

Abstract
Pancreatic cancer (PC) has exceptionally high mortality due to ineffective treatment strategies. Immunotherapy, which mobilizes the immune system to fight against cancer, has been proven successful in multiple cancers; however, its application in PC has met with limited success. In this review, we articulated that the pancreatic tumor microenvironment is immuno-suppressive with extensive infiltration by M2-macrophages and myeloid-derived suppressive cells but low numbers of cytotoxic T-cells. In addition, low mutational load and poor antigen processing, presentation, and recognition contribute to the limited response to immunotherapy in PC. Immune checkpoints, the critical targets for immunotherapy, have high expression in PC and stromal cells, regulated by tumor microenvironmental milieu (cytokine and metabolites) and cell-intrinsic mechanisms (epigenetic regulation, oncogenic signaling, and post-translational modifications). Combining immunotherapy with modulators of the tumor microenvironment may facilitate the development of novel therapeutic regimens to manage PC.
Keywords: Pancreatic cancer, Immunotherapy, Immune checkpoint, Tumor microenvironment, Cancer-associated fibroblast, Regulation network, Cytokine, Combination therapy
1. Introduction
Cancer immunotherapy aims to stimulate the patient’s immune system to fight cancer. The past decade has observed significant advancements in immunotherapy by unleashing the immune system’s potential to combat tumors [1]. Immunotherapy has emerged as a critical strategy to treat cancer in combination with available therapeutic regimens. Despite its success in various cancers, like melanoma, lung cancer, and renal cancer, immunotherapy has been unsuccessful in pancreatic cancer (PC), whihc is projected to account for 49,830 deaths in the United States in 2022 [2]. This lethality can be attributed to late diagnosis due to its asymptomatic nature and the lack of effective therapeutic options.
In the early 1900s, Paul Ehrlich proposed an immunosurveillance theory, which was further refined by Thomas and Burnet [3]. This theory states that the immune system continuously surveys the somatic cells for oncogene-associted molecular alterations and, upon encountering transformed cells, eradicates them [3]. However, some transformed cells escape the surveillance and develop pathological lesions. In 2002, Schreiber introduced the concept of cancer immunoediting: the triple E, including Elimination, Equilibrium, and Escape [4,5]. This concept explains the dual role of the innate and adaptive immune systems in the molding of the tumor cells, which helps them to establish a clinical disease. The immune system identifies and removes the precancerous cells carrying potentially harmful mutations in the first elimination phase. If the cell survives the host immune surveillance, the precancerous cells enter an equilibrium phase in which the immune cells secrete inflammatory cytokines, like interferon-γ (IFN-γ), to exert selective pressure on the transformed cells but not enough to eliminate them [5]. During equilibrium, the cells further mutate and become genetically unstable or undergo selection and survive the immune attack. The resistant tumor cells that develope during equilibrium can escape the immune system and proliferate in the host.
Earlier work to develop therapeutic regimens was generally performed in nude mice. The xenograft tumor models were generated by orthotopically implanting PC cell lines such as MIAPaCa-2 [6], AsPC-1 [7], and PC surgical specimens [6,8]. The xenograft models demonstrated the involvement of natural killer (NK) cells in tumor rejection in the early stages of tumor development [6]. Further, the patient-derived xenograft models helped in studing the proliferation and metastasis of PC and develop personalized drug treatment [8]. However, these tumor models are unsuitable for studying autochthonous tumors’ tumor microenvironment (TME) due to incomplete immune response to tumor development [6]. The advancement in the syngeneic transplantation and genetically engineered mouse models (GEMMs) of cancer led to extensive investigations into disease progression and evolution [9]. GEMMs harboring the mutant Kras (G12D) and p53 mutations in the pancreas were developed to mimic the human disease, as these mutations are most commonly present in PC patients. In both KC (LSL-KrasG12D/+; Pdx-1-Cre) and KPC (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre) models, tumor initiates with the pancreatic intraepithelial neoplasia (PanIN) lesions, which further progress to the invasive tumors, histologically mimicking the human PC. The onset and kinetics of disease progression are more rapid in KPC mice than in KC mice. These models have been extensively characterized for the alterations in the immune system during PC progression [10]. Tumors in the KC model [11] demonstrated a successive increase in the infiltration of CD45+ leukocytes during cancer progression, with nearly 50 % of the total cells from the tumor samples being CD45+ [12]. Further analysis of these leukocytes revealed that more than 15 % of the cells were CD11b+ macrophages present during all stages of PC progression. Another leukocyte subset in the pancreatic TME was the immunosuppressive CD4+ T-regulatory (Treg)-cells with forkhead box P3 (FOXP3) expression, constituting more than 25 % of the total CD4+ cells. These cells were accompanied by myeloid-derived suppressor cells (MDSCs) in the later stages of the disease in the TME as well as in the spleen [12]. Interestingly, there were negligible numbers of cytotoxic CD8+ T-cells present during the early stages of cancer and only slight evidence of their presence during the advanced stages [12]. Therefore, the immunosurveillance and elimination phase of the triple E hypothesis may be restricted in PC due to the presence of highly immunosuppressive cells at the earlier stages, which restrain the cytotoxic immune cells from eliminating the neoplastic lesions [12].
Further, the KPC tumors escape immunosurveillance due to low immunogenicity and lack of neoantigens. Whole-exome sequencing has demonstrated a low mutational burden in PC, and most of the predicted missense mutations encompass epitopes with high affinity for major histocompatibility complex (MHC) molecules [13]. This lack of neoepitopes is not due to selection pressure but can be attributed to immune quiescence [13]. When T-cell antigen OVA was transduced into PC cells, the OVA+ KPC tumor cells were able to evoke an immune response and were rejected post-implantation, as compared to OVA− KPC tumors [13]. Thus, OVA+ tumors were able to reverse the immune quiescence [13]. The patient samples also validated an increased presence of the T-reg population and decreased CD8+ T-cells in the TME [14]. Sixty-eight PC patients’ samples were analyzed for immune infiltration, and more than one-third of the patients had high CD4+ T-cell infiltration[15]. The tumors were deficient in CD8+ T-cells and were dominated by myeloid cell populations, including M2-type macrophages [15]. An upregulation in neutrophilic markers of differentiation, activation, and recruitment such as SRC kinase-associated phosphoproteins 2 (SKAP2) and leucine-rich alpha-2-glycoprotein 1 (LRG1) was also observed in tumor tissues. Survival of PC patients also correlated with immune infiltration. A higher ratio of CD8+ T-cells to CD4+ T-cells associates with prolonged survival and increased T-regs or M2 macrophages in patient samples correspond to poor survival [16]. This review summarizes the immune microenvironment of PC, focusing on the status of various immune checkpoint molecules expressed by immune cells and cancer cells, highlighting the dynamic interplay of checkpoint molecules and cancer cells in shaping up the TME (Figs. 1 and 2).
Fig. 1.
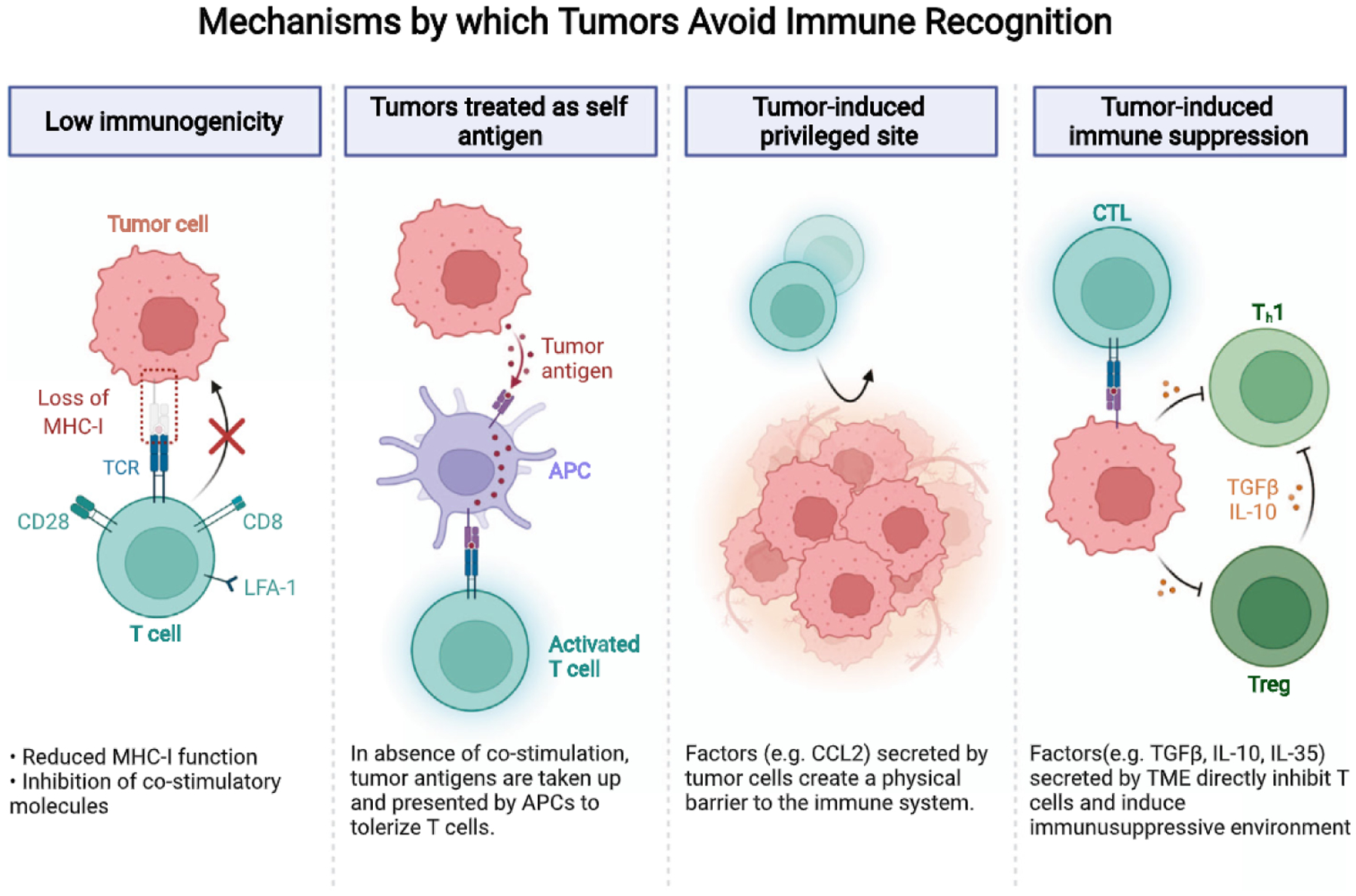
Mechanisms by which tumors avoid immune recognition. Mutations in the Ras signaling impair antigen presentation by decreased expression of MHC I and antigen processing molecules. Regulatory T-cells interact with tumor-associated dendritic cells and hamper their immunogenic function by inhibiting the expression of costimulatoiy ligands required for cytotoxic T-cell activity, thus, creating an immunosuppressive environment. Tumor cells also secrete chemokines such as CCL2 and create a physical barrier that prevents immune infiltration. Immunosuppressive immune cells secrete factors such as TGFβ, IL-10, and IL-35 to inhibit CTL activity.
Fig. 2.
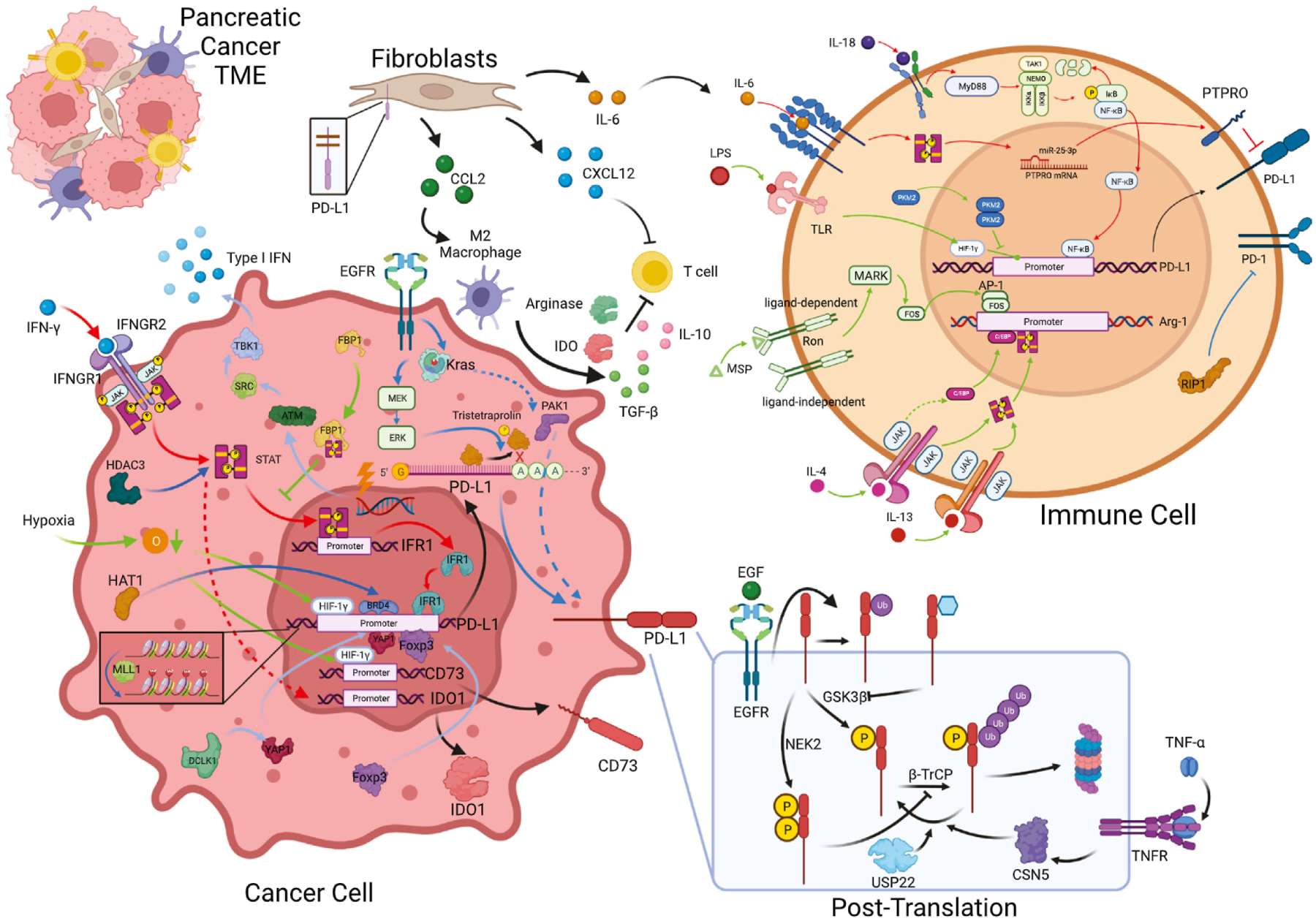
Immune checkpoint regulation network in the pancreatic cancer tumor microenvironment. In the tumor microenvironment of pancreatic cancer, the expression of immune checkpoints were regulated by a comprehensive network, including cytokine (red), tumor microenvironment property and key metabolism enzyme (green), key oncogenic, epigenetic, or other signaling pathways (blue), and post-translation modification (separate box) in both cancer cell and immune cells. Other components, such as fibroblasts and macrophages, also contribute to the regulation of immune checkpoints.
2. PC TME is an immunosuppressed-immune desert
The factors that determine the success of immune checkpoint therapy in cancer include immunogenicity and antigenicity [17]. The term immunogenicity is typically used to refer to the initiation of an immune response by an antigen. In contrast, antigenicity refers to antigen recognition, such as the ability of tumor-associated antigens (TAA) and tumor-specific antigens (TSA) to be presented by MHC molecules and recognized by effector T-cells [17,18]. Another aspect is the activation of the B-cells and T-cells following the recognition of tumor antigens by the B-cell receptor (BCR) and T-cell receptor (TCR). Internalized antigens are processed by the antigen presenting cells (APCs), and the antigenic peptides are presented through MHC II and MHC I molecules to CD4+ and CD8+ T-cells, respectively. In addition to classical APCs (macrophages and dendritic cells), activated B-cells can also present antigen by upregulating MHC and costimulatory molecules such as CD80/ CD86 for T-cell activation [19].
Somatic mutations can sometimes interfere with the cell’s normal function and help drive cancer, but they can also be a source of neoantigens recognized by the T and B-cells [18]. Similarly, epigenetic modifications such as loss of methylation or hypomethylation in the genes encoding for TAA and/ or TSA can lead to limited anticancer immunity, increased proliferation, drug resistance, and immune evasion [20,21]. Hypomethylation leads to genomic instability, which is often accompanied by focal cancer-specific hypermethylation leading to the silencing of tumor suppressor genes located at CpG islands [22]. Therefore, targeting DNA methyltransferases or histone deacetylases can potentially modulate the immunologically cold tumors to a hot state by upregulation of antigens in tumor cells, promoting tumor cell recognition with better MHC I antigen processing and presentation [22]. PC is characterized by a low mutational load as compared to melanoma, and renal cancers; therefore, it responds poorly to checkpoint inhibitors [23–25]. Balachandran et al. demonstrated that neoantigen immunogenicity and quantity are positively correlated with increased CD8+ T-cells and the survival of PC patients. The patients with higher numbers of neoantigens exhibited increased CD3+CD8+ T-cells and polyclonal T-cell repertoires in their tumors, correlating with better survival [26]. However, the survival did not depend entirely on the quantity but also on the quality of neoantigens, i.e., their ability to be recognized by the T-cells. The overall fitness of the neoantigen is important as neoantigens are lost during tumor progression due to reduced gene expression and mutant allelic loss [26], reinforcing PC’s immunologically quiescent characteristics. Thus, it is vital to identify the immunogenic hotspots and promptly target the neoantigens to improve the efficacy of immune checkpoint blockade therapy.
Reduced antigenicity in PC can also be attributed to poor antigen presentation, which dampens immunosurveillance and helps cancer cells escape from the effector T-cells [27,28]. Induction of oncogene expression in mouse fibroblasts led to the locus-specific loss of MHC I expression [29]. Impaired MHC I expression was associated with reduced expression and function of antigen processing components, such as TAP1, TAP2, LMP2, and LMP7 [30]. Targeting oncogenic Ras either by genetic deletion or with an inducible intracellular domain antibody (iDAb) has upregulated MHC I expression on colorectal cell lines [31]. Another factor that causes disseminated cancer cells to lose their MHC I expression is ER stress, as shown by Pommier et al. Pancreatic tumor cells (inhabiting the liver) due to ER stress lost CK19, E-cadherin (Ecad), and MHC I expression, acquired a quiescent (non--proliferative) phenotype and escaped immune surveillance. The disseminated, quiescent cancer cells resolved the ER stress, regained expression of Ecad, CK19, and MHC I, and proliferated to establish metastatic lesions [32].
Based on the gene-expression analysis, PC is classified into various subtypes, and one of them is the immunogenic subtype, which is associated with significant immune infiltration. In the immunogenic subtype, there is upregulation of gene expression related to B- and T-cells (both CTLs and T-regs), along with enrichment of checkpoint molecule such as programmed cell death 1 (PDCD1) PD-1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA4). In PC, there is infiltration of TILs, indicated by the upregulation of lymphocyte-specific protein tyrosine kinase (Lck) [33], which is critical in the early stage of T-cell activation and is the primary kinase in TCR signaling [34]. However, significant downregulation of genes related to T-cell function, such as CD8A, PDCD1, CTLA4, PDL1, and lymphocyte activating 3 (LAG3), suggest the suppression of T-cell activation [33]. Nonetheless, immunogenic PC subtype tumors can resist immune checkpoint blockade by modulating cancer cell-intrinsic immunogenicity [17]. The immunosuppressive environment of PC is also attributed to tumor-associated macrophages (TAMs). The anti-tumor macrophage (M1) infiltration was observed in the early stage of cancer, followed by the pro-tumor macrophages (M2 TAMs) in the later stage [35]. These data indicate that the immune-suppressive components of the TME are dominant and disable the antitumor response. Anti-tumorigenic TAMs release pro-inflammatory cytokines, such as TNF, IL-1, IL-6, IL-8, and IL-12 [36]. However, increased inflammation can expedite M2 polarization due to increased tumor cell death [37]. Other tumor-derived factors related to inflammation, such as TLR (Toll-like receptor) [38], metabolic factors, hypoxia [39,40], lactic acid accumulation in the TME [41], and exosomes enriched in immunomodulatory chemokines including CSF-1, CCL2 and TGF-β [42], can drive M1 macrophages towards the M2 phenotype. According to the gene expression signatures, TAMs can be divided into four sub-clusters, M1-TAMs, M2-TAMs, CD169+ macrophage, and TCR+ macrophage [35]. The TAMs can further enhance the expansion of MDSCs by secreting granulocyte-macrophage colony-stimulating factor (GM-CSF) [43]. The MDSCs further generate an immunosuppressive environment by inhibiting CD8+ T-cell function via the recruitment of T-reg cells. MDSCs can also suppress effector T-cell function in the TME by increased ROS production, which is mediated by upregulation of NADPH oxidase (NOX2) [44]. The highly reactive ROS can induce the nitration of susceptible tyrosine residues in the TCR and alter interaction between TCR and MHC molecules [45]. MDSCs depletion has also resulted in loss of PD-L1 on cancer cells [46]. Another report highlighted the role of Ly6Clow F4/80+ extratumoral macrophages in immune evasion of cancer cells [47]. These macrophages have increased expression of IL4Rα, which regulates production of transforming growth factor β (TGF-β), arginase activity and supports the production of peroxynitrate and ROS, which can suppress T-cell activity [48]. As an experimental therapeutic strategy, TAMs have been targeted by anti-CSF1R antibody, which reduces overall TAM infiltration and enhances immune stimulation by reprograming them towards the anti-tumor phenotype, thereby assisting T-cell-mediated immunity [49]. CSFR1 inhibition can indirectly affect immune checkpoints by upregulating PD-L1 and CTLA-4 expression on tumor and T-cells, respectively, due to chronic exposure to inflammatory cytokines like IFN-γ [49]. Furthermore, a previous study has employed CD40 agonist and gemcitabine treatment in the KPC model to deplete macrophages and enhance T-cell infiltration, leading to tumor regression [50]. However, a similar CTL-mediated immune response was not observed in clinical trials with CD40 agonists in other cancers [51,52]. It was suggested that the CD40 on the APCs is involved in T-cell priming, thereby acting as a co-activator of checkpoint molecules [53]. Therefore, it would be important to target CD40-mediated signaling along with administering checkpoint inhibitors, to enhance the cytotoxic activity of the T-cells.
Chemokines in the TME contribute to PC’s immune desert phenotype. Cancer cells secrete CCL2, which binds to its receptor CCR2 on pro-tumorigenic macrophages, inhibiting CTL infiltration in the tumor [15]. TGF-β, IL-10, and IL-35 from T-regs create an immunosuppressive environment, and these cytokines also interact with myeloid cells and enhance their suppressive function [54]. T-regs also interact with tumor-associated dendritic cells (DCs) and inhibit the expression of costimulatory ligands required for cytotoxic T-cell activation [55]. During the PanIN stage, the microenvironment limits the infiltration of T-cells, and the cells which manage to infiltrate are inhibited by the immunosuppressive cells [12]. Cancer-associated fibroblasts (CAFs) and immunosuppressive immune cells restrict CTL activity at advanced stages. To attain an effective therapeutic T-cell response it is essential to modulate the restrictive stroma to enhance the infiltration of CTLs into the TME. The CAFs secrete chemokine CXCL-12, which is captured by high mobility group box 1 (HMGB1) produced by tumor cells and forms a high-affinity heterodimer complex [56], thereby promoting the CXCL-12/CXCR-4 signaling that impacts T-cell exclusion from pancreatic tumors [57–59]. Targeting with AMD3100, a specific CXCR-4 inhibitor, has been demonstrated to increase tumor infiltration by T-cells, and combination with anti-PD-L1 therapy showed an additive anti-tumor effect in preclinical models [57]. The CAFs also secrete pro-inflammatory cytokines such as IL-6 [60], which play a vital role in cancer progression. Targeting IL-6 and PD-L1 has been shown to decrease α-SMA+ CAFs and improve intratumoral CD3+ T-cell infiltration in KPC mice and transplantation models [61]. However, modulating stroma has its caveats; for example, an increase in the TME T-reg population was observed when the stromal compartment, especially the α-SMA-expressing cells, were targeted in mouse models [62].
The single-cell RNA-sequencing (scRNA-seq) analysis revealed more information about the TME of PC. A recent single-cell RNA sequencing (scRNA-seq) data in PC has shown the presence of more than eleven different T-cell subclusters with unique gene signatures. The anti-tumor immune cells, including but not limited to cytotoxic T-cells, have a more extensive infiltration in the advanced PanIN lesions. However, this larger anti-tumor cell infiltrate was accompanied by more T-regs and exhausted T-cells, which may disable the effector cell function of anti-tumor immune components [35]. Recently, a scRNA-seq analysis identified a significant number of cytotoxic T-cells and NK cells infiltrating the PC TME. While the T-cells expressed ICOS, TIGIT, PD-1, the NK cells expressed LAG3 and CD47, TIGIT, TNFSRF18, and LAG3 [63]. The effector cells in the TME express prominent markers of exhaustion, indicating immune dysfunction with cancer progression [63].
3. Immune checkpoints in PC
After the successful clinical implementation of anti-PD-1/PD-L1 therapy in melanoma, the PD-1/PD-L1 axis received much attention, and its status was explored in PC along with other immune checkpoint molecules. PD-1 protein belongs to the B7-CD28 superfamily of proteins [64]. Various computational analyses and preclinical studies have been done to examine the expression patterns of checkpoint molecules in PC. PD-1 is a 55 kDa protein, mostly expressed on the effector CD4+ and CD8+ T-cells and is associated with T-cell exhaustion [65–67]. The PD-1 ligands, PD-L1 and PD-L2, are expressed by tumor cells, immunosuppressive cells such as MDSCs and TAMS, and tumor-infiltrating DCs [68]. The PD-1 axis is not limited to T-cells and APCs but is also expressed by B-cells and neoplastic cells [15]. In naïve T-cells, the expression of PD-1 is relatively low; chronic exposure to the antigen activates the PD-1/PD-L1 signaling axis leading to T-cell exhaustion [69]. Overexpression of PD-L1 by tumor cells and PD-1/PD-L1 interaction leads to recruitment of SHP2 tyrosine phosphatase, which dephosphorylates CD28, thereby inhibiting T-cell activation [70,71]. Another checkpoint molecule, cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), is expressed primarily by T-reg cells (in the intracellular vesicles and at the cell surface) [72], and T-cell activation results in progressive increase in its expression [73–75]. CTLA-4 expression was observed in the CD25-expressing helper T-cells (Th) subset, where CTLA-4 works intrinsically by inhibiting costimulatory signal, thereby hampering T-cell activation [75]. CTLA-4 also operates extrinsically by eliminating CD80/CD86 from APCs, thus dampening the CD8+ T-cell response [76] and regulating CD4+ T-cell infiltration [77]. Further, the LAG-3 signaling pathway is employed by tumor cells to escape immune surveillance [78]. Galectin-3 binds to LAG-3 on the activated T-cells and suppresses their function. Simultaneously, LAG-3 inhibits the activity of plasmacytoid DCs that present antigen and induce naïve T cell differentiation [79]. LAG-3 modulates T-cell proliferation, inhibits memory and effector T-cell response, and promotes T-reg cell-mediated immunosuppression [80,81]. Another checkpoint molecule, TIM-3, interacts with tumor-associated antigens such as CEACAM-1 and galectin-9 to inhibit the T-cell response. CEACAM-1 forms heterodimers with TIM-3 and inhibits T-cell response by decreasing intracellular IL-2 and TNF-α, which correlates with exhaustion. Antibody-based blocking of TIM- 3 and CEACAM-1 enhanced CD8+ T-cell infiltration and IFN-γ production and inhibited tumor growth [82]. Galectin-9, expressed by the tumor cells, binds to TIM-3 and initiates downstream signaling, causing Ca2+ influx in Th-cells resulting in their death [83]. Further, TIM-3 expressed by tumor-infiltrating DCs interacted with nuclear HMGB1 and reduced the efficacy of chemotherapy by reducing the immunogenicity of nucleic acids derived from the tumor cells [84]. Further, high cytolytic T-cells exhibit increased expression of checkpoint molecules such as CTLA-4, TIGIT, TIM-3, and VISTA; however, PD-L1 expression was found to be low in PC TME [85].
Earlier, the expression of checkpoint molecules was analyzed by staining of patient samples or by in silico analysis. However, with scRNA-seq the expression of checkpoint molecules on many cell types, including ductal cells, immune cells, endothelial cells, stellate cells, acinar cells, endocrine cells, and fibroblasts in the pancreatic TME, has been studied. Over 8,000 cells from the normal pancreas, 46,244 pancreatic tumor cells, over 55,000 peripheral blood mononuclear cells (PBMCs) from PC patients, and over 14,000 PBMCs from healthy donors were analyzed to assess checkpoint molecule heterogeneity [63]. The CD8+ and CD4+ T-cells infiltrating the pancreatic TME were enriched in PD-1, TIGIT, and ICOS. Whereas CD8+ T-cells had increased expression of LAG-3 and decreased expression of CTLA-4, CD4+ T-cells had high CTLA-4 and low LAG-3 expression. There was an intermediate expression of PD-1 on NK cells, but the expression of CD47, TIGIT, and LAG-3 was high. DCs and myeloid cells also expressed some of the immune checkpoint molecules such as SIRPA, PVR, LGALS9, and ICOSLG. TIGIT and TIM3 ligands (PVR and GALECTIN9, respectively) also had heterogeneous expression on the epithelial cells. Interestingly, TIGIT expression was significantly higher in the PC samples as compared to controls, and its expression was observed on both CD8+ and CD4+ CD25+ T- and NK cells. TIGIT was distinct because it was enriched in EOMEShigh CD8+ T-cells, which are exhausted T-cells [63]. In breast cancer models, NK cells expressed elevated levels of TIGIT and were associated with tumor progression, and blockade of TIGIT on NK cells reversed the exhaustion and inhibited tumor growth [86]. The ligands for TIGIT, PVR, and Nectin-2 are also amply expressed by tumor cells and various immunosuppressive cell types. Other immune cells such as B-cells were enriched for CD70, CD40, ICOSLG, and CD80, while DCs are enriched only for TNFRSF4 expression. Similarly, the non-immune cells, such as fibroblasts and endothelial cells, exhibit high expression of TNFSF18 and TNFSF4, respectively. Previous work has shown that TIGIT suppresses T-cell activity by interacting with PVR on DCs, causing upregulation of the inhibitory cytokine IL-10. TIGIT also suppresses T-cell function by competing with CD226 for PVR ligand similar to CD28/CTLA4 pathway [87,88].
Overall, targeting checkpoint molecules or their ligands can potentially improve the immunotherapy response in PC but will require a comprehensive understanding of the regulators involved in their expression.
4. Immune checkpoints regulation in pancreatic cancer
Considering the significant contribution of multiple immune checkpoints in cancer therapy, understanding the regulatory network of immune checkpoint expression, including TME-mediated and cell-intrinsic mechanisms, should provide new targets for combination therapy with immune checkpoint blockade to improve the outcome of PC therapies.
4.1. Tumor microenvironment-mediated regulation of immune checkpoints
4.1.1. Cytokine-mediated regulation of immune checkpoints
CTLs, NK cells, and Th1 cells are major anticancer immune components. These cells secrete IFN-γ to boost their activities and create a pro-inflammatory environment [89]. However, IFN-γ has also been reported to induce the expression of PD-L1 and PD-L2 in PC [90], melanoma [91], colorectal cancer [92], and prostate cancer [93]. IFN-γ treatment of a human PC cell line, PK-45 P, induced the expression of PD-L1 and vimentin, with downregulation of E-cadherin, through activation of STAT1 signaling and upregulation of Interferon Regulatory Factor 1 (IRF1), in a dose-dependent manner [90]. STAT1 knockdown by siRNA blunted the IFN-γ-mediated upregulation of PD-L1, validating the role of STAT1 signaling [90]. Additional details on IFN-γ/PD-L1 expression were revealed in a melanoma study, in which shRNA screening showed that after IFN-γ binds to its receptors, IFNGR1 and IFNGR2 activate the JAK2-STAT1/STAT2/STAT3 signaling axis and induce IRF1 expression. Upregulated IRF1 binds to the promoter region of the PD-L1 gene leading to its upregulation [94]. A similar contribution of IFN-γ-mediated signaling has also been reported in glioma [95]. In addition to PD-L1, IFN-γ also regulates another immune checkpoint, indoleamine 2,3-dioxygenase-1 (IDO1). IDO1, the enzyme catalyzing the transformation of tryptophan into formyl-kynurenine, plays an essential role in folate-dependent one-carbon metabolism and immune invasion [96]. A study showed that a CDK1/2/5 inhibitor, dinaciclib, can inhibit IFN-γ-mediated upregulation of IDO1 [97]. Further analysis indicated that this CDK inhibition blocked the JUN-dependent expression and activation of STAT1 signaling, which is responsible for the transcription and expression of both IDO1 and PD-L1 [97].
As immune checkpoints influence inflammation through negative feedback, multiple inflammation-related cytokines are expected to modulate the expression of immune checkpoints. Cancer cell-derived IL-18 was found to upregulate the expression of PD-L1 and IL-10 by regulatory B-cells in PC. The IL-18-mediated increase in the expression of PD-L1 was associated with MyD88 upregulation and p65 phosphorylation [98]. Another study also validated the contribution of IL-18 to PD-L1 upregulation in NK cells under both in vitro and in vivo conditions [99]. IL-6, one key cytokine secreted by macrophages and pancreatic stellate cells in PC stroma [100,101], can induce PD-L1 expression in immune cells through STAT3 phosphorylation [102]. Even with significant clinical potential in PC treatment, limited studies were performed on the mechanism of IL-6 inducing PD-L1 in PC. One study in hepatocellular carcinoma (HCC) showed that administration of IL-6 to macrophages isolated from HCC murine models induced the expression of PD-L1 at both mRNA and protein levels. Similar findings were verified by using monocyte-like cell lines, U937 and THP-1. A protein tyrosine phosphatase called protein tyrosine phosphatase receptor type O (PTPRO) has been shown to reverse the IL-6-mediated upregulation of PD-L1, as the shRNA-mediated downregulation of PTPRO can enhance IL-6-induced PD-L1 upregulation [102]. PTPRO expression is also regulated by miR-25−3p, which is the downstream effector of IL-6/STAT3/c-Myc signaling. Altogether, IL-6 induces the expression of miR-25−3p through STAT3/c-Myc signaling, resulting in downregulation of PTPRO and upregulation of PD-L1 [102]. Another study blocking the IL-6 receptor with an adenovirus-delivered IL-6 receptor blocking scFv, showed reduced expression of both PD-L1 and PD-1 in DCs, confirming the critical role of IL-6/IL-6R signaling in regulating PD-1/PD-L1 in immune cells [103].
4.1.2. Metabolites-mediated regulation of immune checkpoint
Metabolism-related aspects of the TME, including low oxygen (hypoxia) and higher acidity (acidosis), also influence the expression of immune checkpoints. Hypoxia can induce the expression of PD-L1 in MDSCs, DCs, and tumor cells via HIF-1α-mediated binding to the hypoxia-response elements (HRE) at the proximal promoter of PD-L1 [104]. This finding has been validated using tumor-infiltrating MDSCs and splenic MDSCs from lung cancer (Lewis lung carcinoma, LLC), melanoma (B16-F10), breast cancer (4T1), and colorectal cancer (CT26) cell line-derived subcutaneous mouse models. These findings were also verified in splenic MDSCs from these four models, cultured in hypoxic (0.1 % pO2) conditions [104]. The presence of nucleotides in the TME contributes to the establishment of immune-suppressive conditions. CD73, a surface 5’ nucleosidase that generates adenosine, has been investigated as a potential immune therapy target [105]. Hypoxia induces the expression of CD73 on epithelial cells at both mRNA and protein levels [106]. Further analysis of the CD73 promoter region found at least one HIF-1α binding site. Inhibiting HIF-1α with siRNA resulted in the downregulation of CD73 expression, and a luciferase assay with the CD73 promoter region without the HIF-1α binding site failed to induce the expression under hypoxia [106].
Multiple enzymes in the metabolic pathways are involved in the regulation of immune checkpoints. Pyruvate kinase isoform M2 (PKM2), the rate-limiting enzyme in glycolysis, can form a dimer and translocate into the nucleus to facilitate the transcriptional activity of HIF-1α and p300 recruitment. A PKM2 inhibitor decreased the LPS-induced upregulation of PD-L1 in macrophages by disrupting the direct binding of HIF-1α to HRE at the PD-L1 promoter in these cells [107].
Amino acid metabolism also influences immune surveillance in tumors. For example, arginase, the enzyme catalyzing arginine transformation into ornithine and urea, plays critical functions in MDSC differentiation and macrophage polarization [108]. It has been shown that the receptor tyrosine kinase, recepteur d’origine nantais (RON), participates in regulating arginase1 (Arg1) expression [109]. Treating primary resident peritoneal macrophages with RON ligand, macrophage-stimulating protein (MSP), can induce the expression of Arg1. Overexpression of RON and MSP treatment showed that RON could induce the expression of Arg1 in both ligand-dependent and ligand-independent manners. Arg1 promoter region analysis revealed that AP-1 could be responsible for the RON-mediated upregulation of Arg-1, and luciferase assays verified that an AP-1 binding site truncated construct blunted RON-induced reporter activity. AP-1 is a MAPK downstream effector, and further analysis showed that MSP induced the upregulation of Arg1 through MAPK activation and upregulation of FOS (a component of the AP-1 transcription factor) and its binding to the Arg1 promoter region [109]. Previous studies showed that IL-4 and IL-13, which are inducers of M2 polarization, can also induce the expression of Arg1. IL-4-induced-expression of Arg1 requires the binding of STAT6 and C/EBP to the Arg1 promoter [110], while both IL-4 and IL-13 have been reported to regulate Arg1 expression via cAMP and the JAK/STAT6 pathway in vascular smooth muscle cells [111].
Metabolic changes also contribute to the suppression of immune cells in the PC TME. In the TME, the T-cells have to compete with the cancer cells for nutrient, and therefore glucose uptake by T-cells is limited and hase an impact on T-cell function even in the presence of antigen [112]. Post priming of naïve T-cells, the activated T-cells undergo a metabolic switch from β-oxidation of fatty acids to pyruvate oxidation for quicker ATP production [113,114]. This metabolic switch is accompanied by changes in metabolic transcriptome and induction of endogenous Myc and HIF1α expression [113]. Aerobic respiration can impact PD-1 status on the T-cells, which implies that metabolic regulators play a role in T-cell exhaustion rather than chronic antigen exposure [114,115]. T-cells utilize arginine, but TAMs and MDSCs deplete it due to their increased expression of Arg1. Another enzyme, IDO, produced by cancer cells and other immunosuppressive cells, degrades tryptophan to kynurenine, which acts as an immunosuppressant for T-cells. Another phenomenon that dampens T-cell function is the Warburg effect, where T-cells can aerobically ferment glucose into lactate [116] and decrease the overall pH [117]. T-cell signaling is also inhibited by prostaglandins E2 produced by TAMs and MDSCs [118] and nucleosides such as adenosine expressed by regulatory T-cells and cancer cells [119]. With the modulation of signaling, metabolites produced by immunosuppressive cells and nutrient availability play an essential role in T-cell function and infiltration.
4.2. Cancer cell-intrinsic mechanisms in the regulation of immune checkpoints
4.2.1. Metabolic and epigenetic regulation of immune checkpoint regulation
The metabolic modulation in the TME is closely linked with cancer cell-intrinsic metabolic and epigenetic modulations, which can also influence the expression of the immune checkpoints. The essential enzyme in gluconeogenesis, fructose-1,6-biphosphatase (FBP1), has also been shown to regulate PD-L1 expression [120]. The shRNA-mediated knockdown of FBP1 in PANC-1 and MIAPaCa-2 PC cell lines resulted in the upregulation of PD-L1 expression at both protein and mRNA levels. Concurrently, overexpression of FBP1 reduced the expression of PD-L1. Overexpression of the catalytically inactive mutant (FBP1-G260R) showed a similar downregulation of PD-L1, indicating that the FBP1-mediated downregulation of PD-L1 is through enzyme activity-independent mechanisms. Reciprocal co-immunoprecipitation showed direct interaction between STAT3 and FBP1. Further, co-immunoprecipitation of truncated proteins revealed that the STAT3 and FBP1 interaction is through the SH2 domain in STAT3 and N-terminal domain in FBP1. The dual knockdown of FBP1 and STAT3 blunted the FBP1 knockdown-mediated upregulation of PD-L1 at both protein and mRNA levels. Overexpression of FBP1 and knockdown of STAT3 diminished the FBP1 overexpression-mediated downregulation of PD-L1. The chromatin immunoprecipitation-polymerase chain reaction (ChIP-PCR) analysis showed that the interaction between STAT3 and the PD-L1 promoter region was inversely correlated with FBP1 expression. These experiments illustrated that FBP1-mediated PD-L1 expression regulation was STAT3-dependent [120].
The presence of metabolites like acetyl-CoA and S-adenosyl methionine can influence epigenetic modifications. Histone acetyltransferase-1 (HAT1) catalyzes the acetylation of newly synthesized histone H4 [121]. The shRNA-mediated knockdown of HAT1 was shown to reduce the expression of PD-L1 transcriptionally and translationally. Since BRD4 can bind to the promoter region of PD-L1, knockdown of BRD4 in a HAT1-expressing cell line blunted HAT1-mediated upregulation of PD-L1, indicating HAT1-induced PD-L1 expression in BRD4-dependent manner [121]. Another study reported that an HDAC3-specific inhibitor, RGFP966, reduced the expression of PD-L1 in the MIAPaCa-2 and BxPC-3 PC cell lines in a time- and dose-dependent manner at the mRNA and protein levels [122]. The shRNA-mediated knockdown of HDAC3 further confirmed these findings. The study also illustrated that HDAC3 regulates the expression of PD-L1 through STAT3 activation. In addition, overexpression of Flag-tagged HDAC3 rescued HDAC3 shRNA knockdown-mediated PD-L1 downregulation, while both downregulation and rescue were blunted after STAT3 knockdown. The ChIP analysis of STAT3 binding to the PD-L1 promoter showed that HDAC3 inhibitor treatment and shRNA-mediated knockdown reduced the STAT3 occupancy at the PD-L1 promoter region, confirming that HDAC3-mediated upregulation of PD-L1 is STAT3-dependent [122].
Histone methylation enzymes also participate in the regulation of PD-L1 expression. The H3K4me3 mark was enriched in the proximal region of the PD-L1/CD274 promoter during PD-L1 epigenetic signature screening. Further, ChIP analysis showed the enrichment of H3K4me3 marks at the PD-L1/CD274 promoter in tumor cells compared to the normal pancreas, indicating that the H3K4me3 mark was specific to the tumor cells [123]. Histone methyltransferase analysis identified that MLL1, the histone-lysine N-methyltransferase 2 A, is upregulated in tumor samples and is responsible for the increased H3K4me3 marks [123]. The ChIP analysis also confirmed a direct association between the PD-L1/CD274 promoter and MLL1. The silencing of MLL1 significantly reduced the H3K4me3 marks at the PD-L1 promoter as well as the PD-L1 transcripts, demonstrating that MLL1-mediated regulation of PD-L1 requires histone modification [123]. Similarly, another study observed the increased occupancy of H3K4me3 at the promoter region of PD-L1, osteopontin (OPN), and its receptor CD44 in PC. WD repeat-containing protein 5 (WDR5), the adaptor protein for histone methyltransferase, was responsible for the increased H3K3me presence at OPN/CD44 promoter. The combined targeting by WDR5 inhibitor (WDR5–47) and PD-L1 antibody significantly reduced the tumor weight and volume and sensitized cancer to immune therapy [124]. In addition to histone modification, DNA hypomethylation increases the PD-1 expression, and DNA hypomethylating drug decitabine (5-aza-dC, DAC) in combination with anti-PD-L1 or anti-VISTA significantly increased the survival of the KPC animals (a murine model of PC). Further, DAC treatment increases the tumor infiltration of both CD4+ and CD8+ T-cells and the expression of Chi3l3 (Ym1), suggesting the infiltration of the Argl+ macrophages [125].
4.2.2. Signaling pathways involved in immune checkpoint regulation
Oncogenic signaling can also directly influence the expression of immune checkpoint molecules. Constitutively active Kras mutants are significantly associated with PC, colorectal cancer, and lung cancer [126]. Mutant Kras in lung adenocarcinoma can upregulate the expression of PD-L1 through mitogen-activated protein kinase/extracellular-signal-regulated kinase (MEK-ERK) signaling [127]. Higher MEK-ERK activity phosphorylates serine residues (S52A, S178A) of AU-rich element-binding protein tristetraprolin (TTP) protein, preventing tristetraprolin binding to the 3’-UTR of PD-L1 mRNAs and stabilizing PD-L1 transcripts [127]. EGFR overexpression is also associated with PC tumor progression, metastasis, and poor survival [128]. It has been reported that EGF/EGFR signaling activation is correlated with increased expression of PD-1, CTLA-4, and PD-L1 in microarray analysis and that introducing EGFR mutations into bronchial epithelial cells can upregulate PD-L1 expression in lung cancer [129]. Another study investigating myeloid subsets in PC showed that EGFR signaling in cancer cells contributed to the expression of PD-L1 [46]. The coculture of multiple human primary PC cell lines (UM2, UM5, UM18, and 1319) with mouse bone marrow-derived cells increased the expression of PD-L1 on cancer cells, plausibly through EGFR phosphorylation (at Tyr1068) in tumor cells. Treating the cocultured cells with the EGFR inhibitor erlotinib reduced the expression of PD-L1 at both mRNA and protein levels. A similar result was achieved with the MEK inhibitor GSK1120212, indicating both EGFR and MEK signaling pathways regulate PD-L1 expression [46].
Downstream of Kras, a p21-activated kinase (PAK) family member, PAK1, has also been associated with PD-L1 expression [130]. PD-L1 expression was significantly reduced in the PAK1-knockout KPC murine model compared with the KPC model, and this finding was also validated with the PANC-1 human PC cell line. The treatment of mouse PC cell lines with a PAK1/4 inhibitor, PF-3758309, reduced phosphorylation of PAK1 and PD-L1 expression. Aside from mediating PD-L1 expression in cancer cells, the pancreatic tail injection of mouse cell line TB33117 in PAK1-knockout mice resulted in reduced expression of PD-1 on the surface of infiltrated CD4+ and CD8+ T cells, albeit the difference from control was not statistically significant. These findings indicate that downstream of Kras, PAK1 participated in regulating PD-L1 in cancer cells and possibly PD-1 in T-cells [130].
While cancer stem cells play a vital role in tumorigenesis and resistance to treatment, cancer sternness genes are also involved in regulating PD-L1 expression. Overexpression of the cancer stem cell-associated gene doublecortin-like kinase 1 (DCLK1) in the PD-L1low cell line AsPC-1 significantly upregulated the expression of PD-L1 at both transcriptional and translational levels [131]. Further, in both AsPC-1 and BxPC-3 PC cell lines (expressing low and high PD-L1, respectively), inhibiting the expression of DCLK1 with LRRK2-IN-1 and XMD8–92 significantly downregulated the PD-L1 expression. Analysis of data from The Cancer Genome Atlas (TCGA) and inhibitor-mediated suppression of DCLK1 linked DCLK1 expression with yes-associated protein 1 (YAP1), a hippo signaling pathway component. Subsequently, treatment with verteporfin, a YAP1 inhibitor, mimicked the effects of DCLK1 inhibitor treatment on both AsPC-1 and BxPC-3 cells. Verteporfin treatment also blunted the upregulation of PD-L1 mediated by DCLK1 overexpression, indicating that DCLK1’s regulation of PD-L1 is reliant upon hippo signaling [131].
Cancer is also associated with genomic instability and DNA damage. Silencing of a component of the DNA damage response machinery, ATM, upregulated PD-L1 expression. This study further showed that ATM can regulate type I IFN production in a TBK1/SRC-dependent but cGAS/STING-independent manner, suggesting that ATM modulates the expression of PD-L1 through IFN-STAT signaling [132]. In addition to ATM, other genes have been associated with the regulation of immune checkpoint protein expression. FOXP3, an essential gene in T-reg cells, can induce the expression of PD-L1 [133]. In human PC samples, the expression of FOXP3 and PD-L1 are highly correlated. Overexpression of FOXP3 resulted in increased mRNA transcript and protein expression of PD-L1, whereas shRNA-mediated knockdown of FOXP3 reduced PD-L1 expression at both the mRNA and protein levels. ChIP analysis established that there is a direct interaction between FOXP3 and the PD-L1 promoter region, with two possible binding motifs [133]. Further validation with a luciferase reporter assay identified the approximate promoter region involved in the transcription initiation of PD-L1 upon FOXP3 binding [133]. In addition, receptor-interacting serine/threonine-protein kinase 1 (RIP1), a vital regulator of tumor-associated macrophage differentiation, is also involved in the PD-1 regulation. Treating mice bearing orthotopic KPC murine model with a RIP1 small molecule inhibitor, RIP1i, increased PD-1 expression on both CD4+ and CD8+ T-cells [134].
4.2.3. Post-translational regulation of immune checkpoints
With the advancement in immune therapy, the post-translational modifications of immune checkpoint protein have also been shown to influence the outcomes of immune checkpoint blockade therapy [135]. PD-L1 can go through multiple post-translational modifications (PTMs). For example, EGF treatment increased the PTMs of PD-L1, including tyrosine phosphorylation, acetylation, and ubiquitination. Mono and poly-ubiquitination of PD-L1 were observed before the increased accumulation of PD-L1 after EGF treatment in the epidermoid carcinoma cell line A431, indicating that mono-ubiquitination of PD-L1 might be required for EGF-induced PD-L1 upregulation [136].
Multiple other post-translational modifications can also influence the stability of PD-L1. A study showed that PD-L1 glycosylation could protect PD-L1 from glycogen synthase kinase-3 beta (GSK3β) phosphorylation and sequential beta-transducin repeats-containing protein (β-TrCP)-mediated ubiquitination and degradation by the proteasome [137]. Another study also showed that ubiquitin-specific protease 22 (USP22) could catalyze the de-ubiquitination of PD-L1 via direct binding to the PD-L1 C-terminal domain, extending the half-life of PD-L1 protein [138]. Kinases also influence PD-L1 stability by phosphorylation. For example, never-in mitosis gene A-related kinase 2 (NEK2) deficiency is associated with reduced expression of PD-L1 [139]. Immunoprecipitation showed direct interaction between PD-L1 and NEK2 in human PC cell lines (SW1990 and CFPAC-1) and murine KPC model-derived cell lines. Treatment with MG132, a proteasome inhibitor, restored the NEK2 inhibition-mediated PD-L1 downregulation, indicating that NEK2 interferes with the degradation of PD-L1. This conclusion was further validated by a higher ubiquitination level of PD-L1 in MG132-treated and NEK2 inhibitor-treated cells [139]. Further analysis identified the NEK2-binding motif in the glycosylation-rich region of PD-L1, with two phosphorylation sites identified for NEK2-mediated phosphorylation. Altogether, NEK2 can phosphorylate the PD-L1 glycosylation-rich region, preventing the ubiquitination of PD-L1 and subsequent proteasome-mediated degradation [139]. Pro-inflammatory cytokine signaling is also involved in the stability of PD-L1. A study in breast cancer illustrated that the pro-inflammatory secretome from macrophages could induce the expression of PD-L1 at the protein level, and TNF-α was identified as the primary factor contributing to PD-L1 stabilization [140]. Further analysis showed that TNF-α induces the expression of CSN5 (COP9 signalosome subunit 5), a deubiquitinating enzyme, via nuclear factor-kappa B (NF-κB) signaling facilitating CNS5-mediated de-ubiquitination and thus stabilization of PD-L1 [140]. A follow-up study showed that shikonin, an anticancer drug isolated from the plant Lithospermum, can induce PD-L1 degradation via reducing glycosylation of PD-L1 through NF-κB/STAT3 and NF-κB/CSN5 signaling [141]. These reports indicate that post-translational modifications of the immune checkpoint, PD-L1, are essential to its function, expression, and stability.
5. Strategies to overcome the immune desert phenotype
Multiple targets and strategies have been investigated to eliminate the immune desert/suppressive characteristics of PC during immunotherapy, including physical ablation, neutralizing antibodies, modification of the immune microenvironment, small molecule inhibitors, and selective depletion of specific cell types in the TME.
5.1. Combination of immune checkpoint blockade with physical methods
Irreversible electroporation (IRE), which utilizes a short pulse of high voltage electricity, can induce cell death via membrane disruption [142]. Combining IRE with PD-1 antibody has been shown to increase the survival of mice with orthotopic mutant Kras tumors and improve intratumoral CD8+ T-cell infiltration. Comprehensive analysis within this study also showed that after the IRE and PD-1 antibody treatment, both the CD4+ memory T-cells (2.1-fold, CD4+ CD62L− CD44+) and CD8+ memory T- cells (5.7-fold, CD8+ CD62L− CD44+) increased significantly in the spleen of these animals. PC stromal cells were also impacted since IRE transiently downregulated hypoxia marker carbonic anhydrase-IX (CA-IX), extracellular matrix rigidity marker protein lysyl oxidase (LOX), and PD-L1 after the treatment. The IRE combined with PD-1 antibody also significantly downregulated fibroblast activation protein alpha (FAP-α) expression, a marker of cancer-associated fibroblasts, and hyaluronic acid-binding protein 1 (HABP1), a marker of hyaluronic acid in the stroma [142].
Radiation-induced DNA damage can be sensed by DNA damage repair machinery. One component of this machinery, ataxia-telangiectasia mutated (ATM) protein, one apical kinase, can respond to ionizing radiation-induced DNA damage. The combination of radiation with ATM inhibition, either via small molecule inhibitor or siRNA-mediated knockdown, activated type I IFN signaling. Because IFN-mediated signaling is known to upregulate PD-L1, an anti-PD-L1 antibody was then incorporated into the therapy regimen. After treatment with combined PD-L1 antibody and radiation therapy, the shRNA-mediated ATM-knockdown tumor inoculations had smaller tumor volumes and prevented relapse [132]. Altogether, targeting cancer cells with radiation, blocking DNA damage repair, and neutralizing PD-L1 signaling can contribute to a better PC treatment response.
5.2. Combination of immune checkpoint blockade with neutralizing antibodies
Besides physical methods, antibodies neutralizing important immune regulator cytokines or chemokines can also improve response to conventional immune checkpoint blockade.
IL-1β expression has been reported to be associated with PC tumorigenesis. In an orthotopic model bearing IL-1β-knockdown KrasG12D-derived murine pancreatic ductal epithelial cells (PDEC), there was decreased macrophage recruitment into the tumor stroma (predominantly M2 TAMs), as well as reduced MDSCs, neutrophils, B-regs, and Th17 cells [143]. Concurrently, there was increased infiltration of activated CD8+ T-cells into the tumor site, expressing high levels of IFN-γ and GzmB. The IL-1β-knockdown KrasG12D-PDEC orthotopic model had less pancreatic stellate cell activation and chemokine secretion from CD140a+ CAFs, including CCL2/CCL5/CCL8/CCL12, IL-6, IL-1α, and CXCL12. Co-implantation of immortalized Ly6C+CD140a+ CAFs with IL-1β-KD KrasG12D-PDEC cells in an orthotopic model, rescued IL-1β-knockdown KrasG12D-PDEC cell-mediated M2 macrophage re-education and IFNγ+ CD8+ T-cell infiltration, consistent with the observation that IL-1β-dependent cytokine/chemokine production in CAFs is critical for macrophage infiltration and M2 polarization (CCL2/CCL5) and CD8+ T-cell infiltration (CXCL12). Furthermore, when IL-1β-neutralizing antibody was combined with PD-1 antibody therapy, the tumor weights of the treated mice were significantly reduced, while the intratumoral CD8+ T-cell infiltration was increased [143].
IL-6 expression is enriched in the stroma component in PC and is mainly secreted by pancreatic stellate cells and tumor-associated myeloid cells [144]. Dual blockade against IL-6 and PD-L1 in both subcutaneous and orthotopic models can limit the progression of PC, increasing the survival of the treated mice, with more circulating and tumor-infiltrating effector T-cells and fewer α-SMA+ fibroblasts present after the treatment. Antibody-mediated CD4+ T-cell and CD8+ T-cell depletion demonstrated that IL-6/PD-L1 dual blockade therapy was dependent on CD8+ T- cells but not CD4+ T-cells [61]. As mentioned in the previous section, scFv-aIL-6R-expressing oncolytic adenovirus LOAd713 can also modify the immune environment of PDAC. LOAd713 treatment induced the oncolysis of PC cells, reduced the expression of desmoplasia development factors (such as TGFβ, fibroblast growth factor 5 (FGF5), and hepatocyte growth factor (HGF), prevented myeloid cell transition into MDSCs under IL-6 and GM-CSF treatment, and facilitated DC maturation [103].
IL-17 signaling in PC is associated with Kras-mutation-mediated IL-17 receptor upregulation and inflammation-induced IL-17-secreting immune cell recruitment [145]. An IL-17- neutralizing antibody was able to eliminate the neutrophil extracellular trap-mediated resistance to checkpoint blockade in PC, resulting in more CD8+ T-cell infiltration and better treatment outcomes with PD-1 or CTLA-4 antibody-based immune therapy [146]. In addition, IL-17 inhibits the production of CXCL9/10 and dampens CD8+ T-cell and T-reg infiltration into colorectal cancer [147], indicating that IL-17 may be involved in multiple immune regulation networks. Similarly, IL-18 expression is upregulated in PC patients, and this is associated with regulatory B-cell proliferation and IL-10 secretion [98]. Tail vein injection of IL-18-stimulated B-cells in mice increased the T-reg population while cytotoxic T-cell and NK cell populations decreased. Further, an antibody-dependent cellular cytotoxicity (ADCC) assay showed that coculture of T-cells or NK cells with IL-18-stimulated B-cells could impair the killing effects of T-cells or NK cells to target cells. Blocking IL-18 signaling with IL-18 binding protein (IL-18BP) could release the inhibition of T-cells or NK cells to kill the target cells. As mentioned above, IL-18 also induced the expression of PD-L1. Under dual blockade of IL-18BP and PD-L1/PD-1 inhibitor (PPI) in the orthotopic murine model, the tumor size was significantly reduced in the IL-18BP and PPI groups compared with PBS control. The IL-18BP and PPI combination regimens also prolonged the survival of the mice and reduced the tumor metastasis to the liver [98].
An agonist antibody against glucocorticoid-induced TNF receptor (GITR) was also demonstrated to have potential as a crucial tool for improving PC immunotherapy. It has been reported that the administration of anti-GITR antibodies suppressed the function of T-reg cells, resulting in the suppression of subcutaneous tumor growth and an increase in IFN-γ-producing splenocytes [148]. To improve the immunotherapy outcome, an IFN-α-adenovirus vector was introduced into the combination therapy with the anti-GITR antibody, which not only reduced tumor volumes that received the intratumoral injection of IFN-α-adenovirus but also on the tumor on the contralateral side, suggesting the IFN-α-adenovirus and anti-GITR antibody combination therapy would be capable of successfully treating metastasized tumors. The treatment effects were enhanced with a higher dose of IFN-α-adenovirus, especially on the uninjected side. Detailed immune phenotyping showed that combination therapy could increase the infiltration of IFN-γ-producing CD4+ and CD8+ T-cells and suppress T-reg infiltration, with reduced expression of CCR5 [148].
5.3. Combination of immune checkpoint blockade with small molecule inhibitors
While multiple cytokines modulate the TME, small molecule inhibitors can be utilized with cytokine treatment as one combination therapy strategy. For example, IFN-γ treatment can be combined with a CDK inhibitor [97]. The CDK inhibitor dinaciclib triggered histone-dependent immunogenic cell death. When combining dinaciclib with IFN-γ, the survival of the mutant Kras-driven PC murine model was prolonged, with less lesion formation and more T-cell intratumoral infiltration, Th1 response, and anti-tumor cytokine secretions [97].
Not only can cytokine treatment combine effectively with small molecule inhibitor treatments, but immune checkpoint antibodies can also be integrated with small molecule inhibitors to sensitize the tumor. For example, GSK547, an inhibitor of RIP1, was found to be effective in treating PC. RIP1 expression was upregulated in PC TAMs and targeting RIP1 with GSK547 alone in a murine PC model slowed PanIN lesion initiation, reduced fibrosis and tumor weight, and prolonged survival. RIP1 inhibition altered the infiltrating immune subsets of PC, with more CD4+ and CD8+ T-cells infiltrating into the TME and increased secretion of anti-tumor cytokines (including IFN-γ and TNF-α), along with less TAM infiltration and reduced secretion of TGFβ and IL-10. Elevated secretion of IFN-γ and TNF-α reprogramed the immune-suppressive environment. Combining RIP1 inhibition with a PD-1 antibody in the tumor-bearing murine model significantly reduced tumor weight and prolonged mouse survival, with more IFN-γ and TNF-α-secreting T-cells and fewer TGFβ-secreting T-cells [134].
Because histone methyltransferase (HMTase) regulates PD-L1, a combination of HMTase inhibition with an immune checkpoint inhibitor could potentially enhance the treatment outcome. A study that combined a pharmacological inhibitor of MLL1, verticillin, with anti-PD-L1 antibody therapy demonstrated a significant reduction in tumor volume and weight, with less proliferation and more apoptosis in PANC02-H7 and UN-KC-6141 cells transplanted in orthotopic models [123]. Detailed analysis showed that this combination strategy depends on FasL-mediated apoptosis of cancer cells and the infiltration of CD8+ cytotoxic T-cells. This was validated through transplantation of tumor cells into FasL-knockout mice and CD8+ T-cell depletion with neutralizing antibodies [123]. Histone modifying enzymes can also indirectly increase the efficacy of immunotherapy. A recent study has shown that lysine demethylase 3 A (KDM3A) through KLF5/SMAD4 axis increases the EGFR expression and knockout of KDM3A, and EGFR sensitizes PC to immunotherapy. A similar sensitization was also observed after treatment with the EGFR inhibitor, erlotinib [149].
It has been reported that higher FAK activation in PC was associated with fewer intratumoral CD8+ T-cells but more granulocyte and TAM infiltration, and increased expression of ECM proteins, contributing to the immune-suppressive TME [150]. A selective FAK inhibitor, VS-4718, reduced fibrosis and inhibited tumor progression and metastasis, with reduced immune-suppressive immune cell infiltration. When combined with adoptive cell therapy or immune checkpoint antagonist antibodies, FAK inhibition synergized anti-PD-L1 with other treatments and achieved better outcomes and improved survival, with more CD8+ CTL and fewer intratumoral immune-suppressive immune cells such as T-regs [150].
Heat shock protein 90 (HSP90) is essential in tumor immunity and multiple cellular processes. Using immortalized human PDAC-derived PSC (h-iPSC-PDAC-1) and primary PDAC patient-derived PSC cells (SC37), Zhang et al. demonstrated that inhibiting HSP90 with XL888 alters PSC activation, downregulating ERK and STAT3 activation and reducing IL-6 secretion. The HSP90 inhibitor combined with the PD-1 antibody resulted in reduced PC tumor volume and weight, with fewer α-SMA-positive fibroblasts and more CD4+ and CD8+ T-cell infiltration [151]. The hexosamine biosynthesis pathway (HBP), a branch of the glycolysis pathway, directing the generation of an important substrate for protein glycosylation, plays a critical role in cancer cell survival and remodeling the extracellular matrix. The rate-limiting enzyme of HBP, glutamine-fructose aminotransferase 1(GFAT1), can be targeted by a glutamine analog 6-diazo-5-oxo-L-norleucine (DON). A study reported that the expression of multiple HBP enzymes was upregulated in cerulein-induced pancreatitis and KPC tumors at the mRNA and protein levels and that GFAT1 is predominantly expressed within ductal cells of pancreatic tumors. Targeting GFAT1 with siRNA demonstrated that GFAT1 expression was associated with the expression of sternness-associated genes SOX2, OCT4, NANOG, and KLF4, suggesting its role in self-renewal. Inhibiting GFAT1 with DON reduced the colony formation of tumor epithelial cells and cell viability in vitro and reduced tumor weight and volume in subcutaneous and orthotopic murine models, with reduced expression of hyaluronan and collagen, and higher infiltration of CD68+ macrophages and CD8+ T-cells. Combination therapy with DON and PD-1 antibody significantly improved the treatment outcome in the tumor-bearing murine model, with diminished tumor weight and volume, prolonged survival, and lower expression levels of ECM components [152].
When a small molecule inhibitor is unavailable, shRNA- or siRNA-mediated knockdown can also facilitate PC treatment with immune checkpoint blockade. HAT1 is upregulated in PC and promotes PD-L1 expression, and shRNA-mediated knockdown of HAT1 expression in PANC-1, MIAPaCa-2, and BxPC-3 cell lines curtailed tumor proliferation and colony formation. HAT1-knockdown PANC-1 xenograft tumors were smaller and had fewer Ki-67+ proliferating cells compared to controls. Fan et al. also showed that after PD-1 antibody treatment, immunocompetent mice bearing HAT1-knockdown PC tumors had reduced tumor size, longer survival, more CD8+ T-cell infiltration, and fewer myeloid cells in the tumor, relative to mice with control shRNA-transfected tumors that were treated with PD-1 blockade [121].
5.4. Combining immunotherapy with TME modulation
Targeting cellular or acellular components of TME, both immunological and non-immunological, can modify responses to new treatment regimens, including immune therapy. T-regs, a crucial component for immune tolerance, can significantly influence tumor immunity. T-reg ablation with diphtheria toxin (DT)-mediated inducible gene knockout enhanced antigen presentation by DCs, increased IFN-γ-secreting CD8+ T-cell numbers and improved the treatment efficacy in tumor-bearing mice [55]. Double ablation of DCs and T-regs or combining T-reg ablation with CD8+ T-cell depletion blunted this T-reg-ablation-mediated tumor immunity, indicating that both DC antigen presentation and CD8+ T-cell cytotoxicity contribute to cancer immunity under T-reg depletion. Comprehensive examination showed that T-reg depletion altered DCs migration and trafficking in the tumor-draining lymph node and maturation [55]. On the other hand, another study on T-reg depletion showed different treatment outcomes. Rather than injecting Kras-mutant PC cells into T-reg-depleted mice, this study generated DT-inducible T-reg depletion in the background of the KC murine model of PC. This murine model showed that T-reg depletion decreased the secretion of TGF-β, which reduced the presence of α-SMA-expressing fibroblasts. While the reduction in fibroblast numbers unleashed the restraint on tumor cells, more myeloid cells infiltrated into TME and promoted carcinogenesis through CCR1 [153]. This study suggested that T-reg depletion alone may not be able to modify the immune-suppressive property of the PC TME, and more comprehensive methods to systematically change the TME are needed for better PC treatment regimens.
Other studies have also shown the essential functions of the CAFs in PC. One study showed that DT-induced depletion of fibroblast activation protein (FAP)-expressing CAFs in the KPC murine model could facilitate anti-tumor effects of anti-PD-L1 and anti-CTLA4 antibody therapy for PC. One signature of these FAP-expressing CAFs is CXCL12 expression and inhibiting CXCL12 function with a CXCR4 inhibitor-induced more T-cell infiltration and synergized with anti-PD-L1 therapy [57]. As with T-reg depletion, more studies have explored the fibroblast depletion strategy and clearly showed the functional relevance of fibroblasts in cancer immunity. One study utilized ganciclovir-induced α-SMA-tk mice for CAF depletion in the background of the Ptf1aCre/+, LSL-KrasG12D/+, Tgfbr2flox/flox (PKT) PC model. The α-SMA-myofibroblast deletion in this PC murine model resulted in more invasive tumors and diminished survival, with more hypoxia, T-reg infiltration, EMT, and an expanded number of cancer stem cells [62]. Another study focused on the PC stroma component and targeted sonic hedgehog (shh). Deletion of shh in the KPC murine model resulted in reduced stroma components, including α-SMA+ myofibroblasts, CD45+ myeloid cells, F4/80+ monocytes, and more vascular density and proliferation in the tumor, leading to accelerated tumor progression, metastasis, and worse survival [154]. Rather than targeting α-SMA+ myofibroblasts directly, another recent study combined deletion of type I collagen in α-SMA myofibroblasts with KPC mutation in pancreas acinar cells. This collagen deletion in myofibroblasts augmented immune suppression, promoted tumor progression, and diminished survival, along with reduced B- and T-cell, but increased CD11b+CD206+ myeloid cell and MDSC in TME [155]. Further study into the MDSCs population (CD206+F4/80+ARG1+) showed that these cells were recruited into the TME via increased CXCL5, with elevated expression of CD206, Arg1, F4/80, and chitinase-like protein 3 (Chil3), creating a suppressive environment for B- and T-cells [155]. Altogether, these data suggest that therapeutic approaches targeting fibroblasts involve multiple signaling pathways, T-reg/MSDC infiltration, and angiogenesis. Selective fibroblast targeting and characterizing various populations of fibroblasts may assist in providing novel targets for developing new PC treatments.
Fibroblast functions in PC are strongly associated with myeloid lineage cells, including macrophages. Several studies have examined how myeloid cells act as pro-tumor immune components of PC. For example, CD11b+ myeloid depletion was reported to arrest Kras-mutation-driven tumor growth and reduce the volume of already established subcutaneous pancreatic tumors [46]. Further analysis showed this depletion caused cell apoptosis and increased infiltration of CD8+ T-cells and augmented secretion of effector cytokines, such as IFN-γ, IFN-β1, and perforin-1. Dual deletion of CD11b+ myeloid cells and CD8+ T-cells can rescue the phenotype of CD11b+ myeloid cell depletion, indicating that the CD11b+ myeloid cell depletion-mediated tumor suppression was dependent on CD8+ T-cell functions [46]. Cancer cells express CD47 to deliver a “do not eat me” signal to macrophages. Overexpression of the microRNA miR-340 reduced the expression of CD47, thereby enhancing phagocytosis by macrophages and promoting anticancer immunity [156].
The PC TME is considered to be acidic and hypoxic. Neutralizing the acidity of the TME with oral bicarbonate monotherapy, combined with anti-PD1 or anti-CTLA-4 antibodies, can enhance the anti-tumor response in PC [157]. Hypoxia drives the production of 2-hydroxyglutarate (L-2HG), an epigenetic modifier, inducing H3 hypermethylation and altered global gene expression. Inhibiting the L-2-HG-producing enzyme lactate dehydrogenase (LDH) sensitized PC to anti-PD-1 therapy [158]. As another alternative, enhancing anti-tumor immune signaling can also improve the immune therapy outcome. Anti-OX40 agonist antibodies stimulate OX40 signaling, resulting in T-cell activation and survival. Anti-OX40 agonist and anti-PD-L1 antagonist antibodies eliminated tumor cells in an orthotopic pancreatic tumor model and allowed tumor-free survival of mice for more than 200 days [159]. In KPC murine model, the life span of anti-OX40 and anti-PD-L1 antibody-treated mice was significantly longer compared with IgG control-treated or single antibody-treated mice. Further analysis with antibody-based depletion of CD4+ T-cells showed that this dual-antibody treatment was dependent on CD4+ T-cells, while CyTOF analysis showed that dual-antibody treatment reduced T-reg/exhausted T-cell infiltration and T-cell expression of GATA3 and increased the number of CD127-expressing CD4+ T-cells [159]. A similar design was developed with the CD40 agonist antibody. Triple CD40/PD-L1/CTLA-4 antibody treatment activated anti-tumor T-cell responses to cancer and suppressed tumor growth, independent of innate immune sensor signaling involving the TLR, STING, and IFNAR signaling pathways but dependent on the expression of CD40 and basic leucine zipper ATF-like transcription (Batf3), that functions in priming of T-cells and the cross-presentation of DCs [160].
6. Conclusion and perspective
Projected soon to be the second leading cause of cancer-related deaths, PC still needs the development of novel therapeutic regimens focusing on targeting immunosuppressive TME. With the advancements in the molecular targets for immunotherapy, greater understanding of the immunosuppressive mechanisms in PC TME, including the contribution of stromal components, immunogenic and antigenic properties, and the expression and regulation of immune checkpoints, the stage is set for evaluating novel immunotherapeutic regimens as a mainstay to lay the foundation for developing superior PC treatments. However, as yet, only limited information has been revealed. A more detailed and systematic understanding of the PC TME is needed to analyze the landscape of fibroblast and macrophage heterogeneity. With the development of single-cell RNA sequencing and functional analysis, we have acquired information about the PC TME and an overview of interactions of fibroblasts, macrophages, B-cells, and T-cells. Further, advanced spatial approaches are required to analyze these interactions to gain comprehensive understanding of the pancreatic TME. Exploration of other immune components such as NK cells, mast cells, neutrophils, and tertiary lymphoid structures is required to understand their role in TME and elucidate their crosstalk with other cells. Investigation of other checkpoint molecules, their expression, and signaling is required to understand their role and target them for immune modulation of TME. Novel immunotherapies are being evaluated, focusing on new targets and combining new inhibitors with improved preclinical models, renewing the hope that a cure for PC will soon be possible.
Funding
The authors/work on this manuscript were supported, in parts, by grants from the NIH (R01 CA273349, R01 CA263575, R01 CA256973, R01 CA254036, R01 CA247471, R01 CA210637, R01 CA206444, U01 CA200466, U01 CA210240, P01 CA217798, R44 CA235991, P30 CA036727, T32 CA009476, and U54 GM115458).
Abbreviations:
- APCs
antigen presenting cells
- Arg1
arginase1
- α-SMA
α-smooth muscle actin
- ATM
ataxia telangiectasia mutated
- BCR
B-cell receptor
- β-TrCP
β-transducin repeat-containing proteins
- CAFs
cancer-associated fibroblasts
- CDK
cyclin-dependent kinase
- Chi3l3
chitinase-like 3
- ChIP
chromatin immunoprecipitation
- CSN5
COP9 signalosome subunit 5
- CTL
cytotoxic T lymphocyte
- CTLA-4
cytotoxic lymphocyte-associated antigen-4
- CyTOF
Cytometry by time of flight
- DAC
5-aza-dC
- DCLK1
doublecortin like kinase 1
- EGF
epidermal growth factor
- FAK
focal adhesion kinase
- FAP
fibroblast activation protein
- FBP1
fructose-1,6-biphosphatase
- FOXP3
forkhead box protein 3
- GEMMs
genetically engineered mouse models
- GFAT1
glutamine-fructose amidotransferase 1
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- GSK3β
Glycogen synthase kinase 3β
- HAT1
histone acetyltransferase 1
- HDAC3
histone deacetylase 3
- HIF
hypoxia-inducible factors
- HMTase
histone methyltransferase
- IDO1
indoleamine 2,3-dioxygenase-1
- IFN
interferon
- IRF1
interferon regulatory factor 1
- KDM3A
Lysine demethylase 3A
- KLF5
Kruppel like factor 5
- KPC model
LSL-KrasG12D/+, LSL-Trp53R172H/+, Pdx-1-Cre murine model
- LPS
lipopolysaccharides
- MDSCs
myeloid-derived suppressor cells
- MLL1
myeloid/lymphoid or mixed-lineage leukemia 1
- MSP
macrophage stimulating protein
- NEK2
never-in mitosis gene A-related kinase 2
- NK
natural killer
- OPN
osteopontin
- PAK
p21-activated kinases
- PanIN
pancreatic intraepithelial neoplasia
- PC
pancreatic cancer
- PD-1
programmed cell death-1
- PKM2
pyruvate kinase isoform M2
- PTPRO
protein tyrosine phosphatase receptor type O
- RIP1
receptor-interacting serine/threonine protein kinase
- scFv
single chain fragment variable
- scRNA-seq
single-cell RNA-sequencing
- shh
sonic hedgehog
- SMAD4
SMAD family member 4
- TAA
tumor-associated antigens
- TAMs
tumor-associated macrophages
- Th
helper T cells
- Th1
type 1 T helper cells
- TME
tumor microenvironment
- TNF-α
tumor necrosis factor-α
- Treg
regulatory T cells
- TSA
tumor-specific antigens
- WDR5
WD repeat-containing protein.
Footnotes
Conflicts of Interest
SKB is one of the founders of Sanguine Diagnostics and Therapeutics, Inc. Other authors have no conflicts of interest to declare.
Data Availability
Data will be made available on request.
References
- [1].Couzin-Frankel J, Breakthrough of the year 2013, Cancer Immunother. Sci 342 (6165)(2013) 1432–1433. [DOI] [PubMed] [Google Scholar]
- [2].Siegel RL, Miller KD, Fuchs HE, Jemal A, Cancer statistics, 2022, CA Cancer J. Clin 72 (1) (2022) 7–33. [DOI] [PubMed] [Google Scholar]
- [3].Burnet FM, Immunological surveillance in neoplasia, Transpl. Rev 7 (1971) 3–25. [DOI] [PubMed] [Google Scholar]
- [4].Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD, Cancer immunoediting: from immunosurveillance to tumor escape, Nat. Immunol 3 (11) (2002) 991–998. [DOI] [PubMed] [Google Scholar]
- [5].Dunn GP, Old LJ, Schreiber RD, The three Es of cancer immunoediting, Annu Rev. Immunol 22 (2004) 329–360. [DOI] [PubMed] [Google Scholar]
- [6].Marincola FM, Drucker BJ, Siao DY, Hough KL, Holder WD Jr., The nude mouse as a model for the study of human pancreatic cancer, J. Surg. Res 47 (6) (1989) 520–529. [DOI] [PubMed] [Google Scholar]
- [7].Tan MH, Chu TM, Characterization of the tumorigenic and metastatic properties of a human pancreatic tumor cell line (AsPC-1) implanted orthotopically into nude mice, Tumour Biol. 6 (1) (1985) 89–98. [PubMed] [Google Scholar]
- [8].Fu X, Guadagni F, Hoffman RM, A metastatic nude-mouse model of human pancreatic cancer constructed orthotopically with histologically intact patient specimens, Proc. Natl. Acad. Sci. USA 89 (12) (1992) 5645–5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Westphalen CB, Olive KP, Genetically engineered mouse models of pancreatic cancer, Cancer J. 18 (6) (2012) 502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA, Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice, Cancer Cell 7 (5) (2005) 469–483. [DOI] [PubMed] [Google Scholar]
- [11].Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA, Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse, Cancer Cell 4 (6) (2003) 437–450. [DOI] [PubMed] [Google Scholar]
- [12].Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH, Dynamics of the immune reaction to pancreatic cancer from inception to invasion, Cancer Res. 67 (19) (2007) 9518–9527. [DOI] [PubMed] [Google Scholar]
- [13].Evans RA, Diamond MS, Rech AJ, Chao T, Richardson MW, Lin JH, Bajor DL, Byrne KT, Stanger BZ, Riley JL, Markosyan N, Winograd R, Vonderheide RH, Lack of immunoediting in murine pancreatic cancer reversed with neoantigen, JCI Insight 1 (14) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hiraoka N, Onozato K, Kosuge T, Hirohashi S, Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions, Clin. Cancer Res 12 (18) (2006) 5423–5434. [DOI] [PubMed] [Google Scholar]
- [15].Lenzo FL, Kato S, Pabla S, DePietro P, Nesline MK, Conroy JM, Burgher B, Glenn ST, Kuvshinoff B, Kurzrock R, Morrison C, Immune profiling and immunotherapeutic targets in pancreatic cancer, Ann. Transl. Med 9 (2) (2021) 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, Hiraoka N, Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer, Br. J. Cancer 108 (4) (2013) 914–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Beatty GL, Gladney WL, Immune escape mechanisms as a guide for cancer immunotherapy, Clin. Cancer Res 21 (4) (2015) 687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yarchoan M, Johnson BA, Lutz ER, Laheru DA, Jaffee EM, Targeting neoantigens to augment antitumour immunity, Nat. Rev. Cancer 17 (4) (2017) 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kinker GS, Vitiello GAF, Ferreira WAS, Chaves AS, Cordeiro de Lima VC, Medina TDS, B cell orchestration of anti-tumor immune responses: a matter of cell localization and communication, Front Cell Dev. Biol 9 (2021), 678127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Easwaran H, Tsai HC, Baylin SB, Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance, Mol. Cell 54 (5) (2014) 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Saleh R, Toor SM, Sasidharan Nair V, Elkord E, Role of epigenetic modifications in inhibitory immune checkpoints in cancer development and progression, Front Immunol. 11 (2020) 1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Topper MJ, Vaz M, Marrone KA, Brahmer JR, Baylin SB, The emerging role of epigenetic therapeutics in immuno-oncology, Nat. Rev. Clin. Oncol 17 (2) (2020) 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, Davidson S, Edwards M, Elvin JA, Hodel KP, Zahurancik WJ, Suo Z, Lipman T, Wimmer K, Kratz CP, Bowers DC, Laetsch TW, Dunn GP, Johanns TM, Grimmer MR, Smirnov IV, Larouche V, Samuel D, Bronsema A, Osborn M, Stearns D, Raman P, Cole KA, Storm PB, Yalon M, Opocher E, Mason, Thomas GA, Sabel M, George B, Ziegler DS, Lindhorst S, Issai VM, Constantini S, Toledano H, Elhasid R, Farah R, Dvir R, Dirks P, Huang A, Galati MA, Chung J, Ramaswamy V, Irwin MS, Aronson M, Durno C, Taylor MD, Rechavi G, Maris JM, Bouffet E, Hawkins C, Costello JF, Meyn MS, Pursell ZF, Malkin D, Tabori U, Shlien A, Comprehensive analysis of hypermutation in human cancer, Cell 171 (5) (2017) 1042–1056, e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW, Cancer genome landscapes, Science 339 (6127) (2013) 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, Kurzrock R, Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers, Mol. Cancer Ther 16 (11) (2017) 2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, Herbst B, Askan G, Bhanot U, Senbabaoglu Y, Wells DK, Cary CIO,Grbovic-Huezo O, Attiyeh M, Medina B, Zhang J, Loo J, Saglimbeni J, Abu-Akeel M, Zappasodi R, Riaz N, Smoragiewicz M, Kelley ZL, Basturk O, Gönen M, Levine AJ, Allen PJ, Fearon DT, Merad M, Gnjatic S, Iacobuzio-Donahue CA, Wolchok JD, DeMatteo RP, Chan TA, Greenbaum BD, Merghoub T, Leach SD, Identification of unique neoantigen qualities in longterm survivors of pancreatic cancer, Nature 551 (7681) (2017) 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Beatty GL, Gladney WL, Immune escape mechanisms as a guide for cancer immunotherapy, Clin. Cancer Res 21 (4) (2015) 687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].de Charette M, Marabelle A, Houot R, Turning tumour cells into antigen presenting cells: the next step to improve cancer immunotherapy? Eur. J. Cancer 68 (2016) 134–147. [DOI] [PubMed] [Google Scholar]
- [29].Lohmann S, Wollscheid U, Huber C, Seliger B, Multiple levels of MHC class I down-regulation by ras oncogenes, Scand. J. Immunol 43 (5) (1996) 537–544. [DOI] [PubMed] [Google Scholar]
- [30].Delp K, Momburg F, Hilmes C, Huber C, Seliger B, Functional deficiencies of components of the MHC class I antigen pathway in human tumors of epithelial origin, Bone Marrow Transplant. 25 (2) (2000) S88–S95. [DOI] [PubMed] [Google Scholar]
- [31].El-Jawhari JJ, El-Sherbiny YM, Scott GB, Morgan RS, Prestwich R, Bowles PA, Blair GE, Tanaka T, Rabbitts TH, Meade JL, Cook GP, Blocking oncogenic RAS enhances tumour cell surface MHC class I expression but does not alter susceptibility to cytotoxic lymphocytes, Mol. Immunol 58 (2) (2014) 160–168. [DOI] [PubMed] [Google Scholar]
- [32].Pommier A, Anaparthy N, Memos N, Kelley ZL, Gouronnec A, Yan R, Auffray C, Albrengues J, Egeblad M, Iacobuzio-Donahue CA, Lyons SK, Fearon DT, Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases, Science 360 (6394) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bailey P, Chang DK, Forget M-A, San Lucas FA, Alvarez HA, Haymaker C, Chattopadhyay C, Kim S-H, Ekmekcioglu S, Grimm EA, Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma, Sci. Rep 6 (1) (2016) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Davis SJ, van der Merwe PA, Lck and the nature of the T cell receptor trigger, Trends Immunol. 32 (1) (2011) 1–5. [DOI] [PubMed] [Google Scholar]
- [35].Chen K, Wang Q, Li M, Guo H, Liu W, Wang F, Tian X, Yang Y, Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression, EBioMedicine 66 (2021), 103315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Arango Duque G, Descoteaux A, Macrophage cytokines: involvement in immunity and infectious diseases, Front Immunol. 5 (2014) 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yang S, Liu Q, Liao Q, Tumor-associated macrophages in pancreatic ductal adenocarcinoma: origin, polarization, function, and reprogramming, Front Cell Dev. Biol 8 (2020), 607209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Celhar T, Pereira-Lopes S, Thornhill SI, Lee HY, Dhillon MK, Poidinger M, Connolly JE, Lim LH, Biswas SK, Fairhurst AM, TLR7 and TLR9 ligands regulate antigen presentation by macrophages, Int Immunol. 28 (5) (2016) 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang X, Luo G, Zhang K, Cao J, Huang C, Jiang T, Liu B, Su L, Qiu Z, Hypoxic tumor-derived exosomal miR-301a mediates M2 macrophage polarization via PTEN/PI3Kγ to promote pancreatic cancer metastasis, Cancer Res 78 (16) (2018) 4586–4598. [DOI] [PubMed] [Google Scholar]
- [40].Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL, The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages, Am. J. Pathol 157 (2) (2000) 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Feng R, Morine Y, Ikemoto T, Imura S, Iwahashi S, Saito Y, Shimada M, Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction, Cell Commun. Signal 16 (1) (2018) 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Park JE, Dutta B, Tse SW, Gupta N, Tan CF, Low JK, Yeoh KW, Kon OL, Tam JP, Sze SK, Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift, Oncogene 38 (26) (2019) 5158–5173. [DOI] [PubMed] [Google Scholar]
- [43].Cui R, Yue W, Lattime EC, Stein MN, Xu Q, Tan XL, Targeting tumor-associated macrophages to combat pancreatic cancer, Oncotarget 7 (31) (2016) 50735–50754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI, Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells, J. Immunol 182 (9) (2009) 5693–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI, Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer, Nat. Med 13 (7) (2007) 828–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, Rhim AD, Simeone DM, Beatty GL, Pasca di Magliano M, Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer, Gut 66 (1) (2017) 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, Clendenin C, Gladney WL, Knoblock DM, Guimalda PD, Vonderheide RH, Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6C(low) F4/80(+) extratumoral macrophages, Gastroenterology 149 (1) (2015) 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].L Gabrilovich D, Nagaraj S, Myeloid-derived suppressor cells as regulators of the immune system, Nat. Rev. Immunol 9 (3) (2009) 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG, CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models, Cancer Res. 74 (18) (2014) 5057–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH, CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans, Science 331 (6024) (2011) 1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, Sullivan P, Mahany JJ, Gallagher M, Kramer A, Green SJ, O’Dwyer PJ, Running KL, Huhn RD, Antonia SJ, Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody, J. Clin. Oncol 25 (7) (2007) 876–883. [DOI] [PubMed] [Google Scholar]
- [52].Vonderheide RH, Burg JM, Mick R, Trosko JA, Li D, Shaik MN, Tolcher AW, Hamid O, Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors, Oncoimmunology 2 (1) (2013), e23033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Byrne KT, Vonderheide RH, CD40 stimulation obviates innate sensors and drives T cell immunity in cancer, Cell Rep. 15 (12) (2016) 2719–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sakaguchi S, Yamaguchi T, Nomura T, Ono M, Regulatory T cells and immune tolerance, Cell 133 (5) (2008) 775–787. [DOI] [PubMed] [Google Scholar]
- [55].Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D, Crosstalk between regulatory T cells and tumor-associated dendritic cells negates antitumor immunity in pancreatic cancer, Cell Rep. 20 (3) (2017) 558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Schiraldi M, Raucci A, Muñoz LM, Livoti E, Celona B, Venereau E, Apuzzo T, De Marchis F, Pedotti M, Bachi A, Thelen M, Varani L, Mellado M, Proudfoot A, Bianchi ME, Uguccioni M, HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4, J. Exp. Med 209 (3) (2012) 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, Teichmann SA, Janowitz T, L Jodrell D, Tuveson DA, Fearon DT, Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer, Proc. Natl. Acad. Sci. USA 110 (50) (2013) 20212–20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Liang JJ, Zhu S, Bruggeman R, Zaino RJ, Evans DB, Fleming JB, Gomez HF, Zander DS, Wang H, High levels of expression of human stromal cell-derived factor-1 are associated with worse prognosis in patients with stage II pancreatic ductal adenocarcinoma, Cancer Epidemiol. Biomark. Prev 19 (10) (2010) 2598–2604. [DOI] [PubMed] [Google Scholar]
- [59].Joyce JA, Fearon DT, T cell exclusion, immune privilege, and the tumor microenvironment, Science 348 (6230) (2015) 74–80. [DOI] [PubMed] [Google Scholar]
- [60].Zhang Y, Yan W, Collins MA, Bednar F, Rakshit S, Zetter BR, Stanger BZ, Chung I, Rhim AD, di Magliano MP, Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance, Cancer Res. 73 (20) (2013) 6359–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, Nordquist E, Cruz-Monserrate Z, Yu L, Young G, Zhong X, Zimmers TA, Ostrowski MC, Ludwig T, Bloomston M, Bekaii-Saab T, Lesinski GB, IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer, Gut 67 (2) (2018) 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, De Jesus-Acosta A, Sharma P, Heidari P, Mahmood U, Chin L, Moses HL, Weaver VM, Maitra A, Allison JP, LeBleu VS, Kalluri R, Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival, Cancer Cell 25 (6) (2014) 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Steele NG, Carpenter ES, Kemp SB, Sirihorachai V, The S, Delrosario L, Lazarus J, Amir ED, Gunchick V, Espinoza C, Bell S, Harris L, Lima F, Irizarry-Negron V, Paglia D, Macchia J, Chu AKY, Schofield H, Wamsteker EJ, Kwon R, Schulman A, Prabhu A, Law R, Sondhi A, Yu J, Patel A, Donahue K, Nathan H, Cho C, Anderson MA, Sahai V, Lyssiotis CA, Zou W, Allen BL, Rao A, Crawford HC, Bednar F, Frankel TL, Pasca di Magliano M, Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer, in: Nat. Cancer, 1, 2020, pp. 1097–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Greenwald RJ, Freeman GJ, Sharpe AH, The B7 family revisited, Annu. Rev. Immunol 23 (2005) 515–548. [DOI] [PubMed] [Google Scholar]
- [65].Sfanos KS, Bruno TC, Meeker AK, De Marzo AM, Isaacs WB, Drake CG, Human prostate-infiltrating CD8+ T lymphocytes are oligoclonal and PD-1, Prostate 69 (15) (2009) 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA, Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired, Blood J. Am. Soc. Hematol 114 (8) (2009) 1537–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Keir ME, Butte MJ, Freeman GJ, Sharpe AH, PD-1 and its ligands in tolerance and immunity, Annu. Rev. Immunol 26 (2008) 677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Salmaninejad A, Valilou SF, Shabgah AG, Aslani S, Alimardani M, Pasdar A, Sahebkar A, PD-1/PD-L1 pathway: basic biology and role in cancer immunotherapy, J. Cell. Physiol 234 (10) (2019) 16824–16837. [DOI] [PubMed] [Google Scholar]
- [69].Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, Manos M, Gjini E, Kuchroo JR, Ishizuka JJ, Collier JL, Griffin GK, Maleri S, Comstock DE, Weiss SA, Brown FD, Panda A, Zimmer MD, Manguso RT, Hodi FS, Rodig SJ, Sharpe AH, Haining WN, Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade, Nat. Immunol 20 (3) (2019) 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Liotti F, Kumar N, Prevete N, Marotta M, Sorriento D, Ieranò C, Ronchi A, Marino FZ, Moretti S, Colella R, Puxeddu E, Paladino S, Kano Y, Ohh M, Scala S, Melillo RM, PD-1 blockade delays tumor growth by inhibiting an intrinsic SHP2/Ras/MAPK signalling in thyroid cancer cells, J. Exp. Clin. Cancer Res 40 (1) (2021) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Rota G, Niogret C, Dang AT, Barros CR, Fonta NP, Alfei F, Morgado L, Zehn D, Birchmeier W, Vivier E, Guarda G, Shp-2 is dispensable for establishing T cell exhaustion and for PD-1 Signaling In Vivo, Cell Rep. 23 (1) (2018) 39–49. [DOI] [PubMed] [Google Scholar]
- [72].McCoy KD, Le Gros G, The role of CTLA-4 in the regulation of T cell immune responses, Immunol. Cell Biol 77 (1) (1999) 1–10. [DOI] [PubMed] [Google Scholar]
- [73].Jago CB, Yates J, Camara NO, Lechler RI, Lombardi G, Differential expression of CTLA-4 among T cell subsets, Clin. Exp. Immunol 136 (3) (2004) 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S, Immunologic self-tolerance maintained by CD25+ CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4, J. Exp. Med 192 (2) (2000) 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lang C, Wang J, Chen L, CD25-expressing Thl7 cells mediate CD8(+) T cell suppression in CTLA-4 dependent mechanisms in pancreatic ductal adenocarcinoma, Exp. Cell Res 360 (2) (2017) 384–389. [DOI] [PubMed] [Google Scholar]
- [76].Walker LS, Treg and CTLA-4: two intertwining pathways to immune tolerance, J. Autoimmun 45 (100) (2013) 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Bengsch F, Knoblock DM, Liu A, McAllister F, Beatty GL, CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer, Cancer Immunol. Immunother 66 (12) (2017) 1609–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Qin S, Xu L, Yi M, Yu S, Wu K, Luo S, Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4, Mol. Cancer 18 (1) (2019) 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, Jaffee E, Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells, Cancer Immunol. Res 3 (4) (2015) 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Anderson AC, Joller N, Kuchroo VK, Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation, Immunity 44 (5) (2016) 989–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Workman CJ, Cauley LS, Kim I-J, Blackman MA, Woodland DL, Vignali DA, Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo, J. Immunol 172 (9) (2004) 5450–5455. [DOI] [PubMed] [Google Scholar]
- [82].Huang Y-H, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, Dougan SK, Petersen B-S, Melum E, Pertel T, CEACAM1 regulates TIM-3-mediated tolerance and exhaustion, Nature 517 (7534) (2015) 386–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng XX, Strom TB, Kuchroo VK, The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity, Nat. Immunol 6 (12) (2005) 1245–1252. [DOI] [PubMed] [Google Scholar]
- [84].Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD, Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1, Nat. Immunol 13 (9) (2012) 832–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Balli D, Rech AJ, Stanger BZ, Vonderheide RH, Immune cytolytic activity stratifies molecular subsets of human pancreatic cancer, Clin. Cancer Res 23 (12) (2017) 3129–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, Wang Z, Wu Q, Peng H, Wei H, Sun R, Tian Z, Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity, Nat. Immunol 19 (7) (2018) 723–732. [DOI] [PubMed] [Google Scholar]
- [87].Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA, The TIGIT/CD226 axis regulates human T cell function, J. Immunol 188 (8) (2012) 3869–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, Johnston J, Hammond A, Bontadelli K, Ardourel D, Hebb L, Wolf A, Bukowski TR, Rixon MW, Kuijper JL, Ostrander CD, West JW, Bilsborough J, Fox A, Gao Z, Xu W, Ramsdell F, Blazar BR, Lewis KE, Vstm3 is a member of the CD28 family and an important modulator of T-cell function, Eur. J. Immunol 41 (4) (2011) 902–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ, Interferon-gamma at the crossroads of tumor immune surveillance or evasion, Front. Immunol 9 (2018) 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Imai D, Yoshizumi T, Okano S, Itoh S, Ikegami T, Harada N, Aishima S, Oda Y, Maehara Y, IFN-gamma promotes epithelial-mesenchymal transition and the expression of PD-L1 in pancreatic cancer, J. Surg. Res 240 (2019) 115–123. [DOI] [PubMed] [Google Scholar]
- [91].Thiem A, Hesbacher S, Kneitz H, di Primio T, Heppt MV, Hermanns HM, Goebeler M, Meierjohann S, Houben R, Schrama D, IFN-gamma-induced PD-L1 expression in melanoma depends on p53 expression, J. Exp. Clin. Cancer Res 38 (1) (2019) 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zhao T, Li Y, Zhang J, Zhang B, PD-L1 expression increased by IFN-gamma via JAK2-STAT1 signaling and predicts a poor survival in colorectal cancer, Oncol. Lett 20 (2) (2020) 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Korentzelos D, Wells A, Clark AM, Interferon-gamma increases sensitivity to chemotherapy and provides immunotherapy targets in models of metastatic castration-resistant prostate cancer, Sci. Rep 12 (1) (2022) 6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, Parisi G, Saus CP, Torrejon DY, Graeber TG, Comin-Anduix B, Hu-Lieskovan S, Damoiseaux R, Lo RS, Ribas A, Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression, Cell Rep. 19 (6) (2017) 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Qian J, Wang C, Wang B, Yang J, Wang Y, Luo F, Xu J, Zhao C, Liu R, Chu Y, The IFN-gamma/PD-L1 axis between T cells and tumor microenvironment: hints for glioma anti-PD-1/PD-L1 therapy, J. Neuroinflamm 15 (1) (2018) 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Newman AC, Falcone M, Huerta Uribe A, Zhang T, Athineos D, Pietzke M, Vazquez A, Blyth K, Maddocks ODK, Immune-regulated IDO1-dependent tryptophan metabolism is source of one-carbon units for pancreatic cancer and stellate cells, Mol. Cell 81 (11) (2021) 2290–2302, e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Huang J, Chen P, Liu K, Liu J, Zhou B, Wu R, Peng Q, Liu ZX, Li C, Kroemer G, Lotze M, Zeh H, Kang R, Tang D, CDK1/2/5 inhibition overcomes IFNG-mediated adaptive immune resistance in pancreatic cancer, Gut 70 (5) (2021) 890–899. [DOI] [PubMed] [Google Scholar]
- [98].Zhao Y, Shen M, Feng Y, He R, Xu X, Xie Y, Shi X, Zhou M, Pan S, Wang M, Guo X, Qin R, Regulatory B cells induced by pancreatic cancer cell-derived interleukin-18 promote immune tolerance via the PD-1/PD-L1 pathway, Oncotarget 9 (19) (2018) 14803–14814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Terme M, Ullrich E, Aymeric L, Meinhardt K, Desbois M, Delahaye N, Viaud S, Ryffel B, Yagita H, Kaplanski G, Prévost-Blondel A, Kato M, Schultze JL, Tartour E, Kroemer G, Chaput N, Zitvogel L, IL-18 induces PD-1-dependent immunosuppression in cancer, Cancer Res. 71 (16) (2011) 5393–5399. [DOI] [PubMed] [Google Scholar]
- [100].Nagathihalli NS, Castellanos JA, VanSaun MN, Dai X, Ambrose M, Guo Q, Xiong Y, Merchant NB, Pancreatic stellate cell secreted IL-6 stimulates STAT3 dependent invasiveness of pancreatic intraepithelial neoplasia and cancer cells, Oncotarget 7 (40) (2016) 65982–65992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].van Duijneveldt G, Griffin MDW, Putoczki TL, Emerging roles for the IL-6 family of cytokines in pancreatic cancer, Clin. Sci. (Lond.) 134 (16) (2020) 2091–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Zhang W, Liu Y, Yan Z, Yang H, Sun W, Yao Y, Chen Y, Jiang R, IL-6 promotes PD-L1 expression in monocytes and macrophages by decreasing protein tyrosine phosphatase receptor type O expression in human hepatocellular carcinoma, J. Immunother. Cancer 8 (1) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Eriksson E, Milenova I, Wenthe J, Moreno R, Alemany R, Loskog A, IL-6 signaling blockade during CD40-mediated immune activation favors antitumor factors by reducing TGF-β, collagen Type I, and PD-L1/PD-1, J. Immunol 202 (3) (2019) 787–798. [DOI] [PubMed] [Google Scholar]
- [104].Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S, PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation, J. Exp. Med 211 (5) (2014) 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Harvey JB, Phan LH, Villarreal OE, Bowser JL, CD73’s potential as an immunotherapy target in gastrointestinal cancers, Front Immunol. 11 (2020) 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP, Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia, J. Clin. Invest 110 (7) (2002) 993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Palsson-McDermott EM, Dyck L, Zaslona Z, Menon D, McGettrick AF, Mills KHG, O’Neill LA, Pyruvate kinase M2 is required for the expression of the immune checkpoint PD-L1 in immune cells and tumors, Front Immunol. 8 (2017) 1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Grzywa TM, Sosnowska A, Matryba P, Rydzynska Z, Jasinski M, Nowis D, Golab J, Myeloid cell-derived arginase in cancer immune response, Front. Immunol 11 (2020) 938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Sharda DR, Yu S, Ray M, Squadrito ML, De Palma M, Wynn TA, Morris SM Jr., Hankey PA, Regulation of macrophage arginase expression and tumor growth by the Ron receptor tyrosine kinase, J. Immunol 187 (5) (2011) 2181–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Gray MJ, Poljakovic M, Kepka-Lenhart D, Morris SM Jr., Induction of arginase I transcription by IL-4 requires a composite DNA response element for STAT6 and C/EBPbeta, Gene 353 (1) (2005) 98–106. [DOI] [PubMed] [Google Scholar]
- [111].Wei LH, Jacobs AT, Morris SM Jr., Ignarro LJ, IL-4 and IL-13 upregulate arginase I expression by cAMP and JAK/STAT6 pathways in vascular smooth muscle cells, Am. J. Physiol. Cell Physiol 279 (1) (2000) C248–C256. [DOI] [PubMed] [Google Scholar]
- [112].Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, Tonc E, Schreiber RD, Pearce EJ, Pearce EL, Metabolic competition in the tumor microenvironment is a driver of cancer progression, Cell 162 (6) (2015) 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR, The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation, Immunity 35 (6) (2011) 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG, Pearce EL, Posttranscriptional control of T cell effector function by aerobic glycolysis, Cell 153 (6) (2013) 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R, Restoring function in exhausted CD8 T cells during chronic viral infection, Nature 439 (7077) (2006) 682–687. [DOI] [PubMed] [Google Scholar]
- [116].van der Windt GJ, Pearce EL, Metabolic switching and fuel choice during T-cell differentiation and memory development, Immunol. Rev 249 (1) (2012) 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E, Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation, Int J. Cancer 131 (3) (2012) 633–640. [DOI] [PubMed] [Google Scholar]
- [118].Sreeramkumar V, Fresno M, Cuesta N, Prostaglandin E2 and T cells: friends or foes? Immunol. Cell Biol 90 (6) (2012) 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Ohta A, A metabolic immune checkpoint: adenosine in tumor microenvironment, Front Immunol. 7 (2016) 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Wang B, Zhou Y, Zhang J, Jin X, Wu H, Huang H, Fructose-1,6-bisphosphatase loss modulates STAT3-dependent expression of PD-L1 and cancer immunity, Theranostics 10 (3) (2020) 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Fan P, Zhao J, Meng Z, Wu H, Wang B, Wu H, Jin X, Overexpressed histone acetyltransferase 1 regulates cancer immunity by increasing programmed death-ligand 1 expression in pancreatic cancer, J. Exp. Clin. Cancer Res 38 (1) (2019) 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Hu G, He N, Cai C, Cai F, Fan P, Zheng Z, Jin X, HDAC3 modulates cancer immunity via increasing PD-L1 expression in pancreatic cancer, Pancreatology 19 (2) (2019) 383–389. [DOI] [PubMed] [Google Scholar]
- [123].Lu C, Paschall AV, Shi H, Savage N, Waller JL, Sabbatini ME, Oberlies NH, Pearce C, Liu K, The MLL1-H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion, J. Natl. Cancer Inst 109 (6) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Lu C, Liu Z, Klement JD, Yang D, Merting AD, Poschel D, Albers T, Waller JL, Shi H, Liu K, WDR5-H3K4me3 epigenetic axis regulates OPN expression to compensate PD-L1 function to promote pancreatic cancer immune escape, J. Immunother. Cancer 9 (7) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Gonda TA, Fang J, Salas M, Do C, Hsu E, Zhukovskaya A, Siegel A, Takahashi R, Lopez-Bujanda ZA, Drake CG, Manji GA, Wang TC, Olive KP, Tycko B, DNA A, Hypomethylating drug alters the tumor microenvironment and improves the effectiveness of immune checkpoint inhibitors in a mouse model of pancreatic cancer, Cancer Res. 80 (21) (2020) 4754–4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ, Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov 13 (11) (2014) 828–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Coelho MA, de Carne Trecesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, East P, Spencer-Dene B, Nye E, Barnouin K, Snijders AP, Lai WS, Blackshear PJ, Downward J, Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA, Immunity 47 (6) (2017) 1083–1099, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Oliveira-Cunha M, Newman WG, Siriwardena AK, Epidermal growth factor receptor in pancreatic cancer, Cancers (Basel) 3 (2) (2011) 1513–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, Wilkerson MD, Fecci PE, Butaney M, Reibel JB, Soucheray M, Cohoon TJ, Janne PA, Meyerson M, Hayes DN, Shapiro GI, Shimamura T, Sholl LM, Rodig SJ, Freeman GJ, Hammerman PS, Dranoff G, Wong KK, Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors, Cancer Discov. 3 (12) (2013) 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Wang K, Zhan Y, Huynh N, Dumesny C, Wang X, Asadi K, Herrmann D, Timpson P, Yang Y, Walsh K, Baldwin GS, Nikfarjam M, He H, Inhibition of PAK1 suppresses pancreatic cancer by stimulation of anti-tumour immunity through down-regulation of PD-L1, Cancer Lett. 472 (2020) 8–18. [DOI] [PubMed] [Google Scholar]
- [131].Yan R, Li J, Zhou Y, Yao L, Sun R, Xu Y, Ge Y, An G, Inhibition of DCLK1 down-regulates PD-L1 expression through Hippo pathway in human pancreatic cancer, Life Sci. 241 (2020), 117150. [DOI] [PubMed] [Google Scholar]
- [132].Zhang Q, Green MD, Lang X, Lazarus J, Parsels JD, Wei S, Parsels LA, Shi J, Ramnath N, Wahl DR, Pasca di Magliano M, Frankel TL, Kryczek I, Lei YL, Lawrence TS, Zou W, Morgan MA, Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy, Cancer Res. 79 (15) (2019) 3940–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Wang X, Li X, Wei X, Jiang H, Lan C, Yang S, Wang H, Yang Y, Tian C, Xu Z, Zhang J, Hao J, Ren H, PD-L1 is a direct target of cancer-FOXP3 in pancreatic ductal adenocarcinoma (PDAC), and combined immunotherapy with antibodies against PD-L1 and CCL5 is effective in the treatment of PDAC, Signal Transduct. Target Ther 5 (1) (2020) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Wang W, Marinis JM, Beal AM, Savadkar S, Wu Y, Khan M, Taunk PS, Wu N, Su W, Wu J, Ahsan A, Kurz E, Chen T, Yaboh I, Li F, Gutierrez J, Diskin B, Hundeyin M, Reilly M, Lich JD, Harris PA, Mahajan MK, Thorpe JH, Nassau P, Mosley JE, Leinwand J, Kochen Rossi JA, Mishra A, Aykut B, Glacken M, Ochi A, Verma N, Kim JI, Vasudevaraja V, Adeegbe D, Almonte C, Bagdatlioglu E, Cohen DJ, Wong KK, Bertin J, Miller G, RIP1 kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer, Cancer Cell 34 (5) (2018) 757–774, e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Lee HH, Wang YN, Xia W, Chen CH, Rau KM, Ye L, Wei Y, Chou CK, Wang SC, Yan M, Tu CY, Hsia TC, Chiang SF, Chao KSC, Wistuba II, Hsu JL, Hortobagyi GN, Hung MC, Removal of N-linked glycosylation enhances PD-L1 detection and predicts Anti-PD-1/PD-L1 therapeutic efficacy, Cancer Cell 36 (2) (2019) 168–178, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Horita H, Law A, Hong S, Middleton K, Identifying regulatory posttranslational modifications of PD-L1: a focus on monoubiquitinaton, Neoplasia 19 (4) (2017) 346–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, Khoo KH, Chang SS, Cha JH, Kim T, Hsu JL, Wu Y, Hsu JM, Yamaguchi H, Ding Q, Wang Y, Yao J, Lee CC, Wu HJ, Sahin AA, Allison JP, Yu D, Hortobagyi GN, Hung MC, Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity, Nat. Commun 7 (2016) 12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Huang X, Zhang Q, Lou Y, Wang J, Zhao X, Wang L, Zhang X, Li S, Zhao Y, Chen Q, Liang T, Bai X, USP22 deubiquitinates CD274 to suppress anticancer immunity, Cancer Immunol. Res 7 (10) (2019) 1580–1590. [DOI] [PubMed] [Google Scholar]
- [139].Zhang X, Huang X, Xu J, Li E, Lao M, Tang T, Zhang G, Guo C, Zhang X, Chen W, Yadav DK, Bai X, Liang T, NEK2 inhibition triggers anti-pancreatic cancer immunity by targeting PD-L1, Nat. Commun 12 (1) (2021) 4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, Chang SS, Lin WC, Hsu JM, Hsu YH, Kim T, Chang WC, Hsu JL, Yamaguchi H, Ding Q, Wang Y, Yang Y, Chen CH, Sahin AA, Yu D, Hortobagyi GN, Hung MC, Deubiquitination and stabilization of PD-L1 by CSN5, Cancer Cell 30 (6) (2016) 925–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Ruan Z, Liang M, Shang L, Lai M, Deng X, Su X, Shikonin-mediated PD-L1 degradation suppresses immune evasion in pancreatic cancer by inhibiting NF-κB/STAT3 and NF-κB/CSN5 signaling pathways, Pancreatology 21 (3) (2021) 630–641. [DOI] [PubMed] [Google Scholar]
- [142].Zhao J, Wen X, Tian L, Li T, Xu C, Wen X, Melancon MP, Gupta S, Shen B, Peng W, Li C, Irreversible electroporation reverses resistance to immune checkpoint blockade in pancreatic cancer, Nat. Commun 10 (1) (2019) 899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Das S, Shapiro B, Vucic EA, Vogt S, Bar-Sagi D, Tumor cell-derived IL1beta promotes desmoplasia and immune suppression in pancreatic cancer, Cancer Res. 80 (5) (2020) 1088–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Klöppel G, Yoshimura A, Reindl W, Sipos B, Akira S, Schmid RM, Aigül H, Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer, Cancer Cell 19 (4) (2011) 456–469. [DOI] [PubMed] [Google Scholar]
- [145].McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, Rattigan Y, Roeser JC, Lankapalli RH, Zhang H, Jaffee EM, Drake CG, Housseau F, Maitra A, Kolls JK, Sears CL, Pardoll DM, Leach SD, Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia, Cancer Cell 25 (5) (2014) 621–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Zhang Y, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A, Zoltan M, Arora N, Baydogan S, Home W, Burks J, Xu H, Hussain P, Wang H, Gupta S, Maitra A, Bailey JM, Moghaddam SJ, Banerjee S, Sahin I, Bhattacharya P, McAllister F, Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer, J. Exp. Med 217 (12) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Chen J, Ye X, Pitmon E, Lu M, Wan J, Jellison ER, Adler AJ, Vella AT, Wang K, IL-17 inhibits CXCL9/10-mediated recruitment of CD8(+) cytotoxic T cells and regulatory T cells to colorectal tumors, J. Immunother. Cancer 7 (1) (2019) 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Aida K, Miyakawa R, Suzuki K, Narumi K, Udagawa T, Yamamoto Y, Chikaraishi T, Yoshida T, Aoki K, Suppression of Tregs by anti-glucocorticoid induced TNF receptor antibody enhances the antitumor immunity of interferon-α gene therapy for pancreatic cancer, Cancer Sci. 105 (2) (2014) 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Li J, Yuan S, Norgard RJ, Yan F, Sun YH, Kim IK, Merrell AJ, Sela Y, Jiang Y, Bhanu NV, Garcia BA, Vonderheide RH, Blanco A, Stanger BZ, Epigenetic and transcriptional control of the epidermal growth factor receptor regulates the tumor immune microenvironment in pancreatic cancer, Cancer Discov. 11 (3) (2021) 736–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, Pachter JA, Wang-Gillam A, DeNardo DG, Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy, Nat. Med 22 (8) (2016) 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Zhang Y, Ware MB, Zaidi MY, Ruggieri AN, Olson BM, Komar H, Farren MR, Nagaraju GP, Zhang C, Chen Z, Sarmiento JM, Ahmed R, Maithel SK, El-Rayes BF, Lesinski GB, Heat shock protein-90 inhibition alters activation of pancreatic stellate cells and enhances the efficacy of PD-1 blockade in pancreatic cancer, Mol. Cancer Ther 20 (1) (2021) 150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Sharma NS, Gupta VK, Garrido VT, Hadad R, Durden BC, Kesh K, Giri B, Ferrantella A, Dudeja V, Saluja A, Banerjee S, Targeting tumor-intrinsic hexosamine biosynthesis sensitizes pancreatic cancer to anti-PD1 therapy, J. Clin. Invest 130 (1) (2020) 451–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Zhang Y, Lazarus J, Steele NG, Yan W, Lee HJ, Nwosu ZC, Halbrook CJ, Menjivar RE, Kemp SB, Sirihorachai VR, Velez-Delgado A, Donahue K, Carpenter ES, Brown KL, Irizarry-Negron V, Nevison AC, Vinta A, Anderson MA, Crawford HC, Lyssiotis CA, Frankel TL, Bednar F, Pasca di Magliano M, Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis, Cancer Disco 10 (3) (2020) 422–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Femandez-Barrena MG, Femandez-Zapico ME, Iacobuzio-Donahue C, Olive KP, Stanger BZ, Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma, Cancer Cell 25 (6) (2014) 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Chen Y, Kim J, Yang S, Wang H, Wu CJ, Sugimoto H, LeBleu VS, Kalluri R, Type I collagen deletion in αSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer, Cancer Cell 39 (4) (2021) 548–565, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Xi Q, Zhang J, Yang G, Zhang L, Chen Y, Wang C, Zhang Z, Guo X, Zhao J, Xue Z, Li Y, Zhang Q, Da Y, Liu L, Yao Z, Zhang R, Restoration of miR-340 controls pancreatic cancer cell CD47 expression to promote macrophage phagocytosis and enhance antitumor immunity, J. Immunother. Cancer 8 (1) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, Damaghi M, Wojtkowiak JW, Mule JJ, Ibrahim-Hashim A, Gillies RJ, Neutralization of tumor acidity improves antitumor responses to immunotherapy, Cancer Res. 76 (6) (2016) 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Gupta VK, Sharma NS, Durden B, Garrido VT, Kesh K, Edwards D, Wang D, Myer C, Mateo-Victoriano B, Kollala SS, Ban Y, Gao Z, Bhattacharya SK, Saluja A, Singh PK, Banerjee S, Hypoxia-driven oncometabolite L-2HG maintains stemness-differentiation balance and facilitates immune evasion in pancreatic cancer, Cancer Res. 81 (15) (2021) 4001–4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Ma Y, Li J, Wang H, Chiu Y, Kingsley CV, Fry D, Delaney SN, Wei SC, Zhang J, Maitra A, Yee C, Combination of PD-1 inhibitor and OX40 agonist induces tumor rejection and immune memory in mouse models of pancreatic cancer, Gastroenterology 159 (1) (2020) 306–319, el2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Morrison AH, Diamond MS, Hay CA, Byrne KT, Vonderheide RH, Sufficiency of CD40 activation and immune checkpoint blockade for T cell priming and tumor immunity, Proc. Natl. Acad. Sci. USA 117 (14) (2020) 8022–8031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.