

Abstract
Objective: To investigate the role of Osteopontin (OPN) in mediating macroautophagy, autophagy, and neuroplasticity in the ipsilateral hemisphere after stroke. Methods: Focal stroke was induced by photothrombosis in adult mice. Spatiotemporal expression of endogenous OPN and BECN1 was assessed by immunohistochemistry. Motor function was determined by the grid-walking and cylinder tasks. We also evaluated markers of neuroplasticity and autophagy using biochemical and histology analyses. Results: Herein, we showed that endogenous OPN and beclin1 were increased in the peri-infarct area of stroked patients and mice. Intracerebral administration of OPN (0.1 mg/ml; 3 ml) significantly improved performance in motor behavioral tasks compared with non-OPN-treated stroke mice. Furthermore, the neural repair was induced in OPN-treated stroke mice. We found that OPN treatment resulted in a significantly higher density of a presynaptic marker (vesicular glutamate transporter 1, VgluT1) and synaptic plasticity marker (synaptophysin, SYN) within the peri-infarct region. OPN treatment in stroke mice not only increased protein levels of integrin β1 but also promoted the expression of beclin1 and LC3, two autophagy-related proteins in the peri-infarct area. Additionally, OPN-induced neuroplasticity and autophagy were blocked by an integrin antagonist. Conclusion: Our findings indicate that OPN may enhance neuroplasticity via autophagy, providing a new therapeutic strategy for ischemic stroke.
Keywords: Ischemic stroke, osteopontin, motor functional recovery, neuroplasticity, autophagy
Introduction
Stroke impacts approximately 795,000 people in the U.S. alone [1]. Unfortunately, many people suffer from motor dysfunction post-stroke [2], and it has been difficult to identify drug regimens that promote recovery after stroke. Over the past few decades, numerous reports demonstrate the brain’s ability to rewire itself to rehabilitate lost function in humans and animals [3,4]. This modification of neuronal connections involves several neuroplasticity processes, such as axonal sprouting, the formation of new synapses, and synaptic remodeling [5]. Recent studies suggest that neuroplastic mechanisms reactivate sprouting neurons and induce cellular and molecular processes that promote motor function recovery after ischemic injury [6,7]. Therefore, revealing the mechanisms underlying ischemic-induced neuroplasticity processes should provide new information concerning neural repair that can inform both the prevention and treatment of stroke.
Neuron survival requires a balance between the synthesis and degeneration of cellular proteins and impaired organelles. Autophagy is a critical cellular clearance pathway that is essential for neuronal homeostasis to maintain this balance under stress [8]. However, whether activation of autophagy promotes neuronal survival or increases the rate of neuron death after cerebral ischemia is still under debate. Previously, several studies demonstrated that activation of autophagy has a destructive role after ischemic injury [9,10]. By contrast, growing evidence suggests that cerebral ischemia protects neurons from death by activating autophagy [11,12]. Some reports also indicate that neuronal autophagy influences processes such as synaptic plasticity [13,14] and neurodevelopment [15], demonstrating that autophagy is vital to the regulation of neuronal physiology and survival [16]. However, few investigations have explored the relationship between autophagy and neuroplasticity within neural tissue in response to ischemic insults.
Osteopontin (OPN) is a polyfunctional, inducible, and phosphorylated extracellular matrix glycoprotein that contributes to diverse pathological processes, such as brain injury and cancer [17,18]. Previous studies show that OPN acts in various cell types via an RGD (arginine-glycine-aspartic acid) motif that binds different integrin receptors, including α and β subunits [19,20]. Thus, OPN is a prominent feature of RGD motif-dependent mechanisms. Interestingly, several reports suggest that glycine promotes transcriptional expression of OPN by activating Runx2 and CBFβ [21,22]. These phenomena reveal that OPN itself is regulated by glycine in physiological situations and plays a unique role in amino acid metabolism [23]. In non-central nervous system (CNS)-related cells, several reports demonstrate that OPN is involved in suppressing autophagy [24,25]. Conversely, others found that OPN elicits autophagy through integrin receptors [26,27]. These findings suggest that OPN-integrin signaling is associated with autophagy. OPN is upregulated in sprouting neurons, with a 12-fold increase in expression compared with the expression level of nonsprouting neurons after stroke [28]. Furthermore, OPN positively mediates the repair of retinal ganglion cells and propriospinal and corticospinal neurons by enhancing axon regeneration and synaptogenesis after CNS injury [29-32]. Collectively, these results imply the role of OPN in repairing ischemic brain tissue. However, it remains unclear whether the OPN-integrin signaling pathway mediates endogenous neuroplasticity through autophagy in cortical neurons around the infarct region and improves neurological deficits after ischemic stroke.
Here, we characterized autophagy profiles in neurons and their relationship with neuroplasticity in the peri-infarct area after ischemic stroke. Moreover, we examined the spatial expression patterns of OPN and the integrin β1 subunit (ITGB1), beclin1, and LC3 in the ipsilateral cortex and investigated the potential involvement of OPN-ITGB1 signaling in neural autophagy and neuroplasticity, thereby elucidating the intricate mechanisms that mediate neuron autophagy-neuroplasticity by OPN after stroke.
Materials and methods
Animals and patients ethical statement
Male mice (C57BL/6) aged 2-4 months and weighing 20-25 g were used. All procedures were conducted following U.S. National Institutes of Health guidelines on the care and use of laboratory animals and were approved by the University of Guilin Medical Experimental Animal Ethics Committee (approval ID GLMC202203165). Efforts were made to reduce any pain or discomfort, and the minimum number of animals was used. All animals were housed in a light- and temperature-controlled room with adequate food and water under a 12-hour light/dark cycle. Patients with acute ischemic stroke and non-stroke controls were recruited from Ganzhou People’s Hospital, Ganzhou, Jiangxi, China following a protocol approved by the ethics committee at Ganzhou People’s Hospital (approval ID TY-ZYK2022-010-01).
Photothrombotic model of ischemic cortical stroke
To induce chronic focal cerebral ischemia, photothrombosis was used as previously described. Mice were anesthetized with isoflurane (1.5-3.5% in a 70% N2O/30% O2 mixture) and placed in a stereotactic surgical frame (RWD, China). The skull was uncovered by cutting the skin along the midline. The surface connective tissue was removed, and the bregma was cleared. A cold light source (CL6000LED; Zeiss) attached to a 40× objective provided a 2-mm diameter illumination positioned 1.5 mm lateral from bregma. Rose Bengal (0.2 ml, 15 mg/ml, G8540, Solarbio, China) was injected intraperitoneally. After 5 minutes, the cold light source was turned on to illuminate the skull for 19 minutes to initiate photothrombosis. After surgery, the scalp was sutured, and mice were allowed to recover. Body temperature was kept at 36.9±0.4°C with a heating pad during the operation. Mice in the sham group were treated using the same procedure without light illumination.
Drug administration
Human recombinant OPN (rOPN, GenePharma, China) was prepared following the manufacturer’s instructions. Animals were randomly assigned to one of the following groups: sham, stroke, stroke+saline, and stroke+rOPN (0.1 μg/μl; 3 μl). Briefly, OPN was dissolved in saline (0.1 μg/μl), and a total volume of 3 μl (1 μl/min) was given intracerebrally at 0.3 mg per animal 7 days post-stroke. Next, the mice were fixed on a stereotaxic apparatus (RWD, China). They were continuously anesthetized with isoflurane gas, and a microcranial drill perforated 1.5 mm lateral to the bregma. rOPN was administered with a 10-μl syringe (Hamilton, USA) at the location of the stroke cavity (A/P, 0.0 mm; M/L, 1.5 mm; D/V, 1.0 mm). Sham mice were administered sterile saline. The administration was finished in 5 minutes, and the needle was maintained in the injection position for 2 minutes. Finally, the needle was withdrawn gently out of the brain. The wound was sutured, and mice were allowed to recover. An additional group was intracerebrally injected with 6 μl (1 μl/min) GRGDSP dissolved in saline (0.1 mg/ml, Med Chem Express) 30 minutes before rOPN was administered to evaluate whether integrin receptors were involved.
Human sample collection
Infarcted tissue confirmed by pathology was collected from four patients with large-area cerebral infarction (male, 3; female, 1; age range, 50-58 years; average age, 52.5 years) who underwent craniotomy from June to November 2021 in Ganzhou People’s Hospital. As a control group, normal brain tissue was removed during an internal decompression operation from four patients. These tissues were prepared for immunohistochemistry (IHC) to detect the expression of OPN. Infarcted tissue was also collected from another two large-area cerebral infarction patients (male, 1; female, 1; age range, 65-69 years; average age, 67 years) who underwent an operation from January to February 2022 in Ganzhou People’s Hospital. As a control group, normal brain tissue was removed during internal decompression operation from two patients. These tissues were prepared for IHC to determine the protein levels of beclin1. Exclusion criteria were as follows: patients with other concurrent malignant tumors, incomplete clinical information, serious heart disease, kidney disease, or neurological or psychiatric diseases. No patients underwent chemoradiotherapy before surgery. All patients signed an informed consent form, and the study was approved by the Ganzhou People’s Hospital Medical Ethics Committee (TY-ZYK2022-010-01) and complied with the Declaration of Helsinki.
Immunofluorescence staining
Mice were subjected to conventional anesthesia. After deep anesthesia, mice were sacrificed and perfused with 30 ml of normal saline for 5 minutes until the lungs and liver became grayish-white. Then, the whole brain was removed, and brain tissue was placed in paraformaldehyde and fixed for 48 hours. After removal, the brain was washed three times with PBS, placed in 30% sucrose buffer for 72 hours, embedded in OCT (SKURA, US), and frozen in 40-μm coronal sections (Thermo Fisher Scientific CryoStar NX50). Coronal slices of brain tissue were washed with PBS three times for 5 minutes each time while being gently shaken on a shaker (subsequent washes also employed this method). Blocking buffer (0.1 M PBS+0.05% Triton X-100+5% BSA) was added and sealed at room temperature for 60 minutes. Then, we used rabbit anti-VgluT1 (1:800, ab227805, Abcam, UK), goat anti-OPN (1:400, ab11503, Abcam, UK), mouse anti-NeuN (1:500, MAB377, Merck, US), rabbit anti-integrin-β1 (1:800, bs-0486R, Bioss, China), or ready-to-use DAPI solution (C0065, Solarbio, China) as primary antibodies. At the same time, we prepared a negative control with only the diluent of the primary antibody, which was placed in the refrigerator at 4°C overnight. After removal, brain slices were rinsed with PBST, and we added fluorescent donkey anti-mouse IgG AlexaFluor 488 (1:500, A21202, Invitrogen, US), donkey anti-rabbit IgG/Cy3 (1:500, bs-0295d-Cy3, Bioss, China), donkey anti-goat IgG/CY3 (1:500, bs-0294d-Cy3, Bioss, China), or donkey anti-rabbit IgG/Alexa Fluor 488 (1:500, bs-0295d-af488, Bioss, China) as secondary antibodies in the dark at room temperature for 60 minutes. Finally, brain slices were rinsed with PBST, and slides were incubated with anti-quenching fluorescence (S2110, Solarbio, China). IHC staining of human brain tissue after stroke was performed using primary OPN antibody (1B20, 1:200, NB110-89062, Novus, USA) or anti-beclin1 (1:200, ab210498, Abcam, UK) primary antibodies. Secondary antibodies were from a ready-to-use Express MaxVision HRP Kit (mouse/rabbit; kit-5030, MXB-Biotechnologies). We observed results by fluorescence microscopy (Zeiss Axio Vert.A1). At least three slices from each mouse were analyzed. These slices were sampled from the infarct area, and each slice was collected from the field of view around the infarct area.
Immunoblotting
Normal animals were euthanized at the age of 0, 2, 7, 14, 21, or 28 days (n=3 per group) for endogenous OPN investigation. To assess endogenous OPN expression after stroke, male mice were euthanized 2, 7, 14, 21, or 28 days post-stroke. To evaluate the plasticity effect of OPN, synaptophysin (SYN) was measured 24 h after exogenous OPN delivery. Briefly, mice were sacrificed by decapitation. Twenty milligrams of mouse peri-infarct cortical tissue samples from the experimental and control groups were dissolved using 200 ml RIPA buffer (R0020, Solarbio, China) containing PMSF. After freezing and grinding the tissue, the tissue suspension was centrifuged at 4°C and 12000 r/min for 40 min, and the supernatant was collected. Using BSA as the standard, the supernatant was quantified by the BCA (P0012, Beyotime, China) method. A 20-μg protein sample was separated using 12% SDS-PAGE electrophoresis for 120 min and then transferred to a PVDF membrane for 1 h with 350 mA. The membrane was blocked in blocking buffer at 37°C for 1 hour. Then, we used rabbit anti-SYN (1:1000, bs-8845R, Bioss, China), goat anti-OPN (1:1000, ab11503, Abcam, UK), rabbit anti-integrin-β1 (1:1000, bs-0486R, Bioss, China), rabbit anti-beclin1 (1:1000, bs-1353R, Bioss, China), or rabbit anti-β-actin (1:1000, bs-0061R, Bioss, China) as primary antibodies and incubated the membrane overnight at 4°C. At the same time, the other group was incubated with TBST solution without antibodies as a negative control. After washing the membrane repeatedly, the membrane was incubated with peroxidase-labeled donkey anti-rabbit (1:1000, bs-0295D-HRP, Bioss, China) or anti-goat IgG (1:1000, bs-0294D-HRP, Bioss, China) secondary antibody. The membrane was shaken gently at room temperature for 1 hour, and the color was developed by ECI (PE0010, Solarbio, China). After washing the membrane, it was observed using an Amersham Imager 600. We used ImageJ software (NIH) to determine the absorbance of each band (A) value, with the expression of β-actin protein used as a reference and the gray value of the measured protein band/β-actin gray value as the relative expression level of the measured protein. We repeated the experiment three times.
Grid-walking task
A baseline test was performed 7 days before surgery and 1, 3, or 7 days post-stroke. Each mouse was tested at approximately the same time during the dark cycle. The grid-walking apparatus was constructed using a box with a base made of metal wire mesh with 1×1 cm openings. The grid volume was 32×20×50 cm (length × width × height). A camera was positioned under the apparatus to provide video footage to count the number of foot faults. Each animal was placed individually on top of the high wire grid and allowed to move freely for 5 minutes. The video footage was subsequently analyzed offline to count the total number of foot faults and non-foot fault steps for each limb (stroke+saline and OPN treatment group); evaluators were blinded to the treatment groups. The percentage of foot faults per total steps was calculated as follows: foot faults = number of foot faults/(total number of foot faults+non-foot fault steps) ×100%. Variability in the amount of locomotion between animals and trials was removed by calculating the ratio between foot faults and total steps. If a step did not provide support and the foot went through the grid hole, this was considered a fault. If an animal rested on the grid at the wrist level, this was also defined as a fault. This protocol has often been used to evaluate treatments that enhance behavioral recovery after stroke, with low animal-to-animal differences and high reproducibility [7,33,34].
Forelimb task (cylinder task)
The forelimb test allows the investigation of forelimb use during mouse exploration of the upright walls of a cylinder for 5 min as described previously [7,33,34]. When placed in a Plexiglas cylinder (15 cm in height with a diameter of 10 cm), a mouse spontaneously stands up by pressing the cylinder wall with one or both of its forelimbs. Each mouse was allowed to explore freely for 5 minutes and was videotaped. Video footage was played in slow motion (1/5th real-time speed), and the number of rears using the right forelimb (ipsilateral to the lesion), left forelimb (contralateral to the lesion), or both forelimbs were determined. The number of times each limb was used was calculated. These data were used to derive an asymmetry index as follows: (number of first touches by the right forelimb minus number of first touches by the left forelimb)/(number of right first touches+number of left first touches+number of both right and left touches at the same time). The investigator was blinded to the animal group during quantitative analysis of the footage to determine forelimb preference.
Immunofluorescence analysis
VgluT1 was used to label presynaptic terminal sites. A synapse was defined as the common domain point between the signals generated by these markers. Pre-synapse immune signals in the cerebral cortex peri-infarct area were quantified by a Puncta Analyzer plugin (obtained from c.eroglu@cellbio.duke.edu) using ImageJ software [35].
Statistical analysis
GraphPad Prism 8.0 software was used for statistical analysis and graphing. One-way ANOVA was used for comparison between groups. Data from behavioral experiments were analyzed by two-way ANOVA. All data are presented as mean ± standard error of the mean (SEM). Means of 2 continuous normally distributed variables were compared by independent samples Student’s T test. Mann-Whitney U test and Kruskal-Wallis test were used, respectively, to compare means of 2 and 3 or more groups of variables not normally distributed. To compare proportions of two nominal variables, Pearson’s X2 test and Fisher’s exact test of independence were used. A value of P<0.05 was considered significant.
Results
Patients with acute ischemic stroke show increased OPN and beclin1 expression
Tissue was collected from acute ischemic stroke patients with large-area cerebral infarction who underwent craniotomy surgery (Figure 1A). IHC data confirmed that intracranial decompression control tissue showed very weak OPN staining. By contrast, there was strong OPN staining within the penumbra cortex of acute ischemic stroke patients (Figure 1B).
Figure 1.
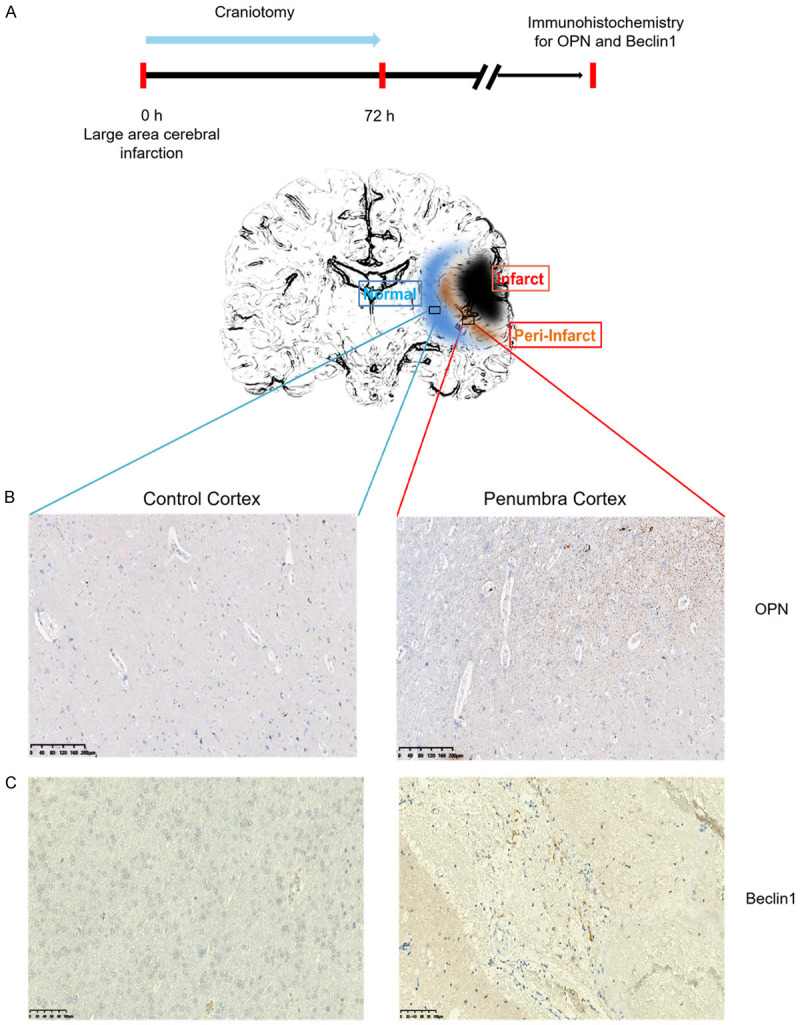
Expression of OPN and beclin1 in acute ischemic stroke patient tissue. A. Experimental timeline. B. Immunohistochemistry for OPN in cortical tissue obtained during intracranial decompression operation as normal control (left) and penumbra cortical tissue (right) (n=4). Original magnification, 4×. Scale bar: 200 μm. C. IHC for beclin1 in cortical tissue obtained during intracranial decompression operation as normal control (left) and penumbra cortical tissue (right) (n=2). Original magnification, 10×. Scale bar: 100 μm.
As autophagy plays a critical role in ischemic stroke [36], we next asked whether autophagy occurs within the penumbra cortex of acute ischemic stroke patients. As shown in Figure 1C, IHC staining indicated a marked elevation in the expression of beclin1, an autophagy-related marker, compared with that in the control group. These data suggest that the expression of genes involved in the modulation of autophagy and OPN are augmented in penumbra tissue after stroke.
Spatial and temporal expression profiles of OPN in the ipsilateral and contralateral hemisphere cortex after ischemic stroke
We assessed changes in OPN expression in the mouse brain after ischemic insult using immunofluorescence and western blotting (Figure 2). There was almost no expression of OPN in the contralateral hemisphere after stroke (Figures 2B and S3). However, endogenous OPN expression in the ipsilateral hemisphere showed significant elevation starting 2 days post-stroke and gradually increased until reaching a peak 7 days after stroke (Figure 2C). To investigate the cell sources and type of OPN expression, we performed double-staining for NeuN/OPN, Iba-1/OPN, and glial fibrillary acidic protein (GFAP)/OPN in ipsilateral brain sections after stroke or sham operation. Interestingly, OPN expression colocalized with neurons and Iba-1 in the peri-infarct cortex in the ipsilateral hemisphere after stroke (Figures 2A and S1), but little expression was observed in astrocytes (Figure S2). Moreover, little expression of OPN was observed in the sham-operated group.
Figure 2.

Increased OPN in ischemic stroke mice. A. Co-staining for OPN (red) and NeuN (green) in sham mice and the peri-infarct area of mice 2, 7, 14, 21, or 28 days post-stroke, n=6 per group. B. Immunoblots of OPN expression in sham mice and the peri-infarct area of mice 2, 7, 14, 21, or 28 days post-stroke, n=3 per group. C. Quantification of OPN protein. Data are expressed as mean ± SEM. ^^P<0.01 vs. sham group, **P<0.01 vs. other experimental groups. Original magnification, 40×. Scale bar: 20 μm.
Neuroplasticity and autophagic activation in the ipsilateral penumbra post-stroke
To evaluate post-stroke changes in neural and axonal plasticity, we visualized presynaptic terminals by immunostaining with antibodies against the glutamatergic synapse marker VgluT1 and used high-content image analysis to quantify VgluT1 immunoreactive puncta in the peri-infarct region [37]. Immunofluorescence results showed that expression of the presynaptic marker VgluT1 was significantly increased in the peri-infarct area 7 days after photothrombotic stroke compared with that in the sham-operated group (Figure 3A). This finding suggests the occurrence of neuroplasticity in the cortex after cerebral ischemia. Furthermore, immunoblotting analysis showed that SYN and beclin1 were significantly upregulated in the cortex in the ipsilateral penumbra 7 days after photothrombotic stroke relative to the sham-operated group (Figures 3B and S4), (Figures 3C and S5). These results imply an association between autophagic activation and neuroplasticity in the ipsilateral penumbra after an ischemic stroke.
Figure 3.
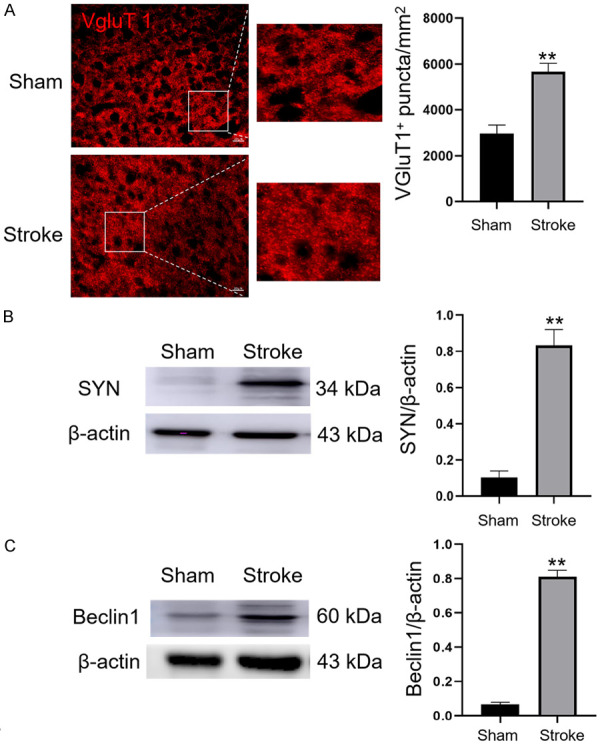
Neuroplasticity and autophagic activation in the ipsilateral cortex after ischemic stroke. A. Immunostaining and quantification of VgluT1 puncta (red) in the penumbra in sham-operated and photothrombotic stroke groups 7 days after stroke, n=6 per group. Original magnification, 40×. Scale bar: 20 μm. B. Immunoblotting and quantification showing expression of SYN relative to that of β-actin in the cortex in the ipsilateral hemisphere from sham-operated and stroke mice, n=3 per group. C. Immunoblotting and quantification showing expression of beclin1 relative to that of β-actin in the cortex in the ipsilateral hemisphere from sham-operated and stroke mice, n=3 per group. Data are expressed as the mean ± SEM. **P<0.01 vs. sham-operated group.
Activating OPN promotes motor function recovery after stroke
To investigate whether OPN enhances neural repair and promotes functional recovery, we tested mice on forelimb motor tasks using the same stroke model in which neural plasticity was measured [37]. The cylinder and grid-walking tasks evaluating exploratory forelimb use and gait were used. All behavioral testing of forelimb motor function was conducted 1-week post-stroke. In addition to clinical intervention, we intracerebrally administered rOPN or saline within 3 hours post-stroke. The cylinder and grid-walking tests were carried out 1, 3, or 7 days after OPN treatment. Baseline was established 1 week before stroke. When comparing stroke+saline and stroke+rOPN groups, we found that 3 days after OPN administration, the movement behavior of mice began to improve. Seven days after injection of rOPN, there were significant improvements in forelimb movement coordination and the foot fault rate of mice (Figure 4). Our data suggest that intracerebral administration of OPN enhances motor recovery and may promote neural remodeling, which may represent the formation of neural circuits that lead to behavioral improvement after stroke.
Figure 4.
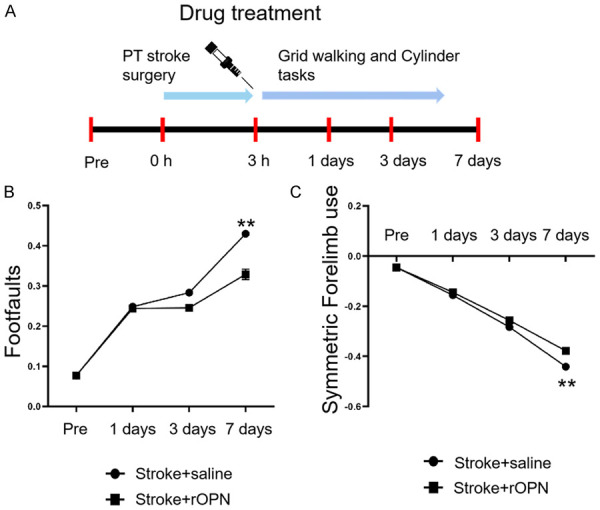
OPN promotes motor function recovery. A. Steps of the motor behavior testing process after stroke and injection of rOPN. B. Statistical analysis of the percentage of foot faults in the grid-walking test for stroke+saline and stroke+rOPN groups (number of mouse steps with errors = number of empty grids/total number of grids walked). C. Cylinder tests for stroke+saline and stroke+rOPN groups to assess the symmetry of forelimb movement. Data are expressed as the mean ± SEM, **P<0.01 vs. stroke+saline group. n=6 per group.
OPN facilitates neuroplasticity and neuronal survival
Underlying neuroplasticity is the primary event of the subacute phase of stroke that leads to recovery and rebuilding of motor and sensor maps (Figure 5A). Therefore, we performed intracranial administration of rOPN 7 days after stroke. Immunostaining further confirmed that the density of VgluT1 puncta was significantly upregulated in the peri-infarct area and contralateral cortex 24 hours after OPN treatment (Figure 5B and 5C), (Tables S1 and S2). These results indicate that a single dose of OPN induces rapid neural remodeling in the ipsilateral and contralateral cortices of mice 7 days after stroke. We also evaluated the protein level of SYN. SYN is a calcium-bound protein expressed in the presynaptic vesicle membrane and a synaptic plasticity marker that plays a critical role in neurorehabilitation [38]. Compared with saline-treated mice, western blot data showed a significant upregulation of SYN in stroke mice treated with rOPN (Figures 5D and S6).
Figure 5.
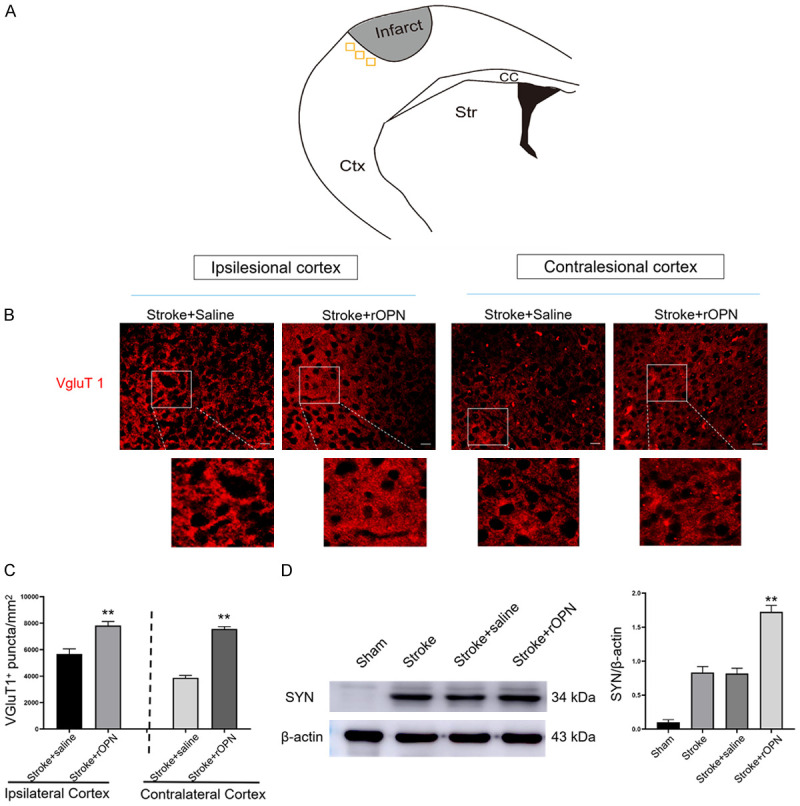
OPN facilitates neural plasticity and neuronal survival. A. Location of visual field acquisition in the peri-infarct area and corresponding regions in the contralesional hemisphere in mice after stroke. B. VgluT1 immunostaining images in the peri-infarct area and contralateral cortex in stroke+saline and stroke+rOPN groups, n=6 per group. C. Quantification of VGluT1 puncta. D. Representative bands and quantification of SYN in the peri-infarct area after rOPN treatment, n=3 per group. Data are expressed as the mean ± SEM, **P<0.01 vs. stroke+saline group. Original magnification, 40×. Scale bar: 20 μm.
OPN improves axon plasticity through the integrin pathway
To verify the mechanism of OPN-induced improvements in neuroplasticity, we performed immunofluorescence staining and western blotting experiments. rOPN treatment significantly increased the expression of ITGB1, which co-located with NeuN in the peri-infarct area (Figure 6A). On the other hand, after administration of GRGDSP, an integrin antagonist, ITGB1 was significantly downregulated (Figure 6A). Western blotting analysis also showed that ITGB1 significantly increased after stroke. Compared with the stroke+saline group, rOPN injection also significantly upregulated the expression of ITGB1. By contrast, GRGDSP decreased the expression of ITGB1 (Figures 6B and S7). To verify whether rOPN can improve axon plasticity in the peri-infarct area after stroke through the integrin pathway, we intracerebrally delivered rOPN in combination with GRGDSP injection in the peri-infarct area and then measured the expression of SYN in the peri-infarct area. We found that SYN significantly increased after stroke. rOPN treatment increased the protein level of SYN around the infarct area compared with that in the stroke+saline group. However, this phenomenon was reversed by GRGDSP (Figures 6C and S8). These results suggest that rOPN promotes SYN expression in the peri-infarct area through the integrin protein pathway and improves axon plasticity.
Figure 6.
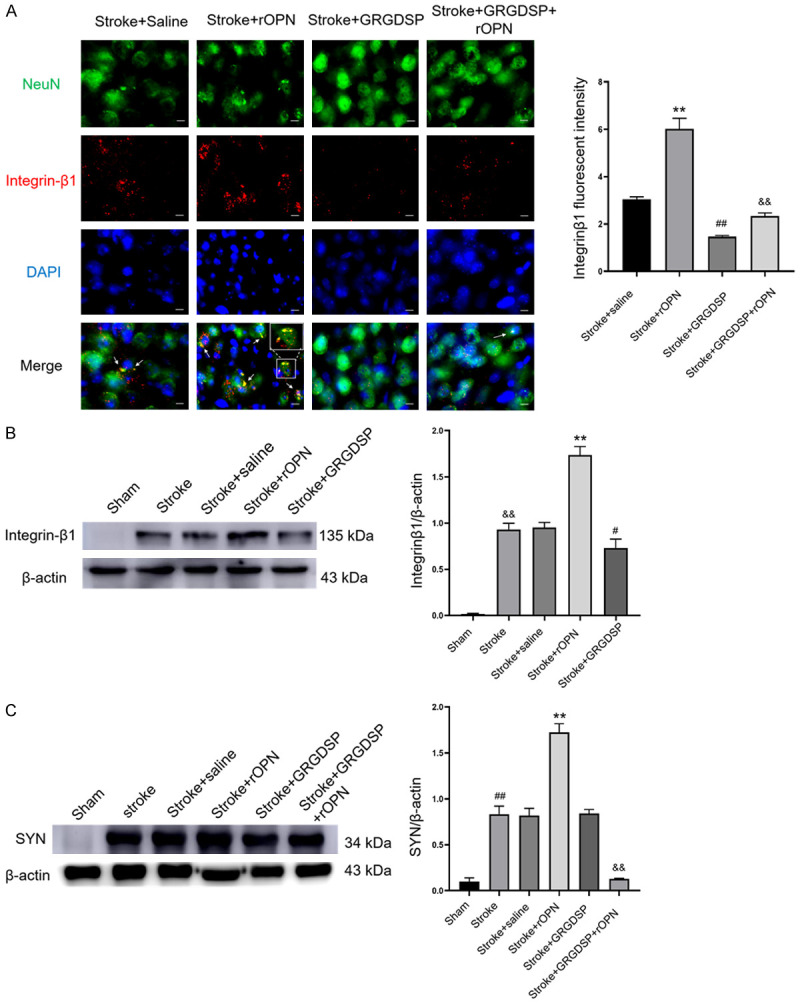
OPN improves axon plasticity through the integrin pathway. A. Immunofluorescence staining images of NeuN and ITGB1 in the peri-infarct area of the stroke+saline, stroke+rOPN, stroke+GRGDSP, and stroke+GRGDSP+rOPN groups, n=6 per group (**P<0.01 vs. stroke+saline group, ##P<0.01 vs. stroke+saline group, &&P<0.01 vs. stroke+rOPN group). B. Western blot of ITGB1 expression in the peri-infarct area after rOPN and GRGDSP injection, n=3 per group (**P<0.01 vs. stroke+saline group, #P<0.05 vs. stroke+saline group, &&P<0.01 vs. sham group). C. Western blot of SYN expression in the peri-infarct area after rOPN, saline, or GRGDSP injection, n=3 per group (**P<0.01 vs. stroke+saline group, ##P<0.01 vs. sham group, &&P<0.01 vs. stroke+rOPN group). Data are expressed as the mean ± SEM. Original magnification, 40×. Scale bar: 20 μm.
OPN enhances integrin-mediated autophagy
We next investigated whether rOPN can promote autophagy in the area around the infarct after stroke through the integrin pathway. After injection of rOPN, we performed immunofluorescence experiments with autophagic markers LC3 and beclin1 in the peri-infarct area. After rOPN injection, immunofluorescence signals of LC3 and beclin1 in the peri-infarct area were significantly increased and colocalized with neurons, which was substantially different from the stroke+saline group (Figure 7A and 7B). However, rOPN was injected after GRGDSP, which does not induce autophagy in the peri-infarct area. Western blot results also showed that the re-injection of rOPN significantly upregulated the expression of beclin1 in the peri-infarct region, and this upregulation disappeared with administration of GRGDSP (Figures 7C and S9). These results indicate that rOPN enhanced the autophagy response of neuronal cells after stroke, and the autophagy response induced by rOPN may proceed through the integrin pathway.
Figure 7.

OPN enhances integrin-mediated autophagy. A. Double-labeled immunofluorescence staining of NeuN/LC3 in the peri-infarct area in stroke+saline, stroke+rOPN, stroke+GRGDSP, and stroke+GRGDSP+rOPN groups, n=6 per group. B. Immunostaining with NeuN/beclin1 in the peri-infarct area in stroke+saline, stroke+rOPN, stroke+GRGDSP, and stroke+GRGDSP+rOPN groups, n=6 per group. C. Western blotting was used to detect the autophagy marker beclin1 in the peri-infarct area in sham, stroke, stroke+rOPN, stroke+GRGDSP, and stroke+GRGDSP+rOPN groups, n=3 per group. Data are expressed as the mean ± SEM. **P<0.01 vs. stroke+saline group, ##P<0.01 vs. stroke+rOPN group. Original magnification, 40×. Scale bar: 20 μm.
Discussion
There are three primary results of our study. First, local administration of rOPN improves functional recovery after ischemic stroke. Second, OPN plays a vital role in stimulating neural plasticity 7 days after stroke instead of having a neuroprotection effect in acute ischemic stroke. We suggest that rOPN greatly stimulates ipsilateral and contralateral hemisphere neuroplasticity by VgluT1 and SYN induction, activating pro-survival signals in synapses. Third, OPN-dependent neuroplasticity is regulated by the OPN/integrin/autophagy signaling pathway. Blockade of integrin receptors via a competitive ligand binding inhibitor of GRGDSP abolished the autophagy-activating and neural plasticity effect of OPN in vivo.
Pathogenic mechanisms following ischemic stroke are very complex. Many studies demonstrate that stroke triggers cell injury and death and leads to neurological dysfunction by inducing excitotoxicity, inflammation, and apoptosis in the acute phase. However, several reports show that endogenous plasticity in the peri-infarct cortex and contralateral hemisphere is associated with recovery in the subacute phase of stroke [3,39]. Stroke triggers a transcriptional regrowth scheme during the subacute phase that stimulates plasticity [28]. This transcriptional scheme is powerful, with hundreds of neuronal genes with different expressions. These neuronal genes originate from sprouting neurons in the peri-infarct cortex, which induces axon guidance, growth factor responses, intracellular growth (e.g., increased expression of adhesion molecules), and cytoskeletal remodeling. These induced signaling pathways in neurons neighboring the infarct result in axonal growth and synapse formation, enabling projections to adjacent cortical areas after an ischemic insult. This phenomenon corresponds to learning and stroke recovery in mouse models [6,7,40]. OPN is an adhesion protein that is a remarkably upregulated gene in sprouting neurons, with a 12-fold increase compared with the control expression level of nonsprouting neurons. Several studies indicate that OPN induces neuroprotection via preconditioning or in the acute phase of stroke [20,41]. However, the function of OPN in the remodeling stage starting several days post-stroke has been underexplored.
The present study shows that OPN was highly expressed in neurons 7 days after stroke (Figure 2). We also observed OPN upregulation in the patients (Figure 1). To confirm the effect of the plasticity process in vivo, mice were intracerebrally injected with rOPN in the ipsilateral hemisphere 7 days after stroke. As expected, we found that OPN delivery induced a characteristic connection network by increasing expression of VgluT1, a presynaptic marker of most glutamatergic synapses [37,42]. Furthermore, OPN also increased neural plasticity in neurons in the sensorimotor cortex of the contralateral hemisphere. This finding is in agreement with previous studies showing that AAV-OPN injected into the unlesioned contralateral cortex induces the projection of corticospinal neurons to the denervated hemisphere after stroke, promoting functional recovery from long-term brain injury [32]. Additionally, SYN is an important synaptic plasticity-relevant protein that plays a critical role in neurorehabilitation after brain injury. We quantitatively measured SYN protein by western blotting to further verify the effect of OPN on synaptic plasticity. The results indicate that OPN upregulated the expression of SYN, a presynaptic marker. These data are consistent with previous publications showing that elevation of SYN is closely related to enhanced synaptic and protective effects after stroke. Notably, Li et al. showed SYN expression downregulation in sprouting neurons [28], which seems paradoxical to the results of our study. This contradiction may be interpreted as follows: SYN is a major small synaptic vesicle protein. It is produced in the neuronal cell body and anterogradely transported in the axon to various synapses. During axonal damage, axon transportation is hindered, and SYN accumulates [43]. When axonal regrowth occurs, transportation is restored, leading to a decrease in SYN. In line with our results, many studies implicate SYN in synapse regrowth after ischemic stroke, and increased SYN expression enhances motor function recovery [44].
So far, treatment of CNS disease is very difficult because of the blood-brain barrier. There are currently three major drug delivery routes to the CNS: intranasal, intracerebroventricular, and intracerebral administration. Previous publications demonstrate that intranasal and intracerebroventricular administration of OPN can alleviate brain injury [20,45,46]. However, there are several limitations to these delivery routes. First, the drug is delivered intraventricularly to cerebrospinal fluid, which is separated from the brain by the ependymal barrier that is similar to the blood-brain barrier and is difficult to absorb. Second, intraventricular and intranasal delivery leads to drug diffusing throughout the whole brain rather than at target areas. Additionally, intraventricular and intranasal delivery may elicit neurologic toxicity due to the wide distribution of the drug in the CNS. Our study shows that OPN protein was delivered to the stroke cavity and contributed to neuronal plasticity in the somatosensory cortex. Furthermore, we showed that rOPN treatment ameliorated functional behavior outcomes in both cylinder and grid-walking tasks. Data from the cylinder task showed improved motor recovery in the rOPN-treated group, and data from the grid-walking task showed improved coordination and sensory function. In agreement with a previous study [7], our findings suggest that intracranial delivery of OPN increased VgluT1 and SYN, which correlated with functional recovery through upregulated synaptic density in the functional area of the cortex and significantly contributed to functional improvement.
How could the effect of OPN on morphological and behavioral outcomes after photothrombosis be regulated? In recent years, accumulating evidence has focused on the role of autophagy and its underlying signaling pathway after stroke [36,47]. Autophagy is necessary for neuronal survival and synaptic plasticity [16]. A previous publication reports that extracellular matrix molecular dysfunction causes neural injury and enhances autophagic deficits in neurodegenerative disease, aggravating neuronal apoptosis [48]. Recent reports demonstrate that OPN attenuates early brain injury (subarachnoid hemorrhage), possibly by promoting autophagy and decreasing apoptosis [49]. On the other hand, inhibiting neuronal autophagy is essential for structural recovery and functional activity-dependent synaptic plasticity in hippocampal neurons [13]. Therefore, we hypothesize that OPN induces autophagy of pro-survival neurons and then stimulates neuroplasticity in delayed brain injury. Here, we show that neuroplasticity and neural autophagic activation occur in the ipsilateral penumbra 7 days after ischemic stroke. Intracranial delivery of OPN promoted neuronal plasticity and survival and increased the expression of autophagy-related proteins beclin1 and LC3 in neurons during the subacute phase of stroke. These results are consistent with previous publications in which researchers demonstrated that autophagy was induced in neurons and downregulated apoptosis in the subacute phase of stroke [50]. Wang et al. demonstrated that appropriate autophagy in neurons during the subacute phase of stroke could cause cell survival, whereas excessive autophagy exacerbates cellular injury following MCAO [51,52]. It is probably due to different ischemic stroke models causing distinct levels of autophagy. Therefore, our data support the conclusion that OPN plays a positive role in neuroplasticity, possibly by activating autophagy in the subacute phase of stroke.
Integrin receptors, including α and β subunits, critically mediate axonal regeneration and synaptic function [53,54]. Moreover, several results suggest that the expression and activity of integrins are associated with a protective effect of autophagy in brain injury [55,56]. The present study shows that OPN can regulate ITGB1 expression. When OPN and GRGDSP were co-injected after stroke, neuroplasticity and autophagy were completely reversed compared with when OPN was injected alone. Combining the results of earlier publications with the present results, we suggest that the ITGB1 receptor may be involved in OPN-integrin-induced autophagy and neuroplasticity.
Our research has some limitations. First, our findings indicate that OPN promotes neural plasticity by activating autophagy. However, we did not inhibit autophagy via 3-methyladenine or spautin-1 to investigate morphological and functional behavior outcomes. Second, many reports demonstrate that ITGB1 is a major integrin involved in OPN-integrin signaling. Thus, our western blotting and immunofluorescence staining results suggest that ITGB1 is engaged in OPN-enhanced autophagy and plasticity. However, we cannot exclude the possibility that blocking ITGB1 by an integrin antagonist (i.e., GRGDSP) may also inhibit other integrin receptors. Moreover, long-term neural repair outcomes and more precise signaling pathways in OPN-enhanced autophagy and neuroplasticity should be investigated in future studies.
Collectively, our research clarifies that OPN contributes to ischemic stroke by stimulating neuroplasticity via activating autophagy, possibly through involving ITGB1 in an upstream modulation pathway. Furthermore, delayed administration of OPN to enhance autophagy may be a potential preventive and therapeutic strategy against ischemic stroke that promotes neuronal survival and plasticity.
Acknowledgements
This work was supported by grants from the Natural Science Foundation of Guangxi, China (2021JJA141110); the Guilin innovation platform and talent project (20210218-6); The project of improving the basic scientific research ability of young and middle-aged teachers in Guangxi Universities (2021KY0491); Guangxi Key Laboratory of Brain and Cognitive Neuroscience, Guilin Medical University (GKLBCN-20200108-01, GKLBCN-20200106); Natural Science Foundation of China (82060268); Natural Science Foundation of Guangxi, China (2020JJA140124).
Disclosure of conflict of interest
None.
Abbreviations
- GRGDSP
Gly-Arg-Gly-Asp-Ser-Pro integrin-competitive inhibitor
- IBA1
Ionized calcium binding adapter molecule 1
- GFAP
Glial fibrillary acidic protein
- ITGB1
Integrin subunit beta 1
- SEM
Standard error of the mean
- OPN
Osteopontin
- SYN
Synaptophysin
- VgluT1
Vesicular glutamate transporter 1
- Ctx
Cortex
- CC
Corpus callosum
- Str
Striatum
- MCAO
Middle cerebral artery occlusion
- BECN1
Beclin1
- IHC
Immunohistochemistry
Supporting Information
References
- 1.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation. 2018;137:e67–e492. doi: 10.1161/CIR.0000000000000558. [DOI] [PubMed] [Google Scholar]
- 2.Carmichael ST, Kathirvelu B, Schweppe CA, Nie EH. Molecular, cellular and functional events in axonal sprouting after stroke. Exp Neurol. 2017;287:384–394. doi: 10.1016/j.expneurol.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carmichael ST. Emergent properties of neural repair: elemental biology to therapeutic concepts. Ann Neurol. 2016;79:895–906. doi: 10.1002/ana.24653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lang CE, Lohse KR, Birkenmeier RL. Dose and timing in neurorehabilitation: prescribing motor therapy after stroke. Curr Opin Neurol. 2015;28:549–555. doi: 10.1097/WCO.0000000000000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joy MT, Carmichael ST. Encouraging an excitable brain state: mechanisms of brain repair in stroke. Nat Rev Neurosci. 2021;22:38–53. doi: 10.1038/s41583-020-00396-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joy MT, Ben Assayag E, Shabashov-Stone D, Liraz-Zaltsman S, Mazzitelli J, Arenas M, Abduljawad N, Kliper E, Korczyn AD, Thareja NS, Kesner EL, Zhou M, Huang S, Silva TK, Katz N, Bornstein NM, Silva AJ, Shohami E, Carmichael ST. CCR5 is a therapeutic target for recovery after stroke and traumatic brain injury. Cell. 2019;176:1143–1157. doi: 10.1016/j.cell.2019.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li S, Nie EH, Yin YQ, Benowitz LI, Tung S, Vinters HV, Bahjat FR, Stenzel-Poore MP, Kawaguchi R, Coppola G, Carmichael ST. GDF10 is a signal for axonal sprouting and functional recovery after stroke. Nat Neurosci. 2015;18:1737–1745. doi: 10.1038/nn.4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 9.Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- 11.Wang P, Guan YF, Du H, Zhai QW, Su DF, Miao CY. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy. 2012;8:77–87. doi: 10.4161/auto.8.1.18274. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Yan H, Yuan Y, Gao J, Shen Z, Cheng Y, Shen Y, Wang RR, Wang X, Hu WW, Wang G, Chen Z. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy. 2013;9:1321–1333. doi: 10.4161/auto.25132. [DOI] [PubMed] [Google Scholar]
- 13.Glatigny M, Moriceau S, Rivagorda M, Ramos-Brossier M, Nascimbeni AC, Lante F, Shanley MR, Boudarene N, Rousseaud A, Friedman AK, Settembre C, Kuperwasser N, Friedlander G, Buisson A, Morel E, Codogno P, Oury F. Autophagy is required for memory formation and reverses age-related memory decline. Curr Biol. 2019;29:435–448. e438. doi: 10.1016/j.cub.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 14.Nikoletopoulou V, Sidiropoulou K, Kallergi E, Dalezios Y, Tavernarakis N. Modulation of autophagy by BDNF underlies synaptic plasticity. Cell Metab. 2017;26:230–242. e235. doi: 10.1016/j.cmet.2017.06.005. [DOI] [PubMed] [Google Scholar]
- 15.Stavoe AK, Hill SE, Hall DH, Colón-Ramos DA. KIF1A/UNC-104 transports ATG-9 to regulate neurodevelopment and autophagy at synapses. Dev Cell. 2016;38:171–185. doi: 10.1016/j.devcel.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hill SE, Colón-Ramos DA. The journey of the synaptic autophagosome: a cell biological perspective. Neuron. 2020;105:961–973. doi: 10.1016/j.neuron.2020.01.018. [DOI] [PubMed] [Google Scholar]
- 17.Rangaswami H, Bulbule A, Kundu GC. Osteopontin: role in cell signaling and cancer progression. Trends Cell Biol. 2006;16:79–87. doi: 10.1016/j.tcb.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 18.Zhou YX, Yao YH, Sheng LS, Zhang JM, Zhang JH, Shao AW. Osteopontin as a candidate of therapeutic application for the acute brain injury. J Cell Mol Med. 2020;24:8918–8929. doi: 10.1111/jcmm.15641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denhardt DT, Giachelli CM, Rittling SR. Role of osteopontin in cellular signaling and toxicant injury. Annu Rev Pharmacol Toxicol. 2001;41:723–749. doi: 10.1146/annurev.pharmtox.41.1.723. [DOI] [PubMed] [Google Scholar]
- 20.Meller R, Stevens SL, Minami M, Cameron JA, King S, Rosenzweig H, Doyle K, Lessov NS, Simon RP, Stenzel-Poore MP. Neuroprotection by osteopontin in stroke. J Cereb Blood Flow Metab. 2005;25:217–225. doi: 10.1038/sj.jcbfm.9600022. [DOI] [PubMed] [Google Scholar]
- 21.Takarada T, Takahata Y, Iemata M, Hinoi E, Uno K, Hirai T, Yamamoto T, Yoneda Y. Interference with cellular differentiation by D-serine through antagonism at N-methyl-D-aspartate receptors composed of NR1 and NR3A subunits in chondrocytes. J Cell Physiol. 2009;220:756–764. doi: 10.1002/jcp.21821. [DOI] [PubMed] [Google Scholar]
- 22.Wang C, Shi Z, Zhang Y, Li M, Zhu J, Huang Z, Zhang J, Chen J. CBFβ promotes colorectal cancer progression through transcriptionally activating OPN, FAM129A, and UPP1 in a RUNX2-dependent manner. Cell Death Differ. 2021;28:3176–3192. doi: 10.1038/s41418-021-00810-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahata Y, Takarada T, Osawa M, Hinoi E, Nakamura Y, Yoneda Y. Differential regulation of cellular maturation in chondrocytes and osteoblasts by glycine. Cell Tissue Res. 2008;333:91–103. doi: 10.1007/s00441-008-0607-7. [DOI] [PubMed] [Google Scholar]
- 24.Huang RH, Quan YJ, Chen JH, Wang TF, Xu M, Ye M, Yuan H, Zhang CJ, Liu XJ, Min ZJ. Osteopontin promotes cell migration and invasion, and inhibits apoptosis and autophagy in colorectal cancer by activating the p38 MAPK signaling pathway. Cell Physiol Biochem. 2017;41:1851–1864. doi: 10.1159/000471933. [DOI] [PubMed] [Google Scholar]
- 25.Lin R, Wu S, Zhu D, Qin M, Liu X. Osteopontin induces atrial fibrosis by activating Akt/GSK-3β/β-catenin pathway and suppressing autophagy. Life Sci. 2020;245:117328. doi: 10.1016/j.lfs.2020.117328. [DOI] [PubMed] [Google Scholar]
- 26.Liu G, Fan X, Tang M, Chen R, Wang H, Jia R, Zhou X, Jing W, Wang H, Yang Y, Yin F, Wei H, Li B, Zhao J. Osteopontin induces autophagy to promote chemo-resistance in human hepatocellular carcinoma cells. Cancer Lett. 2016;383:171–182. doi: 10.1016/j.canlet.2016.09.033. [DOI] [PubMed] [Google Scholar]
- 27.Zheng YH, Tian C, Meng Y, Qin YW, Du YH, Du J, Li HH. Osteopontin stimulates autophagy via integrin/CD44 and p38 MAPK signaling pathways in vascular smooth muscle cells. J Cell Physiol. 2012;227:127–135. doi: 10.1002/jcp.22709. [DOI] [PubMed] [Google Scholar]
- 28.Li SL, Overman JJ, Katsman D, Kozlov SV, Donnelly CJ, Twiss JL, Giger RJ, Coppola G, Geschwind DH, Carmichael ST. An age-related sprouting transcriptome provides molecular control of axonal sprouting after stroke. Nat Neurosci. 2010;13:1496–1504. doi: 10.1038/nn.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson MA, O’Shea TM, Burda JE, Ao Y, Barlatey SL, Bernstein AM, Kim JH, James ND, Rogers A, Kato B, Wollenberg AL, Kawaguchi R, Coppola G, Wang C, Deming TJ, He ZG, Courtine G, Sofroniew MV. Required growth facilitators propel axon regeneration across complete spinal cord injury. Nature. 2018;561:396–400. doi: 10.1038/s41586-018-0467-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bei FF, Lee HHC, Liu XF, Gunner G, Jin H, Ma L, Wang C, Hou LJ, Hensch TK, Frank E, Sanes JR, Chen CF, Fagiolini M, He ZG. Restoration of visual function by enhancing conduction in regenerated axons. Cell. 2016;164:219–232. doi: 10.1016/j.cell.2015.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duan X, Qiao M, Bei FF, Kim IJ, He ZG, Sanes JR. Subtype-specific regeneration of retinal ganglion cells following axotomy: effects of osteopontin and mTOR signaling. Neuron. 2015;85:1244–1256. doi: 10.1016/j.neuron.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu YY, Wang XH, Li WL, Zhang Q, Li Y, Zhang ZC, Zhu JJ, Chen B, Williams PR, Zhang YM, Yu B, Gu XS, He ZG. A sensitized IGF1 treatment restores corticospinal axon-dependent functions. Neuron. 2017;95:817–833. doi: 10.1016/j.neuron.2017.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang L, Han B, Zhang Z, Wang S, Bai Y, Zhang Y, Tang Y, Du L, Xu L, Wu F, Zuo L, Chen X, Lin Y, Liu K, Ye Q, Chen B, Li B, Tang T, Wang Y, Shen L, Wang G, Ju M, Yuan M, Jiang W, Zhang JH, Hu G, Wang J, Yao H. Extracellular vesicle-mediated delivery of circular RNA SCMH1 promotes functional recovery in rodent and nonhuman primate ischemic stroke models. Circulation. 2020;142:556–574. doi: 10.1161/CIRCULATIONAHA.120.045765. [DOI] [PubMed] [Google Scholar]
- 34.Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael ST. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature. 2010;468:305–309. doi: 10.1038/nature09511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ippolito DM, Eroglu C. Quantifying synapses: an immunocytochemistry-based assay to quantify synapse number. J Vis Exp. 2010:2270. doi: 10.3791/2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ajoolabady A, Wang S, Kroemer G, Penninger JM, Uversky VN, Pratico D, Henninger N, Reiter RJ, Bruno A, Joshipura K, Aslkhodapasandhokmabad H, Klionsky DJ, Ren J. Targeting autophagy in ischemic stroke: from molecular mechanisms to clinical therapeutics. Pharmacol Ther. 2021;225:107848. doi: 10.1016/j.pharmthera.2021.107848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stokowska A, Atkins AL, Morán J, Pekny T, Bulmer L, Pascoe MC, Barnum SR, Wetsel RA, Nilsson JA, Dragunow M, Pekna M. Complement peptide C3a stimulates neural plasticity after experimental brain ischaemia. Brain. 2017;140:353–369. doi: 10.1093/brain/aww314. [DOI] [PubMed] [Google Scholar]
- 38.Yang KL, Zhou Y, Zhou LQ, Yan FM, Guan L, Liu HM, Liu W. Synaptic plasticity after focal cerebral ischemia was attenuated by Gap26 but enhanced by GAP-134. Front Neurol. 2020;11:888. doi: 10.3389/fneur.2020.00888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pekna M, Pekny M, Nilsson M. Modulation of neural plasticity as a basis for stroke rehabilitation. Stroke. 2012;43:2819–2828. doi: 10.1161/STROKEAHA.112.654228. [DOI] [PubMed] [Google Scholar]
- 40.Luke LM, Allred RP, Jones TA. Unilateral ischemic sensorimotor cortical damage induces contralesional synaptogenesis and enhances skilled reaching with the ipsilateral forelimb in adult male rats. Synapse. 2004;54:187–199. doi: 10.1002/syn.20080. [DOI] [PubMed] [Google Scholar]
- 41.Hu SL, Huang YX, Hu R, Li F, Feng H. Osteopontin mediates hyperbaric oxygen preconditioning-induced neuroprotection against ischemic stroke. Mol Neurobiol. 2015;52:236–243. doi: 10.1007/s12035-014-8859-6. [DOI] [PubMed] [Google Scholar]
- 42.Micheva KD, Busse B, Weiler NC, O’Rourke N, Smith SJ. Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron. 2010;68:639–653. doi: 10.1016/j.neuron.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gudi V, Gai L, Herder V, Tejedor LS, Kipp M, Amor S, Sühs KW, Hansmann F, Beineke A, Baumgärtner W, Stangel M, Skripuletz T. Synaptophysin is a reliable marker for axonal damage. J Neuropathol Exp Neurol. 2017;76:109–125. doi: 10.1093/jnen/nlw114. [DOI] [PubMed] [Google Scholar]
- 44.Wang CJ, Zhang Q, Yu KW, Shen XY, Wu Y, Wu JF. Enriched environment promoted cognitive function via bilateral synaptic remodeling after cerebral ischemia. Front Neurol. 2019;10:1189. doi: 10.3389/fneur.2019.01189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albertsson AM, Zhang X, Leavenworth J, Bi D, Nair S, Qiao L, Hagberg H, Mallard C, Cantor H, Wang X. The effect of osteopontin and osteopontin-derived peptides on preterm brain injury. J Neuroinflammation. 2014;11:197. doi: 10.1186/s12974-014-0197-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gong L, Manaenko A, Fan R, Huang L, Enkhjargal B, McBride D, Ding Y, Tang J, Xiao X, Zhang JH. Osteopontin attenuates inflammation via JAK2/STAT1 pathway in hyperglycemic rats after intracerebral hemorrhage. Neuropharmacology. 2018;138:160–169. doi: 10.1016/j.neuropharm.2018.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galluzzi L, Bravo-San Pedro JM, Blomgren K, Kroemer G. Autophagy in acute brain injury. Nat Rev Neurosci. 2016;17:467–484. doi: 10.1038/nrn.2016.51. [DOI] [PubMed] [Google Scholar]
- 48.Cescon M, Chen P, Castagnaro S, Gregorio I, Bonaldo P. Lack of collagen VI promotes neurodegeneration by impairing autophagy and inducing apoptosis during aging. Aging (Albany NY) 2016;8:1083–1101. doi: 10.18632/aging.100924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun CM, Enkhjargal B, Reis C, Zhou KR, Xie ZY, Wu LY, Zhang TY, Zhu QQ, Tang JP, Jiang XD, Zhang JH. Osteopontin attenuates early brain injury through regulating autophagy-apoptosis interaction after subarachnoid hemorrhage in rats. CNS Neurosci Ther. 2019;25:1162–1172. doi: 10.1111/cns.13199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang P, Yang L, He H, Deng Y. Differential variations of autophagy and apoptosis in permanent focal cerebral ischaemia rat model. Brain Inj. 2017;31:1151–1158. doi: 10.1080/02699052.2017.1298005. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Cai X, Wu Z, Tang L, Lu L, Xu Y, Bao X. Tetrandrine attenuates ischemia/reperfusion-induced neuronal damage in the subacute phase. Mol Med Rep. 2021;23:297. doi: 10.3892/mmr.2021.11936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mo Y, Sun YY, Liu KY. Autophagy and inflammation in ischemic stroke. Neural Regen Res. 2020;15:1388–1396. doi: 10.4103/1673-5374.274331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chan CS, Weeber EJ, Zong L, Fuchs E, Sweatt JD, Davis RL. Beta 1-integrins are required for hippocampal AMPA receptor-dependent synaptic transmission, synaptic plasticity, and working memory. J Neurosci. 2006;26:223–232. doi: 10.1523/JNEUROSCI.4110-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nieuwenhuis B, Haenzi B, Andrews MR, Verhaagen J, Fawcett JW. Integrins promote axonal regeneration after injury of the nervous system. Biol Rev Camb Philos Soc. 2018;93:1339–1362. doi: 10.1111/brv.12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fang L, Li X, Zhong Y, Yu J, Yu L, Dai H, Yan M. Autophagy protects human brain microvascular endothelial cells against methylglyoxal-induced injuries, reproducible in a cerebral ischemic model in diabetic rats. J Neurochem. 2015;135:431–440. doi: 10.1111/jnc.13277. [DOI] [PubMed] [Google Scholar]
- 56.Ramalingam M, Kim SJ. Insulin on activation of autophagy with integrins and syndecans against MPP(+)-induced α-synuclein neurotoxicity. Neurosci Lett. 2016;633:94–100. doi: 10.1016/j.neulet.2016.09.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.