

Abstract
Background
Skeletal muscle atrophy can occur in response to numerous factors, such as ageing and certain medications, and produces a major socio‐economic burden. At present, there are no approved drugs for treating skeletal muscle atrophy. Arachidonate 5‐lipoxygenase (Alox5) is a drug target for a number of diseases. However, pharmacological targeting of Alox5, and its role in skeletal muscle atrophy, is unclear.
Methods
The potential effects of gene knockdown and pharmacological targeting of Alox5 on skeletal muscle atrophy were investigated using cell‐based models, animal models and human skeletal muscle primary cells. Malotilate, a clinically safe drug developed for enhancing liver regeneration and Alox5 inhibitor, was investigated as a repurposing candidate. Mechanism(s) of action in skeletal muscle atrophy was assessed by measuring the expression level or activation status of key regulatory pathways and validated using gene knockdown and RNA sequencing.
Results
Myotubes treated with the atrophy‐inducing glucocorticoid, dexamethasone, were protected from catabolic responses by treatment with malotilate (+41.29%, P < 0.01). Similar anti‐atrophy effects were achieved by gene knockdown of Alox5 (+30.4%, P < 0.05). Malotilate produced anti‐atrophy effects without affecting the myogenic differentiation programme. In an in vivo model of skeletal muscle atrophy, malotilate treatment preserved muscle force/strength (grip strength: +35.72%, latency to fall: +553.1%, P < 0.05), increased mass and fibre cross‐sectional area (quadriceps: +23.72%, soleus: +33.3%, P < 0.01) and down‐regulated atrogene expression (Atrogin‐1: −61.58%, Murf‐1: ‐66.06%, P < 0.01). Similar, beneficial effects of malotilate treatment were observed in an ageing muscle model, which also showed the preservation of fast‐twitch fibres (Type 2a: +56.48%, Type 2b: +37.32%, P < 0.01). Leukotriene B4, a product of Alox5 activity with inflammatory and catabolic functions, was found to be elevated in skeletal muscle undergoing atrophy (quadriceps: +224.4%, P < 0.001). Cellular transcriptome analysis showed that targeting Alox5 up‐regulated biological processes regulating organogenesis and increased the expression of insulin‐like growth factor‐1, a key anti‐atrophy hormone (+226.5%, P < 0.05). Interestingly, these effects were restricted to the atrophy condition and not observed in normal skeletal muscle cultures with Alox5 inhibition. Human myotubes were also protected from atrophy by pharmacological targeting of Alox5 (+23.68%, P < 0.05).
Conclusions
These results shed new light on novel drug targets and mechanisms underpinning skeletal muscle atrophy. Alox5 is a regulator and drug target for muscle atrophy, and malotilate is an attractive compound for repurposing studies to treat this disease.
Keywords: Skeletal muscle atrophy, Malotilate, Alox5, Drug repurposing, Glucocorticoids, Sarcopenia
Introduction
Skeletal muscle atrophy causes muscle weakness that can progress to disability. It may be produced by a number of different factors, such as ageing, certain medications, degenerative diseases or lifestyle factors. 1 Due to the significant economic impact produced by skeletal muscle atrophy and demographic ageing in industrialized countries, there is a need to develop novel therapeutic options for treating this disorder. 2
Lipoxygenases are a family of non‐haem, iron‐containing enzymes that catalyse the dioxygenation of polyunsaturated fatty acids in lipids possessing a cis,cis‐1,4‐pentadiene hydrocarbon into molecules with diverse autocrine, paracrine and endocrine signalling functions. 3 Arachidonate 5‐lipoxygenase (Alox5) is one of the most widely studied lipoxygenases. Consequently, Alox5 is under investigation as a drug target for numerous diseases. 4 However, the role of Alox5 in skeletal muscle atrophy is unclear. Endoplasmic reticulum (ER) stress was alleviated in myotubes after Alox5 inhibition and improved insulin resistance in a model of type 2 diabetes. 5 In contrast, genetic ablation of Alox5 failed to protect against muscle atrophy caused by denervation, whereas ablation of Alox12 or Alox15 was effective. 6 In addition, a recent study indicated that Alox5 gene expression was decreased, rather than up‐regulated, in a model of ageing‐induced sarcopenia. 7
Drug repurposing has significant advantages compared with traditional drug discovery approaches, because the repositioned compound is already characterized in other disease context(s) and clinical trials. 8 Malotilate (1,3‐dithiol‐2‐ylidenepropanedioic acid bis(1‐methylethyl) ester) is a drug developed for treating liver disease. 9 It was shown to accelerate liver regeneration in rats. 10 Oral delivery in patients with advanced cirrhosis disease showed that malotilate was well tolerated with anti‐fibrotic and hepatoprotective properties, and dry skin was the only possible adverse effect. 11 However, the first‐pass elimination of malotilate was found to be dramatically reduced in cirrhotics, and a smaller amount of drug reached the liver in these patients. 12 Malotilate is included in the FDA‐approved Drug Library (Selleck Chemicals, TX, USA) and is a specific inhibitor of Alox5, which regulates inflammatory responses. 13 In stimulated human peritoneal macrophages, HPLC analysis showed that malotilate concentration dependently decreased the exogenous and endogenous formation of the Alox5 products, 5‐monohydroxy‐eicosatetraenoic acid (5‐HETE) and leukotriene B4 (LTB4) and their metabolites, without decreasing the Alox12 and Alox15 products, 12‐HETE and 15‐HETE, which differed from the properties of known lipoxygenase inhibitors. 13 In light of the therapeutic effects of malotilate treatment in studies of liver degeneration, inflammation and fibrosis, the potential of repurposing this drug for skeletal muscle atrophy was investigated by utilizing myotube‐based models of skeletal muscle atrophy, animal models of muscle atrophy and ageing, and human myotubes induced to undergo atrophy. The mechanism of action of Alox5 inhibition was assessed by measuring the expression level or activation status of major atrophy pathways and validated using gene knockdown and cellular transcriptome analysis.
Materials and methods
Reagents
Dexamethasone (Dex) was purchased from Santa Cruz Biotechnology, TX, USA. Malotilate and zileuton was purchased from Selleckchem, TX, USA. Leukotriene B4 (LTB4) was purchased from Tocris, Bristol, UK.
Cell culture
C2C12 murine skeletal muscle myoblasts were purchased from Koram Biotech. Corp, Republic of Korea, and cultured with growth media (GM), consisting of Dulbecco's Modified Eagle's Medium (DMEM), 10% foetal bovine serum (FBS) and 1% penicillin and streptomycin (PenStrep). Myoblasts were differentiated into myotubes at approximately 90% confluence, by culturing in differentiation media [DM: DMEM, 2% horse serum (HS) and PenStrep] for 96 h. Myotubes were treated with 10 μM Dex for 24 h. To assess myogenesis, differentiating myoblasts were treated with compound of interest at the same time as culture in DM for 24, 48 or 96 h.
Alox5, Alox12 and Alox15 activity assays
5‐HETE ELISA and 12‐HETE ELISA assay kits were purchased from MyBioSource, CA, USA. A 15(S)‐HETE ELISA assay kit was purchased from Cayman Chemical, MI, USA, for analysis of cell lysates. A 15(S)‐HETE ELISA assay kit was purchased from MyBioSource, CA, USA, for analysis of skeletal muscle. The kits were used in accordance with the manufacturers' instructions.
siRNA‐mediated gene knockdown
Knockdown of gene expression using siRNA was carried out in the 12‐well plate format in accordance with the manufacturer's protocol (Thermo Fischer Scientific, Waltham, USA). The transfection step was carried out using the Lipofectamine 3000 agent (Thermo Fischer Scientific, Waltham, USA).
Measurement of protein synthesis
The surface sensing of translation (SUnSET) assay was used to determine protein synthesis, as previously described. 14 In brief, 1 μg/mL puromycin was added to the myotube cultures. The cell lysates were then harvested 10 min later and processed for immunoblotting using an anti‐puromycin 12D10 antibody (MABE343; Millipore). For the measurement of protein synthesis in vivo, the mice were injected with puromycin at 40 nmol/g intraperitoneally and sacrificed in 30 min.
Immunocytochemistry
Myotubes were visualized by myosin heavy chain immunocytochemistry using the previously published protocol. 15 Myotubes were classified as myosin heavy chain positive cells containing more than four nuclei. Myotube diameter was obtained by measuring the midpoint of all visible myotubes within four microscopic images captured using the 10× magnification objective.
Western blotting
Cell protein lysates were quantified using the Bradford reagent (Bio‐Rad, USA, CA) and separated by electrophoresis with 10% or 12% SDS‐PAGE. Blotting and antibody incubation were carried out following the previously published protocol. 15 Details of the primary and secondary antibodies are provided in Tables S1 and S2, respectively.
Real‐time qPCR
mRNA levels of the genes of interest was measured using the StepOnePlus Real Time PCR System (Applied Biosystems, UK) following the previously published method. 16 Details of the primers used in this study are provided in Tables S3 and S4. The expression level of β‐actin was used for normalization while measuring the expression levels of all the other genes.
LTB4 ELISA
An LTB4 ELISA kit was purchased from MyBioSource, CA, USA. The concentrations of LTB4 in skeletal muscles and cell lysates were measured by ELISA according to the manufacturer's instructions. Samples (100 μL) were incubated with polyclonal LTB4 binding in 96‐well plates for 1 h. After stopping the enzymatic reaction, optical density was measured at 450 nm.
Animal studies
Animal studies were carried out under the auspices of the Institute for Laboratory Animal Research Guide for the Care and Use of Laboratory Animals and approved by the Gwangju Institute of Science and Technology Animal Care and Use Committee (study approval number GIST‐2021‐059). Animal studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Animals were purchased from Damool Science, Republic of Korea.
Dexamethasone model of skeletal muscle atrophy
Twelve‐week‐old male C57BL/6J mice were treated with drugs as follows: (1) injection of vehicle (5% DMSO, 5% Tween 80, PBS) alone, (2) 15 mg/kg Dex dissolved in vehicle and (3) injection of 15 mg/kg Dex and 10 mg/kg malotilate, (n = 9 total per group). Mice were treated by intraperitoneal injection every 24 h for 14 d and then assessed for skeletal muscle function, protein synthesis and condition. Liver and gonadal adipose tissue (GAT) mass was obtained after sacrifice.
Ageing model of skeletal muscle atrophy
Twenty‐one‐month‐old male C57BL/6J mice were treated with drugs as follows: (1) vehicle alone (6.73% DMSO in 0.5% carboxylmethyl cellulose) and (2) 50 mg/kg malotilate, (n = 9 per group in total). Mice were treated by oral gavage every 24 h for 4 weeks and then assessed for skeletal muscle function and condition. Liver and GAT mass was obtained after sacrifice.
Measurement of grip strength
Grip strength was measured with the BIO‐GS3 grip strength test meter (Bioseb, FL, USA). Mice were placed onto the grid with all four paws attached and gently pulled backwards to measure grip strength until the grid was released. The maximum grip value used to represent muscle force was obtained using three trials with a 30 s interval.
Muscle fatigue test
Muscle fatigue was measured with two different models using the rotarod. One is the constant model, and the other is the accelerating model that is inherent in the rotarod machine. In brief, the mice were accommodated to training before starting the fatigue task using an accelerating rotarod (Ugo Basile, Italy). Mice were trained with speeds ramping from 10 to 15 rpm. Twenty‐four hours later, the muscle fatigue test was carried out with rotarod running at 5 rpm increments every 5 min up to 15 rpm. Latency to fall off the rotarod for each mouse was then measured. A fatigued mouse was classified as falling off four times within 1 min, which then terminated the test. Maximum rpm was measured by recording the rpm at which the mice fell off from the machine.
Muscle dissection and histological analysis
Mice were anaesthetized with 22 mg/kg ketamine (Yuhan, Republic of Korea) and 10 mg/kg xylazine (Bayer, Republic of Korea) by IP injection before sacrifice. Quadriceps, gastrocnemius, tibialis anterior and soleus muscles were dissected and weighed. For immunohistochemistry, the muscles were then fixed by overnight incubation with 4% paraformaldehyde at 4°C, embedded into paraffin solution and stored −80°C. Sections were obtained using a cryostat. Sectioning and haematoxylin and eosin (H&E) staining were carried out by the Animal Research Facility of the Gwangju Institute of Science and Technology, Republic of Korea. H&E staining was carried out using a kit (Merck, Germany). Muscle fibre cross‐sectional area (CSA) was measured with the ImageJ 1.48 software (National Institutes of Health, USA).
Immunohistochemistry
Immunohistochemistry was carried out using anti‐myosin heavy chain type 2A and 2B antibodies (DSHB, IA, USA) and an anti‐laminin antibody (Abcam, Cambridge, UK). Counterstaining was conducted using 1 μM DAPI solution. Muscle fibre CSA was measured with the ImageJ 1.48 software (National Institutes of Health, USA) after the images were visualized with fluorescence microscopy (LEICA DM 2500).
Oil red O staining of skeletal muscle
An Oil red O stain kit was purchased from Abcam, Cambridge, UK. Staining was carried out in accordance with the manufacturers' instructions and mounted with a coverslip in aqueous mounting medium.
Succinate dehydrogenase histochemistry
A succinate dehydrogenase (SDH) staining kit was purchased from VitroVivo Biotech, MD, USA, and staining was carried out following the manufacturer's instructions. The slides were mounted with a coverslip in Permount medium.
RNA‐Seq
RNA samples were obtained from C2C12 murine myoblasts cultured as follows: (1) differentiation media (DM) for 120 h (Un); (2) DM for 96 h and DM plus 10 μM malotilate for 24 hours (Mal); (3) DM for 96 h and DM plus 10 μM Dex for 24 h (Dex); (4) DM for 96 h and DM plus 10 μM Dex and 10 μM malotilate for 24 h (Dex_Mal); RNA‐Seq was carried out by Macrogen, Republic of Korea. Before proceeding with the sequencing, QC was performed by FastQC v0.11.7 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Illumina paired ends or single ends in sequenced samples were trimmed by Trimmomatic 0.38 (http://www.usadellab.org/cms/?page=trimmomatic) with various parameters. Sequences of samples were analysed and mapped by HISAT2 version 2.1,0, Bowtie2 2.3.4.1 (https://ccb.jhu.edu/software/hisat2/index.shtml). Potential transcripts and multiple splice variants were assembled by StringTie version 2.1.3b (https://ccb.jhu.edu/software/stringtie/). Fifteen thousand six hundred nineteen genes were quantified in total. The heat map in Figures 2C and 6C and 6D were made with GraphPad Prism 7 (GraphPad, CA, USA).
Figure 2.
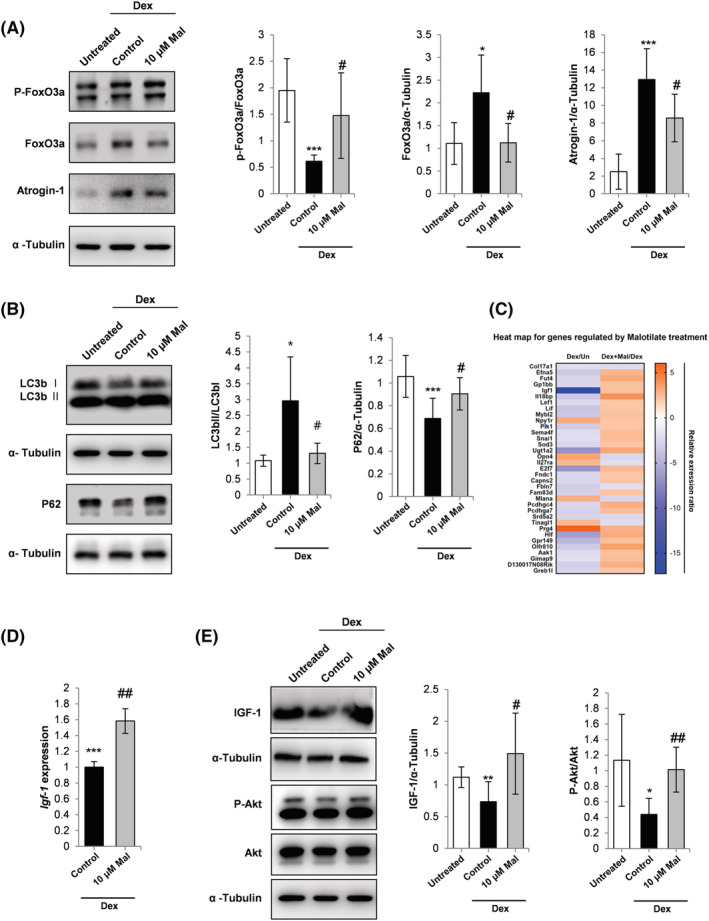
(A) Western blot analysis of FoxO3a phosphorylation, FoxO3a expression and atrogin‐1 expression. α‐Tubulin expression was used as the loading control. (B) Western blot analysis of LC3b I, LC3b II and p62 expression. α‐Tubulin expression was used as the loading control. (C) One‐way hierarchical heat map for the Dex/Un compared to Dex_Mal/Dex treatment groups. (D) qPCR analysis of IGF‐1 expression. C2C12 myoblasts were cultured and treated with Dex and malotilate as in Figure 1. (E) Western blot analysis of IGF‐1 expression, Akt phosphorylation and Akt expression. α‐Tubulin expression was used as a loading control. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with untreated. # P < 0.05, ## P < 0.01 compared with Dex treated.
Figure 6.
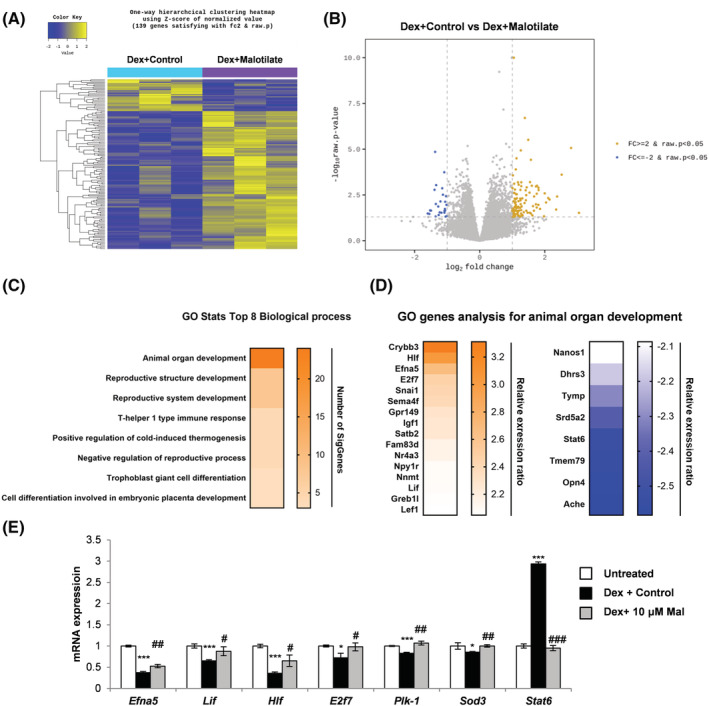
Cellular transcriptome analysis showing total number of upregulated and downregulated genes between C2C12 murine myoblasts cultured as follows: (1) differentiation media (DM) for 120 h (Un); (2) DM for 96 h and DM plus 10 μM malotilate for 24 h (Mal); (3) DM for 96 h and DM plus 10 μM Dex for 24 h (Dex); (4) DM for 96 h and DM plus 10 μM Dex and 10 μM malotilate for 24 h (Dex_Mal). (A) Heat map for genes showing differential expression in Dex and Dex_Mal, compared with Un and Mal. (B) Volcano plot showing gene expression changes in the Dex compared to Dex_Mal treatment groups. (C) Gene ontology (GO) functional analysis for the Dex compared with Dex_Mal treatment groups. (D) GO gene analysis for animal organ development. (E) qPCR analysis of genes linked to skeletal muscle atrophy (Sod3, Plk‐1, Stat6), organ development (Lif, Hif and E2f7) or muscle atrophy and organ development (Efna) in C2C12 myotubes cultured as in part (A). Please note that Fut4 was not detectable in the myotubes. *P < 0.05, ***P < 0.001 compared with untreated. # P < 0.05, ## P < 0.01, ### P < 0.001 compared with Dex treatment alone.
Human skeletal myoblast culture and experimentation
Human skeletal myoblasts were obtained from Thermo Fisher Scientific, USA. The myoblasts were thawed using a 37°C water bath, centrifuged at 180 g for 5 min at RT washed with 20 mL DM. Myoblasts were then re‐suspended in DM and seeded onto 12‐well plates at a density of 4.8 × 104 cells/well. Forty‐eight hours later, the myoblasts were treated with compounds of interest for 24 h. Myotubes were then stained with H&E. Myotube diameter was measured using the ImageJ 1.48 software (National Institutes of Health, USA) after light microscopy analysis of DIC captured images (Olympus CKX41).
Statistical analysis
Statistical significance was determined using the Student's t‐test. A P value of less than 0.05 was considered as significant. Unless otherwise mentioned, all data shown are representative of more than three experimental repeats and the graph error bars are standard deviation.
Results
Malotilate inhibited atrophy in glucocorticoid‐treated murine myotubes
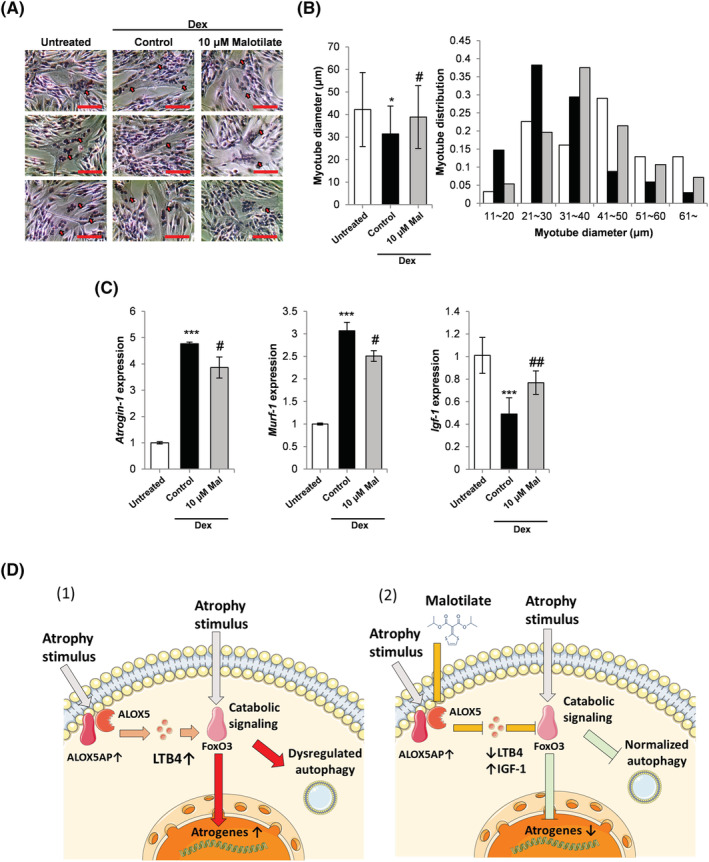
The glucocorticoid, Dex, treatment model was used to investigate the effect of malotilate on myotube atrophy. Malotilate treatment inhibited the effects of Dex on myotube atrophy (Figure 1A and 1B). Malotilate treatment inhibited the upregulation of atrogin‐1 and MuRF‐1 (Figure 1C). Total protein synthesis was decreased by treatment with Dex and recovered by malotilate (Figure 1D).
Figure 1.
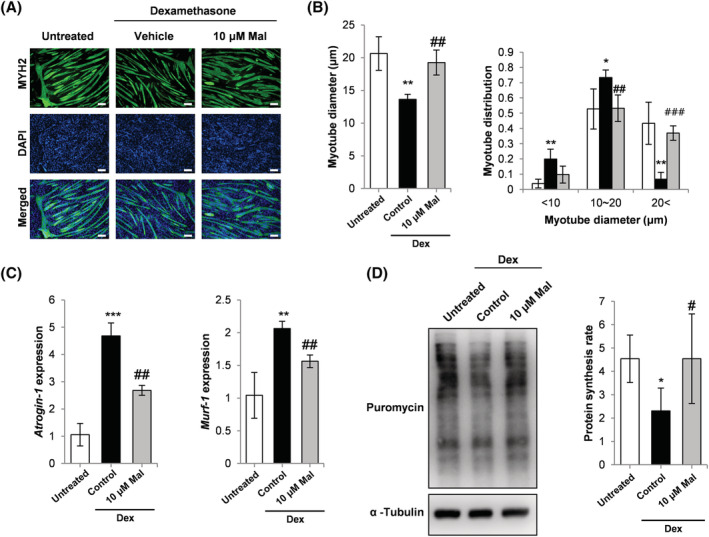
(A) Myosin heavy chain (MYH2) immunocytochemistry of C2C12 myoblasts cultured as follows: (1) differentiation media (DM) for 120 h (untreated); (2) DM for 96 h and DM plus 10 μM Dex for 24 h; (3) DM for 96 h and DM plus 10 μM Dex and 10 μM malotilate (Mal) for 24 h (scale bar = 100 μm). (B) Myotube diameter and myotube diameter distribution. (C) qPCR analysis of atrogin‐1 and MuRF‐1 expression. (D) SUnSET assay of protein synthesis. α‐Tubulin expression was used as the loading control. *P < 0.05, **P< 0.01, ***P < 0.001 compared to untreated. # P < 0.05, ## P < 0.01, ### P < 0.001 compared with Dex treated.
Myotubes were treated with malotilate, and production of the Alox15 product, 15‐HETE, the Alox12 product, 12‐HETE, and the Alox5 product, 5‐HETE, was measured. It was observed that malotilate treatment reduced levels of 5‐HETE, but had no significant effect on 15‐HETE and 12‐HETE in the myotubes (Figure S1).
The transcription factor forkhead box O3 (FoxO3a) is a major regulator of the ubiquitin–proteasome pathway of skeletal muscle atrophy. FoxO3a phosphorylation levels were decreased in myotubes treated with Dex and increased by treatment with malotilate (Figure 2A) and accompanied by a decrease in atrogin‐1 expression (Figure 2A). Dex treatment increased autophagy in the myotubes (increased ratio of LC3bII to LC3bI, and reduced expression of p62). Autophagy was decreased by treatment with malotilate (Figure 2B). The cellular transcriptome was investigated using RNA‐Seq. A heat map for genes showing differential expression indicated that insulin‐like growth factor 1 (IGF‐1) was up‐regulated by malotilate treatment (Figure 2C) and confirmed by qPCR and western blotting (Figure 2D and 2E). Malotilate increased Akt activation in Dex‐treated myotubes (Figure 2E). Malotilate did not affect Akt activation in normal myotubes (Figure S2).
Alox5 gene knockdown prevents myotube atrophy associated with increased levels of the Alox5 product, leukotriene B4
LTB4 levels were shown to be increased in Dex‐treated myotubes and lowered by malotilate (Figure 3A). Treatment with malotilate did not affect Alox5 expression (Figure 3B). LTB4 treatment alone induced myotube atrophy (Figure 3C). Western blot analysis indicated that LTB4 treatment increased phosphorylation of the stress‐activated kinase, c‐Jun N‐terminal kinase (JNK), and increased the phosphorylation of IκBα, which activates the catabolic NF‐κb pathway (Figure 3D).
Figure 3.
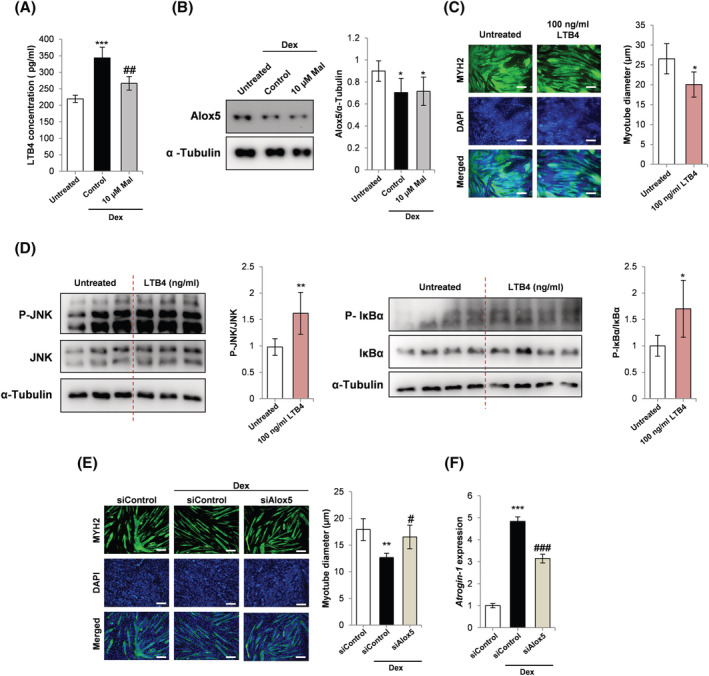
(A) ELISA analysis LTB4 levels in C2C12 myoblasts cultured as follows: (1) 120 h incubation with DM; (2) 96 h incubation with DM and 24 h treatment with 10 μM Dex; (3) 96 h incubation with DM and 24 h treatment with 10 μM Dex and 10 μM malotilate (Mal). (B) Western blot analysis of Alox5 expression in C2C12 myoblasts cultured as in part (A). (C) MYH2 immunocytochemistry and myotube diameter of C2C12 myoblasts cultured as follows: (1) 144 h incubation with DM; (2) 72 h incubation with DM and 72 h incubation with 100 ng/mL LTB4 (scale bar = 100 μm). (D) Western blot analysis of JNK expression, JNK phosphorylation, IκBα expression and IκBα phosphorylation in C2C12 myotubes treated with 100 ng/mL LTB4 for 30 min. α‐Tubulin was used as a loading control. (E) MYH2 immunocytochemistry and myotube diameter of C2C12 myoblasts cultured as follows: (1) 72 h incubation with DM and 48 h incubation with DM plus control, scrambled siRNA; (2) following 72 h incubation with DM, 24 h incubation in with DM plus control, scrambled siRNA and additional 24 h treatment with 10 μM Dex plus scrambled siRNA; (3) following 72 h incubation with DM, 24 h incubation in with DM plus Alox5 siRNA and additional 24 h treatment with 10 μM Dex plus Alox5 siRNA (scale bar = 100 μm). (F) qPCR analysis of atrogin‐1 expression in C2C12 myoblasts cultured as described in part (E). For (A and B): *P < 0.05, ***P < 0.001 compared with untreated. # P < 0.05, ## P < 0.01 compared with Dex treatment alone. For (C and D): *P< 0.01, **P < 0.01 compared with untreated. For (E and F): **P < 0.01, ***P < 0.001 compared with control siRNA. # P < 0.05, ### P < 0.001 compared with control siRNA plus Dex.
The effect of Alox5 gene knockdown on myotube atrophy was assessed in Dex treated myotubes. siRNA‐mediated reduction of Alox5 expression was confirmed by qPCR and western blotting (Figure S3A and S3B). Alox5 gene knockdown inhibited myotube atrophy (Figures 3E and S3C) and was accompanied by a reduction in atrogin‐1 expression (Figure 3F). Application of exogenous LTB4 did not improve atrophy in Dex‐treated myotubes or reduce the expression of atrogin‐1 (Figure S3D and S3E). Zileuton has been validated as a specific Alox5 inhibitor. 5 It was observed that zileuton treatment could prevent Dex‐induced myotube atrophy and reduce the expression of atrogin‐1 and MuRF‐1 (Figure S3F and S3G).
Malotilate does not significantly affect the myogenic program
Murine myotubes were induced to differentiate into myotubes in the presence of malotilate. The expression of myosin heavy chain type 2 (Myh2), a marker of differentiation, was measured at three time points (24, 48, and 96 h). Myh2 expression was not affected by malotilate treatment from the onset of differentiation (Figures S4A and S4B and S2). The phosphorylation level of Akt at 96‐h differentiation was not significantly changed by malotilate treatment (Figure S2). The fusion index was unaffected by Alox5 inhibition or gene knockdown (Figures S4C and S3D). Expression analysis of master genetic regulators the myogenic program (Pax7, Myf5, MyoD and MyoG 17 ) did not show any significant change after 24‐h malotilate treatment (Figure S4E). The 24 h time point was selected because the expression of these genes changes during the early stages of myogenesis. 18 , 19
Pharmacological targeting of Alox5 inhibits skeletal muscle atrophy in a glucocorticoid treatment model
Dex treatment for 14 d did not significantly reduce body weight, and there was no significant effect by malotilate treatment (Figure S5A). Malotilate significantly increased grip strength in the Dex treated mice and reduced latency to fall in the rotarod performance test (Figure 4A and 4B). Maximum running speed was also significantly increased (Figure 4B). Malotilate treatment increased mass in the quadriceps and soleus muscles (Figure 4C). Mean muscle fibre CSA and the proportion of fibres with larger CSA were increased by malotilate (Figures 4D and S5D). Mean GAT mass was increased by Dex and reduced by malotilate, but did not reach statistical significance (Figure S5B and S5C). Liver mass was significantly increased by Dex treatment and unaffected by malotilate (Figure S5B and S5C). ELISA analysis indicated that levels of the Alox5 product, LTB4, were increased by Dex and lowered by malotilate (Figure 4E). Malotilate also decreased expression of atrogin‐1 and MuRF‐1, increased the expression of IGF‐1 and increased the activation of Akt (Figures 4F and S6A and S6B). Protein synthesis rate was also significantly increased in the gastrocnemius muscle of malotilate‐treated mice (Figure S6C). ELISA analysis of 15‐HETE, 12‐HETE and 5‐HETE levels (the products of Alox15, Alox12 and Alox5, respectively) in the gastrocnemius muscle showed that the mean levels of 5‐HETE were increased by Dex and reduced by malotilate, although these values did not reach statistical significance (Figure S6D).
Figure 4.
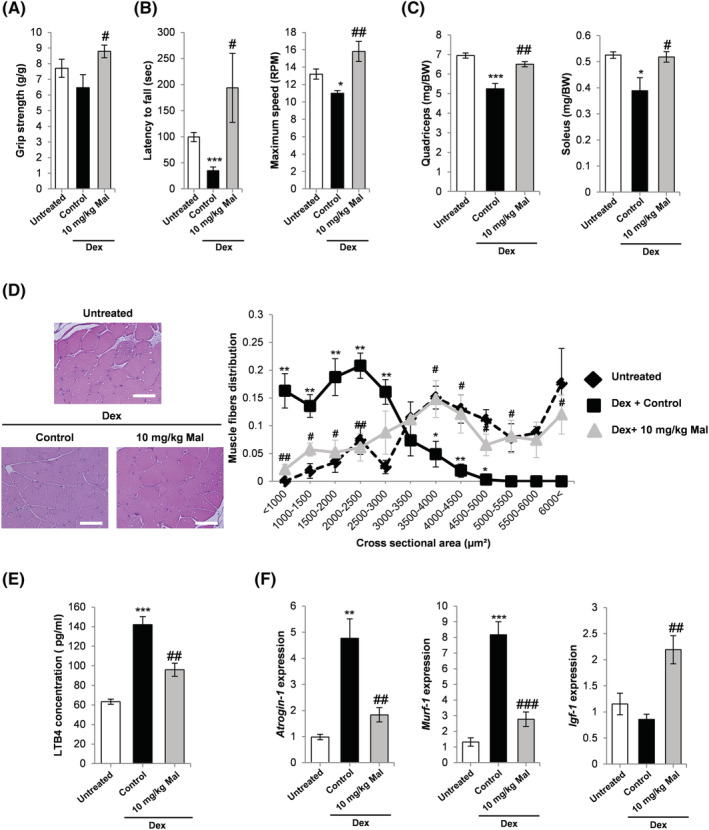
(A) Grip strength in mice treated with vehicle alone, Dex or Dex plus malotilate (Mal) for 14 d. (B) Latency to fall off the rotarod and maximum speed achieved. (C) Quadriceps and soleus mass. (D) Representative H&E staining of the quadriceps (scale bar = 100 μm) and fibre cross‐sectional area. (E) ELISA‐based quantification of the Alox5 product, LTB4, in the gastrocnemius muscle. (F) qPCR analysis of atrogin‐1, MuRF‐1 and IGF‐1 expression in the quadriceps. *P < 0.05, **P < 0.01, *** = p < 0.001 compared with vehicle treated mice. # P < 0.05, ## P < 0.01, ### P < 0.001 compared with Dex‐treated mice (n = 4–5 per experimental group).
Pharmacological targeting of Alox5 inhibits skeletal muscle atrophy in aged mice
Malotilate treatment did not affect body mass in the aged mice (Figure S7A). Grip strength was significantly increased by malotilate (Figure 5A). The mean latency to fall was increased by malotilate, but did not reach statistical significance (Figure 5A). Quadriceps and soleus muscle mass were significantly increased by malotilate (Figure 5B), and GAT mass was significantly decreased (Figures 5C and S7B). Liver mass was unaffected by malotilate (Figure S7B). Histological analysis of quadriceps muscle myofibre diameter distribution indicated a significantly greater proportion of large myofibres (>5000 μm2) in the malotilate treatment group (Figure 5D). Average myofibre CSA was also increased (Figure S7C). Immunohistological analysis of fast‐twitch fibre types IIA and IIB, which preferentially undergo atrophy during aging, 20 indicated that malotilate preserved the CSA of fast fibres (Figures 5E and F and S7D). Malotilate also increased the phosphorylation of Akt in aged muscle (Figure S7E). 5‐Lipoxygenase‐activating protein (Alox5AP) is necessary for Alox5 activation and has been shown to be upregulated in aged human skeletal muscle and after glucocorticoid treatment. 21 , 22 , 23 , 24 Alox5AP expression was upregulated in aged mice and not significantly affected by malotilate, supporting enzyme inhibition as the mechanism by which malotilate reduces Alox5 activity (Figure S7F). Oil red O staining showed that malotilate treatment reduced lipid accumulation in the skeletal muscle of aged mice (Figure S7G). SDH staining indicted that malotilate treatment had no significant effect on the oxidative status of the muscle fibres (Figure S7H).
Figure 5.
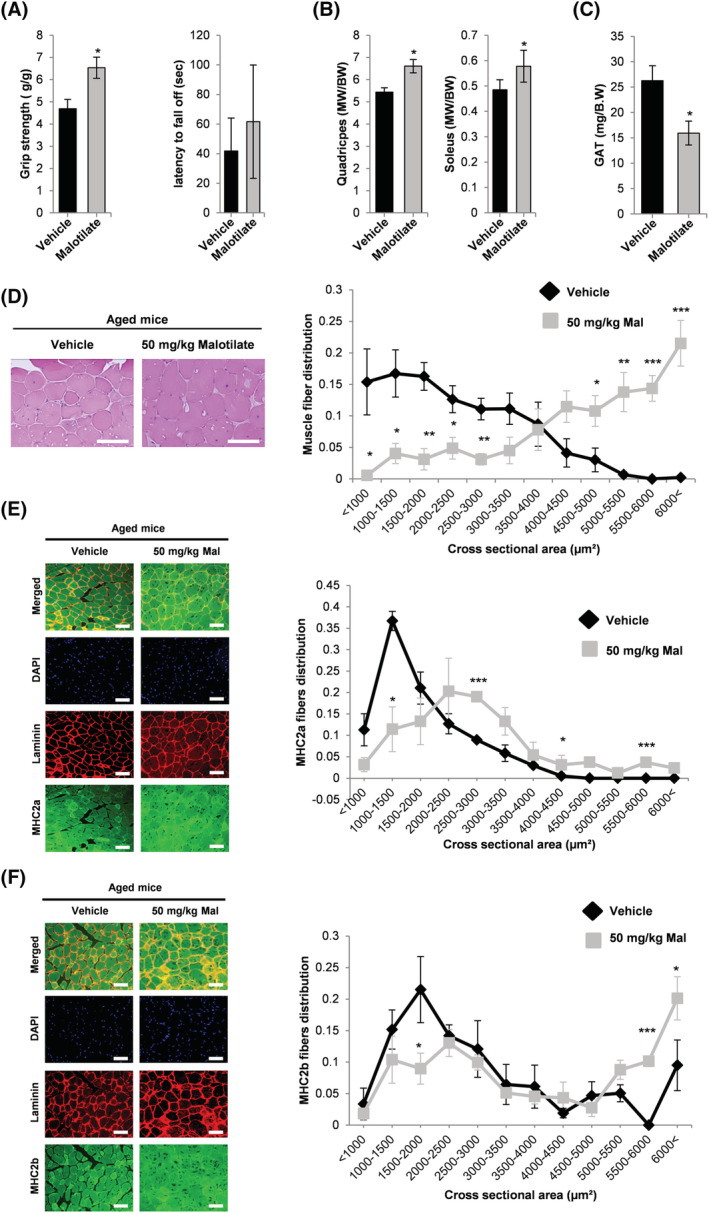
(A) Grip strength and latency to fall off the rotarod in aged (21 months) mice after 4‐week treatment with vehicle or malotilate (Mal). (B) Quadriceps and soleus muscle mass. (C) GAT mass. (D) Representative H&E staining of the quadriceps and muscle fibre area distribution (scale bar = 100 μm). (E and F) Representative images of myosin type 2a and 2b and laminin immunostaining and cross‐sectional area distribution in the quadriceps muscle. DAPI was used to visualize cell nuclei (scale bar = 100 μm). *P < 0.05, ***P < 0.01, ***P < 0.001 compared with vehicle treated (n = 4–5 per experimental group).
Cellular transcriptomic analysis of myotubes with pharmacological targeting of Alox5 in the presence or absence of atrophy
To investigate the biological processes by which Alox5 inhibition by malotilate inhibits muscle atrophy, cellular transcriptome was investigated using RNA‐Seq. Expression patterns were compared with untreated, Dex treated myotubes, normal myotubes or myotubes treated with malotilate alone (Figure S8A). The heat map and volcano plot showed a total of 139 differentially expressed genes (DEGs) between myotubes treated with Dex or Dex plus malotilate (Figure 6A and 6B). KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis showed that metabolic pathways were primarily targeted by malotilate (Figure S8B). Gene ontology (GO) functional analysis indicated that organ development was the top biological process targeted in myotubes treated with Dex plus malotilate compared with Dex alone (Figures 6C and 6D and 8C). Genes linked to skeletal muscle atrophy and/or organ development were selected (Fut4, Sod3, Plk‐1, Stat6 (muscle atrophy), Lif, Hif and E2f7 (organ development), Efna and Igf‐1(muscle atrophy and organ development)) and their normalization of expression by malotilate in Dex‐treated myotubes was validated using qPCR (Figures S8D and 6E). Leukotriene A4 hydrolase (LTA4H), which generates leukotriene A4 (LTA4), an unstable epoxide that is transformed either to LTB4 or a cysteinyl leukotriene LTC4, 25 showed no change in expression by RNA sequencing analysis (data not shown).
Pharmacological targeting of Alox5 prevents atrophy in human myotube cultures
Histological analysis indicated that co‐treatment with malotilate could prevent atrophy in Dex‐treated human myotubes (Figure 7A and 7B). Malotilate treatment also significantly reduced the expression of atrogin‐1 and MuRF‐1, and increased the expression of IGF‐1 (Figure 7C). The muscle atrophy‐related genes identified by RNA‐Seq in murine myotubes were tested for expression levels in the human myotubes. The expression of Plk‐1 was significantly reduced expression after Dex treatment and significantly increased by malotilate (Figure S9A).
Figure 7.
(A) Representative H&E‐stained images of human donor myoblasts cultured as follows: (1) differentiation media (DM) for 72 h (untreated); (2) DM for 48 h and DM plus 10 μM Dex for 24 h; (3) DM for 48 h and DM plus 10 μM Dex and 10 μM malotilate (Mal) for 24 h (scale bar = 100 μm). (B) Mean myotube diameter and diameter distribution. (C) qPCR analysis of atrogin‐1, MuRF‐1 and IGF‐1 expression. *P < 0.05, ***P < 0.001 compared with untreated. # P < 0.05, ## P < 0.01, ### P < 0.001 compared with Dex treatment alone. (D) Working model of the effect on malotilate on skeletal muscle atrophy. (1) Under atrophy‐inducing conditions, such as glucocorticoid treatment or aging, catabolic signalling pathways are activated in skeletal muscle fibres, producing atrophy effector mechanisms including up‐regulated atrogene expression and dysregulated autophagy. Alox5 activating protein (Alox5AP) is also known to be up‐regulated under atrophy‐inducing conditions, and the Alox5 product, LTB4, has been shown to increase the activity of catabolic regulators, such as FoxO signalling [21–23, 40, 41, 49], which would promote muscle fibre atrophy. (2) Malotilate treatment blocks the activity of Alox5, which reduces cellular levels of LTB4 and up‐regulates the expression of anti‐atrophy factors, such as IGF‐1, inhibiting catabolic pathways. This could produce a beneficial effect against the effector pathways of skeletal muscle fibre atrophy.
Discussion
In this study, we investigated the repurposing of malotilate, a clinically safe drug developed for liver regeneration and Alox5 inhibitor, as a muscle atrophy treatment. Our results indicate that malotilate is an attractive candidate for further development to treat muscle atrophy and Alox5 is a novel drug target for this disorder. Malotilate treatment affected both the ubiquitin–proteasome proteolysis system (UPS) pathway and autophagy in myotubes undergoing atrophy. Malotilate inhibited the activation of FoxO3a, which is a major activator of the UPS. It has been reported that 75% of the protein degradation that occurs during skeletal muscle atrophy is contributed by the UPS. 26 Therefore, the protective effect of malotilate on muscle atrophy may be primarily due to the inhibition of FoxO3a. It has been reported that upon autophagy induction, for instance, by nutrient starvation or glucocorticoids, LC3‐I converts to LC3‐II and induces a concurrent decrease in p62. 27 Malotilate treatment normalized the ratio of LC3‐I/LC3‐II and increased expression of p62. It may be concluded that the anti‐atrophy effects of malotilate treatment are mainly via UPS inhibition, along with a prevention of autophagy induction. Malotilate did not significantly affect myoblast differentiation or the expression of key myogenic regulatory genes. Thus, the anti‐atrophy effects of malotilate appear to be related to anti‐atrophy mechanisms in differentiated muscle fibres, such as the inhibition of atrogene expression, rather than enhancing muscle stem cell differentiation. The finding that malotilate does not affect Akt activation in normal differentiated myotubes, compared with conditions of muscle atrophy, also supports this hypothesis.
Malotilate treatment reduced the expression of atrogin‐1 and MuRF‐1, although expression levels remained elevated compared to normal samples. Protein content in muscle is determined by the balance between synthesis and degradation, because the proteins are in constant turnover. 28 , 29 Therefore, increases in synthesis may increase protein content in the presence of breakdown mediated by atrogin‐1 of MuRF‐1. After malotilate treatment, protein synthesis levels were increased in vitro and in vivo, even though atrogin‐1 and MuRF‐1 expression were not completely suppressed. Additionally, dysregulated autophagy contributes to skeletal muscle atrophy. 30 In Dex‐treated myotubes, malotilate treatment normalized autophagic flux, as indicated by p62 expression and the ratio of LC3bII to LC3bI. This may also prevent atrophy in the presence of elevated expression of atrogin‐1 and MuRF‐1. As E3 ubiquitin ligases, the activity of atrogin‐1 and MuRF‐1 would also depend on the expression and/or activity of the E1 ubiquitin‐activating enzymes and E2 ubiquitin‐conjugating enzymes. 31 Although the up‐regulation of atrogin‐1 or Murf‐1 is traditionally used as indicator of increased protein degradation, protein breakdown will be dependent on this E1–E2–E3 enzyme cascade.
Malotilate treatment increased skeletal muscle mass without affecting total body mass. Agents producing muscle atrophy, such as increased glucocorticoid signalling, are also known to increase adiposity. 32 The dual, opposing effects of malotilate on glucocorticoid signalling in muscle and adipose tissues may provide an explanation for the unchanged body mass observed in this study. This is supported by the finding that Dex increased the mean GAT mass value in the mouse model, and this was reduced by malotilate treatment. When studying muscle atrophy, researchers tend to select male mice, because they can live longer than female mice and do not have the high hormone variation observed in female mice. 33 , 34 It should be noted that average serum glucocorticoid concentrations in aged humans are higher than those reported for adult mice, 35 ,[S1] but would be probably be insufficient to produce the degree of muscle atrophy observed in the Dex treatment model. For the model of muscle aging, mice are generally used at 18 months or older, due to the increased expression of aging‐associated biomarkers. 34 Our study used mice at 21 months of age. However, it should be noted that overall body weight does not decline at this stage. 36 It was also observed that the increase in CSA after malotilate treatment was more pronounced than the increase in mass. We speculate that changes in the average skeletal muscle fibre CSA may not necessarily be directly proportional to changes in muscle weight. For example, it has been reported that a major confounding factor affecting the interpretation of CSA is the extent of muscle fibre ‘branching’, which is a common feature of muscle aging. 37 , 38
The results in this study indicate that malotilate‐mediated inhibition of Alox5 is a novel therapeutic approach for treating skeletal muscle atrophy. In addition, Alox5 gene knockdown prevented atrophy in myotubes and treatment with the Alox5 product, LTB4, induced atrophy. LTB4 levels were also found to be elevated in skeletal muscle undergoing atrophy. To our knowledge, there is no report showing that Alox5 is up‐regulated in skeletal muscle fibres undergoing atrophy. Alox5 activity is regulated by arachidonate 5‐lipoxygenase‐activating protein (Alox5AP, also known as 5‐lipoxygenase‐activating protein, or FLAP), which anchors Alox5 to the cell membrane and transfers the arachidonic acid substrate. 39 Interestingly, Alox5AP has been shown to be up‐regulated in aged human skeletal muscle and by glucocorticoid treatment. 21 The increased expression of Alox5AP under conditions of atrophy could explain the effects of malotilate observed in this study. It should also be noted that Alox5 inhibition by malotilate treatment did not affect the expression of Alox5AP, indicating that malotilate reduces Alox5 activity by enzyme inhibition. In addition, the Alox5 product, LTB4, activates the BLT1/2 G‐protein coupled signalling pathway and increases the activity of NF‐κb signalling in atherosclerotic plaques and FoxO signalling in adipocytes. 40 , 41 NF‐κb and FoxO signalling are both master regulatory pathways of skeletal muscle atrophy. 42 Taken together, these previous findings and the results of this study support a mechanism of action wherein atrophy conditions upregulate Alox5AP expression, which enhances the activity of Alox5 and increases LTB4 levels. LTB4 stimulates the activity of atrophy signalling pathways, such as NF‐κb and FoxO, to promote the effectors of myofibre catabolism, including up‐regulated atrogene expression and dysregulated autophagy. Treatment with malotilate reduces LTB4 levels and the activity of atrophy signalling pathways, producing an inhibition of muscle atrophy. A model of the effect of malotilate treatment and Alox5 inhibition on skeletal muscle atrophy is shown in Figure 7D.
RNA‐seq data presented in this study indicate that Alox5 inhibition mediated by malotilate affects metabolic pathways (by KEGG analysis) and animal organ development (by GO analysis) in skeletal muscle cells. The primary metabolic changes associated with muscle atrophy caused by glucocorticoids, such as Dex, are insulin resistance, hyperglycaemia and hyperlipidaemia.[S2] Thus, targeting metabolic pathways using the Alox5 inhibitor malotilate has the potential to correct these changes in metabolism caused by Dex. The molecular mechanisms underlying organ development during embryogenesis have many similarities with the processes regulating tissue regeneration.[S3] Therefore, the major effect on organ development indicated by GO analysis may explain the ability of this inhibitor to ameliorate the progression of skeletal muscle atrophy in the Dex treatment and ageing animal models. IGF‐1, a key modulator of muscle hypertrophy, was up‐regulated by malotilate treatment. IGF‐1 levels are suppressed in many chronic diseases and produce muscle atrophy by the combined effects of suppressed protein synthesis, UPS activity, autophagy and fibre regeneration.[S4] Supplementation with IGF‐1 alone has been shown to be effective for preventing muscle atrophy.[S5] Interestingly, this up‐regulation of IGF‐1 was specific for myotubes undergoing atrophy and not observed in normal myotubes treated with malotilate, indicating a disease‐specific effect of drug treatment. The qPCR results showed that, among these genes linked to skeletal muscle atrophy, Plk‐1 had a conserved expression pattern between murine and human myotubes. Plk‐1 has recently been linked to sepsis‐induced skeletal muscle atrophy in mice.[S6] These findings warrant the further investigation of Plk‐1 as a novel drug target for muscle atrophy in patients.
In summary, malotilate, a previously characterized liver regeneration drug studied in clinical trials, prevented skeletal muscle atrophy in multiple models and was effective using oral delivery. Malotilate treatment produced marked effects on muscle mass and maintained force/strength in the Dex atrophy model, and the preservation of fast‐twitch fibres types in aged muscle, indicating that the Alox5 target may have a major effect on the progression of muscle atrophy. In light of the clinical need to develop new drugs and targets for skeletal muscle atrophy, malotilate and Alox5 inhibitors can be attractive drug candidates for repurposing to treat this disorder.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (NRF‐2020R1A2C2014194) and the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (NRF‐2020M3A9G3080282). This work was partly funded by the Korean government (MSIP) through the Institute for Information and Communications Technology Promotion (IITP) grant (No. 2019‐0‐00567, Development of Intelligent SW systems for uncovering genetic variation and developing personalized medicine for cancer patients with unknown molecular genetic mechanisms), and a 'GIST Research Institute (GRI) IIBR' grant funded by the GIST in 2022. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. Figure 7D was produced under a Creative Commons Attribution 3.0 Unported Licence using https://smart.servier.com/.
Conflict of interest
H‐J.K., D‐W.J. and D.R.W. are named co‐inventors of a pending provisional patent application based in part on the research reported in this paper.
Author contributions
H‐J.K. carried out experiments, analysed the data and wrote the manuscript. S‐H.L. and S‐W.K. carried out experiments. D‐W.J. and D.R.W. acquired funding, supervised the research, and wrote the manuscript.
Supporting information
Figure S1: Related to Figure 1: ELISA of 15 (S) ‐hydroxyeicosatetraenoic acid production (15‐HETE), 12 ‐hydroxyeicosatetraenoic acid production (12‐HETE) and 5 ‐hydroxyeicosatetraenoic acid (5‐HETE) production in C2C12 myotubes treated with 10 μM malotilate (Mal) for 24 h. C2C12 myoblasts were cultured as follows (1) 120 h incubation with DM; (2) 96 h incubation with DM, and 24 h treatment of 10 μM Malotilate. ** = p < 0.01 compared to untreated.
Figure S2: Related to Figure 2: Western blot analysis of Akt phosphorylation, Akt expression, and MYH2 expression in in C2C12 myoblasts cultured as follows (1) 120 h incubation with DM; (2) 96 h incubation with DM, and 24 h treatment of 10 μM Malotilate; α‐Tubulin was used as a loading control. Quantification of MYH2 expression and Akt phosphorylation are shown. * = p < 0.05 compared to untreated.
Figure S3: Related to Figure 3: A) qPCR analysis of Alox5 expression in C2C12 myoblasts cultured as follows: (1) 72 h incubation with DM and 48 h incubation with scrambled siRNA; (2) 72 h incubation with DM and 48 h incubation with Alox5 siRNA. B) Western blot analysis of Alox5 expression in C2C12 myoblasts cultured as described in part (A). C). Myotube diameter distribution of C2C12 myoblasts cultured as follows: (1) 72 h incubation with DM and 48 h incubation with DM plus control, scrambled siRNA; (2) Following 72 h incubation with DM, 24 h incubation with DM plus control, scrambled siRNA and additional 24 h treatment with 10 μM Dex plus scrambled siRNA; (3) Following 72 h incubation with DM, 24 h incubation with DM plus Alox5 siRNA and additional 24 hour treatment with 10 μM Dex plus Alox5 siRNA. D) MYH2 immunocytochemistry and myotube diameter of C2C12 myoblasts cultured as follows: (1) 120 h incubation with DM; (2) 96 h incubation with DM, and 24 h treatment with 10 μM Dex; (3) 96 h incubation with DM, and 24 h treatment with 10 μM Dex plus 50 ng/mL LTB4. E) qPCR analysis of atrogin‐1 expression in C2C12 myoblasts cultured as described in part (D). F) MYH2 immunocytochemistry of C2C12 myoblasts cultured as follows: (1) 120 h incubation with DM; (2) 96 h incubation with DM, and 24 h treatment with 10 μM Dex; (3) 96 h incubation with DM, and 24 h treatment with 10 μM Dex plus 50 μM zileuton (Zil) (scale bar = 100 μm). F‐G) Myotube diameter measurement and qPCR analysis of atrogin‐1 and MuRF‐1 expression in C2C12 myoblasts cultured as described in part (F). * = p < 0.05, ** = p < 0.01, *** = p < 0.001 compared to untreated. # = p < 0.05, ## = p < 0.01, ### = p < 0.001, compared to Dex treatment alone.
Figure S4: Related to Figure 3: A‐B) Western blot analysis of MYH2 expression in C2C12 myoblasts cultured with DM or DM plus 10 μM malotilate (Mal) for 24–48 h. α‐Tubulin was used as a loading control. C) Fusion index of confluent C2C12 myoblasts cultured with DM or DM plus 10 μM malotilate for 48 h. D) Fusion index of confluent C2C12 myoblasts cultured with DM plus control, scrambled siRNA or DM plus Alox5 siRNA. E) qPCR analysis of Pax7, Myf5, MyoD, MyoG, and MYH2 in C2C12 myoblasts cultured with DM or DM plus 10 μM malotilate for 24 h.
Figure S5: Related to Figure 4: A) Body weight in mice treated with vehicle alone, Dex, or Dex plus malotilate (Mal) during 14 d treatment (n = 9). B) GAT and liver mass (n = 4). C) Photographs of dissected GAT and liver tissues. D) Fiber cross sectional area of the quadriceps. *** = p < 0.001 compared to untreated, ### = p < 0.001, compared to Dex treatment alone.
Figure S6: Related to Figure 4: A‐B) Western blot analysis of phosphorylated Akt, Akt, and α‐tubulin in the soleus and gastrocnemius muscle. Quantification of phosphorylated Akt relative to Akt is shown. The red arrow indicates phosphorylated Akt. C) SUnSET assay of protein synthesis in the gastrocnemius muscle. α‐Tubulin expression was used as the loading control. D) ELISA analysis of 15‐HETE, 12‐HETE, and 5‐HETE levels in the gastrocnemius muscle. * = p < 0.05, ** = p < 0.01, compared to untreated, ## = p < 0.01 compared to Dex treatment alone.
Figure S7: Related to Figure 5: A) Body weight in aged (21 months) mice after 4 weeks treatment with vehicle or malotilate (Mal) (n = 9). B) Liver mass. C) Cross sectional fiber area of the quadriceps muscle. D) Mean cross sectional area of type 2a and type 2b fibers in the quadriceps. E) Western blot analysis of phosphorylated Akt, Akt, and α‐tubulin in the soleus muscle. Quantification of phosphorylated Akt relative to Akt is shown. The red arrow indicates phosphorylated Akt. F) Alox5AP expression in the gastrocnemius muscle. G) Oil red O staining of the quadriceps muscle (scale bar = 100 μm). H) SDH staining of the quadriceps muscle. (scale bar = 100 μm). ** = p < 0.01, *** = p < 0.001 compared to vehicle treated (n = 4–5 per experimental group).
Figure S8: Related to Figure 6: A) Cellular transcriptome analysis showing total number of upregulated and downregulated genes between C2C12 murine myoblasts cultured as follows (1) differentiation media (DM) for 120 h (Un); (2) DM for 96 h and DM plus 10 μM Malotilate for 24 hours (Mal); (3) DM for 96 h and DM plus 10 μM Dex for 24 h (Dex); (4) DM for 96 h and DM plus 10 μM Dex and 10 μM malotilate for 24 h (Dex_Mal). B) KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis for the Dex compared to Dex_Mal treatment groups. C) Gene ontology (GO) functional analysis for the Dex compared to Dex_Mal treatment groups. D) Selected genes known to be implicated in skeletal muscle atrophy and/or organ development showing differential expression in Dex and Dex_Mal, compared to Un and Dex.
Figure S9: qPCR analysis of Plk‐1, Sod3, STAT6, and Fut4 and Efna5 in human myotubes cultured as follows: (1) differentiation media (DM) for 72 h (untreated); (2) DM for 48 h and DM plus 10 μM Dex for 24 h; (3) DM for 48 h and DM plus 10 μM Dex and 10 μM malotilate (Mal) for 24 h. * = p < 0.05, *** = p < 0.001 compared to untreated, ## = p < 0.01 compared to Dex treatment alone.
Acknowledgements
The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 43
Kim H.‐J., Kim S.‐W., Lee S.‐H., Jung D.‐W., and Williams D. R. (2022) Inhibiting 5‐lipoxygenase prevents skeletal muscle atrophy by targeting organogenesis signalling and insulin‐like growth factor‐1, Journal of Cachexia, Sarcopenia and Muscle, 13, 3062–3077, doi: 10.1002/jcsm.13092
Contributor Information
Da‐Woon Jung, Email: jung@gist.ac.kr.
Darren R. Williams, Email: darren@gist.ac.kr.
References
- 1. Ding S, Dai Q, Huang H, Xu Y, Zhong C. An overview of muscle atrophy. Adv Exp Med Biol 2018;1088:3–19. [DOI] [PubMed] [Google Scholar]
- 2. Garber K. No longer going to waste. Nat Biotechnol 2016;34:458–461. [DOI] [PubMed] [Google Scholar]
- 3. Veldink GA, Vliegenthart JF. Lipoxygenases, nonheme iron‐containing enzymes. Adv Inorg Biochem 1984;6:139–161. [PubMed] [Google Scholar]
- 4. Sinha S, Doble M, Manju SL. 5‐Lipoxygenase as a drug target: A review on trends in inhibitors structural design, SAR and mechanism based approach. Bioorg Med Chem 2019;27:3745–3759. [DOI] [PubMed] [Google Scholar]
- 5. Kwak HJ, Choi HE, Cheon HG. 5‐LO inhibition ameliorates palmitic acid‐induced ER stress, oxidative stress and insulin resistance via AMPK activation in murine myotubes. Sci Rep 2017;7:5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bhattacharya A, Hamilton R, Jernigan A, Zhang Y, Sabia M, Rahman MM, et al. Genetic ablation of 12/15‐lipoxygenase but not 5‐lipoxygenase protects against denervation‐induced muscle atrophy. Free Radic Biol Med 2014;67:30–40. [DOI] [PubMed] [Google Scholar]
- 7. González‐Hedström D, Priego T, Amor S, de la Fuente‐Fernández M, Martín AI, López‐Calderón A, et al. Olive leaf extract supplementation to old Wistar rats attenuates aging‐induced sarcopenia and increases insulin sensitivity in adipose tissue and skeletal muscle. Antioxidants (Basel) 2021;10:737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim WH, Shen H, Jung DW, Williams DR. Some leopards can change their spots: potential repositioning of stem cell reprogramming compounds as anti‐cancer agents. Cell Biol Toxicol 2016;32:157–168. [DOI] [PubMed] [Google Scholar]
- 9. Ryle PR, Dumont JM. Malotilate: The new hope for a clinically effective agent for the treatment of liver disease. Alcohol Alcohol 1987;22:121–141. [PubMed] [Google Scholar]
- 10. Stenback F, Ala‐Kokko L, Ryhanen L. Morphological, immunohistochemical and ultrastructural changes in dimethylnitrosamine [correction of dimenthylnitrosamine] induced liver injury. Effect of malotilate. Histol Histopathol 1989;4:95–104. [PubMed] [Google Scholar]
- 11. Takase S, Takada A, Yasuhara M, Sato H, Matsuda Y. Effects of malotilate treatment on the serum markers of hepatic fibrogenesis in liver cirrhosis. Gastroenterol Jpn 1988;23:639–645. [DOI] [PubMed] [Google Scholar]
- 12. Bührer M, le Cotonnec JY, Wermeille M, Bircher J. Treatment of liver disease with malotilate. A pharmacokinetic and pharmacodynamic phase II study in cirrhosis. Eur J Clin Pharmacol 1986;30:407–416. [DOI] [PubMed] [Google Scholar]
- 13. Zijlstra FJ, Paul Wilson JH, Vermeer MA, Ouwendijk RJT, Eric Vincent J. Differential effects of malotilate on 5‐, 12‐ and 15‐lipoxygenase in human ascites cells. Eur J Pharmacol 1989;159:291–295. [DOI] [PubMed] [Google Scholar]
- 14. Vinel C, Lukjanenko L, Batut A, Deleruyelle S, Pradère JP, le Gonidec S, et al. The exerkine apelin reverses age‐associated sarcopenia. Nat Med 2018;24:1360–1371. [DOI] [PubMed] [Google Scholar]
- 15. Lee JH, Kim SW, Kim JH, Kim HJ, Um JI, Jung DW, et al. Lithium chloride protects against sepsis‐induced skeletal muscle atrophy and cancer cachexia. Cell 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim HJ, Lee JH, Kim SW, Lee SH, Jung DW, Williams DR. Investigation of niclosamide as a repurposing agent for skeletal muscle atrophy. PLoS ONE 2021;16:e0252135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang YX, Rudnicki MA. Satellite cells, the engines of muscle repair. Nat Rev Mol Cell Biol 2011;13:127–133. [DOI] [PubMed] [Google Scholar]
- 18. Panda AC, Abdelmohsen K, Yoon JH, Martindale JL, Yang X, Curtis J, et al. RNA‐binding protein AUF1 promotes myogenesis by regulating MEF2C expression levels. Mol Cell Biol 2014;34:3106–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rudnicki MA, le Grand F, McKinnell I, Kuang S. The molecular regulation of muscle stem cell function. Cold Spring Harb Symp Quant Biol 2008;73:323–331. [DOI] [PubMed] [Google Scholar]
- 20. Kwak JY, Kwon KS. Pharmacological interventions for treatment of sarcopenia: Current status of drug development for sarcopenia. Ann Geriatr Med Res 2019;23:98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Su J, Ekman C, Oskolkov N, Lahti L, Ström K, Brazma A, et al. A novel atlas of gene expression in human skeletal muscle reveals molecular changes associated with aging. Skelet Muscle 2015;5:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Riddick CA, Ring WL, Baker JR, Hodulik CR, Bigby TD. Dexamethasone increases expression of 5‐lipoxygenase and its activating protein in human monocytes and THP‐1 cells. Eur J Biochem 1997;246:112–118. [DOI] [PubMed] [Google Scholar]
- 23. Uz T, Dwivedi Y, Qeli A, Peters‐Golden M, Pandey G, Manev H. Glucocorticoid receptors are required for up‐regulation of neuronal 5‐lipoxygenase (5LOX) expression by dexamethasone. FASEB J 2001;15:1792–1794. [DOI] [PubMed] [Google Scholar]
- 24. Goppelt‐Struebe M, Schaefer D, Habenicht AJ. Differential regulation of cyclo‐oxygenase‐2 and 5‐lipoxygenase‐activating protein (FLAP) expression by glucocorticoids in monocytic cells. Br J Pharmacol 1997;122:619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Samuelsson B, Dahlén SE, Lindgren JÅ, Rouzer CA, Serhan CN. Leukotrienes and lipoxins: Structures, biosynthesis, and biological effects. Science 1987;237:1171–1176. [DOI] [PubMed] [Google Scholar]
- 26. Khalil R. Ubiquitin‐proteasome pathway and muscle atrophy. Adv Exp Med Biol 2018;1088:235–248. [DOI] [PubMed] [Google Scholar]
- 27. Yoshii SR, Mizushima N. Monitoring and measuring autophagy. Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tipton KD, Hamilton DL, Gallagher IJ. Assessing the role of muscle protein breakdown in response to nutrition and exercise in humans. Sports Med 2018;48:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Breen L, Phillips SM. Skeletal muscle protein metabolism in the elderly: Interventions to counteract the ‘anabolic resistance’ of ageing. Nutr Metab (Lond) 2011;8:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xia Q, Huang X, Huang J, Zheng Y, March ME, Li J, et al. The role of autophagy in skeletal muscle diseases. Front Physiol 2021;12:638983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bodine SC, Baehr LM. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin‐1. Am J Physiol Endocrinol Metab 2014;307:E469–E484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee MJ, Pramyothin P, Karastergiou K, Fried SK. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim Biophys Acta 2014;1842:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baumann CW, Kwak D, Thompson LV. Sex‐specific components of frailty in C57BL/6 mice. Aging (Albany NY) 2019;11:5206–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xie WQ, He M, Yu DJ, Wu YX, Wang XH, Lv S, et al. Mouse models of sarcopenia: classification and evaluation. J Cachexia Sarcopenia Muscle 2021;12:538–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van der Mierden S, Leenaars CHC, Boyle EC, Ripoli FL, Gass P, Durst M, et al. Measuring endogenous corticosterone in laboratory mice ‐ A mapping review, meta‐analysis, and open source database. ALTEX 2021;38:111–122. [DOI] [PubMed] [Google Scholar]
- 36. Miller RA, Harper JM, Galecki A, Burke DT. Big mice die young: early life body weight predicts longevity in genetically heterogeneous mice. Aging Cell 2002;1:22–29. [DOI] [PubMed] [Google Scholar]
- 37. Partridge TA. Enhancing interrogation of skeletal muscle samples for informative quantitative data. J Neuromuscul Dis 2021;8:S257–S269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pichavant C, Pavlath GK. Incidence and severity of myofiber branching with regeneration and aging. Skelet Muscle 2014;4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kennedy BP, Diehl RE, Boie Y, Adam M, Dixon RA. Gene characterization and promoter analysis of the human 5‐lipoxygenase‐activating protein (FLAP). J Biol Chem 1991;266:8511–8516. [PubMed] [Google Scholar]
- 40. Sanchez‐Galan E, Gomez‐Hernandez A, Vidal C, Martin‐Ventura JL, Blanco‐Colio LM, Munoz‐Garcia B, et al. Leukotriene B4 enhances the activity of nuclear factor‐kappaB pathway through BLT1 and BLT2 receptors in atherosclerosis. Cardiovasc Res 2009;81:216–225. [DOI] [PubMed] [Google Scholar]
- 41. Hosooka T, Hosokawa Y, Matsugi K, Shinohara M, Senga Y, Tamori Y, et al. The PDK1‐FoxO1 signaling in adipocytes controls systemic insulin sensitivity through the 5‐lipoxygenase‐leukotriene B4 axis. Proc Natl Acad Sci U S A 2020;117:11674–11684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat Rev Drug Discov 2015;14:58–74. [DOI] [PubMed] [Google Scholar]
- 43. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: Update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Related to Figure 1: ELISA of 15 (S) ‐hydroxyeicosatetraenoic acid production (15‐HETE), 12 ‐hydroxyeicosatetraenoic acid production (12‐HETE) and 5 ‐hydroxyeicosatetraenoic acid (5‐HETE) production in C2C12 myotubes treated with 10 μM malotilate (Mal) for 24 h. C2C12 myoblasts were cultured as follows (1) 120 h incubation with DM; (2) 96 h incubation with DM, and 24 h treatment of 10 μM Malotilate. ** = p < 0.01 compared to untreated.
Figure S2: Related to Figure 2: Western blot analysis of Akt phosphorylation, Akt expression, and MYH2 expression in in C2C12 myoblasts cultured as follows (1) 120 h incubation with DM; (2) 96 h incubation with DM, and 24 h treatment of 10 μM Malotilate; α‐Tubulin was used as a loading control. Quantification of MYH2 expression and Akt phosphorylation are shown. * = p < 0.05 compared to untreated.
Figure S3: Related to Figure 3: A) qPCR analysis of Alox5 expression in C2C12 myoblasts cultured as follows: (1) 72 h incubation with DM and 48 h incubation with scrambled siRNA; (2) 72 h incubation with DM and 48 h incubation with Alox5 siRNA. B) Western blot analysis of Alox5 expression in C2C12 myoblasts cultured as described in part (A). C). Myotube diameter distribution of C2C12 myoblasts cultured as follows: (1) 72 h incubation with DM and 48 h incubation with DM plus control, scrambled siRNA; (2) Following 72 h incubation with DM, 24 h incubation with DM plus control, scrambled siRNA and additional 24 h treatment with 10 μM Dex plus scrambled siRNA; (3) Following 72 h incubation with DM, 24 h incubation with DM plus Alox5 siRNA and additional 24 hour treatment with 10 μM Dex plus Alox5 siRNA. D) MYH2 immunocytochemistry and myotube diameter of C2C12 myoblasts cultured as follows: (1) 120 h incubation with DM; (2) 96 h incubation with DM, and 24 h treatment with 10 μM Dex; (3) 96 h incubation with DM, and 24 h treatment with 10 μM Dex plus 50 ng/mL LTB4. E) qPCR analysis of atrogin‐1 expression in C2C12 myoblasts cultured as described in part (D). F) MYH2 immunocytochemistry of C2C12 myoblasts cultured as follows: (1) 120 h incubation with DM; (2) 96 h incubation with DM, and 24 h treatment with 10 μM Dex; (3) 96 h incubation with DM, and 24 h treatment with 10 μM Dex plus 50 μM zileuton (Zil) (scale bar = 100 μm). F‐G) Myotube diameter measurement and qPCR analysis of atrogin‐1 and MuRF‐1 expression in C2C12 myoblasts cultured as described in part (F). * = p < 0.05, ** = p < 0.01, *** = p < 0.001 compared to untreated. # = p < 0.05, ## = p < 0.01, ### = p < 0.001, compared to Dex treatment alone.
Figure S4: Related to Figure 3: A‐B) Western blot analysis of MYH2 expression in C2C12 myoblasts cultured with DM or DM plus 10 μM malotilate (Mal) for 24–48 h. α‐Tubulin was used as a loading control. C) Fusion index of confluent C2C12 myoblasts cultured with DM or DM plus 10 μM malotilate for 48 h. D) Fusion index of confluent C2C12 myoblasts cultured with DM plus control, scrambled siRNA or DM plus Alox5 siRNA. E) qPCR analysis of Pax7, Myf5, MyoD, MyoG, and MYH2 in C2C12 myoblasts cultured with DM or DM plus 10 μM malotilate for 24 h.
Figure S5: Related to Figure 4: A) Body weight in mice treated with vehicle alone, Dex, or Dex plus malotilate (Mal) during 14 d treatment (n = 9). B) GAT and liver mass (n = 4). C) Photographs of dissected GAT and liver tissues. D) Fiber cross sectional area of the quadriceps. *** = p < 0.001 compared to untreated, ### = p < 0.001, compared to Dex treatment alone.
Figure S6: Related to Figure 4: A‐B) Western blot analysis of phosphorylated Akt, Akt, and α‐tubulin in the soleus and gastrocnemius muscle. Quantification of phosphorylated Akt relative to Akt is shown. The red arrow indicates phosphorylated Akt. C) SUnSET assay of protein synthesis in the gastrocnemius muscle. α‐Tubulin expression was used as the loading control. D) ELISA analysis of 15‐HETE, 12‐HETE, and 5‐HETE levels in the gastrocnemius muscle. * = p < 0.05, ** = p < 0.01, compared to untreated, ## = p < 0.01 compared to Dex treatment alone.
Figure S7: Related to Figure 5: A) Body weight in aged (21 months) mice after 4 weeks treatment with vehicle or malotilate (Mal) (n = 9). B) Liver mass. C) Cross sectional fiber area of the quadriceps muscle. D) Mean cross sectional area of type 2a and type 2b fibers in the quadriceps. E) Western blot analysis of phosphorylated Akt, Akt, and α‐tubulin in the soleus muscle. Quantification of phosphorylated Akt relative to Akt is shown. The red arrow indicates phosphorylated Akt. F) Alox5AP expression in the gastrocnemius muscle. G) Oil red O staining of the quadriceps muscle (scale bar = 100 μm). H) SDH staining of the quadriceps muscle. (scale bar = 100 μm). ** = p < 0.01, *** = p < 0.001 compared to vehicle treated (n = 4–5 per experimental group).
Figure S8: Related to Figure 6: A) Cellular transcriptome analysis showing total number of upregulated and downregulated genes between C2C12 murine myoblasts cultured as follows (1) differentiation media (DM) for 120 h (Un); (2) DM for 96 h and DM plus 10 μM Malotilate for 24 hours (Mal); (3) DM for 96 h and DM plus 10 μM Dex for 24 h (Dex); (4) DM for 96 h and DM plus 10 μM Dex and 10 μM malotilate for 24 h (Dex_Mal). B) KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis for the Dex compared to Dex_Mal treatment groups. C) Gene ontology (GO) functional analysis for the Dex compared to Dex_Mal treatment groups. D) Selected genes known to be implicated in skeletal muscle atrophy and/or organ development showing differential expression in Dex and Dex_Mal, compared to Un and Dex.
Figure S9: qPCR analysis of Plk‐1, Sod3, STAT6, and Fut4 and Efna5 in human myotubes cultured as follows: (1) differentiation media (DM) for 72 h (untreated); (2) DM for 48 h and DM plus 10 μM Dex for 24 h; (3) DM for 48 h and DM plus 10 μM Dex and 10 μM malotilate (Mal) for 24 h. * = p < 0.05, *** = p < 0.001 compared to untreated, ## = p < 0.01 compared to Dex treatment alone.