

Abstract
Adoptive cell therapy (ACT) using tumor-specific tumor-infiltrating lymphocytes (TILs) has demonstrated success in patients where tumor-antigen specific TILs can be harvested from the tumor, expanded, and re-infused in combination with a preparatory regimen and IL-2. One major issue for non-immunogenic tumors has been that the isolated TILs lack tumor specificity and thus possess limited in vivo therapeutic function. An oncolytic virus (OV) mediates an immunogenic cell death for cancer cells, leading to elicitation and dramatic enhancement of tumor-specific TILs. We hypothesized that the tumor-specific TILs elicited and promoted by an OV would be a great source for ACT for solid cancer. In this study, we show that a local injection of oncolytic poxvirus in MC38 tumor with low immunogenicity in C57BL/6 mice, led to elicitation and accumulation of tumor-specific TILs in the tumor tissue. Our analyses indicated that IL-2-armed OV-elicited TILs contain lower quantities of exhausted PD-1hiTim-3+ CD8+ T cells and regulatory T cells. The isolated TILs from IL-2-expressing OV-treated tumor tissue retained high tumor-specificity after expansion ex vivo. These TILs resulted in significant tumor regression and improved survival after adoptive transfer in mice with established MC38 tumor. Our study showcases the feasibility of using an OV to induce tumor-reactive TILs that can be expanded for ACT.
Keywords: oncolytic virus, vaccinia virus, IL-2, antitumor adaptive immunity, tumor-specific T cells, expansion ex vivo, adoptive T cell therapy
INTRODUCTION
Immunotherapy for cancer has become a reality in the last few years, with the approval by the FDA of a therapeutic cancer vaccine for advanced prostate cancer (Provenge), an oncolytic virus T-VEC for advanced melanoma, two types of CAR T cells for leukemia, and multiple antibody-based drugs of immune checkpoint blockade for a variety of cancers. Immunotherapy is rapidly evolving and treating cancer by activating the patient’s immune system presents an attractive therapeutic strategy [1]. One promising strategy is adoptive cell therapy (ACT) using the patient’s own TILs – a form of personalized cancer immunotherapy. This approach consists of excising a tumor, then expanding the lymphocytes from the tumor ex vivo. The patient is then treated with a non-myeloablative preparative chemotherapy regimen followed by TIL infusion and systemic IL-2 [2]. This approach has been successful in patients with cutaneous melanoma where the tumor mutational load (TMB) is high and tumor reactive lymphocytes are abundant in the tumor. Objective response rates (ORR) were observed ranging from 49% to 72%, with long term durable and potentially curative complete response rates of up to 25% [3], ORR was 42% with an attenuated IL-2 dose in another study [4]. However, most human solid tumors are non-immunogenic and tumor-reactive TIL cannot be isolated or expanded for ACT. Recently, however, ACT of autologous TILs, isolated and separated for tumor-reactive TILs (which made up a small percentage of total TILs), was shown to induce objective tumor regression in patients with metastatic uveal melanoma, a low mutational burden, non-immunogenic tumor [5]. In this trial, there was a strong association between the anti-tumor reactivity of the infused TILs and objective clinical response, suggesting that mechanisms to improve recovery of tumor reactive TILs could lead to more effective therapy for other solid tumors.
There are at least two major hurdles in successful ACT for cancers other than melanoma. First, the source of TILs is a major issue as the majority of human solid cancers are considered poorly immunogenic and very few TILs exist in the tumor tissues [6]. Even if it is possible to isolate TILs in non-immunogenic cancers such as uveal melanoma, most TILs isolated were not tumor-reactive [5]. Second, other than TILs from melanoma patients, there have not been many successful studies of isolation and expansion of tumor-specific TILs. The standard operating procedures for isolation and expansion of tumor-specific TILs ex vivo from other types of solid cancers have not been established. This was the case even for relatively high immunogenic renal cell carcinomas until recently [7].
Local immunotherapy, including oncolytic virotherapy, may modulate the tumor immune contexture and convert tumors from immunologically “cold” to “hot” [8–10]. We hypothesized that cytokine armed oncolytic VV could create a local pro-inflammatory environment in otherwise poorly immunogenic tumors leading to tumor reactive T-cells that could be harvested for ACT of cancer. Oncolytic viruses (OVs) selectively infect and/or replicate in cancer cells in vivo leaving normal cells unharmed. The immunogenic cell death induced by VV exposes a natural repertoire of tumor-associated antigens in conjunction with danger signals: damage associated molecular pattern molecules (DAMPs) and virus-derived pathogen-associated molecular pattern molecules (PAMPs), and inflammatory cytokines to reverse the tumor induced immunosuppression and elicit anti-tumor cellular immunity [11, 12]. OVs have been shown in both pre-clinical studies [13–16], and clinical trials with T-VEC and Pexa-Vec [9, 17, 18] to induce potent adaptive antitumor immunity contributing to the overall efficacy of the therapy. To further improve the antitumor immunity, investigators have engineered various oncolytic VV to express tumor antigens, T-cell co-stimulatory molecules and inflammatory chemokines and cytokines, and they have been studied for their efficacy and safety in preclinical tumor models [19–22].
In the current study, we utilized relatively low immunogenic MC38 murine colon tumor, which displays low levels of inflammatory infiltrate and tumor-reactive TILs, as a model for study. We present for the first time that oncolytic VVs can induce local tumor-specific TILs, and these TILs can be harvested, expanded ex vivo, and utilized for ACT to achieve a therapeutic response in a tumor model with low-immunogenicity. We envision that this approach using OV to induce tumor reactive TILs in low- or non-immunogenic tumors could be translated to cancer patients, and such ex vivo expanded TILs could be used for ACT for solid tumors with the potential for long term cure.
MATERIALS AND METHODS
Mice and cell lines.
Female C57BL/6J (B6) mice, age 5–6 weeks old, were obtained from The Jackson Laboratory (Bar Harbor, ME) and housed in specific pathogen-free conditions in the University of Pittsburgh Animal Facility. All animal studies were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Murine colon cancer cell line B16 was originally obtained from American Type Culture Collection (Manassas, VA). Mouse colon cancer cell line MC38-luc was described previously [23]. All cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 1 penicillin/streptomycin (Invitrogen, Carlsbad, CA) in 37°C, 5% CO2 incubator. All cell lines were tested for mycoplasma once every three months or so, to ensure they were free of the contamination.
Viruses.
WR strain-derived recombinant oncolytic VVs, vvDD, vvDD-CCL5 and vvDD-CXC11, [24–26], vvDD-IL15-Rα [27] and vvDD-IL2 expressing a membrane-bound IL-2 [28], have been constructed and studied. These VVs were amplified in HeLa cells and then purified as previously described [23, 26]. Viral titers were determined by plaque assays in CV-1 cells. For intratumoral injection of the viruses, a dose of 1.0e8 pfu per mouse was used unless indicated otherwise.
Rodent Tumor models and virus treatments.
For subcutaneous (s. c.) tumor model, B6 mice were subcutaneously inoculated with 5.0 × 105 MC38 colon cancer cells. When the tumors reached the size ~ 5 × 5 mm, vvDD, vvDD-IL2, vvDD-CCL5, vvDD-IL15Rα, vvDD-CXC11, or PBS was intratumorally injected at 1.0e8 pfu/tumor. In some experiments human IL-2 has been injected intratumorally at 1.0 × 106 IU per tumor (Prometheus, San Diego, CA). The primary tumor size was measured using an electric caliper in two perpendicular diameters followed by measurement every other day. Ten days post virus treatment, tumor tissues or spleens were harvest and then single cell suspension was made for T cell separation and further analysis. For peritoneal (i.p.) tumor models, mice were intraperitoneally inoculated with 5.0 × 105 MC38-luc cancer cells and randomized according to tumor size based on live animal IVIS imaging 7 days post tumor cell injection using a Xenogen IVIS 200 Optical In Vivo Imaging System (Caliber Life Sciences, Hoptikon, MA).
Generation of tumor-reactive TILs for adoptive cell transfer.
B6 mice were s.c. inoculated with 5.0 × 105 MC38 cancer cells at the right hind frank. When the tumor area reached 5 × 5 mm, vvDD-IL2 (1.0e8 pfu per tumor) was intratumorally injected. 10 days later tumors were collected, cut into pieces and incubated at 37°C in digestion buffer (Miltenyi Biotec, San Diego, CA) before being mashed over a 100 μM tissue strainer. Lysis of red blood cells was performed using ACK Lysing buffer (Thermo Fisher Scientific, Waltham, MA). For Leukocyte separation, Percoll gradient centrifuge procedure adapted from the protocol by Liu et al. [29] was used. Leucocytes were collected at the interface between 40% and 80% discontinuous Percoll gradient followed by magnetic separation (CD90.2 beads, Miltenyi Biotec). T cells have been cultured in 24-well plates in a concentration with 1.0 × 106 cells per well in RPMI complete media with 30 IU/ml IL-2 (MiltenyiBiotec) and 5 ng/ml IL-7 (BioLegend, San Diego, CA) over 4 days. As control T cells, spleens from non-tumor bearing untreated mouse have been harvested and proceeded to single cell suspension followed by magnetic separation (CD90.2 beads). The control T cells have been cultured under the same conditions as the virus induced T cells. Prior to adoptive cell transfer, T cells have been analyzed for tumor specificity in a co-culture assay. T cells (2.0 × 104 per well, 96-well plate) were either left unstimulated (medium) or challenged with γ-irradiated MC38 tumor cells (2.0 × 104 per well) or irrelevant target cells as γ-irradiated B16 tumor cells (2.0 × 104 per well) or naïve splenocytes (2.0 × 104 per well) from non-tumor-bearing B6 mice in duplicate for 24 h. The plate setup has been used for IFN-γ ELISPOT assay or flow cytometry analysis as described below.
Tumor model, adoptive cell transfer and cytokine administration.
B6 mice were intraperitoneally inoculated with 5.0 × 105 MC38-luc cancer cells and divided into required groups according to tumor growth condition based on live animal IVIS imaging 7 days post tumor cell injection. Grouped mice received 5.0 × 106 TILs that were isolated from vvDD-IL2-treated tumors and then expanded ex vivo, naïve T cells from control mice or just PBS. All treated mice also received 5 Gy of sublethal irradiation according to clinical protocols prior to cell transfer and exogenous cytokine support of IL-2 (100,000 IU/mouse, i.p.) for 3 days post transfer every 12 h (Prometheus).
Flow cytometry.
Collected tumor tissues were minced and incubated in RPMI 1640 medium containing 2% FBS, 1mg/mL collagenase IV (Sigma: #C5138), 0.1mg hyaluronidase (Sigma: #H6254), and 200U DNase I (Sigma: #D5025) at 37°C for 1 hours to make single cells. In vitro virus-infected cells or single cells from tumor tissues were blocked with α-CD16/32 Ab (clone 93, eBioscience: #14-0161-85; 1:1000) and then cells were stained with 100 μL Zombie Aqua Fixable Viability Kit cell dye (BioLegend, San Diego, CA, USA) at a dilution of 1:1000 and left in room temperature for 15 min in the dark. Cells were stained in 100 μL total stain volume (50 μL BV stain buffer, 50 μl 2% FBS) with antibody at a dilution of 1:200 for 30 min on ice in the dark. The sources of antibodies are listed in Supplementary Table 1. The intracellular staining kit for Foxp3 and IFN-γ was purchased from BioLegend. Cells were then washed once with FACS buffer and fixed in 1% paraformaldehyde (EK Industries, Joliet, IL, USA) before being stored at 4°C overnight and acquired the next day on a BD LSR Fortessa II analyzer (BD Biosciences, San Jose, CA, USA). Data were analyzed using flowJo cytometer software.
The enzyme-linked immunospot (ELISpot) assay.
Collected tumor tissues were cut into pieces and incubated at 37°C in digestion buffer (Miltenyi Biotec, San Diego, CA) before being mashed over a 100 μM tissue strainer. Lysis of red blood cell was performed using ACK Lysing buffer (Thermo Fisher Scientific, Waltham, MA) and single cell suspension was proceeded by straining cell suspension over 40 μM filter. Isolation of CD8+ TILs was performed using negative α-mouse CD8 microbeads isolation protocol (Miltenyi Biotec, San Diego, CA). Ninety-six well plates (MAHAS4510, Millipore, Burlington, MA) were coated with anti-mouse IFN-γ mAb 15mg/ml (clone AN18, Mabtech Inc., Cincinnati, OH). T cells (2.0 × 104 per well) were either left unstimulated (medium) or challenged with γ-irradiated MC38 tumor cells (2.0 × 104 per well in 96-well plate) or irrelevant target cells as γ-irradiated B16 tumor cells (2.0 × 104 per well) or naïve splenocytes (2.0 × 104 per well) from non-tumor-bearing B6 mouse in duplicate for 24 h. After appropriate washes, biotylated secondary antibody (clone R4–6A2-biotin, Mabtech, Inc) was added and incubated at room temperature for 2 h. Spot development followed using Vectastain Elite ABC and AEC peroxidase substrate kit (Vector Laboratories, Inc. Burlingame, CA). Number of IFN-γ spots were analyzed by ImmunoSpot™ (Cellular Technology, Ltd., Shaker Heights, OH).
To determine MC38-reactive responses the average value of spots from control wells were substracted from the number of MC38 challenged wells.
Immunofluorescence staining.
Resected tumors were fixed for 2 h in 2% Paraformaldehyde and incubated in 30% sucrose overnight. Sections were cut (5 μm) and stained with combined primary antibodies CD3 Alexa 488 (100212, BioLegend), CD4 Alexa 594 (100446, BioLegend) and CD8 Alexa 647 (100727, BioLegend) and nuclei were labeled with Hoechst dye (bis benzimide, Sigma B-2283–1 mg/100 ml in dH20). Images were acquired digitally from 9 fields under each condition. Density of positive cells was evaluated by automated image analysis using Nikon Elements (Nikon Instruments Inc, Melville, NY). Percentage of CD3+ T cells, CD3+CD4+, CD3+CD8+ T cells per area has been calculated by number of cells positive for the antibody versus the total number of cells.
Long-term survival of mice.
The health and survival of treated mice was closely monitored. All mice bearing subcutaneous or peritoneal tumors were monitored via caliper measurements for changes in tumor size or abdominal girth. Mice were dead naturally due to the disease or sacrificed when their subcutaneous tumor size exceeded 20 mm in diameter or when abdominal girth exceeded 1.5× the original measurement.
Statistics.
For the majority of experiments, the numbers of mice per group was equal to, or bigger than 5. We have chosen these sample sizes to ensure adequate statistical power either by pilot studies or previous studies we had conducted. For tumor-bearing mice, right before the treatment, they were pooled and then randomly distributed into different groups. For most of the animal experiments, it is not blinded. In rare occasions, if the value of one individual is considered to be an outlier (the value of 2 times of s.d outside of the mean), we might have excluded the value from the presented data.
Statistical analyses were performed using unpaired Student’s t test for two group comparison. For multiple group comparison One-way ANOVA were used where p value is adjusted for multiple test by Dunnett method (GraphPad Prism version 5). Animal survival is presented using Kaplan-Meier survival curves and compared by using log rank test (GraphPad Prism version 5). Value of p < 0.05 is considered to be statistically significant, and all p values were two sided. In the figures, the standard symbols were used: * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001; and NS: not significant.
RESULTS
Oncolytic VVs elicit and attract high numbers of tumor-reactive TILs
We engineered several vvDD-based oncolytic VVs expressing different cytokines/chemokines (vvDD-CCL5, vvDD-CXC11, vvDD-IL15Rα, vvDD-IL2) to enhance the anti-tumor immune response and to attract T cells into the tumor tissue. We first explored a variety of time points after injection and found that 10 days was the optimal time to harvest tumor reactive T-cells from the tumor microenvironment (data not shown). We studied TILs in implanted subcutaneous MC38 tumors 10 days after intratumoral treatment with either PBS, vvDD, vvDD-CCL5, vvDD-CXC11, vvDD-IL15Rα or vvDD-IL2 (Fig. 1A). CD8+ TILs from these tumors were isolated and analyzed using an IFN-γ ELISpot assay against γ-irradiated MC38 tumor cells. From control tumors injected with PBS, there were no tumor-reactive T cells found in the tumor. In contrast, each of the virus-treated tumors demonstrated a significant increase in tumor-reactive CD8+ TILs over control (p < 0.05). vvDD-IL2-treated tumors had high frequencies of tumor-reactive CD8+ TILs compared to control (Fig. 1A). Also, vvDD-IL2 yielded one of the most promising therapeutic results in a separate study with MC38 colon tumor models [28]. Thus, we have picked vvDD-IL2 for the rest of our study here.
Figure 1. Oncolytic VVs elicit tumor-specific antitumor CD8+ T cell response in the tumor tissue.
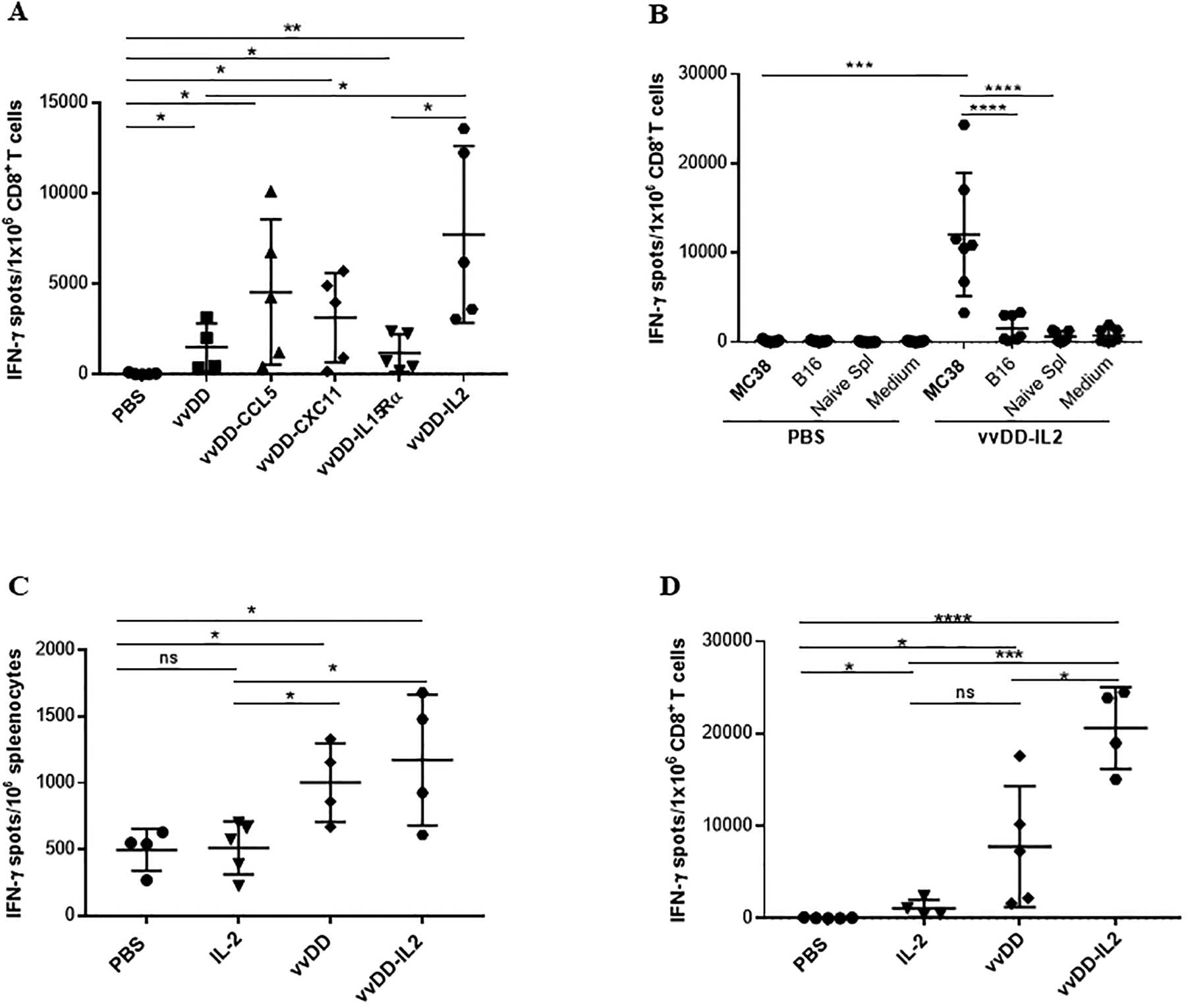
Ten days after viral treatments, subcutaneous MC38 tumors and/or splenocytes were collected and single cell suspensions were made followed by magnetic separation. Then isolated CD8+ T cells or splenocytes were tested by IFN-γ ELISPOT assay. (A). CD8+ T cells isolated from tumors (n = 4 – 5 mice/group) were either left unstimulated or challenged with γ-irradiated MC38 tumor cells for 24 h. Results were shown as individual data points (number of spots in each well) and bars (means ± standard deviation) of IFN-γ+ CD8+ T cells from each mouse evaluated in triplicate. Data from one experiment representing 2 independent experiments are shown. (B). Tumor-specificity of the OV-induced CD8+ TILs (n = 7 mice/group). Data are from one experiment representative of 3 independent experiments. For multiple group comparison, One-way ANOVA was used. ***p < 0.001; ****p < 0.0001. (C). MC38 tumor cell reactivity of splenocytes by IFN-γ ELISPOT assay. Splenocytes from treated mice were either left unstimulated or challenged with γ-irradiated MC38 tumor cells for 24 h. (D). MC38 tumor cell reactivity of isolated CD8+ TILs by IFN-γ ELISPOT assay. One experiment representative of 2 independent experiments is shown (n = 4–5 mice/group). Student’s t-test was used to analyze the statistical significance for data presented in panels A, C and D. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To investigate whether the OV-induced CD8+ TILs were specific for the injected tumor, the experiment was repeated with the murine tumor B16 and naïve splenocytes as additional control targets. Subcutaneous MC38 tumors were injected with vvDD-IL2 or PBS, and 10 days later the TILs were harvested. CD8+ T cells from treated tumors were again analyzed using an IFN-γ ELISPOT assay against γ-irradiated MC38 tumor cells, B16 tumor cells, or naïve splenocytes from a non-tumor-bearing B6 mouse (Fig. 1B). vvDD-IL2-treated tumors again demonstrated a significant increase in MC38 tumor reactive CD8+ T cells. The vvDD-IL2-induced CD8+ TILs were highly reactive and specific against MC38 tumor cells compared to the irrelevant target cells. In summary, vvDD-IL2 injection into MC38 tumors induced high levels of MC38 specific TILs.
We repeated the experiment again with vvDD and IL-2 protein injection as additional controls, using an effective dose based on a prior study [30], and examined both systemic and intratumoral T-cells for tumor reactivity. MC38 s.c. tumor-bearing mice were treated intratumorally with PBS, IL-2, vvDD or vvDD-IL2. Ten days later, both splenocytes and the CD8+ fraction of TILs were isolated and analyzed by IFN-γ ELISPOT assays. Both viruses induced greater frequency of tumor reactive T cells in the spleen compared to controls as shown by IFN-γ release (Fig. 1C). Further, each of the viruses induced a greater frequency of tumor-reactive TILs compared with PBS, and the vvDD-IL-2 was superior to vvDD or IL-2 alone in promoting recovery of antitumor CD8+ lymphocytes from the tumor (Fig. 1D).
While vvDD-IL2 in the above studies induced a higher percentage of tumor-reactive CD8+ TILs amongst recovered CD8+ cells, we also hypothesized that the absolute numbers of CD8+ T cells were increased after viral infection. We determined the quantity of CD4+ and CD8+ T cells in the tumor at day 10 after treatment, using two methods of analyses. First, tumors were analyzed by immunofluorescence staining for total CD3+ infiltrating cells, CD3+CD8+ and CD3+CD4+ T cells (Fig. 2). vvDD-IL2 promoted significant increases of total CD3+ and CD3+CD8+ cell infiltration in the tumor tissue compared to PBS treatment. Oncolytic vvDD-IL2 treatment presented a trend towards increased CD3+CD4+ TILs (% CD3+CD4+ T cells: vvDD-IL2 vs PBS: p = 0.0588). Second, T cells were purified from tumor tissues and quantified as per gram of tumor tissue, and similar patterns were observed (Suppl. Fig. 1). Cumulatively, these results indicate that viral treatment, especially vvDD-IL2, elicits potent adaptive antitumor immunity leading to an increased number of CD3+CD8+ T cells in the tumor tissues, as well as an increased percentage of tumor-reactive CD3+CD8+ T cells in the tumor.
Figure 2. vvDD-IL2 treatments increase the number of TILs.
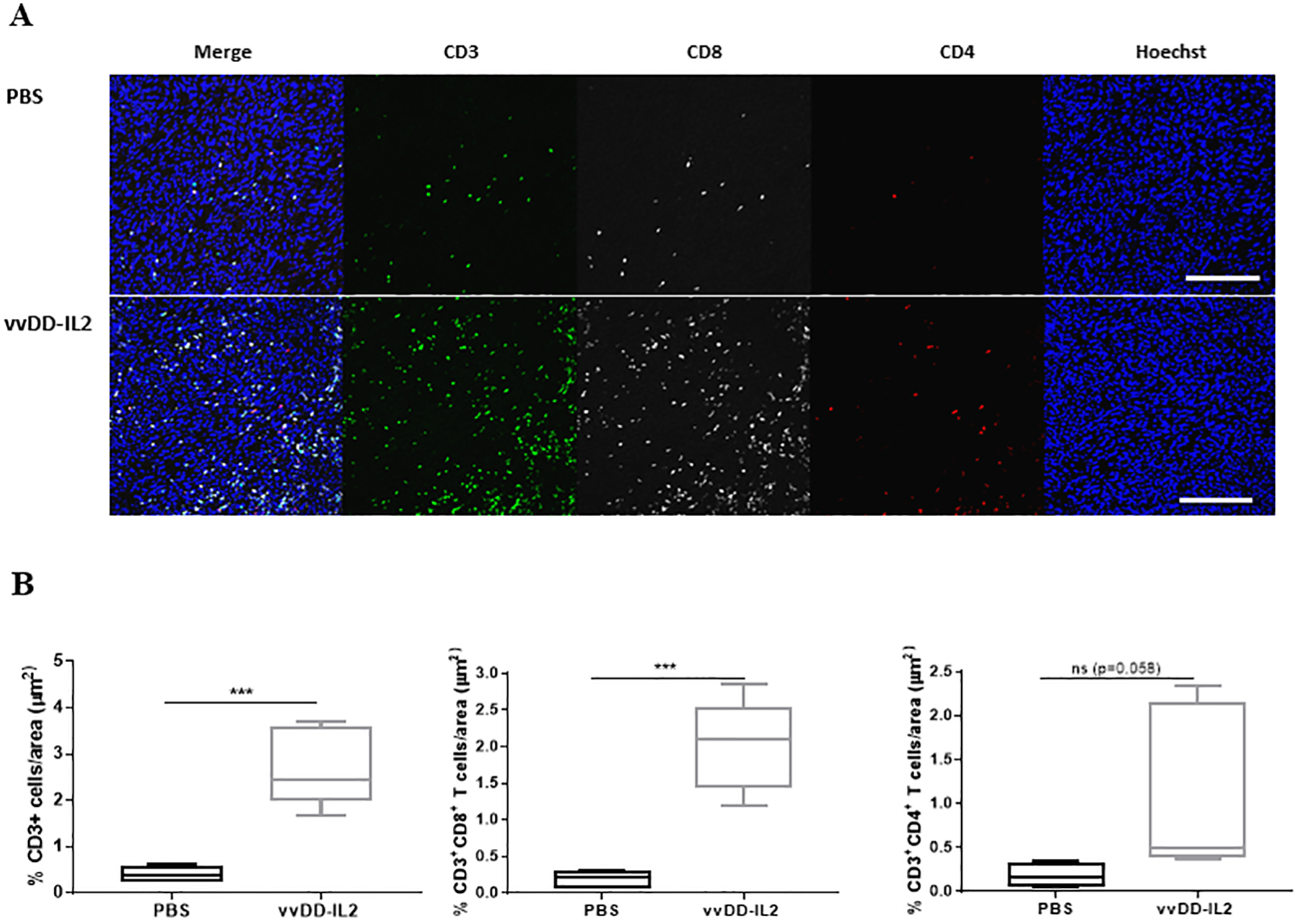
The tumor cell inoculation and tumor harvest were as described previously. Tumors were collected, fixed and stained for Hoechst (blue), CD3 (green), CD4 (red) and CD8 (white). A. Representative immunofluorescence image of one sample from each group (n = 5). For the representative panel, a single representative field was cropped from the original nine field acquisition. Scale bar 25 μm. B. Summary of the percentage of CD3+ T cells and CD3+CD4+ and CD3+CD8+ T cells per area. Student’s t-test was used to analyze the statistical significance. ***p < 0.001.
OV-induced TILs profiling indicated a strong pro-antitumor immunity.
We then analyzed the status of T cells in the tumor tissues by flow cytometry (Fig. 3). To start with, we looked at the splenocytes and set them as control for comparison (Fig. 3A). In the spleen, CTLA-4+CD8+ T cells were slightly reduced in both groups treated with OVs. Tim-3+CD8+ T cells were not changed, while PD-1hiTim-3+ CD8+ T cells were slightly reduced. The PD-1hiTim-3+ CD8+ T cells represent the highly exhausted cell population [31]. Interestingly, Treg (Foxp3+CD4+ T cells) cells were reduced in the mice treated with vvDD-IL2, but not in the group treated with vvDD. Then we analyzed the same set of markers in TILs (Fig. 3B). We observed increased CTLA-4+ or TIM-3+ CD8+ T cells in the virus-treated groups. Surprisingly, when we examined PD-1hiTim-3+ CD8+ T cells, considered to be highly exhausted cells, we saw a very exciting pattern of changes. The group treated with vvDD exhibited a reduction of this exhausted cells, and this reduction was much greater in the mice treated with vvDD-IL2 (p < 0.01). When we analyzed Treg cells, the same patterns of reduction were observed.
Figure 3. Immune status in the TME and spleen post virus treatment.
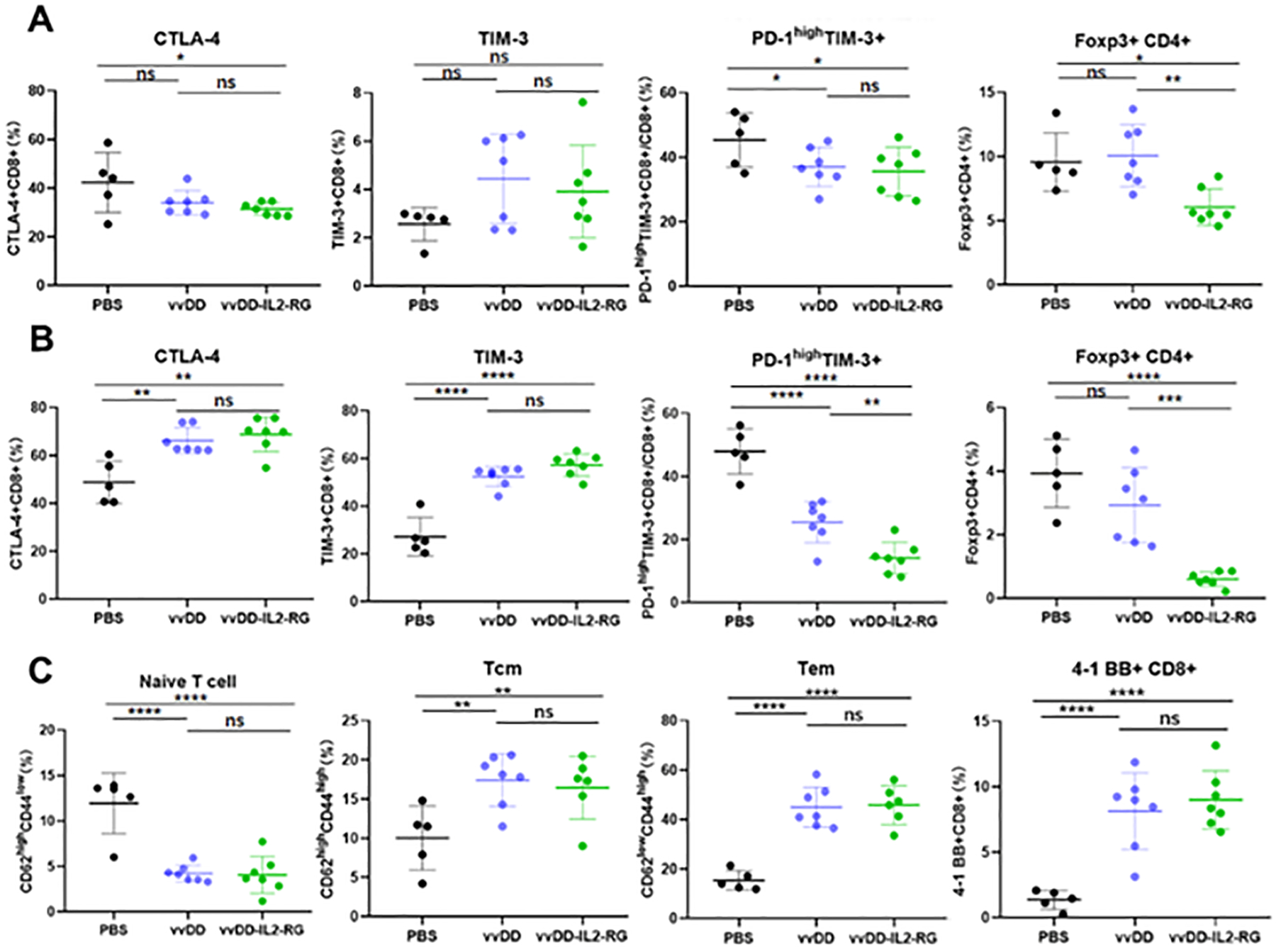
B6 mice were inoculated subcutaneously with 5.0e5 MC38 tumor cells. When tumors reached 5 × 5 mm (about day 9) they were treated intratumorally with PBS, vvDD, vvDD-IL-2 at a dose of 1.0e8 PFU. The mice were sacrificed 10 days post viral treatment and primary tumors and splenocytes were collected and analyzed by flow cytometry to determine the activation and immunosuppressive markers in (A) splenocytes, (B) TILs and (C) subsets of CD8+ T cells in the TILs. Tn for naïve T cells; Tcm for central memory T cells, and Tem for effector memory T cells. The percentages are over total CD8+ T cells, or over total CD4+ T cells for Treg cells. * p < 0.05, ** p < 0.01, *** p < 0.001; **** p < 0.0001.
During T cell activation, there is a dynamic transition from Tn to Tcm to Tem, and finally effector T cells. We therefore examined the CD8+ T cell subsets (Fig. 3C). First, there was a large percentage of Tn (naïve T cells) in the PBS group, while Tn was greatly reduced in the two groups treated with OVs. In the Tcm subset, there was an increase in the groups treated with OVs, and finally, there was a large increase of the Tem, from ~17% in PBS group to ~43% in the groups treated with OVs. When we analyzed the populations of 4–1BB+CD8+ T cells, there were clearly an enhancement in the OV-treated groups (p < 0.001, compared to PBS group). These results showed convincingly that OV activated and promoted differentiation of CD8+ T cells toward effector T cells.
We have also conducted preliminary studies on the specificity of TILs recognizing certain antigens. We pulsed naïve splenocytes with H-2Kb-restricted epitope peptides, p15E604–611 (retrovirally-encoded tumor antigen) [32], B8R20–27 (dominant vaccinia virus antigen) [33], and β-galactosidase-derived epitope (DAPIYTNV), then incubated with TILs isolated from MC38-tumor-bearing mouse treated with vvDD-IL2 for ~12 h. The flow cytometry analysis for IFN-γ+ CD8+ T cells suggested that fractions of TILs were able to recognize p15E and B8R epitopes, respectively (Supplementary Figure 2).
OV-induced TILs can be isolated from tumor tissues and expanded ex vivo
As our eventual aim was to utilize these tumor-reactive TILs for therapeutic ACT, we investigated the ability to expand these TILs while retaining their tumor-specificity. First, we established protocols for TIL isolation with CD90.2 magnetic beads, and then for ex vivo culture and expansion. For induction of tumor-specific TILs, MC38 s. c. tumor-bearing mice were treated as before with intratumoral PBS or vvDD-IL2. Ten days later tumors were harvested and TILs from each tumor were cultured in the presence of IL-2 and IL-7 in RPMI complete media for 3 days. To test if the cultured TILs retained their tumor specificity, these T cells were tested for their tumor recognition using a co-culture assay including irradiated MC38 tumor cells and irrelevant target cells -γ-irradiated B16 tumor cells or naïve splenocytes from non-tumor-bearing B6 mice. After 24 h these T cells were analyzed for tumor specificity using IFN-γ ELISpot or 4–1BB expression by flow cytometry. According to the IFN-γ ELISpot assay, T cells expanded from vvDD-IL2 or PBS-treated tumors demonstrated increased MC-38 specific IFN-γ secretion compared to irrelevant target cells (B16 or naïve splenocytes) (Fig. 4A; Suppl. Fig. 3). To distinguish between CD8+ and CD4+ tumor-specific T cell response, flow cytometry results of 4–1BB expressing CD8+ and CD4+ T cells from each expanded TIL culture are summarized in Fig. 4 (B, C) and Suppl. Fig. 3. The expanded cultures from the vvDD-IL2-induced TILs demonstrated a higher percentage of tumor-specific CD8+ and CD4+ T cells when compared to PBS treatment [% CD8+ 4–1BB+ T cells: vvDD-IL2: 16.4 ± 11; PBS: 1.96 ± 1.6. % CD4+ 4–1BB+ T cells: vvDD-IL2: 6.3 ± 2.4; PBS: 2.4 ± 2.0].
Figure 4. OV-induced TILs can be cultured and expanded ex vivo and retain their tumor specificity.
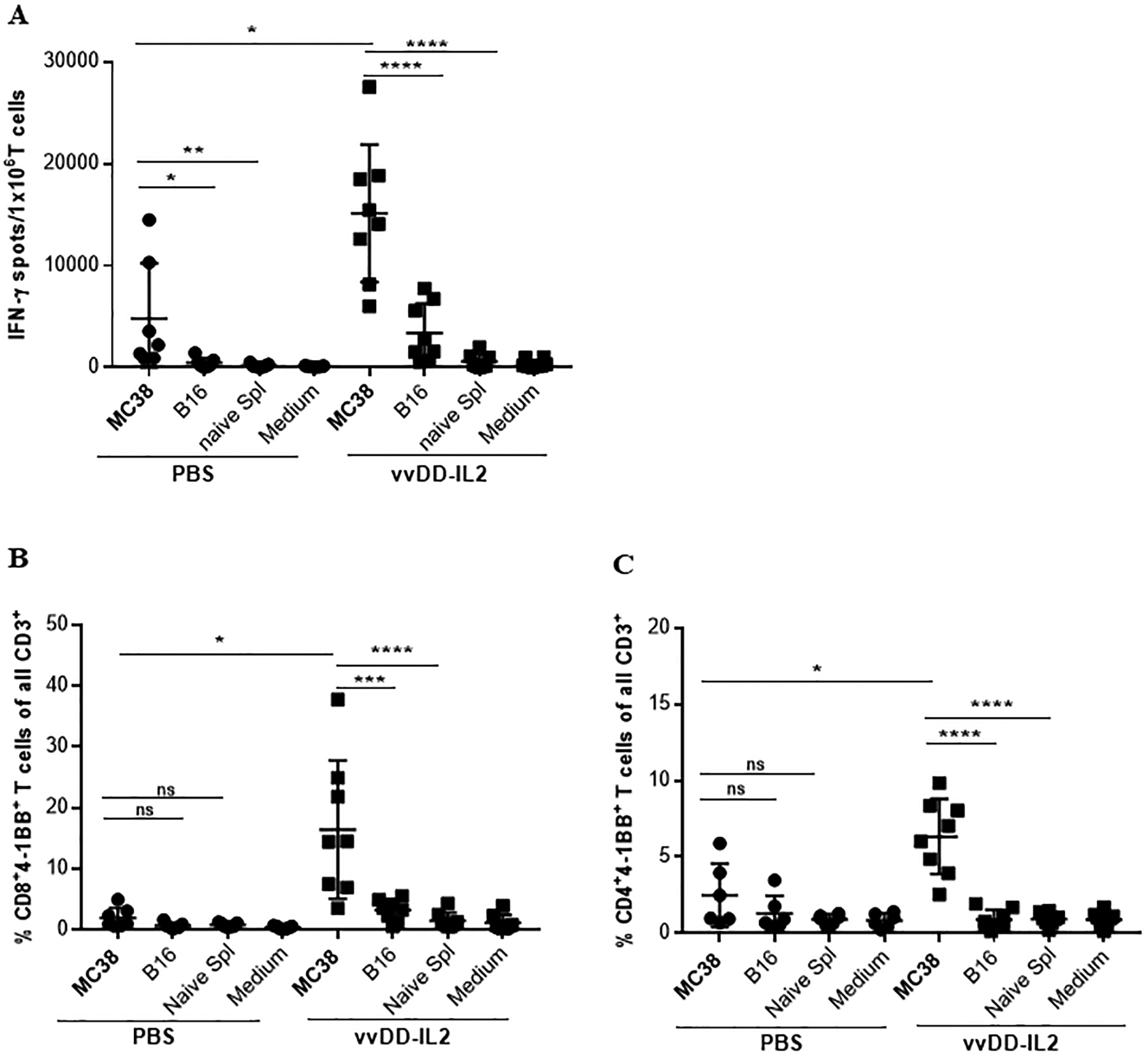
(A). Tumor specificity of the expanded TILs. TILs from each individual mouse have been cultured for 4 days and then tested by IFN-γ ELISPOT assay. (B, C). Analysis of 4–1BB upregulation on (B) CD4+ and (C) CD8+ T cells by flow cytometry. As previously described, T cells were either left unstimulated (medium) or challenged with γ-irradiated MC38 tumor cells or γ-irradiated B16 tumor cells or naïve splenocytes from non-tumor-bearing B6 mouse in duplicate. After 24 h the cells have been stained for flow cytometry analysis for CD3, CD4, CD8, 4–1BB. Results are shown as individual data points (percentage of CD8+4–1BB+ T cells and CD4+4–1BB+ T cells) and bars (means ± standard deviation) of T cells from each mouse evaluated in duplicate. Data are presented as summary from 2 out of 5 independent experiments (n = 3–4 mice/group). For multiple group comparison One-way ANOVA was used. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
ACT of OV-induced TILs led to therapeutic efficacy in a low-immunogenic colon tumor model
We then explored the ability of the vvDD-IL2-induced TILs to be utilized for ACT for cancer treatment. The overall schema is shown in Fig. 5A. We harvested the TILs from MC38 colon cancer-bearing mice treated with vvDD-IL2 as described in Methods. As a control, T cells were isolated from splenocytes from naïve mice. These TILs and control T cells were expanded ex vivo for 4 days as described above and then portions of the cultures were characterized. Expanded TILs from vvDD-IL2 injected tumors had a higher specific reactivity against MC38 compared to naïve splenocytes expanded in an identical manner (Fig. 5, B & C). No IFN-γ was released when cultured control T cells were tested against MC38 cancer cells. We also tested the activation status of the vvDD-IL2-induced TILs using 4–1BB staining and found that 6% of the CD8+ T cells and 5% of CD4+ T cells were 4–1BB positive after co-culture with irradiated MC38 tumor cells, as analyzed by flow cytometry (Fig. 5, D & E; Suppl. Fig. 4). Very low levels of 4–1BB were found when these TILs were co-cultured with B16 tumor cells, naïve splenocytes or growth medium. These results indicated that CD4+ and CD8+ TILs specifically recognized irradiated MC38 cancer cells after expansion.
Figure 5. Analysis of the vvDD-IL2 induced TILs after ex vivo expansion.
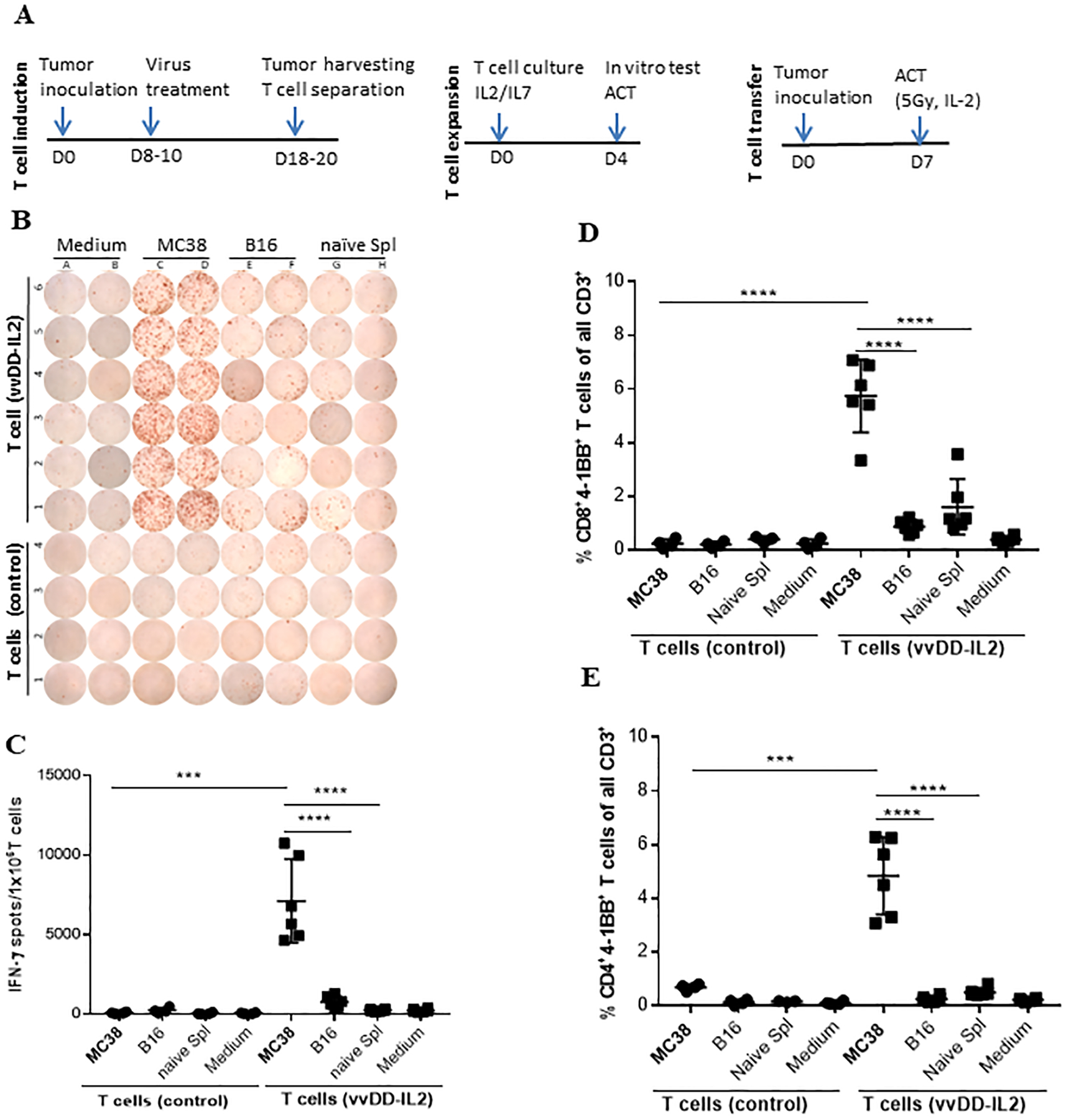
Prior to ACT, samples (T cells from naïve spleens: n = 4; TILs: n = 6) of the ex vivo cultured and expanded TILs were tested for tumor reactivity using IFN-γ ELISpot assay and co-culture assays for 4–1BB expression analyzed by flow cytometry as described previously. (A). Schema of experimental procedure. (B, C). Shown are (B) the ELISpot plate and (C) calculated IFN-γ+ spots per 1.0e6 T cells. (D, E). The cultured and expanded TILs were analyzed for cell surface 4–1BB expression of (D) CD4+ and (E) CD8+ T cells by flow cytometry. For multiple group comparison One-way ANOVA was used. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
We next examined the therapeutic efficacy of the ex vivo expanded TILs via ACT in a peritoneal MC38 colon cancer model (Fig. 6). Seven days after inoculation of 5.0e5 MC38-luc cells i.p. into C57BL/6J mice, the tumor growth was monitored by bioluminescence imaging using the Optical In Vivo Imaging System, and mice were randomly divided into groups with an equivalent range of tumor burden. Prior to ACT, tumor-bearing mice received 5 Gy of sublethal irradiation to mimic lymphodepletion similar to clinical protocols. The mice were injected i.p. with vvDD-IL2-induced and ex vivo expanded TILs, ex vivo expanded control T cells, or PBS saline. All treated mice received exogenous cytokine support with recombinant IL-2. We then followed the therapeutic response by live animal bioluminescence imaging to monitor the kinetics of tumor growth over time (Fig. 6A, B). It is clear that by day 17 post ACT, the group treated with ACT with T cells from vvDD-IL2 mice and radiation showed the best results (p < 0.05, compared to any other group). Mice treated with ACT from vvDD-IL2-induced TILs showed the least amounts of tumor burden and survived the longest when compared to PBS or 5Gy/IL-2 controls (p < 0.001) (Fig. 6C). If we compare the median survival of mice in different groups, the ACT with T cells from VV-IL2 group gave the very best result, with median survival time at 61 days (Fig. 6D; p < 0.01, compared to any other group).
Figure 6. ACT of oncolytic virus-induced TILs led to significant therapeutic efficacy in mice bearing peritoneal carcinomatosis of MC38 colon cancer.
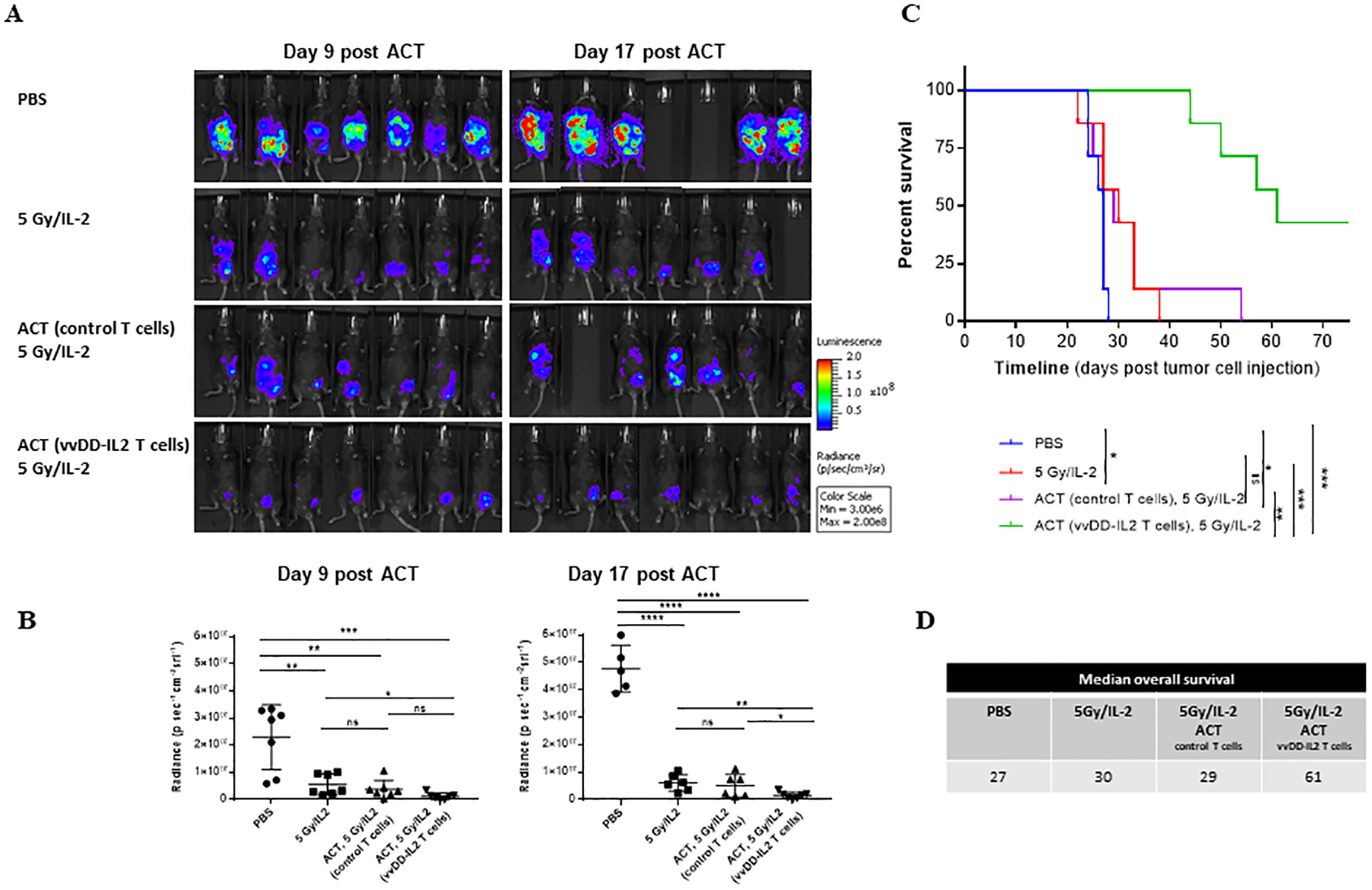
B6 mice were intraperitoneally inoculated with 5 × 105 MC38-luc cancer cells followed in 7 days, these mice were imaged for tumor growth and randomly grouped (n = 7 mice/group). Prior to ACT, mice in the treated groups received 5 Gy of sublethal irradiation. Grouped mice were intraperitoneally injected with PBS, naïve T cells, or vvDD-IL2 induced and ex vivo expanded TILs. All treated mice received exogenous IL-2 (1.0e5 IU/mouse, i.p. injection once every ~12 h for 3 days). One experiment representative of two independent experiments is shown (A, B). (A). Tumor growth has been monitored by live animal imaging 9 days and 17 days post treatment. (B). Radiance data quantified for two time points at days 9 and 17 post ACT. Student’s t-test was used to analyze the statistical significance. The variance was similar between the groups in this experiment. (C). The long-term survival of tumor-bearing mice was monitored by Kaplan-Meier analysis. These data represent one of the two independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001. (D). Summary of median survival in the different treatment groups. This experiment was performed 2 times.
DISCUSSION
ACT using autologous TILs represents a personalized cancer immunotherapy strategy, targeting shared and unique tumor antigens expressed by a patient’s tumor [2, 34]. ACT for cancer using patients’ own TILs has been a successful systemic treatment leading to the cure of a number of advanced melanoma patients, and a few other tumor types. The suppressive TME is addressed with a preparatory non-myeloablative chemotherapy regimen (cytoxan and fludarabine) then ex vivo expanded TILs are delivered intravenously followed by IL-2 to maintain T cell activity. A recent study in uveal melanoma demonstrated a strong association between the anti-tumor reactivity of the infused TILs and objective clinical response. Thus, isolation and delivery of TILs with greater anti-tumor reactivity may result in improved clinical responses. Discovering new methods to improve the recovery of tumor-reactive TILs could have a significant impact on the field of cancer immunotherapy and ultimately patient outcomes.
Immune cell infiltration in the tumor impacts tumor progression and patient survival, and a strong lymphocyte infiltration has been reported to be associated with an antitumor response and improved clinical outcome in a variety of types of cancers, including colorectal cancer [35–39]. The majority of solid tumors, especially those of low tumor mutational burden (TMB), however, lack an inflammatory infiltrate. Cancers develop immune escape mechanisms to avoid detection by effector immune cells. This includes cell surface expression of immune system checkpoint ligands such as PD-L1 [40, 41]; secretion of soluble immunosuppressive factors such as transforming growth factor beta, vascular endothelial growth factor, interleukin-10, galectin-1, indoleamine 2,3-dehydrogenase [42–44]; down-regulation of major histocompatibility complex (MHC) class I expression; and overexpression of receptors such as C-X-C chemokine receptor type 4, basic fibroblast growth factor and epidermal growth factor [45, 46]. The immunosuppressive tumor microenvironment includes immunosuppressive macrophages, myeloid-derived suppressor cells and regulatory T cells interfering with an efficient anti-tumor T cell response. Inhibitory checkpoint molecules such as CTLA-4, PD-1, TIM-3, LAG3, are upregulated in chronically stimulated T cells, promoting T cell anergy.
OVs overcome the immunosuppressive TME as they turn cold tumor ‘hot’ [8–10]. They infect and kill tumor cells in vivo and induce an inflammatory response that clears the virus and some non-infected tumor cells, despite the immunosuppressive TME. The virus is ultimately immunologically cleared from the tumor and tumor reactive T cells are generated. This local cytotoxic and anti-tumor immunologic activity has limited systemic activity. Successful systemic delivery of OVs with adequate tumor infection throughout the body for a meaningful effect on metastatic tumors, remains a challenge. One way to take advantage of local anti-tumor immunity for systemic therapy is to harvest the infected tumor tissues, isolate and expand the tumor-reactive T cells ex vivo, and adoptively transfer them back into the patient. In a clinical study done by Ribas et al., intratumoral injection of talimogene laherparepvec in melanoma patients resulted in an increase in infiltrating T cells independent of the T cell baseline [9]. In our study, we have demonstrated for the first time the feasibility of using these TILs for therapy. When the subsets of T cells in the TIL populations were analyzed, the quality of TILs was much improved in the mice treated with vvDD. They contained much higher percentages of Tcm and Tem cells. Moreover, vvDD-IL2 improved the quality even further, as it contained fewer exhausted PD-1hiTim-3+ CD8+ T cells and Treg cells.
This would effectively expand the utility of both TILs and OV. This immunotherapy would be personalized for each patient’s tumor neoantigens, and in contrast to CAR-T cells would be polyclonal in terms of antigen recognition and immune cell type and unlikely to have reactivity against normal cells (off target toxicity).
Our laboratories have extensive experience with both OVs and T cell-mediated immunotherapy [47, 48]. The anti-tumor immune response induced by oncolytic VV has been studied extensively. Previous depletion studies in animal models indicated the anti-tumor response and long-term survival are attributed to CD8+ T cells, and to a smaller extent CD4+ T cells, NK cells and neutrophils [8, 26, 49]. In the current study, we compared the parental oncolytic VV (vvDD) with those expressing various cytokines and chemokines (IL-2, CCL5, CXCL11 and IL-15) for their capacity to elicit and attract tumor-reactive TILs. Under the experimental conditions, we found that vvDD-IL2 performed the best, leading to the highest levels of tumor-reactive CD8+ TILs. The virus induced a MC38-specific T cell response and the effect was not reproducible by injection of the cytokine without the virus. The cytokine IL-2 was approved to treat metastatic melanoma and renal cancer in the 1990s. Since then different application strategies and combinations have been tested to overcome toxicity and enable a specific anti-tumor immune response [50, 51]. Although the transfer of virus-specific peripheral blood T cells to treat cancer patients have been done [52, 53], our current study has demonstrated, for the first time, a new therapeutic method-ACT using virally induced TILs isolated from solid tumors. The data from immunostaining confirmed that vvDD-IL2 attracts high numbers of total CD3+CD8+ T cells in the tumor with an increased percentage of specific tumor reactivity.
These TILs isolated from tumors treated with vvDD-IL2 were able to be expanded in culture and maintain their activity and specificity. We observed high levels of IFN-γ release and 4–1BB expression from CD8+ and CD4+ TILs after 4 days of in vitro expansion when co-cultured with the same tumor cells. Adoptive transfer of these TILs in a model of peritoneal metastases reduced tumor burden and achieved better survival compared to the control groups. Thus, we have demonstrated the principle of therapeutic efficacy for OV-elicited tumor-reactive TILs in a murine tumor model.
Virally induced tumor-reactive TILs have not previously been tested in ACT for therapeutic effectiveness. In the literature, most studies on TILs have been performed in patients by culturing small pieces of the tumor and expanding the outgrowing T cells [2]. It is well known that isolation of murine TILs is challenging and there exist only a few reports documenting the culturing of murine TILs [54, 55]. Most ACT studies in the murine system have been conducted using T cells derived from splenocytes and universally activated with super-antigens (such as anti-CD3 and CD28 antibodies). A number of investigators have explored the combination of an OV with ACT in cancer models, using the virus to improve trafficking of adoptively transferred cells [56–58], but not as a “pre-TIL” approach to improve the recovery of tumor-reactive T cells. Murine TILs have been isolated with magnetic beads or FACS sorting from tumor tissue-derived single cell suspensions [29, 54, 55]. We utilized magnetic bead sorting (CD90.2 antibody) for isolation, and murine TILs were able to be expanded ex vivo only when clearly separated from the tumor tissue. When assayed for their functionalities after ex vivo short-term expansion they retained their tumor specificity. In our studies MC38 tumor specific 4–1BB expression was enhanced in about 5–20 % of the CD8+ T cells and about 5–6 % of the CD4+ T cells.
We are fully aware that the murine system for ex vivo TIL expansion has limitations and that the i.p. tumor serves only as a model to prove the therapeutic principle. In the future, we will explore the intravenous delivery of TILs to solid tumor models as the homing potential of such ex vivo expanded TILs needs to be analyzed as well. We would like to explore ways to induce, isolate, and expand more optimized TILs for ACT using OVs. For example, it is known that stem cell-like T cells may offer more therapeutic potential [59, 60]. Our comparison of cytokines and chemokines have not been exhaustive, and other agents may have similar or more potent activity as a “pre-TIL” approach. Once these types of questions are addressed, it will be time to translate this improved approach and technology from bench to bedside. In addition, one key issue in ACT is the trafficking of TILs intravenously delivered back to the tumor tissues where they exert their key functions. The study on trafficking of TILs back to tumor tissue is out of scope of this study as many other studies have been done to find the right answers [61].
In summary, we have demonstrated that IL-2 armed OV promotes T cell infiltration and enhances the population of tumor-reactive TILs in a murine colon cancer model. Our report presents a new therapeutic strategy to promote the generation and infiltration of tumor-reactive TILs in lowly or poorly immunogenic tumors, and the expansion of such TILs for ACT. The new strategy may be translated into a clinical application and may allow adoptive T cell therapy for an expanded group of cancer patients.
Supplementary Material
Acknowledgments
We thank the University Imaging Core Facility for technical assistance in immunostaining and imaging, the Hillman Cancer Center Biostatistics facility for consultation, and Prometheus (San Diego, CA) for human recombinant IL-2 used in the study.
Funding
This work was supported in part by David C. Koch Regional Cancer Therapy Center. MF (DFG research fellowship 1655/1-1) and EG were supported by fellowships from German Research Foundation. ED was supported by fellowships from the China Scholarship Council, China. This project has used the University of Pittsburgh shared facilities (Animal Facility, Genomics Research Core, Flow Cytometry) that are supported in part by the NIH grant award P30CA047904.
Footnotes
Conflicts of Interests: MF, ZL, ZSG and BLB filed a patent partly based on this work. Other authors declare that they have no conflict of interest.
References
- 1.Yang Y: Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest 2015; 125:3335–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Restifo NP: Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015; 348:62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ et al. : Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 2011; 17:4550–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Iversen TZ et al. : Long-Lasting Complete Responses in Patients with Metastatic Melanoma after Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes and an Attenuated IL2 Regimen. Clin Cancer Res 2016; 22:3734–3745. [DOI] [PubMed] [Google Scholar]
- 5.Chandran SS, Somerville RPT, Yang JC, Sherry RM, Klebanoff CA, Goff SL et al. : Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: a single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol 2017; 18:792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G: The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol 2017; 14:717–734. [DOI] [PubMed] [Google Scholar]
- 7.Baldan V, Griffiths R, Hawkins RE, Gilham DE: Efficient and reproducible generation of tumour-infiltrating lymphocytes for renal cell carcinoma. Br J Cancer 2015; 112:1510–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Z, Ravindranathan R, Kalinski P, Guo ZS, Bartlett DL: Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat Commun 2017; 8:14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O et al. : Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 2017; 170:1109–1119 e1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gujar S, Pol JG, Kroemer G: Heating it up: Oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade immunotherapies. Oncoimmunology 2018; 7:e1442169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y et al. : Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer 2013; 12:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo ZS, Liu Z, Bartlett DL: Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front Oncol 2014; 4:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Todo T, Rabkin SD, Sundaresan P, Wu A, Meehan KR, Herscowitz HB et al. : Systemic antitumor immunity in experimental brain tumor therapy using a multimutated, replication-competent herpes simplex virus. Hum Gene Ther 1999; 10:2741–2755. [DOI] [PubMed] [Google Scholar]
- 14.Prestwich RJ, Errington F, Ilett EJ, Morgan RS, Scott KJ, Kottke T et al. : Tumor infection by oncolytic reovirus primes adaptive antitumor immunity. Clin Cancer Res 2008; 14:7358–7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gauvrit A, Brandler S, Sapede-Peroz C, Boisgerault N, Tangy F, Gregoire M: Measles virus induces oncolysis of mesothelioma cells and allows dendritic cells to cross-prime tumor-specific CD8 response. Cancer Res 2008; 68:4882–4892. [DOI] [PubMed] [Google Scholar]
- 16.Li X, Wang P, Li H, Du X, Liu M, Huang Q et al. : The efficacy of oncolytic adenovirus is mediated by T-cell responses against virus and tumor in Syrian hamster model. Clin Cancer Res 2017; 23:239–249. [DOI] [PubMed] [Google Scholar]
- 17.Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S: Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol 2010; 17:718–730. [DOI] [PubMed] [Google Scholar]
- 18.Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M et al. : Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med 2013; 19:329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Zeng Z, Fu X, Zhang X: Coadministration of a herpes simplex virus-2 based oncolytic virus and cyclophosphamide produces a synergistic antitumor effect and enhances tumor-specific immune responses. Cancer Res 2007; 67:7850–7855. [DOI] [PubMed] [Google Scholar]
- 20.Chard LS, Maniati E, Wang P, Zhang Z, Gao D, Wang J et al. : A vaccinia virus armed with interleukin-10 is a promising therapeutic agent for treatment of murine pancreatic cancer. Clin Cancer Res 2015; 21:405–416. [DOI] [PubMed] [Google Scholar]
- 21.de Graaf JF, de Vor L, Fouchier RAM, van den Hoogen BG: Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev 2018; 41:28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ylosmaki E, Cerullo V: Design and application of oncolytic viruses for cancer immunotherapy. Curr Opin Biotechnol 2019; 65:25–36. [DOI] [PubMed] [Google Scholar]
- 23.Guo ZS, Parimi V, O’Malley ME, Thirunavukarasu P, Sathaiah M, Austin F et al. : The combination of immunosuppression and carrier cells significantly enhances the efficacy of oncolytic poxvirus in the pre-immunized host. Gene Ther 2010; 17:1465–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCart JA, Ward JM, Lee J, Hu Y, Alexander HR, Libutti SK et al. : Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res 2001; 61:8751–8757. [PubMed] [Google Scholar]
- 25.Li J, O’Malley M, Urban J, Sampath P, Guo ZS, Kalinski P et al. : Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol Ther 2011; 19:650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Z, Ravindranathan R, Li J, Kalinski P, Guo ZS, Bartlett DL: CXCL11-armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. Oncoimmunology 2016; 5:e1091554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kowalsky SJ, Liu Z, Feist M, Berkey SE, Ma C, Ravindranathan R et al. : Superagonist IL-15-Armed Oncolytic Virus Elicits Potent Antitumor Immunity and Therapy That Are Enhanced with PD-1 Blockade. Mol Ther 2018; 26:2476–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Z, Ge Y, Wang H, Ma C, Feist M, Ju S et al. : Modifying the cancer-immune set point using vaccinia virus expressing re-designed interleukin-2. Nat Commun 2018; 9:4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Chen K, Wang C, Gong W, Yoshimura T: Isolation of mouse tumor-infiltrating leukocytes by percoll gradient centrifugation Bio-Protocol 2013; 3:e892. [Google Scholar]
- 30.Jacobs JJ, Sparendam D, Den Otter W: Local interleukin 2 therapy is most effective against cancer when injected intratumourally. Cancer Immunol Immunother 2005; 54:647–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma J, Zheng B, Goswami S, Meng L, Zhang D, Cao C et al. : PD1(Hi) CD8(+) T cells correlate with exhausted signature and poor clinical outcome in hepatocellular carcinoma. J Immunother Cancer 2019; 7:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeh HJ 3rd, Perry-Lalley D, Dudley ME, Rosenberg SA, Yang JC: High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol 1999; 162:989–994. [PubMed] [Google Scholar]
- 33.Oseroff C, Kos F, Bui HH, Peters B, Pasquetto V, Glenn J et al. : HLA class I-restricted responses to vaccinia recognize a broad array of proteins mainly involved in virulence and viral gene regulation. Proc Natl Acad Sci U S A 2005; 102:13980–13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crompton JG, Klemen N, Kammula US: Metastasectomy for tumor-infiltrating lymphocytes: an emerging operative indication in Surgical Oncology. Ann Surg Oncol 2018; 25:565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R et al. : Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med 2005; 353:2654–2666. [DOI] [PubMed] [Google Scholar]
- 36.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C et al. : Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313:1960–1964. [DOI] [PubMed] [Google Scholar]
- 37.Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS et al. : Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol 2007; 25:2586–2593. [DOI] [PubMed] [Google Scholar]
- 38.Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S et al. : Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res 2014; 74:705–715. [DOI] [PubMed] [Google Scholar]
- 39.Turksma AW, Coupe VM, Shamier MC, Lam KL, de Weger VA, Belien JA et al. : Extent and location of tumor-infiltrating lymphocytes in microsatellite-stable colon cancer predict outcome to adjuvant active specific immunotherapy. Clin Cancer Res 2016; 22:346–356. [DOI] [PubMed] [Google Scholar]
- 40.Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H et al. : Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res 2007; 13:2151–2157. [DOI] [PubMed] [Google Scholar]
- 41.Gao Q, Wang XY, Qiu SJ, Yamato I, Sho M, Nakajima Y et al. : Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res 2009; 15:971–979. [DOI] [PubMed] [Google Scholar]
- 42.Martínez-Bosch N, Fernández-Barrena MG, Moreno M, Ortiz-Zapater E, Munné-Collado J, Iglesias M et al. : Galectin-1 drives pancreatic carcinogenesis through stroma remodeling and Hedgehog signaling activation. Cancer Res 2014; 74:3512–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobayashi N, Kubota K, Kato S, Watanabe S, Shimamura T, Kirikoshi H et al. : FOXP3+ regulatory T cells and tumoral indoleamine 2,3-dioxygenase expression predicts the carcinogenesis of intraductal papillary mucinous neoplasms of the pancreas. Pancreatology 2010; 10:631–640. [DOI] [PubMed] [Google Scholar]
- 44.Brandacher G, Perathoner A, Ladurner R, Schneeberger S, Obrist P, Winkler C et al. : Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin Cancer Res 2006; 12:1144–1151. [DOI] [PubMed] [Google Scholar]
- 45.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS et al. : Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 2013; 110:20212–20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chatterjee S, Behnam Azad B, Nimmagadda S: The intricate role of CXCR4 in cancer. Adv Cancer Res 2014; 124:31–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo ZS, Lu B, Guo Z, Giehl E, Feist M, Dai E et al. : Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J Immunother Cancer 2019; 7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dafni U, Michielin O, Lluesma SM, Tsourti Z, Polydoropoulou V, Karlis D et al. : Efficacy of Adoptive Therapy with Tumor-infiltrating Lymphocytes and Recombinant Interleukin-2 in Advanced Cutaneous Melanoma: A Systematic Review and Meta-analysis. Ann Oncol 2019; 30:1902–1913. [DOI] [PubMed] [Google Scholar]
- 49.John LB, Howland LJ, Flynn JK, West AC, Devaud C, Duong CM et al. : Oncolytic virus and anti-4–1BB combination therapy elicits strong anti-tumor immunity against established cancer. Cancer Res 2012; 72:1651–1660. [DOI] [PubMed] [Google Scholar]
- 50.Jiang T, Zhou C, Ren S: Role of IL-2 in cancer immunotherapy. Oncoimmunology 2016; 5:e1163462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pol JG, Caudana P, Paillet J, Piaggio E, Kroemer G: Effects of interleukin-2 in immunostimulation and immunosuppression. J Exp Med 2020; 217:e20191247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berger C, Turtle CJ, Jensen MC, Riddell SR: Adoptive transfer of virus-specific and tumor-specific T cell immunity. Curr Opin Immunol 2009; 21:224–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Houghtelin A, Bollard CM: Virus-Specific T Cells for the Immunocompromised Patient. Front Immunol 2017; 8:1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kodumudi KN, Siegel J, Weber AM, Scott E, Sarnaik AA, Pilon-Thomas S: Immune checkpoint blockade to improve tumor infiltrating lymphocytes for adoptive cell therapy. PLoS One 2016; 11:e0153053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fernandez-Poma SM, Salas-Benito D, Lozano T, Casares N, Riezu-Boj JI, Mancheno U et al. : Expansion of tumor-infiltrating CD8(+) T cells expressing PD-1 improves the efficacy of adoptive T-cell therapy. Cancer Res 2017; 77:3672–3684. [DOI] [PubMed] [Google Scholar]
- 56.Fu X, Rivera A, Tao L, Zhang X: An HSV-2 based oncolytic virus can function as an attractant to guide migration of adoptively transferred T cells to tumor sites. Oncotarget 2015; 6:902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ajina A, Maher J: Prospects for combined use of oncolytic viruses and CAR T-cells. J Immunother Cancer 2017; 5:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klebanoff CA, Gattinoni L, Palmer DC, Muranski P, Ji Y, Hinrichs CS et al. : Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res 2011; 17:5343–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lugli E, Dominguez MH, Gattinoni L, Chattopadhyay PK, Bolton DL, Song K et al. : Superior T memory stem cell persistence supports long-lived T cell memory. J Clin Invest 2013; 123:594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klebanoff CA, Scott CD, Leonardi AJ, Yamamoto TN, Cruz AC, Ouyang C et al. : Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J Clin Invest 2016; 126:318–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Melero I, Rouzaut A, Motz GT, Coukos G: T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov 2014; 4:522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.