

SUMMARY
Red blood cells (RBCs, erythrocytes) are the simplest primary human cells, lacking nuclei and major organelles, and instead employing about a thousand proteins to dynamically control cellular function and morphology in response to physiological cues. In this study, we defined a canonical RBC proteome and interactome using quantitative mass spectrometry and machine learning. Our data reveal an RBC interactome dominated by protein homeostasis, redox biology, cytoskeletal dynamics, and carbon metabolism. We validated protein complexes through electron microscopy and chemical crosslinking, and with these data, built 3D structural models of the ankyrin/Band 3/Band 4.2 complex that bridges the spectrin cytoskeleton to the RBC membrane. The model suggests spring-like compression of ankyrin may contribute to the characteristic RBC cell shape and flexibility. Taken together, our study provides an in-depth view of the global protein organization of human RBCs and serves as a comprehensive resource for future research.
Keywords: Red blood cells, erythrocytes, proteomics, protein complexes, band 3 complex
INTRODUCTION
Red blood cells (RBCs) fill a critical role by transporting oxygen and metabolic waste between the lungs and other cells and tissues. This role has been optimized by the unique features of RBCs, which lack nuclei, mitochondria, golgi, the endoplasmic reticulum, and most other major organelles so as to maximize oxygen carrying capacity. Nonetheless, RBCs respond actively to changing tissue environments and dynamically alter their cell shapes on short time scales in order to thread narrow capillary networks and splenic tissues, pointing to the critical importance of protein interactions, allostery, and post-translational modifications as the likely primary mechanisms to regulate RBC activities. As a result, mutations that disrupt these aforementioned processes lead to various blood disorders such as spherocytosis and hemolytic anemia.
RBCs have been studied extensively for decades and have provided us with essential basic information on cell physiology and molecular biology. However, a consensus on their complete proteome has not been reached, nor is it known how these proteins organize into the large multiprotein assemblies that support all RBC function in the absence of transcriptional and translational activity. While hemoglobin comprises the vast majority of RBC protein content (~98%)(Goodman et al., 2007; Kabanova et al., 2009; Pasini et al., 2006), more than a thousand other distinct proteins are predicted to constitute the remaining 2% of protein, many of which are still largely uncharacterized. Since proteins rarely act alone, building a more complete picture of multiprotein assemblies is key to better understanding RBC functions and diseases, including cell shape control and the “shape-opathies” that result from its disruption.
Although techniques such as affinity purification mass spectrometry (AP-MS) and proximity labeling are available to study protein-protein interactions (PPIs) in other cell types, these approaches are not feasible in RBCs because of the lack of nuclei and transcription, and antibody-based approaches such as immunoprecipitation-mass spectrometry (IP-MS) are prohibitively expensive for large-scale screening. Therefore, we turned to another powerful technique to study protein complexes, co-fractionation mass spectrometry (CF-MS). CF-MS is a high-throughput technique that combines biochemical fractionations, protein mass spectrometry, and machine learning to characterize PPIs, in which the co-elution (co-fractionation) of stably-associated proteins through the course of distinct and orthogonal biochemical separations serves as evidence for the proteins’ physical association (Skinnider and Foster, 2021; Wan et al., 2015). As CF-MS requires neither antibodies nor recombinant epitope tagging of individual proteins, it is uniquely well-suited to study RBCs. The power of this technique comes from the analysis of native proteins’ elution profiles from multiple orthogonal biochemical separations, using machine learning and extensive internal control proteins (known complexes) to distinguish between true PPIs and randomly co-eluting proteins, quantifying the support for the physiologically relevant PPIs in order to maintain strong control over false discovery rates of protein interactions.
In this work, we performed over 30 biochemical fractionations on purified RBCs and applied quantitative mass spectrometry on these fractions (>1,900 mass spectrometry experiments in all) in order to identify a canonical set of approximately 1,200 RBC proteins and to derive an interactome map of RBC protein complexes. Using a rigorous statistical framework, we recovered high confidence PPIs from known complexes as well as novel complexes. Most of the complexes in RBCs are involved in energy metabolism, structural integrity, redox biology, and proteostasis. Furthermore, we performed chemical crosslinking on these native complexes and used integrative structural modeling to shed light on the molecular organization of integral membrane proteins, channels, cytoskeletal proteins, and metabolic enzymes at the RBC membrane. Our findings provide a comprehensive blueprint of the cell surface and subcellular architecture of a key blood cell type, and suggest biophysical mechanisms underlying its adaptability and pathological reorganization in diverse blood disorders.
RESULTS AND DISCUSSION
A large data set of protein abundances and co-purification of RBC proteins
In order to characterize the RBC proteins (Figure 1) and their assemblies (Figure 2), we generated a large proteomics dataset of fractionated soluble and membrane-associated proteins from enriched human RBCs using various methods of biochemical fractionation. Non-denatured protein extracts from hemolysate (soluble proteins) and non-ionic detergent dissolved ghosts (membrane proteins) were separated by discrete biophysical properties such as size, charge, and hydrophobicity (see Zenodo data repository for details of fractionations, donors’ details, detergents used). Each chromatographic fraction was analyzed by high resolution, high sensitivity liquid chromatography/mass spectrometry (LC/MS). In all, we collected 6,255,027 interpretable peptide mass spectra from 1,944 individual chromatographic fractions. Each fraction captures distinct subsets of native RBC proteins and protein assemblies, collectively providing a compendium of informative co-elution profiles.
Figure 1. Defining the canonical red blood cell (RBC) proteome with high accuracy from a synthesis of protein mass spectrometry and mRNA expression data.
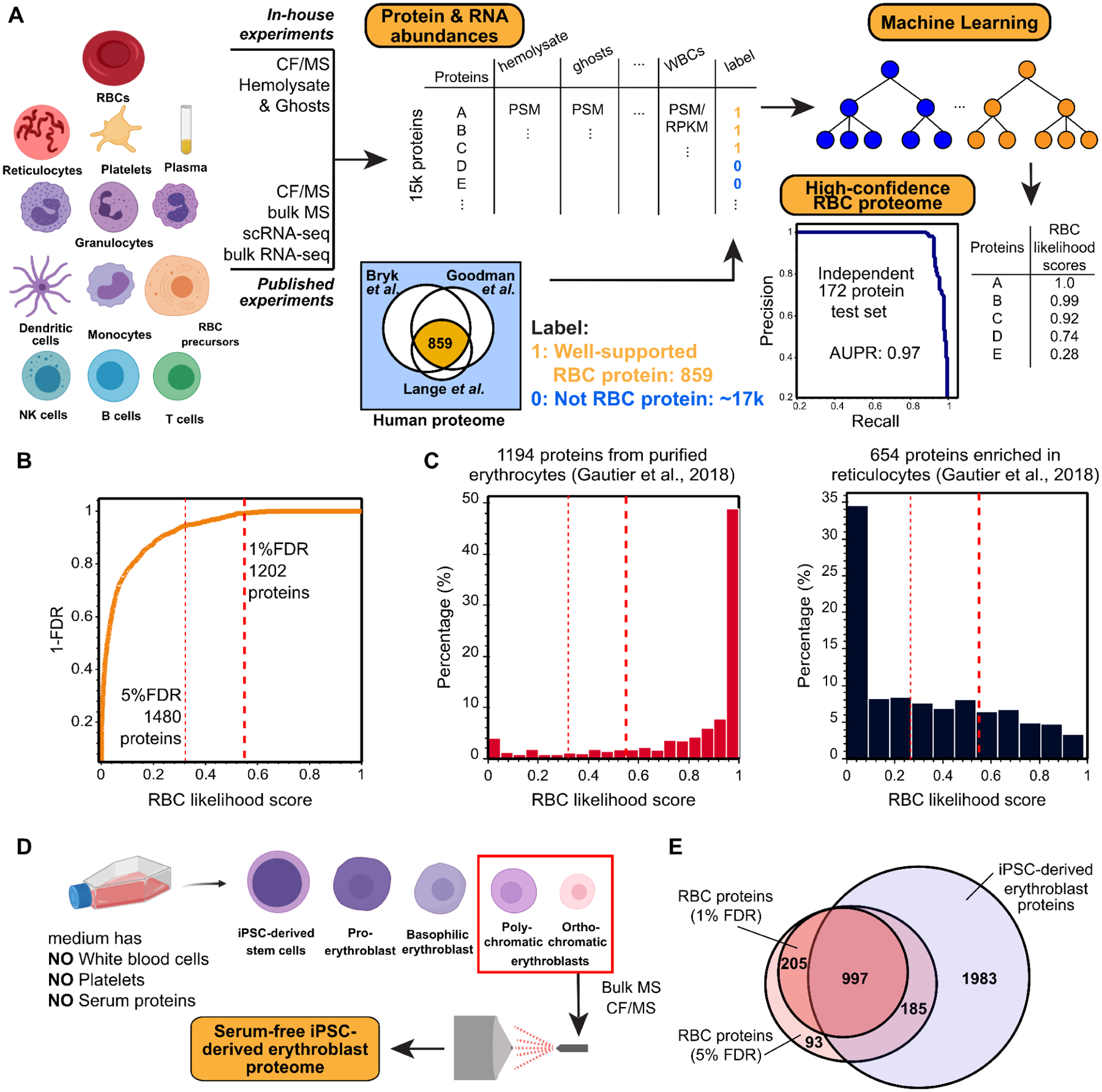
(A) Measured protein and RNA abundances from diverse blood cell types and plasma were used as features for a machine learning classifier to assign confidence scores for proteins whether they belong to the RBCs or rather contaminants contributed by other cells or plasma. The classifier showed high precision and recall (area under the recall-precision curve (AUPR) = 0.97) as assessed on a 172 protein set withheld from the training. Cell images created with Biorender.com.
(B) Applying the classifier and thresholding at a 1% false discovery rate (FDR), we observe the canonical RBC proteome to comprise 1,202 proteins.
(C) The resulting high confidence RBC proteins are highly concordant with proteins previously identified from purified erythrocytes (Gautier et al., 2018). In contrast, the 1% FDR proteome notably excludes proteins known to be strongly enriched in reticulocytes (Gautier et al., 2018).
(D) To further assess the potential for proteins to be contributed from other blood cells or serum, we differentiated iPSC cells into polychromatic and orthochromatic erythroblasts in serum-free medium lacking white blood cells, platelets, and serum proteins, then analyzed the erythroblast proteome using mass spectrometry.
(E) A large majority of high confidence RBC proteins at both the 1% and 5% FDR level (1,202 proteins in total) were also detectable in erythroblasts, consistent with our expectation that mature RBC proteins should generally be detectable in a relevant precursor cell population.
Figure 2. Overview of the integrative Co-Fractionation / Mass Spectrometry (CF-MS) workflow used to determine stable RBC protein complexes.
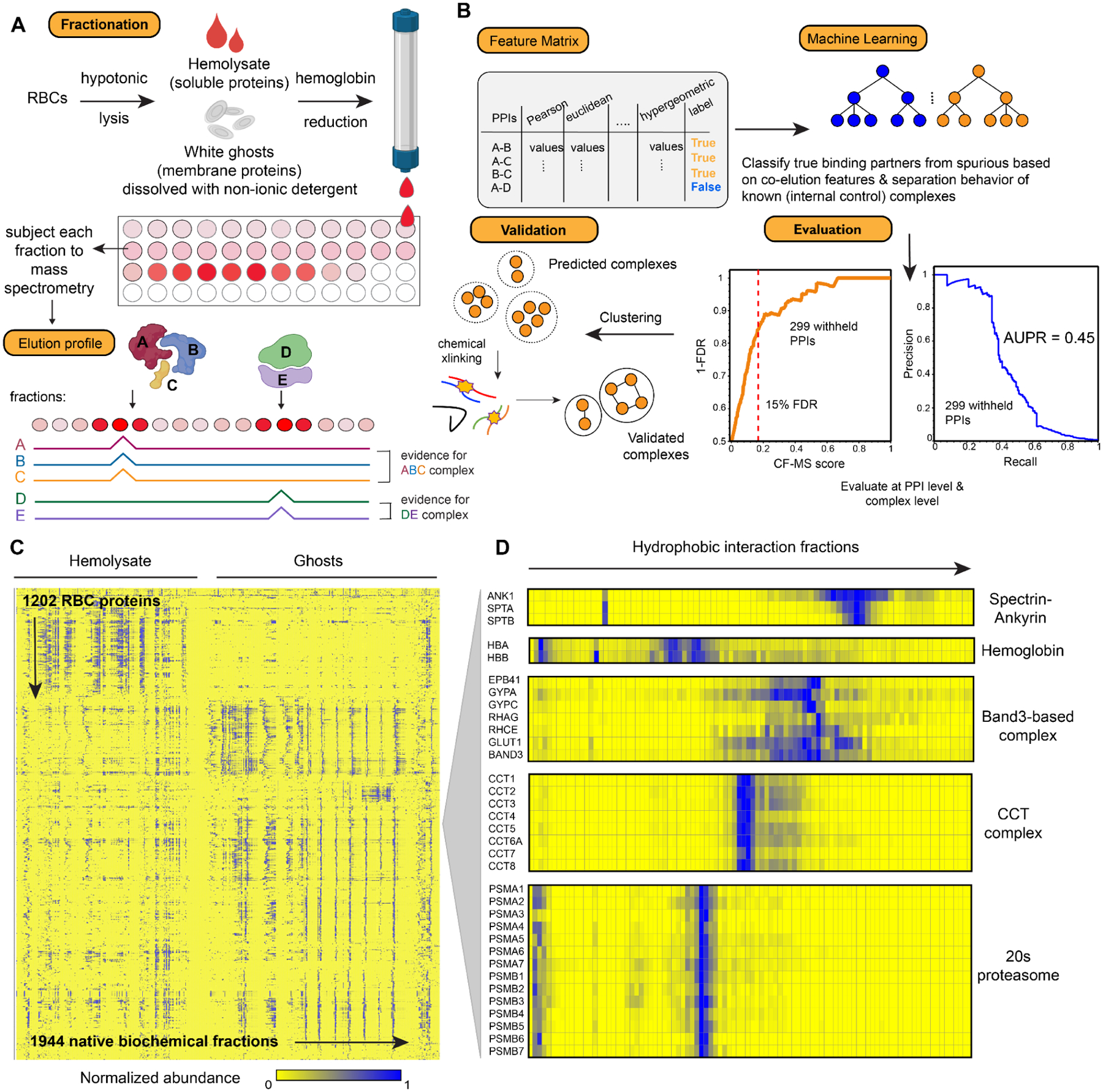
(A) Hemolysate and white ghosts are chromatographically separated and the proteins in each fraction are identified by mass spectrometry. Elution profiles for each protein are graphically represented as ridgelines across multiple separation experiments. Cell/protein complexes images created with Biorender.com.
(B) Different measures of correlation between each pair of proteins are used to construct a feature matrix for machine learning, which computes a score (CF-MS score) indicating how likely the interaction between two proteins would be in RBCs. The classifier showed good precision and recall (area under the recall-precision curve (AUPR) = 0.45) as assessed on 299 PPIs withheld from the training. Further validations were achieved through crosslinking mass spectrometry and direct visualization by electron microscopy.
(C) Heat map of the full dataset of abundance measurements for each of the 1,202 RBC proteins across all fractionations of hemolysate and white ghosts. Blue indicates non-zero signal.
(D) Enlarged portions of (C) showing examples of strong co-elution observed for subunits (gene names on left) of five well-known protein complexes in RBCs (complex names on right). Color intensity (blue is positive signal) depicts abundances for each protein from a representative hydrophobic interaction chromatography experiment (labeled on top) out of the 30 total separations.
Determination of a high-confidence RBC proteome
From this large dataset, we first aimed to comprehensively define a canonical set of RBC proteins. Although multiple previous surveys have identified versions of the RBC proteome (Bryk and Wiśniewski, 2017; Goodman et al., 2007; Lange et al., 2014), these studies only agree on 859 constituent proteins (Figure 1A). Discrepancies might arise from technical variation in the analyses and samples as well as from the presence of contaminating proteins contributed by accompanying reticulocytes, platelets, white blood cells (WBCs), and serum proteins. We attempted to reconcile these published datasets and our own data using a bioinformatic approach: We trained a machine learning classifier on available MS and RNA-seq data from different blood cell types (Figure 1A) in order to recognize the best supported consensus set of RBC proteins. The rationale for this approach is that some proteins detected in previous studies or our fractionation experiments inevitably derive from non-RBC blood cells despite best efforts to enrich RBCs. This issue is exacerbated by the fact that all non-hemoglobin proteins only represent 2% of the RBC proteins, and thus even 2% of contaminant cells could skew protein identifications for RBC proteome. The consideration of data spanning multiple cell types should allow RBC proteins to be better distinguished from proteins from other blood cells and serum.
There are two major issues to consider when trying to identify the comprehensive list of RBC proteins: coverage and contamination. In terms of coverage, we detected >90% of the highly-confident set of 859 previously described proteins among the 2,000 most abundant proteins from our experiments (Figure S1A). This analysis shows that our co-fractionation experiments were well-powered to detect most RBC proteins. Some exceptions are notable, particularly alpha and beta actins (ACTA, ACTB), which are systematically depleted due to our use of mild and non-ionic detergents to extract proteins while preserving stable interactions, and the Duffy antigen ACKR1.
In order to distinguish bona-fide RBC proteins from proteins from other cell types, we employed a supervised machine learning approach to quantify the likelihood of proteins deriving from RBCs. We analyzed protein and RNA abundances (from MS and RNA-seq data) from RBCs, RBC precursor cells, other blood cell types (including white blood cells, platelets, and reticulocytes), and serum (Figure 1A), with the derived likelihood score based solely on these measurements. As confirmed positive training examples, we selected a set of 687 known RBC proteins out of the 859 proteins agreed upon by all three reference studies, and we withheld the remaining 172 positive examples as an independent test set. As confirmed negative training examples, we considered all human proteins observed in our proteomics experiments that were not observed as RBC proteins by the 3 prior studies. We then trained a random forest classifier (with 5-fold cross-validation) to assign a confidence score between 0 and 1 to each protein (see Zenodo data repository for scored proteins), with 1 indicating a high likelihood of the protein existing in mature RBCs, and 0 indicating a likely contaminant.
We assessed the quality of these confidence scores by comparison to the withheld test set of 172 known RBC protein markers and a set of withheld negative test examples. The classifier performed extremely well as indicated by a high area under the precision-recall curve (AUPR) of 0.97 (Figure 1A). Ranking proteins based on their likelihood scores enabled us to measure classifier false discovery rates (FDR) (Figure 1B). At a likelihood score >0.55, we observed 1,202 proteins at 1% FDR (Figure S1B), all of which were directly supported by proteomics evidence (see Table S1 in Zenodo repository). We consider this stringent threshold to define a comprehensive and high confidence set of RBC proteins. Of the 859 gold standard proteins, 785 are recovered in our canonical RBC proteome at 1% FDR and 26 more recovered at the 5% FDR rate, leaving 48 with a substantially lower degree of support from all sources of evidence included in our classifier for being true members of the RBC proteome. Ranking the proteins detected in each of the three prior RBC proteome studies by our RBC likelihood score shows that our classifier efficiently distinguished RBC proteins from non-RBC proteins (Figure S1C–E). Among the reference studies, the preparation by Lange et al. comprised a notably high proportion of mature RBCs.
Besides well-established RBC proteins such as hemoglobin, carbonic anhydrase, RBC-specific membrane proteins (e.g., Band 3, Ankyrin 1, Band 4.2), and blood-type antigens (e.g., Rh antigens, Kell), the high-confidence RBC proteome faithfully reflects the unique biology of mature RBCs in that they lack major organelles, or the apparatus for DNA replication, transcription, and protein synthesis. To further support the proteome, because most proteins in mature RBCs would be already present in RBC precursor cells, we asked if they were additionally present in induced pluripotent stem cell (iPSC)-derived erythroblasts. While nucleated, these RBC precursors were grown in serum-free medium (Vanuytsel et al., 2018) and were devoid of all contaminant proteins from other blood cell types and from serum (Figure 1D). In all, 83% of the high-confidence RBC proteins in our compendium (1% FDR) were also present in the erythroblasts. Similarly, we compared the high confidence set of RBC proteins to those obtained through multi-step RBC purification, including centrifugation, gradient/cellulose purification, and fluorescence activated cell sorting (FACS), by Gautier and colleagues (Gautier et al., 2018). The majority of RBC-specific proteins obtained in this way exhibited high RBC likelihood scores, while the majority of proteins obtained from similarly enriched reticulocytes had markedly lower RBC likelihood scores (Figure 1C). Indeed, the high-confidence RBC proteome excluded CD71 (Transferrin receptor protein 1, TFRC), a well-known reticulocyte marker protein (Gautier et al., 2016, 2018; Goodman et al., 2007). Taken together, these results suggest that our bioinformatic approach effectively captured the proteome of mature RBCs and excluded most major contaminating non-RBC proteins.
Interestingly, some proteins with high RBC likelihood scores consisted of annotated subunits of ribosomes, nuclear transport proteins, and parts of the ER and Golgi machineries, despite RBCs lacking these larger systems. Many of these proteins were also detected as RBC-specific populations by Gautier et al., 2018, and were observed in the iPSC-derived erythroblasts. Thus, they are unlikely to be contaminants. Although we can not rule out the possibility of moonlighting functions in RBCs, another explanation may simply be that such proteins are not eliminated or degraded effectively during the process of enucleation in reticulocytes, in which the nucleus was expelled, major organelles were eliminated, and other superfluous proteins were degraded. Thus, the proteome could at least in part reflect an imperfect cell maturation process.
Systematic identification and scoring of stable RBC protein interactions
Given this high confidence set of RBC proteins, we next sought to determine their association into higher order multi-protein assemblies. In CF-MS, subunits of many well-known complexes coelute with distinct patterns across different types of biochemical separations due to differences in complex sizes, charges, and chemical properties (Figure 2A). From our experience working with different tissues and organisms, detergent-dissolved membrane protein complexes, such as the Band3 complex in Figure 2C, tend to have broader chromatographic peaks compared to discrete soluble complexes such as the proteasome and CCT complexes. Membrane proteins often exist in a variety of forms with different amounts of detergent/lipids/interaction partners, resulting in their detection across multiple biochemical fractions. To analyze such differences and identify co-eluting proteins in a systematic and high-throughput manner, we again used a supervised machine learning approach in order to assign a confidence score to each potential protein-protein interaction based on the mass spectrometry evidence (Figure 2B). Importantly, PPIs were derived solely from the separation behavior of the observed proteins over multiple, orthogonal biochemical fractionation experiments directly from RBCs, with no contribution from external data (see Methods and Zenodo data repository). We focused on abundant RBC proteins with the most robust mass spectrometry evidence (more than 60 independent peptide-spectral matches (PSMs) across 1,944 biochemical fractions). We trained an extra tree classifier (using 5-fold cross-validation) to distinguish between pairs of proteins known to stably interact and random pairs of proteins (details in Methods). The classifier assigned a probabilistic CF-MS score between 0 and 1 to each potential PPI, with 1 indicating a high likelihood of physical association based on observing strongly coordinated protein elution profiles, and 0 indicating no evidence for interaction in RBCs.
We judged the quality of the measured interactions by comparing to a withheld test set of 299 known protein interactions (Figure 2B), which allowed us to measure classifier error rates. The CF-MS scores accurately recapitulated the test PPIs: for interactions with CF-MS scores over 0.17, we observed 85% precision and 35% recall (Figure 2B), with an Area Under the Precision-Recall (AUPR) of 0.45, well above the 0.03 expected by random chance. In all, we observed 3229 PPIs among the high-confidence RBC proteins that scored 0.17 or better, providing an estimate of the RBC interactome.
Direct visualization of biochemically fractionated complexes by electron microscopy
Because CF-MS involves biochemical enrichment and separation of intact endogenous protein complexes, we could use electron microscopy (EM) to directly visualize the larger complexes in a relatively unbiased fashion. This combination of CF-MS with EM on cell extracts can provide rich data for near-native structure determination (Kastritis et al., 2017; Kyrilis et al., 2021; McCafferty et al., 2020; Verbeke et al., 2018) and has been applied to diverse samples (Arimura et al., 2021; Kim et al., 2020; Kirykowicz and Woodward, 2020; Yi et al., 2019), including membrane proteins (Su et al., 2021).
First, to survey the size, shape, and complexity of assemblies across a representative biochemical separation, we performed negative stain EM on pooled, adjacent fractions from a size exclusion chromatography separation (Figure 3A). By comparing the micrographs with proteins identified in the corresponding mass spectrometry experiments and prior knowledge of 3D structures, we identified four distinct protein complexes across the fractions (Figure 3B), three of which were large homo-oligomeric assemblies. Our ability to identify mass spectrometry observations with EM particles was largely due to the lower complexity of the RBC proteome, which is ¼ the complexity of E. coli cells but is dominated by a smaller number of high abundance components that are the major proteins in the MS and EM. The Tripeptidyl-peptidase 2 (TPP2) was the largest observed homo-oligomer and is known to form 5–6 MDa assemblies (Macpherson et al., 1987; Schönegge et al., 2012). The other homo-oligomers identified were the porphyrin-biosynthetic enzyme delta-aminolevulinic acid dehydratase (ALAD) (Mills-Davies et al., 2017), an ~290 kDa homo octamer with D4 symmetry, and the peroxiredoxin PRDX2 (Schröder et al., 2000), an ~218 kDa homo decamer with D5 symmetry. Although the EM reconstructions are at relatively low-resolution, known protein structures could be readily docked into the EM reconstructions, confirming their identities and structural integrity throughout the separations.
Figure 3. Validation of the CF-MS workflow using electron microscopy confirms intact multi-protein complexes.
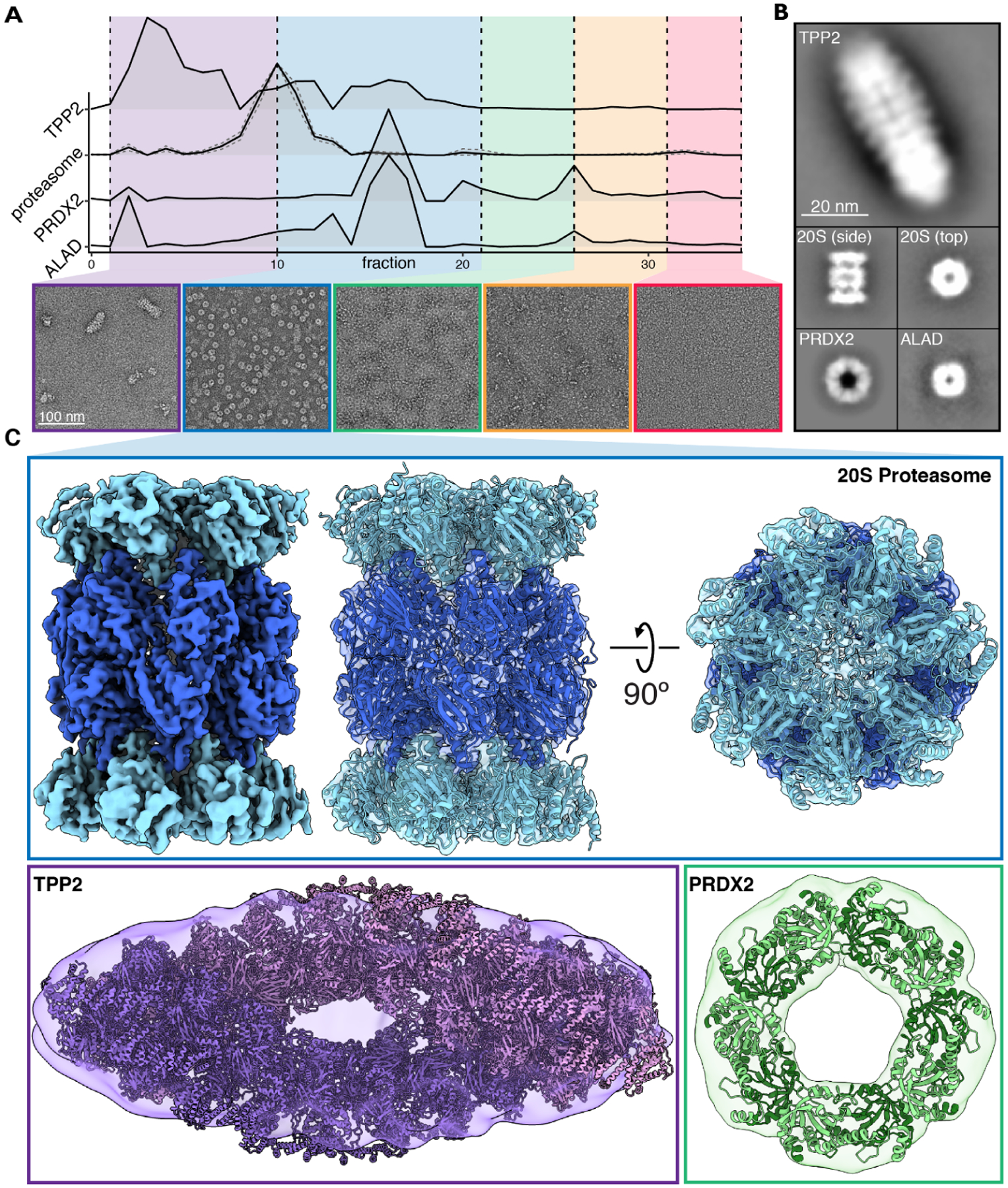
(A) Hemolysate from size exclusion chromatography was partitioned into five groups and visualized with negative stain EM. Elution profiles from corresponding mass spectrometry data were used to assist in identifying abundant protein assemblies.
(B) Reference-free 2D class averages of four protein complexes spanning ~220 – 5,000 kDa were identified in hemolysate.
(C) Cryo-EM reconstruction of the 20S proteasome. Negative stain structures of TPP2 and PRDX2 along with docking of their corresponding atomic structures PDB 3LXU (Chuang et al., 2010) and PDB 1QMV (Schröder et al., 2000), respectively.
These data also clearly showed a high abundance of proteasomes, which provided an opportunity to revisit an outstanding question as to whether proteasomes in mature RBCs are active (i.e., in their regulated 26S forms) or mostly inactive (with separated 20S cores and 19S caps). We first pooled the proteasome containing fractions (fractions 10–21) for higher resolution analysis by cryo-EM. By single particle analysis, we obtained a reconstruction of the 20S proteasome directly from RBCs with a nominal resolution of 3.35 Å, confirming an intact, not visibly modified core proteasome (Figure 3C, Figure S2). To further investigate the proteasome’s activity in RBCs, we used negative stain EM to survey hemolysate after being passed through a 100 kDa filter and quantified different assembly states of the proteasome (Figure S3). We found that ~94% of the proteasomes observed were of the free 20S form, while the remaining ~6% were singly-capped 26S proteasomes (Figure S3). These results agreed with our CF-MS observations of generally separated 19S and 20S proteasomes, and suggested that active 26S proteasomes were present only in relatively low abundances, although we cannot rule out the possibility of the initial cell lysis and hemoglobin depletion processes altering the proteasome assembly states.
The interactome of mature red blood cells
The visualization of known complexes by electron microscopy confirmed that stable multiprotein assemblies were preserved well across our biochemical separations, so we next sought to systematically define protein complexes in mature RBCs by clustering the proteins based on the measured pairwise interactions. To provide a more comprehensive view of protein complexes, we elected to use multiple clustering cutoffs to reflect the hierarchies intrinsic to interacting proteins, the results of which are visualized in Figure 4.
Figure 4. A map of the primary RBC multiprotein complexes.
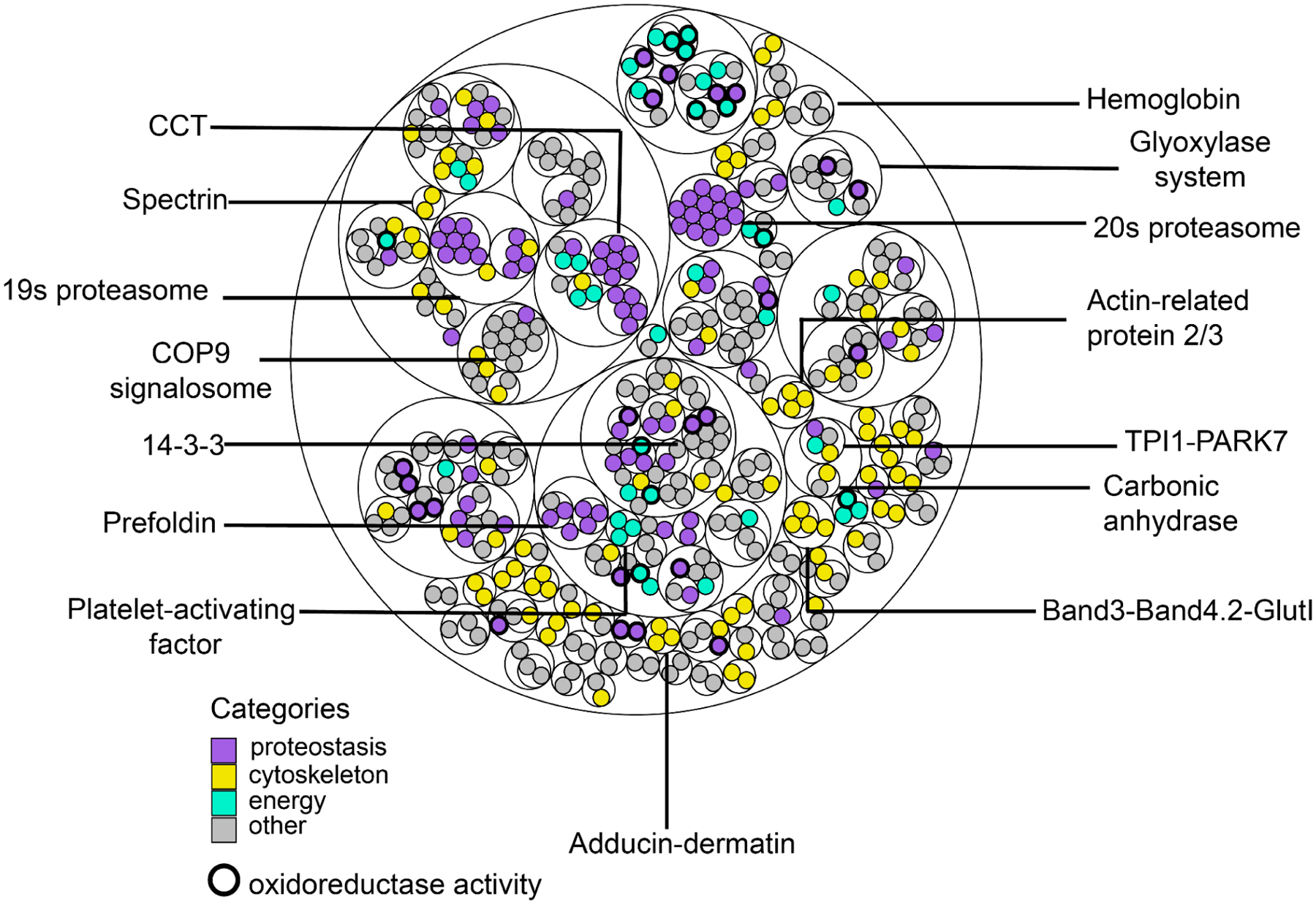
Thin circles show the clustering hierarchy of protein-protein interactions into complexes for each of 3 clustering thresholds (see the Zenodo data repository for complex memberships and annotations). Proteins (filled circles) are colored by broad biological categories (analyzed using the DAVID annotation tool); bold outlines denote proteins with oxidoreductase activity.
In all, the interactome of mature RBCs represents a remarkably compact and minimal assembly of protein machinery. First, we find just over 100 protein complexes in mature RBCs. While this is already apparent at the proteome level (~1,200 total proteins in mature RBCs, or approximately ¼ the complexity of the proteome of the bacterium E. coli and 1/16 of the consensus human proteome), the number of complexes is still striking in comparison to the >400 complexes in E. coli (Hu et al., 2009) and >7,000 human protein complexes known to date (Drew et al., 2021). To gain some insights into the interactome, we analyzed the proteins according to their broad functional categories (Huang et al., 2009a, 2009b), and found most categorized proteins mapped into 4 major categories: proteostasis (purple), cytoskeleton/structural integrity (yellow), energy (mint green), and others (grey). The clustering evident in the map supports the tendency to correctly associate proteins that bind each other and shows the relative balance of the different functions across the interactome.
Proteins of proteostasis dominate the observed RBC interactome, notable among these being the 19S regulatory cap and 20S core of the proteasome. However, consistent with the evidence from EM micrographs (Figure S3), the clustering of PPIs suggests that proteasomes in mature RBC generally exist as distinct 19S and 20S forms. While the 26S proteasome is essential for the enucleation and maturation process as RBCs remove many of their precursor proteins and maximize cellular space for hemoglobin, it is unclear if proteasome activity is essential in RBCs post-maturation. Our detection of higher amounts of the 19S and 20S forms than the 26S form suggests that canonical ubiquitin-dependent degradation might be kept at a minimal level in mature RBCs since this process requires the 26S form. However, it is possible that the 20S proteasome may still act to degrade unfolded and misfolded proteins without the 19S cap, especially when disordered regions are exposed, as seen in a few reports (Alvarez-Castelao and Castaño, 2005; Asher et al., 2005; Sorokin et al., 2005). In addition, the prevalence of 20S and 19S forms over 26S might also result from a more oxidizing cellular environment in RBCs than other cells, such as muscle cells (~11:1 ratio between NAD+/NADH for muscle cells and ~8:1 for RBCs) (Demarest et al., 2019). NADH is known to bind to at least 5 19S subunits and stabilize the 26S form in turn (Tsvetkov et al., 2014). While we observe many proteins related to proteostasis, some of the proteins that fall into this category include chaperones and ubiquitin-related enzymes, which might be remnants of the robust translation and active ubiquitin-dependent degradation during maturation given that most observed proteasomes in mature RBC are 20S.
Besides proteostasis, energy production and redox-related protein complexes were abundant. A survey of the interactome suggests a balance in regulating energy production and managing toxic by-products through detoxification and redox enzymes (bolded circle, Figure 4). RBCs consume energy to maintain proper ion concentrations and appropriate ratios of surface area to cell volume (Lux, 2016; Mohandas and Gallagher, 2008), which are essential to the ability of RBCs to change morphology without lysing. The major source of ATP production in RBCs involves the metabolism of glucose via the Embden-Myerhoff or glycolysis pathway (Brown, 1996). One of the toxic byproducts is methylglyoxal, a highly reactive dicarbonyl compound that reacts with proteins through the Maillard reaction to form glycated proteins, impairing protein function. In order to detoxify methylglyoxal, cells rely on the glyoxalase system/complex, which is recapitulated in the interactome. We find evidence for a novel interaction relevant to methylglyoxal detoxification occurring between triosephosphate isomerase (TPI1) and the Parkinsonism-associated protein (DJ-1/Park7). TPI1 is known to produce a significant amount of methylglyoxal as a byproduct (Richard., 1991), and Park7 is proposed to be a bonafide deglycase (Richarme & Laidrou., 2017). This TPI1-Park7 interaction suggests a physical linkage between energy production and byproduct detoxification in RBCs.
Finally, cytoskeletal complexes are essential to RBCs, as they maintain structural integrity and enable cell morphology changes as RBCs traverse blood vessels. The interactome recapitulates well-known cytoskeletal complexes involving, among other proteins, spectrin, adducin-dermatin, and band 3, and suggests subcomplexes such as Band 3, GlutI, and Band 4.2, as described in more detail below.
Cross-linking and integrative 3D modeling of the major RBC cytoskeletal complexes
As cytoskeletal complexes are essential to many RBC functions, we particularly sought to investigate protein complexes isolated from RBC white ghosts, which are composed almost exclusively of membrane/cytoskeletal components. Thus, we separated detergent-solubilized white ghost protein extracts by size exclusion chromatography and performed chemical crosslinking mass spectrometry (XL-MS) in order to precisely determine amino acid-resolution PPI contacts between membrane and cytoskeletal proteins. We chose disuccinimidyl sulfoxide (DSSO) for crosslinking, as it is MS-cleaveable and thus suitable for highly accurate determination of crosslinked peptides (Kao et al., 2011). In all, we identified 769 high confidence crosslinks among 129 proteins, dominated by interactions among spectrins, ankyrin, and their interaction partners, including the Rh antigens, Band 3, and Band 4.1 (Figure 5A). Crosslinked proteins were more likely to have high CF-MS scores, suggesting that the experiment captured true molecular interactions (Figure S4). In addition to intermolecular crosslinks (Figure 5A), we observed large numbers of intramolecular crosslinks, as highlighted in Figure 5B for spectrin alpha and beta.
Figure 5. Chemical crosslinks confirm mapped interactions and constrain 3D modeling of membrane/cytoskeletal complexes.
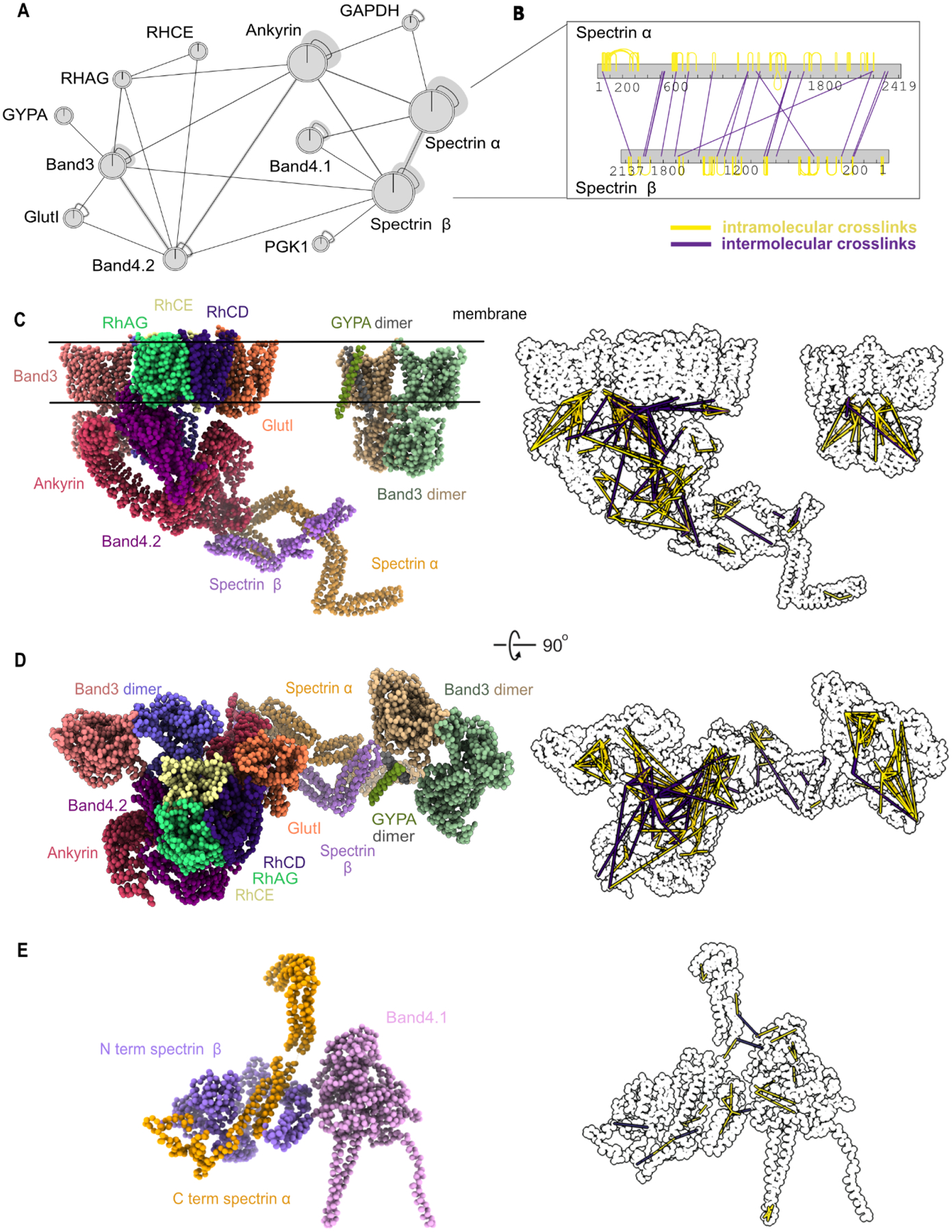
(A) Network plot represents crosslinked interactions among proteins (shown as gene names) in RBC cytoskeletal complexes. Line between each node indicates detected crosslink(s). Shaded lines indicate dense crosslinking.
(B) Bar plot shows extensive crosslinks between spectrin alpha and beta supporting a head-to-tail conformation. Numbers under each bar indicate the amino acid position on each protein. Yellow line indicates intramolecular crosslinks and purple line indicates intermolecular crosslinks.
(C)Side view of integrative structure of band 3-Ank1 complex and band 3-GYPA complex. Our model suggests that GlutI competes with the Rh proteins for binding with band 3 and band 4.2. The outline figure on the right shows intramolecular and intermolecular crosslinks that are overlaid onto the structure.
(D)Top view of integrative structure of the band 3-Ank1 complex and band 3-GYPA complex.
(E) Integrative structure of band 4.1-spectrin complex. Six of these complexes are proposed to link spectrin heterodimers with actin (Lux, 2016).
Each crosslink generally provides evidence for the modified lysines residing within 30 Å of each other. Thus, such data enable initial molecular models to be constructed of the component proteins. For example, the pattern of crosslinks between spectrin alpha and beta strongly supports them associating in a head-to-tail conformation (Figure 5B), consistent with literature reports that the N-terminal domain of spectrin alpha interacts with the C-terminus of spectrin beta (Speicher et al., 1992; Ungewickell and Gratzer, 1978). To further evaluate the quality of the crosslinks, we used known X-ray crystal structures of the proteins band 3 (PDB ID: 1HYN, 4YZF), GAPDH (PDB ID: 1U8F), and PGK1 (PDB ID: 4O33) to calibrate the crosslinks’ accuracies. Of the 24 crosslinks between residue pairs within these X-ray crystal structures, all 24 occurred between amino acids with Cα atoms falling within 30 Å of each other, consistent with the length of the DSSO crosslinker (Figure S4).
Next, we integrated the crosslinks with available structural information in order to build models of several of the major RBC membrane/cytoskeletal complexes. We additionally incorporated knowledge of the positions of transmembrane regions (The UniProt Consortium, 2019), protein structures from comparative modeling, and, where available, X-ray crystal structures. Using integrative modeling (Figure S5) (Russel et al., 2012), we built many (600,000) models in parallel and tested for their agreement (see Methods); the combination of annotated transmembrane regions, chemical crosslinks, and known partial structures provided sufficient spatial restraints to converge on a solution. High-scoring models that satisfied the crosslinking and membrane restraints were clustered based on root-mean-square distance (RMSD), and the largest cluster (Table S2) was selected as the preferred form of each complex, with the centroid model as the representative model for the cluster.
This integrative modeling approach provided us with a detailed molecular view of both the band 3 (Figure 5C–D) and band 4.1 complexes (Figure 5E).Yellow and purple lines to the right of the models in Figure 5C–E highlight positions of intramolecular crosslinks and intermolecular crosslinks, respectively. Surprisingly, 30% of the crosslinks for ankyrin1 violated the 30 Å distance of the crosslinker, far in excess of the typically fewer than 20% of violations expected in such models (Leitner et al., 2016; Liu et al., 2018; Wang et al., 2017) and the 100% concordance we observed with the isolated band 3 and PGK1 structures, and suggesting that the reported X-ray crystal structure of ankyrin1 (Wang et al., 2014) does not match the predominant form found in RBCs. Notably, the reported structure is in an extended (“open”) form. Our crosslinks suggest that the violation could be due to ankyrin adopting a compressed (“closed”) conformation, as the violations are highly directionally correlated in two specific regions of ankyrin (Figure S6). When modeling a closed conformation of the ankyrin complex, 92% of the crosslink violations for ankyrin1 can be satisfied (Figure S6). We speculate that the open and closed conformations could be a result of spring-like behavior of ankyrin repeats; indeed, spring-like behavior has been previously observed for ankyrin by atomic force microscopy (Lee et al., 2006). Given ankyrin’s privileged position connecting the RBC plasma membrane (via interactions with the band 3/Rh membrane proteins) to the spectrin cytoskeleton, it is plausible that spring-like behavior of ankyrin may play an important role in maintaining RBCs’ morphologies as they deform while traversing microvasculature and splenic tissues.
Indeed, the band 3-ankyrin complex has been proposed to locate in the middle of the spectrin tetramer chain to anchor spectrins to the plasma membrane, while the band 4.1 complex connects the end of the tetrameric spectrin chain to one of six binding sites on actin in the actin junctional complex (Lux, 2016; Mohandas and Gallagher, 2008) (Figure 6A). Our model of the band 3-ankyrin complex shows the association of band 3 with band 4.2, Rh proteins, GlutI, ankyrin, spectrin and glycophorin A. The association of band 3 with band 4.2 and GlutI based on crosslinking correspond well with the cluster derived from CF-MS (Figure 4). In addition, our model of the band 3-ankyrin complex is supported by previous co-immunoprecipitation data from RBCs of an individual with almost complete absence of band 3 (Coimbra mutation, homozygous null for band 3), crystallography, in vitro binding assays, and mass spectrometry (Bruce et al., 2003; Ipsaro and Mondragón, 2010; Ipsaro et al., 2009; Jiang et al., 2006; Kumpornsin et al., 2011; Lemmon et al., 1992). A previously observed reduction of Rh proteins and band 4.2 in the absence of band 3 suggests that they also form a complex in RBCs (Bruce et al., 2003). Interestingly, Bruce and colleagues found that GlutI was detected more in the absence of band 3. Our cross-linking evidence and 3D model indicates that GlutI competes with Rh proteins at the same binding sites/regions on band 3 and band 4.2, while band 3 and glycophorin A form a separate complex. These data thus support at least 3 subpopulations of band 3 complexes: those with Rh proteins, GlutI, or glycophorin A.
Figure 6. Reconstructions of band 3-Ank1-accessory protein complexes by integrative 3D modeling suggest Ank1 compression links the membrane to the cytoskeleton.
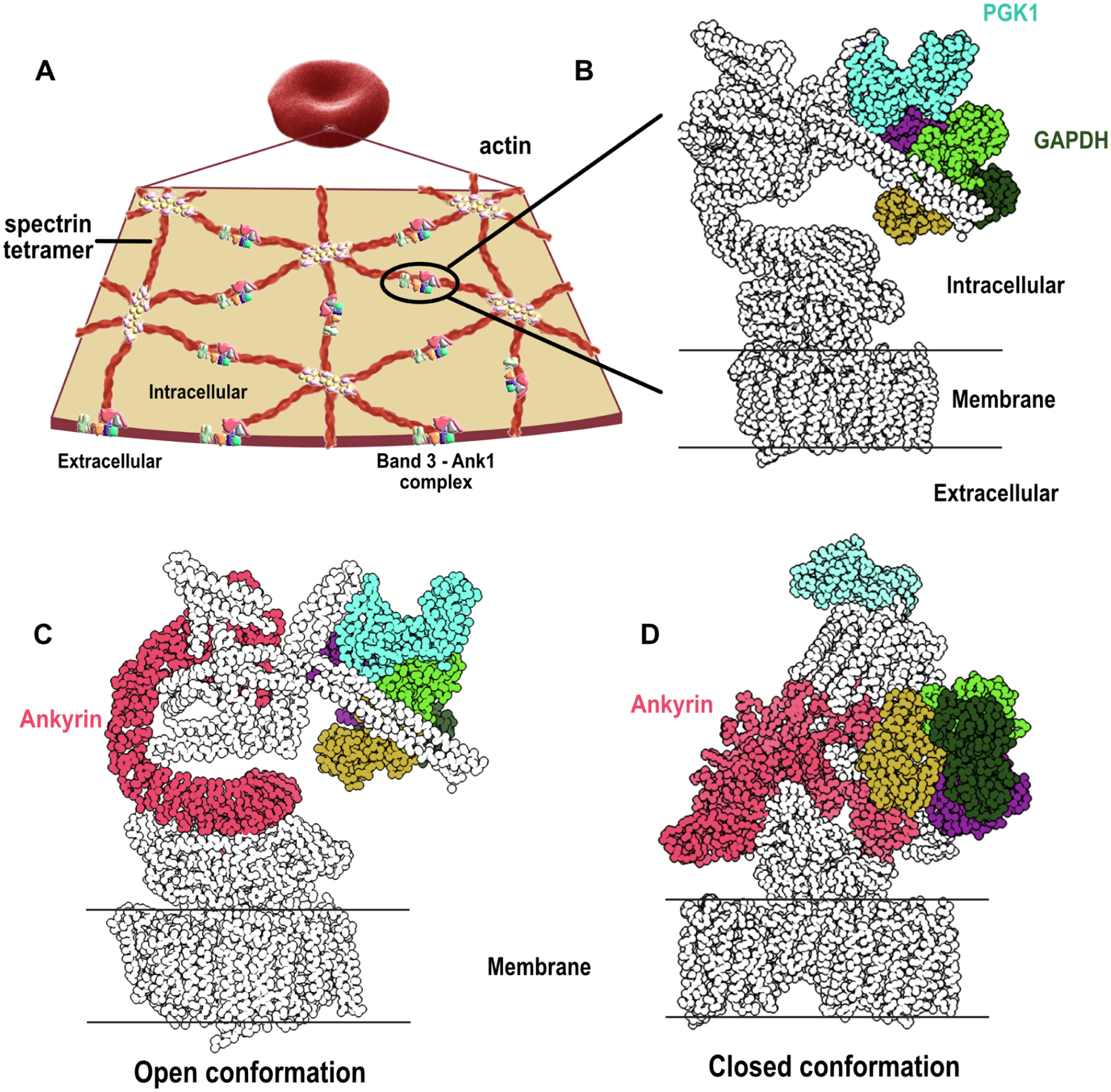
(A) An overview of the cytoskeletal network supporting the membrane of RBC. A pseudohexagonal network of spectrin heterotetramer underlies the membrane and is anchored to the membrane by the band 3-Ank1 complex. The tetramer is attached to actin on the other end through the association of band 4.1, actin, spectrin and other proteins. An actin polymer can interact with 6 spectrin tetramers through band 4.1 (Lux, 2016) (adapted with permission from (Goodman, 2020)).
(B) Glycolytic enzymes such as GAPDH and PGK1 are anchored to the band 3-Ank1 complex which can accommodate these enzymes while ANK1 adopts either open or closed conformations (see Zenodo repository (IMP_supplemt_figures) for more details).
(C) Ank1 in an open form in the band 3-Ank1 complex. Ank1, GAPDH, and PGK1 are colored.
(D) Roughly a third of observed intramolecular Ank1 crosslinks support it adopting a closed form in situ relative to the extended conformation observed for purified ankyrin (Wang et al., 2014), suggesting that Ank1 is capable of adopting either open or closed conformations. (see Zenodo repository (IMP_supplemt_figures) for more details).
In addition to the membrane and cytoskeletal proteins, we found crosslinking evidence for glycolytic enzymes associating with the band 3-Ank1 complex. Based on our modeling, both the potential open and closed forms of ankyrin in the band 3 complex (Figure 6B–D) can accommodate glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and phosphoglycerate kinase 1 (PGK1) binding without any violations of crosslinker distances. Thus, the positions of these enzymes with regards to the band 3-ankyrin complex do not seem to have an impact on the structural integrity of the complex as a whole. Previous experiments confirm the association of these two enzymes with the RBC membrane: GAPDH was found to be associated at the red cell membrane via immunofluorescence and binding assays (Rogalski et al., 1989), and PGK1 activity was detected in RBC white ghosts (Harris and Winzor, 1990; Ronquist and Ågren, 1966; Schrier, 1966). It is possible that localizing energy-producing enzymes to the RBC membrane where ion channels, transporters, and cytoskeletal network are located could be a way for RBCs to rapidly adjust the ratio of cell volume to surface area and change conformational states of cytoskeletal proteins such as ankyrin and spectrin in a timely manner. In turn, these rapid changes would facilitate dynamic morphological changes in response to the movement of RBCs through tissues and microvasculature.
The structure of the Band3-Ank1 complex has recently been investigated using cryo-EM and reported in two preprints (Vallese et al., 2022; Xia et al., 2022). Although the underlying structures have not yet been deposited, the structures are strikingly similar to our integrative model based on cross-linking and transmembrane domain restraints in that Band3 directly binds (i.e. shares a contact interface) with both Band4.2 and Ankyrin1. Consistent with Vallese et al., we find that Band4.2 specifically interacts with the N-terminus of Band3, with the N-terminus of Band4.2 oriented to the membrane. Consistent with Xia et al., we find ankyrin repeats 6–13 directly contact Band4.2. Most notably, multiple variations are evident for the composition of the complex: (1) Vallese et al. observed interactions of the Band3-Ank1 complex to Aquaporin 1, which we also detected by crosslinking, however the number of crosslinks were insufficient for modeling. (2) The cryo-EM structures from both groups of the Band3-Ank1 complexes lack spectrin, a known interactor (Ipsaro and Mondragón, 2010; Ipsaro et al., 2009) and a key component of our model supported by 7 unique intermolecular cross-links between spectrin alpha/beta and Band3-Ank1. We speculate that the absence of spectrin could account for these authors observing open ankyrin conformations rather than the closed conformation evident in our data (Figure S6). (3) GlutI is absent from these cryo-EM structures; in our model, the presence of GlutI is supported by both CF-MS and 2 independent, high confidence cross-links. (4) A variety of alternate subcomplexes are observed by all three groups, likely resulting from the different methods of fractionation employed (size exclusion then crosslinking vs. glycerol gradient) and salt conditions (similar salt concentrations between our work and Vallese et al. vs. low and high NaCl exclusively in Xia et al.). At a minimum, it is clear that the Band3-Ank1 complex possesses multiple, heterogeneous assembly states.
At the junctional complex, actin, non-muscle myosin, and other proteins work together to assert force on the spectrin network and thereby facilitate RBC deformability (Smith et al., 2018). At least under mild detergent conditions, the crosslinking and modeling of Band4.1 and spectrin suggest a stable interaction in the absence of actin. However, we note that the interactions may vary when actin is included as seen in the example of Band3-Ank1 complexes isolated using different salt concentrations or separation methods (Vallese et al., 2022; Xia et al., 2022). Other techniques such as super resolution microscopy might be able to shed more light on these complexes in their native conditions.
Taken together, the band 3-ankyrin complex maintains structural integrity by linking the spectrin network to the membrane, while also serving as the hub for metabolic activities. These metabolic activities include energy metabolism (glycolytic enzymes) and ions and gas exchange (ammonium through Rh proteins and CO2, HCO3−, Cl− through band 3, and carbonic anhydrase (Sterling et al., 2001)). Such elaborate organization of channels, integral proteins, and enzymes makes RBCs a unique and minimalist system which can undergo rapid morphological changes while preventing cell lysis from occurring.
Conclusions
We have used protein mass spectrometry, electron microscopy, chemical crosslinking, and 3D modeling to systematically determine the protein organization of the simplest primary cell in the human body, the erythrocyte. After defining a consensus set of canonical red blood cell proteins, we constructed a reference map of RBC protein interactions and multiprotein complexes, providing a detailed look at the molecular organization of RBC proteins. A 3D model of the band 3-Ank1 complex provides amino acid level detail of a critical protein complex that underlies one of RBCs most fascinating features: the ability of RBCs to undergo rapid dynamic morphological changes, a key aspect of controlling cellular form and activity given RBCs’ absence of gene expression regulation. The detailed molecular organization of proteins in the band 3-Ank1 complex clarifies the structure of a major membrane-cytoskeletal attachment site, supports the possibility of spring-like behavior at the membrane/cytoskeleton connection, and suggests aspects of RBC metabolism are spatially organized. In all, these data represent the first near-complete map of protein-protein interactions in any primary human cell type and thus provide an essential reference for these minimalistic cells, laying the groundwork for future full-cell molecular models.
Materials and Methods
Data and code availability statement
Raw and processed protein mass spectrometry data have been deposited in the MASSIVE/ProteomeXchange database in entry PXD030050, available at doi:10.25345/C5R00B. The 20S proteasome cryo-EM structure was deposited in the Electron Microscopy Data Bank as entry EMD-24822. Electron micrograph datasets have been deposited in EMPIAR as entry EMPIAR-10848. Supporting data files, including descriptions of biochemical fractionations, feature matrices, integrated 3D models, and training and test sets, have been deposited at Zenodo as accession doi:10.5281/zenodo.6465381.
Native protein extraction
All blood cell types and plasma were purchased from Gulf Coast Regional Blood Center (Houston, TX). The most recently drawn blood was requested for each order of cells/plasma, and the orders were shipped on ice overnight to ensure cells’ freshness. Further steps (described below) were performed to improve cell type homogeneity. Typically, 1–4 mg of extract was 0.45μm filtered (Ultrafree-MC HV Durapore PVDF, Millipore) and fractionated by HPLC chromatography. Detergent and salt were minimized where possible to avoid perturbing protein assemblies. The protein concentration was determined by Bio-Rad Protein Assay or Bio-Rad DC Protein Assay (for samples containing detergent). For additional details on types of fractionations and sample-related information, see the Zenodo data repository.
Red blood cells
According to the supplier, leukocytes were reduced via centrifugation of whole blood. However, we took additional steps to further eliminate contaminating plasma proteins and other cell types, namely leukocytes, reticulocytes, and platelets. Each blood unit was stored at 4 °C for 5 days after receipt in order to allow reticulocytes to mature into RBCs before extracting proteins (Urbina and Palomino, 2018). To further reduce leukocytes and platelets, RBCs were washed with PBS pH 7.4 (Gibco, Thermo Scientific, CA, USA) at 200 ×g for 15 minutes for 3 times. At the final wash, the 1 mm top layer of the cells (residual platelets, WBCs, etc) was removed. Cell samples and buffers were kept at 0–4 °C during all steps. Purified RBCs were lysed with 5 volumes of hypotonic lysis solution (5 mM Tris-HCl pH 7.4) containing EDTA-free protease inhibitor cocktail tablets (Sigma) and phosphatase inhibitor tablets (PhosSTOP EASY, Roche). White ghosts and hemolysate were separated from each other by centrifugation at 21 000 ×g for 40 min. The protein concentration of hemolysate was measured, and the samples were snap-frozen with liquid nitrogen until further use.
For ghosts preparation, excess hemoglobin (Hgb) was reduced by washing white ghosts ~10 times with a hypotonic solution until the white ghosts appeared pale pink to almost white. Ghosts were then dissolved with the appropriate detergent (see Zenodo data repository for details), and the protein concentration was measured with a detergent-compatible Bio-Rad DC Protein Assay. For ghosts dissolved in Diisobutylene/Maleic Acid (DIBMA, Cube Biotech) (Oluwole et al., 2017), DIBMA was added to white ghosts to the final concentration 2.5% (50 mM HEPES pH 7.5), and the sample was rotated at 4 °C overnight (Hagn et al., 2018). The ghosts dissolved in 2.5% DIBMA were then treated as for other detergent-dissolved white ghosts. For hemolysate preparations, remnant white ghosts were removed by centrifugation at 21,000 ×g for 40 min at 4 °C, and the supernatant treated with Hemoglobind (Biotech Support Group) in order to bind and remove free Hgb. Protein concentrations of the Hgb-reduced samples were measured by Bradford colorimetric assay (Bio-Rad) prior to biochemical fractionation.
iPSC-derived erythroblasts
Hematopoietic differentiation from iPSCs to hematopoietic stem and progenitor cells (HSPCs) was induced according to (Leung et al., 2018). To induce erythroid differentiation from HSPCs, day-15 HSPCs were specified using a 2-step suspension culture system consisting of Serum-Free Expansion Medium II, 2 mM of L-glutamine, and 100 mg/mL of primocin at 37°C in normoxic, 5% carbon dioxide conditions. Between days 15 and 20, this base was supplemented with 100 ng/mL of human stem cell factor, 40 ng/mL of IGF1, 5×10−7 M of dexamethasone, and 0.5 U/mL of hEPO and between days 20 and 25 with 4 U/mL of hEPO. Protein was extracted from day 25 differentiated cells, at which stage the differentiation cultures consisted of polychromatic and orthochromatic erythroblasts (Vanuytsel et al., 2018).
Platelets
Prostaglandin E1 (PGE1), the aggregation inhibitor, was added to platelet rich plasma (PRP) to the final concentration of 1 mM in order to stop the activation of platelets. To remove contaminating red and white blood cells, contaminating RBCs and WBCs were pelleted at 100 × g for 10 mins at 4 °C with no brake applied. The supernatant (PRP) was then washed two times with 1 volume platelet-wash buffer: 1 volume PRP by centrifugation at 400 × g for 10 min at 4 C with no brake. The wash buffer contains 1 μM PGE1, 10 mM sodium citrate, 150 mM NaCl, 1 mM EDTA, 1% (w/v) glucose pH 7.4. Native protein complexes from platelets were obtained by incubating the washed pallet with Pierce IP lysis buffer (25 mM Tris•HCl pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol) (Thermo Scientific) containing protease and phosphatase inhibitors at 4 °C for 5 minutes. The lysate was clarified at 14,000 × g for 10 mins.
Plasma
Plasma separated from whole blood by centrifugation was purchased and shipped in anticoagulant CPD. The received plasma still contained other blood cell types, so these cells were removed through centrifugation. Plasma was centrifuged at 5,000 × g for 15 mins at 4°C, and only the top part of supernatant was collected. The supernatant was treated with Albuviod (Biotech Support Group) to reduce the presence of serum albumin (HSA). The HSA reduced supernatant was then concentrated in a 3 kD MWCO Amicon Ultra filter unit (MilliporeSigma, Burlington, MA).
White blood cells
In order to broadly assess WBC proteins, we analyzed a buffy coat, consisting of all WBCs (lymphocytes and granulocytes), as well as some RBCs and platelets. WBCs were separated from the other cell types using Histopaque 1077 (Sigma) according to the manufacturer’s protocol. Briefly, the buffy coat was diluted with PBS Gibco at a 1:1 ratio. Buffy coat and Histopaque were held at room temperature to ensure proper cell separation. PGE1 was added to the diluted buffy coat to a final concentration of 1 μM to prevent platelet activation and cell aggregation. WBCs were first purified using Histopaque 1077 where the “ring” of lymphocytes, the layer between plasma and histopaque after centrifugation, and the white layer containing granulocytes on top of the RBC layer at the bottom were kept for further purification steps. Contaminating RBCs were lysed by addition of hypotonic solution, and the mixture of lymphocytes and granulocytes were pelleted at 1,000 g for 10 minutes. Native protein extract of WBCs was prepared by adding Pierce IP lysis buffer, and the extract was clarified by centrifuging at 14,000 × g for 10 mins, prior to biochemical fractionation.
HPLC chromatography
Hgb-reduced hemolysate and detergent-dissolved ghosts were fractionated on a Dionex UltiMate3000 HPLC system consisting of an RS pump, Diode Array Detector, PCM-3000 pH and Conductivity Monitor, Automated Fraction Collector (Thermo Scientific, CA, USA) and a Rheodyne MXII Valve (IDEX Health & Science LLC, Rohnert Park, CA) using biocompatible PEEK tubing and various columns. The columns we used were size exclusion chromatography, ion exchange separations (mixed bed or triple-phase WAXWAXCAT), or hydrophobic interaction chromatography. The sample loaded was 1–4 mg protein as measured by the BioRad Protein Assay (hemolysate) or DC Protein Assay (detergent-dissolved ghosts) as appropriate to the sample buffer. Fractions were collected into 96-deep well plates.
Size exclusion: BioSep-SEC-s4000 600 × 7.8 mm ID, particle diameter 5 μm, pore diameter 500 Å (Phenomenex, Torrance, CA) or higher Mw BioBasic AX HPLC Columns, 600 × 7.8 mm ID 5μm particle size, 300Å pore size were used. Unless otherwise specified the sample was 200 μl, low rate 0.5 ml/min, with fraction collection every 45 seconds, and mobile phase was PBS pH 7.4 (Gibco). For column calibration, molecular weight standards (Sigma -Aldrich, MWGF1000, 2–5 ug each of carbonic anhydrase (C7025), β-amylase (A8781), and bovine serum albumin (A8531)) were fractionated to ensure that the column can separate proteins based on their sizes correctly before the hemolysate/ghosts samples were fractionated.
Mixed bed ion exchange: Poly CATWAX A (PolyLC Mixed-Bed WAX-WCX) 200 × 4.6 mm ID, Particle diameter 5 μm, pore diameter 100 Å (PolyLC Inc., Columbia, MD). The bed contains the cation-exchange (PolyCAT A) and anion-exchange (PolyWAX LP) materials in equal amounts. A 200–250 μl sample was loaded at ≤ 40 mM NaCl, and eluted with a 1-hour salt gradient at 0.5 ml/min with collections of 0.5 ml fractions. Gradient elution was performed with Buffer A (10 mM Tris-HCl pH 7.5, 5% glycerol, 0.01% NaN3), and 0–70% Buffer B (1.5 M NaCl in Buffer A).
Triple phase ion exchange (WWC): Three columns, each 200 × 4.6 mm ID, particle diameter 5 μm, pore diameter 100 Å, were connected in series in the following order: two PolyWAX LP columns followed by a single PolyCAT A (PolyLC, Inc, Columbia, MD). Loading, buffers, and fraction collection were as for mixed bed ion exchange above with slight modifications in low rate and elution from the methods of (Havugimana et al., 2012). The low rate was either 0.25 ml/min with a 120 min gradient from 5–100 %B. For the separation of nuclear extracts, the gradient was modified to a 115-minute multiphasic elution from 5–100% Buffer B.
Hydrophobic interaction: The ProPac HIC-10 hydrophobic interaction chromatography column (Thermo Scientific) had the following characteristics: 4.6 × 250 mm, Amide/Ethyl (phase), 5 μm particle size, and 300 Å pore size. A 200–250 μl sample was loaded at ~250 mM (NH4)2 SO4, and eluted with a 100-min low salt gradient at 0.5 ml/min with collections of 0.5 ml fractions. Gradient elution was performed with Buffer A (2 M (NH4)2 SO4 in 0.1M NaH2PO4 pH 7.0), and 0–70% Buffer B (0.1 M NaH2PO4 pH 7.0).
Mass spectrometry sample preparation
Samples were prepared for mass spectrometry in 96-well plate format using ultrafiltration and in-solution digestion protocols (Wan et al., 2015). Plates were sealed with transparent film during incubation steps.
Ultrafiltration was performed with an AcroPrep Advance 96-filter plate, 3kD MWCO (Pall) using a vacuum manifold (QIAvac 96 or Multiwell, Qiagen) at −0.75 Bar. Before filtering samples, preservatives and remnant polymers that could interfere with MS experiment were removed from the plate by sequential filtration of 100 μl LC/MS quality water and 100μl trypsin digestion buffer (50 mM Tris-HCl, pH 8.0, 2 mM CaCl2). Samples were concentrated to 100 μl, diluted 2-fold with trypsin digestion buffer, and concentrated again to a final volume of 50–100 μl before being transferred back to a 96-deep well plate. 50 μl 2,2,2,-trifluoroethanol (TFE) was added and samples were reduced with TCEP (Bond-Breaker, Thermo) at a final concentration of 5 mM for 30 min. 37°C. Iodoacetamide was added to 15 mM and plates were incubated in the dark 30 min at room temperature. Alkylation was quenched by the addition of DTT to 7.5 mM. TFE was diluted to <5% by the addition of trypsin digestion buffer. 1 μg of trypsin was added to each fraction and the sealed plate was incubated 37°C overnight. Digestion was stopped by adding formic acid to the final concentration of 0.1%, and peptides were desalted using a 5–7 μl C18 Filter Plate (Glygen Corp) with a vacuum manifold, dried, and resuspended for mass spectrometry in 3% acetonitrile, 0.1% formic acid.
Chemical cross-linking
Extract (~2–3 mg hemolysate and detergent-dissolved white ghosts) was fractionated by size exclusion as written above. DSSO (Thermo Scientific) was dissolved immediately before use in dry dimethylformamide or DMSO (stored under nitrogen) at a concentration of 50 mM and then further diluted in PBS for a working stock. Immediately after fractionation, working stock DSSO was added to fractions to a final concentration of 1 mM, and samples were incubated 1 hour at room temperature. Cross-linking was quenched by the addition of 1 M Tris pH 8.0 to a concentration of 24 mM. Cross-linked fractions were immediately frozen and stored at −80°C. Fractions were prepared for mass spectrometry using the ultrafiltration and in-solution digestion methods. Peptides were desalted before being analyzed by mass spectrometry.
Mass spectrometry data acquisition and processing
Acquisition:
Mass spectra were acquired using one of two Thermo mass spectrometers: Orbitrap Fusion or Orbitrap Fusion Lumos. In all cases, peptides were separated using reverse phase chromatography on a Dionex Ultimate 3000 RSLCnano UHPLC system (Thermo Scientific) with a C18 trap to Acclaim C18 PepMap RSLC column (Dionex; Thermo Scientific) configuration. Peptides were eluted using a 3–45% gradient over 60 min. (for Fusion and Lumos) and directly injected into the mass spectrometer using nano-electrospray for data-dependent tandem mass spectrometry.
Mass spectrometry data was acquired for each instrument as follows:
Orbitrap Fusion:
top speed CID with full precursor ion scans (MS1) collected at 120,000 m/z resolution and a cycle time of 3 sec. Monoisotopic precursor selection and charge-state screening were enabled, with ions of charge > + 1 selected for collision-induced dissociation (CID). Dynamic exclusion was active with 60 s exclusion for ions selected once within a 60 s window. For some experiments, a similar top speed method was used with dynamic exclusion of 30 s for ions selected once within a 30 s window and high energy-induced dissociation (HCD) collision energy 31% stepped +/−4%. All MS2 scans were centroid and done in rapid mode.
Orbitrap Lumos:
top speed HCD with full precursor ion scans (MS1) collected at 120,000 m/z resolution. Monoisotopic precursor selection and charge-state screening were enabled using Advanced Peak Determination (APD), with ions of charge > + 1 selected for high energy-induced dissociation (HCD) with collision energy 30% stepped +/− 3%. Dynamic exclusion was active with 20 s exclusion for ions selected twice within a 20s window. All MS2 scans were centroid and done in rapid mode.
For identification of DSSO cross-linked peptides, peptides were resolved using a reverse phase nano low chromatography system with a 115 min 3–42% acetonitrile gradient in 0.1% formic acid. The top speed method collected full precursor ion scans (MS1) in the Orbitrap at 120,000 m/z resolution for peptides of charge 4–8 and with dynamic exclusion of 60 sec after selecting once, and a cycle time of 5 sec. CID dissociation (25% energy 10 msec) of the cross-linker was followed by MS2 scans collected in the orbitrap at 30,000 m/z resolution for charge states 2–6 using an isolation window of 1.6. Peptide pairs with a targeted mass difference of 31.9721 were selected for HCD (30% energy) and collection of rapid scan rate centroid MS3 spectra in the Ion Trap.
Computational analyses of peptide mass spectra
The human reference proteome was downloaded from Uniprot.org (UniProt Consortium, 2019) in August 2018 (20,858 entries). This reference proteome can be found in the MASSIVE/ProteomeXchange database in entry PXD030050, available at doi:10.25345/C5R00B. Mass spectral peptide matching was performed with MSGF+, X!Tandem, and Comet-2013020, each run with 10 ppm precursor tolerance, and allowing for fixed cysteine carbamidomethylation (+57.021464) and optional methionine oxidation (+15.9949). Peptide search results were integrated with MSBlender (Kwon et al., 2011), https://github.com/marcottelab/msblender, https://github.com/marcottelab/run_msblender). For DSSO cross-linked experiments, inter-protein cross-links were identified using the XlinkX (Klykov et al., 2018) node in ProteomeDiscover 2.2 (ThermoScientific). FDR for all analyses was kept at 1%.
RBC proteome
Identification of RBC proteome
In order to identify a consensus proteome of RBCs, we constructed a supervised classifier, trained on proteomics and RNA-seq data from different blood cell types (WBCs, reticulocytes, platelets, plasma proteins, and RBC precursor cells including different stages of erythroblasts). In addition to our own data, MS data from different blood cell types were retrieved from the PRIDE database and several of the datasets were reanalyzed using ProteomeDiscover 2.2 as noted in Table S6 in the Zenodo data repository. A classifier feature matrix was assembled wherein rows corresponded to proteins observed in each of 45 external MS experiments, 12 in-house CF-MS experiments (each summarizing overall protein abundances as the sum of Peptide Spectral Matches (PSMs) for a given protein across all fractions in a fractionation experiment), and 26 RNA-seq experiments (see RBC_proteome_featmat.csv in Zenodo repository), for a total of 83 features (columns).
As gold standard RBC proteins, we used the set of 859 proteins that all three previous MS studies on RBCs identified (Bryk et al., 2017, Goodman et al., 2007 and Lange et al., 2014) as a positive set (i.e., most likely to be true RBC proteins). As a negative gold standard (i.e., most likely not RBC proteins), we selected all proteins found in the set of all MS and RNA-seq experiments used to assemble the feature matrix curated in Uniprot (20,504 proteins) but not found in any of the three prior RBC studies, which identified a total of 3,520 proteins across the three studies. Therefore, the total number of negative gold standard proteins is 16,894. Proteins found in at least one but not all 3 prior RBC studies were considered as unknowns and to be classified.
We divided the gold standard proteins into train and test sets (80:20, train:test). We then used TPOT, an autoML wrapper of scikit-learn machine learning functions (Olson et al., 2016), to perform all training steps, including identifying the best classifier and hyperparameters based on 5-fold cross-validation on the training dataset. All training steps excluded the test set. The best classifier was the RandomForest classifier with TPOT discovered hyperparameters. Application of the classifier to the entire set of proteins resulted in an RBC likelihood score/confidence score for each protein where 1 indicates the highest likelihood that the protein derives from mature RBCs and 0 indicates a non-RBC protein. Precision and recall were calculated from training (687 positive, 13,665 negative) and test (172 positive, 3,229 negative) set proteins for Figure 1A and 1B, respectively. The false discovery rate was calculated from the test set.
In terms of feature importance (measured based on impurity using the scikit-learn function Feature Importance and provided in the Zenodo repository), we observed the strongest 3 features to be our CF-MS experiment on hemolysate and two published MS experiments on CD71− and CD71+ cells (RBCs and reticulocytes, respectively) from (Moura et al., 2018) reporting data from in vitro culture-derived reticulocytes (derived from CD34+ cells) and cultured reticulocytes circulated overnight in an ex vivo circulation system (Moura et al., 2018). Overall, RNA-based features provided some additional power, but made a relatively minor contribution.
Defining RBC complexes based on the CF-MS datasets
Assembly of features for scoring putative protein interactions
We performed 30 fractionation experiments comprising 1,944 total biochemical fractions across all fractionations. For each experiment, we assembled an elution matrix of all identified proteins (rows) by fractions (columns) containing the PSMs for each protein in each fraction, normalizing PSMs to 1 on a per protein basis for each experiment. In addition, we concatenated the elution matrices across all 1,944 fractions as one additional matrix. (Note that cross-linked data from hemolysate and ghosts were not used for training.)
Next, we calculated a series of all-by-all pairwise scores between proteins for individual matrices and the concatenated matrix. We focused our analysis on well-observed proteins, so we additionally filtered for proteins with at least 60 total PSMs observed across the 30 combined fractionations. The scores/features were as follows: (1) Pearson’s r, (2) Spearman’s rho, (3) Euclidean distance, (4) Bray-Curtis similarity, (5) stationary cross-correlation, (6) covariance, and (7) hypergeometric score for the co-occurrence of proteins in fractions with repeated sampling of fractions (Drew et al., 2017). All features/scores were calculated with added Poisson noise as in (Drew et al., 2017). Euclidean distance and Bray-Curtis similarity scores were inverted and normalized to a max score of 1. For each individual matrix from each fractionation, features 1–6 were calculated. Additionally, the average of each type of score/feature was calculated from all individual matrices excluding the concatenated matrix. For the concatenated matrix of all fractionations, features 1–7 were calculated. Features were joined to create a final feature matrix composed of 4,131,128 rows (protein pairs) and 193 features capturing the similarities between the proteins’ elution profiles. Missing values were filled with zeros.
Construction of the gold standard protein complex training and test sets
We used known human protein complexes from the CORUM database (Giurgiu et al., 2019) as a gold standard positive set of stable protein-protein interactions. As most of these CORUM complexes do not exist in RBCs, we further selected those CORUM complexes in which >50% of the protein subunits/members were RBC proteins at the 1% FDR level. The resulting 123 known CORUM complexes with >50% of the protein subunits belonging to the RBC proteome were divided into positive training and test complexes according to the scheme from Drew et al., 2017. In short, we defined each positive protein interaction as a pair of proteins that are part of the same CORUM complex. A negative protein interaction was defined as a pair of proteins that are both found in the set of CORUM complexes but not within the same CORUM complex. Complexes with over 30 members were removed so as not to skew performance measurements as per (Drew et al., 2017). Any overlapping interactions between training and test sets were removed such that the sets were fully disjoint. The final pairwise protein interaction training/test sets consisted of 461/299 and 14,389/11,408 positive and negative interactions, respectively. The final protein complex training/test sets consisted of 62/61 complexes. The complete lists of training/test interactions and complexes are available in the Zenodo repository.
Identification of interacting proteins by supervised machine learning
We again utilized TPOT to train our machine learning model to find pairwise interactions among members of protein complexes. We discovered optimal hyperparameters for an ExtraTree classifier with 5-fold cross-validation of the training interactions, with an area under the precision-recall curve (AUPR) of 0.45. We then trained an ExtraTree with TPOT discovered hyperparameters, and the resulting model was applied to the entire feature matrix to give a CF-MS score to each pair of proteins, with higher scores corresponding to higher confidence in the proteins interacting (specifically, being subunits in the same multi-protein assembly). Precision, recall, and false discovery rates were calculated from the 299 positive and 11,408 negative withheld test set interactions.
Clustering of interacting protein pairs to define multiprotein assemblies Interaction
Interaction scores above a 15% false discovery rate threshold (CF-MS score ≥ 0.17) were input into R igraph cluster_walktrap to define coherent protein complexes. The walktrap algorithm’s reweighted edges between proteins were reformatted to a dendrogram and cut at intervals to obtain a nested hierarchy of complexes as in (McWhite et al., 2020). Cuts closer to the root of the dendrogram result in larger complexes and cuts closer to the tips defined smaller subcomplexes. More details on how to read the hierarchy of complexes can be found in Table S4 - RBC_interactome in the Zenodo repository.
Negative stain electron microscopy
We reduced Hgb from hemolysate which was then passed through 100 kDa filter ultracentrifugation filter (Amicron). 4 uL of hgb-reduced and filtered hemolysate was applied to a glow-discharged 400-mesh continuous carbon grid. After allowing the sample to adsorb for 1 min, the sample was negatively stained with five consecutive droplets of 2% (w/v) uranyl acetate solution, blotted to remove residual stain, and air-dried in a fume hood. Grids were imaged using an FEI Talos TEM (Thermo Scientific) equipped with a Ceta 16M detector. Micrographs were collected manually using TIA v4.14 software at a nominal magnification of x73,000, corresponding to a pixel size of 2.05 Å/pixel. CTF estimation, particle picking, and 2D class averaging were performed using both RELION v3 (Zivanov et al., 2018) and cryoSPARC v2.12.4 (Punjani et al., 2017). Three negative stain datasets were collected. The first dataset collected contained ~220 micrographs of pooled HPLC size exclusion fractions 1–9. ~2,500 particles were manually picked and processed in cryoSPARC to produce the TPP2 structure in Figure 3B–C. Two datasets were collected of hemolysate after being passed through a 100 kDa filter, one as prepared and the other at 1:100 dilution. For the diluted sample, ~400 micrographs were collected and ~42,500 particles were picked using Topaz (Bepler et al., 2019). The resulting particles were used to generate the PRDX2 structure in Figure 3B–C and the 2D class averages in Figure S3B. For the non-dilute sample, ~230 micrographs were collected and ~1,500 proteasome particles were manually picked followed by classification in RELION Figure S3C.
Cryo-EM grid preparation and data collection
C-flat holey carbon grids (CF-1.2/1.3, Protochips Inc.) were glow-discharged for 1 min using a Solarus 950 plasma cleaner (Gatan). 2 μL of 0.2 mg/mL graphene oxide (Sigma-Aldrich) was placed onto the grids for 1 min followed by one wash with water. 3 μL of pooled and concentrated hemolysate from HPLC size exclusion fractions 10–20 was placed onto the grid, blotted for 3.5 sec with a blotting force of 0, and rapidly plunged into liquid ethane using an FEI Vitrobot MarkIV operated at 4 °C and 100% humidity. Data was acquired using an FEI Titan Krios TEM (Sauer Structural Biology Laboratory, University of Texas at Austin) operated at 300 keV with a nominal magnification of ×22,500 (1.045 Å/pixel) and defocus ranging from −1.09 to −2.5 μm. Dose-fractionated movies were collected using 20 frames (0.15 sec/frame) over a total of 3 sec with a dose rate of ~2.13 e−/Å2/sec and a total exposure of 42.58 e−/Å2. A total of 6,606 micrographs were automatically recorded on a K3 detector (Gatan) operated in counting mode using Leginon (Suloway et al., 2005). A full description of the cryo-EM data collection parameters can be found in Table S1.
Cryo-EM data processing
Motion correction, CTF-estimation and particle picking were performed in Warpv1.0.7 (Tegunov and Cramer, 2019). ~1,000,000 extracted particles were imported into cryoSPARC v2.12.4 for 2D classification, 3D classification and non-uniform 3D refinement (Figure S2). ~60,000 particles were used in the final 20S proteasome reconstruction. The nominal resolution of the map using the gold-standard Fourier Shell Correlation (FSC) at 0.143 is 3.35 Å (Figure S2). A previously solved X-ray crystal structure of the human 20S proteasome, PDB 6RGQ (Rêgo and Fonseca, 2019), was aligned by cross-correlation in UCSF Chimera (Pettersen et al., 2004) and used as an initial model for refinement. The model was then refined through two rounds of molecular dynamics flexible fitting and real space refinement using Namdinator (Kidmose et al., 2019), followed by further refinement in Phenix (Adams et al., 2002). The high correlation to a previous structure shows our RBC derived model is similar to the canonical 20S proteasome. However, due to the decreasing quality of the map away from the center, we were unable to build a full and accurate model that might distinguish subtle differences between the structures.
Integrative 3D modeling
Integrative modeling of the red blood cell membrane complexes consisted of four main stages: 1) gathering data, 2) domain representation and configuring of spatial restraints, 3) system sampling and scoring of restraints, and 4) model validation, as previously described in integrative modeling work (Gutierrez et al., 2020; Kim et al., 2018; Russel et al., 2012). The python interface of the Integrative Modeling Platform (IMP) was used to model the complexes (Saltzberg et al., 2019) and all associated data, scripts, and outputs can be found at Zenodo data deposit. The following sections describe the method for modeling the ankyrin complex in the open-spring conformation, the ankyrin complex in the closed-spring conformation, the ankyrin complex in the open-spring conformation with metabolic enzymes bound, the ankyrin complex in the closed-spring conformation with metabolic enzymes bound, and the band 4.1 with spectrin subcomplex.
Data used for modeling
Protein structure representations were constructed from known X-ray crystal structures, modeled using I-TASSER (Yang et al., 2015), or modeled domains from the Swiss-Prot database. Table S3 indicates the source of each of the protein’s structural models. A total of 156 intra- and intermolecular DSSO crosslinks were used to model the ankyrin complex. An additional 21 DSSO crosslinks were used to incorporate the GAPDH and PGK1 enzymes into the model. The subcomplex of spectrin and band 4.1 was modeled using 41 intra- and intermolecular DSSO crosslinks. Transmembrane regions of proteins were determined from Uniprot (The UniProt Consortium, 2019) annotations.
Domain representation and configuring spatial restraints
Protein subunits were represented as rigid bodies, chains of rigid bodies, or beads (Table S3). Band 3 was represented using rigid bodies for the transmembrane region and cytoplasmic domain of the protein. These domains were connected with a chain of flexible beads. The transmembrane region of the band 3 dimer (Arakawa et al., 2015) was represented as a single rigid body. Glut1 and band 4.2 were represented as a single rigid body, due to their high C-score (Table S3) from I-TASSER and agreement with intramolecular crosslinks. The transmembrane regions of the GYPA dimer have a known NMR structure and are represented as a single rigid body. The cytoplasmic and extracellular domains of the GYPA dimer were represented using a flexible chain of beads. RhAG and the two RhCE/D proteins were superimposed on the x-ray crystal structure (PDB 3HD6) and treated as a single rigid body (Gruswitz et al., 2010); these proteins had high C-scores and satisfied intermolecular crosslinks between each other. ANK1 was represented as a chain of rigid bodies (both modeled and X-ray crystal structure) with the end represented as flexible beads. Residues 265 to 790 of SPTA1 were used in the model and were represented as a chain of rigid bodies, with each rigid body beginning/ending at the Uniprot annotated spectrin domains. Residues 1,585 to 2,006 of SPTB were represented similarly to SPTA1 with each annotated spectrin domain being a rigid body connected in a chain. Both GAPDH and PGK1 had available X-ray crystal structures that satisfied all intramolecular crosslinks and were represented as rigid bodies. For the band 4.1-spectrin complex, band 4.1 was represented as a single rigid body, while residues 1,929 to 2,386 of SPTA1 was represented as a chain of rigid bodies by spectrin domain and residues 46 to 741 of SPTB was represented as a chain of rigid bodies by spectrin domain. The flexible chains of beads were coarse-grained to 10 residues per bead.
The DSSO crosslinkers were modeled using a length of 21 Å. The excluded volume restraint was applied to the 10 residue beads, preventing volumes from occupying the same space. The sequence connectivity restraint was applied between beads. Based on Uniprot annotations, segments of proteins were labeled as either inside the membrane (transmembrane), above the membrane (extracellular), or below the membrane (cytoplasmic) scored using a sigmoid potential. All of these restraints were incorporated into the scoring framework for the model.
System sampling and scoring of restraints
The 38 rigid bodies were first randomized in an initial configuration, followed by a steepest descent minimization based on connectivity to ensure that neighboring residues are close together and Monte Carlo sampling. Using integrative modeling (Figure S5) (Russel et al., 2012), we built many (600,000) models in parallel (unique starting positions) and tested for their agreement; the combination of annotated transmembrane regions, chemical crosslinks, and known partial structures provided sufficient spatial restraints to converge on a solution. (Note that detailed explanations of assessing sampling exhaustiveness can be found in previous literature (Viswanath et al., 2017)). These models were constructed using fixed protein compositions based on crosslinking evidence that implicated a limited number of proteins (Figure 5A). The ensembles were clustered based on their scoring parameters, including the clustering precision, which describes the variability between models in the cluster (Table S2). The best scoring cluster was selected to proceed (Table S2).
Model validation
The model cluster was first assessed against the input data. A crosslink is considered to be satisfied if any model in the cluster has the crosslink distance less than 40 Å. The crosslinks that are unsatisfied are believed to represent another conformational state of the complex. The convergence is determined through assessing the exhaustiveness of the sampling (Viswanath et al., 2017). This protocol tests convergence of the model score, whether the model scores were drawn from the same parent distribution, whether the structural clusters include models from each sample proportional to their size (chi-squared and sampling precision), and structural similarity between the model samples (Table S2 CCC between two sample densities).
Supplementary Material
ACKNOWLEDGEMENTS
The authors gratefully acknowledge Dr. Zhongwu Zhou for aid in cryo-EM sample preparation and data collection and Roden Luo for critical reading. Research was funded by grants from the National Institute of General Medical Sciences, Grant/Award Numbers: GM122480, R35GM138348; Cancer Prevention and Research Institute of Texas, Grant/Award Number: RR160088; National Science Foundation, Grant/Award Number: 2019238253; National Institutes of Health, Grant/Award Numbers: HD085901, DK110520; Army Research Office, Grant/Award Number: W911NF-19-1-0021; Welch Foundation, Grant/Award Numbers: W911NF-15-1-0120, F-1515, F-1938. Kevin Drew was supported by NIH R00HD092613 and L40HD096554.
Footnotes
DECLARATION OF INTERESTS
The authors declare no conflicts of interest.
References
- Adams PD, Grosse-Kunstleve RW, Hung L-W, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, and Terwilliger TC (2002). PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr 58, 1948–1954. 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- Alvarez-Castelao B, and Castaño JG (2005). Mechanism of direct degradation of IκBα by 20S proteasome. FEBS Lett. 579, 4797–4802. 10.1016/j.febslet.2005.07.060. [DOI] [PubMed] [Google Scholar]
- Arakawa T, Kobayashi-Yurugi T, Alguel Y, Iwanari H, Hatae H, Iwata M, Abe Y, Hino T, Ikeda-Suno C, Kuma H, et al. (2015). Crystal structure of the anion exchanger domain of human erythrocyte band 3. Science 350, 680–684. 10.1126/science.aaa4335. [DOI] [PubMed] [Google Scholar]
- Arimura Y, Shih RM, Froom R, and Funabiki H (2021). Structural features of nucleosomes in interphase and metaphase chromosomes. Mol. Cell 81, 4377–4397.e12. 10.1016/j.molcel.2021.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G, Bercovich Z, Tsvetkov P, Shaul Y, and Kahana C (2005). 20S proteasomal degradation of ornithine decarboxylase is regulated by NQO1. Mol. Cell 17, 645–655. 10.1016/j.molcel.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Bepler T, Morin A, Rapp M, Brasch J, Shapiro L, Noble AJ, and Berger B (2019). Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160. 10.1038/s41592-019-0575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KA (1996). Erythrocyte Metabolism and Enzyme Defects. Lab. Med 27, 329–333. 10.1093/labmed/27.5.329. [DOI] [Google Scholar]
- Bruce LJ, Beckmann R, Ribeiro ML, Peters LL, Chasis JA, Delaunay J, Mohandas N, Anstee DJ, and Tanner MJA (2003). A band 3–based macrocomplex of integral and peripheral proteins in the RBC membrane. Blood 101, 4180–4188. 10.1182/blood-2002-09-2824. [DOI] [PubMed] [Google Scholar]
- Bryk AH, and Wiśniewski JR (2017). Quantitative Analysis of Human Red Blood Cell Proteome. J. Proteome Res 16, 2752–2761. 10.1021/acs.jproteome.7b00025. [DOI] [PubMed] [Google Scholar]
- Caufield JH, Abreu M, Wimble C, and Uetz P (2015). Protein Complexes in Bacteria. PLOS Comput. Biol 11, e1004107. 10.1371/journal.pcbi.1004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang CK, Rockel B, Seyit G, Walian PJ, Schönegge A-M, Peters J, Zwart PH, Baumeister W, and Jap BK (2010). Hybrid molecular structure of the giant protease tripeptidyl peptidase II. Nat. Struct. Mol. Biol 17, 990–996. 10.1038/nsmb.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarest TG, Truong GTD, Lovett J, Mohanty JG, Mattison JA, Mattson MP, Ferrucci L, Bohr VA, and Moaddel R (2019). Assessment of NAD+metabolism in human cell cultures, erythrocytes, cerebrospinal fluid and primate skeletal muscle. Anal. Biochem 572, 1–8. 10.1016/j.ab.2019.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew K, Lee C, Huizar RL, Tu F, Borgeson B, McWhite CD, Ma Y, Wallingford JB, and Marcotte EM (2017). Integration of over 9,000 mass spectrometry experiments builds a global map of human protein complexes. Mol. Syst. Biol 13, 932. 10.15252/msb.20167490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew K, Wallingford JB, and Marcotte EM (2021). hu.MAP 2.0: integration of over 15,000 proteomic experiments builds a global compendium of human multiprotein assemblies. Mol. Syst. Biol 17, e10016. 10.15252/msb.202010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier E-F, Ducamp S, Leduc M, Salnot V, Guillonneau F, Dussiot M, Hale J, Giarratana M-C, Raimbault A, Douay L, et al. (2016). Comprehensive Proteomic Analysis of Human Erythropoiesis. Cell Rep. 16, 1470–1484. 10.1016/j.celrep.2016.06.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier E-F, Leduc M, Cochet S, Bailly K, Lacombe C, Mohandas N, Guillonneau F, El Nemer W, and Mayeux P (2018). Absolute proteome quantification of highly purified populations of circulating reticulocytes and mature erythrocytes. Blood Adv. 2, 2646–2657. 10.1182/bloodadvances.2018023515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giurgiu M, Reinhard J, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, and Ruepp A (2019). CORUM: the comprehensive resource of mammalian protein complexes—2019. Nucleic Acids Res. 47, D559–D563. 10.1093/nar/gky973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman SR (2020). Goodman’s Medical Cell Biology (San Diego: Academic Press; ). [Google Scholar]
- Goodman SR, Kurdia A, Ammann L, Kakhniashvili D, and Daescu O (2007). The Human Red Blood Cell Proteome and Interactome. Exp. Biol. Med 232, 1391–1408. 10.3181/0706-MR-156. [DOI] [PubMed] [Google Scholar]
- Gruswitz F, Chaudhary S, Ho JD, Schlessinger A, Pezeshki B, Ho C-M, Sali A, Westhoff CM, and Stroud RM (2010). Function of human Rh based on structure of RhCG at 2.1 Å. Proc. Natl. Acad. Sci 107, 9638–9643. 10.1073/pnas.1003587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez C, Chemmama IE, Mao H, Yu C, Echeverria I, Block SA, Rychnovsky SD, Zheng N, Sali A, and Huang L (2020). Structural dynamics of the human COP9 signalosome revealed by cross-linking mass spectrometry and integrative modeling. Proc. Natl. Acad. Sci 117, 4088–4098. 10.1073/pnas.1915542117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagn F, Nasr ML, and Wagner G (2018). Assembly of phospholipid nanodiscs of controlled size for structural studies of membrane proteins by NMR. Nat. Protoc 13, 79–98. 10.1038/nprot.2017.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SJ, and Winzor DJ (1990). Interactions of glycolytic enzymes with erythrocyte membranes. Biochim. Biophys. Acta 1038, 306–314. 10.1016/0167-4838(90)90242-8. [DOI] [PubMed] [Google Scholar]
- Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, et al. (2012). A Census of Human Soluble Protein Complexes. Cell 150, 1068–1081. 10.1016/j.cell.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Janga SC, Babu M, Díaz-Mejía JJ, Butland G, Yang W, Pogoutse O, Guo X, Phanse S, Wong P, et al. (2009). Global Functional Atlas of Escherichia coli Encompassing Previously Uncharacterized Proteins. PLOS Biol. 7, e1000096. 10.1371/journal.pbio.1000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Ipsaro JJ, and Mondragón A (2010). Structural basis for spectrin recognition by ankyrin. Blood 115, 4093–4101. 10.1182/blood-2009-11-255604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipsaro JJ, Huang L, and Mondragón A (2009). Structures of the spectrin-ankyrin interaction binding domains. Blood 113, 5385–5393. 10.1182/blood-2008-10-184358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Ding Y, Su Y, Jiang M, Hu X, and Zhang Z (2006). Interaction of glucose transporter 1 with anion exchanger 1 in vitro. Biochem. Biophys. Res. Commun 339, 1255–1261. 10.1016/j.bbrc.2005.11.138. [DOI] [PubMed] [Google Scholar]
- Kabanova S, Kleinbongard P, Volkmer J, Andrée B, Kelm M, and Jax TW (2009). Gene expression analysis of human red blood cells. Int. J. Med. Sci 156–159. 10.7150/ijms.6.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao A, Chiu C, Vellucci D, Yang Y, Patel VR, Guan S, Randall A, Baldi P, Rychnovsky SD, and Huang L (2011). Development of a Novel Cross-linking Strategy for Fast and Accurate Identification of Cross-linked Peptides of Protein Complexes*. Mol. Cell. Proteomics 10, M110.002170. 10.1074/mcp.M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastritis PL, O’Reilly FJ, Bock T, Li Y, Rogon MZ, Buczak K, Romanov N, Betts MJ, Bui KB, Hagen WJ, et al. (2017). Capturing protein communities by structural proteomics in a thermophilic eukaryote. Mol. Syst. Biol 13, 936. 10.15252/msb.20167412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidmose RT, Juhl J, Nissen P, Boesen T, Karlsen JL, and Pedersen BP (2019). Namdinator – automatic molecular dynamics flexible fitting of structural models into cryo-EM and crystallography experimental maps. IUCrJ 6, 526–531. 10.1107/S2052252519007619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G, Jang S, Lee E, and Song, and J.-J. (2020). EMPAS: Electron Microscopy Screening for Endogenous Protein Architectures. Mol. Cells 43, 804–812. 10.14348/molcells.2020.0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Fernandez-Martinez J, Nudelman I, Shi Y, Zhang W, Raveh B, Herricks T, Slaughter BD, Hogan JA, Upla P, et al. (2018). Integrative structure and functional anatomy of a nuclear pore complex. Nature 555, 475–482. 10.1038/nature26003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirykowicz AM, and Woodward JD (2020). Shotgun EM of mycobacterial protein complexes during stationary phase stress. Curr. Res. Struct. Biol 2, 204–212. 10.1016/j.crstbi.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klykov O, Steigenberger B, Pektaş S, Fasci D, Heck AJR, and Scheltema RA (2018). Efficient and robust proteome-wide approaches for cross-linking mass spectrometry. Nat. Protoc 13, 2964–2990. 10.1038/s41596-018-0074-x. [DOI] [PubMed] [Google Scholar]
- Kumpornsin K, Jiemsup S, Yongkiettrakul S, and Chookajorn T (2011). Characterization of band 3–ankyrin–Protein 4.2 complex by biochemical and mass spectrometry approaches. Biochem. Biophys. Res. Commun 406, 332–335. 10.1016/j.bbrc.2011.02.026. [DOI] [PubMed] [Google Scholar]
- Kwon T, Choi H, Vogel C, Nesvizhskii AI, and Marcotte EM (2011). MSblender: A Probabilistic Approach for Integrating Peptide Identifications from Multiple Database Search Engines. J. Proteome Res 10, 2949–2958. 10.1021/pr2002116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrilis FL, Meister A, and Kastritis PL (2019). Integrative biology of native cell extracts: a new era for structural characterization of life processes. Biol. Chem 400, 831–846. 10.1515/hsz-2018-0445. [DOI] [PubMed] [Google Scholar]
- Kyrilis FL, Semchonok DA, Skalidis I, Tüting C, Hamdi F, O’Reilly FJ, Rappsilber J, and Kastritis PL (2021). Integrative structure of a 10-megadalton eukaryotic pyruvate dehydrogenase complex from native cell extracts. Cell Rep. 34, 108727. 10.1016/j.celrep.2021.108727. [DOI] [PubMed] [Google Scholar]
- Lange PF, Huesgen PF, Nguyen K, and Overall CM (2014). Annotating N Termini for the Human Proteome Project: N Termini and Nα-Acetylation Status Differentiate Stable Cleaved Protein Species from Degradation Remnants in the Human Erythrocyte Proteome. J. Proteome Res 13, 2028–2044. 10.1021/pr401191w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Abdi K, Jiang Y, Michaely P, Bennett V, and Marszalek PE (2006). Nanospring behaviour of ankyrin repeats. Nature 440, 246–249. 10.1038/nature04437. [DOI] [PubMed] [Google Scholar]
- Leitner A, Faini M, Stengel F, and Aebersold R (2016). Crosslinking and Mass Spectrometry: An Integrated Technology to Understand the Structure and Function of Molecular Machines. Trends Biochem. Sci 41, 20–32. 10.1016/j.tibs.2015.10.008. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Flanagan JM, Hunt JF, Adair BD, Bormann BJ, Dempsey CE, and Engelman DM (1992). Glycophorin A dimerization is driven by specific interactions between transmembrane alpha-helices. J. Biol. Chem 267, 7683–7689. 10.1016/S0021-9258(18)42569-0. [DOI] [PubMed] [Google Scholar]
- Leung A, Zulick E, Skvir N, Vanuytsel K, Morrison TA, Naing ZH, Wang Z, Dai Y, Chui DHK, Steinberg MH, et al. (2018). Notch and Aryl Hydrocarbon Receptor Signaling Impact Definitive Hematopoiesis from Human Pluripotent Stem Cells. STEM CELLS 36, 1004–1019. 10.1002/stem.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Lössl P, Rabbitts BM, Balaban RS, and Heck AJR (2018). The interactome of intact mitochondria by cross-linking mass spectrometry provides evidence for coexisting respiratory supercomplexes *. Mol. Cell. Proteomics 17, 216–232. 10.1074/mcp.RA117.000470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux SE IV (2016). Anatomy of the red cell membrane skeleton: unanswered questions. Blood 127, 187–199. 10.1182/blood-2014-12-512772. [DOI] [PubMed] [Google Scholar]
- Macpherson E, Tomkinson B, Bålöw RM, Höglund S, and Zetterqvist O (1987). Supramolecular structure of tripeptidyl peptidase II from human erythrocytes as studied by electron microscopy, and its correlation to enzyme activity. Biochem. J 248, 259–263. 10.1042/bj2480259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCafferty CL, Verbeke EJ, Marcotte EM, and Taylor DW (2020). Structural Biology in the Multi-Omics Era. J. Chem. Inf. Model 60, 2424–2429. 10.1021/acs.jcim.9b01164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhite CD, Papoulas O, Drew K, Cox RM, June V, Dong OX, Kwon T, Wan C, Salmi ML, Roux SJ, et al. (2020). A Pan-plant Protein Complex Map Reveals Deep Conservation and Novel Assemblies. Cell 181, 460–474.e14. 10.1016/j.cell.2020.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills-Davies N, Butler D, Norton E, Thompson D, Sarwar M, Guo J, Gill R, Azim N, Coker A, Wood SP, et al. (2017). Structural studies of substrate and product complexes of 5-aminolaevulinic acid dehydratase from humans, Escherichia coli and the hyperthermophile Pyrobaculum calidifontis. Acta Crystallogr. Sect. Struct. Biol 73, 9–21. 10.1107/S2059798316019525. [DOI] [PubMed] [Google Scholar]
- Mohandas N, and Gallagher PG (2008). Red cell membrane: past, present, and future. Blood 112, 3939–3948. 10.1182/blood-2008-07-161166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura PL, Hawley BR, Mankelow TJ, Griffiths RE, Dobbe JGG, Streekstra GJ, Anstee DJ, Satchwell TJ, and Toye AM (2018). Non-muscle myosin II drives vesicle loss during human reticulocyte maturation. Haematologica 103, 1997–2007. 10.3324/haematol.2018.199083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson RS, Urbanowicz RJ, Andrews PC, Lavender NA, Kidd LC, and Moore JH (2016). Automating Biomedical Data Science Through Tree-Based Pipeline Optimization. In Applications of Evolutionary Computation, Squillero G, and Burelli P, eds. (Cham: Springer International Publishing; ), pp. 123–137. [Google Scholar]
- Oluwole AO, Danielczak B, Meister A, Babalola JO, Vargas C, and Keller S (2017). Solubilization of Membrane Proteins into Functional Lipid-Bilayer Nanodiscs Using a Diisobutylene/Maleic Acid Copolymer. Angew. Chem. Int. Ed 56, 1919–1924. 10.1002/anie.201610778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini EM, Kirkegaard M, Mortensen P, Lutz HU, Thomas AW, and Mann M (2006). In-depth analysis of the membrane and cytosolic proteome of red blood cells. Blood 108, 791–801. 10.1182/blood-2005-11-007799. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- Rêgo AT, and Fonseca PCA da (2019). Characterization of Fully Recombinant Human 20S and 20S-PA200 Proteasome Complexes. Mol. Cell 76, 138–147.e5. 10.1016/j.molcel.2019.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalski AA, Steck TL, and Waseem A (1989). Association of Glyceraldehyde-3-phosphate Dehydrogenase with the Plasma Membrane of the Intact Human Red Blood Cell. J. Biol. Chem 264, 6438–6446. 10.1016/S0021-9258(18)83368-3. [DOI] [PubMed] [Google Scholar]
- Ronquist G, and Ågren G (1966). Formation of Adenosine Triphosphate by Human Erythrocyte Ghosts. Nature 209, 1090–1091. 10.1038/2091090a0. [DOI] [PubMed] [Google Scholar]
- Russel D, Lasker K, Webb B, Velázquez-Muriel J, Tjioe E, Schneidman-Duhovny D, Peterson B, and Sali A (2012). Putting the Pieces Together: Integrative Modeling Platform Software for Structure Determination of Macromolecular Assemblies. PLOS Biol. 10, e1001244. 10.1371/journal.pbio.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzberg D, Greenberg CH, Viswanath S, Chemmama I, Webb B, Pellarin R, Echeverria I, and Sali A (2019). Modeling Biological Complexes Using Integrative Modeling Platform. In Biomolecular Simulations: Methods and Protocols, Bonomi M, and Camilloni C, eds. (New York, NY: Springer; ), pp. 353–377. [DOI] [PubMed] [Google Scholar]
- Schönegge A-M, Villa E, Förster F, Hegerl R, Peters J, Baumeister W, and Rockel B (2012). The Structure of Human Tripeptidyl Peptidase II as Determined by a Hybrid Approach. Structure 20, 593–603. 10.1016/j.str.2012.01.025. [DOI] [PubMed] [Google Scholar]
- Schrier S (1966). Organization of enzymes in human erythrocyte membranes. Am. J. Physiol.-Leg. Content 210, 139–145. 10.1152/ajplegacy.1966.210.1.139. [DOI] [PubMed] [Google Scholar]
- Schröder E, Littlechil JA, Lebedev AA, Errington N, Vagin AA, and Isupov MN (2000). Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7Å resolution. Structure 8, 605–615. 10.1016/S0969-2126(00)00147-7. [DOI] [PubMed] [Google Scholar]
- Skinnider MA, and Foster LJ (2021). Meta-analysis defines principles for the design and analysis of co-fractionation mass spectrometry experiments. Nat. Methods 18, 806–815. 10.1038/s41592-021-01194-4. [DOI] [PubMed] [Google Scholar]
- Smith AS, Nowak RB, Zhou S, Giannetto M, Gokhin DS, Papoin J, Ghiran IC, Blanc L, Wan J, and Fowler VM (2018). Myosin IIA interacts with the spectrin-actin membrane skeleton to control red blood cell membrane curvature and deformability. Proc. Natl. Acad. Sci 115, E4377–E4385. 10.1073/pnas.1718285115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin AV, Selyutina AA, Skabkin MA, Guryanov SG, Nazimov IV, Richard C, Th’ng J, Yau J, Sorensen PH, Ovchinnikov LP, et al. (2005). Proteasome-mediated cleavage of the Y-box-binding protein 1 is linked to DNA-damage stress response. EMBO J. 24, 3602–3612. 10.1038/sj.emboj.7600830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speicher DW, Weglarz L, and DeSilva TM (1992). Properties of human red cell spectrin heterodimer (side-to-side) assembly and identification of an essential nucleation site. J. Biol. Chem 267, 14775–14782. 10.1016/S0021-9258(18)42107-2. [DOI] [PubMed] [Google Scholar]
- Sterling D, Reithmeier RAF, and Casey JR (2001). A Transport Metabolon: FUNCTIONAL INTERACTION OF CARBONIC ANHYDRASE II AND CHLORIDE/BICARBONATE EXCHANGERS*. J. Biol. Chem 276, 47886–47894. 10.1074/jbc.M105959200. [DOI] [PubMed] [Google Scholar]
- Su C-C, Lyu M, Morgan CE, Bolla JR, Robinson CV, and Yu EW (2021). A ‘Build and Retrieve’ methodology to simultaneously solve cryo-EM structures of membrane proteins. Nat. Methods 18, 69–75. 10.1038/s41592-020-01021-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B (2005). Automated molecular microscopy: The new Leginon system. J. Struct. Biol 151, 41–60. 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Tegunov D, and Cramer P (2019). Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 16, 1146–1152. 10.1038/s41592-019-0580-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515. 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov P, Myers N, Eliav R, Adamovich Y, Hagai T, Adler J, Navon A, and Shaul Y (2014). NADH Binds and Stabilizes the 26S Proteasomes Independent of ATP*. J. Biol. Chem 289, 11272–11281. 10.1074/jbc.M113.537175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungewickell E, and Gratzer W (1978). Self-Association of Human Spectrin. Eur. J. Biochem 88, 379–385. 10.1111/j.1432-1033.1978.tb12459.x. [DOI] [PubMed] [Google Scholar]
- Urbina A, and Palomino F (2018). In vitro kinetics of reticulocyte subtypes: maturation after red blood cell storage in additive solution-1 (AS-1). Hematol. Transfus. Cell Ther 40, 143–150. 10.1016/j.htct.2017.12.002. [DOI] [Google Scholar]
- Vallese F, Kim K, Yen LY, Johnston JD, Noble AJ, Calì T, and Clarke OB (2022). Architecture of the human erythrocyte ankyrin-1 complex. 2022.02.10.479914. 10.1101/2022.02.10.479914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanuytsel K, Matte T, Leung A, Naing ZH, Morrison T, Chui DHK, Steinberg MH, and Murphy GJ (2018). Induced pluripotent stem cell–based mapping of β-globin expression throughout human erythropoietic development. Blood Adv. 2, 1998–2011. 10.1182/bloodadvances.2018020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeke EJ, Mallam AL, Drew K, Marcotte EM, and Taylor DW (2018). Classification of Single Particles from Human Cell Extract Reveals Distinct Structures. Cell Rep. 24, 259–268.e3. 10.1016/j.celrep.2018.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanath S, Chemmama IE, Cimermancic P, and Sali A (2017). Assessing Exhaustiveness of Stochastic Sampling for Integrative Modeling of Macromolecular Structures. Biophys. J 113, 2344–2353. 10.1016/j.bpj.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, Xiong X, Kagan O, Kwan J, Bezginov A, et al. (2015). Panorama of ancient metazoan macromolecular complexes. Nature 525, 339–344. 10.1038/nature14877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wei Z, Chen K, Ye F, Yu C, Bennett V, and Zhang M (2014). Structural basis of diverse membrane target recognitions by ankyrins. ELife 3, e04353. 10.7554/eLife.04353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Cimermancic P, Yu C, Schweitzer A, Chopra N, Engel JL, Greenberg C, Huszagh AS, Beck F, Sakata E, et al. (2017). Molecular Details Underlying Dynamic Structures and Regulation of the Human 26S Proteasome *. Mol. Cell. Proteomics 16, 840–854. 10.1074/mcp.M116.065326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Liu S, and Zhou ZH (2022). Structure, dynamics and assembly of the ankyrin complex on human red blood cell membrane. 2022.02.10.480008. 10.1101/2022.02.10.480008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, and Zhang Y (2015). The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8. 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi X, Verbeke EJ, Chang Y, Dickinson DJ, and Taylor DW (2019). Electron microscopy snapshots of single particles from single cells. J. Biol. Chem 294, 1602–1608. 10.1074/jbc.RA118.006686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. ELife 7, e42166. 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed protein mass spectrometry data have been deposited in the MASSIVE/ProteomeXchange database in entry PXD030050, available at doi:10.25345/C5R00B. The 20S proteasome cryo-EM structure was deposited in the Electron Microscopy Data Bank as entry EMD-24822. Electron micrograph datasets have been deposited in EMPIAR as entry EMPIAR-10848. Supporting data files, including descriptions of biochemical fractionations, feature matrices, integrated 3D models, and training and test sets, have been deposited at Zenodo as accession doi:10.5281/zenodo.6465381.