

Abstract
How SARS-CoV-2 penetrates the airway barrier of mucus and periciliary mucins to infect nasal epithelium remains unclear. Using primary nasal epithelial organoid cultures, we found that the virus attaches to motile cilia via the ACE2 receptor. SARS-CoV-2 traverses the mucus layer, using motile cilia as tracks to access the cell body. Depleting cilia blocks infection for SARS-CoV-2 and other respiratory viruses. SARS-CoV-2 progeny attach to airway microvilli 24 h post-infection and trigger formation of apically extended and highly branched microvilli that organize viral egress from the microvilli back into the mucus layer, supporting a model of virus dispersion throughout airway tissue via mucociliary transport. Phosphoproteomics and kinase inhibition reveal that microvillar remodeling is regulated by p21-activated kinases (PAK). Importantly, Omicron variants bind with higher affinity to motile cilia and show accelerated viral entry. Our work suggests that motile cilia, microvilli, and mucociliary-dependent mucus flow are critical for efficient virus replication in nasal epithelia.
Keywords: SARS-CoV-2, COVID-19, organoids, airway epithelial cells, cilia, respiratory virus, proteomics, respiratory synctital virus, protein kinases, microvilli
Graphical abstract
Respiratory viruses, including SARS-CoV-2, bypass the defensive mucus/mucin layer of the airway by entering and exiting epithelial cells via their protruding, motile cilia and microvilli.
Introduction
SARS-CoV-2 caused the COVID-19 pandemic. After the viral spike glycoprotein (SP) binds to its host receptor, the angiotensin-converting enzyme 2 (ACE2)1 and the host transmembrane serine protease 2 (TMPRSS2) prime SP to facilitate viral fusion, and the virus enters the host cells and begins replication.1 , 2 Viral replication has been modeled in tissue culture. However, the primary site of SARS-Cov-2 replication is the upper respiratory tract, which is specialized to create a barrier to virus infection. Indeed, the mechanisms of virus cell entry, exit, and cell-to-cell spread in the airway epithelium are poorly understood.
Nasal airways are pseudostratified epithelia, including multiciliated apical epithelial, basal, and mucus-producing goblet cells. The mucus layer sits atop the ciliary brush, and an underlying periciliary layer (PCL)3 , 4 keeps mucus away from the epithelia but permits ciliary beating.3 , 5 , 6 Mucus traps virus particles that are swept to the laryngopharynx by ciliated epithelial cells (CECs). CECs comprise 80% of nasal epithelium, and each express ∼300 cilia that show coordinated beating.3 , 4 , 5 , 6 Mucociliary clearance (MCC) eliminates infectious particles by coughing or swallowing. The underlying PCL mucin layer is critical to blocking viral entry.6 Small particles (∼25 nm) penetrate the PCL, but larger particles (∼100 nm) cannot.6 , 7 Depleting core PCL components increases influenza infection in various cells.8 , 9 Yet, many large, enveloped viruses (such as influenza and SARS-CoV-2) infect airway epithelial cells. How respiratory viruses bypass the innate barriers and cross back to spread remains unclear.
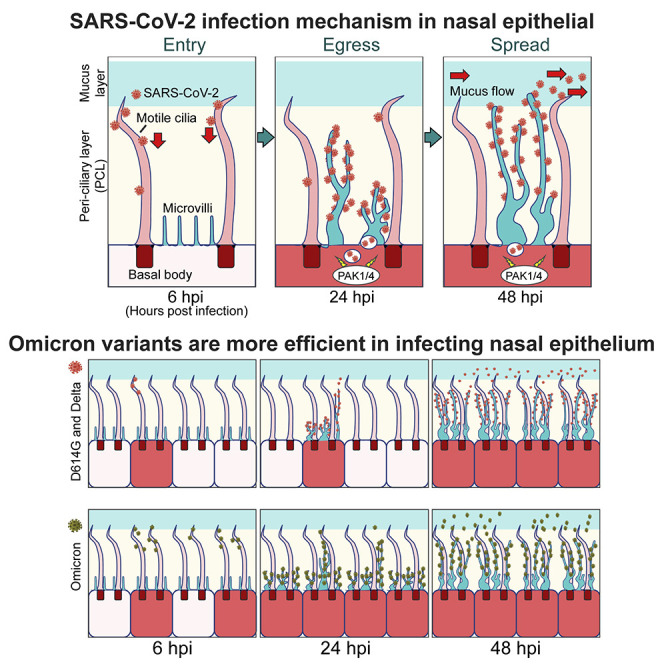
Nasal and upper airway CECs are the main targets for initial COVID-19 infections.2 , 10 , 11 , 12 , 13 Many respiratory viruses, including SARS-CoV-1,14 influenza,15 parainfluenza (PIV),16 rhinovirus (RV),17 , 18 and respiratory syncytial virus (RSV),19 first infect airway CECs. SARS-CoV-2 receptors ACE2 and TMPRSS2 localize to cilia in CECs.20 , 21 Here, we report that SARS-CoV-2 infects nasal mucosa by a two-step process. First, SARS-CoV-2 particles bind the ACE2 receptor on the surface of airway cilia. Cilia then facilitate virus transport through the PCL mucin layer. Initially, only a few ciliated human nasal epithelial cells (HNEs) are productively infected. Within the next 24 h SARS-CoV-2 hijacks the host cell machinery to induce elongated and highly branched microvilli. This rearrangement requires protein kinases, including p21-activated kinases 1 and 4 (PAK1/4). Activating p21-activated kinases (PAK) enables the virus to exit across the PCL layer before lateral spread to other regions, potentially via mucociliary transport (MCT). Omicron variants dramatically accelerate spread via the ciliary transport/microvilli reprogramming pathway, which explains the increase in its attack rate compared to previous variants. This pathway is required for infection by other respiratory viruses, such as PIV and RSV. Understanding how cilia and microvilli are reprogrammed for virus entry and spread may identify candidate therapeutic targets to block airway replication of SARS-CoV-2 and other respiratory viruses.
Results
SARS-CoV-2 infection of nasal epithelium is a two-step process
To understand viral entry, we sought to identify which cell types are initially infected. We differentiated primary HNEs in air-liquid interface (ALI) cultures to form nasal epithelial organoids of ciliated, goblet, and basal cells (see Table S1 for characteristics of the nasal cell donors). These ALI-cultured HNEs recapitulated the morphology and function of normal human upper airway epithelium.22 , 23 , 24 After 28–30 days, fully differentiated nasal epithelial organoids were inoculated with the SARS-CoV-2 D614G variant at a MOI of 0.3 to assess spatial and temporal infection patterns and cell tropism within nasal epithelium. Infected cultures were fixed at various times and stained with antibodies to the SARS-CoV-2 nucleocapsid protein (NP), SP, and markers for ciliated HNEs (acetylated tubulin; ACTUB) or goblet cells (MUC5AC) (Figures 1A, 1B, and S1 ). NP and SP were only seen in ciliated HNEs at 6, 24, and 48 h post infection (hpi), indicating preferential, early infection of ciliated HNEs. SARS-CoV-2 infected few, if any, goblet cells even at 96 hpi. Thus, ciliated HNEs are the primary entry site of SARS-CoV-2 in nasal epithelia.
Figure 1.

SARS-CoV-2 infection of nasal epithelium occurs in two steps
(A and B) SARS-CoV-2 infects ciliated HNEs, not goblet cells. Mock-treated or infected HNEs (MOI 0.3) were stained (6, 24, 48, 96 hpi). Representative IF images of HNEs stained for SP, NP, and either cilia HNE marked by acetylated α-tubulin (ACTUB: A) or goblet cell marker (MUC5AC: B). Quantified percentages of NP- and SP-positive ciliated cells or goblet cells are shown (right panel).
(C) Initial SARS-CoV-2 infections are restricted to few ciliated HNEs. Mock-treated or infected HNEs (MOIs 3, 0.3, 0.03) were stained at 24 and 48 hpi. Representative IF staining of SP and phalloidin (actin) in virus-infected HNEs. Quantified percentages of SP-positive HNEs (24 and 48 hpi: right panel).
(D) Schematic of two-step infection model where SARS-CoV-2 first seeds (0–24 hpi) then spreads (24–48 hpi) during HNE infection. Error bars represent mean ± SD (3,000–4,000 cells quantified from HNEs from Donors 1–4). Each dot represents one donor. Scale bars represent 20 μm. See Figure S1, Table S1.
Figure S1.

SARS-CoV-2 infection in nasal epithelium, related to Figure 1
(A and B) SARS-CoV-2 predominantly infects ciliated HNEs and not goblet cells. Mock-treated or SARS-CoV-2-infected HNEs with MOI of 0.3 were stained after 6-, 24-, 48, and 96 hpi. Representative IF staining of SARS-CoV-2 NP and SP in combination with either the ciliated nasal epithelial cell marker (acetylated α-tubulin [ACTUB]) or goblet cell marker (MUC5AC), and phalloidin in mock or SARS-CoV-2-infected HNEs from Donor 1. Similar results were observed from Donor 2–4, see Table S1. Scale bars represent20 μm.
At 6 and 24 hpi, only ∼3% of ciliated HNEs were SARS-CoV-2 positive; by 48 hpi, positive HNEs increased to ∼80% (Figure 1A). We hypothesized that initial infection and entry are restricted to a few ciliated HENs, and then later, new virions spread laterally into neighboring nasal epithelium. The pattern did not change at different MOIs (MOI: 3, 0.3, and 0.03), suggesting a kinetic bottleneck for infection at viral entry (Figure 1C). These results support a two-step model in which SARS-CoV-2 crosses the airway mucosal barrier in a limited number of ciliated cells, followed by replication and rapid spread of progeny to neighboring cells (Figure 1D).
SARS-CoV-2 attaches to the cilia during initial stages of infection
ACE2 and TMPRSS2 are central to SARS-CoV-2 entry and localize to respiratory cilia.20 , 21 To determine if they also localize to motile cilia in ALI-cultured HNEs, we co-stained ACE2 and TMPRSS2 with ACTUB. Both localized to motile cilia (Figure S2 A) in the ALI-cultured HNEs and human autopsy samples. We hypothesized that SARS-CoV-2 and other respiratory viruses attach to cilia via ACE2 to penetrate the PCL and enable the virus to traffic through the mucin layer.
Figure S2.

SARS-CoV-2 attaches to the cilia during the initial stage of infection, related to Figure 2
(A) ACE2 and TMPRSS2 localize to the motile cilia in ciliated HEN cells. Representative double IF staining of ACE2 and TMPRSS2 in combination with ACTUB in HNEs (left panel) and human nasal tissue (right panel).
(B) SARS-CoV-2 neutralization antibody inhibits attachment of SARS-CoV-2 to cilia. Representative SEM images of SARS-CoV-2 virions attaching to motile cilia after pre-treating HNEs with an unrelated control antibody (left image) or SP neutralizing antibodies (right image) for 2 h before SARS-CoV-2 inoculation with MOI of 0.3 from Donor 7. HNEs were fixed after 6 hpi and observed by SEM Quantification of the average number of virus particles on cilia in infected HNEs. Error bars represent mean ± SD (20–30 cilia were quantified from infected HNEs from Donors 7 and 8, see Table S1).
(C) SARS-CoV-2 RBD binds to the cilia in a ciliary ACE2-dependent manner. ALI-cultured HNEs were labeled by SiR-tubulin, a fluorogenic, cell permeable, and highly specific probe for microtubules and treated with pseudovirion, quantum dot-conjugated SARS-CoV-2 receptor binding domain (QD585-RBD) for 6 h. Live-cell images were taken at the indicated time points (seconds) with control (upper image) or soluble ACE2 (down image) using Marianas spinning disk confocal (SDC) microscopy. Quantification of the percentage of HNEs with cilia-attached QD585-RBD with control or soluble ACE2 was performed on the right panel. Error bars represent mean ± SD (200–300 cells were quantified from HNEs from Donor 5–8, see Table S1). ∗p < 0.05, paired, two-tailed Student’s t test.
(D–F) Depletion of cilia doesn’t affect the epithelium development and the expression level of ACE2 and TMPRSS2. (D) Representative IF staining of FOXJ1 and ACTUB in scrambled control (shControl) and CEP83 knockdown (shCEP83) HNEs from Donor 1, see Table S1. (E) Representative IF staining for MUC5AC (goblet cells) and ACTUB (ciliated HNEs) with phalloidin in shControl and shCEP83 HNEs. Quantified percentages of MUC5AC-positive cells in the right panel. Error bars represent mean ± SD (2,000–3,000 cells were quantified from shCEP83 and shControl HNEs from Donor 1–4, see Table S1). (F) ACE2, TMPRSS2, and CX3CR1 mRNA expression in shControl and shCEP83 HNEs. Data were collected from Donors 1–4, see Table S1.
(G) CX3CR1 localizes to the cilia in HNEs. Representative double IF staining of CX3CR1 and ACTUB in ALI-cultured HNEs (top panel) and normal human nasal tissue (down panel).
(H and I) Depletion of cilia decreases the respiratory syncytial virus (RSV) and human parainfluenza virus (PIV) infection. RSV (H) and PIV (I) infected HNEs with MOI of 0.3 were stained after 24 (left image) and 48 (right image) hpi. Representative IF staining for either RSV fusion protein (H) or PIV fusion glycoprotein (I) in combination with ACTUB and phalloidin in shControl and shCEP83 HNEs. Quantified percentages of RSV fusion protein (H) and PIV fusion glycoprotein (I) positive HNEs are shown on the right panel. Error bars represent mean ± SD (3,000–4,000 cells were quantified from virus-infected shCEP83 and shControl HNEs from Donor 1–4, see Table S1. (B–I) ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, NS represents not significant, paired, two-tailed Student’s t test. Each dot represents one donor. Scale bars represent 500 nm (B), 10 μm (C), and 20 μm (A, C, G, H, and I).
To determine if the mucin network mesh blocks virus infections, we treated HNEs with StcE, a mucin-selective protease, to disrupt the structure of the mucin network mesh.25 This treatment reduced MUC1 levels in mucin network mesh and increased virus entry at 24 hpi (Figure 2 A). Thus, the mucin network mesh blocks SARS-CoV-2 infection of HNE ALI cultures, explaining the delay in virus spread. To test if SARS-CoV-2 specifically binds cilia at the early step, we infected ALI-cultured HNEs with SARS-CoV-2 at an MOI of 0.3 and fixed cells at 6 hpi. Scanning (SEM) and transmission electron microscopy (TEM) confirmed that multiple virions attached to cilia (Figures 2B and 2C), suggesting that cilia are hijacked by SARS-CoV-2 to cross the nasal epithelium barrier. Using confocal immunofluorescence (IF) microscopy and TEM, we found that viral particles were attached to cilia near the mucus layer or to cilia and immersed in the PCL in ∼6% of the ciliated HNEs. An anti-SP monoclonal antibody that neutralizes SARS-CoV-226 inhibited attachment of SARS-CoV-2 to cilia (Figures 2D and S2B) and decreased infected cell numbers (Figure 2E).
Figure 2.

SARS-CoV-2 attaches to the cilia during the initial stage of infection
(A) StcE mucinase reduces MUC1 mucin levels and increases virus infection efficiency. (Upper panel) Representative IF staining of MUC1, phalloidin in control-, and StcE (10 mg/mL)-treated (6 h) HNEs (Donor 6). Quantification of MUC1 from control- or drug-treated HNEs from Donors 6–8 (lower panel). Error bars represent mean ± SD (Lower panel) SARS-CoV-2-treated HNEs were treated with StcE (10 mg/mL, 6 h) before SARS-CoV-2 infection. Representative IF staining of SARS-CoV-2 SP, ACTUB, and phalloidin staining in control versus StcE-treated infected HNEs (Donor 6). Quantified percentages of SP-positive ciliated HNEs. Error bars represent mean ± SD (3,000–4,000 cells quantified from control- or drug-treated SARS-CoV-2-infected HNEs, Donor 6–8). ∗p < 0.05, paired, two-tailed Student’s t test. Each dot represents one donor.
(B and C) SARS-CoV-2 binds to the motile cilia during early viral infection. Mock-treated or SARS-CoV-2-infected HNEs (Donor3) (MOI 0.3) were fixed after 6 hpi. Representative images of SARS-CoV-2 virions attaching to motile cilia as observed by scanning electron microscope (SEM; B) or transmission electron microscopy (TEM; C). Arrowhead: virus particles. Similar results from Donors 4 and 5.
(D and E) SARS-CoV-2 neutralizing antibody inhibits SARS-CoV-2 attachment to cilia and decreases infected cells. (D) Representative IF images of SARS-CoV-2 SP with ACTUB after pre-treating HNEs (2 h) with control antibody (left) versus SP neutralizing antibodies (right) before SARS-CoV-2 inoculation (MOI 0.3). HNEs were fixed 6 hpi and stained for SARS-CoV-2 SP and ACTUB (left panel). Quantification is percentage of ciliated HNEs with cilia-attached SARS-CoV-2 (right panel). Error bars represent mean ± SD (2,000–3,000 cells quantified, HNEs from Donors 5–8). (E) Quantified percentages of SP-positive ciliated HNEs (48 hpi). Error bars represent mean ± SD (3,000–4,000 cells quantified, Donors 6–8). ∗∗p < 0.01, paired, two-tailed Student’s t test. Each dot represents one donor.
(F) Depleting cilia decreases HNE infection. Infected HNEs (MOI 0.3) were stained after 24 (left) and 48 (right) hpi. Representative IF staining for SARS-CoV-2 SP and ACTUB with phalloidin in shControl versus shCEP83 HNEs. Quantified percentages of SP-positive HNEs (right panel). Error bars represent mean ± SD (3,000–4,000 cells quantified from infected shControl and shCEP83 HNEs Donors 1–4).
(G) SARS-CoV-2 viral entry requires TMPRSS2 and ciliary dynein. SARS-CoV-2-infected HNEs were treated: DMSO, Ciliobrevin D (60 μM), camostat mesylate (25 μM), or HCQ (25 μM) 2 h before SARS-CoV-2 infection. Representative IF staining of SARS-CoV-2 SP, ACTUB, and phalloidin staining in DMSO- versus drug-treated infected HNEs at 24 hpi (left) and 48 hpi (right), Donor 6. Quantified percentages of SP-positive ciliated HNEs. Error bars represent mean ± SD (3,000–4,000 control- or drug-treated SARS-CoV-2-infected HNEs, Donor 6–8).
(H) Model for motile cilia during SARS-CoV-2 entry. (A and F) ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, NS represents not significant, paired, two-tailed Student’s t test.
(G) ∗p < 0.05, ∗∗p < 0.01, NS represents not significant, paired, one-way ANOVA with Tukey’s post-test. Scale bars: 200 nm (C), 1 μm (B), 5 μm (A [top figure] and D), 20 μm (F), and 50 μm (A [down figure] and G). Each dot represents one donor. See Figure S2 and Table S1.
To examine SP binding to the ACE2 receptor during early infection in ciliated HNEs, we conjugated the recombinant SP binding domain (RBD) to fluorescent quantum dots to form QD585-RBD pseudovirions. Quantum dots directly visualize RBD binding to ciliary ACE2.27 In time-lapse, live-cell microscopy, pseudovirion QD585-RBD bound to motile beating cilia in ALI-cultured HNEs at 4 hpi. QD585 alone did not (Figure S2C). QD585-RBDs precisely tracked the ciliary beating in multiple time-lapse videos. This interaction was inhibited by competition with soluble ACE2 (Figure S2C), indicating QD585-RBD binds to cilia via ACE2. Our results support that SARS-CoV-2 binds to a ciliary ACE2 receptor to facilitate cell entry.
Cilia facilitate infection by SARS-CoV-2 and other respiratory viruses
We hypothesized that depleting cilia would impede viral infection, so we blocked ciliary assembly by shRNA knockdown of CEP83, a protein critical for motile cilia formation in all ciliated cells, including ALI-cultured HNEs.28 Depleting CEP83 prevented cilia formation (Figure S2D) but had no effect on epithelial or goblet cell differentiation28 or expression of ACE2 and TMPRSS2 receptors (Figures S2E and S2F). Indeed, CEP83 downregulation inhibited SARS-CoV-2 infection of HNEs at 24 and 48 hpi at an MOI of 0.3 (Figure 2F). We observed no apparent toxicity in the cilia-depleted cultures.
We next examined whether the ciliary pathway is used by other respiratory viruses. The RSV and PIV receptors CX3CR1 and α2,3-linked sialic acid localize to motile cilia in human airway tissue sections.29 , 30 , 31 , 32 , 33 CX3CR1 colocalized to cilia in ALI-cultured NHE cells and human autopsy samples (Figure S2G). Depleting cilia with CEP83 shRNA inhibited both infections at 24 and 48 hpi in HNEs, indicating broad exploitation of cilia by respiratory viruses (Figures S2H and S2I).
Inhibiting ciliary trafficking attenuates viral uptake
Retrograde trafficking of ciliary proteins or receptors from the tip of cilia to the bottom is mediated by a ciliary dynein complex.34 We hypothesized that cilia facilitate virus movement from the tip or body of the cilia basally toward the cell body. We treated HNEs with ciliary dynein inhibitor, Ciliobrevin D,35 3 h before SARS-CoV-2 infection and determined the number of infected cells at 24 and 48 hpi. The number of infected cells decreased, indicating that protein trafficking in cilia is important, but not strictly essential, during viral entry (Figure 2G). Next, we examined whether SARS-CoV-2 enters HNEs via endocytosis or cell-surface membrane fusion. We treated HNEs with a TMPRSS2 inhibitor, camostat mesylate,1 or hydroxychloroquine (HCQ), which increases the pH in endosomes, and thereby stops their acidification and maturation,36 for 2 h prior to SARS-CoV-2 infection and then determined the number of infected cells. HCQ had no effect on infection, indicating that endosome acidification is not required for entry in HNEs and that HCQ does not inhibit virus entry into airway cells. Treatment with TMPRSS2 inhibitor, linked to ACE2 entry, decreased the number of infected cells at 24 and 48 hpi (Figure 2G).
These data suggest two models. In model 1, SARS-Co-V2 binds to ACE2, and TMPRSS2 activates SP for ciliary membrane fusion at the cilium surface. Then, the RNA genome is released into the cilium and transported from the cilium into the cytosol by ciliary dynein (Figure 2H-i). Alternatively, an ACE2 and SARS-CoV-2 complex is transported from the tip of the cilia to the cell body by the ciliary dynein dependent, anterograde process. Then, the virus fuses with the cell membrane via an endocytic-independent process (Figure 2H-ii). Future tests will resolve the contributions of binding versus transport to viral uptake.
Newly produced SARS-CoV-2 particles co-localize with microvilli
Our data indicate that the percentage of SARS-CoV-2-infected cells increased from 24 to 48 hpi (Figure 1A). In contrast, the virus rapidly spreads without an evident lag phase in Vero cells. To better understand virus infection in nasal epithelium, we examined the egress of virus from the few initially infected cells. In Caco-2 and Vero cells, SARS-CoV-2 infection promotes actin-based filopodia protrusion with virus particles associated with these structures.37 , 38 Induction of virus-containing filopodia might be important for SARS-CoV-2 egress and cell-to-cell spread of progeny virions in HNE. Whether canonical microvilli exist on primary airway epithelial cells had been unclear. We used SEM and IF staining to systematically characterize microvilli-like structures in primary airway epithelial cells. Two classes of protrusions were noted on the apical surface of ciliated HNEs: long, wide motile cilia and stubby, dome-like microvilli (Figure S3 A). IF staining of multiple core microvilli proteins (EZRIN, EBP50, SLK, and actin [marked by phalloidin])39 , 40 revealed these proteins co-localize in microvilli-like structures in ALI-cultured HNEs and human airway tissue (Figures 3A, S3B, and S3C). This supports that these structures are canonical microvilli. Microvilli were not found on MUC5AC-positive goblet cells (Figure S3D).
Figure S3.

SARS-CoV-2 co-localizes with the microvilli during later stages of virus infection, related to Figure 3
(A) Two different classes of protrusions on the apical surface of HNEs were observed. Long and wide motile cilia (yellow dashed line) and stubby dome-like microvilli (red dashed line). HNEs (Donor 3, see Table S1) were observed by SEM Arrowhead: microvilli.
(B) Representative double IF staining of SLK and phalloidin in ALI-cultured HNEs from Donor 1, see Table S1.
(C) Representative IF staining of EZR and EBP50 in combination with ACTUB in human nasal tissue.
(D) Representative IF staining of ACTUB and MUC5AC in combination with phalloidin in ALI-cultured HNEs from Donor 1, see Table S1.
(E) SARS-CoV-2-infected HNEs with MOI of 0.3 were stained after 48 hpi. Representative IF staining of either SARS-CoV-2 SP or SARS-CoV-2 NP in combination with phalloidin in SARS-CoV-2-infected HNEs from Donor 1 (see Table S1).
(F) Mock-treated or SARS-CoV-2-infected HNEs with MOI of 0.3 were stained after 6, 24, 48, and 96 hpi. Representative IF staining of SARS-CoV-2 SP, CEP164, and phalloidin in mock or SARS-CoV-2-infected HNEs from Donor 3 (see Table S1) is shown.
(G) Small viral vesicles and large viral-containing vesicles could be observed in the infected cells. SARS-CoV-2-infected HNEs (Donor 4–6, see Table S1) with MOI of 0.3 were fixed after 48 hpi. The cells were observed by TEM. Red arrowhead: small viral vesicle. Yellow arrowhead: large viral-containing vesicle.
(H) IHC staining for SARS-CoV-2 in nasal epithelium of infected K18-hACE2 mice. Nasal epithelium tissues were collected at days 3 post-infection, fixed in 4% PFA, embedded in paraffin wax, and cut into 5-μm sections. Representative IF staining of SARS-CoV-2 SP and ACTUB in combination with Phalloidin in mouse nasal tissue. Scale bars represent 1 μm (A), 5 μm (B-F), and 10 μm (H).
Figure 3.

SARS-CoV-2 co-localizes with microvilli in later stages of infection
(A) Representative IF staining of EBP50, phalloidin (microvilli marker), and ACTUB (top) and phosphorylated EZR (pEZR), EZR, and ACTUB (bottom) in ALI-cultured HNEs Donor 1.
(B–D) SARS-CoV-2 attaches to microvilli at 24 hpi. (B) Mock-treated or infected HNEs (MOI 0.3) were stained 6, 24, 48, 96 hpi. Representative IF staining for SARS-CoV-2 NP and ACTUB plus phalloidin in mock or SARS-CoV-2-infected HNEs, Donor 1 (right panel). Quantified percentages of microvilli-attached SARS-CoV-2-positive ciliated HNEs Donors 1–4 are shown. (C and D) Mock-treated versus infected HNEs (Donor 3; MOI 0.3) were fixed after 24 hpi. The cells were observed by TEM (C) or SEM (D). Arrowhead: virus particles. Donors 4–6 showed similar results. Scale bars: 10 μm (A, left), 5 μm (A, right and B), 1 μm (C and D). See Figure S3 and Table S1.
We next sought to test if microvilli have a role in viral infection. We co-stained acetylated tubulin (ACTUB) and actin (phalloidin) with antibodies to SP or NP in infected HNEs at different times, using an MOI of 0.3 to track viral replication and egress. At 6 hpi, the only viral signal was in the cytoplasm, and the microvilli appeared stubby. From 24 hpi onward, NP and SP co-localized with the microvilli (Figures 3B and S3E). To determine if SARS-CoV-2-attached microvilli protrude from infected cell membranes, we co-stained for CEP164, a protein that anchors centrioles to the plasma membrane at the ciliary base and thus marks the apical surface, with spike protein SP and actin (phalloidin) (Figure S3F). Microvilli with attached virions were seen protruding from the membrane as early as 24 hpi. TEM and SEM found abundant viral particles specifically on microvilli rather than motile cilia, indicating that newly generated virus particles accumulate on microvillar structures (Figures 3C and 3D). We also noted that small viral vesicles clustered near or in microvilli and large viral-containing vesicles were in the cytoplasm near the apical side of the cells (Figure S3G). Next, we looked for viral binding to microvilli in infected animals. We infected K18-hACE2 mice with 6 × 104 pfu SARS-CoV-2 (D614G variant) and collected nasal epithelium for immunohistochemistry at 48 hpi. Consistently, SARS-CoV-2 co-localized with microvilli at 48 hpi in vivo (Figure S3H), suggesting that SARS-CoV-2 interacts with microvilli to spread within the respiratory tract in infected animals.
SARS-CoV-2 regulates microvilli dynamics and promotes viral shedding by highly extended microvilli
Based on our initial microscopy, we hypothesized that SARS-CoV-2 infection regulates microvillar structure and function to enable viral egress. Several proteins organize microvilli,41 including the actin-binding protein ezrin (EZR).42 Phosphorylated EZR levels were increased in microvilli in infected HNEs (Figure 4 A). Another microvillar core protein, EBP50, was increased on microvilli and in the cytoplasm of infected HNEs (Figure 4B). TEM showed that infection induced a dome-like structure protruding from the plasma membrane. Multiple SARS-CoV-2 particle-containing vesicles appeared on this dome-like structure (Figures 4C, left panel, and S4A). Infection also induced highly branched microvillar structures with many attached or budded viral particles at 48 hpi, compared to mock infection. Importantly, SARS-CoV-2 infection dramatically increased highly extended microvilli as early as 24 hpi as observed by IF staining (Figure 4D). These microvilli were accompanied by an accumulation of virus particles along its surface as seen by IF (Figure 4D; 48 and 96 hpi) and TEM (Figures 4E and S4B). These results support our model that SARS-CoV-2 infection modulates the activity, structure, and length of microvilli to facilitate viral egress and spread (Figure 4F).
Figure 4.

SARS-CoV-2 regulates the dynamics of microvilli and promotes highly extended microvilli to facilitate viral shedding/secretion
(A and B) SARS-CoV-2 infection increases phosphorylated ezrin (EZR) and EBP50 protein on microvilli.
(A) Mock-treated or infected HNEs (MOI 0.3) were stained 48 hpi. Representative IF staining of pEZR and phalloidin with ACTUB in mock or infected HNEs Donor 3 (left panel). Quantification of pEZR from mock and infected HNEs Donors 3–6 (middle panel). pEZR expression increased in SARS-CoV-2-infected HNEs compared to mock controls. pEZR and EZR immunoblots of in mock or infected HNEs Donor 6 (right panel). Donors 4 and5 showed similar results.
(B) Mock-treated or SARS-CoV-2-infected HNEs (MOI 0.3) were stained 48 hpi. Representative IF staining of EBP50, phalloidin and SARS-CoV-2 SP for mock versus infected HNEs (Donor 3). Quantification of EBP50 on microvilli from mock and infected HNEs Donor 3–6 (right panel). EBP50 expression increased in infected HNEs compared to mock controls.
(C–E) SARS-CoV-2 infection affects microvillar structure. (C and E) Infected HNEs (Donor 3) with MOI of 0.3 were fixed at 24 and 48 hpi. Cells were observed by TEM. (C) Quantification of the number of dome-shaped structures per cell from Donors 3–6 (right panel). Error bars represent mean ± SD (200 ciliated HNEs were counted from 4 donors each).
(A, B, and C) Error bars represent mean ± SD ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, NS represents not significant, paired, two-tailed Student’s t test. Red arrowhead: small viral vesicle.
(D) Mock-treated or infected HNEs with MOI of 0.3 were stained at 6, 24, 48, and 96 hpi. Representative IF staining of SARS-CoV-2 NP, EZR, and phalloidin in mock or SARS-CoV-2-infected HNEs from Donor 3 (right panel). Quantification of the length of EZR from mock and SARS-CoV-2-infected HNEs from Donors 3–550 ciliated HNEs from 4 donors each).
White dotted line: extended microvilli. Red arrowhead: virus particles (E) SARS-CoV-2 infection induce overly long microvilli attached with virus particles at 24 (upper image) and 48 (lower image) hpi. Red dotted line: extended microvilli.
(F) Model of microvilli structures and viral vesicles affected by SARS-Co-V2 infection.
(G) Microvillar inhibitors inhibit microvilli structure. Representative IF staining of pEZR and phalloidin in DMSO-, NSC-668394 (40 μM)-, and SB-633825 (40 μM)-treated HNEs from Donor 5 Quantification of pEZR and phalloidin from DMSO- or drug-treated HNEs (3 h treatment) Donors 5–8 (down panel). pEZR and phalloidin expression showed a decrease in drug-treated HNEs, compared to DMSO-treated cells.
(H) SARS-CoV-2-treated HNEs were treated with NSC-668394 or SB-633825 (see schematic).
(I) Microvilli inhibitors, NSC-668394 (40 μM) and SB-633825 (40 μM), significantly inhibit SARS-CoV-2 infection in HNEs. Representative IF staining of SARS-CoV-2 SP, ACTUB, and phalloidin in DMSO- or drug-treated SARS-CoV-2-infected HNEs at 24 hpi (upper image) and 48 hpi (down image) (Donor 5). Quantified percentages of SP-positive ciliated HNEs (right panel). Error bars represent mean ± SD (3,000–4,000 HNEs were quantified Donors 4–8).
(G and I) Error bars represent mean ± SD ∗p < 0.05, ∗∗∗p < 0.001, NS represents not significant, paired, one-way ANOVA with Tukey’s post-test. Each dot represents one donor. Scale bars: 5 μm (A, B, D, and G), 1 μm (C), 2 μm (E), 20 μm (I). See Figure S4 and Table S1.
Figure S4.

SARS-CoV-2 promotes highly extended and branched microvilli, related to Figure 4
(A and B) SARS-CoV-2 infection induces dome-like structure and highly branched and overly long microvilli. (A) SARS-CoV-2-infected HNEs (Donor 3–6, see Table S1) with MOI of 0.3 were fixed after 48 hpi. The cells were observed by TEM. Arrowhead: virus particles. (B) mock-treated and SARS-CoV-2-infected HNEs (Donor 3–6, see Table S1) with MOI of 0.3 were fixed after 24 hpi. The cells were observed by TEM. Red dotted line represents extended microvilli and dome-like structure.
(C) SARS-CoV-2 viral particles bind to the motile cilia at 6 hpi rather than microvilli. SARS-CoV-2-infected HNEs with MOI of 0.3 were stained after 6 (upper image) and 24 (down image) hpi. Representative IF images of SARS-CoV-2 SP in combination with ACTUB and phalloidin in SARS-CoV-2-infected HNEs from Donor 1. Similar results were observed from Donor 2–4, see Table S1.
(D) ACE2 and TMPRSS2 mainly localize in cilia rather than microvilli. Representative IF images of ACE2 (top panel) and TMPRSS2 (lower panel) in combination with ACTUB and phalloidin in normal human nasal tissue (left panel) and ALI-cultured HNEs (right panel). Scale bars represent 5 μm (D) and 10 μm (C).
We used inhibitors of microvillar core protein to determine if loss of microvilli inhibits infection. HNEs were incubated for 3 h with the EZR inhibitor NSC-668394 or the LOK/STK10 kinase inhibitor SB-633825. Treated cells were fixed and stained with phalloidin and anti-phospho-EZR (pEZR). NSC-668394 and the LOK inhibitor strongly decreased microvillar actin staining and levels of pEZR (Figure 4G). To determine if microvilli have a role in viral entry and/or exit, we treated HNEs before or after SARS-CoV-2 infection (Figure 4H) and counted infected cells at 24 and 48 hpi. The inhibitors had no effect within the first 24 hpi, indicating that they do not inhibit entry. In contrast, infected cell numbers decreased at 48 hpi even when inhibitors were administered after infection at 18 hpi (Figure 4I). SARS-CoV-2 particles attached to the cilia rather than microvilli during the viral entry. In contrast, many SARS-CoV-2 viral particles attached to the microvilli later during viral egress (Figure S4 C). ACE2 and TMPRSS2 were not expressed in the microvilli (Figure S4D), suggesting that microvilli do not participate in SARS-CoV-2 viral entry. Our data suggest that SARS-CoV-2 hijacks the microvillar assembly pathway to facilitate viral egress by promoting formation of highly extended and branched microvilli.
Mucociliary transport assists in spread of SARS-CoV-2
We next hypothesized that, by associating with elongated microvilli, SARS-CoV-2 traffics to the surface mucus layer. In this model, progeny virions use mucus flow to traverse the airway tract to infect other airway cells. To test this, we used ALI cultures from patients with primary ciliary dyskinesia (PCD). PCD patients have little MCT. This rare autosomal recessive disorder results from functional defects that abrogate synchronous ciliary beating and clearance of pathogens.43 , 44 Nonetheless, airway cilia numbers and length are normal,45 , 46 but without MCT, clearance of many pathogens is compromised, and susceptibility to bacterial or viral pathogens is enhanced. Other ciliary functions, including trafficking of the intraflagellar transport complexes (IFT), the microtubule-motor driven transport and signaling system in cilia, are retained.46
To determine if SARS-CoV-2 spreads by MCT, we cultured primary human nasal epithelial cells from three PCD patients and tracked infection (Table S2). Nasal epithelial cells from PCD patients have quantitatively normal cilia density and staining of IFT88 and ARL13B, two proteins essential for normal ciliary protein trafficking (Figure S5 A). The epithelial cells had visible microvilli on their apical surfaces (Figure S5B). The numbers of goblet cells in PCD and healthy samples were similar (Figure S5C), as were levels of ACE2 and TMPRSS2 in nasal epithelial cells (Figure S5D). Percentages of SARS-CoV-2-infected cells at 24 hpi were similar in patients and healthy donors, suggesting that mutations in the dynein axonemal heavy chain do not affect initial infection rates (Figure 5 A). However, at 48 hpi, PCD patients had fewer SARS-CoV-2-infected cells than healthy donors, suggesting MCT is important later in infection (Figure 5A). At low MOI (48 hpi), healthy samples showed long streaks of SP-positive epithelial cells that apparently traced viral infections along the flows in the mucus layer. In PCD nasal epithelial cells, SARS-CoV-2 only infected immediately surrounding cells, forming small local plaques (Figure 5B). Gentle pipetting to induce mechanical mucus flow increased the number of infected cells and infection area in PCD HNEs (Figure 5C). These results provide a mechanism for why individuals with PCD have no increased risk of infection or severe COVID-19 despite defective mucociliary clearance (Figure 5D).47 , 48
Figure S5.

Mucociliary transport assists in the spread of SARS-CoV-2, related to Figure 5
(A–D) Cilia beating defect doesn’t affect the structure of cilia and microvilli, the epithelium development, and the expression level of ACE2 and TMPRSS2. (A) Representative IF staining of ACTUB, IFT88, and ARL13B in HNEs from control (upper panel, Donor 5) and PCD patients (down panel, PCD Donor 1, see Table S2). Similar results were observed from samples harvested from Donor 6–8 and PCD Donor 2 and 3. (B) Representative IF images of ACTUB and EZR with phalloidin in control (upper panel, Donor 5) and PCD (down panel, PCD Donor 1, see Table S2) HNEs. Similar results were observed from samples harvested from Donors 6–8 and PCD Donors 2 and 3. (C) Representative double IF staining of MUC5AC, ACTUB, and phalloidin in control (left image) and PCD (right image) HNEs. Quantified percentages of MUC5AC positive cells (right panel). Error bars represent mean ± SD (2,000–3,000 cells were quantified from control and PCD HNEs from Donor 5–8 and PCD Donor 1–3, see Tables S1 and S2). NS represents not significant, paired, two-tailed Student’s t test. Each dot represents one donor. (D) ACE2 and TMPRSS2 mRNA expression in control and PCD HNEs. Error bars represent mean ± SD (data was collected from Donors 5–8 and PCD Donors 1–3, see Tables S1 and S2).
(E and F) SARS-CoV-2 infection doesn’t affect cell boundaries and cell-cell junctions. SARS-CoV-2-infected HNEs (Donor 3, see Table S1) with MOI of 0.3 were fixed at 24 and 48 hpi. Cells were observed by TEM (E) and IF (F). (F) Representative IF staining of SARS-CoV-2 SP and β-catenin in combination with phalloidin in ALI-cultured HNEs from Donor 1, see Table S1. Scale bars represent 5 μm (A, B, and F) and 20 μm (C).
Figure 5.

Mucociliary transport assists the spread of SARS-CoV-2
(A and B) Cilia beating does not alter viral entry but affects virus spread. (A) Infected HNEs (MOI 0.3) were stained at 24 and 48 hpi. Representative IF staining of SP, ACTUB, and phalloidin in control versus PCD HNEs at 24 hpi (left image) and 48 hpi (right image). Quantified percentages of SP-positive ciliated HNEs (right panels). Error bars represent mean ± SD (3,000–4,000 HNEs were quantified from infected control (Donors 5–8) and PCD HNEs (Donors 1–3, Tables S1 and S2).
(B) Infected HNEs (MOIs 0.3, 0.03) were stained at 48 hpi. Representative IF images of SP in control (Donor 6) and PCD (PCD Donor 2) HNEs. Quantified percentages of SP-positive area (right panel). Error bars represent mean ± SD (data from Donors 5–8 and PCD Donors 1–3).
(C) The simulated mucus flow allows redistribution of virus particles. SARS-CoV-2-treated PCD HNEs were exposed or not treated to mechanical flow by pipetting 50 μL medium at24 hpi and were stained at 48 hpi. Representative IF images of SP in control (left image) and mechanical force (right image) PCD (PCD Donor 1) HNEs. Quantified percentages of SP-positive area (right panel). Error bars represent mean ± SD (PCD Donors 1–3).
(D) Model illustrating mucociliary flow effects during SARS-CoV-2 spread. ∗p < 0.05, ∗∗p < 0.05, NS represents not significant, two-tailed Student’s t test. Each dot represents one donor. Scale bars represent 20 μm (A), 50 μm (C), and 100 μm (B). See Figure S5 and Tables S1 and S2.
We next sought to understand whether cell-to-cell contact is important for SARS-CoV-2 transfer. TEM and IF staining showed that cell boundaries and cell-cell junctions were intact, and no obvious viral particles were present in the intracellular space in SARS-CoV-2-infected nasal epithelium (Figures S5E and S5F). The respiratory epithelium comprises multiple layers of cells. However, until 48 hpi, only the uppermost cells faced to the lumen were infected by the virus, with very few cells in lower layers infected (Figures 1A and S1A). Thus, cell-to-cell contact may not be the only pathway for viral spread in nasal epithelium. Although the virus might spread through cell-to-cell contact in nasal epithelium, we believe spreading depends on mucus flow at the apical surface.
PAK1/4 regulate microvilli to facilitate SARS-CoV-2 budding and spreading
Given that regulatory kinases control cytoskeletal dynamics, including microvilli,49 we asked how infection activates host signaling pathways to modulate microvilli biogenesis, thereby facilitating viral egress. We used global phosphoproteomics to find protein kinases affected by SARS-CoV-2 infection. We infected ALI-cultured HNEs with SARS-CoV-2 at an MOI of 0.3 for 6, 24, 36, and 48 hpi in parallel with mock controls. Cells were harvested, and extracts were prepared for phosphoproteomic mass spectrometry and signaling analysis. We used a substrate-based kinase activity prediction model to determine the activity of kinases from these large-scale phosphoproteomic data. The model assumes that activity can be inferred through by measuring downstream phosphorylation events.50 Using kinase set enrichment analysis (KSEA),51 , 52 we assigned an enrichment score (ES) (weighted Kolmogorov-Smirnov statistic) to each kinase to reflect its activity in a manner analogous to that of gene set enrichment analysis (GSEA).53 These scores reflect a ranked statistical calculation of the most likely strong changes in phosphorylation, not a simple fold-change difference. Differential expression of phosphosites in infected HNEs at 6, 24, 36, and 48 hpi was calculated, compared to controls, and KSEA was performed based upon a kinase-substrate database created using PhosphoSitePlus54 and NetworKin.55 Based on the phosphoproteomics and KSEA, we identified 72 kinases that are potentially highly activated after SARS-CoV-2 infection (Figure S6 A). These activated kinases may serve a variety of functions important for the virus (endocytosis, viral replication, viral assembly, and others) at various times in the viral life cycle. To understand how SARS-CoV-2 modulates signaling pathways that facilitate the viral egress from microvilli, we focused on kinases related to microvilli or cytoskeleton regulation. We focused on five kinases, including cytoskeleton reorganizing p21-activated kinases 1 and 4 (PAK1 and PAK4),56 AKT serine/threonine kinase (AKT1/2),57 , 58 mitogen-activated protein kinase P38 Alpha (p38),59 mitogen-activated protein kinase 3 (ERK1), and Rho-associated coiled-coil containing protein kinase 1 (ROCK1).60 We determined if these kinases are phosphorylated in infected cells. All five were activated in SP-positive HNEs (Figures 6A and S6B). Further, immunoblotting of infected HNE cell lysates confirmed these observations (Figure 6B). These results indicate that SARS-CoV-2 infection activates these five kinases in HNEs.
Figure S6.

PAK1/4 regulate microvilli to facilitate the SARS-CoV-2 budding and spreading, related to Figure 6
(A) SARS-CoV-2 infection-affected kinases. ES values of various kinases from KSEA at 6, 24, 36, and 48 hpi after infection. p values were adjusted using Benjamini-Hochberg procedure. 4 biological replicates of phosphopeptide enriched shotgun proteomic samples (Donors 1–4, see Table S1).
(B) SARS-CoV-2 hijack host signaling pathways. SARS-CoV-2-infected HNEs with MOI of 0.3 were stained after 48 hpi. Representative staining of phospho AKT1/2 (pAKT1/2), phospho ERK1/2 (pERK1/2), and phospho p38 (pp38) in combination with ACTUB and SARS-CoV-2 SP in mock-treated and SARS-CoV-2-infected HNEs from Donor 1 is shown. Similar results were observed from Donor 2–4, see Table S1.
(C–E) Identifying kinases involved in both viral infection and microvilli structure. (C) HNEs were treated with either the PAK1 inhibitor FRAX486, the PAK4 inhibitor LCH-7749944, the CAMK4 inhibitor KN-93, the AKT1/2 inhibitor MK-2206, the ROCK1/2 inhibitor Y27632, the MAPKAPK2 inhibitor MK2IV, or the p38 inhibitor ARRY797 prior to SARS-CoV-2 infection and were fixed after 48 hpi. Quantified percentages of SARS-CoV-2 SP positive ciliated HNEs are shown. Error bars represent mean ± SD (3,000–4,000 cells were quantified from DMSO- or drug-treated SARS-CoV-2-infected HNEs from Donor 3–6, see Table S1). (D) Representative IF staining of pEZR and phalloidin in DMSO-, KN-93 (20 uM)-, MK-2206 (20 μM)-, Y27632 (20 uM)-, MK2IV (20 uM)-, and ARRY797 (20 uM)-treated HNEs from Donor 3, see Table S1. Quantification of pEZR and phalloidin from DMSO- or drug-treated HNEs from Donors 5–8 (lower panel; see Table S1) is shown. Error bars represent mean ± SD Drugs were added for 3 h. (E) Representative IF staining of ACTUB and phalloidin in DMSO-, FRAX486- (20 uM)-, and LCH-7749944 (20 uM)-treated HNEs from Donor 6, see Table S1.
(C and D) ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, NS represents not significant, paired, One-way ANOVA with Tukey’s post-test. Each dot represents one donor.
(F) Cytotoxicity measured in companion uninfected cultures. HNEs were exposed to a dose-response of kinase inhibitors, DMSO, or positive control 2 μM staurosporine in triplicate (Donor 4–6). After 48 h, cytotoxicity was measured using Toxilight Assay, which measures adenylate kinase released into the culture medium from dying cells. Staurosporine was significantly different from vehicle and all kinase inhibitors.
(G and H) PAK4 kinases inhibitor as well as siRNA have partial or no significant viral inhibitory effect on ACE2-expressing A549 cells. (G) ACE2-expressing A549 cells were treated with LCH-7749944 (20 uM) for 2 h before SARS-CoV-2 infection with MOI of 0.5 for 24 h. Representative IF staining of SARS-CoV-2 SP and NP staining in DMSO- or drug-treated cells (left panel). Quantified percentages of NP-positive cells (middle panel). Quantitative analysis of viral titer by plaque assay (right panel). (H) siControl- or siPAK4-treated ACE2-expressing A549 cells were infected with SARS-CoV-2 MOI of 0.5 for 24 h. Representative IF staining of SARS-CoV-2 SP and NP staining in siControl- or siPAK4-treated cells (left panel). Quantified percentages of NP-positive cells (middle panel). Quantitative analysis of viral titer by plaque assay (right panel). Immunoblot showing depletion of PAK4 in ACE2-expressing A549 cells (down panel).
(G and H) Error bars represent mean ± SD (2,000–3,000 cells were quantified from drug- or siRNA treated SARS-CoV-2-infected cells. ∗p < 0.05, NS represents not significant, paired, two-tailed Student’s t test.
(I) Log2 fold change profiles of indicated PAK1 and PAK4 potential substrates during infection in HNEs at 48 hpi.
(J) Infected HNEs with MOI of 0.3 were stained at 48 hpi. Representative IF staining of BAIAP2, PTPN14, ARHGEF2, or MYH14 with ACTUB and SP in infected HNEs from Donor 1. Similar results were observed from Donors 2–4, see Table S1.
(K) Log2 fold change profiles of filopodia- and cytoskeleton-related proteins during SARS-CoV-2 infection in HNEs after 48hpi.
(L) SARS-CoV-2-infected HNEs with MOI of 0.3 were stained after 48 hpi. Shown here are representative IF images of CDK16, PPP1R12A, and ANKRD35 in combination with ACTUB and SARS-CoV-2 SP in SARS-CoV-2-infected HNEs from Donor 1. Similar results were observed from Donor 2–4, see Table S1. Scale bars represent 5 μm (A, D, E, G, H, J, and L) and 20 μm (B).
Figure 6.

PAK1/4 regulate microvilli to facilitate SARS-CoV-2 budding and spreading
(A and B) SARS-CoV-2 infection hijacks host signaling pathways.
(A) Infected HNEs (MOI 0.3) were stained after 48 hpi. Representative images of phospho-PAK1/2 (pPAK1/2, left image), phospho-PAK4 (pPAK4, right image), and ACTUB in virus-infected cells (SP) of mock-treated versus infected HNEs from Donor 1. Donors 2–4 showed similar results.
(B) Immunoblots for pPAK1/2, pPAK4, pAKT1/2, pERK1/2, and pp38 protein expression in mock versus infected HNEs (48 hpi, Donor 6). Donors 7–8 showed similar results.
(C and D) PAK1 and PAK4 kinases reorganize microvilli structure and inhibit viral egress.
(C) Representative IF staining of pEZR and phalloidin in DMSO-, FRAX486 (20 uM)-, and LCH-7749944 (20 μM)-treated HNEs from Donor 3. Quantification of pEZR and phalloidin from DMSO- or drug-treated HNEs from Donors 5–8 (lower panel) is shown. pEZR and phalloidin staining was lower in drug-treated (3 h) HNEs than DMSO. Error bars represent mean ± SD.
(D) SARS-CoV-2-treated HNEs were treated with FRAX486 (20 uM) and LCH-7749944 (20 uM) as in Figure 5H. Representative IF staining of SARS-CoV-2 SP, ACTUB, and phalloidin staining in DMSO- versus drug-treated infected HNEs at 24 hpi (upper image) and 48 hpi (down image), Donor 5. Quantified percentages of SP-positive ciliated HNEs (right panel). Error bars represent mean ± SD (3,000–4,000 HNEs Donors 4–8).
(E–G) Loss of EZR and PAK4 siRNA knockdown doesn’t affect viral entry at 24 hpi but significantly inhibits viral egress at 48 hpi.
(E) Representative IF staining of SARS-CoV-2 SP, ACTUB, and phalloidin staining in siControl versus siEZR- or siPAK4-treated HNEs at 24 hpi (upper image) and 48 hpi (down image), Donor 6.
(F) Quantified percentages of SP-positive ciliated HNEs (right panel). Error bars represent mean ± SD (3,000–4,000 HNEs Donors 6–8).
(G) Immunoblot showing depletion of EZR or PAK4 in HNEs. (C, D, and F) ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, NS represents not significant, paired, one-way ANOVA with Tukey’s post-test. Each dot represents one donor.
(H) Model for SARS-CoV-2 entry, egress, and spread in nasal airway. (1) SARS-CoV-2 binds to cilia to cross the barrier and allow HNEs entry during early stages of infection. (2) SARS-CoV-2 hijacks PAK1 and PAK4 signaling to elongate microvilli and facilitate viral egress. (3) Virus attached to microvilli is carried along extended microvilli from the base to the mucus layer, allowing virus spreads throughout the tissue mucus layer. Scale bars represent 5 μm (C), 20 μm (A and D), and 50 μm (E). See Figure S6 and Table S1.
To understand whether these five kinases regulate microvilli structure, HNEs were pre-treated with kinase inhibitors and infected. Infected ciliated HNEs and cytotoxicity were quantified at 48 hpi (Figures S6C and S6F). PAK1, PAK4, CAMK4, and MAPKAPK2 inhibitors decreased infected cells. To determine which kinases regulate microvilli, we treated HNEs with kinase inhibitors for 3 h. Cells were fixed and stained for pEZR and phalloidin (Figures 6C and S6D). Only FRAX486, a PAK1-selective inhibitor, and LCH-7749944, a PAK4-selective inhibitor, significantly decreased expression of phalloidin and pEZR staining in the microvilli (Figure 6C) and changed the microvilli structure (Figure S6E), indicating a disruption in microvilli. Treatment with these drugs caused no significant cytotoxicity (as measured by adenylate kinase activity; Figure S6F). Interestingly, PAK1 and PAK4 regulate the actin cytoskeleton and filopodia.61 , 62 , 63 , 64 We treated infected HNEs with specific PAK1 and PAK4 kinase inhibitors. Neither affected early viral cell entry. However, each reduced the number of SARS-CoV-2-positive cells during viral egress at 48 hpi (Figure 6D). PAK4 inhibition affected virus replication the most. We used siRNA to knock down EZR and PAK4 in HNEs and obtained similar results (Figures 6E–6G). Conversely, PAK4 kinases inhibitor and PAK4 siRNA showed little or no viral inhibition on ACE2-expressing A549 cells (Figures S6G and S6H). Thus, PAK1/4 signaling seems to regulate microvilli dynamics during SARS-CoV-2 egress in nasal epithelium.
We next investigated downstream targets of PAK1 and PAK4 to identify potential proteins that were phosphorylated at 36–48 hpi. Several phosphorylation sites were found among phosphoproteomic data hits. PAK1 kinase candidates and associated phosphorylation sites included ARHGEF2 (S151 and S174), BAIAP2 (S325), and FLNA (S2152). PAK4 kinase candidates included PAK4 (S181 and T207), MYH14 (T1503), and PTPN14 (S578) (Figure S6I). ARHGEF2, BAIAP2, MYH14, FLNA, PAK4, and PPP1R12A regulate actin cytoskeleton and filopodia.65 , 66 , 67 , 68 , 69 , 70 , 71 , 72 To examine their localization, we co-stained the proteins with a microvillar marker in HNEs. BAIAP2 and PTPN14 localized to the microvilli, and ARHGEF2 and MYH14 localized to the base of the microvilli (Figure S6J). Thus, PAK1 and PAK4 may regulate microvilli by phosphorylating downstream targets during infection. Proteins involved in cytoskeleton and filopodia formation were phosphorylated by unidentified kinases after infection (Figure S6K).73 , 74 , 75 , 76 , 77 , 78 These included PPP1R12A, CDK16, and ANKRD35 localized to the base of microvilli, suggesting pathways in SARS-CoV-2-regulated microvilli elongation and viral egress (Figure S6L). The kinase activities induced by SARS-CoV-2 suggest that SARS-CoV-2 stimulates microvilli-mediated viral egress and spread via PAK1 and PAK4 signaling, suggesting potential therapeutic targets (Figure 6H).
Finally, to confirm the effects of microvilli inhibitors in reducing virus titer in vivo, we applied SLK and PAK4 kinases inhibitors to K18-hACE2 transgenic mice by nasal spray. The PAK4 kinase inhibitor partially repressed infection (Figure S7 A), but the SLK kinase inhibitor was less effective. One possible explanation is that human and mouse nasal cavities differ dramatically. In the mouse, ∼50% of the nasal cavity is olfactory epithelium (OE), and the rest is largely ciliated (respiratory) epithelium (RE). In humans, OE constitutes only ∼3% of the cells in the nasal cavity, and the rest are primarily RE. In the human, ciliated cells in the RE are the main target cell type for SARS-CoV-2 in the nasal cavity.2 , 10 , 11 , 12 , 13 , 79 K18-Ace2 mice express the ACE2 receptor ubiquitously, which can contribute to a different profile of infected cells as compared to humans.
Figure S7.

Higher viral entry and replication of Omicron in human nasal epithelium, related to Figure 7
(A) The effect of microvilli inhibitors in the mouse models. K18-hACE2 mice were infected with SARS-CoV-2 (1000 PFU) intranasally with the inhibitors or vehicle control (solvent) at day 0. Then the inhibitors were delivered intranasally once per day at day 0 and 1. At day 3 (i.e., 3 dpi), the lung were harvested for analysis. Virus titer measurement using plaque assay for the lung. The number of mice is 4 (n = 4) per group. SB-633825 and LCH-7749944 were used as 10 mg/mouse by intranasal inoculation. Data are shown as mean ± SD Kolmogorov-Smirnov test, ∗P < 0.05. The same experiment was repeated 3 times and similar results were obtained.
(B) SARS-CoV-2 D614G and Omicron variants replication kinetics in HNEs. qPCR data were collected with the use of the infected cells derived from Donor 6–8, see Table S1.
(C) Quantitative analysis of viral titer from plaque assays on different strains of SARS-Co-V2-infected Vero E6 cells. ∗∗∗p < 0.001, paired, two-tailed Student’s t test.
(D) Omicron’s RBD has a stronger cilia adhesion ability. ALI-cultured HNEs were labeled by SiR-tubulin and treated with pseudovirion, quantum dot-conjugated SARS-CoV-2 D614G (left image, QD585-SARS-CoV-2 RBD), or Omicron receptor binding domain (right image, QD585-Omicron RBD) for 6 h. Quantification of the intensity of pseudovirion of ciliated HNEs was performed on the right panel. Error bars represent mean ± SD (5–10 cells were quantified from HNEs from Donor 6–8, see Table S1).
(E) Compared to D614G, Omicron has a stronger infectivity. D614G-RFP (MOI 2) or D614G -SpikeOmicron-BA.1-GFP (MOI 0.1) or double-infected (same MOIs) infected HNEs were stained at 48 hpi. Representative IF images of RFP or GFP in HNEs from Donor 6. Quantified percentages of RFP- or GFP-positive area (right panel). Error bars represent mean ± SD (data were collected from Donors 6–8, see Table S1). Scale bars represent 10 μm (D) and 50 μm (E).
Omicron variants are more efficient in infecting nasal epithelia cultures
In 2022, Omicron BA.1 (B.1.1.529) rapidly replaced Delta (B.1.617.2) as the dominant circulating form.80 , 81 Its SP protein has 37 amino acid substitutions, 15 in the receptor binding domain, suggesting that Omicron increased infectivity and transmission by altering virus-receptor binding. More recently, BA.4 and BA.5 displaced BA.1 and BA.2. Thus, Omicron variants may have a replication advantage in the upper respiratory tract from which the virus is transmitted from person to person. Intriguingly, the Omicron virus load from nasal and saliva samples is lower than previous variants.82
To characterize Omicron replication in the nasal epithelium, we determined the spatial and temporal infection pattern of D614G, Delta, and Omicron variants in HNE ALI cultures. D614G and Delta had similar characteristics (1%–3% positive cells at 6 and 24 hpi and 70%–80% at 48 hpi). In contrast, Omicron variants showed a notable increase of infected cells at early timepoints (∼10% at 6 hpi and ∼40% at 24 hpi) (Figures 7A and S7B). Interestingly, the viral titer of infectious D614G in Vero cells was higher than Delta and Omicron, implying that the infection mechanism varies in 2D and 3D cultures (Figure S7C). Omicron SP may bind more strongly to the ACE2 receptor.83 We determined if Omicron variants are more likely to bind to motile cilia. By using time-lapse, live-cell microscopy, we determined that more pseudovirion QD585-Omicron RBDs (BA.1) attached to motile cilia than did D614G RBD in ALI-cultured HNEs at 4 hpi (Figure S7D). Thus, Omicron variants enter and replicate more efficiently in nasal epithelium than D614G and Delta, possibly due to an enhanced interaction with its receptor. To confirm that the entry of Omicron is higher than D614G virus, we used a competition assay with D614G and a chimeric virus harboring Omicron BA.1 SP in the WA1 backbone. Virus was engineered to express RFP and GFP proteins. IF showed that WA1-BA.1-spike-GFP is overwhelmingly superior to D614G-RFP for infecting cells (Figure S7E). Determinants within spike protein seem to be responsible for the increased replication fitness of the Omicron variants. The ability of Omicron to spread within the nasal epithelium without a lag phase suggests a mechanism for how Omicron variants displaced previous variants and became responsible for most global COVID infections.
Figure 7.

Higher viral entry and replication of Omicron in human nasal epithelium
(A) Omicron variants show notable increase of infected cells at early timepoints. Mock-treated or SARS-CoV-2 D614G, Delta, and Omicron (BA.1, BA.4, BA.5) strain-infected HNEs (MOI 0.3) were stained at 6 (left), 24 (middle), and 48 hpi (right). Representative IF images of HNEs stained for SP, NP, and CED marker ACTUB. Quantified percentages of NP- and SP-positive ciliated nasal epithelial cells or goblet cells (right panel).
(B and C) Cilia and microvilli remain critical in Delta and Omicron infections. (B) SARS-CoV-2 variants infected HNEs (MOI 0.3) were stained 48 hpi. Representative IF staining for SP and ACTUB with phalloidin in shControl and shCEP83 Delta (left)- or Omicron BA.1 (right)-infected HNEs. Quantified percentages of SP-positive HNEs (right panel). Error bars represent mean ± SD (3,000–4,000 HNEs Donors 6–8).
(C) SARS-CoV-2 variant-treated HNEs were treated with SB-633825 (40 μM) and LCH-7749944 (20 μM) as in Figure 5H. Representative IF staining of SARS-CoV-2 SP, ACTUB, and phalloidin staining in DMSO- versus drug-treated Delta (left)- or Omicron BA.1 (right)-infected HNEs. Quantified percentages of SP-positive ciliated HNEs (right). Error bars represent mean ± SD (3,000–4,000 NHEs Donors 6–8). (A–C) ∗∗p < 0.01, ∗∗∗p < 0.001, One-way ANOVA with Tukey’s post-test (A and C) or paired, two-tailed Student’s t test (B). Each dot = one donor. Scale bars represent 20 μm.
Finally, we determined if SARS-CoV-2 variants used motile cilia and microvilli for entry and egress. Depletion of cilia with lentivirus expressing CEP83 shRNA inhibited Delta and Omicron infections at 48 hpi in HNEs (Figure 7B). SLK and PAK4 inhibitors also blocked Delta and Omicron viral egress at 48 hpi in HNEs (Figure 7C). Thus, inhibiting viral entry in airway cells is effective even for highly infectious variants.
Discussion
Here, we discovered that SARS-CoV-2 preferentially infects ciliated HNEs and not goblet or basal cells in ALI-cultured human nasal epithelium. We used an ALI airway organoid model with many nuances of the airway in vivo. Strikingly, this culture has a substantial kinetic delay (24–48 h), compared to tissue culture models, which we established as linked to airway barrier function. With EM and IF microscopy, we tracked the detailed steps required for viral entry in the airway. The virus attaches first to airway multicilia via the ACE2 receptor, and that binding and ciliary trafficking are critical for the virus to traverse the mucus-mucin protective barrier. Depleting cilia blocked SARS-Co-2 infection in ALI cultures, but depleting mucins with purified mucinase accelerated viral entry. At 24–48 h, viral numbers increased in parallel with a substantial rearrangement and expansion of microvilli in epithelial cells and activation of PAK1, PAK4, and SLK kinases. Inhibitors of these kinases blocked late viral spread, but not initial binding to cilia. PAK kinase inhibitors attenuated viral spread in mice. The importance of these distinct steps in virus entry is underscored by our finding that recent highly infectious variants of SARS-CoV-2 accelerate infection into the ALI model airway by more than 24 h. Other respiratory viruses require the airway multicilia, suggesting that targeting virus interactions with cilia or microvilli is a broad strategy for blocking airway entry and virus spread for new respiratory viruses.
Only a low percentage of SARS-CoV-2 infects ciliated HNEs at 24 hpi, even at high titers. We suspect the virus cannot penetrate the PCL of most cells and requires a previously unidentified gateway. The mucus and PCL in airway epithelium block entry of pathogens and particles over 40 nm.6 Depleting MUC1 partially increased influenza infection,8 , 9 indicating the importance of the PCL as a physical barrier against viruses.
Using multiple imaging techniques, we showed that SARS-CoV-2 attaches with high efficiency to the distal ends of cilia during early infection (<6 h). Depleting cilia in nasal epithelial cells inhibited infection without affecting ACE2 and TMPRSS2 levels. We propose that cilia facilitate early viral attachment and passage through the PCL layer. Accordingly, the cilia could serve as a high-density binding lattice mediated by ACE2, forming an avidity trap to allow viral particles to find target cells via a multivalent binding “landing pad,” leading to superselectivity.84 Two non-exclusive mechanisms of entry are possible. First, SARS-CoV-2 interacts with ACE2 and TMPRSS2 at the cilia surface to trigger membrane fusion into cilia and release of the viral RNA genome, which could be transported from the cilium to the cytoplasm by ciliary dynein.1 Alternatively, cilia-attached SARS-CoV-2 could be transported along the surface of cilia by cycles of release and rebinding85 and bind to ACE2 on the epithelial cell body surface, directing the virus to undergo TMPRSS2-mediated membrane fusion.86 In this case, the virus-ACE2 complex could be transported by the dynein-dependent retrograde intraflagellar transport down the cilium to the epithelial cell apical surface.
SARS-CoV-2 egress is even less understood. We described the structure of microvilli in ciliated HNEs. SARS-CoV-2 is localized to and exits via the microvilli at 24 hpi, and SARS-CoV-2 infection promotes formation of dome-like structures at the base of microvilli and highly branched and highly extended microvilli over time. Although the mechanisms and consequences of these rearrangements are unknown, SARS-CoV-2 induced the highly extended microvilli to penetrate the PCL layer, and concatenated chains of virus formed on the extended microvillar structure. These viral chains appear to accumulate in the mucus layer and to use MCT to infect other cells. This hypothesis agrees with our findings in cells from PCD patients. PCD patients seem to have no increased risk of infection.47
Disrupting microvilli with two distinct inhibitors had little to no effect on viral entry but severely inhibited viral exit. Thus, cilia may facilitate infection early on, and microvilli may mediate later stages of infection. Our study provides potential valuable targets for drug repurposing against COVID-19. A nasally delivered drug that transiently inhibits microvillar reprogramming in the upper respiratory tract may have the advantage of non-invasive delivery with less systemic toxicity or adverse effects.
Finally, using unbiased phosphoproteomic analysis, we found that SARS-CoV-2 regulates the cytoskeleton and microvilli by activating PAK1 and PAK4 to facilitate viral egress and spread. PAK1 and PAK4 kinase inhibitors inhibited SARS-CoV-2 cell egress and spread but did not affect entry. We identified several undescribed microvilli- or cytoskeleton-associated proteins as downstream targets of PAK1 and PAK4. They regulate filopodia or cytoskeleton and represent critical markers of viral reprogramming and possibly therapeutic targets.71 , 87 , 88 , 89 , 90 Lastly, we identified potential microvilli- or filopodia-associated proteins that are phosphorylated during SARS-CoV-2 infection. Their roles remain to be explored.
Omicron is the dominant variant in the world circa 2022, but the mechanisms for its higher transmissibility are unclear. Our results indicate that, at similar MOIs (as measured in Vero cells), Omicron infected 40% of cells at 24 hpi, compared to 3%–5% for D614G and Delta. The number of cells infected by Omicron also increased at 6 hpi. The more efficient binding of the Omicron variant RBD to ACE2 and motile cilia provides a parsimonious explanation for the increased initial numbers of infected cells.
While cilia facilitate SARS-CoV-2 infection early on, microvilli mediate the later stages of viral infection, allowing particles to spread to other cells. We found several potential targets for antiviral drug therapies with new inhibitors or repurposing of existing inhibitors. Topical administration of kinase inhibitors to the nasal mucosa may modulate microvilli function and avoid undesirable systemic off-target effects for systemic treatment.
Motile cilia-regulated mucociliary clearance is important for the functioning of the human respiratory tract. Therefore, research has focused on how motile cilia beat, but little is known about how motile cilia directly function as a sensory organ and how signaling and airway remodeling are regulated. Specifically, there is very little understanding on whether ciliary proteins and receptors are specifically transported into and out of the motile cilia to regulate and maintain sensory function, and whether motile cilia are regulated by IFT and Bardet–Biedl syndrome complexes that regulate protein trafficking in primary cilia,91 although clearly these pathways have an effect on airway function and morphology.92
Limitations of the study
The sample sizes of HNEs were limited by tissue availability and the challenges of conducting experiments under BSL3. Samples from PCD patients were especially limited, as the estimated prevalence of PCD is 1:10,000 to 1:20,000 live-born children.93 Although patients with PCD share similar defective mucociliary clearance, mutations in >40 genes cause PCD with varying degrees of severity. Therefore, our findings are not necessarily representative of all PCD patients.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-SARS-CoV-2-NP | GeneTex | Cat# GTX135361; RRID: AB_2887484 |
| Mouse anti-SARS-CoV-2-NP | Thermo Fisher | Cat# MA1-7403; RRID: AB_1018420 |
| Mouse anti-SARS-CoV-2-SP | GeneTex | Cat# GTX632604; RRID: AB_2864418 |
| Rabbit anti-ACE2 | Abcam | Cat# ab15348; RRID: AB_301861 |
| Mouse anti-TMPRSS2 | Millipore-Sigma | Cat# MABF2158 |
| Mouse anti-acetylated tubulin (ACTUB) | Santa Cruz | Cat# sc-23950 RRID:AB_628409 |
| Mouse anti-MUC5AC | Abcam | Cat# ab212636 |
| Rabbit anti-MUC1 | Millipore-Sigma | Cat# HPA008855 RRID:AB_1846283 |
| Rabbit anti-CX3CR1 | Abcam | Cat# ab8021 RRID:AB_306203 |
| Mouse anti-FOXJ1 | Thermo Fisher | Cat# 14-9965-82 RRID:AB_1548835 |
| Mouse anti-RSV | Millipore-Sigma | Cat# MAB858-1 RRID:AB_11203839 |
| Mouse anti-PIV type 1 | Novus Biologicals | Cat# NB100-63036 RRID:AB_964926 |
| Mouse anti-PIV type 3 | Novus Biologicals | Cat# MAB10207 RRID:AB_827567 |
| Rabbit anti-EBP50 | Thermo Fisher | Cat# PA1-090 RRID:AB_2191493 |
| Mouse anti-EZR | Sigma-Aldrich | Cat# E8897 RRID:AB_476955 |
| Rabbit anti-phospho-EZR | Cell Signaling | Cat# 3726T RRID:AB_10560513 |
| Rabbit anti-SLK | Millipore-Sigma | Cat# HPA015757RRID:AB_1857236 |
| Rabbit anti-SLK | Millipore-Sigma | Cat# HPA015757 RRID:AB_1857236 |
| Rabbit anti-CEP164 | Sigma-Aldrich | Cat# HPA037605 RRID:AB_10672908 |
| Rabbit anti-IFT88 | ProteinTech | Cat# 13967-1-AP RRID:AB_2121979 |
| Mouse anti-ARL13B | NeuroMab | Cat# 75-287 RRID:AB_2341543 |
| Rabbit anti-β-catenin | Thermo Fisher | Cat# AHO0462 RRID:AB_1500389 |
| Rabbit anti-Phospho-PAK4 | Cell Signaling | Cat# 3241S RRID:AB_2158623 |
| Rabbit anti-PAK4 | Cell Signaling | Cat# 62690S RRID:AB_2827508 |
| Rabbit anti-Phospho-AKT | Abcam | Cat# ab192623 |
| Rabbit anti- Phospho-Erk1/2 | Cell Signaling | Cat# 9101S RRID:AB_331646 |
| Mouse anti- Phospho-Erk1/2 | Cell Signaling | Cat# 9107 RRID:AB_10695739 |
| Rabbit anti-Phospho-p38 | Cell Signaling | Cat# 4511T RRID:AB_2139682 |
| Rabbit anti-BAIAP2 | Millipore-Sigma | Cat# HPA027421 RRID:AB_10602392 |
| Rabbit anti-PTPN14 | Millipore-Sigma | Cat# HPA053864 RRID:AB_2682281 |
| Rabbit anti-ARHGEF2 | Millipore-Sigma | Cat# HPA043437 RRID:AB_2678481 |
| Rabbit anti-MYH14 | Millipore-Sigma | Cat# HPA067889 RRID:AB_2685917 |
| Rabbit anti-CDK16 | Millipore-Sigma | Cat# HPA001366 RRID:AB_1079584 |
| Rabbit anti-PPP1R12A | Millipore-Sigma | Cat# HPA071956 RRID:AB_2732192 |
| Rabbit anti-ANKRD35 | Millipore-Sigma | Cat# HPA035453 RRID:AB_10672864 |
| Rabbit anti-Phospho-JNK1/2 | Cell Signaling | Cat# 4668T |
| Rabbit anti-Phospho-PAK1/2 | Cell Signaling | Cat# 2601S PRID: AB_330220 |
| Mouse anti-DM1α | Santa Cruz | Cat# sc-32293 RRID:AB_628412 |
| Bacterial and virus strains | ||
| SARS-CoV-2 D614G | Joe De-Risi Lab, UCSF | SARS-CoV-2/human/USA/CA-UCSF-0001C/2020 |
| SARS-CoV-2 BA.1 | Melanie Ott Lab, UCSF | B.1.1.529 (California Department of Health) |
| SARS-CoV-2 BA.4 | Melanie Ott Lab, UCSF | Clinical isolates |
| SARS-CoV-2 BA.5 | Melanie Ott Lab, UCSF | Clinical isolates |
| Respiratory Syncytial Virus | Mark Peeples Lab, Rush-Presbyterian-St. Luke’s Medical Center | A2 strain |
| Parainfluenza Virus | Raymond Pickles Lab, University of North Carolina at Chapel Hill | JS strain |
| Biological samples | ||
| Human primary nasal epithelial cells | Jayakar Nayak lab and Carlos Milla Lab, Stanford University | Tables S1 and S2 |
| K18-hACE2 mice | Jackson laboratory | 034860 |
| Chemicals, peptides, and recombinant proteins | ||
| DMSO | Sigma-Aldrich | Cat# 276855 |
| Alexa Fluor™ Plus 647 Phalloidin | Invitrongen | A30107 |
| DAPI | Bio Trend | Cat# 40043 |
| Pen/Strep | Thermo Fisher | Cat# 15140163 |
| Paraformaldehyde | AlfaAesar | Cat# 433689M |
| Normal Donkey Serum | Jackson ImmunoResearch | Cat# 017-000-121 |
| NP40 | Sigma | Cat# 11332473001 |
| SiR-Tubulin | Cytoskeleton Inc. | Cat# CY-SC002 |
| RBD protein | BPS Bioscience | Cat# 100688 |
| Qdot™ 585 Streptavidin Conjugate | Thermo Fisher | Cat# Q10111MP |
| StcE | Carolyn Bertozzi Lab, Stanford University | NA |
| BSA | Sigma-Aldrich | Cat# A6003-25G |
| 1X PBS | Corning | Cat# 46-013-CM |
| Triton X-100 | USB | Cat# 22686 |
| OCT compound | VWR | Cat# 25608-930 |
| Saponin | Sigma-Aldrich | Cat# S7900 |
| SARS-CoV-2 neutralizing antibody | Michael S. Diamond, Washington University in St. Louis | NA |
| Ciliobrevin D | Sigma-Aldrich | Cat# 250401 |
| Camostat | Millipore-Sigma | Cat# SML0057 |
| Hydroxychloroquine | Cayman | Cat# 17911 |
| NSC-668394 | Sigma-Aldrich | Cat# 341216 |
| SB-633825 | MCE | Cat# HY-108333 |
| FRAX486 | Cayman | Cat# 24682 |
| LCH-7749944 | MCE | Cat# HY-125035 |
| KN-93 | Cayman | Cat# 13319 |
| MK-2206 | Santa Cruz | Cat# sc-364537 |
| Y27632 | Santa Cruz | Cat# sc-281642A |
| MK2IV | Cayman | Cat# 14399 |
| ARRY797 | Biovision | Cat# B2462-5 |
| Critical commercial assays | ||
| Adenylate kinase cytotoxicity bioassay | LONZA | Cat# LT17-217 |
| In Situ Cell Death Detection Kit, TMR red | Sigma-Aldrich | Cat# 12156792910 |
| TaqMan 2019-nCoV Control Kit v1 | Thermo Fisher | Cat# A47533 |
| RNAscope® Multiplex Fluorescent Reagent Kit v2 | Bio-Techne | Cat# 323100 |
| TSA Cyanine 3 | Akoya Biosciences | Cat# NEL744001KT |
| High-Select Fe-NTA Phosphopeptide Enrichment Kit | Thermo Scientific | Cat# A32992 |
| Deposited data | ||
| Phosphoproteome of SARS-CoV-2 infected Human Nasal Epithelium | This paper | PRIDE Project ID: PXD028123 |
| Oligonucleotides | ||
| Human CEP83 shRNA | Dharmacon | Cat# SQ-20.0061 |
| Non-Targeting Control Pool | Dharmacon | Cat# D-001910-10-50 |
| Human EZR siRNA | Dharmacon | Cat# E−017370-00-0020 |
| Human PAK4 siRNA | Dharmacon | Cat# E−003615-00-0020 |
| ACE2 (Hs00222343_m1) | Life Technologies | Cat# 4331182 |
| TMPRSS2 (Hs00237175_m1) | Life Technologies | Cat# 4331182 |
| CX3CR1 (Hs01922583_s1). | Life Technologies | Cat# 4331182 |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Zen black | Carl Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| GraphPad Prism 6 | GraphPad | https://www.graphpad.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Peter Jackson (pjackson@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Primary human nasal cell culture
Human tracheobronchial epithelial cells were obtained from patients who underwent bronchoscopy or surgical lung resection during diagnostic procedures for pulmonary diseases at Stanford (Tables S1 and S2). Nasal epithelial cultures were generated using an already well-established protocol in our laboratories.28 After obtaining informed consent (Stanford IRB protocol #42710), subjects underwent brushing of the inferior turbinate from both nasal cavities to obtain a cell sample. The sample was immediately transferred to the laboratory and dissociated in a 1:1 mixture of proteolytic enzymes (Accutase, Thermo Fisher Scientific Inc.) and nonenzymatic cell dissociation solution (C5914, Millipore-Sigma Inc.) for 10 min at 37°C. After gentle agitation the cells were pelleted, resuspended in basal cell proliferation media (PneumaCult-Ex Plus medium, STEMCELL Technologies, Vancouver, Canada) supplemented with antibiotics and plated on collagen-coated Transwell inserts (0.33 cm2) at a density of 20,000 cells per insert with media added to both the basal and apical sides. Once the cells were fully confluent, the media was replaced with PneumaCult-ALI medium (STEMCELL Technologies, Vancouver, Canada) in the basal chamber and the apical surface exposed to provide an air liquid interface (ALI). Monolayers were grown at ALI for an additional 3 weeks to promote differentiation into a nasal epithelium with basal, multiciliated and secretory cells.
Cells lines
Cells used for this study include ACE2-expressing A549 and Vero-E6 cells. ACE2-expressing A549 cells were cultured in DMEM high glucose/F12 medium supplemented with 10% fetal bovine serum (Sigma) and 1x penicillin/streptomycin/glutamine (100xPSG, GIBCO). Vero-E6 cell line (ATCC#1586) was maintained in Minimum Essential Medium (MEM, GIBCO) supplemented with 10% fetal bovine serum (FBS, GIBCO), 1% Penicillin-Streptomycin-Glutamine (GIBCO) at 37°C in a humidified 5% CO2 incubator.
Viruses
A clinical isolate: SARS-CoV-2 strain (D614G) is a gift from Sara Sunshine (UCSF). Clinical isolates: SARS-CoV-2 BA.1, BA.4, and BA.5 are gifts from Melanie Ott (UCSF). SARS-CoV-2 cell culture were performed in the Biosafety level 3 (BSL-3), under the guidance and protocols approved by UCSF Biosafety committees. (See method detail and key resource table).
Mice strains
C57BL/6J mice were purchased from Jackson Laboratory (000664). K18-hACE2 mice (Winkler et al., 2020) were purchased from Jackson Laboratory (034860, B6. Cg-Tg(K18-ACE2)2Prlmn/J, Hemizygous). The K18-hACE2 mice were inbred and housed at the BSL3 level. Male mice between 8 and 10 weeks of age were used for SARS-CoV-2 infection. All mice were maintained under specific pathogen-free conditions at the UCSF and Stanford animal care facility. Mice were cared for and all experiments were approved by the Administrative Panel on Laboratory Care and the Institutional Animal Care and Use Committee (IACUC) of at UCSF and Stanford University.
Method details
Virus production, cell infection, and drug treatment
A549 cells stably expressing ACE2 under the CMV promoter were infected with SARS-CoV-2 clinical isolate of the pandemic D614G variant (SARS-CoV-2/human/USA/CA-UCSF-0001C/2020, kindly provided by Sara Sunshine and Joe De-Risi) with MOI ∼0.05 in MEM medium supplemented with 2% FBS and penicillin/streptavidin (Gibco). 3 days after infection, the medium was collected and cleared from cell debris by centrifugation at 3,000 g for 10 min at 4C. Omicron and Delta variants were isolated with the use of Vero-hACE2-TMPRSS2 cells (BEI NR-54970). Briefly, 200 μL of nasal specimens were added to a well of a 96-well plate and serially diluted 1:1 over five additional wells. 100 μL of freshly trypsinized cells, resuspended in infection media (DMEM with 10% FBS and 200 u/mL penicillin/streptomycin, 5 μg/mL amphotericin B) at 2.5x105 cells/mL, were added to each sample dilution. Cells were cultured at 37°C with 5% CO2 and checked for cytopathic effect from day 2–3. Supernatants were harvested on day 3 after inoculation. 200 μL of P0 was used to infect a confluent T25 flask to generate a P1 culture, harvested after 3 days. Viruses were passaged for 3 times after isolating to get enough higher titer (>107 PFU/mL). RFP-labeled D614G and GFP-labeled chimeric virus with an Omicron spike protein in the backbone of WA1 strain was kindly provided by Dr. Luis Martinez-Sobrido in Texas Biomedical Research Institute. Both fluorescent protein-labeled viruses were propagated in Vero-hACE2-TMPRSS2 cells (BEI NR-54970). Viral titers were quantified with plaque assays. The genotype was confirmed by sequencing. To infect the cells, 100 uL of virus suspension (MOI 0.03–3) was added to the top of the ALI-cultured HNEs and incubated at 37°C for the indicated time. Small molecules were dissolved in DMSO (276855, Sigma-Aldrich). For treatment of HNEs, media containing drugs was added to ALI cultures (100 μL to the apical chamber and 500 μL to the basement chamber). The dose used for each drug and treatment are shown in the figure or figure legend. StcE was kindly provided by Dr. Carolyn R. Bertozzi at Stanford University. ARRY-797 was from Biovision. SB-633825, LCH-7749944, and NSC305787 were from MCE. FRAX486, KN-93, and MK2 IV were from Cayman. MK-2206 and Y-27632 were from Santa Cruz Biotechnology. NSC668394 was from Sigma. Staurosporine was from Tocris. SARS-CoV-2 neutralizing antibody was kindly provided by Michael S. Diamond, Washington University in St. Louis. For the SARS-CoV-2 monoclonal antibody blocking experiment, the virus was preincubated with blocking antibody at the 10 μg/mL final concentration for 1 h at 37°C. SARS-CoV-2 cell culture and animals works were performed in the Biosafety level 3 (BSL-3), under the guidance and protocols approved by UCSF Biosafety committees.
Viral RNA isolation and qPCR
Genomic RNA was extracted from cell pellets using Trizol (Ambion), according to manufacturer instructions. RNA was treated with DNAse I (NEB) and used as a template to reverse-transcribe cDNA by Iscript kit (NEB). qPCR reactions were done using the Luna Universal qPCR Master Mix (NEB) and a CFX connect qPCR Detection System (BioRad). To determine the number of vRNA copies per mL, plasmids containing the nucleocapsid gene of SARS-CoV-2 (cloned from the USAWA1/2020 isolate) were used as standards and diluted serially 10-fold to determine target copy numbers. Threshold cycle (Ct) values were plotted against the number of target copies, and the resultant standard curve was used to determine the number of genome equivalents of vRNA in the samples. For cell pellet samples, the vRNA copy number was normalized to the housekeeping gene HRT1. All samples were within the range of linearity of a standard curve, and the primer efficiencies were 100% +/− 5%. The primer sequences targeting nucleocapsid were: 5′- TCCTGGTGATTCTTCTTCAGG-3′ and 5′-TCTGAGAGAGGGTCAAGTGC-3′. HRT1 primers sequences are: 5′-GGTCCTTTTCACCAGCAAGCT-3′ and 5′-TGACACTGGCAAAACAATGCA-3′.
Mouse experiments for SARS-CoV-2
K18-hACE2 mice94 (The Jackson laboratory, https://www.jax.org/strain/034860, stock number: 034860, B6. Cg-Tg(K18-ACE2)2Prlmn/J, Hemizygous). In all the experiments we performed, the mice were under anesthesia and at the BSL3 level. K18-hACE2 mice were anesthetized and infected with SARS-CoV-2 for72 h. Animals were then sacrificed, and samples were harvested, fixed in 4% PFA, embedded in paraffin wax, and cut into 5-μm sections.
Toxicity test in animals
Natural products were delivered intranasally. Briefly, C57B6/J mice were anesthetized with isoflurane and inoculated with solvent and compounds with different diluted concentrations in 50 μL (6.5, 3.25, 1.63 mg/kg). Mice were treated every 24 h for 3 days. No obvious toxicity was observed up to 6.5 mg/kg/mouse by intranasal inoculation.
Antiviral effect test in mouse models
The SARS-CoV-2 experiments were performed in 8-10-week-old hemizygous K18-hACE2 mice.
For K18-hACE2 mice, at day 0, we administered solvent, SB-633825 and LCH-7749944 and at the same time, 1000 PFU of SARS-CoV-2 in 50 μL (25 μL of virus solution in PBS plus 25 μL of drug solution or solvent) intranasally (I.N.). Then, SB-633825 and LCH-7749944 were administrated I.N. one time per day for 2 days. Mice were anesthetized with isoflurane before intranasal infection. This experiment was repeated twice independently, a total of 4 mice were used in each control or drug-treated group. For virus tissue distribution, at 3 days post-infection (d.p.i.), animals were humanely euthanized. Lungs were harvested. Lungs were weighted and homogenized in 1 mL of 2% FBS MEM medium with gentle MACS - C tubes (Miltenyi Biotec Catalog# 130-093-237). Samples were centrifuged at 3000 rpm for 10 min, and the supernatant was transferred to a new set of tubes and then frozen at −80°C for viral titration via plaque assay. Infectious titers were quantified by plaque assay titration using Vero-E6 cells as described above. Unpaired t test was used for tissue distribution analysis.
Plaque assay
Confluent monolayers of Vero-E6 cells grown in six-well plates were incubated with the serial dilutions of virus samples (250 μL/well) at 37°C for 1 h. Next, the cells were overlayed with 1% agarose (Invitrogen) prepared with MEM supplemented with 2% FBS and 1% penicillin-streptomycin-glutamine (100x, Gibco Invitrogen). After 3 days, cells were fixed with 4% formaldehyde for 2 h, the overlay was discarded, and samples were stained with crystal violet dye.
Lentiviral constructs and lentivirus preparation and infection
Lentiviruses were produced by transfection of HEK293T cells with lentiviral vectors carrying the gene of interest and pMD2.G (12259; Addgene) and psPAX2 (12260; Addgene) packaging constructs. Supernatants were collected and concentrated by PEG-it (System Biosciences). Lentiviral constructs coding for shRNAs targeting human CEP83 were obtained from Dharmacon. Human CEP83 shRNA targeting sequences were: (TGGAGACAGTGGATTGACA, TCAAGCACCTGTTTAATGA, and CCAATGAGAGAACGTTTTA). HNEs were infected with lentivirus on day 2 of culture using spin infection following EGTA treatment to temporarily disrupt epithelial junctions.95 , 96 Briefly, HNEs were treated with 12 mM EGTA in 10 mM HEPES, pH 7.4 to the apical for 25 min at 37°C and centrifuged at 1,500 g for 80 min at 32°C.
siRNA transfection in HNEs
All siRNAs were obtained from Dharmacon/Horizon Discovery: Accell siRNA SMARTPool Human Non-Targeting Control Pool (D-001910-10-50); Accell siRNA SMARTPool Human EZR (E−017370-00-0020); and Accell siRNA SMARTPool Human PAK4 (E−003615-00-0020). siRNAs were resuspended to 100 μM using 1× siRNA buffer (Dharmacon, B-002000-UB-100) and incubated at room temperature for 1 h on a shaker. Passive transfections were performed by adding 1 μM Accell siRNA final concentration to differentiation medium of ALI cultures after 21 days of ALI culture. siRNA concentration was maintained throughout the differentiation protocol with fresh siRNA added with each refeed over the 3 weeks.
IF immunohistochemistry (IF IHC) of cryosections
For cryosections, fully matured HNEs were fixed in 4% paraformaldehyde in 1X PBS for 30 min, followed by three 5 min washes with 1X PBS and equilibration in 30% sucrose/1X PBS overnight. Tissue specimens were processed for OCT embedded, 10-μm cryosections. Cryosections were blocked with 5% normal donkey serum (017-000-121, Jackson ImmunoResearch) in IF buffer (3% BSA and 0.4% saponin in PBS; for all else: 3% BSA and 0.1% NP-40 in PBS) at room temperature for 1 h. HNEs were incubated with primary antibodies in the IF buffer for overnight at 4°C, followed by 3 washes with the IF buffer. Samples were then incubated with fluorescent-labeled secondary antibody at room temperature for 1.5 h, followed by a 5 min incubation with 4′,6-dia-midino-2-phenylindole (DAPI) in PBS at room temperature for 5 min and 3 washes with IF buffer. Coverslips were mounted with Fluoromount-G (0100-01, SouthernBiotech) onto glass slides followed by image acquisition.
IF immunohistochemistry (IF IHC) of FFPE sections
The tissue sections were cut to 4 μm thickness onto frosted glass slides at the Stanford University Histology Service Center and University Hospital Basel. Deparaffinization, rehydration, and heat-induced epitope retrieval (HIER) were performed on an ST4020 small linear stainer (Leica). For deparaffinization, slides were baked at 70°C for 1 h, followed by rehydration in descending concentrations of ethanol (100% twice, 95% twice, 80%, 70%, ddH2O twice; each step for 30 s). HIER was performed in a Lab VisionTM PT module (Thermo Fisher) using Dako Target Retrieval Solution, pH 9 (S236784-2, DAKO Agilent) at 97°C for 10 min and cooled down to 65°C. After further cooling to room temperature for 30 min, slides were washed for 10 min three times in Tris-buffered saline with 0.1% Tween 20 detergent (TBST). Sections were then blocked in 5% normal donkey serum (D9663, Sigma-Aldrich) in TBST at room temperature for 1 h, followed by incubation with primary antibodies in the blocking solution. After one overnight incubation of primary antibodies at 4°C, sections were washed for 10 min three times with TBST and stained with the appropriate secondary antibodies in PBS with 3% BSA, 0.4% saponin, and 0.02% sodium azide at room temperature for 1 h. Following this, sections were washed for 10 min three times each with TBST and mounted with Pro-Long Gold Antifade mounting medium with DAPI (Invitrogen).
Antibodies and reagents
Antibodies used include the following: rabbit anti-SARS-CoV-2-NP (GeneTex, GTX135361, 1:2,000), mouse anti-SARS-CoV-2-NP (Thermo Fisher, MA1-7403, 1:200), mouse anti-SARS-CoV-2-SP (GeneTex, GTX632604, 1:600), rabbit anti-ACE2 (Abcam, ab15348, 1:100), mouse anti-TMPRSS2 (Millipore, MABF2158, 1:50), rabbit anti-Phospho-PAK1/2 (Cell Signaling, 2601S, 1:200), rabbit anti-Phospho-PAK4 (Cell Signaling, 3241S, 1:200), rabbit anti-Phospho-p38 (Cell Signaling, 4511T, 1:200), rabbit anti-Phospho-AKT1 (Cell Signaling, 4060S, 1:100), mouse anti-EZR (Millipore, E8897, 1:600), rabbit anti-Phospho-EZR (Cell Signaling, 4060S, 1:100), rabbit anti-EBP50 (Thermo Fisher, PA1-090, 1:600), rabbit anti-SLK (Millipore, HPA015757, 1:100), rabbit anti-SLK (Millipore, HPA015757, 1:200), rabbit anti-BAIAP2 (Millipore, HPA027421, 1:200), rabbit anti-PTPN14 (Millipore, HPA053864, 1:200), rabbit anti-PTPN14 (Millipore, HPA053864, 1:200), rabbit anti- ARHGEF2 (Millipore, HPA043437, 1:200), rabbit anti-MYH14 (Millipore, HPA067889, 1:200), rabbit anti-PPP1R12A (Millipore, HPA071956, 1:200), rabbit anti-CDK16 (Millipore, HPA067889, 1:200), rabbit anti-ANKRD35 (Millipore, HPA035453, 1:200), rabbit anti-CEP164 (ProteinTech, 22227-1-AP, 1:300), mouse anti-RSV (Sigma-Aldrich, MAB858-1, 1:300), mouse anti-PIV (Millipore, MAB10207, 1:300), rabbit anti-CX3CR1 (Sigma-Aldrich, SAB2900202, 1:200), mouse anti-FOXJ1 (Thermo, 14-9965-82, 1:400), and mouse anti-MUC5AC (Abcam, ab212636, 1:600). SiR-Tubulin Kit (Cytoskeleton, CY-SC002), Qdot 585 Streptavidin (Thermo, Q10113MP), and RBD Biotin-Labeled (BPS Bioscience, 100937-2) were used in the pseudo virion QD585-RBD live images. ToxiLightTM Non-Destructive Cytotoxicity BioAssay Kit was purchased from Lonza.
Live-cell imaging for QD585-RBD
The transwell insert with HNEs was transferred to inner-wells of the glass-bottom 24-well dish (GREINER BIO-ONE, 662892) with 500 μL PneumaCult-ALI medium in the well. The apical surface of HNEs was washed three times with 100 ul calcium-free PBS before the treatment of QD585-RBD. 0.7 μg Biotin-tagged RBD (BPS Bioscience, 100937-2) was dissolved in 100 μL calcium-free PBS and incubated with 1 μL of QD (Thermo Scientific, Q10113MP, 1 μM) for 15 min. Before adding to the HNEs, 1 μL 50nM SiR-tubulin (Cytoskeleton Inc., CY-SC002) was added to the mixture. In the soluble ACE2 treatment group, the same procedure was performed with adding soluble ACE2 (18–615) (Boster, RCOV09) for 1 h before treatment. After the treatment of QD585-RBD, the HNEs were incubated for 4–6 h in a 37°C incubator with 5% CO2. Human Nasal Epithelial cells were imaged on a scanning disk confocal microscope (Zeiss observer Z1, Zeiss; Laser Stack, 3i; CSU-22 confocal scanner unit, Yokogawa) using a Zeiss plan-apochromat 63x 1.4 oil objective on a vibration-dampened table with a Prime 95B Scientific CMOS camera (Teledyne Photometrics). Images and videos are acquired at a resolution of 0.169 um per pixel in an area of 536 X 544 pixels with Slidebook 6 software (3i). Fluorescent videos are obtained with an exposure time of 100 ms for each channel with a frame rate of 1.1 Hz. Bright field videos are obtained with an exposure time of 10 ms with a frame rate of 64 Hz. All imaging is done in a 37°C, and 5% CO2 live supporting chamber that is mounted on the microscope stage.
Microscopy
Fluorescence-immunolabeled images were acquired using a Zeiss AxioImager Z1 microscope or a Marianas spinning disk confocal (SDC) microscopy (Intelligent Imaging Innovations). Post-imaging processing was performed using ZEN (Carl Zeiss). Final figures were organized using Adobe Illustrator.
Quantitative real-time PCR
RNA was extracted using the RNeasy Lipid Tissue Kit (QIAGEN) and cDNA was synthesized using M-MLV Reverse Transcriptase (Invitrogen, 28025-013). Quantitative real time PCR was performed using TaqMan Probes (Invitrogen) and the TaqMan Gene Expression Master Mix (Applied Biosystems, 4369016) in 96-well Micro Amp Optical reaction plates (Applied Biosystems, N8010560). Expression levels were normalized to the average expression of the housekeeping gene. Life Technologies Taqman probe ACE2 (Cat# Hs00222343_m1), TMPRSS2 (Hs00237175_m1), and CX3CR1 (Cat# Hs01922583_s1).
Transmission and scanning electron microscopy
HNEs were grown on transwell insert and fixed in 2% glutaraldehyde (EMS Cat# 16000) and 4% pFormaldehyde (EMS Cat# 15700) in 0.1 M Sodium Cacodylate (EMS Cat# 12300) pH 7.4 in cold for 1 h. For transmission electron microscopy, the fixed cells were replaced with cold/aqueous 1% Osmium tetroxide (EMS Cat# 19100) and were then allowed to warm to room temperature for 2 h in a hood, washed 3 times with ultrafiltered water, then en bloc stained in 1% Uranyl Acetate at room temperature 2 h. Samples were then dehydrated in a series of ethanol. Sample were placed in cold 95% EtOH and allowed to warm to room temperature, changed to 100% 2X, then Propylene Oxide (PO) for 15 min. After 5 min the filter was removed from the transwell frame and placed into 2 mL Eppendorf tubes. Samples are infiltrated with EMbed-812 resin (EMS Cat#14120) mixed 1:2, 1:1, and 2:1 with PO for 2 h each with leaving samples in 2:1 resin to PO overnight rotating at room temperature in the hood. The samples are then placed into EMbed-812 for 2 to 4 h then placed into molds w/labels and fresh resin, cut then orientated face to face and placed into a 65°C oven overnight. Sections were taken around 80 nm using an UC7 (Leica, Wetzlar, Germany) picked up on formvar/Carbon coated 100 mesh Cu grids, stained for 40 s in 3.5% Uranyl Acetate in 50% Acetone followed by staining in Sato’s Lead Citrate for 2 min. Observed in the JEOL JEM-1400 120 kV. Images were taken using a Gatan OneView 4k X 4k digital camera. For scanning electron microscopy analysis, the fixed cells were replaced with cold/aqueous 1% Osmium tetroxide (EMS Cat# 19100) and were then allowed to warm to room temperature for 2 h rotating in a hood, washed 3 times with ultrafiltered water. Samples were then dehydrated in a series of ethanol. Then dried in a Critical Point Dryer (Tousimis 815, Series A), Sputter coated (Leica EM ACE600) with gold. Observed in Zeiss Sigma FESEM. Images were taken using SmartSEM VO5.03.
Phosphopeptide shotgun proteomics
HNEs were infected with SARS-CoV-2 for 6, 24, 36, 48 h or with MOCK. Cells were harvested, lysed, reduce, and alkylated using 100 μL of lysis buffer (6M Guanidine Hydrochloride, 100 mM Tris-HCl pH 8.0, 10 mM TCEP, 10 mM CAA) and boiled 60°C for 1 h. Proteins were precipitated by adding 100 μL methanol, vortexed, 50 μL chloroform, vortexed, 100 μL water, vortexed, and centrifuged at 13,000 g for 2 min. The top aqueous layer was removed, 200 μL of methanol was added, vortexed, and centrifuged at 13,000 g for 3 min. Methanol was removed and dried proteins were resuspended using 200 mM HEPES pH 8.5. Proteins were digested using Trypsin/Lys-C overnight at 37°C 250 RPM. Sample was acidified using TFA and cleaned using stage tips. Stage tips were created using 5 layers of C18 filters packed into a P200 tip. The stage tips were activated using methanol, equilibrated twice with an equilibration buffer (5% ACN, 0.5% TFA). Samples were bound, washed twice with an equilibration buffer, and eluted using an elution buffer (50% ACN, 0.1% FA). Eluted samples were dried using a Speed-Vac and resuspended using Binding/Wash Buffer in High-Select Fe-NTA Phosphopeptide Enrichment Kit (Thermo Scientific, A32992). The samples were enriched for phosphopeptides according to the manufacturer’s instructions. Samples were eluted and resuspended using Solution A (2% ACN, 0.1% FA) and were analyzed using the timsTOF Pro (Bruker Daltonics) (Meier et al., 2018), an ion-mobility spectrometry quadrupole time of flight mass spectrometer. Specifically, a nanoElute (Bruker Daltonics) high pressure nanoflow system was connected to the timsTOF Pro. Peptides were delivered to a reversed phase analytical column (10 cm × 75 μm i.d., Bruker 1866154). Liquid chromatography was performed at 50°C and peptides were separated on the analytical column using a 48 min gradient (solvent A: 2% ACN, 0.1% FA; solvent B: 0.1% FA, in ACN) at a flow rate of 500 nL/min. A linear gradient from 2 to 35% B was applied for 45 min, followed by a step to 95% B for 1.5 and 3 min of washing at 95% B. The timsTOF Pro was operated in PASEF mode with the following settings: Mass Range 100 to 1700 m/z, 1/K0 Start 0.85 V s/cm2, End 1.3 V s/cm2, Ramp time 100 ms, Lock Duty Cycle to 100%, Capillary Voltage 1700, Dry Gas 3 L/min, Dry Temp 200°C, PASEF settings: 4 MS/MS, charge range 0–5, active exclusion for 0.04 min, Scheduling Target intensity 2,0000, Intensity threshold 500, CID collision energy 10 eV. For analysis, Bruker raw data files were processed using Byonic software (Protein Metrics, Inc) to identify peptides and proteins using the NCBI Homo sapiens refseq protein database. Data were searched with 20 ppm error tolerance for precursor and 40 ppm for fragment ions using QTOF/HCD fragmentation type. Besides standard variable modifications, we searched for S/T/Y phosphorylation and set 1% FDR for protein identifications.
Kinase set enrichment analysis (KSEA analysis)
Counts for phosphosites between two technical replicates were summed after the total counts per sample were normalized to the median of total counts. The counts were then log2-transformed, and quantile normalized. Batch correction was done using ComBat function from the sva package from Bioconductor, and the log2-transformation was undone to obtain the counts for differential expression analysis. Differential expression analysis was conducted with the msms.glm.pois function from the msmsTests package from Bioconductor (Josep Gregori, Alex Sanchez, and Josep Villanueva (2020). msmsTests: LC-MS/MS Differential Expression Tests. R package version 1.26.0.). KSEA was calculated with the ksea function from the ksea package from GitHub (David Ochoa (2020). ksea: Kinase Activity Prediction based in Quantitative Phosphoproteomic Data. R package version 0.1.2.) using a kinase substrate database created from PhosphositePlus51 and NetworKin.52 p values were adjusted using Benjamini-Hochberg procedure. GO ORA was conducted using the enrichGO function from the clusterProfiler package from Bioconductor85 and the database org.Hs.eg.db package from Bioconductor (Marc Carlson (2020). org.Hs.eg.db: Genome wide annotation for Human. R package version 3.11.4.).
Quantification and statistical analysis
For signal quantification, samples were stained simultaneously in batch with the primary antibodies (ex. pEZR, EBP50, phalloidin as described above) using the same master mixes and identical incubation times under similar staining conditions described above. Exposure times under confocal microscopy were identical for the quantified samples. Quantification was performed using a custom script developed in the FIJI package of ImageJ as previously described.20 Experimental sample sizes were not predetermined given the exploratory nature of the work and the limited availability of tissue specimens. Mann-Whitney U test was used when the data were not normally distributed by Shapiro Wilk normality test and were not at equal variance by F-test. When the normal distribution and equal variance were confirmed, Student’s t test were used. Analyses were performed with GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA) software and IBM SPSS Statistics version 23 (IBM, Armonk, NY).
Acknowledgments
We thank John Perrino and Ruth Yamawakithe (Stanford Cell Sciences Imaging Facility) for electron microscope sample preparation and image processing. This work was supported by the National Institutes of Health R01DK127665, R01HD085901, R01GM121565, and P30DK116074 to P.K.J.; R01AI149672-01 and U54-CA209971 to G.P.N.; Stanford Diabetes Research Center Pilot and Feasibility Research Grant to P.K.J; Fast Grant Funding for COVID-19 Science to P.K.J. and G.P.N.; NIH (R01 AI36178, AI40085, P01 AI091575), the Bill and Melinda Gates Foundation, and the DARPA Intercept program (Contract No. HR0011-17-2-0027) to R.A.; the Botnar Research Centre for Child Health Emergency Response to COVID-19 Grant and a Bill and Melinda Gates Foundation COVID-19 Pilot Award to S.J. and G.P.N.; the Rachford & Carlotta A. Harris Endowed Chair to G.P.N.; California Institute for Regenerative Medicine (DISC2-09637), Defense Advanced Research Projects Agency (HR001118S0037-PREPARE-FP-001), and The Operndorf Foundation to J.V.N.; Stanford Respond. Innovate. Scale. Empower (RISE) COVID-19 Crisis Response Trainee Seed Grant to C.-T.W., R.C., I.T.L., S.J., and T.N.; Stanford Translational Research and Applied Medicine (TRAM) Pilot Grant and Thrasher Research Fund Early Career Award to I.T.L.; Stanford Maternal and Child Health Research Institute (MCHRI) Clinical (MD) Trainee Support Award to I.T.L., Ernest and Amelia Gallo Endowed Postdoctoral Fellow; Leukemia & Lymphoma Society Career Development Program to S.J.; Cellular and Molecular Biology Training Grant (NIH 5 T32 GM007276) to R.C.; the Mossier Laboratories Research Fund to C.M.; and the ARRA Award Number 1S10RR026780-01 from the National Center for Research Resources.
Author contributions
Concept and study coordination, C.-T.W. and P.K.J. Experimental design/execution, C.-T.W., P.L., Y.X., R.C., I.T.L., T.N., S.J., W.H., R.A., P.K.J., R.E.T., and M.G.K. Microscopy, C.-T.W., IHC, I.T.L., and T.N. Virus production/human nasal epithelial cell infection, C.-T.W., P.L., Y.X., M.G.K., K.C., J.S., and L.M.S. StcE enzyme purification, C.Y. and C.R.B. Patient consent, sample collection/testing, and banking, I.T.L., S.J., T.N., C.M., and J.V.N. Live-cell imaging for QD-RBD, W.H. and C.-T.W. Phosphoproteomics/data analysis, C.-T.W., R.C., and J.D. Statistical analysis, C.-T.W., T.N., and J.D. Human nasal cells, J.V.N., C.M., T.N., and L.S.R.H. Figures, C.-T.W. with help from I.T.L., S.J., and G.P.N. Funding/scientific guidance, R.A. and P.K.J. All authors reviewed the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: December 2, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2022.11.030.
Supplemental information
Data and code availability
The analysis code used to support the findings of this study are available at https://doi.org/10.5281/zenodo.7343831.
References
- 1.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hou Y.J., Okuda K., Edwards C.E., Martinez D.R., Asakura T., Dinnon K.H., 3rd, Kato T., Lee R.E., Yount B.L., Mascenik T.M., et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell. 2020;182:429–446.e14. doi: 10.1016/j.cell.2020.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gallo O., Locatello L.G., Mazzoni A., Novelli L., Annunziato F. The central role of the nasal microenvironment in the transmission, modulation, and clinical progression of SARS-CoV-2 infection. Mucosal Immunol. 2021;14:305–316. doi: 10.1038/s41385-020-00359-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuek L.E., Lee R.J. First contact: the role of respiratory cilia in host-pathogen interactions in the airways. Am. J. Physiol. Lung Cell Mol. Physiol. 2020;319:L603–L619. doi: 10.1152/ajplung.00283.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knowles M.R., Boucher R.C. Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Invest. 2002;109:571–577. doi: 10.1172/JCI15217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Button B., Cai L.H., Ehre C., Kesimer M., Hill D.B., Sheehan J.K., Boucher R.C., Rubinstein M. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science. 2012;337:937–941. doi: 10.1126/science.1223012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kesimer M., Ehre C., Burns K.A., Davis C.W., Sheehan J.K., Pickles R.J. Molecular organization of the mucins and glycocalyx underlying mucus transport over mucosal surfaces of the airways. Mucosal Immunol. 2013;6:379–392. doi: 10.1038/mi.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iverson E., Griswold K., Song D., Gagliardi T.B., Hamidzadeh K., Kesimer M., Sinha S., Perry M., Duncan G.A., Scull M.A. Membrane-Tethered Mucin 1 is stimulated by interferon in multiple cell types and antagonizes influenza A virus infection in human airway epithelium. bioRxiv. 2021 doi: 10.1101/2021.03.11.434997. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McAuley J.L., Corcilius L., Tan H.X., Payne R.J., McGuckin M.A., Brown L.E. The cell surface mucin MUC1 limits the severity of influenza A virus infection. Mucosal Immunol. 2017;10:1581–1593. doi: 10.1038/mi.2017.16. [DOI] [PubMed] [Google Scholar]
- 10.Zou L., Ruan F., Huang M., Liang L., Huang H., Hong Z., Yu J., Kang M., Song Y., Xia J., et al. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N. Engl. J. Med. 2020;382:1177–1179. doi: 10.1056/NEJMc2001737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahn J.H., Kim J., Hong S.P., Choi S.Y., Yang M.J., Ju Y.S., Kim Y.T., Kim H.M., Rahman M.D.T., Chung M.K., et al. Nasal ciliated cells are primary targets for SARS-CoV-2 replication in early stage of COVID-19. J. Clin. Invest. 2021;131:e148517. doi: 10.1172/JCI148517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ravindra N.G., Alfajaro M.M., Gasque V., Huston N.C., Wan H., Szigeti-Buck K., Yasumoto Y., Greaney A.M., Habet V., Chow R.D., et al. Single-cell longitudinal analysis of SARS-CoV-2 infection in human airway epithelium identifies target cells, alterations in gene expression, and cell state changes. PLoS Biol. 2021;19:e3001143. doi: 10.1371/journal.pbio.3001143. ARTN e3001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fiege J.K., Thiede J.M., Nanda H.A., Matchett W.E., Moore P.J., Montanari N.R., Thielen B.K., Daniel J., Stanley E., Hunter R.C., et al. Single cell resolution of SARS-CoV-2 tropism, antiviral responses, and susceptibility to therapies in primary human airway epithelium. PLoS Pathog. 2021;17:e1009292. doi: 10.1371/journal.ppat.1009292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia H.P., Look D.C., Shi L., Hickey M., Pewe L., Netland J., Farzan M., Wohlford-Lenane C., Perlman S., McCray P.B., Jr. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005;79:14614–14621. doi: 10.1128/JVI.79.23.14614-14621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thompson C.I., Barclay W.S., Zambon M.C., Pickles R.J. Infection of human airway epithelium by human and avian strains of influenza a virus. J. Virol. 2006;80:8060–8068. doi: 10.1128/JVI.00384-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L., Bukreyev A., Thompson C.I., Watson B., Peeples M.E., Collins P.L., Pickles R.J. Infection of ciliated cells by human parainfluenza virus type 3 in an in vitro model of human airway epithelium. J. Virol. 2005;79:1113–1124. doi: 10.1128/JVI.79.2.1113-1124.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan K.S., Ong H.H., Yan Y., Liu J., Li C., Ong Y.K., Thong K.T., Choi H.W., Wang D.Y., Chow V.T. In vitro model of fully differentiated human nasal epithelial cells infected with rhinovirus reveals epithelium-initiated immune responses. J. Infect. Dis. 2018;217:906–915. doi: 10.1093/infdis/jix640. [DOI] [PubMed] [Google Scholar]
- 18.Griggs T.F., Bochkov Y.A., Basnet S., Pasic T.R., Brockman-Schneider R.A., Palmenberg A.C., Gern J.E. Rhinovirus C targets ciliated airway epithelial cells. Respir. Res. 2017;18:84. doi: 10.1186/s12931-017-0567-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L., Peeples M.E., Boucher R.C., Collins P.L., Pickles R.J. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J. Virol. 2002;76:5654–5666. doi: 10.1128/jvi.76.11.5654-5666.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee I.T., Nakayama T., Wu C.T., Goltsev Y., Jiang S., Gall P.A., Liao C.K., Shih L.C., Schürch C.M., McIlwain D.R., et al. ACE2 localizes to the respiratory cilia and is not increased by ACE inhibitors or ARBs. Nat. Commun. 2020;11:5453. doi: 10.1038/s41467-020-19145-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakayama T., Lee I.T., Jiang S., Matter M.S., Yan C.H., Overdevest J.B., Wu C.T., Goltsev Y., Shih L.C., Liao C.K., et al. Landscape of SARS-CoV-2 infection in diverse human head & neck and proximal airway mucosal tissues. Cell Rep. Med. 2021;2:100421. doi: 10.1016/j.xcrm.2021.100421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montoro D.T., Haber A.L., Biton M., Vinarsky V., Lin B., Birket S.E., Yuan F., Chen S., Leung H.M., Villoria J., et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature. 2018;560:319–324. doi: 10.1038/s41586-018-0393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitcutt M.J., Adler K.B., Wu R. A biphasic chamber system for maintaining polarity of differentiation of cultured respiratory tract epithelial cells. In Vitro Cell. Dev. Biol. 1988;24:420–428. doi: 10.1007/BF02628493. [DOI] [PubMed] [Google Scholar]
- 24.Wheeler A.H., Nungester W.J. Effect of Mucin on Influenza Virus Infection in Hamsters. Science. 1942;96:92–93. doi: 10.1126/science.96.2482.92. [DOI] [PubMed] [Google Scholar]
- 25.Malaker S.A., Pedram K., Ferracane M.J., Bensing B.A., Krishnan V., Pett C., Yu J., Woods E.C., Kramer J.R., Westerlind U., et al. The mucin-selective protease StcE enables molecular and functional analysis of human cancer-associated mucins. Proc. Natl. Acad. Sci. USA. 2019;116:7278–7287. doi: 10.1073/pnas.1813020116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen R.E., Winkler E.S., Case J.B., Aziati I.D., Bricker T.L., Joshi A., Darling T.L., Ying B., Errico J.M., Shrihari S., et al. In vivo monoclonal antibody efficacy against SARS-CoV-2 variant strains. Nature. 2021;596:103–108. doi: 10.1038/s41586-021-03720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gorshkov K., Susumu K., Chen J., Xu M., Pradhan M., Zhu W., Hu X., Breger J.C., Wolak M., Oh E. Quantum dot-conjugated SARS-CoV-2 spike pseudo-virions enable tracking of angiotensin converting enzyme 2 binding and endocytosis. ACS Nano. 2020;14:12234–12247. doi: 10.1021/acsnano.0c05975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vladar E.K., Nayak J.V., Milla C.E., Axelrod J.D. Airway epithelial homeostasis and planar cell polarity signaling depend on multiciliated cell differentiation. JCI Insight. 2016;1:e88027. doi: 10.1172/jci.insight.88027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson C.S., Chu C.Y., Wang Q., Mereness J.A., Ren Y., Donlon K., Bhattacharya S., Misra R.S., Walsh E.E., Pryhuber G.S., Mariani T.J. CX3CR1 as a respiratory syncytial virus receptor in pediatric human lung. Pediatr. Res. 2020;87:862–867. doi: 10.1038/s41390-019-0677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Punyadarsaniya D., Liang C.H., Winter C., Petersen H., Rautenschlein S., Hennig-Pauka I., Schwegmann-Wessels C., Wu C.Y., Wong C.H., Herrler G. Infection of differentiated porcine airway epithelial cells by influenza virus: differential susceptibility to infection by porcine and avian viruses. PLoS One. 2011;6:e28429. doi: 10.1371/journal.pone.0028429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicholls J.M., Bourne A.J., Chen H., Guan Y., Peiris J.S.M. Sialic acid receptor detection in the human respiratory tract: evidence for widespread distribution of potential binding sites for human and avian influenza viruses. Respir. Res. 2007;8:73. doi: 10.1186/1465-9921-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeong K.I., Piepenhagen P.A., Kishko M., DiNapoli J.M., Groppo R.P., Zhang L., Almond J., Kleanthous H., Delagrave S., Parrington M. CX3CR1 is expressed in differentiated human ciliated airway cells and co-localizes with respiratory syncytial virus on cilia in a G protein-dependent manner. PLoS One. 2015;10:e0130517. doi: 10.1371/journal.pone.0130517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson S.M., McNally B.A., Ioannidis I., Flano E., Teng M.N., Oomens A.G., Walsh E.E., Peeples M.E. Respiratory syncytial virus uses CX3CR1 as a receptor on primary human airway epithelial cultures. PLoS Pathog. 2015;11:e1005318. doi: 10.1371/journal.ppat.1005318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakayama K., Katoh Y. Ciliary protein trafficking mediated by IFT and BBSome complexes with the aid of kinesin-2 and dynein-2 motors. J. Biochem. 2018;163:155–164. doi: 10.1093/jb/mvx087. [DOI] [PubMed] [Google Scholar]
- 35.Firestone A.J., Weinger J.S., Maldonado M., Barlan K., Langston L.D., O'Donnell M., Gelfand V.I., Kapoor T.M., Chen J.K. Small-molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein. Nature. 2012;484:125–129. doi: 10.1038/nature10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Savarino A., Boelaert J.R., Cassone A., Majori G., Cauda R. Effects of chloroquine on viral infections: an old drug against today's diseases? Lancet Infect. Dis. 2003;3:722–727. doi: 10.1016/s1473-3099(03)00806-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bouhaddou M., Memon D., Meyer B., White K.M., Rezelj V.V., Correa Marrero M., Polacco B.J., Melnyk J.E., Ulferts S., Kaake R.M., et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell. 2020;182:685–712.e19. doi: 10.1016/j.cell.2020.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caldas L.A., Carneiro F.A., Higa L.M., Monteiro F.L., da Silva G.P., da Costa L.J., Durigon E.L., Tanuri A., de Souza W. Ultrastructural analysis of SARS-CoV-2 interactions with the host cell via high resolution scanning electron microscopy. Sci. Rep. 2020;10:16099. doi: 10.1038/s41598-020-73162-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pelaseyed T., Bretscher A. Regulation of actin-based apical structures on epithelial cells. J. Cell Sci. 2018;131:jcs221853. doi: 10.1242/jcs.221853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crawley S.W., Mooseker M.S., Tyska M.J. Shaping the intestinal brush border. J. Cell Biol. 2014;207:441–451. doi: 10.1083/jcb.201407015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delacour D., Salomon J., Robine S., Louvard D. Plasticity of the brush border - the yin and yang of intestinal homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2016;13:161–174. doi: 10.1038/nrgastro.2016.5. [DOI] [PubMed] [Google Scholar]
- 42.Gungor-Ordueri N.E., Celik-Ozenci C., Cheng C.Y. Ezrin: a regulator of actin microfilaments in cell junctions of the rat testis. Asian J. Androl. 2015;17:653–658. doi: 10.4103/1008-682X.146103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Solomon G.M., Francis R., Chu K.K., Birket S.E., Gabriel G., Trombley J.E., Lemke K.L., Klena N., Turner B., Tearney G.J., et al. Assessment of ciliary phenotype in primary ciliary dyskinesia by micro-optical coherence tomography. JCI Insight. 2017;2:e91702. doi: 10.1172/jci.insight.91702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McShane D., Davies J.C., Wodehouse T., Bush A., Geddes D., Alton E.W.F.W. Normal nasal mucociliary clearance in CF children: evidence against a CFTR-related defect. Eur. Respir. J. 2004;24:95–100. doi: 10.1183/09031936.04.00097503. [DOI] [PubMed] [Google Scholar]
- 45.Bustamante-Marin X.M., Ostrowski L.E. Cilia and mucociliary clearance. Cold Spring Harb. Perspect. Biol. 2017;9:a028241. doi: 10.1101/cshperspect.a028241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stevens E.M., Vladar E.K., Alanin M.C., Christensen S.T., von Buchwald C., Milla C. Ciliary localization of the intraflagellar transport protein IFT88 is disrupted in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2020;62:120–123. doi: 10.1165/rcmb.2018-0287LE. [DOI] [PubMed] [Google Scholar]
- 47.Pedersen E.S.L., Goutaki M., Harris A.L., Dixon L., Manion M., Rindlisbacher B., Patient Advisory Group C.P., Lucas J.S., Kuehni C.E. SARS-CoV-2 infections in people with PCD: neither frequent, nor particularly severe. Eur. Respir. J. 2021;58:2004548. doi: 10.1183/13993003.04548-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuehni C., Pedersen E., Schreck L. 2022. COVID-19 Infections in People with Primary Ciliary Dyskinesia. [Google Scholar]
- 49.Zhao Z.S., Manser E. PAK and other Rho-associated kinases--effectors with surprisingly diverse mechanisms of regulation. Biochem. J. 2005;386:201–214. doi: 10.1042/BJ20041638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hernandez-Armenta C., Ochoa D., Gonçalves E., Saez-Rodriguez J., Beltrao P. Benchmarking substrate-based kinase activity inference using phosphoproteomic data. Bioinformatics. 2017;33:1845–1851. doi: 10.1093/bioinformatics/btx082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Drake J.M., Graham N.A., Stoyanova T., Sedghi A., Goldstein A.S., Cai H., Smith D.A., Zhang H., Komisopoulou E., Huang J., et al. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc. Natl. Acad. Sci. USA. 2012;109:1643–1648. doi: 10.1073/pnas.1120985109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ochoa D., Jonikas M., Lawrence R.T., El Debs B., Selkrig J., Typas A., Villén J., Santos S.D., Beltrao P. An atlas of human kinase regulation. Mol. Syst. Biol. 2016;12:888. doi: 10.15252/msb.20167295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Linding R., Jensen L.J., Pasculescu A., Olhovsky M., Colwill K., Bork P., Yaffe M.B., Pawson T. NetworKIN: a resource for exploring cellular phosphorylation networks. Nucleic Acids Res. 2008;36:D695–D699. doi: 10.1093/nar/gkm902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bokoch G.M. Biology of the p21-activated kinases. Annu. Rev. Biochem. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- 57.Cenni V., Sirri A., Riccio M., Lattanzi G., Santi S., de Pol A., Maraldi N.M., Marmiroli S. Targeting of the Akt/PKB kinase to the actin skeleton. Cell. Mol. Life Sci. 2003;60:2710–2720. doi: 10.1007/s00018-003-3349-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu L., Zhang L., Zhao S., Zhao X.Y., Min P.X., Ma Y.D., Wang Y.Y., Chen Y., Tang S.J., Zhang Y.J., et al. Non-canonical Notch Signaling Regulates Actin Remodeling in Cell Migration by Activating PI3K/AKT/Cdc42 Pathway. Front. Pharmacol. 2019;10:370. doi: 10.3389/fphar.2019.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guay J., Lambert H., Gingras-Breton G., Lavoie J.N., Huot J., Landry J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J. Cell Sci. 1997;110:357–368. doi: 10.1242/jcs.110.3.357. [DOI] [PubMed] [Google Scholar]
- 60.Amano M., Nakayama M., Kaibuchi K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 2010;67:545–554. doi: 10.1002/cm.20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abo A., Qu J., Cammarano M.S., Dan C., Fritsch A., Baud V., Belisle B., Minden A. PAK4, a novel effector for Cdc42Hs, is implicated in the reorganization of the actin cytoskeleton and in the formation of filopodia. EMBO J. 1998;17:6527–6540. doi: 10.1093/emboj/17.22.6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dharmawardhane S., Sanders L.C., Martin S.S., Daniels R.H., Bokoch G.M. Localization of p21-activated kinase 1 (PAK1) to pinocytic vesicles and cortical actin structures in stimulated cells. J. Cell Biol. 1997;138:1265–1278. doi: 10.1083/jcb.138.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harms F.L., Kloth K., Bley A., Denecke J., Santer R., Lessel D., Hempel M., Kutsche K. Activating mutations in PAK1, encoding p21-activated kinase 1, cause a neurodevelopmental disorder. Am. J. Hum. Genet. 2018;103:579–591. doi: 10.1016/j.ajhg.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang J., Wang J., Guo Q., Wang Y., Zhou Y., Peng H., Cheng M., Zhao D., Li F. LCH-7749944, a novel and potent p21-activated kinase 4 inhibitor, suppresses proliferation and invasion in human gastric cancer cells. Cancer Lett. 2012;317:24–32. doi: 10.1016/j.canlet.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 65.Callow M.G., Zozulya S., Gishizky M.L., Jallal B., Smeal T. PAK4 mediates morphological changes through the regulation of GEF-H1. J. Cell Sci. 2005;118:1861–1872. doi: 10.1242/jcs.02313. [DOI] [PubMed] [Google Scholar]
- 66.Robens J.M., Yeow-Fong L., Ng E., Hall C., Manser E. Regulation of IRSp53-dependent filopodial dynamics by antagonism between 14-3-3 binding and SH3-mediated localization. Mol. Cell Biol. 2010;30:829–844. doi: 10.1128/MCB.01574-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Playford M.P., Nurminen E., Pentikäinen O.T., Milgram S.L., Hartwig J.H., Stossel T.P., Nakamura F. Cystic fibrosis transmembrane conductance regulator interacts with multiple immunoglobulin domains of filamin A. J. Biol. Chem. 2010;285:17156–17165. doi: 10.1074/jbc.M109.080523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zanivan S., Meves A., Behrendt K., Schoof E.M., Neilson L.J., Cox J., Tang H.R., Kalna G., van Ree J.H., van Deursen J.M., et al. In vivo SILAC-based proteomics reveals phosphoproteome changes during mouse skin carcinogenesis. Cell Rep. 2013;3:552–566. doi: 10.1016/j.celrep.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 69.Lee R.S., Zhang L., Berger A., Lawrence M.G., Song J., Niranjan B., Davies R.G., Lister N.L., Sandhu S.K., Rubin M.A., et al. Characterization of the ERG-regulated Kinome in Prostate Cancer Identifies TNIK as a Potential Therapeutic Target. Neoplasia. 2019;21:389–400. doi: 10.1016/j.neo.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Otterpohl K.L., Hart R.G., Evans C., Surendran K., Chandrasekar I. Nonmuscle myosin 2 proteins encoded by Myh9, Myh10, and Myh14 are uniquely distributed in the tubular segments of murine kidney. Physiol. Rep. 2017;5:e13513. doi: 10.14814/phy2.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chinowsky C.R., Pinette J.A., Meenderink L.M., Lau K.S., Tyska M.J. Nonmuscle myosin-2 contractility-dependent actin turnover limits the length of epithelial microvilli. Mol. Biol. Cell. 2020;31:2803–2815. doi: 10.1091/mbc.E20-09-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Belle L., Ali N., Lonic A., Li X., Paltridge J.L., Roslan S., Herrmann D., Conway J.R.W., Gehling F.K., Bert A.G., et al. The tyrosine phosphatase PTPN14 (Pez) inhibits metastasis by altering protein trafficking. Sci. Signal. 2015;8:ra18. doi: 10.1126/scisignal.2005547. [DOI] [PubMed] [Google Scholar]
- 73.Zha J.M., Li H.S., Wang Y.T., Lin Q., Tao M., He W.Q. Characterization of isoform expression and subcellular distribution of MYPT1 in intestinal epithelial cells. Gene. 2016;588:1–6. doi: 10.1016/j.gene.2016.04.048. [DOI] [PubMed] [Google Scholar]
- 74.Iwano S., Satou A., Matsumura S., Sugiyama N., Ishihama Y., Toyoshima F. PCTK1 regulates integrin-dependent spindle orientation via protein kinase A regulatory subunit KAP0 and myosin X. Mol. Cell Biol. 2015;35:1197–1208. doi: 10.1128/MCB.01017-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gabel M., Delavoie F., Demais V., Royer C., Bailly Y., Vitale N., Bader M.F., Chasserot-Golaz S. Annexin A2-dependent actin bundling promotes secretory granule docking to the plasma membrane and exocytosis. J. Cell Biol. 2015;210:785–800. doi: 10.1083/jcb.201412030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grieve A.G., Moss S.E., Hayes M.J. Annexin A2 at the interface of actin and membrane dynamics: a focus on its roles in endocytosis and cell polarization. Int. J. Cell Biol. 2012;2012:852430. doi: 10.1155/2012/852430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Debily M.A., Camarca A., Ciullo M., Mayer C., El Marhomy S., Ba I., Jalil A., Anzisi A., Guardiola J., Piatier-Tonneau D. Expression and molecular characterization of alternative transcripts of the ARHGEF5/TIM oncogene specific for human breast cancer. Hum. Mol. Genet. 2004;13:323–334. doi: 10.1093/hmg/ddh024. [DOI] [PubMed] [Google Scholar]
- 78.Yoshida S., Fukutomi T., Kimura T., Sakurai H., Hatano R., Yamamoto H., Mukaisho K.I., Hattori T., Sugihara H., Asano S. Comprehensive proteome analysis of brush border membrane fraction of ileum of ezrin knockdown mice. Biomed. Res. 2016;37:127–139. doi: 10.2220/biomedres.37.127. [DOI] [PubMed] [Google Scholar]
- 79.Khan M., Yoo S.J., Clijsters M., Backaert W., Vanstapel A., Speleman K., Lietaer C., Choi S., Hether T.D., Marcelis L., et al. Visualizing in deceased COVID-19 patients how SARS-CoV-2 attacks the respiratory and olfactory mucosae but spares the olfactory bulb. Cell. 2021;184:5932–5949.e15. doi: 10.1016/j.cell.2021.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ozono S., Zhang Y., Ode H., Sano K., Tan T.S., Imai K., Miyoshi K., Kishigami S., Ueno T., Iwatani Y., et al. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat. Commun. 2021;12:848. doi: 10.1038/s41467-021-21118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Augusto G., Mohsen M.O., Zinkhan S., Liu X., Vogel M., Bachmann M.F. In vitro data suggest that Indian delta variant B.1.617 of SARS-CoV-2 escapes neutralization by both receptor affinity and immune evasion. Allergy. 2022;77:111–117. doi: 10.1111/all.15065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garcia-Knight M., Anglin K., Tassetto M., Lu S., Zhang A., Goldberg S.A., Catching A., Davidson M.C., Shak J.R., Romero M., et al. Infectious viral shedding of SARS-CoV-2 Delta following vaccination: a longitudinal cohort study. medRxiv. 2022 doi: 10.1101/2022.05.15.22275051. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yin W., Xu Y., Xu P., Cao X., Wu C., Gu C., He X., Wang X., Huang S., Yuan Q., et al. Structures of the Omicron spike trimer with ACE2 and an anti-Omicron antibody. Science. 2022;375:1048–1053. doi: 10.1126/science.abn8863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dubacheva G.V., Curk T., Frenkel D., Richter R.P. Multivalent recognition at fluid surfaces: the interplay of receptor clustering and superselectivity. J. Am. Chem. Soc. 2019;141:2577–2588. doi: 10.1021/jacs.8b12553. [DOI] [PubMed] [Google Scholar]
- 85.Marbach S., Zheng J.A., Holmes-Cerfon M. The nanocaterpillar's random walk: diffusion with ligand-receptor contacts. Soft Matter. 2022;18:3130–3146. doi: 10.1039/d1sm01544c. [DOI] [PubMed] [Google Scholar]
- 86.Bayati A., Kumar R., Francis V., McPherson P.S. SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 2021;296:100306. doi: 10.1016/j.jbc.2021.100306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goh W.I., Lim K.B., Sudhaharan T., Sem K.P., Bu W., Chou A.M., Ahmed S. mDia1 and WAVE2 proteins interact directly with IRSp53 in filopodia and are involved in filopodium formation. J. Biol. Chem. 2012;287:4702–4714. doi: 10.1074/jbc.M111.305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Disanza A., Bisi S., Winterhoff M., Milanesi F., Ushakov D.S., Kast D., Marighetti P., Romet-Lemonne G., Müller H.M., Nickel W., et al. CDC42 switches IRSp53 from inhibition of actin growth to elongation by clustering of VASP. EMBO J. 2013;32:2735–2750. doi: 10.1038/emboj.2013.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abe T., La T.M., Miyagaki Y., Oya E., Wei F.Y., Sumida K., Fujise K., Takeda T., Tomizawa K., Takei K., Yamada H. Phosphorylation of cortactin by cyclin-dependent kinase 5 modulates actin bundling by the dynamin 1-cortactin ring-like complex and formation of filopodia and lamellipodia in NG108-15 glioma-derived cells. Int. J. Oncol. 2019;54:550–558. doi: 10.3892/ijo.2018.4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Matsumura F., Hartshorne D.J. Myosin phosphatase target subunit: Many roles in cell function. Biochem. Biophys. Res. Commun. 2008;369:149–156. doi: 10.1016/j.bbrc.2007.12.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mukhopadhyay S., Wen X., Chih B., Nelson C.D., Lane W.S., Scales S.J., Jackson P.K. TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 2010;24:2180–2193. doi: 10.1101/gad.1966210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shah A.S., Farmen S.L., Moninger T.O., Businga T.R., Andrews M.P., Bugge K., Searby C.C., Nishimura D., Brogden K.A., Kline J.N., et al. Loss of Bardet-Biedl syndrome proteins alters the morphology and function of motile cilia in airway epithelia. Proc. Natl. Acad. Sci. USA. 2008;105:3380–3385. doi: 10.1073/pnas.0712327105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mirra V., Werner C., Santamaria F. Primary ciliary dyskinesia: an update on clinical aspects, genetics, diagnosis, and future treatment strategies. Front. Pediatr. 2017;5:135. doi: 10.3389/fped.2017.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Winkler E.S., Bailey A.L., Kafai N.M., Nair S., McCune B.T., Yu J., Fox J.M., Chen R.E., Earnest J.T., Keeler S.P., et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020;21:1327–1335. doi: 10.1038/s41590-020-0778-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vladar E.K., Brody S.L. Analysis of ciliogenesis in primary culture mouse tracheal epithelial cells. Methods Enzymol. 2013;525:285–309. doi: 10.1016/B978-0-12-397944-5.00014-6. [DOI] [PubMed] [Google Scholar]
- 96.Silva E., Betleja E., John E., Spear P., Moresco J.J., Zhang S., Yates J.R., 3rd, Mitchell B.J., Mahjoub M.R. Ccdc11 is a novel centriolar satellite protein essential for ciliogenesis and establishment of left-right asymmetry. Mol. Biol. Cell. 2016;27:48–63. doi: 10.1091/mbc.E15-07-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The analysis code used to support the findings of this study are available at https://doi.org/10.5281/zenodo.7343831.