

Abstract
Bi‐allelic variants in Iron–Sulfur Cluster Scaffold (NFU1) have previously been associated with multiple mitochondrial dysfunctions syndrome 1 (MMDS1) characterized by early‐onset rapidly fatal leukoencephalopathy. We report 19 affected individuals from 10 independent families with ultra‐rare bi‐allelic NFU1 missense variants associated with a spectrum of early‐onset pure to complex hereditary spastic paraplegia (HSP) phenotype with a longer survival (16/19) on one end and neurodevelopmental delay with severe hypotonia (3/19) on the other. Reversible or irreversible neurological decompensation after a febrile illness was common in the cohort, and there were invariable white matter abnormalities on neuroimaging. The study suggests that MMDS1 and HSP could be the two ends of the NFU1‐related phenotypic continuum.
Introduction
Iron–sulfur [Fe‐S] clusters are important cofactors that play a role in various cellular functions, including electron transfer along the respiratory chain, citric acid cycle, heme biosynthesis, DNA replication and repair, as well as iron homeostasis. 1 , 2 , 3 Iron–Sulfur Cluster Scaffold (NFU1) (MIM: 608100) is an [Fe‐S] cluster biosynthesis factor involved in the last steps of maturation and transfer of [4Fe‐4S] clusters into target proteins, including lipoic acid synthetase, mitochondrial aconitase and some subunits of respiratory chain complexes I and II. 4 , 5 Bi‐allelic variants in NFU1 have previously been associated with multiple mitochondrial dysfunctions syndrome 1 (MMDS1) 6 characterized by early‐onset leukoencephalopathy leading to a fatal outcome, typically before the age of 15 months. 6
Recently, bi‐allelic variants in NFU1 have been reported in two individuals with a milder phenotype, presenting with slowly progressive spastic paraplegia with a relapsing–remitting course, long survival, and intact cognition or mild intellectual disability (ID). 7 , 8 Here, we report 19 affected individuals from 10 independent families with ultra‐rare bi‐allelic NFU1 missense variants associated with a phenotype ranging from early‐onset pure to complex hereditary spastic paraplegia (HSP) characterized by a longer survival (16/19) and neurodevelopmental delay (NDD) with severe hypotonia (3/19).
Methods
Participants and clinical investigations
Exome sequencing (ES) and genome sequencing (GS), data sharing between international genetic centers, and the GeneMatcher platform 9 were employed to identify the 10 families reported here. Clinical data were collected via a uniform proforma. Parents/legal guardians of all affected individuals consented to the publication of genetic and clinical information. The study was approved by The Research Ethics Committee Institute of Neurology University College London (IoN UCL) (07/Q0512/26) and the local Ethics Committees of each participating center.
Exome sequencing and genome sequencing
Research/diagnostic solo or trio‐ES/GS, variant filtering, and variant confirmation by Sanger sequencing and segregation analysis were performed on genomic DNA extracted from blood in different genetic laboratories following the methods previously described (Table 1). Family 10 had a mitochondrial panel test performed for the proband and targeted genotyping for familial variants for the sibling (further details in Table 1 and Supplementary Table S1).
Table 1.
Clinical features of the present cohort and description of NFU1 variants.
| F1 (F1‐II:1,F1‐II:2) | F2 (F2‐II:1) | F3 (F3‐II:1, F3‐II:2) | F4 (F4‐II:4, F4‐II:5,‐F4‐II:6) | F5 (F5‐II:1, F5‐II:2) | F6 (F6‐II:2, F6‐II) | F7 (F7‐II:1, F7‐II:2) | F8 (F8‐II:5, F8‐II) | F9 (F9‐II:1) | F10 (F10‐II:1, F10‐II:2) | Previously published reports with HSP phenotype | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reported in | This study | This study | This study | This study | This study | This study | This study | This study | This study | This study | Tonduti et al., 2015 | Uzunhan et al., 2020 |
| Ethnic origin | Iranian‐Arab | American | Egyptian | Egyptian | Egyptian | Egyptian | Kazakh | Turkish | American | European‐American | Not reported | Not reported |
| Consanguinity | Yes | No | Yes | Yes | Yes | Yes | No | Yes | No | No | No | Yes |
| Genetic investigation | ES | ES | ES | ES | ES | ES | ES | GS | ES | NGS Panel Mitochondrial Disorders | Not reported | ES |
| WES/WGS and variant filtering method | Makrythanasis et al., 2018 10 | Guillen Sacoto et al., 2020 11 | Makrythanasis et al., 2018 10 | Makrythanasis et al., 2018 10 | Sefid et al., 2017 12 ; Cirulli et al., 2010 13 | Guillen Sacoto et al., 2020 11 | Lindy et al., 2018 14 | Tonduti et al., 2015 7 | Uzunhan et al., 2020 8 | |||
| Variant at cDNA level* | c.721G>C | c.298G>C; c.301A>G | c.362T>C | c.295C>G | c.263T>C | c.548C>G; c.629G>T | c.629G>T; c.398T>C | c.146delC; c.565G>A | c.565G>A | |||
| Variant at protein level* | p.(Val241Leu) | p.(Ala100Pro); p.(Arg101Gly) | p.(Val121Ala) | p.(Leu99Val) | p.(Phe88Ser) | p.(Pro183Arg); p.(Cys210Phe) | p.(Cys210Phe); p.(Leu133Pro) | p.(Pro49LeufsTer8); p.(Gly189Arg) | p.(Gly189Arg) | |||
| Sex | 2F | 1M | 1M, 1F | 3F | 1M, 1F | 2M | 2M | 2M | 1M | 1M, 1F | 1M | 1F |
| Age at last examination | 10y and 17 y | 25 y | 9 y and 12 y | 2 y and 6 y | 4,5 m and 6 m | 3 y and 2.2 y | 10 y and 6 y | 7.5 and 6.5 y | 12 y | 1 y and 2 y | 30 y | 3.5 y |
| Age of onset | 6–9 m and 9–10 m | 1 y | 1 y (2) | 0.9 y and 1,2 y | 1,1 y and 0.5 y | 1 y and 0.9 m | 3–4 m (2) | 18 m (2) | 2 y | 6 m and 3 m | 18 m | 23 m |
| Disease duration | 9 y and16 y | 24 y | 8 y and 11 y | 10 m (1), 6 m (1), and 5 y (1) | Died at 6 m (1) and 3y (1) | 2 y and 1.6 y | 10 y and 6 y | 6 y and 5 y | 10 y | Died at 13 m and 25 m | 28.5 y | 21 m |
| Family history | Yes | No | Yes (2) | Yes (3) | Yes (2) | Yes (2) | Yes (2) | Yes (2) | No | Yes (1), No (1) | No | Yes |
| Mortality | Alive (2) | Alive | Alive (2) | Died at 33 m (1) and 30 m (1), alive (1) | Died (1) and alive (1) | Deceased (1) | Alive (2) | Alive (2) | Alive | Died after febrile illness (2) | Alive | Alive |
| Onset with delayed walking/progressive spastic paraplegia | Yes (2) | Yes | Yes (2) | Yes (3) | Yes (1) | Yes (2) | Yes (2) | Yes (2) | Yes | No (2) | Yes (2) | Yes |
| Mild ID/Cogitative impairment | No (2) | No | Yes (2) | Yes (1) | No (2) | No (2) | No (2) | No (2) | No | Yes (2) | Yes | Yes |
| Lower limb spasticity | Yes (2) | Yes | Yes (2) | Yes (3) | Yes (1) | Yes (2) | Yes (2) | Yes (2) | Yes | Yes (1) | Yes | Yes |
| Deterioration with loss of milestones with intercurrent viral/febrile illness | Yes | Yes | No (2) | Yes (2), No (1) | Yes (2) | Yes (2) | No (2) | Yes (2) | Yes | Yes (2) | Yes | Yes |
| Recovery of loss of milestones/previous status after recovery from illness | Yes (1), NA (1) | Yes | NA (2) | No (2), NA (1) | No (1) and Yes (1) | Yes (2) | NAP | No (1), NA (1) | Yes | No (2) | Yes | Yes |
| Mild WMHs in T2WI | Yes (2) | No** | No (2) ** | Yes (2) | Yes (1) | Yes (1), NA (1) | Yes (1), NA (1) | Yes (1) ***, NA (1) | No** | Yes (2) | Yes | Yes |
| Mild T2 hyperintensity in BG | Yes (2) | No** | No (2) ** | Yes (2) | Yes (1), No (1) | Yes (1) | Yes (1), NA (1) | Yes (1) ***, NA (1) | No** | Yes (2) | No | No |
| CC abnormalities | Yes (2) | No** | No (2) ** | Yes (2) | Yes (1), No (1) | Yes (1) | Yes (1), NA (1) | No (1), NA (1) | No** | Yes (2) | Yes | Yes |
| Areas of cystic degeneration/leukomalacia | No (2) | No** | No (2) ** | No (2) | No (2) | No | No (1), NA (1) | No (1), NA (1) | No** | Yes (1), No (1) | No | Yes |
ES, exome sequencing; GS, genome sequencing; WES, Whole exome sequencing; WGS, Whole genome sequencing; NGS, next generation sequencing; F, female; M, male; y, year(s); m, month(s); NAP (no applicable); T2WI, T2 weighted images; BG, basal ganglia; CC, corpus callosum.
Based on the transcripts NM_001002755.4 & NP_001002755.1 respectively.
Reported as normal. Study not independently reviewed.
Questionable, please refer to Supplementary Table S1.
Allele frequencies of the variants, in silico predictions, and ACMG classifications are provided in Supplementary Table S1.
SDS‐PAGE and western blot analysis
Human fibroblasts from an affected individual (F1‐II:1) were obtained and lysed and samples were subjected to SDS‐PAGE and western blotting as described previously 15 and a list of primary antibodies is provided (Supplementary Methods).
Protein modeling and characterization, Fe‐S cluster transfer monitoring, and enzymatic assays
Protein characterization by variable temperature circular dichroism (CD) and analytical ultracentrifugation, Fe‐S cluster transfer monitored by circular dichroism, pyruvate dehydrogenase and α‐ketoglutarate dehydrogenase activity assay studies were performed (Supplementary Methods).
Results
Clinical phenotype
The cohort comprises 19 affected individuals from 10 independent families. There were 8 females and 11 males, 13 of whom are currently alive with a mean age of 9.8 ± 5.7 years (range 2.1–25) at the study recruitment (Fig. 1A). Six affected individuals succumbed to their rapidly progressive disease course triggered by a febrile illness between the ages of 6–36 months. Detailed clinical information is provided in Supplementary Table S1 and Supplemental Case Reports.
Figure 1.
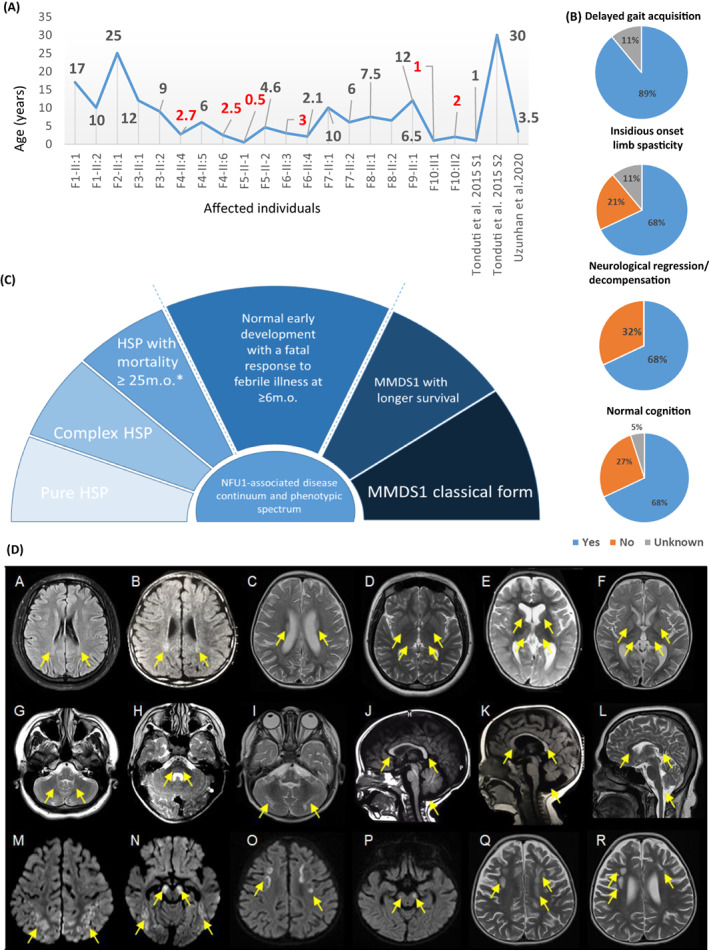
Clinical features of the individuals reported in this study and NFU1‐associated phenotypic continuum. (A) Ages of the affected individuals at the study recruitment. (B) Clinical features of the present cohort. (C) NFU1‐associated phenotypic continuum. HSP, hereditary spastic paraplegia. (D) Representative brain MRI features of the present cohort. Individual F1‐II:2 (A, D, G, L), individual F4‐II:5 (B), individual F10‐II:1 (C, F, I, M, N), individual F5‐II:2 (E, K), individual F7‐II:1 (H), individual F4‐II:6 (J), and individual F10‐II:2 (O, P, Q, R). T2/FLAIR hyperintense signal involving the bilateral posterior centrum semiovale (Q), corona radiata, and periatrial regions (A‐C). T2 hyperintense signal involving the bilateral thalami and basal ganglia (D‐F), pons, and cerebellum (G‐I). Hypoplastic corpus callosum and mega cisterna magna (J‐L). Bilateral cerebral white matter volume loss (A‐F, Q, R). Areas of restricted diffusion involving the bilateral subcortical white matter and cerebral peduncles (M‐P). Areas of cystic degeneration/leukomalacia in the white matter of the bilateral frontal lobes (Q, R). Vermian hypoplasia (K).
Prenatal features and neonatal periods were unremarkable in all subjects. No delay in early developmental milestones including head control, crawling, sitting, and speech was reported in 14/19 individuals. Cognitive function was impaired in 5/18 individuals. Gait acquisition was delayed due to either the insidious onset of limb spasticity in 13/19 individuals or spasticity precipitated by a deterioration in the context of a febrile illness in 4/19 individuals (Fig. 1B). The lower limb spasticity was detected at a mean age of 12 ± 6 months in the cohort. Spasticity was progressive leading to contractures in 13/19 persons and necessitating Achilles' tendon repair surgery in 4/19 affected individuals.
Remarkably, febrile illness leading to metabolic decompensation played a significant role in the disease course of 13/19 individuals. Thus, in F1‐II:1 it led to febrile seizures followed by the onset of lower limb spasticity and reversible cognitive and motor regression, and truncal hypotonia. Similarly, F5‐I:1, F6‐II:3, and F8‐II:5 developed reversible truncal hypotonia and/or progressive lower limb spasticity following several episodes of febrile illness. Intermittent and reversible gait deterioration associated with episodes of febrile illness was reported in F2‐II:1and F9‐II:1, along the course of their disease, which was described as waxing and waning. While the episodes of febrile illness had not been fatal for these persons, the other six affected individuals had regression and developed severe muscular hypotonia leading to early mortality after a febrile illness.
The mean disease duration at the most recent examination for the living individuals was 8.8 ± 5.7 years (range 1.5–24). Neuromuscular examination revealed lower limb spasticity in all persons with increased deep tendon reflexes, upgoing plantar reflexes, and mild symmetrical muscle wasting. Muscle tone from the upper limbs was uniformly intact. While two individuals were wheelchair‐bound, eight could ambulate using crouches or rollator walker, and three could walk independently with spastic gait. Ataxic gait was reported in 2/12 individuals, which was intermittent in one person (F2‐II:1). The oldest individual in the cohort was aged 25 years and had a normal cognitive function and could perform most activities of daily living despite his slowly progressive lower limb spasticity. Seven persons had results available of metabolic screening, which were unremarkable in all.
Brain MRI DICOM files were available for six individuals (F1‐II:1, F1‐II:2, F7‐II:1, F8‐II:5, F10‐II:1, and F10‐II:2), low‐resolution photos of brain MRI studies were available for four individuals (F4‐II:5, F4‐II:6, F5‐II:2, and F6‐II:2), and photos of a head CT in one person (F5‐II:1). All available neuroimaging studies were independently reviewed by a board‐certified neuroradiologist. Four additional individuals had neuroimaging studies that were reported as normal but not independently reviewed (F2‐II:1, F3‐II:1, F3‐II:2, and F9‐II:1). Shared neuroimaging findings included T2 hyperintense signal involving the bilateral posterior centrum semiovale, corona radiata, and periatrial regions (9/9, 100%); T2 hyperintense signal involving the bilateral thalami (9/9, 100%), basal ganglia (7/9, 78%), and pons and cerebellum (7/8, 88%), hypoplastic corpus callosum (9/10, 90%), bilateral cerebral white matter volume loss (8/11, 73%), mega cisterna magna (9/11, 82%), mild prominence of the lateral ventricles and frontoparietal sulci (7/11, 64%), and scattered areas of restricted diffusion which may represent active demyelination, metabolic encephalopathy, or acute/subacute ischemia (3/4, 75%). A single individual (1/11, 9%) had areas of cystic degeneration/leukomalacia in the white matter of the bilateral frontal lobes and another (1/11, 9%) had vermian hypoplasia. Figure 1D shows shared neuroimaging findings and the Supplemental information has case‐based descriptions of the brain MRI/CT interpretations.
Genetic analysis reveals ultra‐rare bi‐allelic variants in NFU1
A total of 9 different NFU1 missense variants were identified in this study (Figs. 2A to C). Exome sequencing carried out in the proband F1‐II:1 revealed a homozygous likely pathogenic NFU1 variant: NM_001002755.4:c.721G>C, p.(Val241Leu). In the third affected individual F2‐II:1, trio‐ES identified compound heterozygous NFU1 variants: a variant of uncertain significance (VUS), c.298G>C, p.(Ala100Pro) inherited from the father and a VUS, c.301A>G, p.(Arg101Gly) inherited from the mother.
Figure 2.
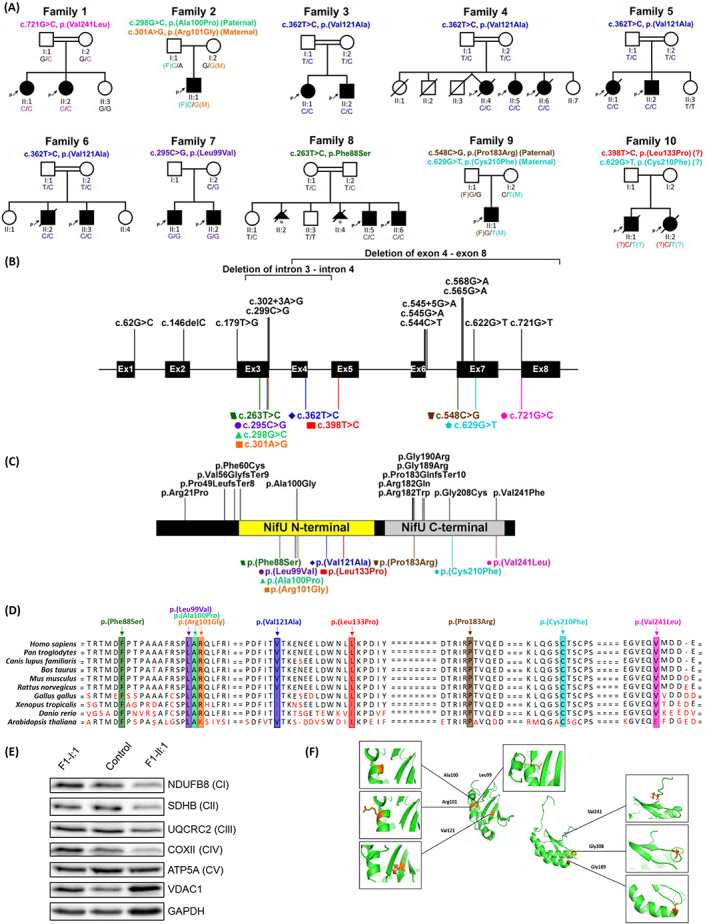
Overview of the genetic and biochemical characteristics of NFU1 variants. (A) Pedigrees and segregation results of the ten families included in this study. (B) NFU1 gene structure with the localization of all previously known mutations (black, above) and mutations reported in this cohort (colored, below). The corresponding amino acid changes is shown in (C) with known protein domains in NFU1 (NifU N‐terminal domain, residue 59 to 155, and NifU C‐terminal domain, residue 162 to 247) depicted in the illustration. The variant, p.(Val241Leu), was the only mutation discovered in this study to have been located outside of the NFU1 protein domains. Regions are not drawn to scales and both illustrations were created from the program. 16 (D) Conservation of the nine mutations reported in the present cohort across 10 species. (E) Western blot analysis of structural subunits from each OXPHOS complex (CI [NDUFB8], CII [SDHB], CIII [UQCRC2], CIV [COXII], and CV [ATP5A]) in fibroblasts from F1‐II:1 (affected individual), F1‐I:1 (mother) and a pediatric control. GAPDH and VDAC1 were used as cell and mitochondrial loading controls, respectively. (F) Protein model of the NifU N‐terminal domain and NifU C‐terminal domain of the NFU1 structure. Positions of amino acids affected by NFU1 missense variants are indicated in orange.
Exome sequencing of four unrelated Egyptian families, F3 to F6, led to the identification of the same pathogenic homozygous NFU1 variant: c.362T>C, p.(Val121Ala). A homozygous VUS, c.295C>G, p.(Leu99Val) was also identified by ES of the proband of family 7. In family 8, a homozygous VUS, c.263T>C, p.(Phe88Ser) was identified by ES of the proband. Trio‐ES in family 9 found compound heterozygous NFU1 variants: a maternally inherited likely pathogenic variant, c.629G>T, p.(Cys210Phe) and a paternally inherited VUS, c.548C>G, p.(Pro183Arg). In family 10, the combination of a mitochondrial gene panel and Sanger sequencing revealed potential compound heterozygous NFU1 variants: a VUS, c.629G>T, p.(Cys210Phe) and a VUS, c.398T>C, p.(Leu133Pro). Apart from, p.(Leu99Val), all the homozygous variants are residing within sizeable regions of homozygosity. All the variants are either absent or observed in extremely low allele frequencies in numerous population variant frequency databases (~2 mln alleles). They affect highly conserved residues across different species (Fig. 2D) and segregated with the disease phenotype in all the families (Fig. 2A). No other relevant variants associated with neurological or neurodevelopmental disorders were identified in the currently known monogenic disease‐causing genes in the ES data.
The characteristics of the variants are further summarized in Table 1 and Supplementary Table S1.
Steady‐state levels of OXPHOS complexes I, II, III, and IV are decreased in homozygous p.(Val241Leu) fibroblasts
Western blot analysis on fibroblast samples showed that subunits of each complex of the electron transport chain (CI‐CIV) were decreased in the affected individual F1‐II:1 compared to the heterozygous mother (F1‐I:1) and a pediatric control, whereas levels of complex V subunit ATP5A remained relatively unchanged (Fig. 2E).
Functional study of the p.(Val241Leu) variant
Protein modeling revealed all nine variants were located in the NFU1 protein domains. Six variants, p.(Phe88Ser), p.(Leu99Val), p.(Ala100Pro), p.(Arg101Gly), p.(Val121Ala), and p.(Leu133Pro), were found in the NifU N‐terminal domain while the remaining three, p.(Pro183Arg), p.(Cys210Phe), and p.(Val241Leu), lie in the NifU C‐terminal [Fe‐S] cluster binding domain (Figs. 2C,F, Supplementary Results). The p.(Val241Leu) variant was further investigated via in vitro [Fe‐S] cluster reconstitution experiments, however, the results showed no significant change in the protein's secondary structure (Supplementary Fig. S1C, Supplementary Results).
Additional discussion of the structural and functional biochemistry of other variants is provided in the Supplementary Material. As patient fibroblasts were available only for the p.(Val241Leu) variant, the impact of the variant on the enzymatic activities of PDH and KGDHC was measured only for this variant. Enzymatic activity assays in fibroblasts derived from F1‐II:1, homozygous for p.(Val241Leu), showed decreased activity for PDH (0.50 nmol/mg protein/min; normal range 0.6–0.9 nmol/mg protein/min), while the mean activity in the heterozygous mother was normal (0.82 nmol/mg protein/min; normal range 0.6–0.9 nmol/mg protein/min). The KGDHC activity was not impaired (Supplementary Fig. S2, Supplementary Results).
Discussion
The bioenergetic function of the mitochondrion is dependent on Fe‐S cluster‐containing proteins. Three distinctly organized biosynthetic pathways are involved in the maturation of Fe‐S clusters in mammals with the last step involving NUBPL, NFU1, BOLA3, IBA57, ISCA2, and ISCA1. Bi‐allelic pathogenic variants in five of them, starting from NFU1 in the above‐mentioned list, are currently associated with MMDS types 1 to 5 respectively, and typically present with severe and fatal early onset encephalopathy with multiple biochemical abnormalities. 6 An atypical presentation of MMDS has been reported in several individuals, mainly encompassing a slightly milder disease course with a longer survival 17 , 18 , 19 for NFU1 and ISCA2, or a complex HSP phenotype for NFU1 and IBA57. 7 , 8 , 20 Our report expands the number of individuals with bi‐allelic NFU1 variants presenting with complex HSP and also describes previously unreported pure HSP phenotype, thereby suggesting that MMDS1 and HSP could be the two ends of the NFU1‐associated phenotypic continuum.
Pyramidal symptoms in the form of spastic tetraparesis have frequently been reported in individuals with typical MMDS1 presentation, highlighting the role of mitochondria and axonal transport in the corticospinal tract function. 6 Currently, due to a large overlap between HSP and other inherited neurological disorders, at least 28/81 genetic forms of HSP are assigned alternative phenotypes on the Online Mendelian Inheritance in Man (OMIM) database, resulting in a diagnostic challenge. 21 Therefore, delineating the full phenotypic spectrum of disease‐associated genes and accurate clinical classification are important for diagnostic rates.
Another remarkable aspect of the present cohort is the presence of neurological decompensation after a febrile illness. Interestingly, three distinct responses to episodes of febrile illness were observed in our cohort: (1) fatal outcome after the first or consecutive episode in six individuals; (2) onset of spasticity with variably reversible motor and cognitive regression in three individuals; and (3) intermittent and fully reversible gait deterioration in four other individuals. The episodes of neurological deterioration, most likely caused by acute illness‐induced metabolic decompensation, is common in mitochondrial diseases 22 but have rarely been reported in the mitochondrial causes of HSP. 23 , 24
A remarkable inter‐and intrafamilial phenotypic variability was observed in the present cohort and the same amino acid substitutions of the NFU1 protein have been observed in both MMDS1 and HSP cases (Supplementary discussion for details). The relatively milder course and phenotypic variability of the most commonly reported MMDS1‐associated NFU1 variant, c.565G>A, p.(Gly189Arg), was further supported in reports by Uzunhan et al. (2020) and Tonduti et al. (2015), 7 , 8 who described individuals with the same NFU1 variant and presenting with HSP, with one individual reaching the age of 30 years. Defects in NFU1 seem to present with a range of phenotype severities suggesting the clinical spectrum of NFU1‐associated disease (Fig. 1C). The constellation of shared neuroimaging findings noted in individuals for whom images were available is similar to the findings reported by Tonduti et al. (2015), 7 including white matter abnormalities of the periventricular regions and thinning of the corpus callosum. Similarly, neuroimaging features present in our cohort overlap with some of those reported by Uzunhan et al. (2020). 8 These include white matter hyperintensity, which was present in most of the individuals in the present series, cystic degeneration and cavitation in the frontal regions, which was noted in individual F10‐II:2, and areas of restricted diffusion involving the bilateral subcortical and periventricular white matter, noted in three out of four individuals with available diffusion‐weighted imaging.
Apart from the possible epigenetic factors, 7 , 8 we suspect that the variants observed in the present study might be causing milder defects for the NFU1 structure and function compared to the MMDS1‐linked variants. The different variants might affect, for example, partner protein binding in a distinct way leading to high phenotypic variability connected to NFU1 variants (Supplementary Discussion for further details). The residual PDH activity and the normal KGDHC activity in F1‐II:1 might be consistent with a less severe form of the disease with prolonged survival. The decreased OXPHOS protein levels and the only slightly affected PDH and normal KGDHC activity results in F1‐II:1 fibroblasts might be explained by the changed affinity of the mutated NFU1 for the different partner proteins (Supplementary Discussion for further details). The disparity between plasma and CSF metabolites levels has been shown in an individual with MMDS, suggesting the manifestation of biallelic NFU1 variants with tissue‐specific phenotypes. 25 In summary, we report 16 affected individuals with an HSP presentation of bi‐allelic NFU1 variants and 3 affected individuals with GDD, hypotonia, longer survival, and fatal response to metabolic decompensation, which contrasts with the typical MMDS1 presentation of NFU1 deficiency and highlights the NFU1‐associated disease continuum.
Conflict of Interest
G.B. is/was a consultant for Passage Bio Inc (2020–2021) and Ionis (2019). She is/was a site investigator for the Alexander's disease trial of Ionis (2021), Metachromatic leukodystrophy of Shire/Takeda (2020–2021), Krabbe and GM1 gene therapy trials of Passage Bio, and Adrenoleukodystrophy/Hematopoietic stem cell transplantation natural history study of Bluebird Bio (2019) and a site sub‐investigator for the MPS II gene therapy trial of Regenxbio (2021). She has received an unrestricted educational grant from Takeda (2021–2022). She serves on the scientific advisory board of the Pelizaeus‐Merzbacher Foundation and is the Chair of the Medical and Scientific Advisory Board of the United Leukodystrophy Foundation. She is on the editorial boards of Neurology Genetics, Frontiers in Neurology – Neurogenetics, and Journal of Medical Genetics. RP, KA, HZE, AL and RB are employees of GeneDx, LLC.
Supporting information
Data S1: Supplemental information including case reports, supplemental methods, supplemental experimental results and discussion
Table S1: Supplemental clinical‐genetic table
Acknowledgments
The authors would like to thank the participants and their families for their essential help with this work. We are grateful to The Spastic Paraplegia Foundation, The Brain Research Trust (BRT), The UK HSP Society, The Medical Research Council (MRC UK MR/J004758/1, G0802760, G1001253), The Wellcome Trust (equipment and the Synaptopathies strategic award (104033)) and the EU FP7/2007‐2013 under grant agreement number 2012‐305121 (NEUROMICS), The Muscular Dystrophy Association (MDA) USA, Muscular Dystrophy UK, Rosetrees Trust, Ataxia UK, and British Neurological Surveillance Unit (BNSU). R.K. was supported by the Research Training Fellowship grant from the European Academy of Neurologists. V.C. is supported by the Association of British Neurologists/MSA Trust Clinical Research Training fellowship (F84 ABN 540868) and J.A.C. by the National Institutes of Health (AI072443). G.B. has received the Clinical Research Scholar Junior 1 award from the Fonds de Recherche du Quebec – Sante (FRQS) (2012‐2016), the New Investigator Salary Award from the Canadian Institutes of Health Research (2017‐2022) and the Clinical Research Scholar Senior from the FRQS (2022‐2025). A.M‐D‐L. is supported by grant R01HD104938 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health. R.W.T. is funded by the Wellcome Centre for Mitochondrial Research (203105/Z/16/Z), the Mitochondrial Disease Patient Cohort (UK) (G0800674), the Medical Research Council International Centre for Genomic Medicine in Neuromuscular Disease (MR/S005021/1), the Medical Research Council (MR/W019027/1), the Lily Foundation, the Pathological Society, the UK NIHR Biomedical Research Centre for Aging and Age‐related disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust and the UK NHS Highly Specialized Service for Rare Mitochondrial Disorders of Adults and Children. For the purpose of Open Access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission.
Funding Statement
This work was funded by Medical Research Council grants G0802760, G1001253, MR/J004758/1, and MR/W019027/1; The Medical Research Council International Centre for Genomic Medicine in Neuromuscular Disease grant MR/S005021/1; The Wellcome Centre for Mitochondrial Research grant 203105/Z/16/Z; The Wellcome Trust (UK) grant 104033.
References
- 1. Yoon T, Cowan JA. Frataxin‐mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J Biol Chem. 2004;279(25):25943‐25946. [DOI] [PubMed] [Google Scholar]
- 2. Tong WH, Rouault TA. Metabolic regulation of citrate and iron by aconitases: role of iron‐sulfur cluster biogenesis. Biometals. 2007;20(3–4):549‐564. [DOI] [PubMed] [Google Scholar]
- 3. White MF, Dillingham MS. Iron‐Sulphur clusters in nucleic acid processing enzymes. Curr Opin Struct Biol. 2012;22(1):94‐100. [DOI] [PubMed] [Google Scholar]
- 4. Lill R, Dutkiewicz R, Freibert SA, et al. The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron‐sulfur proteins. Eur J Cell Biol. 2015;94(7–9):280‐291. [DOI] [PubMed] [Google Scholar]
- 5. Vanlander AV, Van Coster R. Clinical and genetic aspects of defects in the mitochondrial iron‐sulfur cluster synthesis pathway. J Biol Inorg Chem. 2018;23(4):495‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lebigot E, Schiff M, Golinelli‐Cohen MP. A review of multiple mitochondrial dysfunction syndromes, syndromes associated with defective Fe‐S protein maturation. Biomedicine. 2021;9(8):989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tonduti D, Dorboz I, Imbard A, et al. New spastic paraplegia phenotype associated to mutation of NFU1. Orphanet J Rare Dis. 2015;10:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Uzunhan TA, Çakar NE, Seyhan S, Aydin K. A genetic mimic of cerebral palsy: homozygous NFU1 mutation with marked intrafamilial phenotypic variation. Brain Dev. 2020;42(10):756‐761. [DOI] [PubMed] [Google Scholar]
- 9. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36(10):928‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Makrythanasis P, Maroofian R, Stray‐Pedersen A, et al. Biallelic variants in KIF14 cause intellectual disability with microcephaly. Eur J Hum Genet. 2018;26(3):330‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guillen Sacoto MJ, Tchasovnikarova IA, Torti E, et al. De novo variants in the ATPase module of MORC2 cause a neurodevelopmental disorder with growth retardation and variable craniofacial dysmorphism. Am J Hum Genet. 2020;107(2):352‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jalali Sefid Dashti M, Gamieldien J. A practical guide to filtering and prioritizing genetic variants. Biotechniques. 2017;62(1):18‐30. [DOI] [PubMed] [Google Scholar]
- 13. Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole‐genome sequencing. Nat Rev Genet. 2010;11(6):415‐425. [DOI] [PubMed] [Google Scholar]
- 14. Lindy AS, Stosser MB, Butler E, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia. 2018;59(5):1062‐1071. [DOI] [PubMed] [Google Scholar]
- 15. Thompson K, Majd H, Dallabona C, et al. Recurrent De novo dominant mutations in SLC25A4 cause severe early‐onset mitochondrial disease and loss of mitochondrial DNA copy number. Am J Hum Genet. 2016;99(4):860‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu W, Xie Y, Ma J, et al. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics. 2015;31(20):3359‐3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nizon M, Boutron A, Boddaert N, et al. Leukoencephalopathy with cysts and hyperglycinemia may result from NFU1 deficiency. Mitochondrion. 2014;15:59‐64. [DOI] [PubMed] [Google Scholar]
- 18. Ahting U, Mayr JA, Vanlander AV, et al. Clinical, biochemical, and genetic spectrum of seven patients with NFU1 deficiency. Front Genet. 2015;6:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hartman TG, Yosovich K, Michaeli HG, et al. Expanding the genotype‐phenotype spectrum of ISCA2‐related multiple mitochondrial dysfunction syndrome‐cavitating leukoencephalopathy and prolonged survival. Neurogenetics. 2020;21(4):243‐249. [DOI] [PubMed] [Google Scholar]
- 20. Lossos A, Stümpfig C, Stevanin G, et al. Fe/S protein assembly gene IBA57 mutation causes hereditary spastic paraplegia. Neurology. 2015;84(7):659‐667. [DOI] [PubMed] [Google Scholar]
- 21. Saputra L, Kumar KR. Challenges and controversies in the genetic diagnosis of hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2021;21(4):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parikh S, Goldstein A, Karaa A, et al. Patient care standards for primary mitochondrial disease: a consensus statement from the mitochondrial medicine society. Genet Med. 2017;19(12):7‐8. doi: 10.1038/gim.2017.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Casari G, Marconi R. Spastic Paraplegia 7. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews®. University of Washington, Seattle; 2006. [PubMed] [Google Scholar]
- 24. Baple E, Crosby A. Troyer Syndrome. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews®. University of Washington, Seattle; 2004. [PubMed] [Google Scholar]
- 25. Ames EG, McFadden K, Keegan CE, et al. Metabolomic profiling of plasma and cerebrospinal fluid highlight differences and diagnostic utility in multiple mitochondrial dysfunctions syndrome. In: McCabe E, McCabe L, Beitner R, et al., eds. Society for Inherited Metabolic Disorders 41th Annual Meeting. Molecular Genetics and Metabolism; 2019:219‐320. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supplemental information including case reports, supplemental methods, supplemental experimental results and discussion
Table S1: Supplemental clinical‐genetic table