

Abstract
Insulator proteins located at the boundaries of topological associated domains (TAD) are involved in higher‐order chromatin organization and transcription regulation. However, it is still not clear how long‐range contacts contribute to transcriptional regulation. Here, we show that relative‐of‐WOC (ROW) is essential for the long‐range transcription regulation mediated by the boundary element‐associated factor of 32kD (BEAF‐32). We find that ROW physically interacts with heterochromatin proteins (HP1b and HP1c) and the insulator protein (BEAF‐32). These proteins interact at TAD boundaries where ROW, through its AT‐hook motifs, binds AT‐rich sequences flanked by BEAF‐32‐binding sites and motifs. Knockdown of row downregulates genes that are long‐range targets of BEAF‐32 and bound indirectly by ROW (without binding motif). Analyses of high‐throughput chromosome conformation capture (Hi‐C) data reveal long‐range interactions between promoters of housekeeping genes bound directly by ROW and promoters of developmental genes bound indirectly by ROW. Thus, our results show cooperation between BEAF‐32 and the ROW complex, including HP1 proteins, to regulate the transcription of developmental and inducible genes through long‐range interactions.
Keywords: Drosophila, gene regulation, heterochromatin proteins, insulator proteins, topological associated domains (TADs)
Subject Categories: Chromatin, Transcription & Genomics
The transcription regulator ROW, through the cooperation with BEAF‐32 and recruitment of the HP1 complex to promoters of housekeeping genes, is required for the transcription activation of developmental genes via long‐range interactions.

Introduction
Chromosomes are organized at the submegabase scale into domains with a high level of interactions within them, known as topologically associating domains (TADs), separated by sharp boundaries with a lower level of interactions between domains (Dixon et al, 2012; Sexton et al, 2012). TADs are frequently linked with a specific chromatin state and can be divided into active, PcG, HP1, and inactive TADs (Sexton et al, 2012). Most TAD boundaries in Drosophila are located at active promoters; however, it is unclear whether this genomic distribution regulates transcription or is the consequence of transcriptional activity (Oudelaar & Higgs, 2020).
The Drosophila insulator protein BEAF‐32 binds to TAD boundaries located at promoters of housekeeping genes (Ulianov et al, 2016). Those promoters are characterized by having multiple BEAF‐32‐binding motifs surrounded by AT‐rich spacers (Emberly et al, 2008). BEAF‐32 has been implicated in both transcription and genome organization. For example, knockout of BEAF‐32 causes defects in the morphology of the male X polytene chromosome, suggesting that it is involved in chromatin structure and or dynamics (Roy et al, 2007). It is still not clear what is the role of BEAF‐32 in TAD boundaries formation as one study showed that depletion of BEAF‐32 does not affect TAD boundaries (Ramírez et al, 2018), while a more recent study showed that it does change both TAD boundaries and loops (preprint: Chathoth et al, 2021). BEAF‐32 is associated with transcription regulation through multiple mechanisms, including regulation of enhancer‐promoter interactions (Cuvier et al, 2002), through long‐range contacts between promoters bound directly and indirectly by BEAF‐32 (Liang et al, 2014; Heurteau et al, 2020), and by restricting the deposition of H3K9me3, H3K27me3, and the spread of heterochromatin to actively transcribed promoters (Emberly et al, 2008; Heurteau et al, 2020).
A key component in the formation and maintenance of heterochromatin is the protein HP1—a highly conserved protein first identified in Drosophila (now called HP1a; James & Elgin, 1986; Vermaak & Malik, 2009). Drosophila has five HP1 paralogs, three that are ubiquitously expressed (HP1a, HP1b, and HP1c) and two that are germline‐specific (HP1d and HP1e; Vermaak et al, 2005). Most of what we know about the HP1 family is from studies focused on Drosophila HP1a. HP1a is involved mostly in heterochromatin formation and transcription silencing and binds to H3K9me2/3 (Jacobs & Khorasanizadeh, 2002; Eissenberg & Elgin, 2014). The potential functions of HP1b and HP1c are less explored. HP1b is distributed at both euchromatin and heterochromatin, whereas HP1c was found mainly at euchromatin (McNally et al, 2000; Vakoc et al, 2005; Font‐Burgada et al, 2008; Zhang et al, 2011; Mattout et al, 2015). Although HP1c can bind H3K9me2/3 in vitro, it was shown to be localized at active promoters with poised RNA polymerase II (RNA pol II; Font‐Burgada et al, 2008). The molecular mechanisms that determine the different genomic distributions of the HP1 paralogs remain mostly unknown.
The Drosophila HP1c interacts with two zinc‐finger proteins, without children (WOC) and relative‐of‐WOC (ROW), as well as with the ubiquitin receptor protein Dsk2 (Font‐Burgada et al, 2008; Abel et al, 2009; Kessler et al, 2015). Recently, it was suggested that the localization of the HP1c complex at euchromatin is dependent on ROW (Di Mauro et al, 2020). ROW contains protein domains that implicate it in transcription regulation, including multiple zinc‐finger (ZNF) motifs, AT‐hooks, and a glutamine‐rich domain in the C‐terminal, that resemble activation domains found in transcription factors (Font‐Burgada et al, 2008).
Strikingly, the knockdown of row, woc, and HP1c leads to expression changes in a common set of genes (Font‐Burgada et al, 2008). Moreover, several lines of evidence suggest that the complex containing ROW, WOC, and HP1c is involved in transcription activation. First, HP1c interacts with the Facilitates Chromatin Transcription Complex (FACT) to recruit FACT to active genes and the active form of RNA pol II (Kwon et al, 2010). Second, HP1c interacts with the ubiquitin receptor protein Dsk2, which is involved in the positive regulation of transcription (Kessler et al, 2015). Third, results of chromatin immunoprecipitation in Drosophila S2 cells followed by high‐throughput sequencing (ChIP‐seq) of ROW, WOC, and HP1c revealed localization of the complex around the transcription start sites (TSSs) of actively transcribed genes (Kessler et al, 2015). Finally, the depletion of row in S2 cells leads to the downregulation of approximately 80% of the genes that are both differentially expressed and are targets of the complex (Kessler et al, 2015). However, in vivo expression analysis with RNAi lines of row, woc, and HP1c (RNA from whole larvae) showed similar numbers of upregulated and downregulated genes (Font‐Burgada et al, 2008).
ROW is also an ortholog of POGZ—a human risk gene for neurodevelopmental disorders (Stessman et al, 2016) that interacts with heterochromatin proteins (Nozawa et al, 2010). Although row is expressed in most Drosophila tissues and developmental stages, it displays the highest expression level in the larval central nervous system (Stessman et al, 2016). Moreover, neuron‐specific knockdown of row in adult flies affects nonassociative learning (Stessman et al, 2016). Thus, row is similar to POGZ as both interact with heterochromatin proteins and are involved in neurodevelopment and learning (Suliman‐Lavie et al, 2020).
Here, we comprehensively characterized row and its binding partners for the first time in vivo in adult Drosophila. We found that knockdown of row using constitutive promoter results in reduced viability, fertility, and changes in the expression of metabolic‐related genes. Interestingly, we found that in addition to WOC, HP1b, and HP1c, ROW binds to components of the insulator complex, BEAF‐32, and Chromator. ChIP‐seq experiments showed that ROW binds AT‐rich sequences through three AT‐hooks. The binding sites of ROW are located upstream of the transcription start sites of housekeeping genes and flanked by binding motifs of BEAF‐32. Moreover, we found that the genome distribution of ROW was highly correlated with BEAF‐32 and significantly enriched at TAD boundaries. Depleting row and BEAF‐32 in S2 cells resulted in a correlated change in gene expression. The differential expressed genes were more likely to be downregulated, indirect targets of ROW and BEAF‐32 (without binding sequences), and regulated through long‐range contacts. The analysis of Hi‐C data revealed the enrichment of long‐range interactions between promoters of housekeeping genes bound directly by ROW and promoters of developmental and inducible genes bound indirectly by ROW. Thus, our data show that ROW and BEAF‐32 provide a general regulation mechanism depending on the contact between promoters of housekeeping and inducible genes.
Results
Knockdown of row causes a decrease in survival and fertility
To determine row functions in vivo, we utilized two publicly available UAS‐row RNAi transgenic fly lines, hereby referred to as row RNAi‐1 and row RNAi‐2. When combined with the ubiquitous actin5C‐GAL4 driver, the progenies carrying the Gal4 driver and row RNAi construct show a significant decrease in ROW protein levels in fly heads, which was more substantial in row RNAi‐1 relative to row RNAi‐2 (94 and 87%, respectively; P < 0.05; Fig 1A).
Figure 1. Effect of row knockdown on survival and fertility of Drosophila melanogaster .
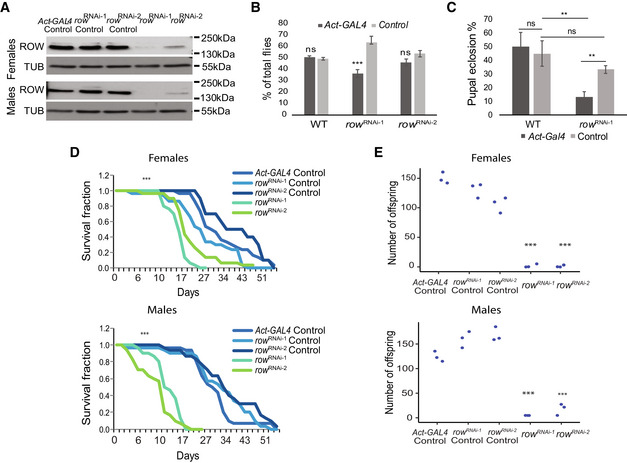
- Levels of ROW protein in row RNAi lines were decreased in both male and female fly heads. Western blot for the two row RNAi lines compared with controls was performed using rat polyclonal αROW and rat αTUBULIN (TUB). Genotype description: row RNAi‐1 (Act‐GAL4/+; UAS‐rowRNAi‐1/+), row RNAi‐2 (Act‐GAL4/+; UAS‐rowRNAi‐2/+), row RNAi‐1 Control (CyO/+; UAS‐rowRNAi‐1/+), row RNAi‐2 Control (CyO/+; UAS‐ rowRNAi‐2) and Act‐GAL4 Control (Act‐GAL4 /+).
- Decrease in the viability of row RNAi lines. To examine the viability of row RNAi flies, we counted the offspring generated from the cross between the heterozygous Act‐GAL4/CyO driver line with the homozygotes row RNAi, or WT (w1118) flies as control. Values are the percentages from the total progeny ± standard error of the mean (SEM). n = 3 (biological replicates). Significance was tested using a two‐sided binomial test.
- Decrease in pupal eclosion of row RNAi line. Pupae were collected from the cross between the heterozygous Act‐GAL4/CyO driver line with the homozygotes row RNAi‐1 or WT (w1118) flies as control. Values are the percentages for each genotype of pupal eclosion ± SEM. n = 3 (biological replicates). Significance was tested using post hoc tests with ANOVA.
- Survival curve for row RNAi flies showing reduced life span compared with controls. n = 3 (biological replicates). Significance was tested using the Kaplan–Meier estimate of survival and log‐rank test.
- Crossing of females/males row RNAi flies with WT males/females (w1118) flies resulted in a strong reduction in offspring number. Values are the number of offspring from crosses between WT (w1118) flies and flies with different genotypes. n = 3 (biological replicates). Statistical tests were performed with ANOVA followed by the Tukey's test.
Data information: Significance is represented by **P < 0.01; ***P < 0.001; ns, nonsignificant. In (D–E), the significance level was received in all comparisons between the row RNAi lines and the different controls.
Source data are available online for this figure.
We examined the viability of row RNAi flies (Fig 1B). Relative to the expected proportion of 50% (the Gal4 driver line is heterozygous for the insertion), there was a small but significant reduction in the progeny carrying both the Gal4 driver and expressing row RNAi‐1 (36.4%; P < 1.1 × 10−4), but the reduction in viability was not significant for row RNAi‐2 (46.2%; P = 0.15; Fig 1B). We observed that the lethality occurred at the pupal stage as row RNAi‐1 pupal eclosion was significantly reduced (13% compared with 33% of row RNAi‐1 control, P = 0.0058; Fig 1C). In addition to developmental phenotypes, knockdown of row also diminished the lifespan of the flies reaching adulthood for both row RNAi lines (P < 0.001; Fig 1D). In males, the effect on lifespan was very pronounced relative to the controls, while in females, we observed a smaller yet still significant lifespan reduction (Fig 1D). To test the fertility of the flies lacking row, we collected virgin males/females row RNAi flies, crossed them with wild‐type (WT) females/males flies, and counted the number of offspring (Fig 1E). Females and males row RNAi flies showed a significant reduction in offspring number compared with control flies (P < 0.001 for both row RNAi). These results demonstrate that row expression is required for normal lifespan and reproduction, in addition to the previously described importance during development. Mutations in the gene woc (the main interactor of ROW) were shown to cause similar phenotypes (Wismar et al, 2000; Warren et al, 2001; Jin et al, 2005; Maimon et al, 2014).
Genes differentially expressed in row knockdown flies are associated with metabolism
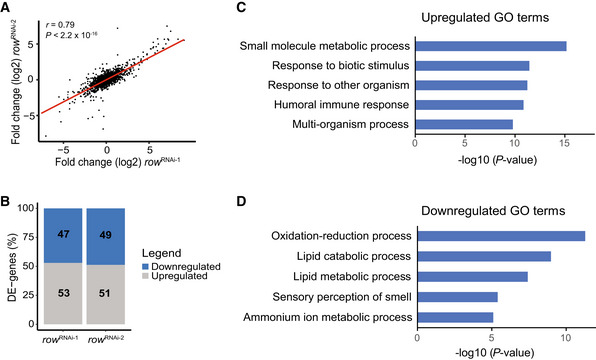
To better understand the mechanisms responsible for the low fitness of the flies due to row knockdown, we generated and sequenced RNA‐seq libraries from control and row knockdown fly heads (3–5 days old). The differential expression analysis showed that the effect of row knockdown on gene expression was consistent between the RNAi lines (r = 0.79, P < 2.2 × 10−16; Fig 2A). However, the change in expression was more substantial for the line with stronger row knockdown (row RNAi‐1, the effect was, on average, 1.5 times larger; Appendix Fig S1A and B). When analyzing the two row RNAi lines together, we found 2035 genes with significant differential expression relative to the control (False Discovery Rate (FDR) < 0.05; Dataset EV1). Despite the suspected role of ROW in transcriptional activations, the number of genes upregulated and downregulated was nearly equal (53% and 51% were upregulated in row RNAi‐1 and row RNAi‐2, respectively; Fig 2B and Appendix Fig S1A and B).
Figure 2. Knockdown of row in fly heads results in misregulation of genes involved in metabolism.
-
ACorrelation between the fold changes (log2) in row RNAi‐1 (n = 3 biological replicates) and row RNAi‐2 (n = 2 biological replicates). Fold changes were calculated relative to Act‐GAL4 control (n = 3 biological replicates; using edgeR). Significance was tested using the Pearson's correlation test.
-
BThe percentage of differentially expressed genes (DE‐genes) upregulated and downregulated in the two row RNAi lines.
-
C, DThe top 5 most significantly enriched biological processes (FDR < 0.05) that are associated with (C) upregulated and (D) downregulated genes. Results are after removing redundant terms using the REVIGO tool. GO—gene ontology.
We tested the enrichment of biological processes to examine the type of differentially expressed genes in the row RNAi lines. The most significant processes for upregulated genes were related to small‐molecule metabolic processes and responses to biotic stimuli (Fig 2C and Dataset EV2). Downregulated genes were enriched with oxidation–reduction processes and lipid catabolic or metabolic processes (Fig 2D and Dataset EV3).
ROW binds in vivo to HP1b/HP1c proteins and components of insulator complexes, BEAF‐32 and Chromator
Our findings suggest a link between the chromatin protein ROW and the regulation of metabolism in vivo. To gain insights into the molecular mechanisms for the expression changes, we first identified the protein interactors of ROW in fly heads. To do so, we first utilized CRISPR technology to FLAG‐tag the endogenous ROW protein in flies. We validated the tagging with western blot (Fig 3A) and sequencing. Importantly, the tagging does not affect the levels of ROW protein (Fig EV1A). We then utilized these flies to identify ROW interacting proteins by performing affinity purification of ROW‐containing protein complexes from fly heads, followed by mass spectrometry analysis. We found five co‐purified proteins with ROW in all three independent experiments that were not detected in the control experiments. The five proteins were the zinc‐finger protein WOC, HP1c, HP1b, the extraproteasomal ubiquitin receptor Dsk2 (also known as Ubqn), and the subunit of the cytoplasmic Dynein, Ctp (Fig 3B). Another 32 proteins were co‐purified with ROW in two of the three experiments (Fig EV1B).
Figure 3. Characterization of the in vivo interactome of ROW in fly heads.
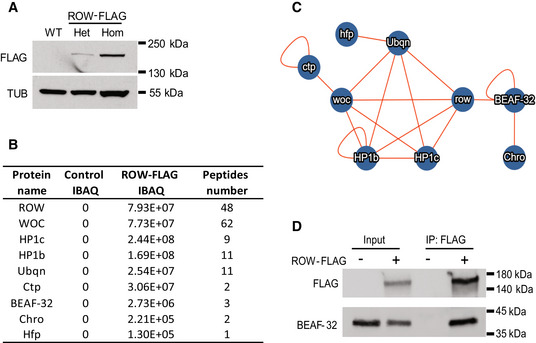
- Western blot for flies expressing endogenous FLAG‐tagged ROW (ROW‐FLAG), with αFlag tag antibody to validate the tagging. αTubulin (TUB) antibody was used as a reference. Het, Heterozygotes; Hom, Homozygotes for tagged ROW.
- Table summarizing the affinity purification–mass spectrometry data. IBAQ (intensity‐based absolute quantification) reflects the protein abundance in the sample. Peptide number is the number of razor and unique peptides. The data are the mean for the control (W1118, n = 3) and ROW‐FLAG flies (n = 3) samples. The proteins presented in the table were co‐purified with ROW in at least two out of the three experiments, none in the control experiments, and are supported by additional evidence from previous studies.
- Network of identified protein–protein interactions associated with ROW in fly heads. The lines represent previously identified interactions.
- BEAF‐32 coimmunoprecipitate with ROW. Lysates from S2 cells not transfected (−) or transfected (+) with ROW‐FLAG tagged plasmid were subjected to immunoprecipitation with αFlag tag beads. The immunoprecipitates and input (5%) were analyzed by western blot with αFlag tag and αBEAF‐32 antibodies.
Source data are available online for this figure.
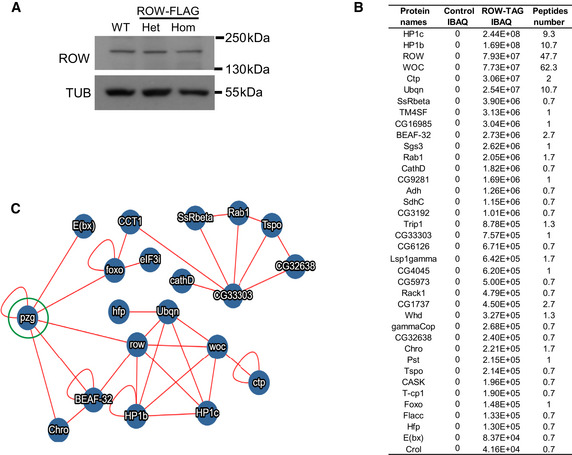
Figure EV1. In vivo interactome of ROW.
-
AWestern blot for flies expressing endogenous FLAG‐tagged ROW (ROW‐TAG) with αROW antibody was used to verify that the tagging does not affect ROW protein levels. αTubulin (TUB) antibody was used as a reference. Het, Heterozygotes; Hom, Homozygotes for tagged ROW.
-
BProteins co‐purified with ROW in at least two of the three experiments that were also not detected in the control experiments. IBAQ (Accurate Label‐Free Protein Quantitation) reflects the protein abundance in the sample. Peptide number is the number of Razor and unique peptides. The data are the mean of control (W1118, n = 3) and ROW‐FLAG flies (n = 3) samples.
-
CProtein–protein interactions that are supported by previous studies. The interactions are based on the molecular interaction search tool (MIST; Hu et al, 2018). The PZG (Z4) protein (circled in green) was not co‐purified with ROW in our study but was found previously to interact with ROW (Guruharsha et al, 2011; Kessler et al, 2015) and may connect the two complexes we identified to interact with ROW.
Source data are available online for this figure.
We used the molecular interaction search tool (MIST; Hu et al, 2018) to identify protein interactions supported by additional evidence from previous studies. The analysis provided evidence for two highly connected complexes (Fig EV1C). The main complex that included ROW was composed of the five proteins that we identified as high‐significant interactors (HP1c, HP1b, WOC, Dsk2, and Ctp), together with the transcription regulator hfp, and two components of an insulator complex: the boundary element BEAF‐32 and the chromodomain protein, chromator (Fig 3B and C). To further confirm the interaction between ROW and BEAF‐32, we performed Co‐immunoprecipitation using S2 cells transfected with a ROW‐FLAG tagged plasmid. Indeed, we found that immunoprecipitation of ROW using αFLAG antibody results in coprecipitation of BEAF‐32 (Fig 3D).
ROW binds upstream to the transcription start sites of housekeeping genes but is less likely to bind genes that are differentially expressed by row knockdown
We assumed that the complex that includes ROW, WOC, HP1c, and HP1b is expected to be responsible for the transcription dysregulation in the fly heads upon row knockdown. To test this possibility, we performed ChIP‐seq for ROW, WOC, HP1c, and HP1b in fly heads to identify the direct targets of the proteins in this complex. The ChIP‐seq signals of ROW in fly heads were substantially reduced when row was knockdown (Fig EV2A and B), indicating high specificity of the ChIP‐seq of ROW.
Figure EV2. In vivo ChIP‐seq results of ROW and its interactors.
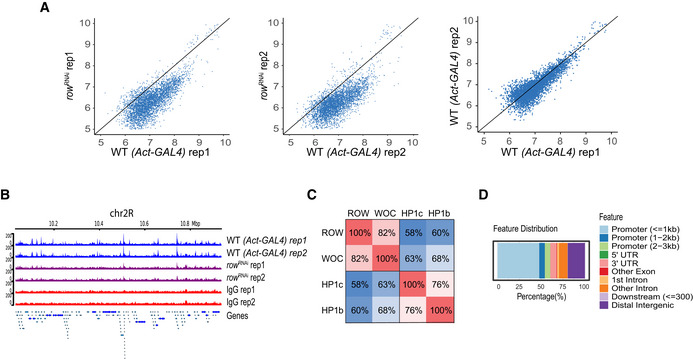
- Scatter plots of the ChIP‐seq signal of ROW in row RNAi flies compared with Act‐GAL4 flies (WT) and the results of the two biological replicates (rep1 and rep2) in WT flies against each other. The result shows that in row RNAi flies, the signal is below the straight line with a slope of 1.
- ChIP‐seq signals for ROW in a representative genomic region. The data are shown for Act‐GAL4 flies, row RNAi‐1 flies, and IgG ChIP‐seq signals (n = 2 biological replicates for each).
- Percentage of overlap between the binding sites of ROW, WOC, HP1c, and HP1b.
- Genomic annotation of the shared binding sites between ROW, WOC, HP1c, and HP1b.
We initially examined the genome distribution relationship between the different proteins in the complex by calculating the pairwise correlation in ChIP‐seq signals in nonoverlapping bins across the genome. We found a very strong and significant correlation between the genome distribution of all the proteins in the complex (P < 2.2 × 10−16; Fig 4A), but the most significant correlation was between ROW and WOC (r = 0.95) and between HP1c and HP1b (r = 0.94). We also examined the overlap between the binding sites that were identified for each protein (MACS2 peak caller, q‐value < 0.05; the number of peaks: ROW = 5,302, WOC = 4,896, HP1c = 4,252, HP1b = 2,508). Similar to the quantitative analysis, we identified the strongest overlap of the binding sites between ROW and WOC (82%) and between HP1c and HP1b (76%; Fig EV2C).
Figure 4. ROW binds in vivo to promoters of housekeeping genes.
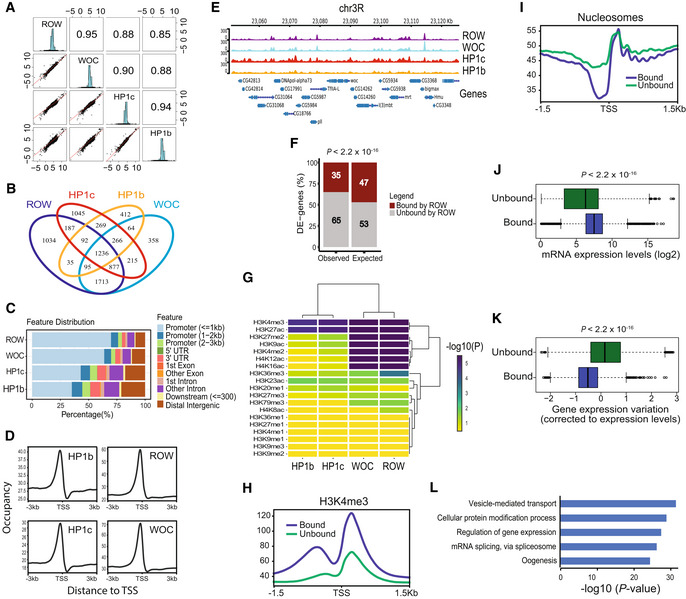
- Pairwise correlation analysis between the ChIP‐seq signals of ROW, WOC, HP1c, and HP1b in nonoverlapping bins of 2,000 bases across the genome. In the boxes of the upper triangle are the Pearson correlation coefficients. In the diagonal boxes are histograms showing the distributions of the ChIP‐seq signal. On the lower triangle boxes are bivariate scatter plots with linear regression lines. The ChIP‐seq signals are an average of three biological replicates for each protein.
- Venn plot showing the overlap between the binding sites of ROW, WOC, HP1c, and HP1b.
- Genomic annotation of ROW‐, WOC‐, HP1c‐, and HP1b‐binding sites.
- Average signal profiles (metagene plot) of ROW, WOC, HP1c, and HP1b over a 6‐kb window around TSSs. The ChIP‐seq signals are an average of three biological replicates for each protein.
- ChIP‐seq signals for ROW, WOC, HP1c, and HP1b at example genomic region. The ChIP‐seq signals are an average of three biological replicates for each protein.
- The percentages of differentially expressed genes in fly heads bound or unbound by ROW and the expected values under an independent assumption. Significance was tested using the Fisher's exact test.
- Heatmap showing the significance of the overlap between different histone modifications and ROW, WOC, HP1c, and HP1b.
- A metagene plot of H3K4me3 over a 3‐kb window around TSSs for genes bound and unbound by ROW with matched expression levels.
- Average nucleosome profile over a 3‐kb window around TSSs for genes bound and unbound by ROW with matched expression levels.
- Expression levels in the fly heads for all genes bound (n = 4,651) and unbound by ROW (n = 9,132). Values are log2 of the normalized reads count based on RNA‐seq from control flies. Within each box, the central band is the median value; boxes represent the range between the 25th to the 75th percentile distribution of values; whiskers denote the minimum/maximum values within 1.5 times the interquartile range of the 25th and 75th percentile; dots are observations outside the whiskers range. Significance was calculated using the Wilcoxon signed‐rank test.
- Gene expression variation (corrected to expression levels, see methods) for genes bound (n = 4,651) and unbound by ROW (n = 9,132) across 30 different developmental stages. Within each box, the central band is the median value; boxes represent the range between the 25th to the 75th percentile distribution of values; whiskers denote the minimum/maximum values within 1.5 times the interquartile range of the 25th and 75th percentile; dots are observations outside the whiskers range. Significance was tested using the Wilcoxon signed‐rank test.
- The top 5 most significantly enriched biological processes (FDR < 0.05) for genes bound by ROW, after removing redundant terms using the REVIGO tool.
We found 1,236 peaks shared between all the proteins in the complex, which include 66% of the peaks overlapping between HP1c and HP1b and 32% between ROW and WOC (Fig 4B). These results indicate that the core proteins of the ROW complex colocalize in vivo at an overlapping set of genomic positions. It also suggests that they may operate at specific binding sites as heterodimers formed by the assembly of ROW/WOC and HP1c/HP1b.
We next characterized the location of ROW‐binding sites and found that most sites (69.8%) overlap promoter regions (≤1 kb to TSS, P = 9.9 × 10−6; Fig 4C). We observed a slightly lower overlap with promoter regions for WOC (63.8%) and a substantially lower overlap for HP1c and HP1b (43.0 and 35.1%, respectively; Fig 4C). 48% of the binding sites shared by all four proteins are in promoter regions (Fig EV2D). The binding profile of all four proteins showed similar enrichment of approximately 150 bases upstream of the TSS (Fig 4D). The ChIP‐seq profile of the four proteins in a representative region is shown in Fig 4E.
As ROW binds upstream to the TSS, we determined which genes are bound by ROW and found 4,784 such genes (with ROW peak between −250 bp and +50 of the TSS; Dataset EV4). We then examined how many differentially expressed genes in row RNAi fly heads are bound by ROW. Surprisingly, out of 2,035 differentially expressed genes, only 713 genes (35%) were bound by ROW, which is significantly lower than what is expected by chance (expected = 959, P < 2.2 × 10−16; Fig 4F). Similar results were obtained when considering the genes bound by both ROW and WOC (observed = 561, expected = 784, P < 2.2 × 10−16) or the genes bound by all four proteins in the complex (observed = 81, expected = 102, P = 0.025). Among the genes bound by all the four proteins, the number of upregulated and downregulated genes was nearly equal (55% were upregulated, P = 0.22). Thus, these results suggest that most differentially expressed genes may represent indirect effects of the knockdown of row.
Given that the differentially expressed genes are mostly not ROW targets, we next asked what type of promoters and genes are bound by ROW. To characterize them, we first tested the overlap between the ChIP‐seq signal of the four proteins and histone marks (using modENCODE data, Riddle et al, 2011). We found that the histone marks of the active transcription (H3K4me3 and H3K27ac) significantly overlapped with all the four proteins in the complex (Fig 4G). However, other histone marks that have been linked to active promoters (H3K4me2, H3K9ac, H4K16ac, and H4K12ac) showed very significant overlap only with ROW and WOC (Fig 4G). To validate the results in fly heads, we performed ChIP‐seq using an anti‐H3K4me3 antibody. We compared the distribution of H3K4me3 near the TSS for genes bound by ROW relative to genes unbound by ROW (with matched gene expression levels) and found a strong H3K4me3 signal flanking the peak of ROW (Fig 4H).
Second, to further characterize the promoters bound by ROW, we performed micrococcal nuclease followed by sequencing (MNase‐seq) from fly heads. The nucleosome structure can be used to distinguish between two promoter types: (i) Promoters of constitutively expressed genes (housekeeping genes) that typically show more defined and clear nucleosome‐free region (NFR) with defined nucleosome positions and spacing upstream to the TSS; (ii) Promoters of regulated genes that typically lack clear NFR and have less organized nucleosome positions and spacing (Juven‐Gershon & Kadonaga, 2010; Rach et al, 2011; Ngoc et al, 2019). We found that the nucleosome organization of promoters bound by ROW was typical for active, housekeeping genes, with a defined nucleosome‐free region near the TSS and well‐spaced nucleosomes upstream to the TSS compared with unbound promoters (with matched gene expression levels; Fig 4I).
Third, to establish that the genes bound by ROW are housekeeping genes, we examined their expression patterns and gene ontologies. Based on the RNA‐seq we performed from fly heads, the ROW‐associated genes show a significantly higher expression level than not‐associated genes (P < 2.2 × 10−16; Fig 4J). ROW‐associated genes also show low variation across developmental stages (P < 2.2 × 10−16; Fig 4K). Using a list of constitutively expressed genes (Corrales et al, 2017), we found that 64% of the genes bound by ROW can be classified as housekeeping genes. Additionally, we found a significant overlap between the binding sites of ROW and enhancers of housekeeping genes (43%; P = 1.0 × 10−5; 58.4% were previously shown to be proximal to TSSs; Zabidi et al, 2015). Gene ontology (GO) analysis found that genes bound by ROW are enriched for multiple essential terms, including regulation of vesicle‐mediated transport, regulation of gene expression, protein modification, mRNA splicing, and oogenesis (Fig 4L and Dataset EV5).
In summary, our in vivo analysis indicates that ROW binds mostly promoters of constitutively active genes, but those are less likely to be the genes that are differentially expressed by row knockdown.
ROW binds AT‐rich sequences through its AT‐hook domains
To identify the DNA sequences responsible for ROW binding, we searched for enriched DNA motifs within the ROW‐binding sites. The most significant enrichment was for AT‐rich sequences (MEME‐ChIP analysis: E‐value = 3 × 10−254), which were located at the center of the ROW peak summit (Figs 5A and EV3A). We found a similar enrichment of AT‐rich sequences for all the other proteins in the complex (WOC, HP1c, and HP1b; Fig EV3A).
Figure 5. ROW binds specifically to AT‐rich sequences by AT‐hooks.
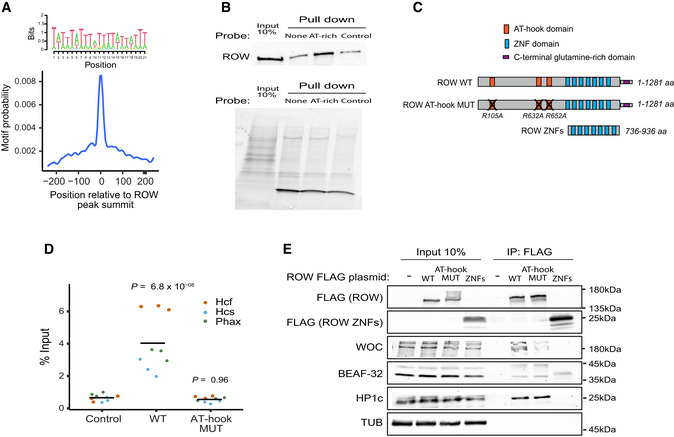
- Central enrichment of AT‐rich sequence in ROW‐binding regions. The logo shows the most significant enriched sequence (based on MEME‐ChIP analysis). The plot shows the motif's probability relative to the ROW ChIP‐seq peaks (calculated with centriMo).
- On the top are the results of DNA affinity pulldown followed by western blot with ROW antibody. None is a pulldown with no probe, AT‐rich is a pulldown with a biotin‐labeled AT‐rich dsDNA probe, and control is a pulldown with a probe composed of 49% A/T bases. On the bottom is the stain‐free gel as a loading control.
- The structure of ROW, the AT‐hook mutant, and the plasmid containing only the ZNF domains of ROW that were used in ChIP–qPCR and Co‐IP experiments. The length of the predicted proteins in numbers of amino acids (aa) and the positions of the mutations in the AT‐hook domains of ROW are shown.
- ChIP–qPCR results using FLAG‐tag antibody for S2 cells transfected with WT or AT‐hook mutant ROW‐FLAG tagged plasmids (n = 3, technical and biological replicates). As a control, cells not transfected with ROW‐FLAG tagged plasmid were used. Input percentages for three biological replicates are shown for three promoters containing AT‐rich sequences (Hcf, Phax, and Hcs). The horizontal line represents the mean. Significance was calculated using a mixed model followed by the Tukey's post hoc test.
- Co‐IP results using αFlag antibody in S2 cells transfected with WT or AT‐hook mutant ROW‐FLAG tagged plasmids or with a plasmid containing only the ZNF domains of ROW (with FLAG‐tag). As a control, cells not transfected with ROW‐FLAG tagged plasmid were used. Western blot using αFLAG, αWOC, αBEAF‐32, αHP1c, and αTUB (loading control) antibodies is shown for input and IP samples.
Source data are available online for this figure.
Figure EV3. ROW and its interactors bind to AT‐rich sequences.
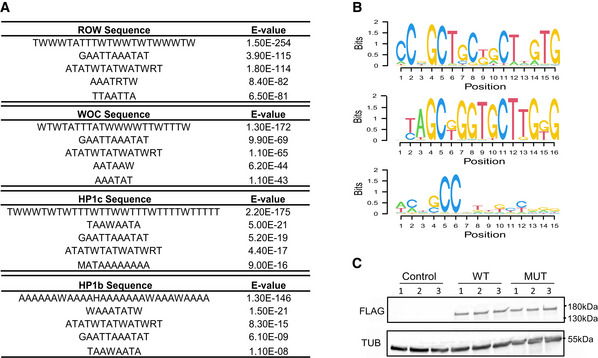
- Top five centrally enriched sequences in ROW‐, WOC‐, HP1b‐, and HP1c‐binding sites in fly heads based on MEME‐ChIP tool.
- Predicted DNA sequences for ROW‐ Cys2His2 zinc fingers under different models.
- Validation of the expression of ROW‐FLAG tagged proteins in S2 cells transfected with WT, or AT‐hook mutant ROW‐FLAG tagged plasmids (MUT) by western blot. Control are cells not transfected with ROW‐FLAG tagged plasmids. Western blot was performed with αFlag tag and αTubulin (TUB) antibodies.
In order to determine whether ROW interacts directly with the AT‐rich motifs, we performed a DNA affinity pulldown assay (Fig 5B). Briefly, we incubated nuclear extract from S2 cells with a biotin‐labeled AT‐rich double‐stranded DNA (dsDNA) probe or a control dsDNA probe (49% are A/T bases). We then performed a pulldown assay utilizing streptavidin beads followed by a western blot using anti‐ROW antibodies. Indeed, we found that ROW is enriched in the fractions pulled down with an AT‐rich probe (4.4 fold) compared with the control experiment with no probe but not in the pulldown with a standard probe (Fig 5B). These results indicate that ROW bind specifically to AT‐rich motifs.
ROW has Cys2His2 zinc fingers (ZNFs) and AT‐hook domains that can mediate the binding to DNA (Fig 5C). Therefore, we used an available tool for predicting DNA‐binding specificities for Cys2His2 ZNFs (Persikov & Singh, 2014) of ROW; however, the predicted sequences (Fig EV3B) did not resemble any motif that was significantly enriched in the binding sites of ROW. Therefore, we thought that the binding of ROW to AT‐rich sequences might not happen through the ZNFs domains but could be mediated by the AT‐hook domains. To test this possibility, we performed ChIP–qPCR using an αFlag tag antibody in Drosophila S2 cells transfected with WT or AT‐hook mutant ROW‐FLAG tagged plasmids at three promoters (Fig 5C and D). The mutated version had a single amino‐acid substitution in each of the 3 AT‐hook domains of ROW (R105A, R632A, and R652A; Fig 5C). The expression levels of WT and AT‐hook mutant ROW in the transfected cells were similarly based on western blot (Fig EV3C). Cells not transfected with ROW‐FLAG tagged plasmid were used as a control. ChIP–qPCR using cells transfected with WT ROW plasmid showed significant enrichment (P = 6.8 × 10−8) at the three promoters containing an AT‐rich motif. However, the AT‐hook mutant showed no significant enrichment at those promoters (P = 0.96; Fig 5D). The results indicate that ROW binds to DNA by its AT‐hook domains.
To exclude the possibility that mutations in the AT‐hook affect the ability of ROW to interact with other proteins, we performed Co‐IP using αFlag antibody in S2 cells transfected with WT or AT‐hook mutant ROW‐FLAG tagged plasmids. We also transfected the cells with a plasmid containing only the ZNF domains of ROW (with FLAG‐tag; Fig 5E). WOC, BEAF‐32, and HP1c were co‐purified with WT and AT‐hook mutant of ROW, but not with the protein containing only the ZNF domains of ROW (Fig 5E). These results demonstrate that the AT‐hook domains of ROW are not required for the protein interactions of ROW and that the interactions are not dependent on the binding of ROW to AT‐rich DNA.
The presence of BEAF‐32 facilitates the bindings of ROW to co‐occupied promoters
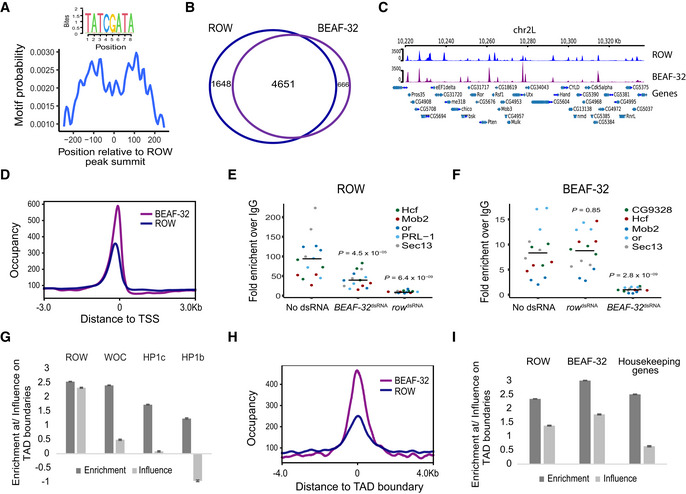
In addition to the AT‐rich motifs, we found significant enrichment for a sequence motif (TATCGA) approximately 100 bp from the peak summit of ROW (E‐value = 8.4 × 10−21; Fig 6A). Notably, BEAF‐32 is known to bind this motif (Zhao et al, 1995; Hart et al, 1997; Cuvier et al, 1998). To test whether ROW and BEAF‐32 share binding sites across the genome, we performed ChIP‐seq for ROW and BEAF‐32 in S2 cells. We replicated in this experiment the enrichment of AT‐rich sequences and BEAF‐32 consensus motifs in the binding sites of both proteins (Fig EV4A and B). We compared the list of genes bound by ROW and BEAF‐32 in S2 cells (Dataset EV6) and found that 87.5% of gene promoters bound by BEAF‐32 were also bound by ROW and 73.8% vice versa (Fig 6B and C for a representative region). We found that 59% of the genes (n = 2,742) bound by both ROW and BEAF‐32 can be defined as housekeeping genes (Fig EV4C).
Figure 6. ROW and BEAF‐32 genomic distribution relative to TSSs and TADs.
-
ABEAF‐32 consensus motif (logo) was enriched within ROW‐binding sites (MEME‐ChIP analysis). The plot (calculated with centriMo) shows that the probability of having the BEAF‐32 motif is highest around 100 bases from the center of the ROW ChIP‐seq peaks.
-
BThe overlap between genes bound by ROW and genes bound by BEAF‐32 (based on ChIP‐seq results in S2 cells; n = 3 biological replicates).
-
CExample of average ChIP‐seq signal of ROW and BEAF‐32 at a representative region with annotation of genes (n = 3 biological replicates).
-
DThe distribution of ROW and BEAF‐32 binding relative to the positions of TSSs. Occupancy is the average ChIP‐seq signal (n = 3 biological replicates).
-
E, FChIP–qPCR results using ROW (E) or BEAF‐32 (F) antibody in S2 cells treated with dsRNA against row or BEAF‐32. Cells not treated with dsRNA were used as a control (n = 3, technical and biological replicates). The enrichment is relative to IgG for three biological replicates and five promoter regions bound by both ROW and BEAF‐32. The horizontal line represents the mean. Significance was calculated using a mixed model followed by the Tukey's post hoc test.
-
GMultiple logistic regression was used to compare the enrichment and independent influence of the proteins (ROW, WOC, HP1c, and HP1b) at TAD boundaries (based on ChIP‐seq results in fly heads). Values are the enrichment and influence beta coefficients ± standard error calculated by the HiCfeat R package (Mourad & Cuvier, 2016).
-
HThe distribution of ROW and BEAF‐32 binding relative to the positions of TAD boundaries (based on ChIP‐seq results in S2 cells).
-
IResults of multiple logistic regression used to compare the enrichment and independent influence at TAD boundaries of BEAF‐32 and ROW (based on ChIP‐seq results in S2 cells) and promoters of housekeeping genes. Values are the enrichment and influence beta coefficients ± standard error calculated by the HiCfeat R package (Mourad & Cuvier, 2016).
Figure EV4. Genomic distribution of ROW and BEAF‐32.
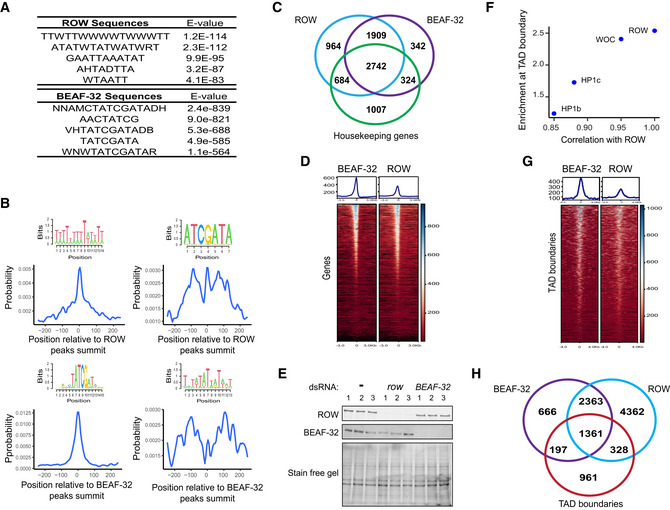
- Top five centrally enriched sequences in ROW‐ and BEAF‐32‐binding sites in S2 cells based on MEME‐ChIP tool.
- Central enrichment of AT‐rich sequences and BEAF‐32 consensus motifs (logo) in ROW‐binding regions (upper figures) and BEAF‐32‐binding regions (lower figures) in S2 cells. The plots show the probability of having the motif relative to ROW or BEAF‐32 ChIP‐seq peak's summits (calculated with centriMo).
- Overlap between the genes bound by ROW, BEAF‐32, and housekeeping genes.
- Heatmap of ROW and BEAF‐32 ChIP‐Seq signals in S2 cells ± 3 kb around TSS (average of n = 3 biological replicates).
- Protein levels of ROW and BEAF‐32 in nontreated S2 cells (−) and cells treated with dsRNA against row or BEAF‐32. The stain‐free gel was used as a loading control.
- Enrichment at TAD boundaries of ROW, WOC, HP1c, and HP1b () as a function of the correlation of the ChIP‐Seq signals in fly heads of the proteins with ROW.
- Heatmap of ROW and BEAF‐32 ChIP‐Seq signals in S2 cells ± 4 kb around TAD boundaries (average of n = 3 biological replicates).
- Overlap of the binding sites of ROW and BEAF‐32 with TAD boundaries.
Source data are available online for this figure.
The association between BEAF‐32 and ROW is not only strong but highly specific, as an unbiased search for overlap between ROW‐associated genes and genes targets of 84 transcription factors (Chen et al, 2013; Kuleshov et al, 2016) found that the most significantly enriched factor was BEAF‐32 (FDR = 0; Dataset EV7). DREF‐binding motif is very similar to BEAF‐32, but the overlap between DREF and ROW binding was not significant (FDR = 0.068; Dataset EV7). These results suggest a substantial overlap between the binding of BEAF‐32 and ROW. However, a metagene plot showed that BEAF‐32 binding displayed a more pronounced peak closer to the TSS, suggesting that the two proteins act in proximity but not precisely in the same DNA location (Figs 6D and EV4D).
Our findings show that ROW and BEAF‐32 interact physically and bind an overlapping set of promoters, but each protein binds a different sequence. Therefore, we wonder if the binding of ROW and BEAF‐32 to the chromatin depend on each other. To test this possibility, we performed ChIP–qPCR with anti‐ROW and BEAF‐32 antibodies in S2 cells treated with dsRNA to knockdown row or BEAF‐32. Untreated cells were used as a control. The reduction in protein levels in the knockdown of row or BEAF‐32 was verified using a western blot (Fig EV4E). We then examined by qPCR five promoter regions bound by both proteins. As expected from a specific ChIP experiment, the knockdown of ROW resulted in a significant reduction in the enrichment of ROW (Fig 6E; P = 6.4 × 10−9), and the knockdown of BEAF‐32 resulted in a significant reduction in the enrichment of BEAF‐32 (Fig 6F; P = 2.8 × 10−9) at the examined promoters. Interestingly, the ChIP enrichment of ROW was significantly reduced when BEAF‐32 was knockdown (Fig 6E; P = 4.5 × 10−5), while the knockdown of ROW did not significantly change the ChIP enrichment of BEAF‐32 (Fig 6F; P = 0.85). It implies that ROW binding to chromatin is facilitated by BEAF‐32 but not vice versa.
ROW and BEAF‐32 are enriched at TAD boundaries
Since BEAF‐32 occupies TAD boundaries in Drosophila (Ulianov et al, 2016), we tested the association of ROW (based on the ChIP‐seq in fly heads) with TAD boundaries (Ramírez et al, 2018). We found that ROW is enriched at the boundaries of TADs (enrichment coefficient = 2.54, P < 1 × 10−20; Fig 6G). The other partners of the ROW complex showed enrichment at TAD boundaries at a lower level ( = 2.41, 1.73, 1.24 for WOC, HP1c, and HP1b, respectively; Fig 6G). Since the proteins in the complex show correlation in their genome distribution (Fig 4A), and this correlation may explain the enrichment at TAD boundaries (Fig EV4F), we used multiple logistic regression to test for the independent influence of each protein, as previously performed (Mourad & Cuvier, 2016). The logistic regression model can distinguish between proteins that are likely to influence the TAD boundaries (influence coefficient ) and proteins that do not influence the boundaries but are enriched because they colocalize with the influential proteins (influence coefficient ). We found a large decrease in the influence coefficients (estimates of the independent influence of the variable on the outcome) for all the proteins except ROW (Fig 6G). These findings suggest a specific and independent role for ROW at TAD boundaries and that the enrichment of WOC, HP1c, and HP1b at TAD boundaries is due to their correlation with ROW.
To compare the binding of ROW and BEAF‐32 relative to TAD boundaries, we used our ChIP‐seq from S2 cells. The signals of ROW and BEAF‐32 were both centered on TAD boundaries (Figs 6H and EV4G). 36% of the sites (n = 1,361) bound by both ROW and BEAF‐32 overlap TAD boundaries (Fig EV4H). The enrichment at TAD boundaries was higher for BEAF‐32 ( = 3.02, P < 1 × 10−20) relative to ROW ( = 2.36 P < 1 × 10−20; Fig 6I). As both ROW and BEAF‐32 occupy promoters of housekeeping genes, we used multiple logistic regression to test their independent influence on TAD boundaries. We observed a proportional reduction in the beta enrichment coefficient for both BEAF‐32 ( = 1.79) and ROW ( = 1.39) but a much stronger reduction for promoters of housekeeping genes ( = 0.65; Fig 6I). This indicates a similar enrichment of BEAF‐32 and ROW at TAD boundaries independent of the occurrence of promoters of housekeeping genes.
ROW and BEAF‐32 regulate the expression of genes that are indirect targets
Since ROW and BEAF‐32 bind most of the same promoters, we wanted to examine whether they also have similar effects on transcription. We treated the S2 cells with dsRNA against row, BEAF‐32, or both genes (Fig EV5A) and analyzed gene expression in treated and untreated cells with RNA‐seq. We found a significant correlation between the changes in expression in the cells with BEAF‐32 knockdown and row knockdown (r = 0.44, P < 2.2 × 10−16; Fig 7A), suggesting an overlap in the genes influenced by ROW and BEAF‐32. The effect on gene expression was the strongest when both genes were knockdown, and it was the weakest in cells with only BEAF‐32 knockdown (P = 0.017; Fig EV5B). Changes in gene expression in cells with knockdown of both row and BEAF‐32 were significantly correlated with the changes in cells with knockdown of row, knockdown of BEAF‐32, and with the additive effect of the two genes (the sum of the fold changes in row and BEAF‐32 separate knockdowns; Fig EV5C).
Figure EV5. Differential expression analysis in S2 cells.
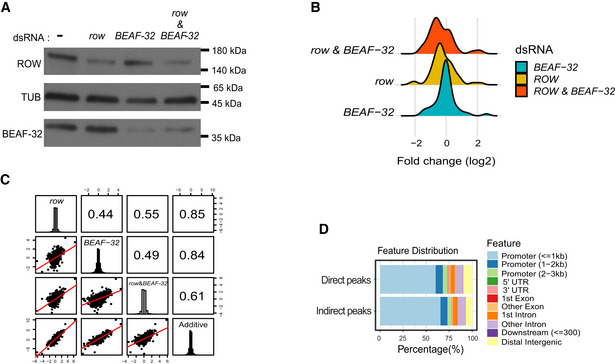
- Protein levels of ROW and BEAF‐32 in nontreated S2 cells (−) and cells treated with dsRNA against row, BEAF‐32, or both. αTubulin (TUB) was used as a loading control.
- Density plot of the gene expression fold changes (log2) in cells treated with row dsRNA, BEAF‐32 dsRNA, and row dsRNA & BEAF‐32 dsRNA, shown for significantly differentially expressed genes.
- Pairwise correlation between the gene expression fold changes (log2) in cells treated with row dsRNA, BEAF‐32 dsRNA, row dsRNA and BEAF‐32 dsRNA, and the additive model (sum of fold change in the separate knockdowns of row and BEAF‐32). Fold changes were calculated relative to control cells (using edgeR). In the boxes of the upper triangle are the Pearson correlation coefficients. In the diagonal boxes are histograms showing the distributions of the fold changes. On the lower triangle, boxes are bivariate scatter plots with linear regression lines.
- Genomic annotation of direct and indirect peaks of ROW shows that most are gene promoters.
Source data are available online for this figure.
Figure 7. Knockdown of row or BEAF‐32 in S2 cells causes downregulation of a common set of long‐range targets.
-
ARelationship between the gene expression fold change (log2) in cells treated with row dsRNA and cells treated with BEAF‐32 dsRNA (n = 3, biological replicates). The fold change was calculated relative to control cells. Significance is based on a Pearson's correlation test.
-
BThe percentage of differentially expressed genes (DE‐genes) upregulated and downregulated in row dsRNA‐, BEAF‐32 dsRNA‐, and row dsRNA‐ and BEAF‐32 dsRNA‐treated cells (n = 3, biological replicates). Significance was calculated using a two‐sided binomial test.
-
C, DThe percentages of differentially expressed genes bound or unbound by (C) ROW or (D) BEAF‐32 and the expected values under an independence assumption. Significance was calculated using the Fisher's exact test.
-
EAssociation between the 51 differentially expressed genes in S2 cells with row and BEAF‐32 knockdown and previously published long‐range targets of BEAF‐32. Values are the odds ratio ± 95% confidence interval of the association between all genes or restricting the analysis to downregulated genes. Significance was calculated using the Fisher's exact test.
-
FIllustration of ROW ChIP direct and indirect peaks and their number.
-
GDensity plots of ROW‐binding signal (ChIP‐seq) at promoters of genes unbound by ROW, promoters bound directly by ROW (with AT‐rich sequences), and promoters bound indirectly by ROW (without AT‐rich sequences). Significance was calculated using a t‐test.
-
HChIP–qPCR results using FLAG‐tag antibody in S2 cells transfected with WT or AT‐hook mutant ROW‐FLAG tagged plasmids. Cells not transfected with ROW‐FLAG tagged plasmid were used as a control. Percentages of input are shown for three biological replicates and three promoters with an indirect binding of ROW (PRL‐1, Mob2, and CG9328). The horizontal line represents the mean. Significant was tested using a mixed model followed by the Tukey's post hoc test.
-
IThe percentages of differentially expressed genes bound by ROW with or without ROW‐binding sequences (15 repeats of A or T) at the promoter regions and the expected values under an independent assumption. Significance was calculated using the Fisher's exact test.
-
JThe percentages of differentially expressed genes with or without a BEAF‐32‐binding motif (TCGATA) at the promoter regions and the expected values under an independence assumption. Significance was calculated using the Fisher's exact test.
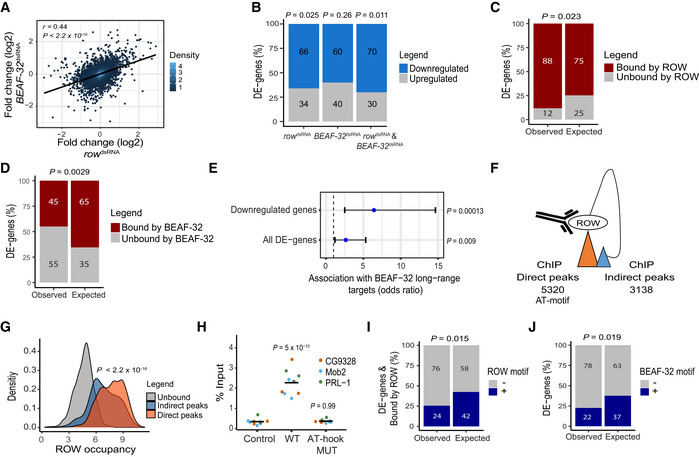
Comparing gene expression in the treated (dsRNA against row, BEAF‐32, or both genes) and untreated cells (no dsRNA), we found 51 differentially expressed genes (FDR <0.1; Dataset EV8). The 51 genes significantly overlap with a previously reported list of differentially expressed genes upon row knockdown in S2 cells (Kessler et al, 2015; OR = 24.1; P = 3.2 × 10−13). The majority of the differential expressed genes in the S2 cells were downregulated (row knockdown: 66%, P = 0.025; BEAF‐32 knockdown: 60%, P = 0.26; knockdown of both: 70%, P = 0.011; Fig 7B), consistent with the role of ROW and BEAF‐32 in transcription activation. We used our ChIP‐seq results to test whether the differentially expressed genes are bound by ROW and BEAF‐32. We found a positive association between the differentially expressed genes and binding by ROW (88% bound by ROW vs. 75% expected by chance, P = 0.023; Fig 7C), but by contrast, for BEAF‐32, the association was significantly lower than expected by chance (45% bound by BEAF‐32 vs. 65% expected, P = 0.0029; Fig 7D).
The negative association between BEAF‐32‐binding and expression changes could result from the involvement of BEAF‐32 in transcription regulation through long‐range contacts between direct ChIP peaks of BEAF‐32 (containing DNA‐binding motifs) and indirect low‐intensity peaks (without BEAF‐32 motifs), as was previously described (Liang et al, 2014). To test this possibility, we compared the list of the 51 differentially expressed genes we identified with a previously published list of genes BEAF‐32 regulates in S2 cells through long‐range contacts (Liang et al, 2014). Those genes were found by introducing a mutation that impairs the interaction between BEAF‐32 and CP190, which abolishes the binding of BEAF‐32 to the indirect peaks (Liang et al, 2014). Remarkably, we found a significant association between the 51 differentially expressed genes and the long‐range targets of BEAF‐32 (OR = 2.6, P = 0.009; Fig 7E), which was more significant for downregulated genes in both datasets (OR = 6.4, P = 0.00013; Fig 7E). This finding implies that the downregulated genes we identified are activated through the long‐range and indirect binding of BEAF‐32.
As an interactor of BEAF‐32, we speculated that ROW might also have indirect low‐intensity peaks associated with changes in expression. These low‐intensity peaks could have been missed in our in vivo ChIP‐seq and could explain why the differentially expressed genes in the fly head were not associated with ROW binding. Therefore, we divided the ROW peaks identified in the S2 cells ChIP‐seq to direct peaks with AT‐rich sequences (15 repeats of A or T) and indirect peaks without AT‐rich sequences, which included 5,320 direct peaks and 3,138 indirect peaks (Fig 7F). The majority of direct and indirect peaks of ROW overlap promoters (60.3% and 65.5%, respectively; Fig EV5D). As predicted, the promoters with indirect peaks had a lower intensity than promoters with direct peaks (P < 2.2 × 10−16; Fig 7G). To confirm that the indirect ROW peaks are specific, we performed ChIP–qPCR on three promoters with indirect peaks using cells transfected with WT ROW plasmid and the AT‐hook mutant (Fig 7H). The three sites were specifically enriched by immunoprecipitation with WT ROW but not with the AT‐hook mutant, showing that indirect peaks result from ROW binding to AT‐rich sequences and probably through long‐range contacts between direct and indirect peaks (Fig 7H).
Next, we tested whether the differentially expressed genes in the S2 cells treated with dsRNA against row and BEAF‐32 are indirect ROW targets. Although most of the differentially expressed genes are bound by ROW, only 24% have the ROW motif (42% are expected by chance; P = 0.015; Fig 7I). We also found that the differentially expressed genes are less likely to have the BEAF‐32 motif (22% have the motif vs. 37% expected by chance; P = 0.019; Fig 7J). These findings suggest that the binding of ROW to the promoter of the differentially expressed genes is the outcome of indirect binding.
ROW binds directly to housekeeping genes and indirectly to developmental genes via long‐range interactions
Our analysis showed that ROW binds housekeeping genes, but it is unclear what type of genes are directly and indirectly bound by ROW. We tested the enrichment of GO terms for genes bound directly and indirectly by ROW. We found that genes bound directly by ROW are enriched with processes involved in the basic maintenance of the cells (i.e., housekeeping genes; Fig 8A and Dataset EV9), while genes bound indirectly by ROW were enriched with GO terms related to developmental and regulated processes (Fig 8B and Dataset EV10).
Figure 8. Long‐range interactions between promoters of housekeeping genes directly bound by ROW and developmental genes bound indirectly by ROW.
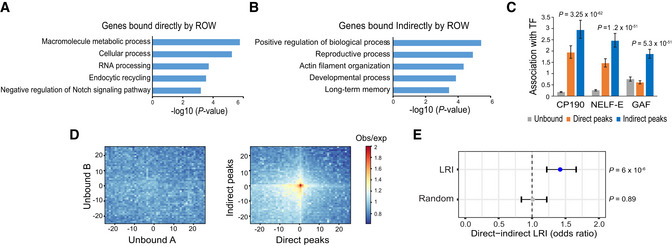
-
A, BThe top 5 most significantly enriched biological processes (P‐value <0.001) for (A) genes bound directly and (B) indirectly by ROW, after removing redundant terms using the REVIGO tool.
-
CThe association of three transcription factors (CP190, NELF‐E, and GAF) with genes unbound (n = 7,378), bound directly (n = 2,335), and bound indirectly by ROW (n = 3,176). Values are the odds ratios ± 95% confidence interval.
-
DPlot of aggregated Hi‐C submatrices of (left panel) random sets of promoters unbound by ROW, and (right panel) promoters bound directly and indirectly by ROW. The plots show the promoters in the center within a region of 50 kb divided into 50 bins (bin size = 1 kb). The values are the mean of observed/expected transformed submatrices (warm colors indicate higher values). The middle region on the right panel shows a high observed/expected value indicating a high level of long‐range interactions between promoters bound directly and indirectly by ROW.
-
ESignificant enrichment of long‐range interactions (LRI) between direct (n = 5,320) and indirect ROW peaks (n = 3,138) but not between randomly generated interactions. The association tests within the interactions identified in Hi‐C data were performed using the PSYCHIC tool (Ron et al, 2017). Values are the odds ratios ±95% confidence interval. Significance was calculated using the Fisher's exact test.
Long‐range targets of BEAF‐32 were previously found to be enriched with factors associated with RNA pol II pausing (GAF and NELF; Liang et al, 2014), and CP190 was found to be required for long‐range interactions of BEAF‐32 (Vogelmann et al, 2014). Promoters with paused RNA pol II tend to be of developmental genes often bound by GAF, which is essential for establishing paused Pol II (Gaertner & Zeitlinger, 2014; Fuda et al, 2015). Consistent with this observation, indirect peaks of ROW were positively associated with CP190 (OR = 2.9, P = 3.2 × 10−62), NELF‐E (OR = 2.5, P = 1.2 × 10−51), and GAF (OR = 1.9, P = 5.3 × 10−32; Fig 8C). Direct peaks of ROW were also enriched, to a lesser extent, with CP190 (OR = 1.9, P = 6.7 × 10−22) and NELF‐E (OR = 1.5, P = 1.1 × 10−9) but were negatively associated with GAF (OR = 0.6, P = 2.6 × 10−17; Fig 8C).
To validate the long‐range interactions between promoters bound directly and indirectly by ROW, we used genome‐wide aggregation of published Hi‐C data in S2 cells (Ramírez et al, 2018). While a random set of promoters unbound by ROW showed no enrichment for long‐range interactions (mean observed/expected = 0.99; Fig 8D, left), promoters bound directly and indirectly by ROW had high levels of long‐range interactions (mean observed/expected = 1.8; Fig 8D, right). Similar findings were obtained with sets of genes with matched expression levels bound and unbound by ROW (Appendix Fig S2A and B). To further confirm the existence of the long‐range interactions, we used a computational approach that identifies over‐represented promoter interactions in Hi‐C data (Ron et al, 2017). There was a significant association between direct and indirect ROW peaks within these identified interactions (OR = 1.4, P = 6 × 10−6; Fig 8E). Thus, our results demonstrate the existence of long‐range interactions between promoters of housekeeping genes, bound directly by ROW, and promoters of developmental genes, bound indirectly by ROW.
Discussion
Long‐range chromatin interactions have an essential role in transcription regulation. Our data strongly indicate that row is required for the transcription regulation of developmental and inducible genes by forming promoter‐promoter interactions with housekeeping genes. Our study uncovers new cooperation between the insulator protein BEAF‐32 and the chromatin‐binding protein ROW (Fig 9). The two proteins interact and bind to overlapping genomic positions, many of which are promoters of housekeeping genes located at the boundaries of TADs. The ROW‐binding sites are AT‐rich sequences flanked by motifs and binding of BEAF‐32. While ROW directly binds promoters of housekeeping genes, we found that the knockdown of row affects the expression of genes that are long‐range targets indirectly bound by ROW. Long‐range interactions between housekeeping genes bound directly by ROW and inducible genes bound indirectly by ROW appear to play an important role in gene regulation, making row an essential gene.
Figure 9. A model for the cooperation of ROW with BEAF‐32 and other chromatin‐bound proteins in transcription and nuclear organization.
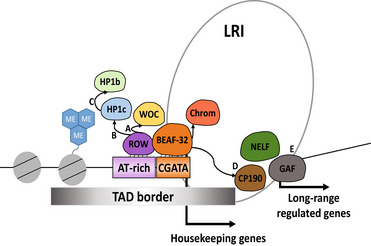
Our proposed model is that ROW and BEAF‐32 provide DNA specificity for the two complexes. ROW recruits the HP1c complex previously shown to be involved in gene activation (Kessler et al, 2015). BEAF‐32 facilitates the binding of ROW to the sites and interacts with CP190 and Chromator, which are required for long‐range interactions (Vogelmann et al, 2014). The model relies on the interaction between ROW and BEAF‐32, the localization of both proteins at most of the same promoters, and the presence of AT‐rich sequences and CGATA motifs in the promoters of housekeeping genes. Those promoters are characterized by a broad nucleosome‐free region and high levels of H3K4me3. The recruitment of proteins such as WOC and HP1c to the site by ROW is proposed to be essential for the transcription activation of developmental genes that have long‐range contact with the active promoters. (A) Our study shows that WOC has the highest overlap in binding with ROW. A previous study showed that ROW is required for WOC binding to chromatin (Di Mauro et al, 2020). Our study shows that ROW binds selectively to AT‐rich sequences using AT‐hook domains. (B) HP1c shows a lower correlation with ROW relative to WOC. HP1c binding to the chromatin also depends on ROW (Font‐Burgada et al, 2008; Di Mauro et al, 2020). (C) The correlation in genome distribution is highest for HP1b with HP1c. Among the proteins tested, HP1b shows the lowest correlation with ROW and with the genomic annotations associated with ROW (active promoters and TAD boundaries). A previous study showed that HP1c and HP1b form heterodimers in vitro and in vivo (Lee et al, 2019). (D) Previously published studies showed that BEAF‐32 regulates the expression of genes not directly bound by BEAF‐32 through long‐range contacts that depend on the interaction of BEAF‐32 with CP190 (Liang et al, 2014; Heurteau et al, 2020). (E) Based on our study, ROW and BEAF‐32 regulate a common set of genes that are indirect targets of the two proteins (promoters that lack the DNA‐binding motifs). We also show the occurrence of long‐range contacts between promoters of housekeeping genes and developmental genes. GAF, NELF, and CP190 bind the long‐range targets of ROW and BEAF‐32.
Our results show that the hierarchical recruitment of the protein complex that includes HP1b/c and WOC to active promoters depends on the sequence‐specific binding of ROW and the interaction with BEAF‐32. BEAF‐32 recruits other insulator proteins to regulate genes through long‐range interactions (Fig 9). Based on our findings, the specificity of the binding of the multiple proteins involved in the long‐range regulation is due to the cooperation of two sequence‐specific DNA‐binding proteins: BEAF‐32 and ROW. BEAF‐32 binds to its motifs (CGATA; Jiang et al, 2009), located near AT‐rich sequences bound by ROW. ROW‐binding to AT‐rich motifs is mediated by its AT‐hook domains and facilitated by BEAF‐32. After ROW and BEAF‐32 bind the DNA, they may recruit other proteins, like WOC and HP1c by ROW and CP190 or Chromator by BEAF‐32. The notion that the sequence‐specific binding of ROW and BEAF‐32 directs the localization of the HP1c complex is consistent with the strongest enrichment of ROW to promoters (relative to WOC, HP1c, and HP1b) and a more significant independent enrichment of BEAF‐32 and ROW to TAD boundaries. Previous studies showed that the recruitment of WOC and HP1c to the chromatin is dependent on ROW (Font‐Burgada et al, 2008; Di Mauro et al, 2020), but our results show a much higher concordance in the binding of WOC and ROW and a much lower correlation between ROW and HP1c. The recruitment of HP1b may depend on the heterodimerization with HP1c, as evident by the high correlation we observed in the genomic distribution between HP1c and HP1b and the lowest correlation of HP1b with ROW. This is consistent with the findings that HP1c/HP1b heterodimers are formed both in vitro and in vivo (Lee et al, 2019).
It was previously found that the ROW complex binds developmental genes associated with RNA pol II pausing (Kessler et al, 2015). We found that the binding of ROW to developmentally regulated genes is established indirectly through long‐range contacts with direct binding sites at promoters of housekeeping genes. Our work defines the ROW protein complex as essential for the transcriptional activation of developmental and inducible genes but with limited effect on housekeeping genes. Housekeeping and developmental genes have different regulation mechanisms, including DNA elements, chromatin architecture, and cofactors. Regulated genes are very sensitive to changes in the levels of activators (Zabidi et al, 2015; Jonge et al, 2017; Haberle et al, 2019), and they rely on long‐distance enhancers. By contrast, housekeeping genes rely on spatial clustering but not on contacts with long‐distance enhancers (Corrales et al, 2017). Future work will be needed to demonstrate the involvement of ROW in regulating the transcription of long‐range inducible genes and why housekeeping genes are less influenced by row knockdown.
The mechanism that may explain how the complex of ROW can promote transcriptional activation of long‐range targets is through stabilizing the NELF complex and stalled Pol II at those promoters. The depletion of Dsk2, a binding partner of ROW, causes a decrease in NELF‐E and Pol II pausing at TSSs (Kessler et al, 2015). Stalled Pol II enhances the expression of developmental genes by maintaining accessible chromatin structure (Zeitlinger et al, 2007; Gilchrist et al, 2008). In addition, the recruitment of the complex FACT can facilitate RNA Pol II elongation. A previous study showed that HP1c recruits FACT to active genes and active forms of RNA polymerase II, and in the absence of HP1c, the recruitment of FACT into heat‐shock genes was altered, and the expression levels were reduced (Kwon et al, 2010).
In conclusion, the above results demonstrate an essential role for ROW and the HP1b/c complex in the transcription activation of developmental genes through long‐range interactions with promoters of housekeeping genes and add a new dimension to our understanding of the relationship between genome organization and transcription.
Materials and Methods
Fly stocks and maintenance
The row RNAi‐1 and row RNAi‐2 are 25,971 and v28196, respectively, from the Bloomington Drosophila Stock Center and VDRC Stock Center. Act‐GAL4/CyO flies (3953) were ordered from the Bloomington Stock Center. All flies were raised at 25°C in a 12:12 light–dark cycle and on standard diets. We generated an isogenic background for the two Drosophila row RNAi strains by crossing females seven times with W1118 male flies. In each cross, female offspring carrying the transgene were selected by the red‐eye phenotype or by genotyping.
Antibodies
Polyclonal rat αROW (Font‐Burgada et al, 2008; WB 1:10000), polyclonal rabbit αROW (Kessler et al, 2015; ChIP‐seq: 1 μl), αHP1c (Kessler et al, 2015; ChIP‐seq: 5 μl) and αHP1b (Kessler et al, 2015; ChIP‐seq: 5 μl) were a gift from the lab of Prof. Fernando Azorin. Rabbit αWOC (Raffa et al, 2005; ChIP‐seq: 1 μl) was a gift from the lab of Prof. Maurizio Gatti. Mouse monoclonal αFlag is SIGMA ALDRICH (F1804, WB 1:500). Rat monoclonal αTubulin (WB: 1:10,000) and rabbit αH3K4me3 (ChIP‐seq: 5 μl) are Abcam (ab6160 and ab8580, respectively). Rabbit igG is Santa Cruz (sc‐2027, ChIP‐seq: 5 μl). Mouse αBEAF‐32 is Developmental Studies Hybridoma Bank (AB_1553420, WB: 1:200, ChIP‐seq: 8 μl).
Western blot
Twenty fly heads per sample were collected on dry ice and homogenized in 200 μl RIPA lysis buffer (50 mM Tris–HCl at pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP‐40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and 1 mM DTT, and protease inhibitor tablets [Roche]) by motorized pestle. For S2 cells, 106 cells were homogenized in 100 μl of RIPA buffer. The lysates were kept on ice for 20 min and then centrifuged at max speed for 30 min. The supernatants were collected, and 20 μl per sample was boiled with protein sample buffer (Bio‐Rad). Criterion XT Bis‐Tris gels (Bio‐Rad) were used for gel electrophoresis.
Viability assay
The UAS‐row RNAi flies or W1118 control flies were crossed with Act‐GAL4/CyO flies. Then, the number of CyO and non‐CyO offspring were counted, and the percentage of the specific genotype from the total progeny was calculated. Each cross was performed three times, and the result's significance was tested using a two‐sided binomial test.
Pupal eclosion
The UAS‐row RNAi‐1 flies or control W1118 flies were crossed with Act‐GAL4/CyO flies (n = 3 for each cross‐type). Seven days after the cross was made, 40 pupae were randomly collected, each pupa to a separate tube with food and a hole for fresh air. The number and the genotype of newly enclosed flies were recorded. Statistical tests were performed with ANOVA followed by the Tukey's test.
Survival assay
From each genotype, three vials containing 20 flies (0–3 days old, 10 males and 10 females) were kept on standard food at 25°C. Once in three days, the number of flies that died was counted, and their gender was recorded. The experiment was continued until all flies died. Tests for the difference between the survival curves were performed with the OIsurv R package using the survdiff function.
Fertility assay
Eight males or 12 females were crossed with w1118 flies from each genotype, and the number of offspring was counted. Three crosses were performed for each genotype. Statistical tests were performed with ANOVA followed by the Tukey's test.
Plasmid generation and transfections for Co‐immunoprecipitation (Co‐IP) and ChIP–qPCR in S2 cells
To overexpress ROW in S2 cells, we generated ROW FLAGx2‐tagged plasmid (WT) controlled by PMT (Metallothionein) promoter (pMT‐ROWx2FLAG). pMT‐V5 plasmid (Invitrogen) was cut using KpnI and XhoI. The gene ROW was amplified by PCR using the RE01954 (BDGP) plasmid as a template. The primers included two FLAG‐TAG sequences. As we used the Gibson Assembly kit, the primers included part of the plasmid backbone as well.
The primers used for the cloning:
5′‐AGGGGGGATCTAGATCGGGGTACAGTTAGCTGTAAGATGACGC‐3′ (F)
5′‐CTTCGAAGGGCCCTCTAGACTCACTTGTCATCGTCATCCTTGTAATCCTTGTCATCGTCATCCTTGTAATCCAATTGCGGATGGTGATGGTG‐3′ (R)
For generating the ROW FLAGx2‐tagged plasmid with mutations in the AT‐hook domains, PCR was performed using the WT plasmid and primers that contain the mutations. Then, two PCR fragments were assembled using the Gibson Assembly kit.
The primers used for the cloning:
5′‐TAGGCACCCCACCACCTCAATTGCCAATTAAAAAGGGTCCAGGTGCTCCGCCGGGCAGTA‐3′ (F)
5′‐GTCGTGGTGGGGCGCCACGACCCCG‐3′ (R)
5′‐CGGGGTCGTGGCGCCCCACCACGAC‐3′ (F)
5′‐ATTGAGGTGGTGGGGTGCCTAATGCATTCGGTGGAGCGCCGCGCTTGACT‐3′ (R)
For generating ROW‐ZNF domains‐tagged plasmid, PCR was performed using the WT plasmid and primers to open the plasmid or primers to include only the ZNF domain region of ROW (736–936 amino acids). Then, two PCR fragments were assembled using the Gibson Assembly kit.
The primers used for the cloning:
5′‐CAATTGGATTACAAGGATGAC‐3′ (F)
5′‐CATCTTACAGCTAACTGTAC‐3′(R)
5′‐ GGTACAGTTAGCTGTAAGATGGAACTGGCCAAGACCGTGGAG‐3′ (F)
5′‐ GTCATCCTTGTAATCCAATTGGCTCACATGATGCTGCACTGC‐3′ (R)
Transection was performed in a 10‐cm dish at 70–80% confluence with 30 μl of TransIT 2020 transfection reagent (Mirus Bio, MIR 5400A) and 10 μg of total DNA (1 μg of pMT‐ROWx2FLAG and 9 μg of bluescript plasmids). 12 h after transection copper (Cu) induction was performed using 500 μM of copper. The cells were collected 24 h after the induction.
Co‐immunoprecipitation (Co‐IP) assay
For each sample, 100 μl of fly heads/~107 S2 cells were homogenized in 500 μl/1 ml lysis buffer (50 mM Tris–HCL 7.4, 150 mM NaCl, 1 mM EDTA, 1% TRITON X100, and protease inhibitor tablets [Roche]). Homogenate was kept on ice for 30 min and then sonicated using a Bioruptor sonication device (Diagenode) for 2 min (10 s on 10 s off). Sonicated lysate was centrifuged at 21,000 g for 10 min, and the supernatant was then collected; 35 μl were removed for input. Preclean: protein A/G PLUS‐Agarose beads (Santa Cruz) were washed three times with 150 μl of lysis buffer, resuspended with 100 μl lysis buffer, and then added to the lysate. The samples were rotated for 30 min at 4°C and centrifuged at 3,000 rpm for 1 min. The lysate was collected in a new tube, and the beads were discarded. IP: 25 μl of Red ANTI‐FLAG M2 Affinity Gel from SIGMA ALDRICH (F2426) were washed three times with TBS (50 mM TRIS pH 7.5,150 mM NaCl and protease inhibitor tablets [Roche]) and then added to samples. The samples were rotated for 3 h at 4°C. Next, the samples were centrifuged at 3,000 rpm for 1 min, and the unbound lysate was collected in a new tube. Beads were washed 3 times using 400 μl washing buffer (300 mM NaCl, 0.1% NP 40, 50 mM Tris–HCl 7.5, and protease inhibitor tablets [Roche]) and twice using 400 μl of TBS. For the western blot, the beads were eluted with SDS–PAGE X1 sample buffer and boiled at 95°C for 5 min.
Mass spectrometry (MS)
Sample preparation: The packed beads were resuspended in 100 μl 8 M urea, 10 mM DTT, 25 mM Tris–HCl pH 8.0 and incubated for 30 min at 22°C. Next, Iodoacetamide (55 mM) was added, and beads were incubated for 30 min (22°C, in the dark), followed by the addition of DTT (20 mM). The Urea was diluted by the addition of 6 volumes of 25 mM Tris–HCl pH 8.0. Trypsin was added (0.3 μg/ sample), and the beads were incubated overnight at 37°C with gentle agitation. The beads were spun down, and the peptides were desalted on C18 Stage tips. Two‐thirds of the eluted peptides were used for MS analysis. NanoLC‐MS/MS analysis: MS analysis was performed using a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) coupled online to a nanoflow HPLC instrument, Ultimate 3000 Dionex (Thermo Fisher Scientific). Peptides were separated over an acetonitrile gradient (3–32% for 45 min; 32–50% for 15 min, 50–80% for 10 min) run at a flow rate of 0.3 μl/min on a reverse‐phase 25‐cm‐long C18 column (75 μm ID, 2 μm, 100 Å, Thermo PepMapRSLC). The survey scans (380–2,000 m/z, target value 3E6 charges, maximum ion injection times 50 ms) were acquired and followed by higher‐energy collisional dissociation (HCD) based fragmentation (normalized collision energy 25). A resolution of 70,000 was used for survey scans. The MS/MS scans were acquired at a resolution of 17,500 (target value 1E5 charges, maximum ion injection times 120 ms). Dynamic exclusion was 60 s. Data were obtained using Xcalibur software (Thermo Scientific). To avoid carryover, the column was washed with 80% acetonitrile and 0.1% formic acid for 25 min between samples.
MS data analysis: Mass spectra data were processed using the MaxQuant computational platform (Tyanova et al, 2016a; version 1.5.3.12). Peak lists were searched against the UniProt Fasta database of Drosophila melanogaster, using both annotated and predicted sequences. The search included cysteine carbamidomethylation as a fixed modification as well as oxidation of methionine as variable modifications and allowed up to two miscleavages. The “match‐between‐runs” option was used. Peptides with a length of at least seven amino acids were considered, and the required FDR was set to 1% at the peptide and protein levels. Protein identification required at least two unique or razor peptides per protein. Relative protein quantification in MaxQuant was performed using the label‐free quantification (LFQ) algorithm and with intensity‐based absolute quantification (IBAQ). Protein contaminants and proteins identified by less than two peptides were excluded. Statistical analysis between control (n = 3) and ROW‐TAG samples (n = 3) was performed using the Perseus statistical package (Tyanova et al, 2016b; computational platform for comprehensive analysis of (prote) omics data). LFQ values were used as the input for Perseus analysis. Ribosomal proteins were excluded from the results.
Tagging row endogenously by CRISPR/Cas9
Tagging row endogenously in Drosophila melanogaster using the CRISPR/Cas9 method was performed based on the approach previously described (Tianfang Ge et al, 2016) with some modifications. Two gRNAs (one targeting row and one targeting the white gene) were cloned into pCFD4d plasmid (Addgene plasmid #83954; as described in the protocol “cloning two gRNAs into plasmid pCFD4”; http://www.crisprflydesign.org/) ‐ (pCFD4dw/row).
Primers used for the cloning are:
5′‐TATATAGGAAAGATATCCGGGTGAACTTCCCGTGGGGCTTGTATCATTGGTTTTAGAGCTAGAAATAGCAAG‐3′ (forward)
5′‐CCAAAGAGCAGGAATGGTATATTTTAACTTGCTATTTCTAGCTCTAAAACCCAAAGAGCAGGAATGGTATCGACGTTAAATTGAAAATAGGTC‐3′ (reverse)
To generate the donor plasmid for homologous recombination (HR) of the gene row, pUC57‐white [coffee] plasmid (Addgene 84006) was digested with SacI and HindIII to exclude the donor template for HR of the white gene. We used w1118 flies genomic DNA to amplify by PCR both ~ 1 kb upstream and ~ 1 kb downstream from the stop codon of the gene row. To add the tag, the primers included the FLAG‐TAG sequence next to the stop codon. As we used the Gibson Assembly kit, the primers included part of the plasmid backbone as well. The vector backbone and the two parts of the amplified homologous arms were assembled using the Gibson Assembly kit (pUC57‐rowtag).
Primers used for the cloning:
Upstream part: 5′‐ACGGCCAGTGAATTCGAGCTCGCGGGTTGAGGTTTATAAGTC‐3′ (F)
5′‐TCACTTGTCATCGTCATCCTTGTAATCTTGCGGATGGTGATGGTGCT‐3′ (R)
Downstream part: 5′‐GATTACAAGGATGACGATGACAAGTGATACAAGCCCCAC.
GGAAA‐3′ (F)
5′‐CTATGACCATGATTACGCCAGGAGCTATGCCTACCCCTTC ‐3′ (R)
pCFD4dw/row, pUC57‐white[coffee], and pUC57‐rowtag plasmids were injected into vas‐Cas9 (y1, M{vas‐Cas9}ZH‐2A) flies (Tianfang Ge et al, 2016) by Rainbow Transgenic Flies, Inc. Individual injected G0 flies were mated with second chromosome balancers flies, and non‐red‐eyed G1‐CyO individual flies were crossed to 2nd chromosome balancers flies again. Screening for the desired row‐FLAG tagged line and elimination of random integration in the G1‐CyO individuals performed by PCR as previously described (Tianfang Ge et al, 2016). For final validation, we sequenced the entire row locus and performed WB as shown in the results section. The flies are available for order at Bloomington Drosophila Stock Center, stock number 94920.
Knockdown experiments in S2 cells
RNAi experiments in S2 cells were performed as previously described (Kessler et al, 2015) with modifications. dsRNA to row and BEAF‐32 were generated using the MEGAscript kit (Ambion). Cells were diluted to 106 ml−1, and 4 μg of dsRNA per 1 ml of cells were added. After two days, the cells were diluted again to 106 ml−1, and 8 μg of dsRNA per 1 ml of cells were added. After two days, the cells were washed twice with PBS, collected, and used for downstream experiments. The primers used for producing dsRNA to row were previously described (Kessler et al, 2015):
Forward row T7: TAATACGACTCACTATAGGGTGATACAGACGCTGAGTGATTG
Reverse row T7: TAATACGACTCACTATAGGGAGGAACCACATCCCAAGATG
Primers used for producing dsRNA to BEAF‐32:
Forward BEAF‐32 T7: TAATACGACTCACTATAGGGGCGAGGATCCACTGTGCTAT
Reverse BEAF‐32 T7: TAATACGACTCACTATAGGGACGCTGATTTGCCCATTTAC
ChIP‐seq using fly heads
The ChIP was performed based on a previous protocol (Menet et al, 2010) with modifications. 100 μl of fly heads were collected using dry ice and homogenized in 1 ml of NEB buffer (10 mM HEPES‐Na at pH 8.0, 10 mM NaCl, 0.1 mM EGTA‐Na at pH 8.0, 0.5 mM EDTA‐Na at pH 8.0, 1 mM DTT, 0.5% Tergitol NP‐10, 0.5 mM Spermidine, 0.15 mM Spermine and protease inhibitor tablets [Roche]) for a total of 2 min. Homogenate was poured into Bio‐Spin chromatography columns (BIO‐RAD) and centrifuged at 1,000 g for 4 min. The filtered homogenate was centrifuged at 6,000 g for 10 min, and the supernatant was discarded. The pellet was resuspended in 1 ml of NEB and centrifuged at 11,000 rpm for 20 min on a sucrose gradient (0.65 ml of 1.6 M sucrose in NEB, 0.35 mL of 0.8 M sucrose in NEB). The Nuclei pellet was then resuspended in 1 ml of NEB, and 11% formaldehyde was added for a final concentration of 1%. Nuclei were cross‐linked for 10 min at room temperature and quenched by adding 1/10 vol of 1.375 M glycine. The nuclei were collected by centrifugation at 6,000 g for 5 min. Nuclei were washed twice in 1 ml of NEB and resuspended in 350 mL of Sonication buffer (10 mM HEPES‐Na at pH 7.5, 2 mM EDTA at pH 8.0, 1% SDS, 0.2% Triton X‐100, 0.5 mM Spermidine, 0.15 mM Spermine). Nuclei were sonicated using a Bioruptor sonication device (Diagenode) for 3 cycles of 7 min (30 s on 30 s off). Sonicated nuclei were centrifuged at 15,000 g for 10 min, and the supernatant was frozen at 80°C in 150 μl aliquots. 50 μl were taken for checking the chromatin quality and the DNA fragment size, which was between 100 and 1,000 bp. For each sample, we used 50 μl from the aliquot diluted with 500 μl of IP buffer (50 mM HEPES/KOH at pH 7.6,2 mM EDTA, 1% Triton, 0.1% Na Deoxycholate in PBS and protease inhibitor tablets [Roche]). Samples were rotated overnight at 4°C after adding antibodies. ChIP‐seq was performed with rabbit αHP1c (n = 3), αHP1b (n = 2), αWOC (n = 3), αROW (n = 3), αH3K4me3 (n = 3) and IgG antibodies (n = 2). 20 μl per sample of Dynabeads Protein G (Invitrogen) was washed twice with 1 ml of IP buffer. Beads were resuspended in 25 μl IP buffer per sample. The beads were added to the samples and rotated for 1 h at 4°C. Chromatin immobilization and barcoding were performed as previously described (Gutin et al, 2018).
ChIP‐seq and ChIP–qPCR using S2 cells
The ChIP was performed based on a previous protocol (Kessler et al, 2015) with modifications. For each sample, a 25 cm flask with 8–10 × 106 S2 cells was collected into a 15 ml tube, and formaldehyde was added to a final concentration of 1.8%. The cells were cross‐linked for 10 min at RT on a shaker. To stop the reaction, glycine was added to a final concentration of 125 mM (from a stock solution of 2.5 M in PBS). The cells were centrifuged for 3 min at 1,500 g at 4°C, the supernatant was discarded, and the cells were resuspended in 2 ml PBS. Then, the cells were centrifuged again (3 min at 1,500 g at 4°C), the supernatant was discarded, and the cells were resuspended in 1 ml washing buffer A (10 mM Hepes pH7.9, 10 mM EDTA 0.5 mM EGTA 0.25% Triton X100m, protease inhibitor). The 1 ml suspension was transferred to an Eppendorf, incubated for 10 min at 4°C on a wheel, and then centrifuged (3 min at 1,500 g at 4°C). The supernatant was discarded, the pellet resuspended in 1 ml washing buffer B (10 mM Hepes pH7.9, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.01% Triton X100) and incubated for 10 min at 4°C on a wheel. The suspension was centrifuged again (3 min at 1,500 g at 4°C), the supernatant was discarded, and the pellet was resuspended in 450 μl TE buffer. 50 μl 10% SDS was added, and the Eppendorf was inverted five times and centrifuged at 1,500 g at 4°C for 3 min. The upper phase was carefully removed with a pipette, 500 μl TE was added, and the Eppendorf was inverted five times, after which it was centrifuged. The last procedure was repeated once again. After the second centrifuge, the upper phase was removed, TE with 1 mM PMSF was added to a final volume of 400 μl, and 4 μl of 10% SDS was added. The suspension was sonicated using Bioruptor plus sonication device (Diagenode) for 20 cycles (30 s on 30 s off).
The following solutions were added in this order to the lysate, and between each addition, the lysate was incubated on the wheel at 4°C for 2 min: 42 μl 10% TRITON (final 1%), 4.2 μl 10% Deoxycholate (final 0.1%), and 15 μl 4 M NaCl (final 140 mM). The lysate was Incubated on the wheel at 4°C for 10 min and centrifuged at full speed at 4°C for 5 min. The supernatant was divided into aliquots and was frozen at −80°C; 45 μl were kept for input and for checking DNA fragment size. For the ChIP‐seq experiment, 50 μl of chromatin aliquot was used and diluted with 450 RIPA buffer (140 mM NaCl, 10 mM Tris–HCl pH, 1 mM EDTA, 1% TritonX100, 0.1% SDS, 0.1% deoxycholate and protease inhibitor). Samples were rotated overnight at 4°C after adding antibodies. Washes were performed as described in the “ChIP‐seq using fly heads” paragraph. Chromatin immobilization and barcoding were performed as previously described (Gutin et al, 2018).
For the ChIP–qPCR in cells transfected with ROW tagged plasmids 100 μl of chromatin aliquot was used and diluted with 1 m of ChIP buffer (10 mM Tris–HCl [pH 8.0], 5 mM EDTA [pH 8.0], 150 mM NaCl, 1% Triton X‐100 and protease inhibitor). Anti‐FLAG M2 magnetic beads (Sigma) were added and samples were rotated overnight at 4°C. Washes were performed with the ChIP buffer (x4) and with TE (x4).
Significant in the ChIP–qPCR experiment was tested using a mixed‐model and post hoc tests.
Primers are shown below:
5′‐TTTGTGTGTTCCATTGCGTA‐3′ (Hcf promoter F)
5′‐TGCCCACTTATTTGACCGCA‐3 (Hcf promoter R)
5′‐CAGTCACGTCACGAGAGCAT‐3′ (Hcs promoter F)
5′‐GGTCATATCTGGCGGGTTTT‐3′ (Hcs promoter R)
5′‐TATGTTGTTCACCCCCACCT‐3′ (Phax promoter F)
5′‐AGCGGCAGAGTGACCATACT‐3′ (Phax promoter R)
5′‐ CAAGCCAAAACCCGATCTAA‐3′ (Sec13 promoter F)
5′‐ GGTTGAGCAGTGTGACCTGA‐3′ (Sec13 promoter R)
5′‐ CGCGAGGTTAAAGGCATTAG‐3′ (or promoter F)
5′‐ GCCAGCTGTTTTGCTTTTTC‐3′ (or promoter R)
5′‐TGATGGTTTTGTGCCATGGAT‐3′ (CG9328 promoter F)
5′‐TGTTGCATCCCAGAGTTTTG‐3′ (CG9328 promoter R)
5′‐GCTTGGTTGCAGTGTCGTAG‐3′ (Mob2 promoter F)
5′‐TAGCCTGCTGTCAGCTGTTG‐3′ (Mob2 promoter R)
5′ACCGATTTCCAAGAGCAAAG‐3′ (PRL‐1 promoter R)
5′‐CCCGCAATCGATGTTTATCT‐3′ (PRL‐1 promoter R)
ChIP‐seq analysis
ChIP‐seq reads were aligned by bowtie2 software to the full genome sequence of Drosophila melanogaster (UCSC version dm3). Duplicate reads were removed using the samtools rmdup function, and uniquely mapped reads were kept (samtools view ‐q 10). Peak calling was performed with the callpeak function of the MACS2 tool (Zhang et al, 2008) with a q‐value < 0.05. An IgG sample was used as a control. Peaks in ChrU and ChrUextra were excluded for subsequent analysis. For visualization, bedGraph files were created by deepTools (a python tool) with bamCoverage function and normalization parameter set to RPKM. The bedGraph file with the average between replicas was generated, and the corresponding bigWig file was created. The bigWig files were uploaded to the integrative genomic viewer (IGV). The ComputeMatrix and plotProfile functions of deepTools were used for plotting the distribution of the ChIP‐seq targets relative to the positions of TSSs or TAD boundaries. For each target, the average across replicates was used. UCSC refGene annotation file was used to determine the TSS locations. Classified TADs locations were obtained from the Chorogenome Navigator (Ramírez et al, 2018). To examine the overlap between TAD boundaries (± 1 kb to the border) and the binding sites of ROW and BEAF‐32, makeVennDiagram function of ChIPpeakAnno R package was used. The correlation of ChIP‐seq signal across the genome was calculated based on nonoverlapping bins of 2,000 bp. The deeptools pyBigWig python extension was used for calculating the average signal in each bin. A pairwise correlation was calculated with the psych R package. The locations of peaks within different genes annotation were determined using the annotatePeak function from the ChIPseeker R package (Yu et al, 2015). The function enrichPeakOverlap from ChIPseeker was used to test the overlap of ChIP‐seq peaks with different histone modifications. Histone modification data were from modENCODE ChIP‐chip data (Riddle et al, 2011). makeVennDiagram function of ChIPpeakAnno R package was used to determine the overlap between the binding sites of ROW, WOC, HP1c, and HP1b (Zhu et al, 2010). Multiple logistic regression implemented in the HiCfeat R package (Mourad & Cuvier, 2016) was used to calculate the enrichment and influence of proteins on TAD boundaries. UCSC refGene annotation was used to define the genes bound by ROW or BEAF‐32. Promoter regions (between − 250 bp and + 50 of TSS) of genes that overlapped with ROW/BEAF‐32 peaks were considered to be bound by the proteins (Datasets EV4 and EV6).
Motif enrichment analysis was performed using the MEME‐ChIP motif analysis tool (Bailey et al, 2009) with the default parameters. Input sequences were taken from an interval that included 250 bp downstream and 250 bp upstream of the peak's summit. CentriMo option (Bailey & MacHanick, 2012) with default parameters was used to calculate the probability to find the motif relative to the peak summit. Genes bound by ROW were defined as direct targets if at least 15 A or T repeats were observed at their promoter and as indirect targets if the motif was not found. Enrichment of GAF, NELF‐E, and CP190 with genes unbound, bound directly and indirectly by ROW was calculated using the Fisher's exact test. Data were downloaded from GSE40646 (Fuda et al, 2015; GAF), GSE116883 (Nazer et al, 2018; NELF‐E), and GSE41354 (Ong et al, 2013; CP190). Gene ontology (GO) enrichment analysis for genes bound by ROW was performed using the FlyEnrichr tool (Chen et al, 2013; Kuleshov et al, 2016). Redundant GO terms were removed using the REVIGO tool (Supek et al, 2011).
The gene expression variation for genes bound or unbound by ROW was calculated using published Drosophila gene expression data from 30 developmental stages (Brown et al, 2014). Correction of the variation to expression levels was performed by calculating the residuals from a loess curve (locally estimated scatterplot smoothing) fitted to a scatterplot between the average genes expression (log2) and the coefficient of variation (log2) in 30 developmental stages. The residuals, which are not correlated with the expression levels, were presented as a corrected variation for genes bound and unbound by ROW. We defined a list of constitutively expressed genes (housekeeping genes) by taking only genes that, in all the developmental stages, the expression was higher than the 40th percentile of expression in this stage, as performed previously (Corrales et al, 2017). The overlap between ROW‐binding sites with housekeeping enhancers was determined with the enrichPeakOverlap function from ChIPseeker.
DNA affinity pulldown
DNA affinity pulldown was performed as previously described with modifications (Ray et al, 2016). Oligonucleotides were annealed by adding equal quantities of 5′ forward biotinylated oligonucleotides and reverse oligonucleotides in an annealing buffer (10 mM Tris, pH 7.5–8.0, 50 mM NaCl and 1 mM EDTA) to the microtube. The microtube was incubated at 95°C for 5 min and slowly cooled to room temperature (turn off and open lid).
AT‐rich sequence oligonucleotides
F biotin 5′‐ ATATTCGCGATTATTATATTTATATTTATTTATATATATCCGG‐3′
R 5′‐ CCGGATATATATAAATAAATATAAATATAATAATCGCGAATAT‐3′
Standard sequence oligonucleotides
F biotin 5′‐CCAAGCGCGACTTCATGAACTAGTACGAACTGAGCACGAACGGAT‐3′
R 5′‐ATCCGTTCGTGCTCAGTTCGTACTAGTTCATGAAGTCGCGCTTGG‐3′
Preclearing nuclear extracts: 20 μl/sample streptavidin‐magnetic beads (Invitrogen, Dynabeads™ MyOne™ Streptavidin C1) were washed three times with (500 μl) buffer B [20 mM Hepes (pH 7.6), 100 mM KCl, 20% glycerol, 1.5 mM MgCl2, 0.5 mM DTT, 0.2 mM EDTA] and (last time with 20 μl per sample) and were added to 500 μg of nuclear extract proteins. The samples were incubated for 1 h at 4°C. Then, beads were discarded, and precleared nuclear extract proteins remained.
Affinity pulldown: Streptavidin‐magnetic beads (30 μl/sample) were washed three times with 500 μl of buffer B and were added to 500 μg of precleared nuclear extract along with 200 pmol of biotin oligomers, 10 μg of poly(dI‐dC), 5 μg/μl BSA, 2 mM spermidine, and 10 μg tRNA, and were incubated at 4°C for 2 h. The beads were washed four times with buffer B and three times with PBS to remove the nonspecific binding.
Elution: 30 μl of buffer B with 1 μl of DNase (Turbo DNase; Ambion) was added to the beads and incubated for 30 min at 37°C. Then, 10 μl of X4 SDS sample buffer was added, the beads were boiled and the supernatant was collected.
Predicting DNA‐binding specificities for Cys2His2 zinc fingers:
ROW protein sequence (download from UCSC) was used as input in the online tool for predicting zinc‐finger DNA‐binding (http://zf.princeton.edu/logoMain.php; Persikov & Singh, 2014).
MNase‐seq
100 μl of fly heads per sample was homogenized for 5 min in a homogenization chamber with 300 μl of ice‐cold Nuc. Buffer I (60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 15 mM Tris–HCl pH = 7.5, 0.5 mM DTT, 0.1 mM PMSF and 1× complete protease inhibitor (Roche)). Next, 200 μl more of Nuc. Buffer I was added. The homogenate liquid was poured into Bio‐Spin chromatography columns (BIO‐RAD) and centrifuged at 1,000 g for 1 min at 4°C. 500 μl of ice‐cold Nuc. Buffer II (0.04% NP‐40 and Nuc. buffer I) was added to the filtered homogenate, mixed by inverting the tube, and incubated for exactly 7 min on ice. During the 7 min incubation, 13 ml tubes with 8 ml ice‐cold Nuc. Buffer III (1.2 M sucrose + Nuc. buffer I) was prepared. The 1 ml filtered homogenate was poured into the 13 ml tube of Nuc. Buffer‐ III. The tube was centrifuged for 10 min at 11,000 rpm. The supernatant was carefully removed so that only the pellet remained (containing the intact purified nuclei). The nuclei pellet was re‐suspend in 500 μl ice‐cold MNase digestion buffer (0.32 M sucrose, 50 mM Tris–HCl pH = 7.5, 4 mM MgCl2, 1 mM CaCl2 and 0.1 mM PMSF). The 500 μl nuclei were split into 2 Eppendorf with 250 μl in each. 5 units (50 Gel Units) of Micrococcal Nuclease (NEB) were added to one tube and 20 units (200 Gel Units) to the other tube (for overdigesting). The tubes were inverted for mixing and incubated at 37°C for 10 min. The reaction was stopped by the addition of 13 μl of 20 mM EDTA and 13 μl of 10% SDS in each tube, followed by inverting and cooling on ice. 2.8 μl of 10 mg/ml Proteinase K was added and mixed by inverting. The nuclei were incubated at 65°C overnight and then centrifuged for 1 min at 15,000 g. The supernatant from each tube was taken and cleaned separately by PCR clean‐up kit (Invitrogen) with 26 μl of elution buffer at the final step. Next, 1 μl of 0.3 μg/μl RNase free DNase (Sigma), 0.75 μl of 0.5 M Tris–HCl pH = 7.4, and 3.5 μl 120 mM NaCl were added to each tube and mixed by pipetting and spin‐down. The tubes were then incubated for 1 h at 37°C, and the mix was cleaned again using a PCR clean‐up kit (Invitrogen) with 12 μl of elution buffer at the final step. Each tube volume was put to run on a 1.5% agarose gel alongside a 100 bp DNA ladder. Mononucleosome bands (150–200 bp) from the gel were excised and cleaned with a gel clean‐up kit (Invitrogen) with 50 μl of DDW as a final step. The purified DNA was quantified by nanodrop, and an equal amount from the two digestion levels was taken for DNA library preparations. DNA libraries were made as previously described (Blecher‐Gonen et al, 2013).
RNA purification and libraries preparation for RNA‐seq using fly heads
We crossed Actin‐GAL4/CyO flies with row RNAi‐1/2 flies or with w1118 control flies and selected 0–3 days old row RNAi‐1 (Act‐GAL4/+; UAS‐row RNAi‐1/+), row RNAi‐2 (Act‐GAL4/+; UAS‐ row RNAi‐2), and Act‐GAL4 Control (Act‐GAL4 /+) flies. The flies were raised for three more days to recover from CO2 exposure and collected using dry ice. RNA was prepared using TRI Reagent (SIGMA) according to the manufacturer's protocol. 3′ RNA‐seq library was performed based on Guttman's lab “RNAtag Seq protocol” (Shishkin et al, 2015) with modifications. Fragmentation of RNA was performed using FastAP Thermosensitive Alkaline Phosphatase buffer (Thermo Scientific) for 3 min at 94°C. Samples were placed on ice, and then the FastAP enzyme was added for 30 min at 37°C. RNA was purified using 2.5× volume of SPRI beads (Agencourt), and then linker ligation was performed with an internal sample‐specific barcode using T4 RNA ligase I (NEB). All RNA samples were pooled into a single Eppendorf and purified using RNA Clean & Concentrator columns (Zymo Research). Poly A selection was performed with Dynabeads Oligo (dt) beads according to the manufacturer's protocol. RT was performed with a specific primer (5′‐CCTACACGACGCTCTTCC‐3′) using AffinityScript Multiple Temperature cDNA Synthesis Kit (Agilent Technologies). RNA degradation was performed by incubating the RT mixture with 10% 1 M NaOH (2 μl of RT mixture, 70°C, 12 min). The pH was then neutralized by AcOH (4 μl for 22 μl mixture). Next, the cDNA was cleaned using Silane beads (Life Technologies). The 3′‐end of the cDNA was ligated to linker2 using T4 RNA ligase I. The sequences of linkers are partially complementary to the standard Illumina read1 and read2/barcode adapters, respectively. The cDNA was cleaned using Silane beads, and PCR was performed using enrichment primers and Phusion HF Master Mix (NEB) for 12 cycles. The library was cleaned with 0.8× volume of SPRI beads.
RNA purification and libraries preparation for RNA‐seq using S2 cells
RNA purification was performed with RNeasy Mini Kit (Qiagen) according to the manufacturer's protocol. 3′ RNA‐seq library generation was performed based on a previous protocol (Klein‐Brill et al, 2019). In brief, 100 ng from each sample was incubated with oligo‐dT reverse transcription (RT) primers containing 7 bp barcode and 8 bp UMI (Unique Molecular Identifier) at 72°C for 3 min and moved directly to ice. This was followed by a Reverse transcription reaction (SmartScribe kit Clontech) for 1 h at 42°C and 70°C for 15 min. Next, samples were pooled and purified using SPRI beads X1.2 (Agencourt AMPure XP, Beckman Coulter). The pooled barcoded samples were then tagmented using Tn5 transposase (loaded with oligos Tn5MEDS‐A) for 8 min at 55°C Followed by the addition of 0.2% SDS to strip off the Tn5, and SPRI X2 purification was performed. Finally, a PCR was performed with primers containing NGS sequences (KAPA HiFi HotStart ReadyMix, Kapa Biosystems, 12 cycles), and the library was cleaned using 0.8x SPRI beads.
Analysis of RNA‐seq data
RNA‐seq reads were aligned by STAR (Dobin et al, 2013) to the full genome sequences of Drosophila melanogaster (UCSC version dm3) and counted per annotated gene using ESET (End Sequencing analysis Toolkit; Derr et al, 2016). Differential expression analysis was performed using the edgeR R package. In the data from fly heads, we compared the two row RNAi lines to the control (Robinson et al, 2009). Only genes with a count per million (CPM) > 1 in at least 3 different samples were included in the analysis. Differentially expressed genes were selected by FDR < 0.05, with no fold‐change cutoff. The ranked list of differentially expressed genes based on FDR was used as an input for the enriched GO terms tool—Gorilla (Eden et al, 2009). The REVIGO tool (Supek et al, 2011) was used to remove redundant GO terms. In the data from S2 cells, we compared all treated samples to untreated samples. The FilterByExpr function in the edgeR R package was used to filter out nonexpressed genes (Dataset EV8). Differentially expressed genes were selected by FDR < 0.1, with no fold‐change cutoff. The associations between genes that were significantly differentially expressed with other lists of genes (For example genes bound by ROW or BEAF‐32 and BEAF‐32 long‐range targets) were tested using the Fisher's exact test.
Analysis of Hi‐C data
Hi‐C data were downloaded from NCBI: GSE97965 (Ramírez et al, 2018) and processed using HiCExplorer (Ramírez et al, 2018; Wolff et al, 2020). Each mate of the paired end was aligned separately with bowtie2 to the full genome sequence of Drosophila melanogaster (UCSC version dm3) with local and reorder parameters. The contact matrix was created using the hicBuildMatrix function of the HiCExplorer tool with a bin size of 1 kb and corrected using the hicCorrectMatrix function with Knight‐Ruiz (KR) method parameter.
To create the aggregation plots hicAggregateContacts function of the HiCExplorer tool was used. In the bed parameter, a bed file with the locations of the direct peaks of ROW was used, and in the BED2 parameter, a bed file with the locations of the indirect peaks of ROW was used. In the OperationType parameter, we chose the mean, and in the transform parameter, we chose obs/exp. The range was set to 10–500 kb. As a control, the same number of promotors not bound by ROW were selected randomly and used as input in the hicAggregateContacts function with the same parameters.
To create the aggregation plots for random sets of promoters unbound by ROW, and random sets of promoters bound directly and indirectly by ROW with matched gene expression levels, the same number of unbound and bound promoters were chosen with a similar distribution of gene expression. The plots represent the average of n = 100 random choices of promoters with matched gene expression levels. The values are Z‐scores of observed/expected values.
To identify over‐represented promoter interactions, we used the Hi‐C analysis tool PSYCHIC (Ron et al, 2017). A symmetric matrix for each chromosome in 5 kb resolution was created and used as input. UCSC refGene annotation file was used as the genes file input. We used the output bed file of over‐represented pairs with FDR value <0.05. The file contains genes and putative long‐range interaction location. Using the Fisher's exact test, we calculated the association between genes with a direct ROW peak (with AT motif) at the promoter region and putative long‐range interactions locations with an indirect ROW peak (without AT motif). Output file of random interactions with genes was used as a control.
Author contributions
Neta Herman: Formal analysis; investigation; methodology; writing—original draft. Sebastian Kadener: Conceptualization; supervision; funding acquisition; writing—review and editing. Sagiv Shifman: Conceptualization; formal analysis; supervision; funding acquisition; writing—review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Dataset EV10
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 5
PDF+
Acknowledgements
This work was supported by the Legacy Heritage Bio‐Medical Program of the Israel Science Foundation (Grant No. 839/10 to SS and SK) and by the Israel Science Foundation (grant no. 575/17 to SS).
EMBO reports (2022) 23: e54720
Contributor Information
Sebastian Kadener, Email: skadener@brandeis.edu.
Sagiv Shifman, Email: sagiv.shifman@mail.huji.ac.il.
Data availability
The datasets supporting the conclusions of this article are included within the article, and its additional files are available in the Gene Expression Omnibus database under accession number GSE169415 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE169415).
References
- Abel J, Eskeland R, Raffa GD, Kremmer E, Imhof A (2009) Drosophila HP1c is regulated by an auto‐regulatory feedback loop through its binding partner Woc. PLoS One 4: e5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME suite: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, MacHanick P (2012) Inferring direct DNA binding from ChIP‐seq. Nucleic Acids Res 40: e128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blecher‐Gonen R, Barnett‐Itzhaki Z, Jaitin D, Amann‐Zalcenstein D, Lara‐Astiaso D, Amit I (2013) High‐throughput chromatin immunoprecipitation for genome‐wide mapping of in vivo protein‐DNA interactions and epigenomic states. Nat Protoc 8: 539–554 [DOI] [PubMed] [Google Scholar]
- Brown JB, Boley N, Eisman R, May GE, Stoiber MH, Duff MO, Booth BW, Wen J, Park S, Suzuki AM et al (2014) Diversity and dynamics of the Drosophila transcriptome. Nature 512: 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chathoth KT, Mikheeva LA, Crevel G, Wolfe JC, Hunter I, Beckett‐Doyle S, Cotterill S, Dai H, Harrison A & Zabet NR (2021) The role of insulators and transcription in 3D chromatin organisation of flies. bioRxiv 10.1101/2021.04.26.441424 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales M, Rosado A, Cortini R, Van Arensbergen J, Van Steensel B, Filion GJ (2017) Clustering of Drosophila housekeeping promoters facilitates their expression. Genome Res 27: 1153–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvier O, Hart CM, Käs E, Laemmli UK (2002) Identification of a multicopy chromatin boundary element at the borders of silenced chromosomal domains. Chromosoma 110: 519–531 [DOI] [PubMed] [Google Scholar]
- Cuvier O, Hart CM, Laemmli UK (1998) Identification of a class of chromatin boundary elements. Mol Cell Biol 18: 7478–7486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derr A, Yang C, Zilionis R, Sergushichev A, Blodgett DM, Redick S, Bortell R, Luban J, Harlan DM, Kadener S et al (2016) End sequence analysis toolkit (ESAT) expands the extractable information from single‐cell RNA‐seq data. Genome Res 26: 1397–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Mauro G, Carbonell A, Escudero‐Ferruz P, Azorín F (2020) The zinc‐finger proteins WOC and ROW play distinct functions within the HP1c transcription complex. Biochim Biophys Acta Gene Regul Mech 1863: 194492 [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissenberg JC, Elgin SCR (2014) HP1a: a structural chromosomal protein regulating transcription. Trends Genet 30: 103–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emberly E, Blattes R, Schuettengruber B, Hennion M, Jiang N, Hart CM, Käs E, Cuvier O (2008) BEAF regulates cell‐cycle genes through the controlled deposition of H3K9 methylation marks into its conserved dual‐core binding sites. PLoS Biol 6: e327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Font‐Burgada J, Rossell D, Auer H, Azorín F (2008) Drosophila HP1c isoform interacts with the zinc‐finger proteins WOC and relative‐of‐WOC to regulate gene expression. Genes Dev 22: 3007–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuda NJ, Guertin MJ, Sharma S, Danko CG, Martins AL, Siepel A, Lis JT (2015) GAGA factor maintains nucleosome‐free regions and has a role in RNA polymerase II recruitment to promoters. PLoS Genet 11: 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaertner B, Zeitlinger J (2014) RNA polymerase II pausing during development. Development 141: 1179–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist DA, Nechaev S, Lee C, Ghosh SKB, Collins JB, Li L, Gilmour DS, Adelman K (2008) NELF‐mediated stalling of pol II can enhance gene expression by blocking promoter‐proximal nucleosome assembly. Genes Dev 22: 1921–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruharsha KG, Rual JF, Zhai B, Mintseris J, Vaidya P, Vaidya N, Beekman C, Wong C, Rhee DY, Cenaj O et al (2011) A protein complex network of Drosophila melanogaster . Cell 147: 690–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutin J, Sadeh R, Bodenheimer N, Joseph‐Strauss D, Klein‐Brill A, Alajem A, Ram O, Friedman N (2018) Fine‐resolution mapping of TF binding and chromatin interactions. Cell Rep 22: 2797–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberle V, Arnold CD, Pagani M, Rath M, Schernhuber K, Stark A (2019) Transcriptional cofactors display specificity for distinct types of core promoters. Nature 570: 122–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart CM, Zhao K, Laemmli UK (1997) The scs' boundary element: characterization of boundary element‐associated factors. Mol Cell Biol 17: 999–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurteau A, Perrois C, Depierre D, Fosseprez O, Humbert J, Schaak S, Cuvier O (2020) Insulator‐based loops mediate the spreading of H3K27me3 over distant micro‐domains repressing euchromatin genes. Genome Biol 21: 193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Vinayagam A, Nand A, Comjean A, Chung V, Hao T, Mohr SE, Perrimon N (2018) Molecular interaction search tool (MIST): an integrated resource for mining gene and protein interaction data. Nucleic Acids Res 46: D567–D574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SA, Khorasanizadeh S (2002) Structure of HP1 chromodomain bound to a lysine 9‐methylated histone H3 tail. Science 295: 2080–2083 [DOI] [PubMed] [Google Scholar]
- James TC, Elgin SC (1986) Identification of a nonhistone chromosomal protein associated with heterochromatin in Drosophila melanogaster and its gene. Mol Cell Biol 6: 3862–3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N, Emberly E, Cuvier O, Hart CM (2009) Genome‐wide mapping of boundary element‐associated factor (BEAF) binding sites in Drosophila melanogaster links BEAF to transcription. Mol Cell Biol 29: 3556–3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Sun X, Song Q (2005) Woc gene mutation causes 20E‐dependent alpha‐tubulin detyrosination in Drosophila melanogaster . Arch Insect Biochem Physiol 60: 116–129 [DOI] [PubMed] [Google Scholar]
- Jonge WJ, O'Duibhir E, Lijnzaad P, Leenen D, Groot Koerkamp MJ, Kemmeren P, Holstege FC (2017) Molecular mechanisms that distinguish TFIID housekeeping from regulatable SAGA promoters. EMBO J 36: 274–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juven‐Gershon T, Kadonaga JT (2010) Regulation of gene expression via the core promoter and the basal transcriptional machinery. Dev Biol 339: 225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler R, Tisserand J, Font‐Burgada J, Reina O, Coch L, Attolini CSO, Garcia‐Bassets I, Azorín F (2015) DDsk2 regulates H2Bub1 and RNA polymerase II pausing at dHP1c complex target genes. Nat Commun 6: 7049 [DOI] [PubMed] [Google Scholar]
- Klein‐Brill A, Joseph‐Strauss D, Appleboim A, Friedman N (2019) Dynamics of chromatin and transcription during transient depletion of the RSC chromatin remodeling complex. Cell Rep 26: 279–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A et al (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SH, Florens L, Swanson SK, Washburn MP, Abmayr SM, Workman JL (2010) Heterochromatin protein 1 (HP1) connects the FACT histone chaperone complex to the phosphorylated CTD of RNA polymerase II. Genes Dev 24: 2133–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Ryu HW, Kim GW, Kwon SH (2019) Comparison of three heterochromatin protein 1 homologs in Drosophila . J Cell Sci 132: jcs222729 [DOI] [PubMed] [Google Scholar]
- Liang J, Lacroix L, Gamot A, Cuddapah S, Queille S, Lhoumaud P, Lepetit P, Martin PGP, Vogelmann J, Court F et al (2014) Chromatin immunoprecipitation indirect peaks highlight long‐range interactions of insulator proteins and pol II pausing. Mol Cell 53: 672–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maimon I, Popliker M, Gilboa L (2014) Without children is required for stat‐mediated zfh1 transcription and for germline stem cell differentiation. Development 141: 2602–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattout A, Aaronson Y, Sailaja BS, Raghu Ram EV, Harikumar A, Mallm JP, Sim KH, Nissim‐Rafinia M, Supper E, Singh PB et al (2015) Heterochromatin protein 1β (HP1β) has distinct functions and distinct nuclear distribution in pluripotent versus differentiated cells. Genome Biol 16: 213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally JC, Müller WG, Walker D, Wolford R, Hager GL (2000) The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 287: 1262–1265 [DOI] [PubMed] [Google Scholar]
- Menet JS, Abruzzi KC, Desrochers J, Rodriguez J, Rosbash M (2010) Dynamic PER repression mechanisms in the Drosophila circadian clock: from on‐DNA to off‐DNA. Genes Dev 24: 358–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourad R, Cuvier O (2016) Computational identification of genomic features that influence 3D chromatin domain formation. PLoS Comput Biol 12: e1004908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazer E, Dale RK, Palmer C, Lei EP (2018) Argonaute2 attenuates active transcription by limiting RNA polymerase II elongation in Drosophila melanogaster . Sci Rep 8: 15685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngoc LV, Kassavetis GA, Kadonaga JT (2019) The RNA polymerase II core promoter in Drosophila . Genetics 212: 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa R‐S, Nagao K, Masuda H‐T, Iwasaki O, Hirota T, Nozaki N, Kimura H, Obuse C (2010) Human POGZ modulates dissociation of HP1alpha from mitotic chromosome arms through Aurora B activation. Nat Cell Biol 12: 719–727 [DOI] [PubMed] [Google Scholar]
- Ong CT, Van Bortle K, Ramos E, Corces VG (2013) Poly(ADP‐ribosyl)ation regulates insulator function and intrachromosomal interactions in Drosophila . Cell 155: 148–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudelaar AM, Higgs DR (2020) The relationship between genome structure and function. Nat Rev Genet 22: 154–168 [DOI] [PubMed] [Google Scholar]
- Persikov AV, Singh M (2014) De novo prediction of DNA‐binding specificities for Cys2His 2 zinc finger proteins. Nucleic Acids Res 42: 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rach EA, Winter DR, Benjamin AM, Corcoran DL, Ni T, Zhu J, Ohler U (2011) Transcription initiation patterns indicate divergent strategies for gene regulation at the chromatin level. PLoS Genet 7: e1001274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffa GD, Cenci G, Siriaco G, Goldberg ML, Gatti M (2005) The putative Drosophila transcription factor woc is required to prevent telomeric fusions. Mol Cell 20: 821–831 [DOI] [PubMed] [Google Scholar]
- Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J, Habermann B, Akhtar A, Manke T (2018) High‐resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun 9: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray P, De S, Mitra A, Bezstarosti K, Demmers JAA, Pfeifer K, Kassis JA (2016) Combgap contributes to recruitment of Polycomb group proteins in Drosophila . Proc Natl Acad Sci U S A 113: 3826–3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle NC, Minoda A, Kharchenko PV, Alekseyenko AA, Schwartz YB, Tolstorukov MY, Gorchakov AA, Jaffe JD, Kennedy C, Linder‐Basso D et al (2011) Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin. Genome Res 21: 147–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2009) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron G, Globerson Y, Moran D, Kaplan T (2017) Promoter‐enhancer interactions identified from hi‐C data using probabilistic models and hierarchical topological domains. Nat Commun 8: 2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Gilbert MK, Hart CM (2007) Characterization of BEAF mutations isolated by homologous recombination in Drosophila . Genetics 176: 801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G (2012) Three‐dimensional folding and functional organization principles of the Drosophila genome. Cell 148: 458–472 [DOI] [PubMed] [Google Scholar]
- Shishkin AA, Giannoukos G, Kucukural A, Ciulla D, Busby M, Surka C, Chen J, Bhattacharyya RP, Rudy RF, Patel MM et al (2015) Simultaneous generation of many RNA‐seq libraries in a single reaction. Nat Methods 12: 323–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stessman HAF, Willemsen MH, Fenckova M, Penn O, Hoischen A, Xiong B, Wang T, Hoekzema K, Vives L, Vogel I et al (2016) Disruption of POGZ is associated with intellectual disability and autism Spectrum disorders. Am J Hum Genet 98: 541–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suliman‐Lavie R, Title B, Cohen Y, Hamada N, Tal M, Tal N, Monderer‐Rothkoff G, Gudmundsdottir B, Gudmundsson KO, Keller JR et al (2020) Pogz deficiency leads to transcription dysregulation and impaired cerebellar activity underlying autism‐like behavior in mice. Nat Commun 11: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T (2011) Revigo summarizes and visualizes long lists of gene ontology terms. PLoS One 6: e21800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tianfang Ge D, Tipping C, Brodsky MH, Zamore PD (2016) Rapid screening for CRISPR‐directed editing of th Drosophila genome using white coconversion. G3 Genes Genomes Genet 6: 3197–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Cox J (2016a) The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat Protoc 11: 2301–2319 [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016b) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Ulianov SV, Khrameeva EE, Gavrilov AA, Flyamer IM, Kos P, Mikhaleva EA, Penin AA, Logacheva MD, Imakaev MV, Chertovich A et al (2016) Active chromatin and transcription play a key role in chromosome partitioning into topologically associating domains. Genome Res 26: 70–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakoc CR, Mandat SA, Olenchock BA, Blobel GA (2005) Histone H3 lysine 9 methylation and HP1γ are associated with transcription elongation through mammalian chromatin. Mol Cell 19: 381–391 [DOI] [PubMed] [Google Scholar]
- Vermaak D, Henikoff S, Malik HS (2005) Positive selection drives the evolution of rhino, a member of the heterochromatin protein 1 family in Drosophila . PLoS Genet 1: 96–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermaak D, Malik HS (2009) Multiple roles for heterochromatin protein 1 genes in Drosophila . Annu Rev Genet 43: 467–492 [DOI] [PubMed] [Google Scholar]
- Vogelmann J, Le Gall A, Dejardin S, Allemand F, Gamot A, Labesse G, Cuvier O, Nègre N, Cohen‐Gonsaud M, Margeat E et al (2014) Chromatin insulator factors involved in long‐range DNA interactions and their role in the folding of the Drosophila genome. PLoS Genet 10: e1004544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren JT, Wismar J, Subrahmanyam B, Gilbert LI (2001) Woc (without children) gene control of ecdysone biosynthesis in Drosophila melanogaster . Mol Cell Endocrinol 181: 1–14 [DOI] [PubMed] [Google Scholar]
- Wismar J, Habtemichael N, Warren JT, Dai J‐D, Gilbert LI, Gateff E (2000) The mutation without childrenrgl causes Ecdysteroid deficiency in third‐instar larvae of Drosophila melanogaster . Dev Biol 226: 1–17 [DOI] [PubMed] [Google Scholar]
- Wolff J, Rabbani L, Gilsbach R, Richard G, Manke T, Backofen R, Grüning BA (2020) Galaxy HiCExplorer 3: a web server for reproducible hi‐C, capture hi‐C and single‐cell hi‐C data analysis, quality control and visualization. Nucleic Acids Res 48: W177–W184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, He QY (2015) ChIP seeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31: 2382–2383 [DOI] [PubMed] [Google Scholar]
- Zabidi MA, Arnold CD, Schernhuber K, Pagani M, Rath M, Frank O, Stark A (2015) Enhancer‐core‐promoter specificity separates developmental and housekeeping gene regulation. Nature 518: 556–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger J, Stark A, Kellis M, Hong JW, Nechaev S, Adelman K, Levine M, Young RA (2007) RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet 39: 1512–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Wang D, Sun F (2011) Drosophila melanogaster heterochromatin protein HP1b plays important roles in transcriptional activation and development. Chromosoma 120: 97–108 [DOI] [PubMed] [Google Scholar]
- Zhao K, Hart CM, Laemmli UK (1995) Visualization of chromosomal domains with boundary element‐associated factor BEAF‐32. Cell 81: 879–889 [DOI] [PubMed] [Google Scholar]
- Zhu LJ, Gazin C, Lawson ND, Pagès H, Lin SM, Lapointe DS, Green MR (2010) ChIPpeakAnno: a bioconductor package to annotate ChIP‐seq and ChIP‐chip data. BMC Bioinformatics 11: 237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]