

Abstract
Sphingosine 1-phosphate (S1P) is a pleiotropic signaling molecule that interacts with five G-protein-coupled receptors (S1P1-5) to regulate cellular signaling pathways. S1P export is facilitated by Mfsd2b and spinster homologue 2 (Spns2). While mouse genetic studies suggest that Spns2 functions to maintain lymph S1P, Spns2 inhibitors are necessary to understand its biology and to learn whether Spns2 is a viable drug target. Herein, we report a structure–activity relationship study that identified the first Spns2 inhibitor 16d (SLF1081851). In vitro studies in HeLa cells demonstrated that 16d inhibited S1P release with an IC50 of 1.93 μM. Administration of 16d to mice and rats drove significant decreases in circulating lymphocyte counts and plasma S1P concentrations, recapitulating the phenotype observed in mice made deficient in Spns2. Thus, 16d has the potential for development and use as a probe to investigate Spns2 biology and to determine the potential of Spns2 as a drug target.
Graphical Abstract
INTRODUCTION
Sphingosine-1-phosphate (S1P, 2) is biosynthesized intra-cellularly from sphingosine (1) and ATP via sphingosine kinases (SphK) 1 and 2 (Figure 1).1 Following phosphorylation, S1P can either revert to sphingosine (via S1P phosphatase) or be degraded to 2-hexadecenal (3) and phosphoethanolamine (4) (via S1P lyase).1 While this pathway is ubiquitous in eukaryotes, in vertebrates, S1P can also be extruded from S1P transporter-expressing cells where it can serve as a ligand for the cell surface G-protein-coupled receptors (GPCRs) S1P1-5. S1P-derived activation of these GPCRs plays essential roles in organism development,2 proving critical to processes such as angiogenesis,3,4 cell survival,5,6 and cytoskeletal arrangement.7,8 The diverse array of pathways affected by these receptors have historically made them valuable pharmaceutical targets in medicinal chemistry campaigns.
Figure 1.

S1P metabolic pathway.
The recognition that the immunomodulatory drug fingolimod (5) is a sphingosine analogue that is converted to an S1P receptor agonist [fingolimod-phosphate (6)] led to the discovery that S1P gradients are required for normal lymphocyte recirculation (Figure 2).9 During clinical trials, it was revealed that fingolimod was effective in reducing relapses in multiple sclerosis patients.10,11 Analysis of blood samples revealed that fingolimod drove reductions in circulating lymphocyte counts (lymphopenia), demonstrating the efficacy of fingolimod as an immunosuppressant and establishing precedence for targeting the S1P pathway.12,13 To date, three additional S1P1/5 receptor agonists [siponimod (7), ozanimod (8), and ponesimod (9)] have received marketing approval in the US for multiple sclerosis indications14,15 and, in some cases, ulcerative colitis.16 All four of these drugs have on-target liabilities including pronounced immunosuppression and first-dose bradycardia.17
Figure 2.

FDA-approved therapies targeting S1P signaling through S1P1 functional antagonism.
An alternative to S1P receptor modulation is inhibiting S1P release, which lies upstream of receptor activation. Two proteins, Spns2 and Mfsd2b, have been validated as S1P transporters. Both transport proteins are members of the major facilitator superfamily (MFS) and extrude S1P in an ATP-independent fashion.18,19 Genetic studies in mice revealed that Mfsd2b, which is expressed only in the erythroid lineage, is required for red blood cell release of S1P into plasma,19-21 while Spns2 is more widely expressed.22 Genetic studies in mice indicate that lymph S1P is primarily generated by Spns2- expressing endothelial cells lining the lymphatic vessels.23-26 While endothelial cells also line blood vessels, S1P transport by Mfsd2b is predominately responsible for maintaining plasma S1P concentrations.19-21 Maintenance of adequate lymph S1P is thought to be essential to promote lymphocyte egress from secondary lymphatic tissue into efferent lymph.23,24,27,28 Inhibition of Spns2 might be a pharmaceutically viable method to direct highly localized reductions in fluid S1P concentrations.23,24 As such, our laboratories concentrated on developing chemical tools for inhibiting Spns2.
Mice made deficient in Spns2 by genetic manipulation have decreased circulating lymphocyte counts and increased cellular localization to lymphatic tissue.23,24,27,28 Indeed, knockout of the Spns2 gene in mice has been shown to be protective against experimental autoimmune encephalomyelitis (EAE), which is the standard model of multiple sclerosis.29,30 Selective Spns2 inhibitors with in vivo activity are required to learn whether such molecules can mimic the efficacy of S1P receptor modulators without the undesirable effects. Herein, we describe the discovery, synthesis, and evaluation of the first reported Spns2 inhibitors, as well as preliminary in vivo data.
RESULTS AND DISCUSSION
Inhibitor Design and Development.
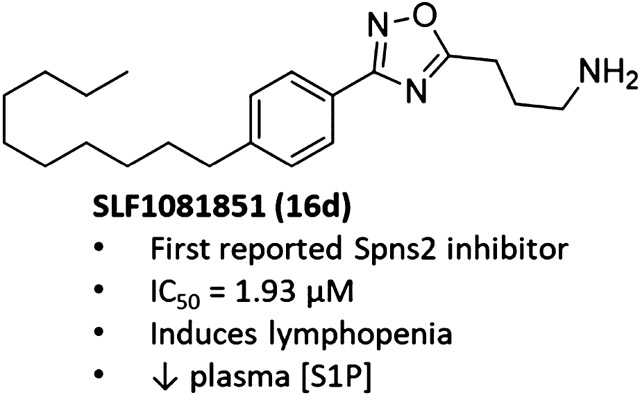
To discover Spns2 inhibitors, we screened an in-house library of sphingosine kinase (SphK)31-39 inhibitors using a yeast (Saccharomyces cerevisiae) assay adapted from a previously reported SphK assay.40 Although the yeast-based assay ultimately proved problematic for screening (nonspecific cytotoxicity of test compounds), we were able to use the assay to identify one hit (11, Figure 3), which is a guanidine-containing oxadiazole derivative.35 Fortunately, as 11 is derived from a β-amino acid and not an α-amino acid (as found in our SphK inhibitors), it displayed minimal activity against either SphK isoform.31 Thus, 11 provided a chemical scaffold for use as a starting point for Spns2 inhibitor development.
Figure 3.

Initial hit generated from our screening of SphK2 inhibitors with proposed structural delineations outlined.
For analogue design, 11 was divided into three distinct regions for chemical modification (Figure 3). Analogous to S1P, 11 contains a positively charged, polar head group moiety, a conformationally restricted 1,2,4-oxadiazole linker, and a hydrophobic alkyl tail. Given that the head group in S1P contains charged ammonium and phosphate groups, we envisioned that inhibitory activity should be particularly sensitive to modifications in these regions. As such, the medicinal chemistry campaign was initially focused on the polar head group and the oxadiazole linker. Once these regions were optimized, we then probed the tail portion of the pharmacophore.
Guanidines, which are strongly basic and hydrophilic, have historically been challenging to incorporate into orally bioavailable drugs as they often fail to be absorbed through the gastrointestinal tract. Thus, we focused on ammonium derivatives of 11. Initial modifications to the structure of 11 centered on replacing β-guanidine with different cyclic or linear amino acid derivatives in accordance with Scheme 1. Synthesis of these derivatives began with a hydroboration of 1-decene (12), followed by a one-pot Suzuki–Miyaura cross-coupling reaction with 4-iodobenzonitrile to provide 13 in 92% yield. Reaction of 13 with hydroxylamine hydrochloride and triethylamine (TEA) in refluxing ethanol afforded the common intermediate amidoxime 14. Subsequent HCTU-mediated coupling of 14 with the corresponding Boc-protected amino acids and Hünig’s base at 100 °C provided the 1,2,4-oxadiazoles 15a–w in moderate-to-good yield. Acetylation of 15w yielded the corresponding protected analogue 15x. Treatment with hydrogen chlorine in dioxane removed the Boc group and generated compounds 16a–x as hydrochloride salts.
Scheme 1. Synthesis of Cyclic and Acyclic Amino Acid Derivatives 16a–xa.
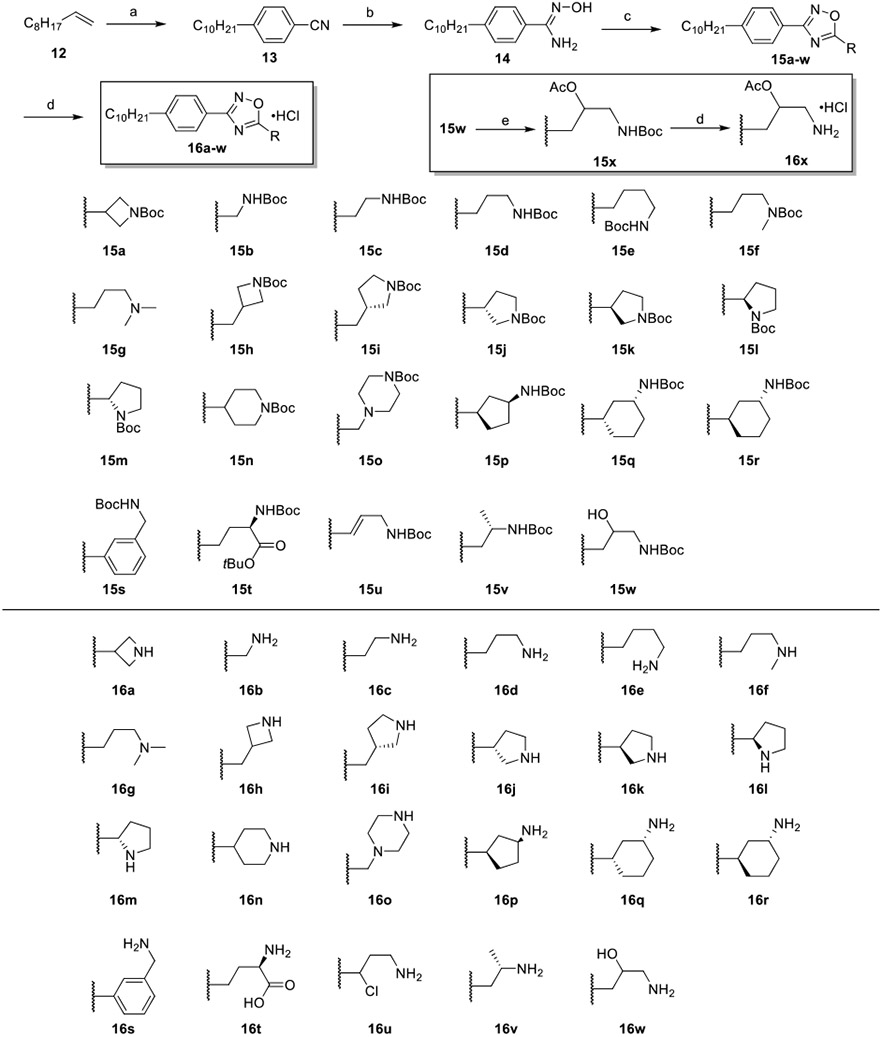
a(a) (i) 9-BBN, THF, 66 °C, 1 h; (ii) 4-iodobenzonitrile, Pd(dppf)Cl2·CH2Cl2, KOH(aq), THF, 66 °C, 4 h, 96%; (b) NH2OH·HCl, EtOH, 78 °C, 2 h, 92%; (c) N-Boc-amino acid or N,N-dimethylglycine, DIEA, HCTU, DMF, 100 °C, 6 h, 46–76%; (d) 4 M HCl/dioxane, DCM, rt, 2 h, 33–89%; (e) Ac2O, TEA, neat, rt, 0.5 h, 82%.
Diamines were synthesized as described in Scheme 2 using the common amidoxime intermediate 14. HCTU-mediated condensation with 2-N-Fmoc terminal N-Boc-protected amino acids afforded the oxadiazole structures 17a–d, which were deprotected using morpholine to yield the monoprotected compounds 18a–d. Subsequent treatment with hydrochloric acid afforded 19a–d. Compound 18d was acetylated using acetyl chloride to yield 20 in excellent yield, which upon treatment with hydrochloric acid produced the ammonium salt 21.
Scheme 2. Synthesis of Diamine Analogues 19a–d and 21a.

a(a) Diprotected amino acid, DIEA, HCTU, THF, 80 °C, 4 h, 40–74%; (b) morpholine, DMF, rt, 18 h, 34–85%; (c) 4 M HCl/dioxane, DCM, rt, 2 h, 37–85%; (d) Ac2O, TEA, neat, rt, 0.5 h, 48%.
Modifications to the ammonium head of 16d and 16w were accomplished according to Scheme 3. The primary amine salts 16d and 16w were treated with dibromoalkanes in the presence potassium carbonate under microwave irradiation to generate the cyclic tertiary amines 22a–f. Secondary amine derivatives were achieved through a two-step one-pot reductive amination reaction. Thus, the corresponding primary ammonium salts were dissolved in methanol and treated with acetaldehyde or paraformaldehyde and acetic acid. The generated iminium intermediate was reduced with sodium cyanoborohydride and quenched with dilute hydrochloric acid, yielding products 23a–b.
Scheme 3. Synthesis of 22a–f and 23a–ba.

a(a) (i) dibromoalkane, K2CO3, H2O, 120 °C, 0.5 h; (ii) 4 M HCl/dioxane, DCM, rt, 2 h, 19–73%; (b) (i) aldehyde, AcOH, MeOH, 0 °C, 0.5 h; (ii) NaBH3CN, MeOH, rt, 4 h, (iii) 4 M HCl/dioxane, DCM, rt, 2 h, 20–27%.
A highly varying approach was undertaken with the linker region of 11 and focused on substituting the 1,2,4-oxadiazole ring with other nitrogen-based heteroaromatics (Scheme 4). Attachment of the decyl tail was achieved through a hydroboration of 12 with 9-borabicyclo[3.3.1]nonane (9-BBN) and a subsequent one-pot Suzuki–Miyaura cross-coupling with 4-bromoacetophenone to yield 24. α-Bromination of 24 with NBS and tosylic acid afforded the monobrominated compound 25 in excellent yield, which upon cyclization with either tert-butyl(4-amino-4-thioxobutyl)-carbamate or tert-butyl(4-amino-4-oxobutyl)carbamate provided the thiazole derivative 27 and the oxazole derivative 29, respectively. Deprotection of the Boc-groups with hydrochloric acid generated the primary ammonium salts 28 and 30. To synthesize the imidazole analogues, compound 25 was converted to keto-ester intermediate 26 with N-Bocgamma aminobutyric acid (GABA) in nearly quantitative yields. Dehydration of 26 with excess ammonium acetate in refluxing toluene formed the imidazole ring and afforded the N-Boc-protected imidazole 31.41 This was then subjected to treatment with either acid to afford the ammonium salt 32 or methyl iodide and sodium hydride to afford the methylated imidazole 33 exclusively, as confirmed via HMBC NMR spectroscopy. Compound 33 was then treated with acid to yield 34 as a hydrochloride salt.
Scheme 4. Heterocyclic Analogues of the Linker Regiona.

a(a) (i) 9-BBN, THF, 66 °C, 1 h; (ii) 1-(4-iodophenyl)ethan-1-one, Pd(dppf)Cl2·CH2Cl2, KOH(aq), THF, 66 °C, 4 h, 70%; (b) NBS, TsOH, MeCN, 82 °C, 18 h, 88%; (c) tert-butyl(4-amino-4-thioxobutyl)carbamate, 2:1 DMF/EtOH, 78 °C, 4 h, 34%; (d) tert-butyl(4-amino-4-oxobutyl)carbamate, NMP, 100 °C, 6 h, 10%; (e) N-Boc-GABA, K2CO3, MeCN, rt, 18 h, 91%; (f) 4 M HCl/dioxane, DCM, rt, 2 h, 60–94%; (g) ammonium acetate, toluene, 110 °C, 5 h, 85%; (h) (i) NaH, THF, 0 °C, 0.5 h; (ii) MeI, THF, 0–25 °C, 18 h, 63%.
Pyrazole analogues were synthesized as described in Scheme 5. Commercially available aryl bromide 35 was reacted with pyrazole boronic acids under Suzuki–Miyaura cross-coupling conditions to afford isomeric pyrazoles 36a and 36b. Deprotonation of the pyrazole ring with sodium hydride in THF generated the sodium amide, which was then alkylated with an N-Boc-protected alkyl bromide. Alkylation of 36a yielded a 90 (39):10 (37) mixture of isomers, which were separable via column chromatography. Due to the symmetric nature of the pyrazole ring in 36b, alkylation exclusively affords 41 as the lone product. Further treatment with hydrochloric acid afforded compounds 38, 40, and 42.
Scheme 5. Synthesis of Pyrazole Analogues 38, 40, and 42a.

a(a) Pyrazole boronic acid, NaHCO3, DMF, 105 °C, 24 h, 46–53%; (b) (i) NaH, THF, 0 °C, 0.5 h; (ii) tert-butyl(3-bromopropyl)carbamate, THF 0–25 °C, 18 h, 16–77%; (c) 4 M HCl/dioxane, DCM, rt, 2 h, 78–89%.
To determine optimal atom arrangement in the linker region, we replaced the 1,2,4-oxadiazole linker with a 1,3,4-oxadiazole (Scheme 6). Hydrazine hydrate was reacted with 4-iodobenzoyl chloride 43 to afford hydrazide 44. This intermediate was coupled to N-Boc-GABA with HCTU to generate an N-acyl intermediate and dehydrated using tosyl chloride in a one-pot fashion, affording 1,3,4-oxadiazole 45. Attachment of the decyl tail was achieved through a tandem hydroboration Suzuki–Miyaura cross-coupling reaction with 1-decene. Deprotection with hydrochloric acid afforded compound 47.
Scheme 6. Synthesis of 1,3,4-Oxadiazole 47a.

a(a) Hydrazine hydrate, EtOH, 80 °C, 20 h, 67%; (b) (i) N-Boc-GABA, DIEA, HCTU, MeCN, rt, 18 h; (ii) DIEA, TsCl, MeCN, rt, 18 h, 79%; (c) (i) 1-decene, 9-BBN, THF, 66 °C, 1 h; (ii) aryl iodide, Pd(dppf)Cl2·CH2Cl2, KOH(aq), THF, 66 °C, 4 h, 79%; (d) 4 M HCl/dioxane, DCM, rt, 2 h, 76%.
Homologation of the linker region was performed as described in Scheme 7. Aryl bromides 48a–b were reacted with hydroxylamine hydrochloride, generating the amidoximes 49a–b. Condensation with N-Boc-GABA was facilitated with HCTU to afford the Boc-protected oxadiazoles 50a–b. Terminal alkenes (1-nonene and 1-octene) were hydroborated with 9-BBN and subjected to Suzuki–Miyaura cross-coupling conditions with the appropriate aryl bromide. Following tail attachment, removal of the Boc groups with acid provided the primary ammonium hydrochloride salts 52a–b.
Scheme 7. Linker Group Homologation Series Synthesisa.

a(a) NH2OH·HCl, EtOH, 78 °C, 2 h, 41–62%; (b) N-Boc-GABA, DIEA, HCTU, DMF, 100 °C, 6 h, 68–76%; (c) (i) alkene, 9-BBN, THF, 66 °C, 1 h; (ii) aryl iodide, Pd(dppf)Cl2·CH2Cl2, KOH(aq), THF, 66 °C, 4 h, 48–50%; (d) 4 M HCl/dioxane, DCM, rt, 2 h, 45–46%.
Ideal tail length and placement were interrogated in accordance with Scheme 8. The iodo-substituted benzonitriles 53a–b were converted to amidoximes 54a–b using hydroxylamine hydrochloride. Cyclization of compounds 54a–b with HCTU and N-Boc-GABA provided the protected oxadiazoles 55a–b. Terminal alkenes were hydroborated with 9-BBN and subsequently attached to 55a–b under Suzuki–Miyaura cross-coupling conditions. The generated alkyl-substituted structures 56a–f were treated with hydrochloric acid to afford the hydrochloride salts 57a–f.
Scheme 8. Synthesis of Compounds 57a–fa.

a(a) NH2OH·HCl, EtOH, 78 °C, 2 h, 61–92%; (b) N-Boc-GABA, DIEA, HCTU, DMF, 100 °C, 6 h, 58–67%; (c) (i) alkene, 9-BBN, THF, 66 °C, 1 h; (ii) aryl iodide, Pd(dppf)Cl2·CH2Cl2, KOH(aq), THF, 66 °C, 4 h, 48–93%; (d) 4 M HCl/dioxane, DCM, rt, 2 h, 27–74%.
Incorporation of ether tails and alterations to the phenyl ring of 16d were accomplished as described in Scheme 9. Starting with either phenols 58a–c or the naphthol 58d, a Williamson ether reaction was performed by employing the appropriate alkyl bromide to afford aryl nitriles 59a–d. Conversion from the nitrile to the amidoximes 60a–d was facilitated with hydroxylamide hydrochloride. As described in earlier schemes, HCTU was employed to condense amidoximes 60a–d with N-Boc-GABA to form the oxadiazoles 61a–d. Removal of the Boc groups afforded products 62a–d as ammonium chloride salts.
Scheme 9. Synthesis of Ether Tail Derivatives 62a–da.

a(a) Alkyl bromide, K2CO3, THF, rt, 2 h, 77–99%; (b) NH2OH·HCl, EtOH, 78 °C, 2 h, 34–93%; (c) N-Boc-GABA, DIEA, HCTU, DMF, 100 °C, 6 h, 57–87%; (d) 4 M HCl/dioxane, DCM, rt, 2 h, 72–88%.
Introduction of amide tails to 16d was performed to increase tail group hydrophilicity and hydrogen bond interactions while simultaneously keeping the overall length consistent at 10 nonhydrogen atoms. Amide bonds were first synthesized through an HCTU-mediated condensation between 4-cyanobenzoic acid and a primary amine (Scheme 10) or 4-cyanoaniline and an aliphatic carboxylic acid (Scheme 11). Treatment of the corresponding benzonitriles with hydroxylamine hydrochloride yielded the amidoximes 65 and 70a–b. Cyclization with HCTU and N-Boc-GABA yielded the 1,2,4-oxadiazole 66 and 71a–b, which were subsequently deprotected using hydrochloric acid to yield the primary ammonium salts 67 and 72a–b.
Scheme 10. Synthesis of the Benzamide Analogue 67a.

a(a) Octylamine, DIEA, HCTU, DCM, rt, 18 h, 94%; (b) NH2OH·HCl, EtOH, 78 °C, 2 h, 52%; (c) N-Boc-GABA, DIEA, HCTU, DMF, 100 °C, 6 h, 48%; (d) 4 M HCl/dioxane, DCM, rt, 2 h, 96%.
Scheme 11. Synthesis of Amide Derivatives 72a–ba.

a(a) Carboxylic acid, DIEA, HCTU, DCM, rt, 18 h, 84–89%; (b) NH2OH·HCl, EtOH, 78 °C, 2 h, 88–95%; (c) N-Boc-GABA, DIEA, HCTU, DMF, 100 °C, 6 h, 22–77%; (d) 4 M HCl/dioxane, DCM, rt, 2 h, 68–80%.
Biological Evaluation.
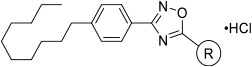
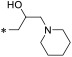
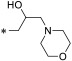
With putative Spns2 inhibitors in hand, we investigated their ability to inhibit S1P release from mouse Spns2-expressing HeLa cells. In this assay, Spns2 inhibitory activity is inversely proportional to S1P concentration in the assay media. Because intracellular S1P metabolism is due to S1P lyase and S1P phosphatase activities, 4-deoxypyridoxine, sodium fluoride, and sodium vanadate were added to retard S1P degradation. Control experiments revealed about a 20-fold increase in S1P extruded into the media compared to Spns2-expressing cells transfected with Spns2Arg200Ser (transport “dead” mutant)28 or nontransfected cells. The results obtained with this assay are presented in Table 1. With focus on the azetidine ring (16a), we determined the effect of acyclic versions with varying carbon spacers (1–4 methylene units, entries 2–5). A three-carbon unit (16d) appeared to be most potent, having 67% inhibition at 2 μM. This activity deteriorated with subsequent N-methylations (entries 6–7). As such, carbocyclic amines with varying methylene spacer, ring size, position, and stereochemistry were synthesized and tested for Spns2 inhibition (entries 8–15). Among these, piperidine bearing analogue 16n had similar activity to 16a. In contrast, exocyclic primaryamines such as 16p, 16q, 16r, and 16s had profound negative impact on Spns2 inhibition. To mimic the negatively charged phosphate group in S1P, we synthesized α-aminobutanoic acid derivative 16t and found no Spns2 inhibitory activity. As 16d bearing a primary amine is the most potent, we introduced additional functional groups such as chloro, amino, N-acetyl, hydroxy, and O-acetyl along the alkyl backbone to improve inhibitory activity (entries 21–29), but these compounds did not result in improved activity. Likewise, pyrrolidine, piperidine, and morpholine tertiary amine analogues with and without an additional hydroxy functionality also had poor activity (entries 30–35). Interestingly, compound 23b with a hydroxy and N,N-dimethyl groups restored Spn2 inhibition, albeit lower than 16d.
Table 1.
Spns2 Inhibitory Activity of Head Group Analoguesa
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Cmpd | R | % Inhibition | Entry | Cmpd | R | % Inhibition |
| 1 | 16a |
|
65 ± 3 | 20 | 16t |
|
5 ± 7 |
| 2 | 16b |
|
62 ± 1 | 21 | 16u |
|
5 ± 4 |
| 3 | 16c |
|
59 ± 3 | 22 | 16v |
|
44 ± 3 |
| 4 | 16d |
|
67 ± 1 | 23 | 16w |
|
59 ± 4 |
| 5 | 16e |
|
55 ± 3 | 24 | 16x |
|
0 ± 6 |
| 6 | 16f |
|
45 ± 0 | 25 | 19a |
|
14 ± 10 |
| 7 | 16g |
|
8 ± 9 | 26 | 19b |
|
40 ± 4 |
| 8 | 16h |
|
63 ± 0 | 27 | 19c |
|
43 ± 3 |
| 9 | 16i |
|
44 ± 4 | 28 | 19d |
|
50 ± 1 |
| 10 | 16j |
|
62 ± 2 | 29 | 21 |
|
40 ± 5 |
| 11 | 16k |
|
26 ± 2 | 30 | 22a |
|
3 ± 8 |
| 12 | 16l |
|
12 ± 5 | 31 | 22b |
|
0 ± 9 |
| 13 | 16m |
|
21 ± 5 | 32 | 22c |
|
2 ± 4 |
| 14 | 16n |
|
66 ± 3 | 33 | 22d |
|
21 ± 7 |
| 15 | 16o |
|
19 ± 1 | 34 | 22e |
|
2 ± 6 |
| 16 | 16p |
|
38 ± 2 | 35 | 22f |
|
0 ± 3 |
| 17 | 16q |
|
29 ± 2 | 36 | 23a |
|
5 ± 2 |
| 18 | 16r |
|
13 ± 2 | 37 | 23b |
|
61 ± 3 |
| 19 | 16s |
|
2 ± 1 | ||||
Spns2 inhibition is presented as percent inhibition relative to the control, during which no inhibitor is introduced. All compounds were assayed with a 2 μM inhibitor. Cell media were extracted, and S1P concentrations were measured by LC/MS. Measurements were performed in duplicate.
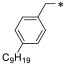
Having identified a propyl amine functionality as an optimal head group in this series, our attention focused toward the tail region (Table 2). Thus, we performed a tail homologation, synthesizing C6─C11 analogues of 16d. As the alkyl chain increased from hexyl to decyl, a corresponding increase in Spns2 inhibitory activity was observed. Because the undecyl chain decreased inhibitory activity, a nonyl or decyl group appears optimal. We then investigated the effect of the substitution pattern on the phenyl ring. In this case, the meta-decyl derivative 57f showed a reduction in activity relative to 16d. Therefore, subsequent studies had the para-substitution pattern and the overall tail length was consistent at 10 atoms.
Table 2.
Spns2 Inhibitory Activity of the Tail and Homologation of the Linker Regiona
| |||||
|---|---|---|---|---|---|
| Cmpd | R | % Inhibition | Cmpd | R | % Inhibition |
| 52a |
|
34 ± 8 | 62a |
|
49 ± 3 |
| 52b |
|
40 ± 5 | 62b |
|
50 ± 2 |
| 57a |
|
55 ± 7 | 62c |
|
24 ± 3 |
| 57b |
|
52 ± 4 | 62d |
|
8 ± 0 |
| 57c |
|
70 ± 4 | 67 |
|
0 ± 14 |
| 57d |
|
61 ± 4 | 72a |
|
35 ± 5 |
| 57e |
|
49 ± 1 | 72b |
|
11 ± 6 |
| 57f |
|
26 ± 4 | |||
Spns2 inhibition is presented as percent inhibition relative to the control, during which no inhibitor is introduced. All compounds were assayed with a 2 μM inhibitor. Cell media were extracted, and S1P concentrations were measured by LC/MS. Compound measurements were performed in duplicate.
To probe the linker region of 16d, we performed a homologation study to gauge the impact of adding additional methylene units between the phenyl and 1,2,4-oxadiazole rings. As a compound length of 21 atoms was identified as the ideal length for Spns2 inhibition in 16d, for each methylene that was added between the two ring systems, one methylene was removed from the tail region. However, compounds 52a–b showed reduced Spns2 inhibition relative to 16d. Insertion of an oxygen atom and additional groups on the ring and amide groups to improve physicochemical properties of the compound also led to diminished Spns2 inhibitory activity (compounds 62a–d, 67, and 72a–b).
As a direct aryl substitution on the 1,2,4-oxadiazole ring in 16d was preferable to flexible alkyl groups, we replaced 1,2,4-oxadiazole with similarly substituted azole rings (Table 3). These modifications included replacement with thiazole (28), oxazole (30), 1H-imidazole (32), N-methylimidazole (34), and N-alkylated pyrazole (38, 40, and 42) rings. Additionally, analogue 47 was synthesized, where the 1,2,4-oxadiazole ring was replaced with a 1,3,4-oxadiazole ring to determine optimal heteroatom location within the ring. While these analogues demonstrated moderate Spns2 inhibition, none had superior activity relative to 16d in the HeLa cell assay.
Table 3.
Spns2 Inhibitory Activity of Heteroaromatic Linkersa
| |||||
|---|---|---|---|---|---|
| Cmpd | R | % Inhibition | Cmpd | R | % Inhibition |
| 28 |
|
45 ± 4 | 38 |
|
45 ± 4 |
| 30 |
|
49 ± 4 | 40 |
|
44 ± 5 |
| 32 |
|
63 ± 3 | 42 |
|
50 ± 4 |
| 34 |
|
51 ± 3 | 47 |
|
59 ± 3 |
Spns2 inhibition is presented as a percent inhibition relative to the control, during which no inhibitor is introduced. All compounds were assayed with a 2 μM inhibitor. Cell media were extracted, and S1P concentrations were measured by LCMS. Compound measurements were performed in duplicate.
Overall, we performed a structure–activity study that interrogated the head, linker, and tail region of initial hit compound 11. The biological screening of this focused library of 60 compounds at 2 and 1 μM (see the Supporting Information) identified several compounds within the margin of error in inhibiting Spns2. Among these, we chose 16d as a potential Spns2 inhibitor for further investigation. Thus, we asked whether 16d inhibited Spns2 in a dose-dependent manner using the HeLa cell assay. As shown in Figure 4A, a robust decrease of extruded S1P as a function of increasing 16d concentration was observed with a calculated IC50 of 1.93 ± 0.04 μM. We then investigated whether 16d inhibits SphK1 and SphK2 using a published protocol.31 As documented in Figure 4B,C, treatment of recombinant mouse SphKs with 16d in a dose–response manner suggests at least 15-fold selectivity (SphK1 IC50 ≥ 30 μM; SphK2 IC50 ≈ 30 μM).
Figure 4.

Concentration–effect assessment of 16d against Spns2 and sphingosine kinases. S1P release decreased as a function of increased concentrations of 16d in Spn2-transfected Hela cells (A). Inhibition of recombinant mSphK1 (B) and mSphK2 (C) inhibition using a standard TLC-based assay. The assays were performed in duplicate.
As previously indicated, results of mouse studies revealed that inactivating the Spns2 gene results in peripheral blood lymphopenia and a modest decrease in plasma S1P compared to litter mate control mice. To determine whether an Spns2 inhibitor recapitulates this phenotype, we dosed 16d into mice and rats. Following administration of 20 mg/kg 16d by intraperitoneal injection, we observed a statistically significant decrease in circulating lymphocytes (Figure 5A) and plasma S1P (Figure 5B) relative to vehicle-treated control mice after 4 h. These results suggest that 16d, in addition to inhibiting S1P release from cultured cells, recapitulates the genetic phenotype of Spns2 null mice. While the activity of 16d has not been tested with Mfsd2b, these pharmacodynamic markers—lymphopenia and a slight decrease in plasma S1P—suggest on-target Spns2 inhibition in vivo.
Figure 5.

Biological evaluation of 16d in mice. Dosing with 16d resulted in both a decrease in circulating lymphocytes (A) and plasma S1P (B). Age- and gender-matched mice (C57BL/6j strain) were injected (intraperitoneal route) with 16d or the vehicle. Blood was drawn 4 h postdose (20 mg/kg). Lymphocyte counts were determined using a Heska Element HT5 blood analyzer, and plasma S1P was quantified via LC/MS. t-test: *≤0.05; ***≤0.001.
To investigate its pharmacokinetic–pharmacodynamic relationship, we administered 16d to rats. A single 10 mg/kg dose (intraperitoneal injection) of 16d into rats revealed a pharmacokinetic profile, reaching a maximum concentration of 5 μM in blood (total plasma) at 2 h with drug levels sustained at ≥ 2 μM for at least 24 h (Figure 6A). This analysis suggested that 16d has a favorable profile and a half-life of over 8 h in rats. We determined also the blood lymphocyte counts in these animals. The appearance of 16d in circulation correlated with a maximal decrease in lymphocyte count at 4 h, which is approximately 25% lower compared to time = 0 (Figure 6B). Taken together, these results suggest that lymphocyte reductions were test article-related.
Figure 6.

Pharmacodynamic and pharmacokinetic analysis of 16d in rats following a 10 mg/kg IP injection. All rats were 4 week old (Sprague-Dawley strain, n = 4) males. (A) Levels of 16d in blood. (B) Blood lymphocyte concentrations reached a minimum 4 h after treatment with 16d. Lymphocyte counts were determined using a Heska Element HT5 blood analyzer, while 16d levels were determined by LC/MS using 57e as an internal standard.
CONCLUSIONS
Inhibition of the S1P transporter Spns2 has the potential to be an alternative to S1P receptor agonists for modulating the immune system. In particular, the ability of Spns2 inhibitors to induce lymphopenia without drastic changes to systemic S1P levels or stimulating the S1P1-5 signaling pathways is advantageous and may provide an alternative therapeutic strategy that precludes side effects associated with S1P1-5 modulators while capturing their efficacy. In this report, we detail the discovery of Spns2 inhibitors using S1P levels in cultured cell media as a readout of S1P export activity. To the best of our knowledge, these are the first compounds reported to inhibit Spns2 activity both in vitro and in vivo. Our discovery of Spns2 inhibitors provides chemical tools that are complementary to genetic manipulation for elucidating the function and underlying biology of Spns2. The structure–activity profile suggests that a positively charged moiety (such as an ammonium) and a lipophilic alkyl tail are necessary for potent Spns2 inhibition. We observed a distinct preference for a sterically unhindered nitrogen in the ammonium head group as most compounds bearing tertiary ammonium head groups were less potent Spns2 inhibitors. While it is surprising that a carboxylic acid mimetic of the phosphate group of S1P was inactive, it is possible that such an analogue, which resembles S1P, serves as a substrate for export rather than an inhibitor. The homologation series wherein the alkyl tail was modified from hexyl to undecyl indicated a lipophilic binding site with a distinct pocket that accommodates a nonyl/decyl chain. Similar to S1P, Spns2 inhibitors appear to prefer long hydrophobic lipid tails. Our work ultimately led to the discovery of 16d with an IC50 of 1.93 μM. Administration of 16d to mice resulted in lymphopenia and concomitant plasma S1P decrease when compared to control animals. These in vivo results recapitulate the phenotype observed in Spns2-deficient mice, suggesting that 16d functions to inhibit Spns2 in vivo. Based on rat pharmacokinetics (PK), 16d has a favorable half-life in vivo, which will allow it to serve as a scaffold for further development and as a tool compound for studying Spns2 biology. The extent of lymphopenia evoked by 16d (ca. 25% reduction) is considerably less than that obtained by the S1P receptor agonist prodrug, fingolimod, or even in an Spns2-null mouse. Determination of the maximum reduction in peripheral blood lymphocytes obtainable with an Spns2 inhibitor awaits the discovery of more potent molecules.
EXPERIMENTAL SECTION
General Materials and Synthetic Procedures.
Reactions were performed using the Schlenk technique under an argon or nitrogen atmosphere, unless otherwise specified. All glassware used was flamedried or oven-dried overnight. Chemicals were obtained from commercial sources and used without further purification, unless otherwise noted. THF, toluene, and DCM were dried using the Innovative Technology Pure SolvMD solvent purification system prior to use. Column chromatography was performed using SiliaFlash P60 40–63 μm, 60 Å. Thin-layer chromatography (TLC) analyses were performed using Silicycle aluminum-backed silica gel F-254 plates. NMR spectroscopic experiments were performed using a Bruker AVANCE II 500 MHz, Agilent 400-MR 400 MHz, or a Varian Inova 400 MHz spectrometer. Chemical shifts are reported in δ ppm and 1H and 13C NMR and referenced using the residual protonated solvent (CHCl3, acetone, or methanol) or an internal standard (TMS). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, dt = doublet of triplets, m = multiplet), coupling constants (Hz), and integration. Minor rotamer peaks are denoted by an asterisk (*). ESI mass spectra were obtained using an Agilent 6220 TOF LC–MS. Purity assessments were performed by Agilent HPLC analysis. HPLC conditions: solvent A: water (0.1% TFA); solvent B: acetonitrile (0.1% TFA); column: Zorbax SB-C18, 5 μM, 4.6 × 150 mm; method: isocratic 95% A, 5% B from 0 to 5 min, then linear gradient from 5 to 95% B by 20 min, hold 95% B by 30 min, and return to 5% B by 40 min; UV wavelength = 254 nm; flow rate: 1.5 mL/min. All compounds tested in biological assays were assessed to have ≥95% purity by HPLC, unless otherwise noted. Compounds evaluated in vivo were HPLC-purified prior to injection into animals and were of ≥95% purity by HPLC.
General Procedure 1: One-Pot Hydroboration-Suzuki–Miyaura Cross-Coupling.
To a round-bottom flask a containing alkene (1.1 equiv) in THF was added 9BBN (1.5 equiv) and then heated to reflux until consumption of alkene, as monitored by TLC (30–60 min). Aryl halide (1.0 equiv) and Pd(dppf)Cl2·CH2Cl2 (0.05 equiv) were then added to the mixture, followed by dropwise addition of a 3M KOH(aq) solution (3.0 equiv). The resulting mixture was then heated to reflux until consumption of aryl iodide, as monitored by TLC (2–6 h). On cooling to room temperature (rt), the reaction mixture was filtered over a pad of Celite, diluted in ethyl acetate, and washed with a brine solution. The organic layer was then dried over sodium sulfate and concentrated in vacuo to afford the crude product as a yellow oil, which was then purified by column chromatography with an appropriate hexanes/ethyl acetate solvent system to afford the pure product.
General Procedure 2: Amidoxime Synthesis.
To a round-bottom flask containing ethanol/water (1:1) were added benzonitrile (1.0 equiv), hydroxylamine hydrochloride (2.0 equiv), and sodium carbonate (5.0 equiv) under ambient air. The reaction mixture was then heated to reflux until complete, as monitored by TLC (1–6 h). The resulting solution was allowed to cool to rt. A white precipitate formed upon cooling, which was vacuum-filtered over a filter frit and washed with water and ethanol to afford the pure product.
General Procedure 3: 1,2,4-Oxadiazole Synthesis.
Amidoxime (1.0 equiv), N-Boc protected β-amino acid (1.1 equiv), and N,N-diisopropylethylamine (DIEA) (1.8 equiv) were added to a round-bottom flask containing DMF at rt. HCTU (1.1 equiv) was then added, and the resulting mixture was heated to 100 °C until completion, as monitored by TLC (6–16 h). Upon cooling to rt, the resulting mixture was diluted in ethyl acetate and washed with a saturated lithium bromide solution. The resulting aqueous layer was then extracted (three times) with ethyl acetate. The organic layers were combined and washed with a brine solution (three times), followed by drying over anhydrous sodium sulfate. Concentration in vacuo afforded the crude product, which was then purified by column chromatography using the appropriate ethyl acetate/hexane solvent system to afford the pure 1,2,4-oxadiazole product.
General Procedure 4: HCl-Assisted Boc Deprotection.
To a 6 dram vial containing Boc-protected amine (1.0 equiv) was added hydrogen chloride (10.0 equiv, 4M in dioxane). The resulting mixture was allowed to stir until consumption of the starting material, as monitored by TLC (0.5–6 h). The solvent was removed under reduced pressure and rinsed with diethyl ether until a thick white precipitate formed. This precipitate was subjected to trituration with an appropriate solvent system to afford the pure product as a hydrochloride salt.
General Procedure 5: Base-Assisted Fmoc Removal.
To a 6-dram vial containing Fmoc-protected amine (1.0 equiv) and DMF was added morpholine (10.0 equiv). The mixture was allowed to stir at rt for 16 h at which point TLC analysis showed complete consumption of the starting material. Following completion, the mixture was diluted with ethyl acetate and portioned with saturated lithium bromide. The resulting aqueous layer was then extracted (three times) with ethyl acetate. The organic layers were combined and washed with a brine solution (three times), followed by drying over anhydrous sodium sulfate. Concentration in vacuo afforded the crude product, which was then purified by column chromatography using the appropriate ethyl acetate/hexane solvent system.
General Procedure 6: Microwave-Assisted Cyclization of Primary Amines.
To a microwave vial containing a stir bar were added primary amine salt (1.0 equiv), potassium carbonate (1.1 equiv), dibromoalkane (1.0 equiv), and acetonitrile. The vial was then heated to 120 °C for 20 min in a CEM Discover SP Microwave Synthesizer. Following completion, the reaction mixture was extracted with ethyl acetate and washed with a 50:50 brine/10% NaOH solution. The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo to afford the crude product as a colorless oil. The crude product was dissolved in a hydrochloric acid solution (4 M in dioxane) and concentrated in vacuo to afford a white solid. The white solid was loaded onto Celite and subjected to silica gel chromatography in an appropriate solvent system to yield the purified the cyclic amine hydrochloride salt.
General Procedure 7: Alkylation of Pyrazole Rings.
To an oven-dried round-bottom flask containing a stir bar were added sodium hydride (60% dispersion in mineral oil, 1.1 equiv) and THF. The mixture was then purged with argon and cooled to 0 °C in an ice bath. The appropriate aryl pyrazole (1.0 equiv) was added to the solution and allowed to stir for 15 min. tert-Butyl(3-bromopropyl)carbamate (3.0 equiv) was slowly added to the flask and the reaction mixture continued to stir at 0 °C for 18 h. Following completion as monitored by TLC, the reaction mixture was concentrated under reduced pressure and purified via column chromatography with an appropriate ethyl acetate/hexane eluent to afford the desired products.
General Procedure 8: Williamson Ether Synthesis.
The appropriate phenol derivative (1.0 equiv) was added to a dried round-bottom flask containing a stir bar and potassium carbonate (3.0 equiv). The appropriate alkyl bromide (1.2 equiv) and dry acetonitrile were added, and the flask was attached to a condenser and heated to reflux for 4 h. Following completion, the crude reaction mixture was concentrated under reduced pressure, diluted with ethyl acetate, and partitioned with brine. The organic layer was rinsed three times with brine, dried over sodium sulfate, and filtered. The product was then either carried forward crude with no additional purification or purified via column chromatography.
General Procedure 9: HCTU Amide Coupling.
To a 6-dram vial containing N-Boc-amino acid (1.1 equiv) were added DMF (0.2 M), DIEA (1.8 equiv), and HCTU (1.1 equiv). The resulting mixture was allowed to stir at rt for 5 min, followed by addition of the amine derivative (1.0 equiv). The resulting mixture was allowed to stir at rt until consumption of amine, as monitored by TLC (1–4 h). The resulting reaction mixture was diluted in ethyl acetate and washed with a saturated lithium bromide solution. The organic layer was then dried over anhydrous sodium sulfate and concentrated in vacuo to afford an orange oil which was then subjected to flash chromatography with an appropriate ethyl acetate in a dichloromethane solvent system to afford the pure product.
4-Decylbenzonitrile (13).
Synthesized according to General Procedure 1. Purified via column chromatography (3% ethyl acetate/hexanes). Off-white solid (96%, 2.05 g). 1H NMR (400 MHz, CdCl3): δ 7.55 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 2.65 (t, J = 7.8 Hz, 2H), 1.61 (p, J = 7.4 Hz, 2H), 1.37–1.18 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 148.7, 132.1, 129.2, 119.2, 109.5, 36.2, 32.0, 31.1, 29.7, 29.6, 29.5, 29.4, 29.3, 22.8, 14.2. Data is consistent with literature.42
4-Decyl-N′-hydroxybenzimidamide (14).
Synthesized according to General Procedure 2. Purified via column chromatography (20% ethyl acetate/hexanes). White solid (92%, 10.56 g). Isolated as a mixture of Z/E (20:1). 1H NMR (400 MHz, CdCl3): δ 7.54 (d, J = 7.8 Hz, 2H), 7.20 (d, J = 7.8 Hz, 2H), 4.89 (br s, 2H), 2.62 (t, J = 7.7 Hz, 2H), 1.61 (p, J = 7.0 Hz, 2H), 1.36–1.20 (m, 14H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 152.8, 145.3, 129.9, 128.8, 125.9, 35.9, 32.0, 31.5, 29.8, 29.7, 29.6, 29.5, 29.4, 22.8, 14.3. HRMS (ESI): [M + H]+ calcd for C17H29N2O, 277.2274; observed, 277.2281.
tert-Butyl 3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)azetidine-1-carboxylate (15a).
Synthesized according to General Procedure 3. Purified via column chromatography (10–30% ethyl acetate/hexanes). Yellow oil (59%, 94 mg). 1H NMR (400 MHz, CdCl3): δ 7.98 (d, J = 8.3 Hz, 2H), 7.29 (d, J = 8.2 Hz, 2H), 4.42–4.28 (m, 4H), 4.04 (tt, J = 8.8, 6.2 Hz, 1H), 2.66 (t, J = 7.7 Hz, 2H), 1.64 (p, J = 6.7 Hz, 2H), 1.46 (s, 9H), 1.37–1.20 (m, 14H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 178.9, 168.8, 156.0, 146.9, 129.1, 127.5, 124.0, 80.3, 53.4, 36.1, 32.0, 31.3, 29.7, 29.7, 29.6, 29.4, 29.4, 28.5, 25.9, 22.8, 14.2. HRMS (ESI): [M + Na]+ calcd for C26H39N3NaO3, 464.2884; observed, 464.2882.
tert-Butyl ((3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)methyl)-carbamate (15b).
Synthesized according to General Procedure 3. Purified via column chromatography (15% ethyl acetate/hexanes). White solid (75%, 281 mg). 1H NMR (400 MHz, CdCl3): δ 7.95 (d, J = 8.0 Hz, 2H), 7.26 (d, J = 8.0 Hz, 2H), 5.59 (br s, 1H), 4.66–4.55 (m, 2H), 2.64 (t, J = 7.7 Hz, 2H), 1.67–1.57 (m, 2H), 1.46 (br s, 9H), 1.36–1.20 (m, 14H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CdCl3): δ 176.5, 168.4, 155.6, 146.7, 128.9, 127.4, 123.9, 80.6, 37.2, 36.0, 32.0, 31.3, 29.7, 29.6, 29.5, 29.4, 29.4, 29.3, 28.3, 22.7, 14.2. HRMS (ESI): [M + H]+ calcd for C24H38N3O3, 416.2908; observed, 416.2925.
tert-Butyl (2-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)ethyl)-carbamate (15c).
Synthesized according to General Procedure 3. Purified via column chromatography (20% ethyl acetate/hexanes). Light-yellow solid (50%, 155 mg). 1H NMR (400 MHz, CdCl3): δ 7.97 (d, J = 8.1 Hz, 2H), 7.27 (d, J = 8.1 Hz, 2H), 5.27 (br s, 1H), 3.69–3.62 (m, 2H) 3.12 (t, J = 6.0 Hz, 2H), 2.65 (t, J = 7.7 hz, 2H), 1.68–1.58 (m, 2H), 1.43 (br s, 9H), 1.37–1.21 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CdCl3): δ 177.8, 168.3, 155.8, 146.6, 129.0, 127.4, 124.1, 79.7, 37.3, 36.0, 32.0, 31.3, 29.7, 29.6, 29.5, 29.4, 29.3, 28.4, 27.6, 22.8, 14.2. HRMS (ESI): [M + H]+ calcd for C25H40N3O3, 430.3064; observed, 430.3076.
tert-Butyl (3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)propyl)-carbamate (15d).
Synthesized according to General Procedure 3. Purified via column chromatography (20% ethyl acetate/hexanes). Yellow oil (65%, 260 mg). 1H NMR (400 MHz, CdCl3): δ 7.97 (d, J = 8.2 Hz, 2H), 7.26 (d, J = 8.2 Hz, 2H), 5.07 (br s, 1H), 3.32–3.22 (m, 2H), 2.97 (t, J = 7.5 Hz, 2H), 2.64 (t, J = 7.7 Hz, 2H), 2.11–2.02 (m, 2H), 1.67–1.57 (m, 2H), 1.43 (br s, 9H), 1.36–1.20 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CdCl3): δ 179.2, 168.2, 156.0, 146.4, 128.9, 127.3, 124.2, 79.2, 39.8, 35.9, 31.9, 31.2, 29.6, 29.6, 29.5, 29.3, 29.3, 28.4, 26.9, 24.1, 22.7, 14.1. HRMS (ESI): [M + Na]+ calcd for C26H41N3NaO3, 466.3040; observed, 466.3034.
tert-Butyl (4-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)butyl)-carbamate (15e).
Synthesized according to General Procedure 3. Purified via column chromatography (20% ethyl acetate/hexanes). Yellow solid (69%, 285 mg). 1H NMR (400 MHz, CdCl3): δ 8.0 (d, J = 8.2 Hz, 2H), 7.3 (d, J = 8.0 Hz, 2H), 4.8 (t, J = 5.7 Hz, 1H), 3.2 (q, J = 6.7 Hz, 2H), 2.9 (t, J = 7.5 Hz, 2H), 2.6 (t, J = 7.7 Hz, 2H), 1.9 (p, J = 7.5 Hz, 2H), 1.7–1.6 (m, 4H), 1.4 (s, 9H), 1.4–1.2 (m, 14H), 0.9 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.4, 168.2, 156.0, 146.4, 128.9, 127.3, 124.2, 79.1, 39.9, 36.0, 31.9, 31.2, 29.6, 29.6, 29.5, 29.4, 29.3, 29.3, 28.4, 26.2, 23.8, 22.7, 14.1. HRMS (ESI): [M + H]+ calcd for C27H44N3O3, 458.3383; observed, 458.3390.
tert-Butyl (3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)propyl)-(methyl)carbamate (15f).
Synthesized according to General Procedure 3. Purified via column chromatography (15% ethyl acetate/hexanes). Yellow oil (54%, 319 mg). 1H NMR (400 MHz, CdCl3): δ 7.96 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 3.38 (t, J = 6.9 Hz, 2H), 2.94 (t, J = 7.6 Hz, 2H), 2.88 (s, 3H), 2.65 (t, J = 7.7 Hz, 2H), 2.10 (p, J = 7.3 Hz, 2H), 1.63 (p, J = 7.5 Hz, 2H), 1.44 (s, 9H), 1.38–1.19 (m, 14H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.0, 168.3, 146.4, 128.9, 127.3, 124.2, 79.3, 47.7, 35.9, 34.2, 31.9, 31.2, 29.6, 29.6, 29.5, 29.3, 29.2, 28.4, 24.7, 23.9, 22.7, 14.1. HRMS (ESI): [M + H]+ calcd for C27H44N3O3, 458.3377; observed, 458.3372.
3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-N,N-dimethylpropan-1-amine (15g).
Synthesized according to General Procedure 3. Crude mixture dried in vacuo and carried forward to the next reaction as a free amine base without further purification.
tert-Butyl 3-((3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)methyl)-azetidine-1-carboxylate (15h).
Synthesized according to General Procedure 3. Purified via column chromatography (16% ethyl acetate/hexanes). Off-white solid (49%, 160 mg). 1H NMR (400 MHz, CdCl3): δ 7.96 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 4.16 (t, J = 8.5 Hz, 2H), 3.79–3.74 (m, 2H), 3.24–3.19 (m, 2H), 3.13–3.03 (m, 1H), 2.65 (t, J = 7.8 Hz, 2H), 1.67–1.58 (m, 2H), 1.44 (br s, 9H), 1.37–1.20 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CdCl3): δ 177.2, 168.4, 156.2, 146.6, 128.9, 127.4, 124.0, 79.6, 53.7, 36.0, 31.9, 31.3, 30.9, 29.6, 29.6, 29.5, 29.4, 29.3, 28.4, 26.4, 22.7, 14.2. HRMS (ESI): [M + Na]+ calcd for C27H41N3O3Na, 478.3046; observed, 478.3029.
tert-Butyl (S)-3-((3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)methyl)-pyrrolidine-1-carboxylate (15i).
Synthesized according to General Procedure 3. Purified via column chromatography (20% ethyl acetate/hexanes). Yellow solid (64%, 273 mg). 1H NMR (400 MHz, CdCl3): δ 7.97 (d, J = 7.6 Hz, 2H), 7.28 (d, J = 7.6 Hz, 2H), 3.77–3.44 (m, 3H), 3.42–3.28 (m, 1H), 3.19–2.94 (m, 3H), 2.84–2.71 (m, 1H), 2.66 (t, J = 7.7 Hz, 2H), 2.19–2.08 (m, 1H), 1.77–1.58 (m, 3H), 1.46 (br s, 9H), 1.38–1.19 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CdCl3): δ 178.0, 168.4, 154.5, 146.6, 129.0, 127.4,124.1, 79.5, 50.8, 45.0, 36.8, 36.0, 32.0, 31.4, 31.3, 30.9, 29.9, 29.7, 29.7, 29.6, 29.4, 29.3, 28.6, 22.8, 14.2. HRMS (ESI): [M + Na]+ calcd for C28H43N3NaO3, 492.3197; observed, 492.3199.
tert-Butyl (R)-3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-pyrrolidine-1-carboxylate (15j).
Synthesized according to General Procedure 3. Purified via column chromatography (16% ethyl acetate/hexanes). Off-white solid (75%, 148 mg). 1H NMR (400 MHz, CdCl3): δ 7.97 (d, J = 8.3 Hz, 2H), 7.28 (d, J = 8.3 Hz, 2H), 3.95–3.41 (m, 6H), 2.65 (t, J = 7.7 Hz, 2H), 2.44–2.29 (m, 2H), 1.68–1.58 (m, 2H), 1.48 (br s, 9H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CdCl3): δ 179.4, 168.4, 154.2, 146.6, 128.9, 127.4, 124.0, 79.8, 49.4, 45.1, 36.6, 36.0, 35.8, 31.9, 30.5, 30.4, 29.6, 29.6, 29.5, 29.4, 29.3, 28.5, 22.7, 14.2. HRMS (ESI): [M + H]+ calcd for C27H41N3NaO3, 478.3040; observed, 478.3065.
tert-Butyl (S)-3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-pyrrolidine-1-carboxylate (15k).
Synthesized according to General Procedure 3. The product was could not be separated from impurity and was carried forward crude to the next reaction.
tert-Butyl (R)-2-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-pyrrolidine-1-carboxylate (15l).
Synthesized according to General Procedure 3. Purified via column chromatography (30% ethyl acetate/hexanes). Yellow oil (61%, 101 mg). 1H NMR (400 MHz, CdCl3): δ 7.97 (d, J = 8.1 Hz, 2H), 7.28 (d, J = 8.1 Hz, 2H), 5.26–5.02 (m, 1H), 3.80–3.44 (m, 2H), 2.65 (t, J = 7.8 Hz, 2H), 2.47–2.33 (m, 1H), 2.22–2.06 (m, 2H), 2.04–1.93 (m, 1H), 1.63 (p, J = 7.1 Hz, 2H), 1.49–1.21 (m, 23H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 180.6, 168.5, 153.7, 146.7, 129.1, 128.9*, 127.6*, 127.5, 124.2, 80.6, 80.4*, 53.9, 46.7*, 46.5, 36.1, 32.5, 32.0, 31.6*, 31.4, 29.7, 29.7, 29.6, 29.5, 29.4, 28.5*, 28.3, 24.5*, 23.8, 22.8, 14.2. HRMS (ESI): [M + H]+ calcd for C27H42N3O3, 456.3221; observed, 456.3219.
tert-Butyl (S)-2-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-pyrrolidine-1-carboxylate (15m).
Synthesized according to General Procedure 3. Purified via column chromatography (30% ethyl acetate/hexanes). Yellow oil (49%, 81 mg). 1H NMR (400 MHz, CdCl3): δ 7.95 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.1 Hz, 2H), 5.21–5.01 (m, 1H), 3.75–3.43 (m, 2H), 2.64 (t, J = 7.7 Hz, 2H), 2.44–2.28 (m, 1H), 2.20–2.04 (m, 2H), 2.03–1.93 (m, 1H), 1.62 (p, J = 7.9 Hz, 2H), 1.47–1.20 (m, 23H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 180.6, 168.5, 153.7, 146.7, 146.4*, 129.1, 128.9*, 127.6*, 127.5, 124.2, 80.5, 80.4*, 53.9, 46.7*, 46.5, 36.1*, 32.5, 32.0, 31.6, 31.4, 29.7, 29.7, 29.6, 29.4, 29.4, 28.5*, 28.3, 24.4*, 23.8, 22.8, 14.2. HRMS (ESI): [M + H]+ calcd for C27H42N3O3, 456.3221; observed, 456.3212.
tert-Butyl 4-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)piperidine-1-carboxylate (15n).
Synthesized according to General Procedure 3. Purified via column chromatography (14% ethyl acetate/hexanes). Yellow oil (76%, 335 mg). 1H NMR (400 MHz, CdCl3): δ 7.97 (d, J = 8.3 Hz, 2H), 7.27 (d, J = 8.3 Hz, 2H), 4.11 (br s, 2H), 3.18–3.09 (m, 1H), 2.98 (t, J = 11.6 Hz, 1H), 2.64 (t, J = 7.7 Hz, 2H), 2.14–2.05 (m, 2H), 1.94–1.82 (m, 2H), 1.67–1.56 (m, 2H), 1.48 (br s, 9H), 1.37–1.19 (m, 14H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CdCl3): δ 181.0, 168.2, 154.5, 146.4, 128.8, 127.3, 124.2, 79.7, 42.8, 35.9, 34.4, 31.9, 31.2, 29.6, 29.6, 29.5, 29.3, 29.2, 29.1, 28.4, 22.7, 14.1. HRMS (ESI): [M + H]+ calcd for C28H44N3O3, 470.3377; observed, 470.3369.
tert-Butyl 4-((3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)methyl)-piperazine-1-carboxylate (15o).
Synthesized according to General Procedure 3. Purified via column chromatography (35% ethyl acetate/hexanes). White solid (63%, 248 mg). 1H NMR (400 MHz, CdCl3): δ 7.99 (d, J = 8.2 Hz, 2H), 7.29 (d, J = 8.2 Hz, 2H), 3.93 (s, 2H), 3.5 (t, J = 5.0 Hz, 4H), 2.66 (t J = 7.7 Hz, 2H), 2.60 (t J = 5.0 Hz, 4H), 1.64 (p, J = 7.3 Hz, 2H), 1.45 (s, 9H), 1.38–1.19 (m, 14H), 0.87 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 175.8, 168.5, 154.7, 146.8, 129.1, 127.6, 124.0, 80.0, 53.2, 52.7, 43.6, 36.1, 32.0, 31.4, 29.7, 29.7, 29.6, 29.5, 29.4, 28.5, 22.8, 14.3. HRMS (ESI): [M + H]+ calcd for C28H45N4O3, 485.3486; observed, 485.3479.
tert-Butyl ((1S,3R)-3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-cyclopentyl)carbamate (15p).
Synthesized according to General Procedure 3. Purified via column chromatography (20% ethyl acetate/hexanes). Yellow solid (63%, 213 mg). 1H NMR (400 MHz, CdCl3): δ 7.98 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.2 Hz, 2H), 5.67 (br s, 1H), 4.23 (br s, 1H), 3.59–3.48 (m, 1H), 2.66 (t, J = 7.7 Hz, 2H), 2.53–2.43 (m, 1H), 2.29–1.91 (m, 4H), 1.85–1.74 (m, 1H), 1.68–1.58 (m, 2H), 1.47 (br s, 9H), 1.38–1.19 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CdCl3): δ 183.6, 168.2, 155.5, 146.7, 129.0, 127.5, 124.1, 79.2, 52.0, 38.1, 36.1, 35.2, 33.4, 32.0, 31.4, 30.1, 29.7, 29.7, 29.6, 29.4, 29.4, 28.6, 28.6, 22.8, 14.3. HRMS (ESI): [M + H]+ calcd for C28H44N3O3, 470.3377; observed, 470.3372.
tert-Butyl ((1R,3S)-3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-cyclohexyl)carbamate (15q).
Synthesized according to General Procedure 3. Purified via column chromatography (12% ethyl acetate/hexanes). Yellow solid (72%, 314 mg). 1H NMR (400 MHz, CdCl3): δ 7.96 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.2 Hz, 2H), 4.58 (s, 1H), 3.61 (s, 1H), 3.10 (dt, J = 11.6, 5.9 Hz, 1H), 2.71–2.58 (m, 2H), 2.48 (d, J = 12.4 Hz, 1H), 2.15 (d, J = 9.4 Hz, 1H), 2.05 (d, J = 12.7 Hz, 1H), 1.99–1.90 (m, 1H), 1.70–1.49 (m, 6H), 1.45 (s, 9H), 1.38–1.19 (m, 14H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 181.5, 168.2, 155.0, 146.4, 128.9, 127.3, 124.2, 79.3, 48.8, 36.6, 35.9, 35.5, 32.6, 31.9, 31.2, 29.6, 29.6, 29.5, 29.3, 29.2, 28.4, 24.1, 22.7, 14.1. HRMS (ESI): [M + Na]+ calcd for C29H45N3NaO3, 506.3353; observed, 506.3337.
tert-Butyl ((1R,3S)-3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-cyclohexyl)carbamate (15q).
Synthesized according to General Procedure 3. Purified via column chromatography (12% ethyl acetate/hexanes). Yellow solid (73%, 318 mg). 1H NMR (400 MHz, CdCl3): δ 7.97 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.3 Hz, 2H), 4.63 (br s, 1H), 3.95 (brs 1H), 3.29 (br s, 1H), 2.65 (d, J = 7.7 Hz, 2H), 2.22–1.54 (m, 10H), 1.45 (s, 9H), 1.39–1.22 (m, 14H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 181.9, 168.4, 155.2, 146.5, 129.0, 127.5, 124.4, 79.5, 45.5, 36.1, 34.5, 32.2, 32.0, 31.4, 30.9, 29.7, 29.7, 29.6, 29.5, 29.4, 29.2, 28.6, 22.8, 20.6, 14.2. HRMS (ESI): [M + Na]+ calcd for C29H45N3NaO3, 506.3353; observed, 506.3336.
tert-Butyl (3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)benzyl)-carbamate (15s).
Synthesized according to General Procedure 3. Purified via column chromatography (17% ethyl acetate/hexanes). Yellow solid (59%, 232 mg). 1H NMR (400 MHz, CdCl3): δ 8.14–8.09 (m, 2H), 8.07 (d, J = 8.2 Hz, 2H), 7.56–7.49 (m, 2H), 7.32 (d, J = 8.3 Hz, 2H), 4.99 (br s, 1H), 4.43 (d, J = 6.2 Hz, 2H), 2.68 (t, J = 7.5 Hz, 2H), 1.65 (p, J = 7.4 Hz, 2H), 1.49 (s, 9H), 1.39–1.20 (m, 14H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ175.5, 169.1, 156.0, 146.7, 140.5, 131.8, 129.6, 129.1, 127.6, 127.2, 127.0, 124.8, 124.4, 80.0, 44.4, 36.1, 32.1, 31.4, 29.8, 29.7, 29.6, 29.5, 29.4, 28.5, 22.8, 14.3. HRMS (ESI): [M + H]+ calcd for C30H42N3O3, 492.3221; observed, 492.3209.
tert-Butyl (R)-2-((tert-Butoxycarbonyl)amino)-4-(3-(4-decylphen-yl)-1,2,4-oxadiazol-5-yl)butanoate (15t).
Synthesized according to General Procedure 3. Purified via column chromatography (20% ethyl acetate/hexanes). Yellow solid (59%, 290 mg). 1H NMR (400 MHz, CdCl3): δ 7.96 (d, J = 7.9 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 5.34 (d, J = 8.1 Hz, 1H), 4.34 (q, J = 7.9 Hz, 1H), 3.11–2.91 (m, 2H), 2.65 (t, J = 7.7 Hz, 2H), 2.49–2.36 (m, 1H), 2.25–2.11 (m, 1H), 1.62 (p, J = 8.5 Hz, 2H), 1.48 (s, 9H), 1.43 (s, 9H), 1.35–1.21 (m, 14H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 178.8, 171.0, 168.4, 155.5, 146.5, 129.0, 127.4, 124.2, 82.6, 80.0, 53.5, 36.0, 32.0, 31.3, 29.8, 29.7, 29.7, 29.6, 29.4, 29.3, 28.4, 28.1, 23.1, 22.8, 14.2. HRMS (ESI): [M + H]+ calcd for C31H50N3O5, 544.3745; observed, 544.3753.
tert-Butyl (E)-(3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)allyl)-carbamate (15u).
Synthesized according to General Procedure 3. Purified via column chromatography (15% ethyl acetate/hexanes). White solid (74%, 143 mg). 1H NMR (400 MHz, CdCl3): δ 7.98 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 8.1 Hz, 2H), 7.09 (dt, J = 16.1, 4.9 Hz, 1H), 6.58 (d, J = 16.1 Hz, 1H), 5.00 (br s, 1H), 4.03 (br s, 2H), 2.65 (t, J = 7.7 Hz, 2H), 1.63 (p, J = 7.1 Hz, 2H), 1.47 (s, 9H), 1.37–1.19 (m, 14H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 174.2, 168.7, 155.7, 146.6, 143.1, 129.0, 127.4, 124.2, 113.5, 80.0, 41.9, 36.0, 32.0, 31.3, 29.7, 29.7, 29.6, 29.4, 29.3, 28.4, 22.8, 14.2. HRMS (ESI): [M + H]+ calcd for C26H40N3O3, 442.3070; observed, 442.3063.
tert-Butyl (S)-(1-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)propan-2-yl)carbamate (15v).
Synthesized according to General Procedure 3. Purified via column chromatography (15% ethyl acetate/hexanes). Yellow solid (46%, 110 mg). 1H NMR (400 MHz, CdCl3): δ 7.98 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 5.02–4.90 (m, 1H), 4.29–4.17 (m, 1H), 3.15 (d, J = 5.8 Hz, 2H), 2.66 (t, J = 7.7 Hz, 2H), 1.68–1.57 (m, 2H), 1.43 (br s, 9H), 1.36–1.20 (m, 17H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CdCl3): δ 177.0, 168.3, 155.0, 146.6, 129.0, 127.5, 124.2, 79.7, 44.5, 36.0, 33.5, 32.0, 31.3, 29.7, 29.7, 29.6, 29.4, 20.3, 28.4, 22.8, 20.30, 14.2. HRMS (ESI): [M + Na]+ calcd for C26H41N3NaO3, 466.3040; observed, 466.3061.
tert-Butyl (3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-2-hydroxypropyl)carbamate (15w).
Synthesized according to General Procedure 3. Purified via column chromatography (40% ethyl acetate/hexanes). Yellow solid (60%, 452 mg). 1H NMR (400 MHz, CdCl3): δ 7.94 (d, J = 8.0 Hz, 2H), 7.26 (d, J = 8.0 Hz, 2H), 5.30 (br s, 1H), 4.30 (br s, 2H), 3.49–3.41 (m, 1H), 3.32–3.22 (m, 1H), 3.14–3.05 (m, 2H), 2.64 (t, J = 7.7 Hz, 2H), 1.66–1.58 (m, 2H), 1.44 (br s, 9H), 1.36–1.20 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CdCl3): δ 177.3, 168.1, 156.9, 146.7, 129.0, 127.4, 123.8, 80.0, 68.5, 45.7, 36.0, 32.0, 31.7, 31.3, 29.7, 29.7, 29.6, 29.4, 29.3, 28.4, 22.8, 14.2. HRMS (ESI): [M + H]+ calcd for C26H42N3O4, 460.3170; observed, 460.3169.
1-((tert-Butoxycarbonyl)amino)-3-(3-(4-decylphenyl)-1,2,4-oxadiazol-5-yl)propan-2-yl Acetate (15x).
2-To a 6-dram vial containing 15w were added acetic anhydride (50.0 equiv) and TEA (9.0 equiv), and the mixture was allowed to stir at rt for 30 min. The mixture was then diluted with ethyl acetate and washed with a saturated sodium carbonate solution, followed by a brine solution. The organic layer was concentrated in vacuo to afford a crude mixture. The desired product was confirmed via HRMS and carried forward as crude with no further purification. Clear oil (82%, 206 mg). HRMS (ESI): [M + H]+ calcd for C28H43N3NaO5, 524.3095; observed, 524.3088.
5-(Azetidin-3-yl)-3-(4-decylphenyl)-1,2,4-oxadiazole Hydrochloride (16a).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (79%, 34 mg). 1H NMR (400 MHz, CD3OD): δ 7.97 (d, J = 8.2 Hz, 2H), 7.31 (d, J = 8.1 Hz, 2H), 4.64–4.46 (m, 5H), 2.66 (t, J = 7.7 Hz, 2H), 1.63 (p, J = 7.5 Hz, 2H), 1.37–1.23 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 178.9, 169.8, 148.1, 130.1, 128.4, 125.0, 50.8, 36.9, 33.1, 32.4, 30.7, 30.7, 30.6, 30.5, 30.3, 29.9, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C21H32N3O, 342.2540; observed, 342.2543.
(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)methanamine Hydrochloride (16b).
Synthesized according to General Procedure 4. Purified via column chromatography (10% methanol/dichloromethane). White solid (86%, 250 mg). 1H NMR (400 MHz, CD3OD): δ 7.99 (d, J = 7.8 Hz, 2H), 7.31 (d, J = 7.8 Hz, 2H), 4.60 (br s, 2H), 2.66 (t, J = 7.7 Hz, 2H), 1.70–1.57 (m, 2H), 1.39–1.20 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CD3OD): δ 174.4, 169.7, 148.4, 130.1, 128.5, 124.7, 36.9, 36.1, 33.0, 32.4, 30.7, 30.6, 30.4, 30.3, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C19H30N3O, 316.2383; observed, 316.2393.
2-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)ethan-1-amine Hydrochloride (16c).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (76%, 110 mg). 1H NMR (400 MHz, CD3OD): δ 7.99 (d, J = 8.2 Hz, 2H), 7.34 (d, J = 8.2 Hz, 2H), 3.52 (t, J = 6.8 Hz, 2H), 3.38 (t, J = 6.7 Hz, 2H), 2.68 (t, J = 7.7 Hz, 2H), 1.70–1.60 (m, 2H), 1.40–1.22 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CD3OD): δ 177.6, 169.6, 148.2, 130.1, 28.4, 125.2, 37.3, 36.9, 33.1, 32.5, 30.7, 30.6, 30.5, 30.3, 25.4, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C20H32N3O, 330.2540; observed, 330.2529.
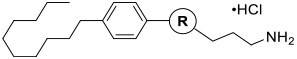
3-(3-(4-Decylphenyl)- 1,2,4-oxadiazol-5-yl)propan-1-amine Hydrochloride (16d).
Synthesized according to General Procedure 4. Purified via column chromatography (10% methanol/dichloromethane). White solid (63%, 160 mg). 1H NMR (400 MHz, CD3OD): δ 7.93 (d, J = 8.1 Hz, 2H), 7.29 (d, J = 8.1 Hz, 2), 3.19–3.07 (m, 4H), 2.64 (t, J = 7.6 Hz, 2H), 2.25 (p, J = 7.5 Hz, 2H), 1.69–1.58 (m, 2H), 1.37–1.20 (m, 14H), 0.87 (t, J = 6.6 Hz, 3H). 13C NMR (100 MHz, CD3OD): δ 180.1, 169.4, 147.9, 130.0, 128.3, 125.3, 39.8, 36.8, 33.0, 32.4, 30.7, 30.6, 30.4, 30.3, 25.1, 24.3, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C21H34N3O, 344.2696; observed, 344.2701.
4-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)butan-1-amine Hydrochloride (16e).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (85%, 190 mg). 1H NMR (400 MHz, CD3OD): δ 7.92 (d, J = 8.2 Hz, 2H), 7.29 (d, J = 8.1 Hz, 2H), 3.04 (t, J = 7.3 Hz, 2H), 2.98 (t, J = 7.8 Hz, 2H), 2.64 (t, J = 7.7 Hz, 2H), 1.95 (p, J = 7.3 Hz, 2H), 1.79 (p, J = 8.3 Hz, 2H), 1.62 (p, J = 7.5 Hz, 2H), 1.35–1.22 (m, 14H), 0.86 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 181.0, 169.4, 148.0, 130.1, 128.3, 125.4, 40.3, 36.8, 33.1, 32.5, 30.7, 30.7, 30.6, 30.5, 30.3, 27.8, 26.6, 24.4, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C22H36N3O, 358.2853; observed, 358.2854.
3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-N-methylpropan-1-amine Hydrochloride (16f).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (79%, 211 mg). 1H NMR (400 MHz, CdCl3): δ 7.95 (d, J = 7.8 Hz, 2H), 7.34 (d, J = 7.7 Hz, 2H), 3.20 (t, J = 7.6 Hz, 2H), 3.13 (t, J = 7.1 Hz, 2H), 2.75 (s, 3H), 2.68 (t, J = 7.6 Hz, 2H), 2.35–2.20 (m, 2H), 1.71–1.59 (m, 2H), 1.42–1.20 (m, 14H), 0.89 (t, J = 6.4 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.9, 169.4, 147.9, 130.0, 128.3, 125.3, 49.7, 36.8, 34.4, 33.0, 32.4, 30.6, 30.5, 30.3, 30.2, 24.6, 24.0, 23.6, 14.4. HRMS (ESI): [M + H]+ calcd for C22H36N3O, 358.2853; observed, 358.2850.
3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-N,N-dimethylpropan-1-amine Hydrochloride (16g).
The amine-free base of the title compound was prepared according to General Procedure 3. The title compound was prepared by dissolving the amine-free base 15g in methanolic HCl, followed by concentration in vacuo. It was purified via column chromatography (10% methanol/dichloromethane). White solid (33%, 80 mg). 1H NMR (400 MHz, CdCl3): δ 7.96 (d, J = 8.2 Hz, 2H), 7.34 (d, J = 7.34 (d, J = 8.2 Hz, 2H), 3.40–3.30 (m, 2H), 3.14 (t, J = 7.3 Hz, 2H), 2.97 (s, 6H), 2.69 (t, J = 7.7 Hz 2H), 2.40–2.29 (m, 2H), 1.66 (p, J = 7.2 Hz, 2H), 1.42–1.22 (m, 14H), 0.90 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.9, 169.5, 148.0, 130.1, 128.3, 125.3, 57.8, 43.6, 36.8, 33.0, 32.4, 30.7, 30.5, 30.4, 30.3, 24.2, 23.7, 22.4, 14.4. HRMS (ESI): [M + H]+ calcd for C23H38N3O, 372.3009; observed, 372.3005.
5-(Azetidin-3-ylmethyl)-3-(4-decylphenyl)-1,2,4-oxadiazole Hydrochloride (16h).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (63%, 164 mg). 1H NMR (400 MHz, CD3OD): δ 7.94 (d, J = 8.3 Hz, 2H), 7.32 (d, J = 8.5 Hz, 2H), 4.34–4.24 (m, 2H), 4.14–4.04 (m, 2H), 3.54 (hept, J = 8.2 Hz, 1H), 3.37 (d, J = 7.6 Hz, 2H), 2.67 (t, J = 7.7 Hz, 2H), 1.64 (p, J = 7.5 Hz, 2H), 1.42–1.21 (m, 14H), 0.89 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 178.5, 169.5, 148.1, 130.1, 128.3, 125.2, 52.1, 36.8, 33.1, 32.4, 30.8, 30.7, 30.6, 30.4, 30.3, 29.9, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C22H34N3O, 356.2696; observed, 356.2729.
(S)-3-(4-Decylphenyl)-5-(pyrrolidin-3-ylmethyl)-1,2,4-oxadiazole Hydrochloride (16i).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (75%, 210 mg). 1H NMR (400 MHz, CD3OD): δ 7.92 (d, J = 8.2 Hz, 2H), 7.29 (d, J = 8.2 Hz, 2H), 3.66–3.59 (m, 1H), 3.47–3.40 (m, 1H), 3.34–3.25 (m, 1H), 3.21–3.14 (m, 2H), 3.09 (dd, J = 11.8, 8.9 Hz, 1H), 3.00–2.83 (m, 1H), 2.65 (t, J = 7.7 Hz, 2H), 2.39–2.30 (m, 1H), 1.83 (dq, J = 13.2, 8.8 Hz, 1H), 1.62 (p, J = 7.2 Hz, 2H), 1.36–1.19 (m, 14H), 0.86 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CD3OD): δ 179.3, 169.5, 148.1, 130.1, 128.3, 125.3, 50.7, 46.3, 36.8, 33.1, 32.4, 31.0, 30.7, 30.6, 30.5, 30.3, 29.7, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C23H36N3O, 370.2853; observed, 370.2856.
(R)-3-(4-Decylphenyl)-5-(pyrrolidin-3-yl)-1,2,4-oxadiazole Hydrochloride (16j).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (72%, 90 mg). 1H NMR (400 MHz, cd3od): δ 7.94 (d, J = 7.9 Hz, 2H), 7.30 (d, J = 7.9 Hz, 2H), 4.04–3.92 (m, 1H), 3.77–3.69 (m, 1H), 3.67–3.60 (m, 1H), 3.49–3.36 (m, 2H), 2.65 (t, J = 7.7 Hz, 2H), 2.61–2.49 (m, 1H), 2.44–2.31 (m, 1H), 1.62 (p, J = 7.5 Hz, 2H), 1.38–1.20 (m, 14H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CD3OD): δ 179.3, 169.5, 163.1, 147.9, 130.0, 128.3, 125.3, 119.6, 116.7, 50.6, 46.2, 36.8, 36.8, 33.0, 32.4, 31.0, 30.7, 30.6, 30.4, 30.3, 29.7, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C22H34N3O, 356.2696; observed, 356.2716.
(S)-3-(4-Decylphenyl)-5-(pyrrolidin-3-yl)-1,2,4-oxadiazole Hydrochloride (16k).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (75%, 210 mg). 1H NMR (400 MHz, cd3od): δ 7.94 (d, J = 8.2 Hz, 2H), 7.30 (d, J = 8.2 Hz, 2H), 4.02–3.90 (m, 1H), 3.75–3.66 (m, 1H), 3.64–3.57 (m, 1H), 3.46–3.32 (m, 2H), 2.65 (t, J = 7.7 Hz, 2H), 2.60–2.47 (m, 1H), 2.43–2.30 (m, 1H), 1.62 (p, J = 7.1 Hz, 2H), 1.37–1.22 (m, 14H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CD3OD): δ 179.3, 169.5, 148.1, 130.1, 128.3, 125.3, 50.7, 46.3, 36.8, 33.1, 32.4, 31.0, 30.7, 30.6, 30.5, 30.3, 29.7, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C22H34N3O, 356.2696; observed, 356.2700.
(R)-3-(4-Decylphenyl)-5-(pyrrolidin-2-yl)-1,2,4-oxadiazole Hydrochloride (16l).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (76%, 88 mg). 1H NMR (400 MHz, CD3OD): δ 8.00 (d, J = 8.3 Hz, 2H), 7.36 (d, J = 8.3 Hz, 2H), 5.21 (t, J = 7.8 Hz, 1H), 3.67–3.49 (m, 2H), 2.75–2.62 (m, 3H), 2.49–2.36 (m, 1H), 2.36–2.20 (m, 2H), 1.64 (p, J = 7.4 Hz, 2H), 1.39–1.24 (m, 14H), 0.89 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 175.9, 169.8, 148.6, 130.2, 128.5, 124.6, 55.6, 47.3, 36.9, 33.0, 32.4, 30.7, 30.7, 30.5, 30.4, 30.3, 30.2, 24.5, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C22H34N3O, 356.2696; observed, 356.2711.
(S)-3-(4-Decylphenyl)-5-(pyrrolidin-2-yl)-1,2,4-oxadiazole Hydrochloride (16m).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (79%, 55 mg). 1H NMR (400 MHz, CD3OD): δ 8.00 (d, J = 8.2 Hz, 2H), 7.36 (d, J = 8.2 Hz, 2H), 5.21 (t, J = 7.7 Hz, 1H), 3.67–3.49 (m, 2H), 2.75–2.62 (m, 3H), 2.49–2.36 (m, 1H), 2.36–2.20 (m, 2H), 1.65 (p, J = 7.6 Hz, 2H), 1.40–1.23 (m, 14H), 0.89 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 175.9, 169.8, 148.6, 130.2, 128.5, 124.6, 55.6, 47.3, 36.9, 33.0, 32.4, 30.7, 30.7, 30.5, 30.4, 30.3, 30.2, 24.5, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C22H34N3O, 356.2696; observed, 356.2698.
3-(4-Decylphenyl)-5-(piperidin-4-yl)-1,2,4-oxadiazole Hydrochloride (16n).
Synthesized according to General Procedure 4. Purified via column chromatography (7% methanol/dichloromethane). White solid (89%, 330 mg). 1H NMR (400 MHz, CD3OD): δ 7.96 (d, J = 8.3 Hz, 2H), 7.33 (d, J = 8.3 Hz, 2H), 3.56–3.43 (m, 3H), 3.27–3.16 (m, 2H), 2.68 (t, J = 7.7 Hz, 2H), 2.48–2.35 (m, 2H), 2.21–2.06 (m, 2H), 1.73–1.57 (m, 2H), 1.42–1.16 (m, 14H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CD3OD): δ 181.5, 169.5, 148.1, 130.1, 128.3, 125.3, 44.0, 36.9, 33.1, 32.9, 32.5, 30.7, 30.6, 30.5, 30.3, 27.2, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C23H36N3O, 370.2853; observed, 370.2826.
3-(4-Decylphenyl)-5-(piperazin-1-ylmethyl)-1,2,4-oxadiazole Hydrochloride (16o).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (49%, 100 mg). 1H NMR (400 MHz, CD3OD): δ 7.91 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 4.03 (s, 2H), 4.03–3.16 (m, 4H), 2.96–2.82 (m, 4H), 2.61 (t J = 7.7 Hz, 2H), 1.66 (p, J = 7.5 Hz, 2H), 1.42–1.45 (m, 14H), 0.84 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, cd3od): δ 175.4, 169.7, 148.4, 130.2, 128.4, 125.0, 52.4, 50.3, 44.0, 36.8, 33.1, 32.4, 30.7, 30.5, 30.4, 30.3, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C23H37N4O, 385.2962; observed, 385.2980.
(1S,3R)-3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)cyclopentan-1-amine Hydrochloride (16p).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (54%, 122 mg). 1H NMR (400 MHz, CD3OD): δ 7.95 (d, J = 8.3 Hz, 2H), 7.32 (d, J = 8.3 Hz, 2H), 3.87–3.78 (m, 1H), 3.72–3.58 (m, 1H), 2.78–2.64 (m, 3H), 2.38–2.07 (m, 4H), 1.98–1.88 (m, 1H), 1.65 (p, J = 7.5 Hz, 2H), 1.38–1.21 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CD3OD): δ 183.1, 169.4, 148.0, 130.1, 128.3, 125.4, 52.6, 36.9, 36.8, 36.8, 36.8, 33.1, 32.5, 31.3, 30.7, 30.6, 30.5, 30.3, 30.3, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C23H36N3O, 370.2853; observed, 370.2859.
(1R,3S)-3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)cyclohexan-1-amine Hydrochloride (16q).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (87%, 226 mg). 1H NMR (400 MHz, CD3OD): δ 7.95 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 8.2 Hz, 2H), 3.71–3.52 (m, 2H), 2.67 (t, J = 7.7 Hz, 2H), 2.58–2.47 (m, 1H), 2.27–2.19 (m, 1H), 2.18–2.09 (m, 1H), 2.08–2.00 (m, 1H), 1.77 (q, J = 12.3 Hz, 1H), 1.71–1.51 (m, 5H), 1.40–1.20 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 182.4, 169.5, 148.0, 130.1, 128.3, 125.4, 48.0, 36.8, 33.3, 33.1, 32.9, 32.4, 30.8, 30.7, 30.5, 30.4, 30.3, 28.5, 23.7, 21.3, 14.4. HRMS (ESI): [M + H]+ calcd for C24H38N3O, 384.3009; observed, 384.2967.
(1R,3R)-3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)cyclohexan-1-amine Hydrochloride (16r).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (74%, 192 mg). 1H NMR (400 MHz, CD3OD): δ 7.93 (d, J = 8.3 Hz, 2H), 7.31 (d, J = 8.3 Hz, 2H), 3.39–3.32 (m, 1H), 3.29–3.19 (m, 1H), 2.66 (t, J = 7.8 Hz, 2H), 2.57–2.50 (m, 1H), 2.28–2.19 (m, 1H), 2.17–2.09 (m, 1H), 2.08–2.01 (m, 1H), 1.77 (q, J = 12.3 Hz, 1H), 1.70–1.41 (m, 5H), 1.39–1.21 (m, 14H), 0.88 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 182.6, 169.5, 148.0, 130.1, 128.3, 125.4, 50.5, 36.8, 35.9, 34.8, 33.0, 32.4, 31.0, 30.7, 30.5, 30.4, 30.3, 30.1, 24.4, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C24H38N3O, 384.3009; observed, 384.2970.
(3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)phenyl)methanamine Hydrochloride (16s).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (91%, 174 mg). 1H NMR (400 MHz, CD3OD): δ 8.37 (t, J = 1.8 Hz, 1H), 8.29 (dt, J = 7.7, 1.5 Hz, 1H), 8.05 (d, J = 8.2 Hz, 2H), 7.79 (dt, J = 7.8, 1.6 Hz, 1H), 7.77–7.68 (m, 1H), 7.38 (d, J = 8.3 Hz, 2H), 4.28 (s, 2H), 2.71 (t, J = 7.5 Hz, 2H), 1.66 (p, J = 7.2 Hz, 2H), 1.44–1.21 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, cd3od): δ 176.46, 170.34, 148.25, 136.04, 134.59, 131.38, 130.18, 129.72, 129.58, 128.41, 126.35, 125.39, 43.86, 36.88, 33.07, 32.45, 30.69, 30.56, 30.44, 30.30, 23.73, 14.43. HRMS (ESI): [M + H]+ calcd for C25H34N3O, 392.2696; observed, 392.2698.
(R)-2-Amino-4-(3-(4-decylphenyl)-1,2,4-oxadiazol-5-yl)butanoic Acid Hydrochloride (16t).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (69%, 145 mg). 1H NMR (400 MHz, CD3OD): δ 7.90 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.1 Hz, 2H), 4.19 (t, J = 6.6 Hz, 1H), 3.24–3.11 (m, 2H), 2.61 (t J = 7.7 Hz, 2H), 2.55–2.35 (m, 2H), 1.59 (p, J = 7.2 Hz, 2H), 1.35–1.15 (m, 14H), 0.83 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 179.7, 171.1, 169.5, 148.0, 130.1, 128.3, 125.3, 53.0, 36.8, 33.1, 32.4, 30.7, 30.6, 30.4, 30.3, 28.1, 28.0, 23.7, 23.5, 14.5. HRMS (ESI): [M + H]+ calcd for C22H34N3O3, 388.2595; observed, 388.2595.
3-Chloro-3-(3-(4-decylphenyl)-1,2,4-oxadiazol-5-yl)propan-1-amine Hydrochloride (16u).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (58%, 54 mg). 1H NMR (400 MHz, CdCl3): δ 7.96 (d, J = 8.2 Hz, 2H), 7.34 (d, J = 8.2 Hz, 2H), 4.84–4.76 (m, 1H), 3.72–3.55 (m, 3H), 3.44 (dd, J = 13.7, 9.9 Hz, 1H), 2.69 (t, J = 7.7 Hz, 2H), 1.66 (p, J = 7.7 Hz, 2H), 1.43–1.18 (m, 14H), 0.89 (t, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 176.9, 169.7, 148.2, 130.1, 128.3, 125.2, 55.6, 46.0, 36.8, 33.9, 33.1, 32.4, 30.7, 30.5, 30.4, 30.3, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C21H33ClN3O, 378.2312; observed, 378.2312.
(S)-1-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)propan-2-amine Hydrochloride (16v).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (67%, 76 mg). 1H NMR (400 MHz, CD3OD): δ 7.98 (d, J = 8.1 Hz, 2H), 7.31 (d, J = 8.1 Hz, 2H), 4.03–3.87 (m, 1H), 3.36 (d, J = 6.3 Hz, 2H), 2.66 (t, J = 7.7 Hz, 2H), 1.71–1.56 (m, 2H), 1.47 (d, J = 6.7 Hz, 3H), 1.39–1.20 (m, 14H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CD3OD): δ 177.0, 169.6, 148.1, 130.1, 128.4, 125.1, 46.6, 36.9, 33.1, 32.4, 32.0, 30.7, 30.6, 30.5, 30.3, 23.7, 18.7, 14.5. HRMS (ESI): [M + H]+ calcd for C21H34N3O, 344.2696; observed, 344.2705.
1-Amino-3-(3-(4-decylphenyl)-1,2,4-oxadiazol-5-yl)propan-2-ol Hydrochloride (16w).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (87%, 298 mg). 1H NMR (400 MHz, CD3OD): δ 7.95 (d, J = 8.3 Hz, 2H), 7.33 (d, J = 8.3 Hz, 2H), 4.43–4.34 (m, 1H), 3.30–3.15 (m, 3H), 3.05 (dd, J = 12.9, 9.5 Hz, 1H), 2.67 (t, J = 7.7 Hz, 2H), 1.64 (p, J = 7.5 Hz, 2H), 1.38–1.21 (m, 14H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CD3OD): δ 178.2, 169.6, 148.1, 130.1, 128.3, 125.4, 66.5, 45.2, 36.9, 33.3, 33.1, 32.5, 30.7, 30.6, 30.5, 30.3, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C21H34N3O2, 360.2646; observed, 360.2654.
1-Amino-3-(3-(4-decylphenyl)-1,2,4-oxadiazol-5-yl)propan-2-yl Acetate Hydrochloride (16x).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (71%, 93 mg). 1H NMR (400 MHz, CdCl3): δ 7.96 (d, J = 7.8 Hz, 2H), 7.35 (d, J = 7.9 Hz, 2H), 5.57 (q, J = 7.5 Hz, 1H), 3.59–3.35 (m, 4H), 2.69 (t, J = 7.6 Hz, 2H), 2.12 (s, 3H), 1.66 (p, J = 7.5 Hz, 2H), 1.43–1.22 (m, 14H), 0.90 (t, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 176.9, 171.9, 169.6, 148.2, 130.1, 128.3, 125.2, 68.8, 43.1, 36.8, 33.1, 32.4, 30.7, 30.5, 30.4, 30.3, 30.0, 23.7, 20.8, 14.4. HRMS (ESI): [M + H]+ calcd for C23H36N3O3, 402.2751; observed, 402.2747.
(9H-Fluoren-9-yl)methyl tert-Butyl (1-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)propane-1,3-diyl)(S)-dicarbamate (17a).
Synthesized according to General Procedure 3. Purified via column chromatography (25% ethyl acetate/hexanes). White solid (0.36 g, 74%). 1H NMR (400 MHz, CdCl3): δ 7.96 (d, J = 7.9 Hz, 2H), 7.77 (d, J = 7.6 Hz, 2H), 7.69–7.59 (m, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.33 (dd, J = 7.1, 2.7 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 6.00 (d, J = 9.0 Hz, 1H), 5.24 (q, J = 7.9 Hz, 1H), 4.93 (t, J = 6.7 Hz, 1H), 4.49 (d, J = 7.0 Hz, 2H), 4.26 (t, J = 7.0 Hz, 1H), 3.59–3.36 (m, 1H), 3.16–2.98 (m, 1H), 2.66 (t, J = 7.7 Hz, 2H), 2.32–2.19 (m, 1H), 2.19–2.02 (m, 1H), 1.64 (p, J = 7.2 Hz, 2H), 1.42 (s, 9H), 1.35–1.19 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 178.8, 168.5, 156.3, 156.2, 146.9, 143.9, 143.7, 141.5, 129.1, 127.9, 127.6, 127.2, 125.2, 123.9, 120.1, 79.8, 67.4, 47.3, 46.8, 36.5, 36.1, 34.4, 32.0, 31.3, 29.7, 29.7, 29.6, 29.5, 29.4, 28.5, 22.8, 14.3.
(9H-Fluoren-9-yl)methyl tert-Butyl (1-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)butane-1,4-diyl)(S)-dicarbamate (17b).
Synthesized according to General Procedure 3. Purified via column chromatography (30% ethyl acetate/hexanes). Yellow solid (0.20 g, 40%). 1H NMR (400 MHz, CdCl3): δ 7.97 (d, J = 8.0 Hz, 2H), 7.76 (d, J = 7.6 Hz, 2H), 7.62 (t, J = 7.3 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.34–7.30 (m, 2H), 7.28 (d, J = 8.2 Hz, 2H), 5.85–5.70 (m, 1H), 5.27–5.09 (m, 1H), 4.64 (s, 1H), 4.51–4.42 (m, 2H), 4.24 (t, J = 7.0 Hz, 1H), 3.27–3.13 (m, 2H), 2.66 (t, J = 7.7 Hz, 2H), 2.14–2.00 (m, 1H), 1.99–1.87 (m, 1H), 1.70–1.62 (m, 2H), 1.61–1.54 (m, 2H), 1.45 (s, 9H), 1.39–1.19 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 178.8, 168.5, 156.2, 155.9, 146.9, 143.9, 143.7, 141.5, 129.1, 127.9, 127.6, 127.2, 125.2, 123.9, 120.1, 79.6, 67.3, 48.9, 47.3, 39.9, 36.1, 32.0, 31.3, 31.3, 29.7, 29.7, 29.6, 29.5, 29.4, 28.5, 26.3, 22.81, 14.2.
(9H-Fluoren-9-yl)methyl tert-butyl (1-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)pentane-1,5-diyl)(S)-dicarbamate (17c).
Synthesized according to General Procedure 3. Purified via column chromatography (30% ethyl acetate/hexane). White solid (0.36 g, 73%). 1H NMR (400 MHz, CdCl3): δ 7.97 (d, J = 7.9 Hz, 2H), 7.76 (d, J = 7.6 Hz, 2H), 7.62 (t, J = 7.0 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.32 (dd, J = 7.2, 2.7 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 5.71 (d, J = 8.6 Hz, 1H), 5.15 (q, J = 7.7 Hz, 1H), 4.62 (t, J = 6.8 Hz, 1H), 4.49 (dd, J = 10.6, 6.9 Hz, 1H), 4.46–4.37 (m, 1H), 4.24 (t, J = 7.0 Hz, 1H), 3.13 (q, J = 7.2 Hz, 2H), 2.66 (t, J = 7.7 Hz, 2H), 2.13–1.89 (m, 2H), 1.65 (p, J = 14.8, 7.4 Hz, 2H), 1.58–1.48 (m, 2H), 1.43 (s, 11H), 1.36–1.18 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.0, 168.4, 156.3, 156.0, 146.8, 143.9, 143.7, 141.4, 129.0, 127.8, 127.6, 127.2, 125.2, 125.2, 123.9, 120.1, 79.4, 67.3, 48.9, 47.3, 40.0, 36.1, 33.6, 32.0, 31.3, 29.7, 29.7, 29.7, 29.6, 29.4, 29.4, 28.5, 22.8, 22.5, 14.3. HRMS (ESI+): calcd for C43H57N4O5 [M + H]+, 709.4323; observed, 709.4322.
(9H-Fluoren-9-yl)methyl tert-Butyl(1-(3-(4-decylphenyl)-1,2,4-ox-adiazol-5-yl)pentane-1,5-diyl)(R)-dicarbamate (17d).
Synthesized according to General Procedure 3. The product could not be separated from the unreacted starting material and was carried forward crude to the next reaction.
tert-Butyl (S)-(3-Amino-3-(3-(4-decylphenyl)-1,2,4-oxadiazol-5-yl)propyl)carbamate (18a).
Synthesized according to General Procedure 5. Purified by silica gel (5% methanol/dichloromethane). Yellow oil (0.02 g, 34%). 1H NMR (400 MHz, CdCl3): δ 7.96 (d, J = 8.0 Hz, 2H), 7.28 (d, J = 8.1 Hz, 2H), 5.06 (s, 1H), 4.28 (dd, J = 8.7, 5.0 Hz, 1H), 3.38–3.20 (m, 2H), 2.65 (t, J = 7.7 Hz, 2H), 2.28–1.90 (m, 2H), 1.63 (p, J = 7.2 Hz, 2H), 1.42 (s, 9H), 1.34–1.13 (m, 14H), 0.87 (t, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 182.1, 168.4, 156.2, 146.8, 129.1, 127.5, 124.1, 79.6, 47.7, 37.6, 36.1, 35.8, 32.0, 31.4, 29.7, 29.7, 29.6, 29.5, 29.4, 28.5, 22.8, 14.3. HRMS (ESI+): calcd for C26H43N4O3 [M + H]+, 459.3330; observed, 459.3325.
tert-Butyl (S)-(4-Amino-4-(3-(4-decylphenyl)-1,2,4-oxadiazol-5-yl)butyl)carbamate (18b).
Synthesized according to General Procedure 5. Purified via column chromatography (5% methanol/dichloromethane). Yellow oil (0.03 g, 83%). 1H NMR (400 MHz, CdCl3): δ 7.97 (d, J =8.1 Hz, 2H), 7.28 (d, J = 8.1 Hz, 2H), 4.69 (br s, 1H), 4.24 (t, J = 6.9 Hz, 1H), 3.18 (q, J = 6.7 Hz, 2H), 2.65 (t, J = 7.7 Hz, 2H), 2.22–1.80 (m, 2H), 1.74–1.63 (m, 2H), 1.63–1.57 (m, 2H), 1.43 (s, 9H), 1.36–1.14 (m, 14H), 0.87 (t, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 182.3, 168.4, 156.1, 146.8, 129.1, 127.5, 124.1, 79.4, 49.1, 40.2, 36.1, 33.1, 32.0, 31.4, 29.7, 29.7, 29.6, 29.4, 29.4, 28.4, 26.4, 22.8, 14.3. HRMS (ESI+): calcd for C27H45N4O3 [M + H]+, 473.3486; observed, 473.3476.
tert-Butyl (S)-(5-Amino-5-(3-(4-decylphenyl)-1,2,4-oxadiazol-5-yl)pentyl)carbamate (18c).
Synthesized according to General Procedure 5. Purified via column chromatography (5% methanol/dichloromethane). Yellow oil (0.06 g, 85%). 1H NMR (400 MHz, CdCl3): δ 7.96 (d, J = 7.9 Hz, 2H), 7.27 (d, J = 8.1 Hz, 2H), 4.58 (s, 1H), 4.19 (t, J = 6.8 Hz, 1H), 3.12 (q, J = 6.5 Hz, 2H), 2.64 (t, J = 7.7 Hz, 2H), 2.01–1.92 (m, 2H), 1.62 (p, J = 7.4 Hz, 2H), 1.58–1.51 (m, 2H), 1.50–1.44 (m, 2H), 1.41 (s, 9H), 1.34–1.20 (m, 14H), 0.86 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 182.5, 168.3, 156.1, 146.7, 129.0, 127.5, 124.1, 79.2, 49.3, 40.4, 36.1, 35.7, 32.0, 31.4, 29.9, 29.7, 29.7, 29.6, 29.4, 29.4, 28.5, 23.0, 22.8, 14.2. HRMS (ESI+): calcd for C28H47N4O3 [M + H]+, 487.3643; observed, 487.3632.
tert-Butyl (R)-(5-Amino-5-(3-(4-decylphenyl)-1,2,4-oxadiazol-5-yl)pentyl)carbamate (18d).
Synthesized according to General Procedure 5. Purified by silica gel (50% ethyl acetate/hexanes). Yellow solid (0.12 g, 44%). 1H NMR (400 MHz, CdCl3): δ 7.96 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.1 Hz, 2H), 4.59 (br s, 1H), 4.19 (t, J = 6.8 Hz, 1H), 3.11 (q, J = 6.4 Hz, 2H), 2.64 (t, J = 7.7 Hz, 2H), 2.05–1.85 (m, 2H), 1.62 (p, J = 7.4 Hz, 2H), 1.58–1.50 (m, 2H), 1.50–1.43 (m, 2H), 1.41 (s, 9H), 1.33–1.16 (m, 14H), 0.86 (t, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 182.5, 168.3, 156.1, 146.7, 129.0, 127.5, 124.2, 79.2, 49.3, 40.4, 36.1, 35.7, 32.0, 31.3, 29.9, 29.7, 29.7, 29.6, 29.4, 29.4, 28.5, 23.0, 22.8, 14.2. HRMS (ESI+): calcd for C28H47N4O3 [M + H]+, 487.3643; observed, 487.3634.
(S)-1-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)propane-1,3-diamine Hydrochloride (19a).
Synthesized according to General Procedure 4. Purified via column chromatography (0–20% methanol/dichloromethane). White solid (0.02 g, 67%). 1H NMR (400 MHz, CD3OD): δ 7.97 (d, J = 8.2 Hz, 2H), 7.34 (d, J = 8.1 Hz, 2H), 4.39 (dd, J = 8.8, 5.1 Hz, 1H), 3.28–3.15 (m, 2H), 2.68 (t, J = 7.7 Hz, 2H), 2.38–2.08 (m, 2H), 1.66 (p, J = 7.5 Hz, 2H), 1.42–1.19 (m, 14H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 182.8, 169.5, 148.1, 130.1, 128.4, 125.3, 48.6, 38.6, 36.8, 33.3, 33.1, 32.4, 30.7, 30.6, 30.4, 30.3, 23.7, 14.4. HRMS (ESI+): calcd for C21H35N4O [M + H]+, 359.2805; observed, 359.2798.
(S)-1-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)butane-1,4-diamine Hydrochloride (19b).
Synthesized according to General Procedure 4. Purified via column chromatography (0–20% methanol/dichloromethane). White solid (0.01 g, 57%). 1H NMR (400 MHz, CD3OD): δ 7.98 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 4.54 (t, J = 6.8 Hz, 1H), 3.02 (t, J = 7.4 Hz, 2H), 2.69 (t, J = 7.7 Hz, 2H), 2.24–1.99 (m, 2H), 1.94–1.75 (m, 2H), 1.65 (p, J = 7.3 Hz, 2H), 1.42–1.19 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 180.8, 169.6, 148.3, 130.2, 128.4, 125.1, 49.3, 40.2, 36.8, 33.1, 32.4, 32.3, 30.7, 30.5, 30.4, 30.3, 24.9, 23.7, 14.4. HRMS (ESI+): calcd for C22H37N4O [M + H]+, 373.2962; observed, 373.2955.
(S)-1-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)pentane-1,5-diamine Hydrochloride (19c).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (0.02 g, 37%). 1H NMR (400 MHz, CD3OD): δ 7.97 (d, J = 8.1 Hz, 2H), 7.34 (d, J =8.1 Hz, 2H), 4.29 (t, J = 6.8 Hz, 1H), 3.01–2.89 (m, 2H), 2.68 (t, J = 7.7 Hz, 2H), 2.09–1.87 (m, 2H), 1.78–1.65 (m, 2H), 1.70–1.59 (m, 2H), 1.62–1.41 (m, 2H), 1.39–1.19 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 183.1, 169.4, 148.1, 130.1, 128.3, 125.4, 49.7, 40.5, 36.8, 35.7, 33.1, 32.5, 30.7, 30.6, 30.5, 30.3, 28.2, 23.7, 23.5, 14.5. HRMS (ESI+): calcd for C23H39N4O [M + H]+, 387.3118; observed, 387.3124.
(R)-1-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)pentane-1,5-diamine Hydrochloride (19d).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (0.09 g, 79%). 1H NMR (400 MHz, CD3OD): δ 8.01 (d, J = 8.0 Hz, 2H), 7.35 (d, J =8.1 Hz, 2H), 4.98 (t, J = 6.9 Hz, 1H), 2.99 (t, J = 7.6 Hz, 2H), 2.68 (t, J = 7.7 Hz, 2H), 2.34–2.14 (m, 2H), 1.80 (p, J = 7.6 Hz, 2H), 1.72–1.63 (m, 2H), 1.63–1.53 (m, 2H), 1.40–1.20 (m, 14H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 176.7, 169.8, 148.5, 130.2, 128.5, 124.6, 48.9, 40.2, 36.8, 33.0, 32.4, 30.6, 30.5, 30.4, 30.3, 27.8, 23.7, 23.1, 14.5. HRMS (ESI+): calcd for C23H39N4O [M + H]+, 387.3118; observed, 387.3106.
tert-Butyl (S)-(5-Acetamido-5-(3-(4-decylphenyl)-1,2,4-oxadia-zol-5-yl)pentyl)carbamate (20).
To a round-bottom flask containing dry DCM and a stir bar were added 18c (1.0 equiv) and TEA (3.0 equiv). Acetyl chloride (1.1 equiv) was added dropwise, and the reaction mixture was stirred at rt for 4 h. The crude reaction mixture was concentrated under reduced pressure and subjected to silica gel chromatography. It was purified via column chromatography (60% ethyl acetate/hexanes). White solid (0.08 g, 48%). 1H NMR (400 MHz, CdCl3): δ 7.93 (d, J = 7.9 Hz, 2H), 7.25 (d, J = 8.4 Hz, 2H), 6.71 (d, J = 8.1 Hz, 1H), 5.36 (q, J = 7.3 Hz, 1H), 4.67 (t, J = 6.2 Hz, 1H), 3.09 (q, J = 6.5 Hz, 2H), 2.63 (t, J = 7.7 Hz, 2H), 2.07 (s, 3H), 2.04–1.84 (m, 2H), 1.61 (p, J = 7.3 Hz, 2H), 1.54–1.46 (m, 2H), 1.41 (s, 9H), 1.39–1.33 (m, 2H), 1.33–1.20 (m, 14H), 0.86 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 178.9, 170.2, 168.4, 156.3, 146.8, 129.0, 127.5, 123.9, 79.3, 46.8, 39.9, 36.0, 33.3, 32.0, 31.3, 29.7, 29.7, 29.6, 29.4, 29.3, 28.5, 23.1, 22.8, 22.4, 14.2.
(S)-N-(5-Amino-1-(3-(4-decylphenyl)-1,2,4-oxadiazol-5-yl)-pentyl)acetamide Hydrochloride (21).
Synthesized according to General Procedure 4. Purified via column chromatography (0–20% methanol/dichloromethane). White solid (0.06 g, 85%). 1H NMR (400 MHz, CD3OD): δ 7.95 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 8.1 Hz, 2H), 5.28 (dd, J = 8.8, 5.9 Hz, 1H), 2.95 (t, J = 7.6 Hz, 2H), 2.67 (t, J = 7.7 Hz, 2H), 2.17–2.07 (m, 1H), 2.05 (s, 3H), 2.04–1.94 (m, 1H), 1.80–1.70 (m, 2H), 1.69–1.60 (m, 2H), 1.60–1.46 (m, 2H), 1.38–1.17 (m, 14H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 180.6, 173.4, 169.6, 148.1, 130.1, 128.3, 125.2, 47.8, 40.4, 36.8, 33.3, 33.1, 32.4, 30.7, 30.6, 30.5, 30.3, 27.9, 23.7, 23.7, 22.4, 14.5. HRMS (ESI+): calcd for C25H41N4O2 [M + H]+, 429.3224; observed, 429.3225.
3-(4-Decylphenyl)-5-(3-(pyrrolidin-1-yl)propyl)-1,2,4-oxadiazole Hydrochloride (22a).
Synthesized according to General Procedure 6. Purified via column chromatography (5–10% methanol/dichloromethane). White solid (68%, 56 mg). 1H NMR (400 MHz, CD3OD): δ 7.95 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 8.2 Hz, 2H), 3.40–3.29 (m, 6H), 3.13 (t, J = 7.3 Hz, 2H), 2.68 (t, J = 7.7 Hz, 2H), 2.37–2.27 (m, 2H), 2.14–2.04 (m, 4H), 1.65 (p, J = 7.3 Hz, 2H), 1.40–1.21 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 180.1, 169.5, 148.1, 130.1, 128.3, 125.4, 55.3, 55.1, 36.8, 33.1, 32.5, 30.7, 30.6, 30.5, 30.3, 24.4, 24.0, 24.0, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C25H40N3O, 398.3166; observed, 398.3155.
3-(4-Decylphenyl)-5-(3-(piperidin-1-yl)propyl)-1,2,4-oxadiazole Hydrochloride (22b).
Synthesized according to General Procedure 6. Purified via column chromatography (5–10% methanol/dichloromethane). White solid (38%, 32 mg). 1H NMR (400 MHz, CD3OD): δ 7.94 (d, J = 8.3 Hz, 2H), 7.31 (d, J = 8.2 Hz, 2H), 3.05 (t, J = 7.3 Hz, 2H), 2.94–2.75 (m, 6H), 2.66 (t, J = 7.7 Hz, 2H), 2.26–2.14 (m, 2H), 1.72 (p, J = 5.7 Hz, 4H), 1.69–1.51 (m, 4H), 1.39–1.18 (m, 14H), 0.89 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 180.7, 169.4, 147.9, 130.1, 128.3, 125.5, 58.2, 55.0, 36.9, 33.1, 32.5, 30.7, 30.6, 30.5, 30.3, 25.5, 24.9, 24.0, 23.8, 23.2, 14.5. HRMS (ESI): [M + H]+ calcd for C26H42N3O, 412.3322; observed, 412.3315.
4-(3-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)propyl)morpholine Hydrochloride (22c).
Synthesized according to General Procedure 6. Purified via column chromatography (5–10% methanol/dichloromethane). White solid (73%, 62 mg). 1H NMR (400 MHz, CD3OD): δ 7.95 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 8.1 Hz, 2H), 4.13–3.76 (m, 4H), 3.63–3.48 (m, 2H), 3.41–3.33 (m, 2H), 3.27–3.12 (m, 4H), 2.68 (t, J = 7.7 Hz, 2H), 2.43–2.32 (m, 2H), 1.65 (p, J = 7.2 Hz, 2H), 1.39–1.21 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 179.9, 169.5, 148.1, 130.1, 128.3, 125.4, 65.1, 57.3, 63.3, 36.8, 33.1, 32.5, 30.7, 30.6, 30.5, 30.3, 24.3, 23.7, 21.6, 14.5. HRMS (ESI): [M + H]+ calcd for C25H40N3O2, 414.3115; observed, 414.3113.
1-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-3-(pyrrolidin-1-yl)-propan-2-ol Hydrochloride (22d).
Synthesized according to General Procedure 6. Purified via column chromatography (5–15% methanol/dichloromethane). White solid (40%, 30 mg). 1H NMR (400 MHz, CD3OD): δ 7.96 (d, J = 8.2 Hz, 2H), 7.34 (d, J = 8.2 Hz, 2H), 4.55–4.47 (m, 1H), 3.39–3.32 (m, 6H), 3.21 (qd, J = 15.5, 6.2 Hz, 2H), 2.68 (t, J = 7.7 Hz, 2H), 2.11–2.03 (m, 4H), 1.65 (p, J = 7.4 Hz, 2H), 1.40– 1.22 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 178.2, 169.6, 148.1, 130.1, 128.3, 125.4, 65.8, 60.7, 55.5, 36.8, 33.6, 33.1, 32.5, 30.7, 30.6, 30.5, 30.3, 24.0, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C25H40N3O2, 414.3115; observed, 414.3133.
1-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-3-(piperidin-1-yl)-propan-2-ol Hydrochloride (22e).
Synthesized according to General Procedure 6. Purified via column chromatography (10% methanol/dichloromethane). White solid (19%, 10 mg). 1H NMR (400 MHz, CD3OD): δ 7.96 (d, J = 8.3 Hz, 2H), 7.33 (d, J = 8.2 Hz, 2H), 4.68–4.59 (m, 1H), 3.68–3.53 (m, 2H), 3.39 (dd, J = 13.3, 3.0 Hz, 1H), 3.33–3.15 (m, 3H), 3.14–2.96 (m, 2H), 2.68 (t, J = 7.7 Hz, 2H), 2.02–1.47 (m, 8H), 1.40–1.19 (m, 14H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 176.5, 168.2, 146.7, 128.7, 126.9, 123.9, 62.5, 60.4, 54.8, 51.7, 35.4, 32.1, 31.6, 31.0, 29.3, 29.1, 29.0, 28.9, 22.4, 22.4, 22.3, 21.2, 13.0. HRMS (ESI): [M + H]+ calcd for C26H42N3O2, 428.3272; observed, 428.3264.
1-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-3-morpholinopropan-2-ol Hydrochloride (22f).
Synthesized according to General Procedure 6. Purified via column chromatography (5–10% methanol/dichloromethane). White solid (64%, 84 mg). 1H NMR (400 MHz, CD3OD): δ 7.96 (d, J = 8.3 Hz, 2H), 7.33 (d, J = 8.2 Hz, 2H), 4.73–4.65 (m, 1H), 4.11–3.98 (m, 2H), 3.93–3.76 (m, 2H), 3.65–3.52 (m, 2H), 3.51–3.63 (m, 2H), 3.34–3.17 (m, 4H), 2.68 (t, J = 7.7 Hz, 2H), 1.64 (p, J = 7.4 Hz, 2H), 1.39–1.21 (m, 14H), 0.89 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 13C NMR (101 MHz, CD3OD): δ 177.9, 169.6, 148.06, 130.1, 128.3, 125.4, 64.7, 63.7, 62.2, 54.8, 52.3, 36.8, 33.5, 33.1, 32.5, 30.7, 30.6, 30.5, 30.3, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C25H40N3O3, 430.3064; observed, 430.3061.
1-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-3-(ethylamino)-propan-2-ol (23a).
To a round-bottom flask containing 16w (1.0 equiv) were added methanol (0.1 M), glacial acetic acid (2.0 equiv), and sodium cyanoborohydride (6.0 equiv) at rt. Acetaldehyde (1.2 equiv) was then added and the mixture stirred for 16 h at rt under argon. The reaction mixture was diluted in dichloromethane and washed with 2 M sodium bicarbonate solution and brine. The aqueous layer was washed with dichloromethane (three times). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure before being subjected to silica gel chromatography. The product was then dissolved in methanolic HCl and concentrated to afford the title compound as a hydrochloride salt. Purified via column chromatography (10% methanol/dichloromethane). White solid (27%, 20 mg). 1H NMR (400 MHz, CD3OD): δ 7.96 (d, J = 7.9 Hz, 2H), 7.34 (d, J = 7.9 Hz, 2H), 4.54–4.41 (m, 1H), 3.39–3.06 (m, 5H), 2.68 (t, J = 7.8 Hz, 2H), 1.65 (p, J = 7.2 Hz, 2H), 1.41–1.20 (m, 17H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 178.1, 169.6, 148.1, 130.1, 128.3, 125.4, 65.7, 52.5, 44.1, 36.8, 33.4, 33.1, 32.5, 30.7, 30.6, 30.5, 30.3, 23.7, 14.5, 11.4. HRMS (ESI): [M + H]+ calcd for C23H38N3O2, 388.2959; observed, 388.2977.
1-(3-(4-Decylphenyl)-1,2,4-oxadiazol-5-yl)-3-(dimethylamino)-propan-2-ol Hydrochloride (23b).
To a round-bottom flask was added 16w (1.0 equiv), followed by paraformaldehyde (10.0 equiv) and methanol (0.2 M). Sodium borohydride (6.0 equiv) was then added, and the mixture was heated to reflux for 16 h. The reaction mixture was diluted in dichloromethane and washed with 2 M sodium bicarbonate solution and brine. The aqueous layer was washed with dichloromethane three times. The combined organic layers were dried over sodium sulfate and concentrated. The product was then dissolved in methanolic HCl and concentrated to afford the title compound as a HCl salt. Purified via column chromatography (0–15% methanol/dichloromethane). White solid (20%, 16 mg). 1H NMR (400 MHz, CD3OD): δ 7.96 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.2 Hz, 2H), 4.53–4.44 (m, 1H), 3.26–3.10 (m, 2H), 3.09–3.04 (m, 2H), 2.73 (s, 6H), 2.68 (t, J = 7.7 Hz, 2H), 1.65 (p, J = 7.6 Hz, 2H), 1.39–1.22 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 178.4, 169.6, 148.0, 130.1, 128.3, 125.4, 65.4 63.7, 44.6, 36.8, 33.6, 33.1, 32.5, 30.7, 30.6, 30.5, 30.3, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C23H38N3O2, 388.2959; observed, 388.2953.
1-(4-Decylphenyl)ethan-1-one (24).
Synthesized according to General Procedure 1. Purified via column chromatography (5% ethyl acetate/hexanes). White solid (70%, 1.15 g). 1H NMR (400 MHz, CdCl3): δ 7.88 (d, J = 8.3 Hz, 2H), 7.26 (d, J = 8.3 Hz, 2H), 2.66 (t, J = 7.7 Hz, 2H), 2.58 (s, 3H), 1.62 (p, J = 8.8 Hz, 2H), 1.38–1.20 (m, 14H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 198.0, 149.0, 135.1, 128.7, 128.6, 36.1, 32.0, 31.3, 29.7, 29.7, 29.6, 29.5, 29.4, 26.7, 22.8, 14.3. HRMS (ESI): [2M + H]+ calcd for C36H57O2, 521.4353; observed, 521.4331.
2-Bromo-1-(4-decylphenyl)ethan-1-one (25).
Compound 24 (1.0 equiv) was dissolved in acetonitrile and added to a round-bottom flask containing a stir bar. NBS (1.0 equiv) and p-toluenesulfonic acid monohydrate (1.6 equiv) were added, and the reaction mixture was heated to reflux for 2 h. The flask was then removed from the heating source and allowed to stir at rt for an additional 18 h. The crude reaction mixture was concentrated under reduced pressure, dissolved in DCM, and partitioned with brine. The organic layer was further rinsed three times with brine, dried over sodium sulfate, filtered, loaded onto Celite, and subjected to silica chromatography. It was purified via column chromatography (5% ethyl acetate/hexanes). Light-pink solid (88%, 2.40 g). 1H NMR (400 MHz, CdCl3): δ 7.90 (d, J = 8.3 Hz, 2H), 7.29 (d, J = 8.2 Hz, 2H), 4.43 (s, 2H), 2.67 (t, J = 7.7 Hz, 2H), 1.63 (p, J = 8.0 Hz, 2H), 1.29 (d, J = 23.7 Hz, 14H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 191.1, 150.1, 131.8, 129.2, 129.0, 36.2, 32.0, 31.1, 31.1, 29.7, 29.7, 29.6, 29.4, 29.4, 22.8, 14.2. HRMS (ESI): [2M + Na]+ calcd for C36H54Br2NaO2, 701.2362; observed, 701.2361.
2-(4-Decylphenyl)-2-oxoethyl 4-((tert-Butoxycarbonyl)amino)-butanoate (26).
Compound 25 (1.0 equiv), N-Boc-GABA (1.1 equiv), and potassium carbonate (3.0 equiv) were added to a round-bottom flask. The flask was purged with argon, and acetonitrile was added. The mixture was allowed to stir overnight at rt. The crude mixture was loaded onto Celite, concentrated under reduced pressure, and subjected to silica gel chromatography. Purified via column chromatography (30% ethyl acetate/hexanes). Off-white solid (91%, 248 mg). 1H NMR (400 MHz, CdCl3): δ 7.82 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.1 Hz, 2H), 5.34 (s, 2H), 4.82 (t, J = 6.1 Hz, 1H), 3.23 (q, J = 6.7 Hz, 2H), 2.66 (t, J = 7.7 Hz, 2H), 2.54 (t, J = 7.2 Hz, 2H), 1.91 (p, J = 7.1 Hz, 2H), 1.62 (p, J = 7.6 Hz, 2H), 1.44 (s, 9H), 1.38–1.18 (m, 14H), 0.86 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 191.9, 172.8, 156.1, 150.0, 131.9, 129.0, 128.0, 79.2, 66.0, 39.8, 36.2, 32.0, 31.3, 31.1, 29.7, 29.6, 29.5, 29.4, 29.3, 28.5, 25.4, 22.8, 14.2. HRMS (ESI): [M + Na]+ calcd for C27H43NNaO5, 484.3033; observed, 484.3032.
tert-Butyl(3-(4-(4-decylphenyl)thiazol-2-yl)propyl)carbamate (27).
To an oven-dried pressure tube containing a stir bar were added 25 (1.0 equiv) and tert-butyl(4-amino-4-thioxobutyl)carbamate (1.2 equiv). The flask was sealed and purged with argon. A 2:1 DMF/EtOH solution was syringed into the flask, and the reaction mixture was heated to 80 °C for 4 h. The resulting solution was concentrated under reduced pressure, loaded onto Celite, and subjected to silica gel chromatography. It was purified via column chromatography (20–30% ethyl acetate/hexanes). Yellow oil (34%, 97 mg). 1H NMR (400 MHz, CdCl3): δ 7.78 (d, J = 8.2 Hz, 2H), 7.26 (s, 1H), 7.21 (d, J = 8.1 Hz, 2H), 4.95 (br s, 1H), 3.26 (q, J = 6.6 Hz, 2H), 3.09 (t, J = 7.4 Hz, 2H), 2.62 (t, J = 7.7 Hz, 2H), 2.03 (p, J = 7.1 Hz, 2H), 1.61 (p, J = 7.3 Hz, 2H), 1.44 (s, 9H), 1.37–1.21 (m, 14H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 170.0, 156.1, 155.4, 143.1, 132.1, 128.9, 126.4, 111.4, 79.3, 40.1, 35.9, 32.0, 31.6, 30.9, 30.0, 29.8, 29.7, 29.7, 29.5, 29.4, 28.6, 22.8, 14.3. HRMS (ESI): [M + H]+ calcd for C27H43N2O2S, 459.3040; observed, 459.3035.
3-(4-(4-Decylphenyl)thiazol-2-yl)propan-1-amine Hydrochloride (28)
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. Off-white solid (94%, 80 mg). 1H NMR (400 MHz, CD3OD): δ 7.86 (s, 1H), 7.80 (d, J = 8.3 Hz, 2H), 7.30 (d, J = 8.3 Hz, 2H), 3.36 (t, J = 7.6 Hz, 2H), 3.13 (t, J = 7.6 Hz, 2H), 2.65 (t, J = 7.7 Hz, 2H), 2.25 (p, J = 7.6 Hz, 2H), 1.64 (p, J = 7.2 Hz, 2H), 1.39–1.21 (m, 14H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 173.7, 153.1, 145.7, 130.2, 130.0, 127.7, 115.5, 39.8, 36.6, 33.0, 32.5, 30.7, 30.7, 30.6, 30.4, 30.3, 29.7, 28.4, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C22H35N2S, 359.2515; observed, 359.2500.
tert-Butyl (3-(4-(4-Decylphenyl)oxazol-2-yl)propyl)carbamate (29)
To an oven-dried pressure tube containing a stir bar were added 25 (1.0 equiv) and tert-butyl(4-amino-4-oxobutyl)carbamate (2.0 equiv). The flask was sealed and purged with argon. NMP was syringed into the flask, and the reaction mixture was heated to 100 °C for 4 h. The resulting solution was diluted with ethyl acetate and portioned with a saturated sodium bicarbonate solution. The organic layer was further rinsed three times with brine, dried over sodium sulfate, and filtered. The concentrated residue was loaded onto Celite and subjected to silica gel chromatography. It was purified via column chromatography (20–30% ethyl acetate/hexanes). Yellow solid (10%, 42 mg). 1H NMR (400 MHz, CdCl3): δ 7.77 (s, 1H), 7.61 (d, J = 8.2 Hz, 2H), 7.19 (d, J = 8.0 Hz, 2H), 5.02 (br s, 1H), 3.24 (q, J = 6.5 Hz, 2H), 2.86 (t, J = 7.3 Hz, 2H), 2.61 (t, J = 7.7 Hz, 2H), 2.00 (p, J = 7.0 Hz, 2H), 1.61 (p, J = 7.0 Hz, 2H), 1.44 (s, 9H), 1.35–1.21 (m, 14H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 164.6, 156.1, 143.0, 140.8, 132.8, 128.9, 128.6, 125.5, 79.3, 40.2, 35.9, 32.0, 31.5, 29.7, 29.7, 29.6, 29.5, 29.4, 28.5, 27.3, 25.9, 22.8, 14.2. HRMS (ESI): [M-Boc]+ calcd for C22H35N2O, 343.2744; observed, 343.2745.
3-(4-(4-Decylphenyl)oxazol-2-yl)propan-1-amine Hydrochloride (30)
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (22 mg, 60%). 1H NMR (400 MHz, CD3OD): δ 8.13 (s, 1H), 7.62 (d, J = 7.8 Hz, 2H), 7.21 (d, J = 7.8 Hz, 2H), 3.10 (t, J = 7.4 Hz, 2H), 2.97 (t, J = 7.2 Hz, 2H), 2.61 (t, J = 7.6 Hz, 2H), 2.18 (p, J = 7.2 Hz, 2H), 1.61 (p, J = 7.0 Hz, 2H), 1.36–1.20 (m, 14H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 165.3, 144.1, 141.8, 135.2, 129.8, 129.5, 126.4, 40.1, 36.6, 33.0, 32.6, 30.7, 30.7, 30.5, 30.4, 30.2, 25.9, 25.6, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C22H35N2O, 343.2744; observed, 343.2744.
tert-Butyl (3-(4-(4-Decylphenyl)-1H-imidazol-2-yl)propyl)-carbamate (31).
To a round-bottom flask attached to a condenser were added alpha acyl ketone 26 (1.0 equiv) and ammonium acetate (20.0 equiv). Dry toluene was then added and the mixture heated to reflux for 5 h. The mixture was then concentrated in vacuo and diluted in ethyl acetate, followed by washing with a saturated sodium bicarbonate solution. The organic layer was then dried over anhydrous sodium sulfate, concentrated in vacuo, and subjected to silica chromatography. Purified via column chromatography (100% ethyl acetate). White solid (85%, 270 mg). 1H NMR (400 MHz, CdCl3): δ 11.32 (s, 1H), 7.58 (s, 2H), 7.46 (d, J = 8.5 Hz, 2H), 7.25 (d, J = 8.9 Hz, 1H), 5.02 (s, 1H), 3.22 (q, J = 6.2 Hz, 2H), 2.83–2.75 (m, 2H), 1.85–1.75 (m, 2H), 1.48 (s, 9H). 13C NMR (101 MHz, CdCl3): δ 157.9, 148.7, 131.8, 126.3, 120.0, 111.8, 80.3, 38.7, 29.7, 28.5, 24.7. HRMS (ESI): [M + Na]+ calcd for C17H22BrN3NaO2, 402.0788; observed, 402.0768.
3-(4-(4-Decylphenyl)-1H-imidazol-2-yl)propan-1-amine Hydrochloride (32).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (86%, 177 mg). 1H NMR (400 MHz, CD3OD): δ 7.76 (s, 1H), 7.67 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 8.2 Hz, 2H), 3.20 (t, J = 7.8 Hz, 2H), 3.09 (t, J = 7.8 Hz, 2H), 2.66 (t, J = 7.6 Hz, 2H), 2.24 (p, J = 7.9 Hz, 2H), 1.72–1.57 (m, 2H), 1.31 (d, J = 21.9 Hz, 14H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 147.9, 146.1, 135.0, 130.5, 126.6, 125.4, 115.1, 39.7, 36.6, 33.0, 32.5, 30.7, 30.7, 30.6, 30.4, 30.3, 26.5, 23.9, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C22H36N3, 342.2904; observed, 342.2905.
tert-Butyl (3-(4-(4-Decylphenyl)-1-methyl-1H-imidazol-2-yl)-propyl)carbamate (33).
Compound 31 was dissolved in a solution of dry THF and added to an oven-dried round-bottom flask containing a stir bar. The flask was purged with argon and cooled to 0 °C in an ice bath. Sodium hydride (1.1 equiv, 60% dispersion in mineral oil) was added, and the reaction mixture was allowed to stir for 15 min. Methyl iodide (1.5 equiv) was added to the reaction mixture dropwise and slowly warmed to rt over the course of 18 h. Following the completion of the reaction, the mixture was loaded onto Celite and subjected to silica gel chromatography. Purified via column chromatography (100% ethyl acetate). White solid (63%, 65 mg). 1H NMR (400 MHz, CD3OD): δ 7.57 (d, J = 8.3 Hz, 2H), 7.22 (s, 1H), 7.14 (d, J = 8.4 Hz, 2H), 3.63 (s, 3H), 3.12 (t, J = 6.7 Hz, 2H), 2.75 (t, J = 7.6 Hz, 2H), 2.58 (t, J = 7.6 Hz, 2H), 1.85 (p, J = 7.6 Hz, 2H), 1.59 (p, J = 7.3 Hz, 2H), 1.44 (s, 9H), 1.36–1.22 (m, 14H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 158.5, 149.8, 142.4, 140.8, 132.8, 129.5, 125.8, 117.9, 79.9, 40.8, 36.6, 33.1, 33.1, 32.7, 30.7, 30.7, 30.6, 30.5, 30.3, 29.4, 28.8, 24.7, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C28H46N3O2, 456.3585; observed, 456.3592.
3-(4-(4-Decylphenyl)-1-methyl-1H-imidazol-2-yl)propan-1-amine Hydrochloride (34).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether and ethyl acetate. White solid (75%, 42 mg). 1H NMR (400 MHz, CD3OD): δ 7.80 (s, 1H), 7.67 (d, J = 8.3 Hz, 2H), 7.33 (d, J = 8.2 Hz, 2H), 3.93 (s, 3H), 3.22 (t, J = 7.9 Hz, 2H), 3.13 (t, J = 7.7 Hz, 2H), 2.66 (t, J = 7.6 Hz, 2H), 2.20 (p, J = 7.9 Hz, 2H), 1.64 (p, J = 6.8 Hz, 2H), 1.38–1.21 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 147.6, 146.2, 134.0, 130.5, 126.6, 125.1, 119.9, 39.7, 36.6, 35.1, 33.0, 32.5, 30.7, 30.7, 30.6, 30.4, 30.3, 26.0, 23.7, 22.7, 14.4. HRMS (ESI): [M + H]+ calcd for C23H38N3, 356.3060; observed, 356.3057.
3-(4-Decylphenyl)-1H-pyrazole (36a).
Compound 35 (1.0 equiv), (1H-pyrazol-3-yl)boronic acid (4.0 equiv), Pd(dppf)Cl2·CH2Cl2 (0.2 equiv), and sodium bicarbonate (6.0 equiv) were added to a two-neck round-bottom flask containing a stir bar and attached to a reflux condenser. The system was purged with argon, and DMF was added (0.2 M). An additional 0.2 mL of DI water was then syringed into the flask and the mixture was heated to 100 °C for 16 h. Upon completion, the flask was cooled to rt, filtered through Celite, and subjected to silica gel chromatography. It was then purified via column chromatography (30–40% ethyl acetate/hexanes). Off-white solid (46%, 1.043 g). 1H NMR (400 MHz, CdCl3): δ 7.64 (d, J = 8.3 Hz, 2H), 7.61 (d, J = 2.2 Hz, 1H), 7.23 (d, J = 8.5 Hz, 2H), 6.58 (d, J = 2.2 Hz, 1H), 2.63 (t, J = 7.7 Hz, 2H), 1.63 (p, J = 7.3 Hz, 2H), 1.40– 1.21 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). HRMS (ESI): [M + H]+ calcd for C19H29N2, 285.2325; observed, 285.2323.
4-(4-Decylphenyl)-1H-pyrazole (36b).
Compound 35 (1.0 equiv), (1H-pyrazol-4-yl)boronic acid (4.0 equiv), Pd(dppf)Cl2·CH2Cl2 (0.2 equiv), and sodium bicarbonate (6.0 equiv) were added to a two-neck round-bottom flask containing a stir bar and attached to a reflux condenser. The system was purged with argon, and DMF was added (0.2 M). An additional 0.2 mL of DI water was then syringed into the flask, and the mixture was heated to 100 °C for 16 h. Upon completion, the flask was cooled to rt, filtered through Celite, and subjected to silica gel chromatography. It was then purified via column chromatography (30–40% ethyl acetate/hexanes). White solid (53%, 761 mg). 1H NMR (500 MHz, CD3OD): δ 8.00–7.78 (m, 3H), 7.47 (d, J = 8.2 Hz, 2H), 7.17 (d, J = 8.2 Hz, 2H), 2.60 (t, J = 7.7 Hz, 2H), 1.61 (p, J = 7.4 Hz, 2H), 1.39–1.23 (m, 14H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (126 MHz, CD3OD): δ 142.2, 131.4, 129.9, 126.5, 123.8, 36.6, 33.1, 32.7, 30.7, 30.7, 30.6, 30.5, 30.3, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C19H29N2, 285.2325; observed, 285.2333.
tert-Butyl (3-(5-(4-Decylphenyl)-1H-pyrazol-1-yl)propyl)-carbamate (37).
Synthesized according to General Procedure 7. Purified via column chromatography (20% ethyl acetate/dichloromethane). Clear oil (16%, 50 mg). 1H NMR (400 MHz, CdCl3): δ 7.52 (d, J = 1.9 Hz, 1H), 7.32–7.22 (m, 4H), 6.25 (d, J = 1.9 Hz, 1H), 4.70 (br s, 1H), 4.20 (t, J = 6.7 Hz, 2H), 3.02 (q, J = 6.4 Hz, 2H), 2.65 (d, J = 7.8, 2H), 1.92 (p, J = 6.6 Hz, 2H), 1.65 (p, J = 7.4 Hz, 2H), 1.41 (s, 9H), 1.37–1.22 (m, 14H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 156.0, 143.9, 143.8, 138.9, 128.9, 128.9, 128.1, 106.3, 79.2, 47.0, 37.8, 35.9, 32.1, 31.5, 30.6, 29.8, 29.7, 29.7, 29.5, 29.5, 28.5, 22.8, 14.3. HRMS (ESI): [M + H]+ calcd for C27H44N3O2, 442.3428; observed, 442.3422.
3-(5-(4-Decylphenyl)-1H-pyrazol-1-yl)propan-1-amine Hydrochloride (38).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (89%, 38 mg). 1H NMR (400 MHz, CD3OD): δ 7.67 (d, J = 2.0 Hz, 1H), 7.44–7.33 (m, 4H), 6.41 (d, J = 2.0 Hz, 1H), 4.32 (t, J = 6.7 Hz, 2H), 2.88 (t, J = 7.7 Hz, 2H), 2.68 (t, J = 7.7 Hz, 2H), 2.11 (p, J = 7.3 Hz, 2H), 1.66 (p, J = 7.1 Hz, 2H), 1.40–1.22 (m, 14H), 0.90 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 146.3, 145.6, 139.6, 130.2, 130.0, 128.2, 107.8, 47.6, 38.4, 36.7, 33.1, 32.6, 30.7, 30.7, 30.6, 30.5, 30.4, 29.2, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C22H36N3, 342.2904; observed, 342.2910.
tert-Butyl (3-(3-(4-Decylphenyl)-1H-pyrazol-1-yl)propyl)-carbamate (39).
Synthesized according to General Procedure 7. Purified via column chromatography (20% ethyl acetate/dichloromethane). Clear oil (77%, 240 mg). 1H NMR (400 MHz, CdCl3): δ 7.69 (d, J = 8.2 Hz, 2H), 7.39 (d, J = 2.3 Hz, 1H), 7.19 (d, J = 8.2 Hz, 2H), 6.49 (d, J = 2.3 Hz, 1H), 5.07 (t, J = 6.2 Hz, 1H), 4.19 (t, J = 6.5 Hz, 2H), 3.15 (q, J = 6.4 Hz, 2H), 2.61 (t, J = 7.7 Hz, 2H), 2.03 (p, J = 6.5 Hz, 2H), 1.61 (p, J = 7.0 Hz, 2H), 1.43 (s, 9H), 1.36–1.21 (m, 14H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 156.2, 151.8, 142.5, 131.0, 130.7, 128.7, 125.5, 102.6, 79.2, 49.8, 38.0, 35.8, 32.0, 31.6, 30.8, 29.7, 29.7, 29.6, 29.4, 29.4, 28.5, 22.8, 14.2. HRMS (ESI): [M + H]+ calcd for C27H44N3O2, 442.3428; observed, 442.3419.
3-(3-(4-Decylphenyl)-1H-pyrazol-1-yl)propan-1-amine Hydrochloride (40).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (83%, 172 mg). 1H NMR (400 MHz, CD3OD): δ 7.73–7.63 (m, 3H), 7.20 (d, J = 8.2 Hz, 2H), 6.60 (d, J = 2.3 Hz, 1H), 4.31 (t, J = 6.5 Hz, 2H), 3.01–2.93 (m, 2H), 2.61 (t, J = 7.6 Hz, 2H), 2.29–2.17 (m, 2H), 1.62 (p, J =7.6 Hz, 2H), 1.39–1.20 (m, 14H), 0.89 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 153.5, 143.9, 133.0, 132.0, 129.7, 126.6, 103.8, 49.8, 38.4, 36.6, 33.1, 32.6, 30.7, 30.7, 30.6, 30.4, 30.3, 29.6, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C22H36N3, 342.2904; observed, 342.2922.
tert-Butyl (3-(4-(4-Decylphenyl)-1H-pyrazol-1-yl)propyl)-carbamate (41).
Synthesized according to General Procedure 7. Purified via column chromatography (20–60% ethyl acetate/hexanes). White solid (64%, 148 mg). 1H NMR (400 MHz, CD3OD): δ 7.90 (s, 1H), 7.76 (s, 1H), 7.41 (d, J = 8.1 Hz, 2H), 7.13 (d, J = 8.0 Hz, 2H), 4.16 (t, J = 6.8 Hz, 2H), 3.04 (d, J = 6.6 Hz, 2H), 2.56 (t, J = 7.6 Hz, 2H), 2.00 (p, J = 6.8 Hz, 2H), 1.59 (p, J = 7.5 Hz, 2H), 1.42 (s, 9H), 1.33–1.23 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 158.4, 142.1, 137.3, 131.1, 129.9, 128.1, 126.3, 124.4, 80.0, 50.5, 38.5, 36.6, 33.1, 32.7, 31.7, 30.7, 30.7, 30.6, 30.5, 30.3, 28.8, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C27H44N3O2, 442.3428; observed, 442.3428.
3-(4-(4-Decylphenyl)-1H-pyrazol-1-yl)propan-1-amine Hydrochloride (42).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (78%, 99 mg). 1H NMR (400 MHz, CD3OD): δ 8.04 (s, 1H), 7.89 (s, 1H), 7.50 (d, J = 8.2 Hz, 2H), 7.23 (d, J = 8.1 Hz, 2H), 4.36 (t, J = 6.6 Hz, 2H), 3.00 (t, J = 7.2 Hz, 2H), 2.66 (t, J = 7.6 Hz, 2H), 2.27 (p, J = 7.2 Hz, 2H), 1.68 (p, J = 7.4 Hz, 2H), 1.44–1.32 (m, 14H), 0.95 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 142.5, 137.8, 131.0, 130.0, 128.3, 126.4, 124.8, 49.9, 38.5, 36.6, 33.1, 32.7, 30.7, 30.7, 30.6, 30.4, 30.3, 29.8, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C22H36N3, 342.2904; observed, 342.2908.
4-Iodobenzohydrazide (44).
To a round-bottom flask were added 4-iodobenzoyl chloride (43, 1.0 equiv) and ethanol to make a 0.24 M solution. Hydrazine (6.0 equiv, 35% in water) was then added, and then the mixture was allowed to stir at 80 °C under ambient air for 20 h. The reaction mixture was removed from heating, cooled to rt, and the product recrystallized from ethanol. The crude product was then filtered and washed with deionized water and cold ethanol to afford the pure product. The material was carried forward as crude without any additional purification. White solid (67%, 1.01 g).
tert-Butyl (3-(5-(4-Iodophenyl)-1,3,4-oxadiazol-2-yl)propyl)-carbamate (45).
To a round-bottom flask containing a stir bar were added N-Boc-GABA (1.0 equiv), 44 (1.0 equiv), DIEA (3.0 equiv), and acetonitrile. The resulting solution was allowed to stir for 10 min before HCTU (1.1 equiv) was added. The reaction mixture was stirred at rt for 2 h. A second portion of DIEA (2.0 equiv) was then added, followed by tosyl chloride (3.0 equiv) addition. The mixture was continued to be stirred at rt for 16 h. Following completion, the reaction mixture was added to a beaker containing 15% ammonium hydroxide solution and stirred for 10 min. The mixture was extracted with dichloromethane (2 × 50 mL), and the organic layer was dried over anhydrous sodium sulfate. The dried organic layer was concentrated under reduced pressure, loaded onto Celite, and subjected to silica gel chromatography. It was then purified via column chromatography (30–50% ethyl acetate/hexanes). White solid (79%, 498 mg). 1H NMR (400 MHz, (CD3)2CO): δ 8.01 (d, J = 8.6 Hz, 2H), 7.84 (d, J = 8.6 Hz, 2H), 6.14 (s, 1H), 3.27 (q, J = 6.5 Hz, 2H), 3.02 (t, J = 7.5 Hz, 2H), 2.10–2.03 (m, 2H), 1.42 (s, 9H). 13C NMR (101 MHz, acetone): δ 167.8, 164.7, 156.7, 139.3, 129.0, 124.9, 98.4, 78.6, 40.3, 28.6, 27.6, 23.3. HRMS (ESI): [M + H]+ calcd for C16H21IN3O3, 430.0622; observed, 430.0620.
tert-Butyl (3-(5-(4-Decylphenyl)-1,3,4-oxadiazol-2-yl)propyl)-carbamate (46).
Synthesized according to General Procedure 1. Purified via column chromatography (30–45% ethyl acetate/hexanes). Light-brown solid (79%, 163 mg). 1H NMR (400 MHz, (CD3)2CO): δ 7.97 (d, J = 8.3 Hz, 2H), 7.45 (d, J = 8.4 Hz, 2H), 6.13 (s, 1H), 3.28 (q, J = 6.6 Hz, 2H), 3.01 (t, J = 7.5 Hz, 2H), 2.75 (t, J = 7.8 Hz, 2H), 2.11–2.04 (m, 2H), 1.70 (p, J = 7.3 Hz, 2H), 1.43 (s, 9H), 1.42–1.23 (m, 14H), 0.91 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, (CD3)2CO): δ 167.2, 165.3, 156.7, 147.7, 130.1, 127.4, 122.8, 78.6, 40.3, 36.4, 32.6, 32.0, 30.3, 30.3, 30.2, 30.0, 29.9, 28.6, 27.6, 23.3, 14.3. HRMS (ESI): [M + H]+ calcd for C26H42N3O3, 444.3221; observed, 444.3219.
3-(5-(4-Decylphenyl)-1,3,4-oxadiazol-2-yl)propan-1-amine Hydrochloride (47).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (76%, 85 mg). 1H NMR (400 MHz, CD3OD): δ 7.94 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 3.20–3.04 (m, 4H), 2.70 (t, J = 7.7 Hz, 2H), 2.24 (p, J = 7.4 Hz, 2H), 1.66 (p, J = 7.2 Hz, 2H), 1.41–1.22 (m, 14H), 0.89 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 167.3, 166.7, 149.1, 130.5, 127.9, 122.2, 39.8, 36.9, 33.1, 32.3, 30.7, 30.7, 30.5, 30.4, 30.3, 24.9, 23.7, 23.3, 14.4. HRMS (ESI): [M + H]+ calcd for C21H34N3O, 344.2696; observed, 344.2693.
2-(4-Bromophenyl)-N′-hydroxyacetimidamide (49a).
Synthesized according to General Procedure 2. The reaction mixture was removed from heating, cooled to rt, and the product recrystallized from ethanol. The crude product was then filtered and washed with deionized water and cold ethanol to afford the pure product. White solid (62%, 542 mg). 1H NMR (400 MHz, CD3OD): δ 7.44 (d, J = 8.4 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 3.34 (s, 2H). 13C NMR (101 MHz, CD3OD): δ 155.5, 137.7, 132.5, 131.6, 121.5, 37.5. HRMS (ESI): [M + H]+ calcd for C8H10BrN2O, 228.9971; observed, 228.9977.
3-(4-Bromophenyl)-N′-hydroxypropanimidamide (49b).
The compound was synthesized according to General Procedure 2. The reaction mixture was removed from heating and cooled to rt, and the product was recrystallized from ethanol. The crude product was then filtered and washed with deionized water and cold ethanol to afford the pure product. White solid (41%, 352 mg). 1H NMR (400 MHz, CD3OD): δ 7.40 (d, J = 8.2 Hz, 2H), 7.15 (d, J = 8.2 Hz, 2H), 2.84 (d, J = 7.7 Hz, 2H), 2.34 (d, J = 7.7 Hz, 2H). 13C NMR (101 MHz, CD3OD): δ 156.4, 141.5, 132.4, 131.5, 120.8, 33.8, 33.7. HRMS (ESI): [M + H]+ calcd for C9H12BrN2O, 243.0128; observed, 243.0135.
tert-Butyl (3-(3-(4-Bromobenzyl)-1,2,4-oxadiazol-5-yl)propyl)-carbamate (50a).
Synthesized according to General Procedure 3. Purified via column chromatography (35% ethyl acetate/hexanes). Yellow oil (68%, 292 mg). 1H NMR (400 MHz, CdCl3): δ 7.44 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 8.5 Hz, 2H), 4.65 (s, 1H), 3.99 (s, 2H), 3.21 (q, J = 6.3 Hz, 2H), 2.89 (t, J = 7.6 Hz, 2H), 1.97 (p, J = 7.0 Hz, 2H), 1.43 (s, 9H). 13C NMR (101 MHz, CdCl3): δ 179.7, 169.1, 156.0, 134.5, 132.0, 130.8, 121.3, 79.6, 39.8, 31.9, 28.5, 27.0, 24.1. HRMS (ESI): [M + H]+ calcd for C17H23BrN3O3, 418.0737; observed, 418.0746.
tert-Butyl (3-(3-(4-Bromophenethyl)-1,2,4-oxadiazol-5-yl)-propyl)carbamate (50b).
Synthesized according to General Procedure 3. Purified via column chromatography (35% ethyl acetate/hexanes). Yellow oil (76%, 322 mg). 1H NMR (400 MHz, CdCl3): δ 7.40 (d, J = 8.3 Hz, 2H), 7.08 (d, J = 8.3 Hz, 2H), 4.71 (s, 1H), 3.22 (q, J = 6.2 Hz, 2H), 3.00 (q, J = 3.1, 2.2 Hz, 4H), 2.90 (t, J = 7.5 Hz, 2H), 1.99 (p, J = 7.1 Hz, 2H), 1.43 (s, 9H). 13C NMR (101 MHz, CdCl3): δ 179.3, 169.6, 156.1, 139.3, 131.7, 130.2, 120.3, 79.6, 39.8, 32.5, 28.5, 27.7, 27.0, 24.0. HRMS (ESI): [M + Na]+ calcd for C18H24BrN3O3Na, 432.0893; observed, 432.0894.
tert-Butyl (3-(3-(4-Nonylbenzyl)-1,2,4-oxadiazol-5-yl)propyl)-carbamate (51a).
Synthesized according to General Procedure 1. Purified via column chromatography (25% ethyl acetate/hexanes). Colorless oil (48%, 149 mg). 1H NMR (400 MHz, CdCl3): δ 7.21 (d, J = 8.0 Hz, 2H), 7.12 (d, J = 8.1 Hz, 2H), 4.69 (s, 1H), 4.00 (s, 2H), 3.20 (q, J = 6.2 Hz, 2H), 2.88 (t, J = 7.6 Hz, 2H), 2.56 (d, J = 7.7 Hz, 2H), 1.97 (p, J = 7.0 Hz, 2H), 1.58 (p, J = 7.4 Hz, 2H), 1.43 (s, 9H), 1.27 (d, J = 14.6 Hz, 12H), 0.86 (t, 3H). 13C NMR (101 MHz, CdCl3): δ 179.4, 169.7, 156.0, 141.9, 132.7, 128.9, 128.9, 79.5, 39.8, 35.7, 32.0, 32.0, 31.6, 29.7, 29.6, 29.5, 29.4, 28.5, 27.0, 24.1, 22.8, 14.2. HRMS (ESI): [M + Na]+ calcd for C26H41N3O3Na, 466.3040; observed, 466.3049.
tert-Butyl (3-(3-(4-Octylphenethyl)-1,2,4-oxadiazol-5-yl)propyl)-carbamate (51b).
Synthesized according to General Procedure 1. Purified via column chromatography (20% ethyl acetate/hexanes). Colorless oil (50%, 166 mg). 1H NMR (400 MHz, CdCl3): δ 7.16–7.06 (m, 4H), 4.72 (s, 1H), 3.23 (q, J = 6.3 Hz, 2H), 3.05–2.98 (m, 4H), 2.91 (t, J = 7.5 Hz, 2H), 2.56 (t, J = 7.7 Hz, 2H), 2.01 (p, J = 6.9 Hz, 2H), 1.58 (p, J = 7.5 Hz, 2H), 1.44 (s, 9H), 1.38–1.20 (m, 10H), 0.87 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.1, 170.1, 156.0, 141.1, 137.6, 128.7, 128.3, 79.5, 39.8, 35.7, 32.8, 32.0, 31.7, 29.6, 29.5, 29.4, 28.5, 28.1, 27.0, 24.0, 22.8, 14.2. HRMS (ESI): [M + Na]+ calcd for C26H41N3O3Na, 466.3040; observed, 466.3044.
3-(3-(4-Nonylbenzyl)-1,2,4-oxadiazol-5-yl)propan-1-amine Hydrochloride (52a).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (45%, 50 mg). 1H NMR (400 MHz, CD3OD): δ 7.19 (d, J = 8.0 Hz, 2H), 7.12 (d, J = 8.0 Hz, 2H), 4.01 (s, 2H), 3.13–2.97 (m, 4H), 2.57 (t, J = 7.8 Hz, 2H), 2.13 (p, J = 7.5 Hz, 2H), 1.58 (p, J = 7.5 Hz, 2H), 1.40–1.22 (m, 12H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 178.8, 169.5, 141.5, 132.7, 128.4, 128.3, 38.4, 35.1, 31.6, 31.3, 31.0, 29.3, 29.2, 29.0, 28.9, 23.6, 22.8, 22.3, 13.0. HRMS (ESI): [M + H]+ calcd for C21H34N3O, 344.2696; observed, 344.2703.
3-(3-(4-Octylphenethyl)-1,2,4-oxadiazol-5-yl)propan-1-amine Hydrochloride (52b).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (46%, 49 mg). 1H NMR (400 MHz, CD3OD): δ 7.13–7.06 (m, 4H), 3.12–2.97 (m, 8H), 2.55 (t, J = 7.7 Hz, 2H), 2.17 (p, J = 7.5 Hz, 2H), 1.59 (p, J = 7.5, 6.6 Hz, 2H), 1.37–1.21 (m, 10H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 179.9, 171.1, 142.1, 138.8, 129.6, 129.2, 39.8, 36.5, 33.5, 33.0, 32.8, 30.6, 30.4, 30.3, 28.7, 25.1, 24.2, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C21H34N3O, 344.2696; observed, 344.2700.
tert-Butyl (3-(3-(4-Iodophenyl)-1,2,4-oxadiazol-5-yl)propyl)-carbamate (55a).
Synthesized according to General Procedure 3. Purified via column chromatography (25% ethyl acetate/hexanes). Off-white solid (67%, 985 mg). 1H NMR (400 MHz, CdCl3): δ 7.84–7.77 (m, 4H), 4.77 (br s, 1H), 3.28 (q, J = 6.5 Hz, 2H), 2.99 (t, J = 7.4 Hz, 2H), 2.07 (p, J = 7.1 Hz, 2H), 1.43 (s, 9H). 13C NMR (101 MHz, CdCl3): δ 179.7, 167.8, 156.1, 138.2, 129.0, 126.5, 98.0, 79.6, 39.9, 28.5, 27.1, 24.2. HRMS (ESI): [M + H]+ calcd for C16H21IN3O3, 430.0622; observed, 430.0614.
tert-Butyl (3-(3-(3-Iodophenyl)-1,2,4-oxadiazol-5-yl)propyl)-carbamate (55b).
Synthesized according to General Procedure 3. Purified via column chromatography (25% ethyl acetate/hexanes). White solid (58%, 333 mg). 1H NMR (400 MHz, CdCl3): δ 8.42 (t, J = 1.7 Hz, 1H), 8.03 (d, J = 7.8 Hz, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.21 (t, J = 7.9 Hz, 1H), 4.75 (s, 1H), 3.28 (q, J = 6.5 Hz, 2H), 2.99 (t, J = 7.5 Hz, 2H), 2.07 (p, J = 7.1 Hz, 2H), 1.43 (s, 9H). 13C NMR (101 MHz, CdCl3): δ 179.8, 167.1, 156.1, 140.2, 136.3, 130.6, 128.8, 126.6, 94.5, 79.6, 39.8, 28.5, 27.1, 24.2.
tert-Butyl (3-(3-(4-Hexylphenyl)-1,2,4-oxadiazol-5-yl)propyl)-carbamate (56a).
Synthesized according to General Procedure 1. Purified by silica chromatography (25% ethyl acetate/hexanes). White solid (86%, 135 mg). 1H NMR (400 MHz, CdCl3): δ 7.94 (d, J = 8.4 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H), 4.79 (s, 1H), 3.26 (q, J = 6.5 Hz, 2H), 2.97 (t, J = 7.5 Hz, 2H), 2.64 (t, J = 7.7 Hz, 2H), 2.11–1.99 (m, 2H), 1.92–1.74 (m, 2H), 1.49 (s, 9H), 1.38–1.20 (m, 6H), 0.86 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.1, 168.1, 155.9, 146.4, 128.9, 127.3, 124.1, 80.9, 39.7, 35.9, 32.0, 31.6, 31.1, 28.9, 28.3, 26.1, 24.0, 22.5, 21.1, 14.0. HRMS (ESI): [M + H]+ calcd for C22H34N3O3, 388.2595; observed, 388.2617.
tert-Butyl (3-(3-(4-Heptylphenyl)-1,2,4-oxadiazol-5-yl)propyl)-carbamate (56b).
Synthesized according to General Procedure 1. Purified by silica chromatography (25% ethyl acetate/hexanes). White solid (93%, 120 mg). 1H NMR (400 MHz, chloroform-d): δ 7.95 (d, J = 8.4 Hz, 2H), 7.26 (d, J = 8.6 Hz, 2H), 4.77 (s, 1H), 3.27 (q, J = 6.6 Hz, 2H), 2.97 (t, J = 7.5 Hz, 2H), 2.64 (t, J = 7.2 Hz, 2H), 2.05 (p, J = 7.0 Hz, 2H), 1.63 (p, J = 7.3 Hz, 2H), 1.42 (s, 9H), 1.31–1.20 (m, 8H), 0.85 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.26, 168.45, 156.08, 146.64, 129.05, 127.49, 124.27, 36.10, 32.22, 31.94, 31.38, 29.36, 29.29, 28.53, 26.37, 22.80, 22.16, 14.24.
tert-Butyl (3-(3-(4-Octylphenyl)-1,2,4-oxadiazol-5-yl)propyl)-carbamate (56c).
Synthesized according to General Procedure 1. Purified by silica chromatography (25% ethyl acetate/hexanes). White solid (48%, 70 mg). 1H NMR (400 MHz, CdCl3): δ 7.95 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 8.4 Hz, 2H), 4.80 (s, 1H), 3.27 (q, J = 6.4 Hz, 2H), 2.97 (d, J = 7.5 Hz, 2H), 2.64 (t, J = 6.9 Hz, 2H), 2.06 (p, J = 7.0 Hz, 2H), 1.68–1.58 (m, 2H), 1.43 (s, 9H), 1.36–1.15 (m, 10H), 0.85 (t, J = 5.5 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.1, 168.2, 155.9, 146.4, 128.8, 127.3, 124.1, 79.3, 39.7, 35.9, 31.8, 31.1, 29.4, 29.2, 29.2, 28.3, 26.9, 24.1, 22.6, 14.0. HRMS (ESi): [M + H]+ calcd for C24H38N3O3, 416.2908; observed, 416.2890.
tert-Butyl (3-(3-(4-Nonylphenyl)-1,2,4-oxadiazol-5-yl)propyl)-carbamate (56d).
Synthesized according to General Procedure 1. Purified by silica chromatography (25% ethyl acetate/hexanes). White solid (67%, 100 mg). 1H NMR (400 MHz, CdCl3): δ 7.96 (d, J = 8.5 Hz, 2H), 7.27 (d, J = 8.5 Hz, 2H), 4.79 (s, 1H), 3.27 (q, J = 6.5 Hz, 2H), 2.98 (t, J = 7.5 Hz, 2H), 2.64 (t, J = 7.8 Hz, 2H), 2.06 (p, J = 7.0 Hz, 2H), 1.68–1.56 (m, 2H), 1.43 (s, 9H), 1.36–1.17 (m, 12H), 0.86 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.0, 168.9, 155.9, 146.4, 128.8, 127.3, 124.1, 79.3, 39.7, 35.3, 31.8, 31.2, 29.5, 29.4, 29.2, 29.2, 28.3, 26.9, 24.0, 22.6, 14.0. HRMS (ESI): [M + H]+ calcd for C25H40N3O3, 430.3064; observed, 430.3050.
tert-Butyl (3-(3-(4-Undecylphenyl)-1,2,4-oxadiazol-5-yl)propyl)-carbamate (56e).
Synthesized according to General Procedure 1. Purified by silica chromatography (20% ethyl acetate/hexanes). White solid (59%, 100 mg). 1H NMR (400 MHz, CdCl3): δ 7.95 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 8.4 Hz, 2H), 4.78 (s, 1H), 3.27 (q, J = 6.5 Hz, 2H), 2.98 (t, J = 7.5 Hz, 2H), 2.64 (t, J = 8.7 Hz, 2H), 2.06 (p, J =7.1 Hz, 2H), 1.62 (p, J = 7.4 Hz, 2H), 1.43 (s, 9H), 1.36–1.18 (m, 16H), 0.87 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.0, 168.2, 155.9, 146.4, 128.8, 127.3, 124.1, 79.5, 39.7, 35.9, 31.8, 31.9, 29.6, 29.5, 29.4, 29.3, 29.2, 28.5, 26.9, 24.0, 22.6, 14.0. HRMS (ESI): [M + H]+ calcd for C27H44N3O3, 458.3377; observed, 458.3359.
tert-Butyl (3-(3-(3-Decylphenyl)-1,2,4-oxadiazol-5-yl)propyl)-carbamate (56f).
The compound was synthesized according to General Procedure 1. The crude mixture was passed through a silica plug (25% ethyl acetate/hexane eluent) and concentrated under reduced pressure. The desired product was confirmed via HRMS and carried forward as crude with no further purification. Yellow oil (60%, 199 mg). HRMS (ESI): [M + H]+ calcd for C26H42N3O3, 444.3221; observed, 444.3205.
3-(3-(4-Hexylphenyl)-1,2,4-oxadiazol-5-yl)propan-1-amine Hydrochloride (57a).
Synthesized according to General Procedure 4. White solid (30%, 50 mg). 1H NMR (400 MHz, CD3OD): δ 7.95 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.3 Hz, 2H), 3.18–3.09 (m, 4H), 2.69 (t, J = 7.7 Hz, 2H), 2.24 (p, J = 7.4 Hz, 2H), 1.66 (p, J = 7.6 Hz, 2H), 1.42–1.25 (m, 6H), 0.90 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 180.1, 169.5, 148.1, 130.1, 128.3, 125.4, 39.9, 36.8, 32.8, 32.4, 30.0, 25.2, 24.3, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C17H26N3O, 288.2070; observed, 288.2125.
3-(3-(4-Heptylphenyl)-1,2,4-oxadiazol-5-yl)propan-1-amine Hydrochloride (57b).
Synthesized according to General Procedure 4. White solid (27%, 70 mg). 1H NMR (400 MHz, CD3OD): δ 7.95 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.2 Hz, 2H), 3.18–3.09 (m, 4H), 2.69 (t, J = 7.7 Hz, 2H), 2.24 (p, J = 7.4 Hz, 2H), 1.65 (p, J = 6.9 Hz, 2H), 1.41– 1.23 (m, 8H), 0.90 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 180.2, 169.5, 148.1, 130.1, 128.3, 125.4, 39.9, 36.8, 33.0, 32.5, 30.3, 30.3, 25.2, 24.3, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C18H28N3O, 302.2227; observed, 302.2230.
3-(3-(4-Octylphenyl)-1,2,4-oxadiazol-5-yl)propan-1-amine Hydrochloride (57c).
Synthesized according to General Procedure 4. White solid (74%, 25 mg). 1H NMR (400 MHz, CD3OD): δ 7.94 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 8.2 Hz, 2H), 3.17–3.08 (m, 4H), 2.67 (t, J = 8.6 Hz, 2H), 2.29–2.17 (m, 2H), 1.70–1.58 (m, 2H), 1.38–1.22 (m, 10H), 0.88 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 178.7, 168.0, 146.6, 128.6, 126.8, 123.9, 38.4, 35.4, 31.5, 31.0, 29.0, 28.9, 28.8, 23.7, 22.8, 22.2, 12.9. HRMS (ESI): [M + H]+ calcd for C19H30N3O, 316.2383; observed, 316.2368.
3-(3-(4-Nonylphenyl)-1,2,4-oxadiazol-5-yl)propan-1-amine Hydrochloride (57d).
Synthesized according to General Procedure 4. White solid (52%, 40 mg). 1H NMR (400 MHz, CD3OD): δ 7.95 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.2 Hz, 2H), 3.18–3.09 (m, 4H), 2.69 (t, J = 7.7 Hz, 2H), 2.24 (p, J = 7.4 Hz, 2H), 1.66 (p, J = 7.3 Hz, 2H), 1.41– 1.24 (m, 12H), 0.89 (t, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 178.7, 168.0, 146.6, 128.6, 126.8, 123.9, 38.4, 35.4, 31.5, 31.1, 29.2, 29.1, 28.9, 28.8, 23.8, 22.3, 22.8, 12.9. HRMS (ESI): [M + H]+ calcd for C20H32N3O, 330.2540; observed, 330.2529.
3-(3-(4-Undecylphenyl)-1,2,4-oxadiazol-5-yl)propan-1-amine Hydrochloride (57e).
Synthesized according to General Procedure 4. White solid (58%, 50 mg). 1H NMR (400 MHz, CD3OD): δ 7.94 (d, J = 8.6 Hz, 2H), 7.32 (d, J = 8.6 Hz, 2H), 3.17–3.08 (m, 4H), 2.67 (t, J = 8.6 Hz, 2H), 2.29–2.17 (m, 2H), 1.71–1.58 (m, 2H), 1.42–1.19 (m, 16H), 0.87 (t, J = 6.1 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 180.2, 169.5, 148.1, 130.1, 128.3, 125.4, 39.9, 36.8, 33.1, 32.4, 30.7, 30.7, 30.5, 30.5, 30.3, 25.2, 24.3, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C22H36N3O, 358.2853; observed, 358.2839.
3-(3-(3-Decylphenyl)-1,2,4-oxadiazol-5-yl)propan-1-amine Hydrochloride (57f).
Synthesized according to General Procedure 4. White solid (53%, 90 mg). 1H NMR (400 MHz, CD3OD): δ 7.87–7.81 (m, 2H), 7.38 (t, J = 7.6 Hz, 1H), 7.32 (d, J = 7.6 Hz, 1H), 3.21–3.09 (m, 4H), 2.64 (t, J = 7.7 Hz, 2H), 2.27 (p, J = 7.5 Hz, 2H), 1.62 (p, J = 7.2 Hz, 2H), 1.39–1.17 (m, 14H), 0.87 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 180.2, 169.5, 144.9, 132.5, 129.9, 128.1, 127.8, 125.7, 39.8, 36.7, 33.0, 32.6, 30.7, 30.6, 30.4, 30.3, 28.8, 25.1, 24.3, 23.7, 14.5. HRMS (ESI): [M + H]+ calcd for C21H34N3O, 344.2696; observed, 344.2685.
4-(Nonyloxy)benzonitrile (59a).
Synthesized according to General Procedure 8. Purified via column chromatography (5% ethyl acetate/hexanes). White solid (80%, 827 mg). 1H NMR (400 MHz, CdCl3): δ 7.57 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 3.99 (t, J = 6.5 Hz, 2H), 1.79 (p, J = 6.6 Hz, 2H), 1.44 (p, J = 7.7 Hz, 2H), 1.38–1.24 (m, 10H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 162.6, 134.1, 119.5, 115.3, 103.8, 68.6, 32.0, 29.6, 29.5, 29.4, 29.1, 26.1, 22.8, 14.2.
3-Fluoro-4-(nonyloxy)benzonitrile (59b).
Synthesized according to General Procedure 8. Purified via column chromatography (7% ethyl acetate/hexanes). White solid (88%, 846 mg). 1H NMR (400 MHz, CdCl3): δ 7.43–7.32 (m, 2H), 6.99 (t, J = 8.3 Hz, 1H), 4.07 (t, J = 6.6 Hz, 2H), 1.84 (p, J = 6.7 Hz, 2H), 1.45 (p, J = 7.3 Hz, 2H), 1.39–1.24 (m, 10H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 152.0 (d, J = 249.9 Hz), 151.6 (d, J = 10.3 Hz), 129.7 (d, J = 3.8 Hz), 119.7 (d, J = 21.4 Hz), 118.3 (d, J =2.6 Hz), 114.5 (d, J = 2.4 Hz), 103.7 (d, J = 8.3 Hz), 69.7, 32.0, 29.6, 29.4, 29.3, 29.0, 25.9, 22.8, 14.2.
4-(Nonyloxy)-3-(trifluoromethyl)benzonitrile (59c).
Synthesized according to General Procedure 8. Purified via column chromatography (7% ethyl acetate/hexanes). White solid (77%, 625 mg). 1H NMR (400 MHz, CdCl3): δ 7.85 (d, J = 1.9 Hz, 1H), 7.77 (dd, J = 8.7, 2.1 Hz, 1H), 7.05 (d, J = 8.7 Hz, 1H), 4.11 (t, J = 6.3 Hz, 2H), 1.84 (p, J = 6.4 Hz, 2H), 1.47 (p, J = 7.0 Hz, 2H), 1.39–1.23 (m, 10H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 160.4, 137.6, 131.5 (q, J = 5.4 Hz), 122.6 (q, J = 273.0 Hz), 120.3 (q, J = 32.1 Hz), 118.1, 113.5, 103.6, 69.6, 32.0, 29.6, 29.3, 29.3, 28.8, 25.8, 22.8, 14.2.
6-(Heptyloxy)-2-naphthonitrile (59d).
Synthesized according to General Procedure 8. Purified via column chromatography (5% ethyl acetate/hexanes). White solid (99%, 1.21 g). 1H NMR (500 MHz, CdCl3): δ 8.12 (d, J =1.6 Hz, 1H), 7.80–7.73 (m, 2H), 7.55 (dd, J = 8.5, 1.7 Hz, 1H), 7.24 (dd, J = 9.0, 2.5 Hz, 1H), 7.13 (d, J = 2.4 Hz, 1H), 4.09 (t, J = 6.6 Hz, 2H), 1.86 (p, J =6.7 Hz, 2H), 1.50 (p, J = 7.1 Hz, 2H), 1.43–1.27 (m, 6H), 0.94–0.87 (m, 3H). 13C NMR (126 MHz, CdCl3): δ 159.7, 136.6, 133.9, 130.0, 127.9, 127.7, 127.1, 121.1, 106.7, 68.4, 31.9, 29.2, 29.2, 26.2, 22.8, 14.2. HRMS (ESI): [M + NH4]+ calcd for C18H25N2O, 285.1961; observed, 285.1962.
N′-Hydroxy-4-(nonyloxy)benzimidamide (60a).
Synthesized according to General Procedure 2. Purified via column chromatography (40% ethyl acetate/hexanes). White solid (35%, 320 mg). 1H NMR (400 MHz, CdCl3): δ 8.76 (s, 1H), 7.64 (d, J = 8.9 Hz, 2H), 6.91 (d, J = 8.8 Hz, 2H), 5.38 (s, 2H), 4.01 (t, J = 6.5 Hz, 2H), 1.82–1.72 (m, 2H), 1.53–1.43 (m, 2H), 1.42–1.22 (m, 10H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 160.9, 152.0, 127.6, 126.8, 114.8, 68.6, 32.6, 30.3, 30.1, 30.0, 30.0, 26.8, 23.3, 14.4. HRMS (ESI): [M + H]+ calcd for C16H27N2O2, 279.2073; observed, 279.2061.
3-Fluoro-N′-hydroxy-4-(nonyloxy)benzimidamide (60b).
The compound was synthesized according to General Procedure 2. The crude mixture was concentrated under reduced pressure, redissolved in ethyl acetate, and partitioned with brine. The aqueous layer was rinsed three times with ethyl acetate. The organic layers were combined, rinsed further three times with brine, dried over sodium sulfate, and concentrated in vacuo. The product was carried forward as crude with no further purification. White solid (78%, 727 mg).
N′-Hydroxy-4-(nonyloxy)-3-(trifluoromethyl)benzimidamide (60c).
Synthesized according to General Procedure 2. Purified via column chromatography (40% ethyl acetate/hexanes).White solid (89%, 616 mg). 1H NMR (400 MHz, (CD3)2CO): δ 9.09 (s, 1H), 8.1 (d, J = 2.3 Hz, 1H), 7.92 (dd, J = 8.7, 2.0 Hz, 1H), 7.22 (d, J = 8.72 Hz, 1H), 5.60 (s, 2H), 4.21 (t, J = 6.3 Hz, 2H), 1.94–1.78 (m, 2H), 1.61–1.50 (m, 2H), 1.39–1.22 (m, 10H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, (CD3)2CO): δ 157.6 (q, J = 1.6 Hz), 150.1, 130.7, 125.5, 124.0 (q, J = 5.4 Hz), 123.9 (q, J = 271.7 Hz), 117.7 (q, J = 30.6 Hz), 112.9, 68.7, 31.7, 29.0, 28.8, 25.6, 22.4, 13.4. HRMS (ESI): [M + H]+ calcd for C17H26N2OF3, 347.1946; observed, 347.1944.
6-(Heptyloxy)-N′-hydroxy-2-naphthimidamide (60d).
Synthesized according to General Procedure 2. Purified via silica plug (dichloromethane wash, followed by elution with 100% ethyl acetate). Gray solid (93%, 2.1 g). 1H NMR (400 MHz, (CD3)2CO): δ 8.98 (s, 1H), 8.13 (d, J = 1.7 Hz, 1H), 7.85 (dd, J = 8.7, 1.8 Hz, 1H), 7.81 (d, J = 8.9 Hz, 1H), 7.74 (d, J = 8.7 Hz, 1H), 7.29 (d, J = 8.7 Hz, 1H), 7.16 (dd, J = 8.9, 2.5 Hz, 1H), 5.56 (s, 2H), 4.12 (t, J = 6.5 Hz, 2H), 1.89–1.79 (m, 2H), 1.57–1.47 (m, 2H), 1.45–1.28 (m, 6H), 0.90 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, (CD3)2CO): δ 158.6, 152.1, 136.1, 130.6, 129.6, 129.3, 127.3, 125.0, 124.8, 120.1, 107.4, 69.6, 32.6, 30.0, 29.8, 26.8, 23.3, 14.4. HRMS (ESI): [M + H]+ calcd for C18H25N2O2, 301.1911; observed, 301.1912.
tert-Butyl (3-(3-(4-(Nonyloxy)phenyl)-1,2,4-oxadiazol-5-yl)-propyl)carbamate (61a).
Synthesized according to General Procedure 3. Purified via column chromatography (15% ethyl acetate/hexanes). White solid (79%, 316 mg). 1H NMR (400 MHz, CdCl3): δ 7.98 (d, J = 9.0 Hz, 2H), 6.96 (d, J = 9.0 Hz, 2H), 4.81 (s, 1H), 4.00 (t, J = 6.6 Hz, 2H), 3.28 (q, J = 6.1 Hz, 2H), 2.97 (t, J = 7.4 Hz, 2H), 2.6 (p, J = 7.0 Hz, 2H), 1.88–1.71 (m, 2H), 1.50–1.41 (m, 11H), 1.36–1.26 (m, 10H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.1, 168.2, 161.7, 156.1, 129.1, 119.1, 114.9, 79.6, 68.3, 39.9, 32.0, 29.7, 29.5, 29.4, 29.3, 28.5, 27.1, 26.2, 24.2, 22.8, 14.2. HRMS (ESI): [M + H]+ calcd for C25H40N3O4, 446.3013; observed, 446.3004.
tert-Butyl (3-(3-(3-Fluoro-4-(nonyloxy)phenyl)-1,2,4-oxadiazol-5-yl)propyl)carbamate (61b).
Synthesized according to General Procedure 3. Purified via column chromatography (20% ethyl acetate/hexanes). Yellow solid (87%, 360 mg). 1H NMR (400 MHz, CD3OD): δ 7.82–7.75 (m, 2H), 7.02 (t, J = 8.6 Hz, 1H), 4.79 (s, 1H), 4.08 (t, J = 6.6 Hz, 2H), 3.28 (q, J = 6.1 Hz, 2H), 2.98 (t, J = 7.4 Hz, 2H), 2.07 (p, J = 7.0 Hz, 2H), 1.85 (p, J = 6.7 Hz, 2H), 1.52–1.42 (s, 11H), 1.40–1.24 (m, 10H), 0.88 (t, 3H). 13C NMR (101 MHz, CdCl3): δ 179.4, 167.5, 156.1, 152.6 (d, J = 246.7 Hz), 149.8 (d, J = 10.7 Hz), 124.0 (d, J = 3.6 Hz), 119.5 (d, J = 7.4 Hz), 115.4 (d, J = 20.7 Hz), 114.5 (d, J = 2.3 Hz), 79.6, 69.5, 39.9, 32.0, 29.6, 29.5, 29.4, 29.2, 28.5, 27.0, 26.0, 24.2, 22.8, 14.2. HRMS (ESI): [M + H]+ calcd for C23H39FN3O4, 464.2919; observed, 464.2910.
tert-Butyl (3-(3-(4-(Nonyloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)propyl)carbamate (61c).
Synthesized according to General Procedure 3. Purified via column chromatography (20% ethyl acetate/hexanes). Yellow solid (57%, 260 mg). 1H NMR (400 MHz, CdCl3): δ 1H NMR (400 MHz, CdCl3): δ 8.27 (d, J = 1.9 Hz, 1H), 8.16 (dd, J = 8.7, 2.0 Hz, 1H), 7.05 (d, J = 8.7 Hz, 1H), 4.74 (s, 1H), 4.10 (t, J = 6.4 Hz, 2H), 3.28 (q, J = 6.1 Hz, 2H), 2.98 (t, J = 7.5 Hz, 2H), 2.07 (p, J = 7.0 Hz, 2H), 1.83 (p, J = 6.4 Hz, 2H), 1.52–1.41 (m, 11H), 1.36–1.24 (m, 10H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.6, 167.4, 159.3, 156.1, 132.5, 126.8 (q, J = 5.3 Hz), 123.4 (q, J = 272.6 Hz), 119.6 (q, J = 31.4 Hz), 118.7, 113.0, 79.6, 69.2, 39.9, 32.0, 29.6, 29.3, 29.3, 29.0, 28.5, 27.1, 25.9, 24.1, 22.8, 14.2. HRMS (ESI): [M + H]+ calcd for C26H39F3N3O4, 514.2887; observed, 514.2881.
tert-Butyl (3-(3-(6-(Heptyloxy)naphthalen-2-yl)-1,2,4-oxadiazol-5-yl)propyl)carbamate (61d).
Synthesized according to General Procedure 3. Purified via column chromatography (20% ethyl acetate/hexanes). White solid (68%, 265 mg). 1H NMR (400 MHz, CdCl3): δ 8.49 (s, 1H), 8.06 (dd, J = 8.6, 1.7 Hz, 1H), 7.82 (d, J = 9.0 Hz, 1H), 7.78 (d, J = 8.6 Hz, 1H), 7.18 (dd, J = 8.9, 2.5 Hz, 1H), 7.14 (d, J = 2.4 Hz, 1H), 4.82 (s, 1H), 4.08 (t, J = 6.6 Hz, 2H), 3.35–3.23 (m, 2H), 3.01 (t, J = 7.5 Hz, 2H), 2.10 (p, J =7.1 Hz, 2H), 1.85 (p, J = 6.7 Hz, 2H), 1.55–1.25 (m, 17H), 0.90 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.3, 168.6, 158.6, 156.1, 136.2, 130.4, 128.4, 127.8, 127.5, 124.4, 121.8, 120.0, 106.7, 79.5, 68.2, 39.9, 31.9, 29.3, 29.2, 28.5, 27.1, 26.2, 24.2, 22.7, 14.2. HRMS (ESI): [M + H]+ calcd for C27H38N3O4, 468.2857; observed, 468.2857.
3-(3-(4-(Nonyloxy)phenyl)-1,2,4-oxadiazol-5-yl)propan-1-amine Hydrochloride (62a).
Synthesized according to General Procedure 4. White solid (88%, 226 mg). 1H NMR (400 MHz, CD3OD): δ 7.96 (d, J = 9.0 Hz, 2H), 7.03 (d, J = 9.0 Hz, 2H), 4.04 (t, J = 6.4 Hz, 2H), 3.17–3.08 (m, 4H), 2.23 (p, J = 7.4 Hz, 2H), 1.79 (p, J = 6.5 Hz, 2H), 1.49 (p, J = 6.7 Hz, 2H), 1.44–1.23 (m, 10H), 0.90 (t, J = 6.5 Hz, 2H). 13C NMR (101 MHz, CD3OD): δ 180.0, 169.3, 163.2, 129.9, 120.0, 115.9, 69.3, 39.9, 33.0, 30.7, 30.5, 30.4, 30.3, 27.1, 25.2, 24.3, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C20H32N3O2, 346.2489; observed, 346.2480.
3-(3-(3-Fluoro-4-(nonyloxy)phenyl)-1,2,4-oxadiazol-5-yl)propan-1-amine Hydrochloride (62b).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (79%, 236 mg). 1H NMR (400 MHz, CD3OD): δ 7.8 (dt, J = 8.6, 1.5 Hz, 1H), 7.7 (dd, J = 11.9,2.0 Hz, 1H), 7.2 (t, J = 8.5 Hz, 1H), 4.1 (t, J = 6.4 Hz, 2H), 3.1 (q, J = 7.4 Hz, 2H), 2.2 (p, J = 7.4 Hz, 2H), 1.9 1.7 (m, 2H), 1.6–1.4 (m, 2H), 1.4–1.2 (m, 10H), 0.9 (d, J = 6.8 Hz, 3H).13C NMR (101 MHz, CD3OD): δ 180.3, 168.6 (d, J = 2.6 Hz), 153.7 (d, J = 245.8 Hz), 151.2 (d, J = 10.7 Hz), 125.1 (d, J = 3.7 Hz), 120.5 (d, J = 7.4 Hz), 115.8, 115.7 (d, J = 23.9 Hz), 70.4, 39.9, 33.0, 30.6, 30.4, 30.4, 30.2, 27.0, 25.2, 24.3, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C20H31FN3O2, 364.2395; observed, 364.2387.
3-(3-(4-(Nonyloxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)propan-1-amine Hydrochloride (62c).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (72%, 155 mg). 1H NMR (400 MHz, CD3OD): δ 8.25–8.20 (m, 2H), 7.30 (d, J = 9.2 Hz, 1H), 4.17 (t, J = 6.2 Hz, 2H), 3.22–3.11 (m, 4H), 2.26 (p, J = 7.5 Hz, 2H), 1.83 (p, J = 6.3 Hz, 2H), 1.52 (p, J = 7.0 Hz, 2H), 1.41–1.26 (m, 10H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 180.5, 168.4, 160.7 (q, J = 1.6 Hz), 133.8, 127.0 (q, J = 5.5 Hz), 124.8 (q, J = 271.8 Hz), 120.2 (q, J = 31.3 Hz), 119.8, 114.8, 70.3, 39.8, 33.0, 30.6, 30.3, 30.3, 30.0, 26.9, 25.1, 24.3, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C21H31F3N3O2, 414.2363; observed, 414.2355.
3-(3-(6-(Heptyloxy)naphthalen-2-yl)-1,2,4-oxadiazol-5-yl)-propan-1-amine Hydrochloride (62d).
Synthesized according to General Procedure 4. White solid (85%, 166 mg). 1H NMR (400 MHz, CD3OD): δ 8.50 (d, J = 1.6 Hz, 1H), 8.03 (dd, J = 8.6 Hz, 1H), 7.88 (d, J = 3.5 Hz, 1H), 7.86 (d, J = 3.8 Hz, 1H), 7.29 (d, J = 2.4 Hz, 1H), 7.21 (dd, J = 9.0 Hz, 2.5 Hz, 1H), 4.13 (t, J = 6.4 Hz, 2H), 3.21–3.11 (m, 4H), 2.27 (p, J = 7.4 Hz, 2H), 1.86 (p, J = 6.7 Hz, 2H), 1.54 (p, J = 6.9 Hz, 2H), 1.47–1.31 (m, 6H), 0.93 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 180.2, 169.7, 160.1, 137.8, 131.2, 129.8, 128.7, 128.6, 125.0, 122.8, 121.1, 107.7, 69.2, 39.9, 33.0, 30.4, 30.2, 27.2, 25.3, 24.3, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C21H31F3N3O2, 414.2363; observed, 414.2355.
4-Cyano-N-octylbenzamide (64).
Synthesized according to General Procedure 9. Purified by silica chromatography (30% ethyl acetate in hexanes). Orange solid (94%, 822 mg). 1H NMR (400 MHz, CdCl3): δ 7.85 (d, J = 8.3 Hz, 2H), 7.72 (d, J = 8.3 Hz, 2H), 6.29 (t, J = 5.9 Hz, 1H), 3.44 (q, J = 7.3 Hz, 2H), 1.61 (p, J = 7.4 Hz, 2H), 1.40–1.22 (m, 10H), 0.86 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 165.8, 138.9, 132.5, 127.7, 118.2, 115.0, 40.5, 31.9, 29.6, 29.4, 29.3, 27.1, 22.8, 14.2. HRMS (ESI): [M + H]+ calcd for C16H23N2O, 259.1805; observed, 259.1804.
4-(N′-Hydroxycarbamimidoyl)-N-octylbenzamide (65).
Synthesized according to General Procedure 2. Purified by silica chromatography (90% ethyl acetate in hexanes). White solid (52%, 555 mg). 1H NMR (400 MHz, CD3OD): δ 7.87 (d, J = 8.3 Hz, 2H), 7.77 (d, J = 8.5 Hz, 2H), 3.41 (t, J = 7.2 Hz, 2H), 1.66 (p, J = 7.1 Hz, 2H), 1.47–1.30 (m, 10H), 0.93 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 169.5, 162.6, 137.2, 136.8, 128.3, 127.3, 41.1, 33.0, 30.5, 30.4, 30.4, 28.1, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C16H26N3O2, 292.2020; observed, 292.2030.
tert-Butyl (3-(3-(4-(Octylcarbamoyl)phenyl)-1,2,4-oxadiazol-5-yl)propyl)carbamate (66).
Synthesized according to General Procedure 3. Purified by silica chromatography (60% ethyl acetate in hexanes). White solid (48%, 188 mg). 1H NMR (400 MHz, CdCl3): δ 8.04 (d, J = 8.0 Hz, 2H), 7.83 (d, J = 8.0 Hz, 2H), 6.68 (t, J = 5.6 Hz, 1H), 4.97 (s, 1H), 3.40 (q, J = 6.8 Hz, 2H), 3.24 (q, J = 6.5 Hz, 2H), 2.95 (t, J = 7.4 Hz, 2H), 2.03 (p, J = 7.0 Hz, 2H), 1.57 (p, J = 7.3 Hz, 2H), 1.39 (s, 9H), 1.36–1.17 (m, 10H), 0.83 (t, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 179.7, 167.6, 166.9, 156.1, 137.2, 129.4, 127.5, 127.5, 79.4, 40.3, 39.8, 31.8, 29.7, 29.3, 29.3, 28.4, 27.1, 26.9, 24.1, 22.7, 14.1. HRMS (ESI): [M + H]+ calcd for C25H39N4O4, 459.2966; observed, 459.2962.
4-(5-(3-Aminopropyl)-1,2,4-oxadiazol-3-yl)-N-octylbenzamide Hydrochloride (67).
Synthesized according to General Procedure 4. Purified via trituration with diethyl ether. White solid (96%, 155 mg). 1H NMR (400 MHz, CD3OD): δ 8.15 (d, J = 8.4 Hz, 2H), 7.95 (d, J = 8.4 Hz, 2H), 3.40 (t, J = 7.2 Hz, 2H), 3.19–3.13 (m, 4H), 2.26 (p, J = 7.5 Hz, 2H), 1.64 (p, J = 7.2 Hz, 2H), 1.44–1.28 (m, 10H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 179.2, 167.7, 167.5, 137.1, 129.3, 127.5, 126.9, 39.7, 38.4, 31.6, 29.0, 29.0, 28.9, 26.7, 23.8, 22.8, 22.3, 13.0. HRMS (ESI): [M + H]+ calcd for C20H31N4O2, 359.2442; observed, 359.2432.
N-(4-Cyanophenyl)nonanamide (69a).
Synthesized according to General Procedure 9. Purified via column chromatography (20% ethyl acetate/hexanes). White solid (84%, 551 mg). 1H NMR (400 MHz, CdCl3): δ 7.67 (d, J = 8.5 Hz, 2H), 7.59 (d, J = 8.7 Hz, 2H), 7.51 (br s, 1H), 2.39 (t, J = 7.6 Hz, 2H), 1.72 (p, J = 7.4 Hz, 2H), 1.40–1.20 (m, 10H), 0.88 (d, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CdCl3): δ 171.9, 142.2, 133.4, 119.6, 119.0, 107.0, 38.0, 31.9, 29.4, 29.4, 29.2, 25.5, 22.8, 14.2. HRMS (ESI): [M + H]+ calcd for C16H23N2O, 259.1805; observed, 259.1802.
N-(4-Cyanobenzyl)octanamide (69b).
Synthesized according to General Procedure 9. Purified via column chromatography (60% ethyl acetate/hexanes). White solid (89%, 873 mg). 1H NMR (400 MHz, CD3OD): δ 7.68 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 8.3 Hz, 2H), 4.42 (s, 2H), 2.25 (t, J = 7.5 Hz, 2H), 1.63 (p, J = 7.5 Hz, 2H), 1.37–1.23 (m, 8H), 0.90 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 176.4, 146.2, 133.4, 129.3, 119.7, 111.9, 43.6, 37.0, 32.9, 30.3, 30.1, 27.0, 23.6, 14.4. HRMS (ESI): [M + H]+ calcd for C16H23N2O, 259.1805; observed, 259.1798.
N-(4-(N′-Hydroxycarbamimidoyl)phenyl)nonanamide (70a).
The compound was synthesized according to General Procedure 2. The crude mixture was concentrated under reduced pressure, redissolved in ethyl acetate, and partitioned with brine. The aqueous layer was rinsed three times with ethyl acetate. The organic layers were combined, rinsed further three times with brine, dried over sodium sulfate, and concentrated in vacuo. The desired product was confirmed via HRMS and carried forward as crude with no further purification. White solid (88%, 545 mg). HRMS (ESI): [M + H]+ calcd for C16H26N3O2, 292.2020; observed, 292.2003.
N-(4-(N′-Hydroxycarbamimidoyl)benzyl)octanamide (70b).
The compound was synthesized according to General Procedure 2. The crude mixture concentrated under reduced pressure, redissolved in ethyl acetate, and partitioned with brine. The aqueous layer was rinsed three times with ethyl acetate. The organic layers were combined, rinsed further three times with brine, dried over sodium sulfate, and concentrated in vacuo. The desired product was confirmed via HRMS and carried forward as crude with no further purification. White solid (95%, 937 mg). HRMS (ESI): [M + H]+ calcd for C16H26N3O2, 292.2020; observed, 292.2010.
tert-Butyl (3-(3-(4-Nonanamidophenyl)-1,2,4-oxadiazol-5-yl)-propyl)carbamate (71a).
Synthesized according to General Procedure 3. Purified via column chromatography (60% ethyl acetate/hexanes). White solid (22%, 191 mg). 1H NMR (400 MHz, CdCl3): δ 7.98 (d, J = 8.7 Hz, 2H), 7.87 (s, 1H), 7.65 (d, J = 8.4 Hz, 2H), 4.92 (t, J = 7.7 Hz, 1H), 3.26 (q, J = 6.6 Hz, 2H), 2.95 (t, J = 7.4 Hz, 2H), 2.37 (t, J = 7.6 Hz, 2H), 2.05 (p, J = 7.1 Hz, 2H), 1.70 (p, J = 7.5 Hz, 2H), 1.42 (s, 9H), 1.37–1.19 (m, 10H), 0.89–0.81 (m, 3H). 13C NMR (101 MHz, CdCl3): δ 179.3, 172.1, 167.9, 156.2, 140.9, 128.4, 122.3, 119.7, 79.6, 39.9, 37.9, 31.9, 29.4, 29.4, 29.2, 28.5, 27.0, 25.7, 24.1, 22.7, 14.2. HRMS (ESI): [M + H]+ calcd for C25H39N4O4, 459.2966; observed, 459.2966.
tert-Butyl (3-(3-(4-(Octanamidomethyl)phenyl)-1,2,4-oxadiazol-5-yl)propyl)carbamate (71b).
Synthesized according to General Procedure 3. Purified via column chromatography (40–60% ethyl acetate/hexanes). White solid (77%, 1129 mg). 1H NMR (400 MHz, CD3OD): δ 8.00 (d, J = 8.4 Hz, 2H), 7.42 (d, J = 8.3 Hz, 2H), 4.43 (s, 2H), 3.20 (t, J = 6.7 Hz, 2H), 2.99 (t, J = 7.5 Hz, 2H), 2.26 (t, J = 7.4 Hz, 2H), 2.04 (p, J = 7.2 Hz, 2H), 1.65 (p, J = 7.5 Hz, 2H), 1.42 (s, 9H), 1.37–1.23 (m, 8H), 0.89 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 181.4, 176.3, 169.2, 158.5, 143.8, 129.0, 128.5, 126.9, 80.1, 43.7, 40.5, 37.0, 32.9, 30.2, 30.1, 28.7, 27.7, 27.0, 24.7, 23.6, 14.4. HRMS (ESI): [M + Na]+ calcd for C25H38N4NaO4, 481.2785; observed, 481.2785.
N-(4-(5-(3-Aminopropyl)-1,2,4-oxadiazol-3-yl)phenyl)-nonanamide Hydrochloride (72a).
Synthesized according to General Procedure 4. Purified via trituration with ethyl acetate and diethyl ether. White solid (68%, 111 mg). 1H NMR (400 MHz, CD3OD): δ 7.98 (d, J = 8.8 Hz, 2H), 7.73 (d, J = 8.7 Hz, 2H), 3.16–3.07 (m, 4H), 2.40 (t, J = 7.5 Hz, 2H), 2.23 (p, J = 7.4 Hz, 2H), 1.71 (p, J = 7.4 Hz, 2H), 1.44–1.25 (m, 10H), 0.89 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 180.3, 174.9, 169.1, 142.9, 129.0, 123.1, 120.9, 40.1, 38.1, 32.9, 30.4, 30.3, 30.3, 26.8 25.7, 24.3, 23.7, 14.4. HRMS (ESI): [M + H]+ calcd for C20H31N4O2, 359.2442; observed, 359.2444.
N-(4-(5-(3-Aminopropyl)-1,2,4-oxadiazol-3-yl)benzyl)-octanamide Hydrochloride (72b).
Synthesized according to General Procedure 4. Purified via trituration with ethyl acetate and diethyl ether. White solid (80%, 782 mg). 1H NMR (400 MHz, CD3OD): δ 8.00 (d, J = 8.4 Hz, 2H), 7.43 (d, J = 8.4 Hz, 2H), 4.42 (s, 2H), 3.18–3.09 (m, 4H), 2.30–2.18 (m, 4H), 1.63 (p, J = 7.3 Hz, 2H), 1.36–1.23 (m, 8H), 0.89 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CD3OD): δ 180.3, 176.3, 169.3, 144.0, 129.1, 128.5, 126.8, 43.7, 39.9, 37.1, 32.9, 30.2, 30.1, 27.0, 25.2, 24.3, 23.6, 14.4. HRMS (ESI): [M + H]+ calcd for C20H31N4O2, 359.2442; observed, 359.2446.
Sphingosine Kinase Assays.
Recombinant baculovirus encoding either mSphK1 or mSphK2 was used to infect Sf9 insect cells, cleared lysates were prepared after 48 h, and 1–2 μL (0.02–0.03 mg protein) was used in each assay. Alternately, plasmids encoding mouse SphK1 or SphK2 were used to transfect HEK293T cells and cleared lysates were prepared after 48 h. SphK activity was measured in kinase assay buffer that consisted of 20 mM Tris–Cl (pH 7.4), 1 mM 2-mercaptoethanol, 1 mM EDTA, 5 mM sodium orthovanadate, 40 mM β-glycerophosphate, 15 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 10 mM MgCl2, 0.5 mM 4-deoxypyridoxine, 10% glycerol, and 0.01 mg/mL each leupeptin, aprotinin, and soybean trypsin inhibitor. To achieve optimal activity of SphK1 or SphK2, the buffer was supplemented with either 0.5% Triton X-100 or 1 M KCl, respectively. To ascertain any inhibitory effect of 16d, the assay was supplemented with the substrate (d-erythro-sphingosine, 10 μM for SphK1 and 5 μM for SphK2), appropriate amount of compounds [(to achieve 10–30,000 nM); γ-[32P]ATP (10 μM, specific activity = 8.3 Ci/mmol)], and recombinant enzyme (0.02–0.03 mg of total protein). After 20 min at 37 °C, the reaction mixture was extracted with 2 volumes of chloroform/methanol/1 N HCl (100:200:1), and the components in the organic phase were separated by TLC using a 1-butanol/glacial acetic acid/water (3:1:1) solvent system. Radio-labeled enzyme products were detected by autoradiography and identified by migration relative to authentic standards. For quantification, the silica gel containing radiolabeled lipid was scraped into a scintillation vial and measured by liquid scintillation counting.
In Vitro Spns2 Assay.
HeLa cells were transfected with pcDNA3.1 plasmids encoding mouse Spns2 or mouse Spns2Arg200Ser (transport dead mutant). Transfected cell pools were selected by inclusion of geneticin (G418) in the cell media. To assess S1P release, HeLa cells were plated onto 12-well plates and grown to near confluence. Growth media were removed by aspiration and 1.5 mL of release assay medium (RPMI 1640 with 0.2% fatty acid-free BSA) supplemented with 4-deoxypyridoxine (to 1 mM), NaF (2 mM), and Na3VO4 (0.2 mM) to retard degradation of S1P by S1P lyase and S1P phosphatases was added to each well. Test articles were introduced into duplicate wells (to 2 μM), and plates were placed in a tissue culture incubator for 18–20 h. Following this incubation, the medium was collected, internal standard (5 μL of 0.5 μM d7-S1P in methanol) and 150 μL of 100% trichloroacetic acid were added, and the mixture was held on ice for 30–60 min. The precipitated material was collected by centrifugation, the pellets washed with water, recentrifuged, and the final pellet mixed vigorously after adding 0.3 mL methanol. After further centrifugation, 0.15 mL of the supernatant fluid was added to UPLC vials and S1P and d7-S1P were quantified by MS/MS by injecting 9 μL into the column. Spns2-expressing cells were found to release S1P into the culture media in ca. 20-fold excess of Spns2R200S-transfected or nontransfected cells.
In Vivo Studies.
Compound 16d (10 or 20 mg/kg) or an equal volume of vehicle (36.1% PEG400/9.1% ethanol/4.6% Solutol/50% H2O) was administered by intraperitoneal injection into mice (C57BL/6j strain) or rats (Sprague-Dawley strain). Blood samples were analyzed via LC–MS/MS as described for analysis using the in vitro Spns2 assay above. Lymphocyte counts were obtained from 20 μL of mouse or rat blood using a Heska HT5 Element blood analyzer. For PK analysis in rats, whole blood collections via tail nick (ca. 70 μL) were performed at 0, 0.5, 1, 2, 4, 6, and 24 h postdose and plasma samples prepared for LC–MS/MS analysis for drug and S1P levels. All animal protocols were approved prior to experimentation by the University of Virginia School of Medicine’s Animal Care and Use Committee.
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Institutes of Health (R01 AI144026) for support of these studies.
ABBREVIATIONS
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- EAE
experimental autoimmune encephalomyelitis
- HCTU
2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate
- MFS
major facilitator superfamily
- MS
mass spectrometry
- mSphK1
mouse sphingosine kinase 1
- mSphK2
mouse sphingosine kinase 2
- S1P
sphingosine-1-phosphate
- S1P1-5
sphingosine-1-phosphate receptors 1–5
- SphK1
sphingosine kinase 1
- SphK2
sphingosine kinase 2
- Spns2
spinster homologue 2
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c02171.
Molecular formula string list (CSV)
Characterizations of compounds and representative HPLC traces (PDF)
The authors declare the following competing financial interest(s): W.L.S. and K.R.L. are among the co-founders of Flux Therapeutics Inc, which was created to commercialize S1P-related discoveries, including Spns2 inhibitors, discovered and characterized in their laboratories.
Contributor Information
Russell Fritzemeier, Department of Chemistry, Virginia Tech, Blacksburg, Virginia 24060, United States.
Daniel Foster, Department of Chemistry, Virginia Tech, Blacksburg, Virginia 24060, United States.
Ashley Peralta, Department of Chemistry, Virginia Tech, Blacksburg, Virginia 24060, United States.
Michael Payette, Department of Chemistry, Virginia Tech, Blacksburg, Virginia 24060, United States.
Yugesh Kharel, Department of Pharmacology, University of Virginia, Charlottesville, Virginia 22908, United States.
Tao Huang, Department of Pharmacology, University of Virginia, Charlottesville, Virginia 22908, United States.
Kevin R. Lynch, Department of Pharmacology, University of Virginia, Charlottesville, Virginia 22908, United States
Webster L. Santos, Department of Chemistry and Virginia Tech Center for Drug Discovery, Virginia Tech, Blacksburg, Virginia 24060, United States
REFERENCES
- (1).Cartier A; Hla T Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, No. eaar5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Liu Y; Wada R; Yamashita T; Mi Y; Deng C-X; Hobson JP; Rosenfeldt HM; Nava VE; Chae S-S; Lee M-J; Liu CH; Hla T; Spiegel S; Proia RL Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Invest 2000, 106, 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Paik J-H; Skoura A; Chae S-S; Cowan AE; Han DK; Proia RL; Hla T Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev. 2004, 18, 2392–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Allende ML; Yamashita T; Proia RL G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood 2003, 102, 3665–3667. [DOI] [PubMed] [Google Scholar]
- (5).Joly S; Pernet V Sphingosine 1-phosphate receptor 1 is required for retinal ganglion cell survival after optic nerve trauma. J. Neurochem 2016, 138, 571–586. [DOI] [PubMed] [Google Scholar]
- (6).Rutherford C; Childs S; Ohotski J; McGlynn L; Riddick M; Macfarlane S; Tasker D; Pyne S; Pyne NJ; Edwards J; Palmer TM Regulation of cell survival by sphingosine-1-phosphate receptor S1P1 via reciprocal ERK-dependent suppression of Bim and PI-3-kinase/protein kinase C-mediated upregulation of Mcl-1. Cell Death Dis. 2013, 4, No. e927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Formigli L; Meacci E; Sassoli C; Chellini F; Giannini R; Quercioli F; Tiribilli B; Squecco R; Bruni P; Francini F; Zecchi-Orlandini S Sphingosine 1-phosphate induces cytoskeletal reorganization in C2C12 myoblasts: Physiological relevance for stress fibres in the modulation of ion current through stretch-activated channels. J. Cell Sci 2005, 118, 1161–1171. [DOI] [PubMed] [Google Scholar]
- (8).Donati C; Bruni P Sphingosine 1-phosphate regulates cytoskeleton dynamics: Implications in its biological response. Biochim. Biophys. Acta, Biomembr 2006, 1758, 2037–2048. [DOI] [PubMed] [Google Scholar]
- (9).Chun J; Hartung H-P Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin. Neuropharmacol 2010, 33, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kappos L; Radue E-W; O’Connor P; Polman C; Hohlfeld R; Calabresi P; Selmaj K; Agoropoulou C; Leyk M; Zhang-Auberson L; Burtin P A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med 2010, 362, 387–401. [DOI] [PubMed] [Google Scholar]
- (11).Calabresi PA; Radue E-W; Goodin D; Jeffery D; Rammohan KW; Reder AT; Vollmer T; Agius MA; Kappos L; Stites T; Li B; Cappiello L; von Rosenstiel P; Lublin FD Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014, 13, 545–556. [DOI] [PubMed] [Google Scholar]
- (12).Kaufmann M; Haase R; Proschmann U; Ziemssen T; Akgun K Real world lab data: Patterns of lymphocyte counts in fingolimod treated patients. Front. Immunol 2018, 9, 2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Rafiee Zadeh A; Parsa S; Tavoosi N; Farshi M; Masaeli MF Effect of fingolimod on white blood cell, lymphocyte and neutrophil counts in MS patients. Am. J. Clin. Exp. Immunol 2019, 8, 9–15. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (14).Cohan S; Lucassen E; Smoot K; Brink J; Chen C Sphingosine-1-phosphate: Its pharmacological regulation and the treatment of multiple sclerosis: A review article. Biomedicines 2020, 8, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Roy R; Alotaibi AA; Freedman MS Sphingosine 1-phosphate receptor modulators for multiple sclerosis. CNS Drugs 2021, 35, 385–402. [DOI] [PubMed] [Google Scholar]
- (16).Sandborn WJ; Feagan BG; D’Haens G; Wolf DC; Jovanovic I; Hanauer SB; Ghosh S; Petersen A; Hua SY; Lee JH; Charles L; Chitkara D; Usiskin K; Colombel J-F; Laine L; Danese S Ozanimod as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med 2021, 385, 1280–1291. [DOI] [PubMed] [Google Scholar]
- (17).Comi G; Hartung H-P; Bakshi R; Williams IM; Wiendl H Benefit-Risk Profile of Sphingosine-1-Phosphate Receptor Modulators in Relapsing and Secondary Progressive Multiple Sclerosis. Drugs 2017, 77, 1755–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Perland E; Bagchi S; Klaesson A; Fredriksson R Characteristics of 29 novel atypical solute carriers of major facilitator superfamily type: evolutionary conservation, predicted structure and neuronal co-expression. Open Biol. 2017, 7, 170142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kobayashi N; Kawasaki-Nishi S; Otsuka M; Hisano Y; Yamaguchi A; Nishi T MFSD2B is a sphingosine 1-phosphate transporter in erythroid cells. Sci. Rep 2018, 8, 4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chandrakanthan M; Nguyen TQ; Hasan Z; Muralidharan S; Vu TM; Li AWL; Le UTN; Thi Thuy Ha H; Baik S-H; Tan SH; Foo JC; Wenk MR; Cazenave-Gassiot A; Torta F; Ong WY; Chan MYY; Nguyen LN Deletion of Mfsd2b impairs thrombotic functions of platelets. Nat. Commun 2021, 12, 2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nguyen TQ; Vu TM; Tukijan F; Muralidharan S; Foo JC; Li Chin JF; Hasan Z; Torta F; Nguyen LN Erythrocytes efficiently utilize exogenous sphingosines for S1P synthesis and export via Mfsd2b. J. Biol. Chem 2021, 296, 100201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).El Jamal A; Briolay A; Mebarek S; Le Goff B; Blanchard F; Magne D; Brizuela L; Bougault C Cytokine-Induced and Stretch-Induced Sphingosine 1-Phosphate Production by Enthesis Cells Could Favor Abnormal Ossification in Spondyloarthritis. J. Bone Miner. Res 2019, 34, 2264–2276. [DOI] [PubMed] [Google Scholar]
- (23).Mendoza A; Breart B; Ramos-Perez WD; Pitt LA; Gobert M; Sunkara M; Lafaille JJ; Morris AJ; Schwab SR The transporter Spns2 is required for secretion of lymph but not plasma sphingosine-1-phosphate. Cell Rep. 2012, 2, 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Fukuhara S; Simmons S; Kawamura S; Inoue A; Orba Y; Tokudome T; Sunden Y; Arai Y; Moriwaki K; Ishida J; Uemura A; Kiyonari H; Abe T; Fukamizu A; Hirashima M; Sawa H; Aoki J; Ishii M; Mochizuki N The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J. Clin. Invest 2012, 122, 1416–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Nagahashi M; Kim EY; Yamada A; Ramachandran S; Allegood JC; Hait NC; Maceyka M; Milstien S; Takabe K; Spiegel S Spns2, a transporter of phosphorylated sphingoid bases, regulates their blood and lymph levels, and the lymphatic network. FASEB J. 2013, 27, 1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Simmons S; Sasaki N; Umemoto E; Uchida Y; Fukuhara S; Kitazawa Y; Okudaira M; Inoue A; Tohya K; Aoi K; Aoki J; Mochizuki N; Matsuno K; Takeda K; Miyasaka M; Ishii M High-endothelial cell-derived S1P regulates dendritic cell localization and vascular integrity in the lymph node. eLife 2019, 8, No. e41239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Nijnik A; Clare S; Hale C; Chen J; Raisen C; Mottram L; Lucas M; Estabel J; Ryder E; Adissu H; Adams NC; Ramirez-Solis R; White JK; Steel KP; Dougan G; Hancock REW The role of sphingosine-1-phosphate transporter Spns2 in immune system function. J. Immunol 2012, 189, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hisano Y; Kobayashi N; Yamaguchi A; Nishi T Mouse SPNS2 functions as a sphingosine-1-phosphate transporter in vascular endothelial cells. PLoS One 2012, 7, No. e38941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Okuniewska M; Fang V; Baeyens A; Raghavan V; Lee J-Y; Littman DR; Schwab SR SPNS2 enables T cell egress from lymph nodes during an immune response. Cell Rep. 2021, 36, 109368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Donoviel MS; Hait NC; Ramachandran S; Maceyka M; Takabe K; Milstien S; Oravecz T; Spiegel S Spinster 2, a sphingosine-1-phosphate transporter, plays a critical role in inflammatory and autoimmune diseases. FASEB J. 2015, 29, 5018–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Patwardhan NN; Morris EA; Kharel Y; Raje MR; Gao M; Tomsig JL; Lynch KR; Santos WL Structure–Activity Relationship Studies and in Vivo Activity of Guanidine-Based Sphingosine Kinase Inhibitors: Discovery of SphK1- and SphK2-Selective Inhibitors. J. Med. Chem 2015, 58, 1879–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Childress ES; Kharel Y; Brown AM; Bevan DR; Lynch KR; Santos WL Transforming sphingosine kinase 1 inhibitors into dual and sphingosine kinase 2 selective inhibitors: Design, synthesis, and in vivo activity. J. Med. Chem 2017, 60, 3933–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Congdon MD; Childress ES; Patwardhan NN; Gumkowski J; Morris EA; Kharel Y; Lynch KR; Santos WL Structure-activity relationship studies of the lipophilic tail region of sphingosine kinase 2 inhibitors. Bioorg. Med. Chem. Lett 2015, 25, 4956–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Raje MR; Knott K; Kharel Y; Bissel P; Lynch KR; Santos WL Design, synthesis and biological activity of sphingosine kinase 2 selective inhibitors. Bioorg. Med. Chem 2012, 20, 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Santos WL; Lynch KR; MacDonald TL; Kenndy A; Kharel Y; Raje MR; Houck J Long Chain Base Sphingosine Kinase Inhibitors. WO 2013119946 A1, August 15, 2013. [Google Scholar]
- (36).Li H; Sibley CD; Kharel Y; Huang T; Brown AM; Wonilowicz LG; Bevan DR; Lynch KR; Santos WL Lipophilic tail modifications of 2-(hydroxymethyl)pyrrolidine scaffold reveal dual sphingosine kinase 1 and 2 inhibitors. Bioorg. Med. Chem 2021, 30, 115941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Congdon M; Fritzemeier RG; Kharel Y; Brown AM; Serbulea V; Bevan DR; Lynch KR; Santos WL Probing the substitution pattern of indole-based scaffold reveals potent and selective sphingosine kinase 2 inhibitors. Eur. J. Med. Chem 2021, 212, 113121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sibley CD; Morris EA; Kharel Y; Brown AM; Huang T; Bevan DR; Lynch KR; Santos WL Discovery of a small side cavity in sphingosine kinase 2 that enhances inhibitor potency and selectivity. J. Med. Chem 2020, 63, 1178–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Mathews TP; Kennedy AJ; Kharel Y; Kennedy PC; Nicoara O; Sunkara M; Morris AJ; Wamhoff BR; Lynch KR; Macdonald TL Discovery, Biological Evaluation, and Structure–Activity Relationship of Amidine Based Sphingosine Kinase Inhibitors. J. Med. Chem 2010, 53, 2766–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kharel Y; Agah S; Huang T; Mendelson AJ; Eletu OT; Barkey-Bircann P; Gesualdi J; Smith JS; Santos WL; Lynch KR Saccharomyces cerevisiae as a platform for assessing sphingolipid lipid kinase inhibitors. PLoS One 2018, 13, No. e0192179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ivashchenko AA; Ivanenkov YA; Aladinskiy VA; Karapetian RN; Koryakova AG; Ryakhovskiy AA; Mitkin OD; Kravchenko DV; Savchuk NP; Zagribelnyy BA; Ivashchenko AV Synthesis, biological evaluation and in silico modeling of novel pan-genotypic NS5A inhibitors. Bioorg. Med. Chem 2020, 28, 115716. [DOI] [PubMed] [Google Scholar]
- (42).Houck JD; Dawson TK; Kennedy AJ; Kharel Y; Naimon ND; Field SD; Lynch KR; Macdonald TL Structural requirements and docking analysis of amidine-based sphingosine kinase 1 inhibitors containing oxadiazoles. ACS Med. Chem. Lett 2016, 7, 487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.