

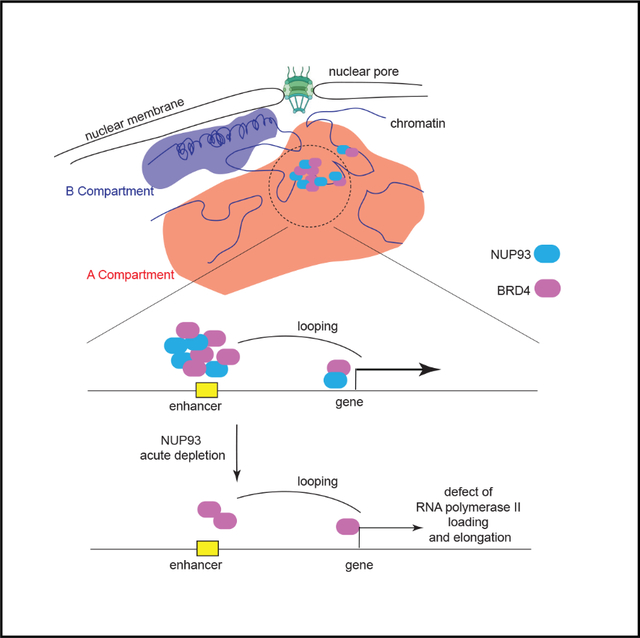
SUMMARY
The nuclear pore complex (NPC) comprises more than 30 nucleoporins (NUPs) and is a hallmark of eukaryotes. NUPs have been suggested to be important in regulating gene transcription and 3D genome organization. However, evidence in support of their direct roles remains limited. Here, by Cut&Run, we find that core NUPs display broad but also cell-type-specific association with active promoters and enhancers in human cells. Auxin-mediated rapid depletion of two NUPs demonstrates that NUP93, but not NUP35, directly and specifically controls gene transcription. NUP93 directly activates genes with high levels of RNA polymerase II loading and transcriptional elongation by facilitating full BRD4 recruitment to their active enhancers. dCas9-based tethering confirms a direct and causal role of NUP93 in gene transcriptional activation. Unexpectedly, in situ Hi-C and H3K27ac or H3K4me1 HiChIP results upon acute NUP93 depletion show negligible changes of 3D genome organization ranging from A/B compartments and topologically associating domains (TADs) to enhancer-promoter contacts.
In brief
Acute depletion of human core nucleoporin, particularly NUP93, elucidates its direct and selective role in transcription activation by facilitating BRD4 recruitment to active enhancers. Zhu et al. report that NUP93 is largely dispensable for direct control of the higher-order 3D genome architecture.
Graphical Abstract
INTRODUCTION
The nuclear pore complex (NPC) is a hallmark of eukaryotes (Akhtar and Gasser, 2007; Buchwalter et al., 2019), which is made up of multiple copies of approximately 30 different protein components, which are collectively called nucleoporins (NUPs). Functionally, the NPC permits regulated nucleocytoplasmic trafficking of all macromolecules in eukaryotes. It can also participate in nuclear activities, including gene regulation, DNA damage repair, and viral infection/integration (Akhtar and Gasser, 2007; Buchwalter et al., 2019; Gordon et al., 2020; Lin and Hoelz, 2019). Pathological alterations of NUPs have been linked to many types of cancer, autoimmune diseases, neurodegeneration, and aging (Buchwalter et al., 2019; Coyne and Rothstein, 2022; Lin and Hoelz, 2019).
Because of the prominent architectural location of NPCs in the nuclear periphery, important efforts have been made to understand their roles in gene regulation and chromatin control (Akhtar and Gasser, 2007; Buchwalter et al., 2019; Lin and Hoelz, 2019). Blobel (1985) proposed that NPC may associate with “transcribable” portions of the genome to facilitate active gene expression or export of their mRNAs, known as the “gene gating” hypothesis. Early studies in yeast (Casolari et al., 2004), HeLa cells (Brown et al., 2008), and Drosophila (Capelson et al., 2010; Kalverda et al., 2010) lend support to this hypothesis. Although “gene gating” mostly pertains to NUPs in gene activation (Blobel, 1985), recent data suggested that they may act as activators and suppressors (Gozalo et al., 2020; Ibarra et al., 2016; Jacinto et al., 2015; Kadota et al., 2020; Raices and D’Angelo, 2017; Toda et al., 2017; Sumner and Brickner, 2022). However, the direct role of NUPs and their selectivity in regulating gene activation or repression and the underlying mechanisms remain elusive.
The mammalian genome is organized into prominent higher-order architectures in three dimensions, encompassing enhancer-promoter loops (several to hundreds of kilobases [kb] in size), topologically associating domains (TADs, about <0.5–1 megabase [Mb]), and A/B compartments (1 to tens of Mb) (Dekker et al., 2013; Kempfer and Pombo, 2020; Yu and Ren, 2017). Because of the notable architectural location of NUPs, there are long-standing hypotheses that NUPs are involved in proper formation of 3D genome architecture (Akhtar and Gasser, 2007; Buchwalter et al., 2019). This has gained support by several reports of specific gene loci in yeast and flies (Pascual-Garcia et al., 2017; Tan-Wong et al., 2009). In mouse embryonic stem cells (ESCs), there is co-localization of NUP153, a nuclear basket NUP, with CTCF and cohesin at TAD boundaries (Kadota et al., 2020). Because of central roles of CTCF/cohesin in 3D genome organization (Dekker et al., 2013; Yu and Ren, 2017), NUPs may affect the 3D genome by affecting CTCF/cohesin.
However, a critical unresolved challenge is that it is difficult to distinguish the direct or indirect roles of NUPs in controlling gene transcription or 3D chromatin organization. First, this is due to the functional essentiality of NUPs that often precludes their genetic deletion (Kim et al., 2018; also see results). Second, many NUPs display high levels of protein stability and half-lives (days to years), often making RNAi knockdown incomplete (Rabut et al., 2004; Toyama et al., 2013). Secondary effects on gene transcription or 3D chromatin organization can be significant after long-term knockdown or knockout of these pleiotropic regulators; for example, because of disrupted nucleocytoplasmic trafficking.
Therefore, there are several important questions concerning NUPs that are still challenging to answer, particularly in human cells: (1) it remains incompletely understood how specific or broad the chromatin association of various members or subcomplexes of NUPs is; (2) it is unclear whether NUPs play direct or causal roles in gene transcription regulation; (3) it is elusive how NUPs participate in transcription regulation; and (4) despite many assumptions as well as supportive data in single loci, it is unknown whether NUPs directly control the high-order 3D genome architecture. In this work, we took advantage of a few advanced techniques, including Cut&Run, auxin-induced rapid degrons, precision run-on sequencing PRO-Seq, dCas9 chromatin tethering, Hi-C, and HiChIP to address these outstanding questions.
RESULTS
Essential NUPs in human cells and their pervasive binding to active enhancers and promoters
NUPs can be divided into six categories: cytoplasmic filaments, outer ring NUPs, inner ring NUPs, nuclear basket NUPs, channel NUPs, and transmembrane NUPs (Figure 1A; Hampoelz et al., 2019a; Lin and Hoelz, 2019). Based on protein function, stability, and dynamics, NUPs can also be categorized as core NUPs (more stable) and peripheral NUPs (more dynamic) (Buchwalter et al., 2019; Gozalo et al., 2020). To study the direct roles of NUPs, we elected to focus on those that are essential for cell survival and health. By analyzing gene essentiality in human cells, we found that SEC13 and NUP93 are the two most essential NUPs; their knockout inhibited survival of more than 99% of cell lines (Meyers et al., 2017; Figure 1B). NUP133, RAE1, NUP85, and NUP43 are also essential (for >90% of cell lines) (Figure 1B). In contrast, NUP210, POM121, and NUP188 are among the least essential (Figure 1B). We also calculated the essentiality scores for other major nuclear and chromatin architectural proteins; i.e., cohesin, condensin, SMC5/6, the nuclear lamina, and CTCF. SEC13 and NUP93 have high essentiality scores comparable with CTCF and some condensins (e.g., NCAPG and SMC2) (Figure 1B). The essentiality of NUP93 is consistent with structural data showing that it is the keystone of NPCs and is also consistent with genetic evidence in yeast showing that loss of Nic96, the NUP93 homology, causes severe growth arrest (Kim et al., 2018).
Figure 1. Core NUPs bind active promoters and enhancers in human cells.
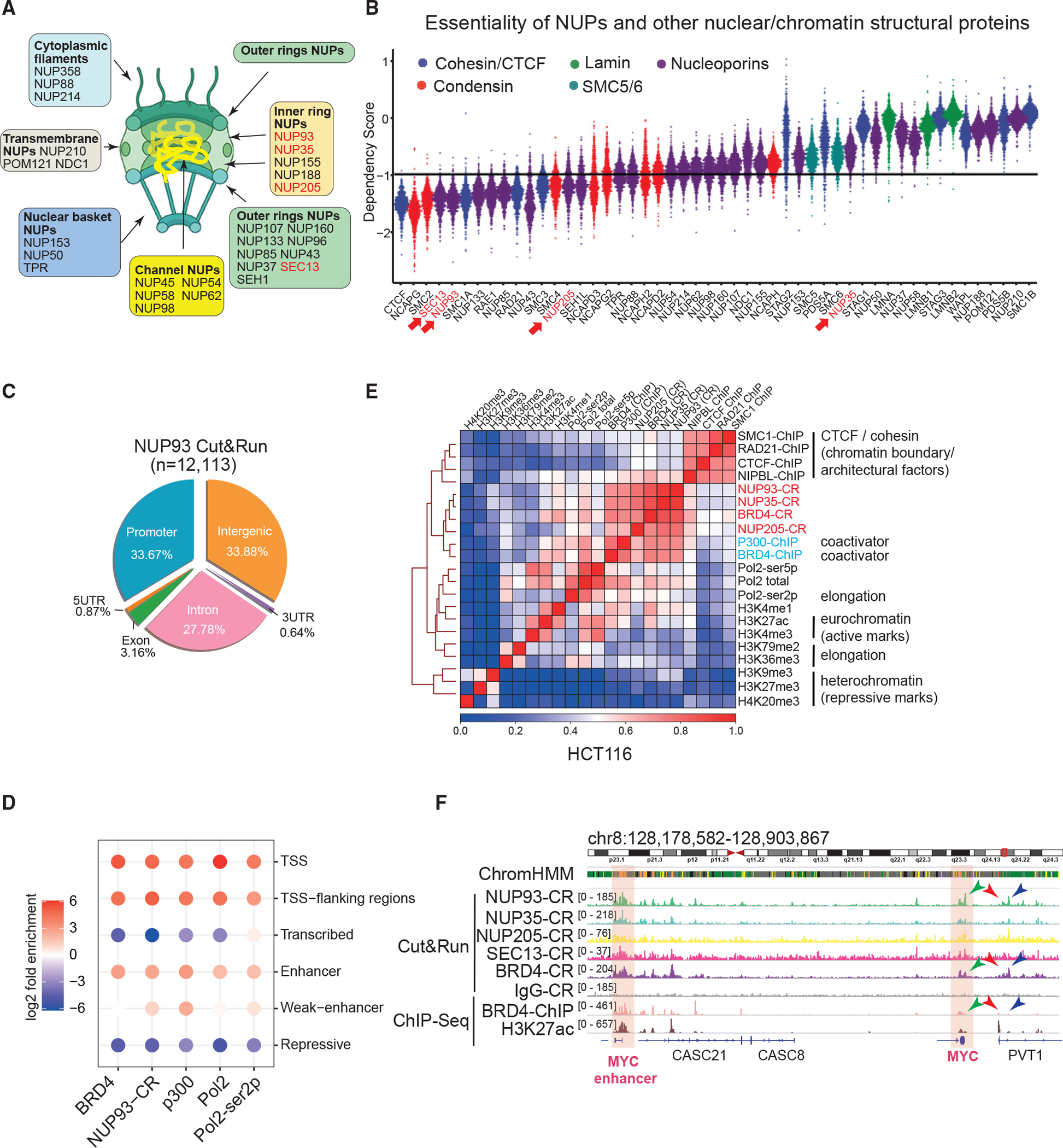
(A) A diagram of the NPC, its subcomplexes, and NUPs.
(B) Dependency score plot for factors. The value of – 1 represents the cutoff to deduce essential genes. Each dot denotes one of the 789 cell lines. NUPs we made using Cut&Run data are marked in red.
(C) The Cut&Run peak distribution of NUP93 in HCT116 cells.
(D) Folds of enrichment of factors in distinctive chromatin states were determined by using observed versus background. 18 states defined by histone marks in Figure S1D were merged into 6 major states. CR, Cut&Run.
(E) Heatmap of pairwise Pearson correlations between ChIP-seq or CR signals for various factors in HCT116 cells. The color scale indicates Pearson correlation coefficients.
(F) Genome browser snapshot of CR and ChIP-seq of core NUPs and BRD4 at the CCAT1-MYC locus. Highlighted areas indicate CCAT1 (i.e., the MYC enhancer) and MYC gene.
We focus on essential NUPs to examine their direct transcription/chromatin roles. Cut&Run is a method to reveal chromatin association of target proteins under native conditions without chemical crosslinking (Skene and Henikoff, 2017). We conducted Cut&Run for a series of NUPs, finding that core NUPs (NUP93, NUP35, and NUP205) as well as SEC13, an outer ring NUP, can be detected to associate with specific chromatin sites (Figures 1C, 1E, S1A–S1C, and S1F). We also conducted Cut&Run for NUPs belonging to other subcomplexes, including NUP98, NUP153, TPR, and NUP210, but failed to find clear chromatin association sites (data not shown). This was possibly because the chromatin dynamics of these proteins were too rapid (Rabut et al., 2004) to be captured by Cut&Run or because of antibody quality. The information can be found in Table S1. Globally, 12,113 peaks of NUP93 were detected in HCT116 cells (Figure 1C), the majority of which overlap promoters and intergenic/intronic regions. For NUP35 and NUP205, we detected 2,549 and 2,324 peaks, respectively, and they were predominantly located in intergenic and intronic regions (Figures S1A and S1B). SEC13 peaks were more often near promoters, but only a small number of peaks was detected (Figure S1C).
Using six histone marks, we applied ChromHMM to divide the genome into 18 chromatin states (Ernst and Kellis, 2012; Figure S1D; see Table S2 for datasets used). We then merged them into 6 major states to examine the enrichment of NUP93 (Figure 1D); we also examined transcriptional coactivators (BRD4 and p300), RNA polymerase II (RNA Pol II), and its serine-2 phosphorylated form (Ser2p). This analysis showed that NUP93 Cut&Run peaks displayed enrichment to active enhancers and promoters (Figure 1D). In contrast, NUP93 and coactivators are excluded from regions of repressive (H3K27me3- and H3K9me3-marked regions) or transcribed chromatin states (with H3K36me3) (Figure 1D). Fewer than 1% of NUP93 peaks (113) were located in the repressive state (heterochromatin and others) defined by ChromHMM.
To analyze NUPs’ chromatin association independent of peak calling, we divided the genome into 1-kb bins and calculated pairwise correlation coefficients between the chromatin binding signals of various factors or histone marks. Hierarchical clustering of pairwise correlations (Figure 1E) showed that core NUPs displayed the best correlation with the binding of BRD4 and p300 (Figure 1E). There are fairly strong correlations between NUPs and active histone marks, such as H3K27ac, or between NUPs and RNA Pol II (Figure 1E). In comparison with the chromatin boundary factors CTCF and cohesin complex, NUPs showed some degrees of correlation (Figure 1E), consistent with a recent report (Kadota et al., 2020). But it is obvious that CTCF/cohesin formed a strong cluster among themselves, quite distinct from the cluster formed by NUP/BRD4/p300 (Figure 1E). Among the cohesin complex, NIPBL is a subunit known to be more enriched to enhancers (Vian et al., 2018), and it showed higher levels of correlation with NUP/BRD4/p300 binding (Figure 1E). The correlation coefficient analysis confirmed that NUPs poorly overlap heterochromatin regions (H3K9me3, H3K27me3, or H4K20me3; Figure 1E). As examples, we show peaks of NUPs in the vicinity of a proto-oncogene, MYC, and its associated enhancer/lncRNA, CCAT1 (McCleland et al., 2016; Xiang et al., 2014) as well as in the loci of KITLG and its associated enhancer (Figures 1F and S1E). Our analyses by peak location or global correlation indicated that NUPs, particularly the inner ring core NUPs, predominantly locate in euchromatin regions. However, although fewer than 1% of NUP93 Cut&Run peaks (113 of 12,113) overlapped with repressed regions in HCT116 cells (and a similar ratio in HeLa cells), chromatin immunoprecipitation sequencing (ChIP-seq) of Nup93 in Drosophila cells found more than 50% of all peaks overlapping heterochromatin or Polycomb regions (Gozalo et al., 2020); ChIP-seq of Npp106, an NUP in Schizosaccharomyces pombe that is homologous to Nic96/NUP93, showed strong signals in pericentromeric heterochromatin (Iglesias et al., 2020). Technical differences between methods, in addition to cell types and species, may contribute to these differences (discussion).
In HeLa cells, we found a consistent distribution of NUP93 Cut&Run peaks mostly at intergenic/intronic regions (Figure S1F) and enrichment similar to active promoters and enhancers (Figure S1G). Its enrichment again mimics that of BRD4 and p300 and was depleted from the repressive or transcribed chromatin (Figure S1G). We identified cell-type-invariant or -specific active enhancers by comparing the ChIP-seq of H3K27ac in HCT116 and HeLa cells (Figure S1H; only non-promoter peaks used) and then plotted the NUP93 binding signals on these sites. This analysis indicated that NUP93 binding exhibits cell type specificity, similar to the patterns of H3K27ac (Figure S1H). Our Cut&Run results validated and extended previous understanding of NUP association with chromatin in human cells (Brown et al., 2008; Ibarra et al., 2016; Kadota et al., 2020), indicating that core NUPs predominantly bind euchromatin and have a strong resemblance to binding of coactivators BRD4 or p300.
Rapid degradation of NUP93 reveals its direct and selective regulation of gene transcription
The high essentiality and protein stability of NUPs often render RNAi strategies insufficient to completely deplete NUPs in a short time, or they may have secondary or indirect effects on gene transcription after prolonged knockdown/knockout. We took advantage of the auxin-inducible OsTir1/mAID degron system to rapidly deplete NUPs in human cells to study their direct functions (Natsume et al., 2016; Figure 2A). We employed CRISPR-Cas9 and homologous recombination to knock in a three-peptide tag to the C terminus of endogenous NUP93 and NUP35, which consists of mAID, mClover, and a tandem hemagglutinin (HA) (mAID-mClover-2HA). We selected NUP93 and NUP35 for knockin because of their strong chromatin association shown by Cut&Run, their critical location in the NPC architecture (Kim et al., 2018; Figure 1A), and their contrasting essentiality for human cells (Figure 1B). Western blots showed that the knockin cells homogeneously expressed NUP93-mAID-mClover-2HA protein (hereafter referred to as tagged NUP93 or NUP93-mAC; Figure 2B), which can be efficiently and rapidly degraded by treatment with IAA (indole-3-acetic acid; Figure 2C). Significant degradation of NUP93 can be seen by about 2–4 h of IAA treatment, but complete loss was achieved by ~8 h (Figures 2C and S2A). This was confirmed by immunoblots using an HA antibody (Figure S2A). We also generated a knockin HCT116 line expressing NUP35-mAID-mClover-2HA, which can be completely degraded in 8 h (Figure S2B). Structural illumination (N-SIM) and confocal microscopy using the mClover tag fused to NUP93 confirmed that endogenous NUP93 predominantly locates to the nuclear periphery (Figures S2C, S2D, and S2F). After IAA treatment, although NUP93 was completely degraded (Figures S2D and S2F), the NPC structure remained unaltered, as shown by staining using Mab414, a marker of pan-NUPs (Davis and Blobel, 1987; Figures S2D and S2E). Staining of Lamin-A/C, the A-type nuclear lamina proteins marking the nuclear periphery, was also unaffected (Figures S2F and S2G).
Figure 2. Acute NUP degradation in human cells deregulates gene and enhancer transcription.
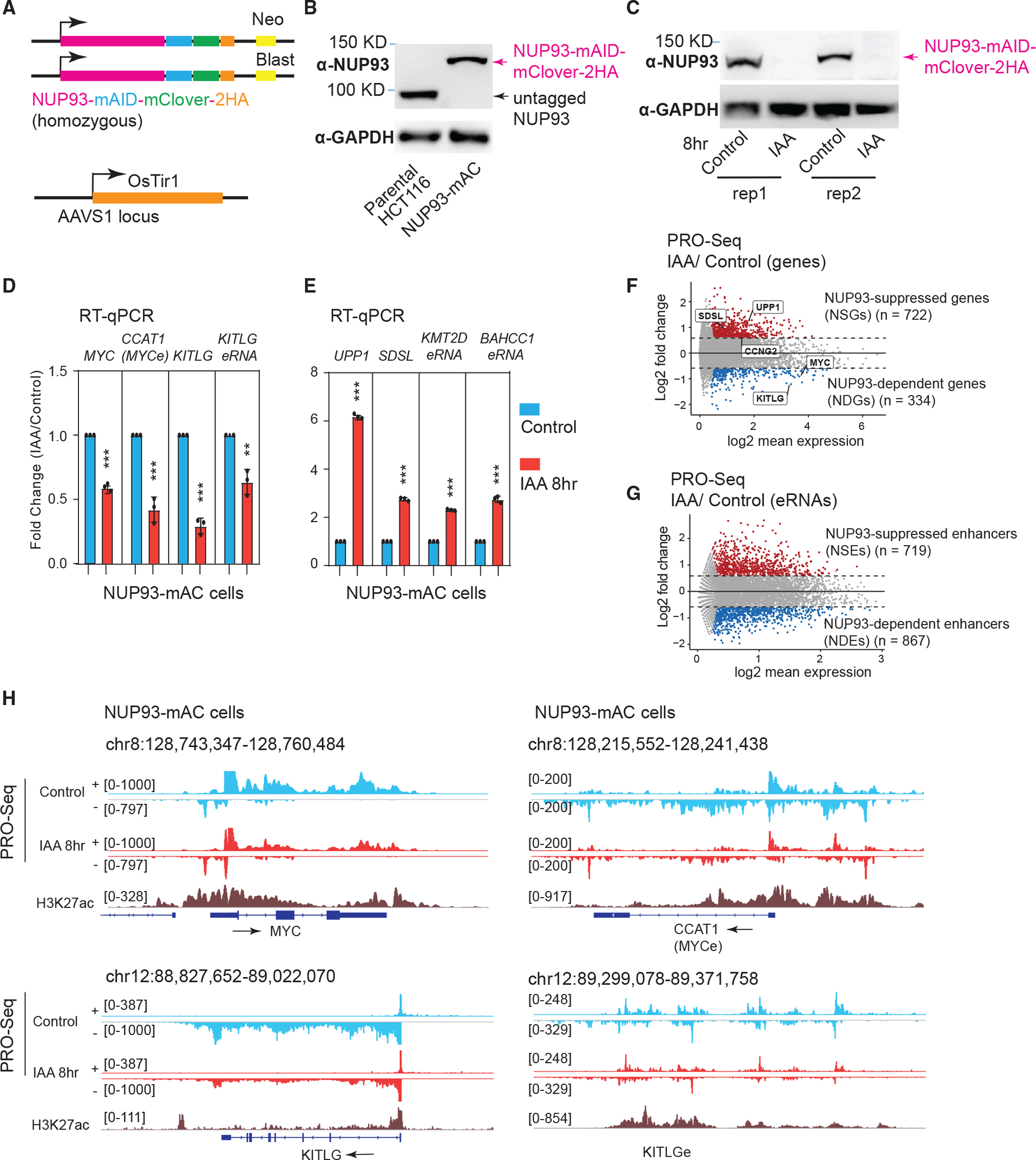
(A) Top: knockin design for generating degron cell lines. Bottom: OsTir1 was inserted into parental HCT116 cells at the AAVS1 locus.
(B) Western blot analysis of NUP93 in parental cells or in degron cells expressing NUP93 tagged by mAID, mClover, and 2HA (NUP93-mAC).
(C) Western blot analysis after 8 h of auxin treatment of NUP93-mAC cells (two sets of samples are shown). IAA, indole-3-acetic acid.
(D and E) qRT-PCR analysis of genes and eRNAs changed by 8 h of IAA treatment to degrade NUP93. The bars represent mean ± SD (n = 3 technical replicates). Each plot is a representative example from more than 3 biological replicates. Student’s t test; **p < 0.01, ***p < 0.001.
(F and G) MA plots depicting differential transcriptional activities (PRO-seq) for genes and enhancers upon acute NUP93 depletion (IAA, 8 h) versus vehicle conditions (control). Upregulated genes and enhancers are highlighted in red and down-regulated ones in blue. The x axis shows log2 transformed RPKM values, and the y axis represents the log2 FC (IAA/control).
(H) PRO-seq tracks for control and acute NUP93 depletion (IAA, 8 h) conditions over representative genomic regions: MYC, CCAT1 (MYC enhancer), and KITLG and KITLG enhancer (KITLGe). + and − indicate Watson and Crick strands, respectively; arrows indicate the transcriptional direction of genes/eRNAs.
The auxin degron system may incur leaky degradation of tagged proteins (Yesbolatova et al., 2020). To investigate this for NUP93/NUP35 degrons, we conducted cell cycle analyses of parental HCT116, NUP93-mAC, and NUP53-mAC cells after vehicle (no IAA) and 8- or 24-h IAA treatment. Prior to IAA treatment, the cell cycle distribution of the two degron cell lines appeared to be similar to the parental HCT116 cells (carrying Os-Tir1) (Figures S2H, S2J, and S2L, leftmost panels), suggesting no significant leaky degradation of NUP93 or NUP35 that may affect cell cycles. After IAA treatment for 8 or 24 h, the cell cycle distributions in the parental and NUP35-mAC cells remained largely unchanged (Figures S2H and S2J). Quantification of cells in each phase of the cell cycle is shown in Figures S2K–S2M. For NUP93-mAC cells, 24-h IAA obviously increased cell numbers in G1 phase and potentially inhibited cell cycle progression from G1 to late phases, but 8-h IAA treatment did not cause this alteration (Figures S2L and S2M). This result indicates that NUP93 is required for proper cell cycle progression, but its degradation will only cause obvious changes after long-term depletion (>8 h). This lends support to the theory that acute depletion of NUP93 is required to study its direct and bona fide roles in gene transcription and chromatin regulation.
To examine the direct roles of NUP93 and NUP35 in gene transcription control, we first examined expression of gene/enhancer pairs that show strong binding by NUPs: MYC-CCAT1 or the KITLG loci (Figures 1F and S1E). Enhancer RNA (eRNA) is a type of noncoding RNA produced by active enhancers and can serve as an indicator of enhancer activity (Li et al., 2016; Sartorelli and Lauberth, 2020). Hereafter, we refer to eRNAs with an “e” after the nearby gene name (e.g., KITLGe). Rapid depletion of NUP93 significantly reduced expression of MYC and KITLG mRNA and their nearby eRNAs (Figure 2D). Prolonged NUP93 depletion by 24 h did not exacerbate the gene expression changes more than those observed after 8 h (Figure S3A), suggesting that its transcriptional effects can be largely revealed by rapid depletion (8 h). Short-term NUP93 depletion (2 h or 4 h of IAA) already elicited significant gene transcription changes (Figure S3B), consistent with significant protein loss by these time points (Figure S2A). These results demonstrate that NUP93 plays a direct role in regulating gene expression. Different from that of NUP93, NUP35 depletion caused no or mild expression changes of genes and eRNAs (Figure S3D), despite the fact that it binds these gene promoters and enhancers (Figures 1F and S1E). One possibility is that rapid NUP35 degradation may be functionally compensated by other NUPs.
In NUP93-mAC cells with or without 8 h of IAA treatment, we interrogated global transcriptional programs by PRO-seq (Mahat et al., 2016). We identified 334 genes significantly decreased because of acute NUP93 depletion, to which we refer as NUP93-dependent genes (NDGs). In contrast, 722 genes showed a transcriptional increase, and we denote them NUP93-suppressed genes (NSGs) (Figure 2F). These selective, rather than massive, changes in gene transcription are consistent with overall comparable levels of 5-ethynyl-uridine (5-EU) labeling of nascent RNAs after acute NUP93 depletion (Figures S3F and S3G), and together they indicate that NUP93 plays a direct role in transcriptional control of only a subset of genes. PRO-seq not only detects transcription from genes but also from noncoding regions, including eRNAs. Acute NUP93 depletion caused ~1,600 eRNAs to show differential transcription, with 719 being induced and 867 decreased (Figure 2G). We refer to enhancers with decreased/increased eRNAs as NUP93-dependent enhancers (NDEs) and NUP93-suppressed enhancers (NSEs), respectively. qRT-PCR validated the changes of randomly selected genes and eRNAs (Figure 2F), which were already detectable by 2 or 4 h of IAA treatment (Figure S3C). These genes or eRNAs were largely unaffected by acute NUP35 depletion (Figure S3E), supporting the theory that NUP35 lacks a direct role in gene transcriptional control. PRO-seq screenshots of example genes/eRNAs are shown in Figure 2H. NUP93-regulated gene and eRNA lists can be found in Table S3.
A causal role and the nuclear periphery action of NUP93 in transcriptional activation
Rapid depletion and PRO-seq results support a direct and causal role of NUP93 as a transcriptional activator for selective genes or enhancers. To test its causal role, we utilized a dCas9-based tethering approach for which a dCas9-NUP93 fusion protein together with specific gRNAs can recruit NUP93 to genomic sites of interest (Figure 3A). To avoid confounding effects of the endogenous NUP93 during tethering, we expressed dCas9-NUP93/gRNA in NUP93-mAC cells to permit removal of the endogenous NUP93 (Figure 3A). qRT-PCR revealed that tethering of NUP93 to the KITLG promoter and enhancer significantly increased the transcriptional activity of the target enhancer and gene (Figure 3B). An irrelevant gene, FTO, was not altered (Figure 3B), indicating the specificity of this effect. Some NSGs and NSEs were transcriptionally upregulated after acute NUP93 depletion, suggesting the possibility that NUP93 directly suppresses their transcription (Figure S4A). However, dCas9 tethering of NUP93 to NSG promoters or NSEs did not suppress or obviously change their transcription (Figure S4B). These data demonstrate that NUP93 is causally important for enhancer and gene activation but appears to not causally suppress genes. We are tempted to speculate that acute NUP93 depletion may have affected NSG transcription indirectly (discussion).
Figure 3. NUP93 acts as a direct transcriptional activator in the nuclear periphery.
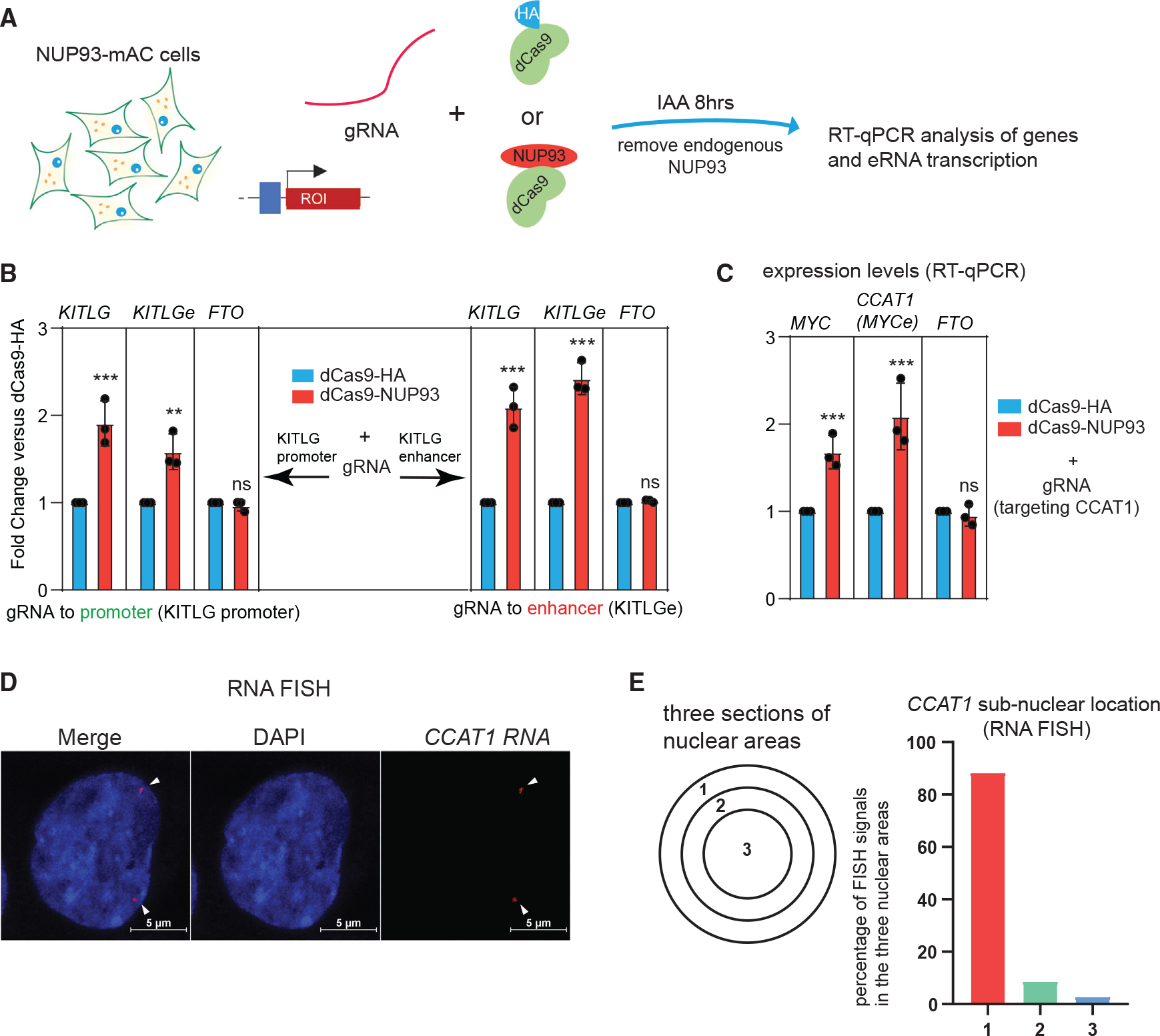
(A) A diagram of the CRISPR-dCas9-based NUP93 tethering strategy in cells with endogenous NUP93-mAC depleted.
(B) qRT-PCR analysis of the target gene and eRNA expression upon CRISPR-dCas9 tethering of NUP93 to the KITLG promoter or enhancer (with endogenous NUP93 depleted).
(C) qRT-PCR analysis of the target MYC gene and eRNA expression upon CRISPR tethering of NUP93 (with endogenous NUP93 depleted).
(B and C) The bars represent mean ± SD (n = 3 technical replicates). Each plot is a representative example from 3 biological replicates. Student’s t test; *p < 0.05, ***p < 0.001; ns, not significant.
(D) Representative RNA FISH images of CCAT1 in HCT116 cells. White arrowheads point to the positions of RNA FISH signals. Scale bar, 5 μm.
(E) A diagram of subnuclear areas defines the nuclear periphery versus nuclear center. Right panel: quantified percentage of CCAT1 FISH signals in each subnuclear area (n > 200 FISH foci counted).
A paradigm in nuclear organization is that the nuclear periphery is often transcriptionally inert in most differentiated mammalian cells (except in rare cases like rod photoreceptor cells), whereas the nuclear center is transcriptionally active (Akhtar and Gasser, 2007; Buchwalter et al., 2019; Van Steensel and Belmont, 2017). mClover-tagged endogenous NUP93 is largely located in the nuclear periphery (Figures S2C, S2D, and S2F). We tested whether NUP93 acts as an activator on its targets near the nuclear periphery. In this case, we focused on CCAT1 because it was strongly bound by NUP93 and other NUPs (Figure 1F), and it can be robustly detected by RNA fluorescence in situ hybridization (FISH) (Xiang et al., 2014). CCAT1 is an enhancer-derived lncRNA with known roles in colorectal cancer growth by promoting MYC gene expression (McCleland et al., 2016; Xiang et al., 2014). We thus sometimes interchangeably refer to it as MYCe in this paper. dCas9 tethering of NUP93 increased expression of CCAT1 RNA and the MYC gene, indicating a direct role of NUP93 on this enhancer (Figure 3C). By RNA FISH, we found that the large majority (>80%) of CCAT1 eRNA signals were located in the nuclear periphery (Figures 3D and 3E). These results demonstrate that NUP93 plays a causal role to transcriptionally activate its targets and that, at least for a subset, this takes place in the nuclear periphery.
NUP93 activates gene transcription by permitting RNA Pol II loading
We ranked all transcribed genes based on their fold change (FC) in PRO-seq after 8 h of IAA treatment (acute NUP93 depletion) versus vehicle (control) (Figures 4A and 4B). This ranked heatmap shows that the genes at the bottom are dependent on NUP93 for transcriptional activation (i.e., NDGs), whereas those at the top are increased by its acute depletion (i.e., NSGs; Figure 4B). We then analyzed the epigenomic features of these genes, including enhancer/promoter histone marks (H3K27ac, H3K4me1, and H3K4me3), transcription elongation mark (H3K36me3), and repressive/heterochromatin marks (H3K27me3 or H3K9me3); we also calculated the ChIP-seq signals of RNA Pol II and Ser2p (Figures 4C, S4C, and S4D). This analysis showed that NDG genes, in comparison with NSGs, exhibit a higher level of RNA Pol II recruitment to their transcriptional start sites (TSSs) and gene bodies and that they also bear stronger signals of transcriptional elongation marks (Figures 4C and 4D). In terms of active histone marks, NDGs displayed lower levels of H3K4me3 and H3K27ac than NSGs on their promoters (Figure S4E). For the repressive histone marks H3K27me3 and H3K9me3, the transcribed genes used in this analysis are of very low signals (i.e., lower than randomly selected non-transcribed genes; Figures S4D and S4F), and there was little difference between NDGs and NSGs (Figure S4F). These results suggest that genes directly promoted by NUP93 tend to bear high levels of RNA Pol II loading and elongation markers; in contrast, NUP93 directly suppresses genes that possess relatively stronger promoters (i.e., H3K27ac and H3K4me3; Figure S4E).
Figure 4. NUP93 is required for RNA Pol II loading and transcriptional elongation of NDGs.
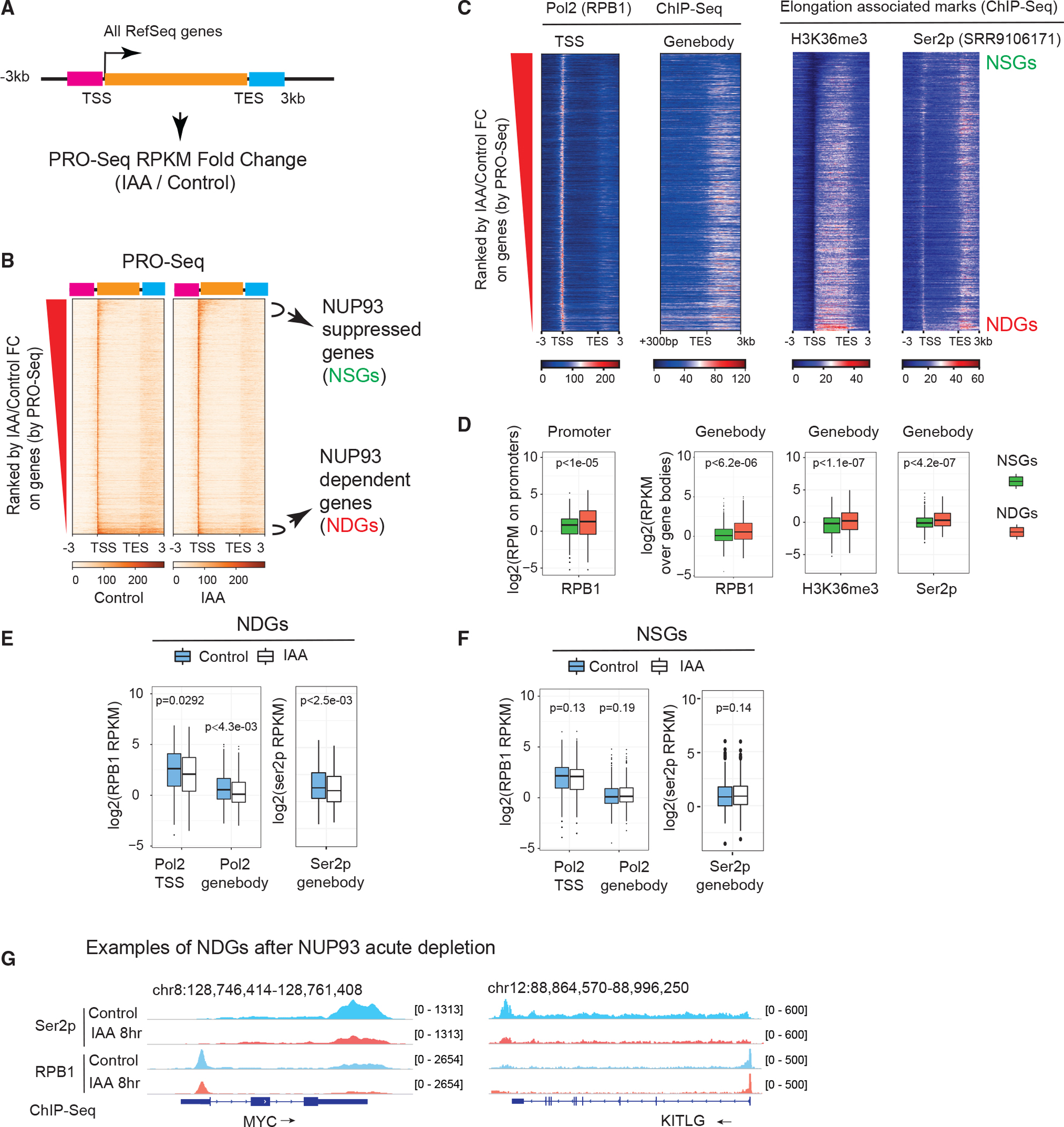
(A) A diagram showing how we ranked transcribed genes by changes in control (vehicle, 8 h) versus NUP93 depletion (IAA, 8 h). Orange bar, gene body; pink bar, 3 kb upstream of transcriptional start sites (TSSs); light blue bar, 3 kb downstream of transcriptional end sites (TESs).
(B) Heatmap displaying all transcribed genes ranked by PRO-seq FC (IAA/control).
(C) Heatmaps of normalized ChIP-seq of total RNA Pol II, the elongation histone mark (H3K36me3), and Ser2p in the same order as in (B).
(D) Boxplots comparing features of NSGs and NDGs over their promoters (TSS ± 300 bp) or gene bodies (+300 bp from the TSS to the gene ends). For Ser2p, we extended gene bodies to 3 kb after TESs. p values: Wilcoxon rank-sum test.
(E and F) Boxplots showing the intensity of RNA Pol II or Ser2p before and after IAA treatment over the TSS regions or gene bodies of NDGs and NSGs.
(D–F) For boxplots, center lines represent medians, box limits indicate the 25th and 75th percentiles, and whiskers extend 1.5 times the interquartile range (IQR) from the 25th and 75th percentiles. p values: Wilcoxon rank-sum test.
(G) Genome browser snapshots over the MYC and KITLG genes; arrows show gene transcription direction.
To test how NUP93 affects gene transcription, we conducted ChIP-Seq for total RNA Pol II and Ser2p. The results showed that, upon NUP93 acute depletion, NDGs displayed a significantly reduced level of RNA Pol II in both their TSS regions and gene bodies; consistently, the level of Ser2p RNA Pol II was reduced in their gene bodies (Figure 4E). By contrast, NSGs displayed no significant changes by meta-analysis (Figure 4F), with individual cases shown in Figure S4G. The MYC and KITLG loci are example NDGs that bear high amounts of RNA Pol II and Ser2p, whose levels were significantly reduced after NUP93 acute depletion (Figure 4G). These results support that NUP93 activates gene transcription via facilitating RNA Pol II loading.
NUP93 licenses transcriptional activation by licensing full BRD4 recruitment to active enhancers
NUP93 association to chromatin resembles that of BRD4 (Figures 1D–1F and S1G), which plays a critical role in transcriptional activation and elongation (Liu et al., 2013; Winter et al., 2017; Yang et al., 2005). This leads us to test whether NUP93 acts in gene activation by affecting BRD4 recruitment (Figure 5A). Although present at active promoters and enhancers (Figure 1F), NUP93 or BRD4 showed higher affinity for enhancers (NDEs) than for promoters (NDGs) (Figure 5B). After acute NUP93 depletion, BRD4 recruitment was significantly reduced on NDEs (Figure 5C). NDG promoters were affected mildly (Figure 5D). These results demonstrate that NUP93 mediates BRD4 enrichment to active enhancers to control target gene activation and RNA Pol II loading/elongation. A diagram summarizing these results is shown in Figure 5E, and example regions are shown in Figures 5F and S5A. We also examined the histone modification H3K27ac by ChIP-seq, finding no detectable changes on any deregulated enhancers or promoters because of acute NUP93 depletion (Figures S5B–S5D).
Figure 5. NUP93 promotes BRD4 recruitment to active enhancers to activate gene transcription.
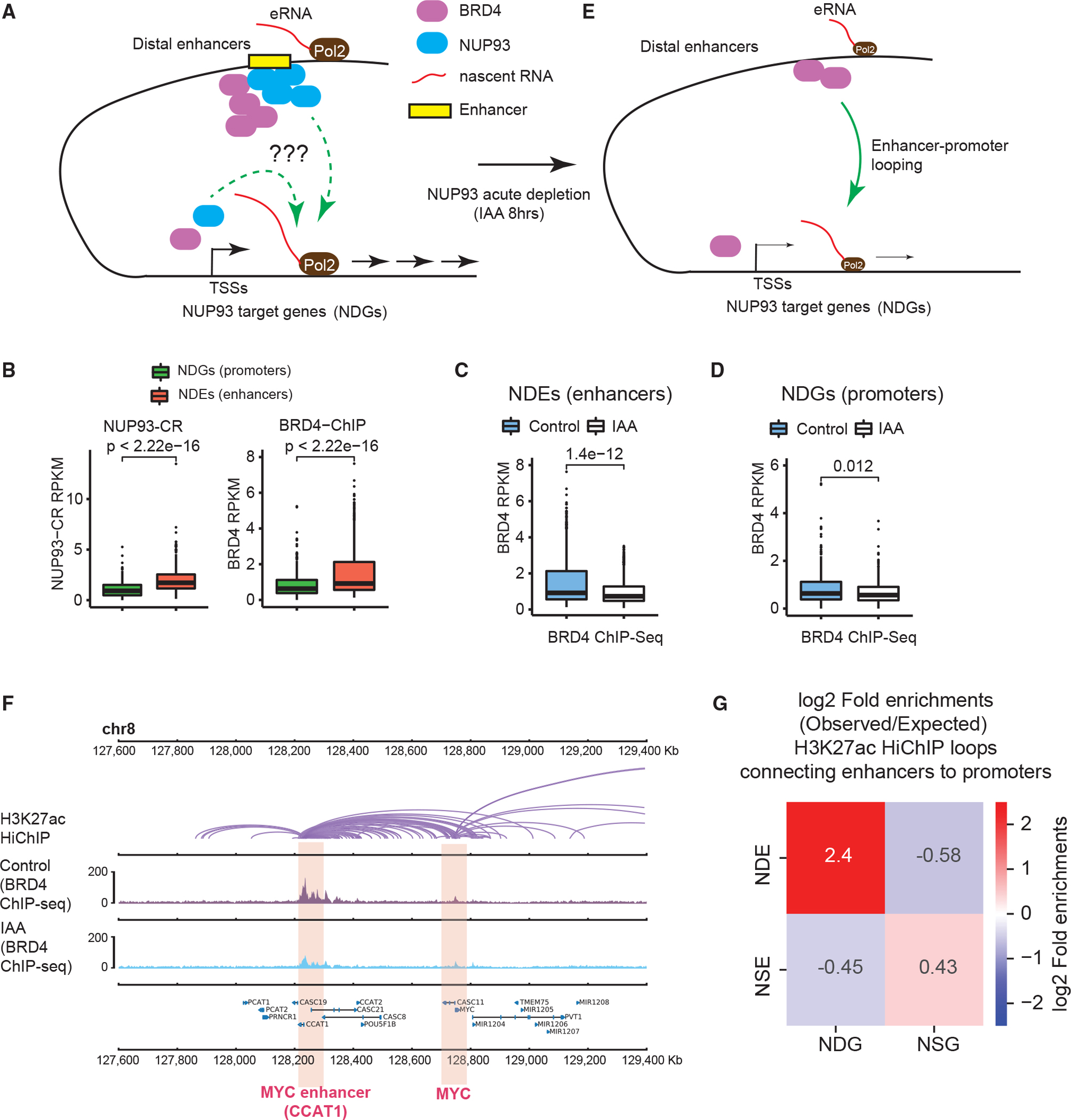
(A) A diagram showing the questions to examine: how does NUP93 regulate RNA Pol II loading and/or target gene elongation, and what is its relationship with BRD4 binding?
(B) Boxplots showing the levels of NUP93 or BRD4 at NDEs or NDG promoters.
(C and D) Boxplots showing the changes of BRD4 ChIP-seq at NDEs or NDG promoters because of acute NUP93 depletion.
(B–D) For boxplots, center lines represent medians, box limits indicate the 25th and 75th percentiles, and whiskers extend 1.5 times the IQR from the 25th and 75th percentiles. p values: Wilcoxon rank-sum test.
(E) A diagram showing BRD4 binding changes after acute NUP93 depletion; the question was also raised whether NDEs act on NDGs via chromatin looping.
(F) A representative region encompassing MYC-CCAT1 showing H3K27ac HiChIP (in wild-type HCT116 cells), BRD4 ChIP-seq under control and IAA (acute NUP93 depletion) conditions; gene annotation is shown at the bottom. Each purple arc in HiChIP denotes a loop called by FitHiChIP.
(G) Heatmaps showing the folds of enrichment of observed enhancer-promoter loops formed between specific enhancer or promoter groups over the expected frequency using randomly shuffled enhancer and promoter lists (see Figure S5E and STAR Methods for additional information). Chromatin loops were defined by H3K27ac HiChIP using FitHiChIP.
NDEs are often located far away from NDGs, as exemplified by the CCAT1-MYC locus (~520 kb; Figure 1F) and the KITLG promoter and enhancer (~400 kb; Figure S1E). We examined whether NDEs may affect NDGs via enhancer-promoter loops (Figure 5E). To examine high-resolution chromatin loops, we conducted H3K27ac HiChIP (Mumbach et al., 2016; also known as PLAC-Seq; Fang et al., 2016) in HCT116 cells and generated ~53 millions of total contact pairs (Table S1), which identified 26,540 chromatin loops by FitHiChIP (Bhattacharyya et al., 2019). These data enabled us to examine the frequency of chromatin loops formed between NUP93-regulated enhancers and promoters in a pairwise manner, and we then calculated the fold enrichment of each “observation” over the “expected background”—the average loop numbers using randomly selected active enhancers or promoters from HCT116 cells (STAR Methods). We found that NDGs and NDEs have a much higher chance to form enhancer-promoter loops than expected (5.3-fold) (Figures 5G and S5E). In contrast, between NDGs/NSEs or NSGs/NDEs, there are lower-than-expected chances to detect chromatin loops. NSGs and NSEs displayed an observed number of loops slightly more than by random chance (Figures 5G and S5E). Examples are shown for two pairs of NDE-NDG loci, MYC-MYCe and KITLG-KITLGe, illustrating H3K27ac HiChIP loops and the reduction of BRD4 recruitment after acute NUP93 depletion (Figures 5F and S5A).
NUP93 is dispensable for direct control of higher-order 3D genome organization
Rapid degradation of chromatin architectural proteins has illuminated critical and specific roles of CTCF and cohesin in organizing TADs (Nora et al., 2017; Rao et al., 2017). Rapid and complete depletion is essential to reveal their bona fide roles in high-order genome organization (Nora et al., 2017). We examined the direct role of NUPs in regulating 3D genome architecture. Triplicates of in situ Hi-C data were sequenced in total to ~570–660 millions of reads per condition (Table S1), and they are of high concordance (Yang et al., 2017; Figure S6A). The P(s) analyses of Hi-C data showed that the overall contact frequency by genomic distance was unaltered with or without acute NUP93 depletion (Figure 6A).
Figure 6. NUP93 is dispensable for the overall organization of higher-order genome architecture.
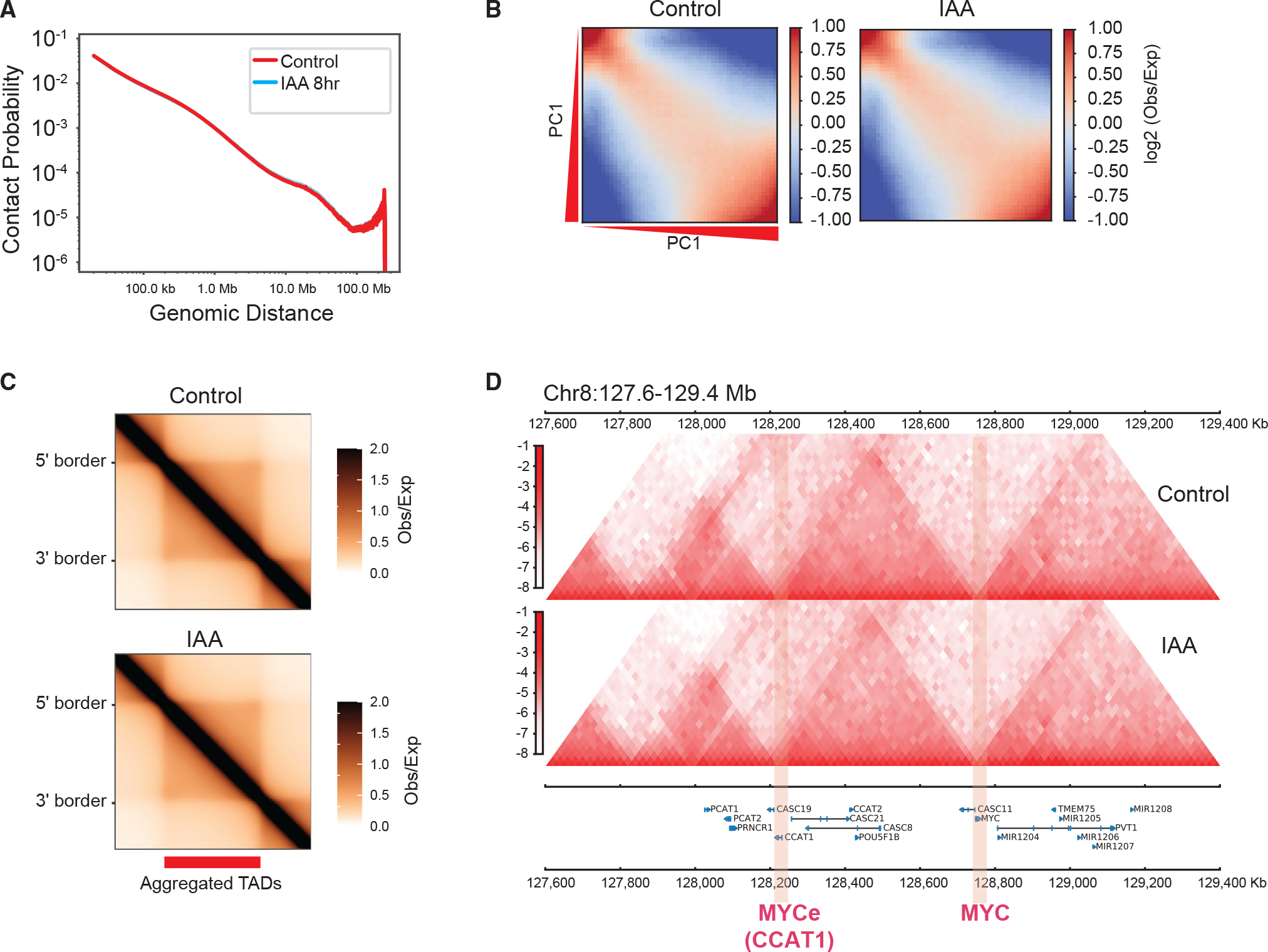
(A) A P(s) curve plot generated from Hi-C, showing the relationship between chromatin contact probability (P) and linear genomic distances (s) under control and 8-h IAA (acute NUP93 depletion) conditions.
(B) Saddle plots of chromatin compartmentalization, showing intrachromosomal interaction frequencies between 20-kb bins, normalized by the expected contact frequency. Bins are ranked by PCA E1 values under the control condition. The top left corner shows B-B interactions, and the bottom right corner shows A-A interactions. The scale indicates interaction strength (observed/expected) in Hi-C.
(C) Aggregated domain analysis (ADA) of all TADs under control (top) and 8-h IAA (bottom) conditions.
(D) An example region encompassing MYC and MYCe (CCAT1), showing Hi-C contact heatmaps under control and 8-h IAA conditions (at 20-kb resolution).
Higher-order chromatin structure in 3D folds into A/B compartments (Lieberman-Aiden et al., 2009; Rao et al., 2014). The eigenvector E1 scores (E1) based on principal-component analysis (PCA) of Hi-C with or without NUP93 depletion were largely identical, suggesting no overall alteration of A/B compartments upon NUP93 loss (Figure S6B); no obvious change was observed for inter-compartmental interactions, as shown by the saddle plot (Figure 6B). By using a loose cutoff to examine A/B compartment changes for every 100-kb bin of the genome (E1 score change of 0.2; Yusufova et al., 2020), we only detected a limited number of bins with weak alteration of compartmental strength (Figure S6C), with almost no bins showing compartmental switching (from B to A or A to B). Unaltered E1 scores across an example region are shown in Figure S6D. Focusing on genes with transcriptional changes, we examined the A/B compartmental scores for NSGs and NDGs, both of which predominantly locate in the A compartment, as shown by their positive E1 scores (Figure S6E). This result is consistent with the fact that NSGs and NDGs predominantly reside in euchromatin and possess active histone marks at their promoters, bearing low levels of H3K27me3 or H3K9me3 marks (Figures S4E and S4F) that are known to be high in the B compartment (Lieberman-Aiden et al., 2009; Rao et al., 2014). After NUP93 depletion, the E1 scores for NSG and NDGs were largely unchanged (Figure S6E).
Through aggregation domain analysis (ADA), we found that TADs, another prominent feature of the 3D genome (Dekker et al., 2013; Yu and Ren, 2017), were also mostly unaffected after acute NUP93 acute depletion (Figure 6C), for which several example regions near MYC, KITLG, and UPP1 are shown (Figures 6D, S7A, and S7B). The strength of TADs, measured by insulation scores of their boundaries (Crane et al., 2015) or by directionality index (Dixon et al., 2012), appeared to be comparable in cells with or without NUP93 depletion (Figure S7C). We also employed HICCUPS to call chromatin loops from Hi-C and examined their strength in cells with or without NUP93. As shown by aggregation peak analysis (APA), the strength of all HICCUPS loops appeared to be almost identical (n = 3,791; Figure S7D), indicating that acute NUP93 acute depletion does not affect chromatin loops in a global manner. Differential loop analysis using DESeq2 (Love et al., 2014) revealed that 49 and 43 HICCUPS loops were quantitatively weakened or strengthened, respectively, after NUP93 depletion (Figures S7E and S7F). However, these altered HICCUPS loops were rarely anchored on transcriptionally deregulated promoters or enhancers (Figures S7E and S7F).
It is possible that transcriptionally deregulated regions after NUP93 depletion may display changes of chromatin contacts that cannot be called as loops in Hi-C data. We thus conducted additional HiChIP in NUP93-mAC cells with or without IAA treatment using two different histone marks, H3K27ac and H3K4me1, to maximize the possibility of detecting chromatin contacts. Focusing on NDGs and NDEs because they have a higher chance to form enhancer-promoter contacts (Figure 5G), we identified 123 and 147 chromatin loops formed between them by H3K27ac and H3K4me1 HiChIP, respectively. The overall strength of the NDE-NDG HiChIP loops did not display a difference in the APAs before or after NUP93 depletion (Figure 7A and 7B). HiChIP contact maps for the two example loci with strong NUP93 binding and transcriptional changes (MYC-MYCe and KITLG-KITLGe) are shown; they illustrated the largely unchanged chromatin loops (Figures 7C, 7D, and S7G). Our results indicate that NUP93 is largely dispensable for organizing the higher-order 3D chromatin architecture at the layers of A/B compartments, TADs, or enhancer-promoter contacts.
Figure 7. Unaltered enhancer-promoter contacts after NUP93 acute depletion and a model figure.
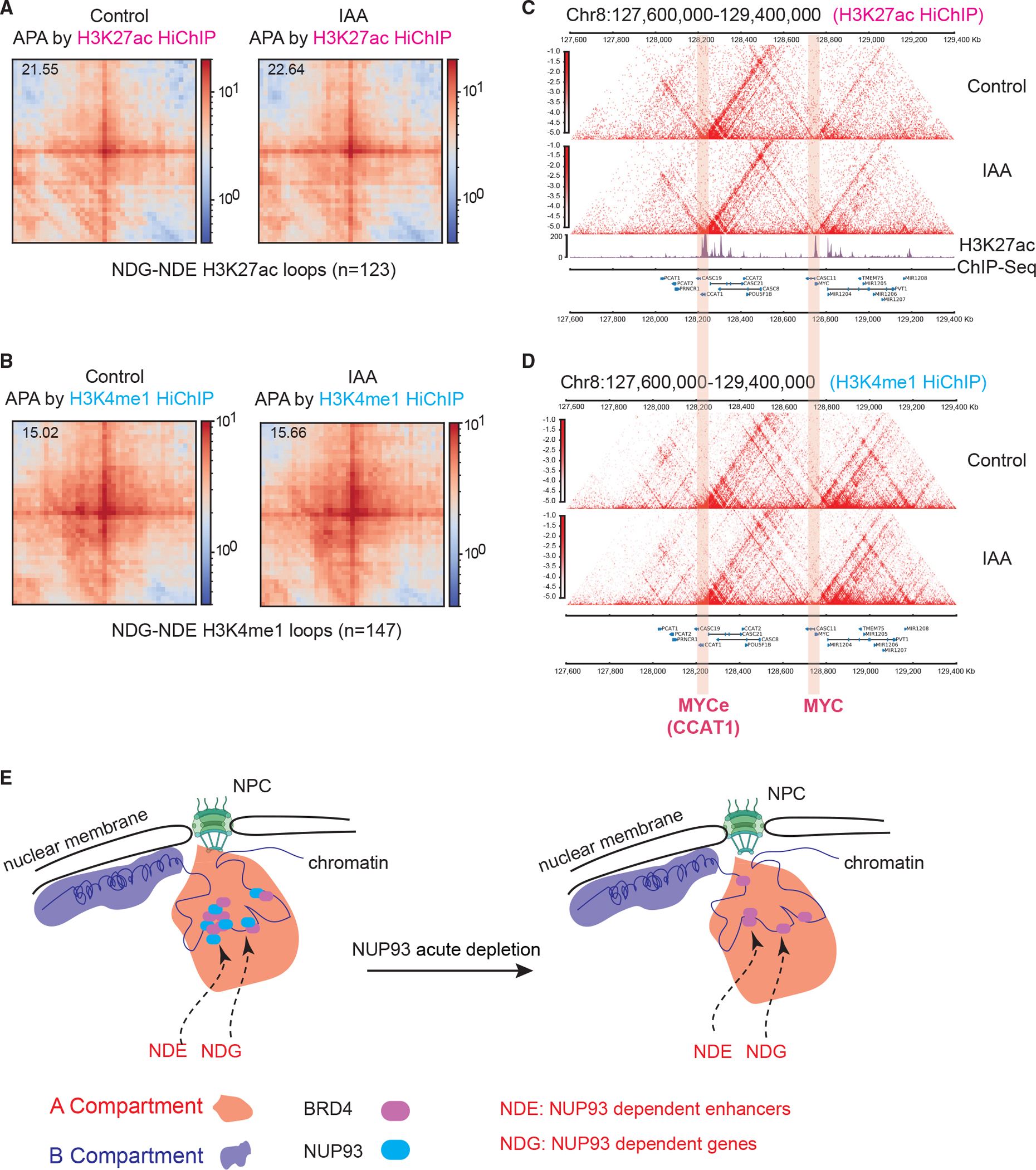
(A and B) Aggregate peak analyses (APAs) of HiChIP loops formed between NDE enhancers and NDG promoters using H3K27ac or H3K4me1 HiChIP data under control and 8-h IAA conditions (5-kb resolution). The top left corner values represent APA scores calculated with respect to the enrichment of the center pixel and neighborhood.
(C and D) HiChIP contact maps from an example region, showing overall unaltered chromatin loops under control and IAA (acute NUP93 depletion) conditions (5-kb resolution).
(E) A model summarizing the results of this study, which revealed the direct roles and nuclear locations of core NUPs, particularly NUP93, in transcription control in human cells. NDE, NUP93-dependent enhancer; NDG, NUP93-dependent gene.
DISCUSSION
The current study applied several advanced methods to study human NUPs in direct control of gene transcription and 3D genome organization. These include Cut&Run, the auxin degron system, PRO-seq, dCas9 chromatin tethering, Hi-C, and HiChIP. Many of these have not been applied previously to study NUPs. Our data provide a rigorous demonstration of a direct and specific role of core NUPs, particularly NUP93, in transcriptional regulation. We also revealed its dispensability for overall 3D chromatin organization. A model of our results is shown in Figure 7E.
Cut&Run detects core NUP association with active chromatin regions
Using Cut&Run, our study revealed, to the largest extent to our knowledge, the chromatin association of NUPs in human cells. Our results indicate that core NUPs preferentially associate with active chromatin regions in HCT116 or HeLa cells but rarely overlap heterochromatin or repressive regions. This is largely consistent with previous ChIP-on-ChIP or DamID data in human cells (Brown et al., 2008; Ibarra et al., 2016). Different from our results, Nup93 ChIP-seq in Drosophila S2 cells revealed its prominent association with heterochromatin and Polycomb regions (>50% of all peaks), with an ~30%–40% of peaks overlapping enhancers/promoters or active chromatin (Gozalo et al., 2020). Such differences may be attributed to distinctions between human and other species (Drosophila) or to cell types of distinct development/differentiation stages (HCT116 and HeLa are carcinoma cells and S2 cells are derived from Drosophila embryos, possibly a macrophage-like line; Rogers and Rogers, 2008). Technical differences between Cut&Run and ChIP-seq or DamID-based methods may also contribute to the differences (e.g., no crosslinking for Cut&Run, accessibility differences of specific genomic regions in native chromatin for Cut&Run versus fragmented chromatin for ChIP). Additional work is warranted to systemically map the chromatin association of NUPs (>30 members) in mammals and other species as well as in various cell lineages or development/differentiation stages. Cut&Run is a powerful method (Skene and Henikoff, 2017) to achieve this goal, with the potential to reach low cell numbers or single-cell resolution (Patty and Hainer, 2021).
Direct and selective roles of NUPs in gene transcriptional control
Binding of NUPs to specific chromatin regions suggested their roles in transcription control (Brown et al., 2008; Buchwalter et al., 2019; Gozalo et al., 2020; Ibarra et al., 2016; Jacinto et al., 2015; Kadota et al., 2020; Toda et al., 2017). However, as described earlier, the essentiality of NUPs and the technical limits of RNAi or genetic knockout prevented our full understanding of their direct roles. Here, the acute depletion system together with a nascent transcriptome assay (PRO-seq; Wissink et al., 2019) provided a definitive answer regarding the direct and causal role of NUPs in transcriptional control. This conclusion was confirmed by our dCas9 tethering approach.
Only selective members of NUPs are directly required for gene transcription. Among the two NUPs we used to made degron lines, NUP93, but not NUP35, plays a direct role in transcription despite both showing chromatin association at many enhancer/promoter regions. Selective functions of NUPs have been described before (Buchwalter et al., 2019; Capelson and Hetzer, 2009), which may in part be due to compensation/redundancy. At least for NUP93 and NUP35, their distinct transcriptional importance seems to correlate with their essentiality for human cells (Figure 1B) or yeast fitness (Kim et al., 2018). NUP93 selectively and causally activates some genes (i.e., NDGs). Although NSGs are increased upon acute NUP93 depletion, this does not seem to be achieved via a direct/causal action, as shown by dCas9 tethering. We speculate that the presence of NUP93 as well as the chromatin environment of the target genes together determine whether these genes are sensitive to its depletion. For example, NDGs are highly loaded with RNA Pol II and bear stronger elongation marks (Figure 4C and 4D), whereas NSGs have stronger promoters (e.g., H3K4me3; Figures S4C and S4E). It is plausible that highly transcribed genes like NDGs are more addicted to transcriptional resources and that NUP93 is required for enriching BRD4 to their enhancers (Figure 5A, 5C, and 5E). For NSGs, we speculate that NUP93 loss may have altered the distribution of critical transcriptional factors/cofactors in the nuclear environment, which can be passively diffused/recruited to their strong promoters (Figure S4E).
The exact mechanism by which NUP93 facilitates BRD4 recruitment to active enhancers will need to be studied further. Our data support the notion that strong enhancers require NUP93 to appropriately recruit a high level of BRD4. This is reminiscent of the properties of highly active enhancers to form liquid-like condensates, in which BRD4 is enriched (Sabari et al., 2018). BRD4 has a C-terminal disordered region, and many NUPs contain FG repeats that are early examples of intrinsically disordered domains (Denning et al., 2003). They have been documented to phase separate into liquid-like condensates (Ahn et al., 2021; Chandra et al., 2021; Hampoelz et al., 2019b; Sabari et al., 2018). We are tempted to speculate that NUP93 may augment BRD4 enrichment to strong enhancers via a process that involves phase separation and transcriptional condensate formation. This will be an important and interesting area to explore in the future.
The dispensability of core NUPs for 3D genome organization
The 3D genome can regulate and can also be regulated by transcriptional activity (van Steensel and Furlong, 2019). Rapid degradation is important to elucidate direct functions of architectural proteins in 3D genome control (Nora et al., 2017; Rowley and Corces, 2018). By in situ Hi-C and HiChIP after acute NUP93 depletion, we found limited changes in the 3D genome, from A/B compartments to TADs, or in enhancer-promoter interaction. This is somewhat surprising because it argues against a proposed critical role of NUPs in genome organization. NUPs represent one major category of architectural components in the nuclear periphery (Akhtar and Gasser, 2007; Buchwalter et al., 2019; Van Steensel and Belmont, 2017). Our results demonstrated that core NUP does not play direct roles in the overall 3D genome organization in human cells. These results reinforce the idea that the 3D genome is rather specifically controlled by a few factors, most prominently CTCF/cohesin, and is not particularly amenable to changes after rapid removal of transcriptional regulators (Rowley and Corces, 2018). Consistent with our data, the BET inhibitor (which inhibits BRD4 and other bromodomain proteins) strongly inhibited gene transcription but caused little change of enhancer-promoter contacts (Crump et al., 2021); similarly, rapid removal of mediator components elicited limited changes of chromatin interactions (Jaeger et al., 2020).
However, it is noteworthy that our data were based on global approaches (Hi-C or HiChIP), which do not exclude quantitative but sufficiently important changes for specific loci and/or in specific cell lineages/species. NUPs have been shown to contribute to the strength of locus-specific chromatin loops (Gozalo et al., 2020; Tan-wong et al., 2009). Long term NUP dysfunction (e.g., because of disease mutations) may still directly or indirectly deregulate the 3D genome, contributing to pathological gene deregulation.
Limitations of the study
The Cut&Run method is useful to study chromatin association of target proteins under native conditions without crosslinking and can potentially be applicable to extremely small cell numbers (Skene and Henikoff, 2017; Patty and Hainer, 2021). Technical differences between Cut&Run and ChIP-Seq or DamID-based methods may contribute to different chromatin binding landscapes of NUPs. The degron system is superior in depleting proteins acutely to study functions, but the mAID may mediate leaky degradation of tagged proteins to affect normal cellular functions even prior to IAA (Yesbolatova et al., 2020). Our results showed little if any leaky degradation of NUP93/NUP35 in our stable lines. We always compared the transcription and chromatin changes after IAA treatment using the same tagged cell line but did not compare NUP35- or NUP93-mAID with the parent HCT116 cells. Thus, even though there may be slight leaky degradation of NUP93, it does not affect our conclusions. Our results suggest that the role of NUP93 at enhancers is, at least in part, to license full transcriptional activation of highly RNA Pol II-loaded genes. However, these results are insufficient to conclude whether NUP93 may also participate in post-transcriptional regulation of enhancer-regulated genes, such as their mRNA transfer to the cytosol.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Wenbo Li, Ph.D., (Wenbo.li@uth.tmc.edu).
Materials availability
This study did not generate new unique reagents. All stable cells generated in this study are available from the lead contact with a completed and fully executed materials transfer agreement.
Data and code availability
All sequencing datasets generated in this paper are available at the Gene Expression Omnibus with accession number GEO: GSE165463.
This paper does not report original code.
Any additional information required to reanalyze the data presented in this work is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
HCT116 cells expressing CMV-OsTir1 in the AAVS1 locus were obtained from Kanemaki lab (Natsume et al., 2016), and were cultured in McCoy’s 5A medium (Corning, 10–050-CV) supplemented with 10% FBS at 37°C with 5% CO2. HeLa cells were gifts from Yubin Zhou lab from Texas A&M University Health Science Center in Houston, which was originally from ATCC. 293T cells were from ATCC.
Transfection and viral transduction
Transfection of plasmids into HCT116 cells was performed using Lipofectamine 2000 or 3000 (Life technologies) following manufacturer’s instructions. To generate lentivirus, 293T cells were transfected with the lentiviral transfer vector DNA, psPAX2 packaging and pMD2.G envelope plasmid DNA at a ratio of 4:3:1, respectively, by lipofectamine 2000. Lentivirus was collected 24 h after transfection. Cells were infected by the lentivirus expressing the dCas9 tethering constructs and gRNAs for 24 h and selected with appropriate antibiotics (e.g., 1 μg/mL puromycin, 10 μg/mL blasticidin or 100 μg/mL Zeocin) for 3–7 days and then were subjected to experiments.
Generation of degron cell lines
For generating AID (Auxin-inducible degron) cells, a parental cell line HCT116 expressing CMV-OsTir1 in AAVS1 locus was obtained from Kanemaki lab (Natsume et al., 2016). A sgRNA targeting the 3′-UTR near the stop codon of NUP93 ORF was cloned into expression vector pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene, #62988). Plasmid-based repair donors we generated with 800–1000bp homologous arms at each side of the gRNA cleavage site, together with the mAID, mClover, HA tags as well as selection mark genes neomycin or blasticidin, which were constructed into pJET1.2 cloning plasmid (Thermo Fisher Scientific, K1232). 600ng of each repair donor was co-transfected into the parental HCT116 cells with 1000ng sgRNA vector by using lipofectamine 2000. To increase the homologous recombination efficiency, 20uM RS-1 was added to treat the cells for two days 20 h post-transfection (Pinder et al., 2015). 24 or more single clones were picked after cells surviving selection with 5μg/mL blasticidin and 700ng/μL neomycin for a week. Homozygotes with mAID tagging were identified by PCR and western blots. Degradation of the mAID-tagged NUP93 was induced by adding 500uM indole-3-acetic acid (IAA, Sigma Aldrich, catalog#: 87,514) for various time points as indicated in the paper. For ChIP-Seq, Hi-C and PRO-Seq experiments, cells were mostly treated for 8 h with fresh medium containing 500uM IAA. Cells treated with ethanol only were used as controls (referred to Control).
METHOD DETAILS
RNA extraction and RT-qPCR
RNA extraction in this paper was mostly performed using Quick-RNA Miniprep Kit (Zymo Research, 11–328) or TRIzol (Thermo Fisher Scientific, 15,596–026) following manufacturer’s instructions. RT-qPCR experiments were carried out as previously described (Lee et al., 2021; Xiong et al., 2021). Reverse transcription was performed by using Superscript™ IV first strand synthesis kit (Thermo Fisher, 18091050), and the random hexamer primer was used. Real-time qPCR (RT-qPCR) was performed using SsoAdvanced™ Universal SYBR® Green Supermix (Bio-rad, 172–5274). Primer sequences are listed in Table S4. Primers for genes are often designed to target introns so they can better measure transcription rather than the transcript abundance of mRNAs.
Western blots
Cell nuclei were collected as previously described (Conrad and Ørom, 2017). Cells were resuspended in Igepal lysis buffer (10 mM Tris pH 7.4, 0.3 M NaCl, 7.5 mM MgCl2, 0.2 mM EDTA, 1% Igepal CA-630) followed by 5 min incubate on ice. Cell nuclei were collected by centrifuge at 1000 × g for 10 min. We then washed the pellet twice by Igepal lysis buffer to ensure purity. Isolated nuclei were washed three times with 1X PBS and lysed in NuPAGE LDS Sample Buffer (Thermo Fisher Scien, NP0007) with 0.1M DTT on ice for 10 min. Lysates were passed through a cell shredder (QIAGEN, 79656) by centrifugation for 4 min at 14,000 rpm and the filtrates were collected. Boiled proteins were separated on a 4%–15% SDS-PAGE gels (Bio-Rad, 4561096) and transferred onto 0.2 μm PVDF membranes (Bio-Rad, 1620177) using the Mini Trans-Blot Cell (Bio-Rad) at 100 V for 60 min (Bio-Rad). Transferred membranes were blocked with blocking buffer (5% milk, 3% BSA and 1% normal donkey serum in Tris-buffered saline containing 0.1% Tween 20 (TBST)) for 1 h and then incubated with primary antibodies at 4°C overnight. Blots were washed with TBST and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h. After washing four times with TBST, proteins were detected using Clarity™ Western ECL Substrate (Biorad, 1705060) in a Bio-Rad ChemiDoc™ Gel Imaging System.
Immunofluorescence microscopy
Fluorescence microscopy was performed on a Confocal Laser Microscope (Nikon A1R) equipped under a 100x oil objective. For the fixed cell images, all the cells were fixed in 4% paraformaldehyde for 10 min followed by quenching in 0.1M Tris-HCl (PH 7.4). 0.5% (v/v, in PBS) Triton X-100 was used to permeabilize cells for 30 min. After blocking in 3% (w/v) bovine serum albumin dissolved in PBST (PBS with 0.1% Tween 20) at room temperature for 1 h, cells were incubated with a 1:500 dilution of primary antibodies at 4°C overnight. After that, cells were washed four times for 5 min in TBST, and Alexa Fluor fluorescence conjugated secondary antibodies (Thermo Fisher Scientific) were used at 1:500 dilution in PBS for 60 min. Cells were washed three times in PBST as before and mounted. Confocal images were obtained on a Nikon microscope as described above. The fluorescence singling intensity of LaminA/C and Mab414 at the nuclear periphery was analyzed by ImageJ software (NIH). DAPI staining was used to create a nuclear rim according to the previous description (Miura, 2020). The mean intensity signal in the nuclear periphery was measured within the nuclear rim. More than 50 cells were analyzed from two independent experiments.
For the 5-EU treatment to cells and nascent RNA labeling experiments, we labeled NUP93-mAC cells (−/+ IAA for 8hrs) with 1 mM EU for 30min. Cells were fixed in a fixation buffer (125 mM PIPES, pH 6.8, 10 mM EGTA, 1 mM magnesium chloride, 0.2% Triton X-100, 3.7% formaldehyde) for 30 min at room temperature. For CLICK chemistry staining, the fixed cells were rinsed with TBS and stained for 30 min at room temperature with staining buffer (100 mM Tris, pH 8.5, 1mM CuSO4, 20 uM fluorescent azide, and 100 mM ascorbic acid (added last from a 0.5 M stock in water)). Cells were then washed several times with TBS with 0.5% Triton X-100 and were stained with DAPI before they were subjected to confocal microscopy imaging. The 5-EU intensity was analyzed by ImageJ software (NIH) using the regions of interest (ROI) manager. DAPI staining was used to create a nuclear mask for nuclear locations. The mean intensity of the 5-EU signal was measured within the nuclear mask. More than 50 cells were analyzed from two independent experiments.
Structured illumination microscopy (N-SIM)
NUP93-mAC cells were fixed by 4% paraformaldehyde for 10 min, similarly as for confocal microscopy. The cells were subjected to N-SIM using a Nikon N-SIM super-resolution microscope system, equipped with a CIF App TIRF 100X objective (NA 1.49, Nikon). The images were captured by an ORCA-Flash4.0 CMOS camera (Hamamatsu). Z-stacks were acquired every 0.2 μm over 10 μm and the resulting 3D-SIM z stack dataset of 15 images per plane were reconstructed using the stack reconstruction method in NIS-Elements (Nikon).
Cell cycle analysis by flow cytometry
Cells were fixed with 70% ethanol for 30 min at 4°C. The cells were then washed twice with cold PBS and stained with 2 μM Hoechst 33342 solution (H0342, Sigma-Aldrich, USA) at room temperature for 30 min. The Hoechst-stained cells were subjected to analyses by BD FACSAria™ II flow cytometry (BD Biosciences, USA) at 450 nm. Data were analyzed with FlowJo Version 10 software (BD Biosciences, USA).
Cut and run
Cut and Run was performed as described in EpiCypher® CUTANA® CUT&RUN protocol with minor modifications. Briefly, 0.5 million live HCT116 or HeLa cells were harvested and resuspended in 100ul wash buffer [20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, supplemented with Protease Inhibitor EDTA-Free tablet (Roche 11836170001)]. Activated Concanavalin A (EpiCypher 21–1401) was incubated with cells at room temperature for 10min to let the cells bind to the beads. For each target protein factor, 0.5μg antibody was added to each sample and incubated in the antibody buffer (wash buffer +0.01% Digitonin and 2 mM EDTA) at 4°C for 4–6hr. The beads were then washed twice with digitonin buffer [wash buffer +0.01% Digitonin], and 2.5 μL pAG-MNase (EpiCypher, 15–1116) was added to each sample. After ten minutes of incubation at room temperature, excessive pAG-MNase was washed out by a two-time digitonin buffer wash. Then targeted chromatin was digested and released from cells by 2 h of incubation with the presence of 2 mM CaCl2 at 4°C, which were collected from the supernatant, were subjected to phenol/chloroform DNA extraction, and finally to sequencing library construction using NEBNext® Ultra™ II DNA Library Prep Kit for Illumina (NEB, E7645L).
Chromatin immunoprecipitation (ChIP-Seq)
ChIP-Seq was performed as previously described (Lee et al., 2021) with minor modifications. HCT116 or HeLa cells growing in about 80–90% confluence from one 10cm dish were crosslinked by 10mL 1% formaldehyde in PBS for 10 min at room temperature. After quenching with 0.125M glycine, we collected cell pellets, and extracted the nuclei by incubating in buffer 1mL LB1 [50 mM HEPES-KOH (pH 7.5), 140 mM NaCl, 1 mM EDTA (pH 8.0), 10% (v/v) glycerol, 0.5% NP-40, 0.25% Triton X-100 and 1×cocktail protease inhibitor] at 4°C for 10 min, after pelted down the cells were resuspended in 1mL LB2 buffer [10 mM Tris-HCl (pH 8.0), 200 mM NaCl, 1 mM EDTA (pH 8.0), 0.5 mM EGTA (pH 8.0) and 1×cocktail protease inhibitor] and rotated at 4°C for 5 min. After centrifuge, cell nuclei were suspended in 350 μL buffer LB3 [10 mM Tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA (pH 8.0), 0.5 mM EGTA (pH 8.0), 0.1% Na-deoxycholate, 0.5% N-lauroyl sarcosine and 1×cocktail protease inhibitor], and fragmented using Q800R3 sonicator (QSONICA) at 4°C for 5 min (10 s on, 20 s off at 25% amplitude). After full speed centrifuge for 15mins at 4°C 100–150μL supernatant containing sheared chromatins were incubated with appropriate 2–3 μg antibodies at 4°C overnight. The next morning, the antibody-protein-chromatin complex was retrieved by incubating with 40μL Protein G Dynabeads (Thermo Fisher Scientific, 10004D) at 4°C for 4 h. Then the Protein G Dynabeads were washed with 1mL RIPA buffer [50mM HEPES pH 7.6, 1mM EDTA, 0.7% Na deoxycholate, 1% NP-40, 0.5M LiCL] 5mins at 4°C for 5 times. Immunoprecipitated DNA was de-crosslinked by 65°C heating overnight, treated by proteinase K, and were harvested by phenol chloroform or by Qiagen Quick DNA extraction kit. The DNAs were subjected to sequencing library construction using NEBNext® Ultra™ II DNA Library Prep Kit for Illumina (NEB, E7645L), following manufacturers’ instructions. We amplified final sequencing libraries by PCR for ~7–12 cycles and purified them by dual size selection using KAPApure beads (0.45X and 0.9X).
PRO-seq experiments
The run-on step of PRO-Seq largely follows a published protocol (Mahat et al., 2016). Briefly, about 10 million cells were washed three times with cold PBS, and then incubated in a hypotonic buffer (10mM Tris-Cl pH 7.5, 2 mM MgCl2, 3 mM CaCl2) for 10mins at 4°C. The cell membrane was lysed by adding 1mL of Lysis buffer (10mM Tris-Cl pH 7.5, 2 mM MgCl2, 3 mM CaCl2, 10% glycerol, 0.5% NP-40, 4U/mL SUPERase•In™ RNase Inhibitor, AM2694), and gentle pipetting using wide-bore tips or vortexing. The nuclei were then washed once more with lysis buffer and resuspended in 100μL freezing buffer (50 mM Tris-HCl pH8.0, 5 mM MgCl2, 0.1 mM EDTA, 40% glycerol, 50U/mL SUPERase•In™ RNase Inhibitor). The nuclei solution was then incubated in equal volume (100μL) of nuclear run-on buffer (10mM Tris-Cl pH 8.0, 5mM MgCl2, 300mM KCL, 1% sarkosyl, 1mM DTT, 500μM of ATP and GTP together with 50μM of biotin-11-CTP and biotin-11-UTP) at 37°C for 5minutes. Total RNAs from the nuclei were extracted immediately using TRIzol LS and were fragmented using 0.2M NaOH hydrolysis on ice for 10 min. After neutralization of the pH using equal volume of 1M Tris-HCL pH 6.8, the fragmented nuclear Run-on RNAs (NRO RNAs) were diluted to 600μL, and then the biotin-NRO RNAs were purified using the Dynabeads® MyOne™ Streptavidin C1 beads following the manufacturer instructions (Thermo Fisher 65002). The purified biotin-NRO RNAs were then extracted from the Streptavidin C1 using TRIzol, and were precipitated overnight. After this, the biotin-NRO RNAs were used for sequencing library making, for which we followed a very detailed protocol used in eCLIP-Seq (Van Nostrand et al., 2016). Briefly, the biotin-NRO RNAs were end-repaired by a fast Alkaline Phosphatase (Thermo Fisher) for and by T4 PNK (NEB) at 37°C for 30mins, cleaned up using Dynabeads MyOne Silane (37002D), and were then ligated to an HPLC purified 3′end RNA linker (synthesized by Integrated DNA Technology): RiL19/5phos/rArGrArUrCrGrGrArArGrArGrCrGrUrCrGrUrG/3SpC3/. The ligated RNA was reverse transcribed using AffinityScript first strand cDNA synthesis kit (Agilent) by using AR17 primer (5′- ACACGACGCTCTTCCGA-3′). The cDNA was then cleaned up and ligated with a DNA linker carrying 10 nucleotides unique molecular index (UMI), rand10–3Tr3 (/5Phos/NNNNNNNNNNAGATCGGAAGAGCACACGTCTG/3SpC3/). This cDNA with both ends carrying illumina adaptors was cleaned up by Dynabeads MyOne Silane (37002D), and PCR was conducted to test the library abundance. Final PCR primers for library amplification were based on PCR_F_D501: 5′- AATGATACGGCGACCACCGAGATCTACACTATAGCCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′ and one of the indexed 3′end primers PCR_Rev_p7x: 5’- CAAGCAGAAGACGGCATACGAGAT XXXXXX GTGACTGGAGTTCAGACGTGTGCTCTTCCGATC -3’ (where XXXXXX indicates 6-nucleotide index sequences based on standard Illumina TruSeq system), and the library was made using NEB Q5 2x PCR master mix by around 9–13 cycles. Final library was size-selected using TBE 10% PAGE gels with a range of ~170–400nt, or by dual size selection using KAPApure beads (0.55X and 0.9X).
In situ Hi-C and HiChIP
In situ Hi-C was performed as previously described with minor modifications (Rao et al., 2014). Briefly, ~5–10 million cells were cross-linked using 1% formaldehyde for 10 min at room temperature, quenched with 0.125M glycine for 10 min, and were washed with cold PBS. Cross-linked cells were resuspended in 1mL iced-cold Hi-C lysis buffer (10 mM Tris-HCl, pH 8.0; 10mM NaCl, 0.2% NP-40 and protease inhibitor cocktail), and rotated at 4°C for 30 min. After pelleting down the nuclei, 100 μL of 0.5% SDS was used to resuspend and permeabilize the nuclei at 62°C for 10 min. Then 260 μL H2O and 50 μL 10% Triton X-100 were added to quench the SDS at 37°C for 15 min. Subsequently, enzyme digestion of chromatin was performed at 37°C overnight by adding an additional 50 μL of 10X NEB buffer 2 and 400U MboI (NEB, R0147M). After overnight incubation, the MboI in solution was inactivated at 62°C for 20 min. To fill-in the DNA overhangs and add biotin, 35U DNA polymerase I (Klenow, NEB, M0210) together with 10 μL of 1mM biotin-dATP (Jeana Bioscience), 1μL 10mM dCTP/dGTP/dTTP were added and incubated at 37°C for 1 h with rotation. Blunt end Hi-C DNA ligation was performed using 5000 U NEB T4 DNA ligase with 10X NEB T4 Ligase buffer with 10mM ATP, 90 μL 10% Triton X-100, 2.2 μL 50 mg/mL BSA at room temperature for four hours with rotation. After ligation, nuclei were pelleted down and resuspended with 440 μL Hi-C nuclear lysis buffer (50mM Tris-HCl pH7.5; 10mM EDTA, 1% SDS and protease inhibitor cocktail), and further sheared using the parameter of 10/20 s ON/OFF cycle, 25% Amp, 5mins by a QSonica 800R sonicator. Hi-C will use 5–10% of the sonicated chromatin in the next section. For HiChIP, the sonicated chromatin was incubated with appropriate antibody (here by 4μg H3K27ac polyclonal antibody) overnight, and the immunocomplex was captured by dynabeads protein G in the next day. The immunocomplex was washed 2 times by low salt wash buffer (20 mM Tris-HCl pH 7.5, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl), 2 times by high salt wash buffer (20 mM Tris-HCl pH 7.5, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 500 mM NaCl), 1 time with Tris-EDTA buffer +0.1% triton-100, and one more time with TE only. The immunocomplex was subjected to overnight decrosslinking at 65°C, protein K treatment, and DNA extraction. For both Hi-C and HiChIP, after DNA extraction, they were used for biotin DNA enrichment by 20 μL Dynabeads MyOne Streptavidin C1 beads (Thermo Fisher 65,002). The biotin bound DNA on beads was used to perform on-beads library making with KAPA Hyper DNA library kit, or using NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB, E7645L).
RNA fluorescent in situ hybridization (RNA-FISH)
CCAT1 5′-end (MYC enhancer in HCT116 cells) FISH probe was designed and synthesized by Biosearch Technologies (https://www.biosearchtech.com/stellaris-designer) with Quasar 570 dye. RNA FISH in HCT116 cells was performed as previously described in Stellaris RNA FISH Protocol for Adherent Cells (Biosearch Technologies), with minor changes. Cells were fixed with 4% PFA for 10 min at room temperature and permeabilized with 70% ethanol overnight at 4°C. The images were taken using a Nikon A1R confocal microscope (Nikon) with 100X oil-immersion objective (and two laser beams (488 nm for DAPI, 560 nm for CCAT1 RNA FISH imaging). For quantification of the RNA FISH signals in specific nuclear areas, we divided the nucleus into three regions (see Figure 3E). The largest concentric circle represents the outline of the DAPI signal. We estimated the sizes of the areas in each ring to be equal by ensuring the diameters of each ring to have ratios of 1.732 vs. 1.414 v.s. 1.
CRISPR/dCas9 mediated tethering
A lentiviral construct (lenti-dCas9-NUP93–2HA-p2A-Hygro) that contains dCas9 and NUP93 fusion protein was generated by replacing the VP64 sequence in the lenti_dCAS9-VP64_Blast plasmid (Addgene, #61425) with the NUP93 protein coding sequence using PCR and gibson cloning. We then replaced the blasticidin selection marker in the backbone by hygromycin in order to select cells using hygromycin. This is needed because we aimed to express this construct in NUP93-mAID-mClover-2HA degron cell line, which already carries puromycin (for OsTir1), neomycin and blasticin (see Figure 2A, because both endogenous alleles of NUP93 coding regions were tagged with mAID-mClover). As a control for dCas9-NUP93 tethering, a tandem HA tagged dCas9 (dCas9–2HA) was generated at the same time. For gRNAs, they were designed by the RGEN designer tool (http://www.rgenome.net/cas-designer/). gRNAs were inserted into lenti sgRNA(MS2)_zeo backbone (Addgene, #61427) at the BsmBI restriction enzyme site. Stable cells expressing dCas9-NUP93_hygro in the NUP93-mAID-mClover-2HA paternal cells were generated by hygromycin selection for >7 days (600μg/mL hygromycin). Then, specific gRNA lentivirus was used to infect these cells individually. After zeocin drug selection for 1 week (200μg/mL), the stable cell lines with NUP93-mAID-mClover-2HA that also express dCas9-NUP93 (or dCas9–2HA) as well as specific gRNAs were treated with IAA for 8 h to degrade the endogenous NUP93-mAC. RNA samples were extracted by using Quick-RNA Miniprep Kit (Zymo Research, 11–328). RT-qPCR experiments were carried out as described earlier in the RNA extraction and RT-qPCR section.
QUANTIFICATION AND STATISTICAL ANALYSIS
Gene essentiality score
For gene dependency scores, we downloaded public genetic screening and dependency data from this website (https://depmap.org). We used the Genetic dependency results (CRISPR (Avana) Public 20Q4) for our analysis in Figure 1B, which was based on 789 human cell lines by CRISPR-Cas9 genetic perturbation. Gene essentiality score was calculated by CERES tool for CRISPR screening to computationally infer and subtract seed effects that arise for each perturbation. Here, value of −1 was used as the cutoff for cell-essential genes, based on previous work (Meyers et al., 2017).
PRO-seq analyses
Adaptor sequences and low-quality reads were trimmed using cutadapt (Martin, 2011), discarding those containing reads shorter than 20 nt (-m 20 -q 20). Remaining reads were aligned to the human genome (RefSeq) using STAR (Dobin et al., 2013). Only unique reads mapped to the hg19 genome were kept for further analysis. For quantifying transcriptional activity of genes, PRO-Seq read counts were calculated from the 300bp downstream of TSSs to the transcriptional end sites (TESs) by htseq-count (Anders et al., 2015). We identified differentially expressed genes or eRNAs by a |log2 (fold change)| greater than 0.58 (NUP93 acute depletion versus Control).
Pairwise correlation analyses
In Figure 1E, we performed genome wide correlation analysis of read coverages for ChIP-Seq and Cut&Run from architectural factors (SMC1, RAD21, CTCF, NIPBL), coactivators (BRD4, p300), NUPs (NUP93, NUP35 and NUP205), total or phosphorylated Pol2 (ser5p and ser2p), histone marks (H3K4me1, H3K27ac, H3K4me3, H3K79me2, H3K36me3, H3K27me3, H3K9me3, H4K20me3) in HCT116 cell line (datasets are listed in Tables S1 and S2). For this, multiBamSummary of the deepTools (Ramínez et al., 2014) package was used. To this end the genome was binned into 1 kb bins and reads mapping to each bin per sample were counted while reads mapping to the ENCODE blacklisted regions were excluded. The resulting table was analyzed with R, with the values (reads per bin) log10 transformed, and a Pearson correlation coefficient was calculated for each pairwise factor comparison. Finally, a correlation plot heatmap was generated with the smoothScatter function.
Cut&Run and ChIP-Seq analyses
Sequencing reads were mapped to hg19 human genome assembly (RefSeq) by using Bowtie2 (Langmead and Salzberg, 2012) with default parameters. Alignments were processed by SAMtools (Li et al., 2009) to remove low quality alignments (“-q 30″), PCR duplicates and mitochondrial reads. Reads that passed this filter were used to call peaks with MACS2 (Zhang et al., 2008). For visualization, each individual library was normalized to RPKM and then the track for each library was generated by bamCoverage. Peaks were annotated with genomic regions and nearby genes using ChIPpeakAnno (Zhu et al., 2010). Overlaps between different Cut&Run or ChIP-Seq were defined as peaks that overlap by at least 1bp. Often, heatmaps of ChIP-Seq signals for the ± 3kb regions centered around peaks were plotted using deepTools (Ramírez et al., 2014).
Genome-wide identification of eRNAs and enhancer transcriptional activity
Peak analysis of p300 and H3K27ac ChIP-Seq data was performed using MACS2 (Zhang et al., 2008). We selected p300 peaks overlapping H3K27ac peaks as active enhancers in HCT116, see Table S2 for public datasets). Next, we removed peaks overlapping GTF-annotated exons/UTRs, rRNAs, miRNAs and GENCODE annotated tRNAs by using the BEDtools (Quinlan and Hall, 2010) (these features (e.g. exons) were extended by 1kb on each side). This analysis resulted in 7,406 enhancer peaks that were examined for their transcriptional response to NUP93 acute depletion. To define enhancer RNA transcription in PRO-Seq, we centered a 2kb window at the midst of the p300 peaks; and for intronic enhancers, we excluded those eRNAs that have the same transcriptional direction with genes. Finally, 9,664 transcribed eRNAs remain for further analysis. We used htseq-count (Anders et al., 2015) to calculate RPKM across the catalog of eRNAs in PRO-Seq data before and after IAA treatment. We identified 1,586 differentially expressed eRNAs.
Chromatin-state analysis by ChromHMM
For chromatin-state analysis we used the ChromHMM algorithm, which has been previously described (Ernst and Kellis, 2012). The input data included ChIP-Seq for H3K4me1, H3K4me3, H3K27ac, H3K36me3, H3K27me3 and H3K9me3 in HCT116, which can be found in the Table S2. The parameter -binsize 200 was used (default) and 18 chromatin states were specified. Emissions parameters were visualized in R. We defined six states of segmentation in Figure S1D: repressive regions; active TSS; TSS flanking regions; transcribed regions; weak enhancers; and enhancers. Additional data used for chromatin state analysis and comparison include ChIP-Seq for NUP93, BRD4, Pol2, Ser2P and p300, or the Cut&Run for NUP93 can be found in Tables S1 and S2. In brief, raw fastq files were downloaded, replicates were combined when available, and adapters were trimmed using cutadapt. Alignment to hg19 was performed using Bowtie2 and duplicates were removed using samblaster (Faust and Hall, 2014). MACS2 was used to identify peak regions for ChIP-Seq and Cut&Run for HeLa cell, Chromatin state bed files (18-state) published by the Roadmap Epigenomics Consortium were downloaded from the following website: (https://egg2.wustl.edu/roadmap/data/byFileType/chromhmmSegmentations/ChmmModels/core_K27ac/jointModel/n18/reordered/) (Roadmap Epigenomics Consortium et al., 2015). The Genomic Association Tester (GAT) tool (Heger et al., 2013) was utilized to determine enrichment or depletion of NUP93 and other factors with respect to the different chromatin states. Enrichment at various chromatin states was then calculated for NUP93 and other factors in each individual state and the heatmap was created using the R ggplot2 function.
In situ Hi-C/HiChIP data processing
Hi-C and H3K27ac and H3K4me1 HiChIP raw data were primarily processed with HiC-Pro (Servant, et al., 2015). The paired-end reads were mapped to the hg19 human reference, and multi-mapped pairs, duplicated pairs, and other unvalid 3C pairs were filtered following the standard procedure of HiC-Pro (Servant, et al., 2015). All valid Hi-C pairs were converted to Juicebox format (Durand, et al. 2016, Abdennur and Mirny, 2019) or cooler format (Abdennur and Mirny, 2019,Abdennur and Mirny, 2019) for visualization and further analyses. Raw and valid read numbers of Hi-C and H3K27ac and H3K4me1 HiChIP are listed in Table S1. HiCRep (Yang et al., 2017) was performed to show the concordance analysis (stratum adjusted correlation coefficient (SCC)) between Hi-C replicates (Figure S6A). The P(s) curve was calculated as a function of contact probability (P) over genomic distances(s) (Figure 6A). Only cis interaction were used to calculate P(s) curve.
A/B compartment analyses
A/B compartments were identified by eigenvalue decomposition from 20kb or 100kb Hi-C contact matrices using cooltools (Abdennur and Mirny, 2019,Abdennur and Mirny, 2019). A/B compartmental scores were further corrected by GC and genes densities within each bin. Saddle plot analyses were performed to measure the inter-compartmentalization strength (A-A, A-B and B-B) at the genome-wide scale using cooltools compute-saddle. Briefly, we ranked the rows and the columns in the order of increasing compartmental scores within observed/expected contact maps, Then we aggregated the rows and the columns of the resulting matrix into 50 equally sized aggregate bins, and plotted the aggregated observed/expected Hi-C matrices as “saddle” plot. The shifts of A/B compartments between Control and NUP93 acute depletion were calculated by compartmental scores (E1) (at 100kb resolution) of replicates, largely referred to recent study (Yusufova et al., 2020). For compartmental scores (E1) of each 100kb bin, a student’s t-test was conducted between Control and IAA (NUP93 acute depletion). Only 100kb bin which show |Control E1 - IAA E1| > 0.2 and p-value < 0.05 was considered as shifting compartments. The cutoff of different categories of compartment shifts (in Figure S6C) were shown below: B to BwA: (IAA E1 - Control E1) ≥0.2, IAA E1< −0.1; B to A: (IAA E1 - Control E1) ≥0.2, IAA E1 >0.1 and Control E1 < −0.1; A to AwA: (IAA E1 - Control E1) ≥0.2, IAA E1 >0.1 and Control E1>0.1; A to AwB: (IAA E1 - Control E1) ≤ −0.2, IAA E1 >0.1 and Control E1>0.1; A to B: (IAA E1 - Control E1) ≤ −0.2, IAA E1 < −0.1 and Control E1>0.1; B to BwB: (IAA E1 - Control E1) ≤ −0.2, IAA E1 < −0.1 and Control E1< −0.1.
TADs and insulation scores and directionality index
The topologically associating domains (TADs) from a previous HCT116 Hi-C data (Rao et al., 2017) were obtained from (http://3dgenome.fsm.northwestern.edu/). The insulation scores of each 20kb bin were measured to quantify the TAD insulation as previously described (Crane et al., 2015). The directionality index, which quantifies the degree of upstream or downstream chromatin interaction bias at the boundaries of TADs was calculated by a Hidden Markov Model (Dixon et al., 2012).
Loop calling, enhancer-promoter contacts analyses
We used HICCUPS loop-calling tool (juicer_tools_1.22.01.jar hiccups –cpu -m 500 -r 5000, 10,000) to identify chromatin loops using public Hi-C dataset in HCT116 (Rao et al., 2017). For the H3K27ac and H3K4me1 HiChIP dataset, FitHiChIP (Bhattacharyya, et al., 2019) was performed to identify putative regulatory chromatin loops at 5kb resolution. The length of loops beyond 2 Mb or less 20 Kb were excluded for further analysis. The APA (Aggregate Peak Analysis) of Hi-C data was performed by superimposing observed/expected Hi-C matrices on HICCUPS loops (n = 3,791) with the coolpuppy tool (Flyamer et al., 2020). For Figures S7D, S7E, S7F, the 5/10kb anchors of HICCUPS loops are used to overlap with NDE, NSE enhancers or NDG, NSG promoters using BedTools (1bp overlap). For Figure 5G, any HiChIP loop with one anchor overlapping NDG/NSG (promoter regions) and with another anchor overlapping NDE/NSE, was considered as an observed loop connecting these two feature sites. We further randomly shuffled genes/enhancers from the list of total active genes/enhancers with numbers matching those of NDGs, NSGs, NDEs, NSEs, respectively. Then the number of HiChIP called loops with one anchor overlapping a shuffled NDG/NSG (promoter regions) and another anchor overlapping a shuffled NDE/NSE was counted as expected loop numbers. The shuffling (permutation) was performed for 1000 times, and we took the average expected loop numbers as final expected numbers to calculate folds of enrichment. p-values were calculated as the portion of the 1000 times that the permuted/expected loop number is greater than the observed loop number. We obtained the total numbers of HiChIP loops for H3K27ac or H3K4me1 after pooling data under control and IAA treatment. For Figure 7A and 7B, H3K27ac or H3K4me1 HiChIP loops (n = 123 or 147) with two anchors respectively overlapping a pair of NDE-NDG by Bedtools were used for the aggregated peak analysis following the above mentioned HiC Aggregate Peak Analysis.
General statistical analysis
For qPCR data, we analyzed them with Prism software and presented them as mean ± SD, which are indicated in figure legends. At least two biological replicates were conducted for all RT-qPCR, and one representative set of data with three technical replicates was shown in the figures. For imaging analyses, quantification was conducted by ImageJ and the cell numbers counted were indicated in figure legends. Student’s t-test (two-tailed) was used to compare means between two groups; p < 0.05 was considered significant, and we labeled the p values with asterisks in each figure panel (*, p < 0.05; **, p < 0.01; ***: p < 0.001). For boxplots, center lines represent medians; box limits indicate the 25th and 75th percentiles; and whiskers extend 1.5 times the interquartile range (IQR) from the 25th and 75th percentiles. Statistical analyses for epigenomics and transcriptome data were performed with Python or Perl or R scripts.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit Polyclonal BRD4 Antibody | Bethyl Laboratories | Cat#A301-985A100, RRID: AB_2620184 |
| Rabbit Polyclonal Anti-RNA polymerase II CTD repeat YSPTSPS (phospho S2) | Abcam | Cat#ab5095, RRID: AB_304749 |
| Rabbit Anti-Histone H3K27ac Antibody | Abcam | Cat# ab4729, RRID: AB_2118291 |
| Mouse Monoclonal Anti-HA antibody | Sigma-Aldrich | Cat# H9658, RRID: AB_260092 |
| Rabbit Polyclonal NUP93 Antibody | Bethyl Laboratories | Cat#A303-979A; RRID: AB_2620328 |
| pan Nuclear Pore Complex Proteins monoclonal antibody [mAb 414] | Abcam | Cat#ab24609, RRID:AB_448181 |
| Mouse Monoclonal LaminA/C Antibody | Millipore | Cat#MAB3211, RRID: AB_94752 |
| Rabbit Polyclonal NUP205 Antibody | Bethyl Laboratories | Cat#A303-935, RRID: AB_2620284 |
| Rabbit Polyclonal NUP35 Antibody | Bethyl Laboratories | Cat#A301-781A, RRID: AB_1211267 |
| Rabbit Polyclonal SEC13 Antibody | Bethyl Laboratories | Cat#A303-980A, RRID: AB_2620329 |
| Rabbit Polyclonal RPB1 Antibody | Cell Signaling Technology | Cat#14958S, RRID: AB_2687876 |
| Rabbit Polyclonal RPB2 Antibody | Genetex | Cat#GTX102535, RRID: AB_1951313 |
| GAPDH Antibody | Proteintech | Cat#60004-1-Ig, RRID: AB_2107436 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| 3-indole acetic acid (IAA) | Sigma Aldrich | I3750-5G-A |
| Lipofectamine 2000 | Thermo Fisher | 11668019 |
| Lipofectamine 3000 | Thermo Fisher | L3000150 |
| RS-1 | Sigma | R9782 |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad Laboratories | 172-5274 |
| cOmplete™, Mini Protease Inhibitor Cocktail | Roche | 11836153001 |
| TRIzol™ reagent | Thermo Fisher | 15596018 |
| TRIzol™ LS reagent | Thermo Fisher | 10296028 |
| Biotin-11-UTP | Jena Bioscience | NU-821-BIOX |
| Biotin-11-CTP | Jena Bioscience | NU-831-BIOX |
| Dynabeads™ MyOne™ Streptavidin C1 | Thermo Fisher | 65002 |
| Protein G Dynabeads™ | Thermo Fisher | 10004D |
| SUPERase In™ RNase Inhibitor | Thermo Fisher | AM2694 |
| Flavopiridol | Sigma | F3055 |
| slip-embedded 35mm dish | MatTek | P35G-1.5-10-C |
| Dynabeads™ Protein G | Thermo Fisher | 10004D |
| Proteinase K | Thermo Fisher | 25530049 |
| RNase A | Thermo Scientific | EN0531 |
| Phenol:Chloroform:Iso-amyl alcohol (125:24:1), pH 4.5 | Thermo Fisher | AM9722 |
| KAPA pure beads | Roche/KAPA | KK8001 |
| Cyanine 3-UTP | Enzo Life Science | ENZ-42505 |
| GlycoBlue™ Coprecipitant | Thermo Fisher | Thermo Fisher |
| Concanavalin A (ConA) Magnetic Beads | EpiCypher | 21-1401 |
| pAG-MNase | EpiCypher | 15-1016 |
| Glycogen | Sigma-Aldrich | Roche 10930193001 |
| Digitonin | Millipore Sigma | 300410 |
| Pure Spermidine (6.4M) | Sigma-Aldrich | S0266 |
| 5-Ethynyl-uridine (5-EU) | Jena Bioscience | CLK-N002-10 |
| MboI | New England Biolabs | R0147M |
| DNA polymerase I | New England Biolabs | M0210 |
| Biotin-14-dATP | Jena Bioscience | NU-835-BIO14-S |
| T4 DNA ligase | New England Biolabs | M0202M |
| ERCC spike in | Thermo Fisher | 4456740 |
| RIPA Lysis and Extraction Buffer | Thermo Fisher | 89901 |
| 2x Laemmli Sample Buffer | Bio-rad | 1610737 |
|
| ||
| Critical commercial assays | ||
|
| ||
| NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® | New England Biolabs | E7760 |
| NEBNext rRNA Depletion Kit | New England Biolabs | E6301 |
| KAPA HyperPrep NGS library kit | Roche | KK8504 |
| NEB Ultra II DNA library kit | New England Biolabs | E7645 |
| 4–20% Mini-PROTEAN® TGX™ Precast Gels | Bio-rad | 4561094 |
| Trans-Blot® Turbo™ Mini PVDF Transfer Packs | Bio-rad | 1704156 |
| MEGAscript™ T7 Transcription Kit | Invitrogen | AM1334 |
| Quick-RNA MiniPrep Kit | Zymo Research | 11-328 |
| Quick-DNA Miniprep Kit | Zymo Research | 11-317C |
| Superscript™ IV First-Strand cDNA Synthesis System | Thermo fisher | 18091050 |
| Pierce™ BCA Protein Assay Kit | Thermo Scientific | 23225 |
| NuPAGE 10% Bis-Tris gel | Life Technologies | WG1201BX1 |
|
| ||
| Deposited data | ||
|
| ||
| Cut&Run, PRO-seq, ChIP-seq, Hi-C and HiChIP data | This Paper | GEO: GSE165463 |
| Published ChIP-seq and Hi-C data | Gene Expression Omnibus (GEO) | See Table S2 for details |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HeLa | ATCC | ATCC CCL-2 |
| 293T | ATCC | ATCC CRL-3216 |
| HCT116, NUP93-mAID-mClover-2HA | Generated in this study | N/A |
| HCT116, NUP35-mAID-mClover-2HA | Generated in this study | N/A |
| HCT116 expressing CMV-OsTIR1 in AAVS1 locus | Laboratory of Masato Kanemaki | (Natsume, et al., 2016). |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers | See Table S4 | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| lenti dCAS-VP64_Blast | (Konermann et al., 2015) | Addgene #61425 |
| lenti sgRNA(MS2)_zeo backbone | (Konermann et al., 2015) | Addgene #61427 |
| pMK286 (mAID-NeoR) | (Natsume, et al., 2016) | Addgene #72824 |
| pMK287 (mAID-Hygro) | (Natsume, et al., 2016) | Addgene #72825 |
| pMK288 (mAID-Bsr) | (Natsume, et al., 2016) | Addgene #72826 |
|
| ||
| Software and algorithms | ||
|
| ||
| BioRender (for some diagram generation) | BioRender | https://biorender.com/ |
| CRISPR RGEN Tools | (Park et al., 2015) | |
| STAR | v2.7.0 | (Dobin et al., 2013) |
| HiC-Pro | v2.8.10 | (Servant et al., 2015) |
| bcl2fastq | v2.2.0 | https://github.com/brwnj/bcl2fastq |
| Bedtools | v2.1.0 | (Quinlan and Hall, 2010) |
| Samtools | v1.9 | (Li et al., 2009) |
| Bowtie2 | v2.2.9 | (Langmead and Salzberg, 2012) |
| Deeptools | v3.5.0 | (Ramírez et al., 2014) |
| Cutadapt | v3.1 | (Martin, 2011) |
| Picard | v2.24.0 | https://github.com/broadinstitute/picard |
| HTSeq | v0.13.5 | (Anders et al.., 2015) |
| MACS2 | v2.2.7 | (Zhang et al.., 2008) |
| ChromHMM | (Ernst and Kellis, 2012) | |
| GAT | v1.0 | (Heger et al., 2013) |
| FitHiChIP | v1.0.0 | (Bhattacharyya et al., 2019) |
| IGV | v2.8.13 | (Robinson et al., 2011) |
| Fiji ImageJ | v2.0.0-rc-69/1.52n | (Schindelin et al., 2012) |
| FlowJo | v10 | (BD Biosciences, USA) |
Highlights.
Cut&Run reveals chromatin association of core NUPs at active enhancers and promoters
Acute depletion of NUPs uncovers direct roles of NUP93 in gene transcriptional control
NUP93 acts as an activator for specific enhancers adjacent to the nuclear periphery
Acute depletion shows that NUP93 is largely dispensable for 3D genome organization
ACKNOWLEDGMENTS
This work is supported by funding from the University of Texas McGovern Medical School, the NIH 4D Nucleome program (U01HL156059), the NCI (K22CA204468), NIGMS (R21GM132778 and R01GM136922), CPRIT (RR160083 and RP180734), the Welch Foundation (AU-2000–20220331), the John S. Dunn Foundation (to W.L.), and the NHGRI (R01HG011633) and NCI (R01CA262623) (to L.H.). J.H.L. and R.W. were partially supported by a UTHealth Innovation for Cancer Prevention Research training program post-doctoral fellowship (RP210042). R.W. received support from a John and Rebekah Harper fellowship. Most of our next-generation sequencing work was conducted with the UTHealth Cancer Genomics Core, which received funding from CPRIT (RP180734). Fluorescence microscopy was performed at the UTHealth Center for Advanced Microscopy, which received support from NIH/NHLBI K01HL143111 (to T.I.M.). We thank the UTHealth Flow Cytometry Service Center for support. Some of the genome sequencing datasets described in this work were derived from the HeLa cell line, which was established from Henrietta Lacks’ tumor cells without her knowledge or consent in 1951. We are grateful to Henrietta Lacks and her family for their contributions to biomedical research. We acknowledge the important contribution of Dr. Masato Kanemaki for making and sharing the HCT116 OsTir1 cell line and degron system.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111576.
REFERENCES
- Abdennur N, and Mirny LA (2019). Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36, 311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn JH, Davis ES, Daugird TA, Zhao S, Quiroga IY, Uryu H, Li J, Storey AJ, Tsai Y-H, Keeley DP, et al. (2021). Phase separation drives aberrant chromatin looping and cancer development. Nature 595, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar A, and Gasser SM (2007). The nuclear envelope and transcriptional control. Nat. Rev. Genet. 8, 507–517. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Chandra V, Vijayanand P, and Ay F (2019). Identification of significant chromatin contacts from HiChIP data by FitHiChIP. Nat. Commun. 10, 4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel G (1985). Gene gating: a hypothesis. Proc. Natl. Acad. Sci. USA 82, 8527–8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Kennedy CJ, Delmar VA, Forbes DJ, and Silver PA (2008). Global histone acetylation induces functional genomic reorganization at mammalian nuclear pore complexes. Genes Dev. 22, 627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwalter A, Kaneshiro JM, and Hetzer MW (2019). Coaching from the sidelines: the nuclear periphery in genome regulation. Nat. Rev. Genet. 20, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capelson M, and Hetzer MW (2009). The role of nuclear pores in gene regulation, development and disease. EMBO Rep. 10, 697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capelson M, Liang Y, Schulte R, Mair W, Wagner U, and Hetzer MW (2010). Chromatin-bound nuclear pore components regulate gene expression in higher eukaryotes. Cell 140, 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casolari JM, Brown CR, Komili S, West J, Hieronymus H, and Silver PA (2004). Genome-wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell 117, 427–439. [DOI] [PubMed] [Google Scholar]
- Chandra B, Michmerhuizen NL, Shirnekhi HK, Tripathi S, Pioso BJ, Baggett DW, Mitrea DM, Iacobucci I, White MR, and Chen J (2021). Phase Separation Mediates NUP98 Fusion Oncoprotein Leukemic Transformation (Cancer discovery). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad T, and Ørom UA (2017). Cellular fractionation and isolation of chromatin-associated RNA. In Enhancer RNAs (Springer; ), pp. 1–9. [DOI] [PubMed] [Google Scholar]
- Coyne AN, and Rothstein JD (2022). Nuclear pore complexes—a doorway to neural injury in neurodegeneration. Nat. Rev. Neurol. 18, 348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, and Meyer BJ (2015). Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump NT, Ballabio E, Godfrey L, Thorne R, Repapi E, Kerry J, Tapia M, Hua P, Lagerholm C, Filippakopoulos P, et al. (2021). BET inhibition disrupts transcription but retains enhancer-promoter contact. Nat. Commun. 12, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis LI, and Blobel G (1987). Nuclear pore complex contains a family of glycoproteins that includes p62: glycosylation through a previously unidentified cellular pathway. Proc. Natl. Acad. Sci. USA 84, 7552–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Marti-Renom MA, and Mirny LA (2013). Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat. Rev. Genet. 14, 390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning DP, Patel SS, Uversky V, Fink AL, and Rexach M (2003). Disorder in the nuclear pore complex: the FG repeat regions of nucleoporins are natively unfolded. Proc. Natl. Acad. Sci. USA 100, 2450–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, and Aiden EL (2016). Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2012). ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Yu M, Li G, Chee S, Liu T, Schmitt AD, and Ren B (2016). Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res. 26, 1345–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust GG, and Hall IM (2014). SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505. 10.1016/j.celrep.2022.111576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyamer IM, Illingworth RS, and Bickmore WA (2020). Coolpup.py: versatile pile-up analysis of Hi-C data. Bioinformatics 36, 2980–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL, et al. (2020). A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozalo A, Duke A, Lan Y, Pascual-Garcia P, Talamas JA, Nguyen SC, Shah PP, Jain R, Joyce EF, and Capelson M (2020). Core components of the nuclear pore bind distinct states of chromatin and contribute to polycomb repression. Mol. Cell 77, 67–81.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampoelz B, Andres-Pons A, Kastritis P, and Beck M (2019a). Structure and assembly of the nuclear pore complex. Annu. Rev. Biophys. 48, 515–536. [DOI] [PubMed] [Google Scholar]
- Hampoelz B, Schwarz A, Ronchi P, Bragulat-Teixidor H, Tischer C, Gaspar I, Ephrussi A, Schwab Y, and Beck M (2019b). Nuclear pores assemble from nucleoporin condensates during oogenesis. Cell 179, 671–686.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heger A, Webber C, Goodson M, Ponting CP, and Lunter G (2013). GAT: a simulation framework for testing the association of genomic intervals. Bioinformatics 29, 2046–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra A, Benner C, Tyagi S, Cool J, and Hetzer MW (2016). Nucleoporin-mediated regulation of cell identity genes. Genes Dev. 30, 2253–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias N, Paulo JA, Tatarakis A, Wang X, Edwards AL, Bhanu NV, Garcia BA, Haas W, Gygi SP, and Moazed D (2020). Native chromatin proteomics reveals a role for specific nucleoporins in heterochromatin organization and maintenance. Mol. Cell 77, 51–66.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto FV, Benner C, and Hetzer MW (2015). The nucleoporin Nup153 regulates embryonic stem cell pluripotency through gene silencing. Genes Dev. 29, 1224–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger MG, Schwalb B, Mackowiak SD, Velychko T, Hanzl A, Imrichova H, Brand M, Agerer B, Chorn S, Nabet B, et al. (2020). Selective Mediator dependence of cell-type-specifying transcription. Nat. Genet. 52, 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadota S, Ou J, Shi Y, Lee JT, Sun J, and Yildirim E (2020). Nucleoporin 153 links nuclear pore complex to chromatin architecture by mediating CTCF and cohesin binding. Nat. Commun. 11, 2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalverda B, Pickersgill H, Shloma VV, and Fornerod M (2010). Nucleoporins directly stimulate expression of developmental and cell-cycle genes inside the nucleoplasm. Cell 140, 360–371. [DOI] [PubMed] [Google Scholar]
- Kempfer R, and Pombo A (2020). Methods for mapping 3D chromosome architecture. Nat. Rev. Genet. 21, 207–226. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Fernandez-Martinez J, Nudelman I, Shi Y, Zhang W, Raveh B, Herricks T, Slaughter BD, Hogan JA, Upla P, et al. (2018). Integrative structure and functional anatomy of a nuclear pore complex. Nature 555, 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-H, Wang R, Xiong F, Krakowiak J, Liao Z, Nguyen PT, Moroz-Omori EV, Shao J, Zhu X, Bolt MJ, et al. (2021). Enhancer RNA m6A methylation facilitates transcriptional condensate formation and gene activation. Mol. Cell 81, 3368–3385.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, and Rosenfeld MG (2016). Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat. Rev. Genet. 17, 207–223. [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E, Van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DH, and Hoelz A (2019). The structure of the nuclear pore complex (an update). Annu. Rev. Biochem. 88, 725–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Ma Q, Wong K, Li W, Ohgi K, Zhang J, Aggarwal A, and Rosenfeld MG (2013). Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 155, 1581–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc. 11, 1455–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12. [Google Scholar]
- McCleland ML, Mesh K, Lorenzana E, Chopra VS, Segal E, Watanabe C, Haley B, Mayba O, Yaylaoglu M, Gnad F, and Firestein R (2016). CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J. Clin. Invest. 126, 639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura K (2020). Measurements of intensity dynamics at the periphery of the nucleus. In Bioimage Data Analysis Workflows (Springer; ), pp. 9–32. [Google Scholar]
- Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, and Chang HY (2016). HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Kiyomitsu T, Saga Y, and Kanemaki MT (2016). Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep. 15, 210–218. [DOI] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Bae S, and Kim J-S (2015). Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics 31, 4014–4016. [DOI] [PubMed] [Google Scholar]
- Pascual-Garcia P, Debo B, Aleman JR, Talamas JA, Lan Y, Nguyen NH, Won KJ, and Capelson M (2017). Metazoan nuclear pores provide a scaffold for poised genes and mediate induced enhancer-promoter contacts. Mol. Cell 66, 63–76.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patty BJ, and Hainer SJ (2021). Transcription factor chromatin profiling genome-wide using uliCUT&RUN in single cells and individual blastocysts. Nat. Protoc. 16, 2633–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinder J, Salsman J, and Dellaire G (2015). Nuclear domain ‘knock-in’ screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 43, 9379–9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabut G, Doye V, and Ellenberg J (2004). Mapping the dynamic organization of the nuclear pore complex inside single living cells. Nat. Cell Biol. 6, 1114–1121. [DOI] [PubMed] [Google Scholar]
- Raices M, and D’Angelo MA (2017). Nuclear pore complexes and regulation of gene expression. Curr. Opin. Cell Biol. 46, 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T (2014). deep-Tools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huang S-C, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon K-R, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. (2017). Cohesin loss eliminates all loop domains. Cell 171, 305–320.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium; Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SL, and Rogers GC (2008). Culture of Drosophila S2 cells and their use for RNAi-mediated loss-of-function studies and immunofluorescence microscopy. Nat. Protoc. 3, 606–611. [DOI] [PubMed] [Google Scholar]
- Rowley MJ, and Corces VG (2018). Organizational principles of 3D genome architecture. Nat. Rev. Genet. 19, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V, and Lauberth SM (2020). Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol. 27, 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, and Barillot E (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, e21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner MC, Brickner J (2022). The Nuclear Pore Complex as a Transcription Regulator. Cold Spring Harb Perspect Biol. 14 (1): a039438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan-Wong SM, Wijayatilake HD, and Proudfoot NJ (2009). Gene loops function to maintain transcriptional memory through interaction with the nuclear pore complex. Genes Dev. 23, 2610–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda T, Hsu JY, Linker SB, Hu L, Schafer ST, Mertens J, Jacinto FV, Hetzer MW, and Gage FH (2017). Nup153 interacts with Sox2 to enable bimodal gene regulation and maintenance of neural progenitor cells. Cell Stem Cell 21, 618–634.e7. [DOI] [PubMed] [Google Scholar]
- Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR 3rd, and Hetzer MW (2013). Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 154, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Steensel B, and Belmont AS (2017). Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Furlong EEM (2019). The role of transcription in shaping the spatial organization of the genome. Nat. Rev. Mol. Cell Biol. 20, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vian L, Pę kowska A, Rao SSP, Kieffer-Kwon KR, Jung S, Baranello L, Huang SC, El Khattabi L, Dose M, Pruett N, et al. (2018). The energetics and physiological impact of cohesin extrusion. Cell 175, 292–294.e1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, et al. (2017). BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol. Cell 67, 5–18.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissink EM, Vihervaara A, Tippens ND, and Lis JT (2019). Nascent RNA analyses: tracking transcription and its regulation. Nat. Rev. Genet. 20, 705–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang JF, Yin QF, Chen T, Zhang Y, Zhang XO, Wu Z, Zhang S, Wang HB, Ge J, Lu X, et al. (2014). Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 24, 513–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong F, Wang R, Lee J-H, Li S, Chen S-F, Liao Z, Hasani LA, Nguyen PT, Zhu X, Krakowiak J, et al. (2021). RNA m6A modification orchestrates a LINE-1–host interaction that facilitates retrotransposition and contributes to long gene vulnerability. Cell Res. 31, 861–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Zhang F, Yardımcı GG, Song F, Hardison RC, Noble WS, Yue F, and Li Q (2017). HiCRep: assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res. 27, 1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Yik JHN, Chen R, He N, Jang MK, Ozato K, and Zhou Q (2005). Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 19, 535–545. [DOI] [PubMed] [Google Scholar]
- Yesbolatova A, Saito Y, Kitamoto N, Makino-Itou H, Ajima R, Nakano R, Nakaoka H, Fukui K, Gamo K, Tominari Y, et al. (2020). The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat. Commun. 11, 5701–5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, and Ren B (2017). The three-dimensional organization of mammalian genomes. Annu. Rev. Cell Dev. Biol. 33, 265–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufova N, Kloetgen A, Teater M, Osunsade A, Camarillo JM, Chin CR, Doane AS, Venters BJ, Portillo-Ledesma S, Conway J, et al. (2020). Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature 589, 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ, Gazin C, Lawson ND, Pagès H, Lin SM, Lapointe DS, and Green MR (2010). ChIPpeakAnno: a bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinf. 11, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing datasets generated in this paper are available at the Gene Expression Omnibus with accession number GEO: GSE165463.
This paper does not report original code.
Any additional information required to reanalyze the data presented in this work is available from the lead contact upon request.