

Abstract
Sulfono-γ-AApeptides recently developed in our group have been proven to be a new class of unnatural foldamer with well-defined helical structure and demonstrated their ability to mimic protein helical domains and disrupt biomedically relevant protein-protein interactions (PPIs). Based the designing concept, in a recent report, we discovered two similar sulfono-γ-AApeptides V2 and V3 which were designed to mimic the VEGF N-terminal helix α1 known to directly interact with VEGFRs. Interestingly, V2 was shown to possess the pro-angiogenic effect, whereas V3 was proved to be a potent inhibitor for angiogenesis. We speculated that the distinct angiogenesis signaling was due to the selective binding of the two molecules toward VEGFR1 and VEGFR2, respectively. Together with their remarkable resistance to proteolytic degradation, relatively small sizes, and amenability to modification with diverse functional groups, V2 and V3 could serve as lead molecules for the development of potential therapeutic agents and molecular probes. These findings highlighted sulfono-γ-AApeptides as an alternative paradigm to mimic the α-helical domain to modulate a wide variety of PPIs in the future.
Keywords: γ-AApeptides, angiogenesis, modulator, VEGF & VEGFR, Peptidomimetic
Graphical Abstract
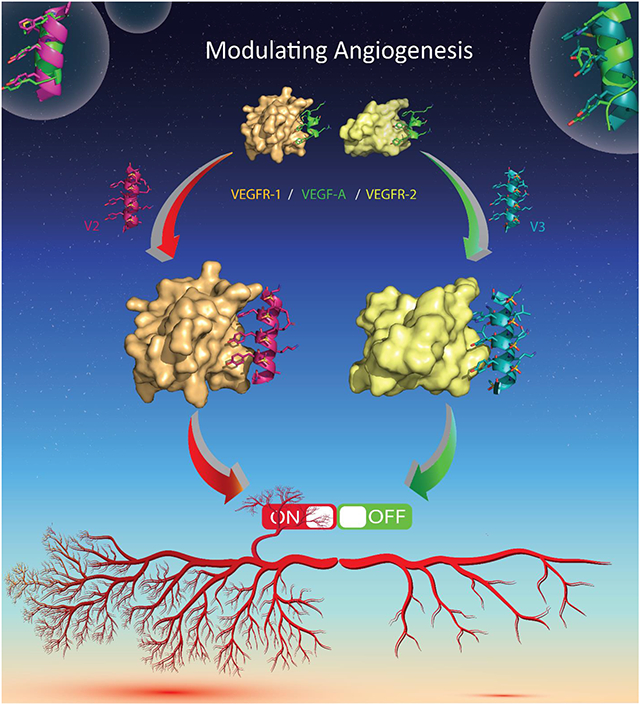
In this concept article, we briefly introduced the structure and application of homogenous sulfono-γ-AApeptides to modulate different PPIs. Most importantly, we described in detail how sulfono-γ-AApeptides mimic the critical binding domain on VEGRA (helix-α1) to modulate VEGF/VEGFR PPI. We have identified two active mimetics V2 and V3 potently activated and inhibited angiogenesis with highly resistant to proteolytic degradation. Therefore, the study further demonstrates the versatility of sulfono-γ-AApeptides to mimic protein helical domain to manipulate various PPIs.
1. Introduction
Peptide and protein play critical roles in virtually all physiological and biological processes, such as function as hormones, enzymatic inhibitors or substrates, growth promoters or inhibitors, and neurotransmitters.[1] Bioactive peptides earn significant interest in diverse areas, however, their low bioavailability and biostability limit their wide biomedical application.[2] Peptidomimetics, which are designed to mimic the structure and function of bioactive peptides and proteins, have demonstrated excellent applications in protein surface mimicry and recognition, modulation of PPIs, catalysis, etc.[3] Oligomeric peptidomimetics attracted considerable attention because they comprise of unnatural amino acids and are generally more resistant to enzymatic hydrolysis.[4] In the past two decades, many peptidomimetic foldamers, such as β-peptides,[5] oligoureas,[6] azapeptides,[7] peptoids,[8] aromatic oligoamides,[9] and others, had been developed.[10] Inspired by the foldamer concept, we designed a new class of helical peptidomimetics, sulfono-γ-AApeptide (Figure 1A), and extended our effort to explore their structure and function relationship.[11]
Figure 1.

(A) Structure of sulfono-γ-AA peptide building block. (B) Schematic representation of distribution of side chains from sulfono-γ-AApeptides. (C) Crystal structure of a sulfono-γ-AApeptide. (D) Top view of (C). Reproduced with the permission of American Chemical Society.[12]
2. The structure of sulfono-γ-AA peptides and their application
2.1. The chemical structure of sulfono-γ-AA peptides
We initially developed γ-AApeptides (γ-substituted-N-acylated-N-aminoethyl amino acids) as a new type of peptidomimetics, which was derived from the γ-chiral PNA scaffold.[13] They demonstrated remarkable resistance to proteolytic degradation as well as amenability to chemical diversification, enabling them suitable candidates for various biological applications.[4, 14] Sulfono-γ-AApeptides (Figure 1A), a subclass of γ-AApeptides, were found to not only maintain the advantage of γ-AApeptides, but also adopt the well-defined helical conformations (Figure 1B).[11]
2.2. The crystal structures of sulfono-γ-AA peptides
We initially discovered that the heterogeneous 2:1 α/D-sulfono-γ-AA hybrid oligomers and heterogeneous 1:1 α/L-sulfono-γ-AA hybrid oligomers adopt right-handed 4.516-14 helical conformation and right-handed 413 helices, respectively, based on their crystal structures.[15] These results showed that peptidomimetics containing sulfono-γ-AApeptides units and α-residues could form distinctive heterogeneous foldamers, which prompted us to believe that homogeneous sulfono-γ-AApeptides comprising completely unnatural sulfono-γ-AApeptides units could also adopt defined folding conformation. Soon later, we successfully obtained the crystal structures of homogeneous L-sulfono-γ-AApeptides and confirmed that this class of foldamers did form the well-defined left-handed 414-helix configuration with a radius of 2.8 Å, a helical pitch of 5.1 Å and four side chains per turn (Figure 1C and 1D).[16] The folding propensity of homogeneous L-sulfono-γ-AApeptides is stable with left-handed 414-helix configuration regardless of side-chain identity. It is noted that helical sulfono-γ-AApeptides bear the similar helical pitch to α-peptides (5.4 Å). Moreover, as the helicity of sulfono-γ-AApeptides is stabilized by both intramolecular hydrogen bonding and intrinsic curvature of sulfonamide moieties on the molecular backbone, the sulfono-γ-AApeptides demonstrated more robust helical stability than α-peptides of the same lengths in solution.[16] Furthermore, the helical handedness of sulfono-γ-AApeptide helices could be manipulated by switching the chirality of chiral side chains in the sulfono-γ-AApeptide sequences.[17] Therefore, sulfono-γ-AApeptide foldamers could be rationally designed to mimic the structure and function of α-helix for biomolecular recognition and modulation of medicinally relevant PPIs.
2.3. The application of homogenous sulfono-γ-AApeptides for the inhibition of PPIs and protein recognition
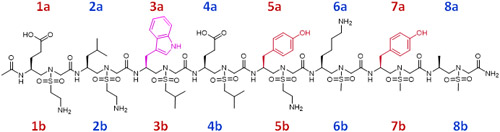
Based on atomic structures, high stability, and the robust helical folding propensity of sulfono-γ-AApeptides, we set out to employ this class of foldamer as the protein helical domain mimetics to modulate a few well-known PPIs.[18] As sulfono-γ-AApeptides demonstrated a similar helical pitch to the α-peptides, and have exactly four side chains per turn which is also analogous to those of α-helix (3.6), the side chains of each helical face (e. g. 1a, 3a, 5a, 7a of Figure 1) of sulfono-γ-AApeptides could mimic the side chains of i, i+4, i+7 on α-helix. To this end, we designed a series of homogenous sulfono-γ-AApeptides which could project similar functional side chains to those crucial residues of p53 helical domain involved in the interaction with MDM2. It turned out that a series of sequences could tightly bind to MDM2 and disrupt MDM2/p53 effectively, with the most potent sequence PS10 causing the analogous chemical shifts of MDM2 upon binding compared to those induced by p53, suggesting that sulfono-γ-AApeptides assumed a similar binding mode to p53.[18a] Encouraged by the findings, we moved to another PPI - β-catenin/BCL9 PPI, in which the α-helical HD2 domain of BCL9 is involved in the binding to β-catenin. The Wnt/β-catenin signaling pathway plays an important role in directing cell proliferation, differentiation, and survival, and this signaling pathway depends on the formation of β-catenin supercomplexes with BCL9 or BCL9-like proteins. Thus, disrupting the interaction between β-catenin and BCL9 is a promising strategy to develop anti-cancer agents. However, this PPI is considerably more challenging to inhibit than p53/MDM2 PPI because both hydrophobic and charged residues in BCL9 are important for the recognition of β-catenin. Nonetheless, We successfully mimicked this α-helix region of BCL9 with sulfono-γ-AApeptides, and demonstrated sulfono-γ-AApeptides were highly cell-permeable and interacted with β-catenin specifically in the cell-based assay.[18b] Not long ago, we adopted the design strategy of sulfono-γ-AApeptides to mimic long helical hormone peptide GLP-1, and showed that certain sequences could reproduce the functionality of GLP-1 on multiple helical faces and effectively activated glucose uptake upon binding to GLP-1R, which could be further developed to treat type 2 diabetes.[18c]
2.4. Homogenous sulfono-γ-AApeptides successfully manipulated the angiogenic response
Angiogenesis is the process of formation of new blood vessels from the existing vasculature.[20] Notably, stimulation of diminished angiogenesis could be therapeutically beneficial for ischemic heart disease, peripheral arterial disease, and wound healing, however, inhibition of elevated angiogenesis is a promising strategy for cancer therapy, ophthalmic conditions, rheumatoid arthritis, and other diseases.[21] Thus, albeit challenging, precise manipulation of contradictory pro-angiogenic and anti-angiogenic signaling is an attractive goal to achieve.
Among the various angiogenic factors, the vascular endothelial growth factor (VEGF) family and its receptor system have been proved to be vital regulators in the cell signaling of angiogenesis.[22] VEGF is a homodimer that binds to three VEGF-specific receptor tyrosine kinases, VEGFR-1, VEGFR-2, and VEGFR-3, and is overexpressed in many cancer cells. VEGF, also known as VEGFR-A, contains four subtypes, such as peptides of 121, 165, 189, and 206 amino acids in humans.[23] Among subtypes of VEGF-A, VEGF-A165 is most essential and sufficient for angiogenesis and could bind to both VEGFR-1 and VEGFR-2 to promote angiogenesis, vascular permeability, cell migration, and gene expression.[24]
2.4.1. Design of sulfono-γ-AApeptides to mimic the structure of the VEGF N-terminal helix α1
The VEGF N-terminal helix α1 (residues 16-25) is known to be directly involved in VEGF/VEGFR PPI.[25] D’Andrea et al.[26] designed a peptide QK (Table 1) derived from helix α1 which was confirmed to have a good binding affinity to VEGFR and activate angiogenesis unexpectedly. However, another peptide, MA (Table 1), having only two amino acids different from QK, potently inhibited angiogenesis.[27] To interrogate this dilemma, we designed a series of VEGF-mimicking sulfono-γ-AApeptides based on the crystal structure of the VEGFR-2/VEGF-A complex (PDB: 3V2A).[12] As shown in Figure 2, there are four major binding residues (Phe17, Met18, Tyr21, and Tyr25) located at the VEGF N-terminal α1 helix positioned on the same face of the helical binding domain. We speculated that the Phe17, Tyr21, and Tyr25 residues could be reproduced by the side chains at 3a, 5a, and 7a positions in sulfono-γ-AApeptides (Table 1), respectively, and Met18 may be mimicked by a hydrophobic and relatively smaller sized sulfono isobutyl group at 4b position. For instance, V3 was shown to perfect overlay with the helix α1 of VEGF (Figure 3).
Table1.
Structures of selected sulfono-γ-AA peptide helical mimics (V2 and V3) a.
| Structure | Kd (μM) | ||
|---|---|---|---|
| VEGFR-1 | VEGFR-2 | ||
| V2 |
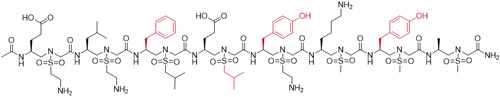
|
0.46 | 2.3 |
| V3 |
|
7.7 | 0.63 |
| QK | Ac-KLTWQELYQLKYKGI | ||
| MA | Ac-KLTWMELYQLAYKGI | ||
Critical residues for binding are shown in red and pink. Binding affinity (Kd) of sequences to VEGFR-1 and VEGFR-2 as determined by SPR. The chemical structures of two reported peptides QK and MA are also included. Reproduced with the permission of American Chemical Society.[12]
Figure 2.

Structure of VEGF-A/VEGFR-2 complex, PDB: 3V2A. Homodimeric VEGF-A (Green) binding to, and dimerization of, VEGFR-2 (Yellow). Key binding residues on VEGF-A helix-α1 (Phe17, Met18, Tyr21 and Tyr25) are highlighted in red. Reproduced with the permission of American Chemical Society.[12]
Figure 3.

(A) Binding interaction of key residues on helix α1 of VEGF-A (green) with VEGFR-2 (yellow), PDB code: 3V2A. (B) Structures of helix α1 of VEGF-A (green), sulfono-γ-AA peptide mimic V3 (teal) and overlay of key binding residues. Reproduced with the permission of American Chemical Society.[12]
2.4.2. Two sulfono-γ-AApeptides V2 and V3 potently manipulate the VEGF/VEGFR PPI
Next, we assessed the activity of V2 and V3 for the modulation of VEGF/VEGFR PPI by multiple in vitro and cellular assays. Firstly, Circular Dichroism (CD) studies suggested that both V2 and V3 adopted a left-handed helical conformation. The surface plasmon resonance (SPR) assays were conducted to assess the binding affinity of V2 and V3 toward the extracellular domains of VEGFR-1 and VEGF-2, respectively. Interestingly, both V2 and V3 exhibited good binding affinity to VEGFRs (Table 1), however, V2 and V3 demonstrated significant selectivity for VEGFR-1 and VEGFR-2 with the only difference between V2 (phenyl group) and V3 (indole group) is one side chain at the position 3a to mimic Phe17. V2 bound to VEGFR1 specifically, whereas V3 demonstrated much higher selectivity for VGFR2 instead.
Following that, we ran a series of assays with human umbilical endothelial cells (HUVECs) to investigate the activity of these two peptidomimetics such as cell migration assay, wound healing assay, capillary tube formation assay, western blot analysis, and immunofluorescence assay, etc. Based on results, both V2 and QK were shown to significantly increase the number of HUVECs migrating and stimulate capillary tube formation, suggesting there are activators of angiogenesis. However, V3 with indole group at 3a position inhibited the cell migration and wound healing, formation of capillary tubes, as well as VEGFR2 mediated signaling pathway, demonstrating that V3 is a potent inhibitor of angiogenesis. We hypothesized that the reason why the sulfono-γ-AApeptides V2 and V3 could switch angiogenesis is due to their selective binding to either VEGFR-1 or VEGFR-2, respectively. V2 and QK specific bound with VEGFR-1, the decoy receptor, amplifying the ability of VEGF to bind with VEGFR2 to enhance angiogenesis (Figure 4). On the contrary, as V3 is directly bound to VEGFR2, it competitively inhibited the access of VEGF to VEGFR2. Since VEGR-2 is the major receptor for mediated angiogenesis signaling, the inhibition of VEGF-A/VEGFR-2 led to the significant diminishment of angiogenic signaling (Figure 4). Taken together, the sulfono-γ-AApeptides could be rationally designed to either activate or inhibit angiogenesis. It should also be mentioned that V2 and V3 were completely resistant to proteolytic degradation, which may augment their potential for further application in vivo.
Figure 4.

Modulation of angiogenic switch with sulfono-γ-AA peptide-based mimics of VEGFR-A. Reproduced with the permission of American Chemical Society.[12]
3. Outlook
The two peptidomimetics V2 and V3 functioned as either the activator or the inhibitor of angiogenesis by mimicking the VEGF N-terminal helix α1 with high stability. To the best of our knowledge, this is the first example to use the helical foldamer with the completely unnatural backbone to target VEGF/VEGFR interface and modulate VEGF/VEGFRs PPIs. A few future perspectives might be taken into consideration.
1. A series of in vivo studies with animal models could be carried out to demonstrate that these two sequences possess the potential to manipulate angiogenesis in vivo. Particularly, it would be intriguing to find out whether V3 could decrease or eradicate the solid tumor in the mice model, and whether V2 could help to treat heart disease, these molecules could advance to clinical trials quickly.
2. The activity of these two sulfono-γ-AApeptides could be further optimized. For instance, backbone stapling might be a promising strategy to enhance the helicity and therefore the activity. The more potent and more selective binders of VEGFR1 and VEGFR2 could be employed to probe mechanisms of angiogenesis more accurately thereby providing more solid evidence for the future design of modulators of angiogenesis with the desired function.
3. There are also other VEGFs in the VEGF family (VEGF-B, VEGF-C and VEGF-D). As our design was based on the mimicry of the helix domain of VEGF, it is anticipated that some sulfono-γ-AApeptides could be manipulated to modulate the interaction of these VEGFs with their receptors. Development of the specific binding probes that interrogate the interactions of these VEGFs with VEGFRs could gain insight into their functional roles.
4. Our design concept of sulfono-γ-AApeptides for the mimicry of protein helical domain could be extended to a wide variety of biomedically relevant PPIs, including both extra- and intra-cellular targets, such as β-catenin/TCF, intrinsically disordered proprotein.
4. Conclusion
Peptidomimetic foldamer offers many opportunities to realize the function of modulating various PPIs, with increased bioavailability and biostability compared with peptide-based strategies. We highlighted our progress on the sulfono-γ-AApeptide foldamer for the modulation of PPI, and particularly we described our effort in the design of a few homogenous sulfono-γ-AApeptides to mimic VEGF N-terminal helix α1 for VEGFR recognition. Although both V2 and V3 were tightly bound to the VEGFRs, in various vitro assays demonstrated that V2 activated the angiogenesis, whereas V3 potently inhibited the angiogenesis. Although the mechanism modulating the VEGF/VEGFR PPI is still unclear, the discovery of V2 and V3 may help to understand this process. We hope that our design of the modulators for the VEGFRs could provide a unique strategy to design the new generation of modulators for PPIs.
Acknowledgement
The work was supported by NIH R01AI149852 (J.C.) and NIH R01AI152416 (J.C.).
Biographies

Songyi Xue
Songyi Xue is a Graduate Student at the University of South Florida. His research interests include the development of novel helical foldamers, and small molecule inhibitors based on structure of γ-AApeptides potentially targeting on various virus.

Lei Wang
Lei Wang is a Graduate Student at the University of South Florida. Her research interests focus on combinatorial screening of γ-AApeptides and design and synthesis of antimicrobial γ-AApeptides.

Jianfeng Cai
Jianfeng Cai is a preeminent professor in the Department of Chemistry at the University of South Florida. His research group focuses on the development and application of AApeptides-based peptidomimetics.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Reference
- [1].Muttenthaler M, King GF, Adams DJ, Alewood PF, Nat Rev Drug Discov 2021, 20, 309–325. [DOI] [PubMed] [Google Scholar]
- [2].Hamman JH, Enslin GM, Kotzé AF, BioDrugs 2005, 19, 165–177. [DOI] [PubMed] [Google Scholar]
- [3].Mabonga L, Kappo AP, International Journal of Peptide Research and Therapeutics 2019, 26, 225–241. [Google Scholar]
- [4].Shi Y, Teng P, Sang P, She F, Wei L, Cai J, Acc Chem Res 2016, 49, 428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Niu J, Cederstrand AJ, Eddinger GA, Yin B, Checco JW, Bingman CA, Outlaw VK, Gellman SH, J Am Chem Soc 2022, 144, 9610–9617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fremaux J, Venin C, Mauran L, Zimmer RH, Guichard G, Goudreau SR, Nat Commun 2019, 10, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chingle R, Proulx C, Lubell WD, Acc Chem Res 2017, 50, 1541–1556. [DOI] [PubMed] [Google Scholar]
- [8].Morimoto J, Fukuda Y, Kuroda D, Watanabe T, Yoshida F, Asada M, Nakamura T, Senoo A, Nagatoishi S, Tsumoto K, Sando S, J Am Chem Soc 2019, 141, 14612–14623. [DOI] [PubMed] [Google Scholar]
- [9].a Barnard A, Long K, Martin HL, Miles JA, Edwards TA, Tomlinson DC, Macdonald A, Wilson AJ, Angew Chem Int Ed Engl 2015, 54, 2960–2965; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kumar S, Hamilton AD, J Am Chem Soc 2017, 139, 5744–5755; [DOI] [PubMed] [Google Scholar]; c Whitby LR, Boger DL, Accounts of chemical research 2012, 45, 1698–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a Goodman CM, Choi S, Shandler S, DeGrado WF, Nat Chem Biol 2007, 3, 252–262; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li Z, Cai B, Yang W, Chen CL, Chem Rev 2021, 121, 14031–14087. [DOI] [PubMed] [Google Scholar]
- [11].Sang P, Shi Y, Huang B, Xue S, Odom T, Cai J, Acc Chem Res 2020, 53, 2425–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Abdulkadir S, Li C, Jiang W, Zhao X, Sang P, Wei L, Hu Y, Li Q, Cai J, J Am Chem Soc 2022, 144, 270–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a Bolarinwa O, Nimmagadda A, Su M, Cai J, Biochemistry 2017, 56, 445–457; [DOI] [PMC free article] [PubMed] [Google Scholar]; b She F, Nimmagadda A, Teng P, Su M, Zuo X, Cai J, Biomacromolecules 2016, 17, 1854–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Teng P, Shi Y, Sang P, Cai J, Chemistry 2016, 22, 5458–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a Teng P, Ma N, Cerrato DC, She F, Odom T, Wang X, Ming LJ, van der Vaart A, Wojtas L, Xu H, Cai J, J Am Chem Soc 2017, 139, 7363–7369; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Teng P, Niu Z, She F, Zhou M, Sang P, Gray GM, Verma G, Wojtas L, van der Vaart A, Ma S, Cai J, J Am Chem Soc 2018, 140, 5661–5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].She F, Teng P, Peguero-Tejada A, Wang M, Ma N, Odom T, Zhou M, Gjonaj E, Wojtas L, van der Vaart A, Cai J, Angew Chem Int Ed Engl 2018, 57, 9916–9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a Sang P, Shi Y, Higbee P, Wang M, Abdulkadir S, Lu J, Daughdrill G, Chen J, Cai J, J Org Chem 2020, 85, 10552–10560; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Teng P, Zheng M, Cerrato DC, Shi Y, Zhou M, Xue S, Jiang W, Wojtas L, Ming L-J, Hu Y, Cai J, Communications Chemistry 2021, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a Sang P, Shi Y, Lu J, Chen L, Yang L, Borcherds W, Abdulkadir S, Li Q, Daughdrill G, Chen J, Cai J, J Med Chem 2020, 63, 975–986; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sang P, Zhang M, Shi Y, Li C, Abdulkadir S, Li Q, Ji H, Cai J, Proc Natl Acad Sci U S A 2019, 116, 10757–10762; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sang P, Zhou Z, Shi Y, Lee C, Amso Z, Huang D, Odom T, Nguyen-Tran VT, Shen W, Cai J, Science advances 2020, 6, eaaz4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Murray JK, Gellman SH, Biopolymers 2007, 88, 657–686. [DOI] [PubMed] [Google Scholar]
- [20].Chung AS, Lee J, Ferrara N, Nat Rev Cancer 2010, 10, 505–514. [DOI] [PubMed] [Google Scholar]
- [21].Carmeliet P, Nature medicine 2003, 9, 653–660. [DOI] [PubMed] [Google Scholar]
- [22].Shibuya M, Cell structure and function 2001, 26, 25–35. [DOI] [PubMed] [Google Scholar]
- [23].Karamysheva A, Biochemistry (Moscow) 2008, 73, 751–762. [DOI] [PubMed] [Google Scholar]
- [24].Rahimi N, Dayanir V, Lashkari K, Journal of Biological Chemistry 2000, 275, 16986–16992. [DOI] [PubMed] [Google Scholar]
- [25].Brozzo MS, Bjelić S, Kisko K, Schleier T, Leppänen V-M, Alitalo K, Winkler FK, Ballmer-Hofer K, Blood, The Journal of the American Society of Hematology 2012, 119, 1781–1788. [DOI] [PubMed] [Google Scholar]
- [26].D'Andrea LD, Iaccarino G, Fattorusso R, Sorriento D, Carannante C, Capasso D, Trimarco B, Pedone C, Proceedings of the National Academy of Sciences 2005, 102, 14215–14220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Basile A, Del Gatto A, Diana D, Di Stasi R, Falco A, Festa M, Rosati A, Barbieri A, Franco R, Arra C, Pedone C, Fattorusso R, Turco MC, D'Andrea LD, J Med Chem 2011, 54, 1391–1400. [DOI] [PubMed] [Google Scholar]