

Abstract
In many groups, sex chromosomes change frequently but the drivers of their rapid evolution are varied and often poorly characterized. With an aim of further understanding sex chromosome turnover, we investigated the polymorphic sex chromosomes of the Marsabit clawed frog, Xenopus borealis, using genomic data and a new chromosome-scale genome assembly. We confirmed previous findings that 54.1 Mb of chromosome 8L is sex-linked in animals from east Kenya and a lab strain, but most (or all) of this region is not sex-linked in natural populations from west Kenya. Previous work suggests possible degeneration of the Z chromosomes in the east population because many sex-linked transcripts of this female heterogametic population have female-biased expression, and we therefore expected this chromosome to not be present in the west population. In contrast, our simulations support a model where most or all of the sex-linked portion of the Z chromosome from the east acquired autosomal segregation in the west, and where much genetic variation specific to the large sex-linked portion of the W chromosome from the east is not present in the west. These recent changes are consistent with the hot potato model, wherein sex chromosome turnover is favoured by natural selection if it purges a (minimally) degenerate sex-specific sex chromosome, but counterintuitively suggest natural selection failed to purge a Z chromosome that has signs of more advanced and possibly more ancient regulatory degeneration. These findings highlight complex evolutionary dynamics of young, rapidly evolving Xenopus sex chromosomes, and set the stage for mechanistic work aimed at pinpointing additional sex determining genes in this group.
Keywords: Recombination suppression, rapid evolution, sex chromosome turnover, amphibians
Graphical Abstract
In most or all frogs, sex is genetically determined but the genes that control this crucial process evolve rapidly. To understand how, we explored an example of intraspecific polymorphism in sex chromosomes in the Marsabit clawed frog throughout its range in Kenya. Genome sequences, a new chromosome-scale genome assembly, and simulations suggest that the large sex-linked region in east Kenya is not present in west Kenya and that most or all of the Z chromosome of the east population segregates autosomally in the west. The sex chromosomes of the west population thus may be recently evolved and/or contain a small region of sex-linked recombination suppression. The findings illustrate how rapid evolution of genetic triggers for sex determination can have genome-wide effects on recombination in structured populations.
Introduction
Sex chromosome turnover and sex-linked recombination suppression
Sex chromosomes evolve from autosomes that acquire genetic variation that initiates sexual differentiation. Sex determination is a developmental gateway for reproduction whose initiation is triggered by genes that are highly conserved in eutherian mammals (Graves, 2008), birds (Zhou et al., 2014), and lepidopterans (Fraisse et al., 2017, Yoshido et al., 2020). However, sex chromosomes of other groups such as squamate reptiles (Gamble et al., 2015, Pennell et al., 2018), teleost fish (Pennell et al., 2018), and amphibians (Evans et al., 2012, Jeffries et al., 2018, Ma & Veltsos, 2021, Pennell et al., 2018) change frequently; some – and perhaps many – of these changes are also associated with origins of novel triggers for sex determination. This contrasts sharply with highly conserved developmental control of several other traits such eyes (Onuma et al., 2002), limbs (Cass et al., 2021), and non-gonadal organs (Irie & Kuratani, 2011), raising the question of why there is such diversity in the genetic basis of sex determination (Bachtrog et al., 2014, Palmer et al., 2019).
Multiple factors may favour sex chromosome turnover and the origin of novel triggers for sex determination. These factors include genetic drift (Bull & Charnov, 1977), sexual antagonism (van Doorn & Kirkpatrick, 2007, van Doorn & Kirkpatrick, 2010), selection against the genetic load of sex-linked mutations (Blaser et al., 2013, Blaser et al., 2014), selection against unbalanced sex ratios (Bull, 1983), including in association with meiotic drive (Jaenike, 2001, Úbeda et al., 2015, Yoshida & Kitano, 2012), and selection against balanced sex ratios under conditions with inbreeding (Werren & Hatcher, 2000).
Some of these factors also could lead to the origin and expansion of recombination arrest in sex-linked regions, including expansion being favored by natural selection or occurring due to neutral evolution (Jeffries et al., 2021, Charlesworth & Wall, 1999, Fisher, 1931, Bull, 1983, Rice, 1987). Natural selection could favour recombination suppression if sex-linkage resolves genomic conflict associated with mutations with sexually antagonistic fitness effects (Fisher, 1931, Charlesworth & Charlesworth, 1980, Bull, 1983, Rice, 1987), or to avoid sterile or intersex progeny (Zou et al., 2021), compartmentalize genes with sex-specific function (Bachtrog et al., 2014), or ward off effects of deleterious recessive mutations in the heterogametic sex (Charlesworth & Wall, 1999). Neutral evolution could lead to expansion of recombination arrest by inversions or nucleotide substitutions (Ironside, 2010, Jeffries et al., 2021). Recombination arrest also could be a pre-existing (ancestral) condition of genomic regions that favors the origin of sex-determining genes (Sun et al., 2017, Sardell et al., 2018). Pre-existing recombination arrest could be linked to natural variation between the sexes in the rates and locations of recombination that stems from fundamental differences in male and female meiosis (Brandvain & Coop, 2012), including the possibility of sex-specific achiasmy (i.e., no recombination in one sex). Factors that counterpose expansion of non-recombining regions include an inability to evolve dosage compensation mechanisms (Adolfsson & Ellegren, 2013) or, in the absence of neutral expansion, a dearth of potential drivers of expansion (e.g., few mutations with sexually antagonistic fitness effects or resolution of genomic conflict by other mechanisms such as sex-biased expression).
African clawed frogs (Xenopus) as models of rapid evolution of sex determination and sex chromosomes
Used in the early 20th century for pregnancy tests (Gurdon & Hopwood, 2000, Weisman & Coates, 1941), African clawed frogs have since been transformed into powerful model organisms for biological inquiry. Being vertebrates, these frogs have extensive homology in genetic function with humans. They are diverse and characterized by frequent allopolyploidization (Supplement), are readily maintained and propagated in captivity, have external fertilization and embryonic development, and a myriad of biological resources have been developed, including chromosome-scale genome assemblies and methods for gene editing (Harland & Grainger, 2011, Mitros et al., 2019, Session et al., 2016, Tandon et al., 2016, Cannatella & de Sá, 1993, Hellsten et al., 2010). Especially in the last decade, another exciting utility of Xenopus has come into focus: this group has high variation in (evolutionarily) young genetic sex determination systems that offer unique insights into how new sex chromosomes and sex determination pathways evolve.
Cytogenetic studies indicate that Xenopus species have cytogenetically indistinguishable (homomorphic) sex chromosomes (Tymowska, 1991). Mapping of genetic variation from reduced representation genome sequencing (RRGS) and microsatellites to high quality genome assemblies (Hellsten et al., 2010, Session et al., 2016, Mitros et al., 2019) has identified sex-linked regions in the diploid X. tropicalis (chromosome 7, hereafter Chr7) and the allotetraploid X. mellotropicalis (one of the homeologous copies of Chr7), X. laevis (Chr2L), and X. borealis (Chr8L; Furman et al., 2016, Furman et al., 2020, Olmstead et al., 2010, Wells et al., 2011, Roco et al., 2015, Cauret et al., 2020, Mitros et al., 2019, Mawaribuchi et al., 2017, Yoshimoto et al., 2008, Furman & Evans, 2018, Song et al., 2020). Using the same approach, the sex chromosomes of two other diploid species from the same family (Pipidae) also have been identified: Hymenochirus bottgeri and Pipa parva (Chr4 and Chr6, respectively; Cauret et al., 2020). With the exception of X. tropicalis, which has a complex system for sex determination discussed below, all Xenopus species thus far have heterogametic (ZW) females; H. bottgeri also has heterogametic females whereas P. parva has heterogametic males (Cauret et al., 2020).
Sex chromosomes of the Marsabit clawed frog, Xenopus borealis
The Marsabit clawed frog, Xenopus borealis, is an allotetraploid species (Supplement) with geographically structured variation in newly evolved sex chromosomes (Song et al., 2020, Furman & Evans, 2016, Furman & Evans, 2018). In X. borealis from east Kenya and a lab strain, females are heterogametic and there is a large female-linked region spanning the first ~54Mb of Chr8L (Furman & Evans, 2016, Furman & Evans, 2018, Song et al., 2020). The lab strain was inferred to have originated from central Kenya based on analysis of ancestry components (Li, 2011) of RRGS data (Song et al., 2020). However, in X. borealis from west Kenya a large sex-linked region is not evident and it is unclear whether males or females are heterogametic (Song et al., 2020). This difference in sex-linkage corresponds with inferred population structure based on analyses of molecular variation in the mitochondrial and nuclear genomes (Song et al., 2020). In multiple tissue types and developmental stages of the lab strain, sex-linked transcripts tend to be more highly expressed in females than males (Song et al., 2020). One of several possible explanations is that the Z chromosome of this strain once was a degenerate Y chromosome (Song et al., 2020).
Several scenarios could account for the intraspecific variation in sex-linkage in X. borealis. Here we consider three scenarios that differ in whether the sex chromosomes in the east are ancestral to or derived from those in the west, whether Chr8L is sex-linked in one or both populations, and whether the sex determining locus is the same in these populations (Fig. 1).
Figure. 1.
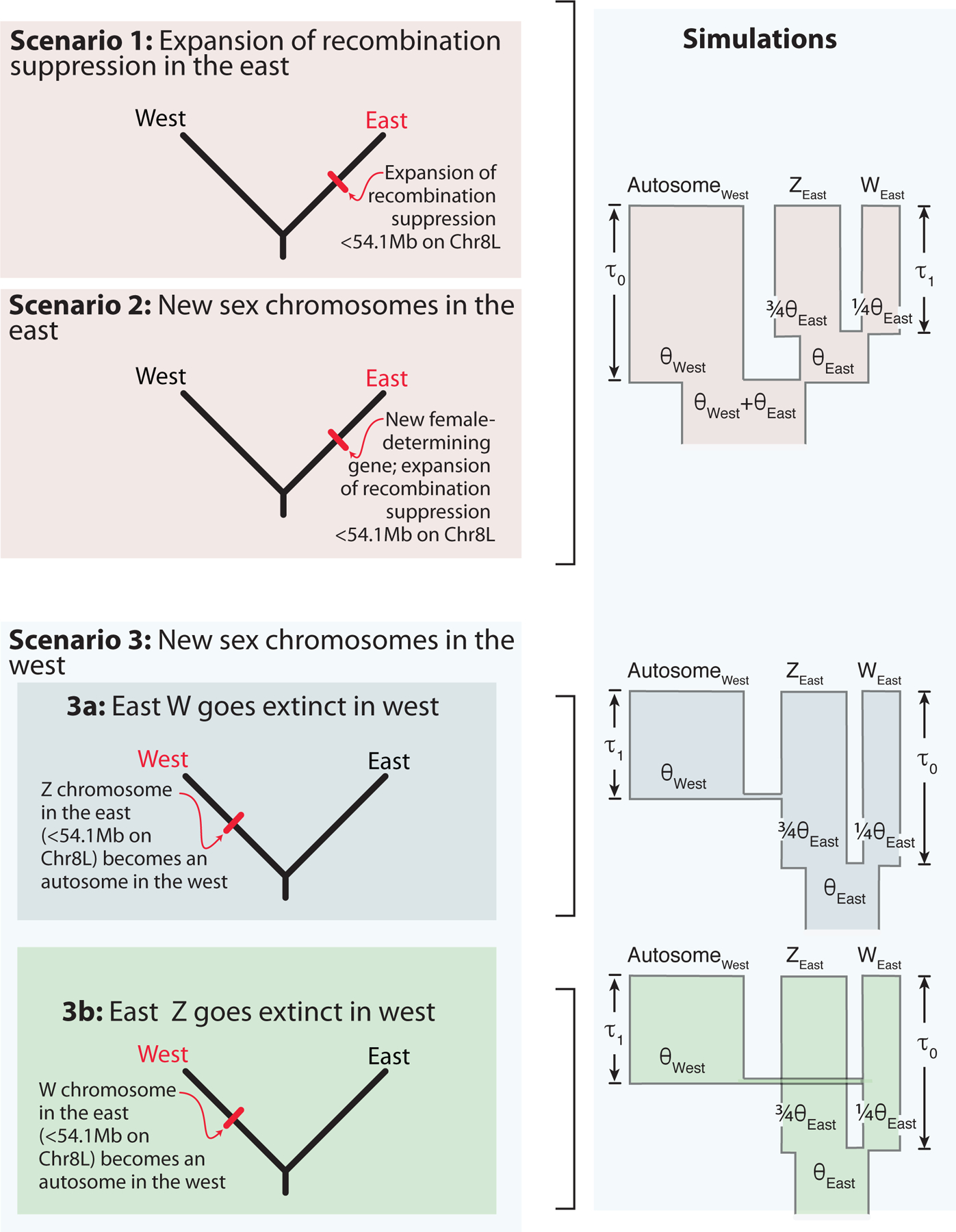
Evolutionary scenarios (left) for the origin of geographically structured variation in X. borealis sex chromosomes with further details provided in the main text. Red font indicates the population with the newest or most recent change in sex chromosomes. Simulations corresponding to these scenarios are depicted on the right with further details provided in the main text and Supplement. θ refers to the population polymorphism parameter of the east (East) and west (West) populations, which are equal to 4Neμ, where Ne is the effective population size and μ is the mutation rate, and τ0 and τ1 refer to divergence times in 4Ne generations.
Scenario 1: the sex determining gene is the same in both populations and the region of suppressed recombination expanded in the east. Both populations have female heterogamy and the large female-specific region in the east is derived from a much smaller region in the west (e.g., due to an inversion on the W chromosome in the east). A prediction associated with Scenario 1 is that W- and Z- linked variants from the east are present in the west but mostly are not sex-linked.
Scenario 2: the sex chromosomes in the east are newly evolved and not homologous to the sex chromosomes in the west. This could happen via “nonhomologous” sex chromosome turnover (van Doorn & Kirkpatrick, 2010), where the location of the ancestral sex chromosomes in the west population was somewhere other than the first ~54 Mb on Chr8L, or via a “homologous” sex chromosome turnover where the location of the ancestral sex chromosomes in the west population is within the first ~54 Mb on Chr8L but involves a mechanism of sex determination in the west (e.g. a male-determining gene or a different female-determining gene) that is different from the one in the east. Heterogametic females would be expected in the west if an ancestral W chromosome from the west was lost in the east population (Bull & Charnov, 1977). Heterogametic males would be expected in the west if an ancestral X chromosome from the west was lost in the east population (Bull & Charnov, 1977). Similar to Scenario 1, a prediction associated with Scenario 2 is that W- and Z-linked variants in the east are present in the west but are not sex-linked.
Scenario 3a: the sex chromosomes in the west are newly evolved, the W chromosome from the east went extinct in the west, and the sex-linked portion of the Z chromosome from the east acquired autosomal segregation in the west. Following the reasoning of (Bull & Charnov, 1977), the new system in the west should have heterogametic females with a dominant female-determining factor on a new W chromosome. This scenario could involve a homologous or non-homologous sex chromosome turnover in the west. A prediction associated with Scenario 3a is that many Z-linked SNPs in the sex-linked region from the east are fixed in both sexes in the west.
Scenario 3b: the sex chromosomes in the west are newly evolved, the Z chromosome from the east went extinct in the west, and the sex-linked portion of the W chromosome from the east acquired autosomal segregation in the west. The new system in the west should have heterogametic males with a male-determining factor on a new Y chromosome that is dominant over the female determining factor on the ancestral W chromosome. This scenario could involve a homologous or non-homologous sex chromosome turnover in the west. A prediction associated with Scenario 3b is that many W-linked SNPs in the sex-linked region from the east are fixed in both sexes in the west.
Each of these scenarios has distinct genomic predictions, and the power to evaluate them with genomic data depends on (i) the extent of recombination suppression surrounding the sex-linked region in the west, (ii) the extent of divergence between regions of recombination suppression, and (iii) the nature of the data (specifically, the genome-wide density of variable positions, the proportion of these genomic positions that are genotyped in each individual, and the number of individuals genotyped).
To evaluate these scenarios, we generated a new draft genome assembly for X. borealis and used it to re-analyze reduced representation genome sequences (RRGS) from a lab strain and wild caught individuals. We also collected and analyzed new genomic data from male and female individuals from the west and east populations of X. borealis and a lab strain, and we followed up inferences using Sanger sequencing of wild caught X. borealis and individuals of X. fraseri, which is closely related to X. borealis. We also performed simulations to test the fit of different evolutionary scenarios to the genomic data. Our results evidence variation between X. borealis populations in the extent of sex-linked recombination suppression. They further support an intraspecific turnover event in which much of the sex-linked portion of the Z chromosome from the east segregates as an autosome in the west, and where much of the genetic variation on the sex-linked portion of the W chromosome from the east is absent in the west. Below we discuss these findings in the context of current understanding of sex chromosome genomics in Xenopus and argue that functional studies of sex-determining genes are a crucial next step for understanding the biology of Xenopus sex chromosomes.
Methods
A draft genome assembly for X. borealis
DNA was extracted from lysed blood of a female frog that was obtained from Nasco (Fort Atkinson, WI, USA). Short insert Nextera and TruSeq libraries were prepared, respectively, by Jessica B. Lyons and by the Functional Genomics Laboratory at the University of California Berkeley, and then sequenced on the Illumina HiSeq 2500 (NCBI-SRA:SRR18802894–SRR18802896) by the Vincent J. Coates Genomics Sequencing Laboratory at the University of California Berkeley (VCGSL). Nextera mate pair libraries were prepared and sequenced on the Illumina HiSeq 2500 (NCBI-SRA:SRR18802888 and SRR18802889) by the HudsonAlpha Institute for Biotechnology. A Chicago in vitro proximity ligation library was prepared by Dovetail Genomics and sequenced on the Illumina HiSeq 2500 (NCBI-SRA:SRR18802892 and SRR18802893) by the VCGSL. Using a liver sample from a male frog, also from Nasco, a DpnII Hi-C library was prepared by Dovetail Genomics and sequenced on the Illumina HiSeq 2500 (NCBI-SRA:SRR18802890 and SRR18802891) by the VCGSL. The short insert data were adapter trimmed with ea-utils fastq-mcf version 1.04.807–18-gbd148d4 (Aronesty, 2013). The mate pair data were adapter trimmed and split with NxTrim version 0.4.1–53c2193 (O’Connell et al., 2015) and filtered using nxtrim_pipeline.sh version 1.0 (Bredeson et al., 2021).
Trimmed data were then assembled with Meraculous version 2.2.4 (Chapman et al., 2011). This assembly was scaffolded with Chicago and Hi-C data using the Dovetail Genomics HiRise algorithm (Putnam et al., 2016). The mitochondrial genome was assembled from adapter trimmed data using organelle_pipeline.py version 1.0 (Bredeson et al., 2021) and NOVOPlasty version 2.6.3 (Dierckxsens et al., 2017), starting with other Pipidae mitochondrial assemblies available on NCBI as input seeds. The assembly was screened with general_decon.sh version 1.0 (Mudd et al., 2020) to identify archaea, bacteria, virus, and vector contaminants using the respective RefSeq and UniVec databases, queried using mt_decon.sh version 1.0 (Mudd et al., 2020) against the mitochondrial assembled sequence, and filtered using nt_decon.sh version 1.0 (Bredeson et al., 2021) against the NT database and other completed frog assemblies. The assembly was then run through align_pipeline.sh version 1.0 (Bredeson et al., 2021) to identify and remove duplicate haplotype sequences. Scaffolds smaller than one kb were removed from the final assembly with Seqtk version 1.3-r106 (https://github.com/lh3/seqtk). Chromosomes were named according to the corresponding chromosomes in X. tropicalis version 9 (Mitros et al., 2019) and X. laevis version 9 (Session et al., 2016) based on alignment using MUMmer version 4.0.0 (Marcais et al., 2018), and scaffolds were numbered in order of decreasing size using SeqKit version 0.7.2-dev (Shen et al., 2016). The above sequencing reads and draft assembly are deposited under NCBI BioProject PRJNA827809. Assembly statistics were calculated using the Genome Assembly Annotation Service (GAAS) (https://github.com/NBISweden/GAAS).
RRGS data and analysis of sex-linkage
RRGS data from a lab strain and wild-caught individuals were obtained from GenBank, including 49 X. borealis lab strain individuals (PRJNA319044; Furman & Evans, 2016) and 54 X. borealis wild-caught individuals (PRJNA616217; Song et al., 2020). These data were trimmed using Trimmomatic version 0.39 (Bolger et al., 2014), and mapped to the draft X. borealis genome assembly using Bwa version 0.7.17. The HaplotypeCaller, CombineGVCFs, and GenotypeGVCF functions of the Genome Analysis Toolkit (GATK) version 4.1 (McKenna et al., 2010) were used to call genotypes for each sample and combine them into a joint genotype file. The VariantFiltration and SelectVariants functions of GATK were used to filter low quality genotypes. Positions with the following attributes were removed: QD > 2.0, QUAL < 20, SOR > 3.0, FS > 60.0, MQ < 30.0, where these acronyms respectively refer to variant confidence/quality by depth (QD), genotype quality (QUAL), Symmetric Odds Ratio of 2×2 contingency table to detect strand bias (SOR), Fisher exact test for strand bias (FS), and map quality (MQ). PLINK version 1.9 (Purcell et al., 2007) was used to assess sex-linkage of single nucleotide polymorphisms (SNPs) from the RRGS data that mapped to any of the 18 chromosome assemblies; data from unplaced scaffolds was excluded.
New whole genome sequencing (WGS) data from geographic isolates
New genomic data were generated from a male and female X. borealis individual from east Kenya (field identification numbers BJE4536 and BJE4515, respectively, both from Wundanyi, Kenya) and a male and female individual from west Kenya (BJE4442 and BJE4441, respectively, both from Lukhome, Kenya). Specimens and genetic samples for these individuals are deposited at the Museum of Comparative Zoology at Harvard University, USA (MCZ Herpetology A-153183, MCZ Herpetology A-153181, MCZ Herpetology A-153148, MCZ Herpetology A-153147, respectively). These data have been deposited in the NCBI-SRA (BioProject PRJNA616217). The new genomic data were obtained using PCR-free library prep and sequencing each on 1/6th of a lane of a Novaseq S4 machine with paired-end 150 base pair reads. We analyzed these new data along with published genomic data from a female and male individual from our lab strain (BioProject PRJNA421481). We mapped these genomic data to the draft X. borealis genome and called genotypes using the same procedures detailed above for the RRGS data, except that a de-duplication step was performed for the genomic data using Picard (Development_team, 2019) before genotyping. Coverage of the six individuals ranged from 30–46X.
Analysis of sex chromosome turnover in the west using genomic data and Sanger sequences
To evaluate the degree to which genetic variation in the sex-linked portion of the W or Z chromosome from the east is present in the west, a Perl script was used to identify W-linked and Z-linked SNPs in the sex-linked region of Chr8L of the two wild individuals from the east population (one female, one male) and the two lab strain individuals (one female, one male), and then evaluate genotypes at these positions in the two west individuals (one female, one male). The females from the east and lab strain have a large sex-linked region spanning the first 54.1 Mb of Chr8L (Results; Furman & Evans, 2016, Furman & Evans, 2018, Song et al., 2020). W-linked SNPs were defined as being present in heterozygous genotypes of both females and not present in homozygous genotypes of the males from the east and lab strain in the first 54.1 Mb of Chr8L; and Z-linked SNPs were defined as being the other nucleotide at positions that had a W-linked SNP. Positions with more than two variants in the six individuals were not considered. For this analysis, we required high quality genotypes that passed our genotype filters for all six individuals. Then, for each homologous position in the west on Chr8L below 54.1 Mb, we tabulated the frequencies of genotypes comprised of W-linked and Z-linked SNPs in the genotypes in the male and female individual from the west. We classified them as being either homozygous for an east W-linked nucleotide, homozygous east Z-linked nucleotide, or heterozygous for these nucleotides. These data were analyzed in 100,000 bp windows for the entire 54.1 Mb region of Chr8L. For comparison we repeated this exercise on the non-sex-linked portion (above 54.1 Mb) of Chr8L.
To explore whether the WGS data provided discernable signals of sex-linkage in other genomic regions, pairwise nucleotide diversity of polymorphic sites (hereafter π) was calculated in 100,000 bp windows using the scripts in the general_genomics repository (Martin, 2021). This statistic was calculated from RRGS data and from variable positions only; consequently, the values of π are higher than the actual pairwise nucleotide diversity (which would also include invariant sites) and standardizing these values by the size of the genomic window (which we do not attempt here) is expected to lead to downward biased estimates (Korunes & Samuk, 2021).
As detailed in the Supplement, Sanger sequencing of wild caught individuals was used to follow up regions of Chr8L that were potentially sex-linked in the west and on Chr7S in the east which, as discussed below, delivered a false signal of sex-linkage in RRGS data from a small number of individuals.
Simulations
We performed coalescent simulations using ms (Hudson, 2002) to explore the plausibility of evolutionary scenarios discussed above and in the Supplement (Supplementary Methods, Fig. 1). For these simulations, the relative effective population sizes of the east and west populations were based on analysis of pairwise nucleotide diversity of polymorphic sites in the genomic data from wild caught individuals from these populations after excluding the region below 54.1 Mb on Chr8L, which is sex-linked in the east population and lab strain (described further in the Supplement). The relative effective populations sizes of the W and Z chromosomes in the east population was assumed to be 1:3. The likelihood of these models was evaluated using a rejection sampling approach (Weiss & von Haeseler, 1998), as described the Supplement.
Results
A new draft genome assembly for X. borealis
The draft genome assembly for X. borealis was 2.75 Gb, including 558 Mb of Ns, in a total of 23,147 contigs that were greater than 1 kb, and 1,481 contigs that were greater than 10 kb. The N50, N90, L50, and L90 statistics were 145,564,450, 105,895,007, 8, and 17 respectively, where these statistics indicates the size of the contig along with all larger contigs that comprise 50% or 90% of the total genome length, and the number of contigs that make up 50% or 90% of the total genome size. These statistics indicate that more than 90% of the genome is contained within 17 chromosome-scale scaffolds. The GC content not counting Ns was 38.7%.
Sex-linkage of Chr8L in the east but not the west based on RRGS data
With the new draft genome assembly as a reference, we were able to replicate previous findings that used the X. laevis genome sequence as a reference (Furman & Evans, 2016, Furman & Evans, 2018, Song et al., 2020), including female heterogamy and sex-linkage below 54.1 Mb of Chr8L in the east population and a lab strain of X. borealis, and a genome-wide absence of a large sex-linked region in the west population of X. borealis (Fig. 2). π of the sex-linked region of Chr8L (<54.1 Mb, Fig. 3 in red) in X. borealis from east Kenya is consistently higher in females compared to the non-sex-linked region of the same chromosome (Fig. 3 in blue) and compared to both of these regions in males (Fig. 3, right side), suggesting divergence between the female-specific and sex-shared portions of the W and Z chromosome, respectively. Within females, π in the sex-linked region is more modest in individuals with lower coverage (Individuals BJE4516, BJE4541; Figs. 3, S1), although still consistently higher than the non-sex-linked region. Divergence between the sex-linked portions of the W and Z chromosomes is also evidenced by higher heterozygosity in the sex-linked region in east females, which are ZW, compared to east males, which are ZZ, but similar levels of heterozygosity in other parts of Chr8L (Fig. S1).
Figure 2.
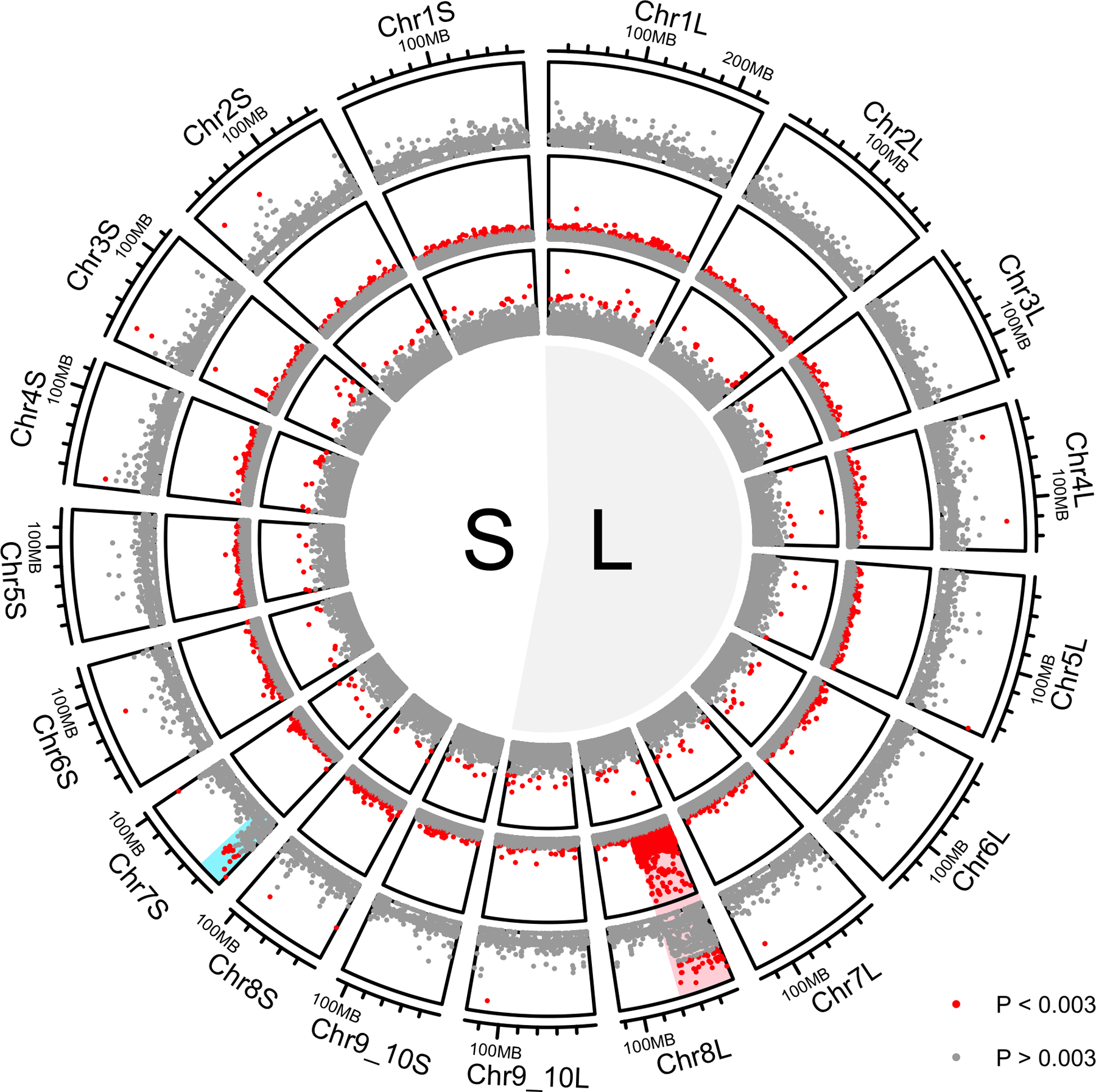
Using the new draft genome assembly for X. borealis and previously published RRGS data, we replicated the finding of sex-linkage of the first 54.1 Mb of Chr8L, highlighted in red, in the east population (outer ring) and lab strain (central ring) (Furman & Evans, 2016, Furman & Evans, 2018) but not the west population (inner ring; Song et al., 2020). We additionally identify a spurious signal of sex linkage below 23 Mb on Chr7S in the east population, highlighted in blue. Colors of dots indicate the negative natural logarithm of the probability of association of genetic variation with sex as indicated in the legend. The sample sizes are five females and five males from east Kenya, 24 females and 22 males from the lab strain, and 16 females and 15 males from the west. S and L refer to X. borealis subgenomes (see Supplement). For the west population, similar results are recovered when RRGS data from four wild caught females and four wild caught males from Njoro (in central Kenya) are added
Figure 3.
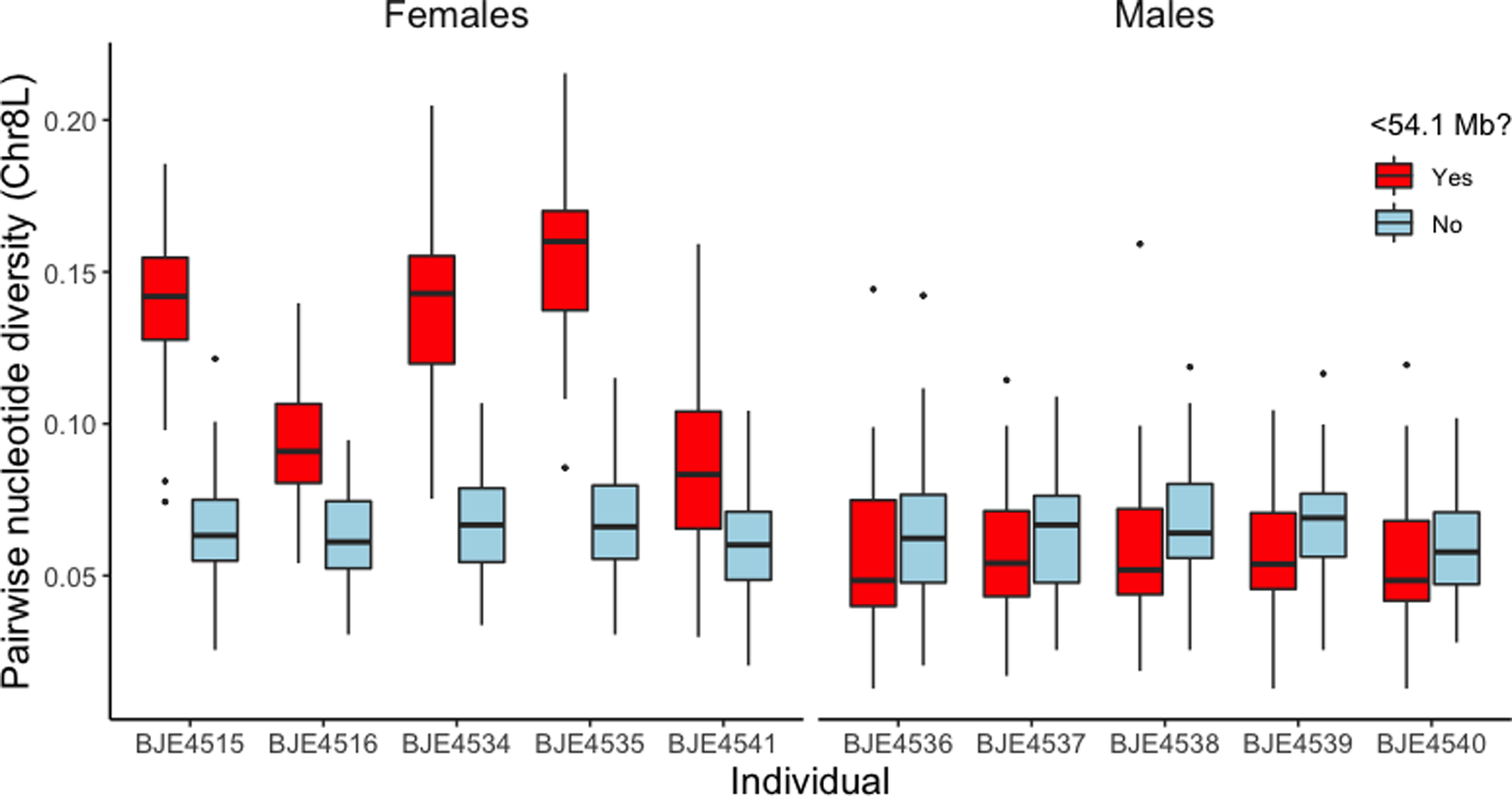
Pairwise nucleotide diversity of polymorphic sites in 2 Mb genomic windows of the sex-linked (<54.1 Mb, in red) and non-sex-linked (blue) portions of Chr8L in X. borealis from east Kenya based on RRGS data. Genomic data were collected from the left-most female and left-most male.
A spurious signature of sex-linkage on Chr7S in the east
Using the new draft genome for X. borealis as a reference, linkage analysis of genotypes from RRGS data from wild-caught X. borealis enabled us to detect a previously unidentified region on the first 23.0 Mb on Chr7S in the east population of X. borealis with an unexpected signature of sex-linkage (Fig. 2). This region of Chr7S has high π in some individuals from each sex (three females, two males; Fig. S2), and is not sex-linked in the laboratory strain or in the west population of X. borealis (Fig. 2).
When a subset of five east individuals with high π were analyzed, there was no evidence of substantial sex-linkage below 23 Mb on Chr7S, but when only the other five individuals with low π below 23 Mb on Chr7S were considered, this region was still strongly sex-linked in the remaining two females and three males (not shown). Inspection of significantly sex-linked sites in the second subset revealed that the signature of sex-linkage was because the two females were mostly homozygous for one haplotype block below 23 Mb on Chr7S, whereas the three males were mostly homozygous for a different diverged haplotype block below 23 Mb on Chr7S. This pattern is evidenced in a plot of RRGS genotypes on Chr7S in the east population of X. borealis (Fig. S3).
We followed up these inferences from RRGS data by Sanger sequencing two variable regions below 23 Mb on Chr7S in wild caught individuals from the east (six females and five males for both regions, Table S1). Genotype inferences were identical to the RRGS data and again were inconsistent with a pattern of sex-linkage where one sex was heterozygous and the other was homozygous. Overall, these results are inconsistent with the possibility that this region is a neo-sex chromosome because we were unable to identify sex-specific SNPs. Instead, it appears that sex-linkage of this portion of Chr7S is a combined consequence of a relatively small sample size (ten individuals) coupled with an unusually large region of linkage disequilibrium that, while carried by both sexes, also happened to have different overall frequencies in a small number of individuals from each sex (two females, three males). Possible explanations for this large haplotype block are discussed further below.
W-specific and Z-specific variation from the east are present in genomic data from both sexes in the west
Previous analyses using RRGS data from wild caught X. borealis and the X. laevis genome assembly as a reference failed to identify a signal of sex-linkage in X. borealis from west Kenya (Song et al., 2020), raising the question of where in the genome the sex determining locus in this population resides. Using genomic data from six individuals (one female and one male from the east, west and a lab strain), we identified W-linked and Z-linked SNPs in the east and lab females and then evaluated how frequently these SNPs and their genotypes occurred in a female and male from the west. For comparison, we also performed this analysis on the non-sex-linked (pseudoautosomal) portion of Chr8L.
In the individuals from the east and lab strain, as expected, far more SNPs had a pattern consistent with W- and Z-linkage in the sex-linked region below 54.1 Mb on Chr8L (n = 101,543) compared to the non-sex-linked region above 54.1 Mb on Chr8L (n = 1,456; Table 1). This is because the region below 54.1 Mb on Chr8L has many divergent sites between the sex-linked portions of the W and Z chromosomes, whereas the region from 54.1 – 123.8 Mb is the pseudoautosomal (recombining) portion of the sex chromosome where SNPs are not sex-linked in the east or lab strain. For convenience hereafter, genomic positions that satisfy our criteria for sex-linkage in the east and lab strain (see Methods) are called “eligible” to distinguish them from other variable positions that did not satisfy our criteria for sex-linkage. We refer to the variants at eligible positions above 54.1 Mb with a W-linked genotype pattern as “pseudo-W-linked”, because these pseudoautosomal variants are not actually sex-linked, and we do the same for the corresponding “pseudo-Z-linked” variants.
Table 1.
Counts of genotypes at eligible positions in WGS data from two west individuals above and below 54.1 Mb on Chr8L. Counts of each genotype are followed by percentages in parentheses.
| Below 54.1 Mb | Homozygous W-linked | Homozygous Z-linked | Heterozygous | Total eligible positions |
|---|---|---|---|---|
| West female (BJE4441) | 17332 (17%) | 78996 (78%) | 5215 (5%) | 101543 |
| West male (BJE4442) | 17606 (17%) | 79263 (78%) | 4674 (5%) | 101543 |
| Above 54.1 Mb | Homozygous pseudo-W-linked | Homozygous pseudo-Z-linked | Heterozygous | Total eligible positions |
|
| ||||
| West female (BJE4441) | 257 (18%) | 857 (59%) | 342 (23%) | 1456 |
| West male (BJE4442) | 265 (18%) | 857 (59%) | 334 (23%) | 1456 |
In principle, eligible positions could have any of three possible genotypes in the genomic data from each of the two individuals from west Kenya: homozygous W-linked, homozygous Z-linked, or heterozygous W/Z-linked. Below 54.1 Mb, the frequencies of each of these three genotypes were essentially identical in the west male and female, which is consistent with no sex-linked variation on most or all of Chr8L in the west. The proportion of eligible positions below 54.1 Mb that were homozygous for east W-linked SNPs in both west individuals (15%) was similar to the proportion of sites above 54.1 Mb on Chr8L that were homozygous for east pseudo-W-linked SNPs in both west individuals (14%; Table S2). However, the proportions of eligible positions below 54.1 Mb that were homozygous for east Z-linked SNPs in both west individuals was far higher (76%) than the proportions of eligible positions above 54.1 Mb on Chr8L that were homozygous for east pseudo-Z-linked SNPs in both west individuals (50%; Table S2). A substantially lower proportion of eligible positions below 54.1 Mb were heterozygous for east W- and Z-linked SNPs in both west individuals (2%) compared to the proportion of genotypes above 54.1 Mb were heterozygous for east pseudo-W- and pseudo-Z-linked SNPs in both west individuals (12%). Similarly, lower proportions of eligible positions were heterozygous for east W- and Z-linked SNPs in one of the two west individuals below 54.1 Mb (range: <1–2%) compared to above 54.1 Mb for east pseudo-W- and pseudo-Z-linked SNPs (range: <1–9%).
The ratio of female to male polymorphism in 100kb genomic windows for each of the three pairs of individuals from the east, lab strain, and west supports several inferences from the RRGS data discussed above (discussed in the Supplement; Fig. S4). Variation in this ratio and principal components analysis of the WGS data (Fig. S5) highlights variation between the male and female lab strain individual that is also evidenced by analysis of ancestry components when the number of components is greater than three (Song et al., 2020). Together, this is consistent with a complex history of the lab strain that may include ancestry from multiple founder populations and episodes of inbreeding (Supplement).
Simulations support autosomal segregation in the west of much of the genetic variation in the sex-linked portion of the east Z chromosome
Using neutral coalescent simulations, we simulated evolution of sex-linkage below 54.1 Mb on Chr8L using three models (Fig. 1) that coarsely match the Scenarios discussed above. We analyzed these simulations in the same way that we did for the WGS data, in that we searched for W-linked and Z-linked variants in simulated individuals from the east and lab strain, and then quantified the frequencies of genotypes at these positions in simulated individuals from west. With several caveats presented below, the simulations provide the strongest support for Scenario 3a (Figs. 1, 4) in which sex-linkage of this region in the east is ancestral, the sex-linked portion of the Z chromosome from the east has autosomal segregation in the west, and the sex-linked portion of the W chromosome from the east went extinct in the west. The natural logarithm of the maximum likelihood values for Scenario 1&2 (Fig. 4, left), Scenario 3a (Fig. 4, center) and Scenario 3b (Fig. 4, right) are −9.115, −7.370, and −8.740. The maximum likelihood parameter values for τ0 and τ1 (in units of 4NA, where NA is the effective population size of the ancestral lineage in each model; Supplement) for Scenario 1&2 are 0.3 and 0.3, for Scenario 3a are 3.1 and 1.3, and for Scenario 3b are 0.5 and 0.4. Based on Akaike Information Criterion weights (Wagenmakers & Farrell, 2004), Scenario 3a is 6.1 times more likely to be the best fitting model than Scenario 3b, and 8.8 times more likely to be the best fitting model than Scenarios 1&2.
Figure 4.
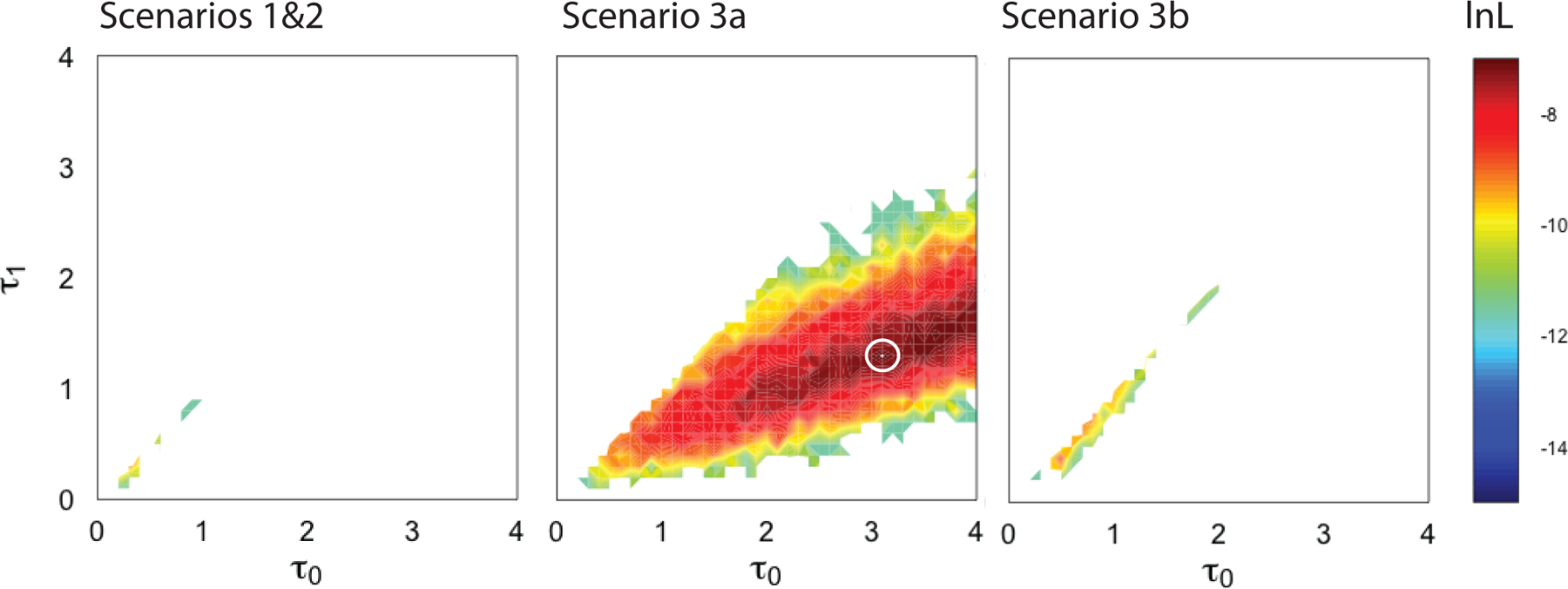
Contour plot of the natural log likelihoods of combinations of parameter settings for Scenario 1&2 (left), Scenario 3a (center) and Scenario 3b (right). Model parameters (τ0 , τ1) correspond to the time of population subdivision between the east and west and recombination cessation between the W and Z chromosome in the east population as defined in Fig. 1. In all models, τ1 ≤ τ0 (hence the triangular contour plots). The parameter combinations with the best fit to the observed data are warmer colors; parameter combinations with no accepted simulations are white and a white circle indicates the maximum likelihood parameter combination of the best fit model.
Sanger sequencing does not identify sex-linked variation on Chr8L in the west population
Although the coalescent simulations provide the strongest support for Scenario 3a, we nonetheless further explored the possibility that X. borealis from east and west Kenya both have a homologous sex-determining region on Chr8L but with differently sized sex-linked regions of recombination suppression, which would be consistent with Scenario 1 (Fig. 1). Using the genomic data as a guide, we attempted to identify a sex-linked portion of Chr8L in the west female that is homologous to the sex-linked regions of the east and lab female. In 100,000 base pair genomic windows we searched for heterozygous genotypes below 54.1 Mb on Chr8L in the genomic data from the west female that were also heterozygous in genomic data from females from the east and the lab strain, but homozygous in the three male genomes (one each from the east, lab strain and the west population). One region was identified (27–30 Mbs) with a high density of genotypes that matched this pattern, and a weaker signal was also present from 37–40 Mbs (Fig. S6). However, Sanger sequencing of six amplicons (Table S3, Fig. S6) in 6–29 wild caught females and 3–19 wild caught males failed to recover female-linked SNPs in the west, suggesting that these regions in fact are not sex-linked in the west population. Instead, these regions appear to be genomic patches that happened to have low heterozygosity in the male individual from the west that we sequenced.
No signal of sex-linkage in one region of Chr8L in X. fraseri
Xenopus borealis is sister to a clade that includes X. fischbergi and X. fraseri (Evans et al., 2015, Evans et al., 2019). If the sex-linked region in the west population were newly evolved relative to the east population (Scenarios 3a or 3b, Fig. 1), it is possible that other species that are closely related to X. borealis might have a sex-linked region on Chr8L that is homologous to the east population of X. borealis. As a preliminary exploration of this possibility, we sequenced a portion of sox3, which is located at 37.85 Mb on Chr8L in the new X. borealis genome assembly, in nine X. fraseri females and four X. fraseri males. We identified four segregating polymorphisms (three SNPs and a one base pair insertion/deletion), but all of these variants were present in both sexes. These preliminary findings do not support sex-linkage of this gene in X. fraseri. Interpretations of these findings are discussed below.
Low variation in depth of coverage on Chr8L and Chr7S
To explore the possibility that portions of Chr8L or Chr7S that were sex-linked or spuriously sex-linked, respectively, in the east population were duplicated or deleted, depth of coverage was quantified for the genomic data for each of the six individuals. The median coverages of each portion of each chromosome were similar within each individual (Figs. S7, S8) and minor variation in depth of coverage was generally population-specific rather than sex-specific (Figs. S9, S10). These results are inconsistent with sex-linked variation in large scale duplications or deletions on either of these chromosomes. Analysis of depth of coverage on unplaced scaffolds also failed to recover evidence of substantially sized, sex-specific differences in the WGS data from the west individuals (Supplement).
Discussion
Xenopus borealis has intraspecific variation in sex chromosomes with small (west population) or large (east population) regions of sex-linked recombination suppression
Using a new chromosome-scale genome assembly for X. borealis, new genomic data, and previously published RRGS data, we confirmed previous findings that used the X. laevis genome assembly as a reference (Furman & Evans, 2016, Furman & Evans, 2018, Song et al., 2020) that a large sex-linked region exists below 54.1 Mb on Chr8L in individuals from east Kenya (Figs. 2, 3) and in a lab strain. Also consistent with previous findings (Song et al., 2020), in the west population we found no evidence of a substantially-sized sex-linked region anywhere in the genome, suggesting that the sex-linked region of this population is small. We detected diverged haplotype blocks below 23 Mb on Chr7S that delivered a false signal of sex-linkage in a small sample of individuals (Fig. 2; S2). This large haplotype block is evidenced by atypically high heterozygosity in individuals that were heterozygous for two diverged blocks and atypically high homozygosity in individuals that were homozygous for one block (Fig. S3). In frogs, 11 Y-chromosome/autosome fusions have been identified, but there are no known examples of a fusion between a W-chromosome and a portion of an autosome (Ma & Veltsos, 2021). Thus, it is perhaps unsurprising that our efforts were unable to authenticate sex-linkage of any region of Chr7S in the east population of X. borealis. Plausible mechanisms behind the haplotype block on Chr7S in the east population of X. borealis include introgression and a segregating inversion. Currently available genomic data reported here include short read WGS and RRGS data from east individuals. There are technical challenges to genotyping large structural variants with short read data, including regions of reference genomes with unknown sequence (and thus no mapped reads) and repetitive regions. Further studies with long reads or cytogenetic approaches are needed to evaluate evidence of inversions on Chr7S and Chr8L.
The ancestral state of X. borealis sex chromosomes
Using genomic data, RRGS data, and Sanger sequencing of wild caught individuals, our efforts to identify a sex-linked region of Chr8L in X. borealis from west Kenya were unsuccessful. This indicates that Scenario 1 (Fig. 1) is only plausible if a sex-linked region on Chr8L is very small. In the genomic data from the two west individuals, genotypes below 54.1 Mb on Chr8L that are homozygous for Z-linked variants from the east are substantially more common (78% of eligible sites) than genotypes above 54.1 Mb on Chr8L that are homozygous for pseudo-Z-linked variants from the east (59% of eligible sites; Table 1). Compared to the other scenarios discussed above (Fig. 1), simulations suggest that these genotype frequencies are far better explained by a coarse model of Scenario 3a (Fig. 1, right-center) in which most genetic variation from the sex-linked portion of the W chromosome of the east is not present in the west and most genetic variation from the sex-linked portion of the Z chromosome from the east population has autosomal (or pseudoautosomal) segregation in the west population (Fig. 4, center).
One question raised by these results asks why so many W-linked variants from the east were found in homozygous genotypes (17%) in the west below 54.1 Mb on Chr8L when Scenario 3a posits that the W chromosome went extinct in the west. The simulations provide an explanation. Many of these positions were initially (ancestrally) invariant across the W and Z chromosome in the east. Many Z-linked variants in the east and lab individuals then arose through new mutations on the east Z chromosome that occurred after the Z-linked portion of this chromosome had already begun segregating as an autosome in the west. Put another way, a substantial portion of the W-linked variants in the east and west are ancestral to derived Z-linked variants in the east. Moreover, the maximum likelihood estimate of the timing of the onset of autosomal segregation of the east Z chromosome in the west (1.3*4NA generations) is ~40% of the estimated timing of the onset of recombination suppression between the W and Z chromosome in the east (3.1*4NA generations; Fig. 4). This indicates that fixation of much of the genetic variation of the Z-linked portion of Chr8L in the west occurred many generations ago and that a substantial proportion of east and lab strain Z-linked variation is specific to the east population and lab strain (and not found in the west population).
There are several caveats to these simulations. One is that recombination in the west population was not simulated. Inclusion of recombination in the west population is expected to reduce the variance in coalescent times of segregating mutations, but not the mean coalescent time (Hudson, 1990). Clearly there are alternative demographic models, especially ones with more parameters, that are possible and that may provide better fits to the data. For example, we did not consider the possibility that recombination suppression on Chr8L arose in an ancestor of the west and east population prior to the origin of population structure and subsequent loss of extensive Chr8L sex-linkage in the west. Additionally, demographic changes in nature generally do not occur instantaneously, the effects of natural selection were not incorporated in these simulations, and the simulations may not capture nuances associated with recombination suppression between the sex-linked portions of the W and Z chromosomes in the east, such as incremental expansion of recombination suppression. We did not attempt to assess the effect of these simplifying assumptions. These caveats in mind, our simulations allowed us to evaluate the relative strengths of the models that we considered, but clearly do not prove that any particular model is correct.
Unfortunately, our efforts to identify a sex-linked region in the west were unsuccessful, which could have provided further evidence for or against the evolutionary scenarios considered here. If there is a very small sex- linked region <54.1 Mb on Chr8L in the west population that is shared with the east population, gene flow between the east and west could influence the distribution of genetic variation in these populations in ways that are not captured by the simulations. If this is the case, the female-linked region in the east is expected to be situated on the end of a chromosome where recombination in females tends to be lower compared to the centers of the chromosomes (discussed further in the Supplement; Furman & Evans, 2018, Sardell & Kirkpatrick, 2020). Further scrutiny of genomic variation in natural populations of X. borealis in the west will permit testing of these expectations, and a more robust evaluation of intraspecific sex chromosome demography in X. borealis. In this study we also report hints that the closely related species X. fraseri may have a different and/or smaller sex-linked region compared to the east population of X. borealis. Additional study of sex-linked regions in X. fraseri and other closely related species (X. muelleri, X. fischbergi) may offer insights origin and age of sex-linked recombination suppression in the east population of X. borealis.
In another Xenopus species – X. tropicalis – there are three functionally distinct sex chromosomes: W, Y, and Z, and the W chromosome is dominant for femaleness over the Z, and the Y chromosome is dominant for maleness over the W and Z (Roco et al., 2015). Female X. tropicalis have either of two sex chromosome genotypes: WZ or WW and males have three: ZZ, WY, or ZY. Analysis of molecular variation suggests that the sex determining regions of the W and Y chromosome are in similar genomic locations on one end of Chr7 (Furman et al., 2020, Mitros et al., 2019). Interestingly, all three X. tropicalis sex chromosomes naturally co-occur in at least two populations in Ghana even though some crosses result in offspring with a skewed sex ratio (Furman et al., 2020, Roco et al., 2015). Further study of X. borealis on a fine geographic scale in nature is needed to identify the sex-linked region in the west population, to assess whether and to what degree the sex chromosome variation in the west and east populations geographically co-occur, and if so, whether there is a dominance hierarchy for sex determination among them, and whether certain crosses produce offspring with a skewed sex ratio.
To the extent that Scenario 3a is indeed a reasonable approximation of the true demography of X. borealis sex chromosomes, why the west population of X. borealis might have experienced a sex chromosome turnover event remains unclear. Under the hot potato model, sex chromosome turnover is favored by natural selection because it purges a “degenerate” sex-chromosome with impaired function due to reduced efficacy of natural selection in non-recombining genomic regions (Blaser et al., 2013, Blaser et al., 2014). Sex chromosome degeneration generally involves sex-specific sex chromosomes (e.g., the W or Y chromosomes) rather than sex-shared sex chromosomes (e.g., the Z or X chromosomes), and the recent loss of the east W chromosome in the west population matches the expectations of this model. Counterintuitively however, sex-linked transcripts of a lab strain of X. borealis tend to have female-biased expression at multiple developmental stages and tissue types, which suggests degeneration of the Z rather than the W chromosome, perhaps when the east Z chromosome previously was a Y chromosome in an ancestor with male heterogamy (Song et al., 2020). In this context, autosomal segregation of the east Z chromosome is not predicted by the hot potato model, and points to other explanations such as genetic drift (Bull & Charnov, 1977), sex-specific advantages of a new system for sex determination (Bull & Charnov, 1977, Vuilleumier et al., 2007), selection on offspring sex-ratio (Kozielska et al., 2006), or sexual antagonism (van Doorn & Kirkpatrick, 2007).
A digression on the sex determining gene of X. laevis, and future directions for Xenopus sex chromosome genomics
In many animals, sex determination is triggered by sex-specific heterozygosity (Bull & Charnov, 1977, Ohno, 1967), such as a dominant-sex specific allele or sex-specific differences in gene dosage (Graves, 2013). A striking discovery in the biology of Xenopus is the female-determining gene dm-w in the allotetraploid species X. laevis (Yoshimoto et al., 2008). Dm-w is a partial duplicate of the male-related gene dmrt1 –specifically the homeolog of dmrt1 that resides in the S-subgenome (dmrt1S) even though dm-w resides in the L subgenome on Chr2L (please see the Supplement for further discussion of Xenopus subgenomes)(Yoshimoto et al., 2008, Bewick et al., 2011). Surveys using PCR and targeted capture sequencing identified orthologs of dm-w in several species in subgenus Xenopus but not species from subgenus Silurana (Bewick et al., 2011, Cauret et al., 2020), indicating that this gene evolved recently in Xenopus. These same assays demonstrate that dm-w was lost in several species in subgenus Xenopus after it arose, including X. borealis (Bewick et al., 2011, Cauret et al., 2020).
Dm-w appears to occur exclusively in females in wild-caught X. laevis and X. gilli, suggesting a homologous dm-w-based mechanism of female sex determination in this pair of species, However, in several Xenopus species dm-w is found in both male and female individuals. In X. itombwensis, dm-w occurs in all individuals of both sexes and appears to have autosomal segregation, and in several other species (X. pygmaeus, X. clivii, and X. victorianus) dm-w occurs relatively frequently in males (>15%), and in at least two species (X. pygmaeus, X. clivii) dm-w does not occur in some females (>10%) (Cauret et al., 2020). This pattern suggests that dm-w had a dynamic functional evolutionary history that includes gene loss (e.g., X. borealis), sidelining on an autosome (X. itombwensis), a potential role as an “influencer” of female sex determination that sometimes segregates as a female-specific allele (e.g., X. pygmaeus, X. clivii, X. victorianus), and eventual empowerment in the ancestor of X. laevis and X. gilli where it is a completely sex-linked master regulator of female differentiation that segregates as a female-specific allele (Cauret et al., 2020). The absence of dm-w in at least 11 species in subgenus Xenopus points to variation in the mechanisms that sexual differentiation in these species that extends beyond that the species whose trigger for sex determination has been mapped to specific sex chromosomes (discussed above; Cauret et al., 2020). New findings reported here suggest that the west population of X. borealis may be added to the list of Xenopus with novel and uncharacterized sex determining systems.
Future work is needed to build on the fascinating discovery of dm-w by mapping sex-linked regions of other Xenopus species (and populations), and by identifying what variation within these regions is responsible for orchestrating sexual differentiation. The first part of this can be accomplished using genomic approaches such as those in this study, but is challenged by the difficulty of obtaining live animals from remote parts of Africa for captive breeding, which is helpful for identifying very small sex-linked regions, such as that of the west population of X. borealis. The second part can be accomplished using gene editing (Tandon et al., 2016), and the best species/populations to work with are ones that have a small region of sex-linkage, such as X. tropicalis, the west population of X. borealis, and possibly others, because these have a small number of candidate sex-determining genes. Insights gained from these efforts are many, including making possible the evaluation that sex-biases in recombination influence the genomic locations of sex-determining genes (Sardell & Kirkpatrick, 2020), genetic mechanisms for developmental systems drift of sex determination (True & Haag, 2001), and evaluation of theoretical expectations associated with selection on sex-ratio in nature (Hamilton, 1967, Charnov, 1982).
Supplementary Material
Acknowledgements
We thank Jacques Robert from the University of Rochester Medical Center School of Medicine and Dentistry for providing animals for genome sequencing and assembly and three reviewers for constructive and comprehensive suggestions on earlier versions of this manuscript.
Funding
This research was supported by the Natural Science and Engineering Research Council of Canada (RGPIN-2017–05770 to BJE), a Resource Allocation Competition award from Compute Canada (to BJE), support from the Museum of Comparative Zoology at Harvard University (to BJE), and the National Institutes of Health (R01HD080708 to DSR; R01GM086321, R01HD065705 to DSR and RMH; R35GM127069 to RMH).
Footnotes
Data accessibility
Genomic data from four wild-caught X. borealis individuals is deposited in the NCBI SRA (BioProject PRJNA616217). Sanger sequences are deposited in GenBank (accession numbers: OM436355 – OM436380; OM489937 – OM490010). The draft genome assembly for X. borealis and the associated sequencing data were deposited into the NCBI Genbank and SRA databases, respectively, under BioProject PRJNA827809. Genotype files are deposited in Dryad: https://doi.org/10.5061/dryad.3tx95×6k0
References
- Adolfsson S & Ellegren H 2013. Lack of dosage compensation accompanies the arrested stage of sex chromosome evolution in ostriches. Mol Biol Evol 30: 806–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronesty E 2013. Comparison of sequencing utility programs. The open bioinformatics journal 7. [Google Scholar]
- Bachtrog D, Mank JE, Peichel CL, Kirkpatrick M, Otto SP, Ashman TL, Hahn MW, Kitano J, Mayrose I, Ming R, Perrin N, Ross L, Valenzuela N, Vamosi JC & Consortium TS 2014. Sex Determination: Why So Many Ways of Doing It? Plos Biology 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewick AJ, Anderson DW & Evans BJ 2011. Evolution of the closely related, sex-related genes DM-W and DMRT1 in African clawed frogs (Xenopus). Evolution 65: 698–712. [DOI] [PubMed] [Google Scholar]
- Blaser O, Grossen C, Neuenschwander S & Perrin N 2013. Sex-chromosome turnovers induced by deleterious mutation load. Evolution: International Journal of Organic Evolution 67: 635–645. [DOI] [PubMed] [Google Scholar]
- Blaser O, Neuenschwander S & Perrin N 2014. Sex-Chromosome Turnovers: The Hot-Potato Model. American Naturalist 183: 140–146. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M & Usadel B 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandvain Y & Coop G 2012. Scrambling Eggs: Meiotic Drive and the Evolution of Female Recombination Rates. Genetics 190: 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredeson JV, Mudd AB, Medina-Ruiz S, Mitros T, Smith OK, Miller KE, Lyons JB, Batra SS, Park J & Berkoff KC 2021. Conserved chromatin and repetitive patterns reveal slow genome evolution in frogs. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull JJ 1983. Evolution of sex determining mechanisms Benjamin Cummings, Menlo Park. [Google Scholar]
- Bull JJ & Charnov EL 1977. Changes in the heterogametic mechanism of sex determination. Heredity 39: 1–14. [DOI] [PubMed] [Google Scholar]
- Cannatella DC & de Sá RO 1993. Xenopus laevis as a model organism. Systematic Biology 42: 476–507. [Google Scholar]
- Cass AN, Elias A, Fudala ML, Knick BD & Davis MC 2021. Conserved Mechanisms, Novel Anatomies: The Developmental Basis of Fin Evolution and the Origin of Limbs. Diversity-Basel 13: Article No.: 384. [Google Scholar]
- Cauret CM, Gansauge M-TT, Tupper A, Furman BLS, Knytl M, Song X, Greenbaum E, Meyer M & Evans BJ 2020. Developmental systems drift and the drivers of sex chromosome evolution. Molecular Biology and Evolution 37: 799–810. [DOI] [PubMed] [Google Scholar]
- Chapman JA, Ho I, Sunkara S, Luo S, Schroth GP & Rokhsar DS 2011. Meraculous: De Novo Genome Assembly with Short Paired-End Reads. PLoS One 6: Article No.: e23501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B & Wall JD 1999. Inbreeding, heterozygote advantage and the evolution of neo-X and neo-Y sex chromosomes. Proceedings of the Royal Society Biological Sciences Series B 266: 51–56. [Google Scholar]
- Charlesworth D & Charlesworth B 1980. Sex differences in fitness and selection for centric fusions between sex chromosomes and autosomes. Genetical Research 35: 205–214. [DOI] [PubMed] [Google Scholar]
- Charnov EL 1982. The Theory of Sex Allocation Princeton University Press. [PubMed] [Google Scholar]
- Development_team (2019) Picard Toolkit. pp. Broad Institute, GitHub Repository https://broadinstitute.github.io/picard/.
- Dierckxsens N, Mardulyn P & Smits G 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Research 45: e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans BJ, Carter TF, Greenbaum E, Gvoždík V, Kelley DB, McLaughlin PJ, Pauwels OSG, Portik DM, Stanley EL, Tinsley RC, Tobias ML & Blackburn DC 2015. Genetics, morphology, adverstisement calls, and historical records distinguish six new polyploid species of African clawed frog (Xenopus, Pipidae) from West and Central Africa. PLoS One 10: e0142823 (51 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans BJ, Gansauge MT, Stanley EL, Furman BLS, Cauret CMS, Ofori-Boateng C, Gvozdik V, Streicher JW, Greenbaum E, Tinsley RC, Meyer M & Blackburn DC 2019. Xenopus fraseri: Mr. Fraser, where did your frog come from? PLoS One 14: e0220892 (14 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans BJ, Pyron RA & Wiens JJ (2012) Polyploidization and sex chromosome evolution in amphibians. In: Polyploidy and Genome Evolution, (Soltis PS & Soltis DE, eds.). pp. 385–410. Springer Verlag. [Google Scholar]
- Fisher RA 1931. The evolution of dominance. Biological Reviews 6: 345–368. [Google Scholar]
- Fraisse C, Picard MAL & Vicoso B 2017. The deep conservation of the Lepidoptera Z chromosome suggests a non-canonical origin of the W. Nature Communications 8: Article No.: 1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman BLS, Cauret CM, Colby G, Measey J & Evans BJ 2016. Limited genomic consequences of hybridization between two African clawed frogs, Xenopus gilli and X. laevis (Anura: Pipidae). Scientific Reports 7: 1091 (11 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman BLS, Cauret CMS, Knytl M, Song XY, Premachandra T, Ofori-Boateng C, C, J. D., Horb, M. E. & Evans, B. J. 2020. A frog with three sex chromosomes that co-mingle together in nature: Xenopus tropicalis has a degenerate W and a Y that evolved from a Z chromosome. Plos Genetics 16: e1009121 (27 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman BLS & Evans BJ 2016. Sequential turnovers of sex chromosomes in African clawed frogs (Xenopus) suggest some genomic regions are good at sex determination. Genes, Genomes, Genetics 6: 3625–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman BLS & Evans BJ 2018. Divergent evolutionary trajectories of two young, homomorphic, and closely related sex chromosome systems. Genome Biol Evol 10: 742–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble T, Coryell J, Ezaz T, Lynch J, Scantlebury DP & Zarkower D 2015. Restriction Site-Associated DNA Sequencing (RAD-seq) Reveals an Extraordinary Number of Transitions among Gecko Sex-Determining Systems. Molecular Biology and Evolution 32: 1296–1309. [DOI] [PubMed] [Google Scholar]
- Graves JAM 2008. Weird animal genomes and the evolution of vertebrate sex and sex chromosomes. Annual Review of Genetics 42: 565–586. [DOI] [PubMed] [Google Scholar]
- Graves JAM 2013. How to evolve new vertebrate sex determining genes. Developmental Dynamics 242: 354–359. [DOI] [PubMed] [Google Scholar]
- Gurdon JB & Hopwood N 2000. The introduction of Xenopus laevis into developmental biology: of empire, pregnancy testing and ribosomal genes. International Journal of Developmental Biology 44: 43–50. [PubMed] [Google Scholar]
- Hamilton WD 1967. Extraordinary sex ratios. Science 156: 477–488. [DOI] [PubMed] [Google Scholar]
- Harland RM & Grainger RM 2011. Xenopus research: Metamorphosed by genetics and genomics. Trends in Genetics 27: 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten U, Harland RM, Gilchrist MJ, Hendrix D, Jurka J, Kaptonov V, Ovcharenko I, Putnam NH, Shu S, Taher L, Blitz IL, Blumberg B, Dichman DS, Dubchak I, Amaya E, Detter JC, Fletcher R, Gerhard DS, Goodstein D, Graves T, Grigoriev IV, Grimwood J, Kawashima T, Lindquist E, Lucas SM, Mead PE, Mitros T, Ogino H, Ohta Y, Poliakov AV, Pollet N, Robert J, Salamov A, Sater AK, Schmutz J, Terry A, Vize PD, C., W. W., Wells D, Wills A, Wilson RK, Zimmerman LB, Zorn AM, Grainger R, Grammer T, Kohokha MK, Richardson PM& Rokhsar DS 2010. The genome of the western clawed frog Xenopus tropicalis. Science 328: 633–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR (1990) Gene genealogies and the coalescent process. In: Oxford Surveys in Evolutionary Biology, Vol. 7 (Futuyma D. & Antoaovics J, eds.). pp. 1–44. [Google Scholar]
- Hudson RR 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Irie N & Kuratani S 2011. Comparative transcriptome analysis reveals vertebrate phylotypic period during organogenesis. Nature Communications 2: Article No.: 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ironside JE 2010. No amicable divorce? Challenging the notion that sexual antagonism drives sex chromosome evolution. BioEssays 32: 718–726. [DOI] [PubMed] [Google Scholar]
- Jaenike J 2001. Sex chromosome meiotic drive. Annual Review of Ecology and Systematics 32: 25–49. [Google Scholar]
- Jeffries DL, Gerchen JF, Scharmann M & Pannell JR 2021. A neutral model for the loss of recombination on sex chromosomes. Philosophical Transactions of the Royal Society of London B Biological Sciences 376: Article No.: 20200096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffries DL, Lavanchy G, Sermier R, Sredl MJ, Miura I, Borzée A, Barrow LN, Canestrelli D, Crochet P-A, Dufresnes C & others 2018. A rapid rate of sex-chromosome turnover and non-random transitions in true frogs. Nature communications 9: 4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korunes KL & Samuk K 2021. pixy: Unbiased estimation of nucleotide diversity and divergence in the presence of missing data. Molecular Ecology Resources 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozielska M, Pen I, Beukeboom LW & Weissing FJ 2006. Sex ratio selection and multi-factorial sex determination in the housefly: a dynamic model. Journal of Evolutionary Biology 19: 879–888. [DOI] [PubMed] [Google Scholar]
- Li H 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27: 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W-J & Veltsos P 2021. The Diversity and Evolution of Sex Chromosomes in Frogs. Genes 12: Article No.: 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL & Zimin A 2018. MUMmer4: A fast and versatile genome alignment system. PLoS Computational Biology 14: Article No.: e1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S (2021) Genomics_general repository. pp. GitHub, San Francisco, CA. [Google Scholar]
- Mawaribuchi I, Takahashi S, Wada M, Uno Y, Matsuda Y, Kondo M, Fukui A, Takamatsu N, Taira M & Ito M 2017. Sex chromosome differentiation and the W- and Z-specific loci in Xenopus laevis. Developmental Biology 426: 393–400. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M & DePristo MA 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitros T, Lyons JB, Session AM, Jenkins J, Shu S, Kwon T, Lane M, Ng C, Grammer TC, Khokha MK, Grimwood J, Schmutz J, Harland RM & Rokhsar DS 2019. A chromosome-scale genome assembly and dense genetic map for Xenopus tropicalis. Dev Biol 452: 8–20. [DOI] [PubMed] [Google Scholar]
- Mudd AB, Bredeson JV, Baum R, Hockemeyer D & Rokhsar DS 2020. Analysis of muntjac deer genome and chromatin architecture reveals rapid karyotype evolution. Communications Biology 3: Article No.: 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell J, Schulz-Trieglaff O, Carlson E, Hims MM, Gormley NA & Cox AJ 2015. NxTrim: optimized trimming of Illumina mate pair reads. Bioinformatics (Oxford) 31: 2035–2037. [DOI] [PubMed] [Google Scholar]
- Ohno S 1967. Monographs on endocrinology. Sex chromosomes and sex-linked genes, 1st Edition ed. Springer-Verlag, Heidelberg. [Google Scholar]
- Olmstead AW, Lindberg-Livingston A & Degitz SJ 2010. Genotyping sex in the amphibian, Xenopus (Silurana) tropicalis, for endocrine disruptor bioassays. Aquatic Toxicology 98: 60–66. [DOI] [PubMed] [Google Scholar]
- Onuma Y, Takahashi S, Asashima M, Kurata S & Gehring WJ 2002. Conservation of Pax 6 function and upstream activation by Notch signaling in eye development of frogs and flies. Proceedings of the National Academy of Sciences of the United States of America 99: 2020–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer DH, Rogers TF, Dean R & Wright AE 2019. How to identify sex chromosomes and their turnover. Molecular Ecology 28: 4709–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennell MW, Mank JE & Peichel CL 2018. Transitions in sex determination and sex chromosomes across vertebrate species. Molecular Ecology 27: 3950–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ & Sham PC 2007. PLINK: A tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam NH, O’Connell BL, Stites JC, Rice BJ, Blanchette M, Calef R, Troll CJ, Fields A, Hartley PD, Sugnet CW, Haussler D, Rokhsar DS & Green RE 2016. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Research 26: 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WR 1987. The accumulation of sexually antagonistic genes as a selective agent promoting the evolution of reduced recombination between primitive sex chromosomes. Evolution 41: 911–914. [DOI] [PubMed] [Google Scholar]
- Roco ÁS, Olmstead AW, Degitz SJ, Amano T, Zimmerman LB & Bullejos M 2015. Coexistence of Y, W, and Z sex chromosomes in Xenopus tropicalis. Proceedings of National Academy of Sciences 112: E4752–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardell JM, Cheng C, Dagilis AJ, Ishikawa A, Kitano J, Peichel CL & Kirkpatrick M 2018. Sex differences in recombination in sticklebacks. G3: Genes, Genomes, Genetics 8: 1971–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardell JM & Kirkpatrick M 2020. Sex differences in the recombination landscape. The American Naturalist 195: 361–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Session AM, Uno Y, Kwon T, Chapman JA, Toyoda A, Takahashi S, Fukui A, Hikosaka A, Suzuki A, Kondo M, van Heeringen SJ, Quigley I, Heinz S, Ogino H, Ochi H, Hellsten U, Lyons JB, Simakov O, Putnam N, Stites J, Kuroki Y, Tanaka T, Michiue T, Watanabe M, Bogdanovic O, Lister R, Georgiou G, Paranjpe SS, van Kruijsbergen I, Shu S, Carlson J, Kinoshita T, Ohta Y, Mawaribuchi S, Jenkins J, Grimwood J, Schmutz J, Mitros T, Mozaffari SV, Suzuki Y, Haramoto Y, Yamamoto TS, Takagi C, Heald R, Miller K, Haudenschild C, Kitzman J, Nakayama T, Izutsu Y, Robert J, Fortriede J, Burns K, Lotay V, Karimi K, Yasuoka Y, Dichmann DS, Flajnik MF, Houston DW, Shendure J, DuPasquier L, Vize PD, Zorn AM, Ito M, Marcotte EM, Wallingford JB, Ito Y, Asashima M, Ueno N, Matsuda Y, Veenstra GJ, Fujiyama A, Harland RM, Taira M & Rokhsar DS 2016. Genome evolution in the allotetraploid frog Xenopus laevis. Nature 538: 336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Le S, Li Y & Hu F 2016. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS One 11: Article No.: e0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X-Y, Furman BLS, Premachandra T, Knytl M, Cauret CMS, Wasonga DV, Measey J, Dworkin I & Evans BJ 2020. Sex chromosome degeneration, turnover, and sex-biased expression of sex-linked transcripts in African clawed frogs (Xenopus). Philosophical Transactions of the Royal Society of London, B 376: 20200095 (15 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Svedberg J, Hiltunen M, Corcoran P & Johannesson H 2017. Large-scale suppression of recombination predates genomic rearrangements in Neurospora tetrasperma. Nature Communications 8: Article No.: 1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon P, Conlon F, Furlow JD & Horb ME 2016. Expanding the genetic toolkit in Xenopus: Approaches and opportunities for human disease modeling. Dev Biol 426: 325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- True JR & Haag ES 2001. Developmental system drift and flexibility in evolutionary trajectories. Evolution and Development 3: 109–119. [DOI] [PubMed] [Google Scholar]
- Tymowska J (1991) Polyploidy and cytogenetic variation in frogs of the genus Xenopus. In: Amphibian Cytogenetics and Evolution, (Green DS & Sessions SK, eds.). pp. 259–297. Academic Press, San Diego. [Google Scholar]
- van Doorn GS & Kirkpatrick M 2007. Turnover of sex chromosomes induced by sexual conflict. Nature 449: 909–912. [DOI] [PubMed] [Google Scholar]
- van Doorn GS & Kirkpatrick M 2010. Transitions Between Male and Female Heterogamety Caused by Sex-Antagonistic Selection. Genetics 186: 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuilleumier S, Lande R, Van Alphen JJM & Seehausen O 2007. Invasion and fixation of sex-reversal genes. Journal of Evolutionary Biology 20: 913–920. [DOI] [PubMed] [Google Scholar]
- Wagenmakers E & Farrell S 2004. AIC model selection using Akaike weights. Psychonomic Bulletin and Review 11: 192–196. [DOI] [PubMed] [Google Scholar]
- Weisman AI & Coates CW 1941. The frog test (Xenopus laevis) as a rapid diagnostic test for early pregnancy. Endocrinology 28: 141–142. [Google Scholar]
- Weiss G & von Haeseler A 1998. Inference of population history using a likelihood approach. Genetics 149: 1539–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells DE, Gutierrez L, Xu Z, Krylov V, Macha J, Blankenburg KP, Hitchens M, Bellot LJ, Spivey M, Stemple DL, Kowis A, Ye Y, Pasternak S, Owen J, Tran T, Slavikova R, Tumova L, Tlapakova T, Seifertova E, Scherer SE & Sater AK 2011. A genetic map of Xenopus tropicalis. Developmental Biology 354: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH & Hatcher MJ 2000. Maternal-zygotic gene conflict over sex determination: Effects of inbreeding. Genetics 155: 1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K & Kitano J 2012. THE CONTRIBUTION OF FEMALE MEIOTIC DRIVE TO THE EVOLUTION OF NEO-SEX CHROMOSOMES. Evolution 66: 3198–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshido A, Sichova J, Pospisilov K, Nguyen P, Volenikov A, Safar J, Provaznik J, Vila R & Marec F 2020. Evolution of multiple sex-chromosomes associated with dynamic genome reshuffling in Leptidea wood-white butterflies 10.1038/s41437-020-0325-9. Heredity 125: 138–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S, Okada E, Umemoto H, Tamura K, Uno Y, Nishida-Umehara C, Matsuda Y, Takamatsu N, Shiba T & Ito M 2008. A W-linked DM-domain gene, DM-W, participates in primary ovary development in Xenopus laevis. Proceedings of the National Academy of Sciences 105: 2469–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Zhang J, Bachtrog D, An N, Huang Q, Jarvis ED, Gilbert MTP & Zhang G 2014. Complex evolutionary trajectories of sex chromosomes across bird taxa. Science 346: 1246338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C, Massonnet M, Minio A, Patel S, Llaca V, Karn A, Gouker F, Cadle-Davidson L, Reisch B, Fennell A & Cantu D 2021. Multiple independent recombinations led to hermaphroditism in grapevine. Proceedings of the National Academy of Sciences 118: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Úbeda F, Patten MM & Wild G 2015. On the origin of sex chromosomes from meiotic drive. Proceedings of the Royal Society B 282: 20141932. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.