhanced memory and elevated hippocampal processing. J. Neurosci. 2011;31:14218–14222. doi: 10.1523/jneurosci.3292-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pawlowski T.L., Huentelman M.J. Identification of a common variant affecting human episodic memory performance using a pooled genome-wide association approach: a case study of disease gene identification. Methods Mol. Biol. 2011;700:261–269. doi: 10.1007/978-1-61737-954-3_17. [DOI] [PubMed] [Google Scholar]
- 19.Yasuda Y., Hashimoto R., Ohi K., Fukumoto M., Takamura H., Iike N., Yoshida T., Hayashi N., Takahashi H., Yamamori H., et al. Association study of KIBRA gene with memory performance in a Japanese population. World J. Biol. Psychiatr. 2010;11:852–857. doi: 10.3109/15622971003797258. [DOI] [PubMed] [Google Scholar]
- 20.Vassos E., Bramon E., Picchioni M., Walshe M., Filbey F.M., Kravariti E., McDonald C., Murray R.M., Collier D.A., Toulopoulou T. Evidence of association of KIBRA genotype with episodic memory in families of psychotic patients and controls. J. Psychiatr. Res. 2010;44:795–798. doi: 10.1016/j.jpsychires.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 21.Makuch L., Volk L., Anggono V., Johnson R.C., Yu Y., Duning K., Kremerskothen J., Xia J., Takamiya K., Huganir R.L. Regulation of AMPA receptor function by the human memory-associated gene KIBRA. Neuron. 2011;71:1022–1029. doi: 10.1016/j.neuron.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heitz F.D., Farinelli M., Mohanna S., Kahn M., Duning K., Frey M.C., Pavenstädt H., Mansuy I.M. The memory gene KIBRA is a bidirectional regulator of synaptic and structural plasticity in the adult brain. Neurobiol. Learn. Mem. 2016;135:100–114. doi: 10.1016/j.nlm.2016.07.028. [DOI] [PubMed] [Google Scholar]
- 23.Vogt-Eisele A., Krüger C., Duning K., Weber D., Spoelgen R., Pitzer C., Plaas C., Eisenhardt G., Meyer A., Vogt G., et al. KIBRA (KIdney/BRAin protein) regulates learning and memory and stabilizes Protein kinase Mzeta. J. Neurochem. 2014;128:686–700. doi: 10.1111/jnc.12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen-Institute_Brain-Atlas © Allen Institute for Brain Science. Allen Mouse Brain Atlas. 2004. https://mouse.brain-map.org/
- 25.Hu J., Ferguson L., Adler K., Farah C.A., Hastings M.H., Sossin W.S., Schacher S. Selective erasure of distinct forms of long-term synaptic plasticity underlying different forms of memory in the same postsynaptic neuron. Curr. Biol. 2017;27:1888–1899.e4. doi: 10.1016/j.cub.2017.05.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johannsen S., Duning K., Pavenstädt H., Kremerskothen J., Boeckers T.M. Temporal-spatial expression and novel biochemical properties of the memory-related protein KIBRA. Neuroscience. 2008;155:1165–1173. doi: 10.1016/j.neuroscience.2008.06.054. [DOI] [PubMed] [Google Scholar]
- 27.Kos M.Z., Carless M.A., Peralta J., Blackburn A., Almeida M., Roalf D., Pogue-Geile M.F., Prasad K., Gur R.C., Nimgaonkar V., et al. Exome sequence data from multigenerational families implicate AMPA receptor trafficking in neurocognitive impairment and schizophrenia risk. Schizophr. Bull. 2016;42:288–300. doi: 10.1093/schbul/sbv135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willsey A.J., Fernandez T.V., Yu D., King R.A., Dietrich A., Xing J., Sanders S.J., Mandell J.D., Huang A.Y., Richer P., et al. De novo coding variants are strongly associated with tourette disorder. Neuron. 2017;94:486–499.e9. doi: 10.1016/j.neuron.2017.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fromer M., Roussos P., Sieberts S.K., Johnson J.S., Kavanagh D.H., Perumal T.M., Ruderfer D.M., Oh E.C., Topol A., Shah H.R., et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016;19:1442–1453. doi: 10.1038/nn.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dev K.K., Henley J.M. The schizophrenic faces of PICK1. Trends Pharmacol. Sci. 2006;27:574–579. doi: 10.1016/j.tips.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lauriat T.L., Dracheva S., Kremerskothen J., Duning K., Haroutunian V., Buxbaum J.D., Hyde T.M., Kleinman J.E., McInnes L.A. Characterization of KIAA0513, a novel signaling molecule that interacts with modulators of neuroplasticity, apoptosis, and the cytoskeleton. Brain Res. 2006;1121:1–11. doi: 10.1016/j.brainres.2006.08.099. [DOI] [PubMed] [Google Scholar]
- 32.Hakak Y., Walker J.R., Li C., Wong W.H., Davis K.L., Buxbaum J.D., Haroutunian V., Fienberg A.A. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. USA. 2001;98:4746–4751. doi: 10.1073/pnas.081071198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hou L., Bergen S.E., Akula N., Song J., Hultman C.M., Landén M., Adli M., Alda M., Ardau R., Arias B., et al. Genome-wide association study of 40, 000 individuals identifies two novel loci associated with bipolar disorder. Hum. Mol. Genet. 2016;25:3383–3394. doi: 10.1093/hmg/ddw181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parikshak N.N., Swarup V., Belgard T.G., Irimia M., Ramaswami G., Gandal M.J., Hartl C., Leppa V., Ubieta L.D.L.T., Huang J., et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature. 2016;540:423–427. doi: 10.1038/nature20612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voineagu I., Wang X., Johnston P., Lowe J.K., Tian Y., Horvath S., Mill J., Cantor R.M., Blencowe B.J., Geschwind D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huganir R.L., Nicoll R.A. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diering G.H., Huganir R.L. The AMPA receptor code of synaptic plasticity. Neuron. 2018;100:314–329. doi: 10.1016/j.neuron.2018.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shepherd J.D., Huganir R.L. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- 39.Tracy T.E., Sohn P.D., Minami S.S., Wang C., Min S.-W., Li Y., Zhou Y., Le D., Lo I., Ponnusamy R., et al. Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy-related memory loss. Neuron. 2016;90:245–260. doi: 10.1016/j.neuron.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Traer C.J., Rutherford A.C., Palmer K.J., Wassmer T., Oakley J., Attar N., Carlton J.G., Kremerskothen J., Stephens D.J., Cullen P.J. SNX4 coordinates endosomal sorting of TfnR with dynein-mediated transport into the endocytic recycling compartment. Nat. Cell Biol. 2007;9:1370–1380. doi: 10.1038/ncb1656. [DOI] [PubMed] [Google Scholar]
- 41.Zhang L., Yang S., Wennmann D.O., Chen Y., Kremerskothen J., Dong J. KIBRA: in the brain and beyond. Cell. Signal. 2014;26:1392–1399. doi: 10.1016/j.cellsig.2014.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fukuda T., Nagashima S., Inatome R., Yanagi S. CAMDI interacts with the human memory-associated protein KIBRA and regulates AMPAR cell surface expression and cognition. PLoS One. 2019;14:e0224967. doi: 10.1371/journal.pone.0224967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duning K., Schurek E.M., Schlüter M., Bayer M., Reinhardt H.C., Schwab A., Schaefer L., Benzing T., Schermer B., Saleem M.A., et al. KIBRA modulates directional migration of podocytes. J. Am. Soc. Nephrol. 2008;19:1891–1903. doi: 10.1681/asn.2007080916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rocca D.L., Martin S., Jenkins E.L., Hanley J.G. Inhibition of Arp2/3-mediated actin polymerization by PICK1 regulates neuronal morphology and AMPA receptor endocytosis. Nat. Cell Biol. 2008;10:259–271. doi: 10.1038/ncb1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kremerskothen J., Kindler S., Finger I., Veltel S., Barnekow A. Postsynaptic recruitment of Dendrin depends on both dendritic mRNA transport and synaptic anchoring. J. Neurochem. 2006;96:1659–1666. doi: 10.1111/j.1471-4159.2006.03679.x. [DOI] [PubMed] [Google Scholar]
- 46.Kremerskothen J., Plaas C., Büther K., Finger I., Veltel S., Matanis T., Liedtke T., Barnekow A. Characterization of KIBRA, a novel WW domain-containing protein. Biochem. Biophys. Res. Commun. 2003;300:862–867. doi: 10.1016/s0006-291x(02)02945-5. [DOI] [PubMed] [Google Scholar]
- 47.Song L., Tang S., Han X., Jiang Z., Dong L., Liu C., Liang X., Dong J., Qiu C., Wang Y., Du Y. KIBRA controls exosome secretion via inhibiting the proteasomal degradation of Rab27a. Nat. Commun. 2019;10:1639. doi: 10.1038/s41467-019-09720-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosse C., Formstecher E., Boeckeler K., Zhao Y., Kremerskothen J., White M.D., Camonis J.H., Parker P.J. An aPKC-exocyst complex controls paxillin phosphorylation and migration through localised JNK1 activation. PLoS Biol. 2009;7:e1000235. doi: 10.1371/journal.pbio.1000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferguson L., Hu J., Cai D., Chen S., Dunn T.W., Pearce K., Glanzman D.L., Schacher S., Sossin W.S. Isoform specificity of PKMs during long-term facilitation in Aplysia is mediated through stabilization by KIBRA. J. Neurosci. 2019;39:8632–8644. doi: 10.1523/jneurosci.0943-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao L., Chen Y., Ji M., Dong J. KIBRA regulates Hippo signaling activity via interactions with large tumor suppressor kinases. J. Biol. Chem. 2011;286:7788–7796. doi: 10.1074/jbc.M110.173468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Erdmann G., Schütz G., Berger S. Inducible gene inactivation in neurons of the adult mouse forebrain. BMC Neurosci. 2007;8:63. doi: 10.1186/1471-2202-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roszkowska M., Krysiak A., Majchrowicz L., Nader K., Beroun A., Michaluk P., Pekala M., Jaworski J., Kondrakiewicz L., Puścian A., et al. SRF depletion in early life contributes to social interaction deficits in the adulthood. Cell. Mol. Life Sci. 2022;79:278. doi: 10.1007/s00018-022-04291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cembrowski M.S., Wang L., Sugino K., Shields B.C., Spruston N. Hipposeq: a comprehensive RNA-seq database of gene expression in hippocampal principal neurons. Elife. 2016;5:e14997. doi: 10.7554/eLife.14997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Habib N., Li Y., Heidenreich M., Swiech L., Avraham-Davidi I., Trombetta J.J., Hession C., Zhang F., Regev A. Div-Seq: single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 2016;353:925–928. doi: 10.1126/science.aad7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Allen-Institute_Cell-Types © 2015 Allen Institute for Brain Science. Allen cell types database. 2015. https://celltypes.brain-map.org/rnaseq/mouse/v1-alm
- 56.Feil S., Valtcheva N., Feil R. Inducible cre mice. Methods Mol. Biol. 2009;530:343–363. doi: 10.1007/978-1-59745-471-1_18. [DOI] [PubMed] [Google Scholar]
- 57.Penn A.C., Zhang C.L., Georges F., Royer L., Breillat C., Hosy E., Petersen J.D., Humeau Y., Choquet D. Hippocampal LTP and contextual learning require surface diffusion of AMPA receptors. Nature. 2017;549:384–388. doi: 10.1038/nature23658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choquet D. Linking nanoscale dynamics of AMPA receptor organization to plasticity of excitatory synapses and learning. J. Neurosci. 2018;38:9318–9329. doi: 10.1523/jneurosci.2119-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chiu S.L., Diering G.H., Ye B., Takamiya K., Chen C.M., Jiang Y., Niranjan T., Schwartz C.E., Wang T., Huganir R.L. GRASP1 regulates synaptic plasticity and learning through endosomal recycling of AMPA receptors. Neuron. 2017;93:1405–1419.e8. doi: 10.1016/j.neuron.2017.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Büther K., Plaas C., Barnekow A., Kremerskothen J. KIBRA is a novel substrate for protein kinase Czeta. Biochem. Biophys. Res. Commun. 2004;317:703–707. doi: 10.1016/j.bbrc.2004.03.107. [DOI] [PubMed] [Google Scholar]
- 61.Matsuo N., Reijmers L., Mayford M. Spine-type-specific recruitment of newly synthesized AMPA receptors with learning. Science. 2008;319:1104–1107. doi: 10.1126/science.1149967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tushev G., Glock C., Heumüller M., Biever A., Jovanovic M., Schuman E.M. Alternative 3' UTRs modify the localization, regulatory potential, stability, and plasticity of mRNAs in neuronal compartments. Neuron. 2018;98:495–511.e6. doi: 10.1016/j.neuron.2018.03.030. [DOI] [PubMed] [Google Scholar]
- 63.Nayak A., Zastrow D.J., Lickteig R., Zahniser N.R., Browning M.D. Maintenance of late-phase LTP is accompanied by PKA-dependent increase in AMPA receptor synthesis. Nature. 1998;394:680–683. doi: 10.1038/29305. [DOI] [PubMed] [Google Scholar]
- 64.Volk L.J., Bachman J.L., Johnson R., Yu Y., Huganir R.L. PKM-zeta is not required for hippocampal synaptic plasticity, learning and memory. Nature. 2013;493:420–423. doi: 10.1038/nature11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bourke A.M., Bowen A.B., Kennedy M.J. New approaches for solving old problems in neuronal protein trafficking. Mol. Cell. Neurosci. 2018;91:48–66. doi: 10.1016/j.mcn.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parkinson G.T., Hanley J.G. Mechanisms of AMPA receptor endosomal sorting. Front. Mol. Neurosci. 2018;11:440. doi: 10.3389/fnmol.2018.00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Widagdo J., Chai Y.J., Ridder M.C., Chau Y.Q., Johnson R.C., Sah P., Huganir R.L., Anggono V. Activity-dependent ubiquitination of GluA1 and GluA2 regulates AMPA receptor intracellular sorting and degradation. Cell Rep. 2015;10:783–795. doi: 10.1016/j.celrep.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moretto E., Passafaro M. Recent findings on AMPA receptor recycling. Front. Cell. Neurosci. 2018;12:286. doi: 10.3389/fncel.2018.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bowen A.B., Bourke A.M., Hiester B.G., Hanus C., Kennedy M.J. Golgi-independent secretory trafficking through recycling endosomes in neuronal dendrites and spines. Elife. 2017;6:e27362. doi: 10.7554/eLife.27362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gerges N.Z., Backos D.S., Rupasinghe C.N., Spaller M.R., Esteban J.A. Dual role of the exocyst in AMPA receptor targeting and insertion into the postsynaptic membrane. EMBO J. 2006;25:1623–1634. doi: 10.1038/sj.emboj.7601065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Diering G.H., Heo S., Hussain N.K., Liu B., Huganir R.L. Extensive phosphorylation of AMPA receptors in neurons. Proc. Natl. Acad. Sci. USA. 2016;113:E4920–E4927. doi: 10.1073/pnas.1610631113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang L., Iyer J., Chowdhury A., Ji M., Xiao L., Yang S., Chen Y., Tsai M.Y., Dong J. KIBRA regulates aurora kinase activity and is required for precise chromosome alignment during mitosis. J. Biol. Chem. 2012;287:34069–34077. doi: 10.1074/jbc.M112.385518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tian M., Chen Q., Graves A.R., Goldschmidt H.L., Johnson R.C., Weinberger D.R., Huganir R.L. KIBRA-PKCγ signaling pathway modulates memory performance in mice and humans. bioRxiv. 2021 doi: 10.1101/2021.10.26.465926. Preprint at. [DOI] [Google Scholar]
- 74.Bradshaw K.D., Emptage N.J., Bliss T.V.P. A role for dendritic protein synthesis in hippocampal late LTP. Eur. J. Neurosci. 2003;18:3150–3152. doi: 10.1111/j.1460-9568.2003.03054.x. [DOI] [PubMed] [Google Scholar]
- 75.Abraham W.C., Williams J.M. LTP maintenance and its protein synthesis-dependence. Neurobiol. Learn. Mem. 2008;89:260–268. doi: 10.1016/j.nlm.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 76.Deller T., Korte M., Chabanis S., Drakew A., Schwegler H., Stefani G.G., Zuniga A., Schwarz K., Bonhoeffer T., Zeller R., et al. Synaptopodin-deficient mice lack a spine apparatus and show deficits in synaptic plasticity. Proc. Natl. Acad. Sci. USA. 2003;100:10494–10499. doi: 10.1073/pnas.1832384100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Volk L., Kim C.-H., Takamiya K., Yu Y., Huganir R.L. Developmental regulation of protein interacting with C kinase 1 (PICK1) function in hippocampal synaptic plasticity and learning. Proc. Natl. Acad. Sci. USA. 2010;107:21784–21789. doi: 10.1073/pnas.1016103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jensen V., Kaiser K.M.M., Borchardt T., Adelmann G., Rozov A., Burnashev N., Brix C., Frotscher M., Andersen P., Hvalby Ø., et al. A juvenile form of postsynaptic hippocampal long-term potentiation in mice deficient for the AMPA receptor subunit GluR-A. J. Physiol. 2003;553:843–856. doi: 10.1113/jphysiol.2003.053637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cao F., Zhou Z., Cai S., Xie W., Jia Z. Hippocampal long-term depression in the presence of calcium-permeable AMPA receptors. Front. Synaptic Neurosci. 2018;10:41. doi: 10.3389/fnsyn.2018.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blanque A., Repetto D., Rohlmann A., Brockhaus J., Duning K., Pavenstädt H., Wolff I., Missler M. Deletion of KIBRA, protein expressed in kidney and brain, increases filopodial-like long dendritic spines in neocortical and hippocampal neurons in vivo and in vitro. Front. Neuroanat. 2015;9:13. doi: 10.3389/fnana.2015.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dutta S., Sengupta P. Men and mice: relating their ages. Life Sci. 2016;152:244–248. doi: 10.1016/j.lfs.2015.10.025. [DOI] [PubMed] [Google Scholar]
- 82.Frederikse P.H., Nandanoor A., Kasinathan C. "Moonlighting" GAPDH protein localizes with AMPA receptor GluA2 and L1 axonal cell adhesion molecule at fiber cell borders in the lens. Curr. Eye Res. 2016;41:41–49. doi: 10.3109/02713683.2014.997886. [DOI] [PubMed] [Google Scholar]
- 83.Jo K., Derin R., Li M., Bredt D.S. Characterization of MALS/Velis-1, -2, and -3: a family of mammalian LIN-7 homologs enriched at brain synapses in association with the postsynaptic density-95/NMDA receptor postsynaptic complex. J. Neurosci. 1999;19:4189–4199. doi: 10.1523/jneurosci.19-11-04189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mathis A., Mamidanna P., Cury K.M., Abe T., Murthy V.N., Mathis M.W., Bethge M. DeepLabCut: markerless pose estimation of user-defined body parts with deep learning. Nat. Neurosci. 2018;21:1281–1289. doi: 10.1038/s41593-018-0209-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
Original code used for behavior analysis has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.