

Abstract
Canonical (H3.1/H3.2) and noncanonical (H3.3) histone 3 K27M-mutant gliomas have unique spatiotemporal distributions, partner alterations, and molecular profiles. The contribution of the cell-of-origin to these differences has been challenging to uncouple from the oncogenic reprogramming induced by the mutation. Here, we perform an integrated analysis of 116 tumors, including single-cell transcriptome and chromatin accessibility, 3D chromatin architecture and epigenomic profiles, and show that K27M-mutant gliomas faithfully maintain chromatin configuration at developmental genes consistent with anatomically distinct oligodendrocyte-precursor-like cells (OPC). H3.3K27M thalamic gliomas map to prosomere 2-derived lineages. In turn, H3.1K27M ACVR1-mutant pontine gliomas uniformly mirror early ventral NKX6-1+/SHH-dependent brainstem OPCs, while H3.3K27M gliomas frequently resemble dorsal PAX3+/BMP-dependent progenitors. Our data suggest a context-specific vulnerability in H3.1K27M-mutant SHH-dependent ventral OPCs, which rely on acquisition of ACVR1 mutations to drive aberrant BMP signaling required for oncogenesis. The unifying action of K27M mutations is to restrict H3K27me3 at PRC2 landing sites, while other epigenetic changes are mainly contingent on the cell-of-origin chromatin state and cycling rate.
Keywords: K27M, EZHIP, OPC, oligodendrocyte development, ACVR1, single cell multiomics, Nkx6-1, Pax3, BMP
Editor summary:
Comprehensive genomic profiling of human H3K27M mutant diffuse midline gliomas, combining single-cell RNA-seq and ATAC-seq with bulk ChIP-seq and other data, proposes distinct oligodendrocyte populations as potential cells-of-origin.
Introduction
Midline high-grade gliomas (HGG) are deadly primary brain tumors of glial origin in children and young adults1. Hallmark alterations2–5 include recurrent somatic mutations on histone 3 variants (oncohistones), substituting lysine 27 (H3K27) to methionine (H3K27M)2,3,5. Mechanistically, oncohistones inhibit the Polycomb Repressor Complex 2 (PRC2)6–8, which deposits mono-, di- or trimethylation on this residue, affecting the global distribution of these and other major chromatin marks. In parallel, PRC2 function is also abrogated in posterior fossa group A ependymomas (PFA-EP) via expression of the Enhancer of Zeste Homologs Interacting Protein4,9–12 (EZHIP), which harbours a K27M-like peptide. Despite convergent mechanisms, H3K27M and EZHIP are mutually exclusive in patients and show an inverse pattern of prevalence: while H3K27M predominates in midline HGG2,3,13 (~87%), EZHIP targets PFA-EP4 (~96%).
A remarkable spatiotemporal specificity13–18 of genetic alterations driving HGG suggests their pathogenesis may stem from aberrant development. K27M in canonical H3.1/H3.2 is restricted to the pons, where it accounts for 20% of K27M-mutant diffuse intrinsic pontine gliomas (DIPGs), affects younger children, and preferentially associates with somatic ACVR1 mutations and phosphatidylinositol-3-kinase (PI3K) pathway activation. ACVR1 mutations, in turn, are also exclusive to the pons13–16,18. They have been suggested to promote tumorigenesis by arresting glial differentiation19 or by inducing a mesenchymal phenotype through Stat3 activation20. In contrast, K27M in noncanonical H3.3 occurs throughout the midline, targets older children, and preferentially associates with TP53 and PDGFRA alterations and, more rarely, gain-of-function of EGFR or FGFR in the thalamus.
Recent studies show that PFA-EP resemble prenatal gliogenic progenitors21, while K27M HGG are presumed to originate in oligodendroglial precursor cells (OPC)22,23. There are, however, several waves of OPC specification with specific epigenomic landscapes and functional properties24–26 that could represent targets for oncohistone alterations and partner mutations. Distinct origins may explain the diverse enhancer landscapes and oncogenic pathways reported in each entity. Alternatively, these distinctive features may reflect downstream effects of oncohistones themselves: several reports indicate these alterations differentially affect PRC2 activity and epigenome remodeling, potentially reprogramming cell lineage27–29. EZHIP is suggested to have a higher inhibitory effect than H3K27M in vitro9–12, as is K27M in replication-dependent H3.1/2 compared to H3.3K27M29. Other studies suggest H3 variant-specific changes in regulatory elements depend on their differential incorporation on chromatin and local enrichment of H3.3K27M at H3.3-rich domains30,31. Thus, the unique molecular profiles of K27M-mutant HGG and PFA-EP could be a product of distinct cells-of-origin, distinct reprogramming of cell identity driven by each oncohistone/EZHIP, or a combination of both.
To identify specific cellular origins and oncogenic mechanisms in gliomas with PRC2 dysfunction, we assembled a multidimensional dataset of 116 tumors (including 4 matched recurrences) and 22 patient-derived cell lines (Supplementary Tables 1–2). All major tumor locations (pons, thalamus, posterior fossa, and cortex) are represented in this cohort. Comprehensive profiling of samples at the single-cell, bulk transcriptomic, 3D chromatin and epigenomic levels identifies specific developmental lineages permissive for the oncogenic potential of H3K27M. Our data also provide a molecular basis for the function of ACVR1 mutations in H3.1K27M DIPG. Last, by uncoupling lineage-of-origin, mutation status and cycling state, we show that oncohistones and EZHIP mainly converge to restrain the spread of H3K27me3 to PRC2 nucleation sites. Location of residual H3K27me3 and the extent of H3K27me2 spread are a consequence of the progenitor chromatin state and its cycling rate, independent of the specific driver alteration.
Results
Unique cell type hierarchies in H3.1 and H3.3K27M HGGs
To define the molecular landscape of HGG and PFA-EP tumors at single-cell resolution, we analyzed 43 patients profiled with Chromium 10x scRNAseq and scATACseq technologies (Figure 1a, Supplementary Tables 3–5). We retained clusters of malignant cells defined based on inferred Copy Number Abnormalities (CNA), filtering out clusters of immune, vascular, or meningeal cells, for a total of 181,282 tumor cells across tumor types. We complemented this data with novel and published3,30,32–38 bulk profiles of patient tumors and cell lines, assessing expression by RNA sequencing; histone marks H3K27ac, H3K27me2 and H3K27me3 by chromatin immunoprecipitation sequencing (ChIPseq); and 3D chromatin architecture by Hi-C chromosome conformation capture (Hi-C) (Figure 1a, Supplementary Protocol 1). Finally, to compare tumors to a normal reference, we expanded our previously reported mouse forebrain and pons developmental dataset34 (Extended Data Figure 1, Supplementary Tables 6–7).
Figure 1. Unique cell type hierarchies in H3.1 and H3.3K27M HGGs.
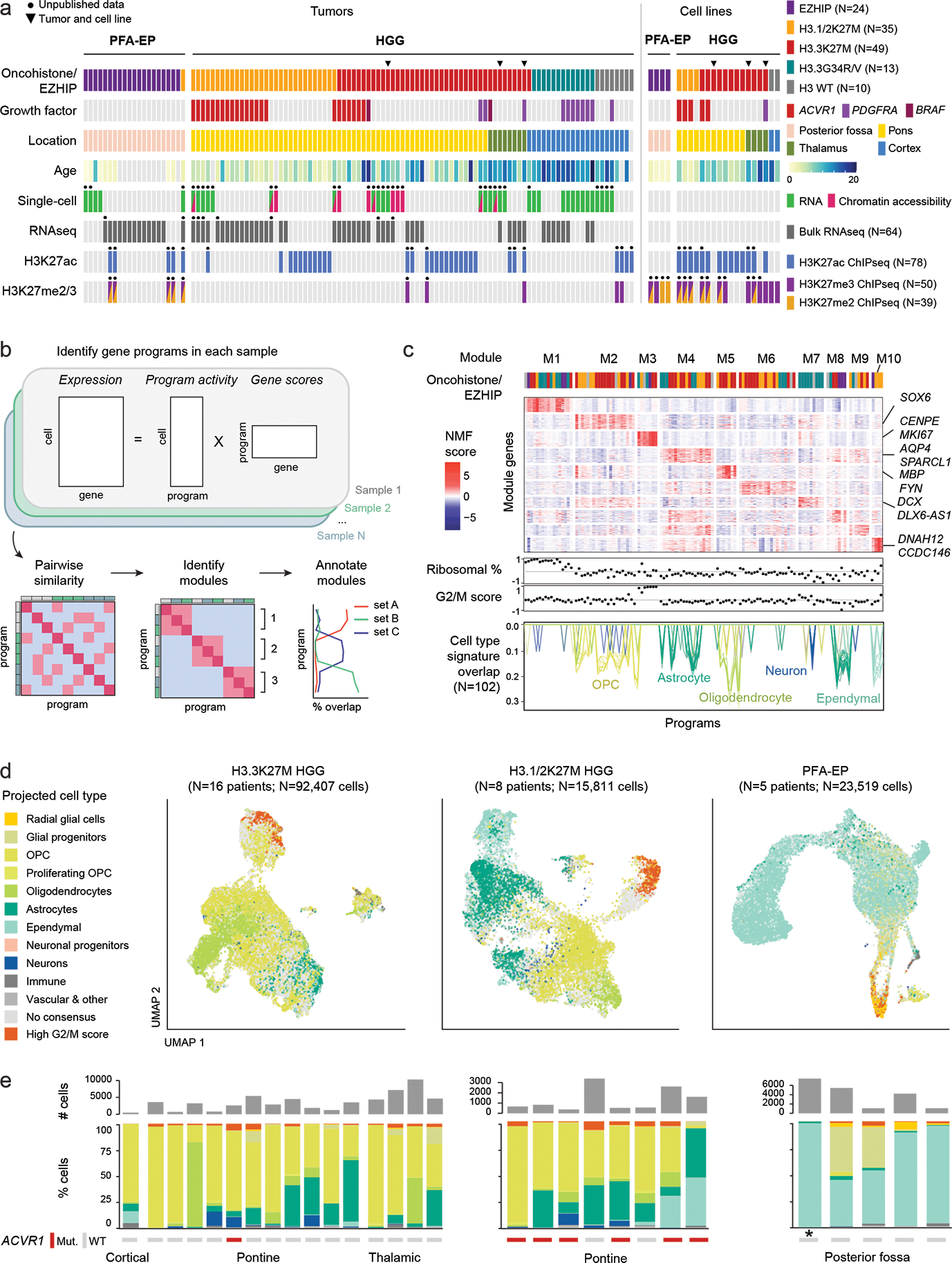
a. Patient tumors (N=116 tumors from 112 patients) and patient-derived cell lines (N=22) included in this study. Black dots indicate that this study provides unpublished data for at least one sample for the corresponding assay/sample. Chromatin accessibility: 10x ATAC or 10x Multiome (ATAC & RNA). HGG: high-grade glioma; PFA-EP: posterior fossa ependymoma.
b. Workflow for unsupervised identification of recurrent gene programs in malignant cells using consensus Non-negative Matrix Factorization (cNMF).
c. Top: heatmap of NMF scores for all module-associated genes, across all programs. Column annotation shows the driver alteration of the sample in which each program was identified, colored as in (a). Middle: Correlation between each program and ribosomal content in each cell, and G2/M cell cycle score in each cell. Bottom: overlap between each program and developmental reference signatures, one line per signature. Only significant overlaps (empirical p-value < 0.001; see Methods and Supplementary Table 10) are shown, with number of significant overlaps indicated in parentheses.
d. UMAP for malignant cells of each tumor type. Projected cell types were obtained by mapping each individual tumor cell to a normal developmental brain reference, using a consensus of automated cell type prediction methods. Cells without a consensus label but with high G2/M cell cycle phase score are shown in orange.
e. Top: number of malignant cells per sample, for each tumor type as in (d). Bottom: quantification of consensus cell type projections among malignant cells. ACVR1 mutation status and tumor location are indicated below. Asterisk (*) denotes the single H3.1K27M-mutant sample among PFA-EP tumors.
To identify recurrent, dominant sources of intratumor variability in an unsupervised manner (Figure 1b, Supplementary Tables 8–10), we performed non-negative matrix factorization39 on the RNA modality at the single-cell level, followed by hierarchical clustering and a recursive algorithm that identifies sets of programs, or modules, with high similarity across samples (Figure 1c, Extended Data Figure 2a). This revealed 10 modules. Two modules (M1, M3), present across all tumor subtypes, were related to protein synthesis and proliferation. Module M6 was partially explained in K27M tumors by hypoxia-related pathways. Strikingly, all remaining modules, amounting to half of intra-tumoral gene programs, were significantly associated with developmental cell type signatures (Figure 1c, Extended Data Figure 2b–c). Module M7, explained by neuronal signatures, was nearly exclusive to H3.3G34R/V gliomas, consistent with their neuronal origins36,40,41. H3K27M gliomas and PFA-EP were associated with distinct OPC, oligodendrocyte, astrocyte, and ependymal programs (Figure 1c). Thus, cell identity is a dominant source of intra-tumoral transcriptional variation in these tumors.
To refine the strong cell identity signal detected in the unsupervised analysis, we determined, for each cell, the most similar cell type in the developmental reference (Figure 1d–e). We validated this cross-species analysis using human prenatal scRNAseq data42,43 (82% concordance, Extended Data Figure 2d–e, Supplementary Tables 11–12). PFA-EP tumors were predominantly ependymal-like, while H3.3K27M gliomas ranged from OPC-like to oligodendrocyte-like and astrocyte-like cells, as previously reported22,23,34. In contrast, H3.1K27M tumors displayed a distinct molecular phenotype. A subset of H3.1K27M gliomas contained large populations of ependymal-like cells (Figure 1e). Inferred CNA profiles confirmed their malignant status (Extended Data Figure 3a–d). These cells uniquely expressed genes encoding dyneins and proteins associated with motile cilia (DNAH6/7/11/12), the ependymal transcription factor FOXJ1, and its targets44, confirming they activate bona fide ependymal transcriptional programs (Extended Data Figure 3e–i). Coexistence of malignant ependymal-like cells with OPC-like populations in the same tumor suggests that H3.1K27M gliomas may originate in a more primitive progenitor, or in OPC populations with capacity to differentiate into all three glial lineages.
HOX architecture points to distinct progenitor domain origins
Distinct epigenomic landscapes associate with each histone variant and location30,33, but to what extent these differences reflect discrete cellular origins remains unclear. We therefore compared tumors bearing the same H3.3K27M mutation in two different locations, the thalamus and pons. We focused on the opposing marks H3K27me3 and H3K27ac, given their key roles in cell type specification and their preferential retention at high affinity PRC2 targets38 and regulatory elements33, despite the global epigenomic remodelling induced by K27M.
Promoter regions displayed strong differential activation of several patterning genes, particularly the HOX family in the pons (Figure 2a, Extended Data Figure 4a). HOX activation could represent a K27M-driven dysregulation of these classical PRC2 targets45,46, or an enduring footprint of cellular origins. We thus assessed expression and chromatin state (H3K27me3, H3K27ac, CTCF, SUZ12, scATAC) in tumor samples across all 39 HOX members. In the normal setting, the order of the HOX genes along the genome determines their sequence of activation along the anterior-posterior axis (spatial co-linearity)47. The 3’ HOX genes in each cluster are expressed most anteriorly, in the hindbrain, while the more 5’ paralogs are progressively activated in posterior tissues (Figure 2b). This highly regulated expression pattern is achieved by evolutionarily conserved, concerted, and redundant epigenetic mechanisms that ensure proper activation of each HOX gene. These include organization of 3D chromatin48, clustered binding of CTCF49, and deposition of histone marks that result in the partition of each HOX cluster into subdomains48,49 (Figure 2c). First, we observed that patient samples, including tumors and patient-derived cell lines, maintained this highly structured genome architecture across epigenomic layers, including bipartite domains delimited by 3D interaction loops, and a high level of H3K27me3 despite its genome-wide depletion (Figure 2d). Second, the overall HOX configuration was consistent with tumor location (Figure 2b,d,e): all clusters were silent in thalamic tumors, while hindbrain tumors maintained co-linearity, activating the 3’ domains and silencing HOXA1 and HOXB1. We conclude that HOX cluster architecture is preserved and a faithful reflection of the cell-of-origin.
Figure 2. Chromatin architecture of HOX clusters implicates distinct progenitor domain origins.
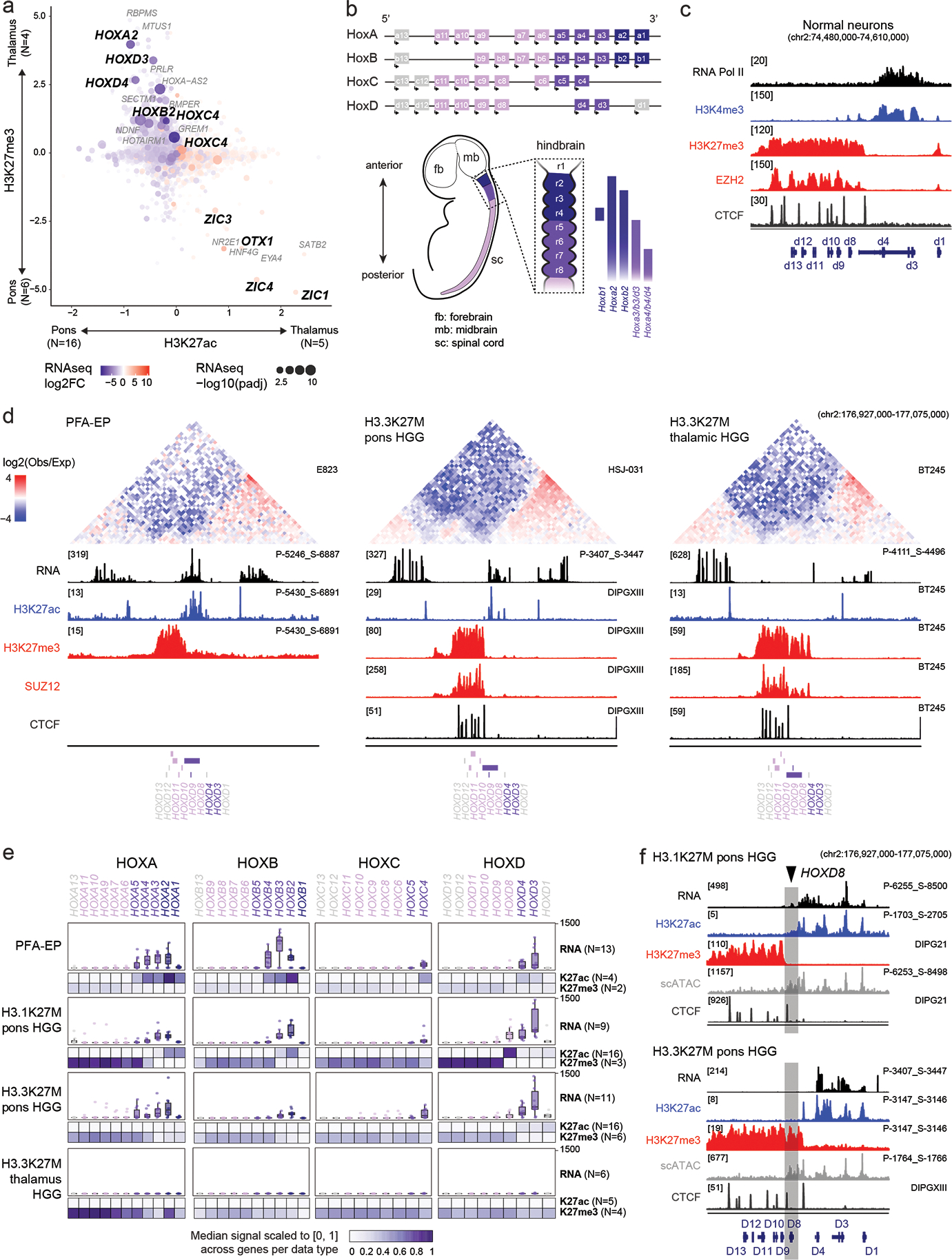
a. Promoter-associated H3K27ac and H3K27me3 over all expressed genes. Axes represent enrichment of each mark in H3.3K27M thalamic vs H3.3K27M pons HGG (Z-score, see Methods and Data Availability). Log2FC: log2 fold-change; padj: adjusted p-value (negative binomial Wald test, Benjamini-Hochberg correction).
b. Top: Schematic of organization of HOX clusters along the linear genome. Bottom: Schematic of HOX expression patterns in the developing embryo (left) and specifically in the hindbrain (right).
c. Epigenomic state at HoxD cluster in mESC-derived cervical motor neurons, data from Narendra et al, Science, 201549). Y-axis limits are indicated in brackets.
d. Top: Hi-C heatmaps depicting chromatin conformation structure at the HOXD cluster in tumors (PFA-EP) or cell lines (H3.3K27M HGG). Heatmaps represent the log2 ratio of observed vs expected chromatin interactions, at 10kb resolution. Bottom: tracks for bulk RNAseq and H3K27ac, H3K27me3, SUZ12, and CTCF ChIPseq data for representative samples. Y-axis limits are indicated in brackets. Sample IDs are indicated at right.
e. Bulk RNAseq levels, promoter H3K27ac, and promoter H3K27me3 at each HOX gene in each tumor type. Heatmaps represent median RPKM in samples of each tumor type; for each data type (H3K27ac and H3K27me3), this median value is scaled to [0,1] across all HOX genes. Sample sizes for each tumor/data type are indicated in parentheses.
f. Epigenomic state at HOXD cluster for representative H3.1K27M pons and H3.3K27M pons HGG, indicating a distinct boundary between active and inactive chromatin states. For scATACseq data, each track represents RPKM-normalized aggregated accessibility for a tumor single-cell population. Y-axis limits are indicated in brackets.
Importantly, we observed a location and variant-specific configuration, suggesting a distinct ontogeny linked to each tumor subtype (Figure 2e, Supplementary Tables 13–14). HOXD had a shifted boundary in H3.3 compared to H3.1K27M hindbrain tumors, with HOXD8 belonging to the active subdomain in H3.1K27M tumors and switching to the silent one in H3.3K27M tumors (Figure 2e–f). Similar boundary differences were observed between PFA-EP and H3.1K27M tumors (HOXA5, HOXB4, HOXD8), as well as between PFA-EP and H3.3K27M tumors (HOXB4). PFA-EP also uniquely activated HOXA4 and HOXB4. Altogether, these results demonstrate that HOX regulation persists in tumors despite extensive oncogenic chromatin perturbation and PRC2 impairment. The specific profile of HOX genes in each tumor entity acts as both an address, mapping these tumors to positional identities in the brain, and a clock, providing a temporal window for specification of the cells where the genetic alteration occurred. Since the earliest-activated HOX genes were silent, OPC specification likely occurred after hindbrain segmentation, establishing a lower temporal bound on the window in which the tumor cellular origins were specified.
H3.3K27M thalamic gliomas arise from the thalamus proper
Thalamic tumors displayed active chromatin states at a different set of developmental genes, OTX1 and ZIC1/4 (Figure 2a). The thalamus is derived from the diencephalon, which, like the hindbrain, is segmented by combinatorial gene expression during embryonal development, in this case into prosomeres (Figure 3a). Prosomere 1 (p1) gives rise to the pretectum, p2 gives rise to the thalamus proper, and p3 gives rise to the pre-thalamus. OTX1 and its homolog OTX2 are involved in positioning the p2/p3 border50. We thus assessed expression, H3K27ac, and H3K27me3 at diencephalon patterning genes. Thalamic tumors and cell lines consistently activated GBX2, IRX3, and LFNG (expressed in p2), but silenced EMX2, SIX3, and FEZF1 (expressed in p3) (Figure 3b, Supplementary Table 15). scATACseq confirmed the patterns occurred in malignant cells, and were not driven by contaminating normal tissue (Figure 3b). While some of these genes have pleiotropic roles in the patterning of other regions, in the context of the diencephalon this profile indicates that H3.3K27M thalamic gliomas arise exclusively from cells of the thalamus proper. In conclusion, despite similar transcriptional profiles as pontine gliomas, epigenome profiles are consistent with an anatomically distinct origin for H3.3K27M thalamic gliomas within p2 or its derivatives.
Figure 3. H3.3K27M thalamic gliomas arise from the thalamus proper.
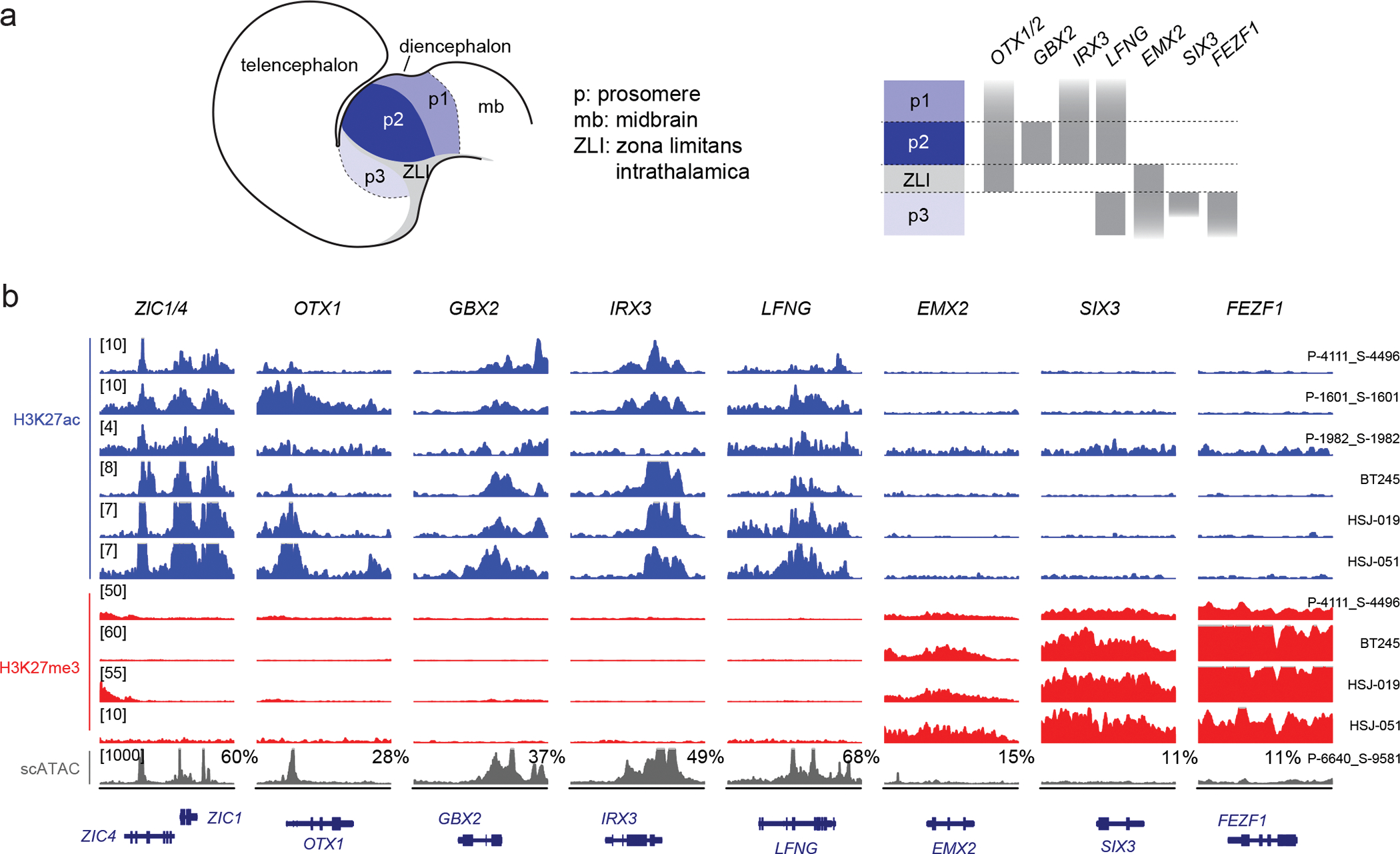
a. Schematic of the developing diencephalon, indicating three embryonal segments (prosomeres, p1–3) and patterning genes that mark each prosomere.
b. H3K27ac and H3K27me3 ChIPseq data and scATACseq for H3.3K27M thalamic HGG primary tumors and cell lines, showing activation of genes marking p2 (the thalamus proper), but silencing of genes marking p3 (the pre-thalamus). Y-axis limit for each sample is indicated in brackets. For scATACseq data, each track represents RPKM-normalized aggregated accessibility for one tumor single-cell population; the percentage of malignant cells where chromatin accessibility in the region was detected (>1 fragment) is indicated for each gene. Chromosome coordinates are indicated in Supplementary Table 15.
H3.1K27M ACVR1 tumors mirror SHH-specified NKX6-1+ lineages
We next asked whether tumors occurring in the same location arise from ontogenetically distinct progenitor cells, comparing molecular profiles of H3.3 and H3.1K27M tumors in the pons. Integration of expression and promoter chromatin marks revealed a strong differential activation of lineage-specific and patterning genes (Figure 4a), including PAX3, FOXB1, ZIC2, and NKX6-1. At the enhancer level, NKX6-1 and PAX3 were among the top differentially acetylated hits (Figure 4b, Supplementary Table 16). We next reconstructed the tumor core regulatory circuits51, by identifying active transcription factors (TF) based on H3K27ac, together with their targets and regulators via a motif analysis. Once again, NKX6-1 and PAX3 were among the TFs with the largest differences in number of regulators and targets between H3.1 and H3.3K27M tumors (Figure 4c, Supplementary Table 17).
Figure 4. H3.1K27M ACVR1-mutant gliomas mirror a SHH-specified NKX6-1+ progenitor.
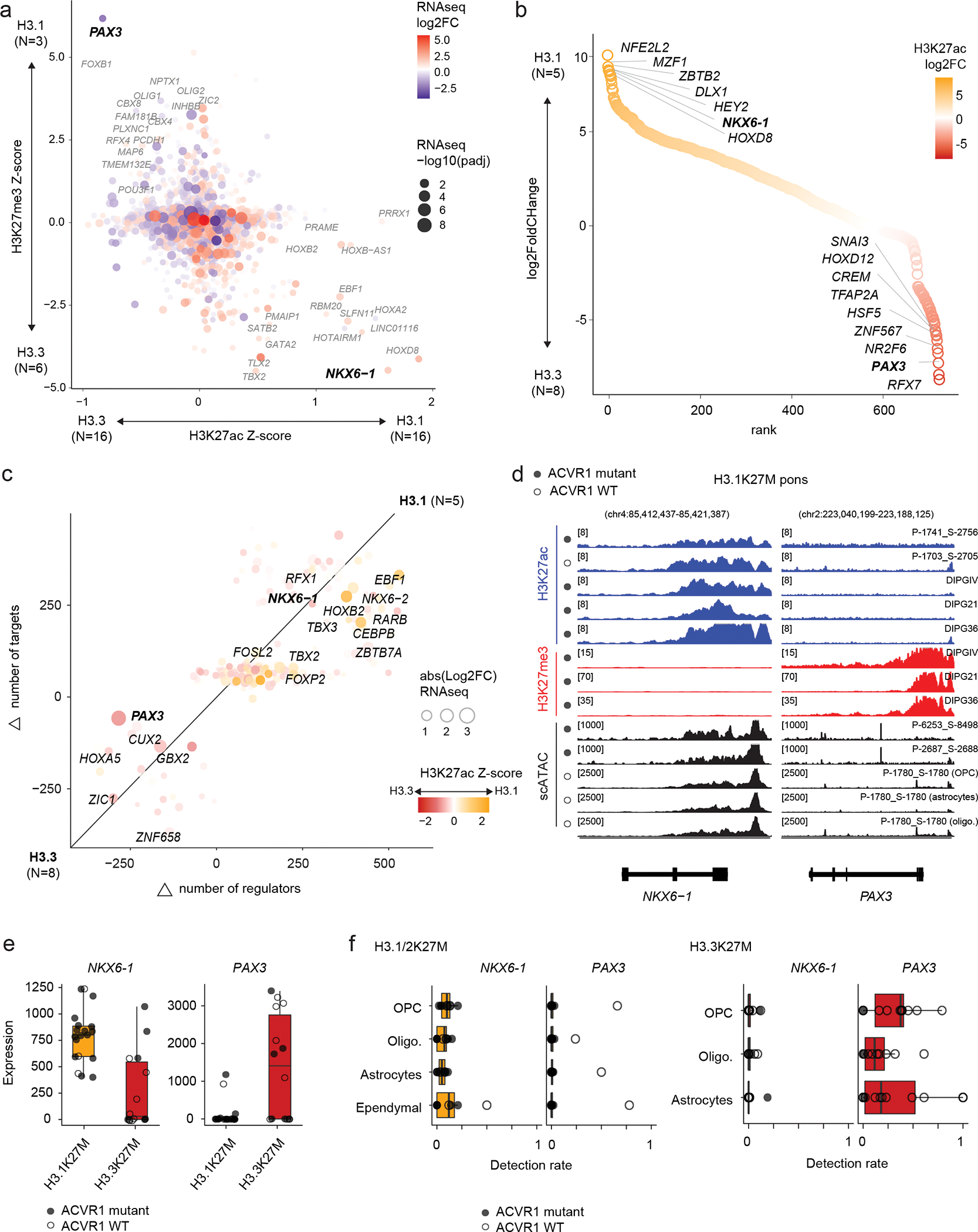
a. Promoter-associated H3K27ac and H3K27me3 over all expressed genes. Axes represent enrichment of each mark in H3.1K27M pons vs H3.3K27M pons HGG (Z-score, see Methods and Data Availability). Log2FC: log2 fold-change; padj: adjusted p-value (negative binomial Wald test, Benjamini-Hochberg correction).
b. Differential enhancer analysis, based on H3K27ac peaks, between H3.1 and H3.3K27M pons HGG. Enhancers are ranked by log2 fold-change of H3K27ac as in (a).
c. Differences in core regulatory circuitry (CRC) between H3.1K27M pons and H3.3K27M pons HGG. X-axis represents differences in number of genes regulating each transcription factor (TF), and y-axis represents differences in number of targets of each TF.
d. Epigenomic state at NKX6-1 and PAX3 in representative H3.1K27M pons tumors and cell lines. For scATACseq data, each track represents RPKM-normalized aggregated accessibility for one malignant single-cell population. Y-axis limits are indicated in brackets.
e. Bulk RNAseq expression levels of NKX6-1 and PAX3 for all pons tumors, by histone 3 variant (H3.1K27M, N=19 patients; H3.3K27M, N=14).
f. Detection levels of NKX6-1 and PAX3 in malignant cells for all pons tumors in the scRNAseq cohort, stratified by projected cell type, for each histone 3 variant (H3.1K27M, N=8 patients; H3.3K27M, N=16). Cells without a consensus projection and cell types comprising less than 5% of the dataset for each tumor type were excluded.
In fact, we observed a particularly strong and opposing signal at the PAX3 and NKX6-1 loci, consistent across samples and modalities (Figure 4d–f). H3.1K27M tumors activated NKX6-1, showing high H3K27ac, low H3K27me3, and open chromatin in malignant cells (Figure 4d), and high expression at the bulk (Figure 4e) and single-cell levels (Figure 4f). In contrast, H3.3K27M tumors showed more variable chromatin conformation (Extended Data Figure 4b) and expressed both NKX6-1 and PAX3, although generally not in the same samples (Extended Data Figure 4c). Importantly, H3.1K27M tumors and xenografts expressed NKX6-1 at the protein level (Figure 5a, Supplementary Table 18). This protein was absent from other HGGs (Figure 5b, Extended Data Figure 5a–d), as well as from several postnatal tissues outside the pancreas where it is normally detected52–54 (Extended Data Figure 5e–h, Supplementary Table 19). Finally, scATACseq revealed open chromatin peaks near NKX6-1, specifically in malignant cells (Figure 5c). These included two developmental cis-regulatory elements: hs680, an enhancer driving expression in the cranial nerve, hindbrain, and midbrain55; and CRMNkx6.1, an evolutionarily conserved NKX6-1 cis-regulatory module with binding sites for the GLI proteins [Sonic hedgehog (SHH) effectors], and for homeodomain TFs participating in cross-repressive interactions with NKX6-1 during dorsal-ventral patterning56. Thus, regulatory elements neighbouring NKX6-1 are consistently active in H3.1K27M tumors.
Figure 5. NKX6-1 is activated in H3.1K27M HGG.
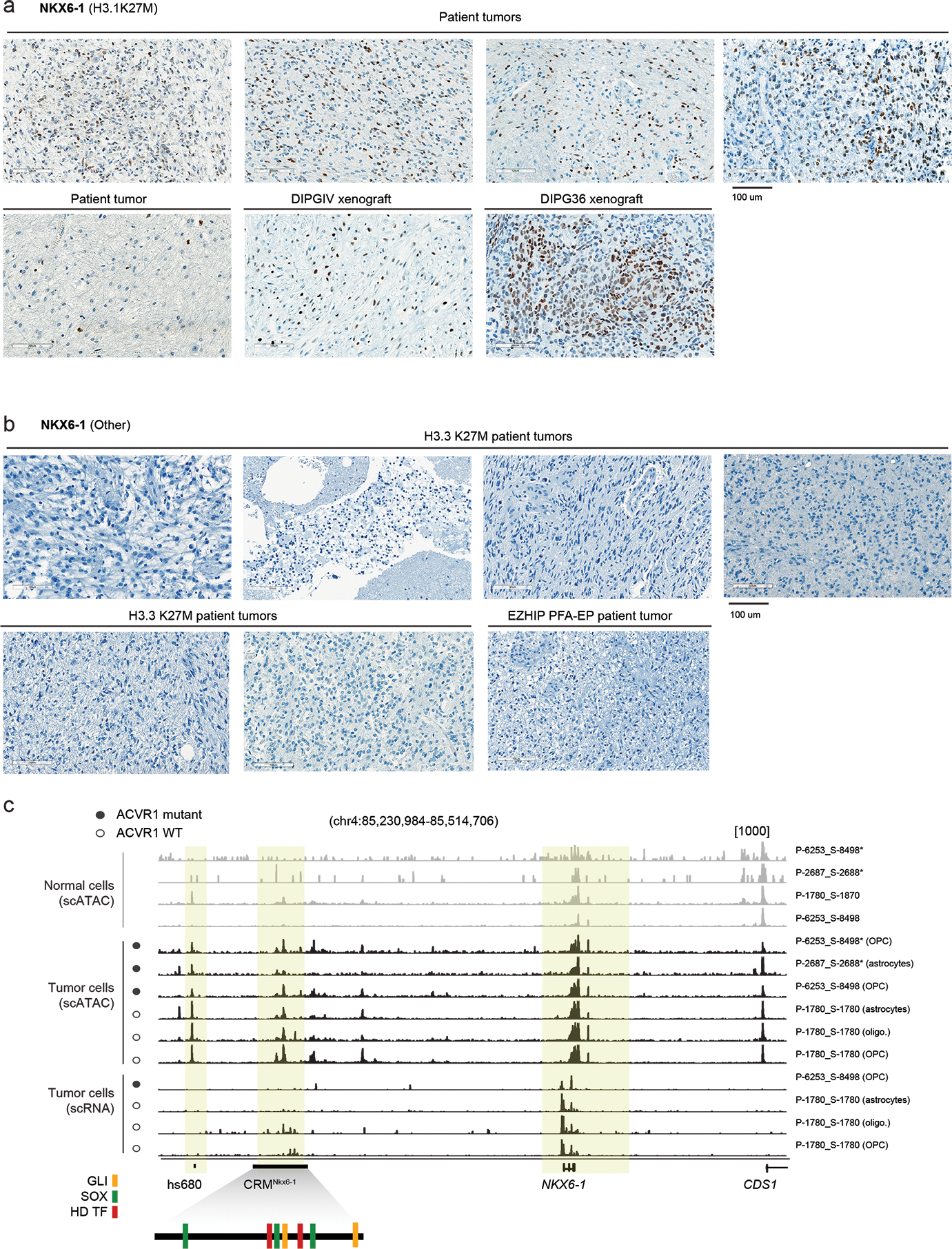
a. Immunohistochemistry staining of NKX6-1 in H3.1K27M HGG samples including patient tumors (N=5) and xenografts derived from the H3.1K27M patient-derived cell lines DIPGIV and DIPG36.
b. Immunohistochemistry for NKX6-1 protein in H3.3K27M HGG patient tumors (N=7).
c. Single-cell chromatin accessibility and RNA tracks for H3.1K27M pons HGG at the NKX6-1 locus. Each track represents RPKM-normalized aggregated accessibility/expression for one single-cell population. Normal cells and malignant cells are indicated. Asterisk (*) denotes tracks from scATAC data, all others are from scMultiome data. VISTA enhancer hs680 and NKX6-1 cis-regulatory module (CRMNkx6-1) are indicated. Schematic of CRMNkx6-1 indicates binding sites for SHH effector GLI transcription factors, SOX transcription factors, and homeodomain transcription factors, identified by Oosterveen et al, Developmental Cell, 201256.
The HOX cluster configuration (Figure 2d–f), the NKX6-1 activation (Figures 4–5), and the potential to differentiate into ependymal-like cells (Figure 1d–e) point to a specific origin for H3.1K27M DIPGs. During hindbrain development, oligodendrocyte specification occurs in distinct, well-characterized waves26,57 (Figure 6a). Migratory OPCs are first born in a ventral NKX6-1+ domain, specified by SHH, with the potential to also generate ependymal and astrocyte cells58. A second wave arises from a dorsal PAX3+ domain, specified by BMPs and WNTs25,26,57. To verify that the NKX6-1/PAX3 dichotomy, well-established in model organisms, is preserved in humans, we obtained human hindbrain snRNAseq data from first and second trimester of gestation42,43. NKX6-1 and PAX3 were mutually exclusive across cell types, co-expressed in less than 1% of cells, both in human and mouse (Figure 6b, Extended Data Figure 6a). Next, we reconstructed the gene regulatory networks (GRNs)59 in the normal developing pons for NKX6-1 and PAX3. Targets of NKX6-1 included other ventral markers (NKX6-2) and members of the SHH and Notch signalling pathways (Figure 6c). Pax3 targets, in turn, included dorsal markers (Pax7, Irx3, Dbx1) and members of the BMP and WNT pathways. NKX6-1 targets were also enriched for ependymal-specific genes (Extended Data Figure 6b–c), including the transcription factor Foxj1 (Extended Data Figure 6c), consistent with the potential of NKX6-1+ progenitors to differentiate into the ependymal lineage58. Importantly, NKX6-1 and Pax3 showed non-overlapping GRNs, sharing only 9% of their target genes. We conclude that, in the normal setting and across multiple developmental stages, cells expressing NKX6-1 and Pax3 maintain distinct, non-overlapping regulatory networks, with SHH/Notch members as intrinsic nodes in the NKX6-1 GRN, and BMP/WNT members in the Pax3 GRN.
Figure 6. ACVR1 mutations confer oncogenic BMP signalling in H3.1K27M HGG.
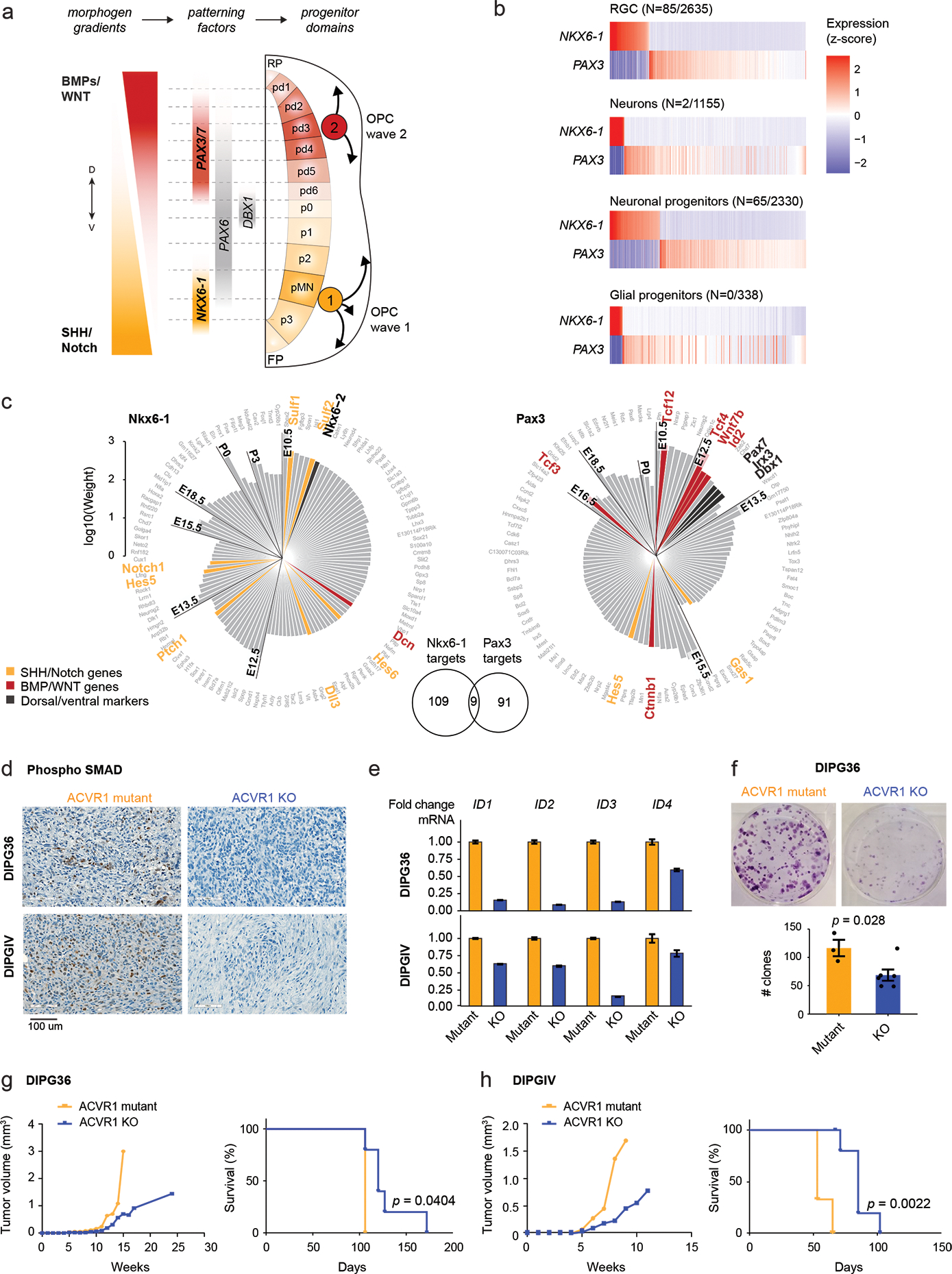
a. Schematic of coronal section of the developing hindbrain/neural tube, depicting ventral (V; NKX6-1+) and dorsal (D; PAX3+) waves of oligodendrocyte generation during development. RP, roof plate. pd, dorsal progenitor domain. p, ventral progenitor domain. pMN, progenitor of motor neurons domain. FP, floor plate.
b. Scaled scRNAseq expression (Z-score across cells) of NKX6-1 and PAX3 in cell types of the normal human fetal hindbrain (N=79,428 cells, 11 donors), showing their expression is largely mutually exclusive. The number of cells where both NKX6-1 and PAX3 are detected out of the total number NKX6-1+ or PAX3+ cells of the cell type is indicated in parentheses.
c. Targets of NKX6-1 and Pax3 extracted from gene regulatory networks inferred from scRNAseq data of E10-P6 mouse pons. Each bar represents one target, and the height of the bar represents the edge weight between the TF and the target. Targets are plotted clockwise from top, in order of the earliest time point at which they are detected as a target.
d. Immunohistochemistry staining of phosphorylated SMAD in mouse xenografts from the H3.1K27M DIPG cell lines DIPG36 and DIPGIV. Left: ACVR1 mutant line, right: isogenic cell line with CRISPR-based removal of ACVR1.
e. ddPCR for ID genes in DIPG36 and DIPGIV ACVR1 mutant and ACVR1 KO lines. Data are represented as the fold change +/− SD, based on N=3 technical replicates per cell line per condition.
f. Clone-formation assay for DIPG36 (ACVR1 mutant) and isogenic ACVR1 KO lines (ACVR1 mutant, N=3 biological replicates; ACVR1 KO, N=6; p-value = 0.028, 2-tailed t-test). Error bars represent mean values +/− SEM.
g-h. Tumor volume evolution and survival of mouse xenograft cohorts generated from DIPG36 (ACVR1 mutant, N=3 mice; ACVR1 KO, N=5 mice; p-value = 0.0404, log-rank test) and DIPGIV (ACVR1 mutant, N=3 mice; ACVR1 KO, N=6 mice; p-value = 0.0022, log-rank test).
ACVR1 mutations result in constitutive BMP signalling60, likely conferring oncogenic properties in the context of SHH-specified NKX6-1+ progenitors, where BMP is uncoupled from cell identity programs. Accordingly, CRISPR-Cas9 removal of ACVR1 mutations in two H3.1K27M patient-derived cell lines reduced BMP signalling, as shown by the marked reduction of downstream SMAD5/8 phosphorylation (Figure 6d, Supplementary Table 20) and decreased expression of ID genes (BMP pathway effectors) (Figure 6e). Furthermore, ACVR1-KO cells showed reduced clonogenic capacity and cell growth in vitro (Figure 6f), and increased survival in patient-derived xenograft models in vivo (Figure 6g–h). Altogether, we propose that the prevalence of ACVR1 mutations in H3.1K27M DIPGs and their restriction to the brain pons is a consequence of context-specific vulnerabilities of the cell-of-origin, where mutant ACVR1 confers the needed oncogenic BMP signalling missing in normal NKX6-1+ cells.
Uncoupling cell-intrinsic properties and oncohistone effects
We next assessed the global impact of H3.1K27M, H3.3K37M, and EZHIP on the epigenome, using histone mass spectrometry (MS) and ChIPseq in patient-derived cell lines and tumors (Figure 7). MS analysis confirmed decreased H3K27me3 compared to wild-type tumors (Figure 7a), with H3.1K27M cell lines showing the lowest and, surprisingly, EZHIP PFA-EP the highest levels. ChIPseq analysis showed similar global H3K27me3 patterns for the three entities, with sharp peaks at CpG islands (CGIs) and evident restriction of spread from these sites (Figure 7a), although CGI occupancy was higher in PFA-EP, in line with MS data (Figure 7a–b). Consistent with restriction of this mark to PRC2 landing sites38, the top H3K27me3 genomic bins preferentially overlapped CGIs and SUZ12 peaks (Extended Data Figure 7a).
Figure 7. H3K27M and EZHIP converge to restrict H3K27me3 to PRC2 nucleation sites.
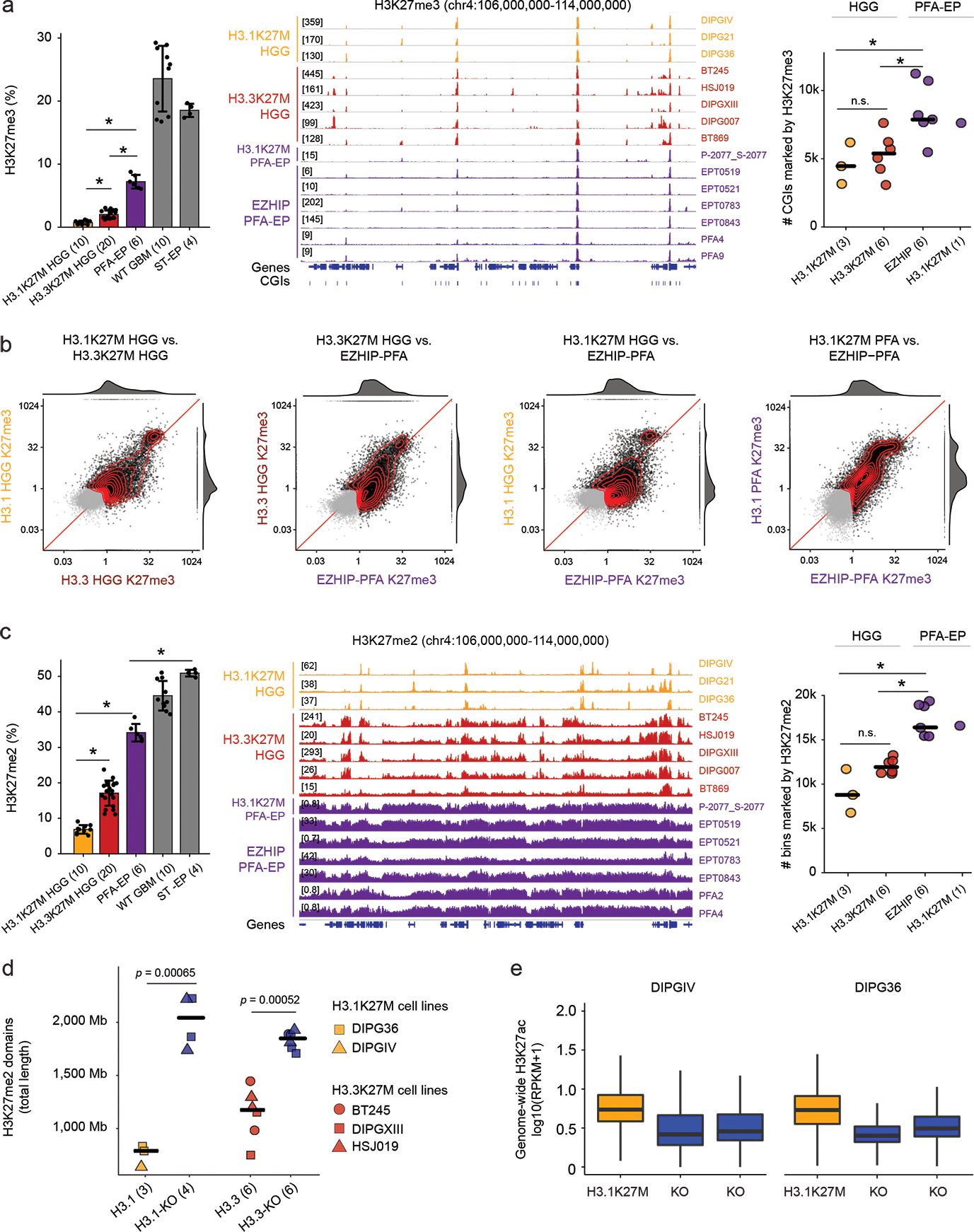
a. Profiling of H3K27me3. Left: Mass spectrometry data of H3K27me3 in cell lines. Number of biologically independent samples per group is indicated in parentheses. WT GBM: H3 wild-type glioblastoma. ST-EP: EZHIP wild-type supratentorial ependymoma. Error bars: mean +/− SD. P-values (Welch two-sample t-test): H3.1K27M vs H3.3K27M, p = 5.3×10−8; H3.1K27M vs PFA-EP, p = 1.6×10−5; H3.3K27M vs PFA-EP, p = 2.6×10−5. Middle: H3K27me3 ChIP-seq enrichment tracks over representative genomic region. Right: Number of H3K27me3-marked CGIs genome-wide. Crossbar indicates the median. P-values (Welch two-sample t-test): H3.1K27M vs. EZHIP PFA, p = 0.022; H3.1K27M vs. H3.3K27M, p = 0.55; H3.3K27M vs EZHIP PFA, p = 0.010.
b. Scatterplots of H3K27me3 signal over CGIs genome-wide in pairwise group comparisons. X- and Y- axes represent log2 mean RPKM value per group, normalized by input. Marked CGIs (mean RPKM > 1 in at least one groups in each comparison) are shown in black, while unmarked CGIs are shown in gray. Joint density and marginal distributions are calculated over marked CGIs only. Red line indicates the diagonal.
c. Profiling of H3K27me2. Left and middle panels: as in (a). P-values (Welch two-sample t-test): H3.1K27M HGG vs H3.3K27M HGG: p = 8.4×10−12; H3.1K27M vs PFA-EP: p = 1.0×10−7; PFA-EP vs ST-EP: p = 1.6×10-6. Right panel: Number of H3K27me2-marked 100kb-bins genome-wide. Crossbar indicates the median. P-values (Welch two-sample t-test without correction): H3.1K27M vs. EZHIP PFA, p = 0.014; H3.1K27M vs. H3.3K27M, p = 0.17; H3.3K27M vs EZHIP PFA, p = 1.8×10-5.
d. Total length of genome covered by H3K27me2 domains in K27M-mutant cell lines and isogenic K27M-KO lines. Domains were identified using a segmentation algorithm (see Methods). Crossbar indicates the median. P-values (Welch two-sample t-test without correction): H3.1K27M vs KO, p = 0.00065; H3.3K27M vs KO; p = 0.00052.
e. Distribution of H3K27ac in 1Mb bins genome-wide in isogenic H3.1K27M HGG cell lines DIPGIV and DIPG36.
We observed, in turn, clear differences between entities for H3K27me2: highest restriction in H3.1K27M HGG, more spread in H3.3K27M HGG lines, and even higher levels and broader domains in PFA-EP (Figure 7c, Extended Data Figure 7b). Removal of H3.1K27M by CRISPR/Cas9 in three DIPG cell lines restored H3K27me2/3 spread on chromatin. Thus, as for H3.3K27M38, the effect of H3.1K27M on chromatin is reversible (Figure 7d, Extended Data Figure 7c). Finally, we examined the H3K27ac mark, known to be increased6,28,38 and pervasively deposited on the silent genome33 in H3.3K27M gliomas. H3K27ac was also increased in H3.1K27M gliomas relative to H3 wild-type tumors (Extended Data Figure 7d), leading to increased deposition across specific classes of repeat elements relative to isogenic H3.1K27M-KO counterparts (Figure 7e, Extended Data Figure 7e). Unexpectedly, PFA-EP tumors showed no increase of H3K27ac (Extended Data Figure 7d), likely explained by the large residual H3K27me2 domains, a mark known to prevent intergenic deposition by acetyltransferase complexes61.
The distinct epigenomic landscape of each entity, including H3K27me3 retention sites, H3K27me2 differential spread, and H3K27ac deposition patterns, may be partially explained by the cell-of-origin chromatin state. To assess this variable, we first analyzed a rare PFA-EP driven by H3.1K27M. This tumor closely mirrored EZHIP PFA-EP both in H3K27me3 enrichment (Figure 7a–b, Extended Data Figure 7a) and H3K27me2 spread (Figure 7c, Extended Data Figure 7b). Furthermore, chromatin configuration at cell type-specific genomic regions derived from single-cell epigenomic data62 was consistent with PFA (Extended Data Figure 8a–b) and ependymal identity (Extended Data Figure 8c), suggesting no major reprogramming of cell identity by H3.1K27M. Next, we profiled two isogenic cell lines with H3.3K27M removed by CRISPR/Cas9, where we overexpressed the mutant canonical histone, H3.1K27M (Figure 8a–e, Extended Data Figure 9). Overexpression of H3.1K27M did not alter H3K27me3 deposition sites, nor did it restrict H3K27me3 and me2 spread compared to the original H3.3K27M parental lines (Figure 8a–e, Extended Data Figure 9e–i). These data suggest that PRC2 recruitment sites and level of spread of these marks are largely independent of EZHIP/H3 variant-specific effects and of the amount of mutant histone present in cells.
Figure 8. Uncoupling the effect of histone variants from cell-of-origin chromatin state and cycling rate.
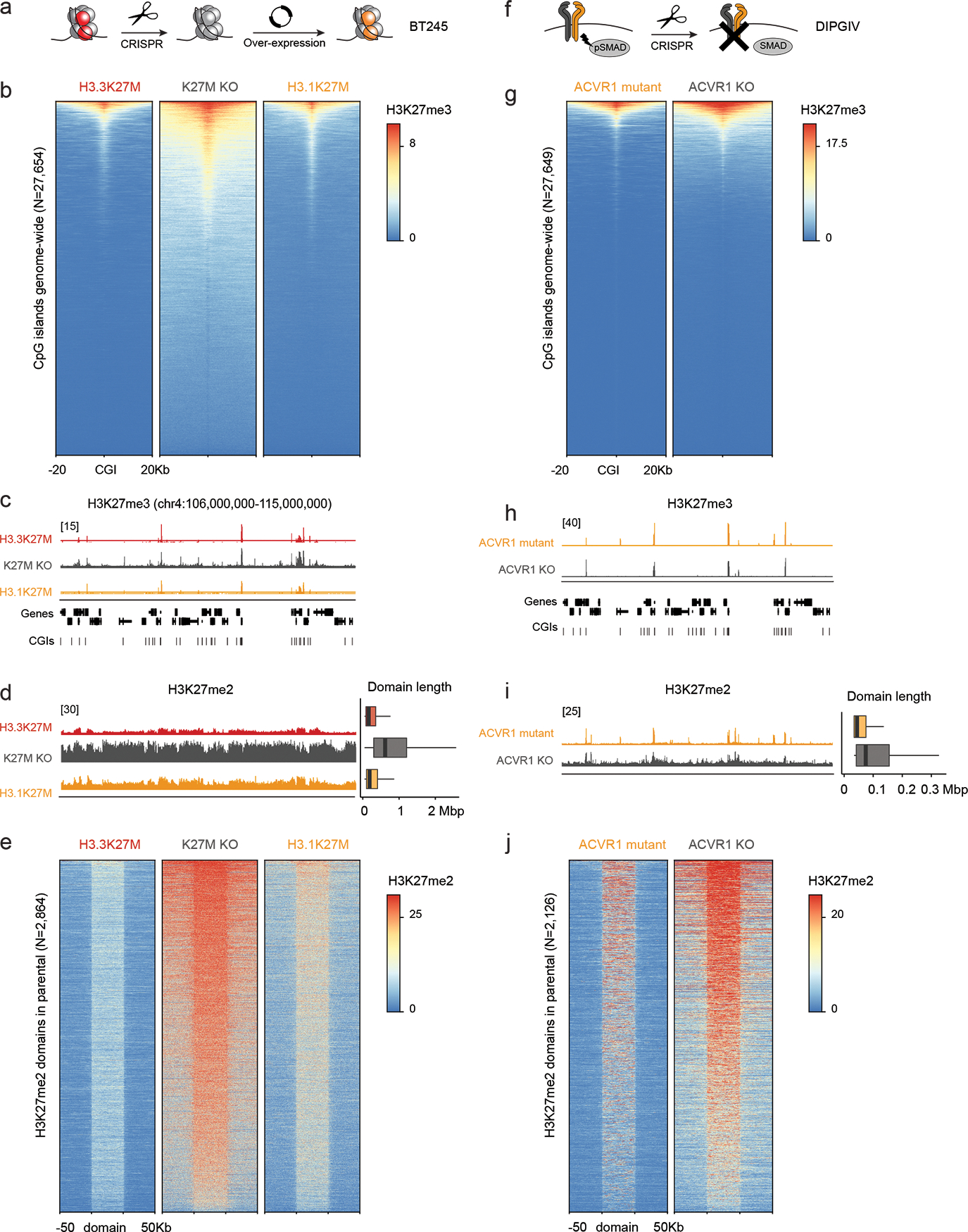
a. Schematic of experimental design.
b. Heatmap showing distribution of Rx-normalized ChIPseq signal for H3K27me3 in BT245 at CpG islands (CGIs), flanked by 20kb on either side.
c. Rx-normalized H3K27me3 tracks in each condition at a representative genomic region. Y-axis limit is indicated in brackets and identical for all tracks.
d. Left: Rx-normalized H3K27me2 tracks in each condition at the same region as in (c). Y-axis limit is indicated in brackets and identical for all tracks. Right: genome-wide distribution of H3K27me2 domain length in each condition (H3.3K27M, N=8436 domains; K27M KO, N=3782; H3.1K27M, N=8200).
e. Heatmap showing distribution of Rx-normalized ChIPseq signal for H3K27me2 in BT245 at H3K27me2 domains across the genome in each condition. Domains are scaled to 50kb and flanked by 50kb on either side. The maximum of the color scale is set to the 90th percentile value across all data points.
f. Schematic of experimental design.
g-j. Analysis of H3K27me3/2 in DIPGIV ACVR1 mutant and KO conditions as in (b-e). For genome-wide distribution of H3K27me2 domains length (ACVR1 mutant, N=11,736 domains; ACVR1 KO, N=10,614).
Finally, we investigated the variable of cell cycling rate. H3K27 methylation by PRC2 is enzymatically demanding, especially for trimethylation which remains proximal to PRC2 nucleation sites and for which EZH2 needs allosteric activation to initiate its spread63. Different OPCs and ependymal progenitors have diverse cycling times, possibly impacting PRC2 kinetics64,65. We observed that H3.3K27M HGG lines had longer doubling times than H3.1K27M (Extended Data Figure 9c), and thus tested the association of H3K27me2 deposition and spread with cycling rates in an isogenic context. We used the H3.1K27M DIPGIV line where we had removed the ACVR1 mutation by CRISPR/Cas9, which slowed down proliferation compared to unedited DIPGIV (Figure 8f, Extended Data Figure 9d). ACVR1-KO DIPGIV, as predicted, showed limited differences in H3K27me3 deposition, with marginally higher levels at CGIs compared to ACVR1-mutant DIPGIV (Figure 8f–h). In contrast, H3K27me2 showed increased spread in the ACVR1-KO cells, consistent with improved PRC2 kinetics due to prolonged cycling time (Figure 8i–j).
In summary, H3.1K27M, H3.3K27M and EZHIP converge to restrain H3K27me3 deposition to PRC2 nucleation sites in the cell-of-origin. The levels and spread of H3K27me2 depend on the cycling rate of the progenitor cell targeted by these genetic alterations. Furthermore, our data suggest that there is no variant-specific PRC2 recruitment and that this complex is free to land on its designated targets on chromatin in the cell-of-origin, but unable to spread H3K27me3 beyond CGIs in the presence of H3K27M or EZHIP.
Discussion
Profiling of tumor transcriptomes, epigenomes, and chromatin architecture indicates that K27M mutations occur in distinct lineages with well-defined positional identities, ruling out a shared cell-of-origin migrating throughout the midline. Each entity preserves a faithful memory of both dorsal-ventral and anterior-posterior patterning of the nervous system; these signatures can be used as a “zipcode” of the developmental window where the lineage-of-origin was specified. In the pons, H3.1K27M DIPG likely occur in cells derived from the earliest waves of OPC specification, NKX6-1+ SHH-dependent ventral progenitors, while H3.3K27M preferentially affects dorsal PAX3+ BMP-reliant progenitors. In the thalamus, H3.3K27M occurs exclusively after commitment to p2 during diencephalon development.
K27M in different H3 variants has been suggested to have differential effects on PRC2 regulation and subsequently on molecular profiles and developmental genes28–30. In turn, aberrant HOX gene activation and disruption of their co-linearity have been reported in multiple cancers, including K27M-mutant DIPGs and other HGG66–68, as has HOX expression heterogeneity in PFA-EP4. Here, we show that the strong signal of patterning genes, including the HOX family in pontine tumors and ZIC4 and OTX1 in thalamic tumors, does not result from oncogenic PRC2 dysfunction. Despite drastic loss of H3K27me3, tumor samples maintain highly structured genome architecture at the HOX clusters across all epigenomic layers, preserving bipartite domains delimited by 3D interaction loops anchored on H3K27me3 and CTCF binding sites. While K27M-induced de-differentiation cannot be completely excluded, this highly structured epigenomic conformation seems unlikely to be acquired after transformation. Furthermore, we observed limited if any novel PRC2 nucleation sites when interchanging the H3K27M mutations in cell lines. Indeed, our data indicate that K27M oncohistones and EZHIP have limited effects on reprogramming cell identity and that the distinct distribution of epigenetic marks in the tumors they drive mainly reflects the cell-of-origin epigenome and cycling rate.
In H3.1K27M gliomas, an NKX6-1+ OPC origin may explain features exclusive to this entity, including the recurrent gain-of-function mutations in the BMP receptor ACVR1. An opposing BMP/SHH morphogen gradient establishes neural tube dorsal-ventral patterning, specifying progenitors along this axis during early development69. In NKX6-1+ ventral OPCs, cell fate is regulated by the SHH pathway, activation of which has been previously reported in DIPG23. BMPs, in turn, are mainly active dorsally. These morphogens have a context-dependent effect60: in vitro, they can inhibit oligodendroglial differentiation and promote stemness or astroglial lineage commitment in neural progenitor cells, while in specific OPCs they can in turn promote differentiation. BMP morphogens lead to SMAD1/5/8 phosphorylation and ID gene expression, which in turn inhibit senescence through CDKN2A repression. Germline ACVR1 mutations result in low-level ligand-independent BMP signaling and enhanced responsiveness to BMPs, and brainstem alterations seen through imaging60, evidence of the role of this receptor in the pons. We propose that somatic acquisition of these mutations in SHH-reliant NKX6-1+ ventral OPCs mutant for H3.1K27M, stalled in their development by the oncohistone mutation, allows progenitor cells to acquire oncogenic BMP signalling. Aberrant BMP signalling would promote in turn the mesenchymal features20,30 and increased stemness observed in these tumors at the expense of differentiation along the oligodendroglial lineage.
Our data indicate that the effects of EZHIP, H3.1 and H3.3 K27M on the epigenome are also largely driven by progenitor state and identity. The prevalence of K27M on cell-cycle dependent canonical H3.1 in ventral OPCs may be due to the rapid developmental expansion of these progenitors64: oncogenic effects in H3.3 would be diluted in these conditions. H3.1K27M has been suggested to lead to a drastic decrease in the deposition of H3K27me329, and by extension H3K27me2. We did not observe this effect in H3.1K27M PFA-EP and, in DIPG, H3K27me3 levels were higher than suggested29. In fact, H3K27me2 seems to spread to the extent allowed by the cell-of-origin context, including epigenome and proliferation kinetics. Last, H3K27M and EZHIP uniformly restrict H3K27me3 deposition to cell-of-origin PRC2 nucleation sites; impaired spread of this repressive mark is possibly at the core of their oncogenic potential. Indeed, H3K27me3 deposition is slower and more enzymatically tasking for EZH2, which evolved to have allosteric activation through EED and auto-phosphorylation to kick-off H3K27me3 spread. We propose that H3.1K27M, H3.3K27M and EZHIP exploit this weakness, converging to block H3K27me3 deposition, restraining its spread to PRC2 landing sites.
In summary, we clarify the etiology of H3K27M gliomas and propose a ventral brainstem progenitor origin for H3.1K27M gliomas. While the precise features making the H3K27M-driven epigenomic landscape permissive to transformation remain unclear, the uniform restriction of H3K27me3 to PRC2 nucleation sites certainly contributes to stall differentiation, which relies on the nimbleness of this complex to spread across the genome to target specific sites based on developmental cues1. We suggest a model in which ACVR1 promotes oncogenesis through ectopic BMP activation in a lineage normally regulated by other signaling pathways. We provide molecular features (NKX6-1, PAX3) to guide subtype-specific modeling of H3K27M gliomas. Our data suggest that, due to context-dependent interpretation of cell-identity signaling, ACVR1 has distinct effects in dorsal versus ventral hindbrain cells. Our hypotheses motivate strategies for modulating cell state and differentiation in H3K27M gliomas based on their intrinsic gene regulatory networks, with important implications for future targeted therapies.
Methods
Statistics and reproducibility
No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment. Analyses in R were performed with versions 3.6 and 4.1. For boxplots throughout the figures, the elements represent the following: center line, median; box limits, upper and lower quartiles; whiskers, 1.5× interquartile range.
Ethics approval and informed consent
This study was approved by the Institutional Review Board of the respective institutions from which the samples were collected. Protocols for this study involving collection of patient samples and information were approved by the Research Ethics and Review Board of McGill University and Affiliated Hospitals Research Institutes and the Research Ethics Board at the Hospital for Sick Children. Informed consent was obtained from all research participants. Animal protocols for orthotopic mouse xenografts were approved by the Animal Compliance Office of the McGill University and Affiliated Hospitals Research Institutes. Animal protocols for mice profiled at developmental timepoints were approved by the Animal Care Committee of The Centre for Phenogenomics, Joseph and Wolf Lebovic Centre.
Single-cell RNA sequencing for normal developing mouse brain reference dataset
Tissue dissections and library preparation
Mouse embryonic brain structures were dissected from C57BL6 mice at embryonic timepoints E10.5, E13.5, E16.5, and E18.5. Both male and female mice were used. For the brainstem, an incision was made between the midbrain and hindbrain boundary, as well as between the medullary hindbrain and spinal cord, to isolate rhombomeres 1 to 11 except for the cerebellar structure that was removed. All mouse dissections were performed under a Leica stereoscope with a pair of Moria ultra fine forceps (Fine Science Tools) in a PBS solution. The tissue was transferred into ice-cold Leibovitz’s medium, followed by single-cell dissociation with the Papain Dissociation System (Worthington Biochemical Corporation). Approximately 10,000 cells per sample were loaded on the Chromium Single Cell 3′ system (10x Genomics). GEM-RT, DynaBeads cleanup, PCR amplification and SPRIselect beads cleanup were performed using Chromium Single Cell 3′ Gel Bead kit. Indexed single-cell libraries were generated using the Chromium Single Cell 3′ Library kit and the Chromium i7 Multiplex kit. Size, quality, concentration and purity of the complementary DNAs and the corresponding 10x library were evaluated by the Agilent 2100 Bioanalyzer system. The 10x libraries were sequenced (multiplexed) on the Illumina HiSeq 4000 sequencing platform.
Data analysis
Cell Ranger (10x Genomics) (v2.2.0 and 3.0.1) was used with default parameters to demultiplex reads and align sequencing reads to the genome, distinguish cells from background, and obtain gene counts per cell. Alignment was performed using the mm10 reference genome build, coupled with the Ensembl transcriptome (v84).
Downstream data processing was performed using Seurat70 (v3.2.1). Cells were filtered using the following quality control (QC) metrics: mitochondrial content (indicative of cell damage), number of genes, and number of unique molecular identifiers (UMIs); exact thresholds were defined for each sample based on its distribution and are specified in Supplementary Table 6. Data was processed consistently without our recently published mouse developmental reference34. Libraries were scaled to 10,000 UMIs per cell and log-normalized. UMI counts and mitochondrial content were regressed from normalized gene counts and the residuals z-scored gene-wise. Dimensionality reduction was performed using principal component analysis (PCA) applied to the top 2000 most variant genes. The first 30 principal components were then used as input for projection to two dimensions, using uniform manifold approximation and projection (UMAP)71, and for clustering, using a shared nearest neighbor (SNN) modularity optimization algorithm72 based on the Louvain algorithm on a k-nearest neighbors graph with k = 20. Gene signatures for each single cell population were derived by taking the top 100 most differentially expressed genes in each single cell cluster compared to all other clusters in the same sample (based on the Wilcoxon rank sum test), sorted by average log2 fold change, after filtering out ribosomal (defined as having gene symbols matching “RPS”, “RPL”, “MRPS”, “MRPL”) and mitochondrial genes (defined as having gene symbol beginning with “MT-”). Gene regulatory network inference was performed using the SCENIC workflow59 (v1.1.1).
Single-cell RNA and ATAC sequencing for human samples
Single-cell RNAseq (scRNAseq), single-nuclei RNAseq (snRNAseq), and single-nuclei ATACseq (scATAC) sample handling and library preparation
Fresh tumors collected after surgery were enzymatically digested and mechanically dissociated using the papain version of the Brain Tumor Dissociation kit (Miltenyi Biotech) or the Worthington Papain dissociation kit. Red blood cells were lysed by ammonium chloride treatment for 5 min on ice. Cell viability was assessed with Trypan Blue. For samples with low viability (< 60%), dissociated cells were enriched for live cells using the Dead Cell Removal kit (Miltenyi Biotech). Approximately 10,000 dissociated cells per sample were loaded on the 10x Genomics Chromium controller. Nuclei were prepared from frozen tissue as follows (also described in ref73). Frozen tissues (5–50mg) were dounced on ice in 3ml of Lysis Buffer (LB: 10mM Tris-HCl pH7.4, 10 mM NaCl, 3 mM MgCl2, 0.05% NP-40, 5 times with the “tight” pestle then 10 times with the “loose” pestle). 2ml of chilled LB were then added and samples were incubated for 5 min on ice. 5 ml of Nuclei Wash and Resuspension Buffer (NWRB :PBS, 5% BSA, 40U/ml RNase Inhibitor, 0.25% Glycerol) were then added and nuclei suspensions were passed through a 30 μm cell strainer to remove clumps and centrifuged (500g for 5 min). Nuclei pellets were washed with 5 ml of NWRB. and centrifuged again. Nuclei pellets were resuspended in a final volume of 1 ml of NWRB., 1 ml of Optiprep 50% (Optiprep + Solution B :150mM KCl, 5 mM MgCl2, 20 mM Tricine, pH7.8, v/v) was added. This 25% Optiprep solution was layered on 29% Optiprep cushion and centrifuge at 10,000g for 30 min at +4°C. For 10X Genomics 3’RNA protocol nuclei pellet was carefully resuspended in NWRB to reach a concentration of 1,000 nuclei/μl. For scATAC, nuclei were resuspended in DNB Buffer (10X Genomics) to reach a concentration of 3,300 nuclei/μl. Nuclei concentrations were assessed with the ReadyProbes Cell Viability Blue/Green kit. Nuclei concentration was assessed using the ReadyProbes Cell Viability fluorescence assay (ThermoFisher Scientific). 20,000 nuclei per sample were loaded on the Chromium controller. Cell capture and library preparation was performed according to the Chromium Single Cell 3’ (v3) protocol for sc/snRNAseq, and according to the Chromium Single Cell ATAQ (v1) protocol for scATACseq. The 10x libraries were then sequenced (multiplexed) on the Illumina HiSeq4000 or NovaSeq sequencing platforms.
Joint single-nuclei RNAseq and ATACseq (scMultiome) sample handling and library preparation
scMultiome nuclei were prepared using either standard manual nuclei preparation as described in the previous section, or automated nuclei preparation. The protocol applied to each sample is specified in Supplementary Table 5. Samples subjected to automated nuclei prep were processed following the Singulator S100 protocol (S2 Genomics). Nuclei were isolated using the Nuclei Isolation kit and Singulator S100 instrument from S2 Genomics. Briefly, 5–20 mg of frozen tissue were put in a pre-cooled nuclei isolation cartridge with RNAse inhibitors. Samples were then processed on the Singulator S100 following the “extended nuclei” protocol.
After nuclei prep, nuclei were centrifuged and washed twice in Diluted Nuclei Buffer (10x Genomics) and counted with the ReadyProbes Cell Viability Blue/Green kit (ThermoFisher Scientific). 20,000 nuclei/sample were loaded on the Chromium Controller (10x Genomics). The 10x libraries (scATAC and sc/snRNA) were then prepared following the manufacturer’s instructions (Next GEM Single Cell Multiome). The 10x libraries were sequenced (multiplexed) on the Illumina HiSeq4000 or NovaSeq sequencing platforms.
Sc/snRNAseq data processing and quality control
Cell Ranger (10x Genomics) (v2.0.0) was used with default parameters to demultiplex reads and align sequencing reads to the genome, distinguish cells from background, and obtain gene counts per cell. Alignment was performed using the hg19 reference genome build, coupled with the Ensembl transcriptome (v75). In the case of snRNAseq, reads mapping to intronic regions were included.
QC and data processing were performed as above for mouse samples, with QC thresholds and metrics indicated in Supplementary Table 3. Cell cycle scores for G2/M and S phases were obtained as implemented in Seurat, by calculating the average expression of G2/M and S phase-associated gene lists74 in each single cell and subtracting the average expression of control gene lists. Control gene lists were derived by binning genes in each input list into 24 bins according to expression levels and randomly selecting 100 control genes from within each expression bin.
scATACseq data processing and quality control
Cell Ranger ATAC (v1.1.0) was used (‘count’ option with default parameters) to filter and align raw reads, identify transposase cut sites, detect accessible chromatin peaks, call cells and generate raw count matrices for the scATAC samples. Sequencing reads were aligned to the hg19 genome, coupled with the Gencode v28 (Ensembl 92) gene annotation.
Quality control and downstream data processing were performed using the Signac75 (v1.3.0) and Seurat (v4.0.9) packages. Cells were filtered using the following QC metrics (Supplementary Table 4): number of peaks detected, total number of transposition sites across peaks, transcription start site enrichment score (fold change of reads across a reference set of transcription start sites, relative to regions flanking those sites), and nucleosome signal (expected fragment length periodicity based on the ratio between mononucleosome-bound fragments to nucleosome-free fragments). Narrow peaks for each sample were called with MACS276 (v2.2.7.1) using the following parameters: -g 2.7e+09 -f BED --nomodel --extsize 200 --shift −100. ATAC reads were quantified in each peak for each cell, and the resulting counts matrix used for downstream analysis. Dimensionality reduction was performed using latent semantic indexing (LSI)70. Nonlinear dimensionality reduction using UMAP and clustering using the SNN algorithm was performed as above for scRNAseq data, using the LSI components as input for each method. Per-gene, per cell accessibility scores were inferred from scATAC data: (i) gene accessibility scores were initialized by counting the total fragments overlapping gene promoters (defined as TSS +/− 2.5kbp) in each cell; (ii) a scaling factor was computed for each sample, defined as the median number of transposition sites in promoters across cells, and (iii) scores were then log-normalized and scaled such that the sum in each cell equals the scaling factor.
scMultiome data processing and quality control
Cell Ranger ARC v2.0.0 (10x Genomics) was used (‘count’ option with default parameters) to filter and align raw reads, identify transposase cut sites, detect accessible chromatin peaks, call cells and generate raw count matrices for scMultiome samples. Sequencing reads were aligned to the hg19 genome, coupled with the Gencode v28 (Ensembl 92) gene annotation.
Quality control and downstream data processing were performed using the Signac75 (v1.3.0) and Seurat (v4.0.9) packages. QC metrics were computed separately for the RNA and ATAC modalities, as described in the above sections sc/snRNAseq data processing and quality control and scATACseq data processing and quality control, and jointly used for filtering (Supplementary Table 5). For joint analysis of RNA and ATAC data, a weighted nearest neighbour (WNN) graph was constructed between all cells using the PCA reduction of the RNA data and the LSI reduction of the ATAC data, with the following default parameters: 20 multimodal neighbors, 200 approximate neighbors, and L2 normalization enabled. This WNN graph was used as input for nonlinear dimensionality reduction (UMAP) and for SNN clustering.
Generation of pseudobulk chromatin accessibility tracks
For visualization of chromatin accessibility in subsets of cells, BAM files from scATAC and scMultiome data were subsetted using the subset-bam utility from 10x Genomics (v1.1). Pseudo-bulk bigwig tracks were generated from subsetted BAMs with a bin size of 1 and RPKM normalization using the deeptools77 bamCoverage functionality (v3.5.0). Bigwig tracks (for all data types) were visualized using plotgardener78 (v0.1.0) or the Integrative Genomics Viewer79 (v2.11.1).
Assembly of normal human fetal brain scRNAseq reference dataset
10x Single-cell RNAseq data for the human fetal hindbrain and thalamus were obtained from the Brain Initiative Cell Census Network (BICCN). Gene counts (Cell Ranger outputs) for datasets from Carnegie Stages 12–2242 and gestational weeks 14–2543 for all hindbrain and thalamic samples were downloaded from the Neuroscience Multi-Omic (NeMO) Archive at http://data.nemoarchive.org/biccn/grant/u01_devhu/kriegstein/transcriptome/scell/10x_v2/human/processed/counts/. Data processing was performed exactly as described in the section sc/snRNAseq data processing and quality control. Thresholds used for filtering and QC metrics are reported on a per-sample basis in Supplementary Table 11.
For human fetal hindbrain samples at Carnegie Stages 12–22, cell type annotations were not available in the original publication42. Therefore, we used the mouse developmental reference to label human fetal hindbrain samples. We computed the Spearman correlation between the expression profile of single human hindbrain cells and the mean expression profile of each cell type in the mouse reference dataset, and each cell was assigned its most highly correlated cell type. Hindbrain cell type labels were aggregated into broad cell classes using the same ontology as described above.
In the case of thalamic samples, cell type labels from the original publication43 were used to label the reprocessed data as follows: each cluster was assigned the most common published label among cells in the cluster, if it was represented by at least 25% of cells in the cluster. Next, clusters without published labels were labelled based on canonical cell type-specific markers (astrocytes: FABP7, S100B, CLU, AQP4; neurons: STMN2, TUBB; microglia: C1QC, LY86, ependymal: FOXJ1, DNAH10). Finally, we annotated OPCs in the dataset based on canonical OPC markers: PDGFRA, OLIG1/2.
Cell type-specificity score
To assess cell type-specificity of individual genes in the developing brain, we derived a score based on gene detection rates in the mouse scRNAseq reference. Detection rate (d) in a cell population is defined as the proportion of cells in which expression of a given gene is detected. For each sample i in the reference, let ci be the cluster with highest expression of gene g, and the set of all other cells in that sample. The cell type specificity score will then be defined as
This score can be interpreted as the largest difference between a gene detection rate in a specific cell type and all other cells, across all samples in the reference.
Integration of single-cell expression data across technologies
For visualization of single-cell gene expression datasets from scRNAseq, snRNAseq and scMultiome technologies in a shared UMAP space as in Figure 1, tumor cells from each group were integrated using Harmony80 (v1.0). Harmony was run using the following parameters: number of input principal components = 30, theta = 2, lambda = 1, sigma = 0.1, and regressing out the differences between technologies and samples. The resulting embedding was used as input to dimensionality reduction using UMAP, and clustering using the shared nearest neighbor modularity optimization algorithm72 using the Louvain algorithm on a k-nearest neighbor graph, with resolution = 0.5 and k = 20.
Identification of normal and malignant cells
To distinguish normal and malignant cells in the scRNAseq tumor data, we combined three strategies: tumor cell type projections to the normal brain, inference of copy-number variations (CNV) on a per-sample basis, and post-clustering quality control of Harmony-integrated data.
First, to identify cell types in scRNAseq tumor data, single cells were projected to their most similar cell type in the normal mouse brain developmental reference. We computed the Spearman correlation between the expression profile of single tumor cells and the mean expression profile of each cell type in the reference dataset, and each cell was assigned its most highly correlated cell type. Next, we defined an ontological relationship between cell type labels to aggregate them into broader cell classes: radial glial cells, glial progenitors, OPC, proliferating OPC, oligodendrocytes, ependymal cells, neuronal progenitors, immune cells, and vascular/other. This ontology is provided in Supplementary Table 7 and used throughout the study. For scMultiome samples, cell type projection was performed based on the RNA modality only. For scATAC samples, all of which had matched scRNAseq data (i.e. scRNAseq generated from the same sample, but not the same cells), we obtained cell type projections by transferring labels to scATAC data from its matched scRNAseq dataset, using the transfer learning method implemented in Seurat V370.
Second, CNV profiles were inferred from expression data for each sample using inferCNV (https://github.com/broadinstitute/infercnv) with window_length = 101 genes, expression_threshold = 0.1, analysis_mode = “samples”, and cluster_by_groups = FALSE. The mitochondrial chromosome (defined as having gene symbols starting with “MT”-), ribosomal genes (defined as having gene symbols matching “MRPS”, “MRPL”, “RPS”, “RPL”), and HLA genes (defined as having gene symbols starting with “HLA-“) were excluded from the CNV inference. For scRNAseq, normal cells previously identified from a collection of pediatric brain tumors34 was used as a reference. For snRNAseq data, normal cells previously identified from a collection of high-grade gliomas36 was used as a reference. In most cases, hierarchical clustering of cells based on their CNV profiles identified clear subtrees of malignant cells with prominent copy-number signal, and normal cells lacking copy-number signal (e.g. Extended Data Figure 3). For 10x Single-cell/Single-nuclei RNA samples, inferCNV (v1.2.1) was used. For 10x Multiome samples, inferCNV (v1.7.2) was used.
Third, post-clustering quality control was performed on the Harmony-integrated datasets. In this step, clusters containing >10% of cells projected as neurons and expressing bona fide neuronal markers were excluded from analysis. Finally, malignant calling was performed as follows: based on the per-sample clustering (see section sc/snRNAseq data processing and quality control), we required that at least 50% of cells in a cluster either belonged to a subtree lacking copy-number alterations or were projected to immune, vascular, or meningeal cells, and then assigned all cells in that cluster as normal. Otherwise, cells were labeled as malignant.
Consensus cell type assignment for tumor cells
Cell type assignment in malignant cells was performed by combining the cell type projections described in the previous section to the normal brain with two additional methods, SciBet81 and Support Vector Machines. SciBet, which, briefly, selects marker genes using an entropy test and then assigns cells to their respective cell types using multimodal distribution models and maximum likelihood estimation, was applied using the default number of marker genes (1,000). In turn, Linear Support Vector Machines82 (SVM), implemented by scikit-learn83, were trained using cross-validation to estimate model parameters (regularization parameter and loss function). For computational tractability, these methods were applied using the mouse developmental reference down-sampled by 35% in a stratified manner (per cell population) order to preserve the cell type distribution of the full dataset. Cells where the projected label obtained by spearman correlation was supported by at least one other method (SciBet, SVM, or both) were considered to have a consensus label. Downstream analyses were then restricted to malignant cells with consensus labels.
Unsupervised identification of gene programs and modules from single-cell expression data
Variable gene programs were inferred from tumor scRNAseq data and from the RNA modality in scMultiome data using non-negative matrix factorization (NMF), as implemented in the consensus NMF method, cNMF39 (v1.1). Briefly, for each value of k, the number of components, this method runs 100 iterations of NMF with different random seeds, clusters the components resulting from each replicate, filters outlier components, and takes the median of each cluster of components as a consensus estimate for that component. For each individual sample, cNMF was applied to raw UMI counts of malignant cells in that sample and run with values of k from 5–9 (Supplementary Table 3). For each value of k, the Silhouette score, measuring the stability of the components, and the Frobenius error were computed, and the k maximizing the Silhouette score and minimizing the Frobenius error was selected for each sample. Outlier components were filtered by retaining only components with mean distance to most similar components of 0.02 (density_threshold = 0.02), resulting in a program activity matrix (the activity of each program in each cell), and a gene scores matrix (reflecting the expected increase in transcripts per million of a given gene for a unit increase of a given program), which was z-scored across genes (see Figure 1b). To identify the genes associated with each program, we selected the top 100 genes with the highest gene score. To avoid rare or noisy programs which were highly specific to small cell populations, for each program in each sample, we calculated the proportion of malignant cells in which that program is the most active one. We then restricted our analysis to programs which were most active in at least 5% of malignant cells.
We next annotated programs based on quality control metrics and prior biological knowledge. To annotate programs with continuous variables, we calculated the Pearson correlation between the per-cell program usage scores, and per-cell metrics: mitochondrial and ribosomal content, number of detected genes, total number of UMIs, and G2/M and G1/S cell-cycle phase scores. To annotate programs using known gene sets and pathways, we computed the overlap between the genes associated with each program and reference gene signatures. Reference gene signatures were obtained from the MsigDB collections84,85, KEGG (N=186), PID (N=196), and Hallmark (N=50), as well our scRNAseq mouse brain developmental dataset, restricted to non-proliferating cell types (N=251). Since reference gene signatures differed in length, we used the percentage of each reference signature overlapping program-associated genes.
To assess statistical significance of the overlaps between tumor program-associated genes and reference signatures, we computed an empirical p-value for the reported overlap in each comparison. For each reference signature S, we obtained a null distribution by repeatedly sampling sets of genes of the same length as S from a background set of genes and computing the number of overlapping genes with each tumor program (N=1,000 iterations). For mouse developmental signatures, the background was the set of all genes detected in the mouse brain dataset. For MsigDB signatures, the background was the set of all genes detected in tumor single-cell gene expression data. Comparisons with P-value < 0.001, and >= 10% overlapping genes between the reference signature and tumor program were considered significant and are displayed in Figure 1/Extended Data Figure 2.
Gene programs identified from each tumor were then used to identify modules, i.e. sets of programs identified recurrently across multiple samples. We generated a pairwise similarity matrix for all programs by counting the number of top genes in common between every pair of programs, following Kinker et al86. This similarity matrix was subjected to hierarchical clustering using complete linkage and Euclidean distance. Finally, we designed a recursive algorithm to traverse the hierarchical clustering dendrogram to define discrete modules: first, a set of subtrees S was arbitrarily initialized by cutting the dendrogram into 5 subtrees. For each subtree t in S, if there were fewer than 4 programs in t, it was dropped from S. If the average inter-program similarity of the programs in t was greater than 10, then t was considered a module. Otherwise, t was cut into 2, and each resulting subtree appended to S. To identify the genes characterizing each module, we selected the 50 genes most frequently associated with programs belonging to the module. Module 11, which correlated with high mitochondrial content and coverage (shown in Extended Data Figure 2), likely reflects technical factors and was thus removed for downstream analyses.
Bulk RNA sequencing
Sample and library preparation
Total RNA was extracted from cell pellets using the AllPrep DNA/RNA/miRNA Universal Kit (Qiagen) according to instructions from the manufacturer. Library preparation was performed with ribosomal RNA (rRNA) depletion according to instructions from the manufacturer (Epicentre) to achieve greater coverage of mRNA and other long non-coding transcripts. Paired-end sequencing (100 bp) was performed on the Illumina HiSeq 4000 platform.
Data analysis
Adapter sequences and the first four nucleotides of each read were removed from the read sets using Trimmomatic87 (v.0.32). Reads were scanned from the 5′ end and truncated when the average quality of a four-nucleotide sliding window fell below a threshold (phred33 < 30). Short reads after trimming (<30 base pairs) were discarded. High-quality reads were aligned to the reference genome hg19 (GRCh37) with STAR88 (v.2.3.0e) using default parameters. Multimapping reads (MAPQ < 1) were discarded from downstream analyses. Gene expression levels were estimated by quantifying reads uniquely mapped to exonic regions defined by ensGene annotation set from Ensembl (GRCh37, N=60,234 genes) using featureCounts89 (v1.4.4). Normalization (mean-of-ratios), variance-stabilized transformation of the data and differential gene expression analysis were performed using DESeq290 (v1.14.1). For the analysis of HOX genes, RNAseq quantification was performed for all HOX transcripts in the Ensembl GRCh37 annotation, to include all annotated promoters. For displaying a single value per gene, the mean expression of each transcript was computed across samples, and for each gene, the transcript with the highest mean expression was selected for display.
ChIP sequencing
Sample and library preparation
Experimental procedures for chromatin immunoprecipitation and sequencing (ChIPseq) are similar to those described in refs33,38. Cells were fixed with 1% formaldehyde (Sigma). Fresh frozen tumor tissue samples were homogenized using mortar and pestle while tissue was still frozen, then fixed with 1% formaldehyde. Fixed cell preparations were washed, pelleted and stored at −80°C. Sonication of lysed nuclei (lysed in a buffer containing 1% SDS) was performed on a BioRuptor UCD-300 for 60 cycles, 10s on 20s off, centrifuged every 15 cycles, chilled by 4°C water cooler. Samples were checked for sonication efficiency using the criteria of 150–500bp by gel electrophoresis. After the sonication, the chromatin was diluted to reduce SDS level to 0.1% and before ChIP reaction 2% of sonicated drosophila S2 cell chromatin was spiked-in the samples for quantification of total levels of histone mark after the sequencing (see below).
ChIP reaction for histone modifications was performed on a Diagenode SX-8G IP-Star Compact using Diagenode automated Ideal ChIPseq Kit. 25ul Protein A beads (Invitrogen) or 70ul of sheep anti-mouse IgG beads (Invitrogen) were washed and then incubated with antibodies (protein A beads with: anti-H3K27ac (1:100, Diagenode C15410196), anti-H3K27me3 (1:40, CST 9733) and anti-H3K27me2 (1:50, CST 9728), and 2 million cells of sonicated cell lysate combined with protease inhibitors for 10 hr, followed by 20 min wash cycle with provided wash buffers. ChIP reaction for DNA binding proteins was performed as follows: antibodies (anti-SUZ12 (1:150, CST 3737), anti-CTCF (1:400, Diagenode C15410210)) were conjugated by incubating with 40ul protein A beads at 4°C for 6 hours, then chromatin from ~4 million cells was added in RIPA buffer, incubated at 4°C o/n, washed using buffers from Ideal ChIPseq Kit (1 wash with each buffer, corresponding to RIPA, RIPA+500mM NaCl, LiCl, TE), eluted from beads by incubating with Elution buffer for 30 minutes at room temperature.
Reverse cross linking took place on a heat block at 65°C for 4 hr. ChIP samples were then treated with 2ul Rnase Cocktail at 65°C for 30 min followed by 2ul Proteinase K at 65°C for 30 min. Samples were then purified with QIAGEN MiniElute PCR purification kit as per manufacturers’ protocol. In parallel, input samples (chromatin from about 50,000 cells) were reverse crosslinked and DNA was isolated following the same protocol.
Library preparation was carried out using Kapa HTP or HyperPrep Illumina library preparation reagents. Briefly, for HTP kit, 25ul of ChIP sample was incubated with 45ul end repair mix at 20°C for 30 min followed by Ampure XP bead purification. A tailing: bead bound sample was incubated with 50ul buffer enzyme mix for 30°C 30 min, followed by PEG/NaCl purification. Adapter ligation: bead bound samples were incubated with 45ul buffer enzyme mix and 5ul of different TruSeq DNA adapters (Illumina) for each sample, at 20°C for 15 min, followed by PEG/NaCl purification (twice). Library enrichment: 12 cycles of PCR amplification. Size selection was performed after PCR using a 0.6x/0.8x ratio of Ampure XP beads (double size selection) set to collect 250–450bp fragments.
For HyperPrep kit, end-repair and A tailing were performed in one reaction: 15ul of ChIP sample was incubated with 45ul end repair+A-tailing mix at 20°C for 30 min, then 65°C for 30 min. Adapter ligation was performed by adding 5ul of IDT for Illumina Unique Dual Indexes (IDT) adapters and 45ul ligation buffer enzyme mix and incubating at 20°C for 15 min, followed by Ampure XP bead purification. Library enrichment: 10 cycles of PCR amplification. Size selection was performed after PCR using a 0.6x/0.8x ratio of Ampure XP beads (double size selection) set to collect 250–450bp fragments. ChIP libraries were sequenced using Illumina HiSeq 2000, 2500 or 4000 at 50bp single reads or Illumina NovaSeq 6000 at 50bp paired-end reads (one read used in the analysis for compatibility).
Read processing and alignment
ChIPseq datasets were processed using the ChIPseq module of GenPipes91 (v3.1.2 for tumors and parental cell lines, v3.1.0 for isogenic cell lines). Briefly, raw reads were trimmed using Trimmomatic87 (v0.32) to remove adapter and sequencing-primer associated reads, then aligned to hg19 or mm10 using bwa-mem92 (v0.7.12) with default parameters. PCR duplicate reads, defined as reads with identical mapping coordinates, were then collapsed by Picard (v2.0.1) to produce uniquely aligned reads. Reads with a mapping quality of 5 or less were then filtered. For single-end (SE) 50bp datasets, reads were extended by 250bp.
Data analysis and signal quantification for tumors and parental cell lines
Bigwig tracks were generated with uniquely aligned reads using Homer93 (v4.9.1). RPKM was calculated using VisRSeq94 (v0.9.40) or SeqMonk (v1.46) at annotated genes. Promoters were defined as 5kb bins centered on transcription start sites. Median values were generated for promoter-associated H3K27me3 and H3K27ac in each tumor group. Z-scores were calculated from the median RPKM as
Data analysis and signal quantification for isogenic cell lines
ChIP-Rx (ChIP with reference exogenous genome) is a technique which applies spike-in Drosophila chromatin as internal control95. For each ChIPseq profile, we calculated the ChIP-Rx ratio (denoted as Rx) as follows:
where s is the percentage of reads mapped to the human genome in the target sample, s_dmel is the percentage of spike-in Drosophila genome in the sample, and similarly i and i_dmel are defined for the input sample.
We observed some variability in ChIP-Rx values obtained from different Drosophila S2 cell batches, and adjusted for these differences between spike-in batches by equalizing the values for replicates of the same conditions (K27M-KO) from different batches:
and then recalculating the values of the other condition (H3.1K27M) within the same Drosophila spike-in batches
ChIPseq RPKM values over CpG islands (CGIs) and 100kb bins genome-wide were calculated using VisRSeq94 (v0.9.40). RPKM values of H3K27me3 were divided by the respective input sample RPKM and averaged for all samples in the same mutation group using a geometric mean.
Rx- and RPKM-normalized BigWig tracks were produced using the bamCoverage functionality of deeptools77 (v3.5.1). Rx ratios were supplied via the ‘--scaleFactor’ option. Reads flagged as duplicates, mapping to random, mitochondrial or sex chromosomes, as well as low-mappability regions according to ENCODE’s blacklist96 were discarded. The resulting tracks were visualized using the IGV79.
To quantify the relative levels of H3K27ac across the genome, the number of reads (scaled using ChIP-Rx ratio) over 1Mb windows was determined using HOMER. The read count over repeat families (DNA, LINE, SINE and LTRs) was determined similarly using HOMER. The Repeatmasker annotation for hg19 was retrieved from UCSC table browser.
H3K27me2 broad domain calling
The broad domain calling procedure was adapted from refs35,97. Briefly, the abundance of H3K27me2 and corresponding input samples was quantified by binning the genome into 1kb bins, counting the number of unique reads in each bin (using the featureCounts function of the Rsubread R package)89 (v2.4.2), and normalizing them to library depth. Enrichment of the mark in each bin was calculated as IP/input, and empty bins were given a score of 0. The bins were then segmented based on mean enrichment scores using the PELT method, SIC penalty, and a Z-test through the changepoint R package98 (v2.2.2). Only segments with sufficient enrichment of the mark (mean score > 1) and length (≥ 50 kb) were retained. To quantify H3K27me2 domains, we calculated, for each sample, the total length of all H3K27me2 domains, as well as the distribution of domain length.
Heatmaps of H3K27me2/H3K27me3 distributions
The deeptools (v3.5.0) package was used to visualize the distributions of the H3K27me2/3 marks genome-wide across isogenic conditions for each cell line. For H3K27me3, the computeMatrix functionality was used in ‘reference-point’ mode with Rx-normalized bigwig tracks as input: briefly, regions of interest were defined as CpG islands (CGIs) with 20kb flanking on either side, and H3K27me3 levels were quantified in non-overlapping 10bp bins across each region. Heatmaps were generated with the plotHeatmap functionality, with regions sorted by mean signal. For H3K27me2, the computeMatrix functionality was used in ‘scale-regions’ mode with Rx-normalized bigwig tracks as input. To define input regions, H3K27me2 domain calls in the parental cell line were first filtered to domains with average signal > 1.2, and domains separated by < 100bp were merged. Then, H3K27me2 domains were scaled to 50kb, with 50kb flanking on either side, and H3K27me2 levels were quantified in non-overlapping 10bp bins across each region. Heatmaps were generated with the plotHeatmap functionality, with regions sorted by mean signal. To avoid outliers, the maximum of the heatmap colour scale was defined as the 90th percentile value across all isogenic conditions for each cell line.
Differential enhancer and core regulatory circuitry analysis
H3K27ac ChIP peaks were identified using MACS276 with a p-value threshold of 1e-9. The ROSE algorithm99 was used to identify enhancers and superenhancers (SEs) .The aggregated H3K27ac binding signals across all H3.1 and H3.3K27M pons samples respectively were determined using ROSE2_META (https://github.com/linlabcode/pipeline/blob/master/ROSE2_META.py). The peaks within +/− 2.5 kb of transcription start site (TSS) were excluded and enhancers within the distance of 12.5 kb were stitched together. The resulting enhancers were ranked by the aggregated H3K27ac signal, and the enhancers above the inflection point were defined as SEs. The SEs were then assigned to the nearest genes. To compare the enhancer landscapes, the fold change of H3K27ac signals between tumor subtypes for each enhancer was calculated and ranked. Core regulatory circuitry (CRC) of SE-associated transcription factors were inferred by scanning for TF motifs in SEs51. A list of expressed genes in each tumor subtype, along with the called SEs were used as input for mapping the regulatory networks using CRC mapper (https://github.com/linlabcode/CRC). This approach used motif scanning, performed using FIMO, to infer number of interacting TF motifs in the proximal SE of a TF (in-degree) and number of SE-associated TFs containing a binding motif for the TF (out-degree). The resulting networks were used to determine the change in in-degree and out-degree between tumor subtypes.
Comparison of tumor epigenomes with single-cell epigenomic data from normal brain
To compare the H3.1K27M PFA-EP tumor tumors with the same mutation (H3.1K27M HGG) or cell-of-origin (EZHIP PFA) as in Extended Data Figure 7, we obtained single-cell Paired-Tag data for OPC and ependymal cells, the posited cells-of-origin of these tumor types from ref62 (GEO accession GSE152020). Cell type labels were used as provided by the authors. ChromHMM calls, as well as bigwig tracks for H3K27ac and RNA, were downloaded from ref62 and used to define genomic regions discriminant for each of the two cell types as follows. First, separately for each cell type, genomic regions were classified as active or inactive based on ChromHMM calls (Active states: promoter-active, promoter-weak, and enhancer-active; Inactive states: heterochromatin-H3K27me3 and heterochromatin-H3K9me3). Next, active and specific regions for each cell type were obtained by requiring that they overlapped with inactive regions in the opposite cell type (minimum 10% overlap). Finally, the nearest gene in the mm10 genome was identified for each region. We then computed H3K27ac levels at promoters of these genes in OPCs and ependymal cells, and for each cell type, we selected the top 20 genes with highest promoter H3K27ac. These genes were used as features for hierarchical clustering over tumors based on their promoter H3K27ac log2(RPKM) levels.
HiC chromatin conformation capture
Sample and library preparation
In situ Hi-C libraries were generated from patient-derived cell lines and murine embryonic brain tissue (1–3 million cells per sample), following ref100 with minor modifications. The full protocol is provided in Supplementary Protocol 1. Briefly, in situ Hi-C was performed in 7 steps: (1) crosslinking cells with formaldehyde, (2) digesting DNA using a 4-cutter restriction enzyme (DpnII) within intact permeabilized nuclei, (3) filling in, biotinylating the resulting 50 overhangs and ligating the blunt ends, (4) shearing the DNA, (5) pulling down bio- tinylated ligation junctions with streptavidin beads, (6) library amplification and (7) analyzing fragments using paired end sequencing. As quality control steps, efficient sonication was checked by agarose DNA gel electrophoresis and for appropriate size selection by Agilent Bioanalyzer profiles for libraries. For final QC, we performed superficial sequencing on the Illumina Hiseq 2500 (30M reads/sample) to assess quality of the libraries using percent of reads passing filter, percent of chimeric reads, and percent of forward-reverse pairs.
Data analysis
Hi-C reads were trimmed and assessed for quality control using the Trim Galore package (TrimGalore v0.6.5, Cutadapt v2.6 and Fastqc v0.11.9). Reads were then mapped to hg19 and filtered for common Hi-C artifacts using HiCUP101 (HiCUP v0.7.2, Bowtie2 v2.3.5). Analysis of Hi-C libraries and downloaded files was performed using Juicer and associated Juicer Tools102 (v1.22.01). Contact maps were generated using Juicer with the following parameter: “-s DpnII”. HiC contact maps were extracted from .hic files using strawr102 (v0.0.1) and visualized with plotgardener78 (v0.1.0). Knight-Ruiz normalized observed/expected ratios were extracted using plotgardener and log2-transformed (using contact data at 10kb resolution).
Histone modification quantification with nLC-MS
The complete workflow for histone extraction, LC/MS, and data analysis was recently described in detail103. Briefly, cell pellets (approx. 1×106 cells) were lysed on ice in nuclear isolation buffer supplemented with 0.3% NP-40 alternative. Isolated nuclei were incubated with 0.4 N H2SO4 for 3 hours at 4°C with agitation. 100% trichloroacetic acid (w/v) was added to the acid extract to a final concentration of 20% and samples were incubated on ice overnight to precipitate histones. The resulting histone pellets were rinsed with ice cold acetone + 0.1% HCl and then with ice cold acetone before resuspension in water and protein estimation by Bradford assay. Approximately 20 μg of histone extract was then resuspended in 100 mM ammonium bicarbonate and derivatized with propionic anhydride. 1 μg of trypsin was added and samples were incubated overnight at 37°C. After tryptic digestion, a cocktail of isotopically labeled synthetic histone peptides was spiked in at a final concentration of 250 fmol/μg and propionic anhydride derivatization was performed a second time. The resulting histone peptides were desalted using C18 Stage Tips, dried using a centrifugal evaporator, and reconstituted using 0.1% formic acid in preparation for nanoLC-MS analysis.
nanoLC was performed using a Thermo ScientificTM Easy nLCTM 1000 equipped with a 75 μm × 20 cm in-house packed column using Reprosil-Pur C18-AQ (3 μm; Dr. Maisch GmbH, Germany). Buffer A was 0.1% formic acid and Buffer B was 0.1% formic acid in 80% acetonitrile. Peptides were resolved using a two-step linear gradient from 5% to 33% B over 45 min, then from 33% B to 90% B over 10 min at a flow rate of 300 nL/min. The HPLC was coupled online to an Orbitrap Elite mass spectrometer operating in the positive mode using a Nanospray Flex™ Ion Source (Thermo Scientific) at 2.3 kV. Two full MS scans (m/z 300–1100) were acquired in the orbitrap mass analyzer with a resolution of 120,000 (at 200 m/z) every 8 DIA MS/MS events using isolation windows of 50 m/z each (e.g. 300–350, 350–400, …, 650–700). MS/MS spectra were acquired in the ion trap operating in normal mode. Fragmentation was performed using collision-induced dissociation (CID) in the ion trap mass analyzer with a normalized collision energy of 35. AGC target and maximum injection time were 10e6 and 50 ms for the full MS scan, and 10e4 and 150 ms for the MS/MS can, respectively. Raw files were analyzed using EpiProfile.
Cell culture
All cell lines used are primary cell lines derived from patient tumors. The sources of these cell lines are as follows: SU-DIPG lines (Michelle Monje, Stanford University), BT245, BT869 (Keith Ligon, Dana-Farber Cancer Institute), HSJD-DIPG007 (Angel Carcaboso, Sant Joan de Déu), HSJ-019 (Nada Jabado, McGill University), PFA lines (Michael Taylor, Hospital for Sick Children). Cell lines SU-DIPGIV, SU-DIPGXXXVI, BT245, SU-DIPGXIII and HSJD-DIPG007 were maintained in NeuroCult NS-A proliferation media (StemCell Technologies) supplemented with bFGF (10ng/mL) (StemCell Technologies), rhEGF (20 ng/mL) (StemCell Technologies) and heparin (0.0002%) (StemCell Technologies) on plates coated in poly-L-ornithine (0.01%) (Sigma) and laminin (0.01 mg/mL) (Sigma). Tumor derived cell lines BT869 and SU-DIPGXXI were maintained in TSM media which contains Neurobasal-A Medium (1X) (Invitrogen Cat no:10888–022), D-MEM/F-12 (Invitrogen Cat no:11330–032), HEPES buffer solution (1M, Invitrogen Cat no:15630–080), MEM Sodium Pyruvate solution (100X, Invitrogen Cat no:11360–070), GlutaPlus, NeuroCultTM SM1 neuronal supplement (StemCell Technologies Cat no: 05711), H-EGF (Shenandoah Biotech Cat no: 100–26), H-FGF-basic-154 (Shenandoah Biotech Cat no: 100–146), H-PDGF-AA (Shenandoah Biotech Cat no: 100–16), H-PDGF-BB (Shenandoah Biotech Cat no: 100–18), Heparin solution (StemCell Technologies Cat no: 07980). All patient derived cell lines were seeded on plates coated in poly-L-ornithine (0.01%) (Sigma) and laminin (0.01 mg/mL) (Sigma). All lines tested negative for mycoplasma contamination, checked monthly using the MycoAlert Mycoplasma Detection Kit (Lonza). Tumor-derived cell lines (Supplementary Table 1) were confirmed to match original samples by STR fingerprinting. Cross linked PFA cell pellets grown in hypoxia were obtained from Michael Taylor. PFA cell lines were cultured in hypoxia (1% O2).
CRISPR/Cas9 genome editing of H3K27M and ACVR1 mutations
SU-DIPGIV, SU-DIPGXXXVI, and SU-DIPGXXI HIST1H3B-K27M cell lines were CRISPR edited for K27M in this study, while the SU-DIPGIV and SU-DIPGXXVI cell lines was CRISPR edited for ACVR1. Additional edited cell lines were generated previously26,27. pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Broad Institute) (Addgene plasmid # 48138). The HIST1H3B-K27M mutation was targeted using the guide sequence GGCTGCTCGCATGAGCGCGC to derive clones from primary HGG cell lines with edited mutant H3.1 K27M allele (knock-out). The ACVR1 guide was designed to target exon 6 of ACVR1 gene overlapping with R206H mutation: GGCTCACCAGATTACACTGT (knock-out). CRISPR-Cas9 editing was carried out following ref104. Eight hundred thousand cells were harvested and electroporated with Amaxa Human Embryonic Stem Cell Nucleofector kit (Lonza) according to the manufacturer’s protocol. Flow cytometry sorted single GFP+ cells In 96 well plates, 72 hours post-transfection. Clones were expanded and the target locus sequenced by Sanger sequencing. Select clones were screened by Illumina MiSeq system for the target exon to confirm complete mutation of the K27M allele. Mass spectrometry confirmed the absence of K27M mutant peptide in these clones. Primer sequences for characterizing ACVR1 clones are provided in Supplementary Table 20.
Cell proliferation assays
Cell lines were plated at low confluency at 15000 cells in 24-well plates. Cell proliferation was assessed using IncuCyte ZOOM System real-time instrumentation (Essen BioScience). Phase contrast was used to assess confluency and 16 images were taken every three hours for each well. Particles smaller than 400 μm2 were removed from analysis. All confluency data were extracted and normalized to starting point. For CRISPR edited clones, at least two clones were compared with parental cell lines with at least three biological replicates.
Clone formation assay
Parental cells and isogenic clones were plated in duplicates on laminin coated 6 well plates at 500 cells per well. Cells were left for 10 days until the appearance of visible clones. Cells were then fixed with 4% formalin, washed in PBS, and stained with 0.5% crystal violet. The number of clones was counted in each well and averaged for each duplicate. Final counts are expressed as the average for three independent experiments.
Droplet digital PCR
RNA was extracted from cells using the Aurum Total RNA Mini Kit (Bio-Rad) and concentration was quantified on the BioDrop uLite (Montreal Biotech). cDNA was generated using iScript Reverse Transcription Supermix for RT-qPCR (Bio-Rad) and 75ng of RNA as starting material. Target concentration was determined using the QX200 ddPCR EvaGreen Supermix assay (Bio-Rad) using 2 uL of cDNA per reaction using manufacturer’s protocol cycling conditions with a 58 degrees Celsius annealing temperature for 40 cycles. Droplets were assayed using the QX200 Droplet Reader (Bio-Rad) and manually scored for positive signal using QuantaSoft Software (Bio-Rad). For each target (run in triplicate), concentration (copies/ul) was normalized to two reference genes (geometric mean of GAPDH and HPRT1). Primer sequences for each target and reference genes are provided in Supplementary Table 20. Significance was determined using normalized non-averaged values using a two-sided t-test.
In vivo mouse xenografts
All mice were housed, bred, and subjected to listed procedures according to the McGill University Health Center Animal Care Committee and in compliance with the guidelines of the Canadian Council on Animal Care. Female NOD.Cg-Prkdcscid mice (4–6 weeks) were used for xenograft experiments (Jackson Laboratory, strain # 005557). For subcutaneous injections, SU-DIPGIV and SU-DIPGXXXVI parental cells and respective isogenic clones, were prepared in Matrigel:PBS (1:1) (Matrigel basement membrane matrix, phenol red free, LDEV free, Corning, #356237, New York, USA), and 4.106 cells were implanted in the left flank of mice: 1) SU-DIPGIV parental cells (n=3), SU-DIPGIV ACVR1 KO clone C103 (n=3), 3) SU-DIPGIV ACVR1 clone C122 (n=3), 4) SU-DIPGXXXVI parental cells (n=3), 5) SU-DIPGXXXVI ACVR1 KO clone C104 (n=3), SU-DIPGXXXVI ACVR1 KO clone C120 (n=3). A maximum tumor volume of 2 cm3 was permitted, and this was not exceeded. Tumor volume was measured twice per week using a caliper and mice were euthanized when tumor volume reached 1.5cm3. Kaplan Meier survival curves were generated using the GraphPad Prism software (v9.3.1). Mice that died due to a tumor are considered as 1. Those that were still surviving at the end of the experiment, or those that were euthanized for different reasons that are related to tumor formation were considered as 0.
Immunohistochemistry (IHC)
Immunohistochemistry (IHC) for patient samples and patient-derived xenografts was performed at the Segal Cancer Centre Research Pathology Facility (Jewish General Hospital). The slides were stained using the Discovery XT Autostainer (Ventana Medical System). All solutions used for automated immunohistochemistry were from Ventana Medical System (Roche) unless otherwise specified. After de-paraffinization and heat-induced epitope retrieval (CC1 prediluted solution Ref: 950–124, standard protocol), sections were incubated with primary antibodies: anti-p-Smad1/5/9 (clone D5B10, CST 13820) and anti-NKX6.1 (clone EPR20405, abcam) diluted at 1:100 and 1:250, respectively. Slides were counterstained with hematoxylin and Bluing Reagent, and finalized with mounting medium (Eukitt, Fluka Analytical). Sections were scanned using the Aperio AT Turbo Scanner (Leica Biosystems).
Lentiviral transduction
Lentiviruses were gifts from Dr. Peter Lewis (University of Wisconsin-Madison). Lentiviruses expressing H3.1WT and H3.1K27M were applied for 24 hours and G418 (Wisent) selection (500 ng/mL) was maintained for the duration of growth, leading to reintroduction of H3.1WT or H3.1K27M in the BT245 and SU-DIPGXIII K27M cell lines after editing out the mutation.
Western blotting
Histone lysates were extracted using Histone Extraction Kit (Abcam, ab113476). Protein amounts were quantified using Micro BCA Protein Assay Kit (Thermo Scientific) Three microgram of histone lysates were separated on home-made 12% gels and transferred onto PVDF membranes (GE Healthcare) for 1 hour. Blocking was then performed using 5% Bovine Serum Albumin (BSA, Multicell) in Tris-buffered saline (50mM Tris, 150mM NaCl, pH 7.4, 0.1% Tween 20) (TBST) for one hour at room temperature. Membranes were incubated in the presence of the relevant antibody overnight at 4 degrees Celsius in 5% BSA in TBST: anti-H3K27M (1:200, Millipore ABE419) and anti-total H3 (1:2000, Abcam 1791). Membranes were washed three times with TBST prior to incubation with Horse Radish Peroxidase-linked secondary antibody (1:20000, Bethyl A120-100P) in 5% BSA in TBST for one hour at room temperature. Membranes underwent a final three-rounds of washes with TBST before the signal was developed with Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare). Finally, signals were visualized using ChemiDoc MP Imaging System (Bio-Rad). Precision Plus Protein™ WesternC™ Blotting Standards (Bio-Rad, #1610376) was used as a molecular weight markers which covers weight ranges from 10 to 250kDa by 10 bands.
Data availability
ChIPseq sequencing data for human cell lines and scRNAseq sequencing data for normal E10, E13, E16, and E18 murine samples have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE188625, while E12, E15, P0, P3, and P6 samples have been previously deposited to GEO under GSE133531. Bulk RNAseq, ChIPseq, HiC, scRNAseq, scATACseq, and scMultiome sequencing data for human tumors have been deposited in the European Genome-phenome Archive (EGA) under accession number EGAS00001005773. Processed data for bulk RNAseq (counts and differential expression analyses), ChIPseq (genome wide H3K27ac/me2/me3 levels), and scRNAseq/scATACseq/scMultiome (counts matrices, cell annotations, and chromatin accessibility bigwig files) have been deposited to GEO under the accession number GSE210568 and Zenodo at https://doi.org/10.5281/zenodo.6773261 105. Accession numbers for previously published data used in this study are provided in Supplementary Tables 1, 2, 6, and 11.
Code availability
Code to reproduce the main results included in the paper is available at https://github.com/fungenomics/HGG-oncohistones and archived on Zenodo at https://doi.org/10.5281/zenodo.6647837 106.
Extended Data
Extended Data Fig. 1. Overview of expanded scRNAseq mouse developmental reference.
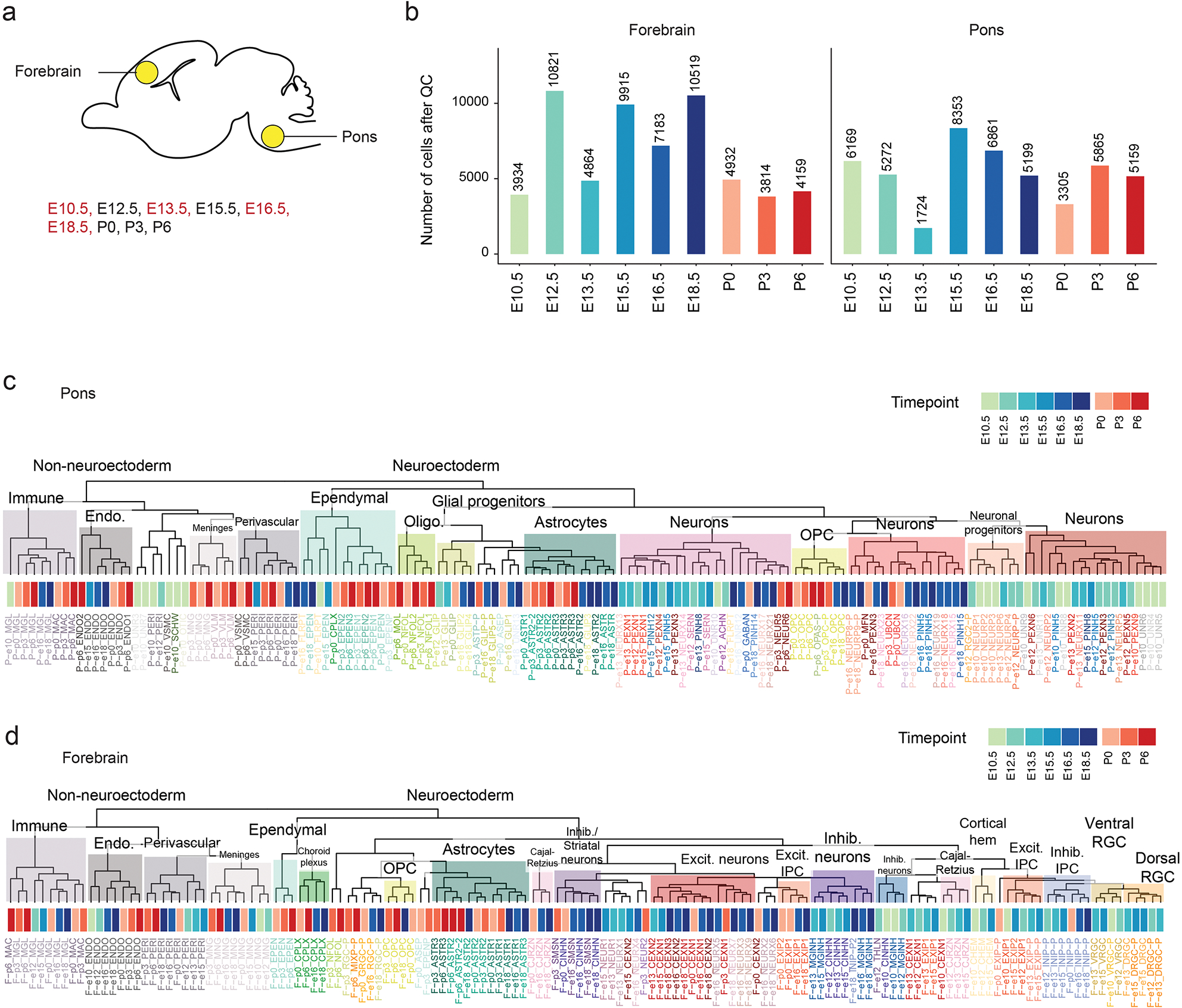
a. Schematic of developing mouse brain, sagittal view, indicating regions and timepoints included in the single-cell reference atlas. Red: data generated in this study; black: data from Jessa et al, Nature Genetics, 201934.
b. Number of cells captured in each time point and brain region after quality control and filtering.
c. Overview of single-cell populations from the mouse pons. Dendrogram constructed based on pairwise Spearman correlations between mean expression profiles in each cluster. Cell class and time point are annotated.
d. Overview of single-cell populations from the mouse forebrain. Dendrogram constructed based on pairwise Spearman correlations between mean expression profiles in each cluster. Cell class and time point are annotated.
Extended Data Fig. 2. Unique cell type hierarchies in H3.1 and H3.3K27M HGGs.
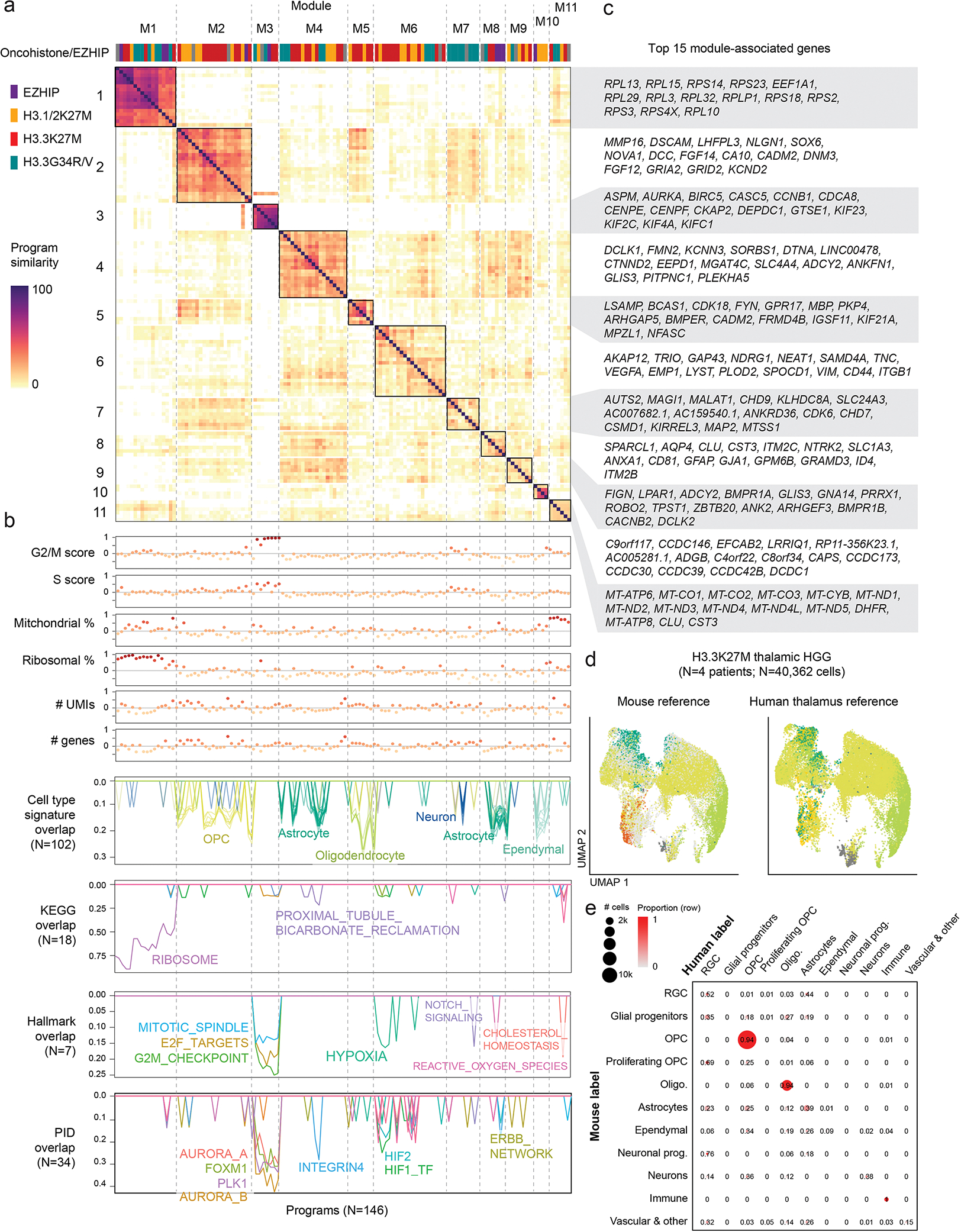
a. Similarity matrix between all Non-negative Matrix Factorization (NMF) programs assigned to modules. Heatmap represents the pairwise overlap (in number of genes) between programs.
b. Annotation of NMF programs. Top: Correlation between each program and QC or biological metrics in each cell. One module (M11) that was explained by technical factors (mitochondrial content and coverage), was consequently removed from further analyses. Bottom: overlap between each program and developmental or MSigDB reference signatures, one line per signature. Only significant overlaps (p-value < 0.001) are shown, and number of significant overlaps is shown in parentheses.
c. Top 15 genes associated with each module. Module-associated genes were selected by identifying the most frequent program-associated genes for all programs contained in the module.
d. UMAP for H3.3K27M thalamic HGG (malignant cells only), with cells coloured by consensus projected cell type based on the normal mouse brain reference (left), or the normal human fetal thalamus reference (right). Cells are colored as in Figure 1d.
e. Confusion matrix comparing projected cell types for H3.3K27M thalamic HGG based on mouse or human reference. Proportions were computed row-wise and represent the fraction of cells from each mouse label which were assigned to each human label. Bubbles are scaled to the number of cells with each combination of labels.
Extended Data Fig. 3. Some H3.1K27M pontine gliomas harbour a malignant ependymal-like component.
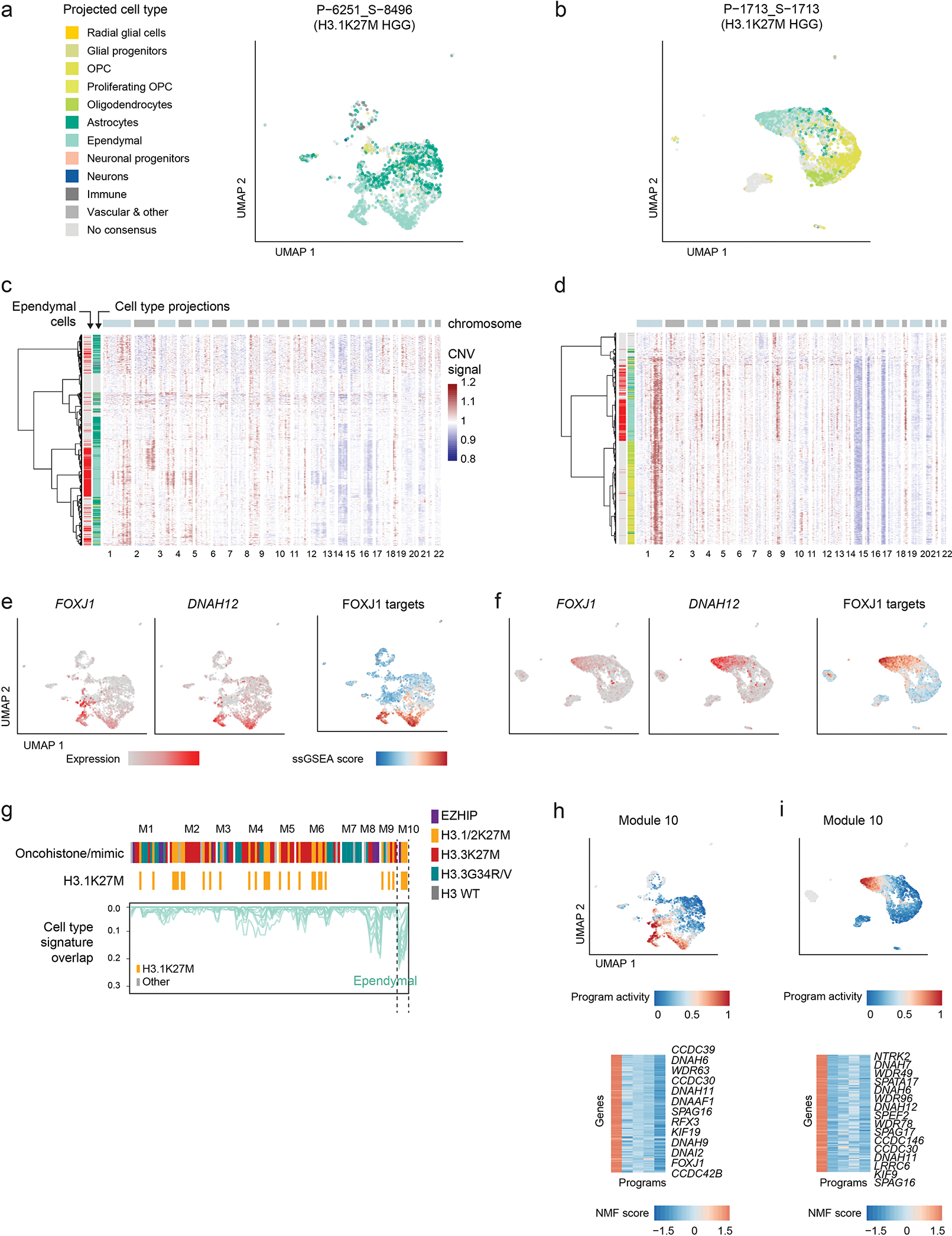
a-b. UMAP plots for two individual H3.1K27M pontine gliomas containing ependymal-like cells. Only malignant cells are shown. Cells are colored by consensus projected cell type.
c-d. Heatmaps of copy-number signal computed for each individual sample using InferCNV. Row annotations correspond to cell type projections, indicating whether they are projected to ependymal cells (left, red), and the overall projected cell class, with colors as in (a) and normal cells colored in gray. Cells lacking a consensus projection were excluded.
e-f. UMAP plots as in (a-b), with cells coloured by expression of FOXJ1 (ependymal transcription factor), DNAH12 (ciliary gene), and single-cell gene set enrichment (ssGSEA) score of candidate FOXJ1 targets in the early postnatal mouse brain. List of FOXJ1 targets was obtained from Jacquet et al, Development, 200944.
g. NMF programs from Figure 1c, displaying only the overlap between program-associated genes with ependymal gene signatures, and filtering out all other developmental signatures. Top column annotation shows the driver alteration of the sample in which each program was identified. A second annotation highlighting H3.1/2K27M tumors is included for clarity. Module 10, significantly overlapping ependymal signatures, is enriched for programs from this tumor entity.
h-i. Activity of the ependymal-related module 10 in individual samples. Top: UMAP plots as in (a-b), cells are coloured by the NMF activity score of the module 10 program from each sample. Bottom: heatmap of NMF score of module 10 program-associated genes; names for selected informative genes are indicated.
Extended Data Fig. 4. H3.1K27M, ACVR1-mutant pontine gliomas arise from an NKX6-1+ ventral brainstem progenitor.
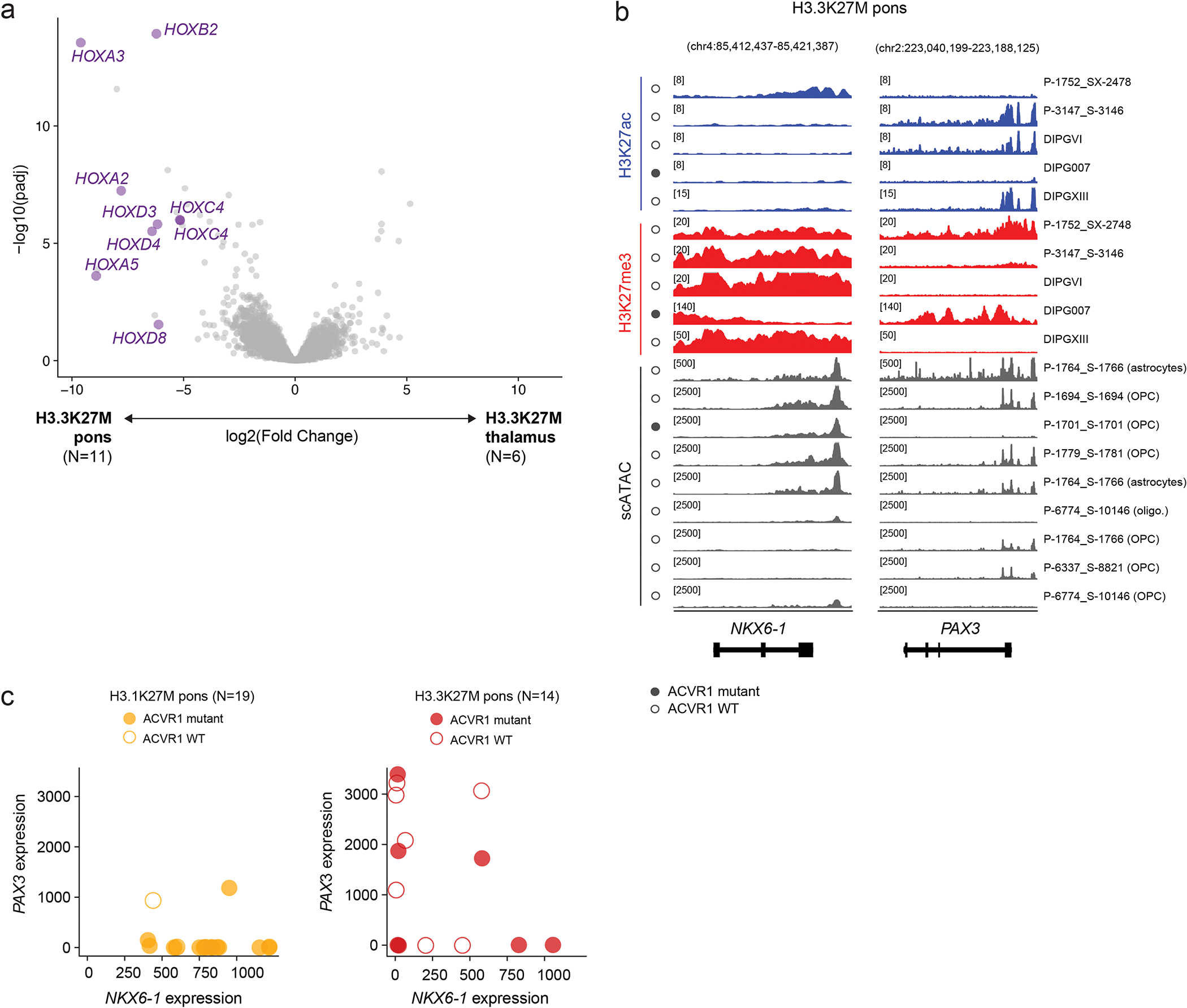
a. Volcano plot of differentially expressed genes between H3.3K27M pons and H3.3K27M thalamus HGG. HOX genes are indicated in purple. Only genes with mean normalized expression > 100 are included.
b. Epigenomic state at NKX6-1 and PAX3 in representative H3.3K27M pons HGG primary tumors and cell lines. For scATACseq data, each track represents RPKM-normalized aggregated accessibility for one malignant single-cell population.
c. Co-expression of NKX6-1 and PAX3 in bulk RNAseq data for pons HGG with each K27M histone variant.
Extended Data Fig. 5. Assessment of NKX6-1 in brain tumors and normal tissues.
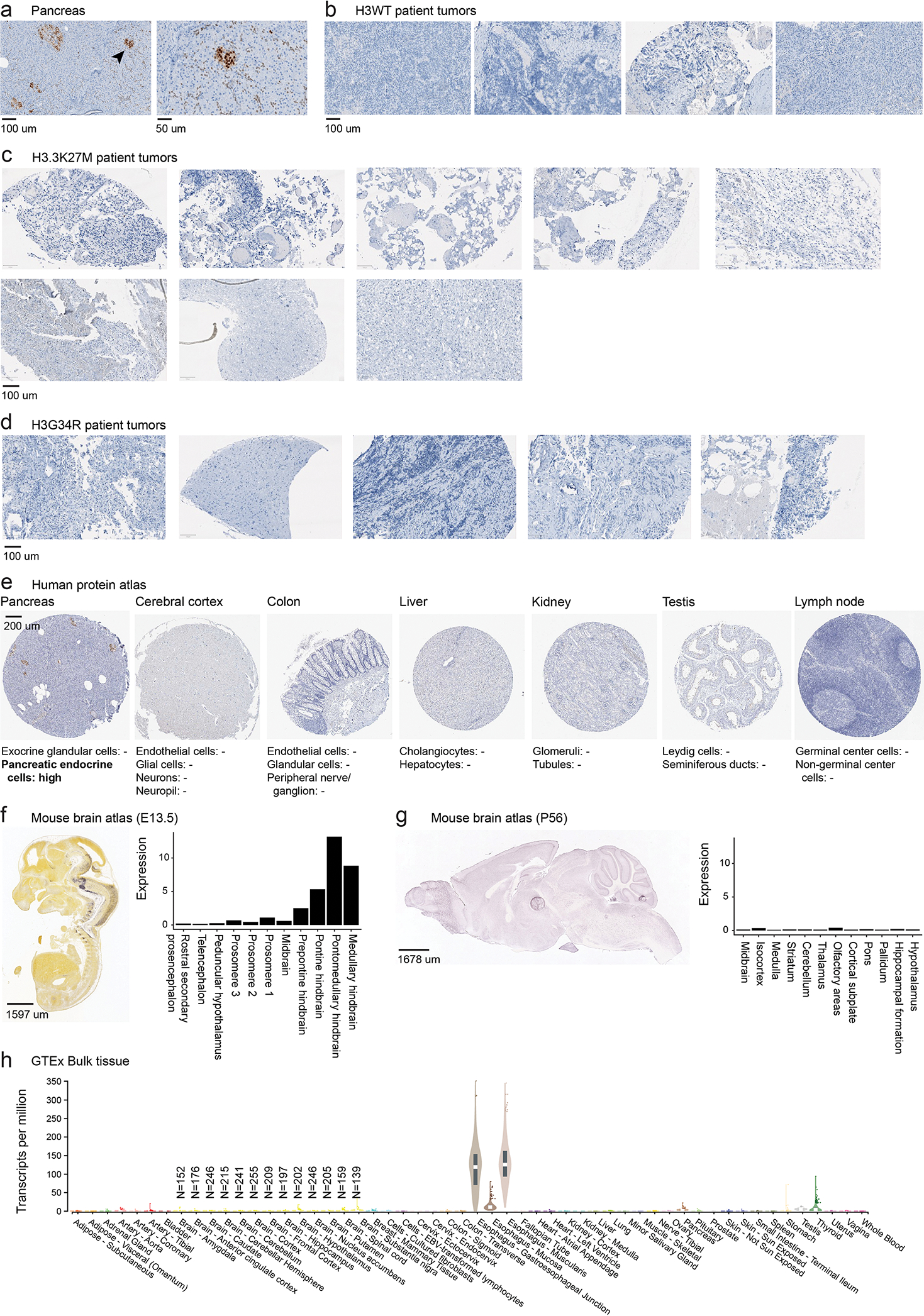
a. Immunohistochemistry staining of NKX6-1 protein in normal pancreas tissue as positive control. Arrowhead in left panel indicates region shown at higher magnification in right panel.
b-d. Immunohistochemistry staining of NKX6-1 protein in Histone 3 WT, H3.3G34R, and H3.3K27M high-grade glioma patient tumors.
e. Antibody staining of NKX6-1 in human tissues from the Human Protein Atlas. Detection levels for each cell type are indicated below, “-” indicates that NKX6-1 was not detected. Image credit: Human Protein Atlas. Images available from http://v21.proteinatlas.org (links provided in Supplementary Table 19).
f. Left: In situ hybridization (ISH) in E13.5 mouse brain from the Allen Brain Atlas (© 2008 Allen Institute for Brain Science. Allen Developing Mouse Brain Atlas. Available from: developingmouse.brain-map.org). Right: quantification of ISH expression levels.
g. Left: In situ hybridization (ISH) in P56 mouse brain from the Allen Brain Atlas (© 2004 Allen Institute for Brain Science. Allen Mouse Brain Atlas. Available from mouse.brain-map.org). Right: quantification of ISH expression levels.
h. Bulk expression levels of NKX6-1 in adult human tissues from GTEx. Sample sizes for brain tissues are indicated.
Extended Data Fig. 6. Nkx6-1/Pax3 expression is mutually exclusive in the normal brain.
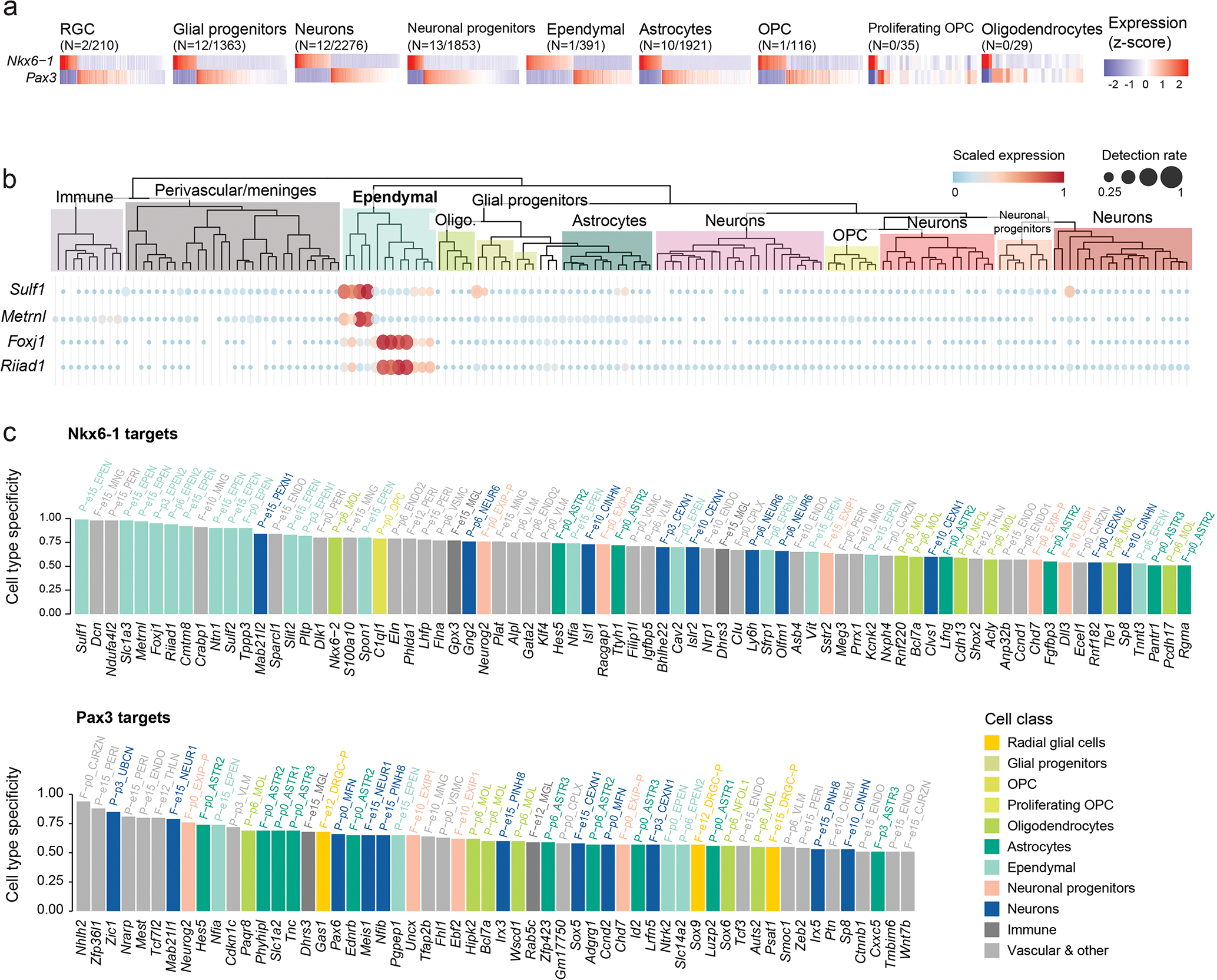
a. Expression of NKX6-1 and Pax3 in cell types of the normal developing mouse pons reference, showing their expression is largely mutually exclusive. The number of cells where both NKX6-1 and Pax3 are detected out of the total number of NKX6-1+ or Pax3+ cells of the cell type is indicated in parentheses.
b. Expression of NKX6-1 target genes with high cell type specificity in ependymal cells. Dendrogram represents cell clusters in the single-cell mouse pons reference, as in Figure S1.
c. Cell type-specificity score for inferred targets of NKX6-1 and Pax3 in the normal mouse pons. For a given gene, score represents the difference between the highest detection rate of the gene in any single-cell cluster in the normal mouse reference, and the detection rate of the gene in all other cells in the same sample (see Methods).
Extended Data Fig. 7. H3K27M and EZHIP converge to restrict H3K27me3 to PRC2 nucleation sites.
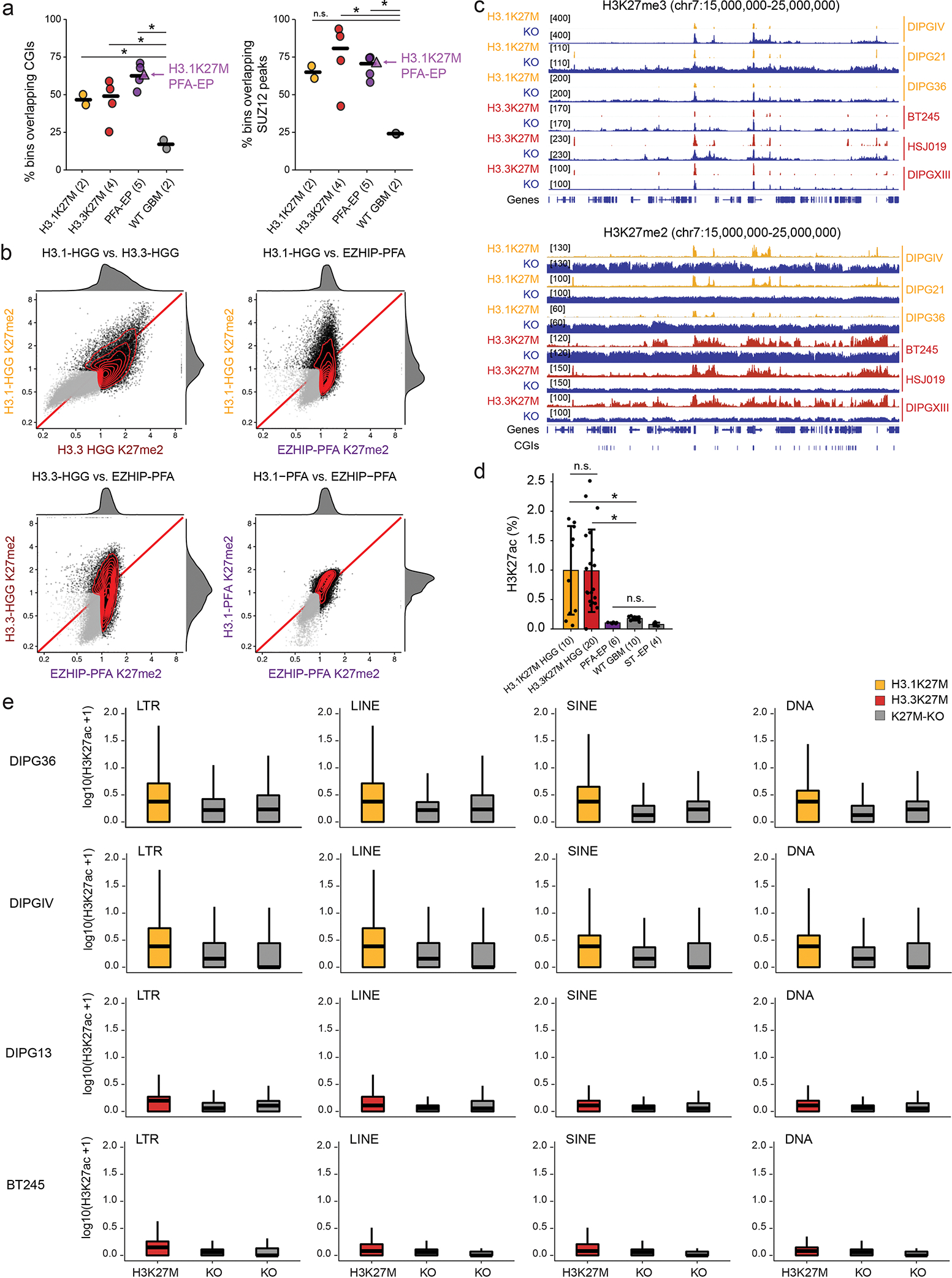
a. Percentage of H3K27me3-marked 10kb bins overlapping CGIs or SUZ12 peaks in cell lines and tumors. Number of biologically independent samples per group is indicated in parentheses. H3K27me3 was quantified in 10kb bins genome-wide and the top 1% bins with highest H3K27me3 in each sample were intersected with CGIs/SUZ12 peaks. For SUZ12, the union of peaks called from SUZ12 ChIPseq in BT245 and DIPGXIII were obtained from Harutyunyan et al, Nature Communications, 201938. Crossbar indicates the median. P-values: left panel (H3.1K27M vs WT GBM, p = 0.024; H3.3K27M vs WT GBM, p = 0.026; PFA-EP vs WT GBM, 0.00099); right panel (H3.1K27M vs WT GBM, p = 0.063; H3.3K27M vs WT GBM, p = 0.023; PFA-EP vs WT GBM, p = 0.0016); n.s., not significant; Welch two-sample t-test.
b. Scatterplots of H3K27me2 signal over 100kb bins genome-wide in pairwise group comparisons. X- and Y- axes represent log2 mean RPKM value per group, normalized by input. Marked bins (mean RPKM > 1 in at least one of the groups in each comparison) are shown in black, while unmarked bins are shown in gray. Joint density and marginal distributions are calculated over marked bins only. Red line indicates the diagonal. RPKM values of H3K27me2 were divided by the respective input sample RPKM and averaged for all samples in the same mutation group using a geometric mean.
c. H3K27me3 (top) and H3K27me2 (bottom) ChIP-seq enrichment tracks, in representative K27M-mutant and isogenic CRISPR-KO cell lines.
d. Mass spectrometry data of H3K27ac in cell. Number of biologically independent samples per group is indicated in parentheses. Error bars represent mean +/− SD. P-values: H3.1K27M vs WT GBM, 0.0074; H3.3K27M vs WT GBM: 5.3×10−5; n.s., not significant; Welch two-sample t-test.
e. Enrichment of H3K27ac over different repeat element families in HGG cell lines and isogenic K27M-KO counterparts.
Extended Data Fig. 8. Cell-of-origin chromatin state contributes to the tumor epigenome.
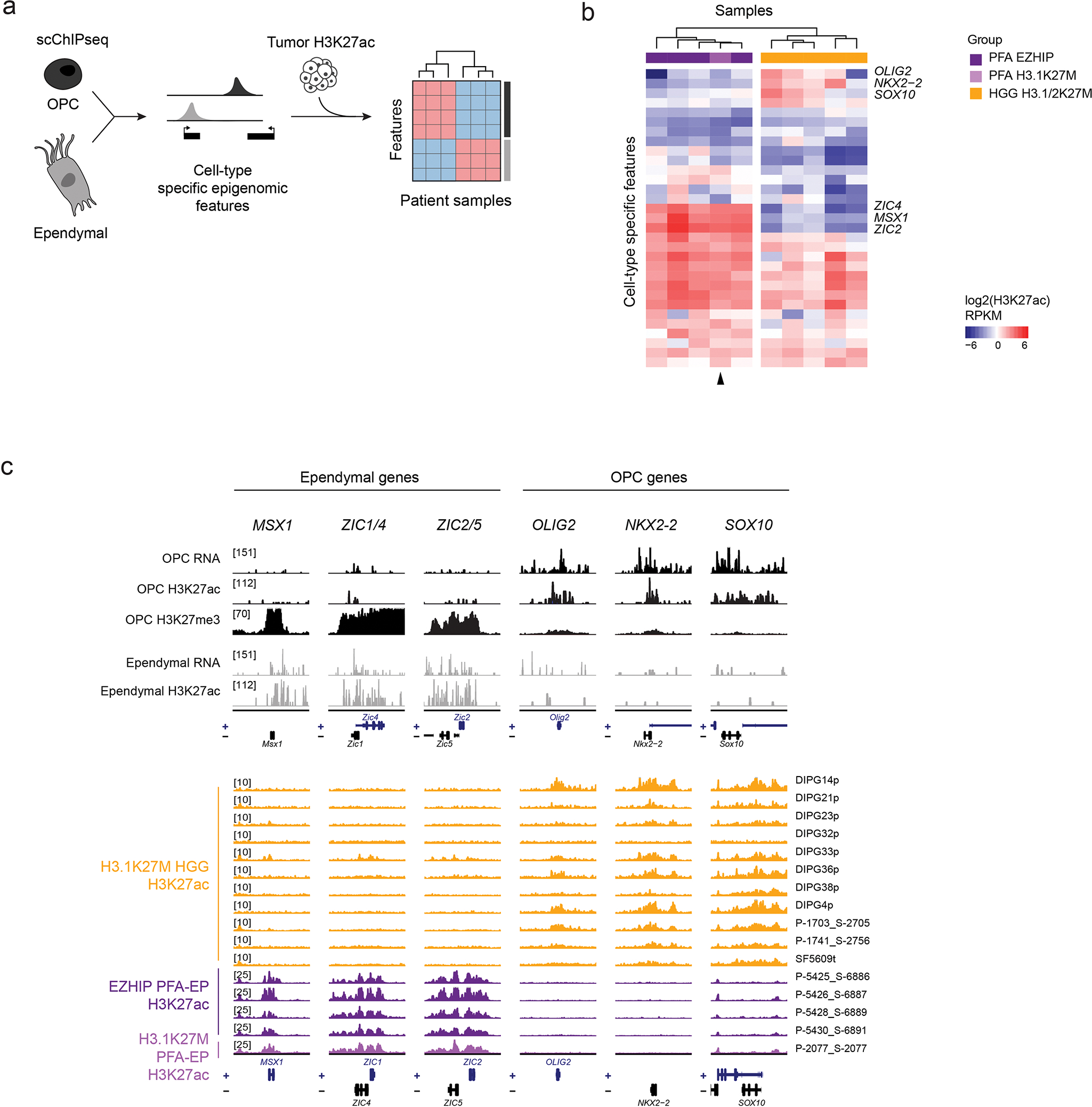
a. Schematic of analysis. Single-cell epigenomic data for normal mouse OPCs and ependymal cells was obtained from Zhu et al, Nature Biotechnology, 202166, and used to extract cell type-specific epigenomic features. Tumors were clustered based on H3K27ac levels at promoters of these genes.
b. Hierarchical clustering of H3.1K27M HGG, H3.1K27M PFA-EP, and EZHIP PFA-EP based on OPC and ependymal-specific epigenomic features. Select features are indicated.
c. Top: RNA and single-cell epigenomic data for normal mouse OPCs and ependymal cells66 at ependymal & OPC genes. Bottom: H3K27ac ChIPseq tracks for H3.1K27M HGG, H3.1K27M PFA-EP, and EZHIP PFA-EP at the same genes as in the top panel. Chromosome coordinates are indicated in Supplementary Table 15.
Extended Data Fig. 9. Uncoupling the effect of histone variants from cell-of-origin chromatin state and cycling rate.
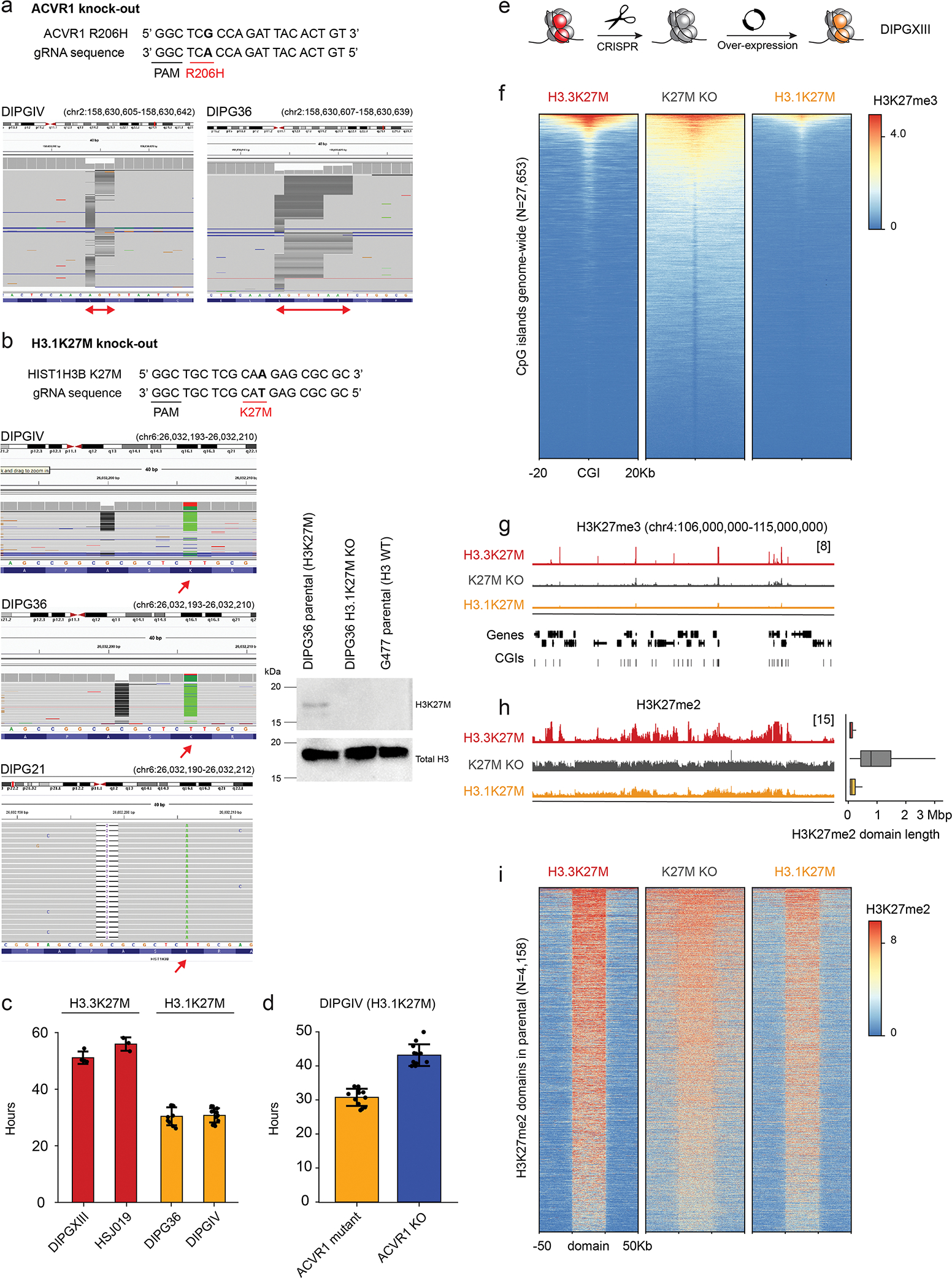
a. Validation of CRISPR removal of ACVR1 in H3.1K27M ACVR1-mutant cell lines by MiSeq (multiple deletions on both alleles (complete KO)).
b. Validation of CRISPR removal of H3K27M in H3.1K27M cell lines DIPGIV and DIPG36 (1bp deletion on K27M allele (frameshift)) and DIPG21 (2bp deletion on K27M allele) by MiSeq and Western Blot. For Western Blot, G477, an H3.1WT HGG patient-derived cell line, was used as control. CRISPR removal of H3K27M in H3.3K27M cell lines has been reported previously for BT245 and DIPGXIII in Krug et al, Cancer Cell, 2019; and for HSJ019 in Harutyunyan et al, Cell Reports, 2020.
c. Doubling time of H3.3K27M and H3.1K27M HGG cell lines (DIPGXIII, N=4 biological replicates; HSJ019, N=3; DIPG36, N=9; DIPGIV, N=12). Error bars represent mean +/− SD.
d. Doubling time of H3.1K27M cell line DIPGIV in ACVR1 mutant and ACVR1-KO conditions. Error bars represent mean +/− SD.
e. Schematic of experimental design.
f. Heatmap showing distribution of Rx-normalized ChIPseq signal for H3K27me3 in DIPGXIII at CpG islands (CGIs), flanked by 20kbp on either side.
g. Rx-normalized H3K27me3 tracks in each condition at a representative genomic region. Y-axis limit is indicated in brackets and identical for all tracks.
h. Left: Rx-normalized H3K27me2 tracks in each condition at the same region as in (g). Y-axis limit is indicated in brackets and identical for all tracks. Right: genome-wide distribution of H3K27me2 domain length in each condition (H3.3K27M, N=16,630 domains; K27M-KO, N=3388; H3.1K27M, N=11,568).
i. Heatmap showing distribution of Rx-normalized ChIPseq signal for H3K27me2 in DIPGXIII H3K27me2 domains across the genome in each condition. Domains are scaled to 50kb, and flanked by 50kb on either side. The maximum of the color scale is set to the 90th percentile value across all data points.
Supplementary Material
Acknowledgements
We thank the patients and their families for their invaluable contributions to this research, without whom it would be impossible. This work was supported by funding from: A Large-Scale Applied Research Project grant from Genome Quebec, Genome Canada, the Government of Canada, and the Ministère de l’Économie, de la Science et de l’Innovation du Québec, with the support of the Ontario Institute for Cancer Research through funding provided by the Government of Ontario to N.J., M.D.T., C.L.K. Fondation Charles Bruneau to N.J., US National Institutes of Health (NIH grant P01-CA196539 to N.J., R01CA148699 and R01CA159859 to M.D.T.); the Canadian Institutes for Health Research (CIHR grant MOP-286756 and FDN-154307 to N.J. and PJT-156086 to C.L.K.); the Canadian Cancer Society (CCSRI grant 705182) and the Fonds de Recherche du Québec en Santé (FRQS) salary award to C.L.K.; NSERC (RGPIN-2016-04911) to C.L.K.; CFI Leaders Opportunity Fund 33902 to C.L.K., Genome Canada Science Technology Innovation Centre, Compute Canada Resource Allocation Project (WST-164-AB); Data analyses were enabled by compute and storage resources provided by Compute Canada and Calcul Québec. N.J. is a member of the Penny Cole Laboratory and the recipient of a Chercheur Boursier, Chaire de Recherche Award from the FRQS. This work was performed within the context of the International CHildhood Astrocytoma INtegrated Genomic and Epigenomic (ICHANGE) consortium with funding from Genome Canada and Genome Quebec. S.J. is supported by a fellowship from CIHR. We also acknowledge support from the We Love You Connie, Poppies for Irini and Kat D Strong Foundations (N.J.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. We acknowledge the contributions of Dylan Marchione and John Wojcik in mass spectrometry work.
Footnotes
Competing Interests Statement
S.P. is a member of the advisory board for Bayer, Novartis and AstraZeneca and has received speaker fees from Bayer and Esai, outside of the submitted work. All other authors declare no competing interests.
Peer review Information:
Nature Genetics thanks Xiao-nan Li and the other, anonymous, reviewer(s) for their contribution to the peer review of this work
References
- 1.Krug B, Harutyunyan AS, Deshmukh S & Jabado N Polycomb repressive complex 2 in the driver’s seat of childhood and young adult brain tumours. Trends Cell Biol. (2021) doi: 10.1016/j.tcb.2021.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khuong-Quang D-A et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 124, 439–447 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwartzentruber J et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Pajtler KW et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 136, 211–226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu G et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis PW et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bender S et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Venneti S et al. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol. 23, 558–564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jain SU et al. PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat. Commun. 10, 2146 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ragazzini R et al. EZHIP constrains Polycomb Repressive Complex 2 activity in germ cells. Nat. Commun. 10, 3858 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hübner J-M et al. EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro. Oncol. 21, 878–889 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jain SU et al. H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Molecular Cell 0, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mackay A et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 32, 520–537.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fontebasso AM et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 46, 462–466 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor KR et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 46, 457–461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu G et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 46, 444–450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sievers P et al. A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro. Oncol. 23, 34–43 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buczkowicz P et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 46, 451–456 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fortin J et al. Mutant ACVR1 Arrests Glial Cell Differentiation to Drive Tumorigenesis in Pediatric Gliomas. Cancer Cell 0, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoeman CM et al. ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nat. Commun. 10, 1023 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vladoiu MC et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature (2019) doi: 10.1038/s41586-019-1158-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Filbin MG et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monje M et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc. Natl. Acad. Sci. U. S. A. 108, 4453–4458 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kessaris N et al. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 9, 173–179 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fogarty M, Richardson WD & Kessaris N A subset of oligodendrocytes generated from radial glia in the dorsal spinal cord. Development 132, 1951–1959 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Cai J et al. Generation of oligodendrocyte precursor cells from mouse dorsal spinal cord independent of Nkx6 regulation and Shh signaling. Neuron 45, 41–53 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Lin GL & Monje M Understanding the Deadly Silence of Posterior Fossa A Ependymoma. Molecular cell vol. 78 999–1001 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Piunti A et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 23, 493–500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarthy JF et al. Histone deposition pathways determine the chromatin landscapes of H3.1 and H3.3 K27M oncohistones. Elife 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagaraja S et al. Histone Variant and Cell Context Determine H3K27M Reprogramming of the Enhancer Landscape and Oncogenic State. Mol. Cell 76, 965–980.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stafford JM et al. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv 4, eaau5935 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mack SC et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 553, 101–105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krug B et al. Pervasive H3K27 Acetylation Leads to ERV Expression and a Therapeutic Vulnerability in H3K27M Gliomas. Cancer Cell 35, 782–797.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jessa S et al. Stalled developmental programs at the root of pediatric brain tumors. Nat. Genet. (2019) doi: 10.1038/s41588-019-0531-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harutyunyan AS et al. H3K27M in Gliomas Causes a One-Step Decrease in H3K27 Methylation and Reduced Spreading within the Constraints of H3K36 Methylation. Cell Rep. 33, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen CCL et al. Histone H3.3G34-Mutant Interneuron Progenitors Co-opt PDGFRA for Gliomagenesis. Cell (2020) doi: 10.1016/j.cell.2020.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dubois FPB et al. Structural variants shape driver combinations and outcomes in pediatric high-grade glioma. Nat. Cancer 3, 994–1011 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harutyunyan AS et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 10, 1262 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kotliar D et al. Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bressan RB et al. Regional identity of human neural stem cells determines oncogenic responses to histone H3.3 mutants. Cell Stem Cell (2021) doi: 10.1016/j.stem.2021.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Funato K, Smith RC, Saito Y & Tabar V Dissecting the impact of regional identity and the oncogenic role of human-specific NOTCH2NL in an hESC model of H3.3G34R-mutant glioma. Cell Stem Cell 0, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eze UC, Bhaduri A, Haeussler M, Nowakowski TJ & Kriegstein AR Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat. Neurosci. (2021) doi: 10.1038/s41593-020-00794-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhaduri A et al. An atlas of cortical arealization identifies dynamic molecular signatures. Nature 598, 200–204 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacquet BV et al. FoxJ1-dependent gene expression is required for differentiation of radial glia into ependymal cells and a subset of astrocytes in the postnatal brain. Development 136, 4021–4031 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee TI et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125, 301–313 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernstein BE et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Philippidou P & Dasen JS Hox genes: choreographers in neural development, architects of circuit organization. Neuron 80, 12–34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noordermeer D et al. The dynamic architecture of Hox gene clusters. Science 334, 222–225 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Narendra V et al. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scholpp S & Lumsden A Building a bridal chamber: development of the thalamus. Trends Neurosci. 33, 373–380 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saint-André V et al. Models of human core transcriptional regulatory circuitries. Genome Res. 26, 385–396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Uhlén M et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Thompson CL et al. A high-resolution spatiotemporal atlas of gene expression of the developing mouse brain. Neuron 83, 309–323 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lein ES et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Visel A et al. A high-resolution enhancer atlas of the developing telencephalon. Cell 152, 895–908 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oosterveen T et al. Mechanistic differences in the transcriptional interpretation of local and long-range Shh morphogen signaling. Dev. Cell 23, 1006–1019 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Vallstedt A, Klos JM & Ericson J Multiple dorsoventral origins of oligodendrocyte generation in the spinal cord and hindbrain. Neuron 45, 55–67 (2005). [DOI] [PubMed] [Google Scholar]
- 58.Masahira N et al. Olig2-positive progenitors in the embryonic spinal cord give rise not only to motoneurons and oligodendrocytes, but also to a subset of astrocytes and ependymal cells. Dev. Biol. 293, 358–369 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Aibar S et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valer JA, Sánchez-de-Diego C, Pimenta-Lopes C, Rosa JL & Ventura F ACVR1 Function in Health and Disease. Cells 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferrari KJ et al. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 53, 49–62 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Zhu C et al. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat. Methods (2021) doi: 10.1038/s41592-021-01060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oksuz O et al. Capturing the Onset of PRC2-Mediated Repressive Domain Formation. Mol. Cell 70, 1149–1162.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lindquist RA et al. Identification of proliferative progenitors associated with prominent postnatal growth of the pons. Nat. Commun. 7, 11628 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nishiyama A, Shimizu T, Sherafat A & Richardson WD Life-long oligodendrocyte development and plasticity. Semin. Cell Dev. Biol. 116, 25–37 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gonçalves CS, Le Boiteux E, Arnaud P & Costa BM HOX gene cluster (de)regulation in brain: from neurodevelopment to malignant glial tumours. Cell Mol Life Sci 77, 3797–3821 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shah N & Sukumar S The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer 10, 361–371 (2010). [DOI] [PubMed] [Google Scholar]
- 68.Le Boiteux E et al. Widespread overexpression from the four DNA hypermethylated HOX clusters in aggressive (IDHwt) glioma is associated with H3K27me3 depletion and alternative promoter usage. Mol. Oncol. (2021) doi: 10.1002/1878-0261.12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bond AM, Bhalala OG & Kessler JA The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Dev. Neurobiol. 72, 1068–1084 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 70.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McInnes L & Healy J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv [stat.ML] (2018). [Google Scholar]
- 72.Waltman L & van Eck NJ A smart local moving algorithm for large-scale modularity-based community detection. The European Physical Journal B 86, 71 (2013). [Google Scholar]
- 73.Nagy C et al. Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat. Neurosci. 23, 771–781 (2020). [DOI] [PubMed] [Google Scholar]
- 74.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stuart T, Srivastava A, Madad S, Lareau CA & Satija R Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ramírez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kramer NE et al. Plotgardener: Cultivating precise multi-panel figures in R. bioRxiv (2021) doi: 10.1101/2021.09.08.459338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thorvaldsdóttir H, Robinson JT & Mesirov JP Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li C et al. SciBet as a portable and fast single cell type identifier. Nat. Commun. 11, 1818 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hearst MA, Dumais ST, Osuna E, Platt J & Scholkopf B Support vector machines. IEEE Intell. Syst. 13, 18–28 (1998). [Google Scholar]
- 83.Pedregosa F et al. Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research 12, 2825–2830 (2011). [Google Scholar]
- 84.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liberzon A et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kinker GS et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat. Genet. 52, 1208–1218 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 90.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bourgey M et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Younesy H, Möller T, Lorincz MC, Karimi MM & Jones SJM VisRseq: R-based visual framework for analysis of sequencing data. BMC Bioinformatics 16 Suppl 11, S2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Orlando DA et al. Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. 9, 1163–1170 (2014). [DOI] [PubMed] [Google Scholar]
- 96.Carroll TS, Liang Z, Salama R, Stark R & de Santiago I Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front. Genet. 5, 75 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Weinberg DN et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature (2019) doi: 10.1038/s41586-019-1534-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Killick R & Eckley IA changepoint: AnRPackage for Changepoint Analysis. J. Stat. Softw 58, (2014). [Google Scholar]
- 99.Lovén J et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rao SSP et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wingett S et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res. 4, 1310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Durand NC et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Karch KR, Sidoli S & Garcia BA Identification and quantification of histone PTMs using high-resolution mass spectrometry. Methods Enzymol. 574, 3–29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ran FA et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jessa S, Hébert S & Kleinman CL HGG-oncohistones processed data. (Zenodo, 2022). doi: 10.5281/zenodo.6773261. [DOI] [Google Scholar]
- 106.Jessa S et al. HGG-oncohistones analysis code. (Zenodo, 2022). doi: 10.5281/zenodo.6647837. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIPseq sequencing data for human cell lines and scRNAseq sequencing data for normal E10, E13, E16, and E18 murine samples have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE188625, while E12, E15, P0, P3, and P6 samples have been previously deposited to GEO under GSE133531. Bulk RNAseq, ChIPseq, HiC, scRNAseq, scATACseq, and scMultiome sequencing data for human tumors have been deposited in the European Genome-phenome Archive (EGA) under accession number EGAS00001005773. Processed data for bulk RNAseq (counts and differential expression analyses), ChIPseq (genome wide H3K27ac/me2/me3 levels), and scRNAseq/scATACseq/scMultiome (counts matrices, cell annotations, and chromatin accessibility bigwig files) have been deposited to GEO under the accession number GSE210568 and Zenodo at https://doi.org/10.5281/zenodo.6773261 105. Accession numbers for previously published data used in this study are provided in Supplementary Tables 1, 2, 6, and 11.