

Abstract
Cilia are ubiquitous eukaryotic organelles impotant for cellular motility, signaling, and sensory reception. Cilium formation requires intraflagellar transport of structural and signaling components and involves 22 different proteins organized into intraflagellar transport (IFT) complexes IFT‐A and IFT‐B that are transported by molecular motors. The IFT‐B complex constitutes the backbone of polymeric IFT trains carrying cargo between the cilium and the cell body. Currently, high‐resolution structures are only available for smaller IFT‐B subcomplexes leaving > 50% structurally uncharacterized. Here, we used Alphafold to structurally model the 15‐subunit IFT‐B complex. The model was validated using cross‐linking/mass‐spectrometry data on reconstituted IFT‐B complexes, X‐ray scattering in solution, diffraction from crystals as well as site‐directed mutagenesis and protein‐binding assays. The IFT‐B structure reveals an elongated and highly flexible complex consistent with cryo‐electron tomographic reconstructions of IFT trains. The IFT‐B complex organizes into IFT‐B1 and IFT‐B2 parts with binding sites for ciliary cargo and the inactive IFT dynein motor, respectively. Interestingly, our results are consistent with two different binding sites for IFT81/74 on IFT88/70/52/46 suggesting the possibility of different structural architectures for the IFT‐B1 complex. Our data present a structural framework to understand IFT‐B complex assembly, function, and ciliopathy variants.
Keywords: AlphaFold, Cilium, IFT‐B structure, Intraflagellar transport, Structural modeling
Subject Categories: Membranes & Trafficking, Organelles, Structural Biology
A combination of alphafold structural modeling and biochemical experiments reveals the structure of the large IFT‐B intraflagellar transport complex and offers insight into IFT‐B complex assembly and cilia formation.

Introduction
Cilia are slim eukaryotic organelles that are conserved from the green alga Chlamydomonas reinhardtii (Cr) to human and protrude from cell surfaces to function in both motility and signaling pathways (Rosenbaum & Witman, 2002). Cilia are organized into an axoneme consisting of microtubule (MT)‐doublets with ninefold symmetry and are surrounded by the ciliary membrane, which is continuous with the plasma membrane but contains a unique composition of lipids and membrane receptors important for signaling (Mourão et al, 2016). Cilium formation and function require the selective ciliary trafficking of both axonemal factors such as tubulin (Bhogaraju et al, 2014) as well as membrane proteins (Long & Huang, 2020). Trafficking along the ciliary axoneme is carried out by intraflagellar transport (IFT; Kozminski et al, 1993), which relies on molecular motors and the 22‐subunit IFT complex that organizes into 6‐subunit IFT‐A and 16‐subunit IFT‐B complexes that loosely associate (Cole et al, 1998; Piperno et al, 1998). IFT‐A and ‐B polymerize into linear assemblies known as IFT trains that associate with the BBSome and move ciliary cargo into and out of cilia (Kozminski et al, 1995; Nachury et al, 2007; Pigino et al, 2009). Anterograde IFT trains move from the base to the tip of cilia powered by the kinesin 2 motor (Kozminski et al, 1995; Wingfield et al, 2017), whereas retrograde IFT trains move from the tip and back to the base of cilia and are powered by the IFT dynein motor (Pazour et al, 1999; Porter et al, 1999). Elegant time‐resolved correlative fluorescence and three‐dimensional electron microscopy revealed that anterograde and retrograde IFT trains drive on different MTs of the MT doublets to avoid head‐on collisions (Stepanek & Pigino, 2016). During kinesin‐driven IFT to the ciliary tip, inactivated IFT dynein motor associates with anterograde IFT trains as a cargo (Jordan et al, 2018).
Interestingly, IFT‐B and IFT‐A assemble at the ciliary base into linear polymers of different repeat lengths (Jordan et al, 2018; van den Hoek et al, 2022). Whereas IFT‐B polymers have a repeat distance of 6 nm and form first, IFT‐A polymers have a repeat distance of 11.5 nm and appear to assemble onto preformed IFT‐B polymers (Jordan et al, 2018; van den Hoek et al, 2022). IFT‐A and IFT‐B thus do not form 1:1 complexes but rather have an approximate 1:2 stoichiometry in IFT trains, which is consistent with mass‐spectrometry (MS) results (Lechtreck et al, 2009). The structures of anterograde IFT trains were determined at 24–37 Å resolution by cryo‐electron tomography (cryo‐ET), which clearly resolved IFT‐A, IFT‐B and inactive IFT dynein complexes (Jordan et al, 2018; van den Hoek et al, 2022). However, the resolution of these studies was insufficient to resolve the position of individual protein subunits. Interestingly, retrograde IFT trains returning from the ciliary tip to the base appear to have very different structures and repeat distances compared to anterograde IFT trains suggesting that significant remodeling of the IFT complexes occurs at or close to the ciliary tip (Jordan et al, 2018).
The IFT‐B complex forms the backbone of IFT trains and is absolutely required for IFT and thus for cilium formation (Taschner & Lorentzen, 2016a). Biochemical studies have provided an architecture of the IFT‐B complex (Boldt et al, 2016; Katoh et al, 2016; Taschner et al, 2016), sometimes with domain resolution, and several high‐resolution crystal structures have been determined for IFT subunits and smaller subcomplexes (Fig 1A; Taschner & Lorentzen, 2016a, 2016b). These include the structures of the IFT27/25 complex (Bhogaraju et al, 2011) involved in BBSome trafficking and hedgehog signaling (Keady et al, 2012; Eguether et al, 2014; Liew et al, 2014; Desai et al, 2020), IFT81N/74N/22 revealing the binding mode of the small GTPase IFT22 on IFT81/74 (Wachter et al, 2019), and the IFT70/52 and IFT52/46 subcomplexes demonstrating how IFT70 wraps around IFT52 as a superhelix (Taschner et al, 2014). The crystal structure of IFT80 revealed the structure of two β‐propellers (BP), suggested an IFT80 homo‐dimer, and allowed for the mechanistic study of ciliopathy disease mutations (Taschner et al, 2018). In addition, crystal structures are available for the N‐terminal IFT54 calponin homology (CH) and IFT52 GIFT domains (Taschner et al, 2016). These studies have established how the IFT‐B complex organizes into approximately equally sized IFT‐B1 and IFT‐B2 complexes that associate via IFT88/52 of IFT‐B1 and IFT57/38 of IFT‐B2 (Katoh et al, 2016; Taschner et al, 2016). However, the high‐resolution structures of IFT proteins cannot be unambiguously fitted to the low resolution cryo‐ET maps thus preventing a structural understanding of the complete IFT‐B complex.
Figure 1. Interaction network of the IFT‐B complex obtained by chemical cross‐linking and mass spectrometry.
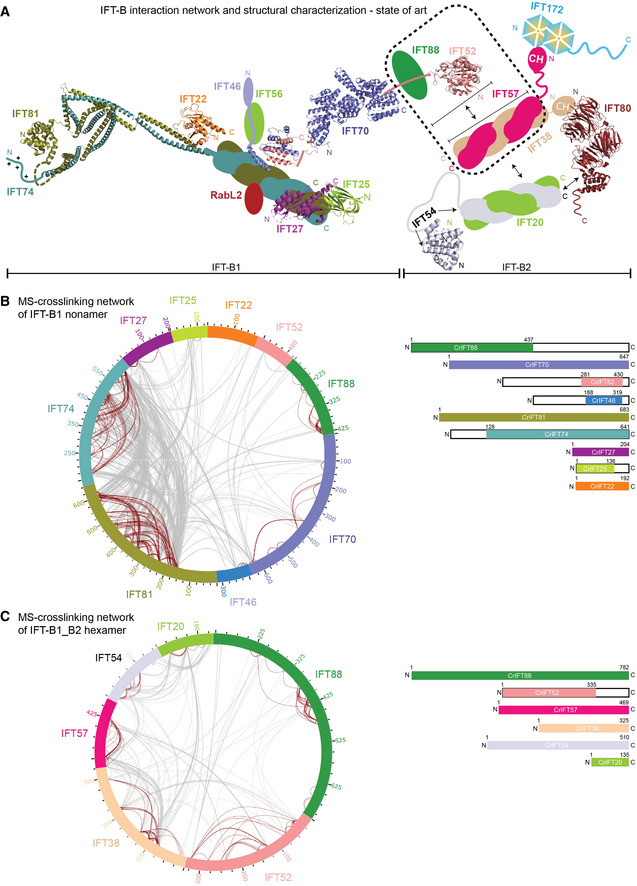
- Schematic representation of IFT‐B complex architecture based on published structural and biochemical data. Structural information is available for Trypanosoma brucei IFT22 and the N‐termini of IFT81/74 (PDB: 6ian), the Chlamydomonas reinhardtii IFT27/25 heterodimer (PDB: 2yc2), the C‐termini of IFT52 and IFT46 from Tetrahymena thermophila (PDB: 4uzz), the IFT70/52 complex from Chlamydomonas reinhardtii (4uzy), the globular N‐terminal GIFT domain of IFT52 from Chlamydomonas reinhardtii (PDB: 5FMR) as well as from Mus musculus (PDB: 5FMS), the N‐terminal CH‐domain of IFT54 from Mus musculus (5FMU) and IFT80 from Chamydomonas reinhardtii (PDB: 5N4A). The IFT‐B complex is subdivided into biochemically salt stable IFT‐B1 (IFT88/81/74/70/56/52/46/27/25/22/RabL2) and IFT‐B2 (IFT172/80/57/54/38/20) complexes.
- The inter‐ and intramolecular cross‐linking network within the Chlamydomonas IFT‐B1 nonamer (IFT881–437/70/52281–430/46188–319/81/74128‐C/27/251–136/22) are depicted as a cartwheel diagram (left panel). The gray lines show intermolecular cross‐linking pairs, and the brown lines show the intramolecular cross‐linking pairs. The protein constructs of the IFT‐B1 nonamer are indicated on the right.
- The inter‐ and intramolecular cross‐linking network within a CrIFT‐B1‐B2 hexamer (IFT88/521–335/57/38/54/20) is displayed as a cartwheel. In this protein complex, only the C‐terminal part of IFT52 is truncated while all other proteins are full length (see schematics on the right panel).
To bridge this gap in our understanding of the IFT‐B structure, we use recent advances in machine learning to model the structure of IFT‐B subcomplexes and assemble an almost complete 15‐subunit structural model of the IFT‐B complex. We use a plethora of biochemical and biophysical methods to validate interactions and interfaces within the IFT‐B complex. Our structural model is consistent with cryo‐ET maps and provides a structural framework to understand IFT and allows for the mapping of ciliopathy variants in context of the IFT‐B complex.
Results
Cross‐linking/MS reveals the interactions within the IFT‐B complex
To obtain a comprehensive map of interactions within the IFT‐B complex, we produced two recombinant Chlamydomonas IFT‐B complexes covering 13 subunits and analyzed these by cross‐linking/mass spectrometry (MS). We enriched an IFT‐B1 complex consisting of the nine Chlamydomonas proteins IFT88, IFT81, IFT74, IFT70, IFT52, IFT46, IFT27, IFT25, and IFT22 by size exclusion chromatography (SEC) and cross‐linked the sample using the MS cleavable disuccinimidyl dibutyric urea (DSBU) cross‐linker (Fig 1B and C; Appendix Fig S1). DSBU is an amine‐ and hydroxy‐specific, homo‐bifunctional cross‐linker with a cross‐linking spacer arm of 12.5 Å (Iacobucci et al, 2018) that connects both closely packed residues within Cα‐Cα Euclidean distances of 27 Å and flexible residues located up to 43 Å apart (Felker et al, 2021). A second protein complex comprising Chlamydomonas IFT‐B2 proteins IFT57, IFT38, IFT54, and IFT20 as well as the IFT‐B1 proteins IFT88 and IFT52N was also subjected to cross‐linking/MS to provide data on the interactions within the IFT‐B2 complex and between the IFT‐B1 and B2 subcomplexes (Fig 1C). The cross‐linking experiments of these two complexes were performed independently and were subsequently digested with both LysC and trypsin. The resulting peptides were enriched by strong cation‐exchange chromatography and subjected to MS/MS analysis. Identification of cross‐linking pairs was performed with the MeroX software (Götze et al, 2015) taking into account all possible cross‐links of DSBU. Only cross‐linking data with false discovery rate (FDR) below 1% and scores above 80 are considered high confidence and used in the analysis below.
Within the IFT‐B1 nonamer, we identified 402 intra‐ and 859 intermolecular cross‐linking pairs (Fig 1B; Dataset EV1). Multiple intramolecular cross‐links were present within IFT88, IFT81, IFT74 and IFT70 (Fig 1B, brown lines). The intramolecular cross‐links of IFT88 show a 34–50 residues periodicity, which agrees with its predicted tetratricopeptide repeat (TPR) structure. The intramolecular cross‐linking network of IFT81 and IFT74 shows a similar pattern with periodicities of 10–25, 50–80, 200–250, and 400–450 residues, suggesting that cross‐links formed within the same helix, and between adjacent coiled coils (CCs). Together with intermolecular cross‐links between the N‐ and C‐terminal halves of IFT81/74, these cross‐links are consistent with a heterodimeric IFT81/74 structure consisting of parallel CCs and agree with the crystal structure of Trypanosoma IFT81N/74N/22 previously published (Wachter et al, 2019). The IFT27/25 hetero‐dimer primarily cross‐links to the C‐terminal part of IFT81/74 while IFT22 cross‐links to the central part of IFT81/74 (Fig 1B). The C‐termini of IFT46 and IFT52 interact in a hetero‐dimer (IFT52C/46C) that was previously shown to mediate the interaction between IFT88/70/52/46 and IFT81/74/27/25/22 subcomplexes (Taschner et al, 2014). In our cross‐linking data, IFT46C/52C cross‐links to the C‐terminal part of IFT81/74 close to the IFT27/25 binding site. In addition, IFT81N/74N cross‐links to IFT88 and IFT70, which may constitute a second interaction site between IFT81/74 and IFT88/70/52/46 subcomplexes within the IFT‐B1 complex. The most N‐terminal 150 residues of IFT88 cross‐link primarily to the 250 most C‐terminal residues of IFT70 indicating an N‐ to C‐interaction, while IFT52 cross‐links to both IFT70 and IFT88.
Analysis of the IFT‐B1_B2 hexamer cross‐linked sample revealed 575 intra‐ and 383 intermolecular high‐confidence cross‐links (Fig 1C; Dataset EV2). 78 intermolecular cross‐links were identified between IFT57 and IFT38 (Fig 1C) in agreement with previous data showing that these two proteins interact via their C‐terminal CC domains (Taschner et al, 2016; Fig 1A). This is also the case for IFT54 and IFT20 (Fig 1C) that form a complex via their C‐terminal helices (Fig 1A). The N‐terminal CH‐domain of IFT54 is linked to the C‐terminal CC domain via a long linker region that is presumably disordered and provides high flexibility in the relative position of these two domains of IFT54. This notion is supported by our cross‐linking analysis where the CH‐domain of IFT54 is cross‐linked to the CCs of IFT54/20 along most of their lengths (Fig 1C). IFT57, like IFT54, contains a long intrinsically disordered central region between the N‐terminal CH‐domain and the C‐terminal CCs. For IFT57, we also observe a cross‐linking pattern where the IFT57 CH‐domain forms cross‐links to the C‐terminal CCs of IFT57 and the binding partner IFT38 (Fig 1C). These results suggest a high degree of flexibility in the position of IFT54 and IFT57 CH‐domains with respect to the CCs. Our MS analysis identified multiple cross‐linking pairs formed between IFT88 and IFT52, IFT57, or IFT38 (25, 13 and 6 high‐confidence cross‐links, respectively). These cross‐links suggest, in agreement with previously published results (Taschner et al, 2016), that IFT88/52 bridges IFT70 of the IFT‐B1 complex to IFT57/38 of the IFT‐B2 complex thus connecting B1 and B2 within IFT‐B.
Prediction and validation of the IFT81/74/27/25/22 structure
IFT81 and IFT74 associate into a hetero‐dimer via CCs and serve as a scaffold onto which the small Rab like GTPases IFT22, IFT27, and RabL2 associate (Taschner et al, 2014; Kanie et al, 2017; Nishijima et al, 2017; Wachter et al, 2019). RabL2 only associates with the IFT trains during the initiation and early steps of anterograde IFT (Kanie et al, 2017) and was not included in the current study. Formation of the IFT81/74 heterodimer is essential for IFT in C. elegans (Kobayashi et al, 2007) and is a prerequisite for IFT train assembly at the ciliary base (Brown et al, 2015). IFT25 is also loaded on the IFT81/74 platform via direct interaction with IFT27 (Bhogaraju et al, 2011). The N‐termini of IFT81 and IFT74 were shown to bind tubulin heterodimers as cargo via a CH‐domain and a positively charged region, respectively (Bhogaraju et al, 2013). The structure of the N‐terminal half of Trypanosoma brucei IFT81/74 in complex with IFT22 was determined by X‐ray crystallography and shows that IFT81N/74N organizes into 6 parallel CCs (CC I‐CC VI), where IFT22 associates with CC VI (Wachter et al, 2019). Although the exact binding site is currently not known, IFT27/25 was shown to bind the C‐terminal half of the IFT81/74 complex in both Chlamydomonas and human cells (Taschner et al, 2014; Zhou et al, 2022).
We made use of the recent advances in protein structure prediction by machine learning as implemented in AlphaFold (AF; Jumper et al, 2021) using a local installation as well as a Colab notebook implementation (Mirdita et al, 2022) to model the structure of Chlamydomonas IFT‐B subcomplexes, which allowed us to assemble a structural model for the 15‐subunit IFT‐B complex. All structural models of protein complexes were modeled using the AF multimer version optimized for the structure prediction of multimeric protein complexes (preprint: Evans et al, 2022). The quality of the resulting AF models was initially assessed using the predicted local distance difference test score (pLDDT), which constitute a per‐residue score reporting on the confidence of the local structure prediction. Structural predictions with pLDDT > 70 indicate confident parts of the model (colored blue in pLDDT figures), whereas low confidence structural segments with pLDDT < 50 likely represent intrinsic disorder (colored orange in pLDDT figures; preprint: Evans et al, 2022; Stevens & He, 2022). To evaluate the accuracy of the relative positions of protein subunits within multimeric structures, the predicted alignment error (PAE) plots were inspected to ensure that protein–protein interface residues have low error scores (for example, see Fig 2D). Importantly, all protein–protein structure models are supported by observations from at least one biochemical or biophysical technique.
Figure 2. The AlphaFold predicted structure of CrIFT81/74/27/251–136/22.
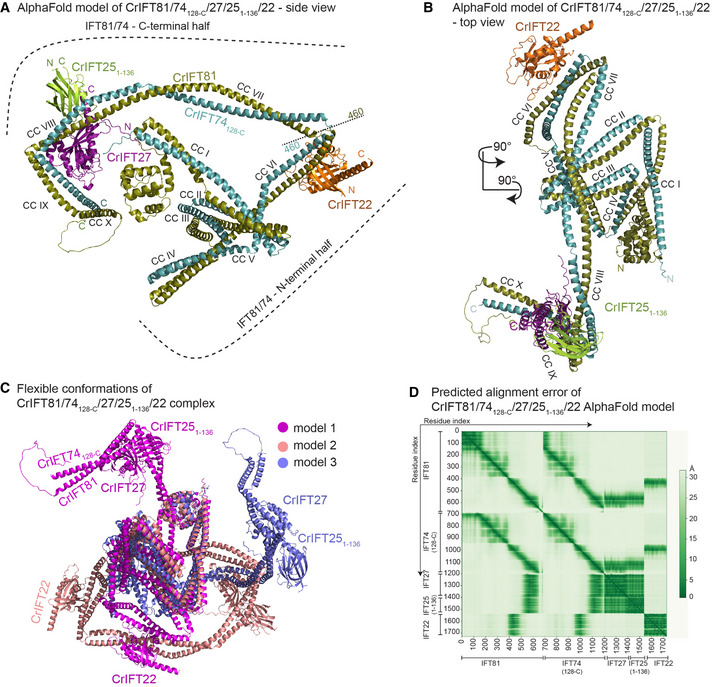
- AlphaFold predicts the structure of IFT81/74128‐C/27/251–136/22 complex as two halves built on the IFT81/74 CC scaffold and hinged by a short loop region around the amino acids 460 of both IFT81 and IFT74. CrIFT22 binds near the hinge region on the CC domain VI (B) while the CrIFT27/25 heterodimer is docked proximal to the C‐termini of IFT81/74 in a ‘L‐shape’ structure formed by CCs VIII and IX.
- The top view of the complex shows the organization of the IFT81/74 CCs and the docking sites for IFT27/25 and IFT22.
- 3 independent AF models of the IFT81/74128‐C/27/251–136/22 complex are superimposed using their N‐terminal halves to illustrate high flexibility between the separate parts of IFT81/74.
- The predicted alignment error (PAE) plot for the model shown in panels A‐B. This plot assesses the confidence in the relative position of subunits within the complex. The Y‐ and the X‐axes show the residues indexed of the corresponding subunits as indicated. The aligned error in angstroms (Å) is color coded according to the bar to the right of the plot. Green color indicates low PAE (high confidence) and white color indicates high PAE (low confidence).
The structural model of the pentameric IFT81/74/27/25/22 complex shows that the IFT81/74 complex folds as 10 parallel CCs (CC I‐CC X) connected by short loops (Fig 2A and B). Although CC VII‐VIII and IX‐X could be considered as single CC segments, resulting in a total of 8 CCs in IFT81/74, we denote these as separate CCs as we observe significant bends and/or breaks in the CC helices. Preceding the CCs are 130 residues of IFT81 that adopt the fold of a CH domain and 100 residues of IFT74 are predicted to be unstructured (Fig 2A and B; Bhogaraju et al, 2013). The local structure of all 10 CCs is predicted with high confidence as highlighted by the coloring of the IFT81/74/27/25/22 model according to the pLDDT score (Appendix Fig S2A). Contrary, the structure of the short linker regions connecting adjacent CCs is predicted with lower confidence implying possible flexibility in the position between connecting CCs (Appendix Fig S2A). In particular, the loop region connecting CC V to CC VI has pLDDT < 50 and may not adopt an ordered structure in solution. Another hinge region formed between CC VI and CC VII divides the IFT81/74 complex into approximate N‐ and C‐terminal halves (Fig 2A). The hinge region and the fact that no interface is observed between the N‐ and C‐terminal halves of IFT81/74 point to a high degree of conformational flexibility. Indeed, different structural models produced by AF vary greatly in the relative positions of N‐ and C‐terminal halves of IFT81/74 (for three different conformations see Fig 2C). This notion is also supported by the PAE plot that shows low errors for IFT81 and IFT74 helices of the same CC but much larger error between residues of N‐ and C‐terminal CCs (Fig 2D). However, 269 cross‐links between residues of the N‐ and C‐terminal halves of IFT81/74 suggest that the complex can adopt a compact conformation in solution where the two halves are in proximity (Fig 1B).
In our structural model, IFT22 and IFT27/25 are positioned on CC VI and CC VIII‐CC IX of IFT81/74, respectively, with high confidence as illustrated by the low PAE for interacting regions (Fig 2D). The Chlamydomonas IFT81/74 model predicted here superimposes well onto the Trypanosoma brucei IFT81N/74N/22 crystal structure (Appendix Fig S2B). The binding site of IFT22 on CC VI of IFT81/74 thus appears to be conserved between Chlamydomonas and Trypanosoma. We further validated the structural model by site directed photo‐cross‐linking using a purified Chlamydomonas IFT81/74/27/251–136/22 complex where the native amino acid E418 of IFT81 located near the IFT22‐binding site was substituted with the UV‐reactive amino acid p‐benzoyl‐L‐phenylalanine (pBpa; Appendix Fig S2C; Young et al, 2010). Upon UV activation, pBpa forms a covalent bond with proteins located in the immediate vicinity allowing the cross‐linked proteins to be resolved on SDS‐PAGE as they migrate slower than their monomeric noncross‐linked counterparts. The IFT81/74/27/251–136/22 complex containing pBpa at position 418 in IFT81 formed a stoichiometric, UV‐dependent cross‐link with IFT22 that migrated on SDS‐PAGE at expected molecular weight of 123 kDa (Appendix Fig S2C). We tested the site‐directed specificity of the method by using another IFT81/74/27/251–136/22 complex that contained pBpa in the CH‐domain of IFT81 (position 68), far away from the predicted IFT22‐binding site. This complex did not form cross‐links with IFT22 upon UV activation. These data provide strong experimental evidence for the position of CrIFT22 on CC VI of CrIFT81/74 as illustrated in Fig 2; Appendix Fig S2.
IFT81534–623/74533–615 encompasses CC VIII and CC IX and adopts an L‐shaped structure that cradles the IFT27/25 heterodimer with IFT27 contacting both CC VIII and CC IX of IFT81/74 and IFT25 contacting only CC VIII (Fig 2A; Appendix Fig S3). This binding site is consistent with the intermolecular cross‐linking data obtained from the IFT‐B1 complex (Fig 1B). A total of 41 cross‐links formed by IFT25 with IFT81/74 were found, of which 20 cross‐links were mapped to the N‐terminal half and 21 to the C‐terminal half of IFT81/74. All IFT25 cross‐links with the C‐terminal half of IFT81/74 were mapped to the IFT81534–623/74533–615 region. IFT27 made 59 cross‐links with IFT81/74 of which 35 were identified within the N‐terminal part and 24 within the C‐terminal part of IFT81/74. Nineteen out of 24 cross‐links between IFT27 and the C‐terminal half of IFT81/74 were mapped to the IFT81534–623/74533–615 region. For a 3D visualization of the cross‐linking network, we labeled the IFT25 (Movie EV1) and IFT27 (Movie EV2) cross‐links onto the IFT81C/74C model. The fact that IFT27/25 also cross‐links with the N‐terminal half of IFT81/74 suggest that the N‐ and C‐terminal halves can be in proximity within the complex consistent with a high degree of conformational flexibility as noted above. In addition to the IFT27/25‐binding site, the C‐termini of IFT81/74 also harbor a binding site for the C‐termini of IFT52/46 located distally to IFT27/25 (Fig 3A). Although IFT27/25 cross‐links to both N‐ and C‐terminal halves of IFT81/74, the main binding site on CC VIII‐IX was verified experimentally as IFT81/74 proteins lacking the N‐terminal 459 residues still associate with IFT27/25 (Fig 4C and D). This notion is in agreement with previous biochemical studies showing that CrIFT27/25 does not bind to the N‐terminal IFT81133–442/74135–475 complex (Taschner et al, 2014). Thus, we conclude that the main docking site of IFT27/25 is on the C‐terminal half of IFT81/74 in agreement with the predicted structural model of the pentameric IFT81/74/27/25/22 complex (Fig 2A).
Figure 3. Structural model of the IFT88/70/52/46 IFT‐B1 subcomplex.
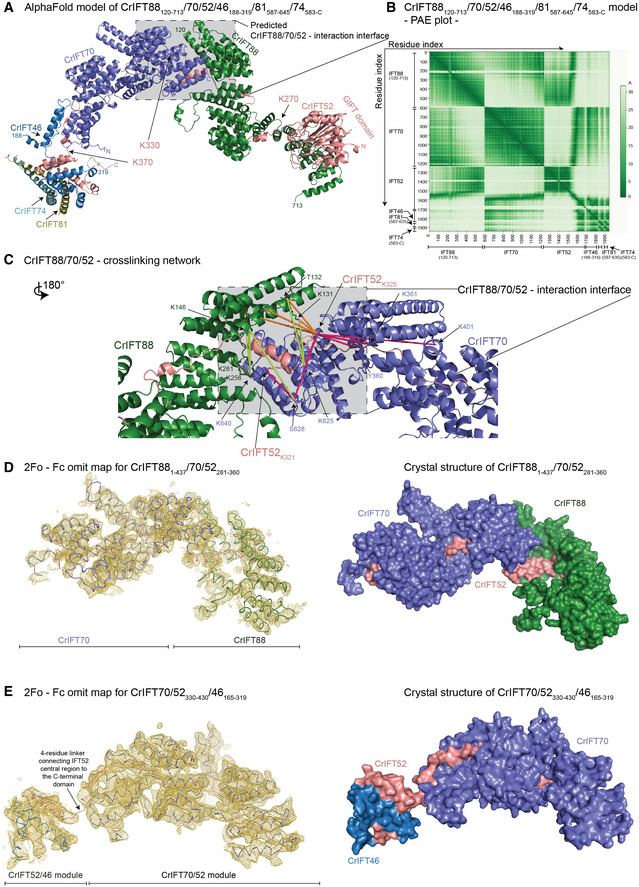
- The AlphaFold predicted model of CrIFT88120–713/70/52/46188–319/81587–645/74583‐C.
- The predicted alignment error plot of the complex from A. The residue indexes are indicated on the X‐ and Y‐axis.
- The CrIFT88/70/52 cross‐linking network validates the interaction interface predicted by AlphaFold. The lime‐green dashed lines are showing cross‐linking pairs formed between IFT88 and IFT70. The orange dashed lines are showing the IFT88/52 cross‐links and the pink lines are showing the cross‐links between IFT52 and IFT70. K321 and K325 of IFT52 make multiple short‐range interactions with both IFT88 and IFT70 residues.
- The crystal structure of Chlamydomonas IFT881–437/70/52281–360 displayed as ribbon and the 2Fo ‐ Fc omit map (3sigma) as a yellow mesh (left panel). Surface representation of the structure is shown on right panel.
- The crystal structure of Chlamydomonas IFT70/52330–430/46165–319 displayed as ribbon and the 2Fo ‐ Fc omit map (3sigma) as a yellow mesh (left panel). Surface representation of the structure is shown on right panel.
Figure 4. IFT81/74 has two separate binding sites on IFT88/70/52/46.
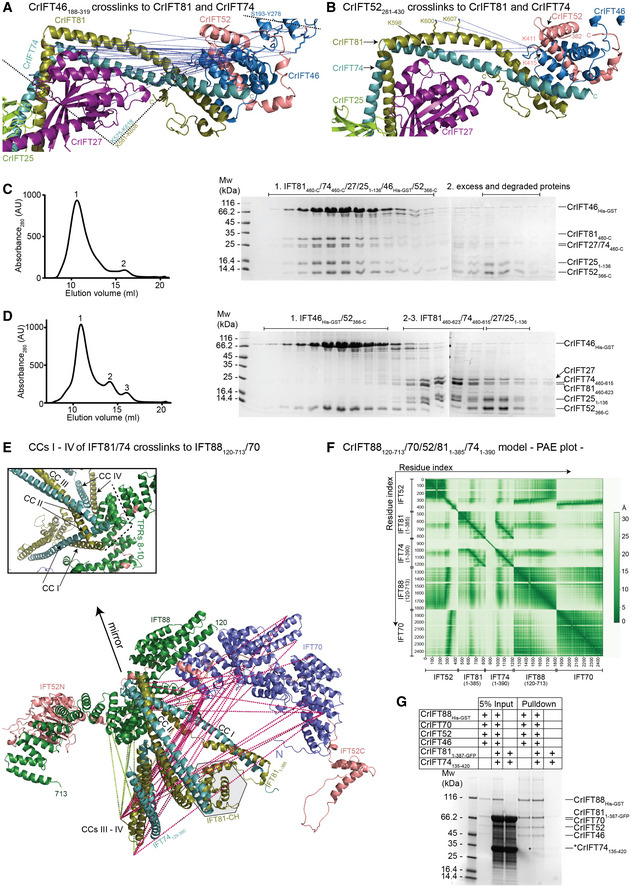
-
AThe blue dotted lines indicate the IFT46 cross‐links to the C‐termini of IFT81 and IFT74. These cross‐links map to a 43 residue stretch from K575 to K618 in IFT81 and a 27 residue stretch in IFT74 bordered by K581 and K608. An additional cross‐linking pair was identified between IFT46 and IFT27.
-
BK411 and K415 of IFT52 cross‐link to K598, K600, and K607 of IFT81. These cross‐links are labeled as in panel (A).
-
C, DThe C‐terminal domains of IFT52 and IFT46 co‐purify with the IFT81460‐C/74460‐C/27/251–136 protein complex (C) but not with a protein complex that is missing residues 623‐C of IFT81 and 615‐C of IFT74 (D). The SEC fractions indicated on the right with 1–3 were analyzed by SDS‐PAGE and stained with Coomassie to evaluate the protein composition.
-
EIFT811–385/74128–390 cross‐linking network with IFT88, IFT70, and IFT52 labeled on the AF predicted model of Chlamydomonas IFT88120–713/70/52/811–385/741–390. The N‐terminal 128 amino acids of IFT74 are predicted to be unstructured and omitted from the figure for clarity. The box shows a zoom‐in on the IFT81/74/88 interaction interface.
-
FThe predicted alignment error plot of the Chlamydomonas IFT88120–713/70/52/811–385/741–390 complex shown in panel (E).
-
GCoomassie stained SDS PAGE gel of a GST‐pull down using the CrIFT88His‐GST/70/521–430/46 complex immobilized on GST beads as bait and the CrIFT811–387‐GFP/74135–420 complex used as prey. The band highlighted by an * corresponds to pulled down CrIFT74135–420. CrIFT811–387‐GFP runs on top of IFT70 and is not visible in the pull down.
Structural model of the IFT88/70/52/46 IFT‐B1 subcomplex
We previously showed that IFT52 functions as a central IFT‐B protein that connects IFT88, IFT70 and IFT46 in a tetrameric IFT‐B1 subcomplex (Taschner et al, 2011). Subsequent structural studies revealed that the TPRs of IFT70 wrap around a proline rich region of IFT52 (residues 330–370) that constitutes the hydrophobic core of IFT70 (Taschner et al, 2014). Proximal to the IFT70‐binding site, IFT88 contacts residue 281–329 of IFT52 (Taschner et al, 2014). In pull‐down assays, residues 118–437 of IFT88 were sufficient for IFT52 interaction (Taschner et al, 2014). In addition, human IFT70 was shown to interact with the IFT88/52 dimer by visual immunoprecipitation assays and this interaction is essential for ciliogenesis (Takei et al, 2018). It is thus firmly established that IFT88/70/52/46 form a tetrameric complex although high‐resolution structures are only available for Chlamydomonas IFT70/52 and Tetrahymena IFT52/46 (Taschner et al, 2014) and it is currently unknown how IFT88 interacts with IFT70/52.
Using AF, we predicted the structure of Chlamydomonas IFT88/70/52/46 in complex with the very C‐terminal helices (CC X) of IFT81/74 (Fig 3A). This structural model is predicted with high confidence, except for a few flexible loops and termini (Appendix Fig S4A), as evident from the high pLDDT score and the low PAE values for interacting residues of all protein–protein interfaces (Fig 3B). Residues 120–713 of IFT88 are predicted to fold into 15 TPRs with the most N‐terminal 119 and the most C‐terminal 67 residues predicted to be intrinsically disordered (Figs 3A and 4E). IFT88 adopts a rather loose and open superhelical structure, in contrast to the tight and closed superhelical structure of IFT70 that buries residues 330–370 of IFT52 (Fig 3A and D). The interaction of IFT88 with IFT521–329 can be divided into two main interfaces. For the first interaction site, the three most C‐terminal TPRs of IFT88 adopt an extended conformation to interact with the N‐terminal GIFT domain of IFT52 (residues 1–270) with 40 predicted high‐confidence close contacts within 5 Å (PAE < 5 Å; Fig 3B). For the second interaction site, residues 271–330 of IFT52 interact in an extended conformation with the most N‐terminal 12 TPRs of IFT88 (Fig 3A). The following amino acids 330–370 of IFT52 snake their way through the interior of the IFT70 superhelix as previously observed in the IFT70/52 crystal structure (Taschner et al, 2014). Finally, the small C‐terminal domain of IFT52 (residues 371‐C) interacts with the C‐terminal domain of IFT46 to form a small hetero‐dimer at the N‐terminal face of the IFT70 superhelix (Fig 4E). In this structural model, the four most N‐terminal TPRs of IFT88 are within interaction distance of the three most C‐terminal TPRs of IFT70 (Fig 3C) in agreement with the direct IFT88‐IFT70 interaction observed in pull‐down experiments (Taschner et al, 2014). However, there appears to be no direct noncovalent interaction to tether IFT52C/46C to the N‐terminal face of IFT70 (Fig 3A and E). We conclude that IFT52 is a central hub that organizes the IFT‐B1 complex, which explains why Chlamydomonas ift52 mutant cells (bld1) contain highly destabilized IFT‐B1 complexes (Richey & Qin, 2012) and display severe ciliogenesis defects (Brazelton et al, 2001).
We validated the structural model of IFT88/70/52/46 using cross‐linking/MS and crystallographic X‐ray diffraction data. Several cross‐links are found at the IFT88/70/52 interaction interface. Lysines 321 and 325 of IFT52 cross‐link to residues of both IFT70 and IFT88 in agreement with a composite interaction interface (Fig 3C; Movie EV3). In addition, lysine 625 and serine 628 of IFT70 make multiple cross‐links to residues in the N‐terminal part of IFT88 (residues 131–262, Fig 3C; Movie EV3). To further validate the predicted structural model, crystals were obtained for a minimal IFT881–437/70/52281–360 complex, and X‐ray diffraction data were collected to 3.8 Å resolution (Appendix Table S1). Molecular replacement with the IFT70/52 crystal structure (Taschner et al, 2014) and the AF model of the IFT88120–437 fragment gave a unique solution (Top LLG of 832) and the resulting omit electron density map clearly identifies the position of IFT70 and IFT88 TPRs and validates the position of the interacting regions of IFT88 and IFT70 within the complex (Fig 3D).
No cross‐links between the IFT52C/46C heterodimer and IFT70 were observed in our cross‐linking/MS data set. Published crystal structures of CrIFT70/52 and Tetrahymena thermophila (Tt)IFT52C/46C (Taschner et al, 2014) and the fact that only 4 residues covalently link the part of IFT52 emerging from the IFT70 superhelix to the C‐terminal domain of IFT52 that interacts with IFT46C effectively restrain the relative position of IFT70 and IFT52C/46C within the complex (Fig 3E). However, to validate the IFT70/52/46 structural model, we crystallized a minimal CrIFT70/52330–430/46165–319 complex and collected X‐ray diffraction data to a resolution of 4 Å. The crystal structure was determined by molecular replacement using the previously determined crystal structure of CrIFT70/52 (Taschner et al, 2014) and an AF generated model of Chlamydomonas IFT52C/46C, which resulted in a unique solution. The resulting omit electron density map clearly positions the IFT52C/46C complex at the N‐terminal face of IFT70 (Fig 3E). However, given that no noncovalent interactions are observed between IFT70 and IFT52C/46C, the position of IFT52C/46C relative to IFT70 is likely quite flexible to accommodate different conformations in solution. In summary, a combination of AF modeling, chemical cross‐linking and X‐ray crystallography support the structural architecture of the IFT88/70/52/46 complex shown in Fig 3.
IFT81/74 can associate with IFT88/70/52/46 via two distinct interaction sites to form the IFT‐B1 complex
With validated structural models of IFT81/74/27/25/22 and IFT88/70/52/46 (Figs 2 and 3) in hand, we wanted to address how these two subcomplexes associate to form the IFT‐B1 complex. We previously showed that the IFT52C/46C complex co‐purified with IFT81ΔN/74ΔN/27/25 for both C. reinhardtii and T. thermophila and mapped the interaction to the C‐terminal half of the IFT81/74 complex (Taschner et al, 2014). Recently, it was shown that human IFT52/46 associates with the C‐terminal part of IFT81/74 (Zhou et al, 2022) demonstrating evolutionary conservation for this interaction. The exact binding site of IFT52C/46C on IFT81C/74C is unknown and no structural information is available for the complex. To this end, we utilized the modeled structures of the C‐terminal part of IFT81/74 together with the C‐terminal domains of IFT52 and IFT46 (IFT81460‐C/74460‐C/27/251–136/46148–328/52382‐C, see Appendix Fig S3) and the modeled structure of the IFT88/70/52/46 tetramer together with the C‐terminal CCs of IFT81/74 (Fig 3A and B). Both complexes are modeled with high confidence as demonstrated by pLDDT and PAE plots (Fig 3B; Appendix Fig S3) and reveal identical binding sites for IFT52C/46C on the most C‐terminal CCs (CC X) of IFT81/74. The interaction interface is distal to the IFT27/25‐binding site, is mostly hydrophobic in nature, and is formed by the residues 623–654 of IFT81, 615–641 of IFT74, 235–319 of IFT46 and 371–454 of IFT52 (Fig 4A and B; Appendix Fig S9C). When mapping the cross‐linking pairs of IFT46 (Fig 4A) or IFT52 (Fig 4B) onto the predicted IFT81/74/27/25 structure, we observe that IFT52C/46C predominantly cross‐links to the IFT81581–608/74575–618 region, which constitute the IFT27‐binding site (Fig 4A and B). The cross‐linking pairs are thus mostly formed proximal to the interaction site predicted by AF suggesting either that the predicted model is inaccurate or is perhaps a high availability of free amine residues that can facilitate cross‐linking formation. We addressed these possibilities experimentally in interaction studies of IFT52/46 and 81/74 complexes either with or without the predicted interacting helices of IFT81/74 (IFT81460‐C/74460‐C/27/251–136 or IFT81460–623/74460–615/27/251–136). The results show that the IFT81623‐C/74615‐C helices predicted to interact with IFT52/46 are indeed required for complex formation on SEC (Fig 4C and D) thus validating the structural model shown in Fig 4A and B.
Interestingly, we observed numerous cross‐links between IFT81N/74N and the opposite end of the IFT‐B1 complex, meaning IFT88, IFT70, and the N‐terminal GIFT domain of IFT52 (Fig 1B; Appendix Fig S8D). This observation suggests a possible second binding site through which IFT81N/74N link more closely to IFT52/88/70. Indeed, AF predictions where the N‐terminal sequences of IFT81 and IFT74 were used as input together with IFT88/70/52 (IFT81N‐385/74N‐390/52/70/88121–713) provided a structural model suggesting a second conformation of the IFT‐B1 complex (Fig 4E; Appendix Fig S4B). In this second IFT‐B1 conformation, the first 4 CCs of IFT81/74 form a tetrahedral structure that interacts directly with IFT88. Specifically, the tips between CCs I – II and CCs III – IV of IFT81/74 binds adjacent to the TPRs 8–10 of IFT88 (residues 468–536) to form an interaction interface of 54 amino acid pairs with high confidence (PAE < 5 Å; Fig 4F). Conservation analysis also corroborates this finding as the region of binding on IFT81/74 is conserved (Appendix Fig S9B). To experimentally investigate this association, we pulled down a complex of IFT811–387/74135–420, which lacks the binding site for IFT52C/46C shown in Fig 4A, using a GST‐tagged IFT88/70/521–430/46 complex to demonstrate a direct interaction (Fig 4G).
To address if the two observed binding modes shown in Fig 4A and E can happen simultaneously or are mutually exclusive, we used AF and IFT‐B1 protein sequences where both binding sites are present (IFT81/74/52251‐C/88120‐C/70/27/25/22/46188–320). We produced 5 AF models of this IFT‐B1 complex as well as two additional quintuples that allow for more flexibility by splitting the IFT81/74 in N‐ and C‐terminus halves either by introducing a break in the polypeptide chains or via a 100‐residue glycine linker at position 458 in both IFT81 and IFT74. Of the 15 resulting models, 5 models have only the IFT81/74‐C‐terminus interaction with IFT52C/46C whereas 1 model identifies only the N‐terminus IFT81/74 interaction with IFT88. A total of 7 models displayed both binding sites simultaneously (Appendix Fig S4C). However, none of the five AF structures, where the native IFT81/74 sequences were used, had properly modeled α‐helical structure of the central CCs but instead, at least some of the CC domains, resemble disordered regions (Appendix Fig S4C). This suggest, that although no steric clashes prevent simultaneous binding of IFT81/74 to IFT88/70/52/46 via the two separate binding sites, it may require unfolding of CC segments and thus be unfavorable.
IFT54/20 and IFT57/38 of the IFT‐B2 complex form an anti‐parallel hetero‐tetramer
Previous studies suggest that IFT57, 54, 38 and 20 form a tetrameric complex composed of IFT54/20 and IFT57/38 hetero‐dimers (Baker et al, 2003; Follit et al, 2006; Omori et al, 2008; Taschner et al, 2016). All four proteins contain C‐terminal CC domains while IFT38, 54 and 57 also harbor N‐terminal CH‐domains. The only experimental structures currently available for the IFT57/38/54/20 complex is of the IFT54 CH‐domain of both Mus musculus and Chlamydomonas reinhardtii. Both the IFT81 and IFT54 CH‐domains, but not the IFT57 or IFT38 CH‐domains, were shown to bind to αβ‐tubulin in vitro (Bhogaraju et al, 2013; Taschner et al, 2016). The N‐terminal CH‐domains of IFT57 and IFT54 are connected to their C‐terminal CC region by long and partly unstructured linkers (Taschner et al, 2012). Using AF, we predicted the structure of the CrIFT57/38/54135‐C/20 tetramer (Fig 5A) and mapped cross‐linking pairs onto the model (Fig 5C and D). The model is predicted with high confidence as revealed by pLDDT scores > 90 (Appendix Fig S5) except for the long unstructured linkers that connect the CH‐ and CC‐domains within IFT57 and IFT54 (these regions were removed in Fig 5C and D for clarity). PAE plots support the structural arrangement of the CC helices of IFT57/38/54/20 and furthermore suggest a well‐defined position for the IFT38 CH‐domain on the IFT54/20 two‐bundle CC close to the tetrameric interface (Fig 5B), which is supported by 26 short‐range (< 32 Å) cross‐linking pairs (Fig 5D). The structural model reveals that the IFT57/38 and IFT54/20 hetero‐dimers are formed by parallel helices of the CC domains (Fig 5A). The IFT57/38 and IFT54/20 hetero‐dimers engage in an anti‐parallel fashion so that the N‐terminal ends of the four CC helices form a four‐helix bundle (Fig 5C). Interestingly, the very N‐terminal part of IFT20 is predicted to fold into a small 2‐stranded anti‐parallel β‐sheet that packs against IFT57/38 to induce a bend in the CC helices and likely constitutes a hinge point for conformational flexibility (Fig 5C; Appendix Fig S5). The structural arrangement of the parallel IFT57/38 and IFT54/20 hetero‐dimers are supported by cross‐linking pairs between numerous residues (Fig 5C and D). In addition, multiple cross‐links between all 4 subunits strongly support the anti‐parallel assembly of the IFT57/38 and IFT54/20 hetero‐dimers into a four‐helix bundle. The association of the IFT38 CH‐domain with IFT54/20 CCs likely strengthens the tetrameric assembly. However, both the low pLDDT score and the high PAE score for the long linker regions connecting IFT54 and IFT57 CH‐domains with their respective CC domains suggest that these are unstructured and likely provide a high degree of flexibility to the position of these CH domains within the IFT‐B complex.
Figure 5. The AF predicted structure of the IFT57/38/54135‐C/20 complex.
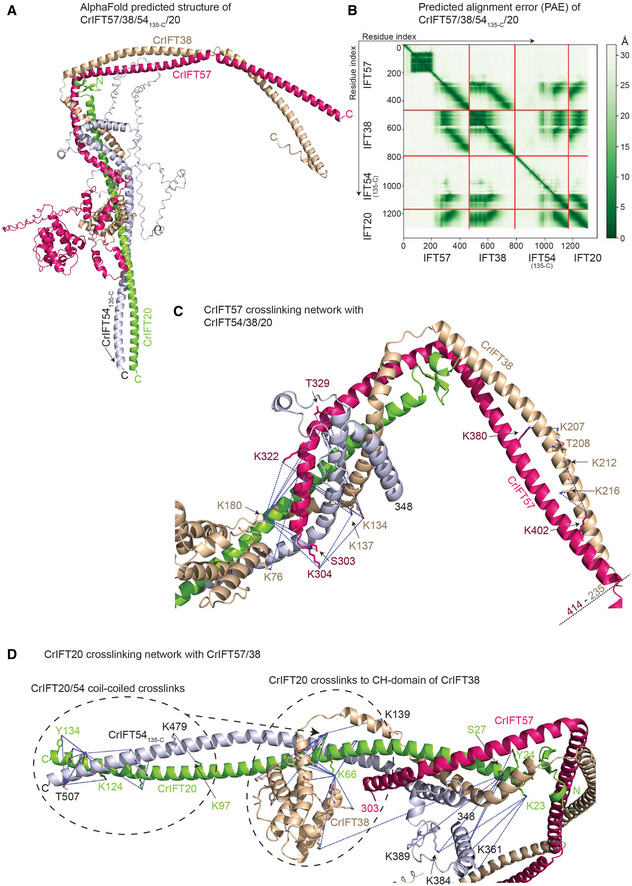
- The AlphaFold predicted model of the Chamydomonas IFT57/38/54135‐C/20 complex colored by chain.
- Predicted alignment error of the AlphaFold model from (A).
- Short‐range cross‐linking pairs formed by the helical domain of IFT57 with IFT54, IFT38 and IFT20 mapped as blue dotted lines between contributing sidechains.
- Short‐range cross‐linking pairs of the IFT20 with IFT57, IFT54 and IFT38 labeled as blue dotted lines between the contributing side chains.
IFT172 and IFT80 of the IFT‐B2 complex interact directly
Previous studies have shown that IFT172 and IFT80 variants can result in skeletal ciliopathies and that these two subunits interact genetically (Halbritter et al, 2013; Boldt et al, 2016). However, IFT172 and IFT80 did not interact physically in sucrose gradients (Lucker et al, 2005) nor did they co‐purify during SEC, which suggest that any direct physical interaction is relatively weak (Taschner et al, 2016). However, given that both IFT172 and IFT80 associate with the IFT57/38 heterodimer (Taschner et al, 2016), they could be in proximity within the IFT‐B2 complex. To test this hypothesis, we used AF with full‐length sequences of CrIFT172 and CrIFT80, which reliably predicted the structure of the heterodimeric IFT172/80 complex (Fig 6A). The structural model of the IFT172/80 complex was predicted with high confidence (Appendix Fig S6A and B) within the respective interacting regions of the proteins and show that the TPR repeats within the residues 626–785 of IFT172 interact with the TPR repeats of IFT80 within residues 583–627 (Fig 6A). Under a conservative 5 Å PAE threshold of the AF‐predicted IFT80‐IFT172 structure, we observed 26 residue pairs at a distance between 2.5‐5 Å and 180 pairs within less than 10 Å apart. Residues 786‐C of IFT172 are predicted to fold into TPRs but as they do not appear to participate directly in the interaction with IFT80 and are predicted with lower confidence, they were omitted from the model shown in Fig 6A. Curiously, we did not observe homo‐dimer formation of IFT80 using AF, which contrasts previous crystallographic analysis (Taschner et al, 2018). To assess the IFT172/80 complex formation experimentally, we monitored the interaction in pulldown assays. Purified Venus‐tagged CrIFT80 was used to pull down purified full length and a N‐terminal deletion of CrIFT172 that lacks the predicted IFT80 interaction domains. The results show that CrIFT80 pulls down full length but not a C‐terminal construct of CrIFT172 (Fig 6B). Venus‐tagged IFT80 clearly pulls down substoichiometric amounts of IFT172 suggesting a weak interaction, which agrees with observation that the two proteins do not associate during SEC (Taschner et al, 2016).
Figure 6. Dissecting the interaction between IFT80, IFT172, IFT57, and IFT38.
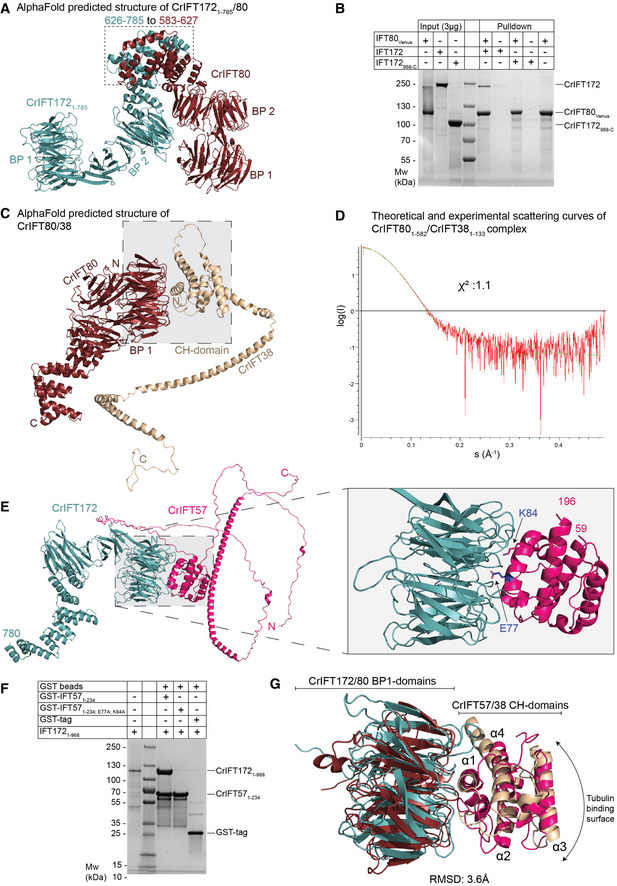
- AphaFold predicted structure of IFT1721–785/80.
- Pull‐down experiments with purified IFT80Venus immobilized on GFP‐binder beads and either IFT172 or IFT172968‐C. IFT80 pulls down full‐length IFT172 but not the truncated version lacking the N‐terminal 967 residues.
- AlphaFold predicted structure of IFT80 in complex with IFT38 maps the interaction at the interface between the BP1 of IFT80 and the CH‐domain of IFT38.
- Comparison of the solution X‐ray scattering curve of CrIFT80/38 as measured by SAXS and the calculated scattering curve for the IFT80/38 structural prediction. A χ2 value of close to 1 indicate an excellent fit between measurement and calculation.
- AlphaFold predicted structure of CrIFT172 and CrIFT57 shows interaction via the BP1 and CH domains. On the right panel, two residues in the CH‐domain of IFT57, which are located at the interface with IFT172, are highlighted.
- GST tagged CrIFT1721–968 pulls down the CH‐domain of CrIFT571–234 but not the mutated CH‐domain of CrIFT57 where E77 and K84 residues were replaced by alanines.
- Superimposition of the structure of BP1 of CrIFT80 (colored in red ruby) in complex with the CH‐domain of 38 (colored in beige) with the BP1/CH domains of CrIFT172/57 (colored in teal and hot pink, respectively) shows a conserved mechanism of interaction different from the canonical tubulin binding mode exhibited by CH domains.
IFT80 and IFT172 associate with the IFT‐B2 complex through conserved β‐propeller/CH‐domain interactions with IFT38 and IFT57
Within the IFT‐B2 complex, IFT80 and IFT172 were shown to associate with the CH‐domains of IFT38 and IFT57, respectively (Taschner et al, 2016, 2018). CH‐domains typically associate with microtubules/tubulin and/or actin along with a few other proteins involved in cellular signaling (Yin et al, 2020) and, to the best of our knowledge there is no structural characterization of how CH‐domains associate with β‐propellers. We thus used AF to model the structure of the respective interacting domains in CrIFT172/57 and CrIFT80/38 and validated the resulting models by small angle X‐ray scattering (SAXS) and structure‐guided mutagenesis.
The structural model of CrIFT80/38 shows that the N‐terminal BP1 of IFT80 interacts with the CH‐domain of IFT38 (Fig 6C). This interaction is mainly mediated by alpha helix α1 of the IFT38‐CH domain that runs across the N‐terminal face of the first β‐propeller domain of IFT80 (Fig 6C and G). The IFT80/38 complex structure is predicted with high confidence as shown by the pLDDT scores > 90 (Appendix Fig S6C) and the low PAE scores for interacting domains (Appendix Fig S6D). On the contrary, the C‐terminal CC helix of IFT38 is folded with low confidence (pLDDT score < 50), which likely reflects the absence of the interacting partner IFT57 in this model. To verify the validity of the model, we purified the IFT801–582/381–133 complex and collected SAXS data (Appendix Table S2). The comparison of the theoretical X‐ray scattering curve of IFT801–582/381–133 structural model with the experimental curve shows an almost perfect fit with a χ2 value of 1.1 (Fig 6D) thus validating the structural model. The fact that the IFT38 CH‐domain associates with the IFT54/20 CCs (Fig 5D) helps position IFT80 along the C‐terminal CCs of IFT54/20 in agreement with previously observed direct interactions between IFT80 and IFT54/20 (Taschner et al, 2018).
As was observed in the CrIFT80/38 structural model, AF predictions of the CrIFT1721–780/57 complex structure revealed an interaction between the first BP of IFT172 and the CH‐domain of IFT57 with high pLDDT and low PAE scores (Fig 6E; Appendix Fig S6E and F). The interaction of IFT172 with the IFT‐B complex was previously shown to be salt labile (Taschner et al, 2016), which agrees with the highly hydrophilic interface observed between IFT172 and IFT57 in our structural model. We used this structural model for mutagenesis designed to disrupt the IFT172/57 interaction interface (Fig 6F). The E77 and K84 residues of the CrIFT57 CH‐domain lie in the interface with IFT172 BP1 and were mutated to alanine and used in pull‐down experiments with purified CrIFT1721–968. The results show that while IFT1721–968 is pulled‐down in stoichiometric amounts by wildtype CrIFT571–234, the interaction is completely lost in the E77A, K84A double point mutant (Fig 6F). Taken together, these data indicates that IFT172 associates with the IFT‐B2 complex through a strong interaction with the CH‐domain of IFT57 and a rather weak interaction with IFT80. The position of the N‐terminal part of IFT172 within the IFT‐B complex is thus fixed via interactions with IFT80, whereas the C‐terminal part of IFT172 does not appear to make contacts with IFT‐B subunits and likely adopts a flexible conformation relative to other IFT‐B subunits.
Interestingly, the interaction involving the first BP1 of IFT172 and the CH‐domain of IFT57 shows striking similarity to the IFT80/38 complex. IFT172/57 and IFT80/38 BP1‐CH structural models superimpose with a root mean square distance (RMSD) of 3.6 Å demonstrating a conserved interaction mechanism (Fig 6G). In both complexes, the interaction is mediated by charged residues of helix α1, which is markedly different from the tubulin‐binding mode exhibited by many CH‐domains, which mainly involves residues in the vicinity of the corresponding helix α3 of the CH‐domain (Hayashi & Ikura, 2003; Bhogaraju et al, 2013; Taschner et al, 2016; Fig 6G). This observation establishes BPs as a new class of interaction partner for CH domains in addition to the well‐characterized tubulin and actin interaction partners.
The IFT‐B1‐B2 connection
Previous biochemical data have shown that IFT88 and the N‐terminal domain of IFT52 (residues 1–335) of IFT‐B1 are sufficient to pull down the IFT‐B2 complex via direct contacts to IFT57/38 (Katoh et al, 2016; Taschner et al, 2016). To gain structural insights into the IFT‐B1‐B2 connection, we used AF to model the structure of the IFT88/52N/57C/38C complex (Fig 7A). The structure of IFT88120–713/521–336/57356‐C/38174–303 was modeled with high confidence as revealed by high pLDDT scores for most of the model (Appendix Fig S7). The PAE plot (Fig 7B) demonstrates high confidence in the relative positions of all 4 proteins within the IFT88/52/57/38 complex. In the structural model, the C‐terminal CC region of IFT57/38 engages IFT88/52, which creates a slightly arched structure where IFT88/57/38 loosely cradles the N‐terminal GIFT domain of IFT52 (Fig 7A). The position of the IFT52 GIFT domain is supported by 11 cross‐linking pairs with IFT88 and IFT57/38 as identified by MS (Fig 7C and D; Movie EV4). Consistent with the structural model of IFT88/52/57/38 shown in Fig 7A, two point mutations in the GIFT domain of IFT52 (K130E and R204E) that were previously published to significantly reduce the IFT‐B1‐B2 interaction (Taschner et al, 2016) lie at the interface with IFT57/38 (Fig 7A; Movie EV4). We also observe multiple cross‐links from IFT88120–713 to IFT38212‐C (Movie EV5) but no cross‐links to the IFT57414‐C region. Taken together, the cross‐linking data supports the predicted structural model of CrIFT88120–713/521–336/57356‐C/38174‐C and reveals how IFT88/52 of IFT‐B1 connect to IFT57/38 of IFT‐B2.
Figure 7. IFT88 links the IFT‐B1 and IFT‐B2 complexes.
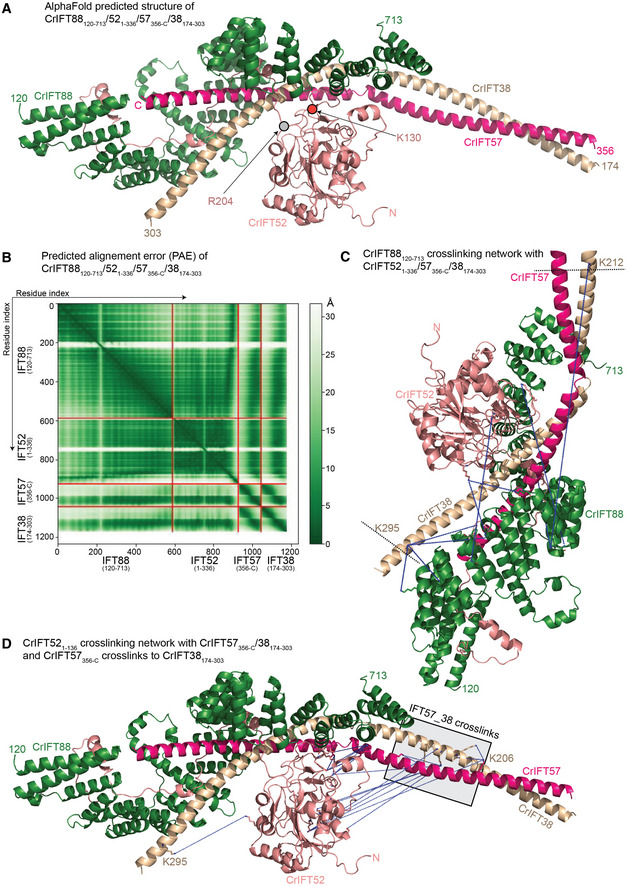
- AlphaFold predicted structure of the CrIFT88120–713/521–336/57356‐C/38174–303 complex.
- Predicted alignment error (PAE) for the complex depicted in (A).
- CrIFT88120–713 cross‐linking network with CrIFT521–336, CrIFT57356‐C and CrIFT38174–303 labeled with blue dotted lines.
- The cross‐linking interaction network of CrIFT521–336 with CrIFT57356‐C/38174–303 and of CrIFT57356‐C with CrIFT38174–303 labeled as in (C).
Structural model of the 15‐subunit Chlamydomonas IFT‐B complex
From the experimentally validated structures of IFT‐B subcomplexes displayed in Figs 2, 3, 4, 5, 6, 7, we assembled the structural model of the 15 subunit Chlamydomonas IFT‐B complex in silico (see Fig 8 and M&M). The structural model contains the proteins IFT81/74/27/25/22/52/46/88/70/57/38/54/20/80/172 but lacks RabL2 and IFT56. RabL2 is not a core member of the IFT complex as it dissociates shortly after departure of the anterograde IFT train from the ciliary base (Kanie et al, 2017; Nishijima et al, 2017). IFT56 is important for recruiting motility complexes to cilia and associates with the IFT‐B complex via IFT46 and possibly IFT70 but does not appear to be essential to ciliogenesis in mice (Ishikawa et al, 2014; Swiderski et al, 2014). AF modeling places IFT56 close to IFT46 and IFT70 but as we were unable to express IFT56 as a soluble protein, we could not experimentally verify its position within the IFT‐B complex and have thus omitted this subunit from our structural modeling. The model reveals an elongated IFT‐B complex with a longest dimension of 430 Å and a shortest dimension of 60 Å (Fig 9A). These measures are consistent with the elongated IFT‐B complexes organized into linear polymers with a repeat distance of 60 Å that were observed in cryo‐ET reconstructions of anterograde IFT trains (Jordan et al, 2018; van den Hoek et al, 2022).
Figure 8. In silico structure of IFT‐B 15‐mer.
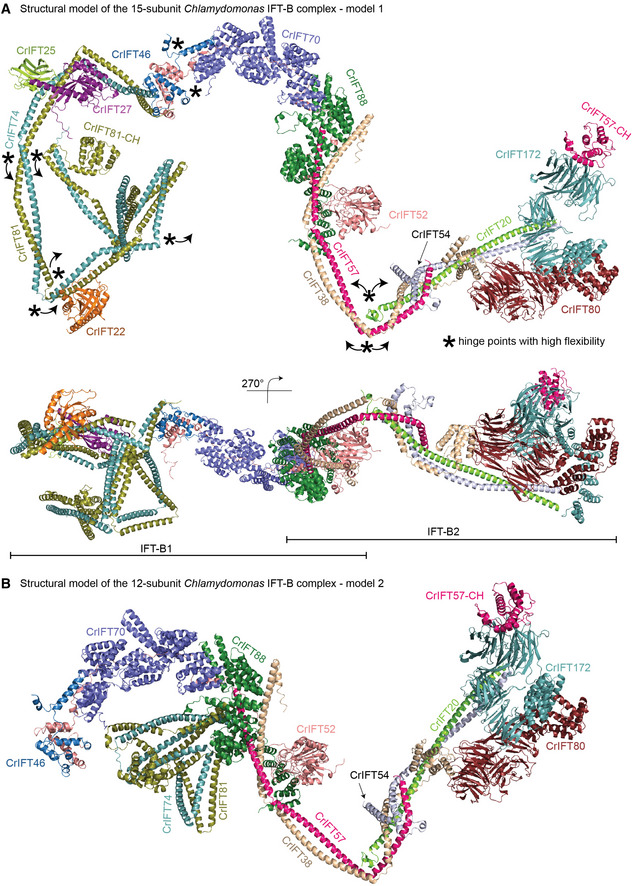
- The model of 15‐subunit Chlamydomonas IFT‐B complex (model 1) assembled in silico as a rigid body from AF predicted subcomplexes using the binding site of IFT81/74 on IFT52/46 shown in Figs 3A and 4A and B. The structural model is assembled from CrIFT81/74128‐C/27/25/22, CrIFT88120–713/70/52/46188–319/81587–645/74583‐C, CrIFT88120‐713/521–336/57360‐C/38174–303, CrIFT57/54135‐C/38/20, CrIFT80/38, and CrIFT1721–780/57.
- The model of 12‐subunit Chlamydomonas IFT‐B complex (model 2) assembled in silico using the IFT81/74‐binding site on IFT88/70 shown in Fig 4E. The rigid body assembly was carried out using AF predicted structures of subcomplexes of CrIFT88120–713/70/52/46188–319/81587–645/74583‐C, CrIFT88120–713/70/52/811–385/741–390, CrIFT88120–713/521–336/57360‐C/38174–303, CrIFT57/54135‐C/38/20, CrIFT80/38, and CrIFT1721–780/57.
Figure 9. The IFT‐B complex in context of anterograde IFT‐B trains.
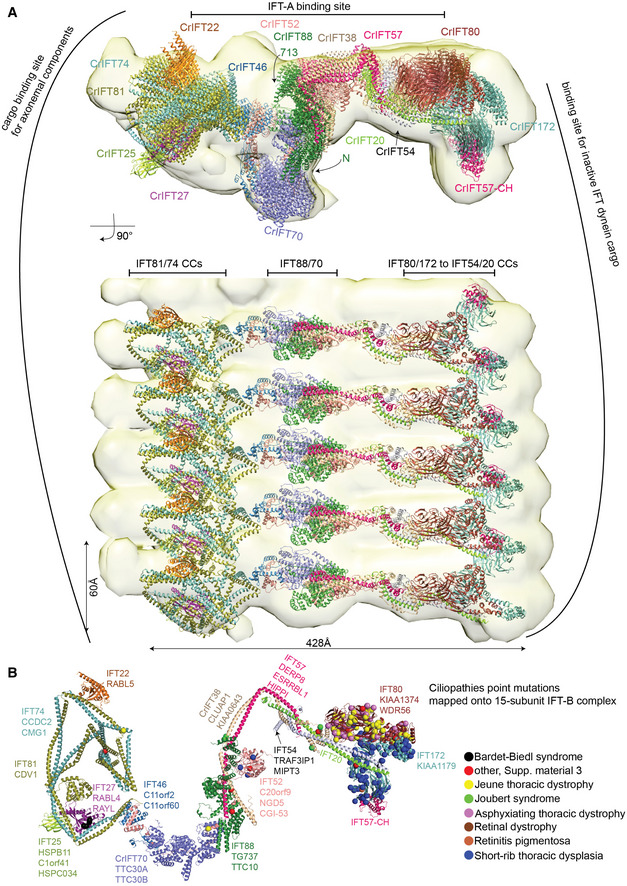
- Molecular dynamic flexible fitting of the IFT‐B 15‐mer into the 25 Å cryo‐electron tomography map of the Chlamydomonas anterograde IFT‐B trains obtained in situ (van den Hoek et al, 2022). The fit is shown in two perpendicular orientations. The highly elongated IFT‐B complex fits with a repeat distance of 60 Å consistent with anterograde IFT trains.
- Single‐point mutations associated with ciliopathies are mapped onto the CrIFT‐B 15‐mer structure as spheres. Noteworthy, mutations that lead to amino acid deletions or additions, as well as frameshifts are not included in the figure. The IFT‐B proteins are annotated according to their corresponding human gene names.
Several potential hinge regions are apparent in the structural model and likely serve as points of conformational flexibility within the IFT‐B complex (marked with * in Fig 8A). Hinges are located between IFT70 and IFT52C/46C as well as between CC segments of IFT81/74 and IFT57/38 (Fig 8). To glean insights into conformational flexibility of the IFT‐B complex in solution from the cross‐linking/MS data, we labeled all intra‐ and intermolecular cross‐linking pairs (Appendix Fig S8A–D; Dataset EV4). We considered cross‐linking pairs with a distance < 32 Å as an indicator of structural rigidity and cross‐linking pairs with distance > 32 Å as an indicator of conformational flexibility. The intramolecular cross‐linking pairs suggest a high degree of conformational flexibility between the N‐ and C‐terminal halves of the IFT81/74 complex (Appendix Fig S8A and B) consistent with our structural modeling (Fig 2C). Similarly, many intramolecular cross‐links with distances beyond 32 Å were found between the N‐ and C‐terminal ends of IFT38 and IFT57 (Appendix Fig S8B), which suggests a high degree of flexibility around the hinge point of the IFT57/38 CCs as indicated in Fig 8A. The intermolecular cross‐links show a similar pattern where numerous cross‐links between the two ends of the IFT57/38–54/20 hetero‐tetramer suggest a significant bendability around the hinge region (Appendix Fig S8C and D). Intermolecular cross‐links are also very pronounced between IFT81N/74N and IFT88/70 and likely capture the second binding site for IFT81/74 on the IFT88/70/52/46 tetramer as highlighted in Fig 4E. The high degree of structural flexibility of the IFT‐B complex is likely important for the polymerization into IFT trains and furthermore may provide a mechanism for the large structural rearrangements that occur when anterograde IFT trains remodel into retrograde IFT trains (Jordan et al, 2018).
The structural model of the 15‐subunit IFT‐B complex presented here was assembled by superposing predicted structures of smaller subcomplexes without the context of the IFT train. It is thus not surprising that a rigid‐body docking of the IFT‐B structural model into the cryo‐ET map of anterograde IFT trains (van den Hoek et al, 2022) resulted in a relatively poor fit with several subunits located outside the density. To obtain a better fit, we made use of the program Namdinator (Kidmose et al, 2019), which is an automatic molecular dynamic flexible fitting algorithm that requires only a structural model and a map as input. Two consecutive rounds of each 400,000 iterations in Namdinator resulted in a relatively good fit of the structural model of the IFT‐B complex to the density of the anterograde IFT trains except for IFT22 and IFT25, which partly sit outside density (Fig 9A). One end of the elongated IFT‐B complex contains the IFT‐B1 complex with previously characterized cargo‐binding sites for tubulin as well as outer‐and inner dynein arms (Hou et al, 2007; Bhogaraju et al, 2013; Kubo et al, 2016; Taschner et al, 2017; Wang et al, 2020). The other end of the IFT‐B complex harbors the IFT‐B2 complex with IFT172, IFT80, IFT57CH, and the C‐terminal part of the IFT54/20 CCs positioned close to the binding site for the IFT dynein cargo on anterograde trains (Fig 9A). In the model, the IFT‐A complex is positioned close to the CCs of IFT81/74, CCs of IFT57/38 and the C‐terminal end of IFT88 (Fig 9A). However, due to the low resolution of the cryo‐ET map (25 Å) and the absence of the C‐terminal domain of IFT172 in our model, it is not possible to accurately pinpoint the IFT‐B proteins involved in the interaction between IFT‐A and IFT‐B.
Our structural modeling and cross‐linking/MS data revealed two separate binding sites of IFT81/74 on the IFT88/70/52/46 (Fig 4). AF was unable to model both sites simultaneously without disrupting and unfolding the CCs connecting the two halves of IFT81/74 and it thus appears likely that the two binding modes are mutually exclusive (for models of the IFT‐B complex containing the alternative IFT81/74‐binding mode see Fig 8B). Both models of the IFT‐B complexes produce a relatively good fit to the 25 Å cryo‐ET map of the anterograde IFT train (using flexible fitting in Namdinator), and it is thus not clear from our data if one of these IFT‐B models better represent the anterograde IFT train conformation.
Discussion
The Chlamydomonas IFT‐B complex in context of IFT trains
We present an experimentally verified structural model of the 15‐subunit IFT‐B complex that is consistent with low‐resolution cryo‐ET reconstructions of anterograde IFT trains (Jordan et al, 2018; van den Hoek et al, 2022). However, given that our experimental validations were carried out on isolated IFT‐B subcomplexes that were not polymerized into IFT‐trains, our IFT‐B structural model could represent a hybrid conformational state capturing conformations of both anterograde and retrograde IFT trains. During the preparation of this manuscript, three preprints using AF and cryo‐EM to elucidate the structures of IFT‐A and IFT‐B complexes were published (preprint: Hesketh et al, 2022; preprint: Lacey et al, 2022; preprint: McCafferty et al, 2022). Preprint: Lacey et al (2022) elucidated the structure of anterograde IFT‐trains at 10‐18 Å resolution and fitted AF generated models of IFT‐A and B complexes into the density to obtain a pseudo‐atomic model for the entire train structure (preprint: Lacey et al, 2022). Overall, the architecture of the IFT‐B complex presented here agrees well with the cryo‐ET structure presented by preprint: Lacey et al (2022). Interestingly, in the anterograde IFT train structure, it is observed that IFT74N/81N associates with IFT88/70 from multiple neighboring complexes of the anterograde IFT train (preprint: Lacey et al, 2022) consistent with the second IFT‐B model described here (Fig 8B). In the cryo‐ET structure, no density is observed for the C‐terminal half of IFT81/74 that associate with IFT27/25 and IFT22 suggesting that this part of IFT‐B adopt flexible conformations and is likely averaged out in the cryo‐ET maps (preprint: Lacey et al, 2022). The interaction between the C‐terminal CC X domain of IFT81/74 and IFT52C/46C shown in Fig 4A and B are not observed in the anterograde IFT‐B Cryo‐ET structure.
If not important to the formation of anterograde IFT trains, what is the functional implication of the IFT81C/74C‐IFT52C/46C interaction highlighted in Figs 4A and B, and 8A? First, IFT81C/74C‐IFT52C/46C is a high affinity interaction (Taschner et al, 2014), which occurs through a conserved hydrophobic interface (Appendix Fig S9C). Indeed, the IFT81C/74C‐IFT52C/46C interaction is evolutionarily conserved and was experimentally observed in Chlamydomonas, Tetrahymena (Taschner et al, 2014) and human (Katoh et al, 2016). Interestingly, our structural modeling by AF showed that significant unfolding of the IFT81/74 CC segments must occur for both IFT81/74‐binding sites on IFT88/70/52/46 to be occupied simultaneously (Appendix Fig S4C). This observation could suggest that the two binding modes are mutually exclusive and may happen separately in different cellular contexts. It is tempting to speculate that the IFT81N/74N‐IFT88/70 structures shown in Figs 4E and 8B represent an anterograde IFT train conformation whereas the IFT81C/74C‐IFT52C/46C structures shown in Figs 4A and B, and 8A represent a retrograde IFT train conformation. In addition, the IFT80 dimerization observed by crystallography (Taschner et al, 2018) may only be relevant in context of retrograde IFT trains as it is not observed in the anterograde IFT train structure. However, these notions can currently not be verified in the absence of cryo‐ET reconstructions of retrograde IFT trains.
Association of IFT‐B with IFT‐A and IFT motors
The IFT‐B complex and its linear polymerization form the backbone of IFT trains onto which IFT‐A polymers, dynein‐1b, and finally kinesin‐2 attach before entering the cilium (van den Hoek et al, 2022). How is IFT complex polymerization into trains and association with motors facilitated? The fitting of the 15‐subunit IFT‐B structural model into a 5‐repeat anterograde IFT‐B train revealed that IFT‐B polymerizes laterally and contains at one end the cargo‐binding sites for axonemal components and at the other end the binding site for inactive IFT dynein‐1b cargo (Fig 9A). The lateral polymerization into trains appears involves 3 contact points provided by adjacent IFT81/74 CCs at one end, IFT88/70 complexes in the middle and the N‐terminal CCs of IFT54/20, the N‐terminal part of IFT172 and IFT80 at the other end (Fig 9A). This arrangement agrees with the recent anterograde IFT structure (preprint: Lacey et al, 2022). However, due to resolution limitations of the cryo‐ET reconstructions that we used in this study, it is yet to be determined which residues within the 3‐point‐junction are essential for lateral polymerization of anterograde IFT trains.
Previous cryo‐EM data have revealed a mismatch between the number of IFT‐B, IFT‐A and dynein‐1b cargo complexes in anterograde IFT trains with approximate 6, 11.5, and 18 nm repeat distances within the trains (Jordan et al, 2018; Toropova et al, 2019). IFT‐A is flexibly tethered to IFT‐B complexes through interactions between IFT139 of IFT‐A and two copies of IFT81/74 of IFT‐B at one end, IFT144/139 of IFT‐A and the C‐terminal TPR domain of IFT172 at the other end (preprint: Lacey et al, 2022). A third IFT‐A/IFT‐B interaction interface was elucidated by preprint: Hesketh et al (2022), who showed that the highly flexible IFT88 C‐terminal extension bridges across to interact with IFT144 (preprint: Hesketh et al, 2022). Although we did not include the C‐terminal part of IFT172 in our structural model of IFT‐B, both IFT81/74 and IFT88 are positioned so that interactions to IFT‐A proteins are favorable (Fig 9A).
Flexible fitting of the IFT‐B model into cryo‐ET maps of anterograde IFT trains suggest that multiple 60 Å wide IFT‐B complexes are required to load the bulky IFT dynein cargo (Fig 9A) in agreement with previous published results (Jordan et al, 2018; Toropova et al, 2019). Lacey and co‐workers showed that dynein cargo is using a composite surface formed by two adjacent IFT‐B2 complexes. In our IFT‐B model, IFT172, IFT80, and the CH domain of IFT57 supported by a shaft formed by the CCs of IFT54 and − 20 are the main contributors for creating this composite binding site for dynein‐1b cargo (Figs 8A and 9A). Interestingly, the platform has a prominent negatively charged groove flanked by two positively charged regions formed by a conserved surface on BP1 of IFT172 and BP2 of IFT80 (Appendix Fig S9A and B).
Upon polymerization of IFT‐A and IFT‐B complexes at the base of cilia, the kinesin 2 motor associates with IFT‐B to drive the anterograde IFT (van den Hoek et al, 2022). Because of its slim and flexible architecture, kinesin‐2 is averaged out in cryo‐ET reconstructions (Jordan et al, 2018; van den Hoek et al, 2022). However, biochemical studies showed that IFT88/57/52/38 (Funabashi et al, 2018) and IFT54 (Zhu et al, 2017) are important IFT‐B interactors of kinesin‐2. Interestingly, we identified two conserved amino acid patches on IFT‐B that likely represent binding sites for kinesin‐2 (Appendix Fig S9A). One patch is composed of IFT88/57/38 where IFT‐B1 and IFT‐B2 connect, while the other patch is contributed by the tetrameric CCs of IFT57/54/38/20 (Appendix Fig S9A). A Chlamydomonas IFT54 deletion mutant that lacks residues 342–356 no longer binds kinesin‐2 in vitro or in vivo (Zhu et al, 2017). In our IFT‐B model, residues 342–356 of IFT54 lie at the tetrameric interface between IFT57, ‐54, ‐38 and ‐20 (Fig 5C and D) and their deletion could disrupt the structure of the tetramer and thus its function as a kinesin‐2 binding platform.
Association of IFT‐B with cargoes
IFT trains carry a variety of cargo into cilia including tubulin, radial spokes, and axonemal motility complexes like outer‐ and inner dynein arms (ODAs and IDAs; Lechtreck et al, 2022). In Chlamydomonas, ODAs are imported into cilia by IFT46 via the cargo adaptor protein ODA16 (Ahmed & Mitchell, 2005; Hou et al, 2007). The N‐terminal part of IFT46 (residues 1–147) interacts with ODA16 while the C‐terminus is important for assembly of IFT‐B complex (Hou & Witman, 2017; Taschner et al, 2017; Wang et al, 2020). The N‐terminal 187 residues of IFT46 are not included in our structural models but are located at the periphery of the IFT‐B complex opposite to the IFT‐A‐binding site and are free to engage binding partners such as ODA16 (Fig 9; Appendix Fig S9). Residues 147–187 of IFT46 do not interact with IFT81/74, IFT52 or ODA16 and are thus free to engage other factors such as IFT56, which is implicated in the ciliary import of certain IDAs (Ishikawa et al, 2014; Xin et al, 2017).
In vitro studies have shown that the CH‐domain of IFT81 and a highly basic amino acid stretch of IFT74N bind αβ‐tubulin and likely constitute the main αβ‐tubulin cargo‐binding site in IFT trains (Bhogaraju et al, 2013). Although most of the αβ‐tubulin that is required for axonemal growth enters cilia by diffusion (Craft Van De Weghe et al, 2020), mutations in the CH‐domain of IFT81 or in IFT74N, while not hampering IFT in general, result in reduction of the frequency of anterograde IFT of αβ‐tubulin to levels of 26 and 11%, respectively, when compared to control Chlamydomonas cells (Kubo et al, 2016). Cells with mutations in both the IFT81 CH‐domain and IFT74N have almost no flagellar assembly highlighting the importance of these domains in ciliogenesis (Kubo et al, 2016). When anterograde IFT trains reach the tip of cilia, αβ‐tubulin cargo must be released for the incorporation into the growing axoneme of the cilium. The release of IFT cargo such as αβ‐tubulin likely employs mechanisms that weaken the interaction with IFT‐trains. This hypothesis is supported by a recent study in C. elegans, which showed that the N‐terminus of IFT74 undergoes phosphorylation by the DYF‐5/MAK kinase (Jiang et al, 2022). Interestingly, the phosphorylation of IFT74N translates into a sixfold reduction in the binding affinity for αβ‐tubulin (Jiang et al, 2022), which likely help facilitate the unloading of αβ‐tubulin cargo from IFT trains. Interestingly, the conformation of the CH‐domain of IFT81 in the second model of IFT‐B presented in Figs 4E and 8B and observed in the structure of anterograde IFT trains (preprint: Lacey et al, 2022) is not compatible with the canonical association of CH domains with αβ‐tubulin. It is possible that αβ‐tubulin associates with both N‐termini of IFT74 and IFT81 at the base of the cilium but that packaging into the anterograde IFT trains results in a re‐positioning of the IFT81 CH‐domain and dissociation from αβ‐tubulin leaving only IFT74N attached. The final dissociation of αβ‐tubulin from anterograde IFT trains may then rely on IFT74N phosphorylation as suggested by Jiang et al, 2022.
Structural mapping of ciliopathy variants onto IFT‐B
IFT is essential for cilium formation and organismic development as highlighted by the Traf3ip1 (encoding the IFT‐B protein IFT54) mutant mice that cannot form cilia and thus fail in proper embryonic development (Berbari et al, 2011). Patients suffering from ciliopathies caused by mutations in IFT‐B genes are thus expected to produce viable IFT particles that support at least some degree of cilium formation and function (Braun & Hildebrandt, 2017). To get insights into the structurally distribution of ciliopathy mutations, we obtained all ciliopathy related data from Uniprot, which comprises 327 distinct variants in the IFT‐B genes discussed here and mapped these onto the IFT‐B complex (Fig 9B; Dataset EV3). The variants are found together with over 15 prominent ciliopathies such as Bardet‐Biedl syndrome, short‐rib thoracic dysplasia, and asphyxiating thoracic dystrophy. In addition, we extracted presumed benign variants (gnomAD mutations from control healthy patients) for human IFT‐B proteins and mapped these onto the Chlamydomonas IFT‐B structural model (Appendix Fig S10). We hypothesized that if benign variants are missing in some regions of the IFT‐B complex, mutation of those regions may disrupt IFT complex formation and thus ciliogenesis. However, we observed a significant enrichment of disease variants for IFT80 and IFT172 relative to their length (Fig 9B), whereas the benign variants were equally distributed within IFT‐B complex (Appendix Fig S10). Although the higher numbers of IFT80/172 disease variants could arise from directed sequencing (and not whole genome/exome data), this observation does suggest that disease variants tend to interfere with cargo loading, particularly for the most severe ciliopathies. The lack of disease variants in certain regions of the complex could signify either lethality or tolerance, though we had insufficient data to distinguish these possibilities.
Ciliopathy variants are distributed along the IFT‐B complex structure with a pronounced clustering of skeletal ciliopathy variants in IFT80 and IFT172 at the IFT‐B2 end of the complex (Fig 9B). These mutations are associated with ciliopathies such as Joubert syndrome, Jeune thoracic dystrophy, asphyxiating thoracic dystrophy, short‐rib thoracic dysplasia (Beales et al, 2007; Halbritter et al, 2013). The short rib polydactyly syndrome and Jeune thoracic dystrophy represent so‐called ciliary chondrodysplasias, with overlapping skeletal and extra‐skeletal presentations (Antony et al, 2021). Interestingly, these ciliopathies are also caused by variants that map to dynein‐2 and are associated with defective retrograde IFT trains resulting in ciliary accumulation of proteins (Antony et al, 2021). This suggests that variants in IFT80 and IFT172 may cause skeletal ciliopathies by affecting the loading of IFT dynein cargo onto anterograde IFT trains.
IFT52 constitutes the backbone of IFT‐B complex (Fig 8A; Taschner et al, 2014), and IFT52 variants destabilize anterograde IFT complex assembly and disrupt ciliogenesis (Zhang et al, 2016). We have mapped 3 ciliopathy mutations associated with short rib thoracic dysplasia and one mutation associated with short rib polydactyly on or in the vicinity of the N‐terminal GIFT domain of IFT52 (Fig 9B, blue and red spheres, respectively; Girisha et al, 2016; Chen et al, 2018; Dupont et al, 2019). These ciliopathy mutations are located close to residues K130 and R204 of IFT52, where mutation to glutamate significantly reduce the IFTB1‐B2 interaction (Taschner et al, 2016) and may thus destabilize IFT‐B integrity (Fig 9B).
The occurrence of Bardet‐Biedl syndrome (BBS) ciliopathy characterized by obesity, polydactyly, retinal degeneration, and mental retardation is typically caused by mutations or knockouts of genes that translates into proteins of the BBSome complex (Forsyth & Gunay‐Aygun, 1993; Nachury et al, 2007). The BBSome complex functions as an IFT adaptor that removes membrane proteins from cilia (Lechtreck et al, 2009, 2013). Within the IFT‐B complex, the IFT27/25 hetero‐dimer was shown to be involved in the regulation of ciliary export of BBSomes and associated cargoes (Eguether et al, 2014; Keady et al, 2012; Liew et al, 2014). Interestingly, mutations or knockouts of IFT27/25 mimic phenotypes associated with BBS (Aldahmesh et al, 2014; Yan & Shen, 2021) or cause foetal lethality (Quélin et al, 2018). In our model, IFT27/25 is placed at the periphery of the IFT‐B complex opposite to the binding site for IFT dynein cargo (Fig 9A). Given the structural flexibility between N‐ and C‐terminal halves of IFT81/74, IFT27/25 could easily be positioned close to the ciliary membrane for BBSome interaction. Four IFT27 variants reported to be associated with BBS map to the interface with IFT74 in our model (Fig 9B, black spheres; Aldahmesh et al, 2014; Schaefer et al, 2019). These variants could disrupt the interaction interface weakening association of IFT27 with IFT74. Recent studies support this notion, as truncations of the C‐terminal region of IFT74, guided by reported missense mutations, abolish the interaction with IFT27 (Zhou et al, 2022). Improper or weakened binding of IFT27/25 on IFT81/74 is thus likely a cause for BBS.
Materials and Methods
Purification and reconstitution of Chlamydomonas IFT‐B complexes
The IFT‐B subcomplexes used for cross‐linking/MS (Fig 1; Appendix Fig S1) were purified according to previously published protocols (Taschner et al, 2014, 2016; Taschner & Lorentzen, 2016a, 2016b). IFT80, IFT172, and the IFT46/52 complex used in interaction studies were purified according to the protocols in (Taschner et al, 2016, 2018; Wang et al, 2018).
The recombinant IFT81460‐C/74460‐C/27/251–136 protein complex (Fig 4C and D) was obtained by co‐transforming the plasmids pEC‐A‐His(6)‐TEV‐IFT81460‐C, pEC‐K‐His(6)‐TEV‐CrIFT74460‐C, pEC‐S‐His(6)‐TEV‐CrIFT251–136 and pEC‐Cm‐CrIFT27 into E. coli BL21 (DE3) cells. IFT81460‐C/74460‐C/27/251–136 were overexpressed in cultures of 6L of terrific broth (TB) medium supplemented with the appropriate antibiotics. The bacterial cultures were grown at 37°C until OD600 reached 0.5, cooled down to 18°C and induced with 0.5 mM of Isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) for 18 h to trigger the expression of recombinant proteins. The cultures were harvested by centrifugation (rotor F9‐6x1000lex, at 7822 RFC (Relative Centrifugal Force), 4°C for 12 min), typically yielding 200 g of wet cell pellet. The cell pellets were dissolved in 200 ml of lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 10% (v/v) glycerol, 1 mM MgCl2, and 5 mM β‐mercaptoethanol (βME)) supplemented with 2 tablets of cOmplete EDTA free protease inhibitor, 0.5 mM of phenylmethylsulfonyl fluoride (PMSF) and DNase 1 (1 U/μl) prior to cell lysis by sonication. The cell lysate was cleared by centrifugation at 30,000 g for 30 min and the supernatant was collected, filtered through 5 μm filters, and circulated through a pre‐equilibrated 5 ml cOmplete Ni2+‐NTA column using a peristaltic pump. The column was further washed with lysis buffer containing 20 mM Imidazole pH 7.5, high salt buffer (50 mM Tris pH 7.5, 1 M NaCl, 10% (v/v) glycerol, 1 mM MgCl2 and 5 mM βME) and low salt buffer (50 mM Tris pH 7.5, 75 mM NaCl, 10% (v/v) glycerol, 1 mM MgCl2, and 5 mM βME). A 5 ml HiTrap Hp Q anion column equilibrated with low salt buffer was mounted below the Ni2+‐NTA column and the protein complexes were eluted from both columns in 5 elution steps each with 25 ml elution buffer (50 mM Tris pH 7.5, 75 mM NaCl, 10% (v/v) glycerol, 600 mM imidazole, 1 mM MgCl2, and 5 mM βME). The elutions were concentrated to 1 ml and loaded onto a HiLoad 16/600 Superdex 200 (GE Healthcare) column equilibrated in SEC buffer (10 mM HEPES pH 7.5, 150 mM NaCl, 1 mM MgCl2, and 1 mM DTT). The fractions containing pure protein complexes were pooled, concentrated, snap cooled in liquid nitrogen, and stored at −70°C until use.
Protein complex prediction with AlphaFold multimer
For predicting the structure of IFT‐B subcomplexes, we used a modified version of AlphaFold v2.1.0 on Colab notebook for protein complexes smaller than 1,200 residues (Mirdita et al, 2022) as well as a local installation of AlphaFold multimer for larger subcomplexes (Jumper et al, 2021; preprint: Evans et al, 2022). AlphaPickle was used to extract the predicted alignment score from AlphaFold runs (mattarnoldbio, 2021). The structural model of the 15 subunit IFT‐B complex was assembled from the structural models of smaller modules using the relevant Chlamydomonas reinhardtii proteins sequences. All sequences used for structure prediction have at least 500 homologs in available sequence databases and all structural predictions shown in the figures have low PAE scores for the interacting regions indicating a high degree of certainty in the relative positions of subunits within the complexes. A total of 10 subcomplexes were predicted using AF and subsequently assembled into the 15 subunit IFT‐B complex in PyMOL v. 2.5 (Schrodinger LLC, https://pymol.org) using the align function. The long regions with pLDDT scores lower than 50 are predicted to be unstructured and were excluded from the assembled model of 15‐subunit IFT‐B complex shown in Figs 8 and 9.
Site directed photo‐cross‐linking
To incorporate pBpa into Chlamydomonas IFT81 we replaced the native DNA sequence at the desired position (E418 or F68) with the TAG sequence that encodes pBpa. As IFT81 can be produced only as part of the IFT81/74/27/25 complex, we co‐transformed the pEC‐A‐His(6)‐TEV‐IFT81, pEC‐K‐His(6)‐TEV‐CrIFT74, pEC‐S‐His(6)‐TEV‐CrIFT27‐RBS‐CrIFT251–136 plasmids carrying the IFT genes together with the pEVol suppression plasmid (Young et al, 2010) into E. coli BL21 (DE3) cells. The bacterial cultures were grown in TB media supplement with 0.5 mM pBpa, at 37°C until OD600 reached 0.5, cooled down to 18°C and induced with 0.5 mM IPTG and 0.2% l‐arabinose for 18 h to trigger the expression of recombinant IFT proteins as well as the production of pBpa‐tRNA. In total, 6 l of pBpa cultures were grown simultaneously with 1 l of IFT22 culture and harvested together to yield a pentameric IFT81/74/27/25/22 complex. Subsequently the protein complex was purified as previously published (Taschner et al, 2011) except that the UV lamp was turned off during SEC to prevent the premature activation of pBpa. Samples containing 7 μM of purified IFT81/74/27/25/22 complexes with or without pBpa were exposed 20 min to 365 nm wavelength UV light (UV chamber: BLX‐365 from Vilber Lourmat) for cross‐linking. 8 μl from each sample were denatured, loaded onto SDS‐PAGE and the resulting gel was stained with Coomassie brilliant blue.
Rigid‐body assembly of the 15‐subunit Chlamydomonas IFT‐B complex
To assemble the IFT‐B complex, the predicted structure of IFT81/74120‐C/27/251–136/22 (Fig 2) was aligned onto the C‐termini of IFT81 and IFT74 of the predicted structure of IFT88120–713/70/52/46188–319/81587–645/74583‐C with an RMSD of 1 Å. Next, the IFT88120–713/521–336/57356‐C/38174–303 structural model was docked onto the IFT88120–713/70/52/46188–319/81/74120‐C/27/251–136/22 complex by superimposition of the IFT88 subunit yielding an RMSD of 3 Å. The N‐terminal globular domain of IFT52 aligned well (although it was not explicitly used in the superpositioning) and preserved its position on the central part of C‐termini of IFT57 and IFT38. In the following step, the structure of IFT57/38/54135‐C/20 was added to the model by superpositioning onto IFT57358–469/38173–303 (RMSD of 2 Å). The predicted IFT80/38 structure had the IFT38 C‐terminal domain removed because of low pLDDT scores in absence of its interacting partner IFT57. The remaining model, including full‐length IFT80 and the CH‐domain of IFT38, was docked onto the IFT88120–713/70/52/46188–319/81/74120‐C/27/251–136/22/57/38/54/20 complex via the CH‐domain of IFT38 (RMSD of 0.2 Å), which resulted in a structural model of the 14‐subunit IFT88120–713/70/52/46188–319/81/74120‐C/27/251–136/22/57/38/54/20/80 complex. Finally, the structural model of IFT1721–785/80 (Appendix Fig S6E) was docked onto IFT80 of the IFT‐B 14mer (RMSD of 1 Å) and the CH‐domain of IFT57 added via superpositioning of the IFT172/57CH model (RMSD of 0.8 Å) yielding a structural model of the 15‐subunit IFT‐B complex as shown in Fig 8. The final model was subsequently subjected to 5 macrocycles of geometric energy minimization in PHENIX (Adams et al, 2010) using geometric, nonbonding, and secondary structure restraints.
Small‐angle X‐ray scattering (SAXS) measurements of CrIFT80/38 complexes
The SAXS experiment on IFT80/38 shown in Fig 6D was performed at the BM29 beamline (ESRF, Grenoble, France) using a Pilatus 1 M detector using the SEC/SAXS protocol described in (Brennich et al, 2017). SAXS data were collected on the purified IFT381–133 protein or the IFT801–582/381–133 complex eluting directly from a Superdex 200 10/300 GL column. Correction for radiation damage, data merging, and buffer subtraction were performed on site. We used GNOME to extract SAXS parameters such as maximum particle size (Dmax; Svergun, 1992) from the ATSAS package software (Petoukhov et al, 2012). The theoretical SAXS curve of IFT801–582/381–133 was calculated from the structural model using CRYSOL (Svergun et al, 1995) and fitted to the experimental data.
Mass spectrometry analysis of DSBU cross‐linked protein complexes
Cross‐linked proteins were digested to generate peptides for MS analysis. Initially, proteins were reduced and alkylated in a buffer containing 5% SDS, 10 mM TCEP, and 11 mM CAA in 100 mM Tris–HCl pH 8.5 for 10 min at 95°C and precipitated on MagResyn HILIC magnetic particles (Resyn Biosciences) in 70% acetonitrile for 20 min and washed with 95% acetonitrile and 70% ethanol before on‐bead digestion with Lys‐C and trypsin in 50 mM ammonium bicarbonate pH 8.0 overnight (Batth et al, 2019). The resulting peptides were desalted on Sep‐Pak tC18 cartridges (Waters Corporation) and subjected to direct MS‐analysis (10% of the sample) or further enriched for multiple charged cross‐linked peptides on mixed‐mode C18/SCX cartridges (Oasis MCX, Waters Corporation) generating four fractions for MS analysis as previously described (Iacobucci et al, 2018). Cross‐linked peptides were analyzed by an Easy nanoLC system coupled to an Orbitrap Exploris 480 mass spectrometer (Thermo Scientific) using data‐dependent acquisition of ions with a charge state between 3 and 8 and HCD fragmentation using stepped normalized collision energy (NCE) of 27, 30 and 33. Interpeptide cross‐links were identified based on their signature doublet signals from the cleavable DSBU cross‐linker as implemented in the RISEUP mode of the Program MeroX version 2.0 (Götze et al, 2019). The identified cross‐links were evaluated by scores for peptide pair identifications and cross‐link position assignments within the peptide sequences.
Labeling of cross‐linking pairs onto the structural models
The MS/cross‐linking results containing residue to residue intra‐ and intermolecular cross‐links with scores > 80 of a recombinant Chlamydomonas IFT‐B1 nonameric complex (IFT881–437/70/52281–430/46188–319/81/74128‐C/27/251–136/22) and of an IFT‐B1_B2 hexameric complex (CrIFT88/521–335‐GST/57/38/54/20 complex) were used. AlphaFold predicted substructures were used as scaffold, but regions predicted to be disordered (containing pLDDT scores < 50) were discarded from the analysis.
Python 3.8 was used to generate a list of commands drawing all the cross‐linking labels simultaneously. As inputs were used the protein‐to‐chain conversion map, the IFT‐B 15mer in pdb format containing the positions of atoms and the amino acid pairs resulting from mass spectrometry. The distances between residue pairs within the IFT‐B 15mer were calculated and classified according to their length as short (< 32 Å) or long (> 32 Å) range cross‐linking pairs. The threshold was chosen based on a previous report, which identified DSBU cross‐linking pairs at a reliable distance of 27 Å (Felker et al, 2021). We decided to increase the threshold to 32 Å to account for a predicted alignment error (PAE) of 5 Å in the AF models. For validation of the automatic labeling procedure, comparison was done with manually labeling using the Wizard tool of PyMOL v. 2.5.
Pull‐down experiments
For CrIFT57‐CrIFT172 pulldown assays, GST‐tagged CrIFT571–234 or free GST tag was immobilized onto GSH resin by incubating 10 μM GST‐IFT571–234 or GST‐tag in 100 μl buffer B1 (10 mM HEPES pH 7.5, 100 mM NaCl, 5% glycerol, and 1 mM DTT) with 30 μl bed volume of GSH resin for 1 h at 4°C. After incubation, the beads were washed 3 times with Buffer B1. CrIFT1721–968 was diluted to 20 μM in Buffer B1 and 100 μl of this sample was incubated with the prepared GSH resin (loaded with GST‐tag or GST‐IFT571–234) for 2 h at 4°C. The beads were washed 3 times with Buffer B1, and bound proteins eluted by incubating the beads with Buffer B1 containing 30 mM reduced glutathione. 3 μg of proteins were loaded as input.
For the CrIFT88‐CrIFT81N/74N pulldown shown in Fig 4G, 2.2 μM of CrIFT88His‐GST/70/52/46 in 100 μl were immobilized onto 30 μl of GSH resin and incubated with 15 μM CrIFT811–387‐GFP/74135–420 for 1 h at 4°C in a total volume of 100 μl of Buffer B1. The beads were washed 4 times with 200 μl of B1 buffer and bound proteins were eluted with 50 μl of Buffer B1 containing 33 mM reduced glutathione.
For the CrIFT80‐CrIFT172 pulldown assays, Venus‐tagged CrIFT80 was immobilized onto GFP‐binder bead resin by incubating 5 μM CrIFT80‐Venus in 100 μl buffer B1 with 20 μl bed volume of GFP binder bead resin for 1 h at 4°C. A control of unbound beads was included. After incubation, the beads were washed 2 times with buffer B1. CrIFT172 or CrIFT172968‐C was diluted to 15 μM in buffer B1 and 100 μl of this sample was incubated with the CrIFT80‐Venus preloaded GFP‐binder bead resin (or unbound beads for control) for 2 h at 4°C. After incubation, beads were washed 4 times with buffer B1, and bound proteins were eluted by incubating the beads with 0.1 M citric acid. 3 μg of proteins were loaded as input. All pull‐down experiments were carried out as at least 3 technical replicates.
X‐ray diffraction analysis of CrIFT881–437/70/52281–360 and CrIFT70/52330–430/46165–319 complexes
CrIFT881–437/70/52281–360 was crystallized by mixing equal volumes of the complex (buffer: 20 mM Tris pH 8.0, 450 mM NaCl, 7.5% glycerol, and 2.5 mM DTT) at a concentration of 33 mg/ml with a crystallization solution of 0.1 M Tris pH 8.5 and 25% PEG 6 k using the sitting drop vapor diffusion method. The CrIFT70/52330–430/46165–319 complex (buffer: 10 mM Hepes pH 7.5, 150 mM NaCl, and 2 mM DTT) at 25 mg/ml was mixed with an equal volume of 50 mM Tris pH 8.0 and 8% PEG3350 for crystallization. Native X‐ray diffraction data were collected at the Swiss Light Source (SLS; Villigen, Switzerland) at the PXII beamline on a Pilatus 6 M detector. Crystals of either complex diffracted to about 4 Å resolution and complete data sets were collected and processed with XDS (Kabsch, 2010) and AIMLESS as part of the CCP4 package (Winn et al, 2011). Molecular replacement was carried out using the CrIFT70/52 crystal structure previously published (Taschner et al, 2014) and AF models of CrIFT52C/46C and IFT88 in the program Phaser (Storoni et al, 2004). Data from both crystal forms were originally processed in orthorhombic space groups, but the true crystal systems were determined as monoclinic during molecular replacement and refinement. Following molecular replacement, the models were manually rebuilt in Coot (Emsley et al, 2010) removing structural elements without clear electron density followed by refinement in PHENIX (Adams et al, 2010). Data and refinement statistics are listed in Appendix Table S1.
Molecular dynamic flexible fitting (MDFF)
The model corresponding to the IFT‐B 15mer was manually fitted into the cryo‐ET density of IFT‐B trains as rigid bodies using the phenix.dock_in_map feature of the PHENIX package (Adams et al, 2010). The approximate rigid‐body fit was used as input for the Namdinator (Kidmose et al, 2019), a locally installed molecular dynamic tool for flexible fitting. After 400,000 reiterations, the resulting models were refitted in the cryo‐ET density and Namdinator was run for another 400,000 cycles to reach final fit. The resulting IFT‐B models were subsequently real‐space refined in PHENIX (Adams et al, 2010) to produce the final model.
Mapping of single‐point mutation causing ciliopathies onto the IFT‐B 15mer structure
The single‐residue variant information for IFT‐B proteins was extracted from the Uniprot variants file homo_sapiens_variation.txt.gz (retrieved on 02.08.2022). Information was preprocessed before aligning the human proteins according to their accession number onto the Chlamydomonas reinhardtii sequences used for modeling in this study. Eight of the 436 variants did not have a corresponding C. reinhardtii residue in the alignment and were thus mapped to the closest preceding amino acid. Disease groupings were introduced according to the first two words in the disease name, specifically to group asphyxiating thoracic dystrophy, Bardet‐Biedl syndrome, retinitis pigmentosa, and short‐rib thoracic dysplasia. Number of variants associated with every group was counted, and those groups/phenotypes associated with more than 3 variants were plotted with separate colors in the model. All the remaining variants are shown in red. The spheres display the C‐alpha atoms of the variants.
Author contributions
Narcis A Petriman: Conceptualization; formal analysis; supervision; validation; investigation; visualization; methodology; writing—original draft; writing—review and editing. Marta Loureiro‐Lopez: Data curation; formal analysis; investigation; methodology; writingȁoriginal draft. Michael Taschner: Conceptualization; investigation; visualization; methodology. Nevin K Zacharia: Investigation; visualization. Magdalena M Georgieva: Software; formal analysis; investigation; visualization; methodology. Niels Boegholm: Formal analysis; investigation; methodology. Andre Mourao: Investigation; methodology. Robert B Russell: Supervision; funding acquisition; visualization; writing—review and editing. Jens S Andersen: Data curation; formal analysis; supervision; funding acquisition; investigation; methodology; project administration; writing—review and editing. Esben Lorentzen: Conceptualization; formal analysis; supervision; funding acquisition; investigation; visualization; methodology; writing—original draft; project administration; writing—review and editing. Jiaolong Wang: Investigation.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix S1
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
PDF+
Acknowledgments
We thank Kathrine Kjærgaard Frederiksen and Anni Christensen for technical assistance with cloning and/or protein purification and Jesper Lykkegaard Karlsen for computational assistance with running Alphafold and Namdinator. We also thank Hugo van den Hoek and Benjamin D. Engel for making their unpublished cryo‐ET maps available to us. This work was funded by grants from the Novo Nordisk Foundation (grant no: NNF15OC00114164) and the Independent Research Fund Denmark (grant no: 1026‐00016B) to E.L. N.A.P. was supported by a postdoc fellowship from the European Commission (H2020, Grant Agreement number 888322). R.B.R. and E.L. have received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No. 861329. M.L.L. and J.S.A. were supported by the Independent Research Fund Denmark (grant no: 8021‐00425B) to J.S.A.
Contributor Information
Narcis A Petriman, Email: narcispetriman@mbg.au.dk.
Esben Lorentzen, Email: el@mbg.au.dk.
Data availability
The structural model is available in ModelArchive at https://modelarchive.org/doi/10.5452/ma‐zqm2q. The cross‐linking/mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez‐Riverol et al, 2022) partner repository with the data set identifier PXD037049 (http://www.ebi.ac.uk/pride/archive/projects/PXD037049).
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L‐W, Kapral GJ, Grosse‐Kunstleve RW et al (2010) PHENIX: a comprehensive python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed NT, Mitchell DR (2005) ODA16p, a Chlamydomonas flagellar protein needed for dynein assembly. Mol Biol Cell 16: 5004–5012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldahmesh MA, Li Y, Alhashem A, Anazi S, Alkuraya H, Hashem M, Awaji AA, Sogaty S, Alkharashi A, Alzahrani S et al (2014) IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with Bardet‐Biedl syndrome. Hum Mol Genet 23: 3307–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony D, Brunner HG, Schmidts M (2021) Ciliary Dyneins and dynein related ciliopathies. Cell 10: 1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SA, Freeman K, Luby‐Phelps K, Pazour GJ, Besharse JC (2003) IFT20 links kinesin II with a mammalian Intraflagellar transport complex that is conserved in motile flagella and sensory cilia*. J Biol Chem 278: 34211–34218 [DOI] [PubMed] [Google Scholar]
- Batth TS, MaximAX T, Rüther P, Gonzalez‐Franquesa A, Prabhakar BS, Bekker‐Jensen S, Deshmukh AS, Olsen JV (2019) Protein aggregation capture on microparticles enables multipurpose proteomics sample preparation*. Mol Cell Proteomics 18: 1027–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beales PL, Bland E, Tobin JL, Bacchelli C, Tuysuz B, Hill J, Rix S, Pearson CG, Kai M, Hartley J et al (2007) IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat Genet 39: 727–729 [DOI] [PubMed] [Google Scholar]
- Berbari NF, Kin NW, Sharma N, Michaud EJ, Kesterson RA, Yoder BK (2011) Mutations in Traf3ip1 reveal defects in ciliogenesis, embryonic development, and altered cell size regulation. Dev Biol 360: 66–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogaraju S, Taschner M, Morawetz M, Basquin C, Lorentzen E (2011) Crystal structure of the intraflagellar transport complex 25/27. EMBO J 30: 1907–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogaraju S, Cajanek L, Fort C, Blisnick T, Weber K, Taschner M, Mizuno N, Lamla S, Bastin P, Nigg EA et al (2013) Molecular basis of tubulin transport within the cilium by IFT74 and IFT81. Science 341: 1009–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogaraju S, Weber K, Engel BD, Lechtreck K‐F, Lorentzen E (2014) Getting tubulin to the tip of the cilium: one IFT train, many different tubulin cargo‐binding sites? Bioessays 36: 463–467 [DOI] [PubMed] [Google Scholar]
- Boldt K, van Reeuwijk J, Lu Q, Koutroumpas K, Nguyen T‐MT, Texier Y, van Beersum SEC, Horn N, Willer JR, Mans DA et al (2016) An organelle‐specific protein landscape identifies novel diseases and molecular mechanisms. Nat Commun 7: 11491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun DA, Hildebrandt F (2017) Ciliopathies. Cold Spring Harb Perspect Biol 9: a028191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazelton WJ, Amundsen CD, Silflow CD, Lefebvre PA (2001) The bld1 mutation identifies the Chlamydomonas osm‐6 homolog as a gene required for flagellar assembly. Curr Biol 11: 1591–1594 [DOI] [PubMed] [Google Scholar]
- Brennich ME, Round AR, Hutin S (2017) Online size‐exclusion and ion‐exchange chromatography on a SAXS beamline. J Vis Exp e54861 10.3791/54861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Cochran DA, Craige B, Kubo T, Witman GB (2015) Assembly of IFT trains at the ciliary base depends on IFT74. Curr Biol 25: 1583–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wang X, Jiang C, Xu M, Liu Y, Qi R, Qi X, Sun X, Xie P, Liu Q et al (2018) IFT52 as a novel candidate for ciliopathies involving retinal degeneration. Invest Ophthalmol Vis Sci 59: 4581–4589 [DOI] [PubMed] [Google Scholar]
- Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL (1998) Chlamydomonas kinesin‐II–dependent Intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J Cell Biol 141: 993–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft Van De Weghe J, Harris JA, Kubo T, Witman GB, Lechtreck KF (2020) Diffusion rather than intraflagellar transport likely provides most of the tubulin required for axonemal assembly in Chlamydomonas. J Cell Sci 133: jcs249805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai PB, Stuck MW, Lv B, Pazour GJ (2020) Ubiquitin links smoothened to intraflagellar transport to regulate hedgehog signaling. J Cell Biol 219: e201912104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont MA, Humbert C, Huber C, Siour Q, Guerrera IC, Jung V, Christensen A, Pouliet A, Garfa‐Traoré M, Nitschké P et al (2019) Human IFT52 mutations uncover a novel role for the protein in microtubule dynamics and centrosome cohesion. Hum Mol Genet 28: 2720–2737 [DOI] [PubMed] [Google Scholar]
- Eguether T, San Agustin JT, Keady BT, Jonassen JA, Liang Y, Francis R, Tobita K, Johnson CA, Abdelhamed ZA, Lo CW et al (2014) IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev Cell 31: 279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R, O'Neill M, Pritzel A, Antropova N, Senior A, Green T, Žídek A, Bates R, Blackwell S, Yim J et al (2022) Protein complex prediction with AlphaFold‐Multimer. bioRxiv 10.1101/2021.10.04.463034 [PREPRINT] [DOI] [Google Scholar]
- Felker D, Zhang H, Bo Z, Lau M, Morishima Y, Schnell S, Osawa Y (2021) Mapping protein‐protein interactions in homodimeric CYP102A1 by crosslinking and mass spectrometry. Biophys Chem 274: 106590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follit JA, Tuft RA, Fogarty KE, Pazour GJ (2006) The Intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol Biol Cell 17: 3781–3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsyth R, Gunay‐Aygun M (1993) Bardet‐Biedl syndrome overview. In GeneReviews®, Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, Amemiya A (eds). Seattle, WA: University of Washington, Seattle; [Google Scholar]
- Funabashi T, Katoh Y, Okazaki M, Sugawa M, Nakayama K (2018) Interaction of heterotrimeric kinesin‐II with IFT‐B–connecting tetramer is crucial for ciliogenesis. J Cell Biol 217: 2867–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girisha KM, Shukla A, Trujillano D, Bhavani GS, Hebbar M, Kadavigere R, Rolfs A (2016) A homozygous nonsense variant in IFT52 is associated with a human skeletal ciliopathy. Clin Genet 90: 536–539 [DOI] [PubMed] [Google Scholar]
- Götze M, Iacobucci C, Ihling CH, Sinz A (2019) A simple cross-linking/mass spectrometry workflow for studying system-wide protein interactions. Anal Chem 91: 10236–10244 [DOI] [PubMed] [Google Scholar]
- Götze M, Pettelkau J, Fritzsche R, Ihling CH, Schäfer M, Sinz A (2015) Automated assignment of MS/MS cleavable cross‐links in protein 3D‐structure analysis. J Am Soc Mass Spectrom 26: 83–97 [DOI] [PubMed] [Google Scholar]
- Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, McInerney‐Leo AM, Krug P, Filhol E, Davis EE et al (2013) Defects in the IFT‐B component IFT172 cause Jeune and Mainzer‐Saldino syndromes in humans. Am J Hum Genet 93: 915–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi I, Ikura M (2003) Crystal structure of the amino‐terminal microtubule‐binding domain of end‐binding protein 1 (EB1)*. J Biol Chem 278: 36430–36434 [DOI] [PubMed] [Google Scholar]
- Hesketh SJ, Mukhopadhyay AG, Nakamura D, Toropova K, Roberts AJ (2022) IFT‐a structure reveals carriages for membrane protein transport into cilia. bioRxiv 10.1101/2022.08.09.503213 [PREPRINT] [DOI] [PubMed] [Google Scholar]
- van den Hoek H, Klena N, Jordan MA, Alvarez Viar G, Righetto RD, Schaffer M, Erdmann PS, Wan W, Geimer S, Plitzko JM et al (2022) In situ architecture of the ciliary base reveals the stepwise assembly of intraflagellar transport trains. Science 377: 543–548 [DOI] [PubMed] [Google Scholar]
- Hou Y, Witman GB (2017) The N‐terminus of IFT46 mediates intraflagellar transport of outer arm dynein and its cargo‐adaptor ODA16. Mol Biol Cell 28: 2420–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Qin H, Follit JA, Pazour GJ, Rosenbaum JL, Witman GB (2007) Functional analysis of an individual IFT protein: IFT46 is required for transport of outer dynein arms into flagella. J Cell Biol 176: 653–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobucci C, Götze M, Ihling CH, Piotrowski C, Arlt C, Schäfer M, Hage C, Schmidt R, Sinz A (2018) A cross‐linking/mass spectrometry workflow based on MS‐cleavable cross‐linkers and the MeroX software for studying protein structures and protein–protein interactions. Nat Protoc 13: 2864–2889 [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Ide T, Yagi T, Jiang X, Hirono M, Sasaki H, Yanagisawa H, Wemmer KA, Stainier DY, Qin H et al (2014) TTC26/DYF13 is an intraflagellar transport protein required for transport of motility‐related proteins into flagella. eLife 3: e01566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Shao W, Chai Y, Huang J, Mohamed MAA, Ökten Z, Li W, Zhu Z, Ou G (2022) DYF‐5/MAK–dependent phosphorylation promotes ciliary tubulin unloading. Proc Natl Acad Sci U S A 119: e2207134119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MA, Diener DR, Stepanek L, Pigino G (2018) The cryo‐EM structure of intraflagellar transport trains reveals how dynein is inactivated to ensure unidirectional anterograde movement in cilia. Nat Cell Biol 20: 1250–1255 [DOI] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010) XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanie T, Abbott KL, Mooney NA, Plowey ED, Demeter J, Jackson PK (2017) The CEP19‐RABL2 GTPase complex binds IFT‐B to initiate Intraflagellar transport at the Ciliary Base. Dev Cell 42: 22–36.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh Y, Terada M, Nishijima Y, Takei R, Nozaki S, Hamada H, Nakayama K (2016) Overall architecture of the Intraflagellar transport (IFT)‐B complex containing Cluap1/IFT38 as an essential component of the IFT‐B peripheral subcomplex*. J Biol Chem 291: 10962–10975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keady BT, Samtani R, Tobita K, Tsuchya M, San Agustin JT, Follit JA, Jonassen JA, Subramanian R, Lo CW, Pazour GJ (2012) IFT25 links the signal‐dependent movement of hedgehog components to Intraflagellar transport. Dev Cell 22: 940–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidmose RT, Juhl J, Nissen P, Boesen T, Karlsen JL, Pedersen BP (2019) Namdinator – automatic molecular dynamics flexible fitting of structural models into cryo‐EM and crystallography experimental maps. IUCrJ 6: 526–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Gengyo‐Ando K, Ishihara T, Katsura I, Mitani S (2007) IFT‐81 and IFT‐74 are required for intraflagellar transport in C. elegans. Genes Cells 12: 593–602 [DOI] [PubMed] [Google Scholar]
- Kozminski KG, Johnson KA, Forscher P, Rosenbaum JL (1993) A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc Natl Acad Sci U S A 90: 5519–5523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozminski KG, Beech PL, Rosenbaum JL (1995) The Chlamydomonas kinesin‐like protein FLA10 is involved in motility associated with the flagellar membrane. J Cell Biol 131: 1517–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo T, Brown JM, Bellve K, Craige B, Craft JM, Fogarty K, Lechtreck KF, Witman GB (2016) Together, the IFT81 and IFT74 N‐termini form the main module for intraflagellar transport of tubulin. J Cell Sci 129: 2106–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey SE, Foster HE, Pigino G (2022) The molecular structure of anterograde Intraflagellar transport trains. bioRxiv 10.1101/2022.08.01.502329 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck K‐F, Johnson EC, Sakai T, Cochran D, Ballif BA, Rush J, Pazour GJ, Ikebe M, Witman GB (2009) The Chlamydomonas reinhardtii BBSome is an IFT cargo required for export of specific signaling proteins from flagella. J Cell Biol 187: 1117–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck KF, Brown JM, Sampaio JL, Craft JM, Shevchenko A, Evans JE, Witman GB (2013) Cycling of the signaling protein phospholipase D through cilia requires the BBSome only for the export phase. J Cell Biol 201: 249–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck KF, Liu Y, Dai J, Alkhofash RA, Butler J, Alford L, Yang P (2022) Chlamydomonas ARMC2/PF27 is an obligate cargo adapter for intraflagellar transport of radial spokes. eLife 11: e74993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew GM, Ye F, Nager AR, Murphy JP, Lee JS, Aguiar M, Breslow DK, Gygi SP, Nachury MV (2014) The Intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev Cell 31: 265–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, Huang K (2020) Transport of ciliary membrane proteins. Front Cell Dev Biol 7: 381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucker BF, Behal RH, Qin H, Siron LC, Taggart WD, Rosenbaum JL, Cole DG (2005) Characterization of THE Intraflagellar transport complex B Core: DIRECT INTERACTION OF THE IFT81 AND IFT74/72 SUBUNITS*. J Biol Chem 280: 27688–27696 [DOI] [PubMed] [Google Scholar]
- mattarnoldbio . 2021. mattarnoldbio/alphapickle: release v1.4.0. 10.5281/zenodo.5708709 [DOI]
- McCafferty CL, Papoulas O, Jordan MA, Hoogerbrugge G, Nichols C, Pigino G, Taylor DW, Wallingford JB, Marcotte EM (2022) Integrative modeling reveals the molecular architecture of the Intraflagellar transport a (IFT‐A) complex. bioRxiv 10.1101/2022.07.05.498886 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M (2022) ColabFold: making protein folding accessible to all. Nat Methods 19: 679–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourão A, Christensen ST, Lorentzen E (2016) The intraflagellar transport machinery in ciliary signaling. Curr Opin Struct Biol 41: 98–108 [DOI] [PubMed] [Google Scholar]
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC et al (2007) A Core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129: 1201–1213 [DOI] [PubMed] [Google Scholar]
- Nishijima Y, Hagiya Y, Kubo T, Takei R, Katoh Y, Nakayama K (2017) RABL2 interacts with the intraflagellar transport‐B complex and CEP19 and participates in ciliary assembly. Mol Biol Cell 28: 1652–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori Y, Zhao C, Saras A, Mukhopadhyay S, Kim W, Furukawa T, Sengupta P, Veraksa A, Malicki J (2008) Elipsa is an early determinant of ciliogenesis that links the IFT particle to membrane‐associated small GTPase Rab8. Nat Cell Biol 10: 437–444 [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Dickert BL, Witman GB (1999) The DHC1b (DHC2) isoform of cytoplasmic dynein is required for flagellar assembly. J Cell Biol 144: 473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Bai J, Bandla C, García‐Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks‐Zipper A, Eisenacher M et al (2022) The PRIDE database resources in 2022: a hub for mass spectrometry‐based proteomics evidences. Nucleic Acids Res 50: D543–D552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV, Svergun DI (2012) New developments in the ATSAS program package for small‐angle scattering data analysis. J Appl Cryst 45: 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Geimer S, Lanzavecchia S, Paccagnini E, Cantele F, Diener DR, Rosenbaum JL, Lupetti P (2009) Electron‐tomographic analysis of intraflagellar transport particle trains in situ . J Cell Biol 187: 135–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piperno G, Siuda E, Henderson S, Segil M, Vaananen H, Sassaroli M (1998) Distinct mutants of retrograde Intraflagellar transport (IFT) share similar morphological and molecular defects. J Cell Biol 143: 1591–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter ME, Bower R, Knott JA, Byrd P, Dentler W (1999) Cytoplasmic dynein heavy chain 1b is required for flagellar assembly in Chlamydomonas. Mol Biol Cell 10: 693–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quélin C, Loget P, Boutaud L, Elkhartoufi N, Milon J, Odent S, Fradin M, Demurger F, Pasquier L, Thomas S et al (2018) Loss of function IFT27 variants associated with an unclassified lethal fetal ciliopathy with renal agenesis. Am J Med Genet A 176: 1610–1613 [DOI] [PubMed] [Google Scholar]
- Richey EA, Qin H (2012) Dissecting the sequential assembly and localization of Intraflagellar transport particle complex B in Chlamydomonas. PLoS One 7: e43118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum JL, Witman GB (2002) Intraflagellar transport. Nat Rev Mol Cell Biol 3: 813–825 [DOI] [PubMed] [Google Scholar]
- Schaefer E, Delvallée C, Mary L, Stoetzel C, Geoffroy V, Marks‐Delesalle C, Holder‐Espinasse M, Ghoumid J, Dollfus H, Muller J (2019) Identification and characterization of known biallelic mutations in the IFT27 (BBS19) gene in a novel family with Bardet‐Biedl syndrome. Front Genet 10: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanek L, Pigino G (2016) Microtubule doublets are double‐track railways for intraflagellar transport trains. Science 352: 721–724 [DOI] [PubMed] [Google Scholar]
- Stevens AO, He Y (2022) Benchmarking the accuracy of AlphaFold 2 in loop structure prediction. Biomolecules 12: 985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ (2004) Likelihood‐enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr 60: 432–438 [DOI] [PubMed] [Google Scholar]
- Svergun DI (1992) Determination of the regularization parameter in indirect‐transform methods using perceptual criteria. J Appl Cryst 25: 495–503 [Google Scholar]
- Svergun D, Barberato C, Koch MHJ (1995) CRYSOL – a program to evaluate X‐ray solution scattering of biological macromolecules from atomic coordinates. J Appl Cryst 28: 768–773 [Google Scholar]
- Swiderski RE, Nakano Y, Mullins RF, Seo S, Bánfi B (2014) A mutation in the mouse Ttc26 gene leads to impaired hedgehog signaling. PLoS Genet 10: e1004689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei R, Katoh Y, Nakayama K (2018) Robust interaction of IFT70 with IFT52–IFT88 in the IFT‐B complex is required for ciliogenesis. Biol Open 7: bio033241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Lorentzen E (2016a) The Intraflagellar transport machinery. Cold Spring Harb Perspect Biol 8: a028092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Lorentzen E (2016b) Recombinant reconstitution and purification of the IFT‐B Core complex from Chlamydomonas reinhardtii. In Cilia: methods and protocols, methods in molecular biology, Satir P, Christensen ST (eds), pp 69–82. New York, NY: Springer New York; [DOI] [PubMed] [Google Scholar]
- Taschner M, Bhogaraju S, Vetter M, Morawetz M, Lorentzen E (2011) Biochemical mapping of interactions within the Intraflagellar transport (IFT) B Core complex IFT52 binds directly to four other IFT‐B subunits. J Biol Chem 286: 26344–26352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Bhogaraju S, Lorentzen E (2012) Architecture and function of IFT complex proteins in ciliogenesis. Differentiation 83: S12–S22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Kotsis F, Braeuer P, Kuehn EW, Lorentzen E (2014) Crystal structures of IFT70/52 and IFT52/46 provide insight into intraflagellar transport B core complex assembly. J Cell Biol 207: 269–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Weber K, Mourão A, Vetter M, Awasthi M, Stiegler M, Bhogaraju S, Lorentzen E (2016) Intraflagellar transport proteins 172, 80, 57, 54, 38, and 20 form a stable tubulin‐binding IFT‐B2 complex. EMBO J 35: 773–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Mourão A, Awasthi M, Basquin J, Lorentzen E (2017) Structural basis of outer dynein arm intraflagellar transport by the transport adaptor protein ODA16 and the intraflagellar transport protein IFT46. J Biol Chem 292: 7462–7473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner M, Lorentzen A, Mourão A, Collins T, Freke GM, Moulding D, Basquin J, Jenkins D, Lorentzen E (2018) Crystal structure of intraflagellar transport protein 80 reveals a homo‐dimer required for ciliogenesis. eLife 7: e33067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toropova K, Zalyte R, Mukhopadhyay AG, Mladenov M, Carter AP, Roberts AJ (2019) Structure of the dynein‐2 complex and its assembly with intraflagellar transport trains. Nat Struct Mol Biol 26: 823–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachter S, Jung J, Shafiq S, Basquin J, Fort C, Bastin P, Lorentzen E (2019) Binding of IFT22 to the intraflagellar transport complex is essential for flagellum assembly. EMBO J 38: e101251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Taschner M, Ganzinger KA, Kelley C, Villasenor A, Heymann M, Schwille P, Lorentzen E, Mizuno N (2018) Membrane association and remodeling by intraflagellar transport protein IFT172. Nat Commun 9: 4684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Taschner M, Petriman NA, Andersen MB, Basquin J, Bhogaraju S, Vetter M, Wachter S, Lorentzen A, Lorentzen E (2020) Purification and crystal structure of human ODA16: implications for ciliary import of outer dynein arms by the intraflagellar transport machinery. Protein Sci 29: 1502–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield JL, Mengoni I, Bomberger H, Jiang Y‐Y, Walsh JD, Brown JM, Picariello T, Cochran DA, Zhu B, Pan J et al (2017) IFT trains in different stages of assembly queue at the ciliary base for consecutive release into the cilium. Elife 6: e26609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A et al (2011) Overview of the ıt CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin D, Christopher KJ, Zeng L, Kong Y, Weatherbee SD (2017) IFT56 regulates vertebrate developmental patterning by maintaining IFTB complex integrity and ciliary microtubule architecture. Development 144: 1544–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Shen Y (2021) Rab‐like small GTPases in the regulation of ciliary Bardet‐Biedl syndrome (BBS) complex transport. FEBS J 10.1111/febs.16232 [DOI] [PubMed] [Google Scholar]
- Yin L‐M, Schnoor M, Jun C‐D (2020) Structural characteristics, binding partners and related diseases of the calponin homology (CH) domain. Front Cell Dev Biol 8: 342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young TS, Ahmad I, Yin JA, Schultz PG (2010) An enhanced system for unnatural amino acid mutagenesis in E. coli . J Mol Biol 395: 361–374 [DOI] [PubMed] [Google Scholar]
- Zhang W, Taylor SP, Nevarez L, Lachman RS, Nickerson DA, Bamshad M, University of Washington Center for Mendelian Genomics Consortium , Krakow D, Cohn DH (2016) IFT52 mutations destabilize anterograde complex assembly, disrupt ciliogenesis and result in short rib polydactyly syndrome. Hum Mol Genet 25: 4012–4020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Qiu H, Castro‐Araya R‐F, Takei R, Nakayama K, Katoh Y (2022) Impaired cooperation between IFT74/BBS22–IFT81 and IFT25–IFT27/BBS19 causes Bardet‐Biedl syndrome. Hum Mol Genet 31: 1681–1693 [DOI] [PubMed] [Google Scholar]
- Zhu X, Liang Y, Gao F, Pan J (2017) IFT54 regulates IFT20 stability but is not essential for tubulin transport during ciliogenesis. Cell Mol Life Sci 74: 3425–3437 [DOI] [PMC free article] [PubMed] [Google Scholar]