

Summary
Although adult pluripotent stem cells (aPSCs) are found in many animal lineages, mechanisms for their formation during embryogenesis are unknown. Here, we leveraged Hofstenia miamia, a regenerative worm that possesses collectively pluripotent aPSCs called neoblasts, and produces manipulable embryos. Lineage-tracing and functional experiments revealed that one pair of blastomeres gives rise to cells that resemble neoblasts in distribution, behavior, and gene expression. aPSCs include transcriptionally-distinct subpopulations expressing markers associated with differentiated tissues; our data suggest that despite their heterogeneity, aPSCs are derived from one lineage, not from multiple tissue-specific lineages during development. Next, we combined single-cell transcriptome profiling across development with neoblast cell lineage tracing and identified a molecular trajectory for neoblast formation that included transcription factors Hes, FoxO, and Tbx. This identification of a cellular mechanism and molecular trajectory for aPSC formation opens the door for in vivo studies of aPSC regulation and evolution.
Graphical Abstract
In Brief
Lineage-tracing and functional experiments in the regenerative worm Hofstenia miamia reveal that a single pair of blastomeres gives rise to cells that resemble neoblasts in distribution, behavior, and gene expression.
Introduction
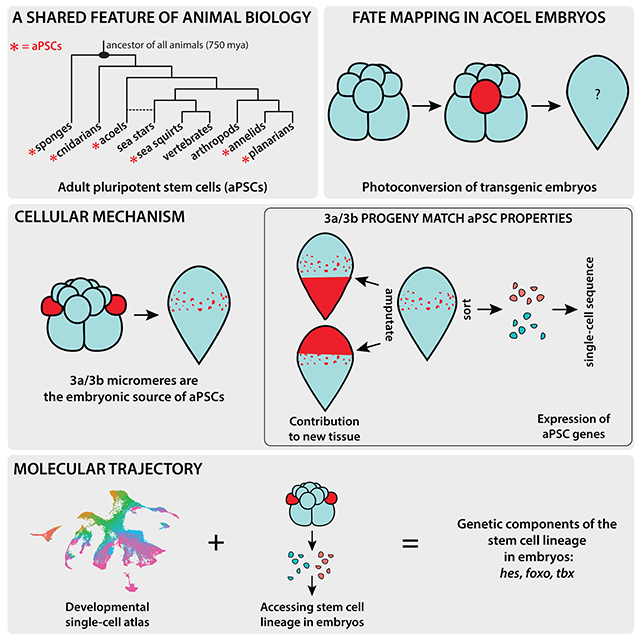
Populations of adult pluripotent stem cells (aPSCs) are found in many distantly-related invertebrate animals, where they facilitate whole-body regeneration and/or homeostatic tissue turnover1 (Fig. 1A). Some examples include the neoblasts of planarians2–4, the i-cells of some cnidarians5,6, and the archaeocytes of sponges7,8. In the majority of these systems, aPSCs express shared markers, including orthologs of Piwi and other proteins associated with germ cells9. Given these shared molecular properties across 750 million years of animal evolution, aPSCs are possibly a fundamental feature of animal biology that is notably absent in mammals. Although the mechanisms underlying the regulation of pluripotency in mammalian embryonic stem cells (ESCs) are known10, these cells lose their potency over the course of embryonic development and aPSCs are absent in the adult body plan. Therefore, studies in animals with aPSCs are needed to uncover how pluripotent stem cells are first specified and then maintained until adulthood in vivo. The timing of emergence of functional stem cells (in planarians)11 and the expression of aPSC marker genes (in cnidarians)12,13 have been investigated, but the underlying cellular and molecular mechanisms for the formation of aPSCs remain unknown. Here, we sought to determine the cellular source of aPSCs and to identify molecular regulators that could be involved in aPSC formation during embryogenesis.
Figure 1: Lineage tracing using photoconvertible transgenic embryos reveals the cellular sources for major tissue types.
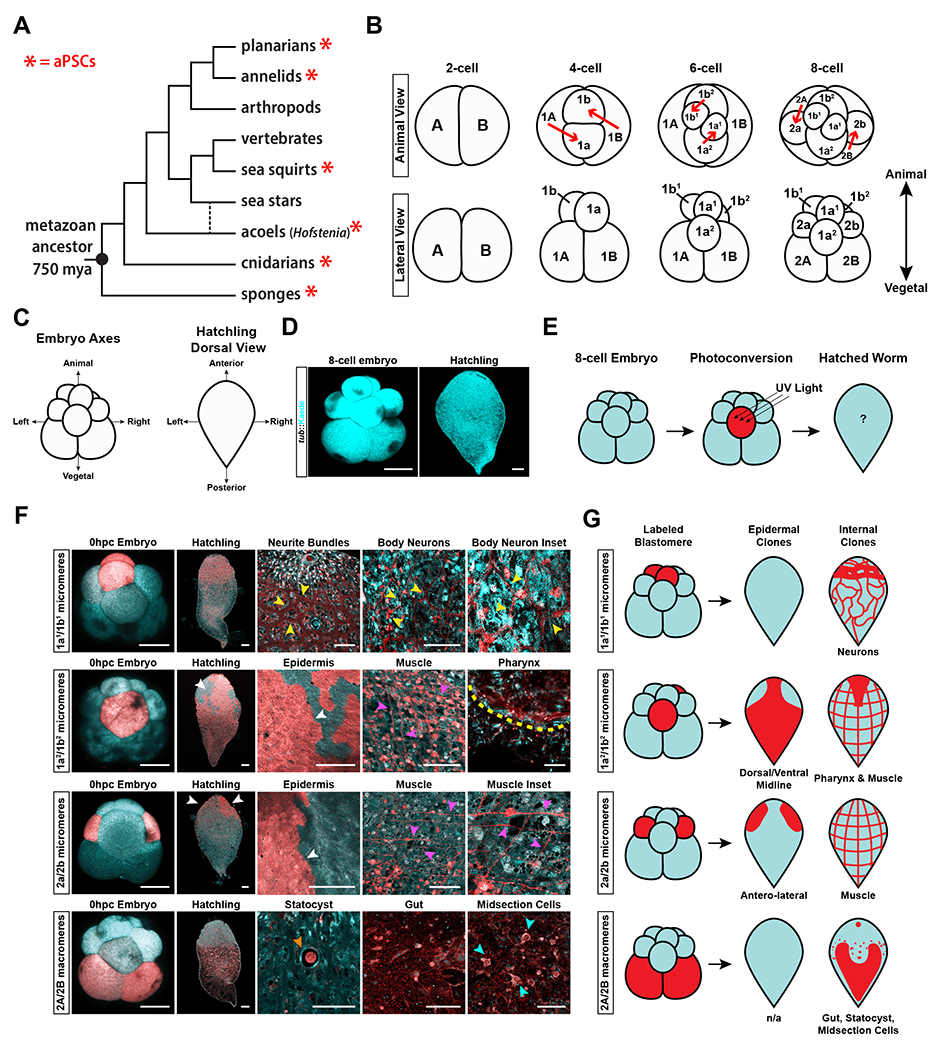
(A) Metazoan tree of life showing the widespread occurrence of adult pluripotent stem cells (aPSCs) (red asterisk) in many lineages and their absence in mammals (vertebrates). The branch for acoels is dotted to indicate two phylogenetic positions currently under debate35–37. (B) Schematic diagram showing the stereotyped duet cleavage pattern of Hofstenia miamia embryos from the 2-cell to 8-cell stage. Top: Animal view of the cleavage pattern. Red arrows show the direction of cleavage to generate blastomeres. Bottom: Lateral view of embryos. The animal and vegetal axis corresponding to the lateral view is denoted in bold arrows (C) Schematic diagram of embryonic and adult axes, relating to D, E, F, and G. Left: Embryonic axes labeled at the 8-cell stage embryo when viewed laterally. Right: Adult body axes labeled on a hatchling worm when viewed from the dorsal side. (D) Images of a tuba::Kaede transgenic 8-cell stage embryo and hatchling worm (pseudo-colored in cyan). (E) Schematic showing the workflow for photoconversion. (F) Fate-mapping of 8-cell state embryos via photoconversion of pairs of blastomeres. Single-cell conversions are shown in Fig. S1D. 1a1/1b1 cells give rise to neurons in the adult, with large neurite bundles being visible in the anterior, and a net-like structure of body neurons throughout the body (yellow arrowheads) (n=10/10). 1a2/1b2 cells give rise to the dorsal-ventral-midline epidermis (white arrowhead), muscle (magenta arrowheads), and the pharynx (yellow dotted line) (n=12/12). 2a/2b cells give rise to the anterior-lateral epidermis (white arrowheads), and muscle (magenta arrowheads) (n=6/6). 2A/2B cells give rise to the statocyst (orange arrowhead), gut, and a population of internal, midsection cells (cyan arrowheads) (n=4/4). The same worms shown here were also followed during embryonic development, shown in Fig. S1C. (G) Schematic diagrams of the cell types generated by each pair of blastomeres. All images of hatchlings were taken with a 10x objective, all embryo images were taken under 20x magnification. Images of specific tissues, regions, or insets were taken at high magnification under a 63x objective. Embryo and hatchling scale bars = 100μm. High magnification image scale bars = 50μm. See also Figure S1, Video S1, Video S2, Video S3, and Videol S4.
The highly regenerative acoel Hofstenia miamia possesses a population of collectively pluripotent stem cells, i.e. aPSCs, referred to as ‘neoblasts’14,15. The embryos of Hofstenia undergo a stereotyped cleavage program (Fig. 1B), enabling an investigation of the cellular source of aPSCs14,15. Hofstenia neoblasts express Piwi orthologs, are required for regeneration14, and enable homeostatic tissue turnover16. These cells are present in the worms upon hatching (Fig. S1A) and therefore must be made during embryogenesis. Single-cell transcriptome profiling suggests that the neoblast population of adult Hofstenia is composed of transcriptionally distinct subsets of cells that express markers associated with differentiated tissues16. Planarian neoblasts17–19 and cnidarian i-cells6 are also known to be transcriptionally heterogeneous. In view of this heterogeneity, two cellular mechanisms could explain the formation of neoblasts in Hofstenia: 1) they are derived from one embryonic lineage and become heterogeneous as they begin to differentiate, 2) they are derived from multiple, tissue-specific embryonic lineages – cell populations first acquire tissue type identity, and within these tissue-specific lineages, corresponding neoblast subtypes are specified. The latter scenario would bear similarity to stem cells in vertebrates, where tissue-associated stem cells originate in specific lineages, e.g. mesodermal progenitors give rise to muscle and muscle satellite cells and endodermal progenitors give rise to intestinal epithelia and intestinal stem cells.
To assess these mechanistic scenarios, we labeled and traced the blastomeres of Hofstenia embryos through development to hatching, and identified one pair of cells at the 16-cell stage as the major embryonic source of neoblasts. We then combined our ability to trace and isolate the neoblast lineage with a single-cell atlas of embryos and identified a molecular trajectory associated with the formation of neoblasts.
Results
Lineage tracing in transgenic embryos reveals the fate map of Hofstenia blastomeres
We utilized transgenic embryos with constitutive presence of Kaede protein, which can be photoconverted in individual cells (blastomeres) to trace each cell’s contribution to tissues in hatched worms, with the ultimate goal of determining the cellular sources of neoblasts (Fig. 1C–E, Fig. S1B), see generation and maintenance of transgenic lines section in Method Details). We found that the photoconverted labeling was stable, and persisted beyond the completion of embryogenesis (Fig. 1F). Hofstenia embryos display a stereotyped cleavage program with clear correspondence of blastomeres across embryos15 (Fig. 1B), which enabled us to systematically photoconvert each blastomere in 8-cell stage embryos and generate a complete fate map by correlating the distribution and morphology of labeled progeny to those of known tissue markers (Fig. 1E–G, S1A, S1C, and S1D). Overall, the highly reproducible and distinct labeling of tissues in these experiments highlighted that the photoconverted Kaede protein had high perdurance and was not leaky, making it an effective tool for lineage tracing.
Photoconversion of embryonic cells followed by time-lapse imaging and visualization of converted progeny in hatched worms identified the embryonic sources of major cell types (Fig. 1E–G, S1C–D, Videos S1–S3). Briefly, early cleavage in Hofstenia proceeds as follows: The first cleavage results in the 2-cell stage consisting of equal-sized cells (A and B). Next, these two cells divide asymmetrically, giving rise to the 4-cell stage with two small cells at the animal pole (1a and 1b) called micromeres, and two large cells at the vegetal pole (1A and 1B) called macromeres. The prefix “1” in the names of these cells reflects that they are the products of the first asymmetric cleavage of the A and B cells. The 1a and 1b micromeres divide, producing cells named 1a1/1a2 and 1b1/1b2 respectively, resulting in the 6-cell stage embryo. Finally, the 1A and 1B macromeres cleave asymmetrically to produce two corresponding daughter cells each (now prefixed with “2”, to reflect that they are produced by the second asymmetric cleavage): micromeres 2a and 2b, and macromeres 2A and 2B to reach the 8-cell stage15 (Fig. 1B). A previous embryonic fate-map study of another acoel had revealed that distinct cell fates were associated with blastomeres at the 8-cell stage27, and therefore, we focused our efforts on understanding the fates of blastomeres in 8-cell stage Hofstenia embryos.
Lineage tracing by photoconversion revealed that the animal-most progeny of 1a/1b micromeres, 1a1/1b1, produced the nervous system whereas the vegetal progeny, 1a2/1b2, gave rise to muscle, epidermis, and pharyngeal tissue (Fig. 1F–G). The second set of micromeres, 2a/2b, gave rise to muscle and the antero-lateral epidermis. The macromeres, 2A/2B, gave rise to multiple distinct tissues including the statocyst, gut, and a population of internal cells in the midsection of the animal along the anterior-posterior axis (Fig. 1F–G). Overall, these data showed that the fates of blastomeres are restricted by the 8-cell stage, and that some tissues such as the nervous system are derived from one source (1a1/1b1) whereas others such as muscle and epidermis are derived from multiple sources (1a2/1b2, 2a/2b).
We followed these blastomeres at the 8-cell stage during development to characterize the timing and modes by which these blastomere progenies internalized. 1a1/1b1 progeny labeled a patch of external cells during the Gastrula stage, but were internalized through delamination at a site adjacent to the “dimple” during the Dimple stage (Fig. S1C and Video S1). The progeny of the 1a2/1b2 pair exhibited a pattern in which half of the progeny internalized at the Dimple stage, whereas the other half remained on the exterior of the embryo until hatching (Fig. S1C). Single photoconversion experiments provided greater resolution into these distinct behaviors – the majority of 1a2 progeny internalized at the Dimple stage through apical constriction (Video S2), while the progeny of 1b2 largely remained on the exterior of the embryo until hatching (Video S3). The 2a/2b progeny labeled bilaterally symmetric patches of cells on the left and right side of the embryo throughout development (likely corresponding to future epidermis), and only a small proportion of them were seen to become internalized at the Dimple stage (likely corresponding to future muscle) (Fig. S1C). The majority of 2A/2B progeny were internalized at the Gastrula stage, with the exception of two bilaterally symmetric patches of cells that were internalized via delamination at the Dimple stage (Fig. S1C and Video S4).
These experiments also allowed us to correlate the broad spatial maps of blastomeres and their progeny in hatched worms (Fig. 1F–G, Figs. S1D–S1H, Videos S1–S4). The progeny of most blastomere pairs (derived from the two cells, A and B, made upon first cleavage), showed symmetry along the left-right axis and their fates can be summarized using data from embryos where both cells of a pair are converted (Figs. 1E–F show conversions of paired cells, Fig. S1D shows single blastomere conversions). Macromeres 2A/2B showed slight differences in left-right symmetry in that both produced gut and midsection cells, but only one, which we designate here as 2B, produced the statocyst. More notably, the 1a2 and 1b2 pair deviated more drastically from this pattern of left-right symmetry. The majority of the 1a2 progeny became internalized at the “dimple”, whereas the small proportion of the progeny that remained at the surface invaginated later during development to form the pharynx, an anterior structure (Fig. S1D, S1E, Video S2). The majority of 1b2 progeny remained on the external periphery of the embryo, and ultimately gave rise to the majority of the epidermis that covered the anterior, posterior, dorsal, and ventral midlines of the animal (Fig. S1D, Video S3). 1b2 progeny also gave rise to muscle, likely from a small proportion of cells that internalized at the Dimple stage (Fig. S1D). Given this distribution of progeny, we inferred that the position of 1a2 at the 8-cell stage corresponds to the anterior of hatched worms, and 1b2 corresponds to the posterior. Furthermore, we correlated the relationship of the animal pole of the embryos to the future adult axis by photoconverting the surface of the embryo at the site where 1a1/1b1 progeny were internalized. This resulted in the labeling of the dorsal epidermis in the hatched worms, suggesting that the animal-vegetal axis of the early embryo corresponds to the dorsal-ventral axis respectively (Fig. S1F). Therefore, in addition to showing the origins of major cell types, our fate map of the 8-cell embryo revealed the correspondence of embryonic and adult axes (Fig. S1G).
Progeny of the 3a/3b blastomeres resemble neoblasts
Among the various blastomere fates observed, the midsection cells derived from the 2A and 2B blastomeres were of interest as their parenchymal, midsection distribution was reminiscent of that of neoblasts (Fig. 1F–G and S1A). Given that the 2A and 2B macromeres also gave rise to the statocyst and gut, we sought further resolution on the origins of these distinct cell populations. We waited for these two blastomeres to cleave once more, and traced the fates of their daughter cells at the 16-cell stage. 2A and 2B cleave asymmetrically to generate micromeres, 3a and 3b, and macromeres, 3A and 3B (Fig. 2A). The change in nomenclature of the blastomeres, i.e. the prefix ‘2’ being replaced with the prefix ‘3’, indicates that 3a/3b and 3A/3B are the product of the third asymmetric cleavage of the macromere lineage. Overall, we found that the distinct cell populations observed in hatchlings derived from labeled 2A/2B blastomeres (statocyst, gut, midsection cells) were made by different daughter cells produced by 2A/2B.
Figure 2: Progeny of 3a/3b, but not of 3A/3B, are midsection cells that resemble neoblasts in spatial distribution.
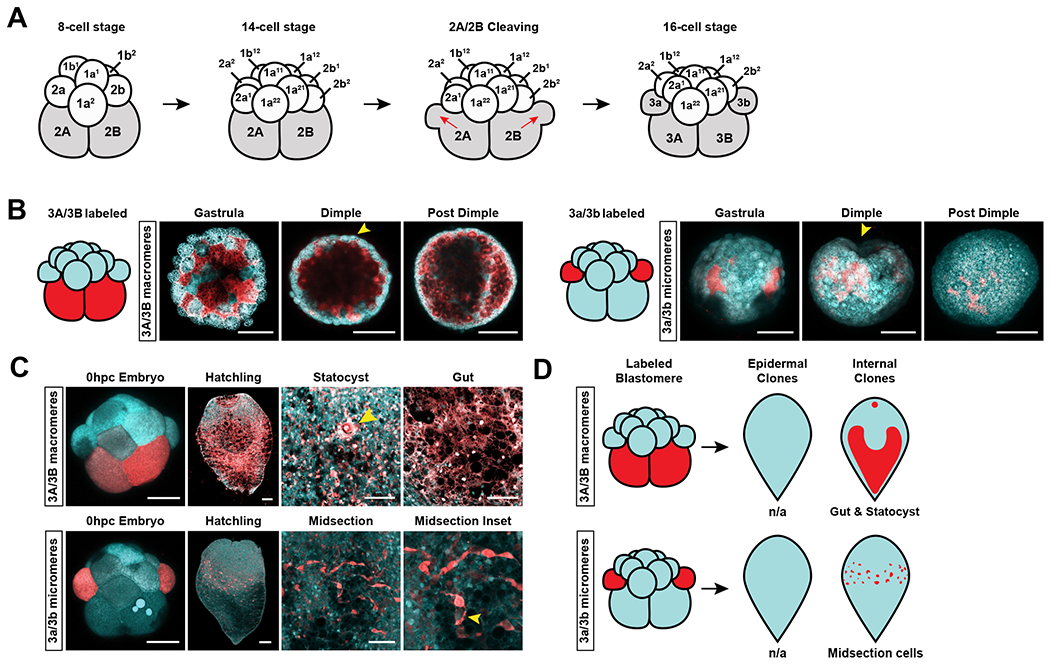
(A) Lateral view schematic of an 8-cell stage embryo cleaving to form the 16-cell stage embryo. 2A/2B blastomeres cleave to form micromeres 3a/3b and macromeres 3A/3B. Red arrows show the direction of cell division. (B) Tracing the fates of 3A/3B and 3a/3b progeny during embryonic development. Yellow arrow heads denote the site of the ”dimple”. Left: Embryonic time series of 3A/3B progeny after photoconversion. All progeny were internalized at the Gastrula stage. Right: Embryonic time series of 3a/3b progeny after photoconversion. Progeny occupied two external patches at the Gastrula stage that were subsequently internalized at the Dimple stage adjacent to the site of the “dimple” (yellow arrowhead) (C) Top row: 3A/3B progeny form the gut and statocyst (yellow arrowhead). Bottom row: 3a/3b cell progeny form a population of midsection cells that were seen amongst the 2A/2B progenies. Midsection inset shows cytoplasmic extensions of the cells (yellow arrowhead). (D) Schematic diagram of the distribution of 3A/3B and 3a/3b progeny in the hatched worm. Embryo and hatchling scale bars = 100μm. High magnification image scale bars = 50μm. See also Figure S2 and Video S4.
The progeny of 3A/3B macromeres became internalized at the Gastrula stage and gave rise to the gut and statocyst (Fig. 2B–D). The lack of muscle progeny from the only cells that generate endodermal progeny (3A/3B) suggests a possible lack of an endomesodermal precursor in Hofstenia, similar to observations in annelids20–22. The progeny of 3a/3b micromeres formed two patches on the exterior at the Gastrula stage that became internalized at the Dimple stage, and gave rise to the internal, midsection cells we had observed in the 2A/2B lineage-trace (Fig. 1F–G, 2B–D). These results gave us increased resolution for the origins of hatchling tissues derived from 2A/2B – the macromere daughters of 2A/2B, 3A/3B, made the gut and statocyst and the micromere daughters of 2A/2B, 3a/3b, made midsection cells. The lineage traces of the daughter cells (3a/3b and 3A/3B) together added up to the fate map of the mother cells (2A/2B), further corroborating our fate-mapping approach.
Importantly, for our objective of identifying the embryonic origins of neoblasts, the increased resolution from tracing the fates of 3a/3b and 3A/3B gave a cleaner label of the midsection population. In hatchlings derived from 3a/3b-labeled embryos, closer inspection of the midsection cells revealed that they possessed large, cytoplasmic extensions, and confirmed their internal, midsection distribution reminiscent of neoblasts (Fig. 2C, S1A, and S2). Single conversions of the 3a/3b cells showed that they both gave rise to these midsection cells, and showed left-right symmetry (Fig. S2). Thus, we next sought to test if the progeny of 3a/3b micromeres exhibited functional characteristics of neoblasts by assaying them together.
Progeny of the 3a/3b blastomeres exhibit functional characteristics of neoblasts
Hofstenia neoblasts exhibit three key functional properties: 1) contribution to newly regenerated tissue, 2) contribution to homeostatic turnover of mature tissue in the animal16, and 3) radiation sensitivity14. We sought to assess whether the progeny of 3a/3b also displayed these characteristics in hatched worms.
To test whether the 3a/3b progeny cells contributed to newly regenerated tissue, we photoconverted 3a/3b, amputated the worms upon hatching, and focused on the outgrowth of new tissue at the wound site, the blastema (Fig. 3A). If 3a/3b progeny cells can contribute to new tissue, we would expect the blastema to possess red, photoconverted cells. As a control, we converted the 1a1/1b1 micromeres that gave rise to neurons, which in theory should not contribute to regenerated tissue, and should not result in red labeling in the blastema. Strikingly, we detected substantial numbers of red, photoconverted cells in the blastema among the animals derived from 3a/3b-labeled embryos. In the 1a1/1b1-labeled animals, only a few red neurons, likely ones that were already present at the wound site, were present in the blastema during both head and tail regeneration (Fig. 3A). Additionally, we assessed the ability of the progeny of all other blastomeres at the 16-cell stage to populate the blastema and did not detect substantial contributions (Fig. S3A, S3B, and S3C), which suggests that the progeny of 3a/3b are the major source of cells in the blastema during regeneration.
Figure 3: 3a/3b micromere progeny exhibit functional characteristics of neoblasts.
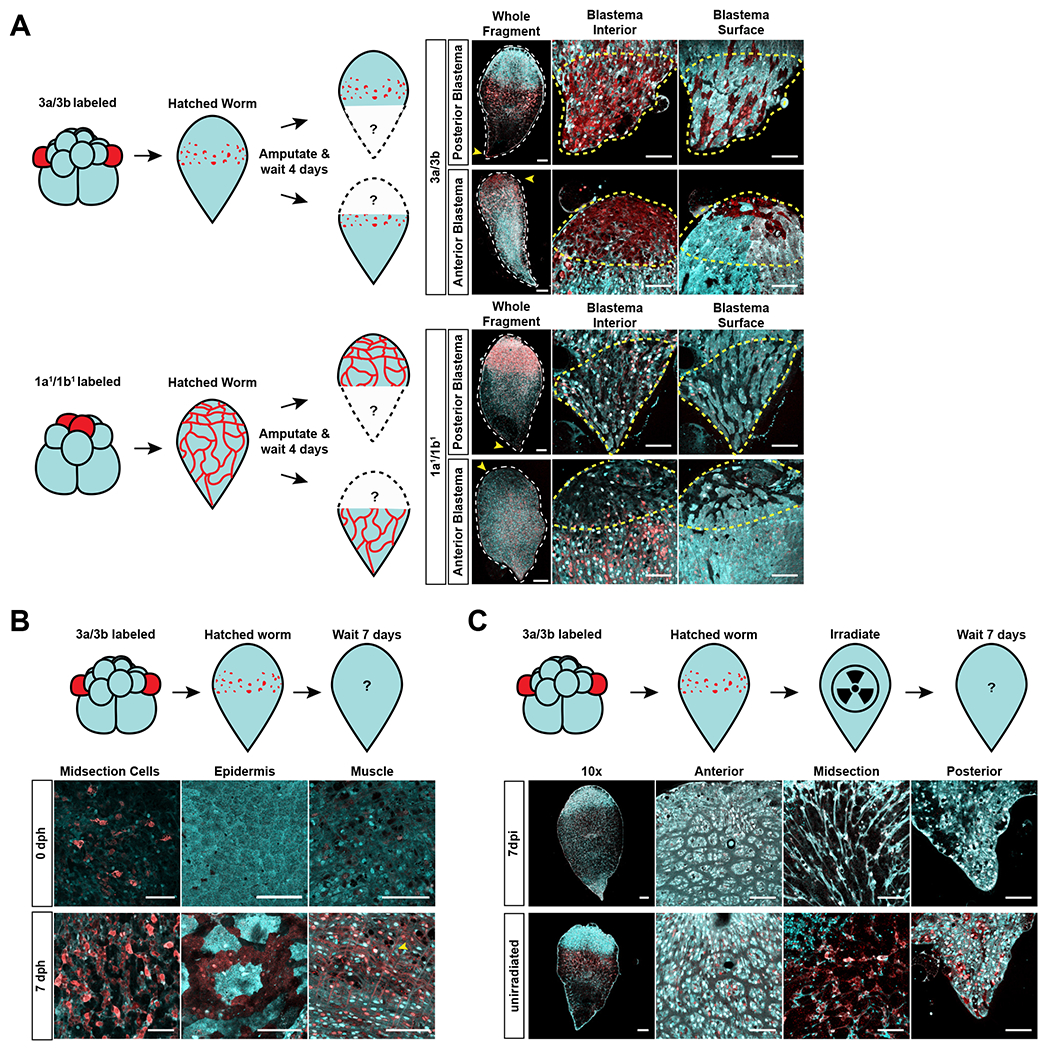
(A) Top Left: Schematic of experimental design for testing the contribution of 3a/3b progeny to the blastema. Top Right: Red labeling from the 3a/3b progeny spread to the blastema (yellow arrowhead in whole fragment images, outlined in yellow dashed lines in high magnification images of blastema) during anterior (n=5/5) and posterior regeneration (n=3/3), showing contribution to newly regenerated tissue. Both interior and surface z-slices of the blastema show the spread of red labeling to new tissue. Bottom Left: Schematic of experimental design for testing the contribution of 1a1/1b1 progeny to the blastema. Bottom Right: Red labeling from the 1a1/1b1 progeny did not spread to the blastema (yellow arrowhead in whole fragment images, outlined in yellow dashed lines in high magnification images of blastema) during anterior (n=3/3) or posterior regeneration (n=3/3), showing absence of contribution to newly regenerated tissue. Both interior and surface z-slices of the blastema do not show the spread of red labeling to new tissue. Images to the right of the hatchling images show high magnification views of relevant areas. Scale bars for hatchling images = 100μm. Scale bars for high magnification images = 50μm. Progeny of other blastomeres at the 16-cell stage also did not contribute to newly regenerated tissue– see Fig. S3B and S3C. (B) Top: Schematic of experimental design for testing the contribution of 3a/3b progeny to homeostatic tissue differentiation. Bottom: At 7 days post hatching (dph), 3a/3b labeled worms showed an increase in the number of midbody cells as well as presence of red labeling to epidermis and muscle (3/3), which were absent in animals that had just hatched (0 dph) (3/3). Scale bars = 50μm. Progeny of other blastomeres that normally do not contribute to epidermis or muscle showed no detectable contribution to these tissues during homeostasis – see Fig. S3D (C) Top: Schematic of experimental design for testing the sensitivity of 3a/3b progeny to radiation. Bottom: 3a/3b progeny are not detectable 7 days post irradiation (dpi) (4/4), in contrast to in unirradiated worms (3/3). Progeny of other blastomeres were not irradiation sensitive - see Fig. S3E. Images to the right of the hatchling images show high magnification views of relevant areas. Scale bars for hatchling images = 100μm. Scale bars for high magnification images = 50μm. See also Figure S3.
Given that neoblasts are the only somatic proliferative cells in Hofstenia, we expect them to contribute to continuous homeostatic turnover of tissues in the adult worm and to be sensitive to radiation14. To study homeostasis, we converted the 3a/3b blastomeres and imaged the worms one week after hatching (Fig. 3B). Compared to newly hatched worms, those imaged a week after hatching exhibited red labeling in epidermal and muscle cells. We also found that the midsection cell population, the putative neoblasts, had expanded in number in comparison to newly hatched, labeled worms (Fig. 3B). We did not, however, detect red labeling in neurons, pharynx, or the gut. This could be because the homeostatic turnover rates for these tissues are low during the first week upon hatching. Alternatively, it could be that there are other, minor sources of neoblasts that are contributing to these tissue types. It is also possible that the label is diluted because of the number of mitoses needed to make these tissues– or, in the case of the gut, because photoconverted protein would spread through a large syncytial tissue23. Meanwhile, the labeling of other blastomeres did not result in the expansion of red color to other differentiated tissue types (e.g., 1a1/1b1 progeny, which label neurons in hatched worms, did not contribute to labeled muscle or epidermis.) (Fig. S3D). This is consistent with the interpretation that other cell populations, derived from blastomeres other than 3a/3b, do not exhibit neoblast-like properties. Additionally, we found that the 3a/3b progeny cells, but not the progeny of other blastomeres, were completely ablated within seven days upon exposure to radiation (Fig. 3C and S3E).
We found that these functional characteristics of neoblasts are likely acquired by 3a/3b progeny late in development. piwi-1 was expressed among all embryonic cells until just before hatching at the Pigmented Prehatchling stage, when the expression of piwi-1 became regionally restricted to resemble a neoblast-like distribution. (Fig. S3F). Double-labeling of Hofstenia embryos with piwi-1 and the mitotic marker H3P showed that mitotic cells were always associated with piwi-1+ cells across development. Mitotic cells did not show regional restriction until the Pigmented Prehatchling stage, the same timepoint when we detected piwi-1 expression in a pattern reminescent of neoblasts (Fig. S3F). This suggested that neoblasts acquire the property of being the only proliferative somatic cells in Hofstenia right before hatching. Consistent with this finding, Pigmented Prehatchling stage embryos were able to regenerate both anterior and posterior structures upon amputation (Fig. S3G). These data suggest that although neoblasts are fated as early as the 16-cell stage, in 3a/3b micromeres, they likely do not acquire adult neoblast function until late in development, similar to planarian neoblasts11.
Given that the functional testing of 3a/3b progeny supported our hypothesis that 3a/3b are the likely source of neoblasts, we next asked if 3a/3b blastomeres are required for the formation of aPSCs in Hofstenia. Laser-ablation of 3a/3b blastomeres resulted in hatchlings where piwi-1 was still detected in its typical expression pattern (Fig. S3H). This could be due to regulative development, where other embryonic sources can compensate for the loss of the normal source of a tissue. To test for regulative capacities of other tissues in Hofstenia, we also ablated the 1a1/1b1 blastomeres, which give rise to the nervous system. In situ hybridization studies of the neural marker gad-1 showed a normal expression pattern in hatchlings derived from 1a1/1b1-ablated embryos (Fig. S3H). Altogether this suggests that Hofstenia embryos may be capable of regulative development where they are able to compensate for the loss of cells that would normally produce specific tissues. Regardless, although embryos can compensate for the loss of 3a/3b, our experiments consistently point to 3a/3b as the source of neoblast-like cells in Hofstenia.
Progeny of the 3a/3b blastomeres exhibit molecular properties of neoblasts
As an independent assessment of neoblast chatacteristics, we next sought to ask if 3a/3b progeny displayed molecular properties associated with neoblasts at the time of hatching. To determine the transcriptomic profile of 3a/3b progeny cells, we photoconverted 3a/3b, and applied fluorescence-activated cell sorting (FACS) to separate the cells based on converted (red) vs. unconverted (green) fluorescence. To obtain sufficient material for this experiment, we pooled hatched worms derived from 18 photoconverted embryos. Stage-matched early embryos in Hofstenia do not hatch perfectly synchronously, which meant 3a/3b progeny in some of the worms could include cells undergoing differentiation for postembryonic growth. This would impact their levels of red fluorescence as well as their transcriptional profiles. In view of this expected heterogeneity, we performed single-cell RNA-sequencing and sought to understand transcriptional diversity among red (3a/3b progeny) and green cells (progeny of other blastomeres) (Fig. 4A, Fig. S4A). Roughly half of the red cells expressed the neoblast marker piwi-1 highly, whereas the majority of green cells did not (p-value = 7.767e-10, Wilcoxon Ranked Sum Test) (Fig. 4B).
Figure 4: Single-cell profiling of 3a/3b micromere progeny shows they exhibit molecular characteristics of neoblasts.
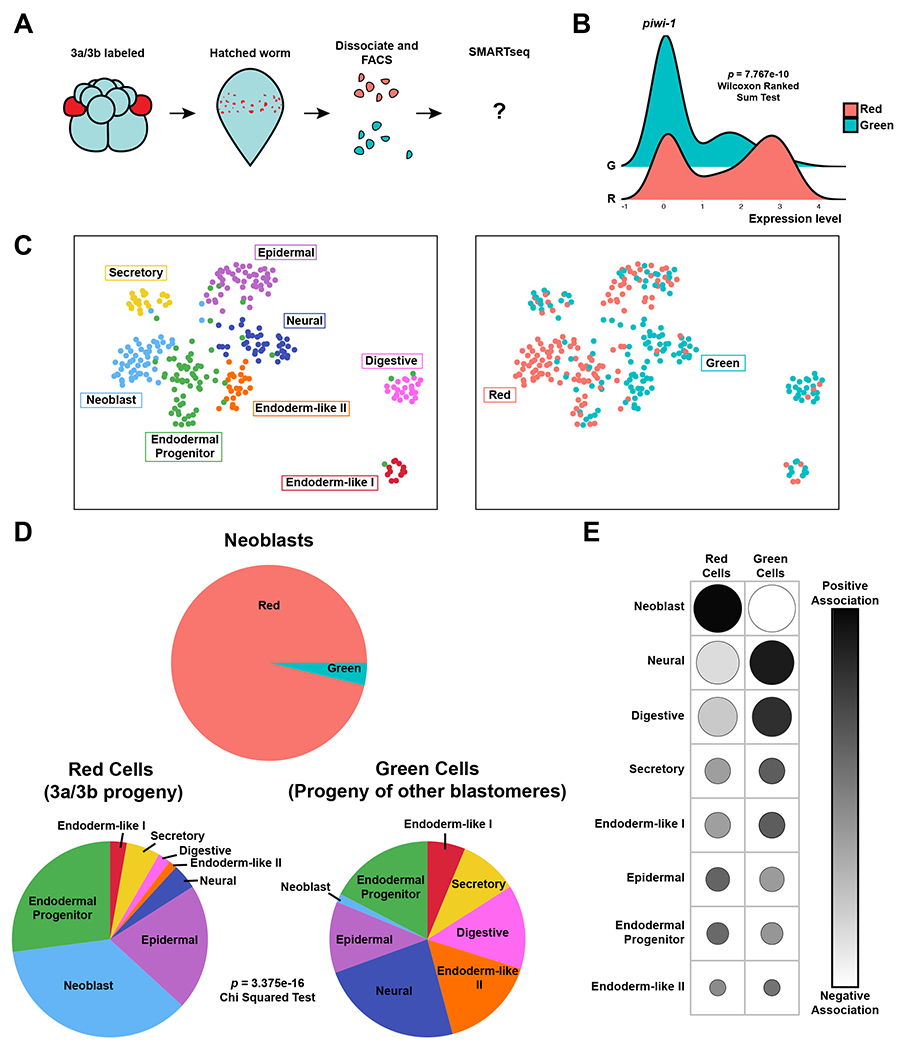
(A) Schematic diagram of experimental design. Worms with labeled 3a/3b progeny were dissociated, and cells sorted based on red vs green (shown in cyan) fluorescence. A total of 288 sorted cells were then sequenced via SMARTseq. (B) Ridgeplot of the relative expression levels of the neoblast marker piwi-1 between red and green cells (p-value = 7.767e-10, Wilcoxon Ranked Sum Test). (C) Left: UMAP of sequenced cells showing cluster identity, which corresponds to clearly identifiable cell types, including neoblasts. Right: Corresponding UMAP with colors showing the photoconversion status of red (3a/3b progeny) and green cells (progeny of other blastomeres). The vast majority of the neoblast cluster is composed of red cells (3a/3b progeny). (D) Top: Pie chart showing the proportion of red (52/54) and green (2/54) cells in the neoblast cluster. Bottom: Pie charts showing the proportion of red (3a/3b progeny) and green (progeny of other blastomeres) cells that belong to different cell clusters. Red cells have a significantly larger proportion of neoblasts compared to green cells. Chi squared test of independence showed statistically significant association of cell types to their red vs green label color (p-value = 3.375e-16). (E) Contingency table showing the Pearson residuals for each cell type and their label color (red vs green). Darker shades show positive association, negative association. Larger dots represent a higher degree of association, while smaller dots represents a smaller degree. Overall, red cells (3a/3b progeny) are largely defined by the presence of neoblasts, whereas green cells (progeny of other cells) are largely defined by the absence of neoblasts and the presence of other cell types. See also Figure S4, Table S1, and Table S3.
Next, we performed unsupervised clustering on a pooled dataset of red and green cells, and assigned identities to these clusters by assessing the expression of known marker genes for cell types determined in the Hofstenia single-cell atlas16 (Fig. S4B, Table S1). We identified seven cell clusters that likely represent epidermal, neural, digestive, endodermal progenitor, endoderm-like II, endoderm-like I, neoblasts (Fig. 4C), and an eighth cell cluster that we found to be present in a previously published single cell RNAseq dataset based on common marker gene expression16. We determined this eighth cluster is likely a secretory cell type based on marker gene expression and GO analysis which included terms such as vesicle mediated transport and protein glycosylation (Fig. 4C, S4C, and Table S1). The neoblast cluster consisted almost entirely of red, photoconverted cells (52/54 cells), suggesting that the majority of isolated cells that exhibited a neoblast-like transcriptomic profile were 3a/3b progeny (Fig. 4D). This was further highlighted when the proportion of cell types were visualized in a pie chart, showing a stark difference in the numbers of red vs. green cells placed in the neoblast cluster (Fig. 4D).
Although imaging of hatchlings derived from 3a/3b-converted embryos did not show clear labeling of other, non-neoblast cells, we found red cells within other cell type clusters in our single-cell analysis (Fig. 4C and 4D). These cells could result from 1) differentiation of 3a/3b-derived neoblasts, or 2) direct contribution of 3a/3b to differentiated tissue. The presence of red cells in the endodermal progenitor cluster, which represents putatively lineage-primed progenitors16, suggests that tissues are undergoing postembryonic differentiation at or soon after hatching. The larger proportion of red cells in the epidermal cluster relative to other differentiated cell clusters could reflect differences in rates of differentiation of tissues, e.g. neoblasts may be contributing to epidermis more than to other tissues upon hatching. Overall, these data suggest that 3a/3b can contribute to differentiated tissues, directly or indirectly (through neoblasts) upon hatching, and further experimentation is needed to distinguish between the relative contributions of these two mechanisms. Here, we were focused on whether bona fide neoblasts are derived from 3a/3b or from other blastomeres, and our analysis strongly indicated that the vast majority of cells that match the transcriptional profile of neoblasts were indeed derived from 3a/3b (Fig. 4D).
We next sought to rigorously assess the significance of the different proportions of cell types found within the red and green cells. A chi-squared test of independence showed that the composition of cell types in the red and green cells is statistically significantly different (p = 3.375e-16) (Fig. 4D). We then determined the degree of association of each cell type to either the red vs. green cells by generating a contingency table of Pearson residuals (Fig. 4E). We found that the number of cells in the neoblast cluster was a key defining factor of both red and green cells, with red cells having a positive correlation with neoblasts and green cells having a negative correlation with neoblasts. Furthermore, the red cells were negatively correlated with the majority of other cell types detected, whereas green cells were positively correlated. Altogether, this suggests that the red cells are largely defined by the presence of neoblasts (as defined based on transcriptome profiles), while the green cells are defined by their absence (Fig. 4E). This evidence suggests that 3a/3b blastomeres are a major source of neoblasts in Hofstenia.
Embryonic single-cell atlas and cell lineage tracing identify a molecular trajectory for neoblast formation
While the acquisition of neoblast functional characteristics occurs late in development (Fig. S3F and S3G), the knowledge that 3a/3b micromeres are fated to produce neoblasts can be leveraged to study the ontogeny of neoblasts. Therefore, we next sought to identify molecular components that are associated with the formation of aPSCs in Hofstenia embryos.
We reasoned that by profiling transcription at the single-cell level over developmental time, we could identify cells whose transcriptional states implicate them as part of the neoblast lineage and ask which molecular players drive their progression towards a bona fide neoblast identity. To generate an embryonic single-cell atlas, we used the droplet-based platform, InDrops24, to obtain the transcriptomes of ~51,000 cells spanning 8 different timepoints of development from 30 hours post laying (hpl) to just before hatching (145 hpl) (Fig. 5A). Uniform Manifold Approximation and Projection (UMAP) analysis of a merged dataset of these embryonic cells and of cells from hatched worms16,25 recovered a graph with a branching pattern (Fig. 5B). Each branch was populated with cells from different stages in correct chronological order, with cells from hatched worms lying at the tips of the branches. Projection of known differentiated cell markers present in the hatchling dataset showed that each of the branch tips corresponded to a differentiated cell type, suggesting that our dataset likely captured cells undergoing differentiation towards these cell fates (Fig. 5B and Fig S5A).
Figure 5: Combining embryo single-cell analysis and neoblast cell-lineage tracing reveals a molecular trajectory for neoblasts during development.
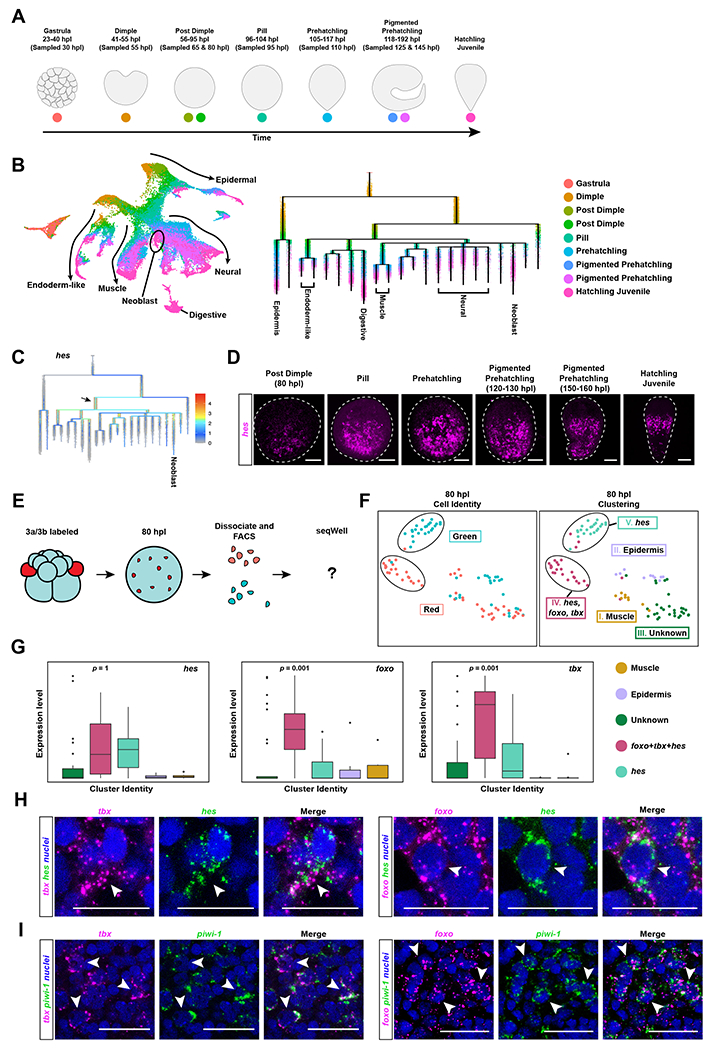
A) Schematic highlighting Hofstenia developmental stages and the developmental timing at which they were sampled to generate the single-cell RNA-seq dataset. (B) Left: UMAP plot of the embryonic dataset merged with the Hatchling Juvenile dataset16 to obtain endpoints in development. Arrows and tissue-type labels highlight the UMAP topology, and the expression of known marker genes of the corresponding tissues as shown in Fig. S5A. Right: URD trajectory inference tree showing the putative molecular trajectories of cells in the form of a tree, with the branch tips annotated with known differentiated cell types16. Dots, representing cells, are colored based on developmental time points shown in A. (C) Expression levels of the gene hes projected onto the URD tree. We see high expression levels of hes at an internode that is populated largely by cells from the Post Dimple stage (80 hours post laying (hpl)) (black arrow) (D) in situ hybridization of hes reveals expression first detectable at the Post Dimple stage (80 hpl) which culminated in a neoblast-like pattern in hatchlings. (E) Schematic workflow for sorting and sequencing a total of 96 cells from Post Dimple stage (80 hpl) embryos with photoconverted 3a/3b progeny at single-cell resolution. Red cells represent 3a/3b progeny that should make neoblasts, whereas green cells are progeny of other blastomeres that give rise to differentiated cell types. (F) Left: UMAP showing photoconversion status (red, 3a/3b progeny; cyan, progeny of other, unconverted blastomeres). Right: UMAP showing cluster identities. Muscle (Cluster I; mustard) and epidermal (Cluster II; light purple) cells were detected. One cluster did not yield any markers with enriched expression, and is labeled as “Unknown” (Cluster III; dark green). Clusters that were predominantly composed of 3a/3b progeny (Cluster IV; cranberry) and predominantly composed unconverted progeny (Cluster V; light green) were detected (circled in black). Both of these clusters expressed hes, but the Cluster IV, composed of 3a/3b progeny, show significantly enriched expression of foxo and tbx. (G) Box plots showing hes was expressed in both of Cluster IV and Cluster V, corroborating the lineage analysis in B that hes marks undifferentiated embryonic progenitors for many cell types, foxo Iand tbx were highly enriched in Cluster V, which consists primarily of 3a/3b progeny. Data represented as mean ± SEM. (H) Double in situ hybridization of foxo and tbx with hes in Pigmented Prehatchling (120 hpl) embryos shows co-localization (white arrowheads). (I) Double in situ hybridization of foxo and tbx with piwi-1 in hatched worms shows co-localization (white arrowheads), confirming that both foxo and tbx mark a subset of neoblasts. Scale bars for embryo and hatchling images = 100μm. Scale bars for high magnification images = 50μm. See also Figure S5, Table S1, Table S2, and Table S3.
To explicitly identify putative differentiation paths and the molecular components underlying them, we applied the trajectory inference tool URD26 and obtained a tree with the Gastrula stage (30 hpl) cells at its root and hatchling cell clusters placed at its tips (Fig. 5B). The branching order in this tree showed the timing of the divergence of molecular trajectories during development for cell types in hatched worms: the epidermal clade diverged first, at the Dimple stage (50 hpl); next, the endodermal trajectory diverged at the Post Dimple stage (65 hpl); muscle, neural, and neoblast branches formed a polytomy at the Pill stage (95 hpl). Surprisingly, the URD topology showed that trajectories for various tissue types diverge transcriptionally at a much later time relative to our fate map, which showed cells had become fated to become neuron, muscle, epidermis, and gut as early as the 8-cell stage. This could either mean that transcriptional differences between embryonic lineages do not become pronounced enough to be detected through single cell RNA-seq until the Gastrula stage, or active differentiation of tissues through transcriptional changes does not occur until then. To determine whether our trajectory analysis captured the transcriptional dynamics of Hofstenia embryos effectively, we next applied differential expression analysis to the terminal branches. This yielded putative marker genes for the corresponding cell types (Table S2). in situ hybridization studies of these marker genes corroborated both the timing of developmental expression and tissue identities of the cells placed along these branches (Fig. S5B and S5C). For example, the gut lineage marker s61a2 was found to be expressed starting at the Post Dimple stage (80 hpl) in an internal region of the embryo. This internal expression pattern persisted until the Hatchling Juvenile stage, where it matched the expected distribution of digestive cells (Fig. S5C and S1A). Overall, these data suggested that the trajectory tree constructed by URD does indeed reflect transcriptional changes in the embryonic cells of Hofstenia. Interestingly, the well-studied marker of neoblasts in hatched worms, piwi-1, was expressed broadly in cells across stages of development, corroborating in expression studies via in situ hybridization in embryos (Fig. S3F, S5A). Our dataset did identify markers that labeled the neoblast trajectory in embryos more specifically, one of which, neo, specifically labeled neoblasts in hatchlings (Fig. S5A, S5B, S5C, and Table S2).
We next probed the URD branching topology to identify genes enriched in cells placed at the internodes leading to the neoblast branch (Table S2). These genes would represent putative regulators of neoblast formation. Among the handful of transcription factors found to be significantly highly expressed at internodes, hes emerged as also enriched in the terminal branch leading to neoblasts in hatched worms (Fig. 5C, Table S2). We found hes to be expressed in cells in the interior of embryos starting at the Post Dimple stage (80hpl), corroborating the timing of expression observed in the URD topology (Fig. 5D). These hes-expressing cells were distributed in a polarized manner in embryos and in hatchlings, where they matched the expression pattern of neoblast marker genes and were co-expressed piwi-1 (Fig. S5D). These data suggest that hes could be expressed in embryonic cells that give rise to neoblasts in hatched worms.
Given that the URD topology infers molecular trajectories and not cellular lineages, the enrichment of hes in deep internodes leading to the neoblast branch could be the result of: 1) hes being expressed in embryonic progenitors for endodermal, muscle, and neuronal tissues in addition to progenitors for neoblasts, 2) hes being expressed only in progenitors for other tissues but not in neoblast progenitors. To distinguish between these possibilities, we isolated neoblast progenitors via FACS from Post Dimple stage (80hpl) embryos where 3a/3b had been photoconverted at the 8-cell stage (Fig. 5E and S5E). Unsupervised clustering of the transcriptomes of converted (red, neoblasts progenitors) and unconverted (green, other embryonic progenitors) cells yielded clusters that were composed of both red and green cells. These included clusters enriched for muscle and epidermal markers, as well as one that did not have any genes with enriched expression, referred to as “unknown” (Fig. 5F and S5F). More strikingly, the data identified two cell clusters – one predominantly populated with red cells, and the other with green cells – that yielded clearly enriched, specific marker genes. Cells in both clusters showed expression of hes (Fig. 5F, 5G, and S5F), corroborating the URD trajectory-based hypothesis that hes is present in progenitors for both neoblasts (red cells; 3a/3b progeny) and other tissues (green cells; progeny of other blastomeres).
Differential expression analysis across these clusters additionally revealed genes that were specifically enriched in a subset of neoblast progenitor cells (red cells), including two transcription factors, foxo and tbx (Fig. 5F, 5G, and Table S2). This suggested that hes may be a marker for progenitor-like states in general during development, but foxo and tbx would be specific to the neoblast lineage. foxo and tbx were co-expressed among 3a/3b progeny at the Post Dimple stage (80 hpl), consistent with having functions in the same lineage of cells (Fig S5G). To further bolster the hypothesis that foxo and tbx are associated with the neoblast lineage in embryos, we performed in situ hybridization studies which showed that both foxo and tbx were expressed in cells that also express hes during embryogenesis (Fig. 5H and S5H). Additionally, association of foxo and tbx with neoblasts was supported by observations in hatchlings: 1) foxo- and tbx-expressing cells were distributed in a neoblast-like pattern in hatchlings (Fig. S5I), 2) Double in situ hybridization of foxo and tbx with piwi-1 in hatchlings showed co-localization, corroborating that both these transcription factors are expressed in neoblasts (Fig. 5I), 3) foxo and tbx RNAi resulted in reduced regeneration and foxo RNAi animals showed diminished piwi-1 expression during homeostasis (Fig. S5J, S5K, and S5L), 4) foxo and tbx were enriched in the neoblast cluster of 3a/3b progeny in hatched worms (Fig. S5M). Altogether, the data show that hes, foxo, and tbx are part of the molecular trajectory for neoblast formation and point to the possibility that foxo and tbx may be specific regulators of neoblast formation during embryonic development.
Discussion
Here we generated a complete fate map of cells in early stage embryos of the highly regenerative acoel worm Hofstenia miamia, which showed important aspects of Hofstenia embryogenesis including the correspondence of embryonic and adult body axes and the lack of a common endomesodermal progenitor. Overall, the fates of Hofstenia blastomeres differ in many aspects from the fate map for another acoel 27, including the notable lack of an endomesodermal progenitor, a departure from the classical definition of gastrulation20,21. However, the possible lack of a common endomesodermal progenitor is not without precedent and has been reported in an annelid22. Most strikingly, our study revealed a cellular source of neoblasts, the aPSCs of acoels. Furthermore, we identified a molecular trajectory associated with the formation of these aPSCs.
A cellular mechanism for neoblast formation
The formation of neoblasts could have occurred through two different mechanisms. In one scenario, multiple embryonic cell lineages could contribute to the neoblast population in adults (Fig. 6A). In this case, tissue-associated neoblast subtypes could be derived from the distinct embryonic sources of their corresponding differentiated tissues. In the alternative scenario, the neoblast lineage could arise from a singular source and acquire heterogeneous, tissue-associated transcriptional profiles over time (Fig. 6A). We found that cells with neoblast characteristics arise from a specific set of early embryonic blastomeres called 3a and 3b, albeit they acquire these properties late in development. Altogether, the lack of detectable contribution to regeneration and homeostatic turnover, insensitivity to lethal doses of irradiation, and extremely low proportion of neoblast-like cells detected among the progeny of all other blastomeres suggests that the 3a/3b blastomeres are the major source of neoblasts during Hofstenia embryogenesis. Thus, our data support the hypothesis that neoblasts likely form from a singular source early during development. Future studies of neoblast dynamics in adult worms will reveal whether the heterogeneous subsets of neoblasts formed in this manner are transcriptionally plastic and interconvertible in adult Hofstenia, as recently demonstrated in planarians28.
Figure 6: Models for the cellular and molecular origins of aPSCs.
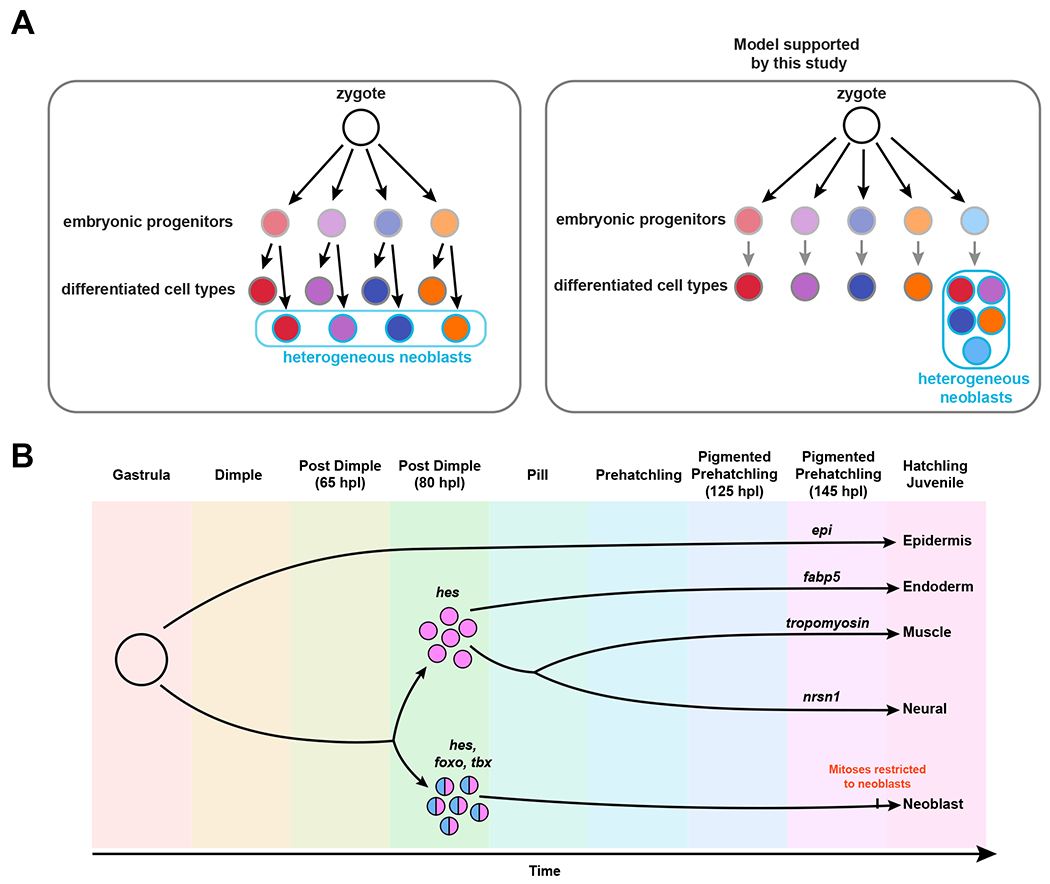
(A) Schematics showing two hypotheses about the formation of heterogeneous aPSCs (neoblasts). Left: Scenario where distinct embryonic progenitors for differentiated tissues give rise to corresponding tissue-associated neoblast subtypes. Right: Model supported by our data, where the neoblast lineage becomes distinct from other cell lineages early in development, and become heterogeneous over time. Light grey outlines represent embryonic cells, dark gray outlines represent differentiated cell types in hatched worms, blue outlines represent neoblasts in hatched worms, fill colors correspond to different cell types: endoderm I (red), epidermis (purple), neural (dark blue), endoderm II (orange), neoblasts (light blue). B) Model depicting transcriptional lineages in development, highlighting hes as a marker of progenitors for multiple cell types, including neoblasts, and foxo and tbx as specific to the neoblast lineage. Although the neoblast lineage is distinct early in development, functional neoblasts, as indicated by restriction of mitoses to piwi-1+ cells likely occurs late, close to hatching.
The origin of aPSCs from a specific set of early embryonic cells is reminiscent of how the germline originates in some species29. In one species of annelid worm, the same early embryonic cell gives rise to the germline and to a putative stem cell population30– although the full potentials of these stem cells remain to be explored. In Hofstenia, neoblasts are expected to ultimately form the germline as animals reach sexual maturity16, which suggests that 3a/3b could be a source both for aPSCs and the germline.
A molecular trajectory for neoblast formation
Our finding that one pair of cells serves as the major source of aPSCs in Hofstenia enabled us to obtain a molecular trajectory specifically associated with neoblast formation (Fig. 6B). Large-scale single-cell transcriptome sequencing can be used to infer molecular trajectories, which serve as a hypothesis for molecular processes that may underlie cellular lineages in development. We generated these hypotheses using an embryonic single-cell atlas for Hofstenia, and then we labeled (by photoconversion of 3a/3b) and isolated the embryonic precursors of neoblasts to directly assess the presence of hypothesized molecular components. The three transcription factors we identified in the neoblasts molecular trajectory all have known roles in regulating pluripotency and differentiation in other species. Homologs of Hes, which appears to be expressed in precursors of neoblasts and other tissues, are known to regulate differentiation of mouse embryonic stem cells (ESCs), where oscillations in the levels of Hes expression result in differential fates of stem cells31. FoxO and Tbx proteins, which appear to be specific to neoblast precursors in Hofstenia, are required for pluripotency in human and mouse ESCs respectively32,33. FoxO proteins are also known to be necessary for continuous self-renewal of hydra i-cells34. Future investigations of the mechanisms of action of these transcription factors in Hofstenia embryos will reveal if the roles they play in neoblast biology are similar to those observed in other systems.
Future directions
Our study reveals the embryonic cellular source of adult pluripotent stem cells, which are present in many lineages across the animal tree of life and therefore are an important feature of animal biology. The identification of an embryonic neoblast lineage and its molecular components serves as an entry point to dissect genetic mechanisms for the formation and maintenance of aPSCs during embryonic development. In addition to deciphering the key regulators of stem cells, these mechanisms will also enable a robust assessment of the putative homology of aPCSs in distantly-related species.
Limitations of the Study
The lineage tracing performed in this study relies on a photoconverted protein, which showed high stability in over the time courses followed, but this is approach is sensitive to differential rates of cleavage across embryonic cell lineages. For example, it is possible that the signal would be too dilute in daughter lineages that underwent high rates of cell proliferation, limiting the ability to detect complete detection of all blastomere progeny. The plate-based sequence analyses alleviate these concerns to some extent, as large discrepancies between photoconversion-based fate-mapping would be detectable via those data.
STAR Methods Text
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mansi Srivastava (mansi@oeb.harvard.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed materials transfer agreement. Plasmids with transgene constructs generated in this study are available from Addgene and the accession number is provided in the key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-H3P | Santa Cruz Biotechnology | Cat#sc-374669 |
| Hofstenia specific Ab Anti-tropomyosin | GenScript | N/A (Available from the Lead Contact upon request) |
| Chemicals, peptides, and recombinant proteins | ||
| Calcein AM | Invitrogen | Cat#65-0853-39 |
| Instant Ocean sea salt | Instant Ocean | Cat#SS15-10 |
| Agarose SeaKem® GTG™ | Lonza | Cat#50070 |
| TOPRO3 | Thermofisher | Cat#T3605 |
| Dextran, Fluorescein, 10,000 MW | Invitrogen | Cat#D1820 |
| 1X SceI Buffer | Benchmade | Cat#15140122 |
| I-SceI enzyme | NEB | Cat#R0694S |
| Cutsmart Buffer | NEB | Cat#B7204 |
| Critical commercial assays | ||
| KAPA Library Quantification kit | Roche | Cat#7960140001 |
| NucleoSpin® Gel and PCR Clean-Up kit | Macherey-Nagel | Cat#740609 |
| Deposited data | ||
| Gene Sequences | NCBI GenBank | hes (OP681623) foxo (OP681624) neo (OP681625) gland (OP681626) tbx (OP681627) grn (OP681628) epi (OP681629) s61a2 (OP681630) nrsn1 (OP681631) abcb6 (OP681632) nfic (OP681633) hexb (OP681634) |
| Sequencing Data | NCBI SRA | PRJNA889328 (FACS sorted 3a/3b progeny at hatching) PRJNA887118 (FACS sorted 3a/3b progeny at 80hpl) PRJNA888438 (Embryonic and Postembryonic datasets) |
| Experimental models: Organisms/strains | ||
| Hofstenia miamia | The animals used in this study are derived from random matings of the progeny of the original 120 worms collected in Bermuda in 2010. | N/A (Available from the Lead Contact upon request) |
| Recombinant DNA | ||
| Plasmid tubr::Kaede | Generated for this paper; deposited to Addgene | Available from the Lead Contact upon request; Addgene plasmid ID: 193054 |
| Software and algorithms | ||
| Seurat | Hao et al. 41 | https://satijalab.org/seurat/ |
| URD | Farrell et al.26 | https://github.com/farrellja/URD |
| R | N/A | https://www.r-project.org/ |
| All R scripts for single cell RNAseq analysis | This manuscript is the source of the R scripts for analysis | https://github.com/JulianKimura/Kimura_2022_Rscripts |
| Code used to generate SMARTseq matrices | This manuscript is the source of the code used to generate SMARTseq matrices | https://github.com/JulianKimura/SMARTseq_Pipeline |
| Other | ||
| quartz needles | Sutter Instrument | Cat#QF100-70-10 |
| Nunc™ bottom glass dishes | Thermo Scientific™ | Cat#150682 |
| Illumina Nextseq 500 | Illumina | Cat#SY-415-1001 |
| Illumina Novaseq 6000 | Illumina | Cat#20012850 |
| Moflo Astrios EQ | Beckman Coulter | Cat#B25982 |
Data and code availability
Single-cell RNA-seq data have been deposited at SRA under Bioproject Accession numbers: PRJNA889328, PRJNA887118, and PRJNA888438 for the FACS sorted 3a/3b progeny dataset at hatching, FACS sorted 3a/3b progeny dataset at 80 hours post laying, and the embryonic and postembryonic datasets respectively. Gene sequences in this study are deposited to GenBank. They are publicly available as of the date of publication. Accession numbers for the gene sequences in Genbank are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at Github (https://github.com/JulianKimura/Kimura_2022_Rscripts for single cell RNAseq analyses in R, and https://github.com/JulianKimura/SMARTseq_Pipeline for generating SMARTseq matrices) and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The laboratory population of H. miamia adults represent many generations derived from 120 sexually reproducing worms collected from Bermuda in 201014,15. The embryos used in this study were the progeny of random matings of these worms, Adult Hofstenia were cultured at 21°C in plastic boxes containing 1 Liter of filtered artificial sea water with 37 parts per thousand Instant Ocean sea salt (AFSW). Twice a week, boxes were cleaned, AFSW renewed, and animals were fed freshly-hatched artemia. Embryos were laid spontaneously, in clutches, on the plastic substrate.
METHOD DETAILS
Generation and maintenance of transgenic lines
We generated a stable transgenic line that expresses the photoconvertible protein Kaede under the control of an alpha-tubulin (tuba) promoter. Identification and cloning of regulatory elements, mRNA injection, and transgenic animal care were done using the protocols developed in Ricci and Srivastava23. The transcript sequence of the tuba gene were identified in the Hofstenia transcriptome and the corresponding genomic locus was determined via BLAST. Primers were designed to amplify regions located 5’ to the ATG and 3’ to the STOP codon from genomic DNA (Genbank Genome accession SCFE00000000, tuba promoter genomic coordinate scaffold1:46,289,791-46,291,668, and tuba 3’UTR coordinate scaffold1:46,293,025-46,293,590). A cassette with Kaede-encoding sequence flanked by the tuba promoter and 3’ UTR and SceI meganuclease sites was cloned into the backbone vector. Following cloning, plasmid isolation and sequencing were performed to assess proper fragment insertion.
Prior to injection, 4 μg of the desired plasmid was fully digested with 4 μl of I-SceI enzyme (Cat#R0694S), in a 50 μl reaction with 1X Outsmart buffer (Cat#B7204), left 1 to 2 hrs at 37 °C, followed by 20 minutes of heat inactivation at 65 °C. The digested plasmid was loaded in a 0.8% agarose SeaKem® GTG™ (Cat#50070, Lonza) gel. The fragment corresponding to the transgenic cassette was cut from the gel, purified with the NucleoSpin® Gel and PCR Clean-Up kit (Cat#740609, Macherey-Nagel), eluted in 15-30 μl of elution buffer and quantified with a NanodropND-1000 spectrophotometer. The injection solution was prepared in a volume of 10 μl, by assembling the following reagents in nuclease-free water, in order to obtain the given final concentration: digested DNA (10-25 ng/μl); Fluorescein Dextran (D1820, Invitrogen; 1.25 μg/μl); 1X I-SceI Buffer; I-SceI enzyme (Cat#R0694S, NEB; 0.375 U/μl).
To inject embryos, injection dishes were prepared, where an injection mold (composed of 300μm x 300μm square pins) was pressed into molten agarose in a 60x15mm culture dish. Individual embryos were pressed into the 300μm x 300μm holes created by the mold to keep in place during the injection process. A quartz needle (Cat#QF100-70-10, Sutter Instrument) was back-loaded with 0.75 μl of the injection solution and manipulated with Narishige MMN1 and MO202U micro-manipulators. Injections were performed under a Leica MZ10F Stereomicroscope, using a Pico-liter Microinjector (Cat# PLI-90A, Warner Instruments). After injection, embryos were placed in artificial seawater (ASW) with penicillin and streptavidin antibiotics (Cat#15140122, Thermofisher Scientific) at 5U/ml and left to grow in a 23°C incubator. The embryos were placed in normal ASW only after 5 days.
Based on data from the Hatchling Juvenile single cell RNAseq dataset, tuba mRNA in Hofstenia is expressed highly among the neoblasts of hatched juvenile worms16. However, in the transgenic embryos, Kaede protein fluorescence was detected in high amounts in the germline and oocytes, resulting in embryos and hatchling juvenile worms that showed ubiquitous fluorescence. This allowed for the fate-mapping experiments to take place.
Photoconversion and imaging for fate mapping
To photoconvert early embryonic cells, we first mounted embryos on a glass slide in artificial seawater (ASW), and lightly compressed them with a glass coverslip with “clay-feet”. We then rolled the embryos underneath the coverslip to orient the blastomere of interest towards the objective. Once embryos were properly mounted, we used a Leica SP8 confocal microscope under the 63x oil objective to photoconvert the blastomere of interest. We found that a laser power of 70% under the 405nm laser was sufficient to photoconvert the blastomeres after a 3-4 seconds of exposure. The length of time spent photoconverting was variable depending on the amount of Kaede fluorescence a particular individual had. After photoconversion, embryos were recovered from their slide by carefully peeling back the coverslip and using a glass pipette. All kaede transgenic embryos were protected from ambient light at all times, as we found that even low levels of ambient light could trigger some levels of photoconversion in the worm. To image photoconverted embryos, we mounted them in between a glass slide and a coverslip with “clay-feet” in ASW. Imaging of embryos was done with the Leica SP8 confocal microscope under the 20x objective. For experiments where tissue contributions of blastomeres were observed, we imaged worms immediately after hatching. To image photoconverted hatchlings, we compressed the animals until they were no longer able to move. To do this, we mounted them in between a glass slide and a coverslip in ASW. We used very minimal amounts of clay for the clay-feet to ensure sufficient compression of the animal. A “whole body” image was taken under the 10x objective, and higher magnification imaging which showcased the different labeled cell types was done using the 63x oil objective. Hatched worms were all imaged live.
Irradiation
Irradiation experiments were performed using a Radiation International, INC. Model B34 machine. All animals were placed in 60 x 15mm culture dishes in ASW and irradiated (10,000 rads) for 30 minutes. Animals were allowed to recover in normal ASW in a 23°C incubator, and were imaged after 7 days.
Regeneration assays
To perform regeneration assays, we placed recently hatched animals into a 70mm petri dish lined with Whatman filter paper. The animals were then cut in half along the transverse plane using a razor blade (Fine Science Tools Item no. 10316-14) and a Leica MZ10F stereomicroscope. The resulting head and tail pieces were then kept in separate wells in a 24-well plate to prevent the head pieces from cannibalizing the tails. Note that the worms were not fed at any point of this experiment. Fragments were then imaged 4 days after cutting. Embryos were cut in half using the same method described above, but were first treated in a previously outlined embryo de-shelling solution (composed of 32mM sodium hydroxide, 0.5mg/ml of sodium thioglycolate, and 1mg/ml of pronase in ASW)15. The egg shells were subsequently removed by shearing the embryos against a sharp, glass pipette.
Blastomere ablation experiments
Early embryo blastomeres were ablated using an IR laser at 1.45μm wavelength (XYClone, Hamilton Thorne Biosciences) that was mounted on a Leica DM5000 compound microscope. Embryos were placed in calcium and magnesium free ASW, and multiple laser pulses at 85% power were applied to the outer edges of blastomeres to be ablated. These pulses were applied in quick succession to different parts of the outer edge of the blastomere, as single pulses resulted in the cell rupture briefly, but close back up again. The quick succession of multiple pulses should result in the majority of the inner cytoplasmic contents to spill out. After ablation, embryos were allowed to recover and develop in 60x55mm culture dishes in fresh ASW at 23°C.
RNAi knockdown experiments
In vitro double-stranded RNA preparation and injection of hatchling worms were done using established protocols14. dsRNA injections were performed using a Drummond Nanoject II. Animals were injected and soaked with dsRNA for 3 days, and then amputated. The fragments were then injected and soaked with dsRNA for 3 days, and then amputated again. We found a phenotype associated with the original tail fragment from which anterior tissue had been removed twice, as depicted in Figure S5K and S5L.
H3P staining
Phosphorylated Histone H3 (H3P) staining was performed using the methods previously published14. Embryos were placed in deshelling solution (composed of 32mM sodium hydroxide, 0.5mg/ml of sodium thioglycolate, and 1mg/ml of pronase in ASW) on a nutator. The incubation time differed depending on the developmental stage. Early cleavage to Pill stages were incubated for 8mins, whereas Prehatchling and Pigmented Prehatchling stages were incubated for 6mins. Embryos were then fixed in 4% Paraformaldehyde in ASW at 4°C overnight. Embryos were stored in 4°C in PBS for a maximum of one week before being used for H3P staining. H3P antibodies (Santa Cruz Biotechnology, Cat#sc-374669) were diluted 1:1000 in phosphate buffer solution (PBS), and samples were incubated in this solution for 1 hour at room temperature. The H3P antibodies were then washed out using four, 20min incubations in PBS.
Cell dissociation and FACS
Cell dissociation was done using previously published protocols23. Animals were placed in calcium and magnesium free ASW with 1% horse serum, and were vigorously pipetted up and down until they dissociated into single cells. After dissociation, the cell suspension was passed through a 40μm strainer into a 2mL tube. Excess debris was then eliminated by centrifuging the cells for 5mins at 500g (4°C) through a 4% BSA solution in calcium magnesium free ASW. All cell debris was expected to be caught at the interface of the 4% BSA solution and the cell suspension. The cell pellet was then resuspended in calcium magnesium free ASW with 1% horse serum. The resuspended cell suspension was then stained with Calcein Blue AM (Thermofisher C1429) to stain live cells. The Calcein Blue AM signal was used to sort for live cells. For our experiments, we dissociated 18 freshly hatched photoconverted worms to determine the transcriptional profile of 3a/3b progeny upon hatching, and 20 photoconverted embryos at 80 hours post laying to determine the transcriptional profile of 3a/3b progeny during embryogenesis. FACS was done using a Beckman Coulter MoFlo Astrios EQ Cell Sorter at the Harvard Bauer Core facility. Cells were sorted based on red vs green fluorescence among those that were positive for calcein (Fig. S4A and S5E). Cells were directly sorted into 96-well plates filled with a lysis buffer, with a total of 288 cells captured for sequencing for the hatchlings, and 96 cells captured for sequencing for the 80hpl embryos.
SMARTseq and SeqWell data collection and single cell analysis
Upon performing FACS, sequencing libraries were prepared by the Harvard Bauer Core facility using the SMARTseq2 protocol38 for the photoconverted hatchling dataset, and the seqWell plexwell library prep (seqWell, sku:pw096) for the photoconverted embryo dataset (Post Dimple 80 hpl). Sequencing was also done by the Harvard Bauer Core facility using the Illumina Novaseq (100bp, paired end). Reads were demultiplexed, and then quantified using Salmon39. The resulting counts were then merged into a single table using a series of custom python scripts that are available on github: https://github.com/JulianKimura/SMARTseq_Pipeline. Once the merged count file was created, it was then imported into Seurat for downstream analysis. All of the code for clustering, plot generation, and statistical tests will be available on github. Cell type identities were assigned to the clusters by projecting marker genes for cell clusters identified in the Hatchling Juvenile dataset16. Specific genes were selected based on their specificity to their respective clusters in the Hatchling Juvenile dataset, and were compiled into a list of diagnostic genes (Table S1 and S2). Justifications for gene names are in Table S3. All raw fastq files for the sequencing data will be deposited into the NCBI sequence read archive (SRA).
Fluorescent in situ hybridization
mRNA localization was visualized using fluorescent in situ hybridization (FISH) described previously15. Embryos were placed in deshelling solution (composed of 32mM sodium hydroxide, 0.5mg/ml of sodium thioglycolate, and 1mg/ml of pronase in ASW) on a nutator. The incubation time differed depending on the developmental stage. Early cleavage to Pill stages were incubated for 8mins, whereas Prehatchling and Pigmented Prehatchling stages were incubated for 6mins. Embryos were then fixed in 4% Paraformaldehyde in ASW at 4°C overnight. Embryos were stored in 4°C in PBS for a maximum of one week before use in in situ hybridization. Embryos were washed out of PBS, and treated with proteinase K for 1 min (Early Cleavage to Pill stage embryos) and 3mins (Prehatchling to Hatched Juveniles). After treatment with proteinase K, embryos were placed in a 8M urea based pre-hybridization and hybridization solution.15 Prehybridization took place for 2 hours, while hybridization took place overnight. After hybridization, samples were taken through a series of wash steps. Two 20min incubations in Prehybridization solution, two 20min incubation in a 50:50 mixture of Prehybridization solution and 2x saline sodium citrate buffer with (SSCT), two 20min incubations in 2x SSCT, and two 20min incubations in 0.2x SSCT. Samples were then placed in either anti-Digoxigenin-POD (1:1500 dilution) or anti-Fluorescein (1:2000 dilution) in blocking solution at 4°C overnight. Samples were then washed ten times for 20mins each in PBST, and were then incubated with Tyramide with rhodamine of Fluorescein (1:1000 and 1:2000 respectively) with hydrogen peroxide (0.002% final concentration) for 10mins on a nutator. Samples were then washed with PBST, and imaged with a Leica SP8 confocal microscope.
Cell dissociation and InDrops encapsulation and library preparation
Embryos at various developmental stages (35, 50, 65, 80, 95, 110, 125, and 145 hours post laying) were placed in a de-shelling solution for various lengths of time using the recipe detailed both above and in a previous publication (See Fluorescent in situ Hybridization)15. Once the embryos were de-shelled, embryos were dissociated and prepped for in drops encapsulation using the methods described above and in a previous study16. Single cell suspensions were encapsulated using the in drops platform at the Harvard University Single Cell Core24. Hofstenia is a marine organism, meaning that their cells needed to be in artificial seawater to maintain osmoregularity as long as possible before being encapsulated. To allow for this, we utilized a specialized microfluidics chip that had a separate channel that supplied phosphate buffer solution (PBS) just prior to the cells being encapsulated16. The subsequent library preparation was done by the Harvard Single Cell Core, following the standard indrops protocol where transcripts are barcoded within individual droplets in oil24. The qPCR quantification and sequencing of libraries was done using the KAPA Library Quantification Kit (Roche, material# 7960140001) and the illumina Nextseq sequencer (75bp, paired end).
Analysis of InDrops single cell data and trajectory inference
Demultiplexing, mapping, quantification, and generation of count matrices were done using the protocols described in the github repository: https://github.com/brianjohnhaas/indrops. All read and count numbers are in Table S1D. The subsequent downstream analysis of clustering was performed using the R package Seurat40–43. Doublets and low quality cells were filtered based on outliers in the number of genes expressed per cell, and the number of UMIs detected. Trajectory inference and differential expression of molecular trajectories of the embryonic data were done using the R package URD26. URD does not allow for datasets above 45,000 cells. Thus, we randomly subsampled our original 51,000 cell dataset into 43,000 cells. The root for the URD tree was defined as cells from the Gastrula stage (35hpl), and the tips for the tree were defined as the cell clusters identified in the Hofstenia Hatchling Juvenile16.
QUANTIFICATION AND STATISTICAL ANALYSIS
Single cell RNA-seq
All differential expression analysis for calling marker genes for clusters was done using the default method in the Seurat package (Wilcoxon rank sum test). A Chi squared test of independence was done using the base R command to test the significance of cell types and their identities (Red vs Green). A Wilcoxon ranked sum test was performed to determine the significance of the expression levels of piwi-1 between the red and green cells. All differential expression for trajectory inference were done using the default area under precision recall (AUPRC) test, as detailed in a previous publication26.
Supplementary Material
Figure S1: Fate map of 8-cell stage Hofstenia embryos reveals embryonic origins of mature tissue types and the correlation of embryonic and adult axes (Related to Figure 1, Video S1, Video S2, and Video S3). (A) Various markers showcase tissue type morphologies and spatial distributions in Hofstenia miamia. Top: Whole-worm images of in situ hybridization detect mRNA for genes that mark specific tissues in Hofstenia miamia. Internal z-slice images for gut and epidermal markers, and maximum intensity projection images of muscle, neoblast, and neural markers are shown. Bottom: High magnification images of transgenic animals and in situ hybridizations showing the morphology of the gut, epidermis, muscle, neoblast, and neurons. Notable cell-type morphologies include the mesh-like structure of muscle, small extensions connecting body neurons (yellow arrowheads), and the mesh-like structure of the anterior condensation is visible among the gad-1 in situ hybridizations. (B) Dorsal and ventral view images of a sexually mature tuba::Kaede transgenic worm (See materials and methods). Kaede protein fluorescence was detected in high amounts in the germline and oocytes (white arrowheads) (C) Confocal imaging of photoconverted embryos during development showing major cell internalization events. The same worms as shown in Fig. 1F were followed until hatching, and thus the same 0 hpc images are shown here. Yellow arrows show the site of the “dimple” where cells have been shown to be internalizing15. (D) Single blastomere labeling via photoconversion showed that, with the exception of 1a2/1b2 and 2A/2B, cells that were a part of the same duet/pair, generated the same fates as shown in Fig. 1F–G, but labeled cells on the left or right sides of the worms. (E) Time series of 1a2-labeled embryos shows that its location is the site of cell internalization at the “dimple”. While the majority of labeled cells were internalized at this stage, a small patch of labeled cells persisted on the exterior (yellow arrowhead), which became invaginated to form the pharynx, an anterior structure. Suggesting that the “dimple” corresponds to the position of the 1a2 blastomere, and to the future anterior. (F) Top: The surface of the embryo where the 1a1/1b1 progeny became internalized, i.e. the animal pole, was photoconverted. Bottom: Images of the hatched worm from the experiment above showed that the dorsal epidermis was labeled, showing the correspondence of the animal pole to the future dorsal. The same embryo used for these images were imaged in Video S1. (G) Schematics summarizing the correspondence of early embryo axes to adult body axes. Top Row: The early 8-cell embryo labeled with the future adult body axes. Show from both the lateral and side views. Bottom: Embryos across different developmental stages oriented to show corresponding axes. Developmental stages named based on15. Whole embryo and hatchling worm image scale bars = 100μm. High magnification tissue image scale bars = 50μm.
Figure S2: Fates of 3a and 3b show bilateral symmetry (Related to Figure 2 and Video S4). Single blastomere labeling via photoconversion for 3a and 3b micromeres showed that they produce the same cell type, midsection cells, in the hatchling with symmetry along the left-right axis. Whole embryo and hatchling worm image scale bars = 100μm. High magnification tissue image scale bars = 50μm.
Figure S3: Blastomeres other than 3a/3b at the 16-cell stage do not possess functional characteristics of neoblasts (Related to Figure 3). (A) Lateral view schematic diagram showing the mother-daughter relationship between 1a/1b and the cells present at the 8-cell stage (gray). (B-C) Given their mother-daughter relationship, 1a/1b-labeled embryos capture the properties of 1a1/1b1 and 1a2/1b2. Therefore, by labeling 1a/1b, 2a/2b, and 3A/3B, the characteristics of all blastomeres, excluding 3a/3b, at the 16-cell stage can be assessed. (B) Progeny of 1a/1b, 2a/2b, and 3A/3B do not contribute to newly regenerated heads. 1a/1b labeled embryos capture the properties of 1a1/1b1 and 1a2/1b2. Left to Right: Schematics showing the blastomeres photoconverted, Images of head fragments regenerating a new tail (yellow arrowhead), High magnification images of newly regenerated tail blastemas showing minimal red labeling. Both internal and surface z-slices of the blastema show minimal spread of red labeling. Surface z-slices of the 1a/1b labeled individuals show red labeling in the epidermis, likely from their expected contribution to the posterior regions of the epidermis during development. Whole fragment images for 1a/1b photoconversion show high levels of red fluorescence in the anterior because their progeny contribute to the nervous system, which labels the anterior brain. (C) Progeny of 1a/1b, 2a/2b, and 3A/3B do not contribute to newly regenerated tails. 1a/1b labeled embryos capture the properties of 1a1/1b1 and 1a2/1b2. Left to Right: Schematics showing the blastomeres photoconverted, Images of whole tail fragments regenerating a new head (yellow arrowhead), High magnification images of newly regenerated head blastemas showing minimal red labeling. Both internal and surface z-slices of the blastema show minimal spread of red labeling. Surface z-slices of the 1a/1b and 2a/2b labeled individuals show red labeling in the epidermis, likely from their expected contribution to the anterior regions of the epidermis during development. (D) The progeny of 1a1/1b1, 1a2, 3A/3B blastomeres do not contribute to homeostatic tissue turnover through contribution to epidermis or muscle. 2a/2b and 1b2 progeny were not tested due to their contribution to both muscle and epidermis normally. Top: Red labeling is not detectable in the epidermis among individuals where blastomeres that do not normally contribute to epidermis were labeled (1a1/1b1, 1a2, 3A/3B blastomeres). Bottom: The spread of red labeling to muscle fibers was not detectable among individuals where blastomeres that do not normally contribute to muscle were labeled (1a1/1b1 and 3A/3B blastomeres). (E) The progeny of 1a1/1b1, 1a2/1b2, 2a/2b, and 3A/3B are not sensitive to irradiation. The labeled progeny of all blastomeres besides 3a/3b did not have any visible changes after irradiation. (F) in situ hybridization of piwi-1 and immunostain of H3P show a restriction of both markers at 170hpl in a neoblast-like pattern, suggesting the distribution of both markers only resemble that of functional neoblast the Pigmented Prehatchling stage. (G) Transverse amputations of embryos show regenerative capacity at the Pigmented Prehatchling stage. In situ hybridizations of the anterior neural marker gad-1 and the posterior marker fz-1 show regeneration of tissues in tail and head fragments respectively. (H) Ablation of early blastomeres. Left: Schematic representation of the ablation of 1a1/1b1 and 3a/3b blastomeres. Right: Ablation of 1a1/1b1 and 3a/3b blastomeres resulted in hatched worms with normal distribution of markers that the corresponding cells were fated to become, showing the ability of these embryos to compensate for the loss of cells during development. All scale bars for images depicting whole hatchlings, fragments, and embryos = 100μm. High magnification tissue image scale bars = 50μm.
Figure S4: FACS and SMARTseq of photoconverted 3a/3b progeny and unconverted blastomere progeny recover a variety of cell types (Related to Figure 4, Table S1, and Table S3). (A) FACS plots showing the gates used to sort red vs green cells. Left: Cells with high levels of calcein fluorescence (indicative of live cells) were then sorted for red vs green fluorescence levels (Right). (B) Projection of tissue-specific marker gene expression on UMAP plots reveal the identities of the clusters determined via unsupervised clustering. Full list of diagnostic genes is shown in Table 2. (C) New cell type identified through unsupervised clustering of SMARTseq data. In situ hybridization of the top marker gene, gland, reveals that it is expressed in cells distributed in a punctate pattern on the periphery of the animal. Left: Maximum intensity z-projection image of an in situ hybridization of gland showing its expression in small, punctate cells. Right: Z-slice of a gland in situ hybridization show that these small, punctate cells are located in the periphery of the hatchling juvenile animal. Whole worm image scale bars = 100μm.
Figure S5: Single-cell analysis of Hofstenia embryos reveal a molecular trajectory of cells as they differentiate into mature cell types (Related to Figure 5, Table S2, and Table S3). (A) Projection of known tissue markers for epidermis, muscle, endoderm, gut, onto a UMAP plot of embryonic development data marks specific branches on the UMAP topology. Projection of the neoblast marker piwi-1 showed high levels of expression throughout all embryonic cells, while neo was found to be more specific to the region of the UMAP with neoblasts. (B) Differential expression analysis of URD branches revealed genes that marked clades of cells corresponding to specific tissue types. (C) in situ hybridization of clade markers identified in (B) across developmental stages. Images for tropomyosin are previously published15. These genes were expressed in regions of the embryo and Hatchling Juvenile worms where the corresponding tissues were expected. The timing at which these transcripts were detectable through in situ hybridization matched the single-cell RNAseq data with the exception of the epidermal clade marker, which was only detectable starting at the Post Dimple stage (80 hpl) despite transcripts being detected in the sequencing data as early as the Dimple stage (55 hpl). (D) Double in situ hybridization of hes and piwi-1 in hatchling worms showing co-expression. (E) FACS plots showing the gates used to sort red vs green cells in Post Dimple stage (80 hpl) embryos with 3a/3b progeny labeled. Cells with high levels of calcein fluorescence (Left) were sorted for red vs green fluorescence levels (Right). (F) Projection of known markers reveal that two of the clusters detected within the Post Dimple stage (80 hpl) FACS dataset corresponded to epidermis and muscle based on the expression of known marker genes. See Table S2 for the full list of diagnostic genes, hes was found to be expressed among the two clusters with red-only and green-only cells. (G) Projection of both tbx and foxo on the 80 hpl SMART-seq dataset showing co-expression of tbx and foxo in cells. (H) High magnification images of double in situ hybridization of tbx (green) and foxo (green) with hes (magenta) respectively show co-localization (white arrowheads) during embryogenesis in Pigmented Prehatchlings (145 hpl) and hatchlings. (I) in situ hybridization of foxo and tbx in hatched worms marked cells with neoblast-like distribution. (J) Schematic diagram showing the work-flow for RNAi knock-down of foxo and tbx during whole-body regeneration. Two successive cuts were made, and the posterior fragment after the second cut showed a phenotype reported in Fig. S5K. (K) RNAi knock-down of foxo and tbx during whole-body regeneration showed a reduction in anterior regeneration, including diminished expression of the neural marker gad-1 and the anterior marker sfrp-1 during head regeneration after the two rounds of amputation. High magnification images show the diminished levels of sfrp-1 present in the anterior blastema among the individuals with foxo and tbx RNAi. (L) RNAi knock-down of foxo during homeostasis resulted in diminished piwi-1 expression (n = 6/6) relative to controls (n = 5/5). (M) Projection of foxo and tbx onto the single-cell RNAseq dataset of 3a/3b progeny in hatchlings shows that they are enriched specifically in the neoblast cluster, further highlighting its potential importance in neoblast biology. Whole embryo and hatchling worm image scale bars = 100μm. High magnification scale bars = 25μm.
Video S1: Internalization of 1a1/1b1 progeny during the Dimple stage (Related to Figure 1 and Figure S1). Time-lapse video of a Dimple stage embryo where the progeny of 1a1/1b1 progeny are labeled in red. The red progeny cells can be seen being internalized at a site that is adjacent to the “dimpling” (arrow). 1 second = 2.5 hours
Video S2: Majority of 1a2 progeny are internalized at the site of the “dimpling” (Related to Figure 1 and Figure S1). Time-lapse video of a Dimple stage embryo where the progeny of 1a2 are labeled in red. As the video progresses, the majority of the red cells could be seen being internalized at the site of the dimple. 1 second = 2.5 hours.
Video S3: Majority of 1b2 progeny are not internalized during the Dimple stage (Related to Figure 1 and Figure S1). Time-lapse video of a Dimple stage embryo where the progeny of 1b2 are labeled in red. As the video progresses, the majority of the red cells are not internalized at the site of the “dimpling” (arrow). 1 second = 2.5 hours.
Video S4: Internalization of two lateral patches of the 3a/3b progeny during the Dimple stage (Related to Figure 2 and Figure S2). Time-lapse video of a Dimple stage embryo where the 2A/2B progeny are labeled in red. The video focuses on the surface to capture the subset of the 2A/2B progeny derived from 3a/3b, which stay on the surface of the embryo at the Gastrula stage. The cells can be seen dividing and ultimately becoming internalized as the video progresses. 1 second = 2.5 hours
Table S1: Cluster markers, diagnostic genes, GO analysis, and single cell statistics (Related to Figure 4, Figure 5, Figure S4, and Figure S5. (A) Marker genes for each of the clusters in Fig. 4C. (B) All diagnostic genes that were used to assign cluster identity to the single cell dataset in Fig. 4C. All genes used in the projections for Fig. S4B are highlighted in yellow. (C) GO terms that were found to be enriched for the secretory cluster described in Fig. 4C, S4B, and S4C. (D) Reads and counts per reads for each of the single cell RNAseq datasets.
Table S2: Marker genes for trajectory inference and diagnostic genes used in Figure 5 (Related to Figure 5 and Figure S5). (A) Marker genes for each of the different tissue-specific clades that were highlighted in Fig. 5B and S5B. All genes shown in Fig. S5B are highlighted in yellow. (B) Marker genes for the internodes that lead up to the neoblast lineage, hes is highlighted in yellow. (C) Diagnostic genes that were used to assign cluster identity to the single cell dataset in Fig. 5F. Genes that were projected onto the dataset in Figure S5F are highlighted in yellow. (D) Marker genes for each of the clusters in Figure 5F and S5F.
Table S3: Justification for the naming of genes (Related to Figure 4, Figure S4, Figure 5, and Figure S5): Table showing the justifications for the naming of genes used in our single cell analyses.
HIGHLIGHTS.
Photoconversion-based lineage tracing reveals fate map for H. miamia embryos
Two micromeres at the 16-cell stage are the likely embryonic source of stem cells
Single-cell developmental atlas provides molecular trajectories for stem cells
Labeling and isolation of stem cell lineage corroborates molecular components
Acknowledgements
We thank Elizabeth D’Haiti for help in generating data showing the distribution of H3P during embryogenesis, and Dr. Yi-Jyun Luo for help in generating the count matrices for the InDrops single cell RNA-seq dataset. We also thank the Harvard Bauer Core Facility for assistance with FACS and with generating, sequencing, and demultiplexing SMARTseq libraries. We also thank members of the Srivastava Lab for intellectual discussions, feedback on the manuscript, and other support in the laboratory. L.R. and M.S. were supported by grants to M.S. by the Searle Scholars Program (SSP-2016-1494), Smith Family Foundation, and the National Institutes of Health (1R35GM128817). D.M.B.R. was supported by National Institutes of Health (1R35GM128817). J.O.K. was supported by the Department of Organismic and Evolutionary Biology, the NSF-Simons Center for Mathematical and Statistical Analysis of Biology at Harvard, and the Harvard Quantitative Biology Initiative (1764269).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare that they do not have any competing interests.
References Cited
- 1.Lai AG and Aboobaker AA (2018). EvoRegen in animals: Time to uncover deep conservation or convergence of adult stem cell evolution and regenerative processes. Dev. Biol 433, 118–131. [DOI] [PubMed] [Google Scholar]
- 2.Randolph H (1897). Observations and Experiments on Regeneration in Planarians. [Google Scholar]
- 3.Wagner DE, Wang IE and Reddien PW (2011). Clonogenic neoblasts are pluripotent adult stem cells that underlie planarian regeneration. Science 332, 811–816 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buchanan JW (1933) Regeneration in Phagocata gracilis (Leidy). Physiol. Zool 6, 185–204. [Google Scholar]
- 5.Müller WA, Teo R and Frank U (2004). Totipotent migratory stem cells in a hydroid. Dev. Biol 275, 215–224. [DOI] [PubMed] [Google Scholar]
- 6.Gahan JM, Bradshaw B, Flici H and Frank U (2016). The interstitial stem cells in Hydractinia and their role in regeneration. Curr. Opin. Genet. Dev 40, 65–73. [DOI] [PubMed] [Google Scholar]
- 7.Funayama N (2013). The stem cell system in demosponges: suggested involvement of two types of cells: archeocytes (active stem cells) and choanocytes (food-entrapping flagellated cells). Dev. Genes Evol 223, 23–38. [DOI] [PubMed] [Google Scholar]
- 8.De Sutter D and Van de Vyver G (1979). Isolation and recognition properties of some definite sponge cell types. Dev. Comp. Immunol 3, 389–397. [DOI] [PubMed] [Google Scholar]
- 9.van Wolfswinkel JC (2014). Piwi and potency: PIWI proteins in animal stem cells and regeneration. Integr. Comp. Biol 54, 700–713. [DOI] [PubMed] [Google Scholar]
- 10.Yeo J-C and Ng H-H (2013). The transcriptional regulation of pluripotency. Cell Res. 23, 20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies EL, Lei K, Seidel CW, Kroesen AE, McKinney SA, Guo L, Robb SM, Ross EJ, Gotting K, and Alvarado AS (2017). Embryonic origin of adult stem cells required for tissue homeostasis and regeneration. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rebscher N, Volk C, Teo R, and Plickert G (2008). The germ plasm component Vasa allows tracing of the interstitial stem cells in the cnidarian Hydractinia echinata. Dev. Dyn 237, 1736–1745. [DOI] [PubMed] [Google Scholar]
- 13.Leclère L, Jager M, Barreau C, Chang P, Le Guyader H, Manuel M, and Houliston E (2012). Maternally localized germ plasm mRNAs and germ cell/stem cell formation in the cnidarian Clytia. Dev. Biol 364, 236–248. [DOI] [PubMed] [Google Scholar]
- 14.Srivastava M, Mazza-Curll KL, van Wolfswinkel JC, and Reddien PW (2014) Whole-body acoel regeneration is controlled by Wnt and Bmp-Admp signaling. Curr. Biol 24, 1107–1113. [DOI] [PubMed] [Google Scholar]
- 15.Kimura JO, Ricci L, and Srivastava M (2021). Embryonic development in the acoel Hofstenia miamia. Development 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hulett RE, Kimura JO, Marcela Bolanos D, Luo Y-J, Ricci L, and Srivastava M (2022). Acoel single-cell atlas reveals expression dynamics and heterogeneity of a pluripotent stem cell population. bioRxiv 2022.02.10.479464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fincher CT, Wurtzel O, de Hoog T, Kravarik KM, and Reddien PW (2018). Cell type transcriptome atlas for the planarian Schmidtea mediterranea. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Wolfswinkel JC, Wagner DE, and Reddien PW (2014). Single-cell analysis reveals functionally distinct classes within the planarian stem cell compartment. Cell Stem Cell 15, 326–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scimone ML, Kravarik KM, Lapan SW, and Reddien PW (2014). Neoblast specialization in regeneration of the planarian Schmidtea mediterranea. Stem Cell Reports 3, 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martindale MQ & Hejnol AA (2009). developmental perspective: changes in the position of the blastopore during bilaterian evolution. Dev. Cell 17, 162–174. [DOI] [PubMed] [Google Scholar]
- 21.Martindale MQ, Pang K & Finnerty JR (2004). Investigating the origins of triploblasty: ‘mesodermal’ gene expression in a diploblastic animal, the sea anemone Nematostella vectensis(phylum, Cnidaria; class, Anthozoa). Development 131 2463–2474. [DOI] [PubMed] [Google Scholar]
- 22.Meyer NP, Boyle MJ, Martindale MQ, and Seaver EC (2010). A comprehensive fate map by intracellular injection of identified blastomeres in the marine polychaete Capitella teleta. EvoDevo 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ricci L and Srivastava M (2021) Transgenesis in the acoel worm Hofstenia miamia. Dev. Cell 56, 3160–3170.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zilionis R, Nainys J, Veres A, Savova V, Zemmour D, Klein AM, and Mazutis L (2017). Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc 12, 44–73. [DOI] [PubMed] [Google Scholar]
- 25.McInnes L, Healy J, Saul N and Großberger L (2018). UMAP: Uniform Manifold Approximation and Projection. Journal of Open Source Software 3, 861. [Google Scholar]
- 26.Farrell JA, Wang Y, Riesenfeld SJ, Shekhar K, Regev A, and Schier AF (2018). Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henry JQ, Martindale MQ and Boyer BC (2000). The unique developmental program of the acoel flatworm, Neochildia fusca. Dev. Biol 220, 285–295. [DOI] [PubMed] [Google Scholar]
- 28.Raz AA, Wurtzel O and Reddien PW (2021). Planarian stem cells specify fate yet retain potency during the cell cycle. Cell Stem Cell 28, 1307–1322.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Extavour CG and Akam M (2003). Mechanisms of germ cell specification across the metazoans: epigenesis and preformation. Development 130, 5869–5884. [DOI] [PubMed] [Google Scholar]
- 30.Özpolat BD, Duygu Özpolat B, Handberg-Thorsager M, Vervoort M and Balavoine G (2017). Cell lineage and cell cycling analyses of the 4d micromere using live imaging in the marine annelid Platynereis dumerilii. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobayashi T and Kageyama R (2010) The cyclic gene Hes1 contributes to diverse differentiation responses of mouse embryonic stem (ES) cells by regulating Notch signaling activation. Neuroscience Research 68, e134. [Google Scholar]
- 32.Zhang X, Yalcin S, Lee D-F, Yeh T-YJ, Lee S-M, Su J, Mungamuri SK, Rimmelé P, Kennedy M, Sellers R, et al. (2011). FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat. Cell Biol 13, 1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russell R, Ilg M, Lin Q, Wu G, Lechel A, Bergmann W, Eiseler T, Linta L, Kumar P P, Klingenstein M, et al. (2015). A Dynamic Role of TBX3 in the Pluripotency Circuitry. Stem Cell Reports 5, 1155–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boehm A-M, Khalturin K, Anton-Erxleben F, Hemmrich G, Klostermeier UC, Lopez-Quintero JA, Oberg H-H, Puchert M, Rosenstiel P, Wittlieb J, et al. (2012). FoxO is a critical regulator of stem cell maintenance in immortal Hydra. Proc. Natl. Acad. Sci. U. S. A 109, 19697–19702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cannon JT, Vellutini BC, Smith J 3rd, et al. (2016) Xenacoelomorpha is the sister group to Nephrozoa. Nature 530(7588): 89–93. [DOI] [PubMed] [Google Scholar]
- 36.Kapli P and Telford MJ (2020). Topology-dependent asymmetry in systematic errors affects phylogenetic placement of Ctenophora and Xenacoelomorpha. Science advances 6(50). DOI: 10.1126/sciadv.abc5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Philippe H, Poustka AJ, Chiodin M, et al. (2019) Mitigating Anticipated Effects of Systematic Errors Supports Sister-Group Relationship between Xenacoelomorpha and Ambulacraria. Current biology: CB 29(11): 1818–1826.e6. [DOI] [PubMed] [Google Scholar]
- 38.Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, and Sandberg R (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098. [DOI] [PubMed] [Google Scholar]
- 39.Patro R, Duggal G, Love MI, Irizarry RA and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butler A, Hoffman P, Smibert P, Papalexi E and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Satija R, Farrell JA, Gennert D, Schier AF and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Fate map of 8-cell stage Hofstenia embryos reveals embryonic origins of mature tissue types and the correlation of embryonic and adult axes (Related to Figure 1, Video S1, Video S2, and Video S3). (A) Various markers showcase tissue type morphologies and spatial distributions in Hofstenia miamia. Top: Whole-worm images of in situ hybridization detect mRNA for genes that mark specific tissues in Hofstenia miamia. Internal z-slice images for gut and epidermal markers, and maximum intensity projection images of muscle, neoblast, and neural markers are shown. Bottom: High magnification images of transgenic animals and in situ hybridizations showing the morphology of the gut, epidermis, muscle, neoblast, and neurons. Notable cell-type morphologies include the mesh-like structure of muscle, small extensions connecting body neurons (yellow arrowheads), and the mesh-like structure of the anterior condensation is visible among the gad-1 in situ hybridizations. (B) Dorsal and ventral view images of a sexually mature tuba::Kaede transgenic worm (See materials and methods). Kaede protein fluorescence was detected in high amounts in the germline and oocytes (white arrowheads) (C) Confocal imaging of photoconverted embryos during development showing major cell internalization events. The same worms as shown in Fig. 1F were followed until hatching, and thus the same 0 hpc images are shown here. Yellow arrows show the site of the “dimple” where cells have been shown to be internalizing15. (D) Single blastomere labeling via photoconversion showed that, with the exception of 1a2/1b2 and 2A/2B, cells that were a part of the same duet/pair, generated the same fates as shown in Fig. 1F–G, but labeled cells on the left or right sides of the worms. (E) Time series of 1a2-labeled embryos shows that its location is the site of cell internalization at the “dimple”. While the majority of labeled cells were internalized at this stage, a small patch of labeled cells persisted on the exterior (yellow arrowhead), which became invaginated to form the pharynx, an anterior structure. Suggesting that the “dimple” corresponds to the position of the 1a2 blastomere, and to the future anterior. (F) Top: The surface of the embryo where the 1a1/1b1 progeny became internalized, i.e. the animal pole, was photoconverted. Bottom: Images of the hatched worm from the experiment above showed that the dorsal epidermis was labeled, showing the correspondence of the animal pole to the future dorsal. The same embryo used for these images were imaged in Video S1. (G) Schematics summarizing the correspondence of early embryo axes to adult body axes. Top Row: The early 8-cell embryo labeled with the future adult body axes. Show from both the lateral and side views. Bottom: Embryos across different developmental stages oriented to show corresponding axes. Developmental stages named based on15. Whole embryo and hatchling worm image scale bars = 100μm. High magnification tissue image scale bars = 50μm.
Figure S2: Fates of 3a and 3b show bilateral symmetry (Related to Figure 2 and Video S4). Single blastomere labeling via photoconversion for 3a and 3b micromeres showed that they produce the same cell type, midsection cells, in the hatchling with symmetry along the left-right axis. Whole embryo and hatchling worm image scale bars = 100μm. High magnification tissue image scale bars = 50μm.
Figure S3: Blastomeres other than 3a/3b at the 16-cell stage do not possess functional characteristics of neoblasts (Related to Figure 3). (A) Lateral view schematic diagram showing the mother-daughter relationship between 1a/1b and the cells present at the 8-cell stage (gray). (B-C) Given their mother-daughter relationship, 1a/1b-labeled embryos capture the properties of 1a1/1b1 and 1a2/1b2. Therefore, by labeling 1a/1b, 2a/2b, and 3A/3B, the characteristics of all blastomeres, excluding 3a/3b, at the 16-cell stage can be assessed. (B) Progeny of 1a/1b, 2a/2b, and 3A/3B do not contribute to newly regenerated heads. 1a/1b labeled embryos capture the properties of 1a1/1b1 and 1a2/1b2. Left to Right: Schematics showing the blastomeres photoconverted, Images of head fragments regenerating a new tail (yellow arrowhead), High magnification images of newly regenerated tail blastemas showing minimal red labeling. Both internal and surface z-slices of the blastema show minimal spread of red labeling. Surface z-slices of the 1a/1b labeled individuals show red labeling in the epidermis, likely from their expected contribution to the posterior regions of the epidermis during development. Whole fragment images for 1a/1b photoconversion show high levels of red fluorescence in the anterior because their progeny contribute to the nervous system, which labels the anterior brain. (C) Progeny of 1a/1b, 2a/2b, and 3A/3B do not contribute to newly regenerated tails. 1a/1b labeled embryos capture the properties of 1a1/1b1 and 1a2/1b2. Left to Right: Schematics showing the blastomeres photoconverted, Images of whole tail fragments regenerating a new head (yellow arrowhead), High magnification images of newly regenerated head blastemas showing minimal red labeling. Both internal and surface z-slices of the blastema show minimal spread of red labeling. Surface z-slices of the 1a/1b and 2a/2b labeled individuals show red labeling in the epidermis, likely from their expected contribution to the anterior regions of the epidermis during development. (D) The progeny of 1a1/1b1, 1a2, 3A/3B blastomeres do not contribute to homeostatic tissue turnover through contribution to epidermis or muscle. 2a/2b and 1b2 progeny were not tested due to their contribution to both muscle and epidermis normally. Top: Red labeling is not detectable in the epidermis among individuals where blastomeres that do not normally contribute to epidermis were labeled (1a1/1b1, 1a2, 3A/3B blastomeres). Bottom: The spread of red labeling to muscle fibers was not detectable among individuals where blastomeres that do not normally contribute to muscle were labeled (1a1/1b1 and 3A/3B blastomeres). (E) The progeny of 1a1/1b1, 1a2/1b2, 2a/2b, and 3A/3B are not sensitive to irradiation. The labeled progeny of all blastomeres besides 3a/3b did not have any visible changes after irradiation. (F) in situ hybridization of piwi-1 and immunostain of H3P show a restriction of both markers at 170hpl in a neoblast-like pattern, suggesting the distribution of both markers only resemble that of functional neoblast the Pigmented Prehatchling stage. (G) Transverse amputations of embryos show regenerative capacity at the Pigmented Prehatchling stage. In situ hybridizations of the anterior neural marker gad-1 and the posterior marker fz-1 show regeneration of tissues in tail and head fragments respectively. (H) Ablation of early blastomeres. Left: Schematic representation of the ablation of 1a1/1b1 and 3a/3b blastomeres. Right: Ablation of 1a1/1b1 and 3a/3b blastomeres resulted in hatched worms with normal distribution of markers that the corresponding cells were fated to become, showing the ability of these embryos to compensate for the loss of cells during development. All scale bars for images depicting whole hatchlings, fragments, and embryos = 100μm. High magnification tissue image scale bars = 50μm.
Figure S4: FACS and SMARTseq of photoconverted 3a/3b progeny and unconverted blastomere progeny recover a variety of cell types (Related to Figure 4, Table S1, and Table S3). (A) FACS plots showing the gates used to sort red vs green cells. Left: Cells with high levels of calcein fluorescence (indicative of live cells) were then sorted for red vs green fluorescence levels (Right). (B) Projection of tissue-specific marker gene expression on UMAP plots reveal the identities of the clusters determined via unsupervised clustering. Full list of diagnostic genes is shown in Table 2. (C) New cell type identified through unsupervised clustering of SMARTseq data. In situ hybridization of the top marker gene, gland, reveals that it is expressed in cells distributed in a punctate pattern on the periphery of the animal. Left: Maximum intensity z-projection image of an in situ hybridization of gland showing its expression in small, punctate cells. Right: Z-slice of a gland in situ hybridization show that these small, punctate cells are located in the periphery of the hatchling juvenile animal. Whole worm image scale bars = 100μm.
Figure S5: Single-cell analysis of Hofstenia embryos reveal a molecular trajectory of cells as they differentiate into mature cell types (Related to Figure 5, Table S2, and Table S3). (A) Projection of known tissue markers for epidermis, muscle, endoderm, gut, onto a UMAP plot of embryonic development data marks specific branches on the UMAP topology. Projection of the neoblast marker piwi-1 showed high levels of expression throughout all embryonic cells, while neo was found to be more specific to the region of the UMAP with neoblasts. (B) Differential expression analysis of URD branches revealed genes that marked clades of cells corresponding to specific tissue types. (C) in situ hybridization of clade markers identified in (B) across developmental stages. Images for tropomyosin are previously published15. These genes were expressed in regions of the embryo and Hatchling Juvenile worms where the corresponding tissues were expected. The timing at which these transcripts were detectable through in situ hybridization matched the single-cell RNAseq data with the exception of the epidermal clade marker, which was only detectable starting at the Post Dimple stage (80 hpl) despite transcripts being detected in the sequencing data as early as the Dimple stage (55 hpl). (D) Double in situ hybridization of hes and piwi-1 in hatchling worms showing co-expression. (E) FACS plots showing the gates used to sort red vs green cells in Post Dimple stage (80 hpl) embryos with 3a/3b progeny labeled. Cells with high levels of calcein fluorescence (Left) were sorted for red vs green fluorescence levels (Right). (F) Projection of known markers reveal that two of the clusters detected within the Post Dimple stage (80 hpl) FACS dataset corresponded to epidermis and muscle based on the expression of known marker genes. See Table S2 for the full list of diagnostic genes, hes was found to be expressed among the two clusters with red-only and green-only cells. (G) Projection of both tbx and foxo on the 80 hpl SMART-seq dataset showing co-expression of tbx and foxo in cells. (H) High magnification images of double in situ hybridization of tbx (green) and foxo (green) with hes (magenta) respectively show co-localization (white arrowheads) during embryogenesis in Pigmented Prehatchlings (145 hpl) and hatchlings. (I) in situ hybridization of foxo and tbx in hatched worms marked cells with neoblast-like distribution. (J) Schematic diagram showing the work-flow for RNAi knock-down of foxo and tbx during whole-body regeneration. Two successive cuts were made, and the posterior fragment after the second cut showed a phenotype reported in Fig. S5K. (K) RNAi knock-down of foxo and tbx during whole-body regeneration showed a reduction in anterior regeneration, including diminished expression of the neural marker gad-1 and the anterior marker sfrp-1 during head regeneration after the two rounds of amputation. High magnification images show the diminished levels of sfrp-1 present in the anterior blastema among the individuals with foxo and tbx RNAi. (L) RNAi knock-down of foxo during homeostasis resulted in diminished piwi-1 expression (n = 6/6) relative to controls (n = 5/5). (M) Projection of foxo and tbx onto the single-cell RNAseq dataset of 3a/3b progeny in hatchlings shows that they are enriched specifically in the neoblast cluster, further highlighting its potential importance in neoblast biology. Whole embryo and hatchling worm image scale bars = 100μm. High magnification scale bars = 25μm.
Video S1: Internalization of 1a1/1b1 progeny during the Dimple stage (Related to Figure 1 and Figure S1). Time-lapse video of a Dimple stage embryo where the progeny of 1a1/1b1 progeny are labeled in red. The red progeny cells can be seen being internalized at a site that is adjacent to the “dimpling” (arrow). 1 second = 2.5 hours
Video S2: Majority of 1a2 progeny are internalized at the site of the “dimpling” (Related to Figure 1 and Figure S1). Time-lapse video of a Dimple stage embryo where the progeny of 1a2 are labeled in red. As the video progresses, the majority of the red cells could be seen being internalized at the site of the dimple. 1 second = 2.5 hours.
Video S3: Majority of 1b2 progeny are not internalized during the Dimple stage (Related to Figure 1 and Figure S1). Time-lapse video of a Dimple stage embryo where the progeny of 1b2 are labeled in red. As the video progresses, the majority of the red cells are not internalized at the site of the “dimpling” (arrow). 1 second = 2.5 hours.
Video S4: Internalization of two lateral patches of the 3a/3b progeny during the Dimple stage (Related to Figure 2 and Figure S2). Time-lapse video of a Dimple stage embryo where the 2A/2B progeny are labeled in red. The video focuses on the surface to capture the subset of the 2A/2B progeny derived from 3a/3b, which stay on the surface of the embryo at the Gastrula stage. The cells can be seen dividing and ultimately becoming internalized as the video progresses. 1 second = 2.5 hours
Table S1: Cluster markers, diagnostic genes, GO analysis, and single cell statistics (Related to Figure 4, Figure 5, Figure S4, and Figure S5. (A) Marker genes for each of the clusters in Fig. 4C. (B) All diagnostic genes that were used to assign cluster identity to the single cell dataset in Fig. 4C. All genes used in the projections for Fig. S4B are highlighted in yellow. (C) GO terms that were found to be enriched for the secretory cluster described in Fig. 4C, S4B, and S4C. (D) Reads and counts per reads for each of the single cell RNAseq datasets.
Table S2: Marker genes for trajectory inference and diagnostic genes used in Figure 5 (Related to Figure 5 and Figure S5). (A) Marker genes for each of the different tissue-specific clades that were highlighted in Fig. 5B and S5B. All genes shown in Fig. S5B are highlighted in yellow. (B) Marker genes for the internodes that lead up to the neoblast lineage, hes is highlighted in yellow. (C) Diagnostic genes that were used to assign cluster identity to the single cell dataset in Fig. 5F. Genes that were projected onto the dataset in Figure S5F are highlighted in yellow. (D) Marker genes for each of the clusters in Figure 5F and S5F.
Table S3: Justification for the naming of genes (Related to Figure 4, Figure S4, Figure 5, and Figure S5): Table showing the justifications for the naming of genes used in our single cell analyses.
Data Availability Statement
Single-cell RNA-seq data have been deposited at SRA under Bioproject Accession numbers: PRJNA889328, PRJNA887118, and PRJNA888438 for the FACS sorted 3a/3b progeny dataset at hatching, FACS sorted 3a/3b progeny dataset at 80 hours post laying, and the embryonic and postembryonic datasets respectively. Gene sequences in this study are deposited to GenBank. They are publicly available as of the date of publication. Accession numbers for the gene sequences in Genbank are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at Github (https://github.com/JulianKimura/Kimura_2022_Rscripts for single cell RNAseq analyses in R, and https://github.com/JulianKimura/SMARTseq_Pipeline for generating SMARTseq matrices) and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.