


Abstract
In-depth analysis of the transcriptomes of several model organisms has revealed that genomes are pervasively transcribed, giving rise to an abundance of non-canonical and mainly antisense RNA polymerase II-derived transcripts that are produced from almost any genomic context. Pervasive RNAs are degraded by surveillance mechanisms, but the repertoire of proteins that control the fate of these non-productive transcripts is still incomplete. Trypanosomes are single-celled eukaryotes that show constitutive RNA polymerase II transcription and in which initiation and termination of transcription occur at a limited number of sites per chromosome. It is not known whether pervasive transcription exists in organisms with unregulated RNA polymerase II activity, and which factors could be involved in the process. We show here that depletion of RBP33 results in overexpression of ∼40% of all annotated genes in the genome, with a marked accumulation of sense and antisense transcripts derived from silenced regions. RBP33 loss does not result in a significant increase in chromatin accessibility. Finally, we have found that transcripts that increase in abundance upon RBP33 knockdown are significantly more stable in RBP33-depleted trypanosomes, and that the exosome complex is responsible for their degradation. Our results provide strong evidence that RBP33 dampens non-productive transcription in trypanosomes.
INTRODUCTION
Recent advances in transcriptome sequencing technologies have uncovered a plethora of non-canonical, non-coding and mainly antisense transcripts that are generated by pervasive (widespread) transcription from almost any genomic context in organisms as diverse as bacteria, yeast and mammals (1,2). Pervasive transcription seems to originate mainly from RNA polymerase II (RNA pol II) transcription termination readthrough, and it is coupled to RNA degradation by the nuclear RNA exosome. In yeast, this is carried out by the Nrd1–Nab3–Sen1 complex, which binds to specific sequences within target transcripts and recruits the nuclear exosome for rapid degradation (3). Chromatin remodeling elements which alter chromatin structure at termination sites are also important to suppress pervasive transcription and subtelomeric silencing in yeast (4). Another important factor is the RNA-binding protein Npl3, whose absence results in widespread transcription readthrough with pervasive effects on gene expression (5). In mammals and Drosophila, the Integrator protein complex has been shown to be essential to attenuate non-productive transcription. This complex cleaves transcripts co-transcriptionally in a sequence-independent manner, and pervasive transcripts are then rapidly degraded in the nucleus by the exosome which associates with Integrator (6,7).
RNA pol II transcription is apparently unregulated in trypanosomatid protozoa, and initiation and termination events occur only at a limited number of sites per chromosome. In these early-branched eukaryotes, protein-coding genes are arranged into polycistronic transcription units (PTUs) that are constitutively transcribed by RNA pol II (8). Mature mRNAs are generated by trans-splicing of a capped spliced leader sequence at the 5′ end, and coupled polyadenylation at the 3′ end. There are no specific polyadenylation signal motifs; instead, polyadenylation sites are chosen depending on the distance to trans-splicing signals present in the cistron immediately downstream (9). Thus, while RNA pol II termination is linked to mRNA polyadenylation in other eukaryotes, this cannot be the case in an organism with polycistronic transcription (10). As mentioned above, pervasive transcription is usually associated with transcription termination, but it is not known whether this process is operative in organisms that regulate gene expression in the absence of RNA pol II transcriptional control.
Trypanosoma brucei is the organism of choice for functional analyses in trypanosomatids. It is responsible for human and animal trypanosomiasis in sub-Saharan Africa. Trypanosomes are transmitted between mammals by tsetse flies, and undergo a profound differentiation process in order to adapt to the different environments they face in the life cycle. Trypanosome cells proliferate as procyclic forms in the insect's midgut, and migrate to the salivary glands to differentiate into quiescent metacyclic forms. Metacyclic trypanosomes are transmitted through tsetse bites to the mammalian bloodstream, where they differentiate first into proliferative bloodstream forms and then into non-dividing stumpy forms that are transferred to the fly during a blood meal and differentiate to procyclic forms, closing the cycle (11). To achieve a successful and persistent infection, bloodstream forms express a dense surface coat consisting of a variant surface glycoprotein (VSG), the basis of antigenic variation that allows the infection to evade the host's immune response (12). Expressed VSG genes are found near telomeres, and a tightly regulated mechanism of monoallelic expression ensures that only one VSG gene is transcribed at a time by RNA pol I (13,14). Metacyclic forms are also covered with a VSG coat that is transcribed from one of several specific metacyclic expression sites (MESs) to pre-adapt parasites for infection of the mammalian host (15). Metacyclic trypanosomes display different VSG variants; however, each individual cell expresses just one specific MES VSG (16). With the exception of the active VSG gene, subtelomeres are mostly silenced in T. brucei (17).
Little is known about RNA pol II transcription initiation and termination in trypanosomatids. PTUs can be divergent or convergent, and are separated by strand switch regions (SSRs). Run-on studies have shown that divergent SSRs (dSSRs) associate with RNA pol II transcription start sites (TSSs), whereas transcription termination sites (TTSs) locate at convergent SSRs (cSSRs) or in the vicinity of genes transcribed by different RNA polymerases (8,10,18,19). TSSs are enriched in the essential epigenetic marks H3K4me3, H4K10ac, H2Az, H2Bv and bromodomain factor 3, have an open chromatin structure and contain sequence-specific promoters able to initiate gene transcription and cause histone variant deposition (18,20–23). In contrast to TSSs, TTSs are enriched in three non-essential chromatin marks: base J (β-d-glucosyl-hydroxymethyluracil, a trypanosomatid-specific DNA modification), H3v and H4v (18,20,24). All three marks have been shown to be important for transcription termination, although H4v seems to be the major epigenetic sign for termination at TTSs in T. brucei (24–27). H4v null trypanosomes are viable and show increased levels of antisense transcripts derived from TTSs (27). H3v and base J, but not H4v, are enriched at telomeric sites (18,20). VSG monoallelic expression is disrupted in H3v null mutants, giving rise to the accumulation of many BES and MES VSG transcripts (17,27).
The fact that readthrough antisense transcripts are detected in the absence of TTS-specific epigenetic marks suggests that pervasive transcription also exists in trypanosomes, but factors that control the fate of these non-productive transcripts are not known at present. A promising candidate is the RNA-binding protein RBP33 (28,29). This protein contains an RNA recognition motif (RRM) close to its N-terminus, and the carboxyl two-thirds have no apparent similarity to any known protein outside trypanosomatids. It is exclusively localized in ∼60 foci in the nucleus outside the nucleolus (29), and it is expressed at similar levels in bloodstream and procyclic T. brucei cells (28). RBP33 depletion leads to rapid cell death in both developmental forms (28), whereas its overexpression results in quick growth arrest in bloodstream trypanosomes (29). RBP33 binds preferentially to RNAs originating from cSSRs and other silent genomic regions in T. brucei (28), which hints at a possible role for this protein in the regulation of the abundance of this type of transcripts. In this work, we have analyzed the transcriptomes of RBP33-depleted bloodstream and procyclic T. brucei cells, and observed that RBP33 loss leads to a strong accumulation of transcripts derived from TTS, subtelomeric genes and MES VSGs. We show using ATAC-seq that RBP33 depletion does not result in an apparent increase in chromatin accessibility at TTSs or MES VSG loci. We also provide evidence that TTS-derived transcripts are significantly more stable in RBP33-depleted trypanosomes and that they are degraded by the exosome complex.
MATERIALS AND METHODS
Trypanosome culture and RNA interference (RNAi)
Trypanosoma brucei 449 procyclic cells (30) were cultured at 28°C in SDM-79 medium (31) supplemented with 10% fetal bovine serum (FBS). ‘Single marker’ T. brucei Lister 427 bloodstream cell line S16 (32) was maintained in HMI-9 medium (33) containing 10% FBS at 37°C with 5% CO2. Trypanosomes were transfected following standard procedures (34). RBP33 expression was silenced by RNAi using bloodstream and procyclic cell lines transfected with plasmid pGR70, which expresses double-stranded RNA (dsRNA) corresponding to the carboxyl-half of RBP33 in a tetracycline-inducible manner (28). Likewise, depletion of exosome subunits RRP44 and RRP6 in bloodstream and procyclic cells was achieved using the dsRNA-expressing plasmids pHD1157 and pHD1165, respectively (35). In all RNAi experiments, cells were incubated for 48 h in the presence of 1 μg/ml tetracycline, and successful depletion of the proteins was confirmed by western blot assays using specific antisera (28,35,36).
RNA-seq
For transcriptome analyses of bloodstream or procylic RBP33-depleted trypanosomes, total RNA was obtained from either untransfected (parental S16 or 449 cells) or RNAi-induced cells using the RNeasy Mini Kit (Qiagen). RNA libraries were prepared from poly(A)-selected transcripts using the standard TruSeq stranded mRNA sample preparation protocol (Illumina). Biological triplicates were sequenced at the Genomics Unit of the IPBLN-CSIC (Granada, Spain) using a NextSeq 500 platform (Illumina). The resulting 76 nt paired-end sequences were checked for quality using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Libraries were subsampled using seqtk (version 1.2, https://github.com/lh3/seqtk) so the differences among sample sizes were <10%, and then mapped to the T. brucei Lister 427 2018 (v4.9) genome using the ‘align’ program of the Subread package [Rsubread version 1.34.7 (37)]; only uniquely mapped reads were considered (option ‘unique’ set to TRUE). Reads assignment to genes was done using the ‘featureCounts’ program of the Subread package [version 1.5.0-p (38)] with options -p -B -C; the annotation file was TriTrypDB-49_TbruceiLister427_2018.gff (downloaded from https://tritrypdb.org/tritrypdb/app/downloads/), which was supplemented with 126 manually annotated SSRs assigned arbitrarily to the Watson (+) strand (Supplementary Table S1). Forward read mapping to forward PTUs, or reverse reads mapping to reverse PTUs, were considered to represent antisense transcription. Antisense and sense count profiles were generated independently (option -s set to 1 or 2 in featureCounts, respectively), and then combined and analyzed for differential expression using edgeR [version 3.36.0 (39)]. Only genes containing more than one count per million mapped reads (CPM) in at least three samples were considered. For Pearson correlation and principal component analysis (PCA), CPM were normalized using the TMM method (39) and transformed to log2 in edgeR. PCA plots were generated using the sklearn.decomposition module [version 0.24.2 (40)]. The breadth of coverage was calculated from sequencing depth values obtained using the ‘depth’ module of samtools [version 1.9 (41)] with options -d 0 -aa. Positional enrichment of RBP33-regulated transcripts was analyzed using right-tailed Fisher's exact tests; P-values were corrected for false discovery rate (FDR) according to the Benjamini–Hochberg method (42). For coverage plots, regions of interest were binned and counted (sliding window, 100 bp; step size, 10 bp) using the ‘countReadsPerBin’ module of the deepTools API [version 3.5.0 (43)], and corrected for library size. To map splice acceptor sites (SASs), forward reads containing at least the last 14 nt of the spliced leader (SL) sequence were selected (44). The SL sequence was removed, reads were aligned to the T. brucei 427 genome as described above and SASs were assigned to the first position of all uniquely mapped SL-containing reads. Only those SASs supported by a minimum of five reads in at least two replicates were considered for further analysis.
External data corresponding to a conditional triple knockout cell line lacking chromatin marks H3v, H4v and base J (27), as well as data from the parental cell line control, were downloaded from the European Nucleotide Archive (project PRJNA727846; samples SRR9088967, SRR9088968, SRR9088969, SRR9088997, SRR9088998 and SRR9088999) and subjected to the same pipeline described above.
Quantitative RT–PCR
We first converted 250 ng of RNA to cDNA using 0.5 μg of hexamers (Invitrogen), 0.25 mM of each dNTP, 20 U of RiboLock (Thermo Scientific) and 200 U of Maxima reverse transcriptase (Thermo Scientific) in a final volume of 20 μl. Reactions were allowed to proceed for 30 min at 50°C followed by incubation at 85°C for 5 min. Quantitative RT–PCRs were carried out in 96-well plates (Thermo Scientific) in a BioRad CFX96 thermal cycler using the following thermocycler steps: 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s alternating with 55°C for 1 min. Melting curve analyses were performed to confirm a single amplicon for each mRNA tested. Reactions were set up in a final volume of 10 μl containing 0.5 μl of cDNA, 1× SYBR Green master mix (Thermo Scientific) and 0.5 μM of each oligodeoxynucleotide. Fold changes in expression were calculated using the 2−ΔΔCT method (45) with actin or ZFP2 mRNAs as references. All quantitative RT–PCR experiments were performed with at least three biological replicates. Oligodeoxynucleotide pairs used are listed in Supplementary Table S2.
Flow cytometry
For enhanced yellow fluorescent protein (eYFP) expression, the eYPF open reading frame (ORF) from p2675 (46) was PCR-amplified and cloned in reverse orientation in the plasmid pGR12 (47) to yield pGR417, where the eYFP ORF is flanked by the EP1 5'-untranslated region (5'-UTR) and the actin 3'-UTR. The rDNA spacer targeting sequence in pGR417 was replaced with DNA fragments corresponding to genes Tb427_110143800 or Tb427_100047800 to yield pGR431 and pGR433, respectively. Plasmids were linearized with EcoRI (pGR431) or MfeI (pGR433) and transfected to bloodstream S16 cells. Stable cell lines were selected in the presence of 5 μg/ml blasticidin, and proper integration of plasmids was checked by PCR of genomic DNA. Cell lines harboring eYFP transgenes were further transfected with pGR70 for tetracycline-inducible RNAi of RBP33. Flow cytometry was carried out with 1 × 106 trypanosomes from uninduced or RNAi-induced cultures. Cells were centrifuged and resuspended in phosphate-buffered saline (PBS). eYFP fluorescence was measured in a FaCSCalibur cytometer (Becton Dickinson) equipped with a 488 nm laser, and 20 000 events were recorded per sample. Data were acquired with CellQuest software and analyzed with FlowJo (version 10) software.
Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq)
We adapted the method described in (17), as follows: 1 × 107 untransfected (S16) or RBP33-depleted bloodstream trypanosomes were washed in cold PBS and resuspended in 100 μl of permeabilization buffer (10 mM Tris, pH 8.0, 100 mM KCl, 25 mM EDTA and cOmplete mini EDTA-free protease inhibitors). Digitonin (Millipore 300410) was added at a final concentration of 40 μM (from a 4 mM stock freshly prepared in permeabilization buffer), and the suspension was incubated for 5 min at room temperature. Cells were then centrifuged at 4°C for 5 min at 1400 g, washed in 600 μl of cold isotonic buffer [10 mM Tris pH 8.0, 100 mM KCl, 5% glycerol, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF)], resuspended in 50 μl of transposition mix (25 μl of 2× TD buffer, 22.5 μl of nuclease-free water, 2.5 μl of Tn5 transposase, Illumina Nextera kit FC-121-1030) and incubated for 30 min at 37°C. As a control, a sample containing 200 ng of genomic DNA was incubated in transposition mix for 5 min at 55°C in the same conditions, except that 1.25 μl of transposase were used. Reactions were purified using the Quiagen MiniElute Kit and eluted in 11 μl of 10 mM Tris pH 8.0. Library amplification was carried out in 2× KAPA HiFi mix and 1.25 μM of Nextera primers during seven cycles; this cycle number was determined by quantitative PCR using the conditions described in (48). Fragment selection was performed using the ‘NucleoMag kit for clean up and size selection’ (Macherey-Nagel 744970) following a double size selection scheme (first round, beads volume = 0.6× sample volume; second round, beads volume = 0.7× sample volume; final elution volume = 17 μl). Size selection for the genomic sample was done in a single step using a volume of beads equal to that of the sample. Biological triplicates were sequenced as described above for RNA-seq. Reads were aligned to the T. brucei Lister 427 2018 (v4.9) genome using bowtie 2.0 (49) with the options –no-unal –no-mixed -5 5 -3 5. Reads corresponding to nucleosome-free regions were selected using the ‘alignmentSieve’ module of deeptools (43) with the options –minMappingQuality 30 –maxFragmentLength 100 –ATACshift –ignoreDuplicates; the -bl option was also included to remove reads corresponding to the maxicircle (mitochondrial) DNA (unitig_851_maxicircle_Tb427v10). For Pearson correlation and PCA, genome-wide (bin size = 100 bp) or gene-wise read coverages were obtained using the ‘multiBamSummary’ module of the deeptools API (43), normalized as for RNA-seq to yield ATAC signals, and transformed to log2; for PCA, the ATAC signal was further normalized to that of the genomic DNA control. Single-base resolution plots were generated using the ‘countReadsPerBin’ module of the deepTools API (43) and normalized for library size. Peak calling on nucleosome-free reads was performed by MACS2 [version 2.2.7.1 (50)] using the module ‘callpeak’ with the following parameters: -g 3.5e7 –nomodel -f BAMPE -B –keep-dup all. Transposed genomic DNA was used as a control for the peak calling. Signal tracks were generated using the module ‘bdgcmp’ of MACS2 with the option -m ppois, and were then converted to bigWig files using the bedGraphtoBigWig module of kenUtils (https://github.com/ENCODE-DCC/kentUtils).
External data corresponding to bloodstream trypanosomes lacking chromatin marks H3v and H4v (17), as well as data from the parental cell line and transposed genomic DNA samples, were downloaded from the European Nucleotide Archive (project PRJNA487259; samples SRR7739654, SRR7739658, SRR7739651, SRR7739655, SRR7739659 and SRR7739660) and analyzed according to the same pipeline as described above.
RNA half-life assays
Untransfected (449 cell line) or RBP33-depleted procyclic trypanosomes were incubated with 10 μg/ml actinomycin D (from a stock of 5 mg/ml in dimethylsulfoxide) and 2 μg/ml sinefungin (from a stock of 2 mg/ml in water). Sinefungin was added 5 min prior to actinomycin D. Total RNA was obtained from samples taken at different times up to 1 h using the NucleoSpin RNA kit (Macherey-Nagel). Transcript abundance was measured by quantitative RT–PCR and normalized to that of ZFP2 (Tb427_110167900/Tb927.11.14950) mRNA, which is stable over the time course of the experiments (51). The rate constant for decay, k, was obtained after fitting data to first-order kinetics using the equation [RNA]t = [RNA]0 × e−kt; half-life values were then calculated as ln(2)/k (52).
Tandem affinity purification (TAP)
A procyclic cell line expressing TAP–RBP33 from the endogenous locus was generated using the strategy described in (53). Protein complexes were purified from 1–2 × 1010 procyclic trypanosomes using the TAP method (54) with the modifications described in (53). TAP-purified material was subjected to liquid chromatography with tandem mass spectrometry (LC-MS/MS) in EASY-nLC equipment (Proxeon) coupled to an amaZon speed ion ETD trap (Bruker). Proteins were identified using ProteinScape (Bruker) and MASCOT (Matrix Science) software. We considered only those proteins showing a score >60 and with two or more detected peptides.
RESULTS
RBP33 depletion leads to a marked increase in the level of antisense transcripts
RBP33 is a nuclear and essential RNA-binding protein (28). The carboxyl two-thirds of the protein are mostly intrinsically disordered, and show no apparent similarity to any known protein outside trypanosomatids [(28), Supplementary Figure S1]. An RBP33 ortholog was readily detected in Bodo saltans, a free-living outgroup species belonging to the Eubodonida order, which is closely related to parasitic trypanosomatids (Supplementary Figure S1), but not in other species from the orders Prokinetoplastida, Parabodonida or Neobodonida. Thus, RBP33 seems to be a trypanosomatid-specific protein with no homologs present even in the most closely related kinetoplastid species.
To gain insight into the function of RBP33, the transcriptomes of bloodstream and procyclic trypanosomes were analyzed upon depletion of the protein by RNAi. Stranded RNA-seq reads were aligned to the Lister 427 (version 2018) genome, and assignment to genes was done separately for sense and antisense reads. Pearson correlation coefficients were >0.980, indicating good reproducibility between replicates (Supplementary Figure S2A). We could detect a notable increase in the abundance of antisense transcripts in both developmental forms when RBP33 expression was silenced, which was readily visible as a ‘hump’ in scatter plots of reads per kilobase of transcript per million mapped reads (RPKM) values for all genes in control versus RNAi-induced cells (Figure 1A). Increased levels of antisense transcripts have also been reported in trypanosomes lacking base J and histone variant H3v (24), in a knockout mutant for histone variant H4v (27) and in a triple knockout (KO) ΔH3vΔH4vΔJ cell line (27). We analyzed the RNA-seq dataset corresponding to the transcriptome of the ΔH3vΔH4vΔJ triple KO mutant (27) to compare the degree of accumulation of antisense transcripts in cells lacking all three histone marks with that observed in RBP33-depleted trypanosomes. As seen in Figure 1A, the increase in abundance of antisense transcripts was more pronounced in the RBP33 RNAi cell line compared with the triple KO mutant. Indeed, antisense mapped reads in RBP33-depleted trypanosomes were 7- to 8-fold higher than those detected in control cells, whereas an increase of just 1.8-fold was observed in the triple KO cell line [Figure 1B and (27)]. Moreover, a PCA of RBP33 and ΔH3vΔH4vΔJ datasets showed that RBP33-depleted replicates cluster more distantly from their corresponding control replicates than triple KO samples do (Supplementary Figure S2B), thus suggesting a larger alteration in gene expression upon RBP33 knockdown.
Figure 1.

Effect of RBP33 depletion on the transcriptome of T. brucei. (A) Scatter plots comparing expression values for control versus RBP33-depleted cells or wild-type versus ΔH3vΔH4vΔJ triple KO (TKO) mutants. Average read counts across replicates were normalized to RPKM and transformed to log2. Each dot represents a transcript. (B) Quantification of antisense reads in RBP33 and histones datasets. The percentage of mapped antisense reads obtained after read assignment to genes relative to total mapped reads is represented as the mean (line) of three RNA-seq biological replicates (dots). BF, bloodstream forms; PF, procyclic forms. (C) Percentage of genes differentially expressed in sense or antisense fashion in RBP33 and histones datasets. (D) Schematic representation of a T. brucei chromosome indicating the different localizations used in this study. (E) Pie chart indicating the relative proportions of up-regulated genes within each localization category in bloodstream trypanosomes. (F) Breadth of coverage of subtelomeric regions in RBP33 and H3vΔH4vΔJ triple KO (Histones) datasets. Box plots represent the percentage of bases in subtelomeres sequenced at 1× depth (bases covered by at least one uniquely mapped read in at least two replicates). Boxes indicate the interquartile range (IQR); whiskers, ±1.5 IQR; waists, medians; dots, individual subtelomeres.
Next, we performed differential expression analysis to investigate which genes are regulated by RBP33. Sense and antisense read count profiles were combined and analyzed using edgeR, and genes with |log2FC| values >1.0 and FDRs <0.01 were considered for further study. We found that 40% (bloodstream forms) or 20% (procyclic forms) of the 17 243 genes annotated in the Lister 427 2018 genome were differentially expressed in RBP33-depleted trypanosomes (Supplementary Tables S3–S8). Among these, the vast majority (>94%) had increased expression, mainly in an antisense fashion (Figure 1C). In ΔH3vΔH4vΔJ trypanosomes, on the other hand, only 6% of genes were differentially expressed (Supplementary Table S9), 96% of which were overexpressed in roughly an equal sense and antisense manner (Figure 1C).
RBP33-regulated transcripts are derived mainly from silenced genomic regions and antisense transcription of core genes
Chromosomes in T. brucei partition into transcribed ‘cores’ and subtelomeric regions, which are thought to be non-expressed (17). A simple glance at the list of differentially expressed genes in RBP33-depleted bloodstream and procyclic trypanosomes revealed a notable abundance of transcripts originating from genomic loci from which no transcripts are detected under physiological conditions, such as SSRs or subtelomeres, and also from antisense reads derived from housekeeping core genes (Supplementary Tables S3 and S6). To quantify this more precisely, we classified T. brucei genes into the following categories according to their localization in the genome: ‘core’, housekeeping protein-coding genes within core regions; ‘subtelomeric’, non-expressed genes located at subtelomeric regions; ‘tRNAs’, tRNA genes within core regions; ‘cSSR’ and ‘dSSR’, convergent and divergent SSRs within core regions; ‘first’, first three genes within a PTU (flanking dSSRs); ‘last’, last three genes within a PTU (flanking cSSRs); and ‘reverse’, non-expressed genes in reverse orientation within a PTU (Figure 1D; Supplementary Tables S3 and S6). We could observe that most RBP33-up-regulated RNAs derive from subtelomeres (sense) or core regions (antisense transcription), as shown in Figure 1E (bloodstream forms) and Supplementary Figure S3A (procyclic forms). We next evaluated the statistical enrichment of RBP33-regulated transcripts within each of the categories using a Fisher's exact test, and observed a significant enrichment (FDR <0.05) of sense transcripts derived from subtelomeres, cSSRs, dSSRs, ‘last’, tRNAs and ‘reverse’ loci, and of antisense transcripts originating from ‘core’, ‘last’, ‘first’, ‘reverse’ and ‘cSSRs’ regions (Supplementary Figure S3B, C). The enrichment in cSSRs was particularly striking, as transcripts arising from 58 (bloodstream forms) or 56 (procyclic forms) of a total of 59 annotated cSSRs were significantly overexpressed in RBP33-depleted trypanosomes (Supplementary Tables S3 and S6).
As mentioned above, most subtelomeres are thought to be transcriptionally silent in physiological conditions, and subtelomeric genes were particularly over-represented in RBP33-depleted datasets, especially in bloodstream forms. To visualize the extent of this phenomenon, we calculated the breadth of coverage for each subtelomere as the percentage of bases that were sequenced at least once in at least two RNA-seq replicates (depth = 1×). Figure 1F shows breadth values for all 33 subtelomeres in control versus RBP33-depleted bloodstream and procyclic trypanosomes; the ΔH3vΔH4vΔJ dataset was also included for comparison (raw data are provided in Supplementary Table S10). A pronounced increase in breadth of coverage was observed upon depletion of RBP33 in bloodstream forms: on average, only 6% of subtelomeric bases were sequenced at least once in control cells, whereas this value increased to 43% in RBP33-depleted bloodstream forms, yielding a 7-fold increase in breadth. Although not as marked, breadth values were also augmented in procyclic and histones datasets (4.5-fold and 2.5-fold, respectively). Distinct increases in breadth values were also observed at different sequencing depths (Supplementary Figure S3D).
The overexpression of transcripts derived from cSSRs and neighbor genes prompted us to study in more detail the distribution of mapped reads around cSSRs. We used sliding windows to analyze genomic regions centered at cSSRs and extended 15–20 kbp on each side. Figure 2A and B shows read coverage profiles corresponding to two representative cSSRs (additional examples are presented in Supplementary Figure S4A to S4C; see also below). Reads per million mapped reads (RPM) values were generated separately for Watson and Crick strands. In control samples, transcripts arising from cSSRs and adjacent regions were barely detected. However, depletion of RBP33 resulted in a clear increase of antisense transcripts that could be readily identified as orange peaks in the upstream (‘left’) PTU or blue peaks in the downstream (‘right’) PTU. This phenomenon was better visualized when plotting the average of RPM across 30 different cSSRs flanked by PTUs at least 50 kbp long (Figure 2C), where it can be observed that antisense transcripts are generated up to 40 kbp away from cSSRs. This phenomenon was also observed in ΔH3vΔH4vΔJ trypanosomes (24,27), although the effect was much less pronounced (Supplementary Figure S4D).
Figure 2.

Coverage plots of representative genomic regions, and effect of RBP33 depletion on VSG transcripts levels. Reads counts were obtained separately from Watson (W, blue lines) or Crick (C, orange lines) strands in bloodstream forms with sliding windows (100 bp bins, 10 bp step in A, B and E; 1000 bp bins and steps of 100 bp in C) and normalized by library size (RPM). Values from control were compared with those from RBP33-depleted samples (RNAi), and represented as the mean (solid lines) ± SEM (shaded area) of the coverage across 30 cSSRs (C) or the mean ± SEM of three RNA-seq biological replicates (A, B and E). Genes are represented as boxes (blue, protein-coding genes in the W strand; orange, protein-coding genes in the C strand), and colored arrows denote the direction of transcription in each PTU. Blue and orange circles indicate trans-splicing acceptor sites assigned to W and C strands, respectively. (A and B) Examples of convergent SSRs, cSSR2:2 and cSSR11:2 (see Supplementary Table S1 for SSR nomenclature details). (C) Mean coverage across 30 cSSRs. (D) Effect of RBP33 depletion and ΔH3vΔH4vΔJ triple KO on the expression of metacyclic (MES) and bloodstream (BES) VSG genes. Read counts were normalized to CPM in edgeR, converted to RPKM and transformed to log2. Average RPKM values across RNA-seq replicates were calculated for 17 BES VSG and 8 MES VSG transcripts and represented in box plots (boxes represent the IQR; whiskers, ±1.5 IQR; waists, medians; dots, individual VSG transcripts). Only VSG transcripts showing values of RPKM ≥1 in at least three replicates were considered. Two-sided Mann–Whitney U-tests were used to assess whether there was significant differential expression between RBP33-depleted or triple KO and their respective control samples; n.s., not significant (P >0.5); *P <0.005; **P <0.0005 (n = 11/13 BES VSGs; n = 8 MES VSGs). (E) Coverage plot corresponding to the metacyclic VSG531 locus.
We showed above that tRNAs and transcripts corresponding to genes located in reverse orientation within a PTU were significantly up-regulated upon depletion of RBP33. Most tRNA genes are located at cSSRs, an explanation for their overexpression in RBP33-depleted conditions (see for example Supplementary Figure S4B and C); some others residing within PTUs also gave rise to increased levels of transcripts upon RBP33 silencing (Supplementary Figure S4E). A similar phenomenon was observed for several protein-coding genes embedded in reverse orientation within PTUs (Supplementary Figure S4F, G), and in boundaries between RNA Pol I and Pol II transcription units (EP1 locus, Supplementary Figure S4H).
Only a small fraction of the differentially regulated transcript set (6% in bloodstream forms) decreased in abundance upon RBP33 depletion. Almost all (98%) were down-regulated in a sense fashion (Supplementary Tables S3 and S6). This could be the result of RNAi triggered by dsRNA formation due to increased levels of antisense transcripts. If that were the case, one would expect down-regulated genes to be located preferentially near cSSRs, where antisense transcripts are prominently produced. To test this, we analyzed the genomic localization of down-regulated genes relative to cSSRs, and observed that indeed most genes were found close to these regions (Supplementary Figure S4I). Interestingly, at least 141 down-regulated genes in bloodstream and 11 in procyclic forms are thought to be essential for trypanosome growth according to genome-wide RNAi screenings [(55), Supplementary Tables S3 and S6].
RBP33-regulated transcripts are trans-spliced
To assess whether RBP33-regulated transcripts were properly processed, we made a global survey of trans-SASs in our RNA-seq dataset. We could identify 8553 SASs in control and 10 426 SASs in RBP33-depleted trypanosomes (Supplementary Tables S11 and S12). A canonical AG acceptor dinucleotide was found at 98% of SASs in both conditions, in agreement with previous reports (56,57). Mapped SASs are indicated as blue (W strand) or orange (C strand) circles in Figure 2A, B and E, and in Supplementary Figure S4, where it is observed that many RBP33-specific peaks are supported by SASs. We detected 42 SASs in control and 707 SASs in RBP33-depleted cells for which the nearest downstream gene was located in the opposite strand, thus being indicative of potential antisense transcripts (Supplementary Tables S11 and S12). We could not address whether these transcripts were also polyadenylated due to the library preparation method used. However, since RNA samples were processed using oligo(dT) chromatography before library construction, it is reasonable to assume that RBP33-regulated transcripts do contain poly(A) tails. In fact, transcripts derived from cSSRs in RBP33-depleted trypanosomes were shown to be polyadenylated by RNase H digestion assays (28).
These results suggest that at least some of the novel transcripts arising from RBP33 depletion are correctly processed and are therefore potentially translatable. Indeed, some of the antisense cSSR-derived transcripts encode peptides with high coding potential (Supplementary Figure S5A). This was directly addressed by inserting a gene encoding eYFP in an antisense orientation at two different cSSR loci in bloodstream trypanosomes (Supplementary Figure S5B; see the Materials and Methods for information on expression constructs). Proper integration of plasmids was assessed by PCR of genomic DNA (Supplementary Figure S5C). The resulting cell lines were further transfected with plasmid pGR70 (28) for tetracycline-inducible RNAi of RBP33, and eYFP expression was measured by flow cytometry. As expected, little fluorescence could be detected in uninduced cultures. However, eYFP expression increased 3- to 10-fold upon depletion of RBP33 (Supplementary Figure S5D, E).
Transcripts encoding metacyclic VSGs are overexpressed in RBP33-depleted trypanosomes
Expression of telomeric regions is strongly repressed in T. brucei (17,58). These loci usually harbor genes encoding VSGs which are organized in BES or MES (see the Introduction). H3v and base J are found at telomeric regions in T. brucei (18,59), and both BES and MES VSGs are overexpressed in ΔH3vΔH4vΔJ trypanosomes (17,27). We next addressed whether depletion of RBP33 had any effect on VSG expression. Normalized read count values corresponding to 17 BES VSG and 8 MES VSG genes were compared between control and RNAi samples. As a reference, the ΔH3vΔH4vΔJ dataset was analyzed in the same manner. All MES VSG transcripts had increased expression in bloodstream RBP33-depleted cells, whereas the levels of mRNAs encoding BES VSGs were barely affected (Figure 2D; Supplementary Figure S6A; Supplementary Tables S13 and S14). Coverage plots of some telomeric regions are shown to illustrate these observations (Figure 2E; Supplementary Figure S6B–D). This was in apparent contrast to the ΔH3vΔH4vΔJ triple KO mutant, where both BES and MES VSG transcripts were strongly up-regulated [(27); see also Figure 2D, Supplementary Figure S6A and Supplementary Tables S13 and S14]. Changes in the expression of two MES VSGs upon RBP33 knockdown were confirmed by quantitative RT–PCR (Supplementary Figure S6E). Some MES VSG transcripts also increased in abundance upon RBP33 depletion in procyclic forms, although their expression levels were generally low compared with those observed in bloodstream forms (Supplementary Tables S13 and S14).
Analysis of chromatin accessibility in RBP33-depleted cells
The up-regulation of transcripts originating from cSSRs and MES VSGs could be explained by an increase in chromatin accessibility caused by RBP33 depletion. In fact, the absence of H3v, H4v and base J, which are enriched in these genomic loci, leads to a somewhat similar phenotype to that observed for RBP33 depletion [(27), see above]. To test this hypothesis, we profiled chromatin accessibility by the ATAC-seq in control and RBP33-depleted trypanosomes, and compared the results with those obtained in an ATAC-seq analysis performed in a ΔH3vΔH4v double KO mutant (17). Nucleosome-free fragments were selected after aligning sequencing reads to the genome (size threshold = 100 bp, Supplementary Figure S7A), and used for further analysis. Pearson correlation coefficients were >0.940, indicating good reproducibility between replicates (Supplementary Figure S7B).
A standard quality control in ATAC-seq experiments involves the analysis of read coverage around known nucleosome-free regions (NFRs) such as TSSs of protein-coding genes (60). However, no defined promoters are associated with individual genes in trypanosomatids, since genes are organized in PTUs. Instead, we measured read coverage at tRNA loci, which are generally nucleosome free in T. brucei and other eukaryotes (61,62). Indeed, a clear NFR was observed when plotting the average of RPM across 10 different isolated (i.e. not clustered) tRNA genes (Figure 3A, blue line; see Supplementary Table S15 for a list of tRNA genes). Identification of open chromatin using the peak caller MACS2 gave essentially the same result (Figure 3A, orange line), whereas neither a transposed genomic DNA control sample (green line) nor the average of 10 randomly selected genomic loci of the same length (black line) showed a noticeable increase in ATAC-seq signal. Similar profiles of open chromatin at tRNA loci were obtained when RBP33 RNAi and ΔH3vΔH4v datasets were analyzed (Supplementary Figure S7C, D). NFRs were also observed at clustered tRNAs loci (Supplementary Figure S7E). A double peak pattern in ATAC signal was consistently observed in most tRNA genes with a valley immediately downstream of the TSS (Figure 3A; Supplementary Figure S7E). This could be interpreted as a footprint caused by an active RNA pol III transcription factor bound to the box A element of the tRNA gene promoter.
Figure 3.

ATAC-seq analysis. (A) Single base coverage plot for ATAC signal at non-clustered tRNA genes in control bloodstream trypanosomes. Black arrow box represents a tRNA gene. Read counts were obtained for genomic regions centered at non-clustered tRNA genes and extended 250 bp on each side, normalized by library size (RPM), and represented as the average of RPM values (blue line) or MACS2 scores (orange line) across 10 different tRNA loci. ATAC signals corresponding to the average of 10 randomly chosen regions of the same size (black line), and to a transposed genomic DNA control (green line) are also shown. (B) Violin plots showing chromatin accessibility in controls (light gray violins), RBP33-depleted and ΔH3vΔH4v double KO trypanosomes (dark gray violins). Average ATAC signal values across ATAC-seq replicates were obtained for cSSRs, ‘last’ and metacyclic VSG genes, and expressed as the log2 ratio relative to transposed genomic DNA control; only loci with CPM values ≥1 in at least two replicates were considered. Miniature box plots are shown inside each violin (boxes represent the IQR; whiskers, ±1.5 IQR; white circles, medians). Two-sided unpaired Student's t-tests were used to assess whether chromatin accessibility was significantly altered in RBP33-depleted or ΔH3vΔH4v double KO samples relative to their respective control samples. n.s., not significant (P >0.5); *P <5 × 10–3; **P <5 × 10–6; ***P <5 × 10–20 (n = 58, n = 279 and n = 8 for cSSRs, ‘last’ genes and metacyclic VSGs, respectively).
We next compared the RBP33 and ΔH3vΔH4v datasets using PCA. Read coverage ratios relative to transposed genomic DNA controls were obtained either gene-wise for all annotated genes in the T. brucei genome, or genome-wide after dividing the whole genome into 100 bp non-overlapping bins. RBP33 control and RNAi replicates clustered together in both analyses, whereas a marked separation was observed between histone controls (wild-type) and ΔH3vΔH4v samples (Supplementary Figure S8A, B). Moreover, strip plots representing coverage ratios between RBP33-depleted and control trypanosomes, or between ΔH3vΔH4v and wild-type cells, showed a much larger effect on chromatin accessibility in the latter dataset (Supplementary Figure S8C, D). These observations already indicate that there are fewer changes in chromatin accessibility upon RBP33 depletion compared with those observed in ΔH3vΔH4v mutants. Indeed, when we looked more specifically at MES VSGs, cSSRs and genes flanking cSSRs (‘last’ genes), where H3v and H4v variants are enriched, we observed that chromatin was significantly more open in ΔH3vΔH4v trypanosomes, as expected, whereas RBP33 depletion did not result in a significant increase in chromatin accessibility (Figure 3B). Coverage plots representing the average of ATAC signals across 47 different cSSRs longer than 1.2 kb confirmed these observations (Supplementary Figure S8E). These results indicate that the observed up-regulation of transcripts derived from these regions does not seem to be due to increased chromatin accessibility in RBP33-depleted cells. In fact, chromatin seemed to be significantly less open at cSSRs in RBP33-depleted cells (Figure 3B), as well as in tRNAs, dSSRs and ‘first’ loci (Supplementary Figure S8F). On the other hand, BES VSG loci did show a small but significant (P = 0.03) increase in accessibility (Supplementary Figure S8F); however, these apparent changes in local chromatin structure did not result in up-regulation of BES VSG transcripts, as shown above.
RBP33 depletion results in the stabilization of regulated transcripts
To assess whether the observed changes in RNA abundance were due to alterations in RNA degradation rates, we analyzed in procyclic trypanosomes the decay of transcripts derived from two different cSSR loci and from a ‘last’ gene, Tb427_060007400, which flanks a third cSSR (see Supplementary Figure S9 for genomic localization and coverage plots). As controls, we measured the degradation of three transcripts encoding housekeeping proteins: AATP6 (Tb427_080052600/Tb927.8.4700), COP9 (Tb427_100084600/Tb927.10.7800) and RBP23 (Tb427_100119800/Tb927.10.11270). AATP6 and COP9 are ‘core’ genes located in the middle of two PTUs, whereas RBP23 is the first gene transcribed within a third PTU. Cells were incubated with actinomycin D and sinefungin to inhibit the synthesis and the maturation of mRNA simultaneously (63), and samples were collected over a period of 1 h. All six transcripts seemed to follow exponential decay patterns, as seen in Figure 4 (R2 >0.95 for all curve fits). We could observe that the half-lives of cSSR- and Tb427_060007400-derived transcripts increased ∼3-fold in RBP33-depleted cells, whereas the decay of housekeeping mRNAs was not significantly altered.
Figure 4.

Effect of RBP33 depletion on transcript stability. Actinomycin D and sinefungin were added to control (gray) or RBP33-depleted (black) procyclic trypanosomes. RNA samples were collected at various time points and analyzed by quantitative RT–PCR. Values in each point represent the mean ± SEM of three independent RNAi inductions. Estimated half-life values are indicated (top, RBP33-depleted; bottom, control). Two-sided unpaired Student's t-tests were used to assess whether half-lives were significantly altered upon RBP33 ablation. n.s., not significant (P >0.4); *P <0.05; **P <0.005.
Interaction of RBP33 with other proteins
We used the TAP method coupled to mass spectrometry to analyze whether RBP33 binds to other proteins in the cell. We first generated a procyclic T. brucei cell line that expressed an N-terminal TAP-tagged version of RBP33 from the endogenous locus (Supplementary Figure S10A). TAP–RBP33 expression was localized to the nucleus (Supplementary Figure S10B). To check whether the fusion protein was functional, we transfected the cell line expressing TAP–RBP33 with an RNAi vector that produced dsRNA corresponding to the endogenous RBP33 5'-UTR. Since the TAP–RBP33 gene contains a heterologous 5'-UTR, the expression of endogenous RBP33, but not TAP–RBP33, was efficiently silenced by RNAi using this strategy (Supplementary Figure S10C). As a control for RNAi efficiency, the RNAi plasmid was transfected in the parental cell line. The viability of the control cell line was severely impaired upon induction of RNAi, as expected, whereas the growth of trypanosomes expressing TAP–RBP33 was unaffected (Supplementary Figure S10D). This indicates that TAP–RBP33 fusion protein is functional.
TAP-purified material from two independent purifications was subjected to LC-MS/MS, and proteins were identified as described in the Materials and Methods. As controls, TAPs were carried out in parallel using protein extracts obtained from cell lines expressing the TAP tag alone (Supplementary Table S16) or the ribonucleoprotein complex PuREBP1/2 (47) using either PuREBP1–TAP or TAP–PuREBP2 as baits (Supplementary Table S17). We also compared the identified RBP33-associated proteins with a list of putative contaminants commonly observed in TAP purifications (64). Proteins were considered as RBP33 partners if they fulfilled the following criteria: (i) were detected in both TAP–RBP33-independent purifications; (ii) were not present more than once in the list of putative contaminants, TAP alone and PuREBP1/2 purifications; and (iii) are considered nuclear proteins based on database annotations or proteomic surveys (65). This gave as a list of six proteins (Table 1; see Supplementary Table S18 for all identified proteins). RBP33 was found associated with other RNA-binding proteins, three of which are known splicing factors: RBSR1 [orthologous to the human splicing factor SRSF7 (66)], the basal splicing factor SF1 (67) and U2AF2/RBSR4 (68). The observed interactions with splicing factors are probably RNA mediated, as has been described for other RNA-binding proteins in T. brucei (53). RBSR1 counterparts are apparently present only in T. cruzi, Bodo saltans and Paratrypanosoma confusum, whereas orthologs corresponding to SF1 and U2AF2/RBSR4 are readily identified in all trypanosomatids for which genome sequences are available (https://tritrypdb.org/).
Table 1.
Proteins associated with RBP33 in procyclic T. brucei cells
| Gene ID | Description |
|---|---|
| Tb927.10.13720 | RNA-binding protein RBP29 |
| Tb927.6.1470 | Hypothetical protein, conserved |
| Tb927.9.6870 | RNA-binding protein RBSR1 |
| Tb927.10.9400 | Splicing factor SF1 |
| Tb927.2.4710 | RNA-binding protein RRM1 |
| Tb927.10.3500 | Splicing factor U2AF2 (RBSR4) |
The T. brucei exosome is involved in the degradation of cSSR-derived transcripts
We showed above that transcripts originating from cSSR loci become stabilized upon depletion of RBP33, which indicates that they are normally degraded in physiological conditions. In other eukaryotes, the nuclear exosome complex plays a central role in RNA surveillance by degrading transcripts produced as a consequence of non-productive transcription (see the Introduction). Trypanosomes also have an exosome complex (35,69) which is localized mainly in the nucleus (36,70). To assess whether the exosome is responsible for the degradation of cSSR-derived transcripts, we generated bloodstream and procyclic cell lines that expressed dsRNA against the exosome catalytic subunits RRP44 and RRP6 [as described in (35)]. Depletion of either subunit was confirmed by immunoblot (Figure 5A). We next analyzed the levels of transcripts derived from four different cSSR loci in control versus exosome-depleted trypanosomes or in control versus RBP33-depleted cells. A marked accumulation of cSSR-derived transcripts was observed for all four loci in both bloodstream (Figure 5B) and procyclic (Figure 5C) cell lines upon exosome knockdown. In procyclic trypanosomes, exosome depletion resulted in even higher levels of cSSR transcripts than those detected upon depletion of RBP33, whereas the effect observed in bloodstream forms was similar in both exosome- and RBP33-depleted trypanosomes. This could be due to a lower RNAi efficiency in the latter case, as some protein could be observed in RNAi-induced bloodstream samples, especially in RRP6-depleted cells (Figure 5A).
Figure 5.
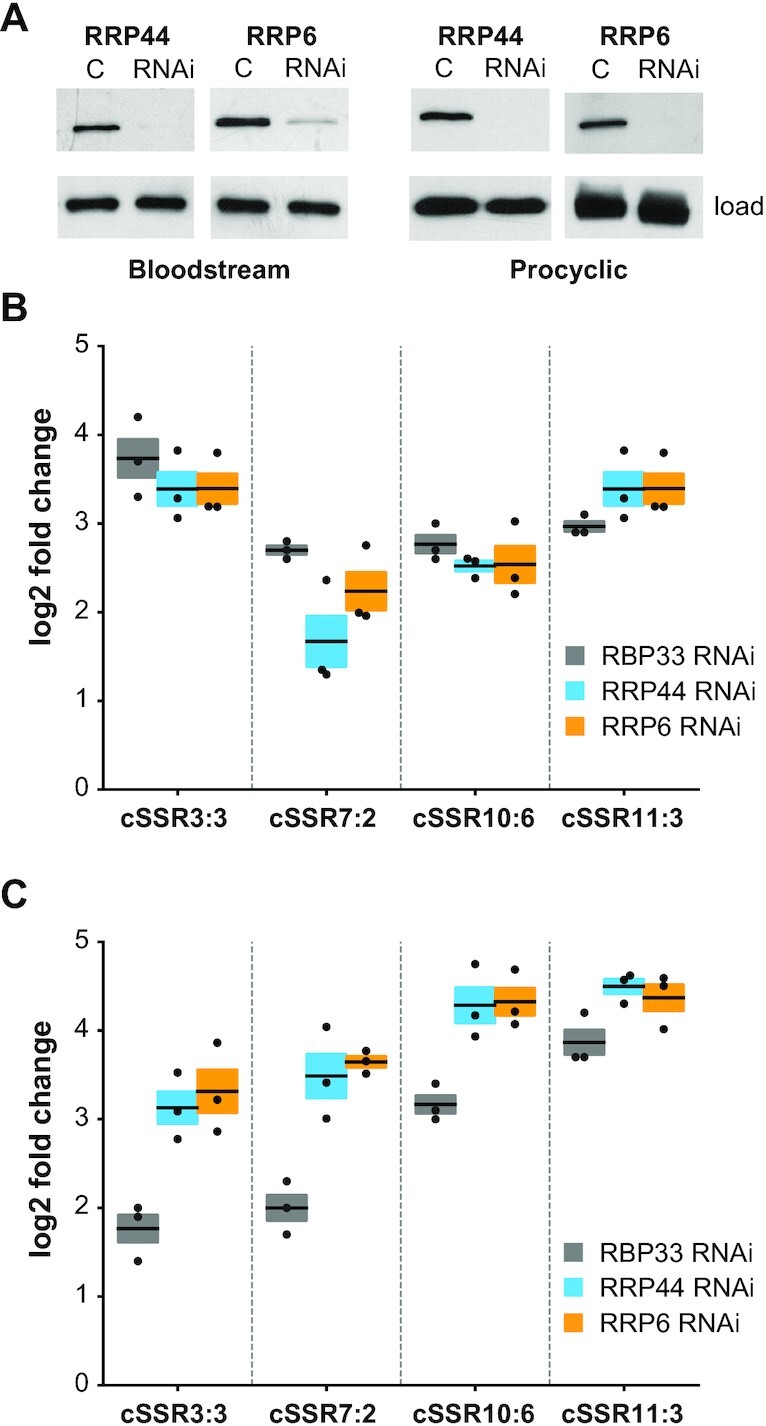
Effect of exosome depletion on the abundance of cSSR-derived transcripts. (A) Bloodstream and procyclic cell lines were generated that expressed dsRNA corresponding to RRP44 or RRP6 exosome subunits in a tetracycline-inducible manner. Depletion of each protein was monitored by immunoblot after 48 h of tetracycline induction (RNAi). C, control (parental) cell lines. RBP33 served as a loading control in all samples except in procyclic blots, where α-tubulin was used instead. (B and C) Quantitative RT–PCR analysis to measure the levels of transcripts derived from four convergent SSRs. Fold changes (log2 converted) relative to control cells are expressed as the mean (horizontal lines) ± SEM (shaded areas) of three independent RNAi inductions in bloodstream (B) or procyclic (C) trypanosomes.
DISCUSSION
Advances in RNA sequencing technologies have revealed that RNA pol II transcription is pervasive, and that RNA pol II-derived transcripts cover almost the entire eukaryotic genome (1). Pervasive transcription gives rise to an abundance of non-coding and apparently non-productive RNAs that are generated mainly from transcriptional readthrough at termination sites, and are degraded by surveillance pathways involving the RNA exosome (6,71). Transcription termination in animals and fungi is linked to RNA polyadenylation, but this cannot be the case in trypanosomatid protozoa. In these organisms, transcription is polycistronic, and individual mRNAs are generated by coupled trans-splicing and polyadenylation reactions (9). Interactions between splicing/polyadenylation and RNA pol II machineries have not been described in trypanosomes; in fact, mRNAs are probably processed once the RNA polymerase is 1–2 kb further downstream (72). RNA pol II transcription appears to be constitutive, and there are few initiation and termination sites per chromosome that are enriched in distinct histone variants (8,18). It is generally assumed that the presence of specific epigenetic marks (H3v, H4v and base J) is enough for efficient transcription termination in trypanosomatids, and that subtelomeres and cSSRs are kept silent simply because they are not transcribed (17,27).
A previous study from our laboratory showed that the RNA-binding protein RBP33 was able to bind specifically to transcripts derived from subtelomeres and cSSRs, and could therefore be an important regulator of the abundance of this type of transcripts (10,28). In this work we have explored this possibility and shown that depletion of RBP33 leads to a 7- to 8-fold increase in the abundance of antisense transcripts arising from TTSs and other silent genomic loci in both bloodstream and procyclic trypanosomes. RBP33 loss also caused overexpression of subtelomeric genes, especially in bloodstream cells; we do not know whether this is due to different RNAi efficiencies or reflects distinct roles of RBP33 in each life form. These changes are not due to cell death associated with RBP33 RNAi, as accumulation of cSSR-derived transcripts was not observed upon knockdown of DRBD3, an unrelated and essential RNA-binding protein (28). Moreover, the trypanosome transcriptome was minimally altered in cells depleted of another essential RNA-binding protein, PuREBP1 (47).
Although apparently similar, the phenotype observed in RBP33-depleted cells presents important differences relative to that exhibited by ΔH3vΔH4v(ΔJ) mutants. RBP33 is essential (28), whereas neither chromatin mark is essential by itself even though their absence results in the accumulation of TSS-derived transcripts (25,27). Importantly, procyclic trypanosomes lack base J, and we could observe a marked accumulation of TSS-derived transcripts in this life form. Moreover, the abundance of BES VSG transcripts was little affected in RBP33-depleted trypanosomes, whereas these RNAs were strongly up-regulated in ΔH3vΔH4v(ΔJ) mutants (17,27) probably due to changes in interchromosomal interactions and to detachment of BESs from specific nuclear sites in the latter case (17). In addition, chromatin accessibility was little altered in RBP33-depleted trypanosomes relative to ΔH3vΔH4v mutants. Lastly, we could observe a much larger effect on antisense transcript abundance and overexpression of subtelomeric genes in RBP33-depleted cells. Even though the ΔH3vΔH4vΔJ RNA-seq samples were obtained from conditional KO cell lines immediately after the knockout, these cell lines are based on ΔH4v mutants (27), and therefore we cannot rule out that the smaller effects seen in ΔH3vΔH4vΔJ mutants are due to unknown events taking place during long periods of culturing that allow trypanosomes to adapt and adjust to readthrough transcription.
Transcripts encoding MES VSGs were overexpressed in RBP33-depleted trypanosomes, while those corresponding to BES VSGs remained unaltered. This suggests that the mechanisms involved in VSG repression are, at least in part, different in MESs and BESs, as proposed previously (73). Intriguingly, the RBP33 transcript was found to be up-regulated in metacyclic cells generated in vitro by inducible expression of a mutant version of the RNA-binding protein RBP6 (74). However, neither RBP33 protein nor transcript levels were altered in metacyclic trypanosomes obtained by the expression of wild-type RBP6 (73). Very little is known about how all MESs are kept silent in bloodstream form T. brucei; whether RBP33 has a role in the establishment of metacyclic forms is an interesting question that awaits further study, and could benefit from the use of single-cell RNA-seq of individual trypanosomes (17).
RBP33 loss also resulted in the down-regulation of many essential genes, probably due to RNAi or antisense silencing. This could readily explain why RBP33 is essential. In fact, antisense transcripts have been shown to alter the expression of nearby genes in other organisms (75,76). Moreover, we have shown that at least some of the antisense transcripts arising from RBP33 depletion seem to be correctly processed, and are therefore potentially translatable. Indeed, an eYFP transgene inserted in the opposite orientation at two different silent loci could be efficiently expressed in an RBP33-dependent fashion. Therefore, RBP33 depletion could lead not only to severe misregulation of numerous essential factors, but also to the appearance of potentially harmful proteins and peptides.
Trypanosomes lacking H3v, H4v and base J overexpress MES and BES VSG transcripts, show high DNA accessibility at repressed BESs and increased levels of transcripts derived from TTSs (17,27). It is known that chromatin opening can result in RNA pol II transcription initiation in trypanosomes (77). It is therefore conceivable that RBP33 depletion could result in chromatin opening at silent regions, which in turn would allow the expression of these silent chromosomal loci. However, no significant increase in ATAC-seq signal was observed at cSSRs and MES VSG loci in RBP33-depleted cells, whereas these regions were significantly more open in ΔH3vΔH4v mutants. Thus, changes in chromatin accessibility do not seem to be responsible for the increased levels of transcripts that are observed upon RBP33 loss.
Importantly, we have shown that transcripts derived from three different cSSR loci become significantly stabilized upon RBP33 depletion, whereas the half-lives of three other, non-regulated transcripts remained unaltered, ruling out a general inhibition of the RNA degradation machinery caused by RBP33 knockdown or cell death. Moreover, we also shown that the exosome seems to be responsible for the degradation of cSSR-derived transcripts. Since RBP33 binds to RNAs derived from silent loci (28), our results strongly suggest that RBP33 is involved in targeting these type of transcripts for selective degradation by the exosome. This is further supported by the observation that overexpression of RBP33 results in an overall decrease of mRNA abundance (29). We cannot rule out, however, that RBP33 could have additional indirect effects on transcription, for example decreasing elongation rates near TTSs. The profound effects of RBP33 depletion on the transcriptome provide evidence for the existence of pervasive transcription in trypanosomes, as has been observed in other organisms. Thus, many genomic loci traditionally considered as ‘non-expressed’ in trypanosomes, such as cSSRs and subtelomeres, would be silent not because they are not transcribed, but because the derived pervasive transcripts are targeted for degradation.
How does RBP33 function in transcript decay? Given the vast repertoire of RBP33-regulated transcripts, it is difficult to envisage a mechanism involving RBP33 targeting mediated by specific RNA sequence motifs. In fact, we could not identify them in the cohort of RBP33-bound transcripts (28), and RBP33 has been shown to have little specificity for RNA sequence in vitro (29). This is in agreement with the fact that, as shown above, RBP33 is able to regulate the expression of a heterologous gene, eYFP. Moreover, nuclear RNA degradation by the exosome does not seem to be selective but rather the default pathway in the competition between RNA destruction and processing in eukaryotes (78). This also holds true for trypanosomes, where it has been shown that competition between mRNA processing and co-transcriptional RNA degradation has an important role in determining steady-state RNA levels (51). In mammalian cells, Integrator-mediated destruction of pervasive transcripts does not seem to be sequence specific, but dependent on RNA pol II velocity instead. As a consequence, RNA degradation is more efficient when RNA pol II is stalled or slowed down, and it is especially noticeable on poorly expressed transcriptional units (6). This mechanism aids in cleansing a considerable fraction of background transcriptional activity that is below a functionally meaningful threshold (6). A similar scenario could occur in trypanosomes, with RBP33 targeting non-productive transcripts expressed at a low level that arise as a consequence of the attenuation of transcription elongation near TTSs or poorly transcribed loci, where RNA precursors are more susceptible to degradation relative to transcripts generated from ongoing transcription. This is supported by the fact that an eYFP transgene could not be expressed under physiological conditions from cSSR loci, but fluorescence was readily detected in RBP33-depleted cells. It is also supported, as mentioned above, by the observation that overall mRNA abundance is decreased in trypanosomes overexpressing RBP33 (29), which is probably the result of RBP33 binding to regular housekeeping transcripts not normally bound when the protein is expressed at physiological levels, targeting them for degradation. Our results are consistent with a scenario in which specific chromatin marks at termination sites slow down RNA pol II but cannot completely prevent readthrough transcription. The resulting antisense transcripts are efficiently turned over by RBP33 and the exosome. Readthrough transcription is increased in the absence of histone marks; as a consequence, higher levels of antisense transcripts are produced, a small fraction of which escape from degradation due to saturation of the surveillance machinery. Upon RBP33 loss, however, antisense transcripts accumulate to toxic levels because they cannot be targeted for degradation. This would imply that RBP33 dosage is very important, as has already been proposed (10).
We hypothesize that RBP33 could act by inhibiting trans-splicing of pervasive transcripts, resulting in unprocessed RNAs that are rapidly degraded by the exosome; conversely, in the absence of RBP33, non-productive transcripts would be properly processed and thus be less prone to degradation. Consistent with this, we showed that RBP33 interacts with several splicing factors. Two of them, SF1 and U2AF2, are essential for early spliceosome formation, suggesting that RBP33 interferes with trans-splicing at this early stage. A role for RBP33 inhibiting trans-splicing is also supported by previous observations suggesting that RBP33 depletion leads to increased maturation or retroposon-derived transcripts (28), and also by the fact that the exosome is responsible for the degradation of misprocessed mRNAs in T. brucei (70). In other eukaryotes there is a direct interaction between proteins that target non-productive transcripts and the exosome complex. However, we could not detect any of the exosome subunits associated with RBP33. Although this could be explained by loose or transient protein–protein interactions, it could also be that trypanosomes possess unique mechanisms to deal with pervasive transcription as a consequence of the peculiar way in which they regulate gene expression. In fact, RBP33 seems to be a protein exclusive to trypanosomatids and, as mentioned above, mRNA processing and RNA pol II termination are not linked in these organisms. The actual mechanism by which RBP33 targets RNAs for destruction remains to be determined.
Our results show that the presence of specific histone marks at TTSs acting as ‘roadblocks’, although important, is not enough to prevent pervasive transcription and subtelomeric silencing, and that a surveillance mechanism involving RBP33 and the exosome exists in trypanosomes to cleanse non-productive transcription.
DATA AVAILABILITY
The RNA-seq and ATAC-seq data discussed in this publication are accessible through GEO Series accession number GSE186570. Flow cytometry raw data are available at FlowRepository with ID FR-FCM-Z55D.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Mark Carrington for eYFP plasmids and critical reading, and Eduardo Andrés, Laura Terrón and José L. Ruiz for helpful advice on bioinformatics and statistics.
Notes
Present address: Sandra M. Fernández-Moya, Gene Regulation of Cell Identity, Regenerative Medicine Program, Bellvitge Institute for Biomedical Research (IDIBELL) and Program for Advancing Clinical Translation of Regenerative Medicine of Catalonia, P-CMR[C], L’Hospitalet del Llobregat, 08908 Barcelona, Spain.
Contributor Information
Claudia Gómez-Liñán, Instituto de Parasitología y Biomedicina ‘López-Neyra’ (IPBLN), CSIC, Parque Tecnológico de Ciencias de la Salud, Avda. del Conocimiento 17, 18016, Armilla, Granada, Spain.
Elena Gómez-Díaz, Instituto de Parasitología y Biomedicina ‘López-Neyra’ (IPBLN), CSIC, Parque Tecnológico de Ciencias de la Salud, Avda. del Conocimiento 17, 18016, Armilla, Granada, Spain.
Gloria Ceballos-Pérez, Instituto de Parasitología y Biomedicina ‘López-Neyra’ (IPBLN), CSIC, Parque Tecnológico de Ciencias de la Salud, Avda. del Conocimiento 17, 18016, Armilla, Granada, Spain.
Sandra M Fernández-Moya, Instituto de Parasitología y Biomedicina ‘López-Neyra’ (IPBLN), CSIC, Parque Tecnológico de Ciencias de la Salud, Avda. del Conocimiento 17, 18016, Armilla, Granada, Spain.
Antonio M Estévez, Instituto de Parasitología y Biomedicina ‘López-Neyra’ (IPBLN), CSIC, Parque Tecnológico de Ciencias de la Salud, Avda. del Conocimiento 17, 18016, Armilla, Granada, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by the Spanish National Research Council (CSIC) [201920E114 to A.M.E.]; the Spanish Ministry of Science and Innovation [PID2019-111109RB-I00 to E.G.D.]; and La Caixa Foundation-Health Research 2020 Call [HR20-00635 to E.G.D]. Funding for open access charge: Spanish Ministry of Science and Innovation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Jensen T.H., Jacquier A., Libri D.. Dealing with pervasive transcription. Mol. Cell. 2013; 52:473–484. [DOI] [PubMed] [Google Scholar]
- 2. Wade J.T., Grainger D.C.. Pervasive transcription: illuminating the dark matter of bacterial transcriptomes. Nat. Rev. Microbiol. 2014; 12:647–653. [DOI] [PubMed] [Google Scholar]
- 3. Tudek A., Porrua O., Kabzinski T., Lidschreiber M., Kubicek K., Fortova A., Lacroute F., Vanacova S., Cramer P., Stefl R.et al.. Molecular basis for coordinating transcription termination with noncoding RNA degradation. Mol. Cell. 2014; 55:467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xue Y., Pradhan S.K., Sun F., Chronis C., Tran N., Su T., Van C., Vashisht A., Wohlschlegel J., Peterson C.L.et al.. Mot1, Ino80C, and NC2 function coordinately to regulate pervasive transcription in yeast and mammals. Mol. Cell. 2017; 67:594–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Holmes R.K., Tuck A.C., Zhu C., Dunn-Davies H.R., Kudla G., Clauder-Munster S., Granneman S., Steinmetz L.M., Guthrie C., Tollervey D.. Loss of the yeast SR protein npl3 alters gene expression due to transcription readthrough. PLoS Genet. 2015; 11:e1005735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lykke-Andersen S., Žumer K., Molska E.Š., Rouvière J.O., Wu G., Demel C., Schwalb B., Schmid M., Cramer P., Jensen T.H.. Integrator is a genome-wide attenuator of non-productive transcription. Mol. Cell. 2021; 81:514–529. [DOI] [PubMed] [Google Scholar]
- 7. Tatomer D.C., Elrod N.D., Liang D., Xiao M.S., Jiang J.Z., Jonathan M., Huang K.L., Wagner E.J., Cherry S., Wilusz J.E.. The integrator complex cleaves nascent mRNAs to attenuate transcription. Genes Dev. 2019; 33:1525–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martínez-Calvillo S., Yan S., Nguyen D., Fox M., Stuart K., Myler P.J.. Transcription of Leishmania major Friedlin chrosomose 1 initiates in both directions within a single region. Mol Cell. 2003; 11:1291–1299. [DOI] [PubMed] [Google Scholar]
- 9. Liang X.H., Haritan A., Uliel S., Michaeli S.. trans and cis splicing in trypanosomatids: mechanism, factors, and regulation. Eukaryot. Cell. 2003; 2:830–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clayton C. Regulation of gene expression in trypanosomatids: living with polycistronic transcription. Open Biol. 2019; 9:190072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matthews K.R. The developmental cell biology of Trypanosoma brucei. J. Cell Sci. 2005; 118:283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horn D. Antigenic variation in African trypanosomes. Mol. Biochem. Parasitol. 2014; 195:123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Figueiredo L.M., Cross G.A.M., Janzen C.J.. Epigenetic regulation in African trypanosomes: a new kid on the block. Nat. Rev. Microbiol. 2009; 7:504–513. [DOI] [PubMed] [Google Scholar]
- 14. Günzl A., Bruderer T., Laufer G., Schimanski B., Tu L.C., Chung H.M., Lee P.T., Lee M.G.S.. RNA polymerase I transcribes procyclin genes and variant surface glycoprotein gene expression sites in Trypanosoma brucei. Eukaryot. Cell. 2003; 2:542–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Graham S.V, Barry J.D. Transcriptional regulation of metacyclic variant surface glycoprotein gene expression during the life cycle of Trypanosoma brucei. Mol. Cell. Biol. 1995; 15:5945–5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tetley L., Turner C.M.R., Barry J.D., Crowe J.S., Vickerman K.. Onset of expression of the variant surface glycoproteins of Trypanosoma brucei in the tsetse fly studied using immunoelectron microscopy. J. Cell Sci. 1987; 87:363–372. [DOI] [PubMed] [Google Scholar]
- 17. Müller L.S.M., Cosentino R.O., Förstner K.U., Guizetti J., Wedel C., Kaplan N., Janzen C.J., Arampatzi P., Vogel J., Steinbiss S.et al.. Genome organization and DNA accessibility control antigenic variation in trypanosomes. Nature. 2018; 563:121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Siegel T.N., Hekstra D.R., Kemp L.E., Figueiredo L.M., Lowell J.E., Fenyo D., Wang X., Dewell S., Cross G.A.M.. Four histone variants mark the boundaries of polycistronic transcription units in Trypanosoma brucei. Genes Dev. 2009; 23:1063–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martínez-Calvillo S., Vizuet-De-Rueda J.C., Florencio-Martínez L.E., Manning-Cela R.G., Figueroa-Angulo E.E.. Gene expression in trypanosomatid parasites. J. Biomed. Biotechnol. 2010; 2010:525241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cliffe L.J., Siegel T.N., Marshall M., Cross G.A.M., Sabatini R.. Two thymidine hydroxylases differentially regulate the formation of glucosylated DNA at regions flanking polymerase II polycistronic transcription units throughout the genome of Trypanosoma brucei. Nucleic Acids Res. 2010; 38:3923–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cordon-Obras C., Gomez-Liñan C., Torres-Rusillo S., Vidal-Cobo I., Lopez-Farfan D., Barroso-del Jesus A., Rojas-Barros D., Carrington M., Navarro M.. Identification of sequence-specific promoters driving polycistronic transcription initiation by RNA polymerase II in trypanosomes. Cell Rep. 2022; 38:110221. [DOI] [PubMed] [Google Scholar]
- 22. Wedel C., Förstner K.U., Derr R., Siegel T.N.. GT-rich promoters can drive RNA pol II transcription and deposition of H2A.Z in African trypanosomes. EMBO J. 2017; 36:2581–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wright J.R., Siegel T.N., Cross G.A.M.. Histone H3 trimethylated at lysine 4 is enriched at probable transcription start sites in Trypanosoma brucei. Mol. Biochem. Parasitol. 2010; 172:141–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schulz D., Zaringhalam M., Papavasiliou F.N., Kim H.S.. Base j and H3.V regulate transcriptional termination in Trypanosoma brucei. PLoS Genet. 2016; 12:e100576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reynolds D., Cliffe L., Förstner K.U., Hon C.C., Siegel T.N., Sabatini R.. Regulation of transcription termination by glucosylated hydroxymethyluracil, base J, in Leishmania major and Trypanosoma brucei. Nucleic Acids Res. 2014; 42:9717–9729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Van Luenen H.G.A.M., Farris C., Jan S., Genest P.A., Tripathi P., Velds A., Kerkhoven R.M., Nieuwland M., Haydock A., Ramasamy G.et al.. Glucosylated hydroxymethyluracil, DNA base J, prevents transcriptional readthrough in Leishmania. Cell. 2012; 150:909–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim H.-S. Genetic interaction between site-specific epigenetic marks and roles of H4v in transcription termination in trypanosoma brucei. Front. Cell Dev. Biol. 2021; 9:744878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fernández-Moya S.M., Carrington M., Estévez A.M.. Depletion of the RNA-binding protein RBP33 results in increased expression of silenced RNA polymerase II transcripts in Trypanosoma brucei. PLoS One. 2014; 9:e107608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cirovic O., Trikin R., Hoffmann A., Doiron N., Jakob M., Ochsenreiter T.. The nuclear RNA binding protein RBP33 influences mRNA and spliced leader RNA abundance in Trypanosoma brucei. Mol. Biochem. Parasitol. 2017; 212:16–20. [DOI] [PubMed] [Google Scholar]
- 30. Biebinger S., Wirtz L.E., Lorenz P., Clayton C.E.. Vectors for inducible expression of toxic gene products in bloodstream and procyclic Trypanosoma brucei. Mol. Biochem. Parasitol. 1997; 85:99–112. [DOI] [PubMed] [Google Scholar]
- 31. Brun R., Jenni L., Tanner M., Schonenberger M., Schell K.F.. Cultivation of vertebrate infective forms derived form metacyclic forms of pleomorphic Trypanosoma brucei stocks. Acta Trop. 1979; 36:387–390. [PubMed] [Google Scholar]
- 32. Wirtz E., Leal S., Ochatt C., Cross G.A.M.. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999; 99:89–102. [DOI] [PubMed] [Google Scholar]
- 33. Hirumi H., Hirumi K.. Continuous cultivation of Trypanosoma brucei bloodstream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989; 75:985–989. [PubMed] [Google Scholar]
- 34. Clayton C., Estevez A.M., Hartmann C., Alibu V.P., Field M.C., Horn D.. Down-regulating gene expression by RNA intereference in Trypanosoma brucei. Methods Mol Biol. 2005; 309:39–60. [DOI] [PubMed] [Google Scholar]
- 35. Estévez A.M., Kempf T., Clayton C.. The exosome of Trypanosoma brucei. EMBO J. 2001; 20:3831–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haile S., Cristodero M., Clayton C., Estévez A.M.A.M.. The subcellular localisation of trypanosome RRP6 and its association with the exosome. Mol. Biochem. Parasitol. 2007; 151:52–58. [DOI] [PubMed] [Google Scholar]
- 37. Liao Y., Smyth G.K., Shi W.. The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013; 41:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liao Y., Smyth G.K., Shi W.. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923–930. [DOI] [PubMed] [Google Scholar]
- 39. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V.et al.. Scikit-learn: machine learning in {P}ython. J. Mach. Learn. Res. 2011; 12:2825–2830. [Google Scholar]
- 41. Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M.et al.. Twelve years of SAMtools and BCFtools. Gigascience. 2021; 10:giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Benjamini Y., Hochberg Y.. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995; 57:289–300. [Google Scholar]
- 43. Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Siegel T.N., Hekstra D.R., Wang X., Dewell S., Cross G.A.M.. Genome-wide analysis of mRNA abundance in two life-cycle stages of Trypanosoma brucei and identification of splicing and polyadenylation sites. Nucleic Acids Res. 2010; 38:4946–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Livak K.J., Schmittgen T.D.. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔ C(T)). Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 46. Kelly S., Reed J., Kramer S., Ellis L., Webb H., Sunter J., Salje J., Marinsek N., Gull K., Wickstead B.et al.. Functional genomics in Trypanosoma brucei: a collection of vectors for the expression of tagged proteins from endogenous and ectopic gene loci. Mol. Biochem. Parasitol. 2007; 154:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rico-Jiménez M., Ceballos-Pérez G., Gómez-Linãn C., Estévez A.M.. An RNA-binding protein complex regulates the purine-dependent expression of a nucleobase transporter in trypanosomes. Nucleic Acids Res. 2021; 49:3814–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J.. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015; 109:21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Langmead B., Salzberg S.L.. Fast gapped-read alignment with bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nussbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of chip-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fadda A., Ryten M., Droll D., Rojas F., Färber V., Haanstra J.R., Merce C., Bakker B.M., Matthews K., Clayton C.. Transcriptome-wide analysis of trypanosome mRNA decay reveals complex degradation kinetics and suggests a role for co-transcriptional degradation in determining mRNA levels. Mol. Microbiol. 2014; 94:307–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen C.Y.A., Ezzeddine N., Shyu A.B.. Messenger RNA half-life measurements in mammalian cells. Methods Enzymol. 2008; 448:335–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fernández-Moya S.M., García-Pérez A., Kramer S., Carrington M., Estévez A.M.. Alterations in DRBD3 ribonucleoprotein complexes in response to stress in Trypanosoma brucei. PLoS One. 2012; 7:e48870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Séraphin B.. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001; 24:218–229. [DOI] [PubMed] [Google Scholar]
- 55. Alsford S., Turner D.J., Obado S.O., Sanchez-Flores A., Glover L., Berriman M., Hertz-Fowler C., Horn D.. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 2011; 21:915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kolev N.G., Franklin J.B., Carmi S., Shi H., Michaeli S., Tschudi C.. The transcriptome of the human pathogen Trypanosoma brucei at single-nucleotide resolution. PLoS Pathog. 2010; 6:e1001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rastrojo A., Carrasco-Ramiro F., Martín D., Crespillo A., Reguera R.M., Aguado B., Requena J.M.. The transcriptome of Leishmania major in the axenic promastigote stage: transcript annotation and relative expression levels by RNA-seq. BMC Genomics. 2013; 14:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cestari I., Stuart K.. Transcriptional regulation of telomeric expression sites and antigenic variation in trypanosomes. Curr. Genomics. 2017; 19:119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cliffe L.J., Kieft R., Southern T., Birkeland S.R., Marshall M., Sweeney K., Sabatini R.. JBP1 and JBP2 are two distinct thymidine hydroxylases involved in J biosynthesis in genomic DNA of African trypanosomes. Nucleic Acids Res. 2009; 37:1452–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yan F., Powell D.R., Curtis D.J., Wong N.C.. From reads to insight: a hitchhiker's guide to ATAC-seq data analysis. Genome Biol. 2020; 21:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Maree J.P., Povelones M.L., Clark D.J., Rudenko G., Patterton H.G.. Well-positioned nucleosomes punctuate polycistronic pol II transcription units and flank silent VSG gene arrays in Trypanosoma brucei. Epigenetics Chromatin. 2017; 10:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nagarajavel V., Iben J.R., Howard B.H., Maraia R.J., Clark D.J.. Global ‘bootprinting’ reveals the elastic architecture of the yeast TFIIIB–TFIIIC transcription complex in vivo. Nucleic Acids Res. 2013; 41:8135–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li C.H., Irmer H., Gudjonsdottir-Planck D., Freese S., Salm H., Haile S., Estévez A.M., Clayton C.. Roles of a Trypanosoma brucei 5′/3′ exoribonuclease homolog in mRNA degradation. RNA. 2006; 12:2171–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ouna B.A., Stewart M., Helbig C., Clayton C.. The trypanosoma brucei CCCH zinc finger proteins ZC3H12 and ZC3H13. Mol. Biochem. Parasitol. 2012; 183:184–188. [DOI] [PubMed] [Google Scholar]
- 65. Goos C., Dejung M., Janzen C.J., Butter F., Kramer S.. The nuclear proteome of Trypanosoma brucei. PLoS One. 2017; 12:e0181884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wippel H.H., Malgarin J.S., Martins S., de T., Vidal N.M., Marcon B.H., Miot H.T., Marchini F.K., Goldenberg S., Alves L.R.. The nuclear RNA-binding protein RBSR1 interactome in Trypanosoma cruzi. J. Eukaryot. Microbiol. 2019; 66:244–253. [DOI] [PubMed] [Google Scholar]
- 67. Gupta S.K., Carmi S., Ben-Asher H.W., Tkacz I.D., Naboishchikov I., Michaeli S.. Basal splicing factors regulate the stability of mature mRNAs in trypanosomes. J. Biol. Chem. 2013; 288:4991–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vazquez M.P., Mualem D., Bercovich N., Stern M.Z., Nyambega B., Barda O., Masiga D., Gupta S.K., Michaeli S., Levin M.J.. Functional characterization and protein–protein interactions of trypanosome splicing factors U2AF35, U2AF65 and SF1. Mol. Biochem. Parasitol. 2009; 164:137–146. [DOI] [PubMed] [Google Scholar]
- 69. Estevez A.M., Lehner B., Sanderson C.M., Ruppert T., Clayton C., Estévez A.M., Lehner B., Sanderson C.M., Ruppert T., Clayton C.. The roles of intersubunit interactions in exosome stability. J. Biol. Chem. 2003; 278:34943–34951. [DOI] [PubMed] [Google Scholar]
- 70. Kramer S., Piper S., Estevez A., Carrington M.. Polycistronic trypanosome mRNAs are a target for the exosome. Mol. Biochem. Parasitol. 2016; 205:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Villa T., Porrua O.. Pervasive transcription: a controlled risk. FEBS J. 2022; 10.1111/febs.16530. [DOI] [PubMed] [Google Scholar]
- 72. Haanstra J.R., Stewart M., Luu V.D., Van Tuijl A., Westerhoff H.V., Clayton C., Bakker B.M.. Control and regulation of gene expression: quantitative analysis of the expression of phosphoglycerate kinase in bloodstream form Trypanosoma brucei. J. Biol. Chem. 2008; 283:2495–2507. [DOI] [PubMed] [Google Scholar]
- 73. Christiano R., Kolev N.G., Shi H., Ullu E., Walther T.C., Tschudi C.. The proteome and transcriptome of the infectious metacyclic form of Trypanosoma brucei define quiescent cells primed for mammalian invasion. Mol. Microbiol. 2017; 106:74–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shi H., Butler K., Tschudi C.. A single-point mutation in the RNA-binding protein 6 generates Trypanosoma brucei metacyclics that are able to progress to bloodstream forms in vitro. Mol. Biochem. Parasitol. 2018; 224:50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Huber F., Bunina D., Gupta I., Khmelinskii A., Meurer M., Theer P., Steinmetz L.M., Knop M.. Protein abundance control by non-coding antisense transcription. Cell Rep. 2016; 15:2625–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xu Z., Wei W., Gagneur J., Clauder-Münster S., Smolik M., Huber W., Steinmetz L.M.. Antisense expression increases gene expression variability and locus interdependency. Mol. Syst. Biol. 2011; 7:468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. McAndrew M., Graham S., Hartmann C., Clayton C.. Testing promoter activity in the trypanosome genome: isolation of a metacyclic-type VSG promoter, and unexpected insights into RNA polymerase II transcription. Exp. Parasitol. 1998; 90:65–76. [DOI] [PubMed] [Google Scholar]
- 78. Schmid M., Jensen T.H.. Controlling nuclear RNA levels. Nat. Rev. Genet. 2018; 19:518–529. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq and ATAC-seq data discussed in this publication are accessible through GEO Series accession number GSE186570. Flow cytometry raw data are available at FlowRepository with ID FR-FCM-Z55D.