

Abstract
Extreme stress can cause long-lasting changes in affective behavior manifesting in conditions such as post-traumatic stress disorder (PTSD). Understanding the biological mechanisms that govern trauma-induced behavioral dysregulation requires reliable and rigorous pre-clinical models that recapitulate multiple facets of this complex disease. For decades, Pavlovian fear conditioning has been a dominant paradigm for studying the effects of trauma through an associative learning framework. However, severe stress also causes long-lasting nonassociative fear sensitization, which is often overlooked in Pavlovian fear conditioning studies. This paper synthesizes recent research on the stress-enhanced fear learning (SEFL) paradigm, a valuable rodent model that can dissociate associative and nonassociative effects of stress. We discuss evidence that the SEFL paradigm produces nonassociative fear sensitization that is distinguishable from Pavlovian fear conditioning. We also discuss key biological variables, such as age and sex, neural circuit mechanisms, and crucial gaps in knowledge. We argue that nonassociative fear sensitization deserves more attention within current PTSD models and that SEFL provides a valuable complement to Pavlovian conditioning research on trauma-related pathology.
Keywords: Stress, Fear, Associative, PTSD, fear sensitization, Nonassociative Learning
Introduction:
Stressful experiences are common in every daily life, and most people will undergo at least one traumatic event throughout their lifetime (as defined by DSM-5 criteria) (Kilpatrick et al., 2013). Although stress-associated fear responses are natural emotional reactions that protect us from imminent danger, exposure to extreme stress sometimes leads to pathological states where fear and anxiety are excessive and inappropriate to a given cue or reminder. PTSD patients, for instance, exhibit sustained (>30 days) multimodal dysfunctions in cognition, attention, and information processing after exposure to extremely traumatic experiences such as assault, accident, natural disaster, or combat, with core symptoms including re-experiencing of trauma, avoidance, and hyperarousal. PTSD is highly prevalent among American adults, with around 12 million cases per year, with women at two times higher risk than men (National Institute of Health) and an estimated 20–30% prevalence among returning war veterans (US Department of Veterans Affairs), with a substantial economic burden associated with healthcare costs and lost wages. Although efforts to uncover neurobiological mechanisms by which severe stress leads to debilitating symptoms of PTSD have been ongoing for years, current treatments have limited effectiveness, and there is a need for additional therapeutic strategies to address the full range of PTSD symptoms.
Mechanisms and treatments of PTSD have commonly been investigated in rodents using Pavlovian fear conditioning, in which animals receive pairings between a neutral cue or context (Conditioned stimulus, CS) and a painful footshock (Unconditioned stimulus, US). Upon re-exposure to the cue or context paired with the US, animals exhibit conditioned fear responses, suggesting that they have formed an association between the CS and the US. Studies employing Pavlovian fear conditioning have yielded many important insights about the neural mechanisms of fear learning and psychological processes—such as fear acquisition, generalization, and extinction—that are often perturbed in PTSD.
A central theme of the present review is that fear learning relevant to PTSD also occurs via a second mechanism often overlooked in Pavlovian fear conditioning studies: fear sensitization, which we define as a nonassociative enhancement of defensive responding. Fear sensitization is nonassociative because it does not depend on a CS and US association. A classic example of fear sensitization is provided in Kandel’s studies in an invertebrate, the Aplysia (Pinsker et al., 1973). Kandel and colleagues observed that repeated shocks applied to the siphon of Aplysia caused a long-term enhancement of the defensive gill withdrawal response. Importantly, the enhancement did not require pairings between a CS and US; repeated delivery of the US (shock) alone was sufficient to induce long-term sensitization. Furthermore, the enhancement of the gill withdrawal response was general in the sense that a variety of different stimuli could elicit it. While much is known about the neural mechanisms of associative fear conditioning, the mechanisms of mammalian nonassociative fear sensitization is just beginning to be unraveled.
This review will focus on a robust rodent model of fear sensitization that is beginning to provide important insights into the mechanisms through which stress exposure can cause long-lasting enhancements in defensive responding and fear learning. Originally developed by Michael Fanselow and colleagues (Rau et al., 2005), stress-enhanced fear learning (SEFL) was designed to examine fear sensitization and has now evolved to model aspects of PTSD. SEFL procedures typically consist of 3 sessions (Fig 1). In the first session, rodents are exposed to a stressor, usually a footshock, in a conditioning chamber, which we will call Context A. In the second session, subjects receive mild contextual fear conditioning in a different conditioning chamber (Context B) that typically consists of a single footshock presentation. Finally, in the third session, freezing to Context B is tested. The typical effect is that stress exposure on the first day enhances subsequent fear conditioning to Context B. As will be discussed below, available evidence suggests that this enhancement of fear conditioning is nonassociative and does not require the formation nor retrieval of an associative fear memory resulting from stress exposure.
Figure 1.
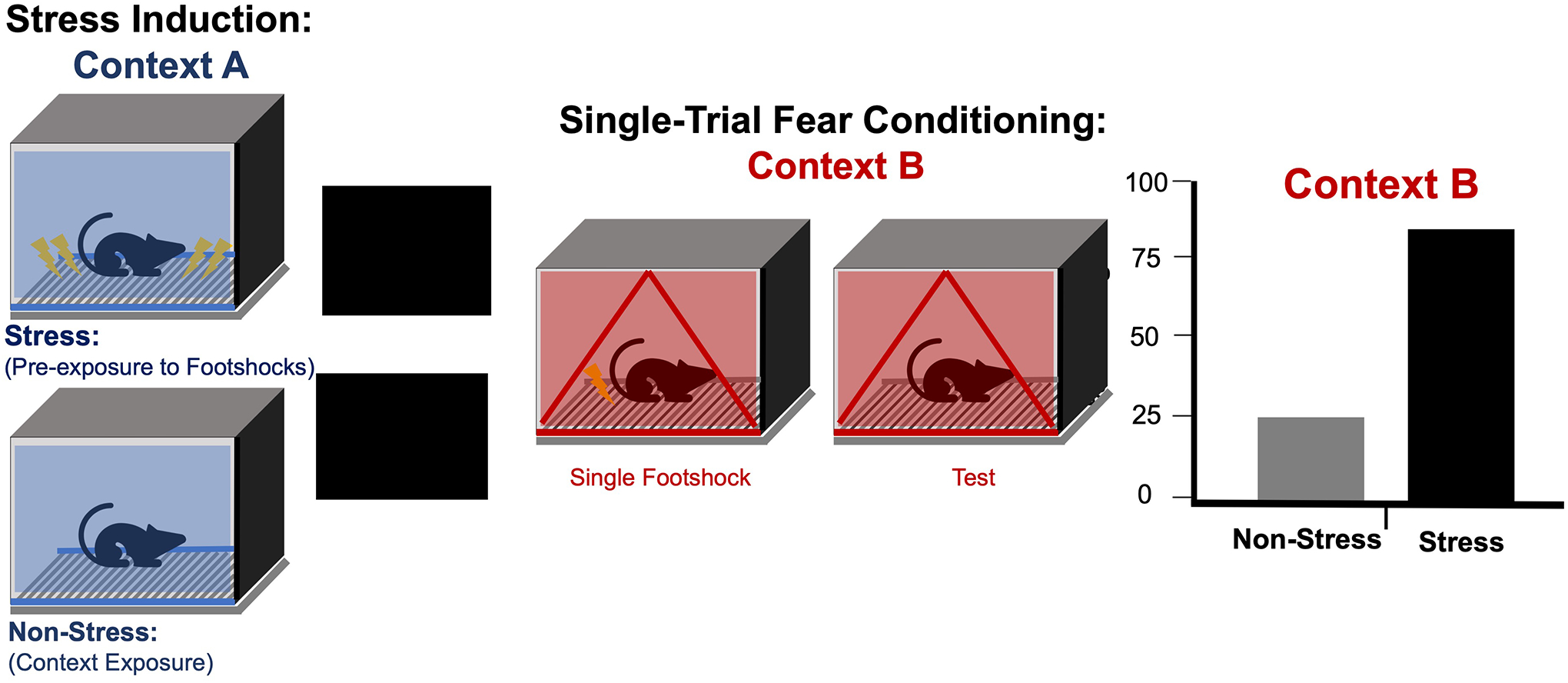
General procedures and outcomes for stress enhancement of fear learning (SEFL). Stress-induction: mice or rats receive repeated or no footshocks in context A. Single-trial fear conditioning: the next day (or weeks later) all subjects, receive a single footshock after a few minutes in a novel context B, and 24 hrs later are returned to context B to assess freezing. As depicted in the simulated bar graph, the group previously exposed to stress exhibits greater freezing than the non-stressed group in context B.
The term “stress-enhanced fear learning” is now used in a variety of ways throughout the literature to describe the original behavioral paradigm, its outcome, or any occurrence of increased fear conditioning resulting from some form of stress. To avoid confusion, we will use the term “SEFL procedure” to refer to the behavioral paradigm developed by Fanselow and colleagues as summarized above. We will use the abbreviation “SEFL” to refer to the behavioral outcome of enhanced fear learning. Other behavioral phenotypes induced by the SEFL procedure will also be described.
Although there is extensive literature on stress effects on fear, anxiety and related behaviors, this review will focus primarily on studies that employed the SEFL procedure. We focus on this SEFL procedure because of our interest in addressing nonassociative mechanisms and because of strong evidence (discussed in subsequent sections) that this procedure differentiates associative and nonassociative consequences of stress. In contrast, these processes are often challenging to dissociate in other stress paradigms. While we recognize that other animal models of stress can enhance fear learning, it is often unknown whether this enhancement is nonassociative in nature. Moreover, there exist excellent reviews broadly examining the influence of stress on affective behavior and fear learning (Aubry et al., 2016; Bangasser & Shors, 2010; Musazzi et al., 2018; Raio & Phelps, 2015).
The goal of this review is to highlight recent progress on the SEFL procedure and address the following key questions: (1) What is the evidence that SEFL can be used to distinguish the associative and nonassociative contributions of stress? (2) What are the neural mechanisms through which footshock stress enhances future fear learning? And (3) to what extent can SEFL be considered a model of PTSD symptoms?
The SEFL procedure
In SEFL, exposure to traumatic stress enhances future fear responses, as measured by Pavlovian fear conditioning to a novel context. SEFL was first demonstrated in studies showing that a history of prior footshocks in a novel context led to enhanced fear conditioning in a different context (Fanselow & Bolles, 1979). The modern SEFL procedure occurs in a fear conditioning apparatus, where mice or rats are given a series of footshocks during a single session for stress induction or “trauma” in Context A (Rajbhandari et al., 2018). This stress group is run alongside a non-stress control group that receives Context A exposure alone. One day or more following the stress session, all groups are given a single footshock in a novel context, termed Context B, where the layout, flooring, scent, lighting, and transport are different from Context A. On the third day, rodents receive a test of conditioned fear in Context B. Rodents that received prior footshock stress show significantly higher levels of conditioned fear to Context B than the non-stress control group, as measured by freezing response. This enhanced or sensitized fear learning is long-lasting: The SEFL effect has been observed when the latency between Context A stress and Context B conditioning is as long as 90 days. One of the advantages of SEFL over other stress procedures is that footshock provides a form of stress that is well-defined, temporally discrete, and can be varied parametrically by the experimenter. In rat studies, using 15 footshocks produces robust SEFL, with almost 92% of rats showing subsequent fear learning enhancement. When stress intensity is reduced by decreasing the number of footshocks to 4 or 5, SEFL exhibits a pronounced bimodal distribution, allowing stress susceptibility factors to be investigated (Gonzalez et al., 2021).
Multifaceted Nature of SEFL
Exposure to stress is believed to induce both associative and nonassociative learning. For instance, during stress exposure in Context A, rodents likely acquire associative fear of Context A. At the same time, this initial stress exposure may alter defensive responses in a nonassociative manner, thereby enhancing fear responses to cues or contexts not associated with the original context A. In this section, we evaluate evidence that these enhanced fear responses reflect nonassociative learning rather than associative.
Potential Associative Accounts of SEFL
Associative accounts of SEFL assume that exposure to repeated footshock stress establishes an associative contextual fear memory that may be partially retrieved during subsequent fear conditioning in a novel context. There are two mechanisms through which this partial retrieval could enhance fear learning: generalization between the stress and novel context or an update in the CS-US associative value.
According to the generalization account, SEFL reflects the summation between associative fear generalized from the stress context and de novo fear conditioning in the new context. While multiple-shock fear conditioning like that used in SEFL produces significant generalized fear (Poulos et al., 2016a), there are several reasons to discount the generalization account. First, the extinction of context fear to the stress context does not mitigate the sensitization of fear to a novel context (Hassien et al., 2020; Long & Fanselow, 2012; Rau et al., 2005). Moreover, the resulting associative memory of stress induction is required for neither the induction nor maintenance of SEFL. Enhanced fear learning persists even after pharmacological disruption of the original associative memory of the stress context via NMDA antagonism or when stress occurs in early life and no associative context fear memory is established (Poulos et al., 2014; Rau et al., 2005). Furthermore, if SEFL resulted simply from the summation of total associative fear, then SEFL should be observed even when the order of the SEFL procedure is reversed, such that 1-shock conditioning (in Context B) precedes footshock stress (in Context A). However, the SEFL effect is not observed under this condition (Rau et al., 2005), indicating that summation of associative strengths between contexts A and B is not sufficient to generate a SEFL-like effect.
Another potential interpretation of SEFL is that exposure to repeated footshock stress could inflate the remembered value (intensity) of the shock US (Rescorla, 1974), thereby enhancing the expression of fear in Context B. US inflation is observed when a CS is paired with a weak footshock and followed (or preceded by) a series of strong unsignaled footshocks, resulting now in the CS eliciting greater fear. If SEFL simply reflected US inflation, then SEFL should not be observed when the footshock stress and 1-shock conditioning US are of the same intensity. However, the standard use of a single shock intensity for stress induction in Context A and 1-shock conditioning in Context B produces the hallmark SEFL effect (Rau et al., 2005). Collectively, this evidence diminishes the potential role of generalization of “trauma” memory or revaluation of the US as mechanisms explaining the SEFL effect.
A related interpretation is that SEFL reflects integration of the original stress context-shock associative memory with a new context-shock association. This interpretation is supported by evidence that even weak footshock can enhance the effects of prior context fear conditioning (Ferrara et al., 2019; Poulos et al., 2015), perhaps through reconsolidation-dependent modification of the original stress memory. In addition, Jarome et al. (2015) showed that novel contexts have a greater ability than previously conditioned contexts to drive the reconsolidation of memory for an auditory cue-footshock association, raising the possibility that exposure to shock in Context B could reactivate the Context A memory through a shared shock representation, leading to memory updating in the group that received prior shock. However, this interpretation is unlikely, given that preventing the formation of original associative memory fails to preclude the stress-enhancement of fear learning (Rau et al. 2005).
Parallels between SEFL and other forms of sensitization
Groves and Thompson characterized sensitization as part of a dual-process account of habituation, where repeated presentations of a strong stimulus can promote a nonassociative short- (minutes-hours) and long-term (weeks) increase in responding to the same or different stimulus modality when applied to identical or distinct bodily locations (Groves & Thompson, 1970)(Groves & Thompson, 1970). Kandel and associates, in several elegant studies in Aplysia, characterized the parameters that promote the long-term sensitization of the gill-withdrawal reflex (Kandel, 2001). When viewed through the lens of sensitization, two key predictions about SEFL can be made: 1) the induction and persistence of stress-enhanced fear learning should be shock intensity- and number-dependent (Dijken et al., 1992; Frost et al., 1985; Pinsker et al., 1973); 2) stimuli different from those used in stress induction should be capable of eliciting a sensitized reaction (Thompson & Spencer, 1966).
It is well-established in Aplysia that the number and intensity of tail shocks modulate the induction of short- and long-term sensitization (Hawkins et al., 2006; Kandel, 2001; Pinsker et al., 1973). For example, a single tail shock of intermediate or strong intensity can induce short-term sensitization, which persists from minutes to an hour (Marcus et al., 1988), while a single session of 4 tail shocks (intermediate intensity) can produce sensitization that persists for 24 hrs. Conversely, four sessions of 4 tail shocks can result in a long-term sensitization that can persist across several days (Frost et al., 1985). Consistent with sensitization in Aplysia, early work on SEFL in rodents identified that a single session of 4 or 15, but not one footshock, can promote a long-lasting enhancement of fear learning when measured 3 days later (Rau & Fanselow, 2009). Furthermore, Rau and Fanselow (2009), using a 15-footshock stress induction procedure with shocks set to 1mA, found SEFL to persist unabatedly across a 3-month interval. The use of a more intense footshock (1.2 mA) has been reported to produce the SEFL effect with a single footshock, but this effect appears to weaken after 4 weeks (Poulos et al., 2016b). More recent work by Gonzalez et al (2021) found that a 4-footshock stress induction procedure set at 1 mA in rats led to patterns of fear learning that followed a bimodal distribution, where nearly half of all subjects failed to exhibit SEFL. In this same study, nearly all subjects that underwent a 15-footshock procedure exhibited SEFL. Further parametric analysis of footshock stress intensity in both rats and mice is warranted and may extend our understanding of the long-term nonassociative nature of SEFL.
A second feature of sensitization is that it is stimulus-general. Early work described sensitization as the dishabituation of a previously habituated response, resulting from a strong or distinct stimulus presentation (Thompson & Spencer, 1966). However, it is now recognized that the dishabituation effect is simply an instance of sensitization elicited by a distinct robust stimulus (Groves & Thompson, 1970). Pre-exposure to repeated footshock sensitizes fear responding to future footshock and potentiates responding to other strong stimuli. For example, footshock stress also potentiates freezing to high decibel tone and white noise stimuli (Hassien et al., 2020; Perusini et al., 2016) and enhances reactivity to white noise in the stress context (Hoffman et al., 2022).
The above findings support the conclusion that the enhancement of fear learning caused by prior footshock stress reflects nonassociative sensitization. However, enhancing fear learning is just one of numerous consequences of footshock stress exposure. Whether these other related consequences of stress reflect associative or nonassociative learning remains to be determined. For instance, the induction of SEFL with repeated footshocks, like other stress procedures, can promote short- and long-term changes in anxiety- and depressive-like phenotypes. Perusini and colleagues (2016) demonstrated that SEFL induction in rats could produce an anxiety-like change in exploratory behaviors in a modified open field (Perusini et al., 2016). Interestingly, Hassien et al (2020) showed in mice that SEFL induction decreased locomotion in the open field, an effect that was abolished by the extinction of conditioned fear to the stress induction context and suggested that this stress-enhanced anxiety-like behavior is mediated by associative memory related to the stress context. However, while the above results were obtained in adult stressed animals, infant stressed rats exhibit long-lasting increases in the elevated plus maze as adults even in the absence of the stress induction memory, suggesting nonassociative contributions (Poulos et al., 2014).
Further parametric analysis of the effects of footshock stress intensity in both rats and mice is warranted and may extend the current understanding of the nonassociative nature of SEFL. For instance, varying the intensity or number of footshocks during stress induction could dissociate potential short-term versus long-term SEFL effects. Indeed, as briefly mentioned above, a recent study showed that using 4 instead of 15 shocks reveals variations in resiliency/susceptibility to the development of SEFL, anxiety-like behavior, and alcohol intake (Gonzalez et al., 2021).
Sex differences and SEFL
As mentioned previously, stress- and trauma-related disorders like PTSD are more common in women than men, yet most studies of SEFL have focused on adult male rats and mice. This is particularly interesting because the initial evidence for SEFL was generated in adult female rats (Fanselow & Bolles, 1979). To date, only a few studies have compared SEFL between adult male and female rodents. In one study, while no sex differences in fear sensitization were evident, the study replicated well-established sex differences in context fear conditioning, whereby males exhibit greater context fear learning than females (Hassien et al., 2020). Subsequently, Gonzalez et al (2021) reported that male and female rats show similar levels of stress susceptibility in SEFL. Hassien et al. (2020), in mice, failed to find evidence of a sex difference in SEFL. Still, they identified in unstressed mice that females exhibit greater nonassociative freezing to a novel robust tone than males (Hassien et al., 2020). The absence of a sex difference in SEFL suggests the possibility that sex differences in PTSD prevalence may be unrelated to nonassociative fear sensitization. However, it is important to recognize that only a small portion of the SEFL parameter space has been explored to date. Further parametric manipulation of features such as shock intensity, shock number, predictability, and testing interval may yield additional information on sex differences in the rodent models.
Developmental Aspects of SEFL
The developmental status of subjects may confer another biological variable of importance in SEFL. The effects of early-life stress are well-documented and can produce life-long alterations in PTSD-related behaviors (Binder et al., 2008; Pratchett & Yehuda, 2011). In the rat, associative aversive memories between olfactory and shock stimuli emerge early in infancy at approximately 10 days of age (Sullivan et al., 2000), while context fear learning does not generally emerge until weaning or >21 days of age (Rudy & Morledge, 1994; Santarelli et al., 2018). Given the specific ontogenic timeline of fear conditioning, important insights may be identified by determining the developmental emergence of SEFL. To date, only a handful of studies have addressed how SEFL manifests in infant, juvenile, and adolescent rodents (Poulos et al., 2014; Quinn et al., 2013). One study revealed that footshock stress in infant male rats produces long-lasting fear sensitization, increased anxiety-like behavior, and disruption of circadian-controlled corticosterone levels that persist into adulthood (Poulos et al., 2014). Similarly, Quinn et. al. demonstrated that fear sensitization that persists into adulthood emerges as early as infancy and pre-adolescence (Quinn et al., 2013) Lastly, adult mice that develop fear sensitization following infantile footshock stress do not exhibit alterations in object recognition, reversal, and discrimination learning, suggesting that infantile trauma does not broadly influence hippocampal- and striatal-dependent tasks but may be restricted to amygdala-related processes (Sneddon et al., 2021). Collectively, these findings reveal that stress exposure during infancy can promote long-lasting adult fear sensitization. Still, to what extent SEFL sensitivity varies across developmental stages remains to be established. Future studies could shed light upon potential sensitive periods for the induction and persistence of SEFL throughout development.
Hormonal outcomes in SEFL
Exposure to a threat, such as during stress induction, can induce a cascade of highly conserved biological processes that facilitate physiological and behavioral adaptation to threat (Joëls & Baram, 2009). This process, referred to as the stress response, is achieved through the coordinated actions of multiple physiological systems interacting with many bodily organs, including the brain (McEwen et al., 2015). Upon encountering a physical stressor, such as footshock, or a psychological stressor, such as a Pavlovian CS, can activate the adrenal glands and engage the autonomic and central nervous system to support adaptive behavioral responses and mental states, such as defensive behavior and cognitive arousal. Stress responses involve the sequential release of hormones corticotrophin-releasing factor (CRF) and adrenocorticotrophic hormone (ACTH) and parallel release of norepinephrine that in turn alter the sympathetic tone, arousal, and peripheral release of other neuromodulators (Sapolsky et al., 2000).
In addition, stress exposure induces secretion of glucocorticoid (GC) stress hormones that exert genomic and non-genomic influence on cellular homeostasis primarily through low affinity glucocorticoid receptors (GR) and high affinity mineralocorticoid receptors (MR) (Reul & Kloet, 1986). Within the brain, GRs are highly expressed in limbic regions. Their activation supports a wide range of essential functions, including learning and memory, emotional processing, neuronal recovery following injury, and termination of the HPA-axis response via a negative feedback loop (Kloet & Reul, 1987; McEwen et al., 2015; Meaney et al., 1985).
Perusini and colleagues examined the role of glucocorticoid stress hormones in SEFL (Perusini et al., 2016). The authors systemically attenuated the endogenous synthesis of corticosterone (CORT) via the administration of metyrapone in rats undergoing footshock stress. Metyrapone dose-dependently reduced SEFL and associative fear memory to the stress context. Thus, CORT synthesis is necessary for both the associative and nonassociative consequences of stress evident in this model. Interestingly, the co-administration of metyrapone and exogenous CORT prior to footshock stress restored SEFL but failed to restore associative fear memory to the stress context. This dissociation suggests that CORT influences associative and nonassociative fear through distinct neural pathways. In addition, administration of CORT in the absence of footshock stress is not sufficient to produce SEFL, indicating that additional mechanisms activated by stress interact with CORT to induce sensitization.
A large body of evidence suggests that exposure to intense chronic or acute stressors causes prolonged sensitization of HPA-axis responses to subsequent novel (heterotypic) threats (Armario et al., 2009; Sabban & Serova, 2009). Given that adrenalectomy attenuates while administration of CORT facilitates Pavlovian fear conditioning, one attractive possibility is that SEFL is mediated by increased CORT secretion during post-stress induction (Monsey et al., 2014; Pugh et al., 1997). However, Perusini and colleagues reported that administration of metyrapone to stressed animals prior to conditioning in a novel context or a context fear test session failed to attenuate SEFL (Perusini et al., 2016). In addition, blood plasma CORT levels measured immediately after the context fear test session in the single-shocked context yielded no significant differences between stressed and unstressed animals, despite differences in freezing behavior. These results corroborate previous reports showing that administration of metyrapone during fear conditioning fails to prevent enhanced fear learning that is typically observed in chronically stressed rat (Kulp et al., 2020). Furthermore, evidence suggests that stress-induced sensitization of the HPA-axis progressively declines over 1 to 2 weeks and is an unlikely mediator of SEFL, which persists unabated for a minimum of 90 days (Belda et al., 2008). These findings suggest that sensitization of CORT responses and enhanced fear learning following stress are likely parallel but independent processes.
Toward a neural circuit understanding of SEFL
Given that maladaptive enhancement of fear learning is the primary behavioral phenotype in the SEFL model and that a great deal is known about the neural circuits underlying associative fear learning, it should not be surprising that studies on the neural mechanisms of SEFL have focused largely on brain regions known to mediate associative fear conditioning—namely the amygdala, hippocampus, and prefrontal cortex (PFC). Thus, we will use this associative fear network as a framework to discuss and review brain mechanisms of fear sensitization. While a comprehensive examination of the neurobiology of fear conditioning is beyond the scope of this review, many excellent reviews are available (Fanselow & Wassum, 2016; Krabbe et al., 2017; LeDoux, 2000; Maren, 2001). Here, we briefly review the roles of amygdala, hippocampus, and PFC in associative fear learning and discuss how these regions may contribute to SEFL.
Basolateral amygdala
The BLA is often considered to be where CS-US associations are formed in Pavlovian fear conditioning (Romanski et al., 1993). This notion is supported by evidence that lesion or reversible inactivation of BLA prior to conditioning impairs contextual and cued fear learning (Maren, 1999b). Moreover, disruption of BLA plasticity mechanisms (via administration of N-methyl D-aspartate (NMDA) receptor antagonist, APV, or protein synthesis inhibitor, anisomycin) during fear conditioning prevents the acquisition of CS-US fear associations (Goosens & Maren, 2004; Miserendino et al., 1990; Nader et al., 2000). Based on these and other findings, the dominant framework for associative fear learning asserts that synaptic connections between LA neurons and inputs representing sensory stimuli are potentiated by pairings between a neutral CS and a strong depolarizing US (Maren & Quirk, 2004). Because of these strengthened synapses, the CS, which was previously neutral, acquires the ability to evoke LA neuronal depolarization and conditioned fear responses (Maren, 1999a; Pape & Pare, 2010).
Although it is well established that the basolateral amygdala (BLA) is required for the formation of associative fear memory, its role in the induction of nonassociative fear sensitization by acute stress is less well-understood. Perusini and colleagues conducted a series of experiments to evaluate the involvement of BLA circuitry in SEFL (Perusini et al., 2016). The authors demonstrated that pharmacological inhibition of BLA during, but not after, footshock stress prevented the induction of SEFL. Thus, BLA is essential for the formation of both associative fear memory and nonassociative fear sensitization. Given that long-term synaptic potentiation of the BLA is a model of associative fear memory and that both are dependent on NMDA receptor function (Lee & Kim, 1998; Maren et al., 1996; Pape & Pare, 2010), one prediction is that similar plasticity mechanisms are responsible for the induction of nonassociative fear sensitization. However, systemic blockade of NMDA receptors during footshock stress fails to prevent induction of SEFL (Rau et al., 2005), whereas such blockade does impair associative fear conditioning (Maren et al., 1996). Additional experiments will be necessary to determine whether BLA or regions downstream of BLA are the critical sites for induction and long-term maintenance of fear sensitization.
Having demonstrated that CORT and BLA activation during stress exposure are individually necessary for SEFL induction (as discussed above), Perusini and colleagues next evaluated the influence of stress hormones, specifically in BLA. They found that blockade of amygdala glucocorticoid receptors during footshock stress prevented the induction of SEFL. Glucocorticoids promote long-lasting changes in neurotransmission and plasticity via the regulation of gene transcription (Gray et al., 2017; Weikum et al., 2017). To begin to probe the structural mechanisms by which glucocorticoid actions in BLA facilitate induction of fear sensitization, the authors examined glutamatergic receptor composition following SEFL induction. Inhibition of CORT synthesis via administration of metyrapone prevented SEFL and the upregulation of GluA1 (AMPA) receptors in BLA. Interestingly, there were no differences in the numbers of GluA2 (AMPA) or GluN1 (NMDA) receptors. Based on the findings, Perusini et al concluded that elevation of GluA1 in BLA is a candidate mechanism by which glucocorticoids induce persistent states of fear sensitization as reflected in SEFL. They further hypothesized that GluA1 upregulation leads to increased BLA excitability that primes synapses for enhanced plasticity in response to subsequent mild threats. However, the causal significance of GluA1 upregulation in SEFL has not been tested directly using gain- or loss-of-function experiments. Furthermore, existing studies do not exclude the possibility that GluA1 upregulation mediates associative fear learning rather than nonassociative sensitization.
Indeed, multiple lines of evidence implicate GluA1 in amygdalar synaptic plasticity and associative fear learning. Slice electrophysiology recordings from GluA1 knockout mice show that LTP is absent at thalamic and cortical inputs to the lateral (LA) and basal (BA) subdivisions of BLA, suggesting that GluA1 would be necessary for associative fear learning, which involves these synapses (Humeau et al., 2007). Consistent with these findings, genetic deletion of GluA1 causes fear memory deficits (Humeau et al., 2007), and GluA1 gene expression and surface distribution are upregulated in the amygdala 24 hours following fear conditioning (Mei et al., 2005; Yeh et al., 2006). Furthermore, Rumpel and colleagues demonstrated that auditory fear conditioning mobilizes GluA1 to LA neuronal synapses and their incorporation into the membrane is necessary for fear conditioning to a tone CS (Rumpel et al., 2005).
Although the precise role of GluA1 and its upregulation in nonassociative fear sensitization remains unclear, there is good evidence that hyperexcitability of BLA neurons mediates stress-induced alterations in affective behavior and fear learning [for review see (Prager et al., 2016; Sharp, 2017; X. Zhang et al., 2018)]. Excitation of BLA pyramidal neurons drives the expression of defensive behavior, and this activity is regulated by a small population of GABAergic interneurons that exert inhibitory control over these cells (Ehrlich et al., 2009; Spampanato et al., 2011; Sun et al., 2020). The balance between these excitatory and inhibitory populations is believed to modulate responses to threats. Exposure to environmental stressors can induce long-term alterations in BLA circuitry that alter the excitatory-inhibitory balance in favor of hyperexcitability (Kavushansky & Richter‐Levin, 2006; Rosenkranz et al., 2010; W. Zhang & Rosenkranz, 2012). For instance, acute immobilization stress leads to a progressive increase in BLA neuronal spine density that positively correlates with the development of anxiety-like behavior (Mitra et al., 2005). Furthermore, exposure to chronic stress enhances fear learning, impairs fear extinction, increases glutamatergic, and reduces GABAergic transmission onto BLA neurons (Chauveau et al., 2012; Kavushansky & Richter‐Levin, 2006; Manzanares et al., 2005). These findings have been thoroughly reviewed by Aubry and colleagues, who outlined evidence for the role of GluA1 as a synaptic placeholder for GluA2 receptors that are inserted during fear acquisition and are responsible for the long-term maintenance of associative fear memory (Aubry et al., 2016) According to this model, the authors hypothesize that stress-induced increases in GluA1 may facilitate the insertion of GluA2, and, in turn, enhance subsequent fear learning, consistent with the hypothesis of (Perusini et al., 2016).
The findings above suggest changes in excitatory-inhibitory balance of the BLA as a potential mechanism through which stress causes lasting enhancements in fear and other threat responses. The extent to which such changes contribute to SEFL, and the mechanisms of such changes, remain unclear. As hypothesized by Perusini et al, upregulation of GluA1 expression by stress could facilitate later fear learning. Still, the causal significance of GluA1 upregulation has not been directly tested in the context of SEFL. Future studies might leverage viral-mediated overexpression or suppression to manipulate BLA GluA1 levels.
Hippocampus
The hippocampal formation is responsible for the encoding, consolidation, and retrieval of episodic, contextual, and spatial memories (Fanselow, 2010; Gewirtz et al., 2000; Wiltgen, 2006). During Pavlovian fear conditioning, the hippocampus is believed to generate multimodal contextual representations, which are signaled to BA via ventral hippocampus (vHP) projections (Tovote et al., 2015). The integration of contextual information and emotional valence that is required for contextual fear learning is thought to occur downstream of the hippocampus in BLA (Fanselow, 2010). Accordingly, post-training hippocampal lesions disrupt the expression of contextual but not auditory-cued fear memory (J. J. Kim et al., 1993; J. J. Kim & Fanselow, 1992; Rudy & Morledge, 1994).
The hippocampus is particularly vulnerable to the effects of stress and is heavily implicated in various stress-induced learning and behavioral changes. Numerous studies have demonstrated that following stress, the hippocampus exhibits reduction in volume, dendritic atrophy, pyramidal neuron spine loss, impaired LTP, and alterations in adult neurogenesis (E. J. Kim et al., 2015; Krugers et al., 2010). These structural alterations have been associated with functional impairments in hippocampal-dependent learning and spatial navigation tasks (Conrad, 2010; Conrad et al., 2017). Interestingly, stress does not uniformly disrupt all hippocampal-mediated functions. Select forms of aversive learning, including contextual fear conditioning, are facilitated following exposure to stress (Conrad et al., 1999; Hoffman et al., 2010; Sandi et al., 2001). Therefore, identifying the specific stress-induced physiological alterations in the hippocampus that sensitize fear learning remains a central question.
A handful of studies have directly examined the role of the hippocampus in SEFL. Hersman et al. demonstrated a role for stress-induced cholinergic activity in the induction of SEFL. The authors report that dorsal hippocampus (dHP) infusion of muscarinic acetylcholine (mAChR) receptor antagonist, scopolamine, prior to stress prevented the acquisition of associative fear memory and sensitization of subsequent context fear learning (Hersman et al., 2019). Notably, the same manipulation failed to block stress-enhanced fear learning to tone, suggesting that dHP cholinergic signaling selectively modulates the contextual component of SEFL. Based on previous findings from this group identifying BLA GluA1 upregulation as a mechanism of fear sensitization, the authors propose that dHP mAChR modulates context memory formation, whereas stress-induced alterations in BLA modulate strength of the context-shock association. (J. J. Kim, 2005; J. J. Kim et al., 2001).
Additional evidence for the role of the hippocampus in SEFL is provided by experiments implicating the pro-inflammatory cytokine interleukin-1ß (IL-1) in the sensitizing effects of footshock stress. Jones et al demonstrated that IL-1ß is elevated in dHP 24–48 hours following footshock stress, and pharmacological blockade of dHP IL-1ß signaling 24 and 48 hours after stress attenuates sensitization of later context fear learning (Jones, Lebonville, et al., 2018). Moreover, the authors identified astrocytes as the primary source of stress-induced IL-1ß signaling in dHP and found that chemogenetic inhibition of dHP astrocytes during stress prevents the development of SEFL (Jones, Paniccia, et al., 2018). Based on these studies, it cannot be determined whether stress-induced dHP IL-1ß specifically mediates enhanced contextual fear learning or is broadly involved in SEFL behavior because cued fear conditioning was not assessed. Nonetheless, the findings suggest that interactions between the immune system and hippocampus that occur during and after stress exposure promote sensitization of contextual fear learning.
The studies reviewed in this section suggest that induction and maintenance of stress-enhanced contextual fear learning depend on stress-induced dHP circuit alterations. An alternative possibility is that the hippocampus directly mediates the sensitizing effects of stress that extend beyond hippocampal-dependent learning and behavior. This idea is consistent with the well-established role of the hippocampus in regulating the emotional and physiological responses to stress. For example, acute stress has been shown to enhance and impair cerebellar-dependent delay eyeblink conditioning in male and female rats respectively, and this modulatory effect of stress on eyeblink conditioning is abolished by hippocampal lesions (Bangasser & Shors, 2007). Importantly, learning was not affected by hippocampal lesions, demonstrating that the hippocampus mediates stress-induced alterations in this form of learning. Similarly, ablation of adult hippocampal neurogenesis enhances anxiety-like behavior in stressed subjects but does not affect anxiety prior to stress exposure (Seo et al., 2015). At present, the contribution of the hippocampus to other nonassociative consequences of stress remains unclear.
Medial Prefrontal cortex
The medial prefrontal cortex (mPFC) is thought to exert top-down control of subcortical regions to regulate emotional processing based on previously learned and higher-order cognitive information (Giustino & Maren, 2015). Within the mPFC, there is significant functional heterogeneity. The prelimbic (PL) subdivision of mPFC is necessary for conditioned fear expression, and activity in this region positively correlates with freezing to an auditory CS+ (Burgos-Robles et al., 2009; Corcoran & Quirk, 2007). In contrast, the infralimbic (IL) subdivision is involved in fear suppression during extinction learning (Do-Monte et al., 2015; Sierra-Mercado et al., 2011). It has been proposed that PL and IL exert opposing influence on fear behavior via output projections to distinct neuronal populations in the amygdala. In particular, activation of IL decreases the responsiveness of CeA output neurons, and this process is thought to underlie fear suppression to an extinguished CS+ (Quirk et al., 2003). In addition, several lines of evidence suggest that IL exerts broad inhibitory influence over fear behavior beyond fear extinction. Both optogenetic (Do-Monte et al., 2015) and electrical stimulation (Milad & Quirk, 2002; Vidal-Gonzalez et al., 2006) of IL paired with previously conditioned tones attenuates fear expression in animals that have not undergone extinction training. Inactivation of IL also disrupts fear discrimination between a CS+ and CS− safety cue (Sangha et al., 2014).
Studies have repeatedly identified IL as a stress-susceptible region. Exposure to chronic stress leads to IL dendritic retraction (Moench et al., 2015), increased synaptic inhibition onto excitatory output neurons (McKlveen et al., 2016), and alterations in IL-BLA and BLA-IL synaptic plasticity (Izquierdo, 2006; Maroun, 2006; Maroun & Richter-Levin, 2003). Based on these findings and the role of the IL in fear suppression, it is reasonable to posit that stress-induced dysregulation of IL could produce fear sensitization. Accordingly, Pennington et al evaluated the contribution of IL to SEFL (Pennington et al., 2017). The authors found pre-stress lesions of ventromedial PFC (vmPFC) that primarily targeted IL attenuated SEFL to contextual but not an auditory cue. Importantly, IL lesions did not disrupt associative fear learning in naïve animals. The results suggest that the intact IL facilitates sensitization of contextual fear learning following stress. Considering the role of IL in fear suppression, the findings were counter to the author’s prediction that IL lesions would enhance SEFL. However, these findings are consistent with other reports that IL lesions attenuate (Sullivan & Gratton, 2002) whereas pharmacological stimulation increases anxiety-like behavior (Bi et al., 2013). Given the apparent heterogeneity of mPFC function—which includes roles in fear suppression, fear expression, and anxiety-like behavior—additional studies, including temporal- and subregion-specific inactivations, will be needed to assess the role of mPFC in the induction and expression of SEFL.
Initial studies investigating the neural mechanisms of SEFL have begun to uncover unique candidate mechanisms within the amygdala, hippocampus, and prefrontal cortex. These studies have identified alterations in BLA GluA1 receptor expression as a potential mechanism by which traumatic stress exerts a lasting influence on subsequent fear learning. However, studies have also shown that manipulations of the hippocampus and vmPFC have distinct effects on cued and contextual SEFL, suggesting that SEFL is mediated by distributed network mechanisms, with different brain regions contributing to specific components of SEFL. Based on the reports discussed here, it is interesting to note that most experimental manipulations that disrupt SEFL also prevent associative fear to the trauma context. A deeper understanding of the various neuromodulators and stress-related peptides involved in SEFL may help elucidate neural mechanisms that are dissociable between fear sensitization and associative fear learning. In addition to clarifying the causal contributions of stress-induced plasticity in the amygdala, hippocampus and PFC, future studies must look beyond these regions. In particular, brainstem regions such as the locus coeruleus (LC), parabrachial nucleus, and periaqueductal gray deserve investigation. These three regions exhibit stress-induced plasticity and modulate Pavlovian fear learning, but their roles in SEFL remain unknown.
Clinical relevance of SEFL
Prior conditioning theories have conceptualized PTSD through an associative learning framework, whereby excessive and inappropriate fear evoked by stimuli or contexts related to the traumatic event underlies this disorder’s behavioral and emotional symptoms (Eysenck, 1979; Mineka & Zinbarg, 2006). Based on this perspective, fear conditioning procedures have emerged as a valuable translational tool for elucidating components of PTSD that are a product of maladaptive outcomes of associative fear learning. Consistent with this account, PTSD patients exhibit higher levels of generalization and greater resistance to the extinction of conditioned fear (Blechert et al., 2007; Milad et al., 2008; Wicking et al., 2016). The associative account of PTSD has primarily focused on treatments designed to reduce conditioned fear responses to trauma-associated stimuli.
Exposure-based therapy is a common treatment for patients with PTSD that is modeled on extinction. The primary goal of exposure therapy is to attenuate conditioned fear responses evoked by stimuli associated with the trauma (Rauch et al., 2012). This is achieved through repeated confrontations with the fear-inducing stimuli during which no aversive outcome is experienced. Briefly, in vivo approaches involve real-life exposure to trauma-associated stimuli in a safe setting and are typically performed gradually (McLean & Foa, 2013). In the case of a patient who develops PTSD after being attacked by a dog, an example treatment plan may involve first viewing pictures of a dog, then interacting with a stuffed dog animal, and finally encountering a real-life dog. Since its early implementation in 1960 by Joseph Wolpe (Wolpe, 1961), several different variations of exposure therapy have been developed, and their efficacy is strongly supported by research (McLean et al., 2021; Rauch et al., 2012; Watkins et al., 2018). Yet, in a meta-analysis comparing studies across two decades, approximately 52% of participants who started treatment and 68% who completed treatment no longer met the criteria for PTSD (Bradley et al., 2005). While exposure therapy is effective in aggregate, it is noteworthy that a meaningful proportion of individuals derive little or no benefit from it, and trauma-focused therapies are associated with higher rates of dropout (Lewis et al., 2020). Furthermore, even among those patients who benefit from exposure therapy, symptoms often persist after therapy, indicating that symptoms are only partially attenuated or that subsets of symptoms are resistant to exposure therapy (Bradley et al., 2005).
One explanation for the limited effectiveness of exposure-based therapies is that subsets of PTSD symptoms reflect nonassociative sensitized fear, independent of Pavlovian or episodic traumatic memories. Indeed, PTSD includes symptoms that resemble fear sensitization. For instance, an early report by Paige et al. showed that Vietnam veterans diagnosed with PTSD exhibited abnormal autonomic hyperactivity in response to novel tone stimuli (Paige et al., 1990). The authors concluded that “PTSD represents a state of central nervous system sensitivity.” In the years following, numerous studies have reported that patients with PTSD exhibit enhanced fear conditioning to novel stimuli, heightened auditory startle responses, and exaggerated physiological and self-reported fear responses to threatening images (Glover et al., 2011; Orr et al., 2000; Peri et al., 2000). Consistent with these observations, the DSM-V criteria for PTSD include an arousal and reactivity category characterized by symptoms of hypervigilance, exaggerated startle response, and irritable behavior. These symptoms often occur in the absence of situational reminders of the trauma. Two longitudinal studies show that hyperarousal strongly influences, but is not influenced by, re-experiencing and avoidance symptom clusters (Marshall et al., 2006; Schell et al., 2004). It is conceivable that these symptoms reflect nonassociative fear sensitization rather than Pavlovian conditioned responses.
Perhaps the most significant advantage of the SEFL model is the ability to distinguish between associative fear memory and nonassociative fear sensitization. We propose that the underlying mechanisms of SEFL are dissociable from those of associative fear memory. This is supported by findings previously discussed in this review demonstrating that in addition to extinction, pharmacological or infantile amnesia-mediated disruption of associative fear memory does not interfere with SEFL development. For this reason, SEFL represents a valuable animal model for investigating the potential nonassociative components of trauma resembling fear sensitization that is resistant to extinction-based therapies.
Conclusions and Future Directions
The studies reviewed above lead to 3 main conclusions: (1) fear sensitization is a robust form of learning that is conserved across species; (2) fear sensitization is both behavioral and biologically distinct from associative fear learning and recruits unique neural mechanisms in regions including amygdala and hippocampus; and (3) SEFL models endophenotypes relevant to stressor-related disorders such as PTSD and can be exploited to better understand and identify treatments for these disorders. In this section, we discuss limitations to the literature reviewed above and highlight relevant future research directions.
Throughout this review, we have argued that associative fear conditioning and nonassociative fear sensitization are mechanistically distinct forms of learning. This distinction is supported by evidence that fear sensitization persists even under conditions that prevent the formation and maintenance of associative fear memory. For instance, pharmacological blockade of NMDARs precludes the formation of an associative context fear memory but fails to prevent induction of SEFL. In addition, extinction of the contextual fear memory generated during stress exposure fails to attenuate SEFL. Still, these and other manipulations designed to attenuate associative fear memories leave some vestige of associative fear memories intact, and we cannot exclude the possibility that fear sensitization depends on these vestiges. For instance, extinction of contextual fear may leave intact other associative fear memories tied to handling cues or other features of the experimental set-up, and these non-extinguished conditioned fear responses could summate to produce an apparent SEFL effect. To firmly establish the independence of fear sensitization and associative fear conditioning, it may be necessary to dig further into the biological mechanisms and clarify whether the two behavioral processes recruit distinct anatomical, molecular, or synaptic pathways.
More research is also needed on how changes to the physiological stress response contribute to SEFL. As discussed above, exposure to intense stressors can sensitize the HPA axis response to stress. However, it remains unknown whether and how this sensitization contributes to SEFL. In addition, little is known about how the autonomic nervous system functions contribute to SEFL. The sympathetic and parasympathetic autonomic nervous systems work in parallel to the HPA-axis in regulating the rapid bodily responses to stressors (Blechert et al., 2007; Cannon, 1932; Fonkoue et al., 2018; Fu, 2022; Roelofs, 2017). Sympathetic-parasympathetic balance is altered in PTSD (Sammito et al., 2015); it is unknown whether this endophenotype is recapitulated in the SEFL model. The LC is a major driver of sympathetic functions in the brain and body and is known to be affected in PTSD patients (Aston-Jones et al., 1994; Morris et al., 2020). Further analysis of the role of LC and its regulation of sympathetic functions in the context of SEFL is warranted. Approaches that increase parasympathetic tone, such as vagal nerve stimulation, can increase the efficacy of extinction in reducing maladaptive fear memories. However, if this approach would reverse nonassociative fear as observed in SEFL remains to be examined (Noble et al., 2019).
Empirical findings from rodent models such as Pavlovian fear conditioning continue to develop and refine theoretical constructs, such as “fear.” With this progress, the field has begun to tease apart the conditions and underlying processes that distinguish adaptive fear responses from those that are maladaptive. SEFL represents an extension of this work and may provide a useful conceptual framework. With mild stressors, learned fear will primarily be of the associative Pavlovian in nature. As such, fear responses should be relatively well-tied to situationally-relevant conditioned stimuli. As stress increases in severity, however, nonassociative sensitization becomes relatively stronger and with this, fear and other threat responses become more generalized.
While PTSD is a human disorder of great complexity, trauma and its persisting effects are not only defining characteristics but also represent essential antecedents that can be further tested using the procedures and variables inherent to SEFL. A detailed discussion of the validity of SEFL as a model of PTSD is beyond the scope of this review. However, as illustrated here, significant progress is being made toward building a rodent model that captures the symptoms of PTSD and conditions and nonassociative mechanisms that underlie stress- and trauma-related disorders. We believe that SEFL represents the best approximation to date of a tractable model system in rodents for the analysis of behavior, cognition, and nervous system mechanisms underlying PTSD.
In summary, the studies reviewed here provide insight into the circuit, hormonal, and autonomic mechanisms of SEFL and illustrate those multiple biological systems acting in coordination contribute to the underlying behavioral phenotype. Although studies to date have identified what appear to be key neural mechanisms, additional research is needed on epigenetic and transcriptomic mechanisms, genetic risk factors, and the role of neuromodulatory systems. Lastly, although studies have not identified differences in SEFL between male and female rodents, additional research is needed, given the sexual dimorphism in the prevalence of disorders like PTSD. The SEFL model should remain a valuable tool for addressing these and other questions about stress-related fear dysregulation.
Acknowledgments:
KJN, AMP, MRD and AKR contributed equally to the writing of this manuscript. AKR is supported by the Brain and Behavior Research Foundation (29227), WhiteHall Foundation, Akira Arimura Foundation and Friedman Brain Institute at Icahn School of Mount Sinai. AMP is supported by the National Institutes of Mental Health (R01MH114961). MRD and KJN are supported by National Institutes of Mental Health R21MH128610 and R01MH117426.
Footnotes
Declaration of Interests: None
References:
- Armario A, Vallès A, Dal-Zotto S, Márquez C, & Belda X (2009). A Single Exposure to Severe Stressors Causes Long-term Desensitisation of the Physiological Response to the Homotypic Stressor. Stress, 7(3), 157–172. 10.1080/10253890400010721 [DOI] [PubMed] [Google Scholar]
- Aston-Jones, Valentino RJ, Bockstaele EJV, & Meyerson AT (1994). Locus coeruleus, stress and PTSD: Neurobiological and clinical parallels. American Psychiatric Press. [Google Scholar]
- Aubry AV, Serrano PA, & Burghardt NS (2016). Molecular Mechanisms of Stress-Induced Increases in Fear Memory Consolidation within the Amygdala. Frontiers in Behavioral Neuroscience, 10, 191. 10.3389/fnbeh.2016.00191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangasser DA, & Shors TJ (2007). The hippocampus is necessary for enhancements and impairments of learning following stress. Nature Neuroscience, 10(11), 1401–1403. 10.1038/nn1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangasser DA, & Shors TJ (2010). Critical brain circuits at the intersection between stress and learning. Neuroscience & Biobehavioral Reviews, 34(8), 1223–1233. 10.1016/j.neubiorev.2010.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belda X, Rotllant D, Fuentes S, Delgado R, Nadal R, & Armario A (2008). Exposure to Severe Stressors Causes Long‐lasting Dysregulation of Resting and Stress‐induced Activation of the Hypothalamic‐Pituitary‐Adrenal Axis. Annals of the New York Academy of Sciences, 1148(1), 165–173. 10.1196/annals.1410.038 [DOI] [PubMed] [Google Scholar]
- Bi L-L, Wang J, Luo Z-Y, Chen S-P, Geng F, Chen Y, Li S-J, Yuan C, Lin S, & Gao T-M (2013). Enhanced excitability in the infralimbic cortex produces anxiety-like behaviors. Neuropharmacology, 72, 148–156. 10.1016/j.neuropharm.2013.04.048 [DOI] [PubMed] [Google Scholar]
- Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, Tang Y, Gillespie CF, Heim CM, Nemeroff CB, Schwartz AC, Cubells JF, & Ressler KJ (2008). Association of FKBP5 Polymorphisms and Childhood Abuse With Risk of Posttraumatic Stress Disorder Symptoms in Adults. JAMA, 299(11), 1291–1305. 10.1001/jama.299.11.1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blechert J, Michael T, Vriends N, Margraf J, & Wilhelm FH (2007). Fear conditioning in posttraumatic stress disorder: Evidence for delayed extinction of autonomic, experiential, and behavioural responses. Behaviour Research and Therapy, 45(9), 2019–2033. 10.1016/j.brat.2007.02.012 [DOI] [PubMed] [Google Scholar]
- Bradley R, Greene J, Russ E, Dutra L, & Westen D (2005). A Multidimensional Meta-Analysis of Psychotherapy for PTSD. American Journal of Psychiatry, 162(2), 214–227. 10.1176/appi.ajp.162.2.214 [DOI] [PubMed] [Google Scholar]
- Burgos-Robles A, Vidal-Gonzalez I, & Quirk GJ (2009). Sustained Conditioned Responses in Prelimbic Prefrontal Neurons Are Correlated with Fear Expression and Extinction Failure. Journal of Neuroscience, 29(26), 8474–8482. 10.1523/jneurosci.0378-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon WB (1932). THE WISDOM OF THE BODY. The American Journal of the Medical Sciences, 184(6), 864. 10.1097/00000441-193212000-00028 [DOI] [Google Scholar]
- Chauveau F, Lange MD, Jüngling K, Lesting J, Seidenbecher T, & Pape H-C (2012). Prevention of Stress-Impaired Fear Extinction Through Neuropeptide S Action in the Lateral Amygdala. Neuropsychopharmacology, 37(7), 1588–1599. 10.1038/npp.2012.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad CD (2010). A critical review of chronic stress effects on spatial learning and memory. Progress in Neuro-Psychopharmacology and Biological Psychiatry, 34(5), 742–755. 10.1016/j.pnpbp.2009.11.003 [DOI] [PubMed] [Google Scholar]
- Conrad CD, Magariños AM, LeDoux JE, & McEwen BS (1999). Repeated restraint stress facilitates fear conditioning independently of causing hippocampal CA3 dendritic atrophy. Behavioral Neuroscience, 113(5), 902–913. 10.1037//0735-7044.113.5.902 [DOI] [PubMed] [Google Scholar]
- Conrad CD, Ortiz JB, & Judd JM (2017). Chronic stress and hippocampal dendritic complexity: Methodological and functional considerations. Physiology & Behavior, 178, 66–81. 10.1016/j.physbeh.2016.11.017 [DOI] [PubMed] [Google Scholar]
- Corcoran KA, & Quirk GJ (2007). Activity in Prelimbic Cortex Is Necessary for the Expression of Learned, But Not Innate, Fears. Journal of Neuroscience, 27(4), 840–844. 10.1523/jneurosci.5327-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijken HHV, Heyden JAMVD, Mos J, & Tilders FJH (1992). Inescapable footshocks induce progressive and long-lasting behavioural changes in male rats. Physiology & Behavior, 51(4), 787–794. 10.1016/0031-9384(92)90117-k [DOI] [PubMed] [Google Scholar]
- Do-Monte FH, Manzano-Nieves G, Quiñones-Laracuente K, Ramos-Medina L, & Quirk GJ (2015). Revisiting the Role of Infralimbic Cortex in Fear Extinction with Optogenetics. The Journal of Neuroscience, 35(8), 3607–3615. 10.1523/jneurosci.3137-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, & Lüthi A (2009). Amygdala Inhibitory Circuits and the Control of Fear Memory. Neuron, 62(6), 757–771. 10.1016/j.neuron.2009.05.026 [DOI] [PubMed] [Google Scholar]
- Eysenck HJ (1979). The conditioning model of neurosis. Behavioral and Brain Sciences, 2(2), 155–166. 10.1017/s0140525x00061653 [DOI] [Google Scholar]
- Fanselow MS (2010). From contextual fear to a dynamic view of memory systems. Trends in Cognitive Sciences, 14(1), 7–15. 10.1016/j.tics.2009.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, & Bolles RC (1979). Triggering of the endorphin analgesic reaction by a cue previously associated with shock: Reversal by naloxone. Bulletin of the Psychonomic Society, 14(2), 88–90. 10.3758/bf03329408 [DOI] [Google Scholar]
- Fanselow MS, & Wassum KM (2016). The Origins and Organization of Vertebrate Pavlovian Conditioning. Cold Spring Harbor Perspectives in Biology, 8(1), a021717. 10.1101/cshperspect.a021717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonkoue IT, Norrholm SD, Marvar PJ, Li Y, Kankam ML, Rothbaum BO, & Park J (2018). Elevated resting blood pressure augments autonomic imbalance in posttraumatic stress disorder. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 315(6), R1272–R1280. 10.1152/ajpregu.00173.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost WN, Castellucci VF, Hawkins RD, & Kandel ER (1985). Monosynaptic connections made by the sensory neurons of the gill- and siphon-withdrawal reflex in Aplysia participate in the storage of long-term memory for sensitization. Proceedings of the National Academy of Sciences, 82(23), 8266–8269. 10.1073/pnas.82.23.8266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q (2022). Autonomic dysfunction and cardiovascular risk in post-traumatic stress disorder. Autonomic Neuroscience, 237, 102923. 10.1016/j.autneu.2021.102923 [DOI] [PubMed] [Google Scholar]
- Gewirtz JC, McNish KA, & Davis M (2000). Is the hippocampus necessary for contextual fear conditioning? Behavioural Brain Research, 110(1–2), 83–95. 10.1016/s0166-4328(99)00187-4 [DOI] [PubMed] [Google Scholar]
- Giustino TF, & Maren S (2015). The Role of the Medial Prefrontal Cortex in the Conditioning and Extinction of Fear. Frontiers in Behavioral Neuroscience, 9, 298. 10.3389/fnbeh.2015.00298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover EM, Phifer JE, Crain DF, Norrholm SD, Davis M, Bradley B, Ressler KJ, & Jovanovic T (2011). Tools for translational neuroscience: PTSD is associated with heightened fear responses using acoustic startle but not skin conductance measures. Depression and Anxiety, 28(12), 1058–1066. 10.1002/da.20880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez ST, Marty V, Spigelman I, Reise SP, & Fanselow MS (2021). Impact of stress resilience and susceptibility on fear learning, anxiety, and alcohol intake. Neurobiology of Stress, 15, 100335. 10.1016/j.ynstr.2021.100335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goosens KA, & Maren S (2004). NMDA receptors are essential for the acquisition, but not expression, of conditional fear and associative spike firing in the lateral amygdala. European Journal of Neuroscience, 20(2), 537–548. 10.1111/j.1460-9568.2004.03513.x [DOI] [PubMed] [Google Scholar]
- Gray JD, Kogan JF, Marrocco J, & McEwen BS (2017). Genomic and epigenomic mechanisms of glucocorticoids in the brain. Nature Reviews Endocrinology, 13(11), 661–673. 10.1038/nrendo.2017.97 [DOI] [PubMed] [Google Scholar]
- Groves PM, & Thompson RF (1970). Habituation: A dual-process theory. Psychological Review, 77(5), 419. 10.1037/h0029810 [DOI] [PubMed] [Google Scholar]
- Hassien AM, Shue F, Bernier BE, & Drew MR (2020). A mouse model of stress-enhanced fear learning demonstrates extinction-sensitive and extinction-resistant effects of footshock stress. Behavioural Brain Research, 379, 112391. 10.1016/j.bbr.2019.112391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Cohen TE, & Kandel ER (2006). Dishabituation in Aplysia can involve either reversal of habituation or superimposed sensitization. Learning & Memory, 13(3), 397–403. 10.1101/lm.49706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersman S, Hoffman AN, Hodgins L, Shieh S, Lam J, Parikh A, & Fanselow MS (2019). Cholinergic Signaling Alters Stress-Induced Sensitization of Hippocampal Contextual Learning. Frontiers in Neuroscience, 13, 251. 10.3389/fnins.2019.00251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman AN, Armstrong CE, Hanna JJ, & Conrad CD (2010). Chronic stress, cyclic 17β-estradiol, and daily handling influences on fear conditioning in the female rat. Neurobiology of Learning and Memory, 94(3), 422–433. 10.1016/j.nlm.2010.08.010 [DOI] [PubMed] [Google Scholar]
- Hoffman AN, Trott JM, Makridis A, & Fanselow MS (2022). Anxiety, fear, panic: An approach to assessing the defensive behavior system across the predatory imminence continuum. Learning & Behavior, 1–10. 10.3758/s13420-021-00509-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Reisel D, Johnson AW, Borchardt T, Jensen V, Gebhardt C, Bosch V, Gass P, Bannerman DM, Good MA, Hvalby O, Sprengel R, & Luthi A (2007). A Pathway-Specific Function for Different AMPA Receptor Subunits in Amygdala Long-Term Potentiation and Fear Conditioning. Journal of Neuroscience, 27(41), 10947–10956. 10.1523/jneurosci.2603-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo A (2006). Brief Uncontrollable Stress Causes Dendritic Retraction in Infralimbic Cortex and Resistance to Fear Extinction in Mice. Journal of Neuroscience, 26(21), 5733–5738. 10.1523/jneurosci.0474-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M, & Baram TZ (2009). The neuro-symphony of stress. Nature Reviews Neuroscience, 10(6), 459–466. 10.1038/nrn2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ME, Lebonville CL, Paniccia JE, Balentine ME, Reissner KJ, & Lysle DT (2018). Hippocampal interleukin-1 mediates stress-enhanced fear learning: A potential role for astrocyte-derived interleukin-1β. Brain, Behavior, and Immunity, 67, 355–363. 10.1016/j.bbi.2017.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ME, Paniccia JE, Lebonville CL, Reissner KJ, & Lysle DT (2018). Chemogenetic manipulation of dorsal hippocampal astrocytes protects against the development of stress-enhanced fear learning. Neuroscience, 388(Cell 2018), 45–56. 10.1016/j.neuroscience.2018.07.015 [DOI] [PubMed] [Google Scholar]
- Kandel ER (2001). The Molecular Biology of Memory Storage: A Dialogue Between Genes and Synapses. Science, 294(5544), 1030–1038. 10.1126/science.1067020 [DOI] [PubMed] [Google Scholar]
- Kavushansky A, & Richter‐Levin G (2006). Effects of stress and corticosterone on activity and plasticity in the amygdala. Journal of Neuroscience Research, 84(7), 1580–1587. 10.1002/jnr.21058 [DOI] [PubMed] [Google Scholar]
- Kilpatrick DG, Resnick HS, Milanak ME, Miller MW, Keyes KM, & Friedman MJ (2013). National Estimates of Exposure to Traumatic Events and PTSD Prevalence Using DSM‐IV and DSM‐5 Criteria. Journal of Traumatic Stress, 26(5), 537–547. 10.1002/jts.21848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Pellman B, & Kim JJ (2015). Stress effects on the hippocampus: a critical review. Learning & Memory, 22(9), 411–416. 10.1101/lm.037291.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ (2005). Amygdalar Inactivation Blocks Stress-Induced Impairments in Hippocampal Long-Term Potentiation and Spatial Memory. Journal of Neuroscience, 25(6), 1532–1539. 10.1523/jneurosci.4623-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, & Fanselow MS (1992). Modality-Specific Retrograde Amnesia of Fear. Science, 256(5057), 675–677. 10.1126/science.1585183 [DOI] [PubMed] [Google Scholar]
- Kim JJ, Lee HJ, Han J-S, & Packard MG (2001). Amygdala Is Critical for Stress-Induced Modulation of Hippocampal Long-Term Potentiation and Learning. The Journal of Neuroscience, 21(14), 5222–5228. 10.1523/jneurosci.21-14-05222.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Rison RA, & Fanselow MS (1993). Effects of amygdala, hippocampus, and periaqueductal gray lesions on short- and long-term contextual fear. Behavioral Neuroscience, 107(6), 1093. 10.1037/0735-7044.107.6.1093 [DOI] [PubMed] [Google Scholar]
- Kloet ERD, & Reul JMHM (1987). Feedback action and tonic influence of corticosteroids on brain function: A concept arising from the heterogeneity of brain receptor systems. Psychoneuroendocrinology, 12(2), 83–105. 10.1016/0306-4530(87)90040-0 [DOI] [PubMed] [Google Scholar]
- Krabbe S, Gründemann J, & Lüthi A (2017). Amygdala Inhibitory Circuits Regulate Associative Fear Conditioning. Biological Psychiatry, 83(10), 800–809. 10.1016/j.biopsych.2017.10.006 [DOI] [PubMed] [Google Scholar]
- Krugers HJ, Lucassen PJ, Karst H, & Joëls M (2010). Chronic Stress Effects on Hippocampal Structure and Synaptic Function: Relevance for Depression and Normalization by Anti-Glucocorticoid Treatment. Frontiers in Synaptic Neuroscience, 2, 24. 10.3389/fnsyn.2010.00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulp AC, Lowden B, Chaudhari S, Ridley C, Krzoska J, Barnard D, Mehta D, & Johnson J (2020). Sensitized corticosterone responses do not mediate the enhanced fear memories in chronically stressed rats. Behavioural Brain Research, 382, 112480. 10.1016/j.bbr.2020.112480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE (2000). Emotion Circuits in the Brain. 23(1), 155–184. 10.1146/annurev.neuro.23.1.155 [DOI] [PubMed] [Google Scholar]
- Lee H, & Kim JJ (1998). Amygdalar NMDA Receptors are Critical for New Fear Learning in Previously Fear-Conditioned Rats. Journal of Neuroscience, 18(20), 8444–8454. 10.1523/jneurosci.18-20-08444.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis C, Roberts NP, Gibson S, & Bisson JI (2020). Dropout from psychological therapies for post-traumatic stress disorder (PTSD) in adults: systematic review and meta-analysis. European Journal of Psychotraumatology, 11(1), 1709709. 10.1080/20008198.2019.1709709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long VA, & Fanselow MS (2012). Stress-enhanced fear learning in rats is resistant to the effects of immediate massed extinction. Stress, 15(6), 627–636. 10.3109/10253890.2011.650251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzanares PAR, Isoardi NA, Carrer HF, & Molina VA (2005). Previous Stress Facilitates Fear Memory, Attenuates GABAergic Inhibition, and Increases Synaptic Plasticity in the Rat Basolateral Amygdala. The Journal of Neuroscience, 25(38), 8725–8734. 10.1523/jneurosci.2260-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus EA, Nolen TG, Rankin CH, Stopfer M, & Carew TJ (1988). Development of behavior and learning inAplysia. Experientia, 44(5), 415–423. 10.1007/bf01940536 [DOI] [PubMed] [Google Scholar]
- Maren S (1999a). Long-term potentiation in the amygdala: a mechanism for emotional learning and memory. Trends in Neurosciences, 22(12), 561–567. 10.1016/s0166-2236(99)01465-4 [DOI] [PubMed] [Google Scholar]
- Maren S (1999b). Neurotoxic Basolateral Amygdala Lesions Impair Learning and Memory But Not the Performance of Conditional Fear in Rats. Journal of Neuroscience, 19(19), 8696–8703. 10.1523/jneurosci.19-19-08696.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S (2001). NEUROBIOLOGY OF PAVLOVIAN FEAR CONDITIONING. Annual Review of Neuroscience, 24(1), 897–931. 10.1146/annurev.neuro.24.1.897 [DOI] [PubMed] [Google Scholar]
- Maren S, Aharonov G, Stote DL, & Fanselow MS (1996). N-methyl-D-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditional fear in rats. Behavioral Neuroscience, 110(6), 1365. 10.1037/0735-7044.110.6.1365 [DOI] [PubMed] [Google Scholar]
- Maren S, & Quirk GJ (2004). Neuronal signalling of fear memory. Nature Reviews Neuroscience, 5(11), nrn1535. 10.1038/nrn1535 [DOI] [PubMed] [Google Scholar]
- Maroun M (2006). Stress reverses plasticity in the pathway projecting from the ventromedial prefrontal cortex to the basolateral amygdala. European Journal of Neuroscience, 24(10), 2917–2922. 10.1111/j.1460-9568.2006.05169.x [DOI] [PubMed] [Google Scholar]
- Maroun M, & Richter-Levin G (2003). Exposure to Acute Stress Blocks the Induction of Long-Term Potentiation of the Amygdala–Prefrontal Cortex Pathway In Vivo. The Journal of Neuroscience, 23(11), 4406–4409. 10.1523/jneurosci.23-11-04406.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall GN, Schell TL, Glynn SM, & Shetty V (2006). The Role of Hyperarousal in the Manifestation of Posttraumatic Psychological Distress Following Injury. Journal of Abnormal Psychology, 115(3), 624–628. 10.1037/0021-843x.115.3.624 [DOI] [PubMed] [Google Scholar]
- McEwen BS, Bowles NP, Gray JD, Hill MN, Hunter RG, Karatsoreos IN, & Nasca C (2015). Mechanisms of stress in the brain. Nature Neuroscience, 18(10), 1353–1363. 10.1038/nn.4086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKlveen JM, Morano RL, Fitzgerald M, Zoubovsky S, Cassella SN, Scheimann JR, Ghosal S, Mahbod P, Packard BA, Myers B, Baccei ML, & Herman JP (2016). Chronic Stress Increases Prefrontal Inhibition: A Mechanism for Stress-Induced Prefrontal Dysfunction. Biological Psychiatry, 80(10), 754–764. 10.1016/j.biopsych.2016.03.2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CP, & Foa EB (2013). Dissemination and implementation of prolonged exposure therapy for posttraumatic stress disorder. Journal of Anxiety Disorders, 27(8), 788–792. 10.1016/j.janxdis.2013.03.004 [DOI] [PubMed] [Google Scholar]
- McLean CP, Levy HC, Miller ML, & Tolin DF (2021). Exposure therapy for PTSD: A meta-analysis. Clinical Psychology Review, 91, 102115. 10.1016/j.cpr.2021.102115 [DOI] [PubMed] [Google Scholar]
- Meaney MJ, Sapolsky RM, & McEwen BS (1985). The development of the glucocorticoid receptor system in the rat limbic brain. II. An autoradiographic study. Developmental Brain Research, 18(1–2), 165–168. 10.1016/0165-3806(85)90260-3 [DOI] [PubMed] [Google Scholar]
- Mei B, Li C, Dong S, Jiang CH, Wang H, & Hu Y (2005). Distinct gene expression profiles in hippocampus and amygdala after fear conditioning. Brain Research Bulletin, 67(1–2), 1–12. 10.1016/j.brainresbull.2005.03.023 [DOI] [PubMed] [Google Scholar]
- Milad MR, Orr SP, Lasko NB, Chang Y, Rauch SL, & Pitman RK (2008). Presence and acquired origin of reduced recall for fear extinction in PTSD: Results of a twin study. Journal of Psychiatric Research, 42(7), 515–520. 10.1016/j.jpsychires.2008.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, & Quirk GJ (2002). Neurons in medial prefrontal cortex signal memory for fear extinction. Nature, 420(6911), 70–74. 10.1038/nature01138 [DOI] [PubMed] [Google Scholar]
- Mineka S, & Zinbarg R (2006). A Contemporary Learning Theory Perspective on the Etiology of Anxiety Disorders. American Psychologist, 61(1), 10–26. 10.1037/0003-066x.61.1.10 [DOI] [PubMed] [Google Scholar]
- Miserendino MJD, Sananes CB, Melia KR, & Davis M (1990). Blocking of acquisition but not expression of conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature, 345(6277), 716–718. 10.1038/345716a0 [DOI] [PubMed] [Google Scholar]
- Mitra R, Jadhav S, McEwen BS, Vyas A, & Chattarji S (2005). Stress duration modulates the spatiotemporal patterns of spine formation in the basolateral amygdala. Proceedings of the National Academy of Sciences, 102(26), 9371–9376. 10.1073/pnas.0504011102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moench KM, Maroun M, Kavushansky A, & Wellman C (2015). Alterations in neuronal morphology in infralimbic cortex predict resistance to fear extinction following acute stress. Neurobiology of Stress, 3, 23–33. 10.1016/j.ynstr.2015.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsey MS, Boyle LM, Zhang ML, Nguyen CP, Kronman HG, Ota KT, Duman RS, Taylor JR, & Schafe GE (2014). Chronic Corticosterone Exposure Persistently Elevates the Expression of Memory-Related Genes in the Lateral Amygdala and Enhances the Consolidation of a Pavlovian Fear Memory. PLoS ONE, 9(3), e91530. 10.1371/journal.pone.0091530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris LS, McCall JG, Charney DS, & Murrough JW (2020). The role of the locus coeruleus in the generation of pathological anxiety. Brain and Neuroscience Advances, 4, 239821282093032. 10.1177/2398212820930321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musazzi L, Tornese P, Sala N, & Popoli M (2018). What Acute Stress Protocols Can Tell Us About PTSD and Stress-Related Neuropsychiatric Disorders. Frontiers in Pharmacology, 9, 758. 10.3389/fphar.2018.00758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader K, Schafe GE, & Doux JEL (2000). Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature, 406(6797), 722–726. 10.1038/35021052 [DOI] [PubMed] [Google Scholar]
- Noble LJ, Souza RR, & McIntyre CK (2019). Vagus nerve stimulation as a tool for enhancing extinction in exposure-based therapies. Psychopharmacology, 236(1), 355–367. 10.1007/s00213-018-4994-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr SP, Metzger LJ, Lasko NB, Macklin ML, Peri T, & Pitman RK (2000). De Novo Conditioning in Trauma-Exposed Individuals With and Without Posttraumatic Stress Disorder. Journal of Abnormal Psychology, 109(2), 290–298. 10.1037/0021-843x.109.2.290 [DOI] [PubMed] [Google Scholar]
- Paige SR, Reid GM, Allen MG, & Newton JEO (1990). Psychophysiological correlates of posttraumatic stress disorder in Vietnam veterans. Biological Psychiatry, 27(4), 419–430. 10.1016/0006-3223(90)90552-d [DOI] [PubMed] [Google Scholar]
- Pape H-C, & Pare D (2010). Plastic Synaptic Networks of the Amygdala for the Acquisition, Expression, and Extinction of Conditioned Fear. Physiological Reviews, 90(2), 419–463. 10.1152/physrev.00037.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington ZT, Anderson AS, & Fanselow MS (2017). The ventromedial prefrontal cortex in a model of traumatic stress: fear inhibition or contextual processing? Learning & Memory, 24(9), 400–406. 10.1101/lm.046110.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri T, Ben-Shakhar G, Orr SP, & Shalev AY (2000). Psychophysiologic assessment of aversive conditioning in posttraumatic stress disorder. Biological Psychiatry, 47(6), 512–519. 10.1016/s0006-3223(99)00144-4 [DOI] [PubMed] [Google Scholar]
- Perusini JN, Meyer EM, Long VA, Rau V, Nocera N, Avershal J, Maksymetz J, Spigelman I, & Fanselow MS (2016). Induction and Expression of Fear Sensitization Caused by Acute Traumatic Stress. Neuropsychopharmacology, 41(1), 45–57. 10.1038/npp.2015.224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinsker HM, Hening WA, Carew TJ, & Kandel ER (1973). Long-Term Sensitization of a Defensive Withdrawal Reflex in Aplysia. Science, 182(4116), 1039–1042. 10.1126/science.182.4116.1039 [DOI] [PubMed] [Google Scholar]
- Poulos AM, Mehta N, Lu B, Amir D, Livingston B, Santarelli A, Zhuravka I, & Fanselow MS (2016a). Conditioning- and time-dependent increases in context fear and generalization. Learning & Memory, 23(7), 379–385. 10.1101/lm.041400.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos AM, Mehta N, Lu B, Amir D, Livingston B, Santarelli A, Zhuravka I, & Fanselow MS (2016b). Conditioning- and time-dependent increases in context fear and generalization. Learning & Memory, 23(7), 379–385. 10.1101/lm.041400.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos AM, Reger M, Mehta N, Zhuravka I, Sterlace SS, Gannam C, Hovda DA, Giza CC, & Fanselow MS (2014). Amnesia for Early Life Stress Does Not Preclude the Adult Development of Posttraumatic Stress Disorder Symptoms in Rats. Biological Psychiatry, 76(4), 306–314. 10.1016/j.biopsych.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager EM, Bergstrom HC, Wynn GH, & Braga MFM (2016). The basolateral amygdala γ‐aminobutyric acidergic system in health and disease. Journal of Neuroscience Research, 94(6), 548–567. 10.1002/jnr.23690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratchett LC, & Yehuda R (2011). Foundations of posttraumatic stress disorder: Does early life trauma lead to adult posttraumatic stress disorder? Development and Psychopathology, 23(2), 477–491. 10.1017/s0954579411000186 [DOI] [PubMed] [Google Scholar]
- Pugh CR, Tremblay D, Fleshner M, & Rudy JW (1997). A Selective Role for Corticosterone in Contextual-Fear Conditioning. Behavioral Neuroscience, 111(3), 503–511. 10.1037/0735-7044.111.3.503 [DOI] [PubMed] [Google Scholar]
- Quinn JJ, Skipper RA, & Claflin DI (2013). Infant stress exposure produces persistent enhancement of fear learning across development: Infant Stress Enhancement of Fear Learning. Developmental Psychobiology, 56(5), 1008–1016. 10.1002/dev.21181 [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Likhtik E, Pelletier JG, & Paré D (2003). Stimulation of Medial Prefrontal Cortex Decreases the Responsiveness of Central Amygdala Output Neurons. The Journal of Neuroscience, 23(25), 8800–8807. 10.1523/jneurosci.23-25-08800.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raio CM, & Phelps EA (2015). The influence of acute stress on the regulation of conditioned fear. Neurobiology of Stress, 1, 134–146. 10.1016/j.ynstr.2014.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajbhandari AK, Gonzalez ST, & Fanselow MS (2018). Stress-Enhanced Fear Learning, a Robust Rodent Model of Post-Traumatic Stress Disorder. Journal of Visualized Experiments, 140. 10.3791/58306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau V, DeCola JP, & Fanselow MS (2005). Stress-induced enhancement of fear learning: An animal model of posttraumatic stress disorder. Neuroscience & Biobehavioral Reviews, 29(8), 1207–1223. 10.1016/j.neubiorev.2005.04.010 [DOI] [PubMed] [Google Scholar]
- Rau V, & Fanselow MS (2009). Exposure to a stressor produces a long lasting enhancement of fear learning in rats. Stress, 12(2), 125–133. 10.1080/10253890802137320 [DOI] [PubMed] [Google Scholar]
- Rauch SAM, Eftekhari A, & Ruzek JI (2012). Review of exposure therapy: A gold standard for PTSD treatment. The Journal of Rehabilitation Research and Development, 49(5), 679. 10.1682/jrrd.2011.08.0152 [DOI] [PubMed] [Google Scholar]
- Rescorla RA (1974). Effect of inflation of the unconditioned stimulus value following conditioning. Journal of Comparative and Physiological Psychology, 86(1), 101. 10.1037/h0035964 [DOI] [Google Scholar]
- Reul JMHM, & Kloet ERD (1986). Anatomical resolution of two types of corticosterone receptor sites in rat brain with in vitro autoradiography and computerized image analysis. Journal of Steroid Biochemistry, 24(1), 269–272. 10.1016/0022-4731(86)90063-4 [DOI] [PubMed] [Google Scholar]
- Roelofs K (2017). Freeze for action: neurobiological mechanisms in animal and human freezing. Philosophical Transactions of the Royal Society B: Biological Sciences, 372(1718), 20160206. 10.1098/rstb.2016.0206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanski LM, Clugnet M-C, Bordi F, & LeDoux JE (1993). Somatosensory and auditory convergence in the lateral nucleus of the amygdala. Behavioral Neuroscience, 107(3), 444–450. 10.1037//0735-7044.107.3.444 [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Venheim ER, & Padival M (2010). Chronic Stress Causes Amygdala Hyperexcitability in Rodents. Biological Psychiatry, 67(12), 1128–1136. 10.1016/j.biopsych.2010.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy JW, & Morledge P (1994). Ontogeny of contextual fear conditioning in rats: Implications for consolidation, infantile amnesia, and hippocampal system function. Behavioral Neuroscience, 108(2), 227–234. 10.1037//0735-7044.108.2.227 [DOI] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, & Malinow R (2005). Postsynaptic Receptor Trafficking Underlying a Form of Associative Learning. Science, 308(5718), 83–88. 10.1126/science.1103944 [DOI] [PubMed] [Google Scholar]
- Sabban EL, & Serova LI (2009). Influence of prior experience with homotypic or heterotypic stressor on stress reactivity in catecholaminergic systems. Stress, 10(2), 137–143. 10.1080/10253890701404078 [DOI] [PubMed] [Google Scholar]
- Sammito S, Thielmann B, Zimmermann P, & Böckelmann I (2015). Influence of post-traumatic stress disorder on heart rate variability as marker of the autonomic nervous system - a systematic review. Fortschritte Der Neurologie ? Psychiatrie, 83(1), 30–37. 10.1055/s-0034-1398779 [DOI] [PubMed] [Google Scholar]
- Sandi C, Merino JJ, Cordero MI, Touyarot K, & Venero C (2001). Effects of chronic stress on contextual fear conditioning and the hippocampal expression of the neural cell adhesion molecule, its polysialylation, and L1. Neuroscience, 102(2), 329–339. 10.1016/s0306-4522(00)00484-x [DOI] [PubMed] [Google Scholar]
- Sangha S, Robinson PD, Greba Q, Davies DA, & Howland JG (2014). Alterations in Reward, Fear and Safety Cue Discrimination after Inactivation of the Rat Prelimbic and Infralimbic Cortices. Neuropsychopharmacology, 39(10), 2405–2413. 10.1038/npp.2014.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarelli AJ, Khan AM, & Poulos AM (2018). Contextual fear retrieval-induced Fos expression across early development in the rat: An analysis using established nervous system nomenclature ontology. Neurobiology of Learning and Memory, 155, 42–49. 10.1016/j.nlm.2018.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, & Munck AU (2000). How Do Glucocorticoids Influence Stress Responses? Integrating Permissive, Suppressive, Stimulatory, and Preparative Actions. Endocrine Reviews, 21(1), 55–89. 10.1210/edrv.21.1.0389 [DOI] [PubMed] [Google Scholar]
- Schell TL, Marshall GN, & Jaycox LH (2004). All Symptoms Are Not Created Equal: The Prominent Role of Hyperarousal in the Natural Course of Posttraumatic Psychological Distress. Journal of Abnormal Psychology, 113(2), 189–197. 10.1037/0021-843x.113.2.189 [DOI] [PubMed] [Google Scholar]
- Seo D, Carillo MA, Lim SC-H, Tanaka KF, & Drew MR (2015). Adult Hippocampal Neurogenesis Modulates Fear Learning through Associative and Nonassociative Mechanisms. The Journal of Neuroscience, 35(32), 11330–11345. 10.1523/jneurosci.0483-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp BM (2017). Basolateral amygdala and stress-induced hyperexcitability affect motivated behaviors and addiction. Translational Psychiatry, 7(8), e1194. 10.1038/tp.2017.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, & Quirk GJ (2011). Dissociable Roles of Prelimbic and Infralimbic Cortices, Ventral Hippocampus, and Basolateral Amygdala in the Expression and Extinction of Conditioned Fear. Neuropsychopharmacology, 36(2), 529–538. 10.1038/npp.2010.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneddon EA, Riddle CA, Schuh KM, Quinn JJ, & Radke AK (2021). Selective enhancement of fear learning and resistance to extinction in a mouse model of acute early life trauma. Learning & Memory, 28(1), 12–16. 10.1101/lm.052373.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampanato J, Polepalli J, & Sah P (2011). Interneurons in the basolateral amygdala. Neuropharmacology, 60(5), 765–773. 10.1016/j.neuropharm.2010.11.006 [DOI] [PubMed] [Google Scholar]
- Sullivan RM, & Gratton A (2002). Behavioral effects of excitotoxic lesions of ventral medial prefrontal cortex in the rat are hemisphere-dependent. Brain Research, 927(1), 69–79. 10.1016/s0006-8993(01)03328-5 [DOI] [PubMed] [Google Scholar]
- Sullivan RM, Landers M, Yeaman B, & Wilson DA (2000). Good memories of bad events in infancy. Nature, 407(6800), 38–39. 10.1038/35024156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Gooch H, & Sah P (2020). Fear conditioning and the basolateral amygdala. F1000Research, 9, F1000 Faculty Rev-53. 10.12688/f1000research.21201.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RF, & Spencer WA (1966). Habituation: A model phenomenon for the study of neuronal substrates of behavior. Psychological Review, 73(1), 16–43. 10.1037/h0022681 [DOI] [PubMed] [Google Scholar]
- Tovote P, Fadok JP, & Lüthi A (2015). Neuronal circuits for fear and anxiety. Nature Reviews Neuroscience, 16(6), nrn3945. 10.1038/nrn3945 [DOI] [PubMed] [Google Scholar]
- Vidal-Gonzalez I, Vidal-Gonzalez B, Rauch SL, & Quirk GJ (2006). Microstimulation reveals opposing influences of prelimbic and infralimbic cortex on the expression of conditioned fear. Learning & Memory, 13(6), 728–733. 10.1101/lm.306106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LE, Sprang KR, & Rothbaum BO (2018). Treating PTSD: A Review of Evidence-Based Psychotherapy Interventions. Frontiers in Behavioral Neuroscience, 12, 258. 10.3389/fnbeh.2018.00258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weikum ER, Knuesel MT, Ortlund EA, & Yamamoto KR (2017). Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nature Reviews Molecular Cell Biology, 18(3), 159–174. 10.1038/nrm.2016.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicking M, Steiger F, Nees F, Diener SJ, Grimm O, Ruttorf M, Schad LR, Winkelmann T, Wirtz G, & Flor H (2016). Deficient fear extinction memory in posttraumatic stress disorder. Neurobiology of Learning and Memory, 136, 116–126. 10.1016/j.nlm.2016.09.016 [DOI] [PubMed] [Google Scholar]
- Wiltgen BJ (2006). Context Fear Learning in the Absence of the Hippocampus. Journal of Neuroscience, 26(20), 5484–5491. 10.1523/jneurosci.2685-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpe J (1961). THE SYSTEMATIC DESENSITIZATION TREATMENT OF NEUROSES. The Journal of Nervous and Mental Disease, 132(3), 189–203. 10.1097/00005053-196103000-00001 [DOI] [PubMed] [Google Scholar]
- Yeh S-H, Mao S-C, Lin H-C, & Gean P-W (2006). Synaptic Expression of Glutamate Receptor after Encoding of Fear Memory in the Rat Amygdala. Molecular Pharmacology, 69(1), 299–308. 10.1124/mol.105.017194 [DOI] [PubMed] [Google Scholar]
- Zhang W, & Rosenkranz JA (2012). Repeated restraint stress increases basolateral amygdala neuronal activity in an age-dependent manner. Neuroscience, 226, 459–474. 10.1016/j.neuroscience.2012.08.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ge T. tong, Yin G, Cui R, Zhao G, & Yang W (2018). Stress-Induced Functional Alterations in Amygdala: Implications for Neuropsychiatric Diseases. Frontiers in Neuroscience, 12, 367. 10.3389/fnins.2018.00367 [DOI] [PMC free article] [PubMed] [Google Scholar]