

Abstract
Pyroptosis provides a new window for relieving the tumor immunosuppressive microenvironment (TIM) and promoting systemic immune responses for tumor treatments. However, gasdermin D (GSDMD), a key protein in the pyroptosis process mediated by caspase-1, is low expressed in the majority of tumor cells and small-molecule inhibitors of DNA methylation suffer from nonspecific or single-function defects. To address these issues, hexahistidine (His6)-metal assembly (HmA) was employed as the drug delivery vector to load nigericin (Nig) and decitabine (DAC) affording a dual-drug delivery system (Nig + DAC)@HmA. The (Nig + DAC)@HmA nanoparticles are efficiently internalized by cells through endocytosis, easily escape from the lysosome, and are highly distributed in the tumor sites. DAC up-regulates the expression of GSDMD which is then cleaved by the nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome and caspase-1 protein activated by Nig. Effective cancer cell pyroptosis is thus achieved and induces a significant systemic antitumor immunity for impressive tumor suppression with negligible side effects in vivo. Our results suggest that such an easy-to-manipulate self-assembled nano-system (Nig + DAC)@HmA provides a new anticancer path by enhancing pyroptosis through reinforced inflammation.
KEY WORDS: Self-assembled nanoparticle, Pyroptosis, NLRP3, GSDMD, Inflammation, IL-8, Immunotherapy responses, Solid tumor
Graphical abstract
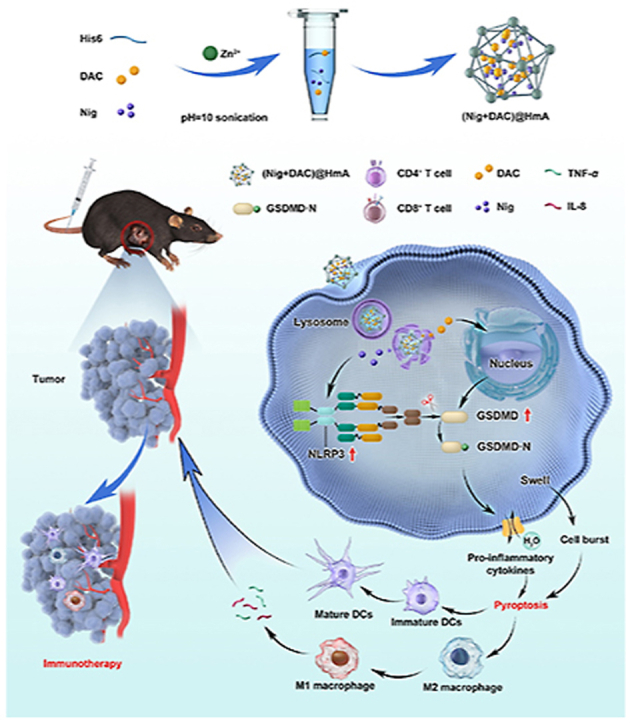
The mechanism of (Nig + DAC)@HmA cell pyroptosis for tumor immunotherapy. Pyroptotic cancer cells in primary tumor secret pro-inflammatory cytokines to reprogram tumor-associated macrophages from M2 to M1 for immunotherapy.
1. Introduction
At present, traditional treatments of cancer such as chemotherapy, surgery, and radiotherapy have been proven to inhibit cancer growth, but they are easy to metastasize and relapse after treatments, which are the main reasons for the high tumor mortality rate. In the latest research progress, immunotherapy has shown that the patient's immune system is invaluable in eradicating tumors, and has exhibited remarkable efficacy in several cancers such as melanoma, leukemia, and lung cancer1, 2, 3, 4. However, the therapeutic effects of immunotherapy have been limited in most tumors due to the down-regulation or inhibition of systemic immune responses by tumor microenvironment immunosuppressive cells such as T regulatory cells, M2 macrophages, and bone marrow-derived inhibitory cells5,6.
To address this, pyroptosis, which is essential for immunity regulation, has aroused researchers’ attention7, 8, 9. Pyroptosis is a powerful cancer immunotherapy strategy, which can relieve the immunosuppression of the tumor microenvironment and induce strong anticancer immunity10. After inflammatory stimulation, cysteine (caspase-1/11/4/5) is activated to decompose gasdermins and release the gasdermin-C and gasdermin-N domains11,12. Then, the gasdermin-N can combine with the cell membrane, making the membrane pore formation and inducing cell swelling with large bubbles, which finally induce cell death13, 14, 15, 16. Nevertheless, gasdermins, the key protein family in the pyroptosis process, are low-expressed for most tumor cells, and thus the pyroptosis pathway is severely limited in the anti-cancer area17, 18, 19, 20. Thus, it is crucial to explore novel pyroptosis inducers to defeat this drawback.
With this in mind, nigericin (C40H67O11, Nig), a polyether ionophore, has attracted a lot of interest in recent years since it exhibits promising cell pyroptosis in macrophages21. Researchers have shown that Nig can form coordinate and transport K+ through the lipid bilayer, reducing the intracellular K+ concentration, which finally induces the activation of nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome. Mariathasan et al.22 and He et al.23 reported that gasdermins were recruited to the NLRP3 inflammasome after the stimulation of LPS and Nig. Gasdermins were then cleaved by caspase-1, which resulted in cell pyroptosis. These studies have convinced us that Nig can induce macrophages pyroptosis mediated by gasdermins. However, in most tumor cells, such as acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS), promoters of tumor suppressor genes are frequently found to be hypermethylated24. Because of the abnormal DNA methylation, gasdermins are low-expressed and the pyroptosis application of Nig in cancer treatments has been severely restricted.
Therefore, DNA demethylation has been considered an important pathway of pyroptosis-mediated therapy for cancer cells25,26. Decitabine (DAC), one of the most commonly used DNA methyltransferase inhibitors, has been widely applied for cancer treatments27, 28, 29, 30. Fan et al.26 showed that low-dose DAC could efficiently elevate gasdermins through demethylation in tumor cells and exhibited tumor-suppressor activity in breast carcinoma cells. Xie et al.31 revealed that the co-administration of doxorubicin (DOX) and DAC led to the demethylation and re-expression of gasdermins for lung cancer treatment. Thus, we hypothesize that the demethylating ability of DAC can be utilized to enhance the expression of gasdermins which is short in tumor cells, and simultaneously the co-administration of DAC and Nig is expected to induce the pyroptosis of tumor cells.
As part of our effort to develop pyroptosis inducers for solid cancers32, we co-encapsulated Nig and DNA methyltransferase inhibitor DAC in hexahistidine (His6)−metal assembly (HmA) particles and researched their anti-cancer efficiency in inducing pyroptosis-mediated cell death. The HmA, as a promising drug delivery vector, is characterized by effective tumor accumulation, low immunogenicity, rapid endocytosis, and lysosomal escape33,34. For the first time, Nig and DAC were co-delivered and their functions of activating NLRP3 inflammasome as well as up-regulating gasdermin D (GSDMD) were integrated into one system, which effectively induced cell pyroptosis of a tumor. The pyroptosis of cancer cells can reprogram tumor-associated macrophages from M2 to M1. M1 macrophages prime the tumor immune and promote tumor rejection by secreting inflammatory factors. Pyroptotic cancer cells secret pro-inflammatory cytokines in the systemic antitumor immunity activation of tumor immunotherapy (Scheme 1). (Nig + DAC)@HmA is highly efficient in activating the cell pyroptosis and is supposed to be a promising therapeutic platform for pyroptosis-mediated tumor immunotherapy.
Scheme 1.

The mechanism of pyroptosis-induced tumor immunotherapeutics by (Nig + DAC)@HmA.
2. Materials and methods
2.1. Materials
Commercial Nig, DAC were purchased from Yuanye Bio-Technology Co., Ltd. (Shanghai, China). His6 was acquired from GL Biochem Co., Ltd. (Shanghai, China). All the ELISA kits were acquired from Beyotime (Shanghai, China). Rabbit anti-mouse antibodies NLRP3, GSDMD, β-actin, and Ki67 were acquired from Affinity Biosciences (Beijing, China). Antibodies CD3, CD4, CD8 CD86, CD11c, CD45, F4/80, CD86, CD206 (MMR) were purchased from BioLegend (Shanghai, China).
2.2. Theoretical details
The binding strength between Nig/DAC and His6 was figured out by AutoDock (Scripps, LA, USA) and DS Visualizer (Accelrys, LA, USA) programs. The three-dimensional structures of the drugs and His6 were obtained from DS 4.5 Client software (Accelrys, LA, USA) and PDB database, respectively. The grid boxes were set as 52, 56, 122 Å for Nig, and 102, 86, and 84 Å for DAC, respectively.
2.3. Synthesis and drug loading
For the preparation of HmA, 10 mg/mL PVPON and 0.05 mol/L HEPES were mixed to prepare PVPON/HEPES stock solution (pH∼10). His6 (50 μL, 40 mg/mL) was added to the solution (250 μL), followed by adding NaOH (28 μL, 0.5 mol/L), Zn(NO3)2 (23.5 μL, 0.1 mol/L) and double distilled water (172 μL). Light blue color was observed in the solution which was kept at 4 °C overnight for the polymerization. The product was centrifuged (Xiangyi, Changsha, China) at 12,000 rpm for 15 min and washed twice with H2O. Replacing pure His6 solution with the mixture of Nig (5 μL, 0.01 mol/L), DAC (7 μL, 0.02 mol/L), and His6 (1.95 mg), (Nig + DAC)@HmA was similarly obtained. In the centrifugation process, the supernatant was collected and the encapsulation efficiency (EE) of Nig or DAC was determined according to the UV-Vis standard curves, as shown in Eq. (1).
| (1) |
The nanoparticles were lyophilized and weighed, and the drug loading efficiency (DL) of Nig or DAC was calculated, as shown in Eq. (2).
| (2) |
2.4. Stability and drug release profile
The stability of (Nig + DAC)@HmA in BSA and FBS buffer was evaluated in a constant temperature shaker at 37 °C. At 0, 6, 14, 24, 36, and 48 h, the sizes of the nanospheres were detected by a particle size analyzer (Mastersizer 3000, Malvern, England).
For the drug release research, a dialysis bag (sized 3.5 kDa) containing 1 mL of (Nig + DAC)@HmA solution was immersed in 50 mL of PBS and shaken continuously at 100 rpm at pH 5 or 7.2 for 4, 8, 24, 48, 72, 96, 120 or 144 h. The accumulated drug release rates at different intervals were evaluated by the UV‒Vis standard curves.
2.5. Cells and animals
Murine bladder tumor MB49, macrophages RAW264.7, and breast cancer 4T1cells were purchased from ATCC and cultured in DMEM/RPMI 1640 medium in an incubator (Thermal Fisher Inc., USA) with 5% CO2 at 37 °C. C57BL/6 and BALB/c mice (18–20 g, 6 weeks old) were supported by the animal center of Wenzhou Medical University and were used based on the Ethics Committee of Wenzhou Medical University.
2.6. Hemolysis evaluation
2 mL of fresh mouse blood was mixed with 4 mL of PBS by the vortex and centrifuged. After removing the supernatant, the red cells were re-suspended and mixed with 20 mL of PBS. The blood cell suspension was mixed with the (Nig + DAC)@HmA working solution and the final concentrations of Nig were 0, 12.5, 25, 50, and 100 μmol/L. Then, the nanoparticles were incubated at 37 °C for 3 h and centrifuged. Deionized water and PBS were used as positive and negative control solutions, respectively. The absorbance at 570 nm of the supernatant was measured and the hemolysis rates were calculated as previously reported1,4.
2.7. Cell internalization
The cell internalization of Sulfo-Cyanine5 (Cy5) and Cy5-labeled HmA (Cy5@HmA) was assessed using a confocal laser scanning microscope (CLSM, AI, Nikon). MB49 cells (5 × 104 per dish) were incubated in the incubator for 12 h, and then fresh medium containing Cy5 and Cy5@HmA was added and incubated for another 2, 4, 6, or 8 h. The cells were fixed with paraformaldehyde, stained with DAPI, and imaged by CLSM.
2.8. Cellular toxicity
The cell cytotoxicity of MB49 cells was detected to screen out the optimum combining concentration of Nig and DAC. MB49 cells (5 × 103 per well) were treated with Nig or DAC for 12 h. The cell cytotoxicity of the drugs was obtained according to the instruction of the CCK8 kit. The cell viability of MB49 cells treated with PBS, HmA, Nig, DAC, the mixture of Nig and DAC (Nig + DAC), and (Nig + DAC)@HmA (Nig: 1 μmol/L, DAC: 2 μmol/L) were comparatively detected with CCK-8.
2.9. RAW264.7 cells stimulation in vitro
MB49 cells pretreated by PBS, HmA, Nig, DAC, Nig + DAC, and (Nig + DAC)@HmA were used to stimulate the RAW264.7 cells which were suspended in a 24-well Transwell® plate and the two cell lines were incubated for 24 h. After treatments, DAPI and TRITC anti-mouse CD86 antibody were employed to stain the RAW264.7 cells and imaged using CLSM. The expression of IL-8 and TNF-α secreted by RAW264.7 cells was assessed by the ELISA kits.
2.10. Biodistribution of Cy5@HmA
Cy5 or Cy5@HmA were intravenously injected in mice transplanted MB49 tumors (1 mg/kg, n = 4) when tumor sizes grew up to 150 mm3. After different intervals of the injection, the NIR fluorescence of Cy5 was imaged by a multimode optical live imaging system (IVIS Lumina XRMS Series III, PerkinElmer, USA). Tumors and the major tissues of the mice were collected for ex vivo NIR fluorescent images.
2.11. Antitumor efficacy evaluation and immune response in vivo
MB49 or 4T1 tumors were established in the C57BL/6 or BALB/c mice at 2 × 106 cells per mouse. PBS, Nig, DAC, Nig + DAC, HmA, and (Nig + DAC)@HmA were injected (dosage: Nig at 1.38 μmol/L·kg, DAC at 2.76 μmol/L·kg, n = 4) every 6 days after tumorigenesis. The blood, major organs, and tumor tissues were obtained after sacrificing the mice on Day 15. The levels of CRE, AST, BUN, and ALT were assessed by a biochemical analyzer (Beckman Coulter, Japan) and the major organs stained with H&E were imaged. The tumor tissues were weighed, photographed, and also stained with H&E and TUNEL. Rabbit anti-mouse Ki67 antibody and Alexa Fluor® 594 labeled goat anti-rabbit IgG(H + L) antibody were applied and imaged by CLSM. For immune response, collected tumors from 4T1 tumor-bearing mice were treated with collagenase I, neutral protease, and hyaluronidase for 30 min. Cells were separated from the treated tumors, filtered, and then blocked by anti-mouse antibodies CD45, F4/80, CD206 (MMR), and CD86. Flow cytometry was then employed for the analysis.
2.12. Statistical analysis
We performed statistical analysis by GraphPad Prism 7.0 software (GraphPad Software, LA, USA). One-way ANOVA or two-tail Student’s t-test was used for the statistical significance analysis. Statistically significance: ∗P < 0.05; extremely significance: ∗∗P<0.01 and ∗∗∗P<0.001; not significant (NS): P > 0.05.
3. Results and discussion
Pyroptosis can relieve the immunosuppression of the tu gasdermins mor microenvironment and induce strong anticancer immunity, and thus is considered a powerful cancer immunotherapeutic strategy. Unfortunately, , the key protein family in the pyroptosis process, are low-expressed in most tumor cells because of the abnormal DNA methylation. Herein, pyroptosis inducer Nig and DNA methyltransferase inhibitor DAC were co-delivered by hexahistidine (His6)-metal assembly with up-regulating GSDMD to achieve pyroptosis-induced immunotherapeutics for cancer treatments. Theoretical molecular docking results indicate that the loaded drugs, Nig and DAC, can intensively bind with His6 mainly through hydrogen bonds and pi-alkyl hydrophobic force (Supporting Information Fig. S1). Considering the proven advantages of HmA such as effective tumor accumulation, rapid endocytosis, and lysosomal escape, we suppose the (Nig + DAC)@HmA nano-system is a promising novel pyroptosis inducer for immunotherapeutics in the anti-cancer field.
3.1. Preparation and characterization
HmA was constructed by the coordination interaction between Zn2+ and His6 side imidazole groups and then Nig and DAC were effortlessly encapsulated (Fig. 1A). The drug loading efficiency of (Nig + DAC)@HmA was calculated to be 2.6% ± 0.2% (DL) and 70.0% ± 2.8% (EE) for Nig, 2.2% ± 0.3% (DL) and 74.0% ± 3.2% (EE) for DAC, respectively. The morphology shows that HmA (Fig. 1B) and (Nig + DAC)@HmA nanoparticles are both irregular and evenly dispersed (Fig. 1C). The average hydrodynamic diameters were 187.5 ± 1.9 nm for HmA (Fig. 1D) and 218.4 ± 3.2 nm for (Nig + DAC)@HmA (Fig. 1E), respectively. The polydispersion index (PDI, 0.30–0.37) suggests narrow distributions. After assembled with Nig and DAC, the zeta potential of HmA increases from 28.9 ± 0.2 to 33.7 ± 0.3 mV for (Nig + DAC)@HmA, indicating the successful drug loading (Fig. 1F). The size of (Nig + DAC)@HmA has no significant change in BSA and FBS for up to 48 h (Supporting Information Fig. S2A), illustrating high stability in blood circulation. The drug release of (Nig + DAC)@HmA at pH 5.0 is more effective than pH 7.2, suggesting that the nano-system can easily unload the drugs in the acid tumor microenvironment without an apparent premature leak in the physical environment (Fig. S2B).
Figure 1.

(A) Schematic representation for the (Nig + DAC)@HmA fabrication. (B, C) SEM images, (D, E) Diameters, scale bar = 200 nm. (F) Zeta potentials of HmA and (Nig + DAC)@HmA. (G, H) Hemolysis evaluation of (Nig + DAC)@HmA (calculated by Nig concentrations at 0, 12.5, 25, 50, and 100 μmol/L and DAC concentrations at 0, 25, 50, 100, and 200 μmol/L). (I) Representative fluorescent images of MB49 cells incubated with Cy5@HmA at 2, 4, 6, and 8 h, scale bar = 20 μm.
To evaluate the biosafety in blood circulation, in vitro hemolysis of the (Nig + DAC)@HmA was assessed at the Nig concentrations ranging from 0 to 100 μmol/L. No obvious hemolysis was observed with the therapeutic dose of (Nig + DAC)@HmA (Fig. 1G) and the hemolysis rates are lower than 5% (Fig. 1H), indicating that the co-delivery system is safe for blood circulation.
For further assessing the drug delivery efficiency of the nanocarrier, the lysosomal escape of HmA was evaluated. After entering cells, nanocarriers are coated by membrane vesicles and transferred into lysosomes. The nanocarriers should rapidly escape from the lysosomes into the cytoplasm to protect their payloads from degradation by various digestive enzymes in the lysosomes. The location of Cy5@HmA in the MB49 cells was observed by CLSM at 2, 4, 6, and 8 h after the treatment (Fig. 1I). In the first 4 h, the yellow fluorescence, which is the overlap between green/red fluorescence, gradually increases in the cells, indicating that the HmA carrier is successfully endocytosed into the cells. The yellow fluorescence becomes stronger at 6 h, which suggests that the Cy5@HmA efficiently enters the lysosomes. The yellow fluorescence separates into green and red after 8 h of the treatment, indicating that the Cy5@HmA escaped from the lysosomes. The rapid lysosomal escape of HmA convinces its potential as an efficient drug carrier by protecting its payloads from degradation in the lysosomes. The endocytic experiment of Cy5 was also carried out for comparison but no statistical difference was observed, indicating that Cy5 molecules are not able to be phagocytosed without HmA carriers (Supporting Information Fig. S3).
3.2. Cell cytotoxicity and pyroptosis stimulated immune response
The in vitro cytotoxicities of HmA, individual and mixture of Nig, DAC drugs, (Nig + DAC)@HmA, were comparatively evaluated using a CCK-8 assay on MB49 cells. The cell viability is negligibly affected by HmA ranging from 0 to 200 μg/mL, which indicates the high biocompatibility of the carrier (Supporting Information Fig. S4). The optimal combination of Nig and DAC for cell inhibitory against MB49 cells was screened in concentrations from 0 to 100 μmol/L, from which we can find that the optimal concentrations for Nig and DAC are 1 and 2 μmol/L, respectively (Fig. 2A). Moreover, as shown in Fig. 2B, compared with the simple drug mixture, (Nig + DAC)@HmA significantly decreases the cell viability (30.9% vs. 45.2%) at the optimum drug concentrations, indicating that the two drugs may have a synergistic effect in the designed drug delivery system.
Figure 2.

(A) Cell viability of MB49 cells treated with Nig and DAC. (B) MB49 cell viability in the presence of individual Nig and DAC, the Nig + DAC mixture and (Nig + DAC)@HmA at 1 μmol/L Nig and 2 μmol/L DAC. (C‒E) NLRP3 and GSDMD gel image and ImageJ quantifications. (F) MB49 cell morphology, scale bar = 20 μm. (G) Fluorescence images of CD86-expressed RAW264.7 cells co-incubated with pretreated MB49 cells, scale bar = 20 μm. (H) IL-8 and (I) TNF-α secretion of RAW264.7 cells co-incubated with pretreated MB49 cells. Data are shown as mean ± SD (n = 5). ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
Taking the severe cell cytotoxicity of (Nig + DAC)@HmA in mind, the expression of pyroptosis-related genes was evaluated by the Western blot method. It is widely accepted that the gasdermin D (GSDMD) and NLRP3 proteins are crucial in the pyroptosis process35,36. As shown in Fig. 2C‒E, Nig, Nig + DAC mixture, and (Nig + DAC)@HmA elevate the expression level of NLRP3 for 2.3, 2.5, and 2.8 fold of the control, respectively. However, the expression of NLRP3 in the presence of DAC and HmA exhibits no significant differences compared with the control. This result is in consistent with the previous reports illustrating that Nig stimulates the expression of NLRP3 inflammasome21, 22, 23. It is notable that (Nig + DAC)@HmA exhibits higher NLRP3 expression than the simple drug mixture, which may result from the cell endocytosis of the nanocarrier. Meanwhile, the expression of GSDMD is enhanced to 1.75-, 1.8-, and 2.1-fold of the control by DAC, Nig + DAC mixture, and (Nig + DAC)@HmA, respectively, while the Nig and HmA groups exhibit no significant difference. As a key protein in the pyroptosis process, GSDMD is often low expressed in tumor cells. The up-regulation of GSDMD by DAC, Nig + DAC mixture, and (Nig + DAC)@HmA indicates that DAC is of great potential value in inducing the pyroptosis of tumor cells. As expected, (Nig + DAC)@HmA enhances the GSDMD expression to a higher extent than the dual drug mixture, confirming the synergistic effect in the co-administration of DAC and Nig by the HmA nanocarrier.
The pyroptosis of cells could be visualized through the change of cell morphology upon different treatments. Pyroptosis is visibly indicated by the membrane pore formation and cell swelling with large bubbles, which results from the assembly of gasdermin-N with the cell membrane37. As shown in Fig. 2F, no obvious difference can be found in the cell morphology with the treatment of HmA, confirming the biocompatibility of the carrier. Individual and the mixture of Nig and DAC induce cell swelling to different extents, while the (Nig + DAC)@HmA-treated group exhibits significant cell swelling with large pores and bubbles. This observation proves the enhanced expression of pyroptosis-related genes, illustrating that pyroptosis is the main pathway of (Nig + DAC)@HmA inducing the programmed cell death.
To analyze the pyroptosis-induced immune stimulation, macrophages RAW264.7 were co-cultured with pretreated MB49 cells. CD86-labeled RAW264.7 cells (red) dramatically increase in the pretreated group (Fig. 2G). What’s more, the expression of CD86 on the surface of cells induced by (Nig + DAC)@HmA is much higher than those treated by Nig, DAC, HmA as well as (Nig + DAC) mixture, suggesting that cancer pyroptosis mediated by the designed nanoparticles is a promising strategy for tumor immunotherapy. Simultaneously, IL-8 and TNF-α secreted by RAW264.7 cells pretreated by (Nig + DAC)@HmA, increase approximately 11 and 2.5 times the control, respectively (Fig. 2H and I). It is well known that in most tumors, the inflamed microenvironment is driven by M2 macrophages. M2 macrophages promote tumor immunosuppression and cancer progression, while M1 macrophages prime the tumor immune and promote tumor rejection by secreting proinflammatory cytokine38. The significantly enhanced expression of IL-8 and TNF-α illustrates that (Nig + DAC)@HmA promotes the tumor-associated macrophages (TAMs) polarized from M2 to M1, which confirms the immune stimulation induced by cell pyroptosis.
3.3. Tumor distribution in vivo
After assessing the anti-tumor efficacy in vitro, the in vivo biodistribution of the nanocarrier was determined by the NIR fluorescence of Cy5 molecules in the administered MB49 tumor-bearing mice. The accumulation of Cy5@HmA in the tumor area gradually increases within the first 12 h and slowly decreases at 24 h after intravenous administration (Fig. 3A and B). Cy5@HmA exhibited significant fluorescence at 72 h post-injection on the tumor site, which is much higher than the bare Cy5. We then compared the distribution of the Cy5 and Cy5@HmA in tumors and major viscera at 72 h post caudal vein injection (Figs. 3C and D). The tumor treated with Cy5@HmA has a 2 fold higher fluorescence intensity than Cy5 itself. Meanwhile, Cy5@HmA is mainly distributed in the liver and can be easily excreted by the kidney. It is worth noting that no obvious drug distribution was found in other major viscera, indicating that Cy5@HmA has no potent toxicity to these viscera. This result illustrates the high tumor accumulation of HmA in vivo, which is crucial for biosafety and effective drug delivery.
Figure 3.

(A) Real-time NIR fluorescence images of tumor-bearing mice after intravenous injection of PBS, Cy5, and Cy5@HmA at different time points. Inside red circles represent the tumors. (B) Quantification of Cy5 fluorescence in the tumor tissues at different time points after indicated treatments. (C) Ex vivo fluorescence images and (D) fluorescent signal quantification in the tumor tissues and main organs of mice at 72 h upon the indicated treatments. Data are presented as mean ± SD (n = 3). ∗∗∗P < 0.001.
3.4. Tumor inhibition effect in vivo
In vivo results from MB49 tumor-bearing models treated with Nig, DAC, Nig + DAC, HmA and (Nig + DAC)@HmA reveal that the (Nig + DAC)@HmA group has a 72.3% tumor size reduction compared with the control group, which is much more efficient than individual Nig (16.7%) and DAC (5.3%) as well as the combined Nig + DAC (38.9%) (Fig. 4A and B, Supporting Information Fig. S5). Encouraging results acquired from the tumor weights suggest that the control group is about 8-fold heavier than that treated with (Nig + DAC)@HmA (Fig. 4C), confirming that the (Nig + DAC)@HmA can significantly inhibit the tumor growth in vivo. The body weight in the (Nig + DAC)@HmA group changes slightly (Supporting Information Fig. S6), demonstrating that it possesses high biocompatibility in vivo.
Figure 4.

(A) MB49 tumors collected on Day 15 upon different treatments. (B) Tumor growth curves and (C) weights of mice transplanted MB49 tumors. (D) Gel image and ImageJ quantification of (E) NLRP3 and (F) GSDMD upon different treatments. (G) Ki67, TUNEL, and H&E staining of MB49 tumor sections, scale bar = 100 μm. Data are presented as mean ± SD (n = 4)∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
By using the Western blot method, the in vivo expression levels of NLRP3 and GSDMD were measured upon different treatments. (Nig + DAC)@HmA enhances the expression of NLRP3 and GSDMD to a greater extent than the individual drugs and mixture groups, which is in high consistent with the in vitro conclusion (Fig. 4D). From the quantitative analysis shown in Fig. 4E and F, (Nig + DAC)@HmA group significantly elevates the expression of NLRP3 and GSDMD by 2.5- and 3.5-fold higher than the control, respectively. The tumor inhibition effects were further investigated by Ki67, TUNEL, and H&E staining experiments (Fig. 4G). The expression of Ki67 is remarkably decreased in the (Nig + DAC)@HmA group, suggesting that the tumor growth is greatly suppressed. The enhanced brownish yellow in the TUNEL experiment indicates the severe cell pyroptosis in this group. From the H&E staining images, one can easily find the dissolution of the cell nucleus, further confirming the thorough tumor inhibition effects of (Nig + DAC)@HmA.
3.5. Systemic immune responses induced by cell pyroptosis in vivo
As discussed above, GSDMD is a key executor of pyroptosis. Bioactive IL-18 and other cellular contents are released from the plasma membrane pores assembled with the N-terminal fragments of GSDMD. The cytokines of the blood serum were evaluated by ELISA. Compared with Nig, DAC, HmA, Nig + DAC mixture exhibits a slight increase while (Nig + DAC)@HmA shows a dramatic enhancement in the cytokine secretion of IL-8 and TNF-α (Fig. 5A and B). This result indicates that (Nig + DAC)@HmA can trigger the antitumor immune response with high efficiency. To further confirm this, the infiltration levels of DCs and macrophages were observed through the expression differentiation of CD11c+ and CD86+, respectively. The expression levels of CD11c and CD86 in the tumor tissues treated with (Nig + DAC)@HmA are markedly enhanced and are more efficient than in other groups (Fig. 5C–E). These results reveal that (Nig + DAC)@HmA can induce macrophages into an anti-tumor M1 phenotype and promote dendritic cell maturation. Moreover, as shown in Fig. 6, the significantly elevated infiltration of CD3+, CD4+, and CD8+ T cells in the tumor sections treated with (Nig + DAC)@HmA identifies that the tumor inhibition is prompted by the enhanced immune effects and the cancer cell pyroptosis is efficient in activating systemic T-cells.
Figure 5.

(A) IL-8 and (B) TNF-α levels in mouse serum on Day 1, 3, and 7. (C) Representative immunofluorescence images of CD86 and CD11c in treated tumor slices, scale bar = 100 μm. (D) ImageJ quantification of CD86 and (E) mature DCs upon different treatments. Data are presented as mean ± SD (n = 3). ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
Figure 6.

(A) Immunofluorescence images and quantitative analysis of (B) CD3+, (C) CD4+, and (D) CD8+ T cells in treated tumor slices of 4T1 tumor-bearing mice. Scale bar = 100 μm. Data are demonstrated as mean ± SD (n = 3). ∗P < 0.05, ∗∗∗P < 0.001.
3.6. Cell pyroptosis-stimulated immune response
The in vivo immune response effect realized by cell pyroptosis was evaluated in 4T1 transplanted mice. The results reveal that the volume of 4T1 tumors decreases by 73.7% when treated with the (Nig + DAC)@HmA group, which is much higher than the other treatments (Fig. 7A and B, and Supporting Information Fig. S7). The average tumor weights of the sacrificed mice treated by (Nig + DAC)@HmA decrease 61.5% compared with the controlled mice (Fig. 7C), confirming that (Nig + DAC)@HmA can significantly inhibit the transplanted tumor growth. What's more, the body weights have a negligible change in the (Nig + DAC)@HmA treatments (Supporting Information Fig. S8), demonstrating its good in vivo biocompatibility.
Figure 7.

(A) 4T1 tumors collected on Day 15 upon different treatments. (B) Tumor growth curves and (C) weights of mice transplanted 4T1 tumors. (D) Quantitative statistical analysis of CD86+/CD206+ level changes in 4T1 tumors detected by flow cytometric analysis. Data are demonstrated as mean ± SD (n = 5). ∗P < 0.05, ∗∗∗P < 0.001.
To identify the in vivo pyroptosis-induced immune stimulation efficiency, the infiltration levels of macrophages in primary tumors were analyzed. The CD86+ and CD206+ expression in differentiation were obtained by flow cytometric analysis of the tumor tissue. As shown in Fig. 7D and Supporting Information Fig. S9, the CD86+ and CD206+ levels apparently increase for the mice treated with (Nig + DAC)@HmA, which are greater than the other treatments. In agreement with the research in vitro, the in vivo results further confirm that pyroptosis can induce macrophages into an anti-tumor M1 phenotype. (Nig + DAC)@HmA can serve as an ideal approach for solid tumor immunotherapy caused by robust immune responses.
Meanwhile, the serum levels of CRE (Fig. 8A), BUN (Fig. 8B), AST (Fig. 8C), and ALT (Fig. 8D) have no obvious difference among these groups, indicating that there is negligible hepatorenal toxicity for all the treatments. After H&E staining, no apparent histopathological lesions were found in the main tissues of mice (Fig. 8E). These results indicate that the drugs and the designed drug delivery system have high biocompatibility and potential clinical application.
Figure 8.

(A) CRE, (B) BUN, (C) AST, and (D) ALT activities of MB49 tumor-bearing mice in different groups. Data are presented as mean ± SD (n = 3). (E) H&E stained photos of the major organs collected from MB49 tumor-bearing mice. Scale bar = 200 μm.
4. Conclusions
Herein, His6‒metal assembly co-loaded with Nig and DAC was effortlessly prepared to achieve pyroptosis-induced tumor immunotherapy. Benefiting from the extraordinary ability of Nig in promoting NLRP3 activation and DAC in up-regulating GSDMD expression, (Nig + DAC)@HmA nanoparticles perfectly solved the critical problems of pyroptosis in tumor immunotherapy that were limited by the low expression of gasdermins resulting from the DNA methylation. (Nig + DAC)@HmA nanoparticles induced the activation of NLRP3, leading to cleavage of the up-regulated GSDMD protein, which in turn induced pyroptosis of cancer cells in vivo. Finally, strong DCs maturation and activated antitumor immunity were caused by inflammatory pyroptosis. Our study provides a new path and inspiration for tumor immunotherapy mediated by pyroptosis. We believe this solution is useful in finding appropriate combination entities to combat the therapeutic defects for a certain disease.
Acknowledgments
This work was financially supported by the National Nature Science Foundation of China (No. 21671150, 52073145, 21877084), and the Key Research Projects of Guangdong Province (NO. 2021ZDZX4019).
Author contributions
Yuna Qian, Ruogu Qi, and Jianliang Shen designed the research. Qiang Niu and Yu Liu carried out the experiments and performed data analysis. Yujing Zheng and Ziwei Tang participated part of the experiments. Qiang Niu wrote the manuscript. Ping Zhao revised the manuscript. All of the authors have read and approved the final manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2022.11.002.
Contributor Information
Ruogu Qi, Email: rqi@njucm.edu.cn.
Jianliang Shen, Email: shenjl@wiucas.ac.cn.
Ping Zhao, Email: pingzhao@gdpu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Zeng Y.P., Li S.F., Zhang S.F., Wang L., Yuan H., Hu F.Q. Cell membrane coated-nanoparticles for cancer immunotherapy. Acta Pharm Sin B. 2022;12:3233–3254. doi: 10.1016/j.apsb.2022.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riley R.S., June C.H., Langer R., Mitchell M.J. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov. 2019;18:175–196. doi: 10.1038/s41573-018-0006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dong S.T., Guo X.N., Han F., He Z.G., Wang Y.J. Emerging role of natural products in cancer immunotherapy. Acta Pharm Sin B. 2022;12:1163–1185. doi: 10.1016/j.apsb.2021.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao Q.Q., Li X.T., Li Y., Wu Z.F., Xu C.J., Chen Z.J., et al. Biological drug and drug delivery-mediated immunotherapy. Acta Pharm Sin B. 2021;11:941–960. doi: 10.1016/j.apsb.2020.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie Q., Ding J., Chen Y. Role of CD8+ T lymphocyte cells: interplay with stromal cells in tumor microenvironment. Acta Pharm Sin B. 2021;11:1365–1378. doi: 10.1016/j.apsb.2021.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J., Anraku Y., Kataoka K. Self-boosting catalytic nanoreactors integrated with triggerable crosslinking membrane networks for initiation of immunogenic cell death by pyroptosis. Angew Chem Int Ed Engl. 2020;59:13526–13530. doi: 10.1002/anie.202004180. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y., Gao W., Shi X., Ding J., Liu W., He H., et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 8.Zhao H., Song Q., Zheng C., Zhao B., Wu L., Feng Q., et al. Implantable bioresponsive nanoarray enhances postsurgical immunotherapy by activating pyroptosis and remodeling tumor microenvironment. Adv Funct Mater. 2020;30:2005747–2005758. [Google Scholar]
- 9.Kovacs S.B., Miao E.A. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017;27:673–684. doi: 10.1016/j.tcb.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu W., Che Y., Zhang Q., Huang H., Ding C., Wang Y., et al. Apaf-1 pyroptosome senses mitochondrial permeability transition. Cell Metab. 2021;33:424–436. doi: 10.1016/j.cmet.2020.11.018. [DOI] [PubMed] [Google Scholar]
- 11.Wu D., Wang S., Yu G., Chen X. Cell death mediated by the pyroptosis pathway with the aid of nanotechnology: prospects for cancer therapy. Angew Chem Int Ed Engl. 2021;60:8018–8034. doi: 10.1002/anie.202010281. [DOI] [PubMed] [Google Scholar]
- 12.Tsuchiya K., Nakajima S., Hosojima S., Thi Nguyen D., Hattori T., Manh Le T., et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat Commun. 2019;10:2091–2110. doi: 10.1038/s41467-019-09753-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broz P., Pelegrin P., Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20:143–157. doi: 10.1038/s41577-019-0228-2. [DOI] [PubMed] [Google Scholar]
- 14.Ding J., Wang K., Liu W., She Y., Sun Q., Shi J., et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 15.Hou J., Zhao R., Xia W., Chang C.W., You Y., Hsu J.M., et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22:1264–1275. doi: 10.1038/s41556-020-0575-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Q., Wang Y., Ding J., Wang C., Zhou X., Gao W., et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature. 2020;579:421–426. doi: 10.1038/s41586-020-2079-1. [DOI] [PubMed] [Google Scholar]
- 17.Brandon E.B., Ashley N.E., Zhu H., Wang S.Z. Gasdermin D in pyroptosis. Acta Pharm Sin B. 2021;11:2768–2782. doi: 10.1016/j.apsb.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panat Y., Chayodom M., Sirawit S., Natthaphat S.A., Siriporn C.C., Nipon C. Gasdermin D-mediated pyroptosis in myocardial ischemia and reperfusion injury: cumulative evidence for future cardioprotective strategies. Acta Pharm Sin B. 2022 doi: 10.1016/j.apsb.2022.08.007. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi J., Kantoff P.W., Wooster R., Farokhzad O.C. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20–37. doi: 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan H., Lin Y., Dou J., Fu Z., Yao Y., Ye S., et al. Wedelolactone facilitates Ser/Thr phosphorylation of NLRP3 dependent on PKA signalling to block inflammasome activation and pyroptosis. Cell Prolif. 2020;53:12868–12880. doi: 10.1111/cpr.12868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antonopoulos C., Russo H.M., El Sanadi C., Martin B.N., Li X., Kaiser W.J., et al. Caspase-8 as an effector and regulator of NLRP3 inflammasome signaling. J Biol Chem. 2015;290:20167–20184. doi: 10.1074/jbc.M115.652321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mariathasan S., Weiss D.S., Newton K., McBride J., O'Rourke K., Roose-Girma M., et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 23.He W.T., Wan H., Hu L., Chen P., Wang X., Huang Z., et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pleyer L., Greil R. Digging deep into "dirty" drugs—modulation of the methylation machinery. Drug Metab Rev. 2015;47:252–279. doi: 10.3109/03602532.2014.995379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao P., Wang M., Chen M., Chen Z., Peng X., Zhou F., et al. Programming cell pyroptosis with biomimetic nanoparticles for solid tumor immunotherapy. Biomaterials. 2020;254:120142–120151. doi: 10.1016/j.biomaterials.2020.120142. [DOI] [PubMed] [Google Scholar]
- 26.Fan J.X., Deng R.H., Wang H., Liu X.H., Wang X.N., Qin R., et al. Epigenetics-based tumor cells pyroptosis for enhancing the immunological effect of chemotherapeutic nanocarriers. Nano Lett. 2019;19:8049–8058. doi: 10.1021/acs.nanolett.9b03245. [DOI] [PubMed] [Google Scholar]
- 27.Sommer S., Cruijsen M., Claus R., Bertz H., Wasch R., Marks R., et al. Decitabine in combination with donor lymphocyte infusions can induce remissions in relapsed myeloid malignancies with higher leukemic burden after allogeneic hematopoietic cell transplantation. Leuk Res. 2018;72:20–26. doi: 10.1016/j.leukres.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y., Tong C., Dai H., Wu Z., Han X., Guo Y., et al. Low-dose decitabine priming endows CAR T cells with enhanced and persistent antitumour potential via epigenetic reprogramming. Nat Commun. 2021;12:409–427. doi: 10.1038/s41467-020-20696-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dhillon S. Decitabine/cedazuridine: first approval. Drugs. 2020;80:1373–1378. doi: 10.1007/s40265-020-01389-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu J., Qin B., Moyer A.M., Nowsheen S., Liu T., Qin S., et al. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J Clin Invest. 2018;128:2376–2388. doi: 10.1172/JCI97924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie B., Liu T., Chen S., Zhang Y., He D., Shao Q., et al. Combination of DNA demethylation and chemotherapy to trigger cell pyroptosis for inhalation treatment of lung cancer. Nanoscale. 2021;13:18608–18615. doi: 10.1039/d1nr05001j. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Z., Zhang Y., Xia S., Kong Q., Li S., Liu X., et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579:415–420. doi: 10.1038/s41586-020-2071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang W., Hao P., Qin J., Luo S., Zhang T., Peng B., et al. Hexahistidine-metal assemblies: a promising drug delivery system. Acta Biomater. 2019;90:441–452. doi: 10.1016/j.actbio.2019.03.058. [DOI] [PubMed] [Google Scholar]
- 34.Zhang T., Zhang L., Wu X., Xu H., Hao P., Huang W., et al. Hexahistidine-metal assemblies: a facile and effective codelivery system of subunit vaccines for potent humoral and cellular immune responses. Mol Pharm. 2020;17:2487–2498. doi: 10.1021/acs.molpharmaceut.0c00212. [DOI] [PubMed] [Google Scholar]
- 35.Song M.Y., Wang J.X., Sun Y.L., Pang J.L., Li X.N., Liu Y., et al. Inhibition of gasdermin D-dependent pyroptosis attenuates the progression of silica-induced pulmonary inflammation and fibrosis. Acta Pharm Sin B. 2021;12:1213–1224. doi: 10.1016/j.apsb.2021.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conos S.A., Chen K.W., De Nardo D., Hara H., Whitehead L., Nunez G., et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci U S A. 2017;114:961–969. doi: 10.1073/pnas.1613305114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding J., Wang K., Liu W., She Y., Sun Q., Shi J., et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 38.Wu T., Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61–68. doi: 10.1016/j.canlet.2016.01.043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.