

Abstract
Excessive fat accumulation in the liver has become a major health threat worldwide. Unresolved fat deposition in the liver can go undetected until it develops into fatty liver disease, followed by steatohepatitis, fibrosis, cirrhosis, and eventually hepatocellular carcinoma. Lipid deposition in the liver is governed by complex communication, primarily between metabolic organs. This can be mediated by hormones, organokines, and also, as has been more recently discovered, metabolites. Although how metabolites from peripheral organs affect the liver is well documented, the effect of metabolic players released from the liver during the development of fatty liver disease or associated comorbidities needs further attention. Here we focus on interorgan crosstalk based on metabolites released from the liver and how these molecules act as signaling molecules in peripheral tissues. Due to the liver’s specific role, we are covering lipid and bile mechanism-derived metabolites. We also discuss the high sucrose intake associated with uric acid release from the liver. Excessive fat deposition in the liver during fatty liver disease development reflects disrupted metabolic processes. As a response, the liver secretes a variety of signaling molecules as well as metabolites which act as a footprint of the metabolic disruption. In the coming years, the reciprocal exchange of metabolites between the liver and other metabolic organs will gain further importance and will help to better understand the development of fatty liver disease and associated diseases.
Keywords: Liver, Fatty liver disease, Metabolomics, Free fatty acids, Bile acids, Uric acid, Hepatokines, Organokines, Metabolites, Interorgan crosstalk, NAFLD, NASH
Introduction
Hepatic fat accumulation, also known as fatty liver disease, is a rising global health problem affecting more than 25% of adults worldwide [1, 2] and creates a substantial burden for our society. The incidence of fatty liver disease will probably increase in the following years if the current trends continue [3]. Fatty liver disease is strongly associated with comorbidities such as obesity, type 2 diabetes (T2D), hyperlipidemia, hypertension, and the metabolic syndrome [2]. Although weight loss improves liver histology and thus has a positive effect on disease progression, patient compliance with strict diets is usually low, with a high relapse rate [4]. Therefore, a better understanding of the molecular mechanisms causing the disease and new targets for therapeutic intervention are urgently needed.
Although the accumulation of lipid droplets (LDs) in hepatocytes has long been recognized as the hallmark of fatty liver disease, it is now widely accepted that the storage of excess lipid molecules in the form of triacylglycerol (TAG) is a protective mechanism against cellular lipotoxicity [5–7]. Lipid accumulation is probably a result of overflow after the adipose tissue expansion is exceeded. Indeed, hepatocytes undergo cellular stress due to lipotoxicity when the excess accumulation of free fatty acids (FAs) cannot be disarmed through the lipid storage mechanism or FA oxidation [8–10]. Untreated fatty liver disease-induced stress can aggravate and lead to hepatocyte injury and eventually cell death, the hallmark of non-alcoholic steatohepatitis [11]. Further progression of steatohepatitis may lead to fibrosis, cirrhosis, and eventually hepatocellular carcinoma, the fourth leading cause of death from cancer worldwide [12]. Recently a group of experts agreed on revising and updating the nomenclature and disease definition for non-alcoholic fatty liver disease (NAFLD) and NASH to metabolic-associated fatty liver disease (MAFLD) [13]. This new definition better reflects the complexity of the disease and its consequences and integrates metabolic dysfunction into the terminology. However, to keep it consistent and easier to follow, we will use the term fatty liver disease throughout the manuscript.
Fatty liver disease is considered a liver-focused disease. However, its strong association with obesity, T2D, and the metabolic syndrome suggests a complex metabolic network between liver and adipose tissue as well as other tissues (e.g., pancreatic islets, muscle, heart, and others). Therefore, we need to consider “interorgan crosstalk”, which can be defined as signaling between different tissues promoted by secreting factors into the bloodstream. The importance of fatty liver disease-associated interorgan crosstalk affecting peripheral tissues via hormones, organokines [14, 15], microRNAs [16], and extracellular vesicles [17] has been well recognized. However, how energy homeostasis byproducts secreted from the liver during the development of fatty liver disease and steatohepatitis orchestrate other metabolic players such as muscle, adipose tissue, and pancreas requires further study. Being the master regulator of lipid metabolism, the liver robustly responds to metabolic dysregulation by fine-tuning the lipid output. Another liver-specific function is the synthesis, secretion, and absorption of bile acids. Bile acids in the plasma act as signaling molecules and are altered in many liver diseases. Therefore, the role of bile acids in fatty liver disease development will also be covered in this review. Finally, the liver is exposed to large amounts of dietary fructose due to unhealthy eating habits and the liver’s coping mechanism is closely associated with uric acid metabolism. Thus, in this review, we intend to focus on how liver-derived lipids, bile acids, and uric acid, are involved in interorgan crosstalk that influences energy homeostasis in the context of the development of fatty liver disease.
Liver-derived lipids as signaling molecules
The human body is evolutionarily well-adapted to control energy homeostasis by storing excess energy in the form of neutral lipids that can be used when nutrients are scarce. Maintaining energy homeostasis is demanding and requires robust orchestration between various tissues. Undoubtedly, the liver is a master regulator of this metabolic network, as it regulates a variety of metabolic functions, including but not limited to the regulation of glucose synthesis (i.e., gluconeogenesis), glycogen storage, and bile acid synthesis. The liver also fine-tunes lipid distribution in the body by plasma lipid uptake, de novo lipid synthesis, and lipid secretion into the bloodstream. Of course, adipose tissue is the main lipid storage of the body and the primary source of plasma FAs. Upon high energy demand (fasting or exercise), adipocyte lipolysis releases FAs from TAGs into the bloodstream for transport to the liver and other organs as an additional source of energy [18]. Hepatocytes then take up plasma FAs either by FA transport proteins (FATP) 2, 5 [19], the scavenger receptor CD36 [20, 21] or to a lesser extent passive diffusion [22]. Moreover, FAs are also synthesized in the liver or taken up from the diet in the form of chylomicron remnants. In the case of obesity-induced insulin resistance, increased adipocyte lipolysis releases more FAs from the TAG stores into circulation [23]. Ironically, this might occur due to endoplasmic reticulum (ER) stress caused by increased dietary FA flux [24] and excessive expansion of adipocytes leading to adipose insulin resistance [25]. Therefore, the net outcome is increased levels of serum FA which are taken up by the liver. The resulting accumulation of FAs in hepatocytes is either used in mitochondria for β-oxidation or esterified into TAG. Hepatic accumulation of TAGs is either utilized to form very low-density lipoproteins (VLDL) or stored in LDs in hepatocytes, the phenotypic trademark of fatty liver disease (Fig. 1). If the elevated FA influx cannot be compensated by intracellular coping mechanisms, further FA accumulation results in lipotoxicity, which eventually leads to cellular damage.
Fig. 1.
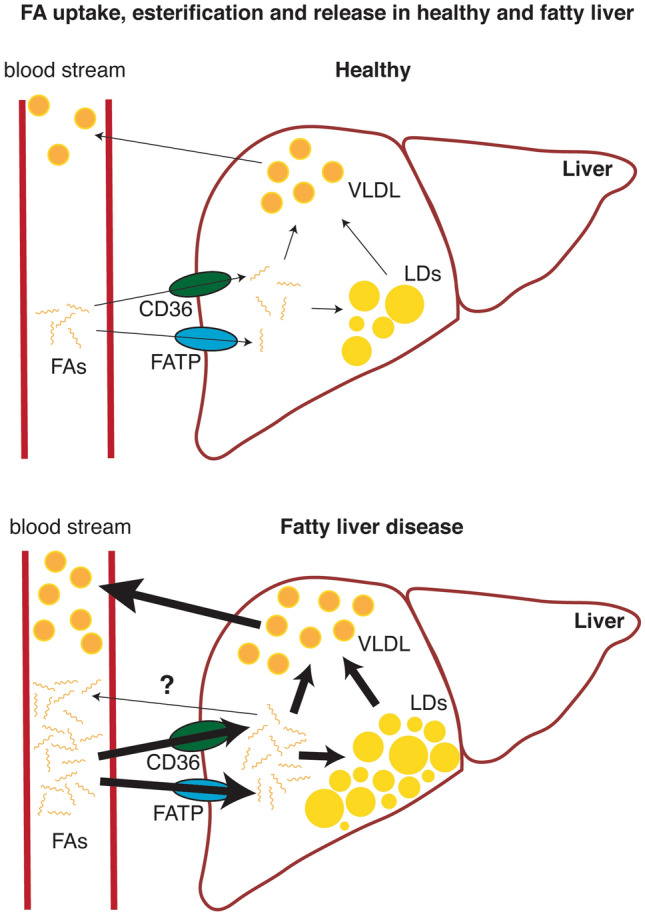
Under physiological conditions, free fatty acids (FA) taken up from plasma by the liver through specific CD36 and FA transport protein (FATP) family receptors can undergo β-oxidation (not shown), Lipid droplet (LD) formation or VLDL formation, depending on the energy needs of the body. However, the metabolic conditions leading to elevated plasma FA concentrations result in increased FA uptake, leading to elevated intracellular FA to which the liver responds by increasing LD deposition, β-oxidation, and VLDL formation. Current evidence also indicates that FAs might be released or transported to the bloodstream as a response to intracellular FA increase
One of the liver’s coping mechanisms for elevated intracellular lipid concentrations is to secrete them in VLDL particles, rich in TAG and cholesteryl esters [26]. VLDL delivers energy-rich TAG to peripheral tissues via the bloodstream, where in endothelial cells lipoprotein lipase (LPL) hydrolyzes TAG to liberate diacylglycerol (DAG) and FA [27]. Adipose tissue, heart, and skeletal muscle express high levels of LPL and are exposed to TAG-derived metabolites. Peripheral tissues take up released FA via FA transporter receptors including CD36 and FATP [28] or passive diffusion (Fig. 2A).
Fig. 2.

A TAG molecules constituting the VLDL particles secreted from the liver can be liberated by lipoprotein lipase (LPL). Free fatty acids (FAs) can then be taken up by CD36 or FA transport protein receptors. Alternatively, FAs can pass through the lipid bilayer by passive diffusion. VLDL particles can also be taken up directly by target tissues via VLDL receptor (VLDLR)-mediated endocytosis. B The amount and the composition of VLDL is altered during the development of fatty liver disease. While LPL-mediated FA uptake is increased in adipose, muscle and heart tissue; receptor-mediated VLDLR uptake is critical for the pancreas. The red arrow indicates the deleterious effect of increased VLDL concentrations. TAG triacylglycerol, PL phospholipids, SPL sphingolipids, CE cholesteryl esters, Cer ceramide
During the development of fatty liver-associated comorbidities such as T2D and obesity, the liver secretes large amounts of VLDL particles [29] that eventually lead to elevated levels of plasma FA uptake through lipase activity (LPL) [30]. Indeed, increased FA uptake largely contributes to tissue insulin resistance, which was shown by muscle-specific deletion of LPL [31]. Lipotoxicity-induced cardiomyopathies are among fatty liver disease-, T2D-, and obesity-associated comorbidities [32, 33]. FAs are the primary energy source of cardiomyocytes and can either be taken up directly from plasma or from lipoproteins via LPL. However, lipoprotein-derived FAs seem to be a limiting factor for cardiac lipid uptake as heart-specific LPL deletion increases plasma TGs but not plasma FAs [34].
The role of the adipose tissue in increasing plasma FAs, thereby increasing liver lipid deposition in the context of obesity and T2D is well acknowledged and has been extensively discussed elsewhere [35, 36]. During fatty liver disease development, the liver also influences the remodeling of the adipose tissue. VLDL, for example, is known to be a major source of lipids for the adipose tissue since the deletion of the VLDL receptor (VLDLR) leads to decreased adipogenesis in high-fat diet-fed mice [37]. Although adipose VLDLR is decreased in morbidly obese patients [38], it mediates excessive VLDL uptake in macrophages and exacerbates adipose tissue inflammation [39, 40].
Although LPL expression in the pancreas is relatively low, evidence indicates that elevated VLDL levels contribute to the development of pancreatitis, as a common condition among obese and T2D patients [41, 42], and are further known to be associated with fatty liver disease [43]. In mice, deletion of the VLDLR in pancreatic stellate cells ameliorates the development of pancreatitis [44].
Ceramides belong to a class of lipids called sphingolipids, essential components of cellular membranes [45] and ceramide synthesis favors saturated long-chain FA over unsaturated or short-chain FA [46]. Ceramides produced in the liver are distributed to the other organs through VLDL and plasma ceramide levels are strong predictors of insulin resistance and cardiovascular diseases [47–49]. Interestingly, unlike in the liver, ceramide levels are associated with insulin resistance in skeletal muscle [50, 51]. Studies have shown that ceramides negatively influence the AKT pathway [52–54], the primary signal transduction mode of the insulin receptor. Studies suggest that this might happen by preventing AKT from translocating to the plasma membrane [55] through PKCζ-mediated inhibition of phosphatidylinositol 3,4,5-trisphosphate binding the AKT pleckstrin homology domain [56, 57] or by augmenting protein phosphatase 2A (PP2A) dephosphorylation of AKT residues [57, 58]. Ceramides may also impair the insulin response in skeletal muscles independent of AKT by inhibiting glucose transporter 4 (GLUT4) translocation to the plasma membrane, thereby reducing muscle glucose uptake [59]. Excess circulating glucose upon skeletal muscle insulin resistance might be taken up by the liver and serves as a source for de novo lipogenesis, of which the primary output is saturated long-chain FA, culminating in further lipid deposition in the liver and secretion in VLDL particles [60]. Recent studies revealed that senescent hepatocytes and adipocytes can promote the development of metabolic diseases. This occurs either by reducing mitochondrial FA oxidation, causing hepatic lipid accumulation and fatty liver disease [61] or by triggering the NFkB-dependent senescence-associated secretory phenotype in adipocytes and causing adipose inflammation [62], a hallmark of diabetes. Ceramides have been shown to promote senescence [63] through PP1 and PP2A-mediated dephosphorylation and inactivation of cyclin-dependent kinase 2 (CDK2) [64]. Taken together, it is not surprising that blocking ceramide synthesis is effective in preventing diet-induced insulin resistance [65]. Conversely, senotherapeutic drugs (inhibiting senescence) may be useful too, since they have recently been shown to improve the regenerative capacity of the adult liver [66]. In summary, these new types of drugs suggest a bright future for treating liver diseases or at least some of their clinical manifestations.
FA are the building blocks of membranes, stored lipids and efficient energy substrates, composed of hydrocarbon chains linked to a carboxylic acid group. FAs that are taken up by the cell, are either re-esterified or activate peroxisome proliferator-activated receptor α (PPARα) to induce mitochondrial and peroxisomal β-oxidation to yield acetyl-CoA [67, 68], with the ultimate goal of generating ATP. Accordingly, PPARα levels are increased in fatty liver disease patients and its deletion leads to the worsening of steatosis, reflecting a compensatory mechanism against excessive FA influx [69]. However, NASH patients exhibit reduced PPARα levels [70] which further exacerbates the condition by impairing FA clearance and causing more FAs to enter the TAG synthesis pathway in a vicious cycle. Overload of cellular FA exceeding the capacity of TAG synthesis leads to increased DAG levels, an intermediate metabolite, which activates protein kinase C (PKC) and blunts insulin receptor signaling [71], resulting in hepatic insulin resistance. It is thus not a coincidence that about 34–70% of diabetic patients also suffer from fatty liver disease [72].
The type of circulating FAs can impact other organs besides the liver. Medium- and long-chain FA are ligands for the receptor GPR40 and act on pancreatic β-cells to enhance the secretion of insulin [73] by inducing the endoplasmic reticulum (ER) to release its calcium stores and triggering exocytosis of insulin vesicles [74]. In addition to the pancreas, FAs activate GPR40 as well as GPR120 on the enteroendocrine cells of the gut to induce the secretion of incretins and indirectly promote pancreatic insulin secretion [75, 76]. Notably, we have observed that the chronic release of excessive amounts of FA into the blood can lead to insulin resistance by sustaining elevated blood insulin levels in a mouse model with dysfunctional hepatic mitochondrial β-oxidation [77]. This FA-mediated organ crosstalk might provide a mechanism by which organ-specific insulin resistance can develop into systemic insulin resistance. Interestingly, GPR40 deficiency protects from insulin resistance [78] while GPR120 deficiency results in obesity and insulin resistance [79]. This discrepancy might be due to a preference for GPR40 for saturated FA and GPR120 for unsaturated FA [80], suggesting a difference in metabolic regulation by saturated and unsaturated FA [81].
Short-, medium- and long-chain FA, can influence insulin secretion indirectly. Short-chain FAs (SCFAs) are ligands of GPR41 and GPR43 and promote the secretion of incretins by enteroendocrine cells of the gut [82]. Paradoxically, SCFAs inhibit insulin signaling only in adipocytes through the activity of GPR43 but increase insulin sensitivity in other organs. This is proposed to prevent excessive storage of energy in the adipose tissue while enhancing energy expenditure in other organs. Of note, this effect was dependent on the composition of the gut microbiota, suggesting microbes are the primary sources of SCFAs [83, 84]. This indicates the importance of metabolite signaling between the gut microbiota and the metabolic organs of our body and will be discussed further below in the context of bile acids. The human body produces SCFAs as a product of β-oxidation and ketogenesis or from ethanol by mitochondrial acetyl-CoA hydrolase (ACOT9) upon ethanol consumption. Free acetate, a type of SCFA, is not used by the liver for ATP generation since hepatic acetyl CoA synthetase has a low affinity for acetate [85]. Acetate is more likely delivered to the heart, as the cardiac acetyl-CoA synthetase has a high affinity for acetate and converts it to acetyl-CoA for energy [85]. On the other hand, excessive ethanol consumption leading to elevated levels of plasma acetate and, correspondingly, acetyl-CoA, can enhance histone acetylation in immune cells to up-regulate the expression of inflammatory cytokines, contributing to acute alcoholic hepatitis [86] or in hippocampal cells to modulate transcriptional responses of alcohol-associated learning and memory [87].
As discussed above, FAs are primarily released from the adipose tissue. Indeed, increased plasma FAs, as in obesity, are scavenged by the liver, leading to elevated lipid deposition. Excessive lipid deposition, namely liver steatosis, triggers compensatory mechanisms by enhancing VLDL formation and secretion, thereby reducing the lipid burden of the liver [26]. Although VLDL secretion is best known as a FA export mechanism, a recent study on liver-specific CDK mutant mice has shown that the liver might directly release FAs into the plasma [77]. CDK family proteins are master regulators of the cell cycle [88]. Among other CDKs, CDK1 drives the cell cycle through G2 to mitosis when bound by the cyclin B1 protein; and its deletion results in mitosis failure, but hepatocytes grow further in size [89]. We have shown that Cdk1 deletion in hepatocytes, thereby preventing mitosis, results in the depletion of liver TAGs and substantially decreased plasma VLDL levels. Partially due to elevated expression of liver adipose triglyceride lipase (ATGL, encoded by PNPLA2), the rate-limiting enzyme releasing FA from cytosolic LDs [90]. Surprisingly, however, plasma FA concentrations were significantly elevated in liver-specific Cdk1 mutant mice [77]. As a result, FAs affected the physiology of the adipose tissue, muscle, and pancreatic islets, resulting in hyperinsulinemia. Over time, these animals developed insulin resistance, and the increased blood glucose levels activated the transcription factor liver X receptor (LXR). The LXR/RXR dimer then promotes the expression of lipogenic genes, resulting in liver steatosis. Interestingly, the phenotype of this mouse model with a mutation in hepatocytes was dependent on interorgan crosstalk, suggesting that more than one organ is at fault in T2D and fatty liver disease [77].
Although the fatty liver disease-associated mechanisms seem to contribute to the total lipid burden of the body (Fig. 2B), there is still a need for a deeper understanding of how the liver remodels peripheral tissues during the development of the disease. Future studies on diverse lipid species secreted from the liver and their effects on target tissues would pave the way to develop more precise therapeutic molecules against fatty liver disease or associated diseases.
Bile acids
In addition to regulating metabolic functions, the liver also plays a critical role, together with the digestive system, to control bile acid production, secretion, and recycling. Although bile acids are well-acknowledged for their function in emulsifying lipids in the intestinal lumen, it has also been shown that plasma bile acid concentrations are dysregulated during the development of fatty liver disease [91] and more importantly, they act as signaling molecules on peripheral tissues [92].
Primary bile acids produced by hepatocytes from cholesterol are chenodeoxycholic acid (CDCA) and cholic acid (CA). These are typically conjugated to glycine or, to a lesser extent, taurine before secretion [93]. Conjugated bile acids are usually referred to as bile salts, stored in the gallbladder and secreted via the bile duct into the duodenum, which is mediated by the ATP-binding cassette subfamily B member 11 (ABCB11) and multidrug resistance-associated protein 2 (MRP2) transporters [94]. Up to 95% of primary bile acids are recycled in the ileum, either via the apical sodium-dependent bile acid transporter (ABST) found on enterocytes or via passive diffusion [95, 96]. Enterocytes secrete bile acids into the portal circulation via the organic solute transporter α (OSTα) and OSTβ proteins [97]. Portal bile acids are then actively transported to hepatocytes by the organic anion transporting polypeptide (OATP) and sodium taurocholate co-transporting polypeptide (NTCP) [98]. A portion of the bile salts, however, becomes deconjugated by microbial hydrolases, and bacterial enzymes further convert these into secondary bile acids: lithocholic acid (LCA) and deoxycholic acid (DCA), which are derived from CDCA and from CA [99], respectively. Therefore, the gut microbiome plays an important role in the recycling of bile acids. Secondary bile acids can then re-enter the enterohepatic system by passive diffusion and serve as signaling molecules [100]. A small portion of bile acids in the portal circulation may escape liver uptake and can be found in the bloodstream. Not surprisingly, serum bile acids are elevated postprandially [101]. However, the liver robustly clears the remaining bile acids from circulation. Therefore, the persistency of high bile acid concentration in the circulation has been associated with liver diseases [102–104] and can be manifested by jaundice.
The presence of circulating bile acids is primarily recognized by two receptors found in peripheral tissues: the farnesoid X receptor (FXR) and G Protein-Coupled Bile Acid Receptor 1 (TGR5). Unconjugated primary bile acids are taken up through the intestines, bind, and activate the transcription factor FXR [105, 106]. This allows for negative feedback on bile acid synthesis on two fronts. Activation of hepatic FXR promotes expression of nuclear receptor subfamily 0 group B member 2 (NR0B2), which binds and represses liver receptor homolog-1 (LRH-1) transcriptional activity, thereby reducing the expression of cholesterol 7 alpha-hydroxylase (CYP7A1), a key enzyme in bile acid synthesis [107, 108]. Alternatively, activation of FXR in the intestine induces fibroblast growth factor 15 (FGF15) production, which travels to the liver via the portal vein and stimulates the FGFR4-JNK-NR0B2 pathway in hepatocytes to inhibit the transcription of CYP7A1 [109]. Primary bile acids also regulate liver lipid metabolism by repressing the transcription factor PPARγ coactivator 1- α (PGC-1α) using the FXR-NR0B2 axis [110], especially given the crucial role of PGC-1α in FA oxidation and utilization [111].
The in vivo data reveals a tissue-specific impact of FXR function on the metabolic outcome. Whole-body FXR-deficiency in mouse models of insulin resistance protects mice from diet-induced obesity and glucose intolerance [112–114], independent of hepatic FXR, as hepatic insulin resistance remains unaffected in the absence of FXR [112]. In this context, intestinal FXR appears to be responsible for promoting systemic insulin resistance, as intestine-specific inhibition of FXR led to a reduced ceramide synthesis and therefore prevented and reversed the development of fatty liver disease [115], possibly by preventing adipose inflammation caused by bile acids produced by the gut microbiota [114]. In contrast, hepatic FXR is protective against fatty liver disease, as liver-specific knockout of Fxr enhanced the incidence of hepatic steatosis [116]. A recent study supports the tissue-specific responses of FXR activation by providing evidence that hepatic FXR modulates lipogenesis, whereas intestinal FXR controls lipid absorption from the gut [117]. Nevertheless, conjugated bile acids might be antagonists of FXR [118, 119], instead of unconjugated bile acids. Since bile acids are released from hepatocytes in the conjugated form and 95% of the conjugated bile acids are absorbed from the ileum [120] (Fig. 3A), hepatic FXR exposed to the extracellular environment in the liver are more likely to encounter conjugated bile acids and become inhibited. On the other hand, intestinal cells are more likely to be exposed to unconjugated bile salts as the gut microbiota deconjugates a small percentage of bile acids that are released into the intestines to promote FXR signaling [119].
Fig. 3.
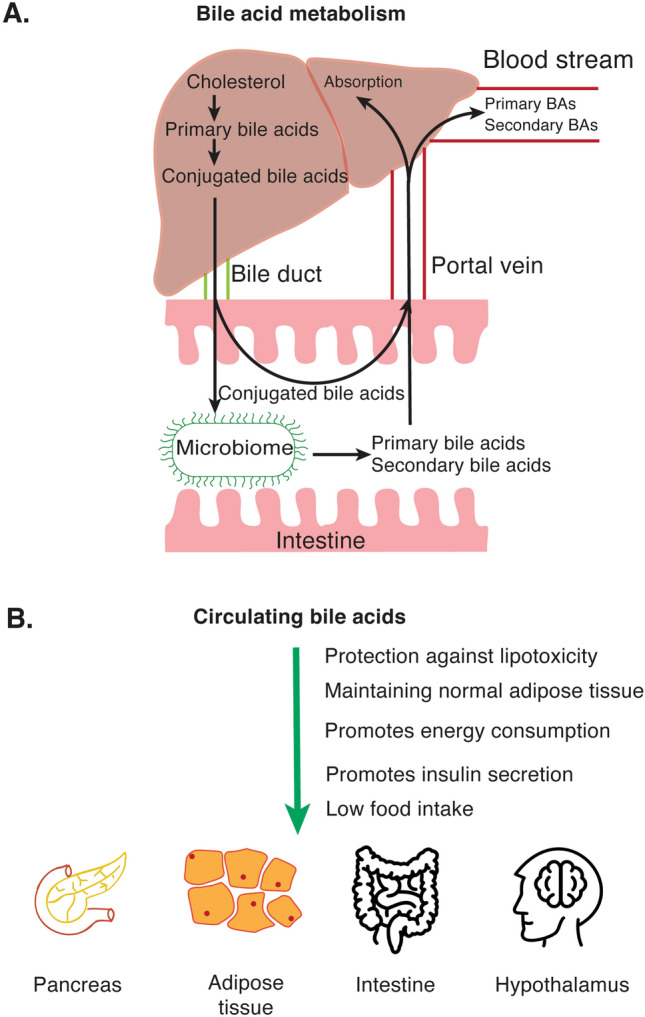
A The liver synthesizes primary bile acids from cholesterol. Bile acids are also conjugated either with glycine and taurine and become conjugated bile acids (CBAs). CBAs are secreted into the intestines through bile ducts. CBAs can be deconjugated and/or turned into secondary bile acids by the microbiome. Primary and secondary bile acids are then absorbed through the ileum and shuttled back to the liver through the portal vein. Although most of the bile acids are taken up by hepatocytes, a small portion escapes from absorption and is released into the bloodstream. Bile acids can directly bind to the nuclear receptor Farnesoid X receptor (FXR) to mediate gene regulation or act on G-protein coupled receptor TGR5 to initiate an intracellular signal cascade. B Although plasma bile acids seem to increase in patients with fatty liver, the effect on peripheral tissue is mostly determined as anti-diabetic (green arrow)
Besides the entero-hepatic system, although expressed at relatively low levels, FXR activation in various tissues significantly impacts energy homeostasis. In the pancreas of obese animals, FXR translocates to the nucleus and protects pancreatic islet cells against lipotoxicity [121]. In another study, FXR activation in β-cells has been shown to stimulate insulin secretion and glucose uptake [122]. Another strong implication of the roles of FXR in energy metabolism is adipose tissue: animals lacking FXR show impaired adipose tissue development and insulin resistance [123]. Interestingly, hypothalamic FXR has been reported to have a residual expression profile [124] but modulates brown adipose tissue through the sympathetic system upon FXR agonist treatment [125].
Obviously, findings of FXR activity are contradictory since fatty liver disease patients show elevated levels of serum bile acids despite the bile acid receptor FXR being mostly associated with improved energy homeostasis [126]. This contradiction could partially be attributed to the bile acid composition instead of the total amount of bile acids. In one clinical study, although serum total bile acid concentration was significantly elevated in fatty liver disease patients compared to the healthy cohort, the amount was not significantly altered between patients suffering from fatty liver or steatohepatitis [127]. Conversely, conjugated bile acid ratios were decreased in NASH compared to fatty liver disease [127] while cholic acid conjugates are increased in NASH. The other plausible explanation could be the differential effect of bile acids on FXR receptors. One such example is that 12α-hydroxylated secondary bile acids, LCA and DCA, increase during T2D and are known to antagonize FXR [128].
Secondary bile acids like taurolithocholic acid (TLCA), LCA, and DCA [129], produced by the gut microbiome from primary bile acids, are preferential ligands for the receptor TGR5 [130]. Unlike FXR, studies on TGR5 conclusively point to TGR5 being anti-diabetic. For example, treatment with bile acids activates TGR5 in brown adipose tissues in mice or skeletal muscle in humans to induce type 2 iodothyronine deiodinase activity, which increases the active thyroid hormone levels and promotes higher energy consumption [131]. Bile acid-mediated activation of TGR5 also regulates glucose homeostasis by enhancing insulin secretion by pancreatic β-cells through cAMP-PKA signaling [132] or triggering intestinal incretin secretion [133]. Accordingly, TGR5 signaling protects against diet-induced obesity [131, 133] and other diabetes-associated pathologies such as fatty liver disease [134], diabetic retinopathy [135], and nephropathy [136]. Notably, observations in humans subjected to CDCA treatment support the findings in vitro and in mice, with elevated brown adipose tissue activity and energy expenditure upon administration of TGR5 agonists but not FXR agonists [137]. In distinct tissues such as the nervous system, activation of TGR5 in the hypothalamus was shown to protect against obesity by reducing food intake [138].
The sensors that secondary bile acids can modulate are not restricted to TGR5; they also bind to FXR receptors with high affinity. It is acknowledged that disruption of the gut microbiome abrogates secondary bile acid formation, disrupting TGR5 and FXR stimuli. Of course, this highlights the importance of maintaining a healthy gut microbiome for proper physiological metabolism. Due to the pleiotropic effect of FXR, long-known inhibitors are of limited use in the clinic. Therefore, in addition to other pharmacological interventions for fatty liver disease, the gut microbiome emerges as an alternative therapeutic option.
The recognition of bile acid metabolism and its contribution to energy homeostasis (Fig. 3B) has enabled the discovery of specific bile acid agonists, which are currently under clinical investigation. Although the results are promising, further safety studies are needed for general use in fatty liver disease patients [94]. This clearly indicates that further investigations on how different bile acid species act on peripheral tissues are needed. In addition to chemical intervention, modulation of microbiota would be a promising tool for adjusting specific plasma bile acid species.
Uric acid
Purine molecules, guanine, and adenine mediate critical cellular functions such as monomers in nucleotide synthesis or energy carriers. Purine catabolism takes place in the liver through nucleotidase, deaminase, and xanthine oxidase enzymes (Fig. 4A). The end product of the purine catabolism in the liver is uric acid which is released into the bloodstream and excreted through the kidneys [139]. Disrupted purine metabolism or uric acid excretion leads to elevated serum uric acid levels, namely hyperuricemia. Hyperuricemia has long been associated with gout, the accumulation of urate crystals in joints, causing painful inflammation. Recent studies have also shown that hyperuricemia is also associated with the development of fatty liver disease.
Fig. 4.
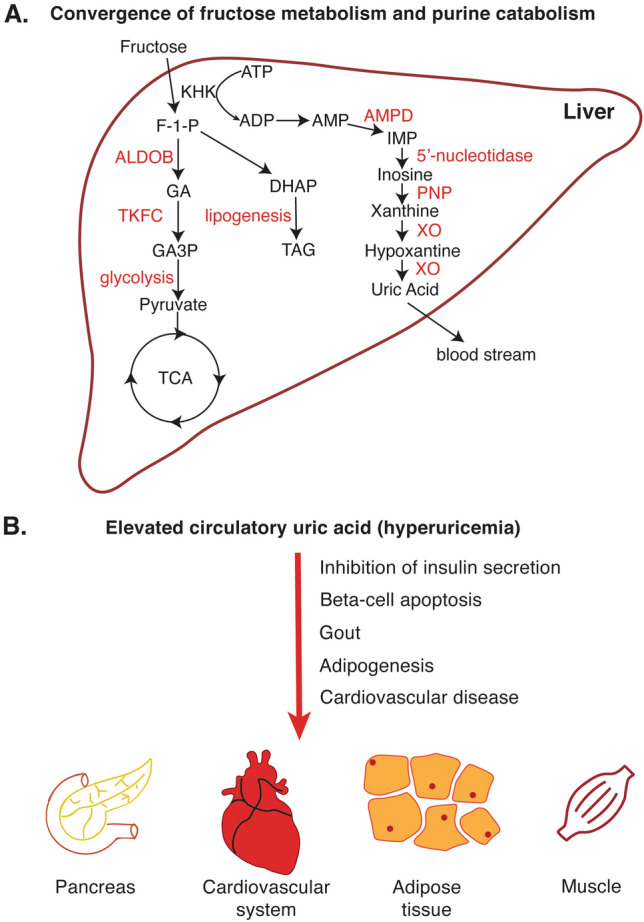
A After digestion, the liver is exposed to large amounts of fructose through the portal vein. Excessive fructose leads to a decrease in ATP levels and an increase in AMP concentration and AMP deaminase. Elevated deaminase activity promotes purine catabolism and eventually leads to a net increase in uric acid formation. B Elevated uric acid is a well-known cause of gout. It was also shown to inhibit insulin secretion, cause β-cell apoptosis, and adipogenesis. (the red arrow indicates the deleterious effect of hyperuricemia) TCA tricarboxylic acid cycle, F-1-P Fructose-1-phosphate, AMPD AMP deaminase, PNP purine nucleotide phosphorylase, XO xanthine oxidase, ALDOB aldolase B, TKFC triokinase/FMN cyclase, GA D-glyceraldehyde, GA3P D-glyceraldehyde-3-phosphase
The lifestyle and feeding habits of human beings have dramatically changed over the last decades, mainly due to the industrialization of food production. Similarly, the composition of the diet has also undergone fundamental changes. One such example is high fructose corn syrup which is heavily used in food and beverages as a sweetener [140]. It is well-documented that fructose has the propensity to promote fatty liver disease and other metabolic diseases [141]. Dietary fructose is taken up in the small intestine either by the transporter GLUT5 into intestinal epithelial cells [142], where it is metabolized by the ubiquitous ketohexokinase-C, or by GLUT2 into the hepatic portal system [143] to be delivered to hepatocytes and broken down by the hepatic ketohexokinase-A. The two ketohexokinases are alternatively spliced isoforms that utilize ATP to convert fructose to fructose-1-phosphate, which is converted to glyceraldehyde and dihydroxyacetone phosphate, after which the fructose metabolic pathway converges with glucose metabolism at the glycolytic step (Fig. 4A) [141]. Interestingly, the different routes of fructose uptake have distinct impacts on the development of fatty liver disease. Intestinal uptake and clearance of fructose protect against fructose-induced fatty liver by reducing the amount of fructose, microbiome, and bacterial toxins taken up by the liver, alleviating liver inflammation [144, 145]. On the other hand, hepatic fructose metabolism is linked to hepatic steatosis [141]. High hepatic ketohexokinase activity, a result of excessive fructose consumption overwhelming the intestinal fructose clearance capacity, can cause a drop in ATP and intracellular phosphate levels [146, 147], thereby triggering the activity of AMP deaminase [148, 149], which promotes purine catabolism. This explains the acute rise in serum uric acid, the end product of purine degradation, following fructose consumption [150]. This raises the question: why is uric acid unfavorable for the liver?
The observation that the fructose-mediated metabolic syndrome can be partially rescued by treatment with allopurinol, a xanthine oxidase inhibitor that blocks uric acid production, suggests that uric acid may be a mediator of fructose-dependent fatty liver development [151, 152]. This is supported by studies showing that treatment with xanthine oxidase inhibitors can rescue the steatotic phenotype in fatty liver disease patients and mouse models [153–155]. Notably, increased uric acid levels have been observed in non-dietary rodent models of insulin resistance by others and ourselves [77, 156], and elevated serum uric acid has been identified as a risk factor for the development of fatty liver disease in humans [157–159], implicating a more generic role for purine catabolism in fatty liver disease pathogenesis. Multiple mechanisms for uric acid-dependent fat accumulation in hepatocytes have been suggested. For example, uric acid induces oxidative stress leading to the accumulation of citrate, which serves as raw material for de novo lipogenesis [160], while promoting the expression of lipogenic enzymes via the JNK-sterol regulatory element-binding protein 1-c (SREBP-1c) pathway [161]. Alternatively, uric acid can cause ER stress, thereby enhancing ER stress-dependent cleavage and activation of SREBP-1c [162]. Uric acid may additionally promote insulin resistance by reducing the expression of the organokines FGF21 through the up-regulation of miR-149-5p [163]. FA oxidation is repressed due to redox inactivation of the β-oxidation enzyme enoyl-CoA hydratase 1 [160]. Besides promoting lipid accumulation, uric acid may also facilitate the progression from hepatic steatosis to steatohepatitis by inducing an inflammatory response through the release of C–C chemokine ligand 2 (CCL2) from endothelial cells [164] and activation of the NRLP3 inflammasome [165] via the ROS-TXNIP pathway [166] to promote cell death in hepatocytes [167].
Besides the liver, uric acid can also affect other organs (Fig. 4B). Perhaps, the most studied clinical manifestations of elevated plasma uric acid (hyperuricemia) in peripheral tissues are crystal formation in joints, namely gout [168]. However, hyperuricemia can also affect other metabolic organs including the adipose tissue and pancreas. Uric acid treatment has been shown to inhibit glucose-stimulated insulin secretion in isolated pancreatic islets and pancreatic β-cell lines while inducing β-cell apoptosis [169–171], a common phenomenon seen in T2D. Uric acid can also promote adipogenesis in mesenchymal stem cells, contributing to fructose-mediated obesity [172]. Interestingly, uric acid may be secreted by adipocytes, through enhanced uric acid production by adipose tissues from obese animals [173]. These findings highlight a positive feedback loop between the liver and the adipose tissue and the interplay between purine catabolism and lipogenesis in these two metabolic organs in cases of metabolic disease. In addition to its direct role on metabolic tissues, uric acid has been shown to act on the central nervous system and modulate energy metabolism. In one study, high-uric acid diet fed rats displayed elevated levels of inflammatory cytokines, activated NF-kB pathway and increased gliosis, a reactive response of glial cells, in the hypothalamus. This lead to dyslipidemia and glucose intolerance [174]. Furthermore, uric acid has also been associated with impaired cognitive functions in rat studies [175].
Despite the well-acknowledged anti-oxidant function in plasma, uric acid has also been described as a pro-oxidant in the cytoplasm or in atherosclerotic plaques, thereby causing/accelerating the development of cardiovascular disease [176].
Conclusions
In the industrialized world, diseases associated with excessive calorie intake have drawn much attention due to chronic but devastating physiological outcomes. In this context, fat accumulation in the liver and the development of fatty liver disease have long been considered associated with obesity and T2D. Here, we discussed selected liver-derived metabolites as a ‘cause’ of direct or indirect actions promoting the development of fatty liver disease.
As one can expect, no single liver-derived molecule has been discovered as the primary contributor to the fatty liver disease itself or the associated diseases. Accordingly, although fatty liver disease biomarkers have been intensively studied, and more metabolites have been covered, the predictive value of the available metabolites is still limited [177]. This is partially due to the current limitation in metabolite coverage of mass spectrometry-based metabolomics [178]. Furthermore, a mere concentration of the plasma metabolites might not reflect true biological functions. For example, plasma lipids derived from the liver are primarily carried in lipoprotein particles [179]. These lipoproteins are able to act as compartments and exchange various lipids (e.g., TAG and cholesteryl ester), which might alter the lipid species delivered via the uptake of lipoproteins in peripheral tissues.
As the number of covered metabolites in untargeted metabolomics studies increases, another issue remains to be tackled: despite primarily being byproducts of physiological processes, many metabolites can enter cells via passive diffusion, while several others are recognized by cellular receptors and taken up by the target tissues. However, many metabolite-specific tissue receptors are yet to be discovered [180].
Indeed, the development of fatty liver disease is a complex process and requires more mechanistic studies to better understand how metabolic disturbances abrogate the lipid compensation mechanisms of the liver. However, it is equally important to elaborate on how the liver responds to the altered physiology.
Acknowledgements
We thank all present and past members of the Kaldis laboratory for discussions, input, and support.
Author contributions
UK, JRO, KBK, LNZ, and PK conceptualized, wrote, and revised this review article.
Funding
Open access funding provided by Lund University. PK was supported by the Swedish Research Council (2021-01331), the Swedish Cancer Society (Cancerfonden; 21-1566Pj), the Crafoord Foundation (Ref. No. 20220628), the Faculty of Medicine, Lund University, the Swedish Foundation for Strategic Research Dnr IRC15-0067, and Swedish Research Council, Strategic Research Area EXODIAB, Dnr 2009-1039. L.N.Z. is supported by the IngaBritt och Arne Lundbergs Forskningsstiftelse LU2020-0013, the Crafoord Foundation (Ref. No. 20210516), and the Åke Wibergs Stiftelse. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Availability of data and material
This does not apply since this is a review article.
Declarations
Conflict of interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Ethics approval and consent to participate
This does not apply since this is a review article.
Consent for publication
All authors provide consent for publication.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Umur Keles and Jin Rong Ow have contributed equally.
References
- 1.Riazi K, Azhari H, Charette JH, Underwood FE, King JA, Afshar EE, et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol. 2022;7(9):851–861. doi: 10.1016/S2468-1253(22)00165-0. [DOI] [PubMed] [Google Scholar]
- 2.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 3.Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11–20. doi: 10.1038/nrgastro.2017.109. [DOI] [PubMed] [Google Scholar]
- 4.Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. 2015;149(2):367–378. doi: 10.1053/j.gastro.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45(6):1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 6.Li ZZ, Berk M, McIntyre TM, Feldstein AE. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl-CoA desaturase. J Biol Chem. 2009;284(9):5637–5644. doi: 10.1074/jbc.M807616200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr, Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA. 2003;100(6):3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geng Y, Faber KN, de Meijer VE, Blokzijl H, Moshage H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol Int. 2021;15(1):21–35. doi: 10.1007/s12072-020-10121-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.González-Rodríguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014;5(4):e1179. doi: 10.1038/cddis.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen X, Li L, Liu X, Luo R, Liao G, Li L, et al. Oleic acid protects saturated fatty acid mediated lipotoxicity in hepatocytes and rat of non-alcoholic steatohepatitis. Life Sci. 2018;203:291–304. doi: 10.1016/j.lfs.2018.04.022. [DOI] [PubMed] [Google Scholar]
- 11.Zhu B, Chan SL, Li J, Li K, Wu H, Cui K, et al. Non-alcoholic steatohepatitis pathogenesis, diagnosis, and treatment. Front Cardiovasc Med. 2021;8:742382. doi: 10.3389/fcvm.2021.742382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2021;18(4):223–238. doi: 10.1038/s41575-020-00381-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eslam M, Sanyal AJ, George J. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 2020;158(7):1999–2014.e1. doi: 10.1053/j.gastro.2019.11.312. [DOI] [PubMed] [Google Scholar]
- 14.Meex RCR, Watt MJ. Hepatokines: linking nonalcoholic fatty liver disease and insulin resistance. Nat Rev Endocrinol. 2017;13(9):509–520. doi: 10.1038/nrendo.2017.56. [DOI] [PubMed] [Google Scholar]
- 15.Watt MJ, Miotto PM, De Nardo W, Montgomery MK. The liver as an endocrine organ—linking NAFLD and insulin resistance. Endocr Rev. 2019;40(5):1367–1393. doi: 10.1210/er.2019-00034. [DOI] [PubMed] [Google Scholar]
- 16.López-Pastor AR, Infante-Menéndez J, Escribano Ó, Gómez-Hernández A. miRNA dysregulation in the development of non-alcoholic fatty liver disease and the related disorders type 2 diabetes mellitus and cardiovascular disease. Front Med (Lausanne) 2020;7:527059. doi: 10.3389/fmed.2020.527059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Y, Zhao MF, Jiang S, Wu J, Liu J, Yuan XW, et al. Liver governs adipose remodelling via extracellular vesicles in response to lipid overload. Nat Commun. 2020;11(1):719. doi: 10.1038/s41467-020-14450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lafontan M, Langin D. Lipolysis and lipid mobilization in human adipose tissue. Prog Lipid Res. 2009;48(5):275–297. doi: 10.1016/j.plipres.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 19.Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol. 2013;48(4):434–441. doi: 10.1007/s00535-013-0758-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pepino MY, Kuda O, Samovski D, Abumrad NA. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu Rev Nutr. 2014;34:281–303. doi: 10.1146/annurev-nutr-071812-161220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koonen DP, Jacobs RL, Febbraio M, Young ME, Soltys CL, Ong H, et al. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes. 2007;56(12):2863–2871. doi: 10.2337/db07-0907. [DOI] [PubMed] [Google Scholar]
- 22.Rajaraman G, Roberts MS, Hung D, Wang GQ, Burczynski FJ. Membrane binding proteins are the major determinants for the hepatocellular transmembrane flux of long-chain fatty acids bound to albumin. Pharm Res. 2005;22(11):1793–1804. doi: 10.1007/s11095-005-7248-2. [DOI] [PubMed] [Google Scholar]
- 23.Arner P, Langin D. Lipolysis in lipid turnover, cancer cachexia, and obesity-induced insulin resistance. Trends Endocrinol Metab. 2014;25(5):255–262. doi: 10.1016/j.tem.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 24.Jiao P, Ma J, Feng B, Zhang H, Diehl JA, Chin YE, et al. FFA-induced adipocyte inflammation and insulin resistance: involvement of ER stress and IKKbeta pathways. Obesity (Silver Spring) 2011;19(3):483–491. doi: 10.1038/oby.2010.200. [DOI] [PubMed] [Google Scholar]
- 25.Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes. 2011;60(10):2474–2483. doi: 10.2337/db11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adiels M, Taskinen MR, Packard C, Caslake MJ, Soro-Paavonen A, Westerbacka J, et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia. 2006;49(4):755–765. doi: 10.1007/s00125-005-0125-z. [DOI] [PubMed] [Google Scholar]
- 27.Mead JR, Irvine SA, Ramji DP. Lipoprotein lipase: structure, function, regulation, and role in disease. J Mol Med (Berl) 2002;80(12):753–769. doi: 10.1007/s00109-002-0384-9. [DOI] [PubMed] [Google Scholar]
- 28.Kazantzis M, Stahl A. Fatty acid transport proteins, implications in physiology and disease. Biochim Biophys Acta. 2012;1821(5):852–857. doi: 10.1016/j.bbalip.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mittendorfer B, Patterson BW, Klein S. Effect of sex and obesity on basal VLDL-triacylglycerol kinetics. Am J Clin Nutr. 2003;77(3):573–579. doi: 10.1093/ajcn/77.3.573. [DOI] [PubMed] [Google Scholar]
- 30.Andersen IR, Søndergaard E, Sørensen LP, Nellemann B, Gormsen LC, Jensen MD, et al. Increased VLDL-TG fatty acid storage in skeletal muscle in men with type 2 diabetes. J Clin Endocrinol Metab. 2016;102(3):831–839. doi: 10.1210/jc.2016-2979. [DOI] [PubMed] [Google Scholar]
- 31.Wang H, Knaub LA, Jensen DR, Young Jung D, Hong EG, Ko HJ, et al. Skeletal muscle-specific deletion of lipoprotein lipase enhances insulin signaling in skeletal muscle but causes insulin resistance in liver and other tissues. Diabetes. 2009;58(1):116–124. doi: 10.2337/db07-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab. 2012;15(6):805–812. doi: 10.1016/j.cmet.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perseghin G, Lattuada G, De Cobelli F, Esposito A, Belloni E, Ntali G, et al. Increased mediastinal fat and impaired left ventricular energy metabolism in young men with newly found fatty liver. Hepatology. 2008;47(1):51–58. doi: 10.1002/hep.21983. [DOI] [PubMed] [Google Scholar]
- 34.Augustus A, Yagyu H, Haemmerle G, Bensadoun A, Vikramadithyan RK, Park SY, et al. Cardiac-specific knock-out of lipoprotein lipase alters plasma lipoprotein triglyceride metabolism and cardiac gene expression. J Biol Chem. 2004;279(24):25050–25057. doi: 10.1074/jbc.M401028200. [DOI] [PubMed] [Google Scholar]
- 35.James DE, Stöckli J, Birnbaum MJ. The aetiology and molecular landscape of insulin resistance. Nat Rev Mol Cell Biol. 2021;22(11):751–771. doi: 10.1038/s41580-021-00390-6. [DOI] [PubMed] [Google Scholar]
- 36.Burhans MS, Hagman DK, Kuzma JN, Schmidt KA, Kratz M. Contribution of adipose tissue inflammation to the development of type 2 diabetes mellitus. Compr Physiol. 2018;9(1):1–58. doi: 10.1002/cphy.c170040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goudriaan JR, Tacken PJ, Dahlmans VE, Gijbels MJ, van Dijk KW, Havekes LM, et al. Protection from obesity in mice lacking the VLDL receptor. Arterioscler Thromb Vasc Biol. 2001;21(9):1488–1493. doi: 10.1161/hq0901.095147. [DOI] [PubMed] [Google Scholar]
- 38.Clemente-Postigo M, Queipo-Ortuño MI, Fernandez-Garcia D, Gomez-Huelgas R, Tinahones FJ, Cardona F. Adipose tissue gene expression of factors related to lipid processing in obesity. PLoS ONE. 2011;6(9):e24783. doi: 10.1371/journal.pone.0024783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen A, Tao H, Metrione M, Hajri T. Very low density lipoprotein receptor (VLDLR) expression is a determinant factor in adipose tissue inflammation and adipocyte-macrophage interaction. J Biol Chem. 2014;289(3):1688–1703. doi: 10.1074/jbc.M113.515320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shin KC, Hwang I, Choe SS, Park J, Ji Y, Kim JI, et al. Macrophage VLDLR mediates obesity-induced insulin resistance with adipose tissue inflammation. Nat Commun. 2017;8(1):1087. doi: 10.1038/s41467-017-01232-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Pretis N, Amodio A, Frulloni L. Hypertriglyceridemic pancreatitis: epidemiology, pathophysiology and clinical management. United European Gastroenterol J. 2018;6(5):649–655. doi: 10.1177/2050640618755002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khatua B, El-Kurdi B, Singh VP. Obesity and pancreatitis. Curr Opin Gastroenterol. 2017;33(5):374–382. doi: 10.1097/MOG.0000000000000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang CL, Wang JJ, Li JN, Yang Y. Nonalcoholic fatty pancreas disease: an emerging clinical challenge. World J Clin Cases. 2021;9(23):6624–6638. doi: 10.12998/wjcc.v9.i23.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang X, Chen J, Wang J, Ma S, Feng W, Wu Z, et al. Very-low-density lipoprotein receptor-enhanced lipid metabolism in pancreatic stellate cells promotes pancreatic fibrosis. Immunity. 2022;55(7):1185–99.e8. doi: 10.1016/j.immuni.2022.06.001. [DOI] [PubMed] [Google Scholar]
- 45.Chaurasia B, Summers SA. Ceramides in metabolism: key lipotoxic players. Annu Rev Physiol. 2021;83:303–330. doi: 10.1146/annurev-physiol-031620-093815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chavez JA, Summers SA. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch Biochem Biophys. 2003;419(2):101–109. doi: 10.1016/j.abb.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 47.Haus JM, Kashyap SR, Kasumov T, Zhang R, Kelly KR, Defronzo RA, et al. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes. 2009;58(2):337–343. doi: 10.2337/db08-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spijkers LJ, van den Akker RF, Janssen BJ, Debets JJ, De Mey JG, Stroes ES, et al. Hypertension is associated with marked alterations in sphingolipid biology: a potential role for ceramide. PLoS ONE. 2011;6(7):e21817. doi: 10.1371/journal.pone.0021817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kasumov T, Li L, Li M, Gulshan K, Kirwan JP, Liu X, et al. Ceramide as a mediator of non-alcoholic Fatty liver disease and associated atherosclerosis. PLoS ONE. 2015;10(5):e0126910. doi: 10.1371/journal.pone.0126910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adams JM, 2nd, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, et al. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes. 2004;53(1):25–31. doi: 10.2337/diabetes.53.1.25. [DOI] [PubMed] [Google Scholar]
- 51.Straczkowski M, Kowalska I, Nikolajuk A, Dzienis-Straczkowska S, Kinalska I, Baranowski M, et al. Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes. 2004;53(5):1215–1221. doi: 10.2337/diabetes.53.5.1215. [DOI] [PubMed] [Google Scholar]
- 52.Ribaux PG, Iynedjian PB. Analysis of the role of protein kinase B (cAKT) in insulin-dependent induction of glucokinase and sterol regulatory element-binding protein 1 (SREBP1) mRNAs in hepatocytes. Biochem J. 2003;376(Pt 3):697–705. doi: 10.1042/bj20031287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang G, Badeanlou L, Bielawski J, Roberts AJ, Hannun YA, Samad F. Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am J Physiol Endocrinol Metab. 2009;297(1):E211–E224. doi: 10.1152/ajpendo.91014.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Summers SA, Garza LA, Zhou H, Birnbaum MJ. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol. 1998;18(9):5457–5464. doi: 10.1128/MCB.18.9.5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hajduch E, Balendran A, Batty IH, Litherland GJ, Blair AS, Downes CP, et al. Ceramide impairs the insulin-dependent membrane recruitment of protein kinase B leading to a loss in downstream signalling in L6 skeletal muscle cells. Diabetologia. 2001;44(2):173–183. doi: 10.1007/s001250051596. [DOI] [PubMed] [Google Scholar]
- 56.Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol Cell Biol. 2003;23(21):7794–7808. doi: 10.1128/MCB.23.21.7794-7808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J Biol Chem. 2004;279(35):36608–36615. doi: 10.1074/jbc.M406499200. [DOI] [PubMed] [Google Scholar]
- 58.Teruel T, Hernandez R, Lorenzo M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes. 2001;50(11):2563–2571. doi: 10.2337/diabetes.50.11.2563. [DOI] [PubMed] [Google Scholar]
- 59.JeBailey L, Wanono O, Niu W, Roessler J, Rudich A, Klip A. Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes. 2007;56(2):394–403. doi: 10.2337/db06-0823. [DOI] [PubMed] [Google Scholar]
- 60.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun. 2017;8:15691. doi: 10.1038/ncomms15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang L, Wang B, Gasek NS, Zhou Y, Cohn RL, Martin DE, et al. Targeting p21(Cip1) highly expressing cells in adipose tissue alleviates insulin resistance in obesity. Cell Metab. 2021;2:2. doi: 10.1016/j.cmet.2021.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Venable ME, Yin X. Ceramide induces endothelial cell senescence. Cell Biochem Funct. 2009;27(8):547–551. doi: 10.1002/cbf.1605. [DOI] [PubMed] [Google Scholar]
- 64.Lee JY, Bielawska AE, Obeid LM. Regulation of cyclin-dependent kinase 2 activity by ceramide. Exp Cell Res. 2000;261(2):303–311. doi: 10.1006/excr.2000.5028. [DOI] [PubMed] [Google Scholar]
- 65.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5(3):167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 66.Ritschka B, Knauer-Meyer T, Goncalves DS, Mas A, Plassat JL, Durik M, et al. The senotherapeutic drug ABT-737 disrupts aberrant p21 expression to restore liver regeneration in adult mice. Genes Dev. 2020;34:489–494. doi: 10.1101/gad.332643.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krey G, Braissant O, L'Horset F, Kalkhoven E, Perroud M, Parker MG, et al. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol Endocrinol. 1997;11(6):779–791. doi: 10.1210/mend.11.6.0007. [DOI] [PubMed] [Google Scholar]
- 68.Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2015;62(3):720–733. doi: 10.1016/j.jhep.2014.10.039. [DOI] [PubMed] [Google Scholar]
- 69.Liss KH, Finck BN. PPARs and nonalcoholic fatty liver disease. Biochimie. 2017;136:65–74. doi: 10.1016/j.biochi.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, et al. PPARalpha gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J Hepatol. 2015;63(1):164–173. doi: 10.1016/j.jhep.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 71.Erion DM, Shulman GI. Diacylglycerol-mediated insulin resistance. Nat Med. 2010;16(4):400–402. doi: 10.1038/nm0410-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tolman KG, Fonseca V, Dalpiaz A, Tan MH. Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease. Diabetes Care. 2007;30(3):734–743. doi: 10.2337/dc06-1539. [DOI] [PubMed] [Google Scholar]
- 73.Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422(6928):173–176. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- 74.Usui R, Yabe D, Fauzi M, Goto H, Botagarova A, Tokumoto S, et al. GPR40 activation initiates store-operated Ca(2+) entry and potentiates insulin secretion via the IP3R1/STIM1/Orai1 pathway in pancreatic beta-cells. Sci Rep. 2019;9(1):15562. doi: 10.1038/s41598-019-52048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes. 2008;57(9):2280–2287. doi: 10.2337/db08-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. 2005;11(1):90–94. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- 77.Ow JR, Caldez MJ, Zafer G, Foo JC, Li HY, Ghosh S, et al. Remodeling of whole-body lipid metabolism and a diabetic-like phenotype caused by loss of CDK1 and hepatocyte division. eLife. 2020;9:e63835. doi: 10.7554/eLife.63835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Steneberg P, Rubins N, Bartoov-Shifman R, Walker MD, Edlund H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. 2005;1(4):245–258. doi: 10.1016/j.cmet.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 79.Ichimura A, Hirasawa A, Poulain-Godefroy O, Bonnefond A, Hara T, Yengo L, et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature. 2012;483(7389):350–354. doi: 10.1038/nature10798. [DOI] [PubMed] [Google Scholar]
- 80.Offermanns S. Free fatty acid (FFA) and hydroxy carboxylic acid (HCA) receptors. Annu Rev Pharmacol Toxicol. 2014;54:407–434. doi: 10.1146/annurev-pharmtox-011613-135945. [DOI] [PubMed] [Google Scholar]
- 81.Luukkonen PK, Sadevirta S, Zhou Y, Kayser B, Ali A, Ahonen L, et al. Saturated fat is more metabolically harmful for the human liver than unsaturated fat or simple sugars. Diabetes Care. 2018;41(8):1732–1739. doi: 10.2337/dc18-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes. 2012;61(2):364–371. doi: 10.2337/db11-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun. 2013;4:1829. doi: 10.1038/ncomms2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci USA. 2008;105(43):16767–16772. doi: 10.1073/pnas.0808567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yamashita H, Kaneyuki T, Tagawa K. Production of acetate in the liver and its utilization in peripheral tissues. Biochim Biophys Acta. 2001;1532(1–2):79–87. doi: 10.1016/S1388-1981(01)00117-2. [DOI] [PubMed] [Google Scholar]
- 86.Kendrick SF, O'Boyle G, Mann J, Zeybel M, Palmer J, Jones DE, et al. Acetate, the key modulator of inflammatory responses in acute alcoholic hepatitis. Hepatology. 2010;51(6):1988–1997. doi: 10.1002/hep.23572. [DOI] [PubMed] [Google Scholar]
- 87.Mews P, Egervari G, Nativio R, Sidoli S, Donahue G, Lombroso SI, et al. Alcohol metabolism contributes to brain histone acetylation. Nature. 2019;574(7780):717–721. doi: 10.1038/s41586-019-1700-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15(6):122. doi: 10.1186/gb4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V, et al. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci USA. 2012;109(10):3826–3831. doi: 10.1073/pnas.1115201109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312(5774):734–737. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 91.Bechmann LP, Kocabayoglu P, Sowa JP, Sydor S, Best J, Schlattjan M, et al. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology. 2013;57(4):1394–1406. doi: 10.1002/hep.26225. [DOI] [PubMed] [Google Scholar]
- 92.Gottlieb A, Canbay A. Why bile acids are so important in non-alcoholic fatty liver disease (NAFLD) progression. Cells. 2019;8(11):1358. doi: 10.3390/cells8111358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Einarsson C, Ellis E, Abrahamsson A, Ericzon BG, Bjorkhem I, Axelson M. Bile acid formation in primary human hepatocytes. World J Gastroenterol. 2000;6(4):522–525. doi: 10.3748/wjg.v6.i4.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stofan M, Guo GL. Bile acids and FXR: novel targets for liver diseases. Front Med (Lausanne) 2020;7:544. doi: 10.3389/fmed.2020.00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ticho AL, Malhotra P, Dudeja PK, Gill RK, Alrefai WA. Intestinal absorption of bile acids in health and disease. Compr Physiol. 2019;10(1):21–56. doi: 10.1002/cphy.c190007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wong MH, Oelkers P, Craddock AL, Dawson PA. Expression cloning and characterization of the hamster ileal sodium-dependent bile acid transporter. J Biol Chem. 1994;269(2):1340–1347. doi: 10.1016/S0021-9258(17)42263-0. [DOI] [PubMed] [Google Scholar]
- 97.Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, et al. The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. J Biol Chem. 2005;280(8):6960–6968. doi: 10.1074/jbc.M412752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Trauner M, Boyer JL. Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev. 2003;83(2):633–671. doi: 10.1152/physrev.00027.2002. [DOI] [PubMed] [Google Scholar]
- 99.Guzior DV, Quinn RA. Review: microbial transformations of human bile acids. Microbiome. 2021;9(1):140. doi: 10.1186/s40168-021-01101-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dowling RH. The enterohepatic circulation of bile acids as they relate to lipid disorders. J Clin Pathol Suppl (Assoc Clin Pathol) 1973;5:59–67. doi: 10.1136/jcp.s1-5.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fausa O. Serum bile acid concentration after a test meal. Scand J Gastroenterol. 1976;11(3):229–232. doi: 10.1080/00365521.1976.12097100. [DOI] [PubMed] [Google Scholar]
- 102.Wu T, Yang M, Xu H, Wang L, Wei H, Ji G. Serum bile acid profiles improve clinical prediction of nonalcoholic fatty liver in T2DM patients. J Proteome Res. 2021;20(8):3814–3825. doi: 10.1021/acs.jproteome.1c00104. [DOI] [PubMed] [Google Scholar]
- 103.Neale G, Lewis B, Weaver V, Panveliwalla D. Serum bile acids in liver disease. Gut. 1971;12(2):145–152. doi: 10.1136/gut.12.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kaplowitz N, Kok E, Javitt NB. Postprandial serum bile acid for the detection of hepatobiliary disease. JAMA. 1973;225(3):292–293. doi: 10.1001/jama.1973.03220300048011. [DOI] [PubMed] [Google Scholar]
- 105.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284(5418):1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 106.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284(5418):1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 107.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6(3):517–526. doi: 10.1016/S1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 108.Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6(3):507–515. doi: 10.1016/S1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 109.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2(4):217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 110.Yamagata K, Yoshimochi K, Daitoku H, Hirota K, Fukamizu A. Bile acid represses the peroxisome proliferator-activated receptor-gamma coactivator-1 promoter activity in a small heterodimer partner-dependent manner. Int J Mol Med. 2007;19(5):751–756. [PubMed] [Google Scholar]
- 111.Cheng CF, Ku HC, Lin H. PGC-1alpha as a pivotal factor in lipid and metabolic regulation. Int J Mol Sci. 2018;19:11. doi: 10.3390/ijms19113447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Prawitt J, Abdelkarim M, Stroeve JH, Popescu I, Duez H, Velagapudi VR, et al. Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes. 2011;60(7):1861–1871. doi: 10.2337/db11-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang Y, Ge X, Heemstra LA, Chen WD, Xu J, Smith JL, et al. Loss of FXR protects against diet-induced obesity and accelerates liver carcinogenesis in ob/ob mice. Mol Endocrinol. 2012;26(2):272–280. doi: 10.1210/me.2011-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Parseus A, Sommer N, Sommer F, Caesar R, Molinaro A, Stahlman M, et al. Microbiota-induced obesity requires farnesoid X receptor. Gut. 2017;66(3):429–437. doi: 10.1136/gutjnl-2015-310283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun. 2015;6:10166. doi: 10.1038/ncomms10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schmitt J, Kong B, Stieger B, Tschopp O, Schultze SM, Rau M, et al. Protective effects of farnesoid X receptor (FXR) on hepatic lipid accumulation are mediated by hepatic FXR and independent of intestinal FGF15 signal. Liver Int. 2015;35(4):1133–1144. doi: 10.1111/liv.12456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Clifford BL, Sedgeman LR, Williams KJ, Morand P, Cheng A, Jarrett KE, et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. 2021;33(8):1671–1684. doi: 10.1016/j.cmet.2021.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dai J, Wang H, Shi Y, Dong Y, Zhang Y, Wang J. Impact of bile acids on the growth of human cholangiocarcinoma via FXR. J Hematol Oncol. 2011;4:41. doi: 10.1186/1756-8722-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17(2):225–235. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 120.Perino A, Demagny H, Velazquez-Villegas L, Schoonjans K. Molecular physiology of bile acid signaling in health, disease, and aging. Physiol Rev. 2021;101(2):683–731. doi: 10.1152/physrev.00049.2019. [DOI] [PubMed] [Google Scholar]
- 121.Popescu IR, Helleboid-Chapman A, Lucas A, Vandewalle B, Dumont J, Bouchaert E, et al. The nuclear receptor FXR is expressed in pancreatic β-cells and protects human islets from lipotoxicity. FEBS Lett. 2010;584(13):2845–2851. doi: 10.1016/j.febslet.2010.04.068. [DOI] [PubMed] [Google Scholar]
- 122.Renga B, Mencarelli A, Vavassori P, Brancaleone V, Fiorucci S. The bile acid sensor FXR regulates insulin transcription and secretion. Biochim Biophys Acta Mol Basis Dis. 2010;1802(3):363–372. doi: 10.1016/j.bbadis.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 123.Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, et al. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem. 2006;281(16):11039–11049. doi: 10.1074/jbc.M510258200. [DOI] [PubMed] [Google Scholar]
- 124.Huang C, Wang J, Hu W, Wang C, Lu X, Tong L, et al. Identification of functional farnesoid X receptors in brain neurons. FEBS Lett. 2016;590(18):3233–3242. doi: 10.1002/1873-3468.12373. [DOI] [PubMed] [Google Scholar]
- 125.Deckmyn B, Domenger D, Blondel C, Ducastel S, Nicolas E, Dorchies E, et al. Farnesoid X receptor activation in brain alters brown adipose tissue function via the sympathetic system. Front Mol Neurosci. 2022;14:2. doi: 10.3389/fnmol.2021.808603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.De Magalhaes Filho CD, Downes M, Evans RM. Farnesoid X receptor an emerging target to combat obesity. Dig Dis. 2017;35(3):185–190. doi: 10.1159/000450909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Caussy C, Hsu C, Singh S, Bassirian S, Kolar J, Faulkner C, et al. Serum bile acid patterns are associated with the presence of NAFLD in twins, and dose-dependent changes with increase in fibrosis stage in patients with biopsy-proven NAFLD. Aliment Pharmacol Ther. 2019;49(2):183–193. doi: 10.1111/apt.15035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Haeusler RA, Astiarraga B, Camastra S, Accili D, Ferrannini E. Human insulin resistance is associated with increased plasma levels of 12α-hydroxylated bile acids. Diabetes. 2013;62(12):4184–4191. doi: 10.2337/db13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278(11):9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 130.Kuhre RE, Wewer Albrechtsen NJ, Larsen O, Jepsen SL, Balk-Moller E, Andersen DB, et al. Bile acids are important direct and indirect regulators of the secretion of appetite- and metabolism-regulating hormones from the gut and pancreas. Mol Metab. 2018;11:84–95. doi: 10.1016/j.molmet.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439(7075):484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 132.Maczewsky J, Kaiser J, Gresch A, Gerst F, Dufer M, Krippeit-Drews P, et al. TGR5 activation promotes stimulus-secretion coupling of pancreatic beta-cells via a PKA-dependent pathway. Diabetes. 2019;68(2):324–336. doi: 10.2337/db18-0315. [DOI] [PubMed] [Google Scholar]
- 133.Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10(3):167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Finn PD, Rodriguez D, Kohler J, Jiang Z, Wan S, Blanco E, et al. Intestinal TGR5 agonism improves hepatic steatosis and insulin sensitivity in Western diet-fed mice. Am J Physiol Gastrointest Liver Physiol. 2019;316(3):G412–G424. doi: 10.1152/ajpgi.00300.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhu L, Wang W, Xie TH, Zou J, Nie X, Wang X, et al. TGR5 receptor activation attenuates diabetic retinopathy through suppression of RhoA/ROCK signaling. FASEB J. 2020;34(3):4189–4203. doi: 10.1096/fj.201902496RR. [DOI] [PubMed] [Google Scholar]
- 136.Wang XX, Edelstein MH, Gafter U, Qiu L, Luo Y, Dobrinskikh E, et al. G Protein-coupled bile acid receptor TGR5 activation inhibits kidney disease in obesity and diabetes. J Am Soc Nephrol. 2016;27(5):1362–1378. doi: 10.1681/ASN.2014121271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Broeders EP, Nascimento EB, Havekes B, Brans B, Roumans KH, Tailleux A, et al. The bile acid chenodeoxycholic acid increases human brown adipose tissue activity. Cell Metab. 2015;22(3):418–426. doi: 10.1016/j.cmet.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 138.Castellanos-Jankiewicz A, Guzmán-Quevedo O, Fénelon VS, Zizzari P, Quarta C, Bellocchio L, et al. Hypothalamic bile acid-TGR5 signaling protects from obesity. Cell Metab. 2021;33(7):1483–1492.e10. doi: 10.1016/j.cmet.2021.04.009. [DOI] [PubMed] [Google Scholar]
- 139.Maiuolo J, Oppedisano F, Gratteri S, Muscoli C, Mollace V. Regulation of uric acid metabolism and excretion. Int J Cardiol. 2016;213:8–14. doi: 10.1016/j.ijcard.2015.08.109. [DOI] [PubMed] [Google Scholar]
- 140.Singh GM, Micha R, Khatibzadeh S, Shi P, Lim S, Andrews KG, et al. Global, regional, and national consumption of sugar-sweetened beverages, fruit juices, and milk: a systematic assessment of beverage intake in 187 countries. PLoS ONE. 2015;10(8):e0124845. doi: 10.1371/journal.pone.0124845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, et al. Fructose and sugar: a major mediator of non-alcoholic fatty liver disease. J Hepatol. 2018;68(5):1063–1075. doi: 10.1016/j.jhep.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Barone S, Fussell SL, Singh AK, Lucas F, Xu J, Kim C, et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J Biol Chem. 2009;284(8):5056–5066. doi: 10.1074/jbc.M808128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kellett GL, Brot-Laroche E, Mace OJ, Leturque A. Sugar absorption in the intestine: the role of GLUT2. Annu Rev Nutr. 2008;28:35–54. doi: 10.1146/annurev.nutr.28.061807.155518. [DOI] [PubMed] [Google Scholar]
- 144.Jang C, Wada S, Yang S, Gosis B, Zeng X, Zhang Z, et al. The small intestine shields the liver from fructose-induced steatosis. Nat Metab. 2020;2(7):586–593. doi: 10.1038/s42255-020-0222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Todoric J, Di Caro G, Reibe S, Henstridge DC, Green CR, Vrbanac A, et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat Metab. 2020;2(10):1034–1045. doi: 10.1038/s42255-020-0261-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Maenpaa PH, Raivio KO, Kekomaki MP. Liver adenine nucleotides: fructose-induced depletion and its effect on protein synthesis. Science. 1968;161(3847):1253–1254. doi: 10.1126/science.161.3847.1253. [DOI] [PubMed] [Google Scholar]
- 147.Kurtz TW, Kabra PM, Booth BE, Al-Bander HA, Portale AA, Serena BG, et al. Liquid-chromatographic measurements of inosine, hypoxanthine, and xanthine in studies of fructose-induced degradation of adenine nucleotides in humans and rats. Clin Chem. 1986;32(5):782–786. doi: 10.1093/clinchem/32.5.782. [DOI] [PubMed] [Google Scholar]
- 148.Van den Berghe G. Fructose: metabolism and short-term effects on carbohydrate and purine metabolic pathways. Prog Biochem Pharmacol. 1986;12:1–32. [PubMed] [Google Scholar]
- 149.Sims B, Mahnke-Zizelman DK, Profit AA, Prestwich GD, Sabina RL, Theibert AB. Regulation of AMP deaminase by phosphoinositides. J Biol Chem. 1999;274(36):25701–25707. doi: 10.1074/jbc.274.36.25701. [DOI] [PubMed] [Google Scholar]
- 150.Perheentupa J, Raivio K. Fructose-induced hyperuricaemia. Lancet. 1967;2(7515):528–531. doi: 10.1016/S0140-6736(67)90494-1. [DOI] [PubMed] [Google Scholar]
- 151.Nakagawa T, Hu H, Zharikov S, Tuttle KR, Short RA, Glushakova O, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2006;290(3):F625–F631. doi: 10.1152/ajprenal.00140.2005. [DOI] [PubMed] [Google Scholar]
- 152.Cho IJ, Oh DH, Yoo J, Hwang YC, Ahn KJ, Chung HY, et al. Allopurinol ameliorates high fructose diet induced hepatic steatosis in diabetic rats through modulation of lipid metabolism, inflammation, and ER stress pathway. Sci Rep. 2021;11(1):9894. doi: 10.1038/s41598-021-88872-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Baldwin W, McRae S, Marek G, Wymer D, Pannu V, Baylis C, et al. Hyperuricemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes. 2011;60(4):1258–1269. doi: 10.2337/db10-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nakatsu Y, Seno Y, Kushiyama A, Sakoda H, Fujishiro M, Katasako A, et al. The xanthine oxidase inhibitor febuxostat suppresses development of nonalcoholic steatohepatitis in a rodent model. Am J Physiol Gastrointest Liver Physiol. 2015;309(1):G42–51. doi: 10.1152/ajpgi.00443.2014. [DOI] [PubMed] [Google Scholar]
- 155.Nishikawa T, Nagata N, Shimakami T, Shirakura T, Matsui C, Ni Y, et al. Xanthine oxidase inhibition attenuates insulin resistance and diet-induced steatohepatitis in mice. Sci Rep. 2020;10(1):815. doi: 10.1038/s41598-020-57784-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wang W, Wang C, Ding XQ, Pan Y, Gu TT, Wang MX, et al. Quercetin and allopurinol reduce liver thioredoxin-interacting protein to alleviate inflammation and lipid accumulation in diabetic rats. Br J Pharmacol. 2013;169(6):1352–1371. doi: 10.1111/bph.12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jensen T, Niwa K, Hisatome I, Kanbay M, Andres-Hernando A, Roncal-Jimenez CA, et al. Increased serum uric acid over five years is a risk factor for developing fatty liver. Sci Rep. 2018;8(1):11735. doi: 10.1038/s41598-018-30267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ma Z, Zhang J, Kang X, Xu C, Sun C, Tao L, et al. Hyperuricemia precedes non-alcoholic fatty liver disease with abdominal obesity moderating this unidirectional relationship: three longitudinal analyses. Atherosclerosis. 2020;311:44–51. doi: 10.1016/j.atherosclerosis.2020.08.006. [DOI] [PubMed] [Google Scholar]
- 159.Wei F, Li J, Chen C, Zhang K, Cao L, Wang X, et al. Higher serum uric acid level predicts non-alcoholic fatty liver disease: a 4-year prospective cohort study. Front Endocrinol (Lausanne) 2020;11:179. doi: 10.3389/fendo.2020.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem. 2012;287(48):40732–40744. doi: 10.1074/jbc.M112.399899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhao H, Lu J, He F, Liu W, Yu W, et al. High uric acid induces liver fat accumulation via ROS/JNK/AP-1 signaling. Am J Physiol Endocrinol Metab. 2021;320(6):1032–1043. doi: 10.1152/ajpendo.00518.2020. [DOI] [PubMed] [Google Scholar]
- 162.Choi YJ, Shin HS, Choi HS, Park JW, Jo I, Oh ES, et al. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Lab Invest. 2014;94(10):1114–1125. doi: 10.1038/labinvest.2014.98. [DOI] [PubMed] [Google Scholar]
- 163.Chen S, Chen D, Yang H, Wang X, Wang J, Xu C. Uric acid induced hepatocytes lipid accumulation through regulation of miR-149-5p/FGF21 axis. BMC Gastroenterol. 2020;20(1):39. doi: 10.1186/s12876-020-01189-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kanellis J, Watanabe S, Li JH, Kang DH, Li P, Nakagawa T, et al. Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension. 2003;41(6):1287–1293. doi: 10.1161/01.HYP.0000072820.07472.3B. [DOI] [PubMed] [Google Scholar]
- 165.Wan X, Xu C, Lin Y, Lu C, Li D, Sang J, et al. Uric acid regulates hepatic steatosis and insulin resistance through the NLRP3 inflammasome-dependent mechanism. J Hepatol. 2016;64(4):925–932. doi: 10.1016/j.jhep.2015.11.022. [DOI] [PubMed] [Google Scholar]
- 166.Zhang X, Zhang JH, Chen XY, Hu QH, Wang MX, Jin R, et al. Reactive oxygen species-induced TXNIP drives fructose-mediated hepatic inflammation and lipid accumulation through NLRP3 inflammasome activation. Antioxid Redox Signal. 2015;22(10):848–870. doi: 10.1089/ars.2014.5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59(3):898–910. doi: 10.1002/hep.26592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dalbeth N, Choi HK, Joosten LAB, Khanna PP, Matsuo H, Perez-Ruiz F, et al. Gout. Nat Rev Dis Primers. 2019;5(1):69. doi: 10.1038/s41572-019-0115-y. [DOI] [PubMed] [Google Scholar]
- 169.Rocic B, Vucic-Lovrencic M, Poje N, Poje M, Bertuzzi F. Uric acid may inhibit glucose-induced insulin secretion via binding to an essential arginine residue in rat pancreatic beta-cells. Bioorg Med Chem Lett. 2005;15(4):1181–1184. doi: 10.1016/j.bmcl.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 170.Jia L, Xing J, Ding Y, Shen Y, Shi X, Ren W, et al. Hyperuricemia causes pancreatic beta-cell death and dysfunction through NF-kappaB signaling pathway. PLoS ONE. 2013;8(10):e78284. doi: 10.1371/journal.pone.0078284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Xin Y, Zhang H, Jia Z, Ding X, Sun Y, Wang Q, et al. Resveratrol improves uric acid-induced pancreatic beta-cells injury and dysfunction through regulation of miR-126. Biomed Pharmacother. 2018;102:1120–1126. doi: 10.1016/j.biopha.2018.03.172. [DOI] [PubMed] [Google Scholar]
- 172.Sodhi K, Hilgefort J, Banks G, Gilliam C, Stevens S, Ansinelli HA, et al. Uric Acid-induced adipocyte dysfunction is attenuated by HO-1 upregulation: potential role of antioxidant therapy to target obesity. Stem Cells Int. 2016;2016:8197325. doi: 10.1155/2016/8197325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tsushima Y, Nishizawa H, Tochino Y, Nakatsuji H, Sekimoto R, Nagao H, et al. Uric acid secretion from adipose tissue and its increase in obesity. J Biol Chem. 2013;288(38):27138–27149. doi: 10.1074/jbc.M113.485094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lu W, Xu Y, Shao X, Gao F, Li Y, Hu J, et al. Uric acid produces an inflammatory response through activation of NF-κB in the hypothalamus: implications for the pathogenesis of metabolic disorders. Sci Rep. 2015;5(1):12144. doi: 10.1038/srep12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tian T, Liu X, Li T, Nie Z, Li S, Tang Y, et al. Detrimental effects of long-term elevated serum uric acid on cognitive function in rats. Sci Rep. 2021;11(1):6732. doi: 10.1038/s41598-021-86279-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ndrepepa G. Uric acid and cardiovascular disease. Clin Chim Acta. 2018;484:150–163. doi: 10.1016/j.cca.2018.05.046. [DOI] [PubMed] [Google Scholar]
- 177.Masoodi M, Gastaldelli A, Hyötyläinen T, Arretxe E, Alonso C, Gaggini M, et al. Metabolomics and lipidomics in NAFLD: biomarkers and non-invasive diagnostic tests. Nat Rev Gastroenterol Hepatol. 2021;18(12):835–856. doi: 10.1038/s41575-021-00502-9. [DOI] [PubMed] [Google Scholar]
- 178.Delvaux A, Rathahao-Paris E, Alves S. Different ion mobility-mass spectrometry coupling techniques to promote metabolomics. Mass Spectrom Rev. 2022;41(5):695–721. doi: 10.1002/mas.21685. [DOI] [PubMed] [Google Scholar]
- 179.Christinat N, Masoodi M. Comprehensive lipoprotein characterization using lipidomics analysis of human plasma. J Proteome Res. 2017;16(8):2947–2953. doi: 10.1021/acs.jproteome.7b00236. [DOI] [PubMed] [Google Scholar]
- 180.Wang Y-P, Lei Q-Y. Metabolite sensing and signaling in cell metabolism. Signal Transduct Target Ther. 2018;3(1):30. doi: 10.1038/s41392-018-0024-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This does not apply since this is a review article.