

ABSTRACT
Immunosuppressive tumor microenvironment (TME) impedes anti-tumor immune responses and contributes to immunotherapy resistance in triple-negative breast cancer (TNBC). ADAM12, a member of cell surface metalloproteases, is selectively upregulated in mesenchymal/claudin-low TNBCs, where its expression is largely restricted to tumor cells. The role of cancer cell-expressed ADAM12 in modulating the immune TME is not known. We show that Adam12 knockout in the T11 mouse syngeneic transplantation model of claudin-low TNBC leads to decreased numbers of tumor-infiltrating neutrophils (TINs)/polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) and increased numbers of tumor-infiltrating B cells and T cells. ADAM12 loss in cancer cells increases chemotaxis of B cells in vitro and this effect is eliminated by inhibition of CXCR4, a receptor for CXCL12, or anti-CXCL12 blocking antibody. Importantly, ADAM12 loss in T11 cancer cells sensitizes tumors to anti-PD1/anti-CTLA4 combination therapy, although the initial responsiveness is followed by acquired therapy resistance. Depletion of B cells in mice eliminates the improved response to immune checkpoint blockade of Adam12 knockout T11 tumors. Analysis of gene expression data for claudin-low TNBCs from the METABRIC patient cohort shows significant inverse correlations between ADAM12 and gene expression signatures of several anti-tumor immune cell populations, as well as a significant positive correlation between ADAM12 and gene expression signature of TINs/PMN-MDSCs. Collectively, these results implicate ADAM12 in immunosuppression within the TME in TNBC.
KEYWORDS: Triple negative breast cancer, mouse models, ADAM, checkpoint inhibition, tumor-infiltrating immune cells, tumor microenvironment, B cells, CXCL12, mesenchymal tumors
Introduction
Triple negative breast cancer (TNBC; estrogen receptor-, progesterone receptor-, and HER2-negative) accounts for ~15% of all breast cancers and is associated with an earlier onset, a more aggressive clinical course, and worse prognosis than other types of breast cancer.1,2 Recently, notable progress has been achieved in applying immune checkpoint blockade (ICB) therapy to treat TNBC.3,4 Two blocking antibodies, pembrolizumab (anti-PD1 mAb) and atezolizumab (anti-PD-L1 mAb), have been FDA-approved for use in combination with chemotherapy in patients with unresectable or metastatic PD-L1-positive TNBC.5–7 Furthermore, the results of several phase 3 and phase 2 clinical trials involving patients with early stage TNBC now indicate that the addition of ICB to neoadjuvant chemotherapy increases the rate of pathologic complete response (pCR) at the time of surgery and improves event free survival (EFS).8–10 Despite these advances, several clinical challenges remain unresolved, including identification of predictive biomarkers for ICB response, optimization of treatment strategies, and preventing disease recurrence after exposure to ICB.11,12
TNBC comprises a heterogenous group of cancers, with differences at the histologic, genomic, and immunologic levels.13,14 The mesenchymal subtype, or the closely related claudin-low subtype, represents one of the molecular subtypes of TNBC.15–17 Claudin-low tumors are characterized by high expression of epithelial-to-mesenchymal transition (EMT) markers and/or stromal genes,15–17 as well as high infiltration with tumor-infiltrating macrophages (TIMs)18 and regulatory T cells (Tregs).19 All these attributes – EMT, fibrotic stroma, TIMs, and Tregs – have been linked to the immunosuppressive tumor microenvironment (TME) and resistance to ICB.20–24 Thus, deciphering molecular mechanisms responsible for the acquisition of these immunosuppressive traits is critical for the success of ICB therapy in TNBC.
ADAM metalloproteases are the family of cell surface proteolytic enzymes expressed in cancer cells and/or in other cells in the TME.25–28 Since ADAMs have the capability to release or post-translationally modify various chemokines and cytokines,29–32 they are well positioned to modulate anti-tumor immune responses. Yet, the role of ADAMs in the recruitment, expansion, or activation of intratumoral immune cells in solid tumors, including TNBC, and in the modulation of ICB responses is poorly understood. In this study, we investigate the role of ADAM12, a prominent member of the ADAM family,33,34 in suppressing anti-tumor immunity and inhibiting ICB responses in the T11 mouse transplantation model of claudin-low breast cancer. T11 is a mouse cell line derived from a Tp53-/- mammary tumor syngeneic transplant.35,36 T11 cells have a mesenchymal appearance. When orthotopically injected into BALB/c mice, T11 cells form triple negative breast tumors with claudin-low features37 which are resistant to ICB treatment.18,19,38
Among breast cancer patients, ADAM12 gene expression is elevated in claudin-low tumors,39,40 where it is upregulated by several pro-tumor signaling pathways/programs, including epithelial-to-mesenchymal transition (EMT),39 hypoxia,41,42 TGF-β,43 and the Notch42,44 pathway. In return, ADAM12 protein stimulates epidermal growth factor receptor (EGFR),39,42 TGF-β,45 and Notch46,47 signaling. We now show that Adam12 knockout in T11 cells, or Adam12 gene editing resulting in the loss of a functional ADAM12 enzyme at the surface, alters the composition of immune cells infiltrating T11 mammary tumors and sensitizes T11 tumors to the ICB therapy, suggesting an important role of ADAM12 in modulating anti-tumor immunity.
Materials and methods
Cell culture
T11 cell line was provided by Dr. Jeffrey M. Rosen (Baylor College of Medicine). Phoenix-Eco cells were a gift from Dr. Garry P. Nolan (Stanford University). 4T1 cells were obtained from American Tissue Culture Collection (ATCC). Cells were cultured in DMEM (T11 and Phoenix-Eco cells) or RPMI-1640 medium (4T1 cells) supplemented with 10% fetal bovine serum (FBS) at 37°C in a humidified incubator containing 5% CO2.
Generation of Adam12 knockout in T11 and 4T1 cells
T11 cells were co-transfected with an Adam12-specific gRNA vector (OriGene Technologies), using Turbofectin transfection reagent. Two different gRNA vectors were used, with Adam12-targeting sequence 5’-GATGACCAAGTACGTAGAGC-3’(gRNA1) or 5’- CCAAGGAACCACCATCGGCA-3’ (gRNA2). To generate Adam12 knockout in 4T1 cells, recombinant S. pyogenes HiFi Cas9 Nuclease V3 (IDT) was mixed with sgRNAs to generate ribonuclear protein complexes, followed by nucleofection into 4T1 cells using the Neon system (Invitrogen). Adam12-targeting sequences were 5’-CCAAGGAACCACCATCGGCA-3’ (gRNA2) or 5’-AGAGCATGACGAACATCCAA-3’ (gRNA3). Single cell clones of T11 and 4T1 cells were isolated using glass cloning cylinders and analyzed by genomic PCR using primers flanking the predicted target sites in the Adam12 gene, Sanger sequencing of the PCR products (GENEWIZ/Azenta), and ADAM12 immunoblotting.
Immunoblotting
Cells were incubated without or with 5 ng/ml of mouse transforming growth factor β (TGF-β; R&D Systems) for 48 hours and then treated with lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% (v/v) Triton X-100, 1 mM 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), 10 μg/ml pepstatin, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 10 mM 1,10-phenanthroline). Cell lysates were centrifuged for 15 minutes at 17,000xg at 4°C. Supernatants were incubated with concanavalin A-agarose (50 µl resin per 1 ml cell lysate) for 4 hours at 4°C to enrich samples for glycoproteins. The resin was then washed three times with lysis buffer and glycoproteins were eluted with 3xSDS gel-loading buffer. Proteins were resolved using SDS-PAGE and transferred to a nitrocellulose membrane. After blocking with 5% milk, membranes were probed with anti-ADAM1247 or anti-β1 integrin antibody and horseradish peroxidase-conjugated IgG secondary antibodies. Signal detection was performed using the SuperSignal West Pico PLUS chemiluminescent detection kit (ThermoFisher) and Azure c500 digital imaging system.
Mutagenesis and stable overexpression of ADAM12
Retroviral expression vector Adam12-pBabePuro was used for the expression of ADAM12-wildtype (ADAM12-WT) protein.48 The Adam12-Δ6 construct was generated using the QuickChange kit (Agilent Technologies). Phoenix-Eco cells were transfected using calcium phosphate precipitation method (20 μg DNA/100-mm plate), in the presence of 25 μM chloroquine. Viral supernatants were harvested 48 hours later, supplemented with 8 μg/ml polybrene, and used without further dilution for infection of T11 cells. After 48 hours, cells with stable overexpression of ADAM12-WT or ADAM12-Δ6 were selected with 2 μg/ml puromycin for 8 days.
Cells surface biotinylation
Cells grown in 6-well plates were washed with Dulbecco’s phosphate-buffered saline (DPBS), incubated at 4 ͦC for 60 minutes with EZ-Link Sulfo-NHS-LC-Biotin (Thermo Scientific), and then washed with ice-cold 100 mM Tris-HCl/DPBS. Cellular proteins were extracted with lysis buffer, as described above. Cell extracts were centrifuged at 17,000xg for 15 minutes at 4 ͦC. The supernatants were incubated for 1 hour at 4 ͦC with NeutrAvidin agarose (Thermo Scientific; 50 μl of resin/0.5 ml cell extract). The resin was washed three times with lysis buffer, eluted with 3xSDS gel-loading buffer. Samples were resolved by SDS-PAGE and transferred to a nitrocellulose membrane for immunoblotting.
ELISA assay
Cells were incubated in 6-well plates for 48 hours. Conditioned media and cell lysates were collected and centrifuged for 15 minutes at 17,000xg. CXCL12 concentrations in the supernatants were measured using mouse CXCL12/SDF-1α ELISA kit (R&D Systems, the kit has some cross-reactivity with CXCL12/SDF-1β) and BioTek H1M microplate reader. CXCL12 concentrations in cell lysates were normalized to equal protein concentrations, measured with the BCA assay kit (ThermoFisher).
Orthotopic T11 cell transplantation
All animal experiments were conducted in accordance with a protocol approved by the Institutional Animal Care and Use Committee at Kansas State University. Female 5-6-week-old BALB/c and NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice were purchased from The Jackson Laboratory. Animals of 6–10 weeks of age were used as the recipients of cell transplantation. Cells were detached, washed, suspended in DPBS, counted, and adjusted to 2 × 105 cells/ml (for tissue harvesting experiments) or 4 × 105 cells/ml (for immunotherapy experiments). Cells (50 μl) were then injected into the left 4th inguinal mammary fat pad of mice under isoflurane sedation (a single injection per animal). Mice were monitored for tumor growth daily; tumor size (length x width) was measured with a caliper.
Isolation of tumor-infiltrating immune cells
In all experiments involving immune-infiltrate analysis, two groups of tumors – with or without Adam12 knockout – were always analyzed side-by-side, on the same day. Each group typically comprised 3–5 different tumors, from different animals. The experiments were repeated several times, and the data obtained for each tumor type were pooled together. Because Adam12 knockout T11 tumors grew slower than control T11 tumors, Adam12 knockout cells were injected first, and control cells were injected 5–7 days later. In any given experiment, mice were euthanized and tumors were harvested when the average tumor size (length x width) was similar for the Adam12 knockout and control groups.
Excised tumors were placed in ice-cold DPBS, weighed, transferred to 5 ml Hank’s balanced salt solution (HBSS) with calcium and magnesium at room temperature, cut into small pieces using razor blades, and transferred to 50 ml tubes containing 10 mg/ml collagenase type IV, 200 units/ml DNase type IV, and 1,500 units/ml hyaluronidase type IV-S (all from Sigma). Tubes were shaken at 130 rpm for 1 hour at room temperature. Single cell suspensions were obtained by filtering through a 70-μm cell strainer. The digestion reaction was stopped by adding 1 mM EDTA/0.1% BSA, followed by centrifugation for 3 minutes at 800 x g, resuspension in 2 ml ACK lysis buffer (Lonza), incubation on ice for 1 minute, centrifugation, and washing with DPBS. Cell suspensions were enriched for lymphocytes by centrifugation in 42%/78% Percoll (GE Healthcare) gradient in DMEM medium, at 1000 x g for 20 minutes with no brakes on deceleration. Approximately 4 ml solution was collected from the interface between the two Percoll layers. Isolated lymphocytes were washed with DPBS and resuspended in FACS buffer (DPBS containing 3% bovine serum albumin).
Flow cytometry
Single-cell suspensions of tumor-infiltrating leukocytes were prepared as described above. Cells were first incubated with 2.5 μg/ml TruStain FcX Plus (BioLegend) in FACS buffer for 10 min on ice. Cells were subsequently stained with fluorochrome-conjugated antibodies (Supplementary Table S1; BioLegend) in FACS buffer for 30 minutes on ice in the dark, followed by a wash with FACS buffer and resuspension in 400 μl of FACS buffer supplemented with 1 μg/ml propidium iodide. In some experiments, the absolute numbers of tumor-infiltrating immune cells were determined using the Precision Count BeadsTM (BioLegend). When staining for FoxP3 in Tregs, cells were first incubated with 1 μl eFluor 450 fixable viability dye (Invitrogen) for 30 minutes on ice in the dark, followed by FcR block and cell surface marker fluorochrome-conjugated antibodies staining. Stained cells were then fixed and permeabilized using BioLegend True Nuclear Transcription Factor Buffer Set and stained with anti-FoxP3 antibody. Samples were analyzed using a LSRFortessa X-20 flow cytometer (BD), and data analysis was performed using FCS Express 7 (DeNovo Software). Gating strategies are shown in Supplementary Figures S1-4.
B cell migration assays
Cancer cells were plated at ~0.5 x 106 cells/ml into lower chambers of Transwell 6-well plates® (Corning) and incubated for 48 hours. Splenocytes were isolated from BALB/c mice by mincing the tissue, passing sequentially through 18, 21, and 26 gauge needles, then through a 70-μm cell strainer, and centrifuging at 800 x g for 3 minutes. Cell pellets were resuspended in T11 or 4T1 cell culture media, cells were counted, and 2.5 × 106 cells were seeded into transwell upper chambers (inserts) containing 3.0-µm pore membrane. After incubation for 4 hours at 37°C, cells migrated into lower chambers were stained with anti-CD45 and anti-CD19 antibodies and analyzed by flow cytometry. In some experiments, splenocytes were pre-incubated with 10 µg/ml of AMD3100, a CXCR4 inhibitor, for 30 minutes prior to the migration assay. Alternatively, cancer cells in the lower compartment were pre-incubated for 1 h with 2.5 µg/ml CXCL12-blocking or isotype-matched antibodies. The initial content of B cells among splenocytes (input) was determined after seeding splenocytes into wells without inserts. The amount of migrated B cells was expressed as a percentage of the input.
Immune checkpoint blockade
Cells were implanted orthotopically, as described above. When tumors reached 10–20 mm2 in size, mice were randomized, and 125 μg anti-CTLA4 (clone 9D9) and 200 μg anti-PD1 (clone RMP1-14) antibodies were delivered intraperitoneally every 3–4 days, for a total of 3–4 doses. Control animals received an equal amount of isotype-matched antibodies (mouse IgG2b clone MPC-11 and rat IgG2a clone 2A3). B cell depletion was achieved by injecting 250 μg anti-CD20 mAb (Ultra-LEAF purified, clone SA271G2, reported depletion of 30 or more days) into a lateral tail vein 3 days prior to ICB; isotype-matched rat IgG2b (clone RTK4530) was injected as control. In a parallel experiment, B cell depletion was confirmed by isolating splenocytes 7 days after anti-CD20 treatment and quantifying B cells by flow cytometry.
Data mining and correlation analyses
Adam12 expression data were retrieved from Gene Expression Omnibus (GEO) at https://www.ncbi.nlm.nih.gov/geo/; GSE series numbers are provided in the text. Expression data for human ADAM12 and immune genes in 106 claudin-low TNBCs from the METABRIC dataset49 were accessed via the cBioPortal for Cancer Genomics50,51 (http://www.cbioportal.org/public-portal/). The list of core claudin-low tumors was obtained from ref. 17; sample IDs for these tumors are listed in Supplementary Table S2. Gene expression signature scores for selected immune cell types were calculated as the average gene expression z-scores for marker genes associated with each cell types (Supplementary Table S3). Correlation analysis between ADAM12 expression and each immune cell signature score was performed using GraphPad Prism 9 software; Pearson r correlation coefficients and P values (two-tailed) are reported for each analysis. Expression data (ultra-low input RNA-Seq) for different human ADAMs in purified immune cell populations were accessed via ImmGen data portal at https://www.immgen.org/
Statistical analyses
Data were analyzed with GraphPad Prism 9 software. When comparing two groups, normality tests were performed first. If the data followed a Gaussian distribution and if the F-test confirmed the same variances, statistical analysis was performed using unpaired two-tailed t-test. When the data did not follow a Gaussian distribution (as some groups in Figure 3 and Figure 4), the Mann-Whitney nonparametric test was used. For survival analysis, Kaplan-Meier curves were generated and compared using the log-rank test.
Figure 3.
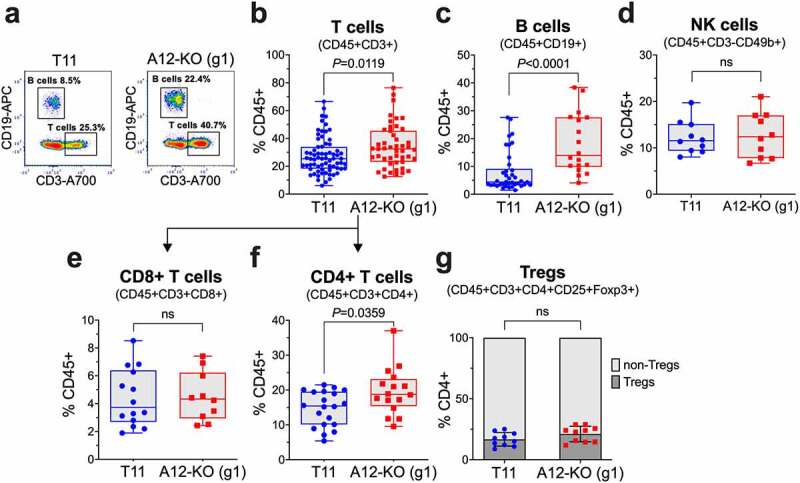
Quantification of tumor-infiltrating lymphocytes (TILs) in A12-KO (g1) and control T11 tumors from BALB/c mice by flow cytometry. Frequencies of TILs were determined after cells enrichment by Percoll gradient centrifugation. (a) Representative FACS analysis of tumor-infiltrating T cells and B cells. Plot is gated on CD45+ cells. Frequencies of T cells (b), B cells (c), NK cells (d), CD8+ T cells (e), and CD4+ T cells (f) among CD45+ cells and frequencies of regulatory T cells (Tregs) among CD4+ T cells (g) are shown. Gating strategies are shown in Supplementary Figures S2 and S3. Data were pooled from two (d,g), four (e,f), seven (c), or fifteen (b) different experiments, each experiment included 3–5 tumors per group. In (b-d, f), the data did not follow a Gaussian distribution and the Mann-Whitney nonparametric test was used to calculate P values; box and whiskers plots are shown for all data sets.
Figure 4.
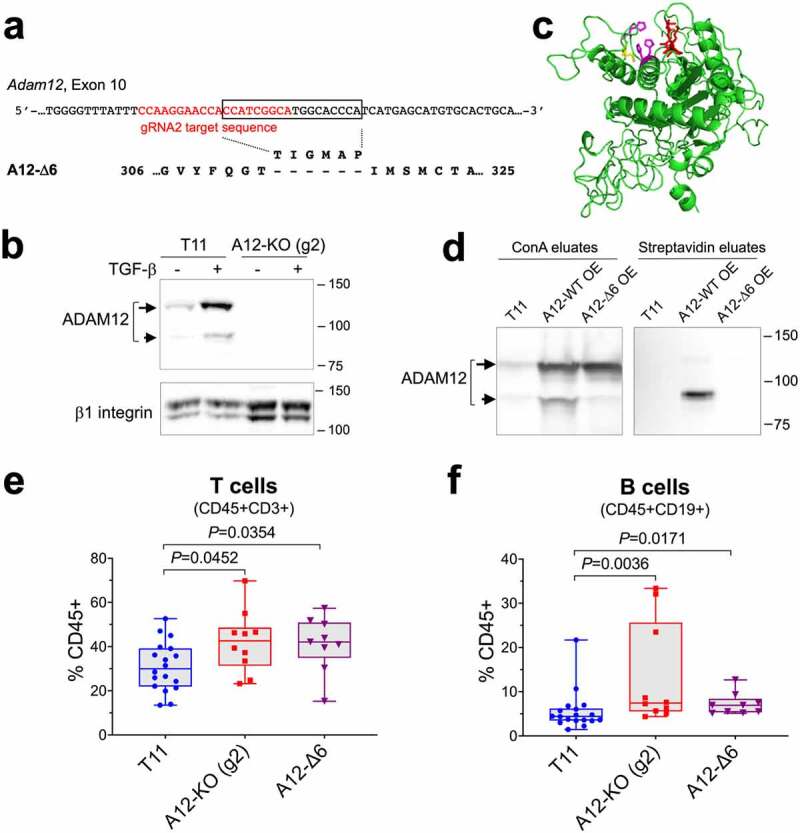
ADAM12 entrapment inside the cell exerts a similar effect on tumor-infiltrating T cells and B cells as Adam12 gene knockout. (a) Position of the gRNA2 target sequence in the Adam12 gene is shown in red. A12-Δ6 is an ADAM12 variant created during CRISPR-Cas9 editing, this variant lacks the T-I-G-M-A-P sequence encoded by the 18-nt fragment in the Adam12 gene (boxed). (b) Detection of ADAM12 in control T11 cells and in T11 cells in which Adam12 was knocked out using gRNA2 (A12-KO (g2)). To facilitate the detection of ADAM12, cells were treated for 2 days with 5 ng/ml TGF-β. Nascent and mature forms of ADAM12 are indicated with long and short arrows, respectively; β1 integrin is gel loading control. (c) A model of the metalloprotease domain of ADAM12 showing the proximity of the TIGMAP peptide (red) and the catalytic residues Glu349 (yellow), His348, His352, and His358 (magenta). The model was generated using the I-TASSER protein structure prediction server at https://zhanggroup.org/I-TASSER/ (d) Cell surface biotinylation of wild-type ADAM12 (A12-WT) and A12-Δ6 deletion mutant. A12-WT or A12-Δ6 were stably overexpressed (OE) in T11 cells. Intact cells were treated with sulfo-NHS-LC-biotin, followed by incubation of cell lysates with concanavalin A agarose (left) or streptavidin agarose (right). The eluates were analyzed by Western blotting. (e, f) Quantification of tumor-infiltrating T cells (e) and B cells (f) in A12-KO (g2), A12-Δ6, and control T11 tumors from BALB/c mice by flow cytometry. Data were pooled from two (A12-KO (g2) and A12-Δ6) or four (T11) different experiments, each experiment included 3–5 tumors per group. The data did not follow a Gaussian distribution and the Mann-Whitney nonparametric test was used to calculate P values.
Results
We analyzed Adam12 microarray gene expression data for 341 unique murine tumors representing different molecular subtypes of breast cancer, including 25 claudin-low tumors, and 18 normal murine mammary gland samples37 (retrieved from Gene Expression Omnibus (GEO) at https://www.ncbi.nlm.nih.gov/geo/, datasets GSE3165, GSE8516, GSE9343, GSE14457, GSE15263, GSE17916, GSE27101, and GSE42640; individual samples are listed in Supplementary Table S4). We noticed that Adam12 expression levels were significantly higher in claudin-low tumors than in other tumor types or in normal mammary glands (Figure 1a). Analysis of RNA sequencing (RNA-seq) results for four murine cell lines representing the mesenchymal/claudin-low subtypes of TNBC, namely 67NR, E0771, PyMT-M, and T1118 (retrieved from GEO:GSE104765) further showed that Adam12 mRNA levels were the highest in T11 cells (Figure 1b), and this cell line was selected for our studies.
Recent investigations have shown that while T11 syngeneic orthotopic tumors are resistant to ICB, they become ICB-sensitive upon increasing the mutation burden in T11 cells via overexpression of Apobec3 or UV exposure.38 We have retrieved Adam12 expression data for T11, T11-Apobec3, and T11-UV cells (GEO:GSE124821) and determined that Adam12 mRNA levels were significantly lower in ICB-sensitive than in ICB-resistant tumors (Figure 1c).
To investigate whether abrogation of Adam12 expression in T11 cells might impact anti-tumor immunity and response to ICB, we performed CRISPR/Cas9-mediated Adam12 gene knockout (KO) in T11 cells. Initially, cells were co-transfected with pCas-Guide CRISPR-gRNA1 vector targeting exon 7 in the Adam12 gene (Figure 1d). Several single cell clones were selected and analyzed by genomic PCR using primers flanking the donor integration site and PCR product sequencing (to validate Adam12 gene KO) and by immunoblotting (to confirm the lack of ADAM12 protein). Since ADAM12 expression is upregulated by TGF-β,43,52 cells were incubated for 48 h with TGF-β to further confirm band identities (Figure 1e). Two individual clones lacking ADAM12 expression (referred to as A12-KO (g1)) were combined and used in further analyses.
There was no difference between proliferation rates of A12-KO and parental T11 cells in vitro. In contrast, breast tumors formed after orthotopic transplantation of A12-KO (g1) cells into immunocompetent BALB/c mice grew significantly slower than tumors formed by parental T11 cells (Figure 1f). The difference in tumor growth was largely reduced in immunodeficient NOD/SCID gamma (NSG) mice (Figure 1g) that lack the adaptive immune system (mature T, B and NK cells are absent) and have deficiencies in the innate branch of the immune system (macrophages and dendritic cells are defective, complement is absent).53 Thus, A12-KO (g1) tumors might be susceptible to anti-tumor attack by tumor-infiltrating lymphocytes (TILs) and/or certain components of the innate immunity. The remaining small, but significant, difference between growth rates of A12-KO (g1) and T11 tumors in NSG mice (Figure 1h) might be linked to the function of residual myeloid cells in NSG mice or to changes in the TME not related to the immune system.
We next compared the numbers and composition of tumor-infiltrating immune cells in A12-KO (g1) versus control T11 tumors. When tumors reached a similar size, they were resected, enzymatically digested, and single cell suspensions were stained and analyzed by flow cytometry. First, we observed that the total number of tumor-infiltrating CD45+ immune cells in A12-KO (g1) tumors was decreased by 25–30% compared to T11 tumors (Figure 2a). This decrease was due to the reduction of the myeloid CD11b+ cell population, from ~2.4x106 to ~1.6x106 cells/g, or from ~90% to ~83% of all CD45+ cells (Figure 2b). In contrast, the absolute number of CD11b- cells was slightly, but not significantly, higher in A12-KO (g1) cells and amounted to ~3x105 cells/g of tumor tissue. The percentage of CD11b- cells among CD45+ cells rose from 10% in T11 tumors to 16% in A12-KO (g1) tumors, and this increase was significant (Figure 2c).
It has been previously reported that the T11 tumor model belongs to a macrophage-enriched subtype (MES) of TNBC, in which the tumor-infiltrating myeloid compartment is dominated by TIMs, with significantly lower frequencies of tumor-infiltrating neutrophils (TINs).18 Here, we have quantified the TIM and TIN populations, defined as CD45+CD11b+Ly6G-Ly6C-F4/80+ and CD45+CD11b+Ly6G+Ly6Cmed/low, respectively.18 Importantly, such designation of the TIN population also comprised polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs), as mouse TINs and PMN-MDSCs are currently not distinguishable based on phenotypic cell surface markers.54 The CD45+CD11b+Ly6G-Ly6Chi population defined monocytic MDSCs (M-MDSCs).54 Representative flow cytometry plots are shown in Figure 2d, and gating strategies are included in Supplementary Figure S1.
The absolute number of TIMs did not differ significantly between T11 and A12-KO (g1) tumors, but TIM frequency among all CD45+ cells increased from ~54% in T11 tumors to 59% in A12-KO (g1) tumors, and this increase was significant (Figure 2e). Interestingly, the absolute number of TINs/PMN-MDSCs dropped from 7.2 × 105 per gram in T11 tumors to 2.8 × 105 per gram in A12-KO (g1) tumors, or from 26% to 15% of CD45+ cells, respectively (Figure 2f). The population of M-MDSCs represented only a small fraction (~1%) of all tumor-infiltrating cells and did not differ between T11 and A12-KO (g1) tumors (Figure 2g).
We next compared tumor-infiltrating lymphocytes (TILs) in control and A12-deficient T11 tumors. Since the CD45+CD11b- population comprising the bulk of TILs amounted to only ~10% of all tumor-infiltrating immune cells in T11 tumors (Figure 2c), TILs were enriched by centrifugation in a discontinuous Percoll gradient55 prior to flow cytometry analysis and quantified as percent of CD45+ cells collected from the 42%/78% Percoll interface. Notably, A12-KO (g1) tumors contained higher percentages of CD3+ T cells (median values 32.4% versus 25.4%; Figure 3a,b) and CD19+ B cells (median values 14% versus 4.4%; Figure 3a,c) than T11 tumors. In contrast, the frequencies of CD3-CD49b+ NK cells, the third major class of lymphoid cells, were similar in A12-KO (g1) and control T11 tumors (Figure 3d).
Figure 1.
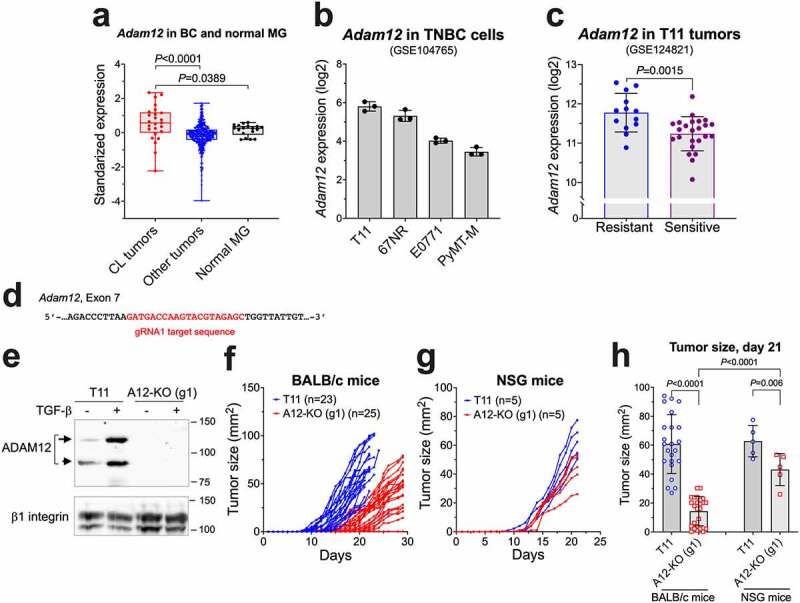
The effect of Adam12 knockout on T11 tumor growth rates in mice. (a) Adam12 expression levels in murine claudin-low (CL) tumors, other tumor types, and normal mammary glands. The data were retrieved from GEO: GSE3165, GSE8516, GSE9343, GSE14457, GSE15263, GSE17916, GSE27101, and GSE42640; the list and classification of individual samples are based on ref. 37 and are included in Supplementary Table S4. The Mann-Whitney nonparametric test was used to calculate P values. (b) Adam12 mRNA levels in four murine mesenchymal breast cancer cell lines, as reported in ref. 18. The data were retrieved from GEO:GSE104765. (c) Expression of Adam12 in ICB-resistant control T11 breast tumors and in ICB-sensitive breast tumors derived from T11 cells with stable overexpression of Apobec3 or treated with UV light, as described in ref. 38. The data were retrieved from GEO:GSE124821. (d) Position of the gRNA1 target sequence in the Adam12 gene. (e) Detection of ADAM12 by Western blotting in control T11 cells and in T11 cells in which Adam12 was knocked out using gRNA1 (A12-KO (g1)). To facilitate the detection of ADAM12, cells were treated for 2 days with 5 ng/ml TGF-β; cell lysates were enriched for glycoproteins on concanavalin A agarose prior to the analysis. Nascent and mature forms of ADAM12 are indicated with long and short arrows, respectively; β1 integrin is gel loading control. (f,g) A12-KO (g1) and control T11 tumor growth rates. 1 × 104 cells were injected into the 4th mammary inguinal mammary fat pad in BALB/c (f) or NSG mice (g). Tumor sizes (length x width) were measured daily with a caliper. In (f), data represent pooled values from five different experiments. (h) Comparison of tumor sizes 21 days after cell injection. Mean values ± S.D. are shown; P values were determined by unpaired two-tailed Student’s t test.
The frequencies of cytotoxic CD8+ T cells did not significantly differ between A12-KO (g1) and T11 tumors (Figure 3e). Accordingly, the increase in T cell frequencies observed in A12-KO (g1) tumors was mainly due to the expansion of CD4+ T cells (median value 19.6% in A12-KO (g1) tumors versus 14.7% in T11 tumors; Figure 3f). Among CD4+ T cells, the relative abundance of immunosuppressive regulatory T cells (Tregs; CD45+CD3+CD4+CD25+Foxp3+) versus non-Tregs remained similar in A12-KO (g1) and T11 tumors and amounted to ~20% (Figure 3g).
Our results so far have suggested that ADAM12 loss in T11 tumor cells may alter certain populations of tumor-infiltrating myeloid and lymphoid cells. In this work, we wanted to further investigate the consequences of ADAM12 loss on TILs, in particular on B cells. First, to validate the results obtained for A12-KO (g1) tumors, we used a different pCas-Guide CRISPR vector to knockout ADAM12 in T11 cells, with gRNA2 targeting exon 10 in the Adam12 gene (Figure 4a). We isolated two different clones: A12-KO (g2), which contained biallelic disruption of the Adam12 gene and lacked ADAM12 protein (Figure 4b), and A12-Δ6, in which one Adam12 allele was disrupted and the second allele contained an in-frame deletion of an 18-nt fragment next to the Cas9 cleavage site, resulting in a 6-amino acid deletion in the metalloprotease domain of the ADAM12 protein (Figure 4a).
Figure 2.
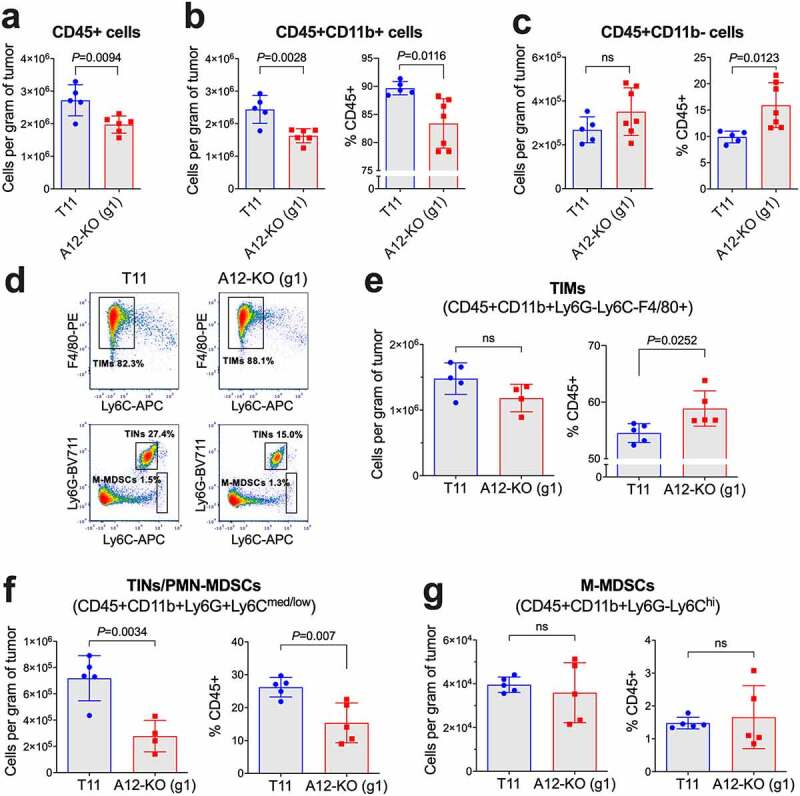
Quantification of tumor-infiltrating immunosuppressive myeloid cells in A12-KO (g1) and control T11 tumors from BALB/c mice by flow cytometry. Absolute numbers of all tumor-infiltrating CD45+ immune cells (a), CD45+CD11b+ cells (b), and CD45+CD11b- cells (c) per gram of tumor tissue were determined using Precision Count beads. In (b, c), percentages of each population among CD45+ cells are also shown. (d) Representative FACS analyses of tumor-infiltrating macrophages (TIMs), tumor-infiltrating neutrophils (TINs)/polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs), and monocytic myeloid-derived suppressor cells (M-MDSCs). Plots are gated on CD45+CD11b+Ly6G– cells (top) and CD45+CD11b+ (bottom). (e-g) Absolute numbers and percentages among CD45+ cells are shown for TIMs (e), TINs/PMN-MDSCs (f), and M-MDSCs (g). Gating strategies are included in Supplementary Figure S1. The unpaired t-test was used to calculate P values; ns, non-significant, with P > 0.05.
In the predicted model of the wild-type ADAM12 metalloprotease domain, the six amino acids missing in A12-Δ6 were positioned close to the catalytic site (Figure 4c). Therefore, we hypothesized that A12-Δ6, generated fortuitously during non-homologous DNA end joining (NHEJ) after Cas9-mediated DNA cleavage, might show defects in folding, post-translational processing, or intracellular trafficking. Typically, nascent full-length forms of ADAMs are catalytically inactive and retained in the intracellular compartments. In contrast, mature processed forms of ADAMs lack the N-terminal inhibitory domain, are catalytically active, and are present at the cell surface.25,56 To examine possible defects in A12-Δ6 processing/trafficking, the wild-type ADAM12 (A12-WT) or A12-Δ6 were overexpressed in T11 cells. Cells were subjected to surface biotinylation using a membrane-impermeable biotinylation reagent, followed by isolation of glycoproteins on concanavalin A agarose (a routine enrichment step for all ADAM12 isoforms) or by isolation of biotinylated proteins on streptavidin agarose. Consistent with previous reports, in A12-WT overexpressing cells, only the ~90-kDa mature form was biotinylated and the ~120-kDa nascent form was not. In A12-Δ6 overexpressing cells, in which the ~120-kDa nascent form was predominant and the ~90-kDa mature form was largely missing, no biotinylated ADAM12 was detected (Figure 4d), confirming defects in the processing/trafficking of A12-Δ6 and the absence of an active A12-Δ6 enzyme at the cell surface.
Despite the different status of ADAM12 in A12-KO (g2) and A12-Δ6 cells (a complete loss of ADAM12 versus the absence of catalytically active ADAM12 at the cell surface), both A12-KO (g2) and A12-Δ6 tumors contained significantly higher frequencies of T cells than T11 tumors (median values 42.6%, 42.1%, and 30.0%, respectively; Figure 4e). The frequencies of tumor-infiltrating B cells in A12-KO (g2) and A12-Δ6 tumors were also significantly higher than in T11 tumors (median values 7.4%, and 6.9%, and 4.4%, respectively; Figure 4f). These results indicate that Adam12 gene knockout in T11 cancer cells increases the frequencies of T cells and B cells among tumor-infiltrating immune cells and this effect is largely due to the lack of functional ADAM12 at the cell surface.
Between tumor-infiltrating T cells and B cells, the relative effect of Adam12 knockout on the frequencies of B cells was particularly prominent (Figure 3c and Figure 4f). Therefore, we wanted to determine whether ADAM12 loss in cancer cells had a direct effect on B cell chemotaxis. We performed transwell cell migration assays, in which cancer cells were plated in lower transwell chambers, freshly isolated splenocytes were seeded in upper chambers, and the amount of B cells migrated into the lower chambers was measured by flow cytometry (Figure 5a). We observed that the presence of T11 cells in the lower chambers increased B cell migration when compared to migration toward empty wells without cancer cells. Importantly, B cell migration toward A12-KO (g1) or A12-KO (g2) T11 cells was significantly higher than toward wild-type T11 cells (Figure 5b and Figure 5c, respectively).
Figure 5.
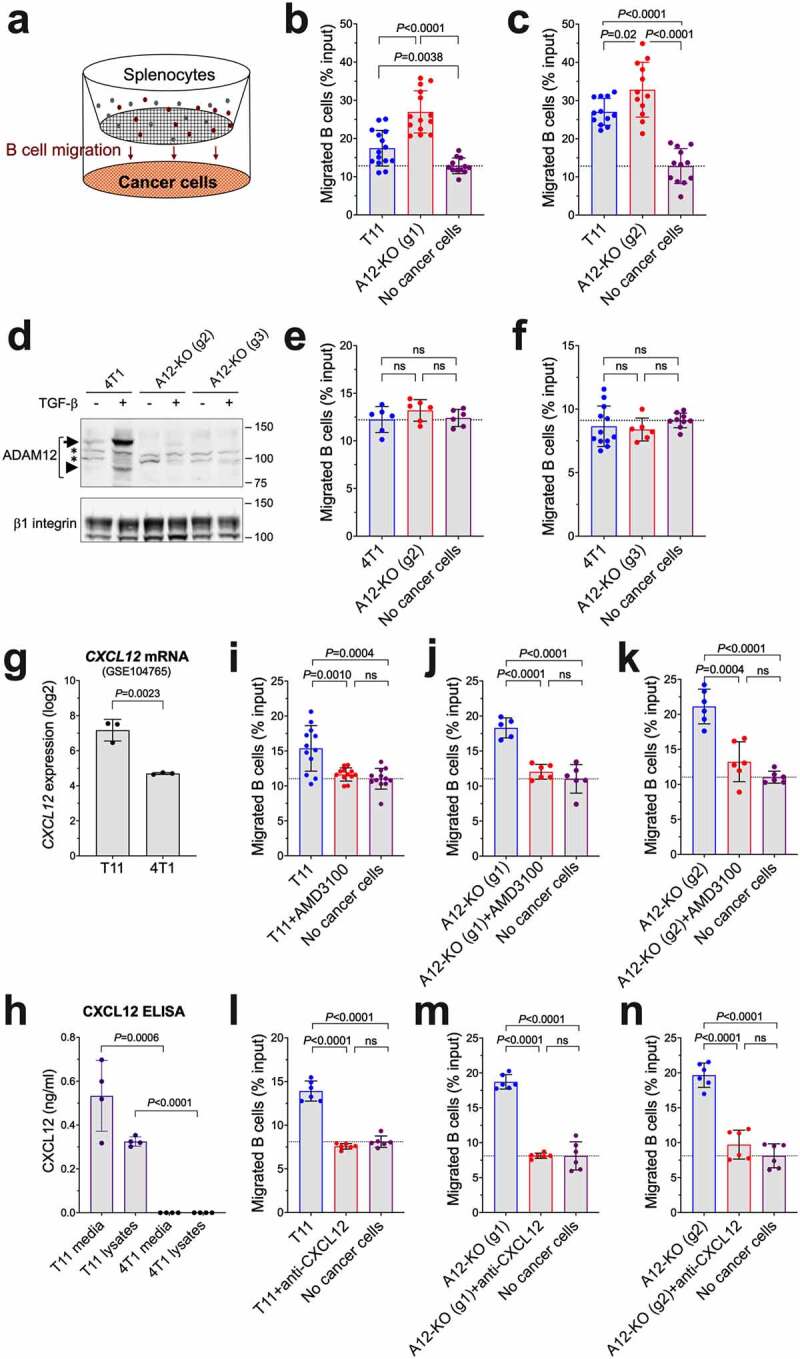
The effect of ADAM12 loss in cancer cells on chemotaxis of B cells in vitro. (a) Schematic diagram of transwell B cell migration assays. B cell content among splenocytes seeded in upper chambers and B cells migrated after 4 hours into lower chambers toward cancer cells were measured by flow cytometry. (b, c) B cell migration toward wild-type T11 cells was compared to A12-KO (g1) T11 cells (b) or A12-KO (g2) cells (c). (d) Detection of ADAM12 by Western blotting in control 4T1 cells and in 4T1 cells in which Adam12 was knocked out using gRNA2 or gRNA3. Glycoprotein enrichment on concanavalin A agarose was as in Figures 1 and 4. Nascent and mature forms of ADAM12 are indicated with long and short arrows, respectively; non-specific bands are marked with asterisks, β1 integrin is gel loading control. (e, f) B cell migration toward wild-type 4T1 cells was compared to A12-KO (g2) 4T1 cells (e) or A12-KO (g3) cells (f). (g) Cxcl12 mRNA levels in T11 and 4T1 cell lines (ref. 18). The data were retrieved from GEO:GSE104765. (h) CXCL12 protein levels in conditioned media and in cell lysates from T11 and 4T1 cells were measured by ELISA. (i-k) Transwell B cell migration assays toward T11 cells (i), A12-KO (g1) (j), or A12-KO (g2) (k) were performed in the absence or presence of 10 μg/ml AMD3100, an inhibitor of CXCR4, added to the upper chambers. (l-n) Transwell B cell migration assays toward T11 cells (l), A12-KO (g1) (m), or A12-KO (g2) (n) were performed in the absence or presence of 2.5 µg/ml CXCL12-blocking antibody added to the lower chambers. For each panel, data were pooled from 2 to 5 independent experiments, each experiment included three different transwells per group. Gating strategies for B cell quantification are shown in Supplementary Figure S4.
We then asked whether Adam12 knockout in a different murine breast cancer cell line would exert a similar effect on directional B cell migration. We tested 4T1 cells, which are commonly used as a model of basal-like triple-negative breast cancer.18 As in T11 cells, ADAM12 expression in 4T1 cells was strongly stimulated by TGF-β. We used two different gRNAs to successfully disrupt the Adam12 gene in 4T1 cells, gRNA2 and gRNA3 (Figure 5d; see also Materials and Methods). Unexpectedly, there was no difference between B cell migration toward wild-type 4T1 cells, Adam12 knockout 4T1 cells, or empty wells (Figure 5e,f).
The CXCL12 chemokine is a known chemoattractant for B cells in vitro and in vivo.57,58 Expression profiling by high throughput RNA sequencing previously revealed a significantly higher Cxcl12 mRNA level in T11 than in 4T1 cells18 (Figure 5g). Here, we performed ELISA assays to measure CXCL12 protein concentrations in conditioned media and cell lysates from T11 and 4T1 cells. Consistent with gene expression profiling, there was a very significant difference between CXCL12 levels in T11 and 4T1 cells, with the latter ones below the ELISA detection limit (Figure 5h). This suggested that CXCL12 secreted by T11 cells might have bound to its receptor CXCR4 in B cells and stimulated directed B cell migration in transwell assays. Also, ADAM12 loss in T11 cells might have potentiated the chemoattractant properties of CXCL12 (see Discussion), and the lack of CXCL12 expression in 4T1 cells might be responsible for the lack of stimulation of B cell migration. In line with this hypothesis, adding AMD3100, a CXCR4 inhibitor, to the upper transwell chambers (Figure 5i-k) or anti-CXCL12 blocking antibody to the lower chambers (Figure 5l-n), efficiently inhibited B cell migration toward T11, A12-KO (g1) T11, and A12-KO (g2) T11 cells. Pre-incubation of cancer cells with an isotype-matched control antibody did not have any effect on B cell migration (Supplementary Figure S5).
We next explored whether alterations in tumor-infiltrating immune cells observed in A12-KO (g1) and A12-KO (g2) tumors translated into improved tumor responses to the ICB therapy. A12-KO (g1), A12-KO (g2), or control T11 tumor-bearing mice were treated with anti-PD1/anti-CTLA4 antibodies (Figure 6a). Control T11 tumors were resistant to ICB (Figure 6b), in agreement with published reports using the same T11 model.18,38 Injection of the same doses of isotype-matched control antibodies did not have any effect on the growth of A12-KO (g1) tumors (Figure 6c). Remarkably, A12-KO (g1) tumors stabilized and did not progress for 7–8 days after the first dose of antibodies (Figure 6d). Median progression-free survival (PFS) was extended from 3 days in untreated mice to 9 days in treated mice (Figure 6d). Progression was defined as a 20% increase in tumor diameter, or 1.44-fold increase in tumor area. Similar effects were observed for A12-KO (g2) tumors, which stabilized for ~5 days after the first dose of antibodies (Figure 6e). Median PFS was extended from 2 days in untreated mice to 5 days in treated A12-KO (g2) mice (Figure 6e). Importantly, while both A12-KO (g1) and A12-KO (g2) tumors showed initial response to ICB, all tumors relapsed after ~8 days (Figure 6d,e) and grew with the rates similar to those before the treatment, which was indicative of acquired resistance to ICB.
Figure 6.
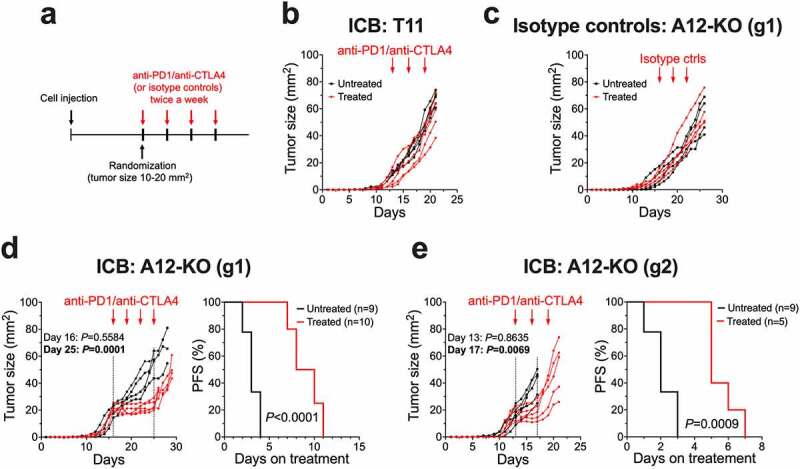
Abrogation of ADAM12 sensitizes T11 tumors to anti-PD1/anti-CTLA4 therapy. (a) Diagram of the treatment. BALB/c mice were orthotopically injected with A12-KO or control T11 cells (2x104 cells/per injection). When tumor sizes reached ~10-20 mm2, mice were randomized and injected intraperitoneally with anti-PD1/anti-CTLA4 antibodies or with isotope control antibodies bi-weekly or remained untreated. (b) The effect of anti-PD1/anti-CTLA4 antibodies on T11 tumor growth. (c) The effect of isotope control antibodies on the growth of A12-KO (g1) tumors. (d) Tumor growth rates and progression free survival of A12-KO (g1) tumors treated with anti-PD1/anti-CTLA4 antibodies versus untreated tumors. (e) Tumor growth rates and progression free survival of A12-KO (g2) tumors treated with anti-PD1/anti-CTLA4 antibodies versus untreated tumors. In d and e, progression was defined as a 20% increase in tumor diameter, or 1.44-fold increase in tumor area.
To explore whether increased frequencies of tumor-infiltrating B cells in ADAM12-deficient tumors contributed to improved tumor responses to the ICB therapy, we performed B cell depletion experiments. Mice bearing A12-KO (g1) or A12-KO (g2) tumors received one dose of B cell-depleting anti-CD20 antibody or an equivalent dose of isotope-matched control antibody, followed by three injections of anti-PD1/anti-CTLA4 antibodies. B cell depletion was confirmed by flow cytometry analysis of splenic B cells (Figure 7a). In both A12-KO (g1) and A12-KO (g2) tumors, B cell depletion significantly reduced the efficacy of anti-PD1/anti-CTLA4 treatment (Figure 7b,c).
Figure 7.
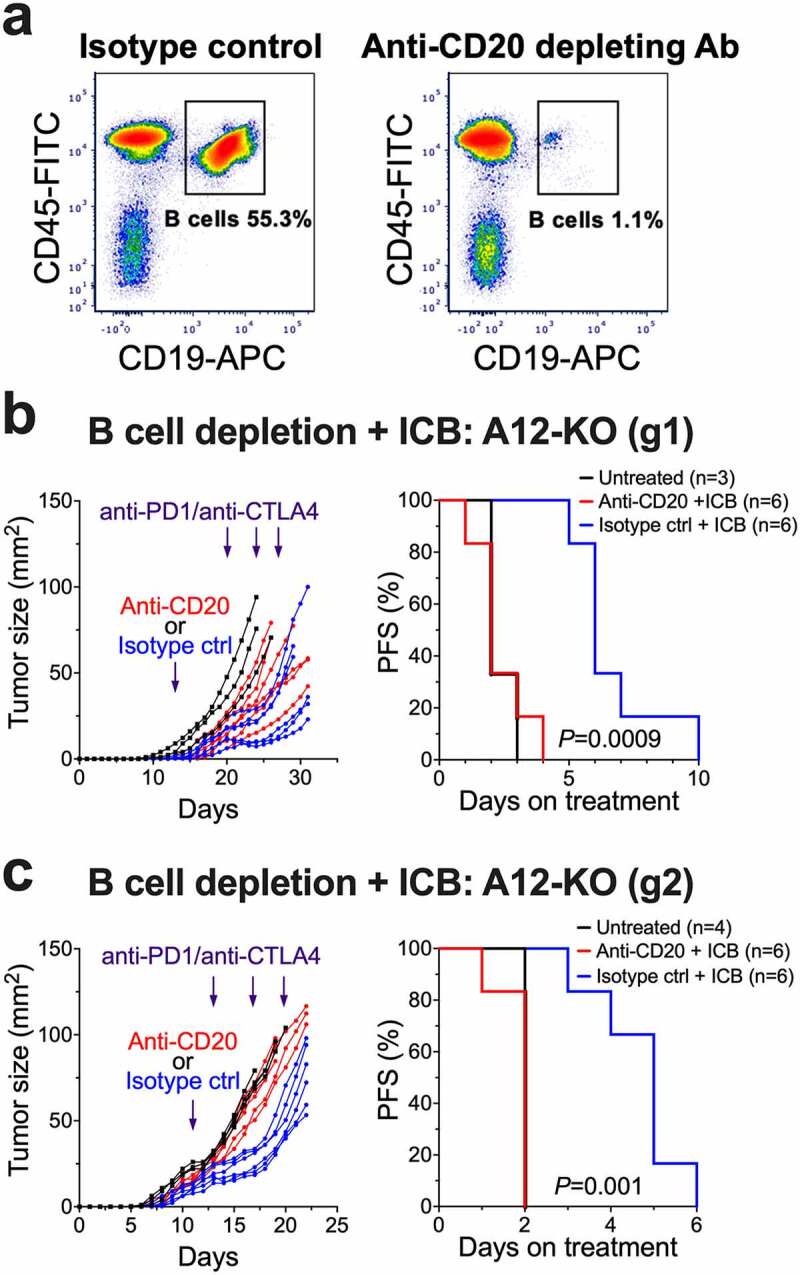
Depletion of B cells eliminates the improved response to anti-PD1/anti-CTLA4 therapy in A12-KO T11 tumors. BALB/c mice were orthotopically injected with A12-KO T11 cells and then either treated with anti-PD1/anti-CTLA4 therapy, as in Figure 6, or untreated. Three days prior to administration of anti-PD1/anti-CTLA4 treatment, mice received tail vein injections of anti-CD20 depleting or isotope control antibodies. (a) Representative FACS analysis of splenic B cells seven days after tail vein injections. (b, c) The effect of anti-PD1/anti-CTLA4 antibodies on tumor growth rates (left) and progression-free survival rates (right) in mice with A12-KO (g1) tumors (b) and A12-KO (g2) tumors (c). Progression was defined as a 20% increase in tumor diameter, or 1.44-fold increase in tumor area.
Using data mining approaches, we then examined whether our results linking the loss of ADAM12 to anti-tumor responses and ICB susceptibility in the T11 mouse model may be applicable to human TNBC patients. First, expression profiling of different immune cell populations by RNA-Seq suggests that ADAM12 is expressed at very low levels in immune cells (Figure 8a, data retrieved at http://www.immgen.org). In contrast, most other catalytically active ADAMs, except for ADAM9, are expressed at high levels in various types of immune cells (Figure 8a). This means that ADAM12 expression values obtained during mRNA profiling of bulk tumors should predominantly reflect ADAM12 expression in cancer cells, with a minimal contribution from tumor-infiltrating immune cells.
Figure 8.
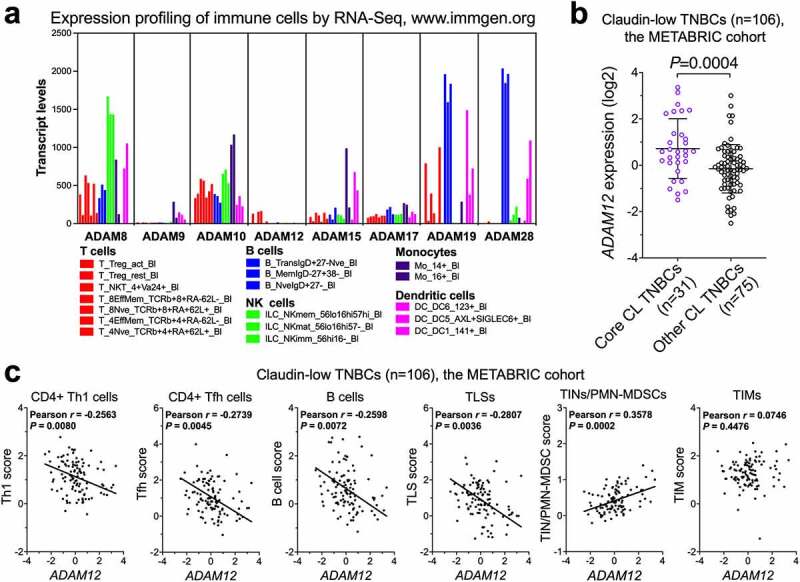
Correlations between human ADAM12 mRNA levels and immune gene expression signatures in claudin-low TNBCs. (a) Population-averaged expression of selected ADAMs in sorted human immune cells. Sample names of individual cell populations are as described at http://immunecellatlas.net/ (b) ADAM12 expression in core claudin-low (core CL, n = 31) TNBC versus other types of CL TNBC (n = 75). Gene expression data for the METABRIC patient cohort were retrieved from cBioPortal at https://www.cbioportal.org The list of core claudin-low tumors was obtained from ref. 17. (c) Relationships between ADAM12 mRNA levels and immune cell signature scores in claudin-low TNBC patients (n = 106) from the METABRIC cohort. Each signature score was calculated as the average gene expression z-score for individual genes in a gene set (Supplementary Table S3).
We then retrieved bulk tumor expression data for ADAM12 and different immune genes from the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) dataset,49 accessed via the cBioPortal for Cancer Genomics50,51 (http://www.cbioportal.org/public-portal/). Among 106 claudin-low TNBC tumors profiled in the METABRIC study, ADAM12 expression was significantly higher in 31 so-called “core claudin-low” tumors,17 with strong features of EMT in carcinoma cells, than in the remaining 75 claudin-low TNBCs (Figure 8b). Importantly, among all 106 claudin-low TNBCs from the METABRIC dataset, there were significant inverse correlations between ADAM12 mRNA levels and immune gene signatures associated with anti-tumor responses (CD4+ Th1 T cells,59 CD4+ Tfh T cells,38,59 B cells,38,60,61 and TLSs62) (Pearson P values <0.01; Figure 8c). In contrast, ADAM12 mRNA levels were positively correlated with the pro-tumor TIN/PMN-MDSC18 gene signature (Pearson P = 0.0002; Figure 8c). There was no significant correlation between ADAM12 mRNA and the TIM signature63 (Figure 8c).
Discussion
Results presented in this study demonstrate that Adam12 knockout in claudin-low T11 breast cancer cells had a significant impact on the composition of tumor-infiltrating immune cells. In particular, the frequencies of B cells were significantly elevated in ADAM12-deficient tumors, and this effect was observed for two different guide RNAs used in CRISPR-Cas9-mediated Adam12 gene knockout. Tumors arising from A12-Δ6 T11 cells lacking the catalytically active ADAM12 at the cell surface also contain higher frequencies of B cells, suggesting that the presence of catalytically active ADAM12 at the surface of cancer cells is vital for its role in modulating B cell infiltration. An inverse correlation between ADAM12 mRNA levels and gene expression signatures for B cells was also detected in claudin-low TNBCs from the METABRIC dataset, further supporting the notion that high expression of ADAM12 limits the accumulation of B cells in the breast TME.
New research indicates that B cells play a prominent role in promoting anti-tumor responses in various types of cancer.64–67 Immunological mechanisms, clinical impact, and therapeutic potential of tumor-infiltrating B cells have been recently discussed in several excellent review articles.58,68–72 For TNBC explicitly, it has been reported that clonally expanded, IgG isotype-biased B cell immunity is associated with improved clinical outcomes.73 Furthermore, using mouse models of claudin-low or basal subtypes of TNBC, it has been demonstrated that increasing the mutation burden in cancer cells by overexpressing APOBEC3B, a cytidine deaminase, or by exposure of cancer cells to short-wave UV radiation prior to transplantation, expanded intratumoral B cells and potentiated the effects of anti-PD1/anti-CTLA4 therapy.38 In our studies, ADAM12-deficient T11 tumors stabilized, albeit transiently, after administration of anti-PD1/anti-CTLA4 antibodies, whereas control T11 tumors did not respond to the ICB treatment. Importantly, B cell depletion prior to ICB eliminated the improved response to ICB, indicating that the susceptibility of ADAM12-deficient tumors to ICB was, at least in part, mediated by increased frequencies of intratumoral B cells.
In addition to B cells, we observed increased frequencies of T cells in ADAM12-deficient tumors, although the effect of ADAM12 loss on tumor-infiltrating T cells was more modest than on B cells. Among T cells, only the CD4+ subset was significantly expanded in ADAM12-deficient tumors, whereas the frequencies of CD8+ cytotoxic T cells were not affected. Finally, among CD4+ cells, the ratio of immunosuppressive Tregs to non-Treg cells was not significantly changed upon the loss of ADAM12, arguing against the release of Treg-mediated immunosuppression in ADAM12-deficient tumors. This conclusion is important, because it has been reported previously that Treg depletion alone reduces immunosuppression and potentiates ICB therapy in claudin-low breast cancer.19,74
Along with changes within the lymphoid compartment, we observed an altered composition of tumor-infiltrating myeloid immune cells in ADAM12-deficient T11 tumors. Specifically, tumors derived from A12-KO (g1) cells contained significantly less TINs/PMN-MDSCs than tumors derived from control T11 cells, and a positive correlation between ADAM12 mRNA levels and the TINs/PMN-MDSC gene expression signature has been detected in claudin-low TNBCs from the METABRIC dataset. Implications of these findings are currently not clear. While PMN-MDCSs are unequivocally immunosuppressive,54 TINs span neutrophil populations with tumor-promoting or tumor-suppressing activities.75,76 Thus, a detailed functional characterization of the TIN/PMN-MDSC population from ADAM12-deficient versus wild-type T11 tumors is needed to obtain a better understanding of the role of ADAM12 in regulating immunosuppression within the myeloid compartment. Of note, lower numbers of tumor-promoting TINs in ADAM12-deficient tumors might explain the fact that A12-KO (g1) tumors grew slightly slower than T11 tumors in NSG mice that retain their neutrophil population, as certain tumor-promoting activities of TINs may be executed even in the absence of the adaptive immune system.76 Furthermore, it has been shown that a conversion of the macrophage-enriched subtype (MES) to the neutrophil-enriched subtype (NES) in several mouse models of TNBCs mediated acquisition of ICB resistance in initially sensitive MES models.18 Since ADAM12-deficient T11 tumors become eventually resistant to ICB after the initial period of therapy response, future enumeration of TIMs and TINs/PMN-MDSCs at different time points after ICB administration will be particularly informative.
TIMs play tumor-promoting functions in breast cancer and are generally associated with a poor prognosis.77 It has been recently demonstrated that TIM inhibition/depletion, combined with a low dose of chemotherapeutic agent cyclophosphamide, induces T cell and B cell infiltration and durable regression in the T11 and related T12 mouse models of TNBC.78 In our experiments, absolute numbers of TIMs did not differ significantly between A12-KO (g1) and control T11 tumors, and the percentage of TIMs among all tumor-infiltrating CD45+ cells was even slightly increased in A12-KO (g1) tumors. Therefore, it seems unlikely that increased B cell infiltration in ADAM12-deficient T11 tumors was mediated via TIMs.
In search for potential mechanisms of increased accumulation of B cells in A12-KO (g1) and A12-KO (g2) tumors, we have discovered that B cells migrated faster toward T11 cells lacking ADAM12 than toward control T11 cells in in vitro transwell assays. Several lines of evidence then suggested a key role for CXCL12 in this process: (i) increased B cell migration toward ADAM12-deficient cells was observed for CXCL12-positive T11 cells but not for CXCL12-negative 4T1 cells, (ii) B cell migration was significantly reduced by a CXCR4 inhibitor, and (iii) B cell migration was significantly diminished by an anti-CXCL12 blocking antibody. Importantly, CXCR4 inhibition or anti-CXCL12 antibody reduced B cell migration toward ADAM12-deficient T11 cells essentially to the basal level, indicating that the CXCL12/CXCR4 interaction was the main driver of B cell migration, at least in our transwell assays. While the contribution of other cytokines to the increased accumulation of B cells in T11 tumors in vivo is also possible, we propose the hypothesis that ADAM12 is a negative regulator of cancer cell-derived CXCL12 and that ADAM12 loss in T11 cells increases the activity of CXCL12.
Of note, CXCL12-α and CXCL12-β, two major splice variants of CXCL12 in mice and humans, can undergo N-terminal truncation by several members of the ADAM-related matrix metalloproteinases (MMPs), including MMP-1, −2, −3, −9, −13 and −14. These MMPs remove the first four amino acids in CXCL12, leading to its complete inactivation.79,80 While ADAM12 activity toward CXCL12 has not been investigated, it is feasible that ADAM12 mediates a similar cleavage and inactivation of CXCL12. In such a case, a combination of ICB with ADAM12 inhibition may represent a rational approach to improve the ICB efficacy in claudin-low TNBC.
In summary, our results establish a paradigm in which a cell surface ADAM in breast cancer cells modulates the repertoire of tumor-infiltrating immune cells and improves ICB responses. This invites a comprehensive examination of ADAM12 and other cancer cell-expressed ADAM metalloproteases as potential regulators of anti-tumor immunity and/or immunosuppression in the TME.
Supplementary Material
Acknowledgments
We thank Kaori Knights and Joel Sanneman (College of Veterinary Medicine, KSU) for their help with flow cytometry experiments. We are grateful to Dr. Tracy Miesner (Comparative Medicine Group, College of Veterinary Medicine, KSU) for training and help with mouse trans-plantation experiments, and to Drs. Susumu Ishiguro and Masaaki Tamura for help with tail vein injections into mice. We thank Dr. Sherry Fleming for valuable discussions and helpful advice.
Funding Statement
This work was supported by NIH grant R03CA235194 and funds from the Flossie West Memorial Trust and KSU Johnson Cancer Research Center to AZ.
Disclosure statement
The authors declare no conflicts of interest.
Data availability statement
The data that support the findings of this study are openly available in Gene Expression Omnibus at https://www.ncbi.nlm.nih.gov/geo/ (GSE3165, GSE8516, GSE9343, GSE14457, GSE15263, GSE17916, GSE27101, GSE42640, GSE104765, and GSE124821), Human Cell Atlas Immune System at http://immunecellatlas.net/, and cBioPortal at https://www.cbioportal.org
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2158006
References
- 1.Derakhshan F, Reis-Filho JS.. Pathogenesis of triple-negative breast cancer. Annu Rev Pathol. 2022;17:181–16. doi: 10.1146/annurev-pathol-042420-093238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zong Y, Pegram M. Research advances and new challenges in overcoming triple-negative breast cancer. Cancer Drug Resist. 2021;4:517–542. doi: 10.20517/cdr.2021.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma P, Siddiqui BA, Anandhan S, Yadav SS, Subudhi SK, Gao J, Goswami S, Allison JP. The next decade of immune checkpoint therapy. Cancer Discov. 2021;11:838–857. doi: 10.1158/2159-8290.CD-20-1680. [DOI] [PubMed] [Google Scholar]
- 4.Emens LA. Immunotherapy in triple-negative breast cancer. Cancer J. 2021;27(1):59–66. doi: 10.1097/PPO.0000000000000497. [DOI] [PubMed] [Google Scholar]
- 5.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im S-A, Shaw Wright G, et al. Investigators IMT. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379(22):2108–2121. doi: 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 6.Adams S, Dieras V, Barrios CH, Winer EP, Schneeweiss A, Iwata H, Loi S, Patel S, Henschel V, Chui SY, et al. Patient-reported outcomes from the phase III IMpassion130 trial of atezolizumab plus nab-paclitaxel in metastatic triple-negative breast cancer. Ann Oncol. 2020;31(5):582–589. doi: 10.1016/j.annonc.2020.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im S-A, Yusof MM, Gallardo C, Lipatov O, Barrios CH, Holgado E, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet. 2020;396(10265):1817–1828. doi: 10.1016/S0140-6736(20)32531-9. [DOI] [PubMed] [Google Scholar]
- 8.Schmid P, Cortes J, Pusztai L, McArthur H, Kummel S, Bergh J, Denkert C, Park YH, Hui R, Harbeck N, et al. KEYNOTE-522 Investigators. Pembrolizumab for early triple-negative breast cancer. N Engl J Med. 2020;382(9):810–821. doi: 10.1056/NEJMoa1910549. [DOI] [PubMed] [Google Scholar]
- 9.Mittendorf EA, Zhang H, Barrios CH, Saji S, Jung KH, Hegg R, Koehler A, Sohn J, Iwata H, Telli ML, et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet. 2020;396(10257):1090–1100. doi: 10.1016/S0140-6736(20)31953-X. [DOI] [PubMed] [Google Scholar]
- 10.Brown LC, Loi S. Immune checkpoint inhibition in the treatment of early stage triple negative breast cancer: 2021 update. Breast. 2022;62(1):S29–S33. doi: 10.1016/j.breast.2021.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol. 2020;20:25–39. doi: 10.1038/s41577-019-0218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanna A, Balko JM. Breast cancer resistance mechanisms: challenges to immunotherapy. Breast Cancer Res Treat. 2021;190:5–17. doi: 10.1007/s10549-021-06337-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prat A, Adamo B, Cheang MC, Anders CK, Carey LA, Perou CM. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist. 2013;18:123–133. doi: 10.1634/theoncologist.2012-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lehmann BD, Jovanovic B, Chen X, Estrada MV, Johnson KN, Shyr Y, Moses HL, Sanders ME, Pietenpol JA. Refinement of triple-negative breast cancer molecular subtypes: implications for neoadjuvant chemotherapy selection. PLoS One. 2016;11:e0157368. doi: 10.1371/journal.pone.0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12:R68.3096954. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pommier RM, Sanlaville A, Tonon L, Kielbassa J, Thomas E, Ferrari A, Sertier AS, Hollande F, Martinez P, Tissier A, et al. Comprehensive characterization of claudin-low breast tumors reflects the impact of the cell-of-origin on cancer evolution. Nat Commun. 2020;11:3431. doi: 10.1038/s41467-020-17249-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fougner C, Bergholtz H, Norum JH, Sorlie T. Re-definition of claudin-low as a breast cancer phenotype. Nat Commun. 2020;11:1787. doi: 10.1038/s41467-020-15574-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, Sheng K, Niu Y, Goldstein A, Zhao N, et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol. 2019;21:1113–1126. doi: 10.1038/s41556-019-0373-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor NA, Vick SC, Iglesia MD, Brickey WJ, Midkiff BR, McKinnon KP, Reisdorf S, Anders CK, Carey LA, Parker JS, et al. Treg depletion potentiates checkpoint inhibition in claudin-low breast cancer. J Clin Invest. 2017;127:3472–3483. doi: 10.1172/JCI90499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL, Weinberg RA. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. 2017;77:3982–3989. doi: 10.1158/0008-5472.CAN-16-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dongre A, Rashidian M, Eaton EN, Reinhardt F, Thiru P, Zagorulya M, Nepal S, Banaz T, Martner A, Spranger S, et al. Direct and indirect regulators of epithelial-mesenchymal transition-mediated immunosuppression in breast carcinomas. Cancer Discov. 2021;11:1286–1305. doi: 10.1158/2159-8290.CD-20-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh S, Zhang XHF, Rosen JM. Time is a great healer-targeting myeloid cells in the tumor immune microenvironment to improve triple-negative breast cancer outcomes. Cells. 2020;10:11. doi: 10.3390/cells10010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taki M, Abiko K, Ukita M, Murakami R, Yamanoi K, Yamaguchi K, Hamanishi J, Baba T, Matsumura N, Mandai M. Tumor immune microenvironment during epithelial-mesenchymal transition. Clin Cancer Res. 2021;27:4669–4679. doi: 10.1158/1078-0432.CCR-20-4459. [DOI] [PubMed] [Google Scholar]
- 24.Kolb HR, Borcherding N, Zhang W. Understanding and targeting human cancer regulatory T cells to improve therapy. Adv Exp Med Biol. 2021;1278:229–256. [DOI] [PubMed] [Google Scholar]
- 25.Seegar TC, Blacklow SC. Domain integration of ADAM family proteins: emerging themes from structural studies. Exp Biol Med (Maywood). 2019;244:1510–1519. doi: 10.1177/1535370219865901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schumacher N, Rose-John S, Schmidt-Arras D. ADAM-mediated signalling pathways in gastrointestinal cancer formation. Int J Mol Sci. 2020;21:5133. doi: 10.3390/ijms21145133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Theret N, Bouezzedine F, Azar F, Diab-Assaf M, Legagneux V. ADAM and ADAMTS proteins, new players in the regulation of hepatocellular carcinoma microenvironment. Cancers (Basel). 2021;13:1563. doi: 10.3390/cancers13071563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lambrecht BN, Vanderkerken M, Hammad H. The emerging role of ADAM metalloproteinases in immunity. Nat Rev Immunol. 2018;18:745–758. doi: 10.1038/s41577-018-0068-5. [DOI] [PubMed] [Google Scholar]
- 29.Mortier A, Van Damme J, Proost P. Regulation of chemokine activity by posttranslational modification. Pharmacol Ther. 2008;120:197–217. doi: 10.1016/j.pharmthera.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Bronger H, Magdolen V, Goettig P, Dreyer T. Proteolytic chemokine cleavage as a regulator of lymphocytic infiltration in solid tumors. Cancer Metastasis Rev. 2019;38:417–430. doi: 10.1007/s10555-019-09807-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cecchinato V, Uguccioni M. Insight on the regulation of chemokine activities. J Leukoc Biol. 2018;104:295–300. doi: 10.1002/JLB.3MR0118-014R. [DOI] [PubMed] [Google Scholar]
- 32.Saw S, Weiss A, Khokha R, Waterhouse PD. Metalloproteases: on the watch in the hematopoietic niche. Trends Immunol. 2019;40:1053–1070. doi: 10.1016/j.it.2019.09.006. [DOI] [PubMed] [Google Scholar]
- 33.Nyren-Erickson EK, Jones JM, Srivastava DK, Mallik S. A disintegrin and metalloproteinase-12 (ADAM12): function, roles in disease progression, and clinical implications. Biochim Biophys Acta. 2013;1830:4445–4455. doi: 10.1016/j.bbagen.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kveiborg M, Albrechtsen R, Couchman JR, Wewer UM. Cellular roles of ADAM12 in health and disease. Int J Biochem Cell Biol. 2008;40:1685–1702. doi: 10.1016/j.biocel.2008.01.025. [DOI] [PubMed] [Google Scholar]
- 35.Jerry DJ, Kittrell FS, Kuperwasser C, Laucirica R, Dickinson ES, Bonilla PJ, Butel JS, Medina D. A mammary-specific model demonstrates the role of the p53 tumor suppressor gene in tumor development. Oncogene. 2000;19(8):1052–1058. doi: 10.1038/sj.onc.1203270. [DOI] [PubMed] [Google Scholar]
- 36.Herschkowitz JI, Zhao W, Zhang M, Usary J, Murrow G, Edwards D, Knezevic J, Greene SB, Darr D, Troester MA, et al. Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Acad Sci USA. 2012;109(8):2778–2783. doi: 10.1073/pnas.1018862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfefferle AD, Herschkowitz JI, Usary J, Harrell JC, Spike BT, Adams JR, Torres-Arzayus MI, Brown M, Egan SE, Wahl GM, et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol. 2013;14(11):R125. doi: 10.1186/gb-2013-14-11-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hollern DP, Xu N, Thennavan A, Glodowski C, Garcia-Recio S, Mott KR, He X, Garay JP, Carey-Ewend K, Marron D, et al. B cells and T follicular helper cells mediate response to checkpoint inhibitors in high mutation burden mouse models of breast cancer. Cell. 2019;179(5):1191–1206. doi: 10.1016/j.cell.2019.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duhachek-Muggy S, Qi Y, Wise R, Alyahya L, Li H, Hodge J, Zolkiewska A. Metalloprotease-disintegrin ADAM12 actively promotes the stem cell-like phenotype in claudin-low breast cancer. Mol Cancer. 2017;16(1):32. doi: 10.1186/s12943-017-0599-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Duhachek-Muggy S, Dubnicka S, Zolkiewska A. Metalloproteinase-disintegrin ADAM12 is associated with a breast tumor-initiating cell phenotype. Breast Cancer Res Treat. 2013;139(3):691–703. doi: 10.1007/s10549-013-2602-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang R, Godet I, Yang Y, Salman S, Lu H, Lyu Y, Zuo Q, Wang Y, Zhu Y, Chen C, et al. Hypoxia-inducible factor-dependent ADAM12 expression mediates breast cancer invasion and metastasis. Proc Natl Acad Sci USA. 2021;118(19):e2020490118. doi: 10.1073/pnas.2020490118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diaz B, Yuen A, Iizuka S, Higashiyama S, Courtneidge SA. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol. 2013;201(2):279–292. doi: 10.1083/jcb.201209151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le Pabic H. ADAM12 in human liver cancers: TGF-β-regulated expression in stellate cells is associated with matrix remodeling. Hepatology. 2003;37(5):1056–1066. doi: 10.1053/jhep.2003.50205. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Solomon E, Duhachek Muggy S, Sun D, Zolkiewska A. Metalloprotease-disintegrin ADAM12 expression is regulated by Notch signaling via microRNA-29. J Biol Chem. 2011;286(24):21500–21510. doi: 10.1074/jbc.M110.207951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atfi A, Dumont E, Colland F, Bonnier D, A A, Prunier C, Ferrand N, Clement B, Wewer UM, Theret N. The disintegrin and metalloproteinase ADAM12 contributes to TGF-β signaling through interaction with the type II receptor. J Cell Biol. 2007;178(2):201–208. doi: 10.1083/jcb.200612046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dyczynska E, Sun D, Yi H, Sehara-Fujisawa A, Blobel CP, Zolkiewska A. Proteolytic processing of Delta-like 1 by ADAM proteases. J Biol Chem. 2007;282(1):436–444. doi: 10.1074/jbc.M605451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang Q, Cao Y, Zolkiewska A. Metalloprotease–disintegrin ADAM 12 binds to the SH3 domain of Src and activates Src tyrosine kinase in C2C12 cells. Biochem J. 2000;352(3):883–892. doi: 10.1042/bj3520883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dyczynska E, Syta E, Sun D, Zolkiewska A. Breast cancer-associated mutations in metalloprotease disintegrin ADAM12 interfere with the intracellular trafficking and processing of the protein. Int J Cancer. 2008;122(11):2634–2640. doi: 10.1002/ijc.23405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signaling. 2013;6(269):l1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Solomon E, Li H, Duhachek Muggy S, Syta E, Zolkiewska A. The role of SnoN in transforming growth factor β1-induced expression of metalloprotease-disintegrin ADAM12. J Biol Chem. 2010;285(29):21969–21977. doi: 10.1074/jbc.M110.133314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, et al. Human lymphoid and myeloid cell development in NOD/LtSz- scid IL2R γnullMice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174(10):6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 54.Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21(8):485–498. doi: 10.1038/s41577-020-00490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Y, Chen K, Wang C, Gong W, Yoshimura T, Liu M, Wang JM. Cell surface receptor FPR2 promotes antitumor host defense by limiting M2 polarization of macrophages. Cancer Res. 2013;73(2):550–560. doi: 10.1158/0008-5472.CAN-12-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cao Y, Kang Q, Zhao Z, Zolkiewska A. Intracellular processing of metalloprotease disintegrin ADAM12. J Biol Chem. 2002;277(29):26403–26411. doi: 10.1074/jbc.M110814200. [DOI] [PubMed] [Google Scholar]
- 57.Luther SA, Bidgol A, Hargreaves DC, Schmidt A, Xu Y, Paniyadi J, Matloubian M, Cyster JG. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J Immunol. 2002;169(1):424–433. doi: 10.4049/jimmunol.169.1.424. [DOI] [PubMed] [Google Scholar]
- 58.Schumacher TN, Thommen DS. Tertiary lymphoid structures in cancer. Science. 2022;375(6576):eabf9419. doi: 10.1126/science.abf9419. [DOI] [PubMed] [Google Scholar]
- 59.Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, Ravoet M, Le Buanec H, Sibille C, Manfouo-Foutsop G, et al. CD4+ follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest. 2013;123(7):2873–2892. doi: 10.1172/JCI67428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Danaher P, Warren S, Dennis L, D’Amico L, White A, Disis ML, Geller MA, Odunsi K, Beechem J, Fling SP. Gene expression markers of tumor infiltrating leukocytes. J Immunother Cancer. 2017;5(1):18. doi: 10.1186/s40425-017-0215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iglesia MD, Vincent BG, Parker JS, Hoadley KA, Carey LA, Perou CM, Serody JS. Prognostic B-cell signatures using mRNA-seq in patients with subtype-specific breast and ovarian cancer. Clin Cancer Res. 2014;20(14):3818–3829. doi: 10.1158/1078-0432.CCR-13-3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crotty S. A brief history of T cell help to B cells. Nat Rev Immunol. 2015;15(3):185–189. doi: 10.1038/nri3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, Soong DYH, Cotechini T, Anur P, Lin EY, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. 2019;35(4):588–602. doi: 10.1016/j.ccell.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun C-M, Calderaro J, Jeng Y-M, Hsiao L-P, Lacroix L, Bougouin A, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577(7791):556–560. doi: 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- 66.Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–565. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 67.Cui C, Wang J, Fagerberg E, Chen P-M, Connolly KA, Damo M, Cheung JF, Mao T, Askari AS, Chen S, et al. Neoantigen-driven B cell and CD4 T follicular helper cell collaboration promotes anti-tumor CD8 T cell responses. Cell. 2021;184(25):6101–6118. doi: 10.1016/j.cell.2021.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fridman WH, Petitprez F, Meylan M, Chen TW, Sun C-M, Roumenina LT, Sautes-Fridman C. B cells and cancer: to B or not to B? J Exp Med. 2021;218(1):e20200851. doi: 10.1084/jem.20200851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Laumont CM, Banville AC, Gilardi M, Hollern DP, Nelson BH. Tumour-infiltrating B cells: immunological mechanisms, clinical impact and therapeutic opportunities. Nat Rev Cancer. 2022;22(7):414–430. doi: 10.1038/s41568-022-00466-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shalapour S, Karin M. The neglected brothers come of age: B cells and cancer. Semin Immunol. 2021;52:101479. doi: 10.1016/j.smim.2021.101479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fridman WH, Meylan M, Petitprez F, Sun C-M, Italiano A, Sautes-Fridman C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat Rev Clin Oncol. 2022;19(7):441–457. doi: 10.1038/s41571-022-00619-z. [DOI] [PubMed] [Google Scholar]
- 72.Downs-Canner SM, Meier J, Vincent BG, Serody JS. B cell function in the tumor microenvironment. Annu Rev Immunol. 2022;40(1):169–193. doi: 10.1146/annurev-immunol-101220-015603. [DOI] [PubMed] [Google Scholar]
- 73.Harris RJ, Cheung A, Ng JCF, Laddach R, Chenoweth AM, Crescioli S, Fittall M, Dominguez-Rodriguez D, Roberts J, Levi D, et al. Tumor-infiltrating B lymphocyte profiling identifies IgG-biased, clonally expanded prognostic phenotypes in triple-negative breast cancer. Cancer Res. 2021;81(16):4290–4304. doi: 10.1158/0008-5472.CAN-20-3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vick SC, Kolupaev OV, Perou CM, Serody JS. Anti–PD-1 checkpoint therapy can promote the function and survival of regulatory T cells. J Immunol. 2021;207(10):2598–2607. doi: 10.4049/jimmunol.2001334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol. 2019;16(10):601–620. doi: 10.1038/s41571-019-0222-4. [DOI] [PubMed] [Google Scholar]
- 76.Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. 2020;20(9):485–503. doi: 10.1038/s41568-020-0281-y. [DOI] [PubMed] [Google Scholar]
- 77.DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19(6):369–382. doi: 10.1038/s41577-019-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Singh S, Lee N, Pedroza DA, Bado IL, Hamor C, Zhang L, Aguirre S, Hu J, Shen Y, Xu Y, et al. Chemotherapy coupled to macrophage inhibition induces T-cell and B-cell infiltration and durable regression in triple-negative breast cancer. Cancer Res. 2022;82(12):2281–2297. doi: 10.1158/0008-5472.CAN-21-3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McQuibban GA, Butler GS, Gong J-H, Bendall L, Power C, Clark-Lewis I, Overall CM. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem. 2001;276(47):43503–43508. doi: 10.1074/jbc.M107736200. [DOI] [PubMed] [Google Scholar]
- 80.Janssens R, Struyf S, Proost P. The unique structural and functional features of CXCL12. Cell Mol Immunol. 2018;15(4):299–311. doi: 10.1038/cmi.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are openly available in Gene Expression Omnibus at https://www.ncbi.nlm.nih.gov/geo/ (GSE3165, GSE8516, GSE9343, GSE14457, GSE15263, GSE17916, GSE27101, GSE42640, GSE104765, and GSE124821), Human Cell Atlas Immune System at http://immunecellatlas.net/, and cBioPortal at https://www.cbioportal.org