

Abstract
The human gastrointestinal tract is home to a dense population of microorganisms whose metabolism impacts human health and physiology. The gut microbiome encodes millions of genes, the products of which endow our bodies with unique biochemical activities. In the context of drug metabolism, microbial biochemistry in the gut influences humans in two major ways: (1) by producing small molecules that modulate expression and activity of human phase I and II pathways; and (2) by directly modifying drugs administered to humans to yield active, inactive, or toxic metabolites. Although the capacity of the microbiome to modulate drug metabolism has long been known, recent studies have explored these interactions on a much broader scale and have revealed an unprecedented scope of microbial drug metabolism. The implication of this work is that we might be able to predict the capacity of an individual's microbiome to metabolize drugs and use this information to avoid toxicity and inform proper dosing. Here, we provide a tutorial of how to study the microbiome in the context of drug metabolism, focusing on in vitro, rodent, and human studies. We then highlight some limitations and opportunities for the field.
OVERVIEW OF THE GUT MICROBIOME
Anatomy of the gastrointestinal tract, succession, and regional localization of gut microbiota
The gut microbiota, from the human infant to the adult
Humans are a composite of both microbial and host cells, and in terms of numbers, microbial cells far outnumber the host cells. Microbial numbers have been estimated to be in the 10–100 trillions, 1 , 2 and a majority of these microorganisms, collectively termed microbiota, are found in the large intestine, especially the colon. The microbiota of the gastrointestinal tract or gut microbiota receives much attention in research; however, it is important to note that of significance are also the microbiota in other compartments of the human body, including the skin microbiota, oral microbiota, airway microbiota, and vaginal microbiota. Unraveling the fundaments underlying the interactions between the microbiota that associate with these various compartments of the human host is critical to understanding the complete physiology of the host, both in good and poor health.
Bacteria colonization of the host occurs immediately after birth, and, as in many ecosystems, this is highly influenced by available nutrients, and, in the case of humans, the mother's milk, with unique nutrients termed human milk oligosaccharides (HMOs). These sugar‐based nutrients have five building blocks, namely glucose (Glc), galactose (Gal), N‐acetylglucosamine (GlcNAc), fucose (Fuc), and the sialic acid N‐acetylneuraminic acid (Neu5Ac). Combinations of these building blocks are linked together in glycosidic bonds to yield diverse oligosaccharides. Based on characterization of greater than 100 different HMOs, it is evident that the composition is highly variable among women. 3 After feeding, the HMOs escape hydrolysis in the acidic stomach and migrate to the small and large intestines where their degradation shapes the microbial and host‐microbe interactions. 3 The microorganisms generally found in the infant gut microbiota are, therefore, well adapted to degrading HMOs to use their glycans as carbon and energy sources, and this observation suggests co‐evolution of this symbiotic relationship.
In similarity to the microbial community structure in many gut systems, the infant microbiota is composed of members from all three domains of life, including the archaea, represented mostly by methanogens, the bacteria, represented by many different lineages, and Eukarya, represented mostly by fungi. Co‐existing with the microbiota are viruses or phages, which are not incorporated in the three‐domain concept, because this classification is based on free‐living organisms and viral life does not meet this criterion. The bacteria are, in general, dominant in the human gut microbiota, and the fungi are present in the lowest numbers. Furthermore, at a given time, the microorganisms present may be native or indigenous (autochtonous) or transients (allochtonous). Under healthy conditions, established microbiota resist colonization by pathogens or parasites due to inability of the “foreign organisms” to compete with the well‐established microbiota (a process known as competitive exclusion). Establishment of the microbial consortia along the gastrointestinal tract is dependent on many environmental factors on the host side, including habitat properties, such as pH, redox state, availability and types of nutrients, water activity, and temperature. 3 In return, the host benefits by the microbiota making available energy from nutrients that are host‐undegradable (i.e., HMOs), promoting cell differentiation (a clear illustration seen with the underdeveloped gastrointestinal tract of a germ‐free compared with microbiota‐colonized animal models), providing protection from pathogens and parasites, stimulating development of the immune system (mucosa education), detoxifying xenobiotics, protecting the integrity of barrier function of the skin and mucosa, and regulating metabolism. 3 , 4
In studies on the infant and its symbiotic microbial population, the recent observation that the infant microbiota structure has consequences on later or adult life has spurred efforts to ensure colonization and presence of the good “bugs” in the microbiota early in life. 5 , 6 The strategy to ensure a good outcome has been two‐fold (i.e., administration of probiotics [live beneficial microbes] and prebiotics [often oligosaccharides that stimulate growth of beneficial bacteria]). This also implies the possibility of achieving the same aim with synbiotics (i.e., administration of a mix of both probiotics and prebiotics). It is anticipated that as our understanding of the beneficial aspects of HMOs and our capacity to synthesize them advance, they will form the foundation of many prebiotics and synbiotics administered to infants, as HMOs represent one of the major differences between human milk and formula milk. In fact, HMOs, such as 2′‐fucosyllactose (2′‐FL) and lacto‐N‐neotetraose (LNnT), have been used to supplement infant formula milk without any adverse effects. 7 This is a promising finding, because HMOs are reported to protect against pathologies such as necrotizing enterocolitis. They also program and modulate the immune system, provide beneficial microbial metabolites, reduce risk of allergies, strengthen and protect intestinal barrier, enhance cognitive development, prevent adhesion of pathogens, and also modulate neonatal viral infection. 8 , 9 , 10 , 11
The type of delivery (i.e., natural birth or cesarean birth [C‐section]) has significant impact on the structure of the infant microbiota, especially during the first few months after birth; thus, we would like to take a brief look at this subject. The differences in the early microbiota in the two delivery systems are ascribed to the observation that naturally delivered infants come into contact with vaginal and fecal microbiota of the mother, with members of the genera Bacteroides and Bifidobacterium dominating in this community. 12 , 13 , 14 , 15 On the other hand, C‐section delivered infants miss these natural sources of microbial inoculation and become more exposed to the microorganisms present on the skin of the mother, the delivery staff, and the delivery environment. Thus, infants delivered through C‐section tend to exhibit gut microbiota with reduced complexity and enriched of organisms often associated with the skin and the environment, such as the genera Enterobacter, Haemophilus, Staphylococcus, Streptococcus, Corynebacterium, and Veillonella. 13 , 16 With time, these differences between vaginally delivered and C‐section delivered babies decrease. A recent comprehensive study in Denmark, however, reported that C‐section delivery babies, compared with vaginal delivery babies, have increased prevalence of four common immune mediated chronic inflammatory diseases (i.e., inflammatory bowel disease, rheumatoid arthritis, celiac disease, and type I diabetes). 17 , 18 , 19 Importantly, there is the potential to partially restore the microbiota of the C‐section babies to that of the vaginally delivered ones through vaginal microbial transfer. 20
Based on the available data, compared to the adult gut microbiota, the human infant gut microbiota is characterized by low diversity, instability of the community structure and therefore a community in a dynamic state. 3 , 21 However, some members, such as the Bifidobacteria, are considered key members, as they are present in high numbers, especially in breast fed infants. On establishment, the core microbiota in general is represented by members of the following (i.e., Bifidobacterium spp., Lactobacillus spp., Streptococcus spp., Faecalibacterium spp., Lachnospiraceae family, Blautia spp., Clostridium spp., Veillonella spp., Escherichia/Shigella spp., Pasteurellales order, and the Bacteroides spp). 3 , 22 During weaning, the microbiota increasingly become more complex and more stable with the community approaching that of the adult, and the Firmicutes and Bacteroidota phyla becoming dominant 23 with their associated properties, such as carbohydrate utilization, vitamin biosynthesis, and xenobiotic degradation. These associated properties occurring early in life are thought to prime the microbiota for an adult diet. 21
Distributions of microbes along the human adult gastrointestinal tract
A summary of the dominant bacterial genera found within the stomach, small intestines, and colon are provided in Figure 1.
FIGURE 1.
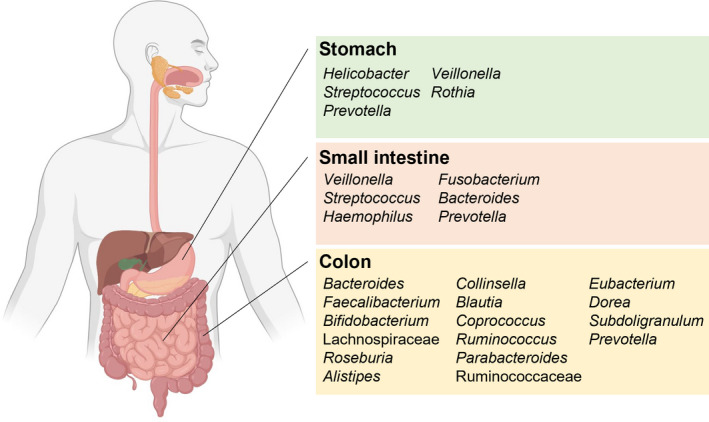
Dominant bacterial genera (or families) across the human gastrointestinal tract. Major bacterial genera identified through 16S rRNA gene surveys and metagenomic studies discussed in the text are highlighted here.
Whereas the stomach was earlier thought to be sterile or only colonized by the bacterium Helicobacter pylori, it has been reported that a number of acid‐resistant strains of Streptococcus, Neisseria, and Lactobacillus were present in this environment, with members of the genera Lactobacillus, Clostridium, and Veillonella seen as transients that derive from the human mouth or refluxed from the duodenum. 24 However, by using 16S rRNA gene sequencing, a more sensitive and reliable approach, it was reported that in 23 healthy adults, five dominant bacterial phyla were present in the gastric mucosa. These were Proteobacteria, Firmicutes, Bacteroidota, Actinobacteria, and Fusobacteria. At the genera level, other than Helicobacter, there were also organisms belonging to Streptococcus, Prevotella, Rothia, and Fusobacterium. 25 Subsequently, another research that sampled from both healthy adults and patients with gastritis, and applied the same molecular approach as above, reported Streptococcus, Prevotella, Neisseria, Haemophilus, and Porphyromonas as the dominant bacteria in the gastric environment. Importantly, as above, sequences reflecting the presence of members of the phyla Firmicutes, Bacteroidota, Actinobacteria, Fusobacteria, and Proteobacteria were also amplified from the samples. 26 Thus, although thought to be colonizable only by the bacterium associated with gastric ulcers (i.e., Helicobacter pylori), it is now clear that other bacteria, such as Streptococcus, Prevotella, Veillonella, and Rothia, most likely their acid‐tolerant strains, are present in the gastric environment, with potential consequences on drug metabolism.
The research on the microbiota in the small intestine pales in comparison to the attention afforded their counterparts in the large intestine, especially the colonic microbiota. It is, however, important that this compartment of the gastrointestinal tract (GIT) is also considered for its capacity to biotransform xenobiotics and drug formulations, as resident microbiota can impact the structure of drugs that traverse or are absorbed in this region. Earlier research analyzed the microbiota present in the small intestine by using culture independent approaches on effluent samples of an individual with an ileostomy. The observation made here was that such effluent samples harbored Escherichia coli, Clostridium sp., and an abundance of bacteria with high G + C DNA contents. Further, using metatranscriptomics to gain insights into what these organisms might be doing, the investigators observed high expression of carbohydrate transporters, especially those assignable to the genus Streptococcus. This observation led the authors to conclude that there is rapid uptake and fermentation of available carbohydrates by the microbiota in the human small intestine. 27 This observation is significant, as the literature abounds with emphasis on the host undegradable polysaccharides being fermented in the large intestine, mostly in the colon. Another study from the same period provided more detailed information on the small intestinal microbiota. Here, it was reported that the microbiota present in the ileal effluent of seven individuals with ileostomies harbored as high as 107–108 bacteria per gram ileal content. On the other hand, archaea, including the methanogens, were present at numbers below the detection limit using quantitative polymerase chain reaction (qPCR). The diversity of the bacterial community was also different from the fecal samples of age‐matched healthy controls analyzed in the study. The authors reported a higher relative abundance of bacterial species in the orders Lactobacillales and Clostridiales that were dominated by Streptococcus bovis‐related species. The bacterium S. bovis can be highly acid resistant or tolerant and strains of this bacterium are found in the cow rumen, where their rapid fermentation of starch leads to accumulation of the end product lactic acid. Their dominance in the cow rumen often culminates in lactic acidosis and bloat, a major economic setback in ruminant animal production. In addition, species of the genus Veillonella and those related to Ruminococcus gnavus, Ruminococcus obeum, and Bacteroides pleibus were also reported. Importantly, the authors noted that there were some interindividual variations of these microbial populations. Furthermore, over a period of 4 weeks, they observed large fluctuations in temporal profiles of the bacterial community in individuals, including between the mornings and the afternoons. 28 A later study that compared the composition of the microbiota associated with the mucosa of healthy individuals and patients of irritable bowel syndrome (IBS), using capsule biopsies from the jejunum, found no differences among the major phyla or genera present in patients with IBS and healthy controls. In all the samples, they found the following phyla, listed in the order of increasing abundance: Fusobacteria, Actinobacteria, Bacteroidota, Proteobacteria, and Firmicutes. Furthermore, the abundant genera in the order of increasing numbers were Fusobacterium, Escherichia, Actinobacillus, Haemophilus, Rothia, Prevotella, Veillonella, and Streptococcus. 29 A more recent report that also compared the small intestinal microbiota of a disease condition to healthy controls discovered that similar to colonic microbiota in dysbiosis due to disease, symptomatic patients of small intestine bacterial overgrowth (SIBO) present lower alpha diversity, richness, and evenness. The symptoms of SIBO are diarrhea, abdominal pain, and bloating, and, in this study, the authors reported the presence of several bacterial genera, including Neisseria, Prevotella, Haemophilus, Porphyromonas, Fusobacterium, and Veillonella in both healthy subjects and patients with symptomatic SIBO, although the proportions were generally lower in the patients. Studies on the microbiota from the small intestine, therefore, suggest that the microbial community in this compartment is less complex and more unstable compared with the microbial community structure in the colon. Importantly, from these studies, bacteria of the genera Veillonella, Streptococcus, Haemophilus, Fusobacterium, and two members of the phylum Bacteriodota (i.e., Bacteroides and Prevotella), are commonly found in the human small intestinal samples, and at the genus level, the organisms do not appear to be very different from those observed in the gastric environment.
Thus, the small intestine is not sterile in terms of microbial presence and metabolic activities, and it is important that future research places emphasis on techniques for their sampling, culturing, and a better understanding and characterization of the community and its potential to impact drug metabolism. Initial assessments could continue to focus on who is present, using the small subunit ribosomal RNA gene approach (16S rRNA and 18S rRNA gene sequencing) or internal transcribed sequence (analysis for fungal populations) analysis, whereas at the same time the extracted community DNA is used for metagenomic sequencing to uncover the genetic potential and diversity. Furthermore, extractions of the associated community RNA should be pursued to determine the metatranscriptome to gain insights to what the observed microbial population is doing in this environment. Attempts to culture strains of the major organisms noted above, for preservation through the culture collections, such as the American Type Culture Collection (ATCC), will also enable a more mechanistic study, through reconstitution of minimal communities to assess small intestine bacterial community impact on drug metabolism.
The human adult colonic microbiota, community structure, stability, and dysbiosis
The succession of different microbes from the early stages of birth to adulthood culminates in a community structure that is dominated by the two phyla Bacteroidota and Firmicutes, which are characterized by many obligate anaerobic bacteria. In a critical report about a decade ago, Arumugam et al. 30 combined 22 stool sample metagenomes of individuals from four different countries and previously published datasets and presented what they referred to as the enterotypes, constituted of three taxonomic clusters found in samples, irrespective of the nation or continent from which the samples originated. These were designated based on the variation of the levels of three genera, the Bacteroides (enterotype 1), Prevotella (enterotype 2), and Ruminococcus (enterotype 3), which was the most frequent among the three classifications. The genera in enterotype 1, 2, and 3 were also noted to co‐occur with the Parabacteroides, Desulfovibrio, and Akkermansia, respectively. Based on detailed analyses of the phylogeny, gene, and pathway levels, the underlying mechanisms underpinning these co‐occurrences were attributed to carbohydrate and protein fermentation for enterotype 1, co‐degradation of mucin glycoproteins in the intestinal mucosa (with Desulfovibrio de‐sulfating the complex substrate structure to enhance degradation by the Prevotella) for enterotype 2, and for enterotype 3, co‐degradation of mucin and uptake of simple sugars. Thus, it appears that energy capture by the three different enterotypes depends on different substrates present in the colon, which should facilitate a harmonious co‐habitation. Importantly, the report showed that at the phylum level, the human colonic microbiota is dominated by three groups (i.e., Firmicutes, followed by the Bacteroidota and then the Actinobacteria). At lower levels were the Proteobacteria, the Synergistetes, Verrucomicrobia, and Fusobacteria. Of note, was also the detection of the Euryarchaeota, which were likely dominated by the methanogens, especially the genus Methanobrevibacter, the most frequently observed in the human gut. At the genus level, the most dominant bacteria were the Bacteroides, followed by the Faecalibacterium, Bifidobacterium, the Lachnospiraceae, Roseburia, Alistipes, Collinsella, Blautia, Coprococcus, Ruminococcus, Parabacteroides, Ruminococcaceae, Eubacterium, Dorea, Subdoligranulum, Prevotella, and then the following at lower abundances (i.e., Anaerostipes, Clostridiales, Akkermansia, Streptococcus, Escherichia/Shigella, Holdemania, Anaerotruncus, Megasphaera, Eggerthella, Peptostreptococcaceae, Dialister, Gordonibacter, and Coprobacillus). In addition to producing diverse metabolites that are absorbed by the host for various metabolic processes, some described below, these enterotypes also produce vitamins, including biotin, riboflavin, pantothenate, ascorbate, thiamine, and folate for absorption by the host. 30 These results were consistent with earlier and later reports that demonstrated that the human colonic microbiome was dominated by the Firmicutes and Bacteroidota phyla with relatively few sequences associated with the Proteobacteria, Actinobacteria, Fusobacteria, and Verrucomicrobia phyla. 31 , 32 , 33 Aside from the bacteria, it is also important to note that the human gut microbiota include eukaryotic cells, such as fungi and also viruses, although most studies fail to consider the two entities during analyses of the microbiota's impact, most likely because the bacterial numbers and functional outputs are so dominant compared to that of their eukaryotic counterparts. Furthermore, it is important for the reader to note that some bacteria detected at a given time could be just transients and not normal inhabitants of the human gut environment.
We will now look at some of the factors that ensure the selection and maintenance of the dominant phyla in the colon. Here, it is important to note that Litvak et al. 34 have nicely outlined how homeostasis of the colonic microbiota is shaped mostly by the colonic epithelial cells (or the colonocytes). In brief, the colonocytes are continually renewed by colonic stem cells derived from the intestinal glands termed crypts of Lieberkühn or simply the crypts. As observed with the gastric pits in the stomach, crypts contain stem cells that can produce different epithelial cell types, including not only the colonocytes, but also enteroendocrine cells and goblet cells. 34 Using a bioassay that measured energy metabolism in mouse colonic crypts and organoids (a 3D culture of colonic crypts), and sorted stem cells, Fan et al. 34 discovered that temporal energy metabolism differed from colonic crypts compared to the sorted leucine‐rich repeat containing G‐protein coupled receptor 5 (Lgr5+) stem cells, which exhibited a Warburg‐like metabolic profile. The Warburg effect, also known as aerobic glycolysis, was discovered by Otto Warburg, and here energy is obtained by cells through glycolysis that converts glucose to lactate, even in the presence of oxygen. 34 , 35 Litvak et al. further note that epithelial cell differentiation requires peroxisome proliferator activated receptor gamma (PPARγ), synthesized by both human and rodent differentiated cells of the colonic epithelium, 34 , 36 to activate fatty acid metabolism, leading to mitochondrial β‐oxidation of long chain and short chain fatty acids, and oxygen consumption through oxidative phosphorylation. 34 , 37 , 38 This energy metabolism of mature colonocytes, characterized by very high oxygen consumption, leads to very low partial pressure of oxygen (<1%) or epithelial hypoxia and ultimately limits free diffusion of oxygen across mucosal surface to the intestinal lumen. This maintenance of very low oxygen partial pressure by the colonocytes ensures the anaerobic environment that supports the growth and function of the colonic microbiota, which is largely constituted of obligate anaerobes 34 that ferment nutrients, such as host undegradable polysaccharides to elicit many beneficial effects, including metabolites such as short chain fatty acids (mostly acetate, butyrate, and propionate) and vitamins to the host. Butyrate, produced by members of the anaerobic microbiota, such as Faecalibacterium prausnitzii, is a major source of energy for the colonic mucosa, especially of the distal colon. 38 Importantly, Litvak et al. note that because dysbiosis is often associated with increased numbers of facultative anaerobic bacteria (organisms that can respire either in the presence or absence of oxygen) in the gut, then many processes that lead to this condition derive from disruption of the capacity of the colonic epithelia to maintain anaerobic conditions in the lumen. Based on this insight, they explain some of the underlying mechanisms associated with dysbiosis and maintenance of gut homeostasis as follows: anaerobic fermentation leads to production of short chain fatty acids, which bind to G‐protein coupled receptors in the intestine to maintain levels of regulatory T‐cells and thereby prevent intestinal inflammation. Furthermore, as noted above, the short chain fatty acid butyrate (a fermentation product) is a preferred energy source of colonocytes, and it activates β‐oxidation to maintain low oxygen partial pressure; however, antibiotics may deplete important anaerobic organisms that produce the short chain fatty acids in the colon to help maintain the anaerobic conditions. The depletion of important anaerobic bacteria due to antibiotic use may lead to silencing of PPARγ signaling, which will result in loss of maintenance of anaerobic conditions, with a concomitant reduction of short chain fatty acids levels and thus impaired regulatory T‐cell activation or reduced circulating regulatory T‐cells, as observed in mice. 34 , 39 , 40 Under such conditions, aerobic or facultative anaerobic bacterial growth may be stimulated and hence lead to a microbial community different to the one dominated by the anaerobes of the Firmicutes and Bacteroidota phyla, a condition often referred to as dysbiosis and associated with many disease states impacted by the gut microbiota.
Interindividual variability in the gut microbiota
Whereas one may expect the microbial community structures described above in the healthy human infant or adult, many factors lead to interindividual variations; therefore, in experiments impacted by the gut microbiota, it is essential that meta‐data, such as the diet, age, sex, and the use of antibiotics and prescription drugs are considered. The existence of such variations may be assessed at the initial stage of the research by determining the baseline microbiota of the cohorts to be studied. However, even where there are consistencies in the community profiles, the experimenter needs to understand that having the same proportions of individual microorganisms at the species level does not necessarily mean that the same organisms are present in the different individuals harboring them. This is because at the strain level, the genomic content may be vastly different, sometimes through acquisition of extra genetic information by the process known as horizontal gene transfer. A simple example here is the extensive genomic differences between two E. coli strains, such as strain K12 which is harmless and the highly pathogenic strain O157:H7. 41 In other words, all E. coli, based only on 16S rRNA gene sequence, are not the same, and this applies to strains of other bacteria.
Below, we would like to look at a few factors (Figure 2) that may lead to interindividual variations in a cohort to be used in a pharmacological study.
FIGURE 2.

Factors affecting the gut microbiota. Multiple factors influence the composition and structure of the gut microbiota. A subset highlighted in the text are diagrammed here. GI, gastrointestinal; HMO, human milk oligosaccharide; NSAID, nonsteroidal anti‐inflammatory drug.
Diet
The diet of an individual can have significant impact on the microbiota present in the GIT, especially the colon. Two major sources of energy are recognized for the proliferation of microbes in the gut. One source is endogenous and derives from both mucopolysaccharides and glycoproteins of the host, 42 , 43 whereas the other is exogenous and derives from host dietary sources, an example being the host undegradable polysaccharides. 44 , 45 , 46 Degradation of the mucin associated nutrients, which has been noted to occur especially during exogenous sugar (or dietary fiber or nutrients) deprivation has pathophysiological implications, as mucin has protective function against pathogens in the GIT. 42 , 43 Under this topic, however, we will briefly look at how the diet may shape the gut microbiota and therefore lead to interindividual variability in microbiota composition. Dietary components that escape degradation in the gastric compartment, due to a lack in the host (human) genome of the right hydrolytic enzymes to release the component sugars, lead to significant fermentation in the colon. The flow of these nutrients to the large intestine or the colon, therefore, in part explains the dense concentration of microorganisms in this compartment of the GIT. Among the undegradable polysaccharides are complex substrates, such as pectins, arabinoxylans, and resistant starch. In vitro studies that have cultured representatives of the human colonic bacteria demonstrate that these recalcitrant nutrients are initially degraded by a group of organisms equipped with the required set of enzymes to depolymerize the polysaccharides to shorter chains. 44 , 45 , 47 Although these organisms transport the shorter chain sugars (oligosaccharides) intra‐cellularly for metabolism, the less complex substrates or oligosaccharides become available to other members of the gut/colonic microbiota to further degrade and ferment for carbon and energy in a process named “cross‐feeding.” 48 , 49 Other forms of cross‐feeding involve some colonic microbiota members using the end products of others, and this could even be gaseous end products, such as hydrogen and carbon dioxide, which serve as energy sources for colonic methanogens and acetogens. The methanogens and acetogens will metabolize these two simple gases to produce methane and acetate, respectively, while generating energy and cellular building blocks for growth.
Both genome sequencing and experimental evidence suggest the Bacteroidota as major contributors to depolymerization of complex polysaccharides. 44 , 45 , 50 , 51 The literature shows that members of this phylum are equipped with gene clusters that allow them to rapidly sense undegraded nutrients, especially polysaccharides, and thus allowing them to efficiently degrade and ferment these sugar‐rich nutrients. These gene clusters, almost uniquely found in the Bacteroidota, have been designated Polysaccharide Utilization Loci (PULs), and each locus or cluster usually targets a unique polysaccharide. 52 Thus, there are PULs for arabinoxylans, starch, pectin, and β‐mannan, to name just a few, and some members of this phylum are reported to contain greater than 80 PULs in the genome, 52 , 53 , 54 , 55 a reflection of the versatility of these bacteria. As noted above, the Bacteroidota are one of the dominant phyla in the colon, and with the ability to carry out degradation of many host‐undegradable energy sources, they likely play a major role in the dynamics of energy availability in the human gut. As such, dietary regimen heavy with plant fiber will have the tendency to enrich for members of this phylum. Members of the phylum Firmicutes also have the capacity to degrade dietary fiber; however, their contributions and mechanisms have not been studied as extensively as the Bacteroidota. The capacity of the Firmicute Ruminococcus bromii to metabolize resistant starch, with an intricate enzymatic machinery known as the amylosome which incorporates diverse enzymes in a complex structure linked to the cell surface, has been well‐studied, 56 , 57 , 58 , 59 and the capacity to degrade xylan and dietary β‐mannan has also been well‐characterized in another Firmicute Roseburia intestinalis. 60 , 61 In line with the foregoing, it is expected that the dietary composition can have a profound impact on the human gut microbiota composition, and Sonnenburg and Sonnenburg noted that pervasiveness of the Western diet, which is low in microbiota‐associated carbohydrates (MACs), especially dietary fiber, has selected for a microbiota that is different from that of human populations living traditional lifestyles. The interaction of this altered microbiota with the host then leads to immune dysregulation, and this may explain many inflammation‐linked diseases associated with this dietary lifestyle. The underlying mechanisms may be attributable to low production of short chain fatty acids by the colonic microbiota of those on a Western diet compared to those on diets high in MACs. As explained above, short chain fatty acids have many beneficial functions, including modulation of inflammation. 62 Furthermore, diets rich in proteins may result in the colonic microbiota enriching for populations with enhanced fermentation of proteins to branched chain fatty acids, ammonia, hydrogen sulfide, phenolic and indolic compounds, polyamines, and amines, and these are metabolites associated with obesity and complications, such as nonalcoholic fatty liver disease and insulin resistance. 48 , 63 Overall, the available data demonstrates that diets rich in MACs promote diversity in the gut microbiota, and this is usually associated with beneficial attributes, whereas diets of the Western type, which is rich in refined sugars, additives, and fat, but low in MACs is associated with low microbiota diversity, missing critical microbial lineages, and very prone to disease. The dietary lifestyle, therefore, plays a critical role in interindividual variability of the microbiome, with its concomitant attributes.
Age
As discussed above, the microbiota in the human infant tends to be unstable, whereas that of the adult is generally found to be stable. In the elderly, however, it is reported that the microbial community in the gut is characterized by instability, as seen in the infant, in addition to becoming prone to recurrent dysbiosis. The underlying causes are not necessarily only genetics, but derive from a combination of factors, some of which were listed above. The physiological component can be assigned to the changes occurring in the GIT and decline in immune function. However, these may be compounded by the diet and lifestyle of the individual. Furthermore, decreased motility of the GIT in the elderly leads to characteristics known to significantly impact microbial proliferation in a gut system. These include transit time or passage rate, which allows longer durations of bacterial accessibility to nutrients and absorption of end products. Other conditions that are thought to impact the microbial community structure in the elderly are reduced diversity and thinning of the mucosal lining, all leading to loss of integrity of the intestinal barrier with its associated frequent inflammation. 64 , 65 , 66 Thus, although there is no unique microbiota described in association with aging, the available data have allowed its description in general terms, as characterized by reduced diversity, shifts in the dominant species, a decline in beneficial bacteria, increased facultative anaerobic bacteria, decreased short chain fatty acids, and changes in the ratios among the short chain fatty acids when compared with younger adults. 67 , 68 , 69 Salazar et al. 66 , 67 , 70 and others note that these changes in the microbiota with aging, with its associated increase in oxidative stress and inflammation, coupled with antibiotics and nonsteroidal anti‐inflammatory drug treatments, predispose the individual to growth and infection with pathogens, such as Clostridioides difficile and Helicobacter pylori. The insights gained based on these various findings, therefore, clearly show that age has an impact on the gut microbiome of individuals; however, it also shows that augmentations such as the use of both probiotics and prebiotics or their combinations may also be harnessed to combat the shift from the stable microbiome of adulthood to the unstable and disease prone microbial community observed in elderly individuals.
Antibiotics
We live in an age of high antibiotic use, and these therapeutics, especially those with broad spectrum activities, can exert a profound effect on the composition of the human gut microbiota. Antimicrobials are of fundamental importance in patients with sepsis, and, in some cases, they are life‐saving. However, their administration may also lead to harm and damage to host‐associated microbiota and hence impair its myriad of functions. For such reasons, cohorts used for studies that are impacted by the microbiota should be carefully screened for their history of antibiotic use. It is noted in the literature that the effect of a given antibiotic is dependent on the initial composition of the gut microbiota, with an individual originally harboring a microbiota of reduced diversity likely to further present community destabilization and increased load of pathogens. A broad‐spectrum antibiotic, therefore, has the potential to impair the stability of the major phyla responsible for maintaining gut microbiota homeostasis, including disrupting the low oxygen partial pressure conditions that support anaerobes in the colon. Such conditions enrich for facultative anaerobes with its associated impairments, including reduced colonization resistance, imparted by the native microbiota. An end result is an increased load of pathogenic organisms. In contrast, a healthy and stable microbiota, as found in the young adult, with its associated high diversity of microorganisms, has been found to be more resilient, likely through modulation of the antibiotic effect by a large library of antibiotic resistant genes in the microbial community. 48
Many of the antibiotics in use are highly potent, and their effect on the microbiota or bacterial diversity is often very rapid (i.e., soon after antibiotic administration), only to begin to rebound to a microbiota composition similar to the original, although complete restoration is often elusive. 48 , 71 However, the effect of antibiotics on the microbiota can also be long lasting, and in a study based on a cohort of 30 young adults in Sweden, Rashid et al. 72 reported that whereas administration of clindamycin and ciprofloxacin, both broad spectrum antibiotics, did not impact the skin anaerobic microbiota and also its proportion of antibiotic‐resistant anaerobic bacteria; among the same group, the relative proportion of ciprofloxacin‐resistant Bifidobacteria increased in the short (day 11) and long‐term (12 months) postdosing compared to the placebo group, and a similar observation was also made for clindamycin‐resistant Bacteroides spp. These finding are examples of the long‐term effects of antibiotic use. One should also note that the extent of the antibiotic disturbances is influenced by several factors, key among them being the spectrum of the drug, the dose, the route of administration, pharmacokinetic and pharmacodynamic properties, and in vivo inactivation. 72 Additionally, some untargeted effects of antibiotics on the microbiota are now leading to new challenges, and some of these are neurological diseases that have posed big challenges to the medical field. As an example, disturbances of the intestinal microbiota due to antibiotics administration are now being linked to the pathogenesis of depression, and this has been attributed to a change in the gut‐brain axis. 73 , 74 Furthermore, dysbiosis induced by broad spectrum antibiotic use is also being considered in the pathophysiology of Alzheimer's disease, 75 and although a study that examined the microbiota of healthy controls compared with several major psychiatric disorders showed no difference with the major depressive disorders, bipolar disorder, and schizophrenia in the number or distribution of bacteria (α‐diversity) in the groups, the study found clear difference in overall community composition (β‐diversity) in people with and without mental disorders. 76 These studies that capture interindividual or inter‐group diversity in the gut microbiota presents the possibility to harness new mechanistic insights to modulate or develop intervention based on the gut microbiota to improve both brain and mental health.
Non‐antibiotic drugs
Whereas it is obvious that antibiotic use will impact the structure of a microbial community and therefore the microbiome in an individual, researchers of microbiome studies in drug metabolism should also be cognizant that non‐antibiotic drugs have also been found to exert extensive impact on the gut microbiota. Some of the human‐ or host‐targeted drugs of note are in the class of proton pump inhibitors, nonsteroidal anti‐inflammatory drugs, and atypical antipsychotics. 77 , 78 In screening the compounds from the Prestwick Chemical Library (off‐patent US Federal Drug Administration‐approved compounds with high chemical and pharmacological diversity), Maier et al. 77 found that many species with high relative abundances in healthy humans were significantly more susceptible to human targeted drugs. Some of the species of note were the butyrate producers Eubacterium rectale, Roseburia intestinalis, and Coprococcus comes and propionate producers including Bacteroides vulgatus, Prevotella copri, and Blautia obeum. The microbial organisms that tended to be the most drug‐resistant in this screening effort were members of the γ‐proteobacteria, especially Bilophila wadsworthia and E. coli strains. In a different report that used entire microbiomes in the screening, instead of individual organisms, Li et al. 79 found that non‐antibiotic compounds, such as berberine, pravastatin, diclofenac, and ibuprofen, decreased the abundances of 10 bacterial genera, especially in the phylum Actinobacteria, including Eggerthella and Gordonibacter, in their in vitro cultures. Furthermore, a more recent report that used fecal samples from over 4000 individuals reported that administration of multiple drugs, polypharmacy, led to a microbiome characterized by a higher abundance of bacterial species more associated with the upper GIT and nosocomial pathobionts, and also reduced short chain fatty acid metabolism. 78
Host genetics
Microbial colonization and proliferation in an environment depend on the chemical and physical landscape, and this is not different from the human host and its microbiota, where the host genetics define these important factors. 80 Hall et al. 80 note that genetic differences could be the underlying factors that define the similarities and differences observed in the microbiomes of different mammalian species, and the observation that the gut microbiome of monozygotic twins is more similar compared to that of dizygotic twins is supportive of this concept. 81 Host factors that have been reported to serve as drivers of the human‐microbiome co‐evolution include diet‐sensing, metabolism, and immune defense, 80 , 82 with antimicrobials peptides, such as the cationic α‐defensins providing some insight to the molecular basis for this observation. These peptides are imbued with antimicrobial, antiviral, antitoxic, and binding properties, 83 and these are attributes known to shape microbial community structures. Buffington et al., 84 using a mouse model, also recently discovered that host genetic variation, the microbiome, and their interactions underlie certain neurodevelopmental disorders. Thus, whereas we do not present an extensive analysis of this topic, it is important that the reader notes that host genetics impact microbiome structure and this in turn may impact drug metabolism.
Enzymology of the gut microbiota
The human gut microbiome encodes numerous enzymes with potential roles in metabolism of xenobiotics and drugs, as previously reviewed. 85 Some of these enzymes have been biochemically characterized 44 , 86 , 87 , 88 and others have functions predicted by bioinformatics, based on sequence similarities to previously characterized enzymes. It is important that the reader notes that assigning function to a polypeptide based only on similarity at the amino acid sequence level can be problematic, as polypeptide sequences may be similar but exhibit different enzymatic activities. 87 , 89 Thus, unless the identity to a characterized enzyme is very high (e.g., 70% or higher), with critical residues including those involved in catalysis and substrate binding conserved, care should be taken in assuming that a polypeptide has the same enzymatic activity as related sequences. If the function of a polypeptide sequence is critical to the purposes of the reader, it is best then to express the protein to determine or confirm the associated activity. As noted above, many enzymes from the human gut microbiota have been functionally characterized and more are discovered each year; thus, here, we will focus only on a few pertinent classes of enzymes, especially those related to our own research (i.e., energy metabolism and bile acid metabolism). The microbiome, however, encodes sets of enzymes essential for all the cellular metabolic processes of the individual cells in the microbiota, including those involved in cell division, DNA transactions, protein synthesis and degradation, and xenobiotic degradation. Many of the enzymatic activities are shared with the host, although the host polypeptide sequences may be different from that observed in the microbiome. If host polypeptide sequences are, however, of the same family as the microbial ones, care must be taken in attempts to modulate either activity, because this may end up impacting both the host and microbial enzymes. Based on the literature, the enzymes of the microbiome that have been most extensively characterized are those involved in carbohydrate metabolism, and this is perhaps because they are the enzymes needed to access the energy required to sustain the microbial community in their symbiotic relationship with the host.
The human infant microbiota, similarly to observations with the adult gut microbiota, have evolved genes coding for the enzymes needed to capture energy from the dietary components undegradable by the host. The microbiome of the infant is enriched with genes encoding fucosidases, β‐hexosaminidases, Lacto‐N‐biose phosphorylases, sialidases, and β‐galactosidases, and these are commonly produced by Bifidobacterium spp., although other organisms, such as Akkermansia, usually described in the adult microbiota but may be detected as early as 1 month in the infant microbiota, are also predicted to possess some of these enzymes. 90 Whereas these unique enzymatic activities are required for the members of the infant microbiota to use HMOs, direct characterization of the enzymes through cloning, expression, and purification of the polypeptides for biochemical characterization are rare, especially compared with counterpart enzymes from the adult microbiome. It is, therefore, common to see the infant microbiota derived bacteria directly grown on HMOs to demonstrate utilization of these substrates and by inference the expression and function of the enzymes noted above. 91 , 92 , 93 , 94 , 95
The human adult gut microbiota encodes diverse enzymes that target the complex undegradable carbohydrates flowing into the colon, and functional homologs of the activities that cleave the different linkages have been biochemically characterized especially in the Bacteroidota. In this phylum, which includes the Bacteroides spp. and Prevotella spp. commonly found in the human microbiota, the genes that target depolymerization of a particular carbohydrate (e.g., arabinoxylan, pectin, β‐mannan, and arabinan) are usually encoded in a cluster on the genome. Each gene cluster is therefore called a PUL. Thus, in a genome, there may be many different loci encoding the enzymes needed to completely hydrolyze the different polysaccharides to their unit sugars for fermentation. Because the host undegradable polysaccharides, such as arabinoxylan and pectin, have complex structures with many different linkages, it means that many different enzymatic activities are required for their complete hydrolysis. These enzymes belong to different families, including glycoside hydrolases (GHs), polysaccharide lyases, carbohydrate esterases, sulfatases, and peptidases. 55 There have been efforts by many groups, including our own group, to demonstrate the function of each enzyme encoded in some of these PULs. As an example, the extensive enzymes harnessed to degrade α‐mannan by the gut Bacteroides spp. have been biochemically characterized. 96 In this report, the authors, using α‐mannan from yeast, find that Bacteroides thetaiotaomicron carries out limited surface cleavage of α‐mannan to generate oligosaccharides that are transported to the periplasm where they are extensively degraded for fermentation. 96 In another study, degradation of pectin, with a backbone of galacturonic acid and rich in fruits and vegetables, was investigated. The authors, using the same model organism as above, demonstrated that different pectin PULs were responsible for the degradation of specific pectin molecules. Furthermore, this research led to the discovery of two hitherto unknown GH enzymes. The enzymatic machinery that depolymerizes the backbone of rhamnogalacturonan I comprised of nine different enzymes, and the action of these enzymes did not only lead to energy accessibility to the organism but also nutrients to cross‐feed other members of the microbial community. 51 Arabinoxylans, the degradation of which we study in our laboratory, is also a complex polysaccharide found in many human foods especially cereals, including rice, wheat, oats, and barley. Arabinoxylans occur in different complexities; however, the backbone is constituted of xylose chains that are linked together in β‐1,4 glycosidic linkages. This chain of the 5‐carbon sugar is decorated with arabinose (also a 5‐C sugar), methyl glucuronic acid, and acetyl group side chains, and the arabinose may be linked frequently to the plant phenolic compound ferulic acid, depending on the complexity of the arabinoxylan. Based on the complex nature of arabinoxylans, it requires multiple enzymes to efficiently depolymerize the polysaccharide to release its sugar in monomeric forms for fermentation by the human gut microbiota. Our research, using bacterial growth, transcriptomics, biochemical, and protein structural studies have demonstrated that several Bacteroides spp., derived from the human gut microbiota, enlist different PULs to sense and degrade simple 44 , 86 , 97 and complex 45 arabinoxylans. Importantly, whereas the polypeptides of the enzymes they use to degrade the two forms of polysaccharides are different, the enzymatic activities are the same. The enzymatic activities utilized to completely hydrolyze these polysaccharides are illustrated in Figure 3a; they include an endoxylanase to cleave the backbone to shorter chains or xylo‐oligosaccharides and a β‐xylosidase to cleave the oligosaccharides into individual xylose sugars. To remove the side‐chains of the polysaccharide, in order to enhance its degradation, the organisms also encode in the PULs α‐glucuronidase activity to cleave the methyl glucuronic acid side‐chain. Furthermore, acetyl‐xylan esterases are encoded to cleave the acetyl groups from the xylose‐backbone, arabinofuranosidases are encoded to cleave the arabinose‐linked to the xylose‐backbone, and ferulic acid esterases are encoded to cleave the ferulic acid linked to the arabinose in complex arabinoxylans. It is also of consequence that we discovered that the human gut Bacteroides spp. that ferment complex arabinoxylans cleave the ferulic acid and that it accumulates in their end products of fermentation. 45 Importantly, because this plant phenolic compound is a potent antioxidant, and the gut epithelial cells are equipped with its transport system, it can be concluded that diets that contain arabinoxylans are a source of antioxidants, which can confer health benefits to the host, including combating reactive oxygen species and therefore inflammation. 98 , 99 To test this hypothesis, we evaluated administration of the fermentation end products of the gut bacterium Bacteroides intestinalis (grown on complex arabinoxylans) both in vitro and in vivo (using mice) and demonstrated that the end products with the accumulated ferulic acid enhanced host immunity. 100
FIGURE 3.

Representative metabolic activities of the gut microbiota. (a) Arabinoxylan metabolism by gut Bacteroidota. Arabinoxylan consists of a beta‐1,4‐linked xylose backbone decorated with 4‐O‐methylglucuronic acid, acetate, and arabinose linkages. Bacteria encode multiple different enzyme activities required to hydrolyze these individual bonds which include endo‐xylanases, β‐xylosidases, α‐glucuronidases, acetyl‐xylan esterases, and arabinofuranosidases. (b) Bile acid 7‐α‐dehydroxylation. Several strains dehydroxylate primary bile acids (e.g., cholic acid), converting them to secondary bile acids (e.g., deoxycholic acid). (c) Digoxin is reduced by cardiac glycoside reductases forming dihydrodigoxin. (d) Bacterial glucuronidases convert the inactive metabolite of irinotecan (SN38G) to the active metabolite (SN38).
The foregoing enzymes are mostly hydrolytic enzymes and, although in this section we do not address how they may impact pharmaceutical drugs, it is important to note that hydrolytic enzymes are known to deconjugate drug metabolites, and such enzymes may include sulfatases, acylesterases, glycosides, and glucuronides. 101 , 102 In subsequent topics, we will discuss some of these enzymes and their interactions with drugs.
We will now turn our attention to a different set of enzymes. Members of the gut microbiota are equipped with the enzymatic activities that convert primary bile acid to secondary bile acids, such as deoxycholic acid (DCA) and lithocholic acid (LCA; Figure 3b). It is reported that both DCA and LCA can accumulate at significant concentrations in the gut environment and have the capacity to block the growth of the significant gut pathogen Clostridioides difficile and further exert other impacts on host physiology. 103 The Fischbach group further reconstituted the enzymatic pathways, including uncovering steps that involve Fe‐S flavoenzymes, for the conversion of the primary to the secondary bile acids. 103 A more recent study reports new bacterial enzymatic activities in bile acid metabolism and its impact on gut health. The activities were from the human adult gut microbiota and demonstrated that 5α reductase (5AR) and 3β‐hydroxysteroid dehydrogenase (3β‐HSDH) were responsible for the production of isoallolithocholic acid (isoalloLCA), a secondary bile acid that exhibits potent antimicrobial activity against Gram‐positive, and not Gram‐negative, multidrug resistant pathogens, including Clostridioides difficile and Enterococcus faecium. 104 The former organism, which is studied extensively in the field of the human gut microbiome, causes diarrhea and severe and sometimes life‐threatening forms of colitis, whereas the latter is known to cause diverse diseases, including endocarditis. The two enzymatic activities, which were present in gut isolates of Odoribacteraceae strains enriched from fecal samples of a centenarian, further demonstrate how metabolites of bacterial bile acid metabolism may aid in reducing the risk of bacterial pathogen infections and therefore help maintain homeostasis in the gut or colonic environment. 104
In the genus Clostridium, Ridlon et al. 105 have demonstrated the expression of the genes in a bile‐acid inducible operon (bai) during growth of Clostridium hylemonae and Clostridium hiranonis on cholic acid (CA) and DCA. This group is systematically unraveling and assembling the enzymes that orchestrate some of the major enzymatic reactions of bile acid metabolism, including deconjugation, oxidation, isomerization, and dehydroxylation. For the bai gene‐encoding clostridia, they propose that expression of bile acid hydroxysteroid dehydrogenase enzymes regulates entrance into and flux through the bile acid 7α‐dehydroxylating pathway, in addition to maintaining adequate levels of the end‐product, DCA, in the gut environment. 106 The group has also recently identified genes encoding enzymes involved in the oxidation 87 , 106 , 107 and epimerization 108 of DCA derivatives, principally in gut Firmicutes and Actinobacteria. Together with others in the field, they have reported novel genes involved in oxidation and epimerization of the C3‐position, 87 , 109 , 110 and in a new twist in bile acid metabolism, this group has hypothesized that in Eggerthella lenta, bile acid oxidation provides reducing equivalents that feed into the Wood–Ljungdahl pathway. 107
MICROBIOME INFLUENCE ON HOST DRUG METABOLISM
Modulation of host metabolism enzymes
Microbes colonizing the GIT influence expression of host metabolic genes expressed by cells lining the GIT as well as those within the liver – a major site for host metabolism. Although there are several different enzyme families that may be regulated by the microbiome (e.g., UDP‐glucuronosyltransferases, sulfatases, flavin monooxygenases, esterases, etc.), most literature has focused on the cytochrome P450 superfamily. Therefore, while we focus on P450s in the following section, we note that the microbiome likely alters expression of a much broader set of host metabolic genes.
Cytochrome P450s
Cytochrome P450s, also designated P450s or CYP, constitute a superfamily of enzymes that are membrane‐bound hemoproteins and play a significant role in the detoxification of xenobiotics, cellular metabolism, and homeostasis. Their induction and inhibition can help explain the mechanisms underlying many drug–drug interactions. The P450s are a very large family of proteins, and Nelson reported that extensive sequencing efforts have provided over 300,000 CYP sequences, 111 and their importance in cellular life is further substantiated by their presence across the three domains of life. 111 , 112 , 113 In humans, the drug metabolizing CYPs are expressed at different levels in different organs, including the liver, intestines, stomach, kidneys, lungs, trachea, and olfactory mucosa. Several P450s have been characterized in humans, and 12 assigned to the CYP1, CYP2, and CYP3 superfamily are responsible for the biotransformation of 70–80% of clinical drugs. 114 , 115 The expression of P450s is impacted by many factors, including genetic polymorphisms, cytokines, hormones, disease state, sex, and age. As we gain a better understanding of how such differences impact expression of P450s, the clinical significance on drug metabolisms, including individual variations in disease susceptibility, adverse drug reactions, therapeutic efficacy of drugs, and dose requirement are becoming more apparent. 114 , 116 The P450s are also involved in the metabolism and biosynthesis of endogenous biomolecules. 115 As an example, the most abundant P450 in the liver CYP3A4 is responsible for the metabolism of numerous xenobiotics and endobiotics. The induction of CYP3A4 is under the influence of many compounds of both exogenous and endogenous origin. In terms of induction by endogenous biomolecules, elevated concentrations of secondary bile acids induce expression of CYP3A4 through the pregnane X receptor (PXR). Gnerre et al. 117 have, however, also shown that physiological concentration of the primary bile acid chenodeoxycholic acid regulates expression of CYP3A4 via the bile acid receptor farnesoid X receptor (FXR). High bile acid concentrations are toxic, and the two receptors, FXR and PXR, are harnessed to tightly regulate their synthesis and degradation, respectively. 118 Metabolites derived from our gut microbiota, however, have the capacity to regulate expression of FXR and PXR, and thus one can infer that our gut resident microorganisms can influence the dynamics of P450s. In fact, LCA, a secondary bile acid, derived from bacterial reductive action on chenodeoxycholic acid, can activate PXR, and this receptor functions as the major xenobiotic nuclear receptor controlling the expression of many xenobiotic metabolizing enzymes. 115 To illustrate the inter‐relationships among these factors, we will draw on a report based on lipopolysaccharide (LPS), a major component of the outer membrane of Gram‐negative bacteria and associated with pro‐inflammatory activity. Here, the investigators observed that injection of LPS in mice resulted in upregulation of the expression of proinflammatory cytokines while downregulating expression of CYP3A11 and CYP3A4 in the liver. By also administering ginsenosides, compounds with reported anti‐oxidation, anti‐inflammation, and anti‐apoptosis properties, they observed a reversal of the downregulated expression of CYP3A11 and CYP3A4 in the mice. These dynamics were confirmed with human HepG2 cells. To determine the underlying mechanism, the investigators used a reporter gene system to demonstrate that the ginsenosides enhanced rifampicin‐induced PXR transactivation of the CYP3A4 promoter, suggesting that the ginsenosides relieved the downregulatory effect of LPS on the PXR promoter. The result was a higher expression of the receptor (PXR) and a concomitant induction and higher expression of CYP3A4 in the liver. 119 This experiment clearly demonstrated how a biomolecule or metabolite from the gut microbiota can interact with a host receptor, and thus act as a signaling molecule to regulate the expression of CYP3A4, a P450 with potent xenobiotic metabolizing activity. 115 Importantly, these effects of microbiota metabolites can be either local (e.g., in the colonic environment) or in a distant organ, such as the liver. 120
Direct modification of therapeutic drugs
The gut microbiota has the capacity to bio‐transform exogenous compounds that originate from the diet, the environment, and also therapeutic drugs. These bio‐transformations can lead to products that are either beneficial or elicit adverse effects on the host metabolism. The inherent potential of the gut microbiota to bio‐transform therapeutic drugs is not new knowledge, as the metabolism of pro‐drugs, such as azo drugs, were known to require microbial processes for their activation. The drug prontosil, for example, was found to be active in vivo, but not in vitro, 121 and Gerhardt Domagk later discovered that the antimicrobial effect of prontosil was activated by its reductive cleavage (azo reduction) to sulfanilamide. Importantly, this activity was found to be associated with microbiota present in both the human intestines and the skin, and also environmental samples and to a lesser extent in human liver azoreductase. 122 The broad spectrum of the microbial species associated with the human gut microbiota and their encoded or elaborated enzymatic reactions; however, suggest that the impact of the microbiota on therapeutic drugs could be profound. Thus, the potential of the microbiome in bio‐transformation reactions should be carefully considered in studies on patient drug responses. Several recent reviews, including those by Wilson and Nicholson 123 and Guthrie and Kelly, 124 present comprehensive views on how the human gut microbiota exerts both direct and indirect pharmacologic and toxicologic drug effects in their symbiotic relationship with the host, and readers needing more detailed insights are encouraged to read these reviews. Underlying the most important biotransformations are those that involve microbial reductive and hydrolytic reactions, but, in addition, those involving, but not limited to, reactions of decarboxylation, dealkylation, dehalogenation, deamination, dehydroxylation, acetylation, deacetylation, deacylation, and demethylation. Examples of these processes are readily found in the literature. Therefore, we will focus only on a few examples to illustrate the consequence of these reactions emanating from the symbiotic relationships with our microbes.
The drug digoxin is one of the oldest medications used to treat various heart conditions. The lactone ring of the drug can be reduced to compounds such as dihydrodigoxin or its aglycone dihydrodigoxigenin, and both reduced forms have diminished activity on the heart. Lindenbaum et al. 125 determined in a bioavailability study that in one third of healthy subjects, digoxin reduction products constituted greater than 5% excretion of digoxin and its metabolites. However, the reduced products were not seen in the first 8 h after a single dose, and maximal excretion was observed in the second day. Furthermore, excretion was reduced on administration of the antibiotic erythromycin, and administration of the drug through the intravenous route also led to less reduced products compared to oral administration. The authors further demonstrated that during oral administration, bioavailability tended to exhibit an inverse relationship with drug preparation, and these critical observations led the investigators to posit that the variabilities are due to differences in intestinal microbiota. In subsequent experiments, the authors reported that in about 10% of patients given digoxin, there was substantial conversion of the drug to the cardio‐inactive reduced metabolites. Further analyses demonstrated that stool samples from three of the “excretors,” and not from the “non‐excretors,” converted digoxin to its reduced metabolites, and this activity could be ablated with the antibiotic tetracycline or erythromycin. The serum concentrations of digoxin also increased about two‐fold after administration of antibiotics, further strengthening the initial hypothesis of the investigators that digoxin inactivation is gut microbiota‐mediated. 126 The bacterial culprit of this reductive process was finally brought into culture and identified as Eggerthella lenta (formerly Eubacterium lentum), a common anaerobe residing in the human colon. Of important significance was the observation that some “non‐excretors,” although harboring large numbers of this bacterium, did not excrete the reduced metabolites when on digoxin administration. The investigators further discovered that growth of E. lenta with the amino acid arginine inhibited inactivation of digoxin. 127 These contributions led others to discover in digoxin‐metabolizing strains of E. lenta, a cytochrome‐encoding operon that is upregulated in expression by digoxin but inhibited by arginine in some strains of the bacterium, and thus unraveling the genetic basis for the inactivation of the cardiac drug (Figure 3c). 128 As expected, this genetic information or operon is absent in non‐digoxin metabolizing strains of E. lenta. Germ‐free mice colonized with E. lenta harboring the identified gene cluster, when used in pharmacokinetic studies, revealed that dietary protein reduces the in vivo metabolism of digoxin, with significant impact on drug concentration in the serum and the urine. As discussed later in this tutorial, the authors concluded that pharmacological considerations should be viewed in the context of both our human and microbial genomes. 128
The gut microbiota is also richly endowed with hydrolytic enzymes, as noted above, and these include glucosidases, glucuronidases, and sulfatases. An example of microbiota effects on drug metabolism involving hydrolytic enzymes can be illustrated with irinotecan hydrochloride (CPT‐11), a derivative of the topoisomerase inhibitor and antitumor drug Camptothecin. In rats, CPT‐11 was found to induce severe chronic diarrhea, loss of weight and anorexia, with the most severe histological damage found in the cecum. Two enzymes are of consequence here (i.e., the intestinal tissue carboxylesterase and the β‐glucuronidase activity present in the intestinal lumen). The former converts CPT‐11 to its active form (7‐ethyl‐10‐hydroxycamptothecin), whereas the activity of the latter (β‐glucuronidase) was found to correlate with the severity of the observed segmental damage noted with CPT‐11. It was also demonstrated that inhibition of the β‐glucuronidase activity in intestinal microbiota with antibiotics significantly abrogated the diarrhea and cecal damage associated with CPT‐11. By analyzing CPT‐11 and its metabolites in the feces, the investigators found that antibiotics completely inhibited the β‐glucuronidase activity that deconjugates the glucuronic conjugate of 7‐ethyl‐10‐hydroxycamptothecin, and therefore further strengthening the attribution of this enzyme to the intestinal microbiota. 129 The investigators later identified the metabolite eliciting the diarrhea and intestinal damage as SN‐38, and this metabolite resulted from the hydrolytic activity of β‐glucuronidase on the glucuronic conjugate of SN‐38 or SN‐38 glucuronide (Figure 3d). Hence, in this process, it was discovered that microbiota‐derived β‐glucuronidase hydrolysis of the detoxified metabolite SN‐38 glucuronide to SN‐38 triggers the severe symptoms linked to CPT‐11. 130
The human host utilizes two main enzyme classes responsible for the majority of steroid biosynthesis. These are cytochrome P450 monooxygenases and hydroxysteroid dehydrogenases (HSDHs), in the short chain dehydrogenase (SDR) aldo‐keto reductase (AKR) families. Pharmaceutical compounds, such as abiraterone acetate, target the host enzyme steroid 17α‐monooxygenase (CYP17A1), which is involved in adrenal production of glucocorticoids and 11‐oxy‐androgen precursors. 131 Abiraterone is prescribed along with synthetic glucocorticoids, such as prednisone, to replace glucocorticoid function during second line treatment of castration‐resistant prostate cancer. 131 Recent evidence indicates that gut bacteria metabolize abiraterone acetate 132 as well as prednisone. 133 , 134 Indeed, in several members of the gut microbiota, a biochemical pathway has been characterized in the conversion of cortisol to 11β‐hydroxyandrostenedione, 88 , 135 , 136 , 137 as well as the side‐chain cleavage of prednisone to a potentially potent androgen capable of driving proliferation of a prostate cancer cell line. 134 Furthermore, beyond the gut, the side‐chain cleavage pathway genes are also found in the bacterium Propionimicrobium lymphophilum, 134 , 136 a normal inhabitant of the urinary tract and previously associated with prostate cancer. 138
Taken together, host‐associated microbial enzymes are important, but so far largely overlooked factors in drug metabolism and therapeutic efficacy. In the examples above, we illustrate direct microbial modification of drugs with four different end results, that is, microbial action on the drug prontosil, which is inactive until activated by reductive cleavage; azo reduction by microbial enzyme to sulfanilamide; the gut bacteria‐derived inactivation of the cardiac drug digoxin and hence decreasing its bioavailability; and the gut microbiota‐induced toxicity of the antitumor drug camptothecin and two prostate cancer drugs, abiraterone and prednisone.
EXPERIMENTAL APPROACHES TO CHARACTERIZING MICROBIOME DRUG INTERACTIONS
A major challenge to defining the role of the microbiome in drug metabolism is the fact that gut microbial communities consist of hundreds of species whose composition varies dramatically across individuals in the population. Identifying the microbes and genes involved in drug metabolism will serve as a cornerstone for integrating the microbiome into pharmacology. Studies are emerging that demonstrate important roles of the gut microbiome in drug metabolism, yet this area of research is still in its infancy. We direct the reader to recent authoritative reviews, 120 , 139 and below we introduce the tools and approaches commonly used. We also highlight a few recent studies that incorporate these approaches to identify drug metabolizing strains, genes, and their relevance to drug metabolism in the host.
In vitro culture systems to characterize drug metabolism
In vitro culture systems provide a convenient way to study microbes and communities, enabling the identification of individual species that metabolize drugs and interrogation of the genes responsible for these activities. However, an unavoidable limitation to these approaches is that in vitro culture systems do not faithfully recapitulate factors of the host gut, such as nutrient composition, inter‐species interactions, and host factors, such as bile acids, pH, and immune products (antimicrobial peptides, secretory IgA, etc.). Nevertheless, they provide a useful starting point to understand the mechanisms that underlie microbial metabolism of drugs. Here, we introduce two commonly used approaches to studying gut microbial metabolism of drugs, (i) individual strain cultures (Figure 4a) and (ii) communities derived from stool donors (Figure 4b).
FIGURE 4.

In vitro cultures of gut microbiota strains or stool to identify microbiota‐drug interactions. (a) Anaerobic gut microbial strains are arrayed into 96‐well plates and cultured in the presence of drugs. After growth, drugs are extracted from culture supernatants and analyzed by LC–MS. Extracted ion chromatograms for drugs present in supernatants of each strain can reveal drug‐microbe interactions. (b) Stool samples from human donors are inoculated into different culture media. After growth, 16S rRNA gene profiling of cultures can be compared to the original stool donor to identify media that best recapitulate the donor community composition. After identifying optimal media, stool is cultured in the presence of drugs and drug remaining in the supernatant after growth is analyzed by LC–MS. Extracted ion chromatograms for drugs present in supernatants of different stool donors can reveal individualized drug‐microbiota interactions. LC–MS, liquid‐chromatography mass spectrometry.
In vitro cultures of strain libraries
The healthy distal GIT, where most microbes reside, is deeply anaerobic and most gut microbes are either obligate or facultative anaerobes. Many obligate anaerobes are exquisitely sensitive to oxygen, thus in vitro studies rely on anaerobic culture conditions. Traditional approaches for ensuring anaerobiosis, such as anaerobic gassing stations and anaerobic jars, are not generally amenable to reasonably high throughput applications, and consequently most laboratories use anaerobic cabinets. 140 These cabinets (available from Coy Laboratory Products, Anaerobe Systems, Sheldon Manufacturing, and Don Whitley Scientific) use palladium catalyst which catalyzes the conversion of hydrogen (provided from premixed gas tanks) and oxygen (present in the atmosphere) to form water. By maintaining consistent hydrogen levels within the chamber atmosphere and routinely baking catalyst to remove built up H2O, these systems can consistently keep oxygen levels below a few ppm. Double‐doored interlocks are used to pass supplies into the chamber with successive vacuum/purge gas cycles limiting the influx of atmospheric oxygen. Specialized media formulations are often required to grow phenotypically diverse gut microbiota strains. These media are prepared outside of the cabinet and brought in to be fully reduced within a day or two. Incubators housed within the cabinet can be used to cultivate organisms at specified temperature, typically 37°C for human gut microbial strains. Plasticware for liquid transfer or microbial culture is also stored within the cabinet, becoming oxygen free within 24–48 h. These anaerobic cabinets enable the culture of diverse microbes in high‐throughput formats, such as 96‐well plates, that are amenable to downstream workflows such as liquid‐chromatography mass spectrometry (LC–MS).
Human microbiome culture collections are not readily available and so many laboratories assemble their own by purchasing strains from commercial strain repositories (e.g., ATCC, The German Collection of Microorganisms and Cell Culture [DSMZ], and Japan Collection of Microorganisms [JCM]), or obtaining them from publicly funded initiatives (e.g., HMP BEI resources). When a decision is made to access a bacterium or bacterial strains from a culture collection or commercial source, it is very critical to note that due to bacterial strain differences, especially at the genetic level, the acquired strains may be very different from the relatives detected by phylogenetic studies in the community of interest. Standard microbiological techniques, such as routinely streaking strains on solid agar plates to evaluate colony morphology and confirm strain identity via 16S rRNA gene sequencing are critical to avoid and monitor contamination when working with multiple different strains. Spore‐forming bacteria are notorious for causing cross‐contamination within culture libraries, so cryo‐archived master stocks confirmed to be pure should always be used and great care should be taken with respect to aseptic technique. Critical factors to consider when culturing gut microbes include (a) matching nutritional requirements through supplementation of culture media, (b) mitigating and testing for cross‐contamination, and (c) synchronizing growth of slow vs. fast dividing strains. Once these conditions are established, microbes can be cultured in the presence of drugs and the parent drug or its metabolites can be measured in cultures or supernatants by analytical methods such as LC–MS (Figure 4a).
A recent study by Zimmermann et al. 133 used a library of 73 phylogenetically diverse human gut microbial strains to test their capacity to metabolize 271 different drugs. Combinatorial pools of the drugs were used to reduce LC–MS time, with each drug‐microbe combination occurring at least in quadruplicate. Remarkably, two thirds of the drugs tested were diminished by greater than 20% in cultures of at least one bacterium, revealing a broad capacity for drug metabolism across bacterial strains. There was a wide variation in the drug metabolizing capacity across strains (11–95 drugs metabolized per strain), with certain organisms, such as Bacteroides dorei and Bacteroides uniformis, each capable of metabolizing 95 different drugs. LC–MS analyses of drug metabolites revealed common drug modifications, which included reduction/oxidation, acetylation/deacetylation, and propionylation. These findings provide species level resolution into the drug metabolizing capabilities of the microbiome and enable mechanistic studies to identify responsible genes that might serve as markers for these activities in metagenomic datasets as described in a later section.
In vitro cultures of human stool samples
Whereas in vitro studies of individual microbes provide foundational insights into the metabolizing activity of the microbiota, this approach suffers from three important limitations: (1) that microbes in the gut do not exist in isolation, rather they are part of a large and complex community; (2) human microbial communities are highly individualized; and (3) metabolic activities of the gut microbiota are often species‐ or even strain‐specific. To complement in vitro studies of individual microbes, approaches to cultivate microbial communities directly from human stool donors may be used. In this strategy, stool is homogenized in pre‐reduced buffer within an anaerobic chamber, then cultivated in several different growth media. To ensure culture conditions best recapitulate the donor community, the 16S rRNA gene profiles from cultured stool communities can be compared with the donor community composition (Figure 4b). After these conditions are identified, the stool cultures can be incubated with various drugs and the metabolizing activity can be assessed by LC–MS (Figure 4b).
Javdan et al. 141 recently used this approach to conduct an individualized survey of drug metabolizing activity within the human gut. They cultured human stool samples from a pilot donor (PD) in 14 different media and using 16S rRNA gene profiling, found one medium (i.e., modified Gifu anaerobic medium [mGAM]), which best recapitulated the microbial composition of donor stool samples. They then cultured PD stool samples in mGAM supplemented with 575 different drugs and quantified drugs before and after culture by LC–MS. Approximately 75% of the drugs were amenable to LC–MS analysis and, of these, 57 drugs were metabolized by PD stool sample cultures. These included known microbiota‐drug interactions such as nitroreductions of dantrolene, 142 clonazepam, 143 , 144 and nicardipine 145 ; hydrolysis of risperidone 146 , 147 ; and azoreduction of sulfasalazine. 148 , 149 However, novel drug‐microbiota interactions were also discovered, such as thioester hydrolysis of spironolactone, nitroreduction/acetylation of tolcapone, deglycosylation of capecitabine, and ester hydrolysis of misoprostol and mycophenolate mofetil. Next, the authors developed a model integrating biomass measurements from stool cultures and 16S rRNA gene profiling to select media compositions that enabled the highest biomass of a culture most similar in composition to the stool donor. Using this approach, a single medium was selected and stool cultures from 20 donors were incubated with a panel of 23 drugs. Although several drugs were consistently depleted from all stool cultures, other drugs showed wide interindividual variability. For example, five of 20 stool cultures depleted digoxin from the media, and of these, three of 20 produced the reduced metabolite, dihydrodigoxin. This mirrors the heterogeneity in urine excretion of reduced digoxin metabolites previously described in human subjects, 125 suggesting that stool cultures faithfully recapitulate roles of the microbiome in drug metabolism.
Approaches to identify microbial genes responsible for drug metabolism
To identify the microbial genes responsible for drug transformations, two strategies are commonly used: (1) loss of function studies using genetic tools (Figure 5a), and (2) gain of function studies by cloning DNA fragments into heterologous hosts, such as E. coli (Figure 5b).
FIGURE 5.
Loss of function and gain of function experiments to identify genes responsible for microbiota‐drug interactions. (a) Loss‐of‐function studies. A transposon library for a bacterium of interest is generated and arrayed into individual wells of microtiter plates. The mutants are then cultured with drugs and levels of drugs in the supernatant are analyzed by LC–MS. Extracted ion chromatograms reveal mutants which do not metabolize drugs as compared to the wild‐type (WT) strain. Sequencing of the transposon insertion site of such mutants can reveal genes responsible for drug metabolism. (b) Gain‐of‐function studies. Genomic DNA isolated from an individual bacterial strain or stool metagenomic DNA is sheared, then fragments are cloned into a plasmid vector and transformed into heterologous hosts (such as Escherichia coli) to make a clone library. Individual clones or pools of clones are cultured with drugs and levels of drugs in the culture supernatants are analyzed by LC–MS. Extracted ion chromatograms reveal clones which deplete drugs in the supernatants as compared to empty vector controls. Sequencing of the DNA within the plasmid can reveal genes responsible for drug metabolism. LC–MS, liquid‐chromatography mass spectrometry.
An important prerequisite for loss of function studies is that the bacterium that performs a drug metabolism of interest is genetically tractable. However, most gut bacteria are genetically intractable, and as such loss of function studies are amenable to only a small number of bacteria. Typically, a transposon mutant library is generated and then individual mutants are screened by LC–MS to identify null mutants. To achieve saturation of a bacterial chromosome, library sizes on the order of 10,000 mutants are required which may be untenable for LC–MS screening if these mutants are individually analyzed. To reduce library size, individual mutants may be sequenced and a library that targets most of the non‐essential genes can be constructed; however, this requires considerable up‐front work. Examples of these arrayed libraries include the Keio collection for E. coli 150 and a library generated by Goodman et al. in B. thetaiotaomicron. 151 Zimmermann et al. 144 recently used this approach to identify drug metabolizing genes in the gut bacterium, B. thetaiotaomicron. The authors found that this bacterium converts the oral antiviral drug brivudine to its inactive metabolite, bromovinyluracil. The authors screened a reduced size arrayed transposon mutant library by LC–MS to identify mutants that in vitro, neither depleted brivudine nor produced bromovinyluracil. This screen identified a single mutant (Tn:bt4554) disrupting a putative purine nucleoside phosphorylase. Follow‐up studies by constructing a clean deletion mutant in BT_4554 and complementation of the gene confirmed that BT_4554 is necessary and sufficient for brivudine metabolism.
Gain of function studies typically involve constructing a clone library in E. coli using DNA fragments from a source organism or microbial community. The E. coli clones then express the exogenous genes and LC–MS can be used to screen for gain of function metabolic activities. Finally, the DNA fragments encoded by gain of function clones can then be sequenced to identify responsible genes. This approach has been used widely in the natural products field to identify new antibiotics or anticancer drugs. 152 However, a major limitation to the approach is whether the source gene is expressed, and an active protein produced by heterologous hosts. 153 Nevertheless, this approach has been used by two recent studies described in the preceding section to identify genes responsible for specific drug metabolisms. Zimmerman et al. 133 generated a clone library of the B. thetaiotaomicron genome in E. coli and screened for drug metabolism by LC–MS. This led to the identification of two putative acyl hydrolases, BT_4096 responsible for deacetylation of the calcium channel‐blocker diltiazem and BT_0569 with promiscuous activity toward multiple structurally diverse drugs. Further loss of functions studies of BT_4096 were conducted by in‐frame gene deletion and complementation, verifying the role of BT_4096 in diltiazem metabolism. In a separate study, Javdan et al. 141 constructed a stool metagenome clone library in E. coli and screened for reduction of hydrocortisone to 20β‐dihydrocortisone. To ensure the clone library was representative of the genetic content of the fecal donor, the authors created a massive library of ~3 × 106 unique clones. Screening individual clones in a library of this size by LC–MS is infeasible, and rather, the authors screened a smaller number of pooled clones, followed by iterative dilution and screening of sub‐pools. Through this work, the authors identified two positive clones which both harbored the same gene, a putative oxidoreductase originating from a Bifidobacterium species. This gene was individually cloned and heterologously expressed in E. coli, and its role in reduction of hydrocortisone to 20β‐dihydrocortisone was verified.
These studies reveal the power of loss of function and gain of function studies in identifying microbial genes responsible for drug metabolism. However, these approaches suffer from important limitations, such as the limited scope of genetically tractable gut bacteria, uncertainty as to whether genes of interest can be expressed in E. coli, and the sheer magnitude of LC–MS screening necessary for identification of clones or mutant strains. As microbiota drug metabolism research expands, new improved strategies to identify microbial drug‐metabolizing genes are likely to follow.
Metagenomics to characterize drug metabolizing genes
Metagenomics is the study of genetic material isolated directly from environmental sources. For microbiota research, genomic DNA is typically isolated from fecal samples, then fragmented and sequenced using next‐generation sequencing techniques. The resulting sequence reads can then be (i) assembled to create gene catalogs; (ii) mapped to reference genes or genomes; or (iii) binned and assembled to create metagenome assembled genomes (MAGs). Metagenomics can identify whether a known drug‐metabolizing gene is present within the gut microbial community which might predict whether an individual's microbiome has the capacity to perform this function. As an example, Zimmerman et al. 133 cultured samples from 28 stool donors in media with drugs and used metagenomics to query the relative abundance of several marker genes they had characterized in in vitro cultures (see above). These results showed a significant positive correlation between the rate of drug metabolism and the abundance of known marker genes for these activities. The implication of this research is that drug metabolism marker genes could be surveyed by stool metagenomics and this might predict whether an individual's microbiome could impact drug metabolism.
Gnotobiotic mouse models
The gut microbiome of mammals is highly individualized, which creates a major challenge for animal research. For example, the same strain of mice obtained from different vendors harbor dramatically different microbial communities 154 , 155 influencing mouse phenotypes which likely include their xenobiotic or drug metabolizing activities. Moreover, there are large taxonomic and functional differences between the microbiomes of mice and humans, 156 , 157 which limits the translatability of findings from conventionally housed animals.
Germ‐free mice, or their colonized (gnotobiotic) derivatives, represent a powerful tool to elucidate host and microbial contributions to drug metabolism. These mice are housed in aseptic flexible film isolators or in racks with HEPA filters at the cage level. The cages, bedding, water, and chow are all autoclaved and transferred into isolators using sterile techniques. Sentinel mice are routinely monitored for sterility by culturing feces and/or extracting fecal DNA and screening for 16S rRNA gene amplicons by PCR with universal primers. In recent years, germ‐free vivaria have become more widely available at universities and a few contract research laboratories. Nevertheless, access to these facilities is relatively limited and the strict aseptic housing conditions limits the types of experiments that can be done with these mice. Notably, genetic models which are so critical to rodent studies require mice to be re‐derived as germ‐free via embryo transfer, a technique performed at relatively few companies or germ‐free core facilities.
To identify the role of gut microbes in drug metabolism, germ‐free mice are often colonized with individual strains of gut bacteria or they are “humanized” via colonization with stool from a human donor. Drugs can then be administered to colonized mice or germ‐free controls and the level of drug in the intestinal contents, serum, and urine can be assessed by LC–MS.
Zimmerman et al. 144 recently used gnotobiotic mice to dissect the host and microbial contributions to metabolism of the oral antiviral drug brivudine. Both microbes and the mammalian host are known to metabolize brivudine (BRV) to bromovinyluracil (BVU), although the relative contributions are not known. When administered orally, BRV achieved similar plasma levels in germ‐free mice compared with their conventionally housed counterparts. However, conventional mice had significantly higher serum levels of the metabolite, BVU, suggesting a contribution of the microbiome to brivudine metabolism. Importantly, BVU inhibits host dihydropyrimidine dehydrogenase with potential toxicity implications for patients receiving therapeutic pyrimidine analogs such as 5‐fluorouracil. 158 , 159 Consistent with inhibition of dihydropyrimidine dehydrogenase, the authors found higher thymine levels in the livers of conventional versus germ‐free mice. To provide a more closely matched comparison, the authors colonized two groups of germ‐free mice with either BRV‐metabolizing wild‐type B. thetaiotaomicron or with a loss of function mutant (bt4554). BRV reached similar serum levels in these two groups of mice, but the BVU levels were significantly lower in cecal contents, serum, and liver of bt4554 colonized mice. Thymine was also elevated in the wild‐type versus bt4554 colonized mice consistent with BVU mediated inhibition of dihydropyrimidine dehydrogenase. The authors then built a pharmacokinetic model to estimate the host and microbial contributions to BRV kinetics. These findings suggest that the microbial contribution to BRV levels in serum (estimated at ~71%) is greater than the host contribution. These mechanistic studies in gnotobiotic mice provide unique insights and enable a quantitative assessment of the contribution of the gut microbiota to host drug responses.
FUTURE PERSPECTIVES FOR THE MICROBIOME AND DRUG METABOLISM
Limitations and opportunities for microbiome drug metabolism research
One of the major challenges to microbiome research is how to establish a causal relationship between microbial functions and disease phenotypes. 160 Knowledge of the mechanisms which drive these interactions is critical to the development of new diagnostics and therapeutics that might be translated to the clinic. Whereas many host phenotypes impacted by the microbiome (e.g., weight gain, response to cancer immunotherapy, cardiovascular health, etc.) are problematic to track and quantify in vitro, drug metabolism is a fundamentally different phenotype that lends itself to mechanistic investigation. By studying drug disposition in pure cultures of bacteria, investigators can identify the individual strains—and even genes—required for drug metabolism by the microbiome. This mechanistic insight can then inform studies in humans by tracking specific microbial functions and establishing how these impact the drug metabolism in the host. To make further gains on an already solid foundation, below we illustrate several key opportunities for growth in the microbiome drug metabolism field.
Building a foundation for microbiome drug metabolism activities
Recent studies summarized in preceding sections have begun to unravel the molecular basis for microbiome influences on drug metabolism. By culturing individual microbes with drugs, strain‐level resolution into drug metabolizing activities have been identified. However, these studies have sampled a relatively minor fraction of the incredible diversity of human‐associated gut microbes. To expand on this already solid foundation, future studies are needed to test drug‐metabolizing activity on a larger scale. Recent advances in high‐throughput culturing technologies (culturomics) 161 will likely be a critical tool to characterize taxonomically and phenotypically diverse gut microbes and their potential roles in drug metabolism. Microbial strain databases that are rigorously assembled, capture taxonomic and functional diversity, and are commercially available will also serve as a critical resource. Work has already progressed on this front, with several microbiome strain libraries currently available (Human Microbiome Project via BEI Resources, Broad Institute Open Biome Microbiome Library, 162 Human Gastrointestinal Bacteria Culture Collection 163 ). Expanding on these strain libraries and making them more widely available will be an important path forward for microbiome drug metabolism research.
The microbiome collectively harbors millions of genes which encode enzymes with diverse biochemical activities. Loss‐of‐function genetic tools are essential to unequivocally assign drug‐metabolizing activities to candidate genes. Yet, genetic tools such as transposon mutagenesis and Clustered Regularly Interspersed Short Palindromic Repeat (CRISPR) technologies are available for only a handful of the most abundant taxa or for specific gastrointestinal pathogens. A recent study by Jin et al. 164 outlined an approach for gene transfer methods that can be applied to a wide variety of genetically intractable gut microbes. This sets the stage for a concerted effort to develop genetic tools that can interrogate new genes involved in drug metabolism by diverse gut microbes.
Large‐scale clinical studies to assess microbiome‐host drug interactions
Mechanistic studies that identify microbial genes involved in specific host‐microbe drug interactions are valuable, but they do not directly address the question of whether an individual's microbiome may influence drug kinetics or toxicity profiles. To answer this, integrated studies are needed that collect and analyze clinical and laboratory data on drug dosage, plasma drug levels, host genotype, drug toxicities, and microbiome metagenomics data. By following known marker genes within the gut microbiome of patients dosed with specific drugs, these studies will likely provide key data on the clinically relevant impact the gut microbiome has on drug metabolism and toxicity in humans. These types of studies could reveal clinically actionable results to enable personalized pharmacology.
Implications of microbiome research in drug metabolism
Increasing evidence suggests that the microbiome plays an important role in influencing drug metabolism in humans and that the microbiome should be considered in personalized medicine. Microbial markers in an individual's gut could be used to predict whether they might require a larger dose of a drug, or whether they are predisposed to accumulating toxic metabolites. This has important implications for hundreds of drugs currently prescribed to patients, but also is highly relevant to future drug development efforts. Here, we highlight two examples of microbiome‐drug interactions and discuss their potential to translate into the clinic.
Irinotecan is a camptothecin analog which targets DNA topoisomerase overexpressed in cancer cells leading to cell death. It is administered as a pro‐drug which is converted to its active metabolite (SN‐38) by host carboxylesterases. The elimination of SN‐38 involves glucuronidation in the liver and subsequent intestinal elimination via the bile. However, bacteria β‐glucuronidases cleave the glucuronic acid moiety in the gut, re‐activating SN‐38 in the intestine which causes dose‐limiting gastrointestinal (GI) toxicity. Decreasing β‐glucuronidase activity in the gut could play an important role in decreasing GI toxicity in patients with cancer receiving irinotecan. To this end, Wallace et al. 165 screened a small molecule library for inhibitors of a bacterial β‐glucuronidase enzymes. They identified several small molecules that specifically inhibited E. coli β‐glucuronidase, binding a region of the E. coli protein not present in the mammalian enzyme. When irinotecan was co‐administered with a β‐glucuronidase inhibitor to mice, the mice experienced less GI toxicity. Further studies by Bhatt et al. 166 revealed that irinotecan administration leads to increased β‐glucuronidase activity in the gut and promotes a bloom in Enterobacteriaceae. Co‐administration of irinotecan with a β‐glucuronidase inhibitor alleviated GI toxicity and enabled irinotecan dose escalation leading to improved therapeutic outcomes in a mouse model of cancer. Together, these findings suggest that specific targeting of bacterial β‐glucuronidases could be a viable approach to improving the safety profile of irinotecan.
The cardiac glycoside, digoxin is used to treat heart failure and has a narrow therapeutic index. About one in 10 individuals excrete high levels of the non‐active reduced metabolite dihydrodigoxin, necessitating dose escalation. Previous studies identified that digoxin is reduced to dihydrodigoxin by a relatively limited group of gut bacteria related to Eggerthella lenta, however, the presence of E. lenta strains isolated from stool samples was not predictive of ex vivo digoxin reduction. 127 In a subsequent study, Haiser et al. 128 identified the cardiac glycoside reduction (cgr) gene cluster encoded by strains of E. lenta that reduced digoxin, and absent in E. lenta strains that did not reduce digoxin. This emphasizes the point made earlier in this tutorial that significant strain to strain differences may exist among members of the same bacterial species, thus identifying microbes at the species level is not sufficient to infer their function. Moreover, these data likely explain why the former study failed to identify a relationship between E. lenta colonization and ex vivo reduction activity. Haiser et al. 53 developed a qPCR assay specific for the E. lenta cgr locus and normalized to the E. lenta 16S rRNA gene (the cgr ratio). The cgr ratio was predictive of the capacity for human stool samples to convert digoxin to dihydrodigoxin in an ex vivo assay. This study identified a genetic biomarker within the human gut microbiota which is predictive of the capacity for microbes to metabolize digoxin. This information might be useful to clinicians to predict who among their patients are likely to require larger doses of digoxin to achieve therapeutic levels, and how antibiotic treatments might affect digoxin levels in their patients.
Concluding thoughts
Recent advances in microbiota research, including the use of large‐scale strain libraries, high‐throughput LC–MS, genomics and metagenomics, and gnotobiotics, now enable the interrogation of microbe‐host drug interactions on an unprecedented scale. These studies have the potential to identify novel ways to predict drug kinetics in patients and may reveal new modalities to improve consistency of therapeutic drug levels in patients and reduce toxicities. Incorporating microbiota research into drug development and preclinical studies could identify important microbiota‐mediated drug interactions, providing insights into clinical trials in humans. However, specialized expertise and facilities are required to study gut microbial activities. Collaborations between experts in both respective fields (microbiota research and pharmacology) will be critical to enhancing the impact microbiota research may have on therapeutic drug modalities.
FUNDING INFORMATION
National Institutes of Health: R35‐GM142873 and R01‐AT011396 for Dylan Dodd, R01‐GM140306 for Isaac Cann.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
ACKNOWLEDGMENTS
The authors would like to thank Jason M. Ridlon and Ahmed M. Abdel‐Hamid of University of Illinois for helpful discussions, suggestions, and reading the manuscript. We also thank Yuanyuan Liu of Stanford University for providing artwork in Figure 2. The funders had no role in the study design, data analyses, decision to publish, or preparation of the manuscript.
Dodd D, Cann I. Tutorial: Microbiome studies in drug metabolism. Clin Transl Sci. 2022;15:2812‐2837. doi: 10.1111/cts.13416
Contributor Information
Dylan Dodd, Email: ddodd2@stanford.edu.
Isaac Cann, Email: icann@illinois.edu.
REFERENCES
- 1. Ursell LK, Clemente JC, Rideout JR, Gevers D, Caporaso JG, Knight R. The interpersonal and intrapersonal diversity of human‐associated microbiota in key body sites. J Allergy Clin Immunol. 2012;129:1204‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14:e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Milani C, Duranti S, Bottacini F, et al. The first microbial colonizers of the human gut: composition, activities, and health implications of the infant gut microbiota. Microbiol Mol Biol Rev. 2017;81:1‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Relman DA. The human microbiome: ecosystem resilience and health. Nutr Rev. 2012;70(Suppl 1):S2‐S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cox LM, Yamanishi S, Sohn J, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell. 2014;158:705‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sarkar A, Yoo JY, Valeria Ozorio Dutra S, Morgan KH, Groer M. The association between early‐life gut microbiota and long‐term health and diseases. J Clin Med. 2021;10:1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Puccio G, Alliet P, Cajozzo C, et al. Effects of infant formula with human milk oligosaccharides on growth and morbidity: a randomized multicenter trial. J Pediatr Gastroenterol Nutr. 2017;64:624‐631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ramani S, Stewart CJ, Laucirica DR, et al. Human milk oligosaccharides, milk microbiome and infant gut microbiome modulate neonatal rotavirus infection. Nat Commun. 2018;9:5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hill DR, Chow JM, Buck RH. Multifunctional benefits of prevalent HMOs: implications for infant health. Nutrients. 2021;13:1‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu RY, Li B, Koike Y, et al. Human milk oligosaccharides increase mucin expression in experimental necrotizing enterocolitis. Mol Nutr Food Res. 2019;63:e1800658. [DOI] [PubMed] [Google Scholar]
- 11. Chichlowski M, De Lartigue G, German JB, Raybould HE, Mills DA. Bifidobacteria isolated from infants and cultured on human milk oligosaccharides affect intestinal epithelial function. J Pediatr Gastroenterol Nutr. 2012;55:321‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Korpela K, Helve O, Kolho KL, et al. Maternal fecal microbiota transplantation in cesarean‐born infants rapidly restores normal gut microbial development: a proof‐of‐concept study. Cell. 2020;183:324‐334 e325. [DOI] [PubMed] [Google Scholar]
- 13. Backhed F, Roswall J, Peng Y, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17:690‐703. [DOI] [PubMed] [Google Scholar]
- 14. Asnicar F, Manara S, Zolfo M, et al. Studying vertical microbiome transmission from mothers to infants by strain‐level metagenomic profiling. mSystems. 2017;2:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Korpela K, Costea P, Coelho LP, et al. Selective maternal seeding and environment shape the human gut microbiome. Genome Res. 2018;28:561‐568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dominguez‐Bello MG, Costello EK, Contreras M, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci USA. 2010;107:11971‐11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fava F, Danese S. Intestinal microbiota in inflammatory bowel disease: friend of foe? World J Gastroenterol. 2011;17:557‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sanz Y. Effects of a gluten‐free diet on gut microbiota and immune function in healthy adult humans. Gut Microbes. 2010;1:135‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andersen V, Moller S, Jensen PB, Moller FT, Green A. Caesarean delivery and risk of chronic inflammatory diseases (inflammatory bowel disease, rheumatoid arthritis, coeliac disease, and diabetes mellitus): a population based registry study of 2,699,479 births in Denmark during 1973‐2016. Clin Epidemiol. 2020;12:287‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dominguez‐Bello MG, de Jesus‐Laboy KM, Shen N, et al. Partial restoration of the microbiota of cesarean‐born infants via vaginal microbial transfer. Nat Med. 2016;22:250‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koenig JE, Spor A, Scalfone N, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4578‐4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Valles Y, Artacho A, Pascual‐García A, et al. Microbial succession in the gut: directional trends of taxonomic and functional change in a birth cohort of Spanish infants. PLoS Genet. 2014;10:e1004406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fallani M, Amarri S, Uusijarvi A, et al. Determinants of the human infant intestinal microbiota after the introduction of first complementary foods in infant samples from five European centres. Microbiology (Reading). 2011;157:1385‐1392. [DOI] [PubMed] [Google Scholar]
- 24. Nardone G, Compare D. The human gastric microbiota: is it time to rethink the pathogenesis of stomach diseases? United European Gastroenterol J. 2015;3:255‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bik EM, Eckburg PB, Gill SR, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci USA. 2006;103:732‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li XX, Wong GLH, To KF, et al. Bacterial microbiota profiling in gastritis without Helicobacter pylori infection or non‐steroidal anti‐inflammatory drug use. PLoS One. 2009;4:e7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zoetendal EG, Raes J, van den Bogert B, et al. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012;6:1415‐1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Booijink CC, El‐Aidy S, Rajilić‐Stojanović M, et al. High temporal and inter‐individual variation detected in the human ileal microbiota. Environ Microbiol. 2010;12:3213‐3227. [DOI] [PubMed] [Google Scholar]
- 29. Dlugosz A, Winckler B, Lundin E, et al. No difference in small bowel microbiota between patients with irritable bowel syndrome and healthy controls. Sci Rep. 2015;5:8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arumugam M, Raes J, Pelletier E, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. The abundance and variety of carbohydrate‐active enzymes in the human gut microbiota. Nat Rev Microbiol. 2013;11:497‐504. [DOI] [PubMed] [Google Scholar]
- 32. Human Microbiome Project, C . Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Takagi T, Inoue R, Oshima A, et al. Typing of the gut microbiota Community in Japanese Subjects. Microorganisms. 2022;10:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Litvak Y, Byndloss MX, Baumler AJ. Colonocyte metabolism shapes the gut microbiota. Science. 2018;362:eaat9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fan YY, Davidson LA, Callaway ES, Wright GA, Safe S, Chapkin RS. A bioassay to measure energy metabolism in mouse colonic crypts, organoids, and sorted stem cells. Am J Physiol Gastrointest Liver Physiol. 2015;309:G1‐G9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lefebvre M, Paulweber B, Fajas L, et al. Peroxisome proliferator‐activated receptor gamma is induced during differentiation of colon epithelium cells. J Endocrinol. 1999;162:331‐340. [DOI] [PubMed] [Google Scholar]
- 37. Duszka K, Oresic M, Le May C, Konig J, Wahli W. PPARgamma modulates long chain fatty acid processing in the intestinal epithelium. Int J Mol Sci. 2017;18:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roediger WE. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut. 1980;21:793‐798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Atarashi K, Tanoue T, Shima T, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short‐chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Welch RA, Burland V, Plunkett G III, et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli . Proc Natl Acad Sci USA. 2002;99:17020‐17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sonnenburg JL, Xu J, Leip DD, et al. Glycan foraging in vivo by an intestine‐adapted bacterial symbiont. Science. 2005;307:1955‐1959. [DOI] [PubMed] [Google Scholar]
- 43. Desai MS, Seekatz AM, Koropatkin NM, et al. A dietary fiber‐deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell. 2016;167:1339‐1353. e1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang M, Chekan JR, Dodd D, et al. Xylan utilization in human gut commensal bacteria is orchestrated by unique modular organization of polysaccharide‐degrading enzymes. Proc Natl Acad Sci USA. 2014;111:E3708‐E3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pereira GV, Abdel‐Hamid AM, Dutta S, et al. Degradation of complex arabinoxylans by human colonic Bacteroidetes. Nat Commun. 2021;12:459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fehlner‐Peach H, Magnabosco C, Raghavan V, et al. Distinct polysaccharide utilization profiles of human intestinal Prevotella copri isolates. Cell Host Microbe. 2019;26:680‐690. e685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dodd D, Mackie RI, Cann IK. Xylan degradation, a metabolic property shared by rumen and human colonic Bacteroidetes. Mol Microbiol. 2011;79:292‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fassarella M, Blaak EE, Penders J, Nauta A, Smidt H, Zoetendal EG. Gut microbiome stability and resilience: elucidating the response to perturbations in order to modulate gut health. Gut. 2021;70:595‐605. [DOI] [PubMed] [Google Scholar]
- 49. Cockburn DW, Koropatkin NM. Polysaccharide degradation by the intestinal microbiota and its influence on human health and disease. J Mol Biol. 2016;428:3230‐3252. [DOI] [PubMed] [Google Scholar]
- 50. Ndeh D, Rogowski A, Cartmell A, et al. Complex pectin metabolism by gut bacteria reveals novel catalytic functions. Nature. 2017;544:65‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Luis AS, Briggs J, Zhang X, et al. Dietary pectic glycans are degraded by coordinated enzyme pathways in human colonic Bacteroides . Nat Microbiol. 2018;3:210‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 2008;4:447‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Grondin JM, Tamura K, Dejean G, Abbott DW, Brumer H. Polysaccharide utilization loci: fueling microbial communities. J Bacteriol. 2017;199:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McNulty NP, Wu M, Erickson AR, et al. Effects of diet on resource utilization by a model human gut microbiota containing Bacteroides cellulosilyticus WH2, a symbiont with an extensive glycobiome. PLoS Biol. 2013;11:e1001637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Terrapon N, Lombard V, Drula É, et al. PULDB: the expanded database of polysaccharide utilization loci. Nucleic Acids Res. 2018;46:D677‐D683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ze X, Duncan SH, Louis P, Flint HJ. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012;6:1535‐1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ze X, Ben David Y, Laverde‐Gomez JA, et al. Unique organization of extracellular amylases into amylosomes in the resistant starch‐utilizing human colonic firmicutes bacterium Ruminococcus bromii . MBio. 2015;6:e01058‐e01015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mukhopadhya I, Moraïs S, Laverde‐Gomez J, et al. Sporulation capability and amylosome conservation among diverse human colonic and rumen isolates of the keystone starch‐degrader Ruminococcus bromii . Environ Microbiol. 2018;20:324‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cockburn DW, Kibler R, Brown HA, et al. Structure and substrate recognition by the Ruminococcus bromii amylosome pullulanases. J Struct Biol. 2021;213:107765. [DOI] [PubMed] [Google Scholar]
- 60. La Rosa SL, Leth ML, Michalak L, et al. The human gut firmicute Roseburia intestinalis is a primary degrader of dietary beta‐mannans. Nat Commun. 2019;10:905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Leth ML, Ejby M, Workman C, et al. Differential bacterial capture and transport preferences facilitate co‐growth on dietary xylan in the human gut. Nat Microbiol. 2018;3:570‐580. [DOI] [PubMed] [Google Scholar]
- 62. Sonnenburg ED, Sonnenburg JL. Starving our microbial self: the deleterious consequences of a diet deficient in microbiota‐accessible carbohydrates. Cell Metab. 2014;20:779‐786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Russell WR, Duncan SH, Scobbie L, et al. Major phenylpropanoid‐derived metabolites in the human gut can arise from microbial fermentation of protein. Mol Nutr Food Res. 2013;57:523‐535. [DOI] [PubMed] [Google Scholar]
- 64. Biagi E, Candela M, Fairweather‐Tait S, Franceschi C, Brigidi P. Aging of the human metaorganism: the microbial counterpart. Age. 2012;34:247‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Salazar N, Valdes‐Varela L, Gonzalez S, Gueimonde M, de los Reyes‐Gavilan CG. Nutrition and the gut microbiome in the elderly. Gut Microbes. 2017;8:82‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Salazar N, González S, Nogacka AM, et al. Microbiome: effects of ageing and eiet. Curr Issues Mol Biol. 2020;36:33‐62. [DOI] [PubMed] [Google Scholar]
- 67. Claesson MJ, Jeffery IB, Conde S, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488:178‐184. [DOI] [PubMed] [Google Scholar]
- 68. Salazar N, López P, Valdés L, et al. Microbial targets for the development of functional foods accordingly with nutritional and immune parameters altered in the elderly. J Am Coll Nutr. 2013;32:399‐406. [DOI] [PubMed] [Google Scholar]
- 69. Salazar N, Arboleya S, Valdés L, et al. The human intestinal microbiome at extreme ages of life. Dietary intervention as a way to counteract alterations. Front Genet. 2014;5:406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Magill SS, Edwards JR, Bamberg W, et al. Multistate point‐prevalence survey of health care‐associated infections. N Engl J Med. 2014;370:1198‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bhalodi AA, van Engelen TSR, Virk HS, Wiersinga WJ. Impact of antimicrobial therapy on the gut microbiome. J Antimicrob Chemother. 2019;74:i6‐i15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rashid MU, Weintraub A, Nord CE. Development of antimicrobial resistance in the normal anaerobic microbiota during one year after administration of clindamycin or ciprofloxacin. Anaerobe. 2015;31:72‐77. [DOI] [PubMed] [Google Scholar]
- 73. Zheng P, Yang J, Li Y, et al. Gut microbial signatures can discriminate unipolar from bipolar depression. Adv Sci (Weinh). 2020;7:1902862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Knudsen JK, Bundgaard‐Nielsen C, Hjerrild S, Nielsen RE, Leutscher P, Sørensen S. Gut microbiota variations in patients diagnosed with major depressive disorder‐a systematic review. Brain Behav. 2021;11:e02177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Angelucci F, Cechova K, Amlerova J, Hort J. Antibiotics, gut microbiota, and Alzheimer's disease. J Neuroinflammation. 2019;16:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. McGuinness AJ, Davis JA, Dawson SL, et al. A systematic review of gut microbiota composition in observational studies of major depressive disorder, bipolar disorder and schizophrenia. Mol Psychiatry. 2022;27:1920‐1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Maier L, Pruteanu M, Kuhn M, et al. Extensive impact of non‐antibiotic drugs on human gut bacteria. Nature. 2018;555:623‐628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nagata N, Nishijima S, Miyoshi‐Akiyama T, et al. Population‐level metagenomics uncovers distinct effects of multiple medications on the human gut microbiome. Gastroenterology. 2022;163:1038‐1052. [DOI] [PubMed] [Google Scholar]
- 79. Li L, Ning Z, Zhang X, et al. RapidAIM: a culture‐ and metaproteomics‐based rapid assay of individual microbiome responses to drugs. Microbiome. 2020;8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hall AB, Tolonen AC, Xavier RJ. Human genetic variation and the gut microbiome in disease. Nat Rev Genet. 2017;18:690‐699. [DOI] [PubMed] [Google Scholar]
- 81. Xie H, Guo R, Zhong H, et al. Shotgun metagenomics of 250 adult twins reveals genetic and environmental impacts on the gut microbiome. Cell Syst. 2016;3:572‐584. e573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Goodrich JK, Davenport ER, Beaumont M, et al. Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe. 2016;19:731‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lehrer RI, Lu W. Alpha‐defensins in human innate immunity. Immunol Rev. 2012;245:84‐112. [DOI] [PubMed] [Google Scholar]
- 84. Buffington SA, Dooling SW, Sgritta M, et al. Dissecting the contribution of host genetics and the microbiome in complex behaviors. Cell. 2021;184:1740‐1756. e1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Koppel N, Maini Rekdal V, Balskus EP. Chemical transformation of xenobiotics by the human gut microbiota. Science. 2017;356:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hong PY, Iakiviak M, Dodd D, Zhang M, Mackie RI, Cann I. Two new xylanases with different substrate specificities from the human gut bacterium Bacteroides intestinalis DSM 17393. Appl Environ Microbiol. 2014;80:2084‐2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mythen SM, Devendran S, Mendez‐Garcia C, Cann I, Ridlon JM. Targeted synthesis and characterization of a gene cluster encoding NAD(P)H‐dependent 3alpha‐, 3beta‐, and 12alpha‐hydroxysteroid dehydrogenases from Eggerthella CAG:298, a gut metagenomic sequence. Appl Environ Microbiol. 2018;84:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Doden HL, Pollet RM, Mythen SM, et al. Structural and biochemical characterization of 20beta‐hydroxysteroid dehydrogenase from Bifidobacterium adolescentis strain L2‐32. J Biol Chem. 2019;294:12040‐12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Dodd D, Kiyonari S, Mackie RI, Cann IK. Functional diversity of four glycoside hydrolase family 3 enzymes from the rumen bacterium Prevotella bryantii B14. J Bacteriol. 2010;192:2335‐2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Luna E, Parkar SG, Kirmiz N, et al. Utilization efficiency of human milk oligosaccharides by human‐associated Akkermansia is strain dependent. Appl Environ Microbiol. 2022;88:e0148721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. LoCascio RG, Desai P, Sela DA, Weimer B, Mills DA. Broad conservation of milk utilization genes in Bifidobacterium longum subsp. infantis as revealed by comparative genomic hybridization. Appl Environ Microbiol. 2010;76:7373‐7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ruiz‐Moyano S, Totten SM, Garrido DA, et al. Variation in consumption of human milk oligosaccharides by infant gut‐associated strains of Bifidobacterium breve . Appl Environ Microbiol. 2013;79:6040‐6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bunesova V, Lacroix C, Schwab C. Fucosyllactose and L‐fucose utilization of infant Bifidobacterium longum and Bifidobacterium kashiwanohense . BMC Microbiol. 2016;16:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Centanni M, Ferguson SA, Sims IM, Biswas A, Tannock GW. Bifidobacterium bifidum ATCC 15696 and Bifidobacterium breve 24b metabolic interaction based on 2'‐O‐fucosyl‐lactose studied in steady‐state cultures in a Freter‐style chemostat. Appl Environ Microbiol. 2019;85:1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sims IM, Tannock GW. Galacto‐ and fructo‐oligosaccharides utilized for growth by cocultures of bifidobacterial species characteristic of the infant gut. Appl Environ Microbiol. 2020;86:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Cuskin F, Lowe EC, Temple MJ, et al. Human gut Bacteroidetes can utilize yeast mannan through a selfish mechanism. Nature. 2015;517:165‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wang K, Pereira GV, Cavalcante JJV, Zhang M, Mackie R, Cann I. Bacteroides intestinalis DSM 17393, a member of the human colonic microbiome, upregulates multiple endoxylanases during growth on xylan. Sci Rep. 2016;6:34360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126‐1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kopustinskiene DM, Jakstas V, Savickas A, Bernatoniene J. Flavonoids as anticancer agents. Nutrients. 2020;12:1‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yasuma T, Toda M, Abdel‐Hamid AM, et al. Degradation products of complex arabinoxylans by Bacteroides intestinalis enhance the host immune response. Microorganisms. 2021;9:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cole CB, Fuller R, Mallet AK, Rowland IR. The influence of the host on expression of intestinal microbial enzyme activities involved in metabolism of foreign compounds. J Appl Bacteriol. 1985;59:549‐553. [DOI] [PubMed] [Google Scholar]
- 102. Akao T, Kawabata K, Yanagisawa E, et al. Baicalin, the predominant flavone glucuronide of scutellariae radix, is absorbed from the rat gastrointestinal tract as the aglycone and restored to its original form. J Pharm Pharmacol. 2000;52:1563‐1568. [DOI] [PubMed] [Google Scholar]
- 103. Funabashi M, Grove TL, Wang M, et al. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature. 2020;582:566‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sato Y, Atarashi K, Plichta DR, et al. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature. 2021;599:458‐464. [DOI] [PubMed] [Google Scholar]
- 105. Ridlon JM, Devendran S, Alves JM, et al. The 'in vivo lifestyle' of bile acid 7alpha‐dehydroxylating bacteria: comparative genomics, metatranscriptomic, and bile acid metabolomics analysis of a defined microbial community in gnotobiotic mice. Gut Microbes. 2020;11:381‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Doden H, Sallam LA, Devendran S, et al. Metabolism of oxo‐bile acids and characterization of recombinant 12alpha‐hydroxysteroid dehydrogenases from bile acid 7alpha‐dehydroxylating human gut bacteria. Appl Environ Microbiol. 2018;84:1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Harris SC, Devendran S, Méndez‐García C, et al. Bile acid oxidation by Eggerthella lenta strains C592 and DSM 2243(T). Gut Microbes. 2018;9:523‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Doden HL, Wolf PG, Gaskins HR, Anantharaman K, Alves JMP, Ridlon JM. Completion of the gut microbial epi‐bile acid pathway. Gut Microbes. 2021;13:1‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Paik D, Yao L, Zhang Y, et al. Human gut bacteria produce Th17‐modulating bile acid metabolites. Nature. 2022;603:907‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Devlin AS, Fischbach MA. A biosynthetic pathway for a prominent class of microbiota‐derived bile acids. Nat Chem Biol. 2015;11:685‐690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Nelson DR. Cytochrome P450 diversity in the tree of life. Biochim Biophys Acta, Proteins Proteomics. 2018;1866:141‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Finnigan JD, Young C, Cook DJ, Charnock SJ, Black GW. Cytochromes P450 (P450s): a review of the class system with a focus on prokaryotic P450s. Adv Protein Chem Struct Biol. 2020;122:289‐320. [DOI] [PubMed] [Google Scholar]
- 113. Nishida CR, Ortiz de Montellano PR. Thermophilic cytochrome P450 enzymes. Biochem Biophys Res Commun. 2005;338:437‐445. [DOI] [PubMed] [Google Scholar]
- 114. Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138:103‐141. [DOI] [PubMed] [Google Scholar]
- 115. Dempsey JL, Cui JY. Microbiome is a functional modifier of P450 drug metabolism. Curr Pharmacol Rep. 2019;5:481‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Manikandan P, Nagini S. Cytochrome P450 structure, function and clinical significance: a review. Curr Drug Targets. 2018;19:38‐54. [DOI] [PubMed] [Google Scholar]
- 117. Gnerre C, Blattler S, Kaufmann MR, Looser R, Meyer UA. Regulation of CYP3A4 by the bile acid receptor FXR: evidence for functional binding sites in the CYP3A4 gene. Pharmacogenetics. 2004;14:635‐645. [DOI] [PubMed] [Google Scholar]
- 118. Jung D, Mangelsdorf DJ, Meyer UA. Pregnane X receptor is a target of farnesoid X receptor. J Biol Chem. 2006;281:19081‐19091. [DOI] [PubMed] [Google Scholar]
- 119. Sun HY, Yan YJ, Li YH, Lv L. Reversing effects of ginsenosides on LPS‐induced hepatic CYP3A11/3A4 dysfunction through the pregnane X receptor. J Ethnopharmacol. 2019;229:246‐255. [DOI] [PubMed] [Google Scholar]
- 120. Spanogiannopoulos P, Bess EN, Carmody RN, Turnbaugh PJ. The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat Rev Microbiol. 2016;14:273‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. van Miert AS. The sulfonamide‐diaminopyrimidine story. J Vet Pharmacol Ther. 1994;17:309‐316. [DOI] [PubMed] [Google Scholar]
- 122. Chung KT. Azo dyes and human health: a review. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2016;34:233‐261. [DOI] [PubMed] [Google Scholar]
- 123. Wilson ID, Nicholson JK. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl Res. 2017;179:204‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Guthrie L, Kelly L. Bringing microbiome‐drug interaction research into the clinic. EBioMedicine. 2019;44:708‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Lindenbaum J, Tse‐Eng D, Butler VP Jr, Rund DG. Urinary excretion of reduced metabolites of digoxin. Am J Med. 1981;71:67‐74. [DOI] [PubMed] [Google Scholar]
- 126. Lindenbaum J, Rund DG, Butler VP Jr, Tse‐Eng D, Saha JR. Inactivation of digoxin by the gut flora: reversal by antibiotic therapy. N Engl J Med. 1981;305:789‐794. [DOI] [PubMed] [Google Scholar]
- 127. Saha JR, Butler VP Jr, Neu HC, Lindenbaum J. Digoxin‐inactivating bacteria: identification in human gut flora. Science. 1983;220:325‐327. [DOI] [PubMed] [Google Scholar]
- 128. Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta . Science. 2013;341:295‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Takasuna K, Hagiwara T, Hirohashi M, et al. Involvement of beta‐glucuronidase in intestinal microflora in the intestinal toxicity of the antitumor camptothecin derivative irinotecan hydrochloride (CPT‐11) in rats. Cancer Res. 1996;56:3752‐3757. [PubMed] [Google Scholar]
- 130. Takasuna K, Hagiwara T, Hirohashi M, et al. Inhibition of intestinal microflora beta‐glucuronidase modifies the distribution of the active metabolite of the antitumor agent, irinotecan hydrochloride (CPT‐11) in rats. Cancer Chemother Pharmacol. 1998;42:280‐286. [DOI] [PubMed] [Google Scholar]
- 131. Pretorius E, Arlt W, Storbeck KH. A new dawn for androgens: novel lessons from 11‐oxygenated C19 steroids. Mol Cell Endocrinol. 2017;441:76‐85. [DOI] [PubMed] [Google Scholar]
- 132. Daisley BA, Chanyi RM, Abdur‐Rashid K, et al. Abiraterone acetate preferentially enriches for the gut commensal Akkermansia muciniphila in castrate‐resistant prostate cancer patients. Nat Commun. 2020;11:4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zimmermann M, Zimmermann‐Kogadeeva M, Wegmann R, Goodman AL. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature. 2019;570:462‐467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ly LK, Rowles JL III, Paul HM, et al. Bacterial steroid‐17,20‐desmolase is a taxonomically rare enzymatic pathway that converts prednisone to 1,4‐androstanediene‐3,11,17‐trione, a metabolite that causes proliferation of prostate cancer cells. J Steroid Biochem Mol Biol. 2020;199:105567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Ridlon JM, Ikegawa S, Alves JMP, et al. Clostridium scindens: a human gut microbe with a high potential to convert glucocorticoids into androgens. J Lipid Res. 2013;54:2437‐2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Devendran S, Mendez‐Garcia C, Ridlon JM. Identification and characterization of a 20beta‐HSDH from the anaerobic gut bacterium Butyricicoccus desmolans ATCC 43058. J Lipid Res. 2017;58:916‐925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Devendran S, Mythen SM, Ridlon JM. The desA and desB genes from Clostridium scindens ATCC 35704 encode steroid‐17,20‐desmolase. J Lipid Res. 2018;59:1005‐1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Shrestha E, White JR, Yu SH, et al. Profiling the urinary microbiome in men with positive versus negative biopsies for prostate cancer. J Urol. 2018;199:161‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zimmermann M, Patil KR, Typas A, Maier L. Towards a mechanistic understanding of reciprocal drug‐microbiome interactions. Mol Syst Biol. 2021;17:e10116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Holland KT, Knapp JS, Shoesmith JG. Techniques in anaerobic microbiology. Anaerobic Bacteria. Blackie; 1987:48‐67. [Google Scholar]
- 141. Javdan B, Lopez JG, Chankhamjon P, et al. Personalized mapping of drug metabolism by the human gut microbiome. Cell. 2020;181:1661‐1679. e1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kuroiwa M, Inotsume N, Iwaoku R, Nakano M. Reduction of dantrolene by enteric bacteria. Yakugaku Zasshi. 1985;105:770‐774. [DOI] [PubMed] [Google Scholar]
- 143. Elmer GW, Remmel RP. Role of the intestinal microflora in clonazepam metabolism in the rat. Xenobiotica. 1984;14:829‐840. [DOI] [PubMed] [Google Scholar]
- 144. Zimmermann M, Zimmermann‐Kogadeeva M, Wegmann R, Goodman AL. Separating host and microbiome contributions to drug pharmacokinetics and toxicity. Science. 2019;363:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kuroiwa M, Inotsume N, Nakano M. Reduction of nicardipine, calcium antagonist, with enteric bacteria. Yakugaku Zasshi. 1986;106:698‐702. [DOI] [PubMed] [Google Scholar]
- 146. Mannens G, Huang ML, Meuldermans W, Hendrickx J, Woestenborghs R, Heykants J. Absorption, metabolism, and excretion of risperidone in humans. Drug Metab Dispos. 1993;21:1134‐1141. [PubMed] [Google Scholar]
- 147. Meuldermans W, Hendrickx J, Mannens G, et al. The metabolism and excretion of risperidone after oral administration in rats and dogs. Drug Metab Dispos. 1994;22:129‐138. [PubMed] [Google Scholar]
- 148. Azadkhan AK, Truelove SC, Aronson JK. The disposition and metabolism of sulphasalazine (salicylazosulphapyridine) in man. Br J Clin Pharmacol. 1982;13:523‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Peppercorn MA, Goldman P. The role of intestinal bacteria in the metabolism of salicylazosulfapyridine. J Pharmacol Exp Ther. 1972;181:555‐662. [PubMed] [Google Scholar]
- 150. Baba T, Ara T, Hasegawa M, et al. Construction of Escherichia coli K‐12 in‐frame, single‐gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Goodman AL, McNulty NP, Zhao Y, et al. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe. 2009;6:279‐289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Clardy J, Fischbach MA, Walsh CT. New antibiotics from bacterial natural products. Nat Biotechnol. 2006;24:1541‐1550. [DOI] [PubMed] [Google Scholar]
- 153. Wenzel SC, Muller R. Recent developments towards the heterologous expression of complex bacterial natural product biosynthetic pathways. Curr Opin Biotechnol. 2005;16:594‐606. [DOI] [PubMed] [Google Scholar]
- 154. Long LL, Svenson KL, Mourino AJ, et al. Shared and distinctive features of the gut microbiome of C57BL/6 mice from different vendors and production sites, and in response to a new vivarium. Lab Anim (NY). 2021;50:185‐195. [DOI] [PubMed] [Google Scholar]
- 155. Alegre ML. Mouse microbiomes: overlooked culprits of experimental variability. Genome Biol. 2019;20:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Beresford‐Jones BS, Forster SC, Stares MD, et al. The mouse gastrointestinal bacteria catalogue enables translation between the mouse and human gut microbiotas via functional mapping. Cell Host Microbe. 2022;30:124‐138 e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Kieser S, Zdobnov EM, Trajkovski M. Comprehensive mouse microbiota genome catalog reveals major difference to its human counterpart. PLoS Comput Biol. 2022;18:e1009947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Nishiyama T, Ogura K, Okuda H, et al. Mechanism‐based inactivation of human dihydropyrimidine dehydrogenase by (E)‐5‐(2‐bromovinyl)uracil in the presence of NADPH. Mol Pharmacol. 2000;57:899‐905. [PubMed] [Google Scholar]
- 159. Desgranges C, Razaka G, de Clercq E, et al. Effect of (E)‐5‐(2‐bromovinyl)uracil on the catabolism and antitumor activity of 5‐fluorouracil in rats and leukemic mice. Cancer Res. 1986;46:1094‐1101. [PubMed] [Google Scholar]
- 160. Fischbach MA. Microbiome: focus on causation and mechanism. Cell. 2018;174:785‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Lagier JC, Dubourg G, Million M, et al. Culturing the human microbiota and culturomics. Nat Rev Microbiol. 2018;16:540‐550. [DOI] [PubMed] [Google Scholar]
- 162. Poyet M, Groussin M, Gibbons SM, et al. A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research. Nat Med. 2019;25:1442‐1452. [DOI] [PubMed] [Google Scholar]
- 163. Forster SC, Kumar N, Anonye BO, et al. A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat Biotechnol. 2019;37:186‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Jin WB, Li TT, Huo D, et al. Genetic manipulation of gut microbes enables single‐gene interrogation in a complex microbiome. Cell. 2022;185:547‐562 e522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Wallace BD, Wang H, Lane KT, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330:831‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Bhatt AP, Pellock SJ, Biernat KA, et al. Targeted inhibition of gut bacterial beta‐glucuronidase activity enhances anticancer drug efficacy. Proc Natl Acad Sci USA. 2020;117:7374‐7381. [DOI] [PMC free article] [PubMed] [Google Scholar]