

Abstract
The incidence of oropharyngeal squamous cell carcinoma (OPSCC) has increased rapidly in the United States, driven by rising human papillomavirus (HPV) infections in the U.S. population. HPV-positive OPSCC patients have a better prognosis than HPV-negative patients. To gain insights into the unique biology of HPV(+) OPSCC that may contribute to its clinical behaviors, we performed a multi-stage epigenome-wide methylation profiling of leukocyte and tumor DNA in OPSCC patients and compared the methylation levels of CpG sites between HPV(+) and HPV(−) OPSCC patients. We identified and validated a significantly differentially methylated region (DMR) of 1,355 bp encompassing non-coding RNA 886 (nc886) gene and its promoter region. Nc886 is hypermethylated in both leukocytes and tumor DNA of HPV(+) OPSCC patients. Homozygous knockout of nc886 by CRISPR-Cas9 in head and neck cell lines was lethal, but nc886 could be knocked out on the background of protein kinase R (PKR) knockout. Our data suggest that HPV induces nc886 hypermethylation, and nc886 acts as both a viral sensor and a tumor sensor in OPSCC patients and contribute to the better prognosis of HPV(+) OPSCC patients. Nc886 may become a therapeutic target in OPSCC.
Keywords: MT: Non-coding RNAs, oropharyngeal cancer, OPSCC, HPV, DNA methylation, non-coding RNA, nc886, PKR, leukocyte
Graphical abstract
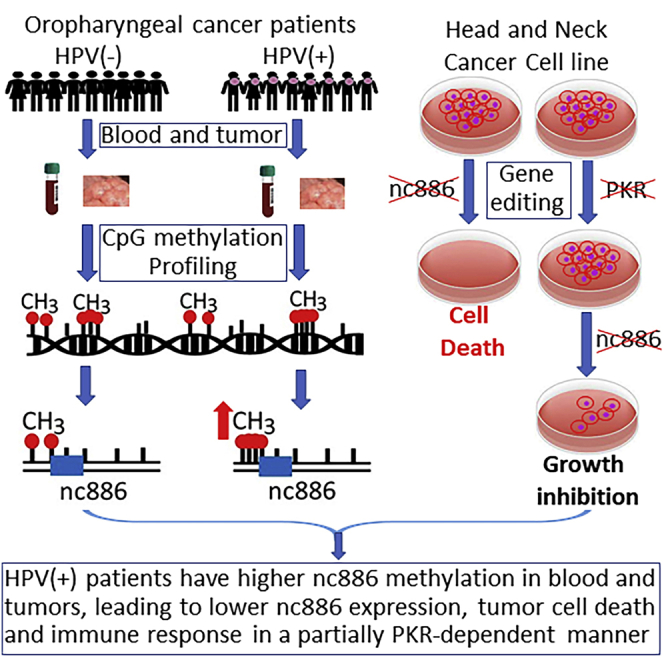
HPV-positive oropharyngeal squamous cell carcinoma (OPSCC) patients exhibit hypermethylation of non-coding RNA nc886 in both immune cells and tumors. Nc886 knockout causes growth inhibition and apoptosis. Nc886 hypermethylation acts as a defense mechanism in HPV-positive patients and contributes to their better prognosis. Nc886 may become a therapeutic target in OPSCC.
Introduction
Head and neck squamous cell carcinoma (HNSCC), a group of cancers in the upper aerodigestive tract including the oral cavity, larynx, oropharynx, hypopharynx, and nasopharynx, has traditionally been strongly associated with tobacco and alcohol use.1,2,3 In the United States and some Western countries, the incidence rate of overall HNSCC has been steadily decreasing in the past four decades because of decreased tobacco use; however, the incidence of OPSCC, a distinct subtype of HNSCC, has been increasing by about 2.5% per year.4 This increase in OPSCC incidence was driven by the rising HPV infections in the general population in these countries.4,5,6 The percentage of HPV-positive cases among all newly diagnosed OPSCC patients increased from approximately 20% in the 1980s to more than 70% currently, and OPSCC has surpassed cervical cancer as the most frequently diagnosed cancer caused by HPV.7 Although HPV vaccines are highly efficacious for the prevention of anogenital HPV infections and associated malignancies,8 the population-level effect of HPV vaccination on the burden of oral HPV infections in U.S. young adults was estimated to be only 25% in women and 7% in men because of low vaccine uptake.9 Given the high prevalence of HPV in U.S. population, poor HPV vaccine uptake in men, the male predominance of OPSCC, and the long latency of developing OPSCC following HPV exposure, it was estimated that the incidence of HPV(+) OPSCC will continue to rise until 2060.5
HPV(+) and HPV-negative OPSCC are distinct entities with unique epidemiological, clinical, and biological characteristics.10 Clinically, HPV(+) OPSCC patients have a significantly better therapeutic response and survival than HPV(−) patients.11,12 Biologically, although HPV(+) and HPV(−) OPSCC tumors have similar overall mutation rate and mutational burden,13,14 HPV(+) tumors have a significantly higher number of aberrant DNA methylations than HPV(−) OPSCC tumors.15,16 DNA methylation at CpG dinucleotides plays important roles in cancer development and progression.17 Hypermethylation in the promoter regions of tumor suppressor genes leads to their transcriptional silencing, and global hypomethylation causes chromosomal instability and/or activate the expression of proto-oncogenes.17 HPV oncoproteins E6 and E7 can directly target methyltransferases and modulate DNA methylation.18,19 Many studies have compared DNA methylations between HPV(+) and HPV(−) HNSCC tumors and identified potential differentially methylated CpG sites (DMCs) and differentially methylated regions (DMRs).15,16,20,21,22,23,24 Differential DNA methylations contribute to the clinical and biological differences between HPV(+) and HPV(−) tumors.
Besides inducing methylation changes in tumor tissues, HPV infection also induces extensive systemic DNA methylation alterations, particularly in the immune system. CpG methylation in leukocyte DNA has profound, long-standing effects on inflammation and immune responses.25 Leukocyte DNA methylation profile has been used to derive systemic inflammation indices26,27,28,29 and immune cell lineages.25 This may be particularly relevant to HPV-associated OPSCC, as HPV-induced immune activation has been shown to be a critical driver of OPSCC prognosis.30,31,32,33 No study has specifically compared DNA methylations in leukocytes from HPV(+) OPSCC patients and HPV(−) patients.
In this study, we performed a multi-stage epigenome-wide methylation profiling to leukocytes and tumor tissues from HPV(+) and HPC(−) OPSCC patients to identify DMCs and DMRs between HPV(+) and HPV(−) OPSCC patients. The most notable observation was that nc886 was specifically hypermethylated in both leukocyte and tumor DNA of HPV(+) compared with HPV(−) patients. Moreover, functional studies suggest that hypermethylation of nc886, leading to down-regulation of nc886, may contribute to the better prognosis of HPV(+) OPSCC patients.
Results
Hypermethylation of nc886 gene in leukocyte DNA from HPV(+) OPSCC patients compared with HPV(−) patients
We conducted a pilot epigenome-wide association analysis (EWAS) of DNA methylation with the HPV status of OPSCC patients. We used a three-phase case-control design: the discovery phase included 5 pairs of HPV(+) and HPV(−) OPSCC patients, the internal validation phase consisted of 15 HPV(+) and 13 HPV(−) OPSCC patients, and the external validation phase had 79 HPV(+) and 78 HPV(−) OPSCC patients (Table 1). A total of 790,462 CpG sites passed quality control and were analyzed for their differential methylation between HPV(+) and HPV(−) OPSCC. In individual CpG site analysis, 1,107 CpG sites reached a false discovery rate (FDR) q value of <0.05, among which 25 sites had test statistic scores (d) of >5 and were defined as highly significant DMCs (Table S1). Most of the DMCs are in open seas, although some are located in potential enhancer regions and DNase I hypersensitivity sites, and two CpG sites are located in CpG islands (Table S1). Gene Ontology (GO) enrichment analyses using genes containing these 1,107 DMCs found the top enriched biological processes included cytoskeleton organization, negative regulation of transcription by polymerase II, cell cycle process, cellular localization, and organelle organization, and the top enriched molecular functions were kinase binding, enzyme binding, ion binding, and protein binding (data not shown). Reactome pathway enrichment analysis revealed that three pathways, chromatin-modifying enzymes, chromatin organization, and olfactory signaling pathway, were enriched, with an FDR < 0.05 (Table S2).
Table 1.
Clinical characteristics of study populations
| Discovery stage, N (%) |
Internal validation, N (%) |
External validation, N (%) |
||||
|---|---|---|---|---|---|---|
| HPV(+) | HPV(−) | HPV(+) | HPV(−) | HPV(+) | HPV(−) | |
| N | 5 (50.0) | 5 (50.0) | 15 (53.6) | 13 (46.4) | 79 (50.3) | 78 (49.7) |
| Age (years) | ||||||
| Mean (SD) | 58 (6.6) | 58.8 (5.2) | 58.1 (10.6) | 61.6 (10.6) | 55 (8.6) | 58 (10.12) |
| Median (range) | 55 (51–66) | 58 (52–64) | 54 (46–83) | 59 (48–83) | 56 (39–78) | 58 (42–86) |
| Gender | ||||||
| Female | 1 (20.0) | 1 (20.0) | 0 | 7 (53.8) | 12 (15.2) | 14 (20.6) |
| Male | 4 (80.0) | 4 (80.0) | 15 (100.0) | 6 (46.2) | 67 (84.8) | 64 (79.4) |
| Smoking status | ||||||
| Never | 0 | 0 | 5 (33.3) | 0 | 24 (30.8) | 23 (30.6) |
| Former | 3 (60.0) | 3 (60.0) | 5 (33.3) | 4 (30.8) | 50 (64.1) | 35 (46.7) |
| Current | 2 (40.0) | 2 (40.0) | 5 (33.3) | 9 (69.2) | 4 (5.1) | 17 (22.7) |
Because DMRs, particularly those within or encompassing CpG islands, are more likely to be functionally important, we focused on analyses on DMRs rather than individual CpG sites. We used the Bumphunter method34 to identify DMRs between HPV(+) and HPV(−) patients. At the discovery phase, there were 86 DMRs at p value area < 0.05; at the internal validation stage, there were 32 DMRs with the same testing criteria. Four DMRs were consistent in both the discovery and internal validation phases (Table 2). Each DMR contained at least one CpG island or was part of a CpG island. All the four replicated DMRs are located near genes and in the DNase I hypersensitivity region on the basis of University of California, Santa Cruz (UCSC), annotation. The most significant one was is located on chromosome 5 covering the gene of nc886 and its promoter region (1,500 bp of transcription start site [TSS1500]). The methylation levels of all the 15 CpG sites in the nc886 DMR were consistently higher in HPV(+) OPSCC patients than in HPC(−) patients (Figure 1A).
Table 2.
Differentially methylated regions in leukocyte DNA between HPV(+) and HPV(−) OPSCC patients
| Chromosome | Gene | Discovery |
Internal validation |
External validation |
|||
|---|---|---|---|---|---|---|---|
| Δβ valuea | p value | Δβ valuea | p value | Δβ valuea | p value | ||
| Chr5 | Nc886 | 0.130 | 0.003 | 0.100 | 0.001 | 0.046 | 0.042 |
| Chr11 | CAT | −0.091 | 0.006 | −0.033 | 0.020 | −0.012 | 0.020 |
| Chr12 | GLIPR1L2 | −0.120 | 0.010 | −0.062 | 0.009 | 0.008 | 0.321 |
| Chr6 | PTCHD4 | −0.054 | 0.049 | −0.034 | 0.032 | −0.002 | 0.759 |
Δβ value is the difference of the mean β of all the CpG sites in the DMR between HPV(+) and HPV(−) patients.
Figure 1.

CpG sites in the Nc886 genomic region are significantly hypermethylated in HPV(+) OPSCC patients compared with HPV(−) patients
(A) Leukocytes (discovery and interval validation). (B) Tumor tissues.
We then used a large independent external validation population to validate these four DMRs. Two of the four DNRs were validated, nc886 region and CAT gene on chromosome 11, which was hypomethylated in HPV(+) OPSCC patients (Table 2). Figure 2 and Table 3 show the detailed distribution of the 15 CpG sites within this DMR and their differential methylation in the validation population. One CpG site, cg04481923, sits in the gene body of nc886. This CpG site and its close neighboring CpG sites exhibited >20% higher methylation in HPV(+) OPSCC patients than HPV(−) patients.
Figure 2.

Detailed distribution of the 15 differentially methylated CpG sites in the DMR of nc886 gene and vicinity in leukocytes of the external validation patient population
Table 3.
Differentially methylated region of nc886 on 5q31.1 and its encompassing CpG sites
| CpG ID | β value |
Δβ valuea | Chromosome positions | Gene locations | p value | |
|---|---|---|---|---|---|---|
| HPV (−) | HPV (+) | |||||
| cg15837280 | 0.242 | 0.265 | 0.024 | 135415258 | 0.280 | |
| cg07158503 | 0.381 | 0.453 | 0.072 | 135415693 | 0.042 | |
| cg04515200 | 0.177 | 0.202 | 0.025 | 135415762 | 0.113 | |
| cg13581155 | 0.104 | 0.123 | 0.019 | 135415781 | 0.074 | |
| cg11978884 | 0.159 | 0.171 | 0.012 | 135415819 | 0.558 | |
| cg11608150 | 0.267 | 0.327 | 0.060 | 135415948 | 0.021 | |
| cg06478886 | 0.283 | 0.339 | 0.056 | 135416029 | 0.018 | |
| cg04481923 | 0.298 | 0.359 | 0.062 | 135416205 | Gene body | 0.038 |
| cg18678645 | 0.270 | 0.321 | 0.051 | 135416331 | TSS200 | 0.061 |
| cg06536614 | 0.384 | 0.456 | 0.072 | 135416381 | TSS200 | 0.036 |
| cg25340688 | 0.379 | 0.454 | 0.075 | 135416398 | TSS200 | 0.035 |
| cg26896946 | 0.353 | 0.417 | 0.065 | 135416405 | TSS200 | 0.042 |
| cg00124993 | 0.288 | 0.342 | 0.054 | 135416412 | TSS200 | 0.049 |
| cg08745965 | 0.263 | 0.313 | 0.050 | 135416529 | TSS1500 | 0.052 |
| cg18797653 | 0.352 | 0.411 | 0.059 | 135416613 | TSS1500 | 0.060 |
Δβ value is the difference of the mean β of all the CpG sites in the DMR between HPV(+) and HPV(−) patients.
nc886 DMR is also hypermethylated in HPV(+) compared with HPV(−) OPSCC tumors
To determine whether nc886 DMR is also hypermethylated in HPV(+) OPSCC tumors, we performed the same MethylationEPIC methylation array in tumor DNA from the 5 pairs of HPV(+) and HPV(−) OPSCC patients in the discovery phase. Like leukocyte DNA, nc886 region was hypermethylated in HPV(+) tumors compared with HPV(−) tumors (mean β value 0.613 vs. 0.525, p = 0.0387). We further confirmed nc886 hypermethylation in HPV(+) OPSCC tumors using a Cancer Genome Atlas (TCGA) methylation dataset of 28 HPV(+) tumors and 40 HPV(−) tumors.35 Because TCGA used Illumina Methylation450K arrays, there were slightly fewer CpG sites covered in this region than MethylationEPIC (12 vs. 15 sites). Again, the mean β values of all CpG sites in nc886 were significantly higher in HPV(+) tumors than HPV(−) tumors (0.632 vs. 0.540, p < 0.001) in the TCGA dataset. All the 12 CpGs showed significantly higher methylation in HPV(+) tumors than in HPV(−) tumors (Figure 1B), similar to the observations in leukocytes. These data from independent patient cohorts provided compelling evidence that HPV infection induces nc886 promoter CpG island hypermethylation in both leukocytes and tumor DNA.
Functional consequence of nc886 knockout on HNSCC cell growth and apoptosis
We next determined the functional impact of nc886 knockout (KO) on HNSCC cell proliferation and apoptosis. We used the CRISPR-Cas9 HDR method to replace the nc886 with GFP in FaDu cells. GFP-positive cells were sorted using flow cytometry and cultured in 96-well plates before moving to 6-well plates for expansion. The DNA modification of nc886 sequence area was verified using Sanger sequencing (Figure 3A). After moving to 6-well plates, all the selected GFP(+) cell colonies died during expansion (Figure 3B). The inability to establish stable nc886 KO cells suggests that nc886 is essential for cell growth and knocking out nc886 leads to growth inhibition and cell death. We could not produce nc886 KO in another HNSCC cell line, SCC-47, either (data not shown).
Figure 3.

Knockout of nc886 in FaDu cells is lethal
Nc886 gene was replaced by GFP using CRISPR technology. (A) Sanger sequencing of nc886 gene in nc886KO cells showing replacement by GFP gene. (B) Representative images of two nc886KO colonies after moving from 96-well plates to 6-well plates. All the selected GFP(+) cell colonies died during expansion.
Previous studies have shown that nc886 binds and inhibits PKR activity.36 Nc886 KO is believed to induce apoptosis mainly through PKR activation, resulting in blockade of translation.37,38,39 We then tested whether we could generate nc886 KO in PKR KO FaDu cells. We first generated PKR KO cells (Figure 4A) and then introduced GFP into PKRKO cells to replace nc886. We were able to amplify GFP-positive colonies (Figure 4B). Sequencing confirmed nc886 sequence modification and real-time reverse transcription PCR showed dramatic reduction of nc886 expression (Figure 4C). Interestingly, the nc886KO/PKRKO cells grew slowly than the PKRKO cells (Figure 4D), suggesting that there might be additional pathway(s) that mediate nc886’s effect on cell proliferation and apoptosis besides PKR.
Figure 4.

Double knockout of PKR and nc886 in FaDu cells
(A) Confirmation of PKRKO by western blot. (B) Representative images of two PKRKO/nc886KO colonies. (C) Confirmation of nc886KO by real-time quantitative reverse transcription PCR. (D) CCK-8 cell proliferation assays of PKRKO and PKRKO/nc886KO cells.
Discussion
In this study, we applied epigenome-wide DNA methylation profiling to leukocytes and tumor tissues of OPSCC patients and identified a large number of DMCs and four DMRs between HPV(+) and HPV(−) patients. Most interestingly, we found that the methylation levels of nc886 were significantly higher in both leukocytes and tumors tissues in HPV(+) than HPV(−) OPSCC patients. Furthermore, we found that knockout of nc886 resulted in growth inhibition and apoptosis, suggesting that hypermethylation of nc886, leading to down-regulation of nc886, may contribute to the better prognosis of HPV(+) OPSCC patients. Methylation is generally tissue specific and rarely is there common methylation changes between immune cells and tumor cells. To our knowledge, this is the first report linking nc886 hypermethylation to HPV infection and the first report of a molecule that is hypermethylated in both leukocytes and tumors of cancer patients.
HPV(+) OPSCC differs from HPV(−) OPSCC in etiology, treatment, prognosis, and biology.40 HPV(+) OPSCC patients are more sensitive to radiotherapy and have significantly better survival than HPV(−) patients.11 The biological mechanisms underlying the better therapeutic response and survival of HPV(+) OPSCC patients are not fully understood. Although the mutational spectrums of HPV(+) and HPV(−) tumors are understandably different because of the different etiologic agents (HPV infection vs. smoking and alcohol exposure), the overall mutation rate and mutational burden of HPV(+) and HPV(−) tumors are similar13,14 On the other hand, HPV infection causes widespread DNA methylation alterations, as HPV oncoproteins E6 and E7 can directly target methyltransferases.18,19 HPV(+) HNSCC tumors have been shown to have higher amount of CpG methylation aberrations than HPV(−) HNSCC tumors.41,42 Several studies have compared DNA methylations between HPV(+) and HPV(−) OPSCC tumors and identified potential DMCs and DMRs.15,16,20,21,22,23,24 However, the results are highly heterogeneous, and there were very few consistent DMCs and DMRs between different studies. For example, CDKN2A and RASSF1A, two tumor suppressor genes, were reported to have hypermethylation, hypomethylation, or no significant methylation changes in HPV(+) compared with HPV(−) OPSCC in different studies.20,21,22,23,24 The inconsistent results can be attributed to multiple factors, including tumor heterogeneity, different assay techniques, different tissues (frozen or formalin-fixed, paraffin-embedded [FFPE]), reliability of HPV tests, confounding environmental exposures, and small sample sizes. Biologically, the differences of methylation levels between HPV(+) and HPV(−) OPSCC tumors are not expected to be as large as the differences between tumors and normal tissues. In the present study, for the first time, we found consistent nc886 hypermethylation in HPV(+) compared with HPV(−) OPSCC tumors in our own tumor samples and TCGA samples. Interestingly, when we queried nc886 methylation level in HPV16(+) and HPV16(−) cervical tumor tissues in the TCGA database, we found that nc886 methylation level was about 10% higher in HPV16(+) than in HPV16(−) cervical tumors (mean β value 0.500 vs. 0.455), although the difference did not reach statistical significance (p = 0.319), because more than 90% of cervical tumors are HPV16(+) and there were only nine HPV19(−) cervical tumors in the dataset. The fact that nc886 is also hypermethylated in both tumors and leukocytes of HPV(+) OPSCC patients, and suggestive evidence that it is hypermethylated in HPV16(+) cervical tumors, provides compelling evidence that nc886 is a bona fide HPV-regulated non-coding RNA modulated by DNA methylation.
Nc886 is a 101-nt-long, medium-sized non-coding RNA that is transcribed by RNA polymerase III.37 Nc886 controls gene expression by binding to and inhibiting the activity of PKR.36 PKR was originally identified as a double-stranded RNA-activated viral sensor, a serine-threonine kinase that phosphorylates the translation initiation factor eIF2 in response to viral infections. eIF2 phosphorylation blocks global translation of both cellular and viral mRNAs, as an efficient defense against virus replication.38,43 In addition to viral sensing, PKR is involved in many cellular signaling pathways.38 Analogous to its role in viral sensing, PKR was proposed to play a “tumor sensing” role because activation of PKR in cancers leads to translation blockade and apoptosis.37,38,39 The roles of nc886 in cancers are cell type and context dependent, playing a tumor suppressor role in some cancers such as leukemia and lung, esophageal, gastric, and prostate cancers44,45,46,47,48 and an oncogenic role in other cancers, including thyroid, renal, endometrial, cervical, and ovarian cancers.49,50,51,52,53 No previous study has evaluated nc886 in HNSCC broadly or OPSCC specifically. We showed that nc886 was hypermethylated in HPV(+) OPSCC patients. Single knockout of nc886 (mimicking hypermethylation) was lethal to FaDu cells and SCC-47 cells because of growth inhibition and apoptosis, supporting a pro-growth and oncogenic role of nc886 in OPSCC. Cells with double knockout of PKR and nc886 were viable, with a slower growth rate, supporting that PKR is a major mediator of nc886’s growth-promoting effect and that there are additional pathway(s) involved in nc886’s growth-promoting activity. In this regard, it is interesting to note that a recent publication showed nc886 promoted adenoviral gene expression and replication and its pro-adenoviral activity is not dependent on its function to inhibit PKR but via facilitating the nuclear entry of adenovirus.54 Consistent with our data, the above study also showed that it was not possible to generate nc886 knockout cell lines, but double-knockout cells of nc886 and PKR were viable.54 Given that the pro-growth nc886 is inhibited in HPV(+) OPSCC tumors through hypermethylation, it is tempting to speculate that nc886 inhibition and resulting increased apoptosis may at least partially contributes to the higher radiotherapy response and better prognosis of HPV(+) OPSCC patients.
In addition to directly inhibit tumor growth and promote apoptosis through hypermethylation in HPV(+) OPSCC tumors, nc886 could also have profound impact on immune system, because it is also hypermethylated in leukocytes of HPV(+) OPSCC patients. Golec et al.55 recently reported that nc886 controls PKR and NF-κB signaling during T cell activation and regulates cytokine production in human cells. A more recent study showed that nc886 suppresses the activation of interferon regulatory factor 3 (IRF3) and inhibits NF-κB and AP-1 via PKR pathway, leading to reduced expression of IFN-β and its downstream genes.56 Interferons, especially IFN-β, are essential players in the innate immune response against pathogens and also have strong anti-tumor activities.57 Hypermethylation of nc886 in leukocytes may therefore attenuate the inhibitory effect of nc886 on IFN-β production and enhance immune response. Figure 5 shows a simplified model of PKR-dependent nc886 activity in HPV(+) OPSCC. HPV infection increases nc886 gene methylation, leading to reduced nc886 expression and lessening of its inhibition on PKR activity. Increased PKR activity results in increased phosphorylation of translation initiation factor eIF2 and blockage of cellular and viral protein translation, thus exhibiting anti-tumor and anti-viral effects. In immune cells, increased PKR activity can increase IFN-β production and enhance immune response. The anti-tumor activity of nc886 hypermethylation in local epithelial cells and systemic immune cells both contribute to the better prognosis of HPV(+) compared with HPV(−) OPSCC patients. Nc886 hypermethylation acts as both a viral and a tumor sensor.
Figure 5.

A simplified model of PKR-dependent nc886 activity in HPV(+) OPSCC
Nc886 hypermethylation acts as both a viral and a tumor sensor. HPV infection increases nc886 gene methylation, leading to reduced nc886 expression and lessening of its inhibition on PKR activity. Increased PKR activity results in increased phosphorylation of translation initiation factor eIF2 and blockage of cellular and viral protein translation, thus exhibiting anti-tumor and anti-viral effects. Additionally, in immune cells, increased PKR activity can increase IFN-β production and enhance immune response. The anti-tumor activity of nc886 hypermethylation in local epithelial cells and systemic immune cells both contribute to the better prognosis of HPV(+) compared with HPV(−) OPSCC patients. There are other PKR-independent activities downstream of nc886, which are not depicted in this diagram.
The second consistent DMR observed in our study encompasses CAT gene and its promoter region on chromosome 11p13. CAT gene encodes catalase, a key antioxidant enzyme that converts reactive oxygen species hydrogen peroxide to water and oxygen, thus preventing the accumulation of oxygen species and impedes tumor initiation and progression.58 CAT gene promoter contains several CpG islands and previous reports have implicated DNA methylation as a regulatory mechanism of CAT gene expression.58,59,60 We found lower promoter methylation level of CAT gene in HPV(+) than HPV(−) OPSCC patients, indicating increased expression of catalase and elevated antioxidant activity, likely acting as a host defense mechanism against exogeneous HPV infection and tumor progression.
The major strengths of this study include the multi-stage design of discovery, internal and external validation, the integration of blood, tumor tissues, and cell line studies, and the novel, consistent, biologically plausible, and clinically significant observation of nc886 hypermethylation in HPV(+) OPSCC patients. This is the first report linking nc886 hypermethylation to HPV infection and the first report of a molecule that is hypermethylated in both blood and tumors. The major weakness of this study is the small sample size, which limits our ability to identify individual DMCs and DMRs with smaller effect sizes between HPV(+) and HPV(−) patients. Further larger studies are needed to validate individual DMCs and additional DMRs.
In summary, we performed an epigenome-wide CpG methylation profiling of leukocyte DNA in OPSCC patients and compared the methylation levels CpG sites between HPV(+) and HPV(−) patients. We found that nc886 exhibited consistent hypermethylation in both leukocyte and tumor DNA in HPV(+) OPSCC patients. Knockout of nc886 causes growth inhibition and apoptosis, and PKR pathway at least partially mediates the apoptotic effect of nc886 inhibition. Our study suggests that HPV caused nc886 hypermethylation, which acts as a viral and tumor sensor in OPSCC patients and may also partially contribute to the better survival of HPV(+) patients.
Materials and methods
Study patients and data collection
All patients in the discovery and internal validation were newly diagnosed, histopathologically confirmed, and untreated OPSCC patients recruited from the University of Texas MD Anderson Cancer Center (MDACC) as part of a case control study of HNSCC.61,62 All patients completed an epidemiological questionnaire to provide information on demographic and risk factors. Clinical and follow-up data were abstracted from the electronic medical record. The details of case recruitment and data collection were described in our previously studies.63,64 HPV16 status in tumor tissues was determined by in situ hybridization or specific RT-PCR, as well as p16 immunohistochemical analysis as a standard clinical practice at MDACC. The patients in the external validation stage were derived from a case control study of HNSCC in the greater Boston area, and the patient recruitment and data collection process was described in detail previously.65,66 This study was approved by the institutional review board of MDACC. All patients provided informed consent.
Epigenome CpG methylation profiling, bioinformatic, and quality control
The whole-epigenome CpG site methylation profiling for both the discovery and the internal and external validation stages was performed using Illumina human MethylationEPIC BeadChip as previously described.66,67 The MethylationEPIC BeadChip contains more than 850,000 CpG sites across the human epigenome, which is enriched for CpG sites located in gene promoter regions (54%), gene bodies (30%), and CpG islands (19%).68 Briefly, genomic DNA was treated with sodium bisulfite using the EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA) and hybridized to the BeadChips. BeadChips were scanned and the fluorescence intensities were extracted using the built-in Genome Studio Methylation Module. Raw fluorescence intensity data (idat files) were processed and normalized using Noob and Functional Normalization from Minfi R package.69 The detection p value, an indicative of the quality of the signal, for every CpG in every individual was calculated that compared the total signal (methylated + unmethylated) for each probe to the background signal level. The CpG sites that failed (p > 0.01) in more than 10% of the total samples were filtered out. Poor-quality samples on the basis of detection p value (mean p > 0.05) were also removed. The probes on the sex chromosomes, the probes for CpGs that overlaps with known SNPs, and the probes that are cross-reactive were excluded. The batch effect was removed using the ComBat function from R package.70 The variation in peripheral blood white cell proportions was controlled using cell proportion estimates generated by the estimateCellCounts function in Minfi R package.
Identification of differentially methylation CpG sites and regions
The significantly DMCs between HPV(+) and HPV(−) OPSCC patients were identified using the SAM method.71 This approach was based on analysis of random fluctuations in the data that accounted for the signal-to-noise ratio. In brief, the statistic was a modified t-test statistic that was based on the ratio of change in methylation level to SD in the data for that probe. The false discovery rate q value was estimated by 2,000 permutations with the modified t-test statistic d, which takes both the effect size and the SD of each CpG site into account. CpG sites with q value < 0.05 and the test score d > 5 were regarded as significant DMCs. To identify DMRs between HPV(+) and HPV(−) OPSCC patients, we applied the Bumphunter method.34 Data-driven clusters (methylation regions) were identified with at least 7 CpG sites in each cluster and the largest distance between each CpG site within one cluster being 500 bp. We used the loess curve to smooth the summary statistics of every consecutive CpG sites regressed on the HPV status and the unmeasured confounders. The cluster for which smoothed estimate passed the cutoff threshold (99% of all estimates) was classified as a candidate methylation region. Then each methylation region was assessed by 2,000 permutation tests to assign statistical uncertainty of the candidate region. DMRs with p value area < 0.05 were defined as significant DMRs. All statistical analyses were carried out using R software.
Knockout of nc886 using the CRISPR-Cas9 system
We attempted to knock out nc886 gene and replace with GFP reporter gene using the CRISPR-Cas9 homology-directed repair (HDR) method. Briefly, two partially complementary oligos containing guide RNA (gRNA) sequences for nc886 (gRNA-F: CACCGCGGGTCGGAGTTAGCTCAAG; gRNA-R: AAACCTTGAGCTAACTCCGACCCGC) were annealed and subcloned into the BsmBI sites of LentiCRISPR V2 (plasmid #52961; Addgene). The GFP-HDR fragment was constructed by connecting the DNA sequences upstream and downstream of nc886 and the GFP coding sequence using overlap PCR. To generate the overlap PCR constructs, two fragments corresponding to ∼500 bp upstream and downstream sequences of nc886 were amplified by PCR from human genomic DNA (nc886-upstream-F: TGCGTAACAGCTCCCTTTTT; nc886-upstream-R: AAGTCCCGTTGATTTTGGTGGCCTGATCAAAGGTGCGTAT; nc886-downstream-F: AAAACCTCCCACACCTCCCTTTTAAGCAAGACAGGCAGACA; nc886-downstream-R: CCTGCTAACGTGTCCTGGAG). The upstream-R and downstream-F primers incorporated 25 bases complementary to the GFP coding sequence. The GFP coding sequence was amplified by PCR using the primers (GFP-F: ATACGCACCTTTGATCAGGCCACCAAAATCAACGGGACTT; GFP-R: TGTCTGCCTGTCTTGCTTAAAAGGGAGGTGTGGGAGGTTTT). All three PCR amplicons were purified and mixed in a 1:1:1 ratio for a second round PCR with the nc886-up-F and nc886-down-R primers to construct the HDR fragment. The nc886 gRNA expression LentiCRISPR V2 vector and the HDR fragment were co-transfected into FaDu cells using the Lipofectamine 3000 Transfection Reagent (Thermo Fisher Scientific, Waltham, MA). At 48 h post-transfection, cells were selected by puromycin (1 μg/mL) for six days. The GFP-positive cells were then sorted using the MoFlo Astrios Cell Sorter (Beckman Coulter). The knockout of nc886 and insertion of GFP was verified by PCR amplification and Sanger sequencing.
Double knockout of protein kinase RNA-activated (PKR) and nc886 in FaDu cells
Because PKR acts downstream of nc886 and nc886 knockout is lethal, we hypothesize that PKR knockout may reverse the lethal phenotype of nc886 knockout. We therefore attempted to knock out PRK first followed by nc886 KO. The PKR gene on 2p22.2 was first knocked out using the CRISPR-Cas9 non-homologous end-joining (NHEJ) method. The PKR gRNA and Cas9 expression and control plasmids were purchased from Addgene (lentiCas9-Blast #52962; PKR gRNA 1 #75637; PKR gRNA 2 #75638). The gRNA and Cas9 plasmids were introduced into FaDu cells via lentivirus transduction using the 3rd-generation packaging plasmids pMD2.G (plasmid #12259; Addgene), pMDL/RREg/p (plasmid #12251; Addgene), and pRSV-Rev (plasmid #12253; Addgene). At 48 h post-infection, cells were selected by treating with 10 μg/mL Blasticidin. The knockout of PKR was verified by western blot. After we obtained PKR-KO cells, we then used the same approach as above to knockout nc886 and replace with GFP in PKR-KO background. The nc886 KO single colonies were verified using Sanger sequencing.
Data availability
The data presented in this study are available on request from the corresponding author upon signed material transfer form (MTA). The data are not publicly available because of patient privacy information and ethical consideration.
Acknowledgments
We thank the Biospecimen Extraction Facility of MD Anderson Cancer Center for DNA extraction and the Population Genomics Core of the Department of Epidemiology for performing Illumina methylation arrays. This study was financially supported by a National Cancer Institute grant (R01CA236859) and an institutional faculty incentive award from MD Anderson Cancer Center.
Author contributions
J.G. and G.L. conceived and planned the experiments. Z.W., P.W., and R.G. analyzed performed bioinformatics analysis and data analysis. Y.X. performed laboratory experiments. K.T.K. and A.G.S. provided study materials and thoughtful discussions. Y.X., Z.W., and J.G. drafted the manuscript. All the authors reviewed and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.11.012.
Contributor Information
Guojun Li, Email: gli@mdanderson.org.
Jian Gu, Email: jiangu@mdanderson.org.
Supplemental information
References
- 1.Argiris A., Karamouzis M.V., Raben D., Ferris R.L. Head and neck cancer. Lancet. 2008;371:1695–1709. doi: 10.1016/S0140-6736(08)60728-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marur S., Forastiere A.A. Head and neck squamous cell carcinoma: update on epidemiology, diagnosis, and treatment. Mayo Clin, Proc. 2016;91:386–396. doi: 10.1016/j.mayocp.2015.12.017. [DOI] [PubMed] [Google Scholar]
- 3.Rettig E.M., D'Souza G. Epidemiology of head and neck cancer. Surg. Oncol. Clin. 2015;24:379–396. doi: 10.1016/j.soc.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Mourad M., Jetmore T., Jategaonkar A.A., Moubayed S., Moshier E., Urken M.L. Epidemiological trends of head and neck cancer in the United States: a SEER population study. J. Oral Maxillofac. Surg. 2017;75:2562–2572. doi: 10.1016/j.joms.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gillison M.L., Chaturvedi A.K., Anderson W.F., Fakhry C. Epidemiology of human papillomavirus-positive head and neck squamous cell carcinoma. J. Clin. Oncol. 10 2015;33:3235–3242. doi: 10.1200/JCO.2015.61.6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taberna M., Mena M., Pavon M.A., Alemany L., Gillison M.L., Mesia R. Human papillomavirus-related oropharyngeal cancer. Ann. Oncol. 2017;28:2386–2398. doi: 10.1093/annonc/mdx304. [DOI] [PubMed] [Google Scholar]
- 7.Senkomago V., Henley S.J., Thomas C.C., Mix J.M., Markowitz L.E., Saraiya M. Human papillomavirus-attributable cancers - United States, 2012-2016. MMWR Morb. Mortal. Wkly. Rep. 2019;68:724–728. doi: 10.15585/mmwr.mm6833a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehtinen M., Dillner J. Clinical trials of human papillomavirus vaccines and beyond. Nat. Rev. Clin. Oncol. 2013;10:400–410. doi: 10.1038/nrclinonc.2013.84. [DOI] [PubMed] [Google Scholar]
- 9.Chaturvedi A.K., Graubard B.I., Broutian T., Pickard R.K.L., Tong Z.Y., Xiao W., Kahle L., Gillison M.L. Effect of prophylactic human papillomavirus (HPV) vaccination on oral HPV infections among young adults in the United States. J. Clin. Oncol. 2018;36:262–267. doi: 10.1200/JCO.2017.75.0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan C., Issaeva N., Yarbrough W.G. HPV-driven oropharyngeal cancer: current knowledge of molecular biology and mechanisms of carcinogenesis. Cancers Head Neck. 2018;3:12. doi: 10.1186/s41199-018-0039-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ang K.K., Harris J., Wheeler R., Weber R., Rosenthal D.I., Nguyen-Tan P.F., Westra W.H., Chung C.H., Jordan R.C., Lu C., et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010;363:24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ragin C.C., Taioli E. Survival of squamous cell carcinoma of the head and neck in relation to human papillomavirus infection: review and meta-analysis. Int. J. Cancer. 2007;121:1813–1820. doi: 10.1002/ijc.22851. [DOI] [PubMed] [Google Scholar]
- 13.Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seiwert TY, Zuo Z, Keck MK, Khattri A, Pedamallu CS, Stricker T, Brown C, Pugh TJ, Stojanov P, Cho J, et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. Feb 1 2015;21:632-641. 10.1158/1078-0432.CCR-13-3310. [DOI] [PMC free article] [PubMed]
- 15.Nakagawa T., Kurokawa T., Mima M., Imamoto S., Mizokami H., Kondo S., Okamoto Y., Misawa K., Hanazawa T., Kaneda A. DNA methylation and HPV-associated head and neck cancer. Microorganisms. 2021;9 doi: 10.3390/microorganisms9040801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Camuzi D., Buexm L.A., Lourenco S.Q.C., Esposti D.D., Cuenin C., Lopes M.S.A., Manara F., Talukdar F.R., Herceg Z., Ribeiro Pinto L.F., et al. HPV infection leaves a DNA methylation signature in oropharyngeal cancer affecting both coding genes and transposable elements. Cancers. 2021;13 doi: 10.3390/cancers13143621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baylin S.B., Jones P.A. A decade of exploring the cancer epigenome - biological and translational implications. Nat. Rev. Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burgers W.A., Blanchon L., Pradhan S., de Launoit Y., Kouzarides T., Fuks F. Viral oncoproteins target the DNA methyltransferases. Oncogene. 2007;26:1650–1655. doi: 10.1038/sj.onc.1209950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sen P., Ganguly P., Ganguly N. Modulation of DNA methylation by human papillomavirus E6 and E7 oncoproteins in cervical cancer. Oncol. Lett. 2018;15:11–22. doi: 10.3892/ol.2017.7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Kempen P.M., Noorlag R., Braunius W.W., Stegeman I., Willems S.M., Grolman W. Differences in methylation profiles between HPV-positive and HPV-negative oropharynx squamous cell carcinoma: a systematic review. Epigenetics. 2014;9:194–203. doi: 10.4161/epi.26881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ekanayake Weeramange C., Tang K.D., Vasani S., Langton-Lockton J., Kenny L., Punyadeera C. DNA methylation changes in human papillomavirus-driven head and neck cancers. Cells. 2020;9 doi: 10.3390/cells9061359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren S., Gaykalova D., Wang J., Guo T., Danilova L., Favorov A., Fertig E., Bishop J., Khan Z., Flam E., et al. Discovery and development of differentially methylated regions in human papillomavirus-related oropharyngeal squamous cell carcinoma. Int. J. Cancer. 2018;143:2425–2436. doi: 10.1002/ijc.31778. [DOI] [PubMed] [Google Scholar]
- 23.Gougousis S., Mouchtaropoulou E., Besli I., Vrochidis P., Skoumpas I., Constantinidis I. HPV-related oropharyngeal cancer and biomarkers based on epigenetics and microbiome profile. Front. Cell Dev. Biol. 2020;8:625330. doi: 10.3389/fcell.2020.625330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Degli Esposti D., Sklias A., Lima S.C., Beghelli-de la Forest Divonne S., Cahais V., Fernandez-Jimenez N., Cros M.P., Ecsedi S., Cuenin C., Bouaoun L., et al. Unique DNA methylation signature in HPV-positive head and neck squamous cell carcinomas. Genome Med. 2017;9:33. doi: 10.1186/s13073-017-0419-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelsey K.T., Wiencke J.K. Immunomethylomics: a novel cancer risk prediction tool. Ann. Am. Thorac. Soc. 2018;15(Suppl 2):S76–S80. doi: 10.1513/AnnalsATS.201706-477MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koestler D.C., Usset J., Christensen B.C., Marsit C.J., Karagas M.R., Kelsey K.T., Wiencke J.K. DNA methylation-derived neutrophil-to-lymphocyte ratio: an epigenetic tool to explore cancer inflammation and outcomes. Cancer Epidemiol. Biomarkers Prev. 2017;26:328–338. doi: 10.1158/1055-9965.EPI-16-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ambatipudi S., Langdon R., Richmond R.C., Suderman M., Koestler D.C., Kelsey K.T., Kazmi N., Penfold C., Ho K.M., McArdle W., et al. DNA methylation derived systemic inflammation indices are associated with head and neck cancer development and survival. Oral Oncol. 2018;85:87–94. doi: 10.1016/j.oraloncology.2018.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grieshober L., Graw S., Barnett M.J., Thornquist M.D., Goodman G.E., Chen C., Koestler D.C., Marsit C.J., Doherty J.A. Methylation-derived neutrophil-to-lymphocyte ratio and lung cancer risk in heavy smokers. Cancer Prev. Res. (Phila). 2018;11:727–734. doi: 10.1158/1940-6207.CAPR-18-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ligthart S., Marzi C., Aslibekyan S., Mendelson M.M., Conneely K.N., Tanaka T., Colicino E., Waite L.L., Joehanes R., Guan W., et al. DNA methylation signatures of chronic low-grade inflammation are associated with complex diseases. Genome Biol. 2016;17:255. doi: 10.1186/s13059-016-1119-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saber C.N., Gronhoj Larsen C., Dalianis T., von Buchwald C. Immune cells and prognosis in HPV-associated oropharyngeal squamous cell carcinomas: review of the literature. Oral Oncol. 2016;58:8–13. doi: 10.1016/j.oraloncology.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 31.Andersen A.S., Koldjaer Solling A.S., Ovesen T., Rusan M. The interplay between HPV and host immunity in head and neck squamous cell carcinoma. Int. J. Cancer. 2014;134:2755–2763. doi: 10.1002/ijc.28411. [DOI] [PubMed] [Google Scholar]
- 32.Subbarayan R.S., Arnold L., Gomez J.P., Thomas S.M. The role of the innate and adaptive immune response in HPV-associated oropharyngeal squamous cell carcinoma. Laryngoscope Invest. Otolaryngol. 2019;4:508–512. doi: 10.1002/lio2.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hladikova K., Koucky V., Boucek J., Laco J., Grega M., Hodek M., Zabrodsky M., Vosmik M., Rozkosova K., Vosmikova H., et al. Tumor-infiltrating B cells affect the progression of oropharyngeal squamous cell carcinoma via cell-to-cell interactions with CD8(+) T cells. J. Immunother. Cancer. 2019;7:261. doi: 10.1186/s40425-019-0726-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaffe A.E., Murakami P., Lee H., Leek J.T., Fallin M.D., Feinberg A.P., Irizarry R.A. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int. J. Epidemiol. 2012;41:200–209. doi: 10.1093/ije/dyr238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller D.L., Davis J.W., Taylor K.H., Johnson J., Shi Z., Williams R., Atasoy U., Lewis J.S., Jr., Stack M.S. Identification of a human papillomavirus-associated oncogenic miRNA panel in human oropharyngeal squamous cell carcinoma validated by bioinformatics analysis of the Cancer Genome Atlas. Am. J. Pathol. 2015;185:679–692. doi: 10.1016/j.ajpath.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee K., Kunkeaw N., Jeon S.H., Lee I., Johnson B.H., Kang G.Y., Bang J.Y., Park H.S., Leelayuwat C., Lee Y.S. Precursor miR-886, a novel noncoding RNA repressed in cancer, associates with PKR and modulates its activity. RNA. 2011;17:1076–1089. doi: 10.1261/rna.2701111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee Y.S. A novel type of non-coding RNA, nc886, implicated in tumor sensing and suppression. Genom. Inf. 2015;13:26–30. doi: 10.5808/GI.2015.13.2.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marchal J.A., Lopez G.J., Peran M., Comino A., Delgado J.R., Garcia-Garcia J.A., Conde V., Aranda F.M., Rivas C., Esteban M., et al. The impact of PKR activation: from neurodegeneration to cancer. FASEB J. 2014;28:1965–1974. doi: 10.1096/fj.13-248294. [DOI] [PubMed] [Google Scholar]
- 39.Jeon S.H., Johnson B.H., Lee Y.S. A tumor surveillance model: a non-coding RNA senses neoplastic cells and its protein partner signals cell death. Int. J. Mol. Sci. 2012;13:13134–13139. doi: 10.3390/ijms131013134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nogues J.C., Fassas S., Mulcahy C., Zapanta P.E. Human papillomavirus-associated head and neck cancer. J. Am. Board Fam. Med. 2021;34:832–837. doi: 10.3122/jabfm.2021.04.200588. [DOI] [PubMed] [Google Scholar]
- 41.Sartor M.A., Dolinoy D.C., Jones T.R., Colacino J.A., Prince M.E., Carey T.E., Rozek L.S. Genome-wide methylation and expression differences in HPV(+) and HPV(-) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics. 2011;6:777–787. doi: 10.4161/epi.6.6.16216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lleras R.A., Smith R.V., Adrien L.R., Schlecht N.F., Burk R.D., Harris T.M., Childs G., Prystowsky M.B., Belbin T.J. Unique DNA methylation loci distinguish anatomic site and HPV status in head and neck squamous cell carcinoma. Clin. Cancer Res. 2013;19:5444–5455. doi: 10.1158/1078-0432.CCR-12-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia M.A., Meurs E.F., Esteban M. The dsRNA protein kinase PKR: virus and cell control. Biochimie. 2007;89:799–811. doi: 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 44.Treppendahl M.B., Qiu X., Sogaard A., Yang X., Nandrup-Bus C., Hother C., Andersen M.K., Kjeldsen L., Mollgard L., Hellstrom-Lindberg E., et al. Allelic methylation levels of the noncoding VTRNA2-1 located on chromosome 5q31.1 predict outcome in AML. Blood. 2012;119:206–216. doi: 10.1182/blood-2011-06-362541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao J., Song Y., Bi N., Shen J., Liu W., Fan J., Sun G., Tong T., He J., Shi Y., et al. DNA methylation-mediated repression of miR-886-3p predicts poor outcome of human small cell lung cancer. Cancer Res. 2013;73:3326–3335. doi: 10.1158/0008-5472.CAN-12-3055. [DOI] [PubMed] [Google Scholar]
- 46.Lee H.S., Lee K., Jang H.J., Lee G.K., Park J.L., Kim S.Y., Kim S.B., Johnson B.H., Zo J.I., Lee J.S., et al. Epigenetic silencing of the non-coding RNA nc886 provokes oncogenes during human esophageal tumorigenesis. Oncotarget. 2014;5:3472–3481. doi: 10.18632/oncotarget.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee K.S., Park J.L., Lee K., Richardson L.E., Johnson B.H., Lee H.S., Lee J.S., Kim S.B., Kwon O.H., Song K.S., et al. nc886, a non-coding RNA of anti-proliferative role, is suppressed by CpG DNA methylation in human gastric cancer. Oncotarget. 2014;5:3944–3955. doi: 10.18632/oncotarget.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fort R.S., Matho C., Geraldo M.V., Ottati M.C., Yamashita A.S., Saito K.C., Leite K.R.M., Mendez M., Maedo N., Mendez L., et al. Nc886 is epigenetically repressed in prostate cancer and acts as a tumor suppressor through the inhibition of cell growth. BMC Cancer. 2018;18:127. doi: 10.1186/s12885-018-4049-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee E.K., Hong S.H., Shin S., Lee H.S., Lee J.S., Park E.J., Choi S.S., Min J.W., Park D., Hwang J.A., et al. nc886, a non-coding RNA and suppressor of PKR, exerts an oncogenic function in thyroid cancer. Oncotarget. 2016;7:75000–75012. doi: 10.18632/oncotarget.11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lei J., Xiao J.H., Zhang S.H., Liu Z.Q., Huang K., Luo Z.P., Xiao X.L., Hong Z.D. Non-coding RNA 886 promotes renal cell carcinoma growth and metastasis through the Janus kinase 2/signal transducer and activator of transcription 3 signaling pathway. Mol. Med. Rep. 2017;16:4273–4278. doi: 10.3892/mmr.2017.7093. [DOI] [PubMed] [Google Scholar]
- 51.Hu Z., Zhang H., Tang L., Lou M., Geng Y. Silencing nc886, a non-coding RNA, induces apoptosis of human endometrial cancer cells-1A in vitro. Med. Sci. Monit. 2017;23:1317–1324. doi: 10.12659/msm.900320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li J.H., Wang M., Zhang R., Gao W.L., Meng S.H., Ma X.L., Hou X.H., Feng L.M. E2F1-directed activation of nc886 mediates drug resistance in cervical cancer cells via regulation of major vault protein. Int. J. Clin. Exp. Pathol. 2017;10:9233–9242. [PMC free article] [PubMed] [Google Scholar]
- 53.Ahn J.H., Lee H.S., Lee J.S., Lee Y.S., Park J.L., Kim S.Y., Hwang J.A., Kunkeaw N., Jung S.Y., Kim T.J., et al. nc886 is induced by TGF-beta and suppresses the microRNA pathway in ovarian cancer. Nat. Commun. 2018;9:1166. doi: 10.1038/s41467-018-03556-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saruuldalai E., Park J., Kang D., Shin S.P., Im W.R., Lee H.H., Jang J.J., Park J.L., Kim S.Y., Hwang J.A., et al. A host non-coding RNA, nc886, plays a pro-viral role by promoting virus trafficking to the nucleus. Mol. Ther. Oncolytics. 2022;24:683–694. doi: 10.1016/j.omto.2022.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Golec E., Lind L., Qayyum M., Blom A.M., King B.C. The noncoding RNA nc886 regulates PKR signaling and cytokine production in human cells. J. Immunol. 2019;202:131–141. doi: 10.4049/jimmunol.1701234. [DOI] [PubMed] [Google Scholar]
- 56.Lee Y.S., Bao X., Lee H.H., Jang J.J., Saruuldalai E., Park G., Im W.R., Park J.L., Kim S.Y., Shin S., et al. Nc886, a novel suppressor of the type I interferon response upon pathogen intrusion. Int. J. Mol. Sci. 2021;22:2003. doi: 10.3390/ijms22042003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arico E., Castiello L., Capone I., Gabriele L., Belardelli F. Type I interferons and cancer: an evolving story demanding novel clinical applications. Cancers. 2019;11:1943. doi: 10.3390/cancers11121943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Galasso M., Gambino S., Romanelli M.G., Donadelli M., Scupoli M.T. Browsing the oldest antioxidant enzyme: catalase and its multiple regulation in cancer. Free Radic. Biol. Med. 2021;172:264–272. doi: 10.1016/j.freeradbiomed.2021.06.010. [DOI] [PubMed] [Google Scholar]
- 59.Min J.Y., Lim S.O., Jung G. Downregulation of catalase by reactive oxygen species via hypermethylation of CpG island II on the catalase promoter. FEBS Lett. 2010;584:2427–2432. doi: 10.1016/j.febslet.2010.04.048. [DOI] [PubMed] [Google Scholar]
- 60.Galasso M., Dalla Pozza E., Chignola R., Gambino S., Cavallini C., Quaglia F.M., Lovato O., Dando I., Malpeli G., Krampera M., et al. The rs1001179 SNP and CpG methylation regulate catalase expression in chronic lymphocytic leukemia. Cell. Mol. Life Sci. 2022;79:521. doi: 10.1007/s00018-022-04540-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neumann A.S., Lyons H.J., Shen H., Liu Z., Shi Q., Sturgis E.M., Shete S., Spitz M.R., El-Naggar A., Hong W.K., et al. Methylenetetrahydrofolate reductase polymorphisms and risk of squamous cell carcinoma of the head and neck: a case-control analysis. Int. J. Cancer. 2005;115:131–136. doi: 10.1002/ijc.20888. [DOI] [PubMed] [Google Scholar]
- 62.Shete S., Liu H., Wang J., Yu R., Sturgis E.M., Li G., Dahlstrom K.R., Liu Z., Amos C.I., Wei Q. A genome-wide association study identifies two novel susceptible regions for squamous cell carcinoma of the head and neck. Cancer Res. 2020;80:2451–2460. doi: 10.1158/0008-5472.CAN-19-2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tao Y., Sturgis E.M., Huang Z., Wang Y., Wei P., Wang J.R., Wei Q., Li G. TGFbeta1 genetic variants predict clinical outcomes of HPV-positive oropharyngeal cancer patients after definitive radiotherapy. Clin. Cancer Res. 2018;24:2225–2233. doi: 10.1158/1078-0432.CCR-17-1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tao Y., Sturgis E.M., Huang Z., Sun Y., Dahlstrom K.R., Wei Q., Li G. A TGF-beta1 genetic variant at the miRNA187 binding site significantly modifies risk of HPV16-associated oropharyngeal cancer. Int. J. Cancer. 2018;143:1327–1334. doi: 10.1002/ijc.31530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Langevin S.M., McClean M.D., Michaud D.S., Eliot M., Nelson H.H., Kelsey K.T. Occupational dust exposure and head and neck squamous cell carcinoma risk in a population-based case-control study conducted in the greater Boston area. Cancer Med. 2013;2:978–986. doi: 10.1002/cam4.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang B., Eliot M., McClean M.D., Waterboer T., Pawlita M., Butler R., Nelson H.H., Langevin S.M., Christensen B.C., Kelsey K.T. DNA methylation-derived systemic inflammation indices and their association with oropharyngeal cancer risk and survival. Head Neck. 2022;44:904–913. doi: 10.1002/hed.26981. [DOI] [PubMed] [Google Scholar]
- 67.Han Y., Zhang M., Xu J., Li J., Xu Y., Thompson T.C., Logothetis C.J., Sun D., Gu J. Genome-wide DNA methylation profiling of leukocytes identifies CpG methylation signatures of aggressive prostate cancer. Am. J. Cancer Res. 2021;11:968–978. [PMC free article] [PubMed] [Google Scholar]
- 68.Pidsley R., Zotenko E., Peters T.J., Lawrence M.G., Risbridger G.P., Molloy P., Van Djik S., Muhlhausler B., Stirzaker C., Clark S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17:208. doi: 10.1186/s13059-016-1066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aryee M.J., Jaffe A.E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A.P., Hansen K.D., Irizarry R.A. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leek J.T., Johnson W.E., Parker H.S., Jaffe A.E., Storey J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tusher V.G., Tibshirani R., Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this study are available on request from the corresponding author upon signed material transfer form (MTA). The data are not publicly available because of patient privacy information and ethical consideration.