

Abstract
The human APOBEC family of eleven cytosine deaminases use RNA and single-stranded DNA (ssDNA) as substrates to deaminate cytosine to uracil. This deamination event has roles in lipid metabolism by altering mRNA coding, adaptive immunity by causing evolution of antibody genes, and innate immunity through inactivation of viral genomes. These benefits come at a cost where some family members, primarily from the APOBEC3 subfamily (APOBEC3A-H, excluding E), can cause off-target deaminations of cytosine to form uracil on transiently single-stranded genomic DNA, which induces mutations that are associated with cancer evolution. Since uracil is only promutagenic, the mutations observed in cancer genomes originate only when uracil is not removed by uracil DNA glycosylase (UNG) or when the UNG-induced abasic site is erroneously repaired. However, when ssDNA is present, replication protein A (RPA) binds and protects the DNA from nucleases or recruits DNA repair proteins, such as UNG. Thus, APOBEC enzymes must compete with RPA to access their substrate. Certain APOBEC enzymes can displace RPA, bind and scan ssDNA efficiently to search for cytosines, and can become highly overexpressed in tumor cells. Depending on the DNA replication conditions and DNA structure, RPA can either be in excess or deficient. Here we discuss the interplay between these factors and how despite RPA, multiple cancer genomes have a mutation bias at cytosines indicative of APOBEC activity.
INTRODUCTION
APOBEC enzymes are cytosine deaminases that deaminate cytosine to form uracil in single-stranded (ss)DNA or RNA. Although these promutagenic enzymes are usually beneficial, having roles in lipid metabolism, antibody affinity maturation, and restriction of viral replication, their unregulated activity can also cause mutations in the human genome that cause cancer evolution. However, in order to access genomic DNA, the APOBEC enzymes need to compete with replication protein A (RPA) that normally binds ssDNA to avoid unwanted degradation or recombination. Here we discuss the biochemistry of RPA–DNA and APOBEC–DNA interactions. These fields have largely stayed separate, despite the growth of the APOBEC/cancer field that has examined APOBEC enzymes and how they access transient genomic single-stranded (ss)DNA substrates, which would also be bound by RPA. The details of the competition of APOBECs and RPA for ssDNA has yet to be fully examined and put into the context of DNA replication and repair and cancer. Here we discuss the biochemistry of each of these DNA binding proteins and based on their biochemistry and the few studies that examined them together we suggest mechanisms and areas of future research.
Overview of the AID/APOBEC family
Apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like (APOBEC) enzymes are a family of evolutionary conserved, zinc-dependent polynucleotide deaminases that deaminate cytosines and consist of Activation Induced Deaminase (AID), APOBEC1 (A1), APOBEC2 (A2), APOBEC3A-H, except E (A3) and APOBEC4 (A4) (Figure 1) (1). A3H is the most variable as it has seven haplotypes (hap), but only A3H hap II, A3H hap V and A3H hap VII are stable in cells (2). All A3 enzymes contain at least one cytosine deaminase domain that is defined by a consensus amino acid (aa) sequence motif (HxEx25–30PCx2–4C) that is embedded in a conserved fold comprised of five beta sheets and six α-helices (Figure 1A and B) (3–5). This aa sequence is a zinc-dependent deaminase (ZDD) domain that is crucial for deamination activity on ssDNA and RNA. A3A, A3B and A3G have been shown to bind the ssDNA in a U-shaped conformation through primarily contacts on loops 1 and 7 (Figure 1C) (6–8). Several members of the A3 subfamily, namely, A3B, A3D, A3G and A3F, all contain two zinc coordinating domains, however only the C-terminal domain is catalytically functional (Figure 1A, D). Despite the lack of catalytic activity in the N-terminal domain, this domain still retains RNA- and DNA-binding (9–11). Additionally, it confers processivity enabling these enzymes to deaminate consecutive cytosines in a single enzyme-substrate encounter, rather than deaminating and then dissociating from ssDNA in a non-processive manner (12). The protein sequences within these ZDD domains enable the APOBECs to be grouped further into three sub-families: AID, A1, A3 and the A3s can be further sub-divided into three sub-groups based on their Z1, Z2 and Z3 domains (Figure 1A) (5,13). A3H uniquely forms the Z3 group and dimerizes using a double-stranded (ds) RNA molecule with no protein-protein contacts in the dimer molecule (Figure 1E) (14). These three zinc domains have been predicted to have undergone a minimum of eight duplication events due to positive selection as a protective measure against rapidly evolving pathogenic retroelements and retroviruses (15). This is supported by the discovery of ancestral AID genes that were first identified in jawless fish present over 500 million years ago, which ultimately led to the existence and expansion of today's APOBEC genes in amphibians, birds and mammals (1,16).
Figure 1.

Protein domains of APOBECs and RPA. (A) The eleven human APOBEC enzymes have either a single or double Zn2+-binding domain. In the double domain enzymes, the N-terminal domain (NTD) mediates processivity and the C-terminal domain (CTD) catalyzes cytosine deaminations (8). The amino acid sequences surrounding the catalytic residues (underlined) for the three zinc domains (Z1–Z3) of the A3 subfamily that originated from the ancestral ZDD are illustrated below the NTD and CTD domain schematic. The colored labels for the domains indicate which NTD and CTD contain which amino acid motifs. (B) Crystal structure of A3A demonstrates the conserved APOBEC structure that is comprised of a five stranded β-sheet (labeled β1–5) surrounded by six α-helices (labeled α1–6). Loop 1 (L1) and Loop 7 (L7) control access to the active site and make key interactions with ssDNA that determine deamination motif specificity, respectively. The Zinc molecule is represented by a blue sphere (14). (C) Crystal structure of A3A as in (B), but bound to an ssDNA. The DNA takes a U-shaped conformation, which was also found with A3B and A3G (not shown). A3A structures in (B, C) are from PDB 5SWW (8). (D) Full length crystal structure of Rhesus Macaque A3G is used to represent the structure of double domain enzymes such as A3B, for which a full length structure is not yet available. The NTD and CTD are connected by a flexible linker. PDB 6P40 is shown (175). (E) The human A3H crystal structure revealed that it formed an obligate dimer with dsRNA. There are no protein-protein contacts in the dimer. PDB 6B0B is shown (14). (F) Human RPA is a heterotrimer consisting of RPA70, RPA32 and RPA14 (176). RPA contains six oligonucleotide binding domains called DNA-binding domains (DBD). RPA70 contains DBD-A, DBD-B, DBD-C and DBD-F. RPA32 contains DBD-D and a C-terminal winged-helix (WH) domain. RPA14 contains DBD-E. DBD-D, DBD-C and DBD-E from each subunit form the trimerization core to enable formation of the stable heterotrimer. (G) Structure of the RPA trimerization core demonstrates how the DBD-C of RPA70 (pink) interacts with the DBD-D of RPA32 (teal) and RPA14 (green). The interaction forms through three C-terminal α-helices that arrange in parallel. PDB 1L1O is shown (176). Structural figures were made using PyMol 2.3.1.
Diverse functions of AID/APOBECs
APOBEC-mediated cytosine deamination can have different consequences depending on whether it occurs in RNA or DNA. While RNA deaminations are modifications due to the coding functionality of uracils, DNA deaminations are promutagenic lesions that can lead to undesired permanent alterations in the genome if unrepaired or repaired erroneously. Although it may be detrimental to have uracil in DNA under normal circumstances, different cell types use these uracils in a variety of positive ways which emphasizes the importance of regulating these enzymes. In addition, genome engineering technologies have taken advantage of the ability of certain APOBECs to modify genomic DNA by fusing them to Cas9 to allow for programmable CRISPR/Cas9 base editing (17).
The founding member, A1 was originally discovered as an RNA deaminase that plays an important role in lipid metabolism (Figure 2) (18). AID, the ancestral member of the APOBECs is responsible for initiating mutagenesis of immunoglobulin genes to allow production of antibodies with strong affinities (somatic hypermutation; SHM) and different classes, e.g. IgG or IgA (class switch recombination; CSR) (Figure 2) (19,20). When AID is dysregulated it has been found to contribute to inflammation and autoimmunity (Figure 2) (21). The A3 subfamily consists of seven members, A3A-A3H (except E) and are host restriction factors that are constitutively expressed in T cells, macrophages and germ cells and are expressed in epithelial cells in response to viral infection (22–24). The A3 enzymes enable suppression of ssDNA intermediates of retroviruses, retrotransposons, herpesviruses, and other foreign DNA through ssDNA deamination as a means of mutagenesis (Figure 2). A3A and A3G have also been shown to deaminate RNA. A3A RNA editing occurs in response to interferons and hypoxic conditions, but the cell type and function of A3G editing is unknown (Figure 2) (25,26). Retroviral restriction occurs when relevant A3s avoid antagonism from the viral Vif protein and are packaged into viral particles along with the viral genome (27,28). Like other DNA viruses, replication of herpes viruses takes place in the nucleus and Epstein Barr virus encodes the protein BORF2 that depletes A3B from the nucleus to avoid host restriction (Figure 2) (23). A similar antagonism occurs with Herpes Simplex Virus Type 1 ICP6 protein, a BORF2 ortholog and A3A (29,30). The ssDNA of the viral genome is the substrate for deamination and results in hypermutations that lead to inactivation or degradation of the viral DNA (31). Because the expansion of the A3 locus correlates with the expansion of retrotransposons in primate genomes, these genomic elements are thought to be their original target (Figure 2) (32,33).
Figure 2.

The diverse functions of the AID/APOBEC family. APOBECs are expressed in different cell types and have many diverse functions. A1 and A3A can undergo RNA editing in the small intestine and in monocytes, respectively. A3G (not shown) can catalyze RNA editing in 293T cells, but the physiological role is not known. In B cells, AID plays an essential role in immunoglobulin diversification through somatic hypermutation and class switch recombination. When dysregulated, AID can cause inflammation and autoimmunity. In CD4+ T cells, A3s supress retroviruses through deamination of ssDNA intermediates and inhibition of reverse transcriptase. A3A and A3B can also restrict dsDNA viruses during viral replication in epithelial cells. In CD4+ T cells and monocytes, restriction of foreign DNA by A3s can suppress the DNA-induced inflammatory response. In germ and somatic cells, restriction of endogenous retroelements occurs through deamination of ssDNA intermediates or RNA binding. Aberrant expression of APOBECs can lead to ‘off-target’ deaminations in genomic DNA that could result in cellular transformation or cancer.
Biochemical pathways of transcriptional regulation have not been fully elucidated for the AID/APOBEC family, but subcellular localization is an important factor in maintaining proper function. Studies have shown that in cancer cells A3B and A3A transcription correlates with upregulation of other cell cycle and DNA damage response genes (34,35). For other A3s, including A3H hap I and A3A, transcription in cancer cells correlates with immune related genes (35). Overall, further research is needed to fully elucidate the transcriptional regulators of A3 activity. Cytoplasmic A3s (A3D, A3F, A3G) appear to not have access to genomic DNA even during mitosis in normal cells, but there have been reports of A3G in the nucleus in cancer cells (36–38). These A3s have a dominant cytoplasmic retention signal, even with creation of a chimeric NLS, which may be a preventative measure against off-target deaminations (39). The localization of A3H appears to be haplotype dependent where the stable A3H hap II is actively retained in cytoplasm whereas the less stable A3H hap I enters the nucleus (40,41). RNA-binding is required for A3H hap II cytoplasmic localization and HIV-1 restriction. RNA binding mutants showed disrupted cytoplasmic localization and poor encapsidation into viral particles, leading to lower antiviral activity (14). Interestingly, it was also found that A3H hap II is actively retained in the nucleolus in addition to the cytoplasm suggesting A3H may form two different ribonucleoprotein complexes to regulate subcellular localization. Like A1, A3B has N-terminal positively charged residues that mediates nuclear localization through interactions with importin proteins (42,43). A3A that is endogenously expressed in monocytic cells is retained in the cytoplasm whereas expression in other cell types appears cell wide (44,45). The varying localizations of A3s highlight the importance of regulation as nuclear A3s have been implicated in various cancers (46–49).
AID/APOBECs as genomic mutators
The roles of A3s in viral restriction have been the focus of research for many years until it was discovered that there is a cost to this defense mechanism. Current evidence suggests that aberrant A3 expression can lead to the formation of promutagenic uracils on genomic DNA (Figure 2). The fact that normal expression of A3s in their respective cell types does not usually lead to cancer suggests that they have developed their own regulatory mechanisms in addition to the individual intrinsic enzyme properties to prevent aberrant deamination. A3-catalyzed deaminations are one of the most common mutational processes in cancer and were identified in > 50% of cancer genomes (50,51). The APOBEC signature in cancer genomes was identified as part of 21 single base substitution (SBS). The two APOBEC-associated signatures are SBS2 (C > T) and SBS13 (C > G and C > A) within a TCN (underlined C denotes target cytosine) nucleotide context (50–53). With more extensive studies on the mutation signatures of A3A and A3B since the original identification of the SBS types, it has become apparent that there are specific extended signatures attributable to each A3. A3A prefers deamination at 5' YTCA (Y = C or T) and A3B prefers deamination at 5' RTCA (R = A or G) (54). A3A also can efficiently deaminate cytosine in hairpin loops, unlike A3B and A3H hap I (55). In these hairpins, A3A has been found to be able to deaminate 5'VC (V = A, C, or G) sequences (56).
However, it is unknown as to what stage APOBECs exert their mutagenic potential, namely whether they are involved at the early stages of cellular transformation or the late stages and provide tumor genetic diversity. Additionally, we are lacking an understanding of the effect of these mutations. It is unclear whether they are deleterious, neutral or advantageous to tumor growth or evolution but it is likely that these effects are cancer type specific. Although this has yet to be tested, it has been suggested that APOBECs provide a ‘just right’ amount of mutagenesis for tumor diversity, meaning that too little diversity results in the inability of a tumor to adapt to selective pressures but in contrast, too much can be detrimental due to genomic instability (57). For instance, A3 activity has been observed to be a contributing factor in branched evolution as well as the accumulation of subclonal mutations in breast cancer, esophageal squamous cell carcinomas, lung adenocarcinomas, and head and neck squamous cell carcinoma (48,58–60). Conversely, there is also evidence suggesting that A3s can generate ‘driver’ mutations that are associated with human papillomavirus (HPV)-associated cancers (61). Further studies on the outcomes of A3 deamination activity in tumorigenesis is needed.
Despite the role of A3s in cancer being a relatively new topic of research, the role of AID in cancer had already been discovered years prior. In the early 1980’s researchers had already found that genomic rearrangements such as translocations were present in many B-cell lymphomas, specifically between an immunoglobulin (Ig) locus and an oncogene. A number of different oncogenes including Bcl-2, Bcl-6, c-myc have been characterized in B-cell malignancies (62–64). Analysis of breakpoints of c-myc/IgH breakpoints revealed that most of the translocations involved the immunoglobulin switch region, a long, repetitive sequence required for CSR. The recombination between two switch regions in the immunoglobulin gene locus allows a B cell to switch from IgM to a more specialized immunoglobulin, e.g. IgG. This is initiated by excision of AID-catalyzed uracils that lead to dsDNA breaks and enable recombination (65). The involvement of AID was confirmed when translocations were not observed in the absence of AID in vivo even when artificial breaks were introduced to recapitulate the c-myc/IgH translocation (66,67).
Since the discovery of APOBEC-associated mutational signatures, the major focus has been trying to determine which A3s are active during cancer as well as investigate their biological effects. This has been a focus of research because the predominant in silico studies have only characterized mutation signatures retroactively or used RNA-seq data from databases like The Cancer Genome Atlas (TCGA) to demonstrate the presence of mRNA, but not catalytically active protein. Additionally, A3 protein levels may differ depending on the type or grade of cancer, which may or may not affect the number and outcome of A3-induced mutations. Determining which A3s are responsible for the SBS2 and SBS13 mutation signatures has been hindered in part because many of the studies use comparisons of normal and tumor tissues and there is evidence of some tumor samples being a mixed population with immune cells that infiltrated the tumor microenvironment (35). Since A3s are also highly expressed in immune cells, the analysis of their mRNA expression in tumor samples needs to be corroborated by other evidence of their involvement in somatic mutagenesis (22,24). Most of the studies revolved around A3B since it was found to be highly expressed in a number of cancers including breast, ovarian, cervical, lung, head and neck and bladder (47,68). It was suspected to play a major role in A3 mutagenesis as there was evidence demonstrating the correlation between A3B expression and A3 mutation signatures unlike some of the other A3s (47). Conversely, one study found that A3B expression could occur in breast tumors in the absence of an A3 mutational footprint, but A3A expression only occurred in breast tumors that had an A3 mutational footprint suggesting that A3A expression is more indicative of APOBEC deamination (69). As previously described, APOBECs deaminate cytosines within a preferred nucleotide sequence context and in the case of A3B it is 5'A/GTCA (where underlined C is mutated) (47,54,57,70). A3B has also been demonstrated to be a biomarker of poor prognosis for estrogen receptor positive breast cancers, again strongly suggesting that A3B induced mutations contribute to breast cancer progression (71). However, because A3B-null cancers still have a bias of 5'TC cytosine mutations, it meant that there must be at least one other APOBEC family member contributing to the overall mutation load (48,72). Studies have identified mutation footprints of 5'TTCA or 5'CTCA in tumors, which are the preferred context for A3A-catalyzed deaminations, but despite this, a corresponding upregulation of A3A mRNA has not always been detected (73,74).
Nonetheless, A3A can cause genomic damage in cell and tissue experiments. A3A been suggested to be the most active deaminase as it has been shown to translocate to the nucleus and induce dsDNA breaks that can lead to cell-cycle arrest and death (44,46,70). Due to the inability of some studies to find a correlation between upregulation of A3A mRNA and its mutational footprints, it is difficult to determine if A3A-induced mutagenesis is an ongoing event. It may just be more stable than the other A3s and therefore does not require high amounts of upregulation to see activity (69). Several studies propose that A3A expression is an early event that is later shut off because long exposure to A3A-catalyzed deaminations could be detrimental to cells rather than providing a selective advantage (44,46,54). A study has provided evidence that A3 activity occurs episodically and is difficult to pinpoint specific times since A3s appear to go through periods of inactivity (52). It is unknown what controls the episodic nature of A3s but viral infections and other oncogenic events may result in transient upregulation. Despite the high activity of A3A, there is emerging evidence suggesting a deamination independent role of A3A in tumorigenesis. One study did not find a correlation with mutations and A3A expression in pancreatic cancer and suggested that A3A expression without deamination when in combination with a KRAS mutation or alterations in p53 was sufficient in causing chromosomal instability (CIN), which is a hallmark of cancer (75). Additionally, another study demonstrated that catalytically active A3A can drive tumorigenesis in two murine systems where low expressing A3A enhanced polyp formation in an adenomatous polyposis coli multiple intestinal neoplasia murine model for colon cancer. High expressing A3A resulted in elevated frequencies of hepatocellular carcinoma in a tp53-depleted system. Although A3 mutation signatures were detected in both systems, no recurring driver mutations were evident in either system (76). This suggests that A3s may have a possible deamination independent mechanism when in combination with additional genetic alternations that result in CIN. The role of a deamination independent mechanisms remains to be characterized as the mechanistic basis is not yet understood (76). A3A also has RNA editing capabilities and in one study, researchers were able to detect mutation signatures in RNA in the absence of corresponding DNA mutations and this correlated with A3A but not A3B expression (73). Detection of RNA editing by A3A may be a way of measuring ongoing A3A activity as these mutations are not inherited in the genome and are transient (73). However, the impact of RNA editing by A3A in cancer remains to be determined.
It has become apparent that one of the main caveats of past studies was not only relying on mRNA expression, but also studying the A3 enzymes in isolation. Petljak et al. used breast cancer cell lines that had the ability to express both A3B and A3A and made knockout cell lines to only express A3B or A3A (74). Interestingly, although in vitro deamination assays using cell lysates demonstrated that A3B had more activity than A3A, when the somatic mutations were analyzed, the A3A preferred 5'YTCA signature was more prominent that the A3B preferred 5'RTCA signature and deletion of A3A rather than A3B more significantly reduced SBS2 and SBS13 mutations in the tested cell lines (74). Further, upon A3B deletion, there appeared to be an increase in A3A activity in some cell lines that correlated with increased A3A protein levels/deamination activity, suggesting some type of related transcriptional regulation of A3A and A3B (74). Overall, the authors concluded that A3A is the main driver of SBS2 and SBS13 mutations in breast cancer and B-cell leukemia cell lines.
Despite A3H having seven haplotypes, only A3H hap I has been demonstrated to be able to induce mutations in breast and lung cancer cells in A3B-null cancers despite being far less stable and antiviral compared to the other haplotypes (48,77,78). This is surprising as there is evolutionary evidence suggesting that the unstable A3H hap I was the result of one of two independent polymorphic events that lead to increased ubiquitination and degradation, resulting in reduced antiviral activity (2,77). The majority of the human population have less functional A3H alleles like A3H hap I, whereas the stably expressed proteins, like A3H hap II, are primarily found in African populations perhaps due to ongoing pathogenic pressure to maintain activity (77). This suggests the loss of activity may be an evolutionary protective measure against off-target deaminations. In one study, an A3H hap I K121E single nucleotide polymorphism (SNP) was discovered to correlate with lung cancer (79). Surprisingly, the SNP resulted in active site disruption and destabilization of the RNA-mediated dimer interface (80). This unexpected result suggests that A3H hap I induced mutations in lung cancer may be too mutagenic and lead to death of tumor cells, suggesting that A3H hap I expression in lung cancer is detrimental for the tumor (80). Conversely, there has been another SNP that was identified that had a R18L substitution that had deleterious effects on both the structure and function of the protein and potentially interfered with transcriptional regulation but it was suggested to be associated with lowered risk of lung cancer (81). Further studies are warranted to determine the roles of SNPs of A3H and their association with lung cancer.
Computational analysis identified the potential role of A1 to be a genomic mutator. Evidence of a mutational signature correlating with the expression of A1 was found in esophageal adenocarcinomas (82). These data suggested that aberrant A1 expression outside of intestinal epithelial cells could lead to undesired somatic mutations and potentially cellular transformation (82). Mutator assays in both bacterial and human cells have also demonstrated its ability to induce mutations in DNA as well as produce a mutator phenotype (82,83). A1 mRNA editing targets the 3'UTRs of transcripts (84). This indicates that both RNA and DNA can be deaminated by A1 and suggests that A1 can both deregulate mRNAs as well as hypermutate cancer genomes. However, measurement of γH2AX foci as a marker for dsDNA breaks or slowed replication in the presence of A1 showed that it can only cause a low-level of DNA damage (85–87). Although A1 has similar specific activity to A3H hap I and A3A on naked ssDNA, biochemical studies showed that A1, in contrast to A3H hap I and A3A, was unable to compete with replication protein A (RPA) for ssDNA (85). Thus, these data suggested how the ssDNA conditions in cells can modulate APOBEC-induced damage.
Replication protein A
RPA is a ssDNA binding protein that is essential for all aspects of DNA metabolism including DNA replication, transcription, recombination, DNA damage checkpoints and repair pathways such as base excision, nucleotide excision, and double-stranded break (DSB) repair (88,89). Its main role is to protect it from unwanted nuclease activity, secondary structure formation and to promote DNA processing (88,89). RPA interacts with numerous proteins involved in these pathways and ensures the proper proteins are able to access the DNA while still protecting and maintaining the integrity of the DNA (90–93).
Human RPA is a hetero-trimer consisting of RPA70, RPA32 and RPA14 (70, 32 and 14 kDa, respectively) (94) (Figure 1F, G). All three subunits contain at least one DNA-binding domain (DBD) designated from A-F and have oligonucleotide/oligosaccharide binding (OB) folds (Figure 1F, G). RPA70 contains four OB-folds where the DBD-A and DBD-B central folds are the DNA-binding core and are necessary for high affinity binding (89). DBD-F is at the N-terminus and is required for binding partially duplex DNA and checkpoint activation through phosphorylation. DBD-C is located at the C-terminus and has roles in DNA interaction, trimer formation and DNA damage recognition (Figure 1F, G). RPA32 contains a DBD-D fold, an N-terminal phosphorylation domain to signal DNA damage and modulate activity, and a C-terminal winged-helix (WH) domain that is primarily involved in protein-protein interactions (Figure 1F, G). RPA14 only has a DBD-E domain and interacts weakly with DNA but is required for complex formation (Figure 1F-G) (95). RPA is very flexible due to its modular structure and can adopt multiple conformations with varying modes of interaction with DNA nucleotides (nt): a low affinity mode (binds ∼8 nt, Kd ∼100 nM), a moderate mode (binds 12–23 nt, Kd ∼ 5 nM) and high affinity mode (binds 30 nt, Kd ∼ 0.05 nM) (96).
DISCUSSION.
INTERSECTIONS OF RPA AND A3 ENZYMES
DNA replication
Since APOBECs catalyze deaminations on ssDNA, these ‘off-target’ deamination events only occur when ssDNA intermediates are present such as during transcription, replication, or DSB repair (34,97–99). During DNA replication initiation, RPA is recruited by the helicase to bind on emerging ssDNA to stabilize ssDNA regions formed by origin unwinding and to help recruit proteins such as DNA polymerase alpha/primase to the nascent replication fork (100–102). A3-mediated mutations frequently occur during DNA replication and are most enriched in early-replicating, gene-dense and regions with active chromatin (98). Despite the protection provided by RPA during leading and lagging strand synthesis, there is a lagging-strand bias of clustered and sporadic A3-induced mutations (Figure 3A). Additionally, A3A-induced DNA damage has been demonstrated to lead to cell-cycle arrest at early S-phase further indicating that replication forks are susceptible to A3 activity and that A3-induced DNA damage has the potential to lead to stalled replication forks (44). During stalled replication, it is known that the helicase can become uncoupled from the polymerase where it can continue unwinding DNA without synthesis, generating long stretches of DNA and creating an abundance of substrate for A3s to deaminate (Figure 3B) (103). Altogether, this strongly suggests that there is a correlation between the number of mutations and the amount of time DNA remains single-stranded (97,104,105). Furthermore, A3A, A3B, and A3H can deaminate both methylated and non-methylated cytosines and study found that there were 2-fold higher number of mutations on unmethylated compared to methylated DNA, further illustrating that these off-target deaminations are occurring on newly synthesized DNA when DNA methyltransferases have not had the opportunity to methylate the DNA (104,106–108).
Figure 3.

Models of the RPA/APOBEC interface during DNA replication, transcription, and repair. (A) During normal replication, APOBEC enzymes compete with RPA primarily for access to the lagging strand where there is more ssDNA. (B) Replication stress can cause uncoupling of the DNA polymerase with helicase and can result in larger regions of ssDNA that are bound by RPA on both the leading and lagging strands. APOBECs can compete with the RPA that accumulates at these sites for access to ssDNA. (C) RPA can bind the non-template DNA strand of a stalled transcription bubble that has formed an R-loop. The R-loop is intentionally depicted larger than what would occur in a cell to illustrate potential protein–DNA interactions. Although there does not appear to be a major role of RPA during normal transcription, it does accumulate when transcription stalls or on R-loops. The APOBEC enzyme AID is more likely to deaminate during transcription than A3 enzymes, although some A3 activity during transcription has been demonstrated. (D) DNA repair resulting from DSB can lead to end-resection of DNA exposing ssDNA that is bound by RPA. APOBECs compete with RPA for access to this DNA, which may be facilitated if there is an RPA deficiency in the cell due to excessive firing of replication forks or high amounts of DNA damage. (E) BIR gives extended access to ssDNA. The migrating bubble replication during BIR creates ssDNA that persists and is thought to be a major source of A3-induced kataegis.
In addition to recruiting proteins necessary for DNA replication, RPA also functions as a sensor for ssDNA at stalled replication forks and DNA damage. Transformed cells with pre-existing genomic instability have been shown to experience higher levels of replication stress and fork stalling leading to more ssDNA exposure and opportunities for A3-mediated mutagenesis (97,99,103,104,109). There are several factors that can cause replication fork stalling and collapsing to prevent erroneous replication. Exposure to chemicals such as hydroxyurea (HU), which causes dNTP pool depletion, has been shown to enhance A3 activity (34). Reduced or depleted dNTPs can lead to reduced DNA synthesis due to a lack of substrate for DNA polymerases. DNA synthesis arrest may occur before nucleotide pools are exhausted to preserve dNTPs for DNA repair or prevent synthesis under these suboptimal conditions. Low levels of DNA polymerases have also been shown to affect the formation of mutations. It has been demonstrated in yeast that low levels of DNA polymerase causes replication stress, which can result in stalled replication forks that are prone to breakage or uncoupling of leading- and lagging-strand synthesis (110). This leads to high levels of ssDNA, suggesting that the high mutation rates observed arise from the instability of replication forks that have long stretches of ssDNA. Both DNA breaks and uncoupling of strand synthesis results in large ssDNA regions at the fork, which are susceptible to A3-induced mutagenesis.
Transcription
Unlike DNA replication and repair, there is little evidence suggesting the involvement of RPA during normal transcription. However, one study suggested that it may be involved in elongation and stabilizing the non-transcribed strand because Yeast Rfa1 (equivalent to RPA70 in humans) was found to associate with the RNA polymerase II (RNAPII) complex, interact with elongation factors Spt4 and Bur2, and in a proteomic screen in budding yeast be present at active genes transcribed by RNAPII and RNAPIII (111). Further studies are required to elucidate the role of RPA during normal transcription. A3 enzymes have not been found to significantly target ssDNA in transcription bubbles, in contrast to AID (97,104,112). Due to the known and specific role of AID deaminating cytosines in the immunoglobulin gene locus during transcription, the role of A3 deaminase activity during transcription was investigated when they were discovered to induce somatic mutagenesis (Figure 3C). In yeast cells, A3A can deaminate during transcription, but to such a lesser amount than during DNA replication that the authors of the study concluded this was not a source of A3-induced mutations (97). The reasons for this are not known, however in vitro studies with T7 transcription systems have raised some possibilities, an obvious caveat being that a phage RNA polymerase is not similar to a human transcription complex. A3s have been demonstrated to have the ability to deaminate the non-transcribed strand during an in vitro phage T7 transcription system (113). A3A and A3H were both able to deaminate during T7 RNA polymerase mediated transcription whereas A3B was unable (113). If we take into consideration that A3H hap I and A3B deamination activity decreases in the presence of RPA-saturated ssDNA, it is highly probable that in the tight confines of the transcription bubble their activities would be further reduced (113). In contrast, A3A would be the most probable candidate to deaminate during transcription, however it is unknown why this is not observed to be significant in human cells (97). Data from Brown et al. found that even A3A cannot deaminate ssDNA in a transcription bubble generated by T7 RNA polymerase if the polymerase is stalled (114). Rather their data suggest that A3s can only deaminate in R-loops behind the RNA polymerase and not actually during active transcription (114).
Stalled transcription can trigger the stress response signal that is driven by RPA and Ataxia-telangiectasia mutated and Rad3-related serine/threonine kinase (ATR) as they monitor elongation during transcription. This leads to the phosphorylation of Ser-15 of p53 in a replication-independent matter (115). In contrast to DNA polymerases, RNAPII is unaffected by DNA lesions but will stall at bulky DNA adducts such as thymidine dimers and cisplatin-induced crosslinks and trigger transcription-coupled repair (116). Stalling of RNAPII by blocking agents can trigger the stress response signal. Since ATM serine/threonine kinase (ATM) that is activated by DNA DSB is not involved in Ser-15 phosphorylation, it is unlikely that transcription stalling results in this type of damage (115). Transcription can also be impaired if the RNA transcript re-anneals to the template strand leading to formation of an R-loop. These RNA-DNA hybrids called R-loops are formed when the newly synthesized RNA transcript is transiently paired to the coding strand. The non-coding strand is looped out and remains single-stranded (Figure 3C). This can trigger a DNA damage response since this can impair transcription and DNA replication leading to replication stress and DSB formation (117). RNase H is required to degrade the RNA of the RNA-DNA hybrid in a sequence independent manner. Absence of RNase H during transcription-associated DSB results in stabilization of the DNA–RNA hybrids around the DSBs and prevents RPA recruitment whereas overexpression of RNase H results in the opposite where enhanced resection and recruitment of RPA is observed (118). RNase H1 activity on R-loops is enhanced by RPA. The loss of interaction between RPA and RNase H1 results in the inability to supress R-loops leading to genomic instability (119). Therefore RPA is also important for sensing R-loops and regulating RNase H1 activity, further building on its role for sensing DNA damage and replication stress. A3A, A3B and A3H hap I can all deaminate an in vitro R-loop suggesting that they are able to access the small segment of ∼8–20 nt ssDNA available albeit with lower activity than fully ssDNA (113). Further analysis suggested that the larger oligomeric state of A3B may hinder its ability to stabilize itself on an active transcription bubble (113). Despite this, R-loops can span 0.1–2 kb and so depending on the length of the R-loop, A3B may be able to compete with RPA for binding on bigger R-loops (120).
DNA repair
DNA repair processes are more likely to leave ssDNA exposed for longer periods than DNA replication enabling higher levels of deamination by A3 enzymes (Figure 3D-E). These higher levels of deamination are seen as clustered mutations called kataegis (meaning thundershowers in Greek), but the mechanism enabling this extended access to ssDNA was not initially known (121). Kataegis are clustered C-to-T and/or C-to-G mutations that are enriched at TCN motifs (N denotes any nucleotide) located on the same DNA strand (51). In addition, several studies found that A3 expression can cause γH2AX foci (80,122,123). These foci represent dsDNA breaks and/or slowed replication (86,87). Most studies have focused on the DSB formation, but the distribution of γH2AX foci due to DNA breakage or simply just slowed replication is not known. Nonetheless, when DSB are induced after uracil removal, usually by overexpressing A3A, several repair pathway options ensue (Figure 3D) (123). However, one type of repair that gives extended access to ssDNA is DSB repair break-induced replication (BIR) (Figure 3E) (124). BIR is used to repair DSB with only one repairable end, such as would occur at collapsed replication forks. The DNA synthesis during BIR is a migrating bubble where lagging strand synthesis is significantly delayed in comparison to leading strand synthesis (125). This accumulation of ssDNA behind the replication bubble is thought to be a major substrate for A3 enzymes, leading to kataegis (Figure 3E) (126,127).
In response to DNA damage, DNA checkpoints are activated to arrest cell cycle progression and enable repair. RPA is essential for a number of DNA repair pathways including base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), and DSB repair because it is needed for signalling damage and recruiting and positioning various repair factors (94). The N-terminal domain of RPA32 is phosphorylated in response to DNA damage by ATM, ATR and DNA-dependent protein kinase (DNA-PK). The amount of phosphorylation of RPA depends on the type of DNA damage where RPA32 S33 is primarily phosphorylated by ATR and S4/S8 and Thr21 by ATM and DNA-PK (128). This phosphorylation happens sequentially by first ATR at S33 followed by DNA-PK at S4/S8. During replication catastrophe caused by the absence of ATR and accumulation of ssDNA by HU, T21 is phosphorylated as well (129). DNA damaging agents that result in DSB only result in modest phosphorylation whereas UV, HU and camptothecin that result in generation of large amounts of ssDNA from fork stalling can induce hyperphosphorylation where the N-terminal domain of RPA32 is phosphorylated at five or more distinct sites, which serves as a marker for DNA damage (130). It has been suggested that the addition of negative charges through phosphorylation will generate electrostatic repulsive forces between hyperphosphorylated RPA32 and negatively charged ssDNA that will encourage dissociation to enable DNA repair (89,131). If this was the case, then it would mean that A3s would be able to gain easier access to ssDNA when RPA is phosphorylated because of its weakened interaction with ssDNA. However, this decreased ssDNA binding affinity of phosphorylated RPA for ssDNA was only observed for purine-rich substrates (132). In general, phosphorylated RPA has a decreased ability to bind and destabilize dsDNA (132–134).
Mechanisms of competitive binding of ssDNA
Although RPA binds very tightly to ssDNA, it must be displaced from ssDNA to allow access by downstream proteins during replication or repair processes. The rate and extent of RPA dissociation are dependent upon the concentration of free RPA (135,136). RPA can remain bound to ssDNA for hours at an infinite dilution limit but when there is an excess of free RPA present in solution, the protein exchanges between free and bound states (137). This mechanism ensures that RPA remains bound and protects ssDNA while still maintaining the flexibility to be displaced when necessary. Interestingly, one study observed that a small subpopulation of RPA remained resistant to facilitated exchange, which could suggest that different binding modes exist when RPA binds DNA and that it may be dependent on the number of DBDs interacting with ssDNA (135,138). Another study proposed a different model suggesting that all DBDs transiently bind ssDNA with high affinities but differ in their dynamic interactions on ssDNA therefore enabling other proteins to compete with DBDs for binding (138). Adjacent RPAs can also contact each other via the DBD-A of one RPA and DBD-E of the other suggesting that this interaction could stabilize a conformation that linearizes ssDNA (139). However, this interaction is weak and occurs after ssDNA binding since no cooperative binding was observed. Based on this tight interaction on ssDNA and protein-protein interaction between adjacent RPAs, the likelihood that facilitated exchange is sufficient to provide enough access to ssDNA for repair proteins is low. A mechanism through phosphorylation was proposed as a means to facilitate more exchange. The phosphomimetic mutant, Rfa1 S178D (equivalent to RPA70 S180 in humans), was observed to have a weakened interaction with ssDNA (mediated by DBD-A and DBD-B) but enhanced affinity with adjacent RPA (mediated by DBD-A and DBD-E). This shift in binding association between ssDNA and adjacent RPA molecules suggests that is may be a switch mechanism that RPA allows for displacement and exchange with other ssDNA-binding proteins. (139).
In addition to facilitated exchange between RPA proteins, RPA has been demonstrated to diffuse rapidly on ssDNA while remaining bound to invade and melt hairpin structures as well as coordinate the assembly and disassembly of other proteins (140). This mechanism of diffusion may involve sequential local dissociation and rebinding of two or more of its OB-folds since bound RPA can undergo a concentration dependent exchange with free RPA. This ability to diffuse on ssDNA enables RPA to melt DNA hairpins (140). However, another study has proposed that at the replication fork RPA is unable to efficiently bind ssDNA that forms strong hairpins, suggesting that in some situations RPA cannot melt DNA hairpins (56). This has implications for A3 deamination of structured ssDNA at replication forks.
Similar to RPA, A3s use facilitated diffusion as a means of moving on DNA to search for a specific target motif since they do not use an energy source (12,141,142) (Figure 4). Electrostatic interactions with the DNA enables enzymes to undergo Brownian motion driven diffusion which facilitates the enzyme binding longer to the DNA rather than being released into the bulk solution (Figure 4A) (12,141,142). Diffusion can be further classified depending on whether the A3s diffuse along the negatively charged phosphate backbone; termed sliding, diffuse along the DNA while intermittently making contact with the phosphate backbone; termed jumping or hopping, or move through a doubly bound state; termed intersegmental transfer (12,141–143) (Figure 4B-D). The ability for the enzyme to undergo these types of motions increases efficiency as they are able to processively catalyze deaminations in a single enzyme-substrate encounter. Sliding movements encompass <20 nucleotides (nt) of DNA and are essential for locating the deamination motif. Each A3 studied to date is processive except A3A (144). The other A3 enzymes each have a unique processive mechanism with some relying primarily on sliding and jumping (A3G, A3H), jumping (A3F), or are very efficient at intersegmental transfer in addition to sliding and jumping (A3B) (145–147). Intersegmental transfer is movement of enzymes on DNA by an intermediate doubly bound state to a distal segment of the same or a different DNA (141–143). With intersegmental transfer enzymes are more likely to bind a different DNA than the same DNA as the local DNA concentration increases (145). As a result, this mechanism can be processive or non-processive depending on the conditions.
Figure 4.

Mechanisms of ssDNA scanning by facilitated diffusion. (A) Electrostatic interactions between APOBECs and the negatively charged region (blue shaded region) of ssDNA are essential for facilitated diffusion. (B) One-dimensional sliding of the enzyme enables short-range localized searching (≤20 nt). (C) Three-dimensional scanning of the enzyme by jumping enables larger translocations through micro-dissociations and reassociations on ssDNA without diffusion into the bulk solution, but lacks a local search process. (D) Three-dimensional scanning by intersegmental transfer enables even larger translocations mediated through a doubly bound state. Enzymes with two binding domains can simultaneously bind two different regions of ssDNA before dissociating from one region to move onto the other.
Since it was initially thought that A3s could not displace RPA, the original theory was that A3s only had limited access to ssDNA and could only deaminate the transient ssDNA gaps not bound by RPA that may be present normally or during RPA depletion (148,149). While several studies have demonstrated that A3 processivity and staying bound to viral DNA is important to increase deamination efficiency, it is not the main driving factor for A3 activity on genomic ssDNA (144). Rather, it was found that the ability to undergo rapid cycling between ssDNA substrates, either by non-processive cycling or intersegmental transfer, increased the deamination activity of A3s on RPA coated ssDNA and ability to induce γH2AX foci (85,113) (Figure 5). A1, A3B, and A3H have been shown to be able to cycle between ssDNA substrates and maintain processivity whereas A3A can only cycle on and off rapidly (113). In contrast, A3G, which is the most processive A3 enzyme, is unable to cycle as frequently as the others (113). A3G is primarily a cytoplasmic enzyme, although one study has observed A3G in the nucleus and others have correlated genomic instability to A3G expression (36,150). Nonetheless, A3G is still a useful control for studying the role of processivity and access to ssDNA in the presence of RPA. Biochemical assays performed in the presence of RPA showed that while processivity decreased approximately 2-fold for A3B and A3H hap I, the specific activities of A3A, A3B and A3H hap I were fairly similar. In contrast, A1 and A3G exhibited 7- and 10- fold decreases in specific activity (85,113). The reasoning for this is their inability to cycle as effectively prevents them from displacing RPA (Figure 5). For example, A3G binds ssDNA as a monomer and then oligomerizes, which stabilizes its binding and it can remain bound for 5–10 min (151,152). Without oligomerization, A3G cannot form a stable enough association to remain on the DNA and presumably RPA blocks that oligomerization, even if A3G can initially compete with RPA to bind the DNA. A1, which is a multimer off or on ssDNA, may not have enough room to stay bound (85). This suggests that the combination of the constant on and off of A3A, A3B and A3H hap I and ability to bind without further oligomerization can actively displace RPA and enable ssDNA binding, with this cycle repeating itself. A3s must be able to bind to the ssDNA quickly before a free RPA can bind as RPA has a very fast kon = 1.1 M−1 s−1 (138). Ultimately, the data suggests that the faster the A3 on/off cycling, the better the competition for ssDNA with RPA during facilitated exchange (Figure 5). It is likely due to rapid cycling that A3A is not found to colocalize with RPA (153). A3s other than A3A have not been directly studied for colocalization with RPA, but it is likely that they also do not colocalize with RPA. Even though A3B and A3H hap I are processive enzymes, they use intersegmental transfer to different degrees, which means that the enzymes can easily move from one DNA segment to another distal DNA segment and that high local concentrations of DNA promote enzyme dissociation and rebinding at another location (145). As a result, the A3 enzyme dissociation rate on ssDNA is faster than RPA (138,152).
Figure 5.

Model of competitive binding of APOBECs and RPA. The tight binding of RPA on ssDNA acts as a roadblock, limiting APOBECs from catalyzing processive deaminations. (Top panel) This effectively prevents competitive exchange of RPA with APOBECs that can only slide along the DNA backbone, leading to lowered APOBEC specific activity. (Bottom panel) Bound RPA can undergo exchange with free RPA through facilitated dissociation allowing an opportunity for APOBECs to bind. Three-dimensional rapid cycling on/off ssDNA enables APOBECs to compete with RPA for ssDNA as both proteins exchange between bound and free states. The displacement of RPA enables deamination activity from A3s that do not require processive sliding, but instead locate cytosines by cycling on/off the ssDNA.
Although the ability of A3s to compete with RPA for linear ssDNA has been established, it has also become apparent that A3A-induced mutations frequently occur in the loops of hairpin structures that can form from ssDNA during replication (55). Although A3A has a higher deamination rate in vitro on hairpin loops than A1, A3B, or A3H hap I, when comparing A3A deamination on hairpin loops and linear ssDNA in the absence of RPA, the difference in specific activity is marginal to none, depending on the study, and cannot account for observations in cells (55,85,114). Since RPA is proposed to be unable to bind ssDNA that can form strong hairpins during replication, different groups have proposed that A3A has a higher rate of deamination of hairpin loops simply because there is no competition with RPA (56,114). Brown et al., found that RPA bound hairpin loops ∼7.5- (0.5 RPA:1 DNA) to 3.6- (2 RPA:1DNA) fold less than linear ssDNA (114). Often these loops have small ssDNA regions, e.g. 4–6 nt, and a single RPA trimer binds 8–30 nt of ssDNA, suggesting that RPA is excluded or is less able to bind these regions (55,56,96,114). However, Brown et al. did find a 3.5-fold decrease of A3A activity on hairpin loops in the presence of RPA, compared to 6.3-fold on linear ssDNA (114). In contrast, Wong et al. found that there was no in vitro inhibition of A3A specific activity on linear ssDNA in the presence of RPA (85). Wong et al. used a high salt buffer which makes the in vitro DNA binding ability of proteins more competitive than Brown et al.’s reaction conditions which had no salt. However, Wong et al. did not test inhibition of A3A by RPA on hairpin loops, precluding any direct comparisons (85,114). In addition to the different reaction conditions on linear ssDNA between these two studies, the different sequences of the DNA substrates are potential reasons for the different results. More studies using the same DNA substrates and reaction conditions are needed to resolve these different in vitro observations. Overall, these studies highlight that more direct experiments need to be carried out to determine the effect of RPA on A3 deamination activity in cells, and the extent to which RPA localizes to hairpin regions as cellular conditions with multiple proteins can be difficult to mimic in vitro.
A similar protective mechanism by an ssDNA binding protein is observed in HIV-1 replication. In HIV-1, nucleocapsid, a nucleic acid chaperone protein, rapidly binds and unbinds the RNA or ssDNA to remodel secondary structure and protect ssDNA. A3s must also compete with nucleocapsid to access viral (-) ssDNA. However, unlike RPA, nucleocapsid binds non-specifically at every 5–7 nt on RNA or DNA only at high concentrations (154). Unlike genomic DNA deamination, cycling is not a required characteristic to deaminate HIV-1 (–) ssDNA and instead, studies have shown that processivity directly correlates to the number of mutations (146,155). The restrictive potential of the enzyme is determined by a balance of sliding mechanisms to perform local searches and jumping mechanisms to scan larger distances. In the case of A3F which lacks the sliding mechanism, but can jump, it is unable to perform localized searches and therefore induces less mutations than A3G which is capable of performing both scanning mechanisms (146). These data demonstrate how A3 activity is highly specialized. While A3G does not restrict any viruses that replicate in the nucleus or have dsDNA intermediates, A3B and A3A do restrict dsDNA viruses during DNA replication in the nucleus, highlighting how although the somatic mutagenesis during cancer is considered an off-target event, the involved enzymes are specialized to access this type of substrate (23,29,156,157).
The significance of APOBECs and involvement in somatic mutagenesis
RPA is essential for ssDNA protection during replication and is estimated to be at a 6- to 10-fold excess to deal with perturbations that result in fork stalling (129). The fact that cells will accumulate such high amounts of a specific protein highlights how crucial is RPA for maintaining genomic integrity. During replication fork stalling, RPA-coated ssDNA activates ATR which in turn will phosphorylate RPA to facilitate recruitment of repair factors. Additionally, ATR activates checkpoint signalling to supress dormant origin firing to limit the number of active forks and reduce potential sources of ssDNA (129). This is meant to keep RPA in sufficient supply to ensure the stalled forks remain stable. Cells with supra-physiological RPA levels have been demonstrated to sustain longer durations of checkpoint inhibition, replication stress and tolerated high quantities of ssDNA without fork breakage, suggesting that as long as there is a surplus of RPA the active or stalled replication forks remain stable (129). These data make RPA critical for managing the replication stress and recruiting the necessary proteins to protect the surrounding area, facilitating repair, and preventing catastrophic collapse of the replication fork. Additionally, fork breakage was also observed to be a sudden occurrence rather than a gradual accumulation of isolated events at active replicons (129).
Another competition for A3 enzymes, perhaps not for ssDNA per se, but for activity is with DNA repair enzymes. This is because the WH domain of RPA is known to bind multiple proteins involved in DNA repair and genome maintenance, such as SMARCAL1, TIPIN, ETAA1, RAD52, XPA and UNG (90–93). The binding of RPA to UNG enables UNG to efficiently excise uracils from RPA-bound ssDNA (158). The normal function of this interaction is to remove any dUMPs incorporated by the DNA polymerases (159). Thus, each uracil the A3s catalyze may be easily and subsequently removed. This could mean that cancers would be biased with SBS13 (C→G mutations), which results from insertion of a C opposite abasic sites by Rev1 during replication (74,121). However, there are still C→T mutations (SBS2) present in cancer genomes, which result from using uracil as a template during replication. It appears that somehow A3 enzymes induce enough uracils to overwhelm DNA repair. A similar conundrum was recently solved for AID where it was found that a protein of previously unknown function, Fam72A, induces degradation of UNG in order to promote AID-induced mutations during SHM (160,161). Although Fam72A is expressed in cancer cells, it has no associated function as of yet (161). This overwhelming of DNA repair with excessive uracils could occur during BIR, which is thought to enable kataegis (126,127). Although RPA would still be involved in binding these ssDNA intermediates, the WH domain of RPA may be bound by other proteins such as RAD52, excluding UNG. Although A3 induced mutations can occur during lagging strand synthesis, considering these factors, it is likely that the original number of deaminations on the ssDNA far exceed the mutation frequency. Thus, the timing of A3 catalyzed deaminations and the type of ssDNA substrate appear to be very important in determining the mutation frequency and these types of analyses need to be an area of future research.
Another condition that needs to be considered is the level of free RPA in the cells. If excessive ssDNA accumulates due to replication fork stalling or excessive replication fork firing, the free RPA may become depleted if it is all bound to the ssDNA. Depletion of RPA can also leave significant amounts of ssDNA exposed and if combined with A3s, the normally less active enzymes can have the perfect opportunity to deaminate ssDNA that is usually not easily accessible. Assuming A3s are present during this RPA depleted state, the exposed ssDNA would be subjected to high amounts of deamination and would allow for A3s to undergo all possible processive scanning mechanisms as it would no longer be restricted to rapid cycling on/off ssDNA. This may tip the balance of the effect of A3-induced mutations to catastrophe, killing the cancer cell. This scenario also depends on the availability of the nuclease Mre11 since in the absence of RPA, the ssDNA is sensitive to Mre11-dependent degradation, which would remove any A3 substrate, but also likely lead to elevated levels of cell death in itself (162). However, the likelihood of this happening is very low since RPA depletion only occurs when ATR signalling is inhibited and would likely cause cell death due to the high number of mutations from unrepaired deaminations, if the cell was not forced prematurely to enter into mitosis (129,163). Accordingly, ATR defective cancers are rare (164). ATM is a similar checkpoint kinase that plays a pivotal role in the detection of DSB, signalling, and regulating DSB resection to activate homologous recombination (HR) (165). ATM is also activated when there is an excess of single-stranded breaks (SSB) generated during BER since ligation of SSB is slower than DNA incision by APE. By delaying S-phase entry, ATM ensures SSB intermediates generated by BER have sufficient time for repair, thereby preventing DSB formation (166). In contrast to ATR, ATM defective cancers are more common as they are resistant to apoptosis and despite having defective homologous recombination, can rely on the redundant DSB repair pathway and non-homologous end-joining (NHEJ) for repair (167). Unlike HR, NHEJ can be activated anytime to repair DSB and therefore does not appear to pose the same risk since RPA depletion occurs only during replication stress. Under these conditions, ssDNA is limited and RPA concentration is high and therefore A3s are reliant on their ability to compete with RPA. There are effects of other proteins on RPA, such as RECQ1, a helicase that can suppress firing of dormant origins of replication to ensure a normal replication rate in cells (168). In the presence of RECQ1 defects, which have been found in tumors, excess replication fork firing results in sequestration of RPA on the nascent ssDNA (168). The sequestration of RPA also inhibits NER, further contributing to genomic instability (162). Altogether the genetic background of a cancer cell also determines the effect of A3s and this has not been analyzed in previous studies. Some cancers, such as bladder or breast cancer, have a slight bias for more SBS13 than SBS2 mutations and it would be interesting to determine if this bias correlates with the availability of RPA when compared to other cancers that have equal amounts of SBS2 and SBS13 mutations (50).
One caveat to consider in the RPA depletion theory and how it relates to A3 deamination activity is that the scarcity of RPA also means that there is a higher propensity for secondary structure formation. RPA has strand melting abilities that resolve DNA secondary structure, perhaps not during replication, but during processes such as BIR or stalled replication that create tracts of ssDNA that persist for longer times (Figure 3E) (103,124,169). Biochemical studies have demonstrated that RPA acts as a physical roadblock for certain A3s and prevents A3s from 1D sliding thereby reducing processivity and specific activity (85,113,114). Although RPA depletion would appear to circumvent this issue and allow enzymes such as A1 that are less able to compete with RPA to induce similar amounts of damage as A3A, A3B or A3H hap I, only A3A has the ability deaminate hairpin loops efficiently, although A1, A3B, and A3H hap I can to a lesser degree (55,85,113). If there is enough secondary structure to block A3s other than A3A remains to be determined. What is also not yet known is if the cell can recover from RPA depletion in the presence APOBECs and what effect this has on the cell post recovery. Depending on the amount of APOBEC-induced mutagenesis, there can be different results. Fewer deaminations by less active A3s may have a lesser effect if the cell is able to recover. Alternatively, a more active enzyme such as A3A which has a rapid on/off rate and can deaminate large stretches of DNA may lead to replication catastrophe or induce synthetically lethal mutations. Due to the uncertainty of APOBEC induced mutagenesis and dependence on the cellular state, the effect of their mutations result in a spectrum of beneficial or detrimental outcomes for the host (Figure 6). The amount of APOBEC-induced DNA damage strongly correlates with their ability to displace RPA as this is a determinant for deamination activity. Further biochemical characterization can provide insights as to how A3 enzymes would manage an RPA depletion scenario. Overall, RPA depletion appears to be a rare event, seemingly more often encountered in laboratory experiments than in vivo, suggesting that for the most part, A3s will need to compete for ssDNA with RPA (149,170).
Figure 6.
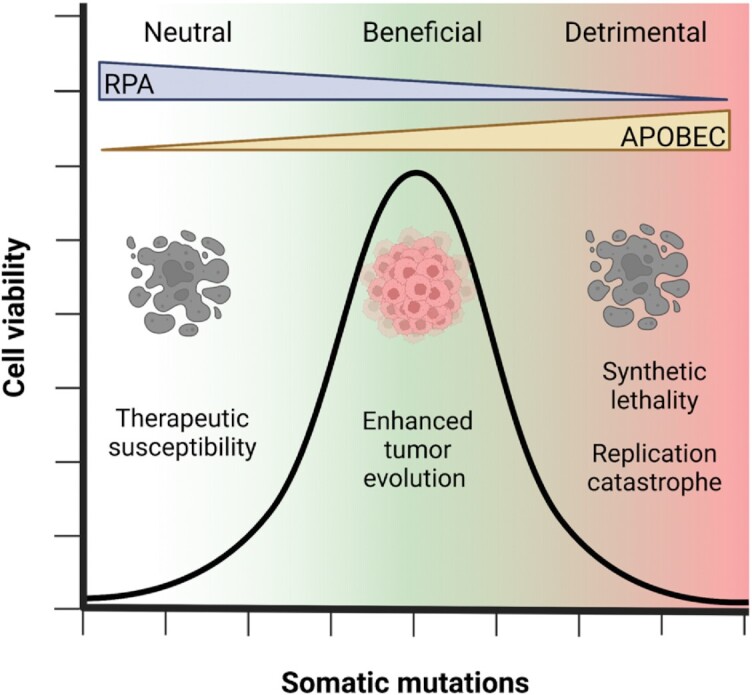
Spectrum of APOBEC induced mutagenesis. The effects of APOBEC induced mutagenesis is dependent on the cellular state and can be either neutral, beneficial or detrimental to the host. APOBEC-induced DNA damage strongly correlates with their ability to displace RPA and the amount of RPA available to protect ssDNA. Low levels of APOBEC-induced mutagenesis may not have an effect in the development of drug resistance in cancer cells and as a result, cancer cells are susceptible to therapies. An optimal amount of APOBEC-induced mutagenesis may provide genomic diversity to cancer cells that can enhance tumor evolution and growth. High levels of APOBEC-induced mutagenesis can lead to cell death as a result of synthetic lethality and replication catastrophe due to RPA exhaustion.
In the context of chemotherapy, RPA depletion and DNA repair can be beneficial. ATR inhibitors (ATRi) are in development but there are concerns of possible severe side effects in highly proliferative normal tissues since it is an essential gene. This issue could be circumvented by using ATRi only in cancers expressing high levels of A3s since they exhibit a synthetic lethal relationship with ATRi (171). A3s induce a unique type of replication stress where the resulting abasic sites from UNG are susceptible to ATRi which leads to greater accumulation of ssDNA substrate for A3s, ultimately leading to RPA depletion and cell death (Figure 6). Another example of where A3-induced replication stress is beneficial is during the use of platinum based chemotherapeutic drugs such as cisplatin that induces interstrand crosslinks (ICL). Although overexpression of A3s in several cancer types have been associated with poor clinical outcomes and drug resistance, studies have investigated the combinatorial effect of A3s and DNA repair pathways and observed that A3 deamination activity can confer sensitivity to ICL agents (172,173). BER and MMR repair of A3-catalyzed uracils physically inhibits NER and HR from repairing ICLs and leads to cell death (174). This approach of using cancers that are overexpressing A3s to exploit synthetic lethal interactions is a way to ensure that A3-induced mutations do not enhance tumor genetic diversity and growth.
CONCLUSIONS
Since the discovery that A3 enzymes are involved in somatic mutagenesis during cancer, significant knowledge has been acquired on the A3-induced mutation types, their distribution across the genome, and the different cancers affected. However, since most of the studies in the A3 cancer field are in silico analysis of cancer genomes or animal studies, the mechanistic details on how they access DNA are lacking. The sources of ssDNA are known, but the details of how A3s intersect with RPA and DNA repair enzymes are not as well studied (Figure 6). The DNA repair field has several methodologies to study the efficiency of specific DNA repair pathways. Using these methods without or with the addition of A3s would enable A3 researchers to test directly how A3s effect different DNA repair outcomes and the effect of the DNA structure, e.g. overhangs, on A3 deamination activity. Merging existing DNA repair biochemical assays with purified A3 enzymes and A3 biochemical knowledge would be useful to answer many of the questions on how uracils become heritable mutations. Although there has been more of a clinical focus that has resulted in more broad-based genomic analyses, the mechanistic details of how mutations arise can be highly useful for designing potential therapies. The synthetic lethality of A3 activity with ATRi is one example, and there may be many more if we understood their dynamics on ssDNA with the multiple other proteins that gather at replication forks and sites of repair.
DATA AVAILABILITY
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
ACKNOWLEDGEMENTS
We thank Milaid Granadillo Rodríguez for critical comments on the manuscript.
Contributor Information
Lai Wong, University of Saskatchewan, College of Medicine, Department of Biochemistry, Microbiology, and Immunology, Saskatoon, Saskatchewan, S7N 5E5, Canada.
Alina Sami, University of Saskatchewan, College of Medicine, Department of Biochemistry, Microbiology, and Immunology, Saskatoon, Saskatchewan, S7N 5E5, Canada.
Linda Chelico, University of Saskatchewan, College of Medicine, Department of Biochemistry, Microbiology, and Immunology, Saskatoon, Saskatchewan, S7N 5E5, Canada.
FUNDING
Canadian Institutes of Health Research [PJT#159560 to L.C.]; College of Medicine Graduate Award (to L.W.); National Sciences and Engineering Research Council of Canada Undergraduate Student Research Award (to A.S.); College of Medicine Summer Biomedical Award (to A.S.). The open access publication charge for this paper has been waived by Oxford University Press – NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement. None declared.
REFERENCES
- 1. Rogozin I.B., Iyer L.M., Liang L., Glazko G.V., Liston V.G., Pavlov Y.I., Aravind L., Pancer Z.. Evolution and diversification of lamprey antigen receptors: evidence for involvement of an AID-APOBEC family cytosine deaminase. Nat. Immunol. 2007; 8:647–656. [DOI] [PubMed] [Google Scholar]
- 2. Chesarino N.M., Emerman M.. Polymorphisms in human APOBEC3H differentially regulate ubiquitination and antiviral activity. Viruses. 2020; 12:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Salter J.D., Smith H.C.. Modeling the embrace of a mutator: APOBEC selection of nucleic acid ligands. Trends Biomed. Sci. 2018; 43:606–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jarmuz A., Chester A., Bayliss J., Gisbourne J., Dunham I., Scott J., Navaratnam N.. An anthropoid-specific locus of orphan c to u RNA-editing enzymes on chromosome 22. Genomics. 2002; 79:285–296. [DOI] [PubMed] [Google Scholar]
- 5. LaRue R.S., Jonsson S.R., Silverstein K.A., Lajoie M., Bertrand D., El-Mabrouk N., Hotzel I., Andresdottir V., Smith T.P., Harris R.S.. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol. Biol. 2008; 9:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maiti A., Myint W., Kanai T., Delviks-Frankenberry K., Sierra Rodriguez C., Pathak V.K., Schiffer C.A., Matsuo H.. Crystal structure of the catalytic domain of HIV-1 restriction factor APOBEC3G in complex with ssDNA. Nat. Commun. 2018; 9:2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kouno T., Silvas T.V., Hilbert B.J., Shandilya S.M.D., Bohn M.F., Kelch B.A., Royer W.E., Somasundaran M., Kurt Yilmaz N., Matsuo H.et al.. Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun. 2017; 8:15024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shi K., Carpenter M.A., Banerjee S., Shaban N.M., Kurahashi K., Salamango D.J., McCann J.L., Starrett G.J., Duffy J.V., Demir O.et al.. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 2017; 24:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Navarro F., Bollman B., Chen H., Konig R., Yu Q., Chiles K., Landau N.R.. Complementary function of the two catalytic domains of APOBEC3G. Virology. 2005; 333:374–386. [DOI] [PubMed] [Google Scholar]
- 10. Feng Y., Chelico L.. Intensity of deoxycytidine deamination of HIV-1 proviral DNA by the retroviral restriction factor APOBEC3G is mediated by the noncatalytic domain. J. Biol. Chem. 2011; 286:11415–11426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chelico L., Prochnow C., Erie D.A., Chen X.S., Goodman M.F.. Structural model for deoxycytidine deamination mechanisms of the HIV-1 inactivation enzyme APOBEC3G. J. Biol. Chem. 2010; 285:16195–16205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feng Y., Baig T.T., Love R.P., Chelico L.. Suppression of APOBEC3-mediated restriction of HIV-1 by vif. Front. Microbiol. 2014; 5:450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Conticello S.G. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008; 9:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shaban N.M., Shi K., Lauer K.V., Carpenter M.A., Richards C.M., Salamango D., Wang J., Lopresti M.W., Banerjee S., Levin-Klein R.et al.. The antiviral and cancer genomic DNA deaminase APOBEC3H is regulated by an RNA-Mediated dimerization mechanism. Mol. Cell. 2018; 69:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sawyer S.L., Emerman M., Malik H.S.. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004; 2:E275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salter J.D., Bennet R.P., Smith H.C.. The APOBEC protein family: united by structure, divergent in function. Trends Biomed. Sci. 2017; 41:578–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R.. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016; 533:420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen S.-H., Habib G., Yang C.-Y., Gu Z.-W., Lee B.R., Weng S.-A., Silberman S.R., Cai S.-J., Deslypere J.P., Rosseneu M.et al.. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science. 1987; 238:363–366. [DOI] [PubMed] [Google Scholar]
- 19. Peled J.U., Kuang F.L., Iglesias-Ussel M.D., Roa S., Kalis S.L., Goodman M.F., Scharff M.D.. The biochemistry of somatic hypermutation. Annu. Rev. Immunol. 2008; 26:481–511. [DOI] [PubMed] [Google Scholar]
- 20. Stavnezer J., Guikema J.E., Schrader C.E.. Mechanism and regulation of class switch recombination. Annu. Rev. Immunol. 2008; 26:261–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zan H., Casali P.. Regulation of aicda expression and AID activity. Autoimmunity. 2013; 46:83–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Refsland E.W., Stenglein M.D., Shindo K., Albin J.S., Brown W.L., Harris R.S.. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 2010; 38:4274–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cheng A.Z., Yockteng-Melgar J., Jarvis M.C., Malik-Soni N., Borozan I., Carpenter M.A., McCann J.L., Ebrahimi D., Shaban N.M., Marcon E.et al.. Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nat. Microbiol. 2019; 4:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koning F.A., Newman E.N., Kim E.Y., Kunstman K.J., Wolinsky S.M., Malim M.H.. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 2009; 83:9474–9485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sharma S., Patnaik S.K., Taggart R.T., Baysal B.E.. The double-domain cytidine deaminase APOBEC3G is a cellular site-specific RNA editing enzyme. Sci. Rep. 2016; 6:39100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sharma S., Patnaik S.K., Taggart R.T., Kannisto E.D., Enriquez S.M., Gollnick P., Baysal B.E.. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun. 2015; 6:6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gaba A., Flath B., Chelico L.. Examination of the APOBEC3 barrier to cross species transmission of primate lentiviruses. Viruses. 2021; 13:1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Desimmie B.A., Delviks-Frankenberrry K.A., Burdick R.C., Qi D., Izumi T., Pathak V.K.. Multiple APOBEC3 restriction factors for HIV-1 and one vif to rule them all. J. Mol. Biol. 2014; 426:1220–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stewart J.A., Holland T.C., Bhagwat A.S.. Human herpes simplex virus-1 depletes APOBEC3A from nuclei. Virology. 2019; 537:104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng A.Z., Moraes S.N., Shaban N.M., Fanunza E., Bierle C.J., Southern P.J., Bresnahan W.A., Rice S.A., Harris R.S.. APOBECs and herpesviruses. Viruses. 2021; 13:390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Refsland E.W., Harris R.S.. The APOBEC3 family of retroelement restriction factors. Curr. Top. Microbiol. Immunol. 2013; 371:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koito A., Ikeda T.. Intrinsic immunity against retrotransposons by APOBEC cytidine deaminases. Front. Microbiol. 2013; 4:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Uriu K., Kosugi Y., Suzuki N., Ito J., Sato K.. Elucidation of the complicated scenario of primate APOBEC3 gene evolution. J. Virol. 2021; 95:e00144-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kanu N., Cerone M.A., Goh G., Zalmas L.P., Bartkova J., Dietzen M., McGranahan N., Rogers R., Law E.K., Gromova I.et al.. DNA replication stress mediates APOBEC3 family mutagenesis in breast cancer. Genome Biol. 2016; 17:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ng J.C.F., Quist J., Grigoriadis A., Malim M.H., Fraternali F.. Pan-cancer transcriptomic analysis dissects immune and proliferative functions of APOBEC3 cytidine deaminases. Nucleic Acids Res. 2019; 47:1178–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Talluri S., Samur M.K., Buon L., Kumar S., Potluri L.B., Shi J., Prabhala R.H., Shammas M.A., Munshi N.C.. Dysregulated APOBEC3G causes DNA damage and promotes genomic instability in multiple myeloma. Blood Cancer J. 2021; 11:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Botvinnik A., Shivam P., Smith Y., Sharma G., Olshevsky U., Moshel O., Manevitch Z., Climent N., Oliva H., Britan-Rosich E.et al.. APOBEC3G rescues cells from the deleterious effects of DNA damage. FEBS J. 2021; 288:6063–6077. [DOI] [PubMed] [Google Scholar]
- 38. Lackey L., Law E.K., Brown W.L., Harris R.S.. Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle. 2013; 12:762–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bennett R.P., Presnyk V., Wedekind J.E., Smith H.C.. Nuclear exclusion of the HIV-1 host defense factor APOBEC3G requires a novel cytoplasmic retention signal and is not dependent on RNA binding. J. Biol. Chem. 2008; 283:7320–7327. [DOI] [PubMed] [Google Scholar]
- 40. Salamango D.J., Becker J.T., McCann J.L., Cheng A.Z., Demir O., Amaro R.E., Brown W.L., Shaban N.M., Harris R.S.. APOBEC3H subcellular localization determinants define zipcode for targeting HIV-1 for restriction. Mol. Cell. Biol. 2018; 38:e00356-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li M.M., Emerman M.. Polymorphism in human APOBEC3H affects a phenotype dominant for subcellular localization and antiviral activity. J. Virol. 2011; 85:8197–8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lackey L., Demorest Z.L., Land A.M., Hultquist J.F., Brown W.L., Harris R.S.. APOBEC3B and AID have similar nuclear import mechanisms. J. Mol. Biol. 2012; 419:301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang Y., Sowden M.P., Yang Y., Smith H.C.. Intracellular trafficking determinants in APOBEC-1, the catalytic subunit for cytidine to uridine editing of apolipoprotein b mRNA. Exp. Cell. Res. 2001; 267:153–164. [DOI] [PubMed] [Google Scholar]
- 44. Landry S., Narvaiza I., Linfesty D.C., Weitzman M.D.. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011; 12:444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Land A.M., Law E.K., Carpenter M.A., Lackey L., Brown W.L., Harris R.S.. Endogenous APOBEC3A DNA cytosine deaminase is cytoplasmic and nongenotoxic. J. Biol. Chem. 2013; 288:17253–17260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mussil B., Suspene R., Aynaud M.M., Gauvrit A., Vartanian J.P., Wain-Hobson S.. Human APOBEC3A isoforms translocate to the nucleus and induce DNA double strand breaks leading to cell stress and death. PLoS One. 2013; 8:e73641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Burns M.B., Lackey L., Carpenter M.A., Rathore A., Land A.M., Leonard B., Refsland E.W., Kotandeniya D., Tretyakova N., Nikas J.B.et al.. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013; 494:366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Starrett G.J., Luengas E.M., McCann J.L., Ebrahimi D., Temiz N.A., Love R.P., Feng Y., Adolph M.B., Chelico L., Law E.K.et al.. The DNA cytosine deaminase APOBEC3H haplotype i likely contributes to breast and lung cancer mutagenesis. Nat. Commun. 2016; 21:12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leonard B., Hart S.N., Burns M.B., Carpenter M.A., Temiz N.A., Rathore A., Vogel R.I., Nikas J.B., Law E.K., Brown W.L.et al.. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013; 73:7222–7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Tian Ng A.W., Wu Y., Boot A., Covington K.R., Gordenin D.A., Bergstrom E.N.et al.. The repertoire of mutational signatures in human cancer. Nature. 2020; 578:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Dale Borresen-et al.. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Petljak M., Alexandrov L.B., Brammeld J.S., Price S., Wedge D.C., Grossmann S., Dawson K.J., Ju Y.S., Iorio F., Tubio J.M.C.et al.. Characterizing mutational signatures in human cancer cell lines reveals episodic APOBEC mutagenesis. Cell. 2019; 176:1282–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Alexandrov L.B., Stratton M.R.. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014; 24:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chan K., Roberts S.A., Klimczak L.J., Sterling J.F., Saini N., Malc E.P., Kim J., Kwiatkowski D.J., Fargo D.C., Mieczkowski P.A.et al.. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat. Genet. 2015; 47:1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Buisson R., Langenbucher A., Bowen D., Kwan E.E., Benes C.H., Zou L., Lawrence M.S.. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science. 2019; 364:eaaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Langenbucher A., Bowen D., Sakhtemani R., Bournique E., Wise J.F., Zou L., Bhagwat A.S., Buisson R., Lawrence M.S.. An extended APOBEC3A mutation signature in cancer. Nat. Commun. 2021; 12:1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Swanton C., McGranahan N., Starrett G.J., Harris R.S.. APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov. 2015; 5:704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rosenthal R., McGranahan N., Herrero J., Taylor B.S., Swanton C.. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016; 22:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. de Bruin E.C., McGranahan N., Mitter R., Salm M., Wedge D.C., Yates L., Jamal-Hanjani M., Shafi S., Murugaesu N., Rowan A.J.et al.. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014; 346:251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Venkatsesan S., Rosenthal R., Kanu N., McGranahan N., Bartek J., Quezada S.A., Hare J., Harris R.S., Swanton C.. Perspective: APOBEC mutagenesis in drug resistance and immune escape in HIV and cancer evolution. Ann. Oncol. 2018; 29:563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Henderson S., Chakravarthy A., Su X., Boshoff C., Fenton T.R.. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 2014; 7:1833–1841. [DOI] [PubMed] [Google Scholar]
- 62. Tsujimoto Y., Finger L.R., Yunis J., Nowell P.C., Croce C.M.. Cloning of the chromosome breakpoint of neoplastic b cells with the t(14;18) chromosome translocation. Science. 1984; 226:1097–1099. [DOI] [PubMed] [Google Scholar]
- 63. Ye B.H., Lista F., Lo Coco F., Knowles D.M., Offit K., Chaganti R.S., Dalla-Favera R.. Alterations of a zinc finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science. 1993; 262:747–750. [DOI] [PubMed] [Google Scholar]
- 64. Taub R., Kirsch I., Morton C., Lenoir G., Swan D., Tronick S., Aaronson S., Leder P.. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. U.S.A. 1982; 79:7837–7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Saha T., Sundaravinayagam D., Di Virgilio M.. Charting a DNA repair roadmap for immunoglobulin class switch recombination. Trends Biochem. Sci. 2021; 46:184–199. [DOI] [PubMed] [Google Scholar]
- 66. Ramiro A.R., Jankovic M., Eisenreich T., Difilippantonio S., Chen-Kiang S., Muramatsu M., Honjo T., Nussenzweig A., Nussenzweig M.C.. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004; 118:431–438. [DOI] [PubMed] [Google Scholar]
- 67. Jankovic M., Robbiani D.F., Dorsett Y., Eisenreich T., Xu Y., Tarakhovsky A., Nussenzweig A., Nussenzweig M.C.. Role of the translocation partner in protection against AID-dependent chromosomal translocations. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Burns M.B., Temiz N.A., Harris R.S.. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013; 45:977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cortez L.M., Brown A.L., Dennis M.A., Collins C.D., Brown A.J., Mitchell D., Mertz T.M., Roberts S.A.. APOBEC3A is a prominent cytidine deaminase in breast cancer. PLoS Genet. 2019; 15:e1008545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Taylor B.J., Nik-Zainal S., Wu Y.L., Stebbings L.A., Raine K., Campbell P.J., Rada C., Stratton M.R., Neuberger M.S.. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife. 2013; 2:e00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Siewerts A.M., Willis S., Burns M.B., Look M.P., Meijer-Van Gelder M.E., Schlicker A., Heideman M.R., Jacobs H., Wessels L., Leyland-Jones B.et al.. Elevated APOBEC3B correlates with poor outcomes for estrogen-receptor-positive breast cancers. Horm. Cancer. 2014; 5:405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Caval V., Suspene R., Shapira M., Vartanian J.P., Wain-Hobson S.. A prevalent cancer susceptibility APOBEC3A hybrid allele bearing APOBEC3B 3'UTR enhances chromosomal DNA damage. Nat. Commun. 2014; 9:5129. [DOI] [PubMed] [Google Scholar]
- 73. Jalili P., Bowen D., Langenbucher A., Park S., Aguirre K., Corcoran R.B., Fleischman A.G., Lawrence M.S., Zou L., Buisson R.. Quantification of ongoing APOBEC3A activity in tumor cells by monitoring RNA editing at hotspots. Nat. Commun. 2020; 11:2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Petljak M., Dananberg A., Chu K., Bergstrom E.N., Striepen J., von Morgen P., Chen Y., Shah H., Sale J.E., Alexandrov L.B.et al.. Mechanisms of APOBEC3 mutagenesis in human cancer cells. Nature. 2022; 607:799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wormann S.M., Zhang A., Thege F.I., Cowan R.W., Rupani D.N., Wang R., Manning S.L., Gates C., Wu W., Levin-Klein R.et al.. APOBEC3A drives deaminase domain-independent chromosomal instability to promote pancreatic cancer metastasis. Nat Cancer. 2021; 2:1338–1356. [DOI] [PubMed] [Google Scholar]
- 76. Law E.K., Levin-Klein R., Jarvis M.C., Kim H., Argyris P.P., Carpenter M.A., Starrett G.J., Temiz N.A., Larson L.K., Durfee C.et al.. APOBEC3A catalyzes mutation and drives carcinogenesis in vivo. J. Exp. Med. 2020; 217:e20200261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. OhAinle M., Kerns J.A., Li M.M., Malik H.S., Emerman M.. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe. 2008; 4:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Harari A., Ooms M., Mulder L.C., Simon V.. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J. Virol. 2009; 83:295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Silvestrov P., Maier S.J., Fang M., Cisneros G.A.. DNArCdb: a database of cancer biomarkers in DNA repair genes that includes variants related to multiple cancer phenotypes. DNA Repair (Amst.). 2018; 70:10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hix M.A., Wong L., Flath B., Chelico L., Cisneros G.A.. Single-nucleotide polymorphism of the DNA cytosine deaminase APOBEC3H haplotype i leads to enzyme destabilization and correlates with lung cancer. NAR Cancer. 2020; 2:zcaa023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhu M., Wang Y., Wang C., Shen W., Liu J., Geng L., Cheng Y., Dai J., Jin G., Ma H.et al.. The eQTL-missense polymorphisms of APOBEC3H are associated with lung cancer risk in a han chinese population. Sci. Rep. 2015; 5:14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Saraconi G., Severi F., Sala C., Mattiuz G., Conticello S.G.. The RNA editing enzyme APOBEC1 induces somatic mutations and a compatible mutational signature is present in esophageal adenocarcinomas. Genome Biol. 2014; 15:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Harris R.S., Petersen-Mahrt S.K., Neuberger M.S.. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell. 2002; 10:1247–1253. [DOI] [PubMed] [Google Scholar]
- 84. Rosenberg B.R., Hamilton C.E., Mwangi M.M., Dewell S., Papavasiliou F.N.. Transcriptome-wide sequencing reveals numerous APOBEC1 mRNA-editing targets in transcript 3' UTRs. Nat. Struct. Mol. Biol. 2011; 18:230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wong L., Vizeacoumar F.S., Vizeacoumar F.J., Chelico L.. APOBEC1 cytosine deaminase activity on single-stranded DNA is suppressed by replication protein A. Nucleic Acids Res. 2021; 49:322–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ward I.M., Chen J.. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001; 276:47759–47762. [DOI] [PubMed] [Google Scholar]
- 87. Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M.. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998; 273:5858–5868. [DOI] [PubMed] [Google Scholar]
- 88. Zou Y., Liu Y., Wu X., Shell S.M.. Functions of human replication protein a (RPA): from DNA replication to DNA damage and stress responses. J. Cell. Physiol. 2006; 208:267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Dueva R., Iliakis G.. Replication protein A: a multifunctional protein with roles in DNA replication, repair and beyond. NAR Cancer. 2020; 2:zcaa022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nagelhus T.A., Haug T., Singh K.K., Keshav K.F., Skorpen F., Otterlei M., Bharati S., Lindmo T., Benichou S., Benarous R.et al.. A sequence in the N-terminal region of human uracil-DNA glycosylase with homology to XPA interacts with the C-terminal part of the 34-kDa subunit of replication protein A. J. Biol. Chem. 1997; 272:6561–6566. [DOI] [PubMed] [Google Scholar]
- 91. Bansbach C.E., Betous R., Lovejoy C.A., Glick G.G., Cortez D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009; 23:2405–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ali S.I., Shin J.S., Bae S.H., Kim B., Choi B.S.. Replication protein a 32 interacts through a similar binding interface with TIPIN, XPA, and UNG2. Int. J. Biochem. Cell Biol. 2010; 42:1210–1215. [DOI] [PubMed] [Google Scholar]
- 93. Park M.S., Ludwig D.L., Stigger E., Lee S.H.. Physical interaction between human RAD52 and RPA is required for homologous recombination in mammalian cells. J. Biol. Chem. 1996; 271:18996–19000. [DOI] [PubMed] [Google Scholar]
- 94. Wold M.S. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997; 66:61–92. [DOI] [PubMed] [Google Scholar]
- 95. Fanning E., Klimovich V., Nager A.R.. A dynamic model for replication protein a (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006; 34:4126–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Fan J., Pavletich N.P.. Structure and conformational change of a replication protein a heterotrimer bound to ssDNA. Genes Dev. 2012; 26:2337–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hoopes J.I., Cortez L.M., Mertz T.M., Malc E.P., Mieczkowski P.A., Roberts S.A.. APOBEC3A and APOBEC3B preferentially deaminate the lagging strand template during DNA replication. Cell Rep. 2016; 14:1273–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kazanov M.D., Roberts S.A., Polak P., Stamatoyannopoulos J., Klimczak L.J., Gordenin D.A., Sunyaev S.R.. APOBEC-Induced cancer mutations are uniquely enriched in early-replicating, gene-dense, and active chromatin regions. Cell Rep. 2015; 13:1103–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Haradhvala N.J., Polak P., Stojanov P., Covington K.R., Shinbrot E., Hess J.M., Rheinbay E., Kim J., Maruvka Y.E., Braunstein L.Z.et al.. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell. 2016; 164:538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Jiang X., Klimovich V., Arunkumar A.I., Hysinger E.B., Wang Y., Ott R.D., Guler G.D., Weiner B., Chazin W.J., Fanning E.. Structural mechanism of RPA loading on DNA during activation of a simple pre-replication complex. EMBO J. 2006; 25:5516–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Yeeles J.T.P., Deegan T.D., Janska A., Early A., Diffley J.F.X.. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature. 2015; 519:431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Melendy T., Stillman B.. An interaction between replication protein a and SV40 t antigen appears essential for primosome assembly during SV40 DNA replication. J. Biol. Chem. 1993; 268:3389–3395. [PubMed] [Google Scholar]
- 103. Techer H., Koundrioukoff S., Nicolas A., Debatisse M.. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet. 2017; 18:535–550. [DOI] [PubMed] [Google Scholar]
- 104. Seplyarskiy V.B., Soldatov R.A., Popadin K.Y., Antonarakis S.E., Bazykin G.A., Nikolaev S.I.. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016; 26:174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bhagwat A.S., Hao W., Townes J.P., Lee H., Tang H., Foster P.L.. Strand-biased cytosine deamination at the replication fork causes cytosine to thymine mutations in escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:2176–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Gu J., Chen Q., Xiao X., Ito F., Wolfe A., Chen X.S.. Biochemical characterization of APOBEC3H variants: implications for their HIV-1 restriction activity and mC modification. J. Mol. Biol. 2016; 428:4626–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Fu Y., Ito F., Zhang G., Fernandez B., Yang H., Chen X.S.. DNA cytosine and methylcytosine deamination by APOBEC3B: enhancing methylcytosine deamination by engineering APOBEC3B. Biochem. J. 2015; 471:25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Carpenter M.A., Li M., Rathore A., Lackey L., Law E.K., Land A.M., Leonard B., Shandilya S.M., Bohn M.F., Schiffer C.A.et al.. Methylcytosine and normal cytosine deamination by the foreign DNA restriction enzyme APOBEC3A. J. Biol. Chem. 2012; 287:34801–34808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Gaillard H., García-Muse T., Aguilera A.. Replication stress and cancer. Nat. Rev. Cancer. 2015; 15:276–289. [DOI] [PubMed] [Google Scholar]
- 110. Sui Y., Qi L., Zhang K., Saini N., Klimczak L.J., Sakofsky C.J., Gordenin D.A., Petes T.D., Zheng D.Q.. Analysis of APOBEC-induced mutations in yeast strains with low levels of replicative DNA polymerases. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:9440–9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Sikorski T.W., Ficarro S.B., Holik J., Kim T., Rando O.J., Marto J.A., Buratowski S.. Sub1 and RPA associate with RNA polymerase II at different stages of transcription. Mol. Cell. 2011; 44:397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Chandra V., Bortnick A., Murre C.. AID targeting: old mysteries and new challenges. Trends Immunol. 2015; 36:527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Adolph M.B., Love R.P., Feng Y., Chelico L.. Enzyme cycling contributes to efficient induction of genome mutagenesis by the cytidine deaminase APOBEC3B. Nucleic Acids Res. 2017; 45:11925–11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Brown A.L., Collins C.D., Thompson S., Coxon M., Mertz T.M., Roberts S.A.. Single-stranded DNA binding proteins influence APOBEC3A substrate preference. Sci. Rep. 2021; 11:21008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Derheimer F.A., O’Hagan H.M., Krueger H.M., Hanasoge S., Paulsen M.T., Ljungman M.. RPA and ATR link transcriptional stress to p53. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:12778–12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Brueckner F., Cramer P.. DNA photodamage recognition by RNA polymerase iI. FEBS Lett. 2007; 581:2757–2760. [DOI] [PubMed] [Google Scholar]
- 117. Helmrich A., Ballarino M., Nudler E., Tora L.. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013; 20:412–418. [DOI] [PubMed] [Google Scholar]
- 118. Ohle C., Tesorero R., Schermann G., Dobrev N., Sinning I., Fischer T.. Transient RNA-DNA hybrids are required for efficient double-strand break repair. Cell. 2016; 167:1001–1013. [DOI] [PubMed] [Google Scholar]
- 119. Nguyen H.D., Yadav T., Giri S., Saez B., Graubert T.A., Zou L.. Functions of replication protein a as a sensor of r loops and a regulator of RNaseH1. Mol. Cell. 2017; 65:832–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Rinaldi C., Pizzul P., Longhese M.P., Bonetti D. Sensing R-Loop-Associated DNA damage to safeguard genome stability. Front. Cell Dev. Biol. 2020; 8:618157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Nik-Zainal S., Alexandrov L.B., Wedge D.C., Van Loo P., Greenman C.D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L.A.et al.. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012; 149:979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Burns M.B., Lackey L., Carpenter M.A., Rathore A., Land A.M., Leonard B., Refsland E.W., Kotandeniya D., Tretyakova N., Nikas J.B.et al.. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013; 494:366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Green A.M., Landry S., Budagyan K., Avgousti D.C., Shalhout S., Bhagwat A.S., Weitzman M.D.. APOBEC3A damages the cellular genome during DNA replication. Cell Cycle. 2016; 15:998–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kockler Z.W., Osia B., Lee R., Musmaker K., Malkova A.. Repair of DNA breaks by break-induced replication. Annu. Rev. Biochem. 2021; 90:165–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Osia B., Twarowski J., Jackson T., Lobachev K., Liu L., Malkova A.. Migrating bubble synthesis promotes mutagenesis through lesions in its template. Nucleic Acids Res. 2022; 50:6870–6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Sakofsky C.J., Roberts S.A., Malc E., Mieczkowski P.A., Resnick M.A., Gordenin D.A., Malkova A.. Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep. 2014; 7:1640–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sakofsky C.J., Saini N., Klimczak L.J., Chan K., Malc E.P., Mieczkowski P.A., Burkholder A.B., Fargo D., Gordenin D.A.. Repair of multiple simultaneous double-strand breaks causes bursts of genome-wide clustered hypermutation. PLoS Biol. 2019; 17:e3000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Oakley G.G., Patrick S.M.. Replication protein A: directing traffic at the intersection of replication and repair. Front. Biosci. (Landmark Ed.). 2010; 15:883–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Toledo L.I., Altmeyer M., Rask M.B., Lukas C., Larsen D.H., Povlsen L.K., Bekker-Jensen S., Mailand N., Bartek J., Lukas J.. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013; 155:1088–1103. [DOI] [PubMed] [Google Scholar]
- 130. Vassin V.M., Wold M.S., Borowiec J.A.. Replication protein a (RPA) phosphorylation prevents RPA association with replication centers. Mol. Cell. Biol. 2004; 24:1930–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Fried L.M., Koumenis C., Peterson S.R., Green S.L., van Zijl P., Allalunis-Turner J., Chen D.J., Fishel R., Giaccia A.J., Brown J.M.et al.. The DNA damage response in DNA-dependent protein kinase-deficient SCID mouse cells: replication protein a hyperphosphorylation and p53 induction. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:13825–13830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Patrick S.M., Oakley G.G., Dixon K., Turchi J.J.. DNA damage induced hyperphosphorylation of replication protein a. 2. Characterization of DNA binding activity, protein interactions, and activity in DNA replication and repair. Biochemistry. 2005; 44:8438–8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Binz S.K., Lao Y., Lowry D.F., Wold M.S.. The phosphorylation domain of the 32-kDa subunit of replication protein a (RPA) modulates RPA–DNA interactions. Evidence for an intersubunit interaction. J. Biol. Chem. 2003; 278:35584–35591. [DOI] [PubMed] [Google Scholar]
- 134. Oakley G.G., Patrick S.M., Yao J., Carty M.P., Turchi J.J., Dixon K.. RPA phosphorylation in mitosis alters DNA binding and protein-protein interactions. Biochemistry. 2003; 42:3255–3264. [DOI] [PubMed] [Google Scholar]
- 135. Ma C.J., Gibb B., Kwon Y., Sung P., Greene E.C.. Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res. 2017; 45:749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Gibb B., Silverstein T.D., Finkelstein I.J., Greene E.C.. Single-stranded DNA curtains for real-time single-molecule visualization of protein-nucleic acid interactions. Anal. Chem. 2012; 84:7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Gibb B., Ye L.F., Gergoudis S.C., Kwon Y., Niu H., Sung P., Greene E.. Concentration-dependent exchange of replication protein a on single-stranded DNA revealed by single-molecule imaging. PLoS One. 2014; 9:e87922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Pokhrel N., Caldwell C.C., Corless E.I., Tillison E.A., Tibbs J., Jocic N., Tabei S.M.A., Wold M.S., Spies M., Antony E.. Dynamics and selective remodeling of the DNA-binding domains of RPA. Nat. Struct. Mol. Biol. 2019; 26:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Yates L.A., Aramayo R.J., Pokhrel N., Caldwell C.C., Kaplan J.A., Perera R.L., Spies M., Antony E., Zhang X.. A structural and dynamic model for the assembly of replication protein a on single-stranded DNA. Nat. Commun. 2018; 9:5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Nguyen B., Sokoloski J., Galletto R., Elson E.L., Wold M.S., Lohman T.M.. Diffusion of human replication protein a along single-stranded DNA. J. Mol. Biol. 2014; 426:3246–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Berg O.G., Winter R.B., von Hippel P.H.. Diffusion-driven mechanisms of protein translocation on nucleic acids. Biochemistry. 1981; 20:6929–6948. [DOI] [PubMed] [Google Scholar]
- 142. von Hippel P.H., Berg O.G.. Facilitated target location in biological systems. J. Biol. Chem. 1989; 264:675–678. [PubMed] [Google Scholar]
- 143. Stanford N.P., Szczelkun M.D., Marko J.F., Halford S.E.. One- and three-dimensional pathways for proteins to reach specific DNA sites. EMBO J. 2000; 19:6546–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Adolph M.B., Love R.P., Chelico L.. Biochemical basis of APOBEC3 deoxycytidine deaminase activity on diverse DNA substrates. ACS Infect Dis. 2018; 4:224–238. [DOI] [PubMed] [Google Scholar]
- 145. Adolph M.B., Love R.P., Feng Y., Chelico L.. Enzyme cycling contributes to efficient induction of genome mutagenesis by the cytidine deaminase APOBEC3B. Nucleic Acids Res. 2017; 45:11925–11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ara A., Love R.P., Chelico L.. Different mutagenic potential of HIV-1 restriction factors APOBEC3G and APOBEC3F is determined by distinct single-stranded DNA scanning mechanisms. PLoS Pathog. 2014; 10:e1004024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Feng Y., Love R.P., Ara A., Baig T.T., Adolph M.B., Chelico L.. Natural polymorphisms and oligomerization of human APOBEC3H contribute to Single-stranded DNA scanning ability. J. Biol. Chem. 2015; 290:27188–27203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Mertz T.M., Harcy V., Roberts S.A.. Risks at the DNA replication fork: effects upon carcinogenesis and tumor heterogeneity. Genes (Basel). 2017; 8:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Toledo L., Neelsen K.J., Lukas J.. Replication catastrophe: when a checkpoint fails because of exhaustion. Mol. Cell. 2017; 66:735–749. [DOI] [PubMed] [Google Scholar]
- 150. Nowarski R., Wilner O.I., Cheshin O., Shahar O.D., Kenig E., Baraz L., Britan-Rosich E., Nagler A., Harris R.S., Goldberg M.et al.. APOBEC3G enhances lymphoma cell radioresistance by promoting cytidine deaminase-dependent DNA repair. Blood. 2012; 120:366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Morse M., Naufer M.N., Feng Y., Chelico L., Rouzina I., Williams M.C.. HIV restriction factor APOBEC3G binds in multiple steps and conformations to search and deaminate single-stranded DNA. Elife. 2019; 8:e52649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Chelico L., Sacho E.J., Erie D.A., Goodman M.F.. A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. J. Biol. Chem. 2008; 283:13780–13791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Stewart J.A., Schauer G., Bhagwat A.S.. Visualization of uracils created by APOBEC3A using UdgX shows colocalization with RPA at stalled replication forks. Nucleic Acids Res. 2020; 48:e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Bazzi A., Zargarian L., Chaminade F., De Rocquigny H., René B., Mély Y., Fossé P., Mauffret O.. Intrinsic nucleic acid dynamics modulates HIV-1 nucleocapsid protein binding to its targets. PLoS One. 2012; 7:e38905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Love R.P., Xu H., Chelico L.. Biochemical analysis of hypermutation by the deoxycytidine deaminase APOBEC3A. J. Biol. Chem. 2012; 287:30812–30822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Warren C.J., Westrich J.A., Doorslaer K.V., Pyeon D. Roles of APOBEC3A and APOBEC3B in human papillomavirus infection and disease progression. Viruses. 2017; 9:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Warren C.J., Xu T., Guo K., Griffin L.M., Westrich J.A., Lee D., Lambert P.F., Santiago M.L., Pyeon D. APOBEC3A functions as a restriction factor of human papillomavirus. J. Virol. 2015; 89:688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Kavli B., Iveland T.S., Buchinger E., Hagen L., Liabakk N.B., Aas P.A., Obermann T.S., Aachmann F.L., Slupphaug G.. 2021) RPA2 winged-helix domain facilitates UNG-mediated removal of uracil from ssDNA; implications for repair of mutagenic uracil at the replication fork. Nucleic Acids Res. 49:3948–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Sousa M.M., Krokan H.E., Slupphaug G.. DNA-uracil and human pathology. Mol. Aspects Med. 2007; 28:276–306. [DOI] [PubMed] [Google Scholar]
- 160. Feng Y., Li C., Stewart J.A., Barbulescu P., Seija Desivo N., Alvarez-Quilon A., Pezo R.C., Perera M.L.W., Chan K., Tong A.H.Y.et al.. FAM72A antagonizes UNG2 to promote mutagenic repair during antibody maturation. Nature. 2021; 600:324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Guo C., Zhang X., Fink S.P., Platzer P., Wilson K., Willson J.K., Wang Z., Markowitz S.D.. Ugene, a newly identified protein that is commonly overexpressed in cancer and binds uracil DNA glycosylase. Cancer Res. 2008; 68:6118–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Belanger F., Fortier E., Dube M., Lemay J.F., Buisson R., Masson J.Y., Elsherbiny A., Costantino S., Carmona E., Mes-Masson A.M.et al.. Replication protein a availability during DNA replication stress is a major determinant of cisplatin resistance in ovarian cancer cells. Cancer Res. 2018; 78:5561–5573. [DOI] [PubMed] [Google Scholar]
- 163. Ruiz S., Mayor-Ruiz C., Lafarga V., Murga M., Vega-Sendino M., Ortega S., Fernandez-Capetillo O.. A Genome-wide CRISPR screen identifies CDC25A as a determinant of sensitivity to ATR inhibitors. Mol. Cell. 2016; 62:307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. de Klein A., Muijtjens M., van Os R., Verhoeven Y., Smit B., Carr A.M., Lehmann A.R., Hoeijmakers J.H.. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol. 2000; 10:479–482. [DOI] [PubMed] [Google Scholar]
- 165. Shibata A., Jeggo P.A.. ATM’s role in the repair of DNA double-strand breaks. Genes (Basel). 2021; 12:1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Khoronenkova S.V., Dianov G.L.. ATM prevents DSB formation by coordinating SSB repair and cell cycle progression. Proc. Natl. Acad. Sci. U. S. A. 2015; 112:3997–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Riabinska A., Daheim M., Herter-Sprie G.S., Winkler J., Fritz C., Hallek M., Thomas R.K., Kreuzer K.A., Frenzel L.P., Monfared P.et al.. Therapeutic targeting of a robust non-oncogene addiction to PRKDC in ATM-defective tumors. Sci. Transl. Med. 2013; 5:189ra178. [DOI] [PubMed] [Google Scholar]
- 168. Banerjee T., Sommers J.A., Huang J., Seidman M.M., Brosh R.M.,Jr. Catalytic strand separation by RECQ1 is required for RPA-mediated response to replication stress. Curr. Biol. 2015; 25:2830–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Bartos J.D., Willmott L.J., Binz S.K., Wold M.S., Bambara R.A.. Catalysis of strand annealing by replication protein a derives from its strand melting properties. J. Biol. Chem. 2008; 283:21758–21768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Givalos N., Gakiopoulou H., Skliri M., Bousboukea K., Konstantinidou A.E., Korkolopoulou P., Lelouda M., Kouraklis G., Patsouris E., Karatzas G.. Replication protein a is an independent prognostic indicator with potential therapeutic implications in colon cancer. Mod. Pathol. 2007; 20:159–166. [DOI] [PubMed] [Google Scholar]
- 171. Buisson R., Lawrence M.S., Benes C.H., Zou L.. APOBEC3A and APOBEC3B activities render cancer cells susceptible to ATR inhibition. Cancer Res. 2017; 77:4567–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Conner K.L., Shaik A.N., Ekinci E., Kim S., Ruterbusch J.J., Cote M.L., Patrick S.M.. HPV induction of APOBEC3 enzymes mediate overall survival and response to cisplatin in head and neck cancer. DNA Repair (Amst.). 2020; 87:102802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Serebrenik A.A., Argyris P.P., Jarvis M.C., Brown W.L., Bazzaro M., Vogel R.I., Erickson B.K., Lee S.H., Goergen K.M., Maurer M.J.et al.. The DNA cytosine deaminase APOBEC3B is a molecular determinant of platinum responsiveness in clear cell ovarian cancer. Clin. Cancer Res. 2020; 26:3397–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Kothandapani A., Dangeti V.S., Brown A.R., Banze L.A., Wang X.H., Sobol R.W., Patrick S.M.. Novel role of base excision repair in mediating cisplatin cytotoxicity. J. Biol. Chem. 2011; 286:14564–14574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Yang H., Ito F., Wolfe A.D., Li S., Mohammadzadeh N., Love R.P., Yan M., Zirkle B., Gaba A., Chelico L.et al.. Understanding the structural basis of HIV-1 restriction by the full length double-domain APOBEC3G. Nat. Commun. 2020; 11:632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Bochkareva E., Korolev S., Lees-Miller S.P., Bochkarev A.. Structure of the RPA trimerization core and its role in the multistep DNA-binding mechanism of RPA. EMBO J. 2002; 21:1855–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.