

Abstract
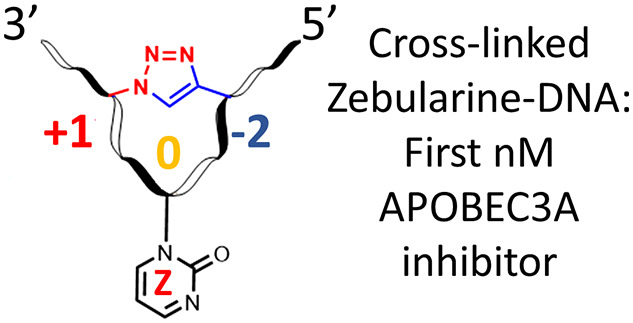
Drug resistance is a major problem associated with anticancer chemo- and immunotherapies. Recent advances in the understanding of resistance mechanisms have revealed that enzymes of the APOBEC3 (A3) family contribute to the development of drug resistance in multiple cancers. A3 enzymes are polynucleotide cytidine deaminases that convert cytosine to uracil (C→U) in single-stranded DNA (ssDNA) and in this way protect humans against viruses and mobile retroelements. On the other hand, cancer cells use A3s, especially A3A and A3B, to mutate human DNA, and thus by increasing rates of evolution, cancer cells escape adaptive immune responses and resist drugs. However, as A3A and A3B are non-essential for primary metabolism, their inhibition opens up a strategy to augment existing anticancer therapies and suppress cancer evolution. To test our hypothesis that pre-shaped ssDNA mimicking the U-shape observed in ssDNA–A3 complexes can provide a better binder to A3 enzymes, a Cu(I)-catalyzed azide–alkyne cycloaddition was used to cross-link two distant modified nucleobases in ssDNA. The resultant cytosine-containing substrate, where the cytosine sits at the apex of the loop, was deaminated faster by the engineered C-terminal domain of A3B than a standard, linear substrate. The cross-linked ssDNA was converted into an A3 inhibitor by replacing the 2′-deoxycytidine in the preferred TCA substrate motif by 2′-deoxyzebularine, a known inhibitor of single nucleoside cytidine deaminases. This strategy yielded the first nanomolar inhibitor of engineered A3BCTD and wild-type A3A (Ki = 690 ± 140 and 360 ± 120 nM, respectively), providing a platform for further development of powerful A3 inhibitors.
Introduction
Nucleic acid editing by members of the APOBEC3 (apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3; A3) family is an important part of the innate immune system involved in combating pathogens but often having detrimental consequences in cancers.1−4 Unlike single-nucleoside cytidine deaminases (CDA), which act on individual cytidines, A3 enzymes deaminate cytosine to uracil (C→U) (Figure 1A) only on single-stranded DNA (ssDNA), although some activity has been reported on RNA.5,6
Figure 1.

(A) Deamination of dC to dU in ssDNA by A3 enzymes. C0 represents target cytosine; nucleobases (thymine T and adenine A) at 5′ relative to C0 are represented as T–1, T–2, and so forth and those at 3′ relative to C0 as A+1, T+2, and so forth. (B) Inhibitors of cytidine deamination used in this work, 2′-deoxyzebularine (dZ) and 5-fluoro-2′-deoxyzebularine (FdZ), incorporated into ssDNA (left) and their proposed hydrates in the active site of A3 (right), which mimic an intermediate of C-to-U conversion. (C) Crystal structure of an engineered C-terminal domain of A3B, A3BCTD (light brown ribbon), bound to ssDNA, showing C0 flipped out (yellow) to project into the Zn2+-containing active site (gray ball), +1 2′-deoxyadenosine (red), and −2 thymidine (blue) are close together in the U-shaped conformation in ssDNA (PDB: 5TD5).7
There are seven human A3 enzymes (A3A-A3H, excluding A3E). Although A3 enzymes are a part of the human immune system, they can also mutate human genomic DNA and thus contribute to cancer genesis, cancer mutagenesis, and drug resistance.1−4,8−12 Single-domain A3A and double-domain A3B have been identified as the major contributors to cancer mutagenesis; their mutational signatures have been found in different cancers including bladder, breast, cervix, head/neck, and lung cancers.13−15 A3B is also associated with poor survival prognosis in some cancers.4,8,16 Since A3A and A3B are not essential to primary metabolism and A3B deletion is prevalent in some populations,17 their inhibition (especially of nucleus-targeted A3B) offers a potent strategy to suppress cancer evolution and thereby make existing anticancer therapies more efficient.11,18 As A3 enzymes deaminate predominantly ssDNA, we have been exploring the possibilities of ssDNA-based inhibitors that can be used as conjuvants to existing cancer therapies, i.e. be used in combination with existing therapies to extend their efficacy.19−21
The enzymes A3 and CDA share a structurally homologous Zn2+-containing active site; consequently, they are proposed to have a similar mechanism of cytosine deamination. We have recently developed the first rationally designed competitive inhibitor of A3 by incorporating a cytidine analogue 2′-deoxyzebularine, dZ, a known inhibitor of CDA (Figure 1B), into ssDNA fragments.19 We have also demonstrated that dZ as a single nucleoside does not inhibit A3, but once embedded into ssDNA, it becomes a micromolar competitive inhibitor of A3 enzymes.19 By analogy to the binding of dZ to CDA, this key observation supports a mechanism in which ssDNA delivers dZ to the active site of A3 for inhibition. In addition, we and others7,22,23 have shown that ssDNA bound to both A3A and to an engineered mutant of the catalytically active C-terminal domain of A3B, A3BCTD (PDB: 5TD5 and 5SWW), is not linear but adopts a distinctive U-shape, projecting the target dC (or dZ) into the active site (Figure 1C). We hypothesized that locking DNA sequences into the observed U-shape may provide not only better substrates but also, appropriately modified, better inhibitors of A3 enzymes.
Methodology
To test our hypothesis, we evaluated two different approaches for pre-shaping DNA using covalent cross-links between distant nucleotides in the sequence, that is, terminal and internal cross-linking strategies (Figure 2A,B). For pre-shaping ssDNA (both strategies), we used copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC), which is compatible with DNA synthesis and does not change the chemical structure of native nucleotides.24−27 For terminal cross-linking strategy, terminal alkyne and azide moieties were incorporated at 5′ and 3′-ends of ssDNA, respectively. Commercially available alkyne Y(28) (Figure 2A) was used and in-house synthesized azido-sugar dRN3 was attached to the solid support (controlled-pore glass, CPG). ssDNAs of different lengths were synthesized by standard DNA phosphoramidite chemistry. The second strategy used detailed structural information from the A3–ssDNA complexes. Relative to the central 5′-TCA motif of the ssDNA, the positions of the alkyne and azide for the internal cross-linking strategy were selected based on the observation that 2′-deoxyadenosine at the +1 position immediately next to the target dC (position 0) and thymidine at the −2 position are close together and form hydrogen bond interactions with each other through a water molecule for the bound ssDNA in A3BCTD and A3A crystal structures. This makes dA in the +1 position and dT in the −2 position suitable for cross-linking (Figure 1C).7,29−31 We employed an in-house synthesized azide-containing 2′-deoxyadenosine (dAN3) phosphoramidite to place it in position +1 and commercially available 5-ethynyl-2′-deoxyuridine (dUE, Figure 2D) phosphoramidite32 to mimic dT in position −2 for the internal cross-linking strategy.
Figure 2.
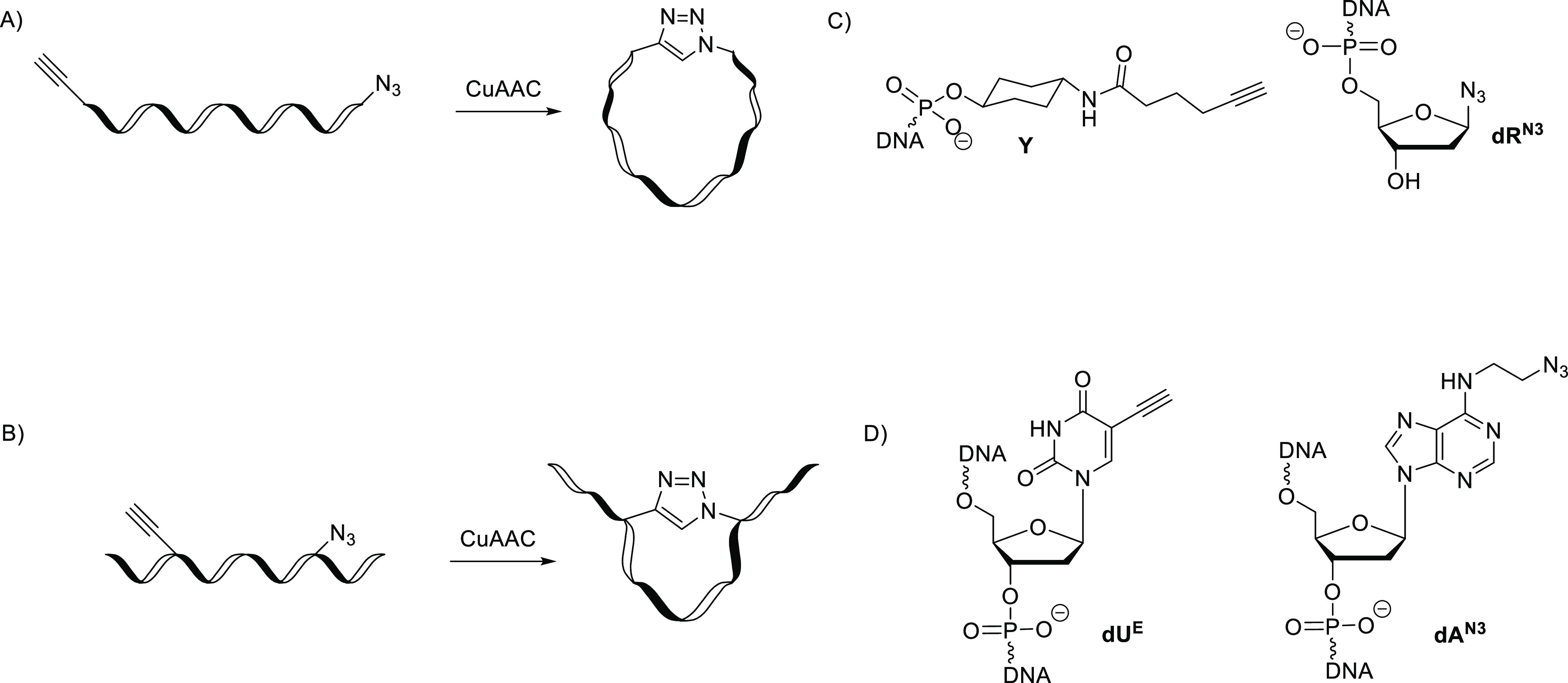
Terminal (A) and internal (B) cross-linking strategies by CuAAC to pre-shape a linear ssDNA as a substrate (dC-containing oligo) and as an inhibitor of A3BCTD (dZ-containing oligo). (C) Alkyne (Y) and azide (dRN3) modifications used in the sequence for terminal cross-linking strategy. (D) Alkyne (dUE) and azide (dAN3) modifications used for internal cross-linking strategy.
After the oligonucleotide synthesis, for terminal and internal cross-links, we employed CuAAC as a post-synthetic step, monitored by analytical reverse-phase high-performance liquid chromatography (HPLC) and mass spectrometry. The cross-linked oligonucleotides (oligos) were then evaluated as A3 substrates and compared to a standard linear substrate by a nuclear magnetic resonance (NMR)-based assay. Real-time NMR assays are advantageous because they are direct, utilizing only A3 enzymes and oligos in a suitable buffer system, unlike many fluorescence-based assays where a secondary enzyme and a fluorescently modified oligo are used.33 The NMR-based assay yields directly the initial velocity of deamination of various ssDNA substrates, including the modified ones, in the presence of A3 enzymes. Consequently, the Michaelis–Menten kinetic model is used to characterize substrates and inhibitors of A3. Therefore, currently, the NMR-based direct assay is best suited to study modified ssDNA-based substrates and inhibitors of A3.
The engineered A3 enzyme used in our experiments is the well-characterized and very active A3BCTD-QM-ΔL3-AL1swap,19 where loop 3 is deleted and loop 1 is replaced with the corresponding loop 1 from A3A. This is the same enzyme used in the crystal structure of the A3–ssDNA complex (except that for the X-ray structure, the active-site Glu255 was mutated to Ala to prevent deamination of the substrate, dC-containing ssDNA).7 Our recent work on A3G has shown that inhibition of the C-terminal catalytic domain is mirrored by the inhibition of full-length enzyme, validating our approach to use catalytic C-terminal domains for inhibitor development. ssDNA substrates were converted into A3 inhibitors by incorporating dZ in place of the target dC. The evaluation and optimization strategy is summarized in Figure 3. The inhibitory potential of dZ-containing oligonucleotides was evaluated by the NMR-based assay in the presence of a linear substrate. We also characterized by isothermal titration calorimetry the thermodynamics of binding of our cross-linked oligos with A3 enzymes in comparison with linear ssDNA. The inactive E72A mutant of A3A (the same as in A3A–ssDNA crystal structure, PDB: 5SWW) was chosen to evaluate substrate binding, and the active A3BCTD-QM-ΔL3-AL1swap was chosen for inhibitor binding. Finally, we demonstrated that the cross-linked DNA inhibitor suppressed the activity of wild-type A3A.
Figure 3.

Methodology for the design and evaluation of cross-linked substrates and inhibitors.
Our strategy has led to the design, synthesis, and evaluation of the first nanomolar inhibitor of A3 enzymes.
Results
In the past 15 years, alkyne- and azide-modified oligos have been widely used for the synthesis of a vast variety of bioconjugates via azide–alkyne “click chemistry”. Usually, terminal alkynes can be introduced into oligos using phosphoramidite chemistry, and several reagents are commercially available, such as the Y and dUE modifications used in the current work. However, there are limited options for the introduction of the azide functionality into oligos, especially in the middle of the DNA sequence. Usually, isolation of azido-containing phosphoramidite and its use in the DNA synthesis is problematic34,35 due to plausible Staudinger reaction between organic azide and a trivalent phosphorus in the phosphoramidite function. Indeed, the Staudinger reaction has been used recently for modification of a phosphate backbone.36−41 Interestingly, a Staudinger reaction does not occur on the azide group in the support-bound oligonucleotide chain during subsequent elongation using nucleoside phosphoramidites.42 This led to the development of azide-containing solid supports to produce oligos bearing organic azides at the 3′-end.43−46 In 2014, Fomich et al.47 demonstrated that azide-carrying phosphoramidite can be obtained and used in DNA synthesis to produce oligos having up to three organic azides in the middle of the sequence. This is achievable if the azide-carrying phosphoramidite is not concentrated and is used directly in automated DNA synthesis after aqueous work-up and drying of the solution over sodium sulfate. These advancements in the field were used in the current work to produce oligos containing an organic azide at the 3′-end as well as in the middle of the sequence using azide-modified 2-deoxyribose and dA described below, respectively.
Synthesis of DNA Precursors Containing Organic Azides
A method reported recently48 described the synthesis of compound 2 from Hoffer’s chlorosugar 1 (Scheme 1) using a Lewis acid and trimethylsilyl azide, but low β/α selectivity and moderate yields were reported. In contrast, we employed a phase-transfer protocol49 with minor changes using NaN3 and Bu4NHSO4 under vigorous stirring in saturated aqueous NaHCO3 and chloroform followed by addition of Hoffer’s chlorosugar 1. The reaction was finished in 20 min, and after work-up resulted in almost pure azide 2 with high yield (88%) and β/α ratio of 16:1 (6% of α-anomer). Subsequent recrystallization from EtOH slightly increased the β/α ratio to 19:1 (5% of α-anomer). This protocol was optimized to a multi-gram scale synthesis of azide 2 in our laboratory. The CPG conjugated to azido-sugar 4 was prepared from a 5′-dimethoxytrityl (DMT) protected azido-sugar 3 by a slight modification of reported methods44,50 (Scheme 1). The load on CPG was found to be 39 μmol/g by UV absorption of the DMT cation released upon treatment of a sample of the modified support 4 with a 3% solution of dichloroacetic acid in dichloromethane.
Scheme 1. Synthesis of Azido Sugar dRN3 Modified CPG-Support.

Reagents and conditions: (i) NaN3, Bu4NHSO4, satd. NaHCO3/CHCl3 (1:1), rt, 20 min; (ii) 30% aq. NH3, MeOH, rt, 3 days; (iii) 4,4′-dimethoxytrityl chloride (DMTCl), pyridine, 0°C to rt, overnight; (iv) long-chain aminoalkyl CPG (LCAA-CPG-CO2H), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC), Et3N, 4-(dimethylamino)pyridine (DMAP), pyridine/DMF (1:1), rt, 3 days.
The second strategy relied on an internal cross-link created between azide-containing dAN3 and commercially available alkyne-modified dUE (Figure 2B,D).
Here, we decided to introduce an azidoalkane linker into the N6 position of dA, due to close proximity of an exocyclic amine of dA+1 and 5-methyl group of thymidine–2 in the crystal structure (Figure 1C). In the past, various methods have been employed for N6 alkylation of dA, which usually relied upon alkylation in the N1 position followed by subsequent Dimroth rearrangement to the N6-alkylated product.51,52 In contrast to previous reports, we observed a direct alkylation of the N6 position in triacetylated dA (7) using 2-azidoethyl-4-tosylate (5)53 and Cs2CO3 as the base (Scheme 2). The alkylation at the N6 position of dA providing compound 8 was confirmed by 1H and 13C heteronuclear multiple-bond correlation (HMBC) NMR experiments, in which three-bond correlations were detected. We observed that proton signals of the CH2 group of the azidoalkyl linker at 4.31 ppm had a cross-peak not only with C6 of adenine at 152.58 ppm but also with the carbonyl carbon of the acetyl group at 170.06 ppm (Figure S49, see Supporting Information). This is only possible to observe for N6 and not for N1 alkylated dA (three versus five bonds between hydrogen of CH2 and carbon of CO, respectively). After the selective deprotection of 3′- and 5′-acetyl groups, the 5′-hydroxy group was converted into the 4,4′-dimethoxytrityl derivative 10 under standard conditions. Next, a pilot phosphitylation reaction was performed on compound 10 using previously described conditions47 in dry CDCl3, producing phosphoramidite 11 with a purity of more than 70% as shown by 1H, 13C, and 31P NMR spectra (Figures S59–S61, Supporting Information). This suggests that compound 11 can be used for automated DNA synthesis, with a limitation that the freshly prepared phosphoramidite should be used immediately after synthesis because the storage of this solution at room temperature for 24 h results in significant degradation of compound 11 as evidenced by NMR spectroscopy (Figure S62, Supporting Information).
Scheme 2. Synthesis of Modified 2′-Deoxyadenosine Phosphoramidite Containing a Two-Carbon Linker with Azide.

Reagents and conditions: (i) acetic anhydride, pyridine, rt overnight then 60°C for 6 h; (ii) compound 5, Cs2CO3, CH3CN, 60°C, overnight; (iii) Et3N in MeOH/H2O, rt, 15 min; (iv) 4,4′-dimethoxytrityl chloride, pyridine, 0°C to rt, overnight; (v) N,N-diisopropylamino-2-cyanoethoxychlorophosphine (CEP-Cl), Et3N, dry DCM, 0°C.
For automated DNA synthesis, the reaction was performed under the same conditions using dry dichloromethane as the solvent. To prevent a Staudinger reaction between the azide and phosphoramidite present in one molecule, we did not purify the product or concentrate it. Instead, it was used directly for the oligonucleotide synthesis. The concentration of reagent 11 in DCM was found to be 0.2 M, determined by UV absorption of the 4,4′-dimethoxytrityl cation released upon treatment of an aliquot of this sample with a 3% solution of dichloroacetic acid in dichloromethane.
Synthesis of Cross-Linked Oligonucleotides by CuAAC
Oligodeoxynucleotides (oligos) were synthesized by the phosphoramidite method on a MerMade-4 DNA/RNA synthesizer (BioAutomation) on a 5 μmol scale using the standard manufacturer’s protocol for native nucleotides, whereas increased coupling time from 2 to 10 min was used for modified alkyne and azide phosphoramidites. Detailed procedures are given in Supporting Information.
The purified linear oligos were cross-linked by CuAAC following the protocol implemented in our laboratory (see Supporting Information). Progress of the reaction was monitored by reverse-phase HPLC. Gratifyingly, a new peak appeared in the chromatogram for all cross-linking experiments over a period of 16 h, characterized by shorter retention times than those of the starting materials (Table 1). These results agree with previously published properties of circularized DNA, which were also prepared using CuAAC.44 After separation by reverse-phase HPLC, the products were desalted and analyzed by mass-spectrometry, showing, as expected, the same mass as for the starting materials, confirming the composition of monomeric cross-linked oligos (Table 1). Abbreviation of individual oligos is explained in the caption of Table 1.
Table 1. List of Linear Oligos with Azide/Alkyne Modifications as well as Cross-Linked Oligos Having 1,4-Disubstituted 1,2,3-Triazole Modification.
| DNA sequence, 5′→3′ | namea | retention time (min) | ESI–MS [Da] found/calculated | |
|---|---|---|---|---|
| YTTTCATTdRN3 | linear | dC-9-mer | 17.5 | 2552.47/2552.48 |
| cross-linked | dC-9-mer-X | 14.1 | 2552.47/2552.48 | |
| YTTCATdRN3 | linear | dC-7-mer | 18.0 | 1944.37/1944.37 |
| cross-linked | dC-7-mer-X | 13.6 | 1944.37/1944.37 | |
| YTCAdRN3 | linear | dC-5-mer | 17.8 | 1336.29/1336.30 |
| cross-linked | dC-5-mer-X | 13.5 | 1336.29/1336.30 | |
| AdUETTCdAN3TTT | linear | dC[UE(−3), AN3(+1)] | 16.7 | 2757.46/2757.49 |
| cross-linked | dC[UE(−3), AN3(+1)]X | 15.6 | 2757.47/2757.49 | |
| ATdUETCdAN3TTT | linear | dC[UE(−2), AN3(+1)] | 16.3 | 2757.46/2757.49 |
| cross-linked | dC[UE(−2), AN3(+1)]X | 15.3 | 2757.46/2757.49 | |
| AdUETTdZdAN3TTT | linear | dZ[UE(−3), AN3(+1)] | 16.9 | 2742.45/2742.48 |
| cross-linked | dZ[UE(−3), AN3(+1)]X | 15.7 | 2742.46/2742.48 | |
| ATdUETdZdAN3TTT | linear | dZ[UE(−2), AN3(+1)] | 16.9 | 2742.45/2742.48 |
| cross-linked | dZ[UE(−2), AN3(+1)]X | 15.7 | 2742.45/2742.48 |
d stands for DNA backbone, C or Z denotes substrate (dC) or inhibitor (dZ) of A3 present in position 0 in the DNA sequence. The length of terminally cyclized oligos is described as being 9-, 7-, or 5-mer in relation to the length of the native DNA sequence having the same number of phosphate groups. In other words, modifications Y and dRN3 are counted as modified nucleotides. X stands for a cross-linked oligo containing 1,4-disubstituted 1,2,3-triazole, that is, dC-9-mer-X refers to a cross-linked oligo. For internally cross-linked oligos, the abbreviation of alkyne- and azido-containing nucleotides is presented in square brackets, and the position of each modification relative to the position 0 being dC or dZ is shown in brackets.
Additionally, to prove that our cross-linking strategy works for internal modifications, we performed NMR experiments on linear and cross-linked oligos (see Supporting Information, Section S5.1). Differences in chemical shifts between the starting material and the product in 1H NMR were observed for all methyl peaks of thymidines located between 1.6 and 2.0 ppm, suggesting the formation of a new product (cross-linked oligo) as the result of CuAAC reaction (Figure S13 in the Supporting Information). In addition, acetylenic proton at 2.44 ppm in the linear oligo that was assigned by HMBC NMR having a cross peak with the CH2 (18.30 ppm in 13C NMR) immediately next to it (Figures S11 and S12 in the Supporting Information) was absent in the cross-linked oligo, which provided additional evidence of the successful cross-linking by CuAAC.
Evaluation of DNA Substrates by NMR-Based Activity Assay
Using the 9-mer oligonucleotide 5′-AT3CAT3 as the standard substrate, we compared the deamination of both terminally and internally cross-linked oligos using our previously described real-time NMR assay.19,20 For the terminally cross-linked oligos, we observed decreased rates of deamination from 9-mer to 5-mer. One should note that the number of phosphates in the modified 9-mer sequence is the same as for the 9-mer DNA control (Figure 4A). This means that terminally cross-linked oligos are poor mimics of the DNA shape seen in the complex, although the flexibility associated with the longer oligos allows some deamination to occur. This agrees with earlier observations that longer linear oligos bind A3A better than shorter oligos.30 In marked contrast, a faster deamination rate was observed for the internally cross-linked oligo dC[UE(−2), AN3(+1)]X compared to the standard, linear substrate (Figure 4B). The linked −2 and +1 positions were spatially close in the X-ray structure of the inactive A3BCTD-QM-ΔL3-AL1swap(E255A)/DNA complex.7 Moreover, an extremely slow deamination rate was detected for dC[UE(−3), AN3(+1)]X oligo, which we attribute to a shape and conformation that together hinder proper binding with the enzyme compared to the dC[UE(−2), AN3(+1)]X oligo. We did not create a cross-link between dA at position +1 and T at position −1 as the X-ray structure (pdb: 5TD5) shows a clear binding pocket for T in the −1 position, from which we infer that cross-linking in this position will disrupt the DNA–protein interaction.
Figure 4.

Initial speed of A3BCTD-QM-ΔL3-AL1swap catalyzed deamination of terminally (A) and internally (B) cross-linked oligos in comparison to the linear substrate. Substrate concentration: 100 μM in (a) and 800 μM in (b). Enzyme concentration: 50 nM in (A) and 300 nM in (B). Buffer: 50 mM citrate–phosphate (pH 5.5), 200 mM NaCl, 2 mM β-mercaptoethanol, 200 μM 4,4-dimethyl-4-silapentane-1-sulfonic acid, and 10% D2O at 25°C. Error bars are estimated standard deviations from triplicate measurements.
Evaluation of Inhibitors by NMR Assay
The increased speed of deamination of the dC[UE(−2), AN3(+1)]X oligo prompted us to investigate the inhibition potential of this oligo by changing dC to dZ. We used the NMR assay to compare the internally cross-linked dZ-containing oligos with a linear DNA inhibitor containing dZ (dZ-linear, 5′-AT3dZAT3) that was characterized earlier.19 The NMR assay was performed using unmodified oligo (5′-T4CAT) as a substrate in the presence of A3BCTD-QM-ΔL3-AL1swap and a known concentration of dZ-containing inhibitors (linear and cross-linked).
The results revealed that the internally cross-linked dZ-containing oligo derived from the faster deaminated substrate, dZ[UE(−2), AN3(+1)]X, inhibited the A3-catalyzed deamination by greater than an order of magnitude more potently than our best linear dZ oligo (Figure 5). On the other hand, dZ[UE(−3), AN3(+1)]X was found to be a much weaker inhibitor, in line with the earlier observation that dC[UE(−3), AN3(+1)]X was a very poor substrate. Interestingly, its inhibitory potential is comparable with that of the linear oligo, probably because of dZ interacting strongly with the active site of the enzyme.
Figure 5.

Inhibition of A3BCTD-QM-ΔL3-AL1swap-catalyzed deamination of 5′-T4CAT by dZ-containing linear and cross-linked oligos. Experimental conditions are 400 μM of 5′-T4CAT, 8 μM of dZ-containing oligos, 300 nM of A3BCTD-QM-ΔL3-AL1swap in a 50 mM sodium phosphate buffer (pH 6.0) containing 100 mM NaCl, 2.5 mM β-mercaptoethanol, 0.1 M EDTA, and 50 μM 3-(trimethylsilyl)-2,2,3,3-tetradeuteropropionic acid at 25°C. Ki value of linear dZ-oligo on A3BCTD-QM-ΔL3-AL1swap has been established previously by our group.19
For determination of inhibition constants (Ki) of inhibitors, we first evaluated kinetic parameters of A3BCTD-QM-ΔL3-AL1swap on two DNA substrates (Table 2), which were used to detect the residual deamination rate in the presence of inhibitors (Table 3). Fitting the experimental data by non-linear least-squares using a global fit provided values of kinetic parameters similar to those obtained by the linearized Lineweaver–Burk plot (Table 2). Then, the inhibition constant (Ki) for our cross-linked inhibitor was calculated by several methods assuming a competitive mode of inhibition19 (Table 3), based on the obtained apparent Km and experiments in which the concentration of the individual inhibitor was varied, and the residual deamination initial rate was measured.
Table 2. Kinetic Parameters of A3BCTD-QM-ΔL3-AL1swap on Two DNA Substrates Used in This Work.
| oligo | parameters | linear analysis (Lineweaver–Burk plot) | non-linear regression analysis |
|---|---|---|---|
| 5′-AT3CAT3 | Km, μM | 200 ± 30 | 150 ± 30 |
| Vmax, μM/s | 0.014 ± 0.002 | 0.0126 ± 0.0008 | |
| kcat, s–1 | 0.29 ± 0.04 | 0.26 ± 0.02 | |
| kcat/Km, s–1 μM–1 | 0.0014 | 0.0018 | |
| 5′-T4CAT | Km, μM | 350 ± 70 | 290 ± 70 |
| Vmax, μM/s | 0.056 ± 0.008 | 0.051 ± 0.005 | |
| kcat, s–1 | 0.19 ± 0.03 | 0.171 ± 0.015 | |
| kcat/Km, s–1 μM–1 | 0.00054 | 0.00059 |
Table 3. Inhibition Constants (Ki) for dZ-Containing Oligo Inhibitors of A3BCTD-QM-ΔL3-AL1swap Obtained by Various Calculation Methodsa.
| oligo name | parameters | Lineweaver–Burk plot for substrate/Dixon plot for inhibitor | non-linear regression analysis for both substrate and inhibitor |
|---|---|---|---|
| dZ-linear | Ki, μM | 7.5 ± 1.7 | 7.0 ± 1.3 |
| Vmax, μM/s | 0.0126 ± 0.0008 | ||
| dZ[UE(−2), AN3(+1)]X | Ki, μM | 0.66 ± 0.14 | 0.69 ± 0.14 |
| Vmax, μM/s | 0.052 ± 0.006 |
dZ-linear inhibitor was evaluated against 5′-AT3CAT3 substrate using 50 nM of A3BCTD-QM-ΔL3-AL1swap; dZ[UE(−2), AN3(+1)]X was evaluated against 5′-T4CAT substrate using 200 nM of A3BCTD-QM-ΔL3-AL1swap.
The Ki for dZ[UE(−2), AN3(+1)]X is 0.69 ± 0.14 μM, a value around 10 times lower than that for the linear dZ-oligo (7.0 ± 1.3 μM).19 The nanomolar inhibitory potential exhibited by our internally cross-linked oligo shows that properly pre-shaped, cross-linked DNA can become a better inhibitor than the linear one and that the better substrate becomes the better inhibitor, which supports our original hypothesis.
Binding of Cross-Linked Oligos to A3 Enzymes
To understand if the improved inhibition and quality of substrate is due to enthalpic or entropic contributions, we used ITC to determine the thermodynamics of binding. We tested how the cross-linked oligos bind to A3 enzymes and compared this binding to that of linear oligos. For oligos containing dZ, catalytically active A3BCTD-QM-ΔL3-AL1swap was used, as it has the active-site glutamic acid required for protonation of N3 in dZ. This protonation activates C4 in dZ to accept nucleophilic –OH/H2O coordinated to the Zn2+, which converts dZ into a transition-state analogue of cytidine deamination (Figure 1B).54 In contrast, A3A-E72A, in which the active-site glutamic acid is replaced with alanine, was used particularly for oligos containing dC so that the complications of deamination can be avoided. This mutation has no change to the active-site structure but does cause a small but significant diminution in ligand binding19 through loss of hydrogen bonding to the ligand. We expect that the binding of inactivated A3BCTD-QM-ΔL3-AL1swap (as the E255A mutant) to the substrate will be close to or even worse than Km of 200 μM, which places substrate binding outside ITC applicability. Results of ITC experiments are presented in Table 4. The raw titration data and binding curves are available in Supporting Information (Figures S23–S32). One should emphasize that the compositions of buffers used for NMR kinetic and ITC binding experiments are different, mainly because proteins in ITC experiments need to be at much higher concentration than for kinetic assays (10–100 μM for ITC versus 50–300 nM in kinetic experiments). This required a search for buffer components that prevent protein precipitation during ITC titrations. Our optimized ITC buffers were also different for A3A(E72A) and A3BCTD-QM-ΔL3-AL1swap, which means that Kd and Ki values cannot be directly compared. However, our aim was to compare the binding of different oligos to A3 to ascertain if general trends can be established.
Table 4. Thermodynamic Parameters of Oligos with A3 Enzymes Obtained by ITCa.
| oligo | protein | ITC bufferb | Kd (μM) | ΔH(kcal/mol) | TΔS(kcal/mol) |
|---|---|---|---|---|---|
| 5′-AT3CAT3 | A3A-E72A | 1 | 0.20 ± 0.04 | –22.9 ± 1.2 | –13.4 ± 1.2 |
| 5′-T4CAT | A3A-E72A | 1 | 0.27 ± 0.04 | –19.8 ± 0.9 | –13.1 ± 0.9 |
| dC-9-mer-X | A3A-E72A | 1 | 0.63 ± 0.13 | –18.1 ± 0.9 | –9.7 ± 0.9 |
| dC-7-mer-X | A3A-E72A | 1 | 0.48 ± 0.10 | –14.1 ± 1.4 | –5.4 ± 1.4 |
| dC-5-mer-X | A3A-E72A | 1 | 4.5 ± 0.9 | –10.9 ± 1.2 | –3.8 ± 1.1 |
| 5′-AT3CAT3 | A3A-E72A | 2 | 0.15 ± 0.03 | –53 ± 3 | –44 ± 3 |
| 5′-T4CAT | A3A-E72A | 2 | 0.75 ± 0.15 | –45 ± 2 | –36 ± 2 |
| dC[UE(−2), AN3(+1)]X | A3A-E72A | 2 | 0.030 ± 0.006 | –67 ± 3 | –57 ± 4 |
| dZ-linear | A3BCTD-QM-ΔL3-AL1swap | 3 | 11.4 ± 2.3 | –13.0 ± 0.6 | –6.2 ± 0.6 |
| dZ[UE(−2), AN3(+1)]X | A3BCTD-QM-ΔL3-AL1swap | 3 | 2.5 ± 0.5 | –24.6 ± 1.2 | –16.7 ± 1.7 |
Kd is the dissociation constant for the ligand from A3; ΔH and TΔS are for association of the ligand to A3; the temperature is 25°C.
ITC buffer 1: 50 mM MES, pH 6.0, 100 mM NaCl, 200 μM EDTA, and 1 mM β-mercaptoethanol; ITC buffer 2: 50 mM Na+/K+ phosphate, pH 6.0, 50 mM NaCl, 50 mM choline acetate, 2.5 mM TCEP, and 200 μM EDTA with 30 mg/mL bovine serum albumin; ITC buffer 3: 50 mM Na+/K+ phosphate, pH 6.0, 200 mM trimethylamine N-oxide dihydrate, and 2.5 mM TCEP, 200 μM EDTA.
The dissociation constants (Kd) obtained by ITC showed no gain in affinity of A3A-E72A for terminally cross-linked oligos in comparison with the standard linear oligos (5′-AT3CAT3 and 5′-T4CAT). The dC-5-mer-X had noticeably weaker binding to A3A-E72A and lower deamination rate as a substrate to A3BCTD-QM-ΔL3-AL1swap compared to its 7-mer and 9-mer analogues; this is partly consistent with previous observations on linear substrates of varying lengths19,30 but also with not enough flexibility to achieve the right conformation, when a short oligo is cyclized. A reduced entropic component to the free energy of binding is not sufficient to overcome a markedly depressed enthalpic component resulting in relatively weak interactions of ligand with protein.
The internally cross-linked oligo dC[UE(−2), AN3(+1)]X, which gave higher substrate activity for A3BCTD-QM-ΔL3-AL1swap, has bound five times more strongly (Kd = 0.030 ± 0.006 μM) in comparison to the linear substrate oligo (5′-AT3CAT3, Kd = 0.15 ± 0.03 μM) in the same buffer for A3A-E72A. Both unfavorable entropic and favorable enthalpic contributions increased in magnitude, indicating that affinity was improved by the better fitting of active site (enthalpic contribution). The entropic contribution to the Gibbs free energy of binding became more negative (more unfavorable), despite the pre-shaped DNA. This somewhat unexpected observation is attributed to the decrease in protein conformational flexibility upon protein–oligo complex formation as the main source of the entropic contribution, with this decrease being greater for the cross-linked oligo. In contrast, the terminally linked oligos had less favorable enthalpic and less unfavorable entropic contributions upon binding to A3A-E72A.
Next, we evaluated the binding data of dZ-containing oligos toward catalytically competent A3BCTD-QM-ΔL3-AL1swap. The dissociation constant of dZ[UE(−2), AN3(+1)]X (Kd = 2.5 ± 0.5 μM) was around 4.5 times smaller (i.e., stronger binding) than that of dZ-containing linear oligo (Kd = 11.4 ± 2.3 μM), similar to the difference seen in substrates. This trend is also consistent with the trend observed in inhibition constants (Ki) determined by our real-time NMR assay. This effect is smaller in ITC measurements, most likely due to the differences in experimental conditions, as the ITC experiments were conducted in a different buffer and in the presence of trimethylamine N-oxide (TMAO) to stabilize the enzyme.
Inhibition of Wild-Type A3A
To evaluate inhibitory potential of cross-linked hairpin oligo against wild-type A3, we expressed in E. coli and purified wild-type A3A possessing a (his)6 tag at the C-terminal end. Recently, we have shown that linear DNA containing 5-fluoro-2′-deoxyzebularine (FdZ-linear)21 is superior to the corresponding DNA containing dZ in the inhibition of A3BCTD and A3A.21 The best FdZ-linear and cross-linked oligo dZ[UE(−2), AN3(+1)]X were tested in the NMR assay of A3A activity, where the hairpin DNA 5′-T(GC)2TTC(GC)2T (dC-hairpin, deaminated C is bold) was used as a substrate. (Table 5).55 The kinetic data were measured over time until exhaustion of the substrate and analyzed by global regression using Lambert’s W function, which provides superior estimates for Km and Vmax than non-linear regression analysis of initial rate data or any of the known linearized transformations of the Michaelis–Menten equation, such as Lineweaver–Burk, Hanes–Woolf, and the Eadie–Hofstee transformations.56
Table 5. Ki Values of Inhibitors of Wild-Type A3Aa.
| inhibitor | Ki (nM) |
|---|---|
| FdZ-linear,215′-AT3FdZAT3 | 2400 ± 940 |
| cross-link, dZ[UE(−2), AN3(+1)]X | 360 ± 120 |
Conditions: 50 mM Na+/K+ phosphate buffer, pH 7.4 supplemented with 100 mM NaCl, 1 mM TCEP, 100 μM DSS, and 10% D2O; enzyme concentration: 140 nM; substrate concentration (dC-hairpin): 500 μM. Km for dC-hairpin against A3A is 21 μM, kcat is 0.13 s–1, and kcat/Km is 6.2 s–1 mM–1.
Obtained Ki values show that dZ-containing cross-linked oligo is a superior inhibitor of the wild-type A3A in comparison with the best FdZ-linear oligo. This is consistent with the results obtained for A3BCTD-QM-ΔL3-AL1swap.
Discussion
Recently, we have developed the first selective A3 inhibitors19−21 based on incorporation of cytidine-like 2′-deoxyzebularine57,58 and 5-fluoro-2′-deoxyzebularine (dZ and FdZ on Figure 1B) into short, linear ssDNA, achieving low micromolar inhibition constants (Ki). Recent structural studies7,22,23,59 have revealed that flexible ssDNA adopts a more rigid U-shaped loop upon binding to A3, projecting cytosine into the active-site pocket (Figure 1C). One should note, however, that these studies were conducted on A3A, chimeric A3BCTD (as A3BCTD-QM-ΔL3-AL1swap), and modified A3GCTD. Chimeric A3BCTD had four mutations distant from the active site, loop 3 was truncated, and loop 1 of A3BCTD was swapped with loop 1 of A3A; this loop 1, constitutes the main differences between the amino acid sequences of A3A and A3BCTD. This A3B construct has the substrate specificity of A3A, at least in the context of the CCC motif,60 which has been used by us recently to develop A3B-specific ssDNA-based inhibitors.20 Strictly speaking, there is no A3BCTD crystal or NMR structure with ssDNA bound that would support a notion that DNA adopts a U-shape in complex with wild-type A3BCTD as well. However, A3A and, to a lesser extent, A3BCTD have been reported recently to deaminate cytosine in short 3–4 nucleotide loops of DNA hairpins.55 This reassures that both A3A and A3BCTD accept U-shaped loops. Interestingly, A3H haplotype I (A3H-I) does not deaminate cytosine in DNA hairpins at all,55 suggesting that U-shaped DNA may have some specificity toward A3 Z1 sequence signatures present in A3A, A3B, and A3G.61 Here, we demonstrate that by cross-linking dA+1 and T–2, mimicking the close proximity observed in the crystal structure,7 a faster deaminated substrate and a better inhibitor can be obtained in comparison with linear ssDNA of the same length and nucleotide content. These results obtained in our NMR-based activity assay have been corroborated in ITC binding experiments. It is interesting that creating a covalent cross-link between the same dA+1 but with the T in position −3 completely abolished substrate activity. The cytosine-containing circularized (terminally cyclized) oligos showed lower deamination rates compared to the linear DNA substrate and for that reason were not converted to inhibitors.
The overall inhibition effect was improved from 25-fold (dZ-linear) to nearly 200-fold (dZ[UE(−2), AN3(+1)]X) when the apparent inhibition constants (Ki) of oligos are compared with the Km of the linear ssDNA substrate 5′-ATTTCATTT (Km = 150 μM) on A3BCTD-QM-ΔL3-AL1swap. The same trend was observed for inhibition of wild-type A3A. As Km (and Ki) depends strongly on the surrounding environment (e.g., buffer composition, pH, and ionic strength—see Table 4), inhibition with even lower Ki may be observed in the cellular environment, because Km and Kd values in the low μM30,31 and nM62 range have been reported. During our study, two articles have been published on drug discovery efforts to obtain small molecule inhibitors of A3.63,64 None approaches the potency of our inhibitors. This highlights the interest and need for powerful A3 inhibitors.
In future, we plan to test if selectivity can be achieved, analogous to that observed with linear ssDNA inhibitors,20 by varying the nucleobases adjacent to the dZ or FdZ in the context of the cross-linked oligos. The pre-shaping of oligos, using our “click coupling” of alkyne and azide, may become a useful general synthetic strategy for oligonucleotide-based inhibitors, as U-shaped nucleic acids feature in complexes with several cellular proteins. So far, the U-shape has been observed in APOBEC-related tRNA adenosine deaminase TadA (PDB 2B3J)65 as well as in functionally and structurally unrelated proteins, such as telomere protein Pot1pc (PDB 4HIO)66−68 and bacterial cold-shock protein Bs-CspB (PDB 2ES2 and 3PF4).69,70 More generally, the structural information can be harvested to design pre-shaped DNA/RNA inhibitors for any DNA/RNA-interacting protein, if a significant change in polynucleotide shape is found upon complex formation.
Conclusions
Inhibition of APOBEC enzymes, especially A3A and A3B, is clinically relevant as these enzymes are involved in cancer mutagenesis and development of drug resistance. In this study, we developed pre-shaped ssDNAs as potentially better substrates for A3 enzymes using Cu(I)-catalyzed “click chemistry” to cross-link alkyne and azide moieties embedded in a ssDNA fragment. This has led to the first nM inhibitor of A3BCTD-QM-ΔL3-AL1swap and A3A by incorporating dZ in place of the target dC in the cross-linked DNA fragment. This work provides the stepping stone for further development of modified ssDNAs as potential A3 inhibitors.71
Acknowledgments
We gratefully acknowledge NMR and mass spectrometry facilities at Massey University and the assistance of Dr Patrick J. B. Edwards and David Lun. We thank Prof. Reuben S. Harris (HHMI and University of Minnesota, USA) and members of his cancer research program for many helpful discussions. We are grateful for the financial support provided by the Worldwide Cancer Research (grant 16-1197), Palmerston North Medical Research Foundation, Massey University Research Fund (MURF 2015, 7003, and RM20734), and the School of Natural Sciences, Massey University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.2c00449.
Experimental details about the synthesis of azido-2-deoxyribose CPG 4, adenosine 11, modified oligos, cross-linking protocol, and NMR analysis of linear versus cross-linked oligo; protein expression and purification; ITC and NMR-based kinetic assays; examples of the calculation of inhibition of A3 enzyme by dZ-containing cross-linked oligos; sequence alignment of proteins used in this study; 1H, 13C, 31P NMR, IR, and HRMS (ESI) spectra of new compounds synthesized; and RP-HPLC profiles and HRMS (ESI) spectra of linear and cross-linked oligos (PDF).
The authors declare the following competing financial interest(s): The authors have filed a patent application on cross-linked ssDNA as enzyme inhibitors (WO 2022162536).
Supplementary Material
References
- Swanton C.; McGranahan N.; Starrett G. J.; Harris R. S. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov. 2015, 5, 704–712. 10.1158/2159-8290.cd-15-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi T.; Shirakawa K.; Takaori-Kondo A. Cytidine deaminases as a weapon against retroviruses and a new target for antiviral therapy. Mini-Rev. Med. Chem. 2008, 8, 231. 10.2174/138955708783744047. [DOI] [PubMed] [Google Scholar]
- Henderson S.; Fenton T. APOBEC3 genes: retroviral restriction factors to cancer drivers. Trends Mol. Med. 2015, 21, 274–284. 10.1016/j.molmed.2015.02.007. [DOI] [PubMed] [Google Scholar]
- Roberts S. A.; Lawrence M. S.; Klimczak L. J.; Grimm S. A.; Fargo D.; Stojanov P.; Kiezun A.; Kryukov G. V.; Carter S. L.; Saksena G.; Harris S.; Shah R. R.; Resnick M. A.; Getz G.; Gordenin D. A. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S.; Patnaik S. K.; Taggart R. T.; Baysal B. E. The double-domain cytidine deaminase APOBEC3G is a cellular site-specific RNA editing enzyme. Sci. Rep. 2016, 6, 39100. 10.1038/srep39100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S.; Patnaik S. K.; Taggart R. T.; Kannisto E. D.; Enriquez S. M.; Gollnick P.; Baysal B. E. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun. 2015, 6, 6881. 10.1038/ncomms7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K.; Carpenter M. A.; Banerjee S.; Shaban N. M.; Kurahashi K.; Salamango D. J.; McCann J. L.; Starrett G. J.; Duffy J. V.; Demir O.; Amaro R. E.; Harki D. A.; Harris R. S.; Aihara H. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 2017, 24, 131–139. 10.1038/nsmb.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M. B.; Lackey L.; Carpenter M. A.; Rathore A.; Land A. M.; Leonard B.; Refsland E. W.; Kotandeniya D.; Tretyakova N.; Nikas J. B.; Yee D.; Temiz N. A.; Donohue D. E.; McDougle R. M.; Brown W. L.; Law E. K.; Harris R. S. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S.; Alexandrov L. B.; Wedge D. C.; VanLoo P.; Greenman C. D.; Raine K.; Jones D.; Hinton J.; Marshall J.; Stebbings L. A.; Menzies A.; Martin S.; Leung K.; Chen L.; Leroy C.; Ramakrishna M.; Rance R.; Lau K. W.; Mudie L. J.; Varela I.; McBride D. J.; Bignell G. R.; Cooke S. L.; Shlien A.; Gamble J.; Whitmore I.; Maddison M.; Tarpey P. S.; Davies H. R.; Papaemmanuil E.; Stephens P. J.; McLaren S.; Butler A. P.; Teague J. W.; Jönsson G.; Garber J. E.; Silver D.; Miron P.; Fatima A.; Boyault S.; Langerød A.; Tutt A.; Martens J. W. M.; Aparicio S. A. J. R.; Borg A.; Salomon A. V.; Thomas G.; Børresen-Dale A.-L.; Richardson A. L.; Neuberger M. S.; Futreal P. A.; Campbell P. J.; Stratton M. R. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S. A.; Sterling J.; Thompson C.; Harris S.; Mav D.; Shah R.; Klimczak L. J.; Kryukov G. V.; Malc E.; Mieczkowski P. A.; Resnick M. A.; Gordenin D. A. Clustered Mutations in Yeast and in Human Cancers Can Arise from Damaged Long Single-Strand DNA Regions. Mol. Cell 2012, 46, 424–435. 10.1016/j.molcel.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law E. K.; Sieuwerts A. M.; LaPara K.; Leonard B.; Starrett G. J.; Molan A. M.; Temiz N. A.; Vogel R. I.; Gelder M. E. M.; Sweep F. C. G. J.; Span P. N.; Foekens J. A.; Martens J. W. M.; Yee D.; Harris R. S. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci. Adv. 2016, 2, e1601737 10.1126/sciadv.1601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law E. K.; Levin-Klein R.; Jarvis M. C.; Kim H.; Argyris P. P.; Carpenter M. A.; Starrett G. J.; Temiz N. A.; Larson L. K.; Durfee C.; Burns M. B.; Vogel R. I.; Stavrou S.; Aguilera A. N.; Wagner S.; Largaespada D. A.; Starr T. K.; Ross S. R.; Harris R. S. APOBEC3A catalyzes mutation and drives carcinogenesis in vivo. J. Exp. Med. 2020, 217, e20200261 10.1084/jem.20200261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M. B.; Temiz N. A.; Harris R. S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan S.; Rosenthal R.; Kanu N.; McGranahan N.; Bartek J.; Quezada S. A.; Hare J.; Harris R. S.; Swanton C. Perspective: APOBEC mutagenesis in drug resistance and immune escape in HIV and cancer evolution. Ann. Oncol. 2018, 29, 563–572. 10.1093/annonc/mdy003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J.; Wang C.; Ma X.; Wang E.; Peng G. APOBEC3B, a molecular driver of mutagenesis in human cancers. Cell Biosci. 2017, 7, 29. 10.1186/s13578-017-0156-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieuwerts A. M.; Willis S.; Burns M. B.; Look M. P.; Gelder M. E.; Schlicker A.; Heideman M. R.; Jacobs H.; Wessels L.; Leyland-Jones B.; Gray K. P.; Foekens J. A.; Harris R. S.; Martens J. W. M. Elevated APOBEC3B Correlates with Poor Outcomes for Estrogen-Receptor-Positive Breast Cancers. Horm. Cancer 2014, 5, 405–413. 10.1007/s12672-014-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd J. M.; Newman T. L.; Tuzun E.; Kaul R.; Eichler E. E. Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet. 2007, 3, 584–592. 10.1371/journal.pgen.0030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson M. E.; Harris R. S.; Harki D. A. APOBEC Enzymes as Targets for Virus and Cancer Therapy. Cell Chem. Biol. 2018, 25, 36–49. 10.1016/j.chembiol.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvach M. V.; Barzak F. M.; Harjes S.; Schares H. A. M.; Jameson G. B.; Ayoub A. M.; Moorthy R.; Aihara H.; Harris R. S.; Filichev V. V.; Harki D. A.; Harjes E. Inhibiting APOBEC3 Activity with Single-Stranded DNA Containing 2’-Deoxyzebularine Analogues. Biochemistry 2019, 58, 391–400. 10.1021/acs.biochem.8b00858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzak F. M.; Harjes S.; Kvach M. V.; Kurup H. M.; Jameson G. B.; Filichev V. V.; Harjes E. Selective inhibition of APOBEC3 enzymes by single-stranded DNAs containing 2’-deoxyzebularine. Org. Biomol. Chem. 2019, 17, 9435–9441. 10.1039/c9ob01781j. [DOI] [PubMed] [Google Scholar]
- Kvach M. V.; Barzak F. M.; Harjes S.; Schares H. A. M.; Kurup H. M.; Jones K. F.; Sutton L.; Donahue J.; D’Aquila R. T.; Jameson G. B.; Harki D. A.; Krause K. L.; Harjes E.; Filichev V. V. Differential Inhibition of APOBEC3 DNA-Mutator Isozymes by Fluoro- and Non-Fluoro-Substituted 2’-Deoxyzebularine Embedded in Single-Stranded DNA. ChemBioChem 2020, 21, 1028–1035. 10.1002/cbic.201900505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouno T.; Silvas T. V.; Hilbert B. J.; Shandilya S. M. D.; Bohn M. F.; Kelch B. A.; Royer W. E.; Somasundaran M.; Yilmaz N. K.; Matsuo H.; Schiffer C. A. Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun. 2017, 8, 15024. 10.1038/ncomms15024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harjes S.; Jameson G. B.; Filichev V. V.; Edwards P. J. B.; Harjes E. NMR-based method of small changes reveals how DNA mutator APOBEC3A interacts with its single-stranded DNA substrate. Nucleic Acids Res. 2017, 45, 5602–5613. 10.1093/nar/gkx196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sagheer A. H.; Brown T. Click chemistry with DNA. Chem. Soc. Rev. 2010, 39, 1388–1405. 10.1039/b901971p. [DOI] [PubMed] [Google Scholar]
- Gierlich J.; Burley G. A.; Gramlich P. M. E.; Hammond D. M.; Carell T. Click chemistry as a reliable method for the high-density postsynthetic functionalization of alkyne-modified DNA. Org. Lett. 2006, 8, 3639–3642. 10.1021/ol0610946. [DOI] [PubMed] [Google Scholar]
- Bouillon C.; Meyer A.; Vidal S.; Jochum A.; Chevolot Y.; Cloarec J. P.; Praly J. P.; Vasseur J. J.; Morvan F. Microwave assisted ″click″ chemistry for the synthesis of multiple labeled-carbohydrate oligonucleotides on solid support. J. Org. Chem. 2006, 71, 4700–4702. 10.1021/jo060572n. [DOI] [PubMed] [Google Scholar]
- Géci I.; Filichev V. V.; Pedersen E. B. Stabilization of parallel triplexes by twisted intercalating nucleic acids (TINAs) incorporating 1,2,3-triazole units and prepared by microwave-accelerated click chemistry. Chem.—Eur. J. 2007, 13, 6379–6386. 10.1002/chem.200700053. [DOI] [PubMed] [Google Scholar]
- Astakhova I. K.; Kumar T. S.; Campbell M. A.; Ustinov A. V.; Korshun V. A.; Wengel J. Branched DNA nanostructures efficiently stabilised and monitored by novel pyrene–perylene 2′-α-L-amino-LNA FRET pairs. Chem. Commun. 2013, 49, 511–513. 10.1039/c2cc37547h. [DOI] [PubMed] [Google Scholar]
- Roberts S. A.; Gordenin D. A. Hypermutation in human cancer genomes. Nat. Rev. Cancer 2014, 14, 786–800. 10.1038/nrc3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvas T. V.; Hou S.; Myint W.; Nalivaika E.; Somasundaran M.; Kelch B. A.; Matsuo H.; Kurt Yilmaz N.; Schiffer C. A. Substrate sequence selectivity of APOBEC3A implicates intra-DNA interactions. Sci. Rep. 2018, 8, 7511. 10.1038/s41598-018-25881-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S.; Silvas T. V.; Leidner F.; Nalivaika E. A.; Matsuo H.; Kurt Yilmaz N.; Schiffer C. A. Structural Analysis of the Active Site and DNA Binding of Human Cytidine Deaminase APOBEC3B. J. Chem. Theory Comput. 2019, 15, 637–647. 10.1021/acs.jctc.8b00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham D.; Parkinson J. A.; Brown T. DNA duplexes stabilized by modified monomer residues: synthesis and stability. J. Chem. Soc., Perkin Trans. 1 1998, 1131–1138. 10.1039/a707031d. [DOI] [Google Scholar]
- Grillo M. J.; Jones K. F. M.; Carpenter M. A.; Harris R. S.; Harki D. A. The current toolbox for APOBEC drug discovery. Trends Pharmacol. Sci. 2022, 43, 362–377. 10.1016/j.tips.2022.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T.; Mochizuki A.; Higashiya S.; Tsuruoka H.; Kawahara S.-i.; Ishikawa M.; Sekine M. Synthesis and properties of 2-azidodeoxyadenosine and its incorporation into oligodeoxynucleotides. Tetrahedron Lett. 2001, 42, 9215–9219. 10.1016/s0040-4039(01)02028-7. [DOI] [Google Scholar]
- Jawalekar A. M.; Meeuwenoord N.; Cremers J. G. O.; Overkleeft H. S.; van der Marel G. A.; Rutjes F. P. J. T.; van Delft F. L. Conjugation of Nucleosides and Oligonucleotides by [3+2] Cycloaddition. J. Org. Chem. 2008, 73, 287–290. 10.1021/jo702023s. [DOI] [PubMed] [Google Scholar]
- Heindl D.Preparation of LightCycler Red 640-labeled DNA. WO 2007059912 A1, 2007; p 27.
- Kupryushkin M. S.; Pyshnyi D. V.; Stetsenko D. A. Phosphoryl guanidines: a new type of nucleic acid analogues. Acta Naturae 2014, 6, 116–118. 10.32607/20758251-2014-6-4-116-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokhorova D. V.; Chelobanov B. P.; Burakova E. A.; Fokina A. A.; Stetsenko D. A. New oligodeoxyribonucleotide derivatives bearing internucleotide N-tosyl phosphoramidate groups: Synthesis and complementary binding to DNA and RNA. Russ. J. Bioorg. Chem. 2017, 43, 38–42. 10.1134/s1068162017010071. [DOI] [Google Scholar]
- Su Y.; Fujii H.; Burakova E. A.; Chelobanov B. P.; Fujii M.; Stetsenko D. A.; Filichev V. V. Neutral and Negatively Charged Phosphate Modifications Altering Thermal Stability, Kinetics of Formation and Monovalent Ion Dependence of DNA G-Quadruplexes. Chem.—Asian J. 2019, 14, 1212–1220. 10.1002/asia.201801757. [DOI] [PubMed] [Google Scholar]
- Miroshnichenko S. K.; Patutina O. A.; Burakova E. A.; Chelobanov B. P.; Fokina A. A.; Vlassov V. V.; Altman S.; Zenkova M. A.; Stetsenko D. A. Mesyl phosphoramidate antisense oligonucleotides as an alternative to phosphorothioates with improved biochemical and biological properties. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 1229–1234. 10.1073/pnas.1813376116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y.; Edwards P. J. B.; Stetsenko D. A.; Filichev V. V. The Importance of Phosphates for DNA G-Quadruplex Formation: Evaluation of Zwitterionic G-Rich Oligodeoxynucleotides. ChemBioChem 2020, 21, 2455–2466. 10.1002/cbic.202000110. [DOI] [PubMed] [Google Scholar]
- Kiviniemi A.; Virta P.; Lönnberg H. Utilization of intrachain 4’-C-azidomethylthymidine for preparation of oligodeoxyribonucleotide conjugates by click chemistry in solution and on a solid support. Bioconjugate Chem. 2008, 19, 1726–1734. 10.1021/bc800221p. [DOI] [PubMed] [Google Scholar]
- Brown S. D.; Graham D. Conjugation of an oligonucleotide to Tat, a cell-penetrating peptide, via click chemistry. Tetrahedron Lett. 2010, 51, 5032–5034. 10.1016/j.tetlet.2010.07.101. [DOI] [Google Scholar]
- Pourceau G.; Meyer A.; Vasseur J. J.; Morvan F. Azide Solid Support for 3’-Conjugation of Oligonucleotides and Their Circularization by Click Chemistry. J. Org. Chem. 2009, 74, 6837–6842. 10.1021/jo9014563. [DOI] [PubMed] [Google Scholar]
- Santner T.; Hartl M.; Bister K.; Micura R. Efficient Access to 3’-Terminal Azide-Modified RNA for Inverse Click-Labeling Patterns. Bioconjugate Chem. 2014, 25, 188–195. 10.1021/bc400513z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentzsch E.; Mokhir A. A Fluorogenic, Nucleic Acid Directed ″Click″ Reaction. Inorg. Chem. 2009, 48, 9593–9595. 10.1021/ic9006795. [DOI] [PubMed] [Google Scholar]
- Fomich M. A.; Kvach M. V.; Navakouski M. J.; Weise C.; Baranovsky A. V.; Korshun V. A.; Shmanai V. V. Azide Phosphoramidite in Direct Synthesis of Azide-Modified Oligonucleotides. Org. Lett. 2014, 16, 4590–4593. 10.1021/ol502155g. [DOI] [PubMed] [Google Scholar]
- Yang H.; Tang J. A.; Greenberg M. M. Synthesis of Oligonucleotides Containing the N(6)-(2-Deoxy-α,β-D-erythropentofuranosyl)-2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapydG) Oxidative Damage Product Derived from 2’-Deoxyguanosine. Chem.—Eur. J. 2020, 26, 5441–5448. 10.1002/chem.201905795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Štimac A.; Kobe J. Stereoselective synthesis of 1,2-cis- and 2-deoxyglycofuranosyl azides from glycosyl halides. Carbohydr. Res. 2000, 329, 317–324. 10.1016/S0008-6215(00)00186-5. [DOI] [PubMed] [Google Scholar]
- Damha M. J.; Giannaris P. A.; Zabarylo S. V. An improved procedure for derivatization of controlled-pore glass beads for solid-phase oligonucleotide synthesis. Nucleic Acids Res. 1990, 18, 3813–3821. 10.1093/nar/18.13.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottria R.; Casati S.; Baldoli E.; Maier J. A. M.; Ciuffreda P. N6-Alkyladenosines: Synthesis and evaluation of in vitro anticancer activity. Bioorg. Med. Chem. 2010, 18, 8396–8402. 10.1016/j.bmc.2010.09.030. [DOI] [PubMed] [Google Scholar]
- Tararov V. I.; Kolyachkina S. V.; Alexeev C. S.; Mikhailov S. N. N6-Acetyl-2′,3′,5′-tri-O-acetyladenosine; A Convenient, ‘Missed Out’ Substrate for Regioselective N6-Alkylations. Synthesis 2011, 2483–2489. 10.1055/s-0030-1260090. [DOI] [Google Scholar]
- Sugai N.; Heguri H.; Ohta K.; Meng Q.; Yamamoto T.; Tezuka Y. Effective Click Construction of Bridged- and Spiro-Multicyclic Polymer Topologies with Tailored Cyclic Prepolymers (kyklo-Telechelics). J. Am. Chem. Soc. 2010, 132, 14790–14802. 10.1021/ja103402c. [DOI] [PubMed] [Google Scholar]
- Borchers C. H.; Marquez V. E.; Schroeder G. K.; Short S. A.; Snider M. J.; Speir J. P.; Wolfenden R. Fourier transform ion cyclotron resonance MS reveals the presence of a water molecule in an enzyme transition-state analogue complex. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 15341–15345. 10.1073/pnas.0406781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R.; Langenbucher A.; Bowen D.; Kwan E. E.; Benes C. H.; Zou L.; Lawrence M. S. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science 2019, 364, eaaw2872 10.1126/science.aaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paar M.; Schrabmair W.; Mairold M.; Oettl K.; Reibnegger G. Global Regression Using the Explicit Solution of Michaelis-Menten Kinetics Employing Lambert’s W Function: High Robustness of Parameter Estimates. ChemistrySelect 2019, 4, 1903–1908. 10.1002/slct.201803610. [DOI] [Google Scholar]
- Barchi J. J.; Haces A.; Marquez V. E.; McCormack J. J. Inhibition of Cytidine Deaminase by Derivatives of 1-(β-D-Ribofuranosyl)-Dihydropyrimidin-2-One (Zebularine). Nucleosides Nucleotides 1992, 11, 1781–1793. 10.1080/07328319208017823. [DOI] [Google Scholar]
- Marquez V. E.; Barchi J. J. Jr.; Kelley J. A.; Rao K. V.; Agbaria R.; Ben-Kasus T.; Cheng J. C.; Yoo C. B.; Jones P. A. Zebularine: a unique molecule for an epigenetically based strategy in cancer chemotherapy. The magic of its chemistry and biology. Nucleosides Nucleotides Nucl. Acids 2005, 24, 305–318. 10.1081/ncn-200059765. [DOI] [PubMed] [Google Scholar]
- Maiti A.; Myint W.; Kanai T.; Delviks-Frankenberry K.; Sierra Rodriguez C.; Pathak V. K.; Schiffer C. A.; Matsuo H. Crystal structure of the catalytic domain of HIV-1 restriction factor APOBEC3G in complex with ssDNA. Nat. Commun. 2018, 9, 2460. 10.1038/s41467-018-04872-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon I.-J. L.; Byeon C.-H.; Wu T.; Mitra M.; Singer D.; Levin J. G.; Gronenborn A. M. Nuclear Magnetic Resonance Structure of the APOBEC3B Catalytic Domain: Structural Basis for Substrate Binding and DNA Deaminase Activity. Biochemistry 2016, 55, 2944–2959. 10.1021/acs.biochem.6b00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRue R. S.; Andrésdóttir V.; Blanchard Y.; Conticello S. G.; Derse D.; Emerman M.; Greene W. C.; Jónsson S. R.; Landau N. R.; Löchelt M.; Malik H. S.; Malim M. H.; Münk C.; O’Brien S. J.; Pathak V. K.; Strebel K.; Wain-Hobson S.; Yu X.-F.; Yuhki N.; Harris R. S. Guidelines for Naming Nonprimate APOBEC3 Genes and Proteins. J. Virol. 2009, 83, 494–497. 10.1128/jvi.01976-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.; Ito F.; Zhang G.; Fernandez B.; Yang H.; Chen X. S. DNA cytosine and methylcytosine deamination by APOBEC3B: enhancing methylcytosine deamination by engineering APOBEC3B. Biochem. J. 2015, 471, 25–35. 10.1042/bj20150382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King J. J.; Borzooee F.; Im J.; Asgharpour M.; Ghorbani A.; Diamond C. P.; Fifield H.; Berghuis L.; Larijani M. Structure-Based Design of First-Generation Small Molecule Inhibitors Targeting the Catalytic Pockets of AID, APOBEC3A, and APOBEC3B. ACS Pharmacol. Transl. Sci. 2021, 4, 1390–1407. 10.1021/acsptsci.1c00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-H.; Guo X.-C.; Zhong J.-B.; Zhong D.-X.; Huang X.-H.; Fang Z.-Y.; Zhang C.; Lu Y.-J. Discovery of APOBEC Cytidine Deaminases Inhibitors Using a BspH1 Restriction Enzyme-Based Biosensor. ChemistrySelect 2022, 7, e202201456 10.1002/slct.202201456. [DOI] [Google Scholar]
- Losey H. C.; Ruthenburg A. J.; Verdine G. L. Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat. Struct. Mol. Biol. 2006, 13, 153–159. 10.1038/nsmb1047. [DOI] [PubMed] [Google Scholar]
- Dickey T.H.; McKercher M.A.; Wuttke D. S. Nonspecific Recognition Is Achieved in Pot1pC through the Use of Multiple Binding Modes. Structure 2013, 21, 121–132. 10.1016/j.str.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey T. H.; Wuttke D. S. The telomeric protein Pot1 from Schizosaccharomyces pombe binds ssDNA in two modes with differing 3’ end availability. Nucleic Acids Res. 2014, 42, 9656–9665. 10.1093/nar/gku680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal A.; Levy Y. Structure, stability and specificity of the binding of ssDNA and ssRNA with proteins. PLoS Comput. Biol. 2019, 15, e1006768 10.1371/journal.pcbi.1006768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Max K. E. A.; Zeeb M.; Bienert R.; Balbach J.; Heinemann U. T-rich DNA Single Strands Bind to a Preformed Site on the Bacterial Cold Shock Protein Bs-CspB. J. Mol. Biol. 2006, 360, 702–714. 10.1016/j.jmb.2006.05.044. [DOI] [PubMed] [Google Scholar]
- Sachs R.; Max K. E. A.; Heinemann U.; Balbach J. RNA single strands bind to a conserved surface of the major cold shock protein in crystals and solution. RNA 2012, 18, 65–76. 10.1261/rna.02809212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzak F. M. Y.; Filichev V. V.; Harjes E.; Harjes S.; Jameson G. B.; Kurup H. M.; Kvach M. V.; Su Y.. Single stranded DNA enzyme inhibitors. WO 2022162536, 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.