

Abstract
We previously demonstrated that the Neisseria IgA1 protease cleaves LAMP1 (lysosome-associated membrane protein 1), a major integral membrane glycoprotein of lysosomes, thereby accelerating its degradation rate in infected A431 human epidermoid carcinoma cells and resulting in the alteration of lysosomes in these cells. In this study, we determined whether the IgA1 protease also affects the trafficking of Neisseria gonorrhoeae across polarized T84 epithelial monolayers. We report that N. gonorrhoeae infection of T84 monolayers, grown on a solid substrate or polarized on semiporous membranes, also results in IgA1 protease-mediated reduction of LAMP1. We demonstrate that iga mutants in two genetic backgrounds exited polarized T84 monolayers in fewer numbers than the corresponding wild-type strains. Finally, we present evidence that these mutants have a statistically significant and reproducible defect in their ability to traverse T84 monolayers. These results add to our previous data by showing that the IgA1 protease alters lysosomal content in polarized as well as unpolarized cells and by demonstrating a role for the protease in the traversal of epithelial barriers by N. gonorrhoeae.
Neisseria gonorrhoeae (i.e., the gonococcus [GC]) causes gonorrhea in humans, its only host, and gains entry into the body via the mucosal surfaces. There is no animal model for GC disease. Studies on the molecular requirements of GC interactions with the epithelium rely on human fallopian tube organ cultures (hFTOC), immortalized human epithelial cells grown on solid substrates, and the human challenge model of urethral infection. Such studies reveal that GC attach to and invade nonciliated cells of the mucosal epithelium through the coupling of several bacterial adhesins (type IV pili, PilC, and certain Opa variants) with their cognate host cell receptors (CD46, CD66, and heparan sulfate proteoglycans), which are present in a large number of human tissues (3, 4, 7, 8, 11–13, 17, 18, 21–24, 27, 30, 35, 40–44, 47; J. G. Cannon, D. Johannsen, J. D. Hobbs, N. Hoffman, J. A. F. Dempsey, D. Johnston, H. Koyman, and M. S. Cohen, presented at the Eleventh Int. Pathog. Neisseria Conf., 1998).
The trafficking of GC within the epithelial cell was initially examined using hFTOC (20, 29). However, the nature of the system limits the type and scale of trafficking studies that can be performed. Experimentation on Neisseria (GC and Neisseria meningitidis [MC]) transepithelial trafficking has more recently relied on polarized T84 human colorectal epithelial cell monolayers (32, 33, 36, 37, 46). Similar to cells in native epithelia, T84 cells have the capacity to polarize and form impermeable barriers with high electrical resistance when grown on semiporous membranes (10, 26). This latter attribute permits the detection of small changes in barrier integrity. T84 cells are derived from human colorectal epithelia and thus are a model system for a site that is infected by GC in vivo. CD66 receptors have been demonstrated on the apical membranes of polarized T84 monolayers, and Opa-mediated binding of these receptors allowed rapid traversal of nonpiliated GC across the barrier (46).
Neisserial infections of polarized T84 monolayers share key features with infections of human organ cultures. GC invade, traverse, and exit hFTOC and T84 monolayers in identical time courses (28, 32). Very similar results were reported in studies comparing MC transcytosis in T84 and infected human nasopharyngeal organ cultures (32, 36, 39). Furthermore, the process of transepithelial trafficking does not disrupt monolayer barrier functions (32, 36, 46). The trafficking of GC across polarized T84 monolayers is influenced by a number of factors. Piliation modulates the speed of transepithelial trafficking in a manner that is independent of its role in attachment (32). Certain Opa variants also influence transcytosis. Nonpiliated (P−) strains traverse T84 monolayers very quickly, provided they express Opa variants that bind CD66 (46).
The pathogenic neisseriae, like a number of other mucosal pathogens, secrete immunoglobulin A1 (IgA1) protease, an enzyme that cleaves the hinge of human IgA1 (hIgA1) (34, 38). Numerous functions have been ascribed to the IgA1 protease, but its role in pathogenesis remains enigmatic. The protease has been proposed to promote bacterial colonization through cleavage of hIgA1 on the mucosal surface. IgA1 protease activity, hIgA1 cleavage fragments (2), and anti-IgA1 protease antibodies (16) have been found in the cervical mucus of infected women. A recent human challenge study showed that an iga (IgA1 protease gene) mutant was not impaired in its ability to initiate an infection in the human male urethra (19).
The IgA1 protease also cleaves LAMP1 (15, 25), a major integral membrane glycoprotein of lysosomes with an hIgA1-like hinge in its luminal domain (5). Proteolysis accelerates the LAMP1 degradation rate (25) and results in multiple alterations in the lysosomes of infected cells (1). An iga mutant is defective in intracellular growth, compared to the wild-type (WT) parent strain (25), and this phenotype is likely to be due to the inability of the mutant to cleave LAMP1 and alter lysosomes.
Studies on the effects of the IgA1 protease on host cell lysosomes were performed using the A431 human epidermoid carcinoma cell line grown on a solid support and the GC clinical isolate GCM740 and its isogenic iga derivative, GCM740Δ4 (38). To further our understanding of the virulence function(s) of the IgA1 protease, we sought to determine whether the protease affects GC interactions with T84 monolayers, including transepithelial trafficking. We report that infection of T84 cells, either grown on a solid substrate or polarized on semiporous membranes, by GC strain MS11 variant A (MS11A) also resulted in reduced steady-state levels of LAMP1. In contrast, T84 cells infected with MS11A500, an isogenic iga mutant of MS11A, had near-normal LAMP1 levels. We show that this iga mutant exited polarized T84 monolayers in significantly fewer numbers than the WT strain. Finally, we demonstrate that two iga mutants have a reproducible and statistically significant defect in their traversal across polarized T84 monolayers.
Construction and characterization of an iga mutant in strain MS11A.
In previous studies, we used GC strain GCM740Δ4, a P+ Opa− clinical isolate with a well-defined mutation in iga, the type 2 IgA1 protease gene (38). The iga mutant and GCM740, its WT parent, have identical outer membrane protein profiles and adhered to and invaded cells equally well (25). We wished to perform further studies on IgA1 protease interactions with T84 cells using an iga mutant in the MS11A background (14), as this laboratory strain has been used extensively for genetic studies and studies on cell adhesion, invasion, and transcytosis. Both strains are type 2 IgA1 protease producers (25).
To construct an iga null mutant in MS11A, the 4.2-kb HindIII fragment within the iga gene (codon 140 to beyond the stop codon) was deleted and the 1.6-kb HindIII fragment containing the kanamycin and bleomycin resistance cassette from pKIXX (Pharmacia) was inserted at this site. This construct, pKanΔiga2 (pBR322 background), contains ∼2.7 kb of 5′ iga sequence, the first 139 codons, and ∼2.5 kb of 3′ downstream sequence. The WT iga gene in strain MS11A was replaced with the mutated iga gene in pKanΔiga2 by transformation and allelic exchange. In the resulting mutant, MS11A500, >95% of the iga coding sequence has been exchanged for the kanamycin and bleomycin resistance cassette, while the 5′ upstream and 3′ downstream flanking sequences are intact (data not shown).
MS11A500 was tested for IgA1 protease activity. hIgA1 (4 μg; Calbiochem) was incubated with the supernatant (5.5 μl) from a 7.5-h culture of MS11A or MS11A500 grown in supplemented GCB broth as described previously (45). As a control, hIgA1 was incubated with purified recombinant Neisseria IgA1 protease (0.1 μg; Boehringer) or sterile medium. The proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and stained with Coomassie blue. Results show that hIgA1 was cleaved by the MS11A supernatant and by purified IgA1 protease, but not by GCB medium or by the MS11A500 supernatant (Fig. 1). MS11A500 therefore has no detectable IgA1 protease activity.
FIG. 1.

IgA1 protease activities of WT MS11A and its isogenic iga mutant MS11A500. Human IgA1 was incubated for 4 h at 37°C in growth medium (lane 1) or with purified recombinant Neisseria type 2 IgA1 protease (lane 2), the supernatant from a broth culture of MS11A (lane 3), or the supernatant from a broth culture of MS11A500 (lane 4), as described in the text. The reactions were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and stained with Coomassie blue. The arrow indicates the position of the IgA1 cleavage products of the hIgA1 heavy chain. The arrowhead indicates the position of the full-length hIgA1 heavy chain.
Next, MS11A500 was assessed for its ability to adhere to cells. T84 cells from the American Type Culture Collection were propagated as described previously (10) and infected with MS11A500 or the WT parent MS11A strain at a multiplicity of infection (MOI) of 10. At 2 and 4 h postinfection, the adhesion index of the infecting strains was determined as described before (45). WT MS11A adhered to T84 (Table 1) and A431 (32) cells comparably. MS11A500 adhered slightly better to T84 cells than MS11A (Table 1), although the difference is statistically significant only for the 2-h time point (P < 0.05, two-tailed t test for unpaired samples). MS11A500 also adhered slightly better to A431 human epithelial cells early after infection (data not shown).
TABLE 1.
Adhesion and invasion indices of MS11A and MS11A500a
| Strain | Cell line | Hours PI | Adhesion index | Invasion index |
|---|---|---|---|---|
| MS11A | T84 | 2 | 0.30 ± 0.11 | ND |
| MS11A500 | T84 | 2 | 0.56 ± 0.12b | ND |
| MS11A | T84 | 4 | 0.45 ± 0.27 | ND |
| MS11A500 | T84 | 4 | 0.60 ± 0.27c | ND |
| MS11A | A431 | 6 | 0.11 ± 0.01 | (2.16 ± 0.28) × 10−4 |
| MS11A500 | A431 | 6 | 0.09 ± 0.03c | (1.85 ± 0.72) × 10−4 |
Determined as described previously (45). The means and standard deviations of six replicates from a representative experiment are shown. PI, postinfection; ND, not determined.
P < 0.05 (two-tailed t test for unpaired samples).
Difference not statistically significant.
Attempts to compare the invasion indices of MS11A and MS11A500 using T84 cells were unsuccessful and yielded variable data. In these experiments, a significant number of extracellular bacteria survived gentamicin treatment, even at an antibiotic concentration of 400 μg/ml. The inability of gentamicin to kill all extracellular bacteria in T84 cultures may be due to the fact that T84 cells, like many epithelial cells (48), secrete large amounts of heavily glycosylated mucins to the luminal (apical) surface. This mucous layer is likely to reduce access of gentamicin to adherent bacteria. The invasiveness of MS11A500 was therefore assessed using the A431 human epithelial cell line as described previously (25). In these experiments, MS11A500 and MS11A invaded A431 cells equally well (Table 1), indicating that the null mutation in the MS11A iga gene does not affect the invasiveness of the strain.
LAMP1 levels in infected T84 cells.
Previous studies from our laboratory indicated that Neisseria infection of A431 cells resulted in reduced steady-state levels of LAMP1 (25). To determine whether T84 cells respond similarly to GC infection, LAMP1 levels were quantitated in GC-infected T84 cultures as described for A431 cells (25). T84 monolayers were infected with MS11A for 8 h at an MOI of 50, and total cell proteins from these cultures were immunoblotted with monoclonal antibodies against LAMP1 (H4A3) and β-tubulin (E7) (Developmental Studies Hybridoma Bank, University of Iowa [6]). The blots were developed using anti-mouse alkaline phosphatase antibodies (Boehringer) by a colorimetric reaction (Boehringer), and the signals from the blots were scanned and quantitated using the NIH Image version 1.61 program. β-Tubulin levels served as our internal control (1, 25). Results indicate that LAMP1 levels in cultures infected with WT MS11A were noticeably reduced compared to those in uninfected cells (Fig. 2A, compare lanes 1 and 2 with lanes 5 and 6). Normalization of LAMP1 signals to their respective internal β-tubulin signals revealed a 71% decrease in LAMP1 signals in MS11A-infected cultures. LAMP1 levels were higher in cultures infected with lower MOIs (S. Hopper, P. Ayala, and M. So, unpublished data), and the 71% reduction in steady-state LAMP1 levels calculated in this study is very similar to the value reported for A431 cultures infected with WT Neisseria at lower MOIs for longer periods of time (1, 25). Thus, the degree of LAMP1 reduction is dependent in part on the exposure of infected cells to the IgA1 protease.
FIG. 2.

LAMP1 levels in infected T84 cultures grown on a solid substrate (A) or polarized on semipermeable membranes (B). (A) Lanes 1 and 2, uninfected cultures; lanes 3 and 4, cultures infected with iga MS11A500; lanes 5 and 6, cultures infected with WT MS11A. (B) Lanes 1 and 2, uninfected monolayers; lanes 3 and 4, monolayers infected with WT MS11A.
To determine whether LAMP1 reduction is due to the IgA1 protease, MS11A500 was used to infect T84 cells under the same conditions as described above, and LAMP1 levels in these cultures were quantitated by immunoblotting. As shown in Fig. 2A, LAMP1 levels in MS11A500-infected cultures (lanes 3 and 4) were nearly identical to those in uninfected cultures (lanes 1 and 2). This is consistent with previous observations that A431 cells infected with the iga mutant GCM740Δ4 have nearly normal LAMP1 levels (25).
LAMP1 levels were next quantitated in polarized T84 monolayers. T84 cultures were maintained and polarized as described previously (32) and infected with MS11A for 17 h at an MOI of 10. Total proteins from infected and uninfected monolayers were quantitated by immunoblotting as described before (25). LAMP1 levels in infected polarized T84 monolayers were also reduced (Fig. 2B, compare lanes 1 and 2 with lanes 3 and 4). In this representative example, there was a 61% decrease in LAMP1.
Double immunofluorescence confocal microscopy of infected polarized T84 monolayers supported these immunoblot results. Polarized T84 monolayers were infected with MS11A at an MOI of 1 and processed for microscopy after 12 h as described before (Fig. 3) (31, 33). The monolayer was infected at a low MOI to allow comparison of LAMP1 levels in adjacent infected and uninfected cells. At this time, P+ Opa− MS11A organisms were mostly observed in the apical region of the cell. The panels in Fig. 3 represent successive ∼1-μm-thick optical cross sections of one such monolayer, beginning at the apical plasma membrane (A) and ending near the basal membrane (I). These micrographs illustrate that cells infected with bacteria (red) have few LAMP1 signals (green) throughout their entire length and demonstrate the extent of LAMP1 reduction in individual infected T84 cells. Taken together, these results demonstrate that GC infection of T84 cells, grown on solid supports and polarized on microporous membranes, also results in reduced LAMP1 levels and that this reduction is due to the bacterial IgA1 protease.
FIG. 3.
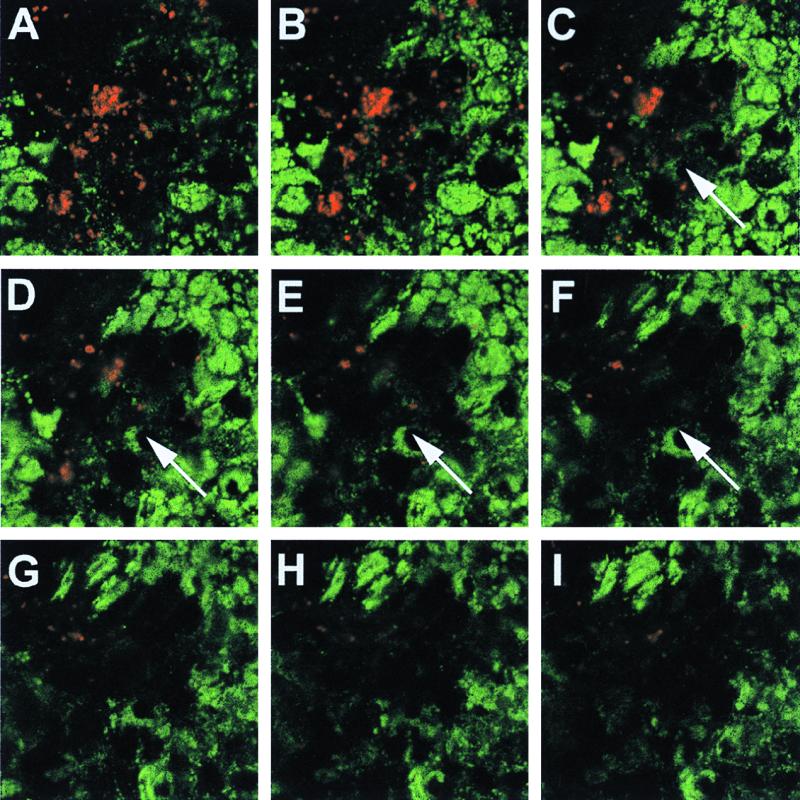
Double immunofluorescence laser scanning confocal microscopy of GC-infected T84 monolayers. Polarized T84 monolayers were infected with MS11A and fixed and stained for bacteria (red) and LAMP1 (green) using polyclonal anti-total GC protein antibody 8547 (33) and monoclonal anti-LAMP1 antibody H4A3 and secondary BODIPY anti-mouse and Texas red anti-rabbit antibodies (Molecular Probes). Each panel represents an ∼1-μm-thick optical cross section of an infected monolayer, starting at the apical membrane (A) and ending near the basal membrane (I). The arrows mark an infected region of the monolayer. Images were acquired with a Leica (L900) confocal laser scanning microscope and were compiled and processed using Adobe Photoshop version 4 software.
Transcytosis of WT MS11A and MS11A500 across polarized T84 monolayers.
The influence of the IgA1 protease on GC transcytosis was next examined. T84 monolayers were polarized as described before (32) and infected with MS11A or MS11A500 at an MOI of 10 (12 filters per strain). Between 22 and 34 h postinfection, the basal medium of each infected filter was plated every 2 h on GCB agar for determination of total bacterial counts (Fig. 4). In complete infection medium, the doubling time of the two GC strains is approximately 35 min (data not shown). The bacterial counts in the basal well are therefore not an accurate indication of the actual numbers of bacteria that have exited the monolayers between two time points. However, since the extracellular growth rates of these two GC strains are identical (data not shown), the relative bacterial counts should be the same, allowing a comparison of the WT and iga strains. In such an experiment, the number of exocytosed bacteria cannot be normalized to the total number of infecting bacteria, as to quantitate the latter would require lysing of the monolayers for determination of cell-associated bacterial counts.
FIG. 4.

Means and standard deviations of MS11A and MS11A500 exiting polarized T84 monolayers. Polarized T84 monolayers were infected with MS11A (black bars) or its isogenic iga mutant MS11A500 (white bars) (12 filters per strain). At various times, the basal medium from each infected filter was plated for determination of total bacterial counts. The values are the averages of all 12 filters. ∗, P < 0.05, two-tailed t test for unpaired samples.
At 24 h postinfection, no bacteria were recovered from the basal well of any infected filter; at 26 and 28 h, only a small number of MS11A and MS11A500 organisms had crossed their monolayers. At 30, 32, and 34 h, sizeable numbers of MS11A were recovered from the basal wells. In contrast, significantly fewer MS11A500 organisms were recovered. For instance, at the 34-h time point, there were 4.8 times as many MS11A organisms in the basal well as MS11A500 organisms (P < 0.05, two-tailed t test for unpaired samples). At the last time point, the electrical resistance of all infected monolayers was high, indicating that infection did not noticeably affect barrier integrity. An experiment comparing GCM740 and GCM740Δ4 yielded similar results (data not shown). Thus, fewer iga mutants than WT parental strains exited polarized T84 monolayers.
Finally, the transcytosis times of WT and iga mutants were examined. The apical wells of polarized monolayers were inoculated with WT bacteria (GCM740 or MS11A) or their isogenic iga mutants (GCM740Δ4 or MS11A500). At each time point, each infected filter was transferred to a new well containing sterile medium in another microtiter plate, and this new plate and the plate of basal medium from the previous incubation period were incubated further. The presence of bacteria in the basal medium from the previous time point was determined by the turbidity and the yellow color of the medium after overnight incubation. The basal medium was scored as either positive or negative for bacteria. A positive score for a particular basal medium was taken as an indication that by that time point bacteria had crossed the filters. For each bacterial strain, the number of wells yielding bacteria in the basal medium was expressed as the percentage of the total number of wells infected. In all, 67 experiments were performed, using a total of 3,180 polarized T84 filters; 786 filters were infected with MS11A, 845 filters were infected with MS11A500, 772 filters were infected with GCM740, and 777 filters were infected with GCM740Δ4. Results are shown in Fig. 5. For each strain, each time point represents an average of the sum of filters from all experiments. The electrical resistance of all monolayers was determined before and after each experiment. The measurements were consistently high for all monolayers up to the 36-h time point (data not shown). Thus, the bacteria were not destroying the cellular tight junctions and entering the basal medium through intercellular spaces or damaged cells.
FIG. 5.

Transcytosis times of WT GC strains and their isogenic iga mutants across polarized T84 monolayers. At defined intervals after inoculation, the basal medium was removed and incubated further to determine the presence of viable bacteria. At each time point, the number of infected filters yielding bacteria in the basal medium is expressed as a percentage of the total number of filters infected. The least-likelihood-ratio test of each pair of strains yielded a P value of <0.005 in both cases.
Results indicate that in both the GCM740 and MS11A genetic backgrounds the iga mutants were 3 to 5 h slower in crossing the monolayers than the WT strains at earlier stages of infection. The differences in transcytosis times between WT and iga mutants are statistically significant: the least-likelihood-ratio test yielded a P value of < 0.005 for each pair of strains assayed. In this experiment, bacteria were detected in the basal medium as early as 10 h postinfection. In contrast, results from the previous experiment indicate that bacteria did not enter the basal medium until 26 h postinfection. The discrepancy in these two results probably lies in the fact that in this experiment the presence of one bacterium in the basal well will result in a positive score for that filter. This method is therefore more sensitive than the preceding experiment (Fig. 4), which requires plating a portion of each basal well for determination of bacterial counts.
In the present study, we have demonstrated that GC infection of T84 cells, either grown on solid substrates or polarized on semiporous membranes, results in reduced steady-state levels of LAMP1 and that this reduction is due to the IgA1 protease (Fig. 2). We have also provided evidence that the MS11A500 iga mutant exited polarized T84 monolayers in fewer numbers than its parental WT MS11A strain (Fig. 4). This defect cannot be due to a reduced adhesion or invasion ability, as MS11A500 and MS11A invaded epithelial cells equally well and, in fact, MS11A500 was slightly more adherent than MS11A at 2 h postinfection (Table 1). It was reported previously that the iga mutant is defective in its ability to replicate within A431 cells (25). This traversal phenotype of the MS11A500 iga mutant may reflect a similar growth defect in T84 cells, although the hypothesis cannot be confirmed: intracellular growth assays cannot be performed in T84 cells due to the intrinsic limitations of the cell line. Alternately, the traversal phenotype may be due to an undefined trafficking defect.
We have also provided evidence that in the earlier stages of bacterial transcytosis (up to 36 h postinfection), iga mutants in the GCM740 and MS11A genetic backgrounds were 3 to 5 h slower than the WT strain in crossing polarized T84 monolayers (P < 0.005 for each pair of strains [Fig. 5]). These results strongly support the notion that the IgA1 protease influences GC transcytosis across polarized T84 monolayers. The partial transcytosis defect of these iga mutants may be related to the defect in intracellular replication observed for the GCM740 iga mutant in A431 cells (25), although we cannot offer an explanation for how fewer intracellular bacteria might result in a slower-trafficking phenotype. Alternatively, the slow-transcytosis phenotype of the iga mutants may be due to the defect in IgA1 protease-mediated processing of bacterial (38) and/or host cell membrane proteins.
That an iga mutant is affected in its intracellular trafficking is not at variance with the results from a previous study which concluded that an iga mutant and its isogenic WT parent behaved identically in infected hFTOC (9). The nature of the hFTOC assay precluded the detailed examination of bacterial transcytosis and exocytosis. Nor can our results be compared to those from a recent study which demonstrated that expression of the CD66-binding Opa variants by GC and Escherichia coli recombinants resulted in rapid bacterial transcytosis across polarized T84 monolayers (46). That study compared P− Opa− with P− Opa52-expressing MS11, while all our strains were P+ Opa−. A P+ Opa− MS11 strain in that study did not cross polarized T84 monolayers within the time frame of the experiment (24 h), in contrast to the results from this and other studies (32; S. Hopper and M. So, unpublished data; S. Clary and M. So, unpublished data), which indicate that P+ Opa− MS11A transcytoses T84 monolayers at a predictable time course of 36 to 48 h. We believe that the discrepancy in these results is likely to be due to the pilin variants expressed by these strains (S. D. Gray-Owens, personal communication).
Recent studies indicated that an iga mutant of GC strain FA1090 was able to colonize the urethra in male volunteers and cause gonococcal urethritis (19). Thus, the IgA1 protease is not strictly required for establishing a urethral infection. In relating these observations to our findings, it must be borne in mind that the human challenge model, although powerful in many respects, has practical limitations that do not permit a full examination of the role of the IgA1 protease in gonococcal disease. As stated by the authors of reference 19, the challenge model does not detect slight differences in bacterial infectivity. Our findings that the IgA1 protease has a mild effect on GC traversal across T84 monolayers would likely be missed in the challenge study. For ethical reasons, the human challenge study permits examination only of early stages of an infection and thus cannot address issues pertinent to later stages of a gonorrhea infection, such as persistence. GC is able to colonize and infect a number of sites besides the urethra, all of which have distinguishing characteristics. It also causes disease in women. If the IgA1 protease plays a role in the infectivity of bacteria for these other sites, this would be missed by the challenge model.
In summary, our studies strongly suggest that the IgA1 protease plays a role in GC transepithelial trafficking. The ability to detect such subtle defects illustrates the power and sensitivity of the T84 system for such studies. It will be interesting to determine the exact molecular basis for these trafficking defects and the significance of these findings to gonococcal disease.
Acknowledgments
This work was supported in part by NIH grant RO1 AI32493 awarded to M. So.
REFERENCES
- 1.Ayala P, Lin L, Hopper S, Fukuda M, So M. Infection of epithelial cells by pathogenic neisseriae reduces the levels of multiple lysosomal constituents. Infect Immun. 1998;66:5001–5007. doi: 10.1128/iai.66.10.5001-5007.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blake M, Holmes K K, Swanson J. Studies on gonococcus infection. XVII. IgA1-cleaving protease in vaginal washings from women with gonorrhea. J Infect Dis. 1979;139:89–92. doi: 10.1093/infdis/139.1.89. [DOI] [PubMed] [Google Scholar]
- 3.Bos M P, Grunert F, Belland R J. Differential recognition of members of the carcinoembryonic antigen family by Opa variants of Neisseria gonorrhoeae. Infect Immun. 1997;65:2353–2361. doi: 10.1128/iai.65.6.2353-2361.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buchanan T M, Pearce W A, Schoolnik G K, Arko R J. Protection against infection with Neisseria gonorrhoeae by immunization with outer membrane protein complex and purified pili. J Infect Dis. 1977;136(Suppl.):S132–S137. doi: 10.1093/infdis/136.supplement.s132. [DOI] [PubMed] [Google Scholar]
- 5.Carlsson S R, Roth J, Piller F, Fukuda M. Isolation and characterization of human lysosomal membrane glycoproteins, h-lamp1 and h-lamp-2. J Biol Chem. 1988;263:18911–18919. [PubMed] [Google Scholar]
- 6.Chen J W, Chen G L, D'Souza M P, Murphy T L, August J T. Lysosomal membrane glycoproteins: properties of LAMP-1 and LAMP-2. Biol Soc Symp. 1986;51:97–112. [PubMed] [Google Scholar]
- 7.Chen T, Gotschlich E C. CGM1a antigen of neutrophils, a receptor of gonococcal opacity proteins. Proc Natl Acad Sci USA. 1996;93:14851–14856. doi: 10.1073/pnas.93.25.14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen T, Grunert F, Medina-Marino A, Gotschlich E C. Several carcinoembryonic antigens (CD66) serve as receptors for gonococcal opacity proteins. J Exp Med. 1997;185:1557–1564. doi: 10.1084/jem.185.9.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooper M D, McGee Z A, Mulks M H, Kooney J M, Hindman T L. Attachment to and invasion of human fallopian tube mucosa by an IgA1 protease-deficient mutant of Neisseria gonorrhoeae and its wild type parent. J Infect Dis. 1984;150:737–744. doi: 10.1093/infdis/150.5.737. [DOI] [PubMed] [Google Scholar]
- 10.Dharmsathaphorn K, Madara J L. Established intestinal cell lines as model systems for electrolyte transport studies. Methods Enzymol. 1990;192:354–389. doi: 10.1016/0076-6879(90)92082-o. [DOI] [PubMed] [Google Scholar]
- 11.Grassme H U, Ireland R M, van Putten J P. Gonococcal opacity protein promotes bacterial entry-associated rearrangements of the epithelial cell actin cytoskeleton. Infect Immun. 1996;64:1621–1630. doi: 10.1128/iai.64.5.1621-1630.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gray-Owen S D, Dehio C, Haude A, Grunert F, Meyer T F. CD66 carcinoembryonic antigens mediate interactions between Opa-expressing Neisseria gonorrhoeae and human polymorphonuclear phagocytes. EMBO J. 1997;16:3435–3445. doi: 10.1093/emboj/16.12.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gray-Owen S D, Lorenzen D R, Haude A, Meyer T F, Dehio C. Differential Opa specificities for CD66 receptors influence tissue interactions and cellular response to Neisseria gonorrhoeae. Mol Microbiol. 1997;26:971–980. doi: 10.1046/j.1365-2958.1997.6342006.x. [DOI] [PubMed] [Google Scholar]
- 14.Hagblom P, Segal E, Billyard E, So M. Intragenic recombination leads to pilus antigenic variation in Neisseria gonorrhoeae. Nature. 1985;315:156–158. doi: 10.1038/315156a0. [DOI] [PubMed] [Google Scholar]
- 15.Hauck C R, Meyer T F. The lysosomal/phagosomal membrane protein h-lamp-1 is a target of the IgA1 protease of Neisseria gonorrhoeae. FEBS Lett. 1997;405:86–90. doi: 10.1016/s0014-5793(97)00163-4. [DOI] [PubMed] [Google Scholar]
- 16.Hedges S R, Mayo M S, Kallman L, Mestecky J, Hook III E W, Russell M W. Evaluation of immunoglobulin A1 (IgA1) protease and IgA1 protease-inhibitory activity in human female genital infection with Neisseria gonorrhoeae. Infect Immun. 1998;66:5826–5832. doi: 10.1128/iai.66.12.5826-5832.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.James J F, Swanson J. Studies on gonococcus infection. XIII. Occurrence of color/opacity colonial variants in clinical cultures. Infect Immun. 1978;19:332–340. doi: 10.1128/iai.19.1.332-340.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jerse A E, Cohen M S, Drown P M, Whicker L G, Isbey S F, Seifert H S, Cannon J G. Multiple gonococcal opacity proteins are expressed during experimental urethral infection in the male. J Exp Med. 1994;179:911–920. doi: 10.1084/jem.179.3.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johannsen D B, Johnston D M, Koymen H O, Cohen M S, Cannon J G. A Neisseria gonorrhoeae immunoglobulin A1 protease mutant is infectious in the human challenge model of urethral infection. Infect Immun. 1999;67:3009–3013. doi: 10.1128/iai.67.6.3009-3013.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson A P, Taylor-Robinson D, McGee Z A. Species specificity of attachment and damage to oviduct mucosa by Neisseria gonorrhoeae. Infect Immun. 1977;18:833–839. doi: 10.1128/iai.18.3.833-839.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kallstrom H, Liszewski M K, Atkinson J P, Jonsson A B. Membrane cofactor protein (MCP or CD46) is a cellular pilus receptor for pathogenic Neisseria. Mol Microbiol. 1997;25:639–647. doi: 10.1046/j.1365-2958.1997.4841857.x. [DOI] [PubMed] [Google Scholar]
- 22.Kellogg D S, Jr, Cohen I R, Norins L C, Schroeter A L, Reising G. Neisseria gonorrhoeae. II. Colonial variation and pathogenicity during 35 months in vitro. J Bacteriol. 1968;96:596–605. doi: 10.1128/jb.96.3.596-605.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kellogg D S, Jr, Peacock W L, Jr, Deacon W E, Brown L, Pirkle C I. Neisseria gonorrhoeae. I. Virulence genetically linked to clonal variation. J Bacteriol. 1963;85:1274–1279. doi: 10.1128/jb.85.6.1274-1279.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kupsch E M, Knepper B, Kuroki T, Heuer I, Meyer T F. Variable opacity (Opa) outer membrane proteins account for the cell tropisms displayed by Neisseria gonorrhoeae for human leukocytes and epithelial cells. EMBO J. 1993;12:641–650. doi: 10.1002/j.1460-2075.1993.tb05697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin L, Ayala P, Larson J, Mulks M, Fukuda M, Carlsson S R, Enns C, So M. The Neisseria type 2 IgA1 protease cleaves LAMP1 and promotes survival of bacteria within epithelial cells. Mol Microbiol. 1997;24:1083–1094. doi: 10.1046/j.1365-2958.1997.4191776.x. [DOI] [PubMed] [Google Scholar]
- 26.Madara J L, Stafford J, Dharmsathaphorn K, Carlson S. Structural analysis of a human intestinal epithelial cell line. Gastroenterology. 1987;92:1133–1145. doi: 10.1016/s0016-5085(87)91069-9. [DOI] [PubMed] [Google Scholar]
- 27.Makino S, van Putten J P, Meyer T F. Phase variation of the opacity outer membrane protein controls invasion by Neisseria gonorrhoeae into human epithelial cells. EMBO J. 1991;10:1307–1315. doi: 10.1002/j.1460-2075.1991.tb07649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGee Z, Stephens D, Hoffman L, Schlech III W, Horn R. Mechanisms of mucosal invasion by pathogenic Neisseria. Rev Infect Dis. 1983;5(Suppl. 4, Sept.-Oct.):S708–S714. doi: 10.1093/clinids/5.supplement_4.s708. [DOI] [PubMed] [Google Scholar]
- 29.McGee Z A, Woods M L., Jr Use of organ cultures in microbiological research. Annu Rev Microbiol. 1987;41:291–300. doi: 10.1146/annurev.mi.41.100187.001451. [DOI] [PubMed] [Google Scholar]
- 30.McGee Z A, Johnson A P, Taylor-Robinson D. Pathogenic mechanisms of Neisseria gonorrhoeae: observations on damage to human fallopian tubes in organ culture by gonococci of colony type 1 or type 4. J Infect Dis. 1981;143:413–422. doi: 10.1093/infdis/143.3.413. [DOI] [PubMed] [Google Scholar]
- 31.Merz A J, Enns C A, So M. Type IV pili of pathogenic neisseriae elicit cortical plaque formation in epithelial cells. Mol Microbiol. 1999;32:1316–1332. doi: 10.1046/j.1365-2958.1999.01459.x. [DOI] [PubMed] [Google Scholar]
- 32.Merz A J, Rifenbery D B, Arvidson C G, So M. Traversal of a polarized epithelium by pathogenic Neisseriae: facilitation by type IV pili and maintenance of epithelial barrier function. Mol Med. 1996;2:745–754. [PMC free article] [PubMed] [Google Scholar]
- 33.Merz A J, So M. Attachment of piliated, Opa− and Opc− gonococci and meningococci to epithelial cells elicits cortical actin rearrangements and clustering of tyrosine-phosphorylated proteins. Infect Immun. 1997;65:4341–4349. doi: 10.1128/iai.65.10.4341-4349.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plaut A G, Gilbert J V, Artenstein M S, Capra J D. Neisseria gonorrhoeae and Neisseria meningitidis: extracellular enzyme cleaves human immunoglobulin A. Science. 1975;190:1103–1105. doi: 10.1126/science.810892. [DOI] [PubMed] [Google Scholar]
- 35.Prall F, Nollau P, Neumaier M, Haubeck H D, Drzeniek Z, Helmchen U, Loning T, Wagener C. CD66a (BGP), an adhesion molecule of the carcinoembryonic antigen family, is expressed in epithelium, endothelium, and myeloid cells in a wide range of normal human tissues. J Histochem Cytochem. 1996;44:35–41. doi: 10.1177/44.1.8543780. [DOI] [PubMed] [Google Scholar]
- 36.Pujol C, Eugene E, de Saint Martin L, Nassif X. Interaction of Neisseria meningitidis with a polarized monolayer of epithelial cells. Infect Immun. 1997;65:4836–4842. doi: 10.1128/iai.65.11.4836-4842.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pujol C, Eugene E, Marceau M, Nassif X. The meningococcal PilT protein is required for induction of intimate attachment to epithelial cells following pilus-mediated adhesion. Proc Natl Acad Sci USA. 1999;96:4017–4022. doi: 10.1073/pnas.96.7.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shoberg R J, Mulks M H. Proteolysis of bacterial membrane proteins by Neisseria gonorrhoeae type 2 immunoglobulin A1 protease. Infect Immun. 1991;59:2535–2541. doi: 10.1128/iai.59.8.2535-2541.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stephens D S, Farley M M. Pathogenic events during infection of the human nasopharynx with Neisseria meningitidis and Haemophilus influenzae. Rev Infect Dis. 1991;13:22–33. doi: 10.1093/clinids/13.1.22. [DOI] [PubMed] [Google Scholar]
- 40.Swanson J. Studies on gonococcus infection. IV. Pili: their role in attachment of gonococci to tissue culture cells. J Exp Med. 1973;137:571–589. doi: 10.1084/jem.137.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Putten J P, Duensing T D, Cole R L. Entry of OpaA+ gonococci into HEp-2 cells requires concerted action of glycosaminoglycans, fibronectin and integrin receptors. Mol Microbiol. 1998;29:369–379. doi: 10.1046/j.1365-2958.1998.00951.x. [DOI] [PubMed] [Google Scholar]
- 42.Virji M, Everson J S. Comparative virulence of opacity variants of Neisseria gonorrhoeae strain P9. Infect Immun. 1981;31:965–970. doi: 10.1128/iai.31.3.965-970.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Virji M, Makepeace K, Ferguson D J, Watt S M. Carcinoembryonic antigens (CD66) on epithelial cells and neutrophils are receptors for Opa proteins of pathogenic Neisseriae. Mol Microbiol. 1996;22:941–950. doi: 10.1046/j.1365-2958.1996.01551.x. [DOI] [PubMed] [Google Scholar]
- 44.Virji M, Watt S M, Barker S, Makepeace K, Doyonnas R. The N-domain of the human CD66a adhesion molecule is a target for Opa proteins of Neisseria meningitidis and Neisseria gonorrhoeae. Mol Microbiol. 1996;22:929–939. doi: 10.1046/j.1365-2958.1996.01548.x. [DOI] [PubMed] [Google Scholar]
- 45.Waldbeser L S, Ajioka R S, Merz A J, Puaoi D, Lin L, Thomas M, So M. The opaH locus of Neisseria gonorrhoeae MS11A is involved in epithelial cell invasion. Mol Microbiol. 1994;13:919–928. doi: 10.1111/j.1365-2958.1994.tb00483.x. [DOI] [PubMed] [Google Scholar]
- 46.Wang J, Gray-Owen S D, Knorre A, Meyer T F, Dehio C. Opa binding to cellular CD66 receptors mediates the transcellular traversal of Neisseria gonorrhoeae across polarized T84 epithelial cell monolayers. Mol Microbiol. 1998;30:657–671. doi: 10.1046/j.1365-2958.1998.01102.x. [DOI] [PubMed] [Google Scholar]
- 47.Weel G F L, Hopman C T P, Van Putten J P M. In situ expression and localization of Neisseria gonorrhoeae opacity proteins in infected epithelial cells: apparent role of Opa proteins in cellular invasion. J Exp Med. 1991;173:1395–1405. doi: 10.1084/jem.173.6.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang K, Baeckstrom D, Brevinge H, Hansson G C. Comparison of sialyl-Lewis a carrying CD43 and MUC1 mucins secreted from a colon carcinoma cell line for E-selectin binding and inhibition of leukocyte adhesion. Tumor Biol. 1997;18:175–187. doi: 10.1159/000218028. [DOI] [PubMed] [Google Scholar]