

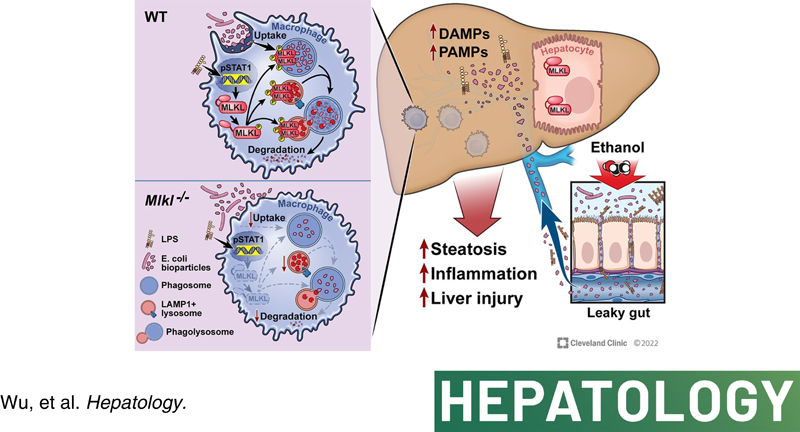
Background and Aims:
Mixed lineage kinase domain‐like pseudokinase (MLKL), a key terminal effector of necroptosis, also plays a role in intracellular vesicle trafficking that is critical for regulating liver inflammation and injury in alcohol‐associated liver disease (ALD). Although receptor interacting protein kinase 3 (Rip3) −/− mice are completely protected from ethanol‐induced liver injury, Mlkl −/− mice are only partially protected. Therefore, we hypothesized that cell‐specific functions of MLKL may contribute to ethanol‐induced injury.
Approach and Results:
Bone marrow transplants between Mlkl −/− mice and littermates were conducted to distinguish the role of myeloid versus nonmyeloid Mlkl in the Gao‐binge model of ALD. Ethanol‐induced hepatic injury, steatosis, and inflammation were exacerbated in Mlkl −/− →wild‐type (WT) mice, whereas Mlkl deficiency in nonmyeloid cells (WT→Mlkl −/− ) had no effect on Gao‐binge ethanol‐induced injury. Importantly, Mlkl deficiency in myeloid cells exacerbated ethanol‐mediated bacterial burden and accumulation of immune cells in livers. Mechanistically, challenging macrophages with lipopolysaccharide (LPS) induced signal transducer and activator of transcription 1–mediated expression and phosphorylation of MLKL, as well as translocation and oligomerization of MLKL to intracellular compartments, including phagosomes and lysosomes but not plasma membrane. Importantly, pharmacological or genetic inhibition of MLKL suppressed the phagocytic capability of primary mouse Kupffer cells (KCs) at baseline and in response to LPS with/without ethanol as well as peripheral monocytes isolated from both healthy controls and patients with alcohol‐associated hepatitis. Further, in vivo studies revealed that KCs of Mlkl −/− mice phagocytosed fewer bioparticles than KCs of WT mice.
Conclusion:
Together, these data indicate that myeloid MLKL restricts ethanol‐induced liver inflammation and injury by regulating hepatic immune cell homeostasis and macrophage phagocytosis.
INTRODUCTION
Alcohol‐associated liver disease (ALD) is a common nonviral cause of high liver‐related morbidity and mortality associated with chronic, excessive alcohol consumption.1 ALD is a spectrum of disorders ranging from steatosis to alcohol‐associated hepatitis (AH), cirrhosis, and hepatocellular carcinoma. Development of ALD is a complex process involving communications between hepatic parenchymal and nonparenchymal cells, recruitment of peripheral immune cells into the liver,2 and interorgan crosstalk between liver and other organs, including intestine and adipose tissue.3 Importantly, activation of innate immunity not only contributes to progression of ALD but is also critical for resolution of injury. Appropriate regulation of innate immune responses to multiple challenges faced in response to chronic, excessive alcohol consumption is critical to maintain liver homeostasis.4
Necroptosis, one form of programmed cell death, is widely implicated in diverse human diseases. The classical necroptotic pathway depends on receptor interacting protein kinase 3 (RIP3)–mediated phosphorylation of mixed lineage kinase domain‐like pseudokinase (MLKL), leading to its translocation to the plasma membrane, where it oligomerizes and forms pores that mediate necroptosis.5,6 MLKL also translocates to multiple intracellular compartments in a stimulus‐ and cell‐specific manner; recent evidence indicates that many of MLKL's non‐necroptotic functions involve the regulation of intracellular and extracellular vesicle trafficking.7–10 However, the impact of the necroptosis‐independent functions of MLKL in multiple disorders, including ALD, remains largely unknown. It is also unclear whether these non‐necroptotic functions of MLKL involve RIP3‐dependent or ‐independent mechanisms.
Recent work has revealed differential roles of RIP3 and MLKL in liver diseases of varied etiologies.11,12 For example, although phospho‐MLKL (c) is detected in livers of patients with drug‐induced liver injury,13 Mlkl deficiency does not prevent acetaminophen‐induced cell death and injury.14 In a murine model of autoimmune hepatitis, interferon (IFN)‐triggered MLKL‐dependent death contributes to the pathogenesis of concanavalin A–induced injury independently of RIP3.15 We and others have reported that Mlkl −/− mice are protected from liver injury and insulin resistance in murine models of obesity,9,16 associated with a RIP3‐independent regulation of autophagic flux and inhibition of necroptosis.8,9 In contrast, Mlkl deficiency only partially prevents Gao‐binge and chronic ethanol‐induced injury.17 Here, we hypothesized that cell‐specific functions of MLKL may contribute to and/or protect from injury in response to ethanol. In liver, both hepatocytes and nonparenchymal cells express MLKL, with somewhat higher expression in nonparenchymal cells.14 However, the role of MLKL in different cell types in ALD has not been investigated. In this study, we demonstrated that Mlkl deficiency in myeloid cells increased ethanol‐mediated bacterial burden and inflammatory response, thereby contributing to ethanol‐induced liver injury. Mechanistically, challenging macrophages with lipopolysaccharide (LPS)‐induced expression and phosphorylation, as well as oligomerization and translocation of MLKL to intracellular compartments, including phagosomes and lysosomes. RIP3 dependent/independent mechanisms are cooperatively involved in ethanol/LPS‐mediated phosphorylation of MLKL in vivo and in vitro. Further, pharmacological and genetic inhibition of MLKL impaired uptake and degradation of bioparticles in primary mouse Kupffer cells (mKCs) at baseline and in response to LPS. Importantly, in vivo studies revealed that hepatic macrophages in Mlkl −/− mice phagocytosed fewer bioparticles than those in wild‐type (WT) mice. Together, these findings identify an unexpected non‐necroptotic role for macrophage‐derived MLKL in the pathogenesis of ethanol‐induced liver injury through regulating the phagocytic capability of macrophages.
METHODS AND MATERIALS
Animals, bone marrow transplants, and Gao‐binge ethanol feeding
All procedures using animals were approved by the Cleveland Clinic Institutional Animal Care and Use Committee. Mlkl −/− mice were purchased from Taconic Biosciences (#TF2780) and backcrossed to a C57BL/6J background. WT controls were either C57BL/6J purchased from the Jackson Laboratory or WT littermates. WT and Mlkl −/− mice were lethally irradiated at 5 weeks of age and then transplanted with bone marrow from Mlkl −/− or WT mice and allowed to recover for 4 weeks. Mice were then treated with clodronate containing liposomes to deplete resident macrophages in the liver 7 days prior to the start of the Gao‐binge protocol. Mice were allowed free access to a Lieber‐DeCarli liquid diet (Dyets; Cat#710260) containing ethanol at 5% (vol/vol)/28% of total calories or pair‐fed control diet that isocalorically substituted maltose dextrin for ethanol for 10 days. On the final day of the experiment, pair‐fed mice were gavaged with 5 g/kg maltose and ethanol‐fed mice were gavaged with 5 g/kg ethanol in water. Mice were euthanized 6 h after gavage. Additional details of the feeding protocol and tissue collection can be found in Supporting Materials.
Human peripheral blood mononuclear cells and monocyte enrichment
The study protocol was approved by the Institutional Review Board for the Protection of Human Subjects at the Cleveland Clinic. All procedures were performed according to the IRB guidelines and all subjects provided written informed consent. Enrolled patients had confirmed diagnosis of AH by clinicians at the Cleveland Clinic based on medical history, physical examination, and laboratory results, according to the guidelines of the American College of Gastroenterology (https://gi.org/clinical‐guidelines/) (Table S1). Healthy controls (HC) were recruited from the Clinical Research Unit at the Cleveland Clinic. CD14+/CD16+ monocytes were isolated from cryopreserved peripheral blood mononuclear cells (PBMCs) from patients with AH and age‐matched HC using the protocol from Easysep human monocyte enrichment kit (#19359, Stemcell). Cryopreserved PBMCs have highly correlated expression patterns to fresh cells.18 Cells were placed in culture and treated with an MLKL inhibitor, GW806742X, for up to 24 h.
Biochemical assays, immunohistochemistry, and immunofluorescence
Detailed methods can be found in Supporting Materials.
Statistical analysis
Values shown in all figures represent the means ± SEM. Analysis of variance was performed using the general linear models procedure (SAS). Data were log‐transformed as necessary to obtain a normal distribution. Follow‐up comparisons were made by least square means testing. p values of less than 0.05 were considered significant.
RESULTS
Myeloid MLKL restricts ethanol‐induced steatosis and injury
We recently reported that although Rip3 −/− mice are protected from ethanol‐induced liver injury, Mlkl −/− mice were not protected from overall ethanol‐induced injury. However, Mlkl deficiency did ameliorate Gao‐binge–induced production of inflammatory cytokines.17 This led us to hypothesize that there might be cell‐specific contributions of MLKL in the response to ethanol. pMLKL (human, S358) immunoreactivity was increased in livers of patients with AH and detected in both hepatocytes and mononuclear immune cells (Figure 1A). Similarly, MLKL was colocalized with macrophage marker F4/80 in livers of mice exposed to Gao‐binge ethanol (Figure 1B). In order to ascertain the functional roles of MLKL in hepatocytes and immune cells, bone marrow transplant between Mlkl −/− and WT mice was conducted, and mice were then exposed to Gao‐binge ethanol (Figure S1A). As expected, macrovesicular and microvesicular steatosis was increased in WT→WT mice after ethanol feeding; WT→Mlkl −/− mice had a similar degree of steatosis, but the response was exacerbated in Mlkl −/− →WT mice (Figure 1C). Mlkl −/− →WT mice also had increased presence of inflammatory foci in response to Gao‐binge (blue arrows, Figure 1C). Consistent with the histology, Gao‐binge ethanol increased plasma aminotransferases and hepatic triglycerides to a greater extent in Mlkl −/− →WT mice, compared to WT→WT mice (Figure 1D). Few apoptotic (M30 positive) cells were detected in both WT→WT and Mlkl −/− →WT chimeras after ethanol feeding (Figure S1D). Taken together, Mlkl deficiency in myeloid cells, but not in nonmyeloid cells, exacerbated ethanol‐induced steatosis and hepatocyte injury.
FIGURE 1.
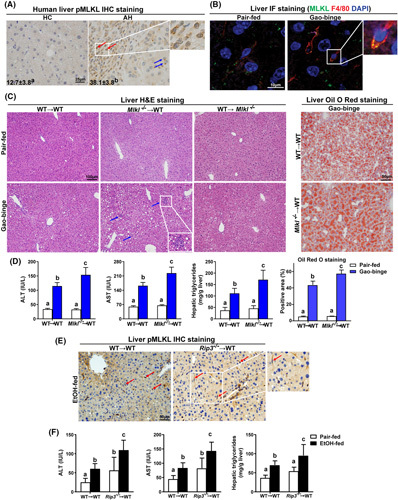
Differential roles of myeloid and nonmyeloid mixed lineage kinase domain‐like pseudokinase (Mlkl) and receptor interacting protein kinase 3 (Rip3) on Gao‐binge ethanol‐induced liver steatosis and injury. (A) Paraffin‐embedded human liver samples from healthy controls (HC; n = 10) and alcohol‐associated hepatitis (AH; n = 10) were stained for phospho‐MLKL (pMLKL; 40×) using standard immunohistochemistry (IHC) technique. Nuclei were counterstained with hematoxylin. pMLKL‐positive areas (%) were quantified using Image Pro‐Plus. pMLKL was localized in hepatocytes (blue arrows) and mononuclear immune cells (red arrows). (B) Double‐immunofluorescence (IF) staining for MLKL (green) and F4/80 (red) in murine liver from pair‐fed and Gao‐binge ethanol wild‐type (WT) mice. The magnified image illustrates colocalization (yellow). (C–F) Bone marrow chimeras were created as described in Materials and Methods and Figure S1A. Chimeras were allowed free access to Gao‐binge ethanol or pair‐fed control diets. (C) Hematoxylin and eosin (H&E) (10×) and Oil Red O (ORO; 20×) staining of livers from chimeric mice following the Gao‐binge protocol. n = 5–12 per group. (D) ALT and AST concentration in plasma, hepatic triglyceride content in whole liver homogenate, and quantification of ORO staining in Mlkl chimeras. (E) IHC staining for pMLKL (20×) in liver sections from WT→WT and Rip3 −/− →WT mice after ethanol (EtOH) feeding. Red arrows indicate positive staining in mononuclear immune cells. (F) ALT and AST concentration in plasma and hepatic triglyceride content in whole liver homogenate were detected in Rip3 chimeras, n = 3–7 per group. Values represent means ± SEM. Values with different superscripts are significantly different from each other. p < 0.05, assessed by t test (group = 2) or ANOVA (group ≥ 3).
MLKL is classically activated via phosphorylation by RIP3, but evidence indicates that MLKL can also be activated via alternative, RIP3‐independent mechanisms.19 Thus, we further investigated whether ethanol‐induced phosphorylation of MLKL (mouse, S345) in myeloid cells was RIP3‐dependent. Surprisingly, immunohistochemistry staining and morphological analysis revealed that ethanol exposure increased the phosphorylation of MLKL in mononuclear immune cells in both WT→WT and Rip3 −/− →WT mice (red arrows, Figure 1E), suggesting that phosphorylation of MLKL in response to ethanol is, at least partially, independent of RIP3. On the other hand, Rip3 −/− →WT mice phenocopied the increase in ethanol‐induced injury (Figure 1F) observed in Mlkl −/− →WT mice (Figure 1D), indicating that RIP3 kinase may also be involved in regulating with phosphorylation of MLKL in ethanol (EtOH)‐fed mice. Taken together, these data suggest that RIP3‐dependent and independent mechanisms are involved in regulation of phosphorylation of MLKL in response to ethanol.
Mlkl deficiency in myeloid cells exacerbated bacterial burden and inflammatory response in the liver of ethanol‐fed mice
Ethanol exposure increases bacterial and pathogen‐associated molecular pattern (PAMP) translocation from the gut into liver, stimulating an inflammatory response in the liver and contributing to liver injury.20,21 Bacterial DNA (16S ribosomal RNA [rRNA]) in the liver was moderately increased in WT→WT mice in response to the Gao‐binge; this response was exacerbated in Mlkl −/− →WT mice (Figure 2A). Consistent with the histological evidence for increased inflammatory foci after Gao‐binge ethanol feeding to Mlkl −/− →WT mice (blue arrows, Figure 1C), expression of messenger RNA (mRNA) for proinflammatory cytokines, including Tnf‐α and Il‐1β, and chemokines such as monocyte chemoattractant protein (Mcp‐1) and C‐X‐C motif chemokine (Cxcl5) in liver was also higher in Mlkl −/− →WT chimeric mice compared to WT→WT mice (Figure 2B). Mlkl deficiency in myeloid cells also increased accumulation of immune cells in the liver in response to ethanol, with increased staining for F4/80 (resident macrophage marker) (Figure 2C), NIMP‐R14 (neutrophil marker) (Figure 2D), and Ly6C (infiltrating monocytes) (Figure 2E).
FIGURE 2.

Effect of mixed lineage kinase domain‐like pseudokinase (Mlkl) deficiency in myeloid cells on Gao‐binge ethanol‐induced bacterial burden and inflammatory responses. (A) Accumulation of 16S ribosomal RNA (rRNA) bacterial DNA in livers from wild‐type (WT)→WT and Mlkl −/− →WT mice was assessed by quantitative real‐time polymerase chain reaction (qRT‐PCR). (B) Expression of messenger RNA for cytokines, including Tnf‐α, Il‐1β, and monocyte chemoattractant protein (Mcp‐1), and C‐X‐C motif chemokine (Cxcl5) in livers from WT→WT and Mlkl −/− →WT mice was assessed by qRT‐PCR and normalized to 18S rRNA. (C) Immunohistochemistry (IHC) staining (10×) for F4/80 in livers from transplanted mice, n = 5–7. Paraffin‐embedded livers were deparaffinized followed by (D) NIMP‐R14 (neutrophil) IHC and (E) LY6C (monocyte) immunofluorescence (IF) staining, n = 5–7. Representative images are shown. Images were acquired using 10× and 20× objectives, respectively. Red arrows indicate positive staining. Positive numbers were quantified using Image Pro‐Plus. Values represent means ± SEM; values with different superscripts are significantly different from each other; p < 0.05.
Accumulation of bacteria in the liver is likely due, at least in part, to disruption of the gut mucosal barrier, allowing for increased bacterial translocation to the liver. However, it is also possible that clearance of bacteria is impaired due to dysfunctional phagocytosis of hepatic macrophages. Although ethanol is known to disrupt the integrity of the gut barrier4; much less is known about the impact of ethanol on the phagocytic capability of macrophages. Analysis of a published RNA‐sequencing (RNA‐seq) dataset (GSE143318) revealed 3458 genes that were significantly changed in livers of patients with severe AH (sAH) compared to HC. Among them, differentially expressed genes (DEGs) were enriched in biological processes involving intracellular vesicle trafficking, especially phagocytosis and endocytosis (Figure 3A). This was further confirmed by an upset plot analysis (Figure 3B) that illustrates the intersection between DEGs enriched in intracellular vesicle trafficking in patients with sAH from two published liver RNA‐seq datasets and genes enriched in four gene ontology biological process classes, including phagocytosis, endocytosis, and lysosomes. Principal component analysis (Figure S2A) of the DEGs enriched in intracellular vesicle trafficking clearly separated patients with AH (sAH and expl_AH: explants from patients with AH who underwent early transplantation) and HC in the Pitt_RNA_seq dataset whereas patients with early alcohol‐associated steatohepatitis (ASH) clustered close to HC. Heatmaps illustrating DEGs involved in intracellular vesicle trafficking (Figure S2B) are consistent with these data on dysregulated expression of multiple genes in livers of patients with sAH and expl_AH compared to those with early ASH or healthy subjects. Consistent with the DEG data, analysis of a proteomics dataset22 revealed that 2349 proteins were significantly changed in livers of patients with sAH. Multiple differentially expressed proteins were enriched in intracellular vesicle trafficking pathways, especially phagocytosis and endocytosis (Figure 3C,D).
FIGURE 3.
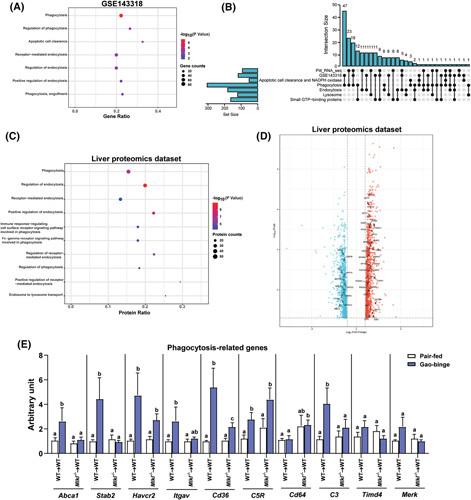
Bioinformatic analysis of liver RNA‐sequencing and proteomics data from patients with alcohol‐associated hepatitis (AH) and effect of mixed lineage kinase domain‐like pseudokinase (Mlkl) deficiency in myeloid cells on expression of phagocytosis‐related genes in liver in response to ethanol. (A) Gene ontology biological process (GO‐BP) enrichment analysis was performed based on differentially expressed genes (DEGs) involved in intracellular vesicle trafficking pathways between healthy controls (HC; n = 5) and patients with severe AH (sAH; n = 5) in GSE143318 dataset. (B) An upset plot visualized the intersection between DEGs enriched in intracellular vesicle trafficking in HC and patients with sAH from Pitt_RNA_seq and GSE143318 datasets and genes in multiple GO‐BP classes and key protein systems, including apoptotic cell clearance, phagocytosis, lysosome, small guanosine triphosphate (GTP)‐binding proteins, and NADPH oxidase. (C) GO‐BP enrichment analysis was performed based on differentially expressed proteins (DEPs) involved in intracellular vesicle trafficking pathways between HC (n = 12) and patients with sAH (n = 6) in the global proteomics dataset.22 (D) The volcano plot revealed upregulated and downregulated proteins that were enriched in intracellular vesicle trafficking in the proteomics dataset.22 (E) Expression of messenger RNA (mRNA) for phagocytic genes including ATP binding cassette subfamily A member (Abca1), stabilin (Stab2), hepatitis A virus cellular receptor (Havcr2), intergrin subunit alpha V (Itgav), Cd36, C5r, Cd64, C3, T cell immunoglobulin and mucin domain containing (Timd4), and MER proto‐oncogene, tyropsine kinase(Merk) in livers from wild‐type (WT)→WT and Mlkl −/− →WT mice was assessed by quantitative real‐time polymerase chain reaction and normalized to 18S ribosomal RNA. n = 4–6. Values represent means ± SEM; values for each mRNA with different superscripts are significantly different from each other; p < 0.05.
Because myeloid Mlkl restricts ethanol‐induced liver injury and phagocytic pathways were dysregulated in patients with AH, we hypothesized that Mlkl deficiency in myeloid cells might affect the phagocytosis process in macrophages, exacerbating hepatic injury. Surprisingly, ethanol feeding markedly upregulated expression of mRNA for multiple phagocytosis‐related genes (Abca1, Stab2, Havcr2, Itgav, Cd36, C5r, and C3) in WT→WT mice (Figure 3E). Interestingly, these ethanol‐induced increases (Abca1, Stab2, Cd36, and C3) were prevented in Mlkl −/− →WT mice (Figure 3E), which is associated with greater accumulation of 16S rRNA in Mlkl −/− →WT (Figure 2A). Together, these data indicate that Mlkl deficiency in myeloid cells exacerbates ethanol‐mediated bacterial burden and inflammatory response in the liver, associated with impaired phagocytic capacity of hepatic macrophages.
Challenging macrophages with LPS‐induced expression, phosphorylation, and nuclear accumulation of MLKL
In order to understand the mechanistic contributions of MLKL to macrophage function in the context of ethanol, we exposed macrophage‐like RAW 264.7 cells and bone marrow–derived macrophages (BMDMs) to LPS, one of the major contributors to ALD progression. Exposure of RAW cells and BMDMs to LPS increased expression of Mlkl, but not Rip3, mRNA (Figures 4A and S3A). LPS induced expression of MLKL protein in a time‐dependent manner in cultured RAW cells (Figure 4B), whereas EtOH did not further increase LPS‐induced expression of MLKL (Figure S3B). Treatment of BMDMs with LPS for 24 h increased both expression and phosphorylation of MLKL (Figure 4C).
FIGURE 4.

Expression and phosphorylation of mixed lineage kinase domain‐like pseudokinase (MLKL) in lipopolysaccharide (LPS)‐treated macrophages. (A) Expression of messenger RNA (mRNA) for receptor interacting protein kinase (RIP) 1‐RIP3‐MLKL‐axis in bone marrow–derived macrophages (BMDMs) after challenge with 10 ng/ml LPS for 24 h. (B) MLKL protein in lysates of LPS‐treated RAW cells over 24 h was assessed by western blot and normalized to HSC70. (C–E) Protein expression of phospho‐MLKL (pMLKL), MLKL, RIP3, phospho‐STAT1 (pSTAT1), and signal transducer and activator of transcription 1 (STAT1) in (C) BMDM lysates, (D) RAW cell lysates, and (E) RAW cell cytosolic and nuclear fractions after LPS challenge assessed by western blot and normalized to HSC70 or GAPDH. (D,E) RAW cells were pretreated with or without 25 μM Fludarabine (Fludara) for 2 h prior to challenge with LPS. (E) Lamin B1 and GAPDH were employed as loading controls for nuclear and cytoplasmic fractions, respectively. (F) RAW cells were pretreated with or without 5 μM GSK872 for 2 h prior to challenge with LPS and phosphorylation of MLKL assessed by western blot. Values represent means ± SEM. Values with different superscripts are significantly different from each other; n = 3–6, p < 0.05.
MLKL transcription in hepatocytes is mediated, at least in part, by IFN‐γ–dependent signal transducer and activator of transcription 1 (STAT1) activation15; STAT1 is also an important mediator for LPS‐mediated macrophage activation.23 Challenge of BMDMs or RAW cells with LPS increased phospho‐STAT1 (Figure 4C,D). Pretreatment of RAW cells with fludarabine, an inhibitor of STAT1 activation, decreased LPS‐mediated MLKL expression (Figure 4D). Interestingly, although the majority of MLKL remained in non‐nuclear fraction, both MLKL and pMLKL protein were detected in the nuclei in response to LPS. Inhibition of STAT1 activation with fludarabine suppressed the accumulation and phosphorylation of MLKL in both cytosolic and nuclear fractions in response to LPS (Figure 4E). LPS and fludarabine had no effect on RIP3 protein in either whole cell lysates or subcellular fractions (Figure 4D,E).
To test if RIP3 mediates LPS‐triggered phosphorylation of MLKL in vitro, an inhibitor of RIP3 kinase, GSK872, was employed to suppress RIP3 kinase activity. GSK872 only partially reduced MLKL phosphorylation in LPS‐treated macrophages (Figure 4F), consistent with the concept that both RIP3‐dependent and independent mechanisms work cooperatively to regulate phosphorylation of MLKL in macrophages. Taken together, these data demonstrate that LPS induces expression, phosphorylation, and nuclear accumulation of MLKL in macrophages through activation of STAT1 in RIP3‐dependent and independent mechanisms.
LPS‐triggered translocation of MLKL to multiple intracellular vesicular compartments in macrophages
Given the critical role of MLKL in necroptotic death signaling, we sought to determine if MLKL translocated to the plasma membrane of macrophages and mediated cell death in response to LPS. MLKL did not colocalize with phalloidin staining for F‐actin on the cell surface in BMDMs challenged with LPS (Figure 5A). In line with the absence of MLKL at the plasma membrane, challenge with LPS alone did not trigger death in BMDMs (Figure S3C).
FIGURE 5.

Lipopolysaccharide (LPS)‐trigged translocation of mixed lineage kinase domain‐like pseudokinase (MLKL) in macrophages. (A) Bone marrow–derived macrophages (BMDMs) isolated from C57BL/6J mice were exposed to LPS for 24 h. Colocalization of MLKL with Alexa Fluor–labeled phalloidin, which stains cell surface–associated F‐actin, was examined by confocal microscopy. (B,D) P10 (10,000 g pellet) fraction was isolated from LPS‐treated RAW cells in response to 1 μM GW806742X or 5 μM GSK872. (C) P10 and plasma membrane fractions were isolated from LPS‐treated RAW cells. (B–D) Proteins were resolved by nonreducing plPAGE and probed with antibody to MLKL. Images are representative on n = 3 independent experiments. (E,F) BMDMs were exposed to LPS, as described in Figure 4. BMDMs were incubated with CellLight BacMam 2.0 dye for overnight to label early endosomes, late endosomes, and Golgi. (E) Colocalization of MLKL and (F) quantification with phagosomes, lysosomes, early endosomes, late endosomes, and Golgi was examined by confocal microscopy. All images were obtained using a 40× objective (Zoom 4). Representative images are shown. Single‐channel images are shown in Figure S4. Values represent means ± SEM. Values with different superscripts are significantly different from each other; n = 3–4 independent BMDM isolations, p < 0.05. IF, immunofluorescence; IB, immunoblotting.
The classical pathway of necroptosis is mediated by the formation of oligomeric, channel‐like MLKL structures at the plasma membrane. Although LPS did not trigger movement of MLKL to the cell surface, we next investigated whether MLKL also forms oligomers in the cytosolic cell fraction. Crude intracellular membrane fractions (P10) were isolated from RAW cells treated with or without GW806742X, an MLKL inhibitor. At baseline, MLKL oligomers were not detected. However, MLKL oligomers were detected in the P10, but not in the plasma membrane, fractions when macrophages were challenged with LPS; this response was completely suppressed by GW806742X (Figure 5B,C). The RIP3 inhibitor, GSK872, only partially reduced MLKL oligomerization in response to LPS (Figure 5D), consistent with the in vivo data indicating that the activation of MLKL is only partially dependent on RIP3 (Figure 1E,F).
Using markers for multiple intracellular compartments, we found that LPS induced translocation of MLKL to multiple intracellular vesicles, including phagosomes, lysosomes, early endosomes, late endosomes, and Golgi (Figure 5E,F) but not mitochondria (Figure S3D). These data suggest that non‐necroptotic functions of MLKL mediate responses to LPS in macrophages via interactions with intracellular vesicles in mechanisms that are only partially dependent on RIP3 kinase activity.
Genetic and pharmacological inhibition of MLKL impaired phagocytic capability of primary mKCs and cultured RAW cells
Macrophages are the major phagocytes in the innate immune system, engulfing microbial pathogens as well as cellular debris and dead cells into specialized vacuoles called phagosomes that fuse with lysosomes for particle degradation. Because LPS stimulated intracellular translocation of MLKL to phagosomes/lysosomes in macrophages, we hypothesized that MLKL contributes to the regulation of the phagocytic process in macrophages. To test this hypothesis, we performed a phagocytosis assay with pHrodo red fluorescence conjugated Escherichia coli bioparticles, which fluoresce brightly red in acidic compartments, including lysosomes and phagolysosomes. We first evaluated the effect of LPS and/or EtOH on phagocytosis activity of isolated primary mKCs. Interestingly, compared to nontreated cells, more bioparticles were detected in LPS‐treated primary mKCs, whereas fewer bioparticles accumulated in cells exposed to EtOH alone (Figure 6A). LPS still stimulated bioparticle uptake in EtOH‐treated cells but to a lesser extent than in the absence of ethanol, with some bioparticles accumulating on the cell surface (Figure 6A). These findings demonstrate that LPS increased bacterial uptake in primary mKCs; in contrast, EtOH impaired basal and LPS‐mediated uptake of bioparticles.
FIGURE 6.
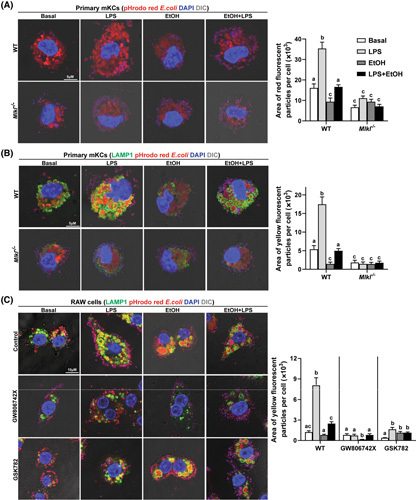
Effect of genetic and pharmacological inhibition of mixed lineage kinase domain‐like pseudokinase (MLKL) on phagocytosis by macrophages. (A,B) Primary mouse Kupffer cells (mKCs) isolated from C57BL/6J and Mlkl −/− mice were challenged with 10 ng/ml lipopolysaccharide (LPS), 50 mM ethanol (EtOH), or LPS + EtOH for 24 h following incubation with 50 μg/ml pHrodo red fluorescence conjugated Escherichia coli bioparticles for 3 h. Representative confocal images and quantification of (A) engulfed bioparticles and (B) particles colocalized with lysosomal associated membrane protein 1 (LAMP1); n = 4. (C) RAW cells were pretreated with GW806742X or GSK872 for 2 h and then exposed to LPS, EtOH, or LPS + EtOH for 24 h, as described above, followed by incubation with bioparticles for 3 h. Representative confocal images and quantification of colocalization with LAMP1 are shown. All images were obtained using a 63× objective (Zoom 4). Values represent means ± SEM. Values with different superscripts are significantly different from each other within the same color bars; n = 3, p < 0.05. WT, wild type. DIC, differential interference contrast.
Lysosomes are the terminal degradative compartment of phagocytosis.24 To further assess if LPS and/or ethanol affects the intracellular degradativsse process after macrophages phagocytose foreign bioparticles, we assessed the colocalization of engulfed particles with lysosomal associated membrane protein 1 (LAMP1). Exposure of primary mKCs to LPS increased the number of LAMP1+ lysosomes with engulfed bioparticles. This response was inhibited by ethanol. Primary mKCs challenged only with ethanol also had fewer particles engulfed by LAMP1+ lysosomes than untreated mKCs (Figure 6B). Collectively, these data demonstrate that in addition to the uptake step, LPS enhanced bioparticle association with lysosomes. Importantly, both stages were impaired by ethanol.
Next, we explored the role of MLKL in mediating phagocytosis in the context of LPS and/or EtOH. Mlkl‐deficient primary mKCs phagocytosed fewer E. coli bioparticles than primary mKCs from WT mice at baseline (Figure 6A). In response to LPS, primary mKCs from Mlkl −/− mice were unable to phagocytose many bioparticles; instead, they were accumulated at the cell surface (Figure S5B). Mlkl deficiency also prevented LPS‐mediated increase of bacterial uptake in the presence of ethanol (Figure 6A). Similarly, Mlkl deficiency reduced the number of bioparticles engulfed by LAMP1+ lysosomes at baseline and in response to LPS with or without EtOH (Figure 6B). Pharmacological inhibition of MLKL with GW806742X and RIP3 with GSK872 also reduced uptake and degradation of bioparticles in LPS‐treated RAW cells with/without EtOH (Figure 6C).
Pharmacological inhibition of MLKL decreased bacterial uptake of peripheral monocytes from HC and patients with AH
Using our published single‐cell RNA‐seq data from PBMCs from HC and patients with AH,18 we further found that mRNA levels of genes for membrane trafficking including Lamp1/2 and Rab5a/Rab7a/Rab2b, reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (p22‐phox/gp91‐phox/p40‐phox/p47‐phox) and phagocytic receptors (Itgav/Cd36/Abca1), and components of endosomal sorting complexes required for transport (ESCRT) complex were dysregulated in different clusters of monocytes from patients with AH (Figure S6). To explore the functional impact of these changes in gene expression, we assessed the phagocytic capability of PBMCs isolated from HC and patients with AH. Using flow cytometry, we found that the uptake of fluorescent bioparticles per cell was lower in CD14+ cells from patients with AH compared to HC (Figure 7B). Confocal analysis of isolated CD14+/CD16+ monocytes also revealed that AH monocytes phagocytosed fewer bioparticles than HC monocytes (Figure 7C).
FIGURE 7.

Mixed lineage kinase domain‐like pseudokinase (MLKL) inhibitor, GW806742X, impaired uptake of bioparticles in circulating monocytes from healthy controls (HC) and patients with alcohol‐associated hepatitis (AH). (A) Expression of messenger RNA (mRNA) for receptor interacting protein kinase (RIP) 1‐RIP3‐MLKL‐axis in peripheral blood mononuclear cells (PBMCs) isolated from HC and patients with AH. n = 5–9. (B) Human PBMCs were pretreated with or without GW806742X and then incubated with Escherichia coli bioparticles; n = 3. Mean fluorescent intensity (MFI) of bioparticles in CD14+ monocytes was assessed by flow cytometry, and MFI per cell was then calculated. (C) Engulfed bioparticles in CD14+/CD16+ monocytes isolated from human PBMCs were visualized by confocal microscopy. All images were obtained using a 63× objective (Zoom 4); n = 3. Values represent means ± SEM. Values with different superscripts are significantly different from each other within the same color bars; p < 0.05. hRIP1, human RIP1. hRIP3, human RIP3. hMLKL, human MLKL. DIC, differential interference contrast.
We next investigated if MLKL was involved in regulating phagocytosis of circulating monocytes. mRNA expression of Mlkl, but not Rip1 and Rip3, was higher in PBMCs from patients with AH, compared to HC (Figure 7A). Importantly, GW806742X decreased accumulation of bioparticles in CD14+ cells from PBMCs of HC and patients with AH (Figure 7B). Confocal analysis showed similar results, with fewer bioparticles in monocytes isolated from both HC and patients with AH after GW806742X treatment (Figure 7C). Taken together, GW806742X suppresses phagocytosis of peripheral monocytes from HC and patients with AH.
Mlkl deficiency impaired phagocytic capability of hepatic Kupffer cells in vivo
To confirm the role of MLKL in regulating phagocytosis activity of hepatic Kupffer cells (KCs) in vivo, pHrodo red E. coli bioparticles were injected into WT and Mlkl −/− mice. The radiant efficiency of hepatic fluorescence was measured at 15, 30, and 60 min after injection. The fluorescent signal was strongest at 30 min (Figure 8A); therefore, subsequent experiments were performed at this time point. Compared to WT mice, fluorescence intensity of the entire liver was decreased in Mlkl −/− mice after injection (Figure 8B). Importantly, Gao‐binge ethanol lowered the fluorescence intensity in livers of WT mice compared to controls; intensity was further decreased in Mlkl −/− mice (Figure 8B). When liver sections collected at the 30 min time point were analyzed ex vivo by microscopy, accumulation of engulfed bioparticles in the liver was reduced by Mlkl deficiency at baseline and in response to ethanol (Figure 8C). Importantly, colocalization (yellow) of bioparticles (red) with F4/80+ macrophages (green) indicates that the bioparticles were taken by hepatic macrophages. Hepatic KCs of Mlkl −/− mice phagocytosed fewer bioparticles than KCs of WT mice both at baseline and in response to ethanol (Figure 8D), demonstrating that Mlkl deficiency impaired phagocytic capability of hepatic KCs in vivo.
FIGURE 8.
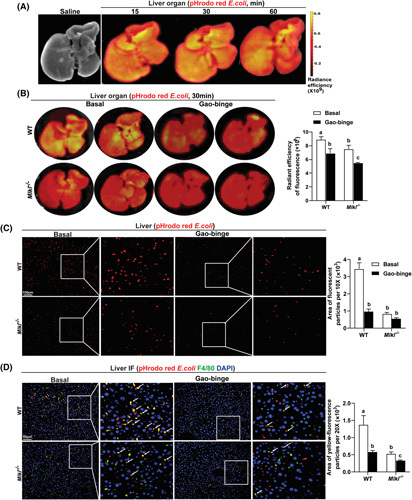
Mixed lineage kinase domain‐like pseudokinase (Mlkl) deficiency inhibited phagocytosis by hepatic macrophages in vivo. (A) pHrodo red conjugated Escherichia coli bioparticles (50 mg/kg) were injected into C57BL/6J mice by tail vein and the radiant efficiency of fluorescence in the entire liver organ was measured by in vivo imaging system (IVIS) at 15, 30, and 60 min after injection. Saline was employed as the vehicle control. (B) E. coli bioparticles were injected into wild‐type (WT) and Mlkl −/− mice on chow diet or Gao‐binge ethanol diet. The radiant efficiency of hepatic fluorescence was measured by IVIS after 30 min. Images from individual animals are shown, n = 3–4 per group. (C,D) Microscopic analysis for (C) E. coli bioparticles only (red) or (D) colocalization (yellow) of pHrodo red E. coli bioparticles with F4/80 (green) in liver sections from both genotypes. Values represent means ± SEM. Values with different superscripts are significantly different from each other within the same color bars; n = 3–4, p < 0.05.
DISCUSSION
The canonical role of MLKL as the executor of necroptosis is relatively well understood; however, the non‐necroptotic functions of MLKL in homeostasis and disease, including ALD, have not been extensively investigated. Here, we identified a non‐necroptotic function in which MLKL translocates to phagosomes and lysosomes and regulates phagocytosis in myeloid cells. Regulation of phagocytosis by MLKL contributed to the maintenance of hepatic immune homeostasis and immune cell function in ethanol‐induced liver injury. These findings expand our current understanding of the noncanonical functions of MLKL in macrophages.
Alcohol consumption results in translocation of microbiota, microbial components, and metabolites through the damaged gut barrier into the circulation and liver, resulting in both systemic and local inflammatory responses that contribute to liver injury.25 Translocation of viable bacteria to the liver contributes to hepatic injury in murine models of ALD.21,26 In a model of high fat diet‐induced liver injury, E. coli strain NF73‐1 aggravated disease progression by translocating into the liver and increasing proinflammatory responses.27 Given this increased exposure to bacteria and bacterial products, bacterial clearance becomes an important means to prevent inflammation and injury. Bacterial clearance is primarily mediated by myeloid cells, notably macrophages and monocytes. EtOH dampens alveolar macrophage phagocytosis in vivo and in vitro,28–31 likely associated with modifications in actin organization32 and changes in the activity of downstream regulators of actin, such as small guanosine triphosphatases (GTPases).33,34 However, the role of EtOH in phagocytosis of hepatic macrophages and the underlying regulatory mechanisms are understudied. Our bioinformatics analysis revealed that genes and proteins enriched in phagocytic process were dysregulated in livers of patients with AH. Recently, Schnabl's group reported that CRIg, a complement receptor, mediates KC phagocytosis of gut‐derived Enterococcus faecalis and protects mice from ethanol‐induced liver injury.20 Here, we also found that ethanol impaired uptake of E. coli bioparticles in primary mKCs and circulating monocytes from patients with AH. Further, ethanol inhibited lysosome‐mediated degradation of bioparticles, consistent with a previous report showing that ethanol affects transcription factor EB‐mediated lysosome biogenesis.35 Importantly, hepatic macrophages from ethanol‐fed mice phagocytosed fewer bioparticles than cells from control mice; phagocytosis was further reduced by Mlkl deficiency. However, challenging macrophages with ethanol in vitro had no effect on MLKL expression. Thus, MLKL, is necessary, but not sufficient, to regulate ethanol‐mediated phagocytosis.
MLKL‐mediated necroptosis is an important mechanism by which MLKL functions to limit bacterial dissemination and regulate the inflammatory response in different scenarios. For example, bacterial burden and inflammation are exacerbated in Mlkl −/− mice in response to infection with Staphylococcus aureus.36 Further, in acute and chronic models of methicillin‐resistant S. aureus infection, MLKL‐dependent necroptosis prevented bacterial replication in the blood and organs via facilitation of the formation of neutrophil extracellular trap.37 However, it is unknown whether non‐necroptotic functions of MLKL also contribute to susceptibility to bacterial infection or bacterial clearance.
In the current manuscript, transplant of bone marrow from Mlkl −/− mice to WT mice increased accumulation of bacterial DNA in liver and production of inflammatory cytokines in response to Gao‐binge ethanol exposure. We recently reported that MLKL oligomers are detectable in intracellular membrane fractions, but not in the plasma membrane fraction, in livers from EtOH‐fed mice.17 Challenge of macrophages with LPS‐induced translocation and oligomerization of MLKL to specific intracellular membrane compartments but not to plasma membrane. Together, these data suggest that noncanonical functions of MLKL, independent of its ability to disrupt plasma membrane integrity, are important for maintaining functionality of myeloid cells. Indeed, our studies revealed that the protective effect of myeloid MLKL in a Gao‐binge ethanol model was associated with regulation of phagocytosis in macrophages, rather than necroptosis. Intriguingly, cell‐specific roles of MLKL are also implicated in other disease models. For example, nonmyeloid MLKL protected intestinal defenses against Salmonella infection via inflammasome activation in intestinal epithelial cells, thereby limiting early bacterial colonization, in bone marrow chimeras.38 Interestingly, we found no protective role of MLKL in the nonmyeloid compartment in ethanol‐induced injury. An additional non‐necroptotic mechanism for MLKL action in host defense involves direct binding of MLKL to Listeria; this interaction disrupted their plasma membranes and inhibited intracellular replication without triggering cell death.39 More recently, Yoon et al. found that a sequential process of phosphorylation and ubiquitination of MLKL in HT‐29 and L929 cells enhanced bacterial translocation from endosomes to lysosomes, resulting in reduced infection.40 This work is consistent with MLKL localization with phagosomes and lysosomes and regulation of bacterial clearance in hepatic macrophages. Collectively, these findings suggest that, in addition to the ability of necroptotic functions of MLKL to regulate host defense, noncanonical functions of MLKL also contribute to host defense against microorganisms. It is also possible that MLKL contributes to efferocytosis which serves to remove apoptotic cells. However, efferocytosis is not likely a contributor to Gao‐binge ethanol‐induced injury because only a few apoptotic hepatocytes were detected in chimeras (Figure S1D). Studies on the role of MLKL in efferocytosis will be of interest for future studies utilizing disease models with more predominant hepatocyte apoptosis, such as hepatic fibrosis.
Accumulating evidence indicates that MLKL can be activated via alternative RIP3‐independent mechanisms.15,41 Other kinases, such as TAM (Tyro3, Axl, and Mer) family members, promote necroptotic cell death with RIP3‐dependent phosphorylation of MLKL,42 suggesting the possibility that other kinases cooperate with RIP3 to facilitate MLKL‐mediated necroptosis. Our data suggest that such cooperative phosphorylation is likely taking place in myeloid cells in response to ethanol/LPS. For example, although Rip3 −/− →WT mice phenocopied Gao‐binge–induced injury seen in the Mlkl −/− →WT mice; Gao‐binge ethanol increased phosphorylation of MLKL in mononuclear immune cells even in Rip3 −/− →WT mice. Moreover, the RIP3 kinase inhibitor GSK872 only partially suppressed LPS‐induced phosphorylation and oligomerization of MLKL in macrophages. Together, these in vivo and in vitro data suggest that RIP3‐dependent and independent mechanisms are involved in ethanol‐induced activation of the regulatory pathways controlling phosphorylation of MLKL. A phospho‐ or ubiquitin‐proteomics approach paired with targeted mechanistic strategies will be beneficial to investigate RIP3‐independent mechanisms by which ethanol activates MLKL signaling.
In addition to the translocation of MLKL to multiple intracellular vesicles, MLKL was also detected in the nucleus in macrophages challenged with LPS. This finding is consistent with previous work showing that the MLKL kinase domain has a nuclear localization sequence motif, which is required for nuclear shuttling.43 MLKL translocates to the nucleus prior to executing necroptosis in diverse human and mouse cell types.43 For example, MLKL translocated to the nucleus of endothelial cells challenged with TNF, where it interacted with RNA‐binding motif 6 and promoted expression of adhesion molecules by regulating their mRNA stability.44 Although the precise function of MLKL in the nucleus is not understood, it is most likely not directly involved in necroptosis. Nuclear accumulation of MLKL might be associated with the regulation of phagocytic gene expression through interacting with RNA‐binding proteins. This will be an interesting future direction to understand the mechanism for nuclear accumulation of MLKL in macrophages.
In the last decade, numerous advances have illuminated the important involvement of MLKL in intracellular and extracellular vesicle trafficking, including regulation of autophagy7–9 and exosome generation.10 Our data add macrophage phagocytosis to the growing list of important roles that MLKL plays in regulating membrane trafficking. Our data suggest that accumulation of bacterial DNA in the liver is due, at least in part, to impaired phagocytosis and clearance by hepatic macrophages in Mlkl −/− →WT mice in response to Gao‐binge ethanol exposure. However, we cannot exclude the possibility that Mlkl −/− →WT mice have exacerbated disruption of the gut barrier and/or bacterial overgrowth in response to ethanol, allowing for increased bacterial translocation. Future studies, by making use of cell‐specific knockouts, can explore more precisely the role of MLKL in specific cell types, such as intestinal epithelial cells and specific immune cells.
In summary, our work identifies for the first time the role of macrophage MLKL in hepatic innate immune homeostasis and function and investigates the contributions of myeloid and nonmyeloid MLKL to ALD progression. These findings provide a mechanistic insight into the pathophysiology of ALD and advance our knowledge of necroptosis‐independent functions of MLKL in regulation of macrophage phagocytosis via interaction with phagosomes and lysosomes.
Supplementary Material
Acknowledgments
AUTHOR CONTRIBUTIONS
Concept and design: Xiaoqin Wu, Laura E. Nagy. Acquisition of data; analysis and interpretation of data: Xiaoqin Wu, Megan R. McMullen, Xiude Fan, Tatsunori Miyata, Adam Kim, Jianguo Wu, Vai Pathak, Le Z. Day, Josiah E. Hardesty, Jon M. Jacobs, Daniela S. Allende, Daniel M. Rotroff, Laura E. Nagy. Collection of patient samples: Nicole Welch, Jaividhya Dasarathy, Arthur J. McCullough, Srinivasan Dasarathy. Drafting of the manuscript: Xiaoqin Wu, Laura E. Nagy. Critical revision of the manuscript for important intellectual content: Xiaoqin Wu, Laura E. Nagy. Statistical analysis: Xiaoqin Wu, Laura E. Nagy. Obtained funding: Laura E. Nagy, Xiaoqin Wu, Srinivasan Dasarathy, Daniela S. Allende, Xiude Fan, Tatsunori Miyata, Adam Kim, Jianguo Wu, Nicole Welch, Daniel M. Rotroff, Jon M. Jacobs.
ACKNOWLEDGMENTS
This work was supported in part by National Institutes of Health (NIH) grants P50 AA024333 (Laura E. Nagy, Srinivasan Dasarathy, Daniel M. Rotroff, Daniela S. Allende, Jaividhya Dasarathy), R01 AA027456 (Laura E. Nagy), U01 AA026938 (Laura E. Nagy), R01 AA023722 (Laura E. Nagy), K99 AA029146 (Xiaoqin Wu), R21 AR071046, R01 GM119174, R01 DK113196 and U01 AA021890 (Srinivasan Dasarathy), K99 AA028048 (Adam Kim), K01 AA029474, P30 DK097948 (pilot project) (Jianguo Wu), K08 AA028794 (Nicole Welch), and R21AA028117 (Jon M. Jacobs) and by China Scholarship Council (File:201806280215) (Xiude Fan) and JSPS Overseas Research Fellowship 201960331 (Tatsunori Miyata). Collection of patient samples used for the proteomics dataset was supported by NIH grants P50AA024337 (Craig J. McClain) and R24AA025017 (ZhaoLi Sun). This work utilized the IVIS Spectrum Pre‐clinical In Vivo Imaging System that was supported by NIH Shared Instrument Grant Award S10OD018205. We thank Jeannette Wat for her help in conducting the RIP3 bone marrow transplant studies.
CONFLICT OF INTEREST
Daniel M. Rotroff owns stock in Clarified Precision Medicine. Daniela S. Allende advises Incyte.
Footnotes
Funding information China Scholarship Council, Grant/Award Number: File:201806280215; JSPS Overseas Research Fellowship, Grant/ Award Number: 201960331; National Institute of Arthritis and Musculoskeletal and Skin Diseases, Grant/Award Number: R21 AR071046; National Institute of Diabetes and Digestive and Kidney Diseases, Grant/Award Number: P30 DK097948 and R01 DK113196; National Institute of General Medical Sciences, Grant/Award Number: R01 GM119174; National Institute on Alcohol Abuse and Alcoholism, Grant/Award Number: K01 AA029474, K08 AA028794, K99 AA028048, K99 AA029146, P50 AA024333, P50 AA024337, R01 AA023722, R01 AA027456, R21 AA028117, R24 AA025017, U01 AA021890 and U01 AA026938
Abbreviations: ABCA1, ATP binding cassette subfamily A member; AH, alcohol‐associated hepatitis; ALD, alcohol‐associated liver disease; BMDM, bone marrow–derived macrophage; DEG, differentially expressed gene; EtOH, ethanol; HAVCR2, hepatitis A virus cellular receptor 2; HC, healthy control; KC, Kupffer cell; IHC, immunohistochemistry; ITGAV, intergrin subunit alpha V; LAMP1, lysosomal associated membrane protein 1; LPS, lipopolysaccharide; MERK, MER proto‐oncogene, tyropsine kinase; mKC, mouse Kupffer cell; MLKL, mixed lineage kinase domain‐like pseudokinase; mRNA, messenger RNA; NADPH, reduced nicotinamide adenine dinucleotide phosphate; NIH, National Institutes of Health; PBMC, peripheral blood mononuclear cell; pMLKL, phospho‐MLKL; RIP3, receptor interacting protein kinase 3; RNA‐seq, RNA‐sequencing; rRNA, ribosomal RNA; sAH, severe alcohol‐associated hepatitis; STAB2, stabilin 2; STAT1, signal transducer and activator of transcription 1; TIMD4, T cell immunoglobulin and mucin domain containing 4; WT, wild type.
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.hepjournal.com.
REFERENCES
- 1.Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol. 2019;70:151–71. [DOI] [PubMed] [Google Scholar]
- 2.Nagy LE, Ding WX, Cresci G, Saikia P, Shah VH. Linking pathogenic mechanisms of alcoholic liver disease with clinical phenotypes. Gastroenterology. 2016;150:1756–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang H, Mehal W, Nagy LE, Rotman Y. Immunological mechanisms and therapeutic targets of fatty liver diseases. Cell Mol Immunol. 2021;18:73–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao B, Ahmad MF, Nagy LE, Tsukamoto H. Inflammatory pathways in alcoholic steatohepatitis. J Hepatol. 2019;70:249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galluzzi L, Kepp O, Chan FK, Kroemer G. Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol. 2017;12:103–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frank D, Vaux DL, Murphy JM, Vince JE, Lindqvist LM. Activated MLKL attenuates autophagy following its translocation to intracellular membranes. J Cell Sci. 2019;132:jcs220996. [DOI] [PubMed] [Google Scholar]
- 8.Wu X, Nagy LE. MLKL contributes to western diet‐induced liver injury through inhibiting autophagy. Autophagy. 2020;16:1351–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu X, Poulsen KL, Sanz‐Garcia C, Huang E, McMullen MR, Roychowdhury S, et al. MLKL‐dependent signaling regulates autophagic flux in a murine model of non‐alcohol‐associated fatty liver and steatohepatitis. J Hepatol. 2020;73:616–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity. 2017;47:51–65.e7. [DOI] [PubMed] [Google Scholar]
- 11.Dara L. The receptor interacting protein kinases in the liver. Semin Liver Dis. 2018;38:73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyata T, Nagy LE. Programmed cell death in alcohol‐associated liver disease. Clin Mol Hepatol. 2020;26:618–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain‐like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–46. [DOI] [PubMed] [Google Scholar]
- 14.Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D, et al. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology. 2015;62:1847–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gunther C, He GW, Kremer AE, Murphy JM, Petrie EJ, Amann K, et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J Clin Invest. 2016;126:4346–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu H, Du X, Liu G, Huang S, Du W, Zou S, et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol Metab. 2019;23:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyata T, Wu X, Fan X, Huang E, Sanz‐Garcia C, Ross CKC, et al. Differential role of MLKL in alcohol‐associated and non‐alcohol‐associated fatty liver diseases in mice and humans. JCI Insight. 2021;6:e140180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim A, Bellar A, McMullen MR, Li X, Nagy LE. Functionally diverse inflammatory responses in peripheral and liver monocytes in alcohol‐associated hepatitis. Hepatol Commun. 2020;4:1459–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meng Y, Sandow JJ, Czabotar PE, Murphy JM. The regulation of necroptosis by post‐translational modifications. Cell Death Differ. 2021;28:861–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duan Y, Chu H, Brandl K, Jiang L, Zeng S, Meshgin N, et al. CRIg on liver macrophages clears pathobionts and protects against alcoholic liver disease. Nat Commun. 2021;12:7172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duan Y, Llorente C, Lang S, Brandl K, Chu H, Jiang L, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature. 2019;575:505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hardesty J, Day L, Warner J, Warner D, Gritsenko M, Asghar A, et al. Hepatic protein and phosphoprotein signatures of alcohol‐associated cirrhosis and hepatitis. Am J Pathol. 2022; S0002‐9440: 121–3. 10.1016/j.ajpath.2022.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang Y, et al. Macrophage polarization and its role in liver disease. Front Immunol. 2021;12:803037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009;10:623–35. [DOI] [PubMed] [Google Scholar]
- 25.Wang R, Tang R, Li B, Ma X, Schnabl B, Tilg H. Gut microbiome, liver immunology, and liver diseases. Cell Mol Immunol. 2021;18:4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Llorente C, Jepsen P, Inamine T, Wang L, Bluemel S, Wang HJ, et al. Gastric acid suppression promotes alcoholic liver disease by inducing overgrowth of intestinal Enterococcus. Nat Commun. 2017;8:837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Jiang W, Xu J, Wu N, Wang Y, Lin T, et al. E. coli NF73‐1 isolated from NASH patients aggravates NAFLD in mice by translocating into the liver and stimulating M1 polarization. Front Cell Infect Microbiol. 2020;10:535940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaphalia L, Srinivasan MP, Kakumanu RD, Kaphalia BS, Calhoun WJ. Ethanol exposure impairs AMPK signaling and phagocytosis in human alveolar macrophages: role of ethanol metabolism. Alcohol Clin Exp Res. 2019;43:1682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karavitis J, Murdoch EL, Deburghgraeve C, Ramirez L, Kovacs EJ. Ethanol suppresses phagosomal adhesion maturation, Rac activation, and subsequent actin polymerization during FcgammaR‐mediated phagocytosis. Cell Immunol. 2012;274:61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karavitis J, Murdoch EL, Gomez CR, Ramirez L, Kovacs EJ. Acute ethanol exposure attenuates pattern recognition receptor activated macrophage functions. J Interferon Cytokine Res. 2008;28:413–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rimland D, Hand WL. The effect of ethanol on adherence and phagocytosis by rabbit alveolar macrophages. J Lab Clin Med. 1980;95:918–26. [PubMed] [Google Scholar]
- 32.Szabo G, Dolganiuc A, Dai Q, Pruett SB. TLR4, ethanol, and lipid rafts: a new mechanism of ethanol action with implications for other receptor‐mediated effects. J Immunol. 2007;178:1243–9. [DOI] [PubMed] [Google Scholar]
- 33.Martinez SE, Lazaro‐Dieguez F, Selva J, Calvo F, Piqueras JR, Crespo P, et al. Lysophosphatidic acid rescues RhoA activation and phosphoinositides levels in astrocytes exposed to ethanol. J Neurochem. 2007;102:1044–52. [DOI] [PubMed] [Google Scholar]
- 34.Rothenfluh A, Threlkeld RJ, Bainton RJ, Tsai LT, Lasek AW, Heberlein U. Distinct behavioral responses to ethanol are regulated by alternate RhoGAP18B isoforms. Cell. 2006;127:199–211. [DOI] [PubMed] [Google Scholar]
- 35.Chao X, Wang S, Zhao K, Li Y, Williams JA, Li T, et al. Impaired TFEB‐mediated lysosome biogenesis and autophagy promote chronic ethanol‐induced liver injury and steatosis in mice. Gastroenterology. 2018;155:865–79.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitur K, Wachtel S, Brown A, Wickersham M, Paulino F, Penaloza HF, et al. Necroptosis promotes staphylococcus aureus clearance by inhibiting excessive inflammatory signaling. Cell Rep. 2016;16:2219–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D'Cruz AA, Speir M, Bliss‐Moreau M, Dietrich S, Wang S, Chen AA, et al. The pseudokinase MLKL activates PAD4‐dependent NET formation in necroptotic neutrophils. Sci Signal. 2018;11:eaao1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu SX, Chen W, Liu ZZ, Zhou FH, Yan SQ, Hu GQ, et al. Non‐hematopoietic MLKL protects against salmonella mucosal infection by enhancing inflammasome activation. Front Immunol. 2018;9:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sai K, Parsons C, House JS, Kathariou S, Ninomiya‐Tsuji J. Necroptosis mediators RIPK3 and MLKL suppress intracellular Listeria replication independently of host cell killing. J Cell Biol. 2019;218:1994–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoon S, Bogdanov K, Wallach D. Site‐specific ubiquitination of MLKL targets it to endosomes and targets Listeria and Yersinia to the lysosomes. Cell Death Differ. 2022;29:306–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH, 3rd, Tait SWG, et al. RIPK3 restricts viral pathogenesis via cell death‐independent neuroinflammation. Cell. 2017;169:301–13.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Najafov A, Mookhtiar AK, Luu HS, Ordureau A, Pan H, Amin PP, et al. TAM kinases promote necroptosis by regulating oligomerization of MLKL. Mol Cell. 2019;75:457–68.e4. [DOI] [PubMed] [Google Scholar]
- 43.Weber K, Roelandt R, Bruggeman I, Estornes Y, Vandenabeele P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun Biol. 2018;1:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dai J, Zhang C, Guo L, He H, Jiang K, Huang Y, et al. A necroptotic‐independent function of MLKL in regulating endothelial cell adhesion molecule expression. Cell Death Dis. 2020;11:282. [DOI] [PMC free article] [PubMed] [Google Scholar]