

ABSTRACT
In proliferating cells and tissues a number of checkpoints (G1/S and G2/M) preceding cell division (M-phase) require the signal provided by growth factors present in serum. IGFs (I and II) have been demonstrated to constitute key intrinsic components of the peptidic active fraction of mammalian serum. In vivo genetic ablation studies have shown that the cellular signal triggered by the IGFs through their cellular receptors represents a non-replaceable requirement for cell growth and cell cycle progression. Retroactive and current evaluation of published literature sheds light on the intracellular circuitry activated by these factors providing us with a better picture of the pleiotropic mechanistic actions by which IGFs regulate both cell size and mitogenesis under developmental growth as well as in malignant proliferation. The present work aims to summarize the cumulative knowledge learned from the IGF ligands/receptors and their intracellular signaling transducers towards control of cell size and cell-cycle with particular focus to their actionable circuits in human cancer. Furthermore, we bring novel perspectives on key functional discriminants of the IGF growth-mitogenic pathway allowing re-evaluation on some of its signal components based upon established evidences.
KEYWORDS: GF, growth factor, trophic (effect), property of a GF to increase growth (hypertrophy) and number (hyperplasia) of a target cell or tissue, GH, growth hormone, RTK, receptor tyrosine kinase, IGF-I/II: insulin like growth factor peptides, igf1/2, insulin like growth factor genes, igf1r, IGF-Type I receptor gene, IGF1R, Insulin-like growth factor receptor, type I (protein), insr, insulin receptor gene, IR (also InsR), insulin receptor protein, IR-A, IR isoform A (exon 11-), HybR, IGF1R/IR hybrid receptor, HybR-A, IGF1R/IR-A, igf2r/m6pr/SpI2-6, (scavenger protein for IGF2 & mannose-6-phosphate), trans-membrane high affinity IGF2 scavenger protein, IRS, insulin receptor substrate, mTOR, mammalian target of rapamycin, mTORC, mTOR complex. additional acronyms are clarified in the text
Introduction
Three and an half decades ago, while the receptors for insulin and insulin-like growth factors were being discovered and found to be part of a then growing family of trans-membrane receptor tyrosine kinases [1-6], it became clear that most viral oncogenic proteins were indeed constitutive activated counterparts of a pre-existing cellular gene product (like AKT, RAF and RAS, among others) which also encoded for a kinase or another type of signaling factor triggering a growth-promoting enzymatic activity ultimately hijacking the proliferative program of the cell [7]. Since then, it became clear that the known biological actions of growth factors were intrinsically linked to their activated kinase receptors and the intracellular signals triggered by them. It took more than three decades to start making sense of the actors, the hierarchies and the mechanistic features of activation of the enzymatic signaling network targeted and recruited by specific growth factors. It became apparent that such cascade(s) of post-translational modifications (PTM) driven-by protein phosphorylation were translated into the mechanical events governing cell growth and mitosis. When the parallel discoveries surrounding the cell division components in S. Cerevisiae, Sea Urchin and other cellular models started shedding light on the phylogenetically conserved features shared by the mammalian cell cycle machinery it was clear that the language used by the cell to regulate cell division was based upon sequential targeted phosphorylation events. It was also evident that each cell-cycle phase was driven by a defined set of cyclins with their respective kinases counteracted by specific phosphatases regulating individual checkpoints and transitions [8]. Indeed a further key step towards understanding how growth factors regulate cell growth and proliferation took place when the role of another post-translational modification, namely selective protein degradation by ubiquitin tagging, was shown to be an intrinsic additional mechanism in proliferating cells towards affecting the timing for each cell cycle phase. Interestingly, it soon become clear that phosphorylation and ubiquitination are coordinated events common to many cellular proteins affecting both their function as well as their lifespan. While our cumulative knowledge about the structure/function of each gene product involved in cell growth and proliferation is still growing, so is also our understanding of basic growth/ division pathways and their redundant/complementary/compensatory circuits under non-physiological circumstances such as in the cancer cell. In this context, the biological role of the Insulin-like growth factors, as key intrinsic components of the cellular program driving growth and division, has been further consolidated by a number of discoveries which have drastically modified the historical view surrounding this ligand/receptor system. Such scientific work has brought to the growing realization of the relevance of specific isoforms and paralog variants of known signaling molecules towards supporting physiological versus cancer-promoting effects [9,10]. The present review provides an indispensable summary our current knowledge of the Insulin/IGF ligand/receptor signal transduction circuitry specifically involved in the regulation of growth and cell cycle. Special emphasis has been given on the differential mechanistic actions mediated by the individual signal transduction components with emphasis to their effect in fetal and cancer cells compared to differentiated cells and tissues.
Revisiting the role of endocrine, paracrine and autocrine actions of the IGF ligands in growth and cell cycle regulation
The establishment of the trophic effect of insulin growth factors as major cell-, organs- and body- size regulators originates both from established experimental and clinical observations. While fundamental animal studies using gene ablation for each of the IGF ligands and receptors have proven the central role for the Insulin/IGF family in body growth regulation (summarized in Table 1), the evidence of such role in humans comes from a plethora of longstanding observations in the field of endocrinology which identified clinical conditions per each of the occurrences in which at least one of these factors was either pathologically over-produced or deficient. Specifically, IGF-I overproduction during the pre-adult developmental stage is responsible for the condition known as Acromegaly (“gigantism”). IGF-II over-production has been observed in the rare Beckwith-Wiedemann syndrome where it has been demonstrated to functionally associate with a macrosomic newborn phenotype (abnormal enlargement of a number of internal organs) [11,12]. Insulin can also act as a growth factor, although in a self-limiting fashion. Its growth-promoting effect can become apparent in chronic hyperinsulinemic states, such as seen in obese subjects or in metabolically compensated Diabetes type 2 patients, in which increased and sustained insulin secretion sets in as a compensatory mechanism to counteract insulin-resistance, a metabolic hallmark of these conditions. Another compelling example of the ability of sustained insulin levels to act as a growth factor relate to fetal overgrowth observed in gestational diabetes where excess of abdominal fat, a highly insulin-sensitive tissue, can be already detected in the fetus of diabetic pregnant mothers at 20–24 gestational weeks due to the fetal exposure to high circulating insulin levels originated by the mother [13]. Opposite clinical disturbances due to lack of IGF physiologic growth effects have also been described depending upon their defective availability or altered physiologic signals in conditions such as prepuberal growth retardation due to low circulating levels of IGF1 [14,15]. An important difference between Insulin compared to the related IGF-I and IGF-II ligands in terms of direct physiological involvement in cell growth and cell cycle regulation under physiological requirements stands on the acute versus chronic tissue availability. This is linked to their biologically diversified secretion type. Specifically, the availability of insulin secretion in the bloodstream and its clearance are both acute and tightly regulated mechanisms ultimately able to make insulin blood levels negligible within 30–90 minutes from the hormonal surge to decrease blood glucose and amino acids as a result of food digestion until insulin-induced cellular uptake of these nutrients removes them from the extracellular environment. As for IGF-I blood levels, they do not undergo the rapid spike and the acute clearance observed for circulating insulin levels but, on the contrary, its secretion from the main tissue storage (the liver) is kept relatively constant in response to Growth Hormone (GH) under a negative feedback mechanism with the GH-producing cells of the pituitary gland in order to fulfil the somatic growth of the developing body. Similarly, but with a different prenatal and post-natal type of control, IGF-II retains both metabolic and proliferative effects with a growing number of evidences suggesting a specific roles (not shared with IGF-I) during embryonic growth, neural development, adult stem cells maintenance and metabolism [16–19]. It is worth noticing that, while under physiological conditions insulin displays exquisite endocrine functions and its paracrine and autocrine effects are restricted to the pancreatic tissue where it is produced. On the contrary, in the adult organism IGF-I and IGF-II are produced by stromal and connective tissue (fibroblast and derived component) allowing them to exert physiological paracrine effects as well as contributing to the established serum growth factor trophic activity. This IGF-mediated endocrine effect is essential for all cells in the body towards the development, maturation, and structural maintenance of tissues and body organs. Although both IGF-I and IGF-II are able to trigger the activation of a specific cell signal in normal and cancer cells via activation of their RTK in vitro, it has become evident that the majority of solid cancer cell lines express and secrete IGF-II [20–26]. In fact, retrospective and current cumulative experimental evidences support that while the paracrine actions of IGF-I and IGF-II are functional to both physiological and pre-cancerous stages, the autocrine IGF-II stimuli becomes the predominant IGF loop towards gaining and maintenance of the malignant phenotype [27–29]. The increased levels of IGF-II secondary to loss of heterozygosity (LOH) for its gene parental imprinting, by affecting its presence in the tissue microenvironment or specific organs, are compatible with the overall increased tumorigenic effect described in the literature [30]. A remodulated view of the actual role of the growth/proliferative-linked effects of Insulin and IGFs under the discussed physio-pathological contexts is provided in Table 1.
Table 1.
Mode of action of the insulin/IGF ligands.
 |
Physiology Pathology (Cancer)
* As suggested by scientific cumulative translational and clinical research and reviewed in [29] under growth/proliferative physio-pathological conditions in mammalians*
The insulin/IGF receptors system in cell growth and proliferation: a retrospective view on the changing paradigm
The previous view and understanding of the role of IGF-I and IGF-II [31] and their receptorial system towards promoting cellular growth and proliferation have significantly changed in the light of the discoveries of the last two decades displaying a more complex network between the insulin/IGF ligands and their cellular targets. In first place, the old assumption of the functional compartmentalization of the insulin receptor and IGF1 receptor effects has been fully revisited. Basically, throughout the mid-nineties, the generally accepted view was that insulin, through exclusive high-affinity binding to its receptor, would exert its exquisite metabolic functions while the IGF-I receptor would mediate the growth and proliferative effects of both IGF-I and IGF-II [32], which were formerly known as Somatomedin-C and Somatomedin-A, respectively [33]. Indeed, the search for an high-affinity IGF-II transducing receptor mediating its growth effects had initially brought to the identification of a kinase-less gene referred as IGF2 “receptor” (igf2r) [34,35]. Both biochemical purification- and sequence/structural studies for this transmembrane protein established that it does not carry any kinase activity nor growth/proliferative-promoting effects like any other GF-activated RTK [36,37]. On the contrary, as discussed herein this assumed igf2 “receptor” has been found to exert TK-independent tumor-suppressing functions based on cellular expression and LOH studies [38–42]. Nonetheless, the demonstration of the ability of IGF-II to bind and activate the IGF1R along with IGF-1 [32,43] reinforced the general view that the IGF1R receptor could be indeed transducing all the IGFs growth/proliferative signals and cellular effects. This forced exemplification came to light following a number of basic and translational studies contradicting this mechanistic scenario. Indeed, a few earlier studies had already shown an increase of insulin receptors in cancer tissues and cell lines compared to their normal counterparts along with the demonstration that the IR overexpression carries pro-oncogenic potential [44–46]. However, the biological relevance of such findings had been underscored due to the assumption that such increase in IR content in cancer would purely reflect the opportunistic need of cancer cells to enhance nutrients uptake to fulfil their increased metabolic needs. This concept assigning a pure metabolic function to the IR started being challenged by the identification of a number of co-existing “atypical” insulin and IGFs receptors in the same cancer cells and tissues, supporting the possibility that the IGFs could also activate unknown variants of insulin- and IGF receptors to exert their cellular functions [47–49]. Indeed, these observations were not in contrast with experimental findings showing IGF-I and IGF-II to bear those insulin metabolic (anabolic) effects in cultured cancer cells [50] raising the question on the biochemical nature and function of such receptorial entities in vivo. The answer on the nature and composition of such receptors toward mediating the IGFs’ growth, proliferative and metabolic effects came in the late nineties from the identification of the previously referred atypical IGF receptors. Part of such “atypical” activity was in fact found to depend on the presence of IR-IGF1R hybrids which behave like IGF1Rs but with differential IGF1 vs IGF2 stimulated activity based upon the involved IR isoforms [10,23,51]. However, at the time of these studies a major question related to the “atypical” insulin receptor with high affinity with IGF-II also supported by the in vivo demonstration of a fetal IGF-II-InsR growth axis [52] was still left unanswered. Namely, what was the nature of such atypical InsR mediating developmental growth and cancer cell proliferation so effectively? The discovery that a specific IR isoform variant (IR exon11-) displayed a previously uncharacterized high affinity for IGF-II binding and that such isoform was highly expressed in fetal and cancer cells provided the long missing IGF-II TK receptor able to diversify its biological proliferative effects from those exerted via the IGF1R [21,53]. As a result, the IGF-II/IR-A autocrine loop is currently a compelling new model for understanding the role of IGFs in cancer, especially at the light of the commonly secreted IGF-II feature in cancer cells [54]. Indeed, the apparent mechanistic complexity of the insulin/IGF system, which has launched the search for molecular determinants to clarify the role of its individual components in mediating their cellular effects in physiology and disease, can now be re-evaluated in the light of their different contextual expression in terms of isoform content as well as potential involvement in novel signaling complexes under specific conditions such as those observed under hypoxic states [10]. The additional findings that the very same receptor variants can mediate ligand-specific effects in terms of downstream signaling components [25,55,56] further support the importance of the contextual expression of IGF ligands, receptors and downstream signaling components in individual cell types and tissues as a determinant to understand their role both at the cellular and whole body level. Specifically, the observation that all IGFs, growth factors and receptors, can promote both cell growth and cell division in isolated cellular models (although in diversified cellular and physio-pathological contexts) requires additional attention when designing modulatory pharmacological strategies. In particular, and as previously suggested [10], any molecular-targeting strategy meant to inhibit the IGF-dependent proliferative effect calls for routine profiling approaches to pinpoint the co-expressed isoform variants and signaling circuits in vivo. Ultimately, the need to adopt integrated experimental approaches based upon omics profiling in the field of IGFs and cancer has become a logical requirement [57]. The advantage for such profiling approach is further supported by the number of naturally occurring variants in the IGF family in humans [58]. Indeed, the failure of the IGF1R single targeting in the clinical setting [59], has painstakingly shown the importance of fully embracing the molecular complexity and the contextual physio-pathologic functions of this distinctive ligands/receptor family (Figure 1), as an opportunity towards developing sounder strategies to therapeutically modulate IGF-triggered growth and proliferative effects without affecting their central physiologic role.
Figure 1.

Growth/proliferative-related effects of the Insulin/IGF ligands/receptor system in mammalians*. *ased on predominant effects observed in isolated/optimized ex-vivo models or net clinical study end-point.
Role of IGF binding- and scavenger proteins in IGF ligands/TK receptors-mediated growth and proliferation: a case for the adoption of the new functional acronym for the igf2/m6p-“receptor”
A broad number of studies have been generated over time on the extracellular binders known as IGF binding proteins. Interestingly, a surge of IGFBP studies has preceded the current understanding of the IGF ligands and relative TK-mediated intracellular signal. In the context of the present review we deemed relevant to focus on the specific loss-of-function studies potentially connecting the IGFBPs to the Insulin/IGF-family mediated growth/proliferative effects. Such studies, reviewed elsewhere [60], did not find this family of proteins to have an effect on growth and proliferation mediated by the IGF ligands/RTK system. Due to the claimed independent IGF binding effects of IGFBPs (namely, IGFBP1- IGFBP6 along with mac2/IGFBP-7) for which opposite or paradoxical effects to IGF-neutralization can be found in the literature, we will not review such factors for the scope of this work. With the same token, among the bona-fide IGFBPs based upon presence of an IGF binding motif (found in IGFBP 1–6) (reviewed in [61] along with other known IGF binders, the ones for which we found in vivo loss-of-function studies addressing their potential effect on growth are: (a) IGFBP1, (b) IGFBP2, (c) IGFBP3, (d) the so called IGF2 “Receptor” (herein referred as SpI2-6), and, (e) the IR secreted form.
IGFBP1
Despite some studies have shown a relationship between IGFBP1 and IGF1-dependent growth/proliferative effects in its phosphorylated form [62], its role as a general modulator of IGF growth/proliferative signals remains uncertain [63]. This is in part due to the demonstrated IGF-1-binding independent actions exerted by this protein in cancer along with the inconsistent effects observed by IGFBP1 gene ablation in a cancer model [64].
IGFBP2
Although a growth limiting effect was observed upon IGBP2 gene ablation in zebrafish
[65] following constitutive loss-of-function study in mice [66] has revealed a different biological mode of action for this gene product besides its potential IGF-binding interference in IGF-mediated growth. These findings along with other cellular-based observations showing opposite biological effects have raised the question on IGFBP2 mechanism of action towards promote and/ or inhibiting IGFs proliferative actions [67].
IGFBP3
In the case of IGFBP3 the relationship between its mild growth suppressing effect in loss-of-function studies in cancer has been reported by a few authors [68,69]. However, also for IGFBP3, the actual IGF-binding dependent effects compared to the IGF-binding independent ones are still a matter of open investigation.
SpI2-6 (igf2/m6p-“receptor”)
The role of the igf2/m6p-“receptor” gene product to date stands as the major functional IGF-binding factor among all studied bona-fide IGF binding proteins. Indeed, the overgrowth phenotype observed in the Igf2/m6p-“receptor” gene knockout nude mouse [70] provides to date the most remarkable link between a physiological IGF binder and IGF-mediated growth. A further demonstration that such tumor suppressing activity is linked exquisitely to the modulation of IGF-II levels comes from the evidence that the igf2/m6p-“receptor” genetic ablation in an igf1/igf1r double ko mice background is able to rescue the igf/igfr-linked growth defect by reconstituting IGF2 bioavailability [52,53,71,72]. Based upon these established findings, supporting (a) a tumor-suppressor role for this transmembrane protein upon its ability to effectively interfere with the autocrine IGF-II loop active in fetal and cancer cells, and (b) in absence of structural/functional evidences of this IGF-II binder to trigger a second messenger intracellular signal upon ligand interaction, besides its internalization/degradation, we here propose the more suitable acronym of “SpI2-6” (Scavenger Protein for IGF2 & mannose-6-phosphate). The new acronym, we believe, better fits the experimentally-tested effects observed upon its gene ablation in animal studies (reviewed herein and summarized in Table 2). Furthermore, the acronym of “SpI2-6” rules out the longstanding misleading concept that this transmembrane protein may actively mediate a signal transduction mechanism comparable to that of the tyrosine kinase receptors used by IGF-II to trigger its growth and proliferative effects in vivo (through activation of the IR-A and the IGF1R in their homodimeric and heterodimeric combinations).
Table 2.
Effects of constitutive gene KO for the insulin/IGF-receptors signal components on body/organ size.
| Protein (gene)KO/Silencing | Species/models | Bodysize | Effect of gene ablation | References |
|---|---|---|---|---|
| IGF-I (igf1) | Mouse, drosophila | ✓ | Decrease | [320,321] |
| IGF-II (igf2) | Mouse, drosophila | ✓ | Decrease | [52,320,321] |
| IGF-IR (igf1r) | Mouse, drosophila | ✓ | Decrease | [320,322] |
| IR (insr) | Mouse, drosophila | ✓ | Decrease | [323,324] |
| M6PR(igf2r) | mouse | ✓ | Overgrowth | [70] |
| IRS-1 (irs1) | Mouse, drosophila | ✓ | Decrease | [99, 325, 326] |
| IRS-2 (irs2) | Mouse, drosophila | ✓ | Decrease | [326] |
| H/N/K Ras | Mouse | ✓ | Normal for H/N Ras KOdelayed embryo growth for KRas | [327] |
| PIK3C1A | Drosophila, mouse | ✓ | Decrease | [133] |
| PDK1 | Mouse | ✓ | Decrease | [328–330] |
| AKT1/2/3 | Mouse, drosophila | ✓ | Decrease | [147,331] |
| PTEN | Mammalian cells,drosophila | ✓n.a* | Cell/tissue-specific overgrowth | [161,164,332] |
| TSC1/TSC2 | Mouse, drosophila | ✓ | Overgrowth | [166,333] |
| mTORC1(h-raptor,dTOR) | Mouse cells and tissues,drosophila | n.a*✓ | Decrease | [166] |
| mTORC2 (rictor) | Mouse cells and tissues | n.a* | Decrease | [215,334,335] |
| GSK3β | Mouse cells and tissues | n.a* | Perilethal;GSK3b KORescued pancreatic growth defectcaused by IR-/- | [204,205] |
| 4EBP | Mouse | ✓ | Overgrowth not observed due to increased protein metabolism | [185,336] |
| S6K1 | Mouse, drosophila | ✓ | Decrease | [279,337] |
| FOXO1/3α | Mouse, drosophila | ✓ | Foxo1-/- mice die perinatallyFoxo3a KO does not cause overgrowth | [320,321] |
Secreted InsR
A less canonical bona-fide IGF protein binder with yet undefined biological role but demonstrated in vitro capability to bind Insulin/IGF ligands relates to the finding of a cellular-secreted Insulin receptor form [73]. Despite this secreted IR variant displays conserved high affinity and Insulin/IGF binding ability, its role in modulating Insulin/IGFs mitogenic signals in vivo is unknown. We speculate that the IGF-binding activity derived by secreted and/or ectoshed receptorial components could bear yet undefined biological significance under those conditions potentially favored by an increase of secreted canonical receptors and/or proteolytically-generated IGF-binding moieties (including SpI2-6) such as in conditions of general or localized tissue over-expression in cancer.
The IGFs-regulated intracellular signaling circuitry involved in growth and cell cycle regulation: an up-to-date actionable summary
An essential overview of the established signaling components involved in the insulin and IGFs-regulated pathways underlying their growth and mitogenic effects is provided herein with emphasis to their phosphorylative network. Although a number of additional targets and circuits relate to the Insulin/IGF signal (eg in differentiated cells and tissues under physiological contexts), these known signaling features and targets not related to growth and cell cycle-related effects (eg towards migration, morphogenesis, vasculogenesis, and differentiation) displayed by the same signaling components have not been included for the scope of this review. The established circuitry for the IGFs-growth/cell cycle promoting signal is summarized in Figure 2. The effect of loss-of-function of the key IGF signaling components on in vivo growth has been conveyed in Table 2, whereas their oncogenic versus tumor-suppressing actions have been conveyed in Table 3.
Figure 2.
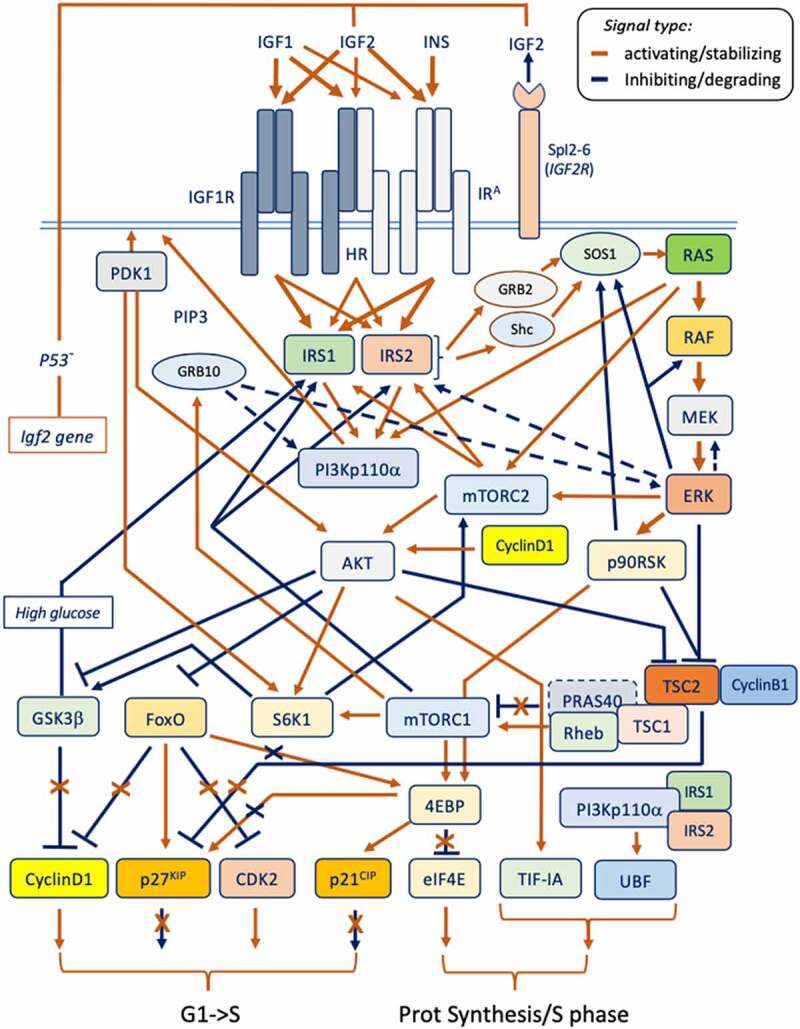
The essential circuitry for the Insulin/IGFs system involved in growth and proliferation signal (refer to text and Table IV).
Table 3.
Summary of insulin/IGF signal transducers acting as oncogenic factors versus tumor suppressors.
| Oncogenic | Tumor-suppressing |
|---|---|
| IGF-I/IGF-II* | M6PR/Igfr |
| IGF-1 R | PTEN |
| IR-A | GSK3β |
| PIK3CA(p110) | 4EBP |
| PDK | TSC1 (Hamartin) |
| AKT | TSC2 (Tuberin) |
| RAS | FOXO |
| RAF | P21CIP |
| CyclinD1/D2 | P27kip |
*Only IGF-II has been confirmed to be broadly overexpressed in cancer [29]
IRS 1/2
IRS1 and IRS2 belong to a family of highly conserved non-enzymatic adaptor proteins (IRS1-4) of which the first two have been widely implicated in the growth and proliferative effects induced by insulin and the IGF ligands (as summarized in Table 2). Indeed, the discovery of insulin receptor substrate (IRS) 1 with its paralog IRS2 [74,75] and the studies following their identification have accelerated our understanding of how Insulin and IGFs mediate their cell growth and proliferative effects. With few exceptions, the IRS proteins are a unique class of adaptors used by the Insulin receptor [76], the IGF-IR [77] and by their hetero-dimeric hybrid receptors counterparts [56] in response to their tyrosine kinase (TK) activation. Although two additional paralogs (IRS3 and IRS4) have been identified, current evidences suggest that their effect on growth and proliferative signals is respectively minor for IRS3 (even interfering with cell cycle circuitry according to some authors [78] or bearing a growth antagonistic effect under certain conditions such as in retroviral infections in which IRS4 has been found to highjack IRS1/2 signals [79]. A common feature of all IRS adaptors responsible for physically conveying the RTK-initiated tyrosine-phosphorylation to downstream signal transducers is the presence of the YMXM docking motif that is targeted by their upstream associated RTK allowing recruitment of other adaptor molecules containing a src-homology SH2 domain such as Shc and Grb2 [80–83] and initiate a phosphorylation-dependent dynamic complex formation involving both SH3 containing proteins (such as the above cited Grb2, Shc adaptors and p85, the PI3K regulatory component) ultimately leading to the coordinated activation of enzymatic proteins with instrumental signaling, synthetic and mitotic activities. Among these enzymatic transducers, to date, a well-characterized Lipid kinase (using PIP3 as messenger) [84] a number of Serine/Threonine kinases [85–93], GTPases, and GTP-GDP exchange factors [94] have all been associated to the insulin/IGFs signal through the mechanistic and functional involvement of the IRS proteins. IRS adaptors have been found to be both positively and/or negatively modulated by feedback signals targeting them on specific Serine/Threonine residues to modulating their stability and protein-protein interaction [95,96]. Indeed a number of studies have already started exposing the specific different roles exerted by IRS1 and IRS2 towards cell growth, proliferation and other cancer-related effects [97,98]. Our understanding of the role of IRS1 and 2 in growth and proliferation relies on the loss-of-function animal studies associated with growth defects as shown by their small phenotype [99–102]. Such studies have cumulatively consolidated the general view that IRS1 mediates body/organ growth and cell size effects predominantly via involvement with the IGF-IR/ IRS1/PI3K/ AKT/ mTORC1/ S6K-related signal. Furthermore, these studies have shown that the two IRS adaptors display a differential subsets of downstream target kinases, such as PDHK1, MAP3K and PKD1 for IRS1, and PIN1 and PKCβ for IRS2, where the proliferative/cell-cycle components seem to be affected by both IRS proteins in complementary ways. In this context, differential interactions and PTMs found in IRS1, IR, IGF-IR, respectively [103], the ability of IRS1 to sustain prolonged IGF-1 R-mediated activation of AKT-FOXO [96], the reciprocal stability effect of IRS2 and FoxO1 [104], and the ability of mTORC21 and mTORC2 to differentially regulate IRS1 and IRS2 stability [105–107], provide just few known examples of the complementary, but different, roles played by these two key insulin/IGFs signal transducers towards the control of cell growth and proliferation. In further support of a specific role of IRS1 towards control of cell size upon insulin and IGFs receptors activation, some studies have demonstrated the ligand-induced nuclear translocation and nucleolar localization of IRS1 along with PIK3p110a and their ability to promote cellular growth by directly activating ribosomal DNA transcription via association with the Polymerase-I core transcription factor UBF [108]. Furthermore, nuclear IRS1 has been found to form complexes with small nucleolar RNAs [109]. Interestingly, two independent studies conducted in different cellular models suggest a parallel/alternative activation of ribosomal DNA transcription via an IGF1R/IRS1 signal versus an IR-A/IRS2 -generated one [97,98], in which the IR-A/IRS2 axis has been also confirmed to associate with the Pol-I- transcription factor UBF. This concept has been conveyed in Figure 3. It is worth noticing that most of the initial knowledge about IRS proteins has been obtained in highly differentiated tissues (namely, muscle, liver and beta-pancreatic cells) for their role in mediating the insulin metabolic actions in mammalians. Since a common feature of pre-cancerous cells, independently from their embryological tissue of origin, is to de-differentiate and start expressing genes out of the tissue/organ functional context (what in pathology is referred as “ectopic expression”), therefore, when considering the growth and proliferative signals mediated by IRS proteins (as for any other IGF activated signaling molecule) in contexts such as cancer, it becomes important to confirm the underlying signaling protein variants under which context they operate. Additional differences between IRS1- and IRS2-mediated signals supporting the above scenario have been shown by studies focusing on the malignant features associated to cellular proliferation in cancer cells, supporting a specific role of IRS2 towards the invasive behavior mediated by the IGF system in Non-Small Cell Lung Cancer (NSCLC) lines and in Head and Neck cancer cell lines [110,111]. An interesting finding specifically linking IRS2 to cell cycle comes from the recent observation that IRS2 is an APC substrate and promotes cell cycle proteins expression and spindle assembly during M-phase [112] disclosing further interest in the mechanisms by which direct IGF-mediated signal transducers affect pathophysiological growth and proliferation.
Figure 3.
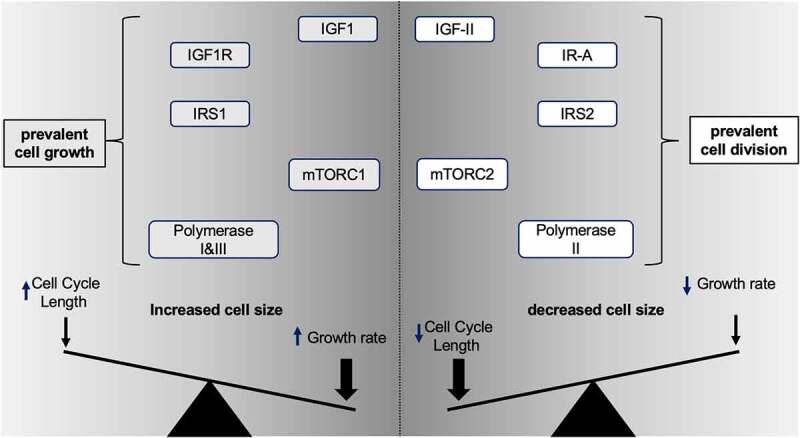
Essential IGF ligands/receptor signal determinants for cell size net effect (refer to text for further explanation).
Grb and Shc protein adaptors and the SOS GTP exchange factor
Grb2
Grb2 was originally cloned as an EGFR signal transduction adaptor protein containing SH2 and SH3 domains and involved in inducing RAS activation and cell proliferation upon ligand stimulation [113]. GRB2 specific involvement in insulin and IGF signal transduction upstream of RAS activation came from the demonstration of the physical interaction between Grb2 and IRS along with the Ras activating effect of such interaction upon insulin stimuli [81]. The GTP exchange factor (GEF) SOS1 was soon identified as a common functional link connecting Grb2 to Ras activation [114] as further mentioned below.
Grb10
Grb10 belongs to the Grb7 family of molecular adaptors. It inhibits insulin receptor biological actions via both inhibiting the IR interaction with IRS1/2 and consequent block of PI3K activation by insulin [115]. Furthermore, it allows IR protein degradation upon sustained insulin stimuli [116] indicating that the Grb10-mediated degradation effect is exerted on the IRK activated form. An additional key finding comes from a study placing Grb10 as a direct target of mTORC1 as part of a negative feedback loop enabling its inhibitory actions [117].
Grb14 also belongs to the Grb7 gene family. Grb14 was found to bind activated Insulin receptors and inhibits their kinase activity [118]. Maximal inhibitory effect of Grb14 on the IRK has been shown to require binding with PDK1 [119]. GRB14 has been also shown to bear inhibitory effects on Insulin receptor mediated mitotic signals. In particular, it has been shown that GRB14 inhibits the insulin receptor-induced cell division through (a) insulin-induced binding to the ubiquitin ligase Chfr, a factor involved in the control of the G2/M checkpoint, and (b) the further targeting and degradation of Aurora A and Polo-like kinase which ultimately leads to growth arrest [120].
Shc
Similarly to Grb2, Shc SH2/SH3 adaptors have been shown to be phosphorylated upon activation of the insulin/IGF tyrosine kinases and to convey their proliferative signal to RAS upon IRS recruitment and SOS1 binding [83,121,122]. Besides functionally and physically linking to the RAS pathway a number of RTK proliferative signals initiated by GFs, Shc proteins have also been shown to convey to RAS a variety of other extracellular stimuli linked to matrix proteins towards exerting migration effects [123,124].
SOS
SOS paralogs (acronym of son of seven-less) belongs to a family of phylogenetically conserved GTP exchange factors found to translated RTK phosphorylative signals towards activation of RAS and its growth promoting pathway [125]. SOS1, the most studied and common member of this family, has widely been shown to exert such function through direct binding to Grb2- and Shc SH3 domains and consequent binding to RAS [122]. More recently, SOS1 has been also shown to act as an extracellular proliferative signals sensor and a retrograde signal regulator of the RAS-MEK-ERK-RSK pathway since both ERK and RSK have been shown to phosphorylate and inhibit SOS-mediated RAS activation [126,127] therefore establishing a negative feedback checkpoint towards protecting the cell from excessive exposure to extracellular originated growth stimulation.
PI3K
Although the enzymatic phosphatidylinositol-3 kinase activity is shared by the homonymous family of lipid kinases with associated Serine/Threonine kinase capability, of all the three characterized sub-classes identified, each displaying class-specific regulatory factors and catalytic product, the PIK3C1A class and specifically the isoform characterized by the catalytic p110α subunit and the use for the regulatory adapters p85α/β and p55α/γ constitutes the PI3K variant found to mediate most of the insulin and IGFs-mediated growth, and proliferative effects [128–130]. A critical finding related to PI3K upstream regulation contributing to its proliferative effects has come from the finding that PI3K is also a direct downstream target of RAS [131]. Indeed, the confirmation of PI3K110α role in cell size and proliferation has been further confirmed by the recent identification of a naturally occurring group of rare overgrowth syndromes linked to an activating mutation in the PIK3CA gene referred as PROS (from PIK3CA-related overgrowth spectrum) (reviewed in [132]. The key downstream mediator of the Insulin/IGF-activated PI3K Class I heterodimer is the AKT/PKB serine threonine kinase which is a key transducer of the growth and proliferative effects of this family of ligands and receptors, as further reviewed herein.
PDK1
PDK1, together with the PIK3C1A catalytic subunit, is part of the upstream Serine/Threonine Kinase cascade stimulated by the insulin and IGFs signal. Its membrane recruitment requires the PIP3 signal generated by the PI3K Lipid Kinase in response to its binding with the InsR/IGF1R-activated IRS adapters [86] and is necessary for the activation of the PH-motif membrane-anchored AKT on Threonine 308 [85]. Important to the context of the present review is the demonstration by loss-of-function in vivo that PDK1 contributes to cell and body size ([133] and Table 1). PDK1 exerts its main growth/proliferative effects by priming phosphorylation events directly enabling other transducers which are key components of the PI3K-AKT-TSC1/2-Rheb1-mTORC1-S6K signaling axis. In fact, PDK1 has been found to be a direct kinase activator for AKT [86], S6K [134], PKC [135],and SGK [136]. This has more recently triggered a specific interest for the development and clinical use of PDK1-specific anti-cancer compounds [136,137]. Indeed, PDK1-blocking compounds have been shown to re-sensitize cancer cells which become resistant upon PI3K inhibition via a PDK1-SGK axis which, in such resistant cells, compensate the deficient growth/proliferative signal initially caused by PI3K block at the level of mTORC1 [136]. This provides an example on how the clinical use of specific signaling kinases inhibitors in cancer treatments is speeding up our understanding of the redundant signaling pathways which can then be further scrutinized at the molecular level for optimization of their use in a personalized setting. It can, therefore, be stated that a contextually designed block of key Insulin/IGF-signal transducers is expected to constitute the mainstay of cancer therapy either as a first line of biological treatment or a mandatory combination following expected single target block recurrence.
PKB/AKT
AKT is an oncogenic Serine/Threonine kinase of the AGC family which involvement as mediator of insulin and IGF growth and proliferation-related effects has been widely established [85,138]. Its full activation requires dual phosphorylation on Thr308 by PDK1 [86] and on Ser473 by mTORC2 [139,140]. On a mechanistic standpoint, ins/IGF-induced PIK3CA lipid kinase activation causes local membrane increase in phosphatidylinositol-3,4,5-triphosphate (PIP3) levels allowing AKT membrane recruitment via its PH domain [141] which is instrumental for its activation by PDK1 and mTORC2 [86,139]. In particular, its phosphorylation on Ser473 by mTORC2 also requires membrane translocation via its core factor mSIN1 [142]. Lysine-mediated ubiquitination [143] has been found to play a key role for AKT activity, sub-cellular localization and protein lifespan upon ligand stimuli [144]. In further support of the direct role of a IRS1-PIK3CA-AKT axis in the insulin and IGFs-stimulated control of ribosomal biogenesis, a critical checkpoint for cell growth and size regulation, a study has also described a direct axis between AKT, CK2 and the Pol-I core transcription factor TIF-IA (also known as RRN3) towards direct activation of the ribosomal gene transcription [145]. In regards to the AKT regulation, which is central to the control of cell growth and cell proliferation, a growing number of evidences connect this serine/threonine kinase to the machinery involved in regulating such effects. Among such evidences, besides the above finding of an direct PI3K-AKT-CK2-TIF-IA(Pol-I) axis increasing ribosomal gene transcription, it has been recently demonstrated a direct regulation of AKT1 on Serine 473 by CyclinD1 (the same Serine residue phosphorylated by mTORC2) towards promoting mitogenesis [146]. These findings provide additional mechanistic details to the previously established circuitry by which AKT promotes growth and proliferation.
PTEN is the acronym of Phosphatase/Tensin homolog on chromosome 10. PTEN is a key negative regulator of PI3K, AKT and S6K1 growth-promoting/proliferative activities and as such it bears tumor-suppressor activity [147,148]. This is consistent with PTEN negative regulatory role on growth and body size observed in vivo [149]. Any of the insulin and IGFs biological actions, including cellular growth and proliferation, promoted by generation of the PIP3 messenger through the activation of the PI3-lipid kinase, requires a biologic counter-inhibitor to attenuate and/or modulate such signal. This role in the cell is exerted by PTEN, bearing both lipid and serine/threonine dual phosphatase activity [150,151]. Since PTEN phosphorylation by GSK3β and CK2 stabilize its activity [152] insulin/IGFs-mediated inhibition of GSK3β and CK2 constitutive activities favors PTEN rapid cellular turnaround. The discovery of PTEN fine regulation by ubiquitination dependent-degradation through WWP1-E3 ligase [153] creates interest on the yet undefined mechanistic landscape by which PTEN is regulated in response to cellular stimuli. A further layer of PTEN regulation potentially affecting its anti-proliferative signals regards the identification and role of new isoforms and/or pathological variants towards dimer formation and signaling [154].
TSC1/2
The role of Tuberous Sclerosis Complex (TSC) proteins is constitutive part of a key heterodimer complex acting as a negative regulator of growth and proliferation under GF-unstimulated conditions and which dimer resolution and single protein effects are critical for the stimulation of their targets (reviewed in Saxton and Sabatini [155]). TSC2 dimer partner TSC1, originally known as Hamartin, acts as direct brake for mTORC1 which is specifically released from the above complex upon Insulin and IGFs-induced phosphorylation of TSC2 (Tuberin). Specifically, the TSC1-TSC2 dimer is dissociated upon direct phosphorylation of TSC2 by AKT [156–158], which ultimately leads to activation of mTORC1 allowing release of the TSC1 inhibition on the mTORC1-activator Rheb1 [159,160]. The intrinsic role of such mTORC1-inhibitory factors downstream to the insulin/IGFs pathway has been shown in Drosophila where their gene over-expression counteracts the insulin and AKT growth promoting effects and is able to reduce cell growth, cell proliferation and organ size [161]. The cellular growth- and proliferation promoting role of TSC2 phosphorylation by Insulin and IGFs activation of the IRS1/2-PI3K-AKT axis is further synergized by the direct phosphorylation of TSC2 by ERK/MAPK and its effector p90RSK [162,163] which is also able to cause dissociation of the TSC1/TSC2 dimer by unleashing the TSC1-Rheb1-mTORC1-S6K1 signal (graphically summarized in Figure 1). TSC1 is also regulated by GSK3β phosphorylation, which inhibition is relieved by AKT-mediated phosphorylation in response to Insulin/IGFs stimuli [164]. The growth/proliferative inhibitory role in absence of the above growth factor dependent signal ultimately depends on TSC1 negative effect on the p70S6K (S6K1) actions dependent on the PI3K-AKT axis [89]. However, studies on TSC2−/− MEF demonstrate that TSC2 also potentiates this TSC1 inhibitory effect on S6K1 due to TSC2 antagonistic effect on IRS1 phosphorylation by S6K1 on S302 (and correspondent Serine on IRS2) as well as by repression of S6K1-mediated IRS1 (but not IRS2) gene expression [88]. IRS1 phosphorylation on S302 by S6K1 in TSC2-/- MEF in the same study is acutely reduced by rapamycin treatment along with IRS1 binding to PI3Kp85. It is also shown that sustained Rapamycin treatment in the same cell line reverses such trend and inhibits IRS1 binding to the insulin receptor ultimately supporting the key role of S6K1 phosphorylation on its growth signal antagonized by TSC2.
mTORC1/2
Despite mTOR has been demonstrated to be a phosphorylative target of insulin antagonized by rapamycin treatment since the late nineties [165], the discovery and characterization of the two mTOR complexes known as mTORC1 and mTORC2 have provided a critical puzzle piece for the understanding of a central mechanism by which insulin and IGFs exert their cellular metabolic and mitogenic functions. Following extensive biochemical and functional characterization of mTOR and its two complexes (mTORC1 and mTORC2) we now know that complex-specific constitutive factors confer mTOR Serine/Threonine kinase its specific rapamycin-sensitivity, specificity and underlying functions. In particular, the combination of constitutive factors known as Raptor and mLST8 are constitutive elements of mTORC1 [166,167], while Rictor, mSIN1(formerly also known as MAPK associated protein 1) and mLST8 are distinctive components of mTORC2 [142,166,168]. Furthermore, other non-core factors can also associate to and modulate mTORC1 and mTORC2 such as Deptor which binds both mTORC1 and mTORC2, and Proctor1/2 as specific binder of mTORC2. mTORC1 has been established to be directly activated by the GTPase Rheb1 upon GF-signal induced release of TSC1 and PRAS40 inhibition [169]. Indeed, the discovery of mTOR as the invoked direct molecular target of the immunosuppressant/ antiproliferative agent Rapamycin, which competes with the insulin- and IGFs-induced activation of 4EPB1and p70S6K1 [170,171] and for causing G1 cell cycle arrest, further supports the specific functions exerted by insulin and IGF-I/II towards G1/S cell-cycle transition [172]. The critical function of mTORC1 in response to insulin and IGF stimuli towards promoting cell growth and metabolism relates to its direct promotion of protein, lipid, nucleic acid synthesis and glucose metabolism [171]. Such effects have been shown to rely upon the PI3K-dependent activation of AKT which acts upstream of mTORC1 via a pathway that involved TSC2 phosphorylation, TSC2/TSC1 dimer dissociation, and PRAS40 and Rheb1, respective unstimulated activities reversal [160,173]. Worth noticing that the mTORC1-depedent growth/proliferative signal is activated also by ERK2 upstream phosphorylation of TSC2 [163]. This is an alternative switch to the TSC2-/TSC1-PRAS40-Rheb1-mTORC1 axis activation by AKT supporting the synergistical, contextual and, partially redundant possibilities by which the Insulin/IGF-inducible growth/proliferative signal takes place in the cell. The growth and proliferative effect of mTORC1 are mostly mediated by its Rheb1-dependent activation of S6K1 and 4EPB1 [169] which, through respective direct targets, regulate key synthetic- and cell-cycle events (see Figure 2 and Table 4). mTORC2 has been found to be insensitive to the acute inhibitory effect of rapamycin [174,175]. Nonetheless, prolonged exposure of cells to rapamycin is able to determine a delayed inhibition also on mTORC2 which is considered to be caused by depletion/ pool exhaustion of available mTOR enzyme for its complex formation [176]. mTORC2 has been shown to control proliferation and survival via at least three known direct targets. First, via mTORC2 phosphorylation of ATKS473 which allows its full activation [139] and reverberates on key substrates and proliferative mediators such as GSK3β, FoxO1 and SGK1 (reviewed by [155]. Second, via positive feedback phosphorylation at the level of the Insulin/IGF receptor kinases [95], and, third, via phosphorylative cross-talk with RSK [177] and PKC family members [178,179]. mTORC1 and mTORC2 are also responsible for establishing negative feedback loops causing the degradation of the IRS proteins towards down-modulating sustained upstream growth/ proliferative- stimulation. In particular, they exert such feedback effects via mTORC1-S6K1-mediated phosphorylation of IRS1 [106] and via IRS1 and IRS2 phosphorylation and proteolytic degradation by mTORC2 [107,180]. The targetability of mTORC1 and mTORC2 to block their growth and proliferative effects in cancer has been considered since the first introduction of rapamycin (Serolimus) in the clinical setting. Using later generation mTOR inhibitors targeting both cellular complexes, it has been established that such block, although overcoming the limitations of rapalogs (rapamycin analogues) still causes a rebound or compensatory upstream hyperactivation of RTKs and PI3K leading to AKT re-phosphorylation on T308 (caused by via PI3K-PDK1 compensatory activation) following an initial transient inhibition [181] and a compensatory increased activity RAS-ERK pathway [182,183]. Therefore, the general current view for the use of mTOR blockers as anti-proliferative/anti-cancer agents in the clinical setting is to combine them with RTK inhibitors to prevent AKT re-activation [181].
Table 4.
Downstream signal targets and relevant PTM sites for the insulin/IGF signal transduction components implicated in cell growth and cell-cycle control.(a) www.phosphositeplus.com DB, (b) UniProt, [301].
| Signaling component/transducer | Y/S/T/K PTM residuesModulated by Ins/IGF* | Downstream direct targetsInvolved in growth/proliferation | |
|---|---|---|---|
| Insulin | n/a | Insulin receptor (InsR) | |
| Insulin Receptor and isoform A (InsR-A) | Y989, Y999,Y1101, Y1139, Y1185, Y1189/90, S1132/33, S1354, Y1351, Y1355, Y1361, T1375 | IRS1, IRS2, (IRS3), IRS4 | |
| IGF-I | n/a | IGF1R > Hybrid InsR/IGF1R ≫ InsR | |
| IGF-II | n/a | InsR-A = IGF1R > Hybrid InsR/IGF1R | |
| IGF-I receptor (IGF-IR) | Y974, Y981, Y988, K1055, Y1127, K1130, Y1161, Y1253, Y1281, Y1283, S1310/11/12, Y1346, Y1352 | IRS1, IRS2, IRS4 | |
| IRS-1 | S24, Y47, Y107, S270, S303, S307, S312, S323, S330, S348, S362, T446, Y460, T495, S527, S531, Y546, Y608, S616, S629, S632, S639, Y658, Y760, Y891, Y935, Y1006, S1078, S1100/1101, S1142/45, Y1220 | P85PI3K, GRB2, GRB10, SHC, UBF | |
| IRS-2 | Y136, Y191, Y214, S306, S365, S577, Y628, Y649, Y671, Y734, Y758, Y814, Y911, Y970, Y1061, S1149, Y1242, Y1303 | P85PI3K, GRB2, GRB10, SHC, 14.3.3, UBF, APC | |
| Shc1p52 | Y427 | SOS1/2 | |
| Grb2Grb10Grb14 | GRB2: (Y209)GRB10: Y67, S150, S428 S476GRB14: S366 |
|
|
| PIK3R1 (PI3Kp85) | S608 | PIK3CA Type I class (p110 isoforms) | |
| PIK3CA (PI3K110α) | TBD | AKT1/2/3, | |
| PDK1 | Y9, S25, S64, S241, T354, Y373, Y376, S394, S396/98, T513 | AKTS308, PKC, S6K, RSK, SGK | |
| PKB/AKT-1/2/3 | AKT1:S129,Y176, K276sm, S308, T450, S473, S477, T479AKT2: S126, S131, T309,T451,S474,AKT3: T305, S472 | S6K1/2, GSK3a/b, mSIN1T89(mTORC2)TSC2, FOXO1/3a | |
| PKC-α,β,δ,β,ζ | PKCa: T638PKCb: TBDPKCd: T507, S645, S664PKCg: TBDPKCz: T410 | S6K1, mTORC1/2?, c-Fos,Negative feedback target:IRS1 | |
| PTEN | Y27, Y174, S380 | PIP3 dephosphorylation (PI3K) | |
| RAS (H, K, N) | HRAS: Y32, T144, T148KRAS: Y32, Y64 | Raf, PI3KCA, PKC, mTORC2 | |
| B-Raf | S365, S429, S446 | MEK1/2 | |
| MEK1/2 | MEK1: S218, S222MEK2: S222, S226 | ERK1/2 | |
| ERK1/2 | ERK1: T202, Y204, Y210ERK2: T185, Y187 | RSK1, TSC2, mTORC2, cFOS, cJUN, ETS1/2, ELK1, egr-1, Cyclin D1Negative feedback direct targets:MEK1, RAF, SOS1, IRS2 | |
| RSK1/2 | RSK1: S221, T573, T359, S380RSK2: S19, T577 | GSK3, S6K1S235/236, c-FOS, p27KIP1, TP53Negative feedback direct target:SOS1 | |
| S6K1/S6K2 | S6K1: T252, S394, T412, S427, S434, T444, S447, K516acS6K2: T228, S473 | GSK3β, eIF3, Ribosomal biogenesis nucleolar proteins (Nop56, Nop14, Gar1, Rrp9, Rrp12, Rrp15, Pwp2)Negative feedback targets:IRS1, mTORC2S1135 | |
| 4EBP1 | T37, T41, S44, T46, S65, T68, T70, S101, S112 | eIF4E, p21CIP1, p27KIP1 | |
| GSK3β | S9, T43, Y216 | CyclinD1, 4EBP1Negative feedback targets:HRAS, PTEN | |
| TSC1TSC2 | TSC1: not directly targetedTSC2: S939, S981, S1387, S1452, T1462 | TSC1: TSC2, Rheb1, PRAS40, p27KIP1TSC2: TSC1, p27KIP1, CyclinB1 | |
| RHEB1 | S130 | mTORC1 | |
|
mTOR: S1261, S1415, T2446, S2448, S2481Raptor: S696, S722, S855, S859, S863, T865, S877, S881/82, T883, S884, S886/87, T889LST8: TBDDeptor:, T241, S244, S258, T259, S263, S265, S282/83, S286/87, S291, S293, T297/98/99 | S6K1S389,S235/236,S240/244, 4EBP1S37/46, TIF1A/RRN3Negative feedback target:IRS1, mTORC2 (via S6K1) | |
| mTORC2:mTORRictorLST8SIN1 | mTOR: see aboveRICTOR: T1135, S1177LST8: TBDSIN1: T86, S128 | AKTS473, RSKS380,E2F->cMyc (transcription)Upstream positive feedbackIGF1R, IR,Negative feedback target:IRS1, IRS2 | |
| Casein Kinase-2 (CK2)CK2A1:Catal. Sub A1CK2A2:Catal. Sub A2CK2B: regulat. Sub B | CK2A1: T13CK2A2: TBDCK2B: S2, S3, | PKC, UBF, TIFI, MBM-cMYC, PTEN | |
| CyclinD1/D2 | CCND1: T286CCND2: T280 | AKTS473 | |
| Cyclin E1/E2 | CCNE1: T395CCNE2: (S21) | Rb1/2, Frap1, Mybl1, Dmrtc2 | |
| P21CIP1 | T145, S146 | PCNA, TAF1 | |
| P27KIP1 | T157 | AURORA-K, RB2p130 | |
| RbRb2 (p107, p130) | RB1: S780, S807, T821RB2: (S639, T642, T986, S1035) | SL1(RPol-I), SNAc(RPol-III)E2F, Pin1 | |
| TP53 | (S15, S20, S33, S46, T55) | Suppresses IGF-I, IGFII, IGFIR and InsR genes transcriptionSuppresses RPol-I transcription | |
| MDM2 | S166, S186, S188 | p53, IGF1R | |
| RPol-I (190 subunit) | TBD | rDNA gene(UBF/TIF1A-RRN3, TIFI63, TIFI110) | |
| UBF1 (RPol-I core TF) | (T9, S23, K61, K71, T117, K132, K144, K160, T201, K216, K232, K266, S273, K279, K352, S389, S412, S433, S449, K480, S484, S495, S546, S584, S638) | rDNA gene promotervia RPol-I and other associated TFs | |
| RRN3/TIF-IA(RPol-I core TF) | (S44, K75, S170, S172, S199, T200, K610, S633, S635, S636, S640, S649) | rDNA gene promotervia RPOL-I and other associated TFs | |
| TAF1 (TAFII250) | TBD | TFIID (RPol-II)UBF (RPol-I) | |
| TAFII150 (RPol-II) | TBD | CyclinD1 transcription via SP1 | |
| RPol-III (220 subunit) | TBD | ERK activates tRNA synthesis by RPOL-III by phosphorylating TFIIIB | |
| MAF1 (RPol-III) | S60, T64, S65, S68, S70, S73, K74, S75, S85, S89, T212, S214 | RPol-III regulated genes | |
| JunFos | Jun: S63Fos: (T232,, S362) | CyclinD, sox6, jun-d, gadd45a, and tob1, YAP/TAZ | |
| MycMax | MYC: T58, S62MAX: (S2, S11) | Myc: rDNA gene/Pol-I transcription, E2FMyc/Max: p27kip1 CyclinE, CDK2, CDK4 | |
| FOXO1AFOXO3A | FOXOA1: T24, S256, S319, S322, S325, S329FOXO3A: T32, S253, S315, S318, S321 | CyclinD1/D2, p21CIP1, p27KIP1,MDM2, 4EBPNegative feedback target:InsR |
S6K
The S6K family of Serine/Threonine kinases is part of a wider AGC kinase super-family that include S6K1 with its two paralogs referred as to p70 and p85, and the homolog S6K2 (p54 and p56) with which it shares 80% of their amino-acid sequence, along with generally conserved domains (but the C-terminal PDZ found only in S6K1, and a proline rich motif in the intermediate C-terminal portion present only in S6K2) and partially conserved activation sites (reviewed by [184]. S6K1 role in promoting growth and proliferative signals is consistent with the small size of animals with ablation of its gene (Table 2) [185]. S6K1 mechanistic role towards promoting protein and ribosomal synthesis has been conveyed in Table 4 and it has been reviewed elsewhere [186]. In here we summarized S6K1 regulatory aspects. S6K activity is activated by insulin and IGFs via direct AKT and mTORC1 phosphorylation [170]. Indeed, S6K1 constitutes one of the two known effectors along with 4EBP by which mTORC1 promotes protein synthesis [173,187–190]. S6K1 is regulated by PDK1 which phosphorylation is required for its full activation. S6K1 has been shown also to be a growth/proliferative target of GSK3β [91] despite being in its turn a reciprocal phosphorylative proliferative target of S6K in cells lacking TSC1 and TSC2 [89]. S6K1 also negatively regulates mTORC2 via phosphorylation of Rictor [191–193] establishing a parallel mTORC1->S6K1->mTORC2 negative feedback. S6K1 is also part of the mTORC1-mediated negative feedback regulation on IRS1 discussed above. In addition to the classically described S6 protein, additional 7 nucleolar targets of S6K1 involved in ribosome biogenesis have been described (Chauvin et al., 2014 [194]), Table 4. p70S6K1 known actions towards regulation of protein translation and ribosomal proteins biogenesis [171,194] involve its ordered translocation between cytoplasm and the nucleus. In this context, the exact mechanistic roles of the other paralog and homologs, p85S6K1 and S6K2, also activated by the Ins/IGF stimuli, is an open topic of investigation at the light of the prevalent and/or diversified nuclear localization of such homologs underlying a different subset of targets and regulated nuclear activities [195].
4EBP1
4EBP1 (formerly reported also as PHAS-1) and its paralog 4EBP-2 play a growth-inhibiting and anti-proliferative role by binding, phosphorylating and suppressing the activity of their cellular targets [196]. Their phosphorylation by insulin and IGFs, by removing their inhibition, unleashes the growth and proliferative effects of their targets among which a major role is played by eIF4E towards driving protein translation [197]. Specifically, 4EBP1 is directly phosphorylated by mTORC1 in response to the insulin and IGFs activation of the IRS1-PI3K-pathway towards exerting its growth and mitogenic actions [198,199]. Phosphorylation of 4EBP1 drives its cellular re-localization and inhibits eIF4E [200] which causes an increase in its mRNA capping activity by enhancing the protein translation process [201]. 4EBP1 also plays a key role in cell cycle regulation by (a) stabilizing p21CIP1 [202] as well as upregulating p27KIP1 activity [203]. 4EBP1 phosphorylation by mTORC1 upon insulin and IGF stimuli mitigates the activation of these downstream cell cycle natural inhibitors resulting in a cell cycle promoting effect. Despite 4EBP1 established tumor-suppressing actions at the cellular level such effect has been hidden in loss-of-function studies in animal models due to an observed hypercatabolic state caused by it gene ablation resulting in animals with apparent normal size [204,205].
GSK3β
Mammalian Glycogen synthase kinase-3 (GSK3) beta along with its paralog GSK3α belongs to a family of constitutively activated serine/threonine kinases with inhibitory functions and preferential phosphorylation of their primed targets [206]. GSK3β is a well-established inhibited target of the Insulin, IGF-I and IGF-II stimuli via AKT-mediated phosphorylation on its Ser9 [207,208]. Its role in mediating Insulin/IGFs-induced cell growth and proliferative effects is associated to GSK3β control of anabolic/synthetic functions [209,210] and of growth/cell cycle gene products [211,212]. This triggers an increase in glucose homeostasis and microtubules formation, on which processes depend both on cellular growth and division. Specific targets of the GSK3β inhibitory control towards regulation of growth and cell cycle include MYC [211], the AP1 transcription factor JUN [213], and CyclinD1 [212]. In regards to the control of parallel synthetic metabolic requirements GSK3β regulates eIF2B [209] and Glycogen Synthase [210]. As for the underlying mechanistic features, GSK3β promotes CyclinD1 proteasome-mediated degradation by phosphorylation on Thr286 [212]. Similarly, GSK3β phosphorylation of MYC on Thr58 has been found to antagonize its proteasome-dependent degradation via SCFFbw7 [211]. Interestingly, GSK3β can mediate the S6K1 growth/proliferative signal by direct phosphorylation/kinase inhibition in TSC1/2−/− MEF when AKT is suppressed by mTORC1 [89]. To date GSK3α has been less studied compared to its GSK3β homolog. Indeed it constitutes a multifunctional Ser/Thr protein kinase activated by insulin and IGFs [210] which has been implicated in the control of several regulatory proteins [214]. Due to its bona fide tumor suppressing actions a few studies have addressed the in vivo phenotype under constitutive loss-of-function conditions. However, the lethality of such mouse model has delayed to strengthen the evidences obtained in cellular models. Nonetheless, a tissue-specific knockout model, namely a GSK3-/- mouse, has confirmed the GSK expected growth rescue function since its gene deletion was able to rescue the major proliferative and metabolic defect caused by the parallel block of the insulin receptor towards rescuing normal pancreatic cells and organ growth [215].
The RAS-RAF-MEK-ERK-RSK Axis
RAS
represents the most known cellular GTPase ([216,217] which, under any of its known paralogs functional/structural composition (H-RAS, N-RAS, and K-RAS) is activated by the Insulin/IGF signal [94]. An established effect of oncogenic RAS expression in cancer cells is its ability to drive serum/growth factor independence by inducing growth factors expression and autocrine stimulation towards autonomous growth and proliferation [218]. Gain-of-function studies in immortalized non-tumorigenic IG1R or InsR expressing cells in presence of oncogenic RAS have been shown to produce a robust growth and proliferative signal leading to malignant phenotypic features [46,219]. The study by Sell et al has also established the permissive function of the signals mediated by the IGF1R towards allowing RAS transformation [219]. As for the underlying signal connecting ligand-induced activation of the Insulin/IGF membrane receptor tyrosine kinases to RAS, it has been established that the activated receptors recruit the IRS proteins via their YXMX motifs and allow the sequential recruitment of an additional SH2/SH3 adaptors, namely GRB2 and/or Shcp52. These activated targets can then bind the GTP/GDP exchange factor SOS1 which is responsible for the positive feedback loop causing an exponential number of membrane-linked RAS molecules to acquire their active complex conformational state underlying its intracellular activation cascade (reviewed in [216]. Since RAS protein activation plays a critical role in the regulation of the growth and proliferative potential of any eukaryotic organism via the activation of its downstream cellular targets, it has been at the center of intense investigation from its original discovery [220]. It is established that the RAS gene products are differentially regulated by polyisoprenylation, palmitoylation, methylation and coordinated proteolysis [221–223]. The canonical RAS-Raf-MEK-ERK-RSK signaling cascade has been found to be shared by both the Insulin/IGF ligand/receptor system as well as by other GFs/RTK systems with the unique difference being in the main upstream circuitry due to the almost exclusive use of the IRS adaptor proteins by the IR and IGF1R system (see also under chapter 4, Figure 2 and Table 4). These other GF/RTK systems (such as EGFR, PDGFR, and others) have been shown to directly recruit either Grb or Shc proteins towards triggering the SOS1-mediated activation of membrane-linked RAS [81,224,225]. The canonical RAS pathway has been further enriched by the demonstration of the direct RAS-induced activation of PIK3CA (the catalytic subunit of PI3K) [131] and very recently by mTORC2 [226] disclosing newer scenarios on the mechanisms by which RAS proteins trigger their established growth and proliferative effects (see Figure 2 and Table 4). Noteworthy, K-RAS is the most represented variant in cancer with the only exception of glioblastoma in which N-Ras is the predominant variant [217]. Despite recognizable differences in their biochemical activity, mutant RAS isoforms within cells have displayed similar ranges of ERK output [227] suggesting that the qualitative aspects of the RAS circuitry induced by extracellular signals plays a major role in the modulation of this pathway as well as on its intrinsic biological message towards directing growth and mitotic events. Interestingly, loss-of-function studies in mice have shown that H-RAS and N-RAS constitutive individual and double gene deletion does not affect normal growth while K-RAS KO mice were perinatally lethal [228]. Nonetheless, by comparing K-RAS−/− versus K-RAS-/+ mice intrauterine growth Johnson at al were able to confirm K-RAS growth/proliferative physiologic role since RAS−/− mice fetuses displayed a smaller phenotype starting E10.5 compared to their RAS−/+ counterparts [229].
Raf
B-Raf with its Raf1 (C-Raf), and A-Raf paralogs is the direct downstream activated component of the RAS pathway [230]. The molecular mechanism shared by RAS isoforms to activate RAF is now better understood at the light of the recognition of the RAS-GTP/RAF/14-3-3 intramolecular complex dynamics in order to allow full RAS activation [231]. This, especially at the light that B-Raf isoforms can be differentially regulated as shown in isoforms containing exon 8b which are phosphorylated on S365 corresponding to the Raf/14-3-3- binding site, causing enhanced intramolecular interaction between the regulatory domain and the kinase domain of B-Raf and reduction of this isoform kinase activity [232]. As for Raf paralogs role in mediating specific growth and cell-cycle promoting signals, it is worth mentioning that the three Raf (A-/B- and C-Raf) kinases differ in their ability to activate MEK in vitro and the ERK pathway in vivo, and to transform NIH 3T3 cells. in fact, B-Raf is the paralog which outperforms the others in terms of both basal activity an responsivity to RAS and extracellular stimuli (reviewed in [233]. The dimerization of Raf and its paralogs (Raf1 or C-Raf, B-Raf and A-Raf) via homologous and heterologous dimers formation has been shown to play a key permissive role towards downstream targets activation [234]. In this context, despite C-Raf (Raf-1) has been shown to play a key role in normal embryo growth and development with embryonal lethality timing depending upon the underlying genetic background [235], another loss-of-function study in mice found that MEK activity was not required for Raf-1 physiologic growth/proliferative function in [236]. This can find explanation on the fact that B-Raf, as functionally central paralog is still be able to dimerize with other isoforms and compensate for Raf-1 genetic or Y340/Y341 activation sites ablation. Nonetheless, the basic role of Raf-1 (C-Raf) and A-Raf has been specifically addressed in another loss-of-function study [237] confirming the relevance of these Raf paralog genes towards normal embryological growth both individually and by double null ablation as well as for the need of such genes for G1/S cell cycle transition, MEK-ERK phosphorylation and c-Fos and CyclinD1 induction as shown in MEF cells obtained from these mice embryos. Indeed, a degree of C-Raf dependency has also been described in a KRAS mutant lung cancer cell (A549) where C-Raf ablation triggers A-Raf mediated hyperactivation of ERK [238]. Raf is also target of the ERK negative feedback loop towards downmodulating hyperactivation of the RAS-Raf-MEK-ERK pathway [239].The biological and mechanistic meaning of RAF dimers towards downstream activation of MEK and the modulation of the underlying proliferative effect has been further clarified upon studying B-Raf naturally occurring mutation, BRAF-V600E, which constitutes the most common type of B-Raf mutation detected in melanoma (86% of all B-Raf mutations in this cancer type) and in a relevant percentage of other solid cancers. This B-Raf mutant, differently from its wild type counterpart and paralogs does not require dimerization for MEK activation and is insensitive also to native negative feedbacks as shown by its inability to bind to Sprouty2 [240,241], mechanistically and clinically explaining the type of response and observed resistance mechanism to the anti-proliferative agent vemurafenib along with B-Raf central role in Raf biology (reviewed in [242]. Altogether, the current findings on Raf paralogs suggest the presence of a broader and integrated signal regulation both in the context of the RAS-ERK canonical pathway as for yet undefined cross-talk circuits leading to growth/proliferative effects.
MEK1/2
are Serine/Threonine and Tyrosine dual phosphorylation kinases directly targeted by RAF proteins in response to extracellular proliferative signals [243]. They also integrate signals from other kinases activated in response to other types of stimuli. MEKs have been found to phosphorylate ERK/MAPK both on Serine/Threonine and Tyrosine residues (summarized in Table 4) and their upstream activation requires phosphorylation on two specific serine sites [243]. A MEK1 associated protein (MP1) in a complex with a 14KDa protein has been found to provide a scaffold to increase both MEK1 and ERK activation and enhancing its downstream targeting specificity towards ELK1 [244,245]. Interestingly, till recently the almost exclusive downstream target of MEK1 and MEK2 was ERK/MAPK. This has changed with the demonstration of the ability of MEK1 and MEK2 to interact with AKT through their Proline-rich domain and promote the activation of FOXO [246]. This finding is further demonstration of the cross talk between the RAS-Raf-MEK-ERK-RSK axis and the PI3K-AKT main alternative proliferation signal in response IGFs under permissive integrated conditions towards controlling the growth and proliferation status of the cell. Loss-of-function studies in mice have shown the MEK1 absolute dependency for normal embryological and post-natal growth [247]. On the opposite, constitutive MEK2 somatic ablation in mice leads to viable and normal organisms [248] suggesting a full compensatory function of MEK1 in mammalians.
ERK1/2
(MAP2K-p42/p44) are Serine/Threonine kinases long known to be Insulin/IGF-signal targets and mediators of their proliferative signals [249]. As for other kinases involved in growth and cell-cycle regulation, ERKs’ phosphorylation of their downstream targets releases their inhibited conformations allowing proliferation and cancer promoting effects [250,251]. As a result, growth-factor-mediated sustained activation of ERKs can downregulate antiproliferative genes involved in G1 phase unleashing cell cycle progression, in part through involvement of AP1 [250]. Among such G1/S promoting targets of ERKs is FOXO3a, known repressor of Cyclin D and activator of p27KIP1 [252,253]. The removal of the FOXO inhibitory effect on G1 Cyclins/CDKI targets by ERK has indeed been shown to play a constitutional role in ERK mediated proliferative effect [254]. Another ERK target triggered by the IGFs proliferative signal is TSC2, which is also targeted by AKT as cited herein (Table 4). TSC2 targeting by ERK2 determines TSC2 dissociation from its dimer partner, TSC1, and consequent repression of Rheb1, a key step in mTORC1 activation triggering proliferation and growth signals during both developmental growth and in tumorigenesis [163]. ERK activation can be transient or sustained, depending upon the involved cell stimuli. Part of the mechanism for controlling the duration of ERK activation is exerted through ERKs-mediated inhibition of upstream components of the signaling pathway through direct feedback phosphorylation following ERK activation. This ERK negative feedback control so far has been demonstrated at four levels. Specifically, by (a) ERK feedback phosphorylation of MEK1 (Ser 292 and Thr386) which inhibits MEK1 activity [255,256] and decrease duration of MEK1 activation in newly adherent cells [239]; (b) ERK phosphorylation of Raf (Ser 29, 289, 296, 301 and 642) inhibiting RAF/RAS interaction and the overall RAF activity [257,258]. This feedback phosphorylation is highly reduced for A-Raf in cells bearing the B-Raf V600E and G469A mutants showing how such mutation can overcome ERK negative feedback mechanisms and maintain increased kinase-dependent signal activation; (c) ERK phosphorylation of SOS1, within a proline-rich SH3 region responsible for its interaction with GRB2 (S1132, S1167, S1178, S1193) and shown to downregulate upstream signals mediated by a number of membrane RTKs using GRB2 or Shc to activate the RAS-ERK pathway [127,259] and, lastly (d) ERK phosphorylation of IRS2 (on Ser907) [260] which interferes with ERK activity and which functional role still awaits elucidation. Interestingly, in a cancer prostate cell IRS2 interacted with deubiquitinylase (DUB) USP9X resulting in increased IRS2 ubiquitination/degradation and suppressing basal ERK levels. These were restored by IRS2 exogenous expression suggesting that stabilization of IRS2 is critical for basal Erk1/2 activation [261]. As for the requirement for each ERK paralog in growth and proliferation, loss-of-function studies in KO mice have shown that ERK2 function is essential for normal growth and proliferation both embryonically and post-embryonically while ERK1 constitutive gene ablation allows normal viable growth of the null organisms supporting full functional compensation by ERK2 [262,263].
RSK
has been described since the late nineteen eighties as one of the kinases active against the S6 ribosomal protein upon insulin and IGF signals [249,264,265]. Two isoforms have been described and they are parallel phosphorylative targets of ERK1/2 and PDK1 and phosphorylate a number of cytoplasmic and nuclear proteins (reviewed by [266] part of which support their proliferative function. Specifically, RSK1 and 2 phosphorylate p27KIP1 promoting 14-3-3 binding and cytoplasmic retention [267]. Both isoforms also phosphorylate, stabilize c-FOS therefore increasing its growth promoting effects [264,268]. RSK2 phosphorylates and inhibits GSK3β negative regulatory activity allowing downstream signal activation [269]. As for ERK1/2, RSK2 does also display a negative feedback by phosphorylating SOS1, therefore downregulating RAS/ERK activation [270]. An interesting finding relates to the observation that ERK signaling integrates extracellular signals with p53 activity to determines the duration of the G2 mitotic arrest such as that determined by DNA breakage [271]. A previous study had already pointed at the relevance of asynchronous ERK activation pulses for the transmission of proliferative signals triggered by extracellular stimuli [272]. In agreement with the previous findings, this recent study further uncovers that under G2 phase arrest, ERK displays a pulsatile activity (such as that expected after prolonged serum starvation) paralleling the frequency pulses of p53 expression under the same circumstances. Upon sustained ERK activation, such as that observed upon continuous growth factors availability (eg autocrine stimuli in cancer cells) or by oncogenic constitutive activation of an ERK-activating pathway, CDC25C becomes a target of ERK-dependent phosphorylation and p53 expression pulses are inhibited leading to the accumulation of pro-mitotic factors (namely, CyclinB1 and PLK1). Worth noticing that a smaller phenotype associates with RSK2 null mice [273] independently from the neurological symptoms (Coffin-Lowry syndrome-related) appearing after 36 weeks of age suggesting that the other mammalian paralogs, namely RSK1, RSK3 and RSK4 can compensate RSK2 functions towards normal growth and proliferative embryonal and post-embryonic functions.
These known mechanisms have been summarized in Figure 2 and conveyed in Tables 2, 3 and 4.
RNA Polymerase-II-dependent transcription factors activated by the IGF-growth/cell cycle-related signal
The transcription factors included below provide a selected well established set of players linking the IGF signal involved in the control of cell size and cell division. The general knowledge and mechanistic details on the type of signal network and feedback mechanisms are expected to grow consistently in time. All of them depend on the transcriptional activity of Polymerase-II (Pol-II) and are typically considered co-activators since they increase the transcription rate of a specific set of genes bearing their respective binding motifs and act by physically associating the regulated gene to the polymerase complex, namely, the polymerase II enzyme along with its constitutive (“core”) transcription factors. As for the Pol-II core transcription factors involved in the growth and mitogenic effect of insulin and IGFs these are discussed in chapter 5.
FOXO
FOXO genes belong to the Forkhead box O family of transcription factors, (TF) (co-activators) targeting a set of genes directly involved in cell cycle regulation [274]. They also modulate Insulin/IGF growth and proliferative actions such as providing specific feedback phosphorylation loops that modulate their upstream signal [275] (see Figure 2 and Table 4). In particular, FOXO1 (FKHR), FOXO3a (FKHR-L1) when over-expressed or in insulin/IGF-deprived conditions induce cell cycle arrest in agreement with their ability to upregulate p27KIP1 and downregulate CDK2, CyclinD1 and PCNA [276,277]. Insulin/IGFs stimuli determines both their phosphorylation by AKT as well as their transcriptional repression [274,276] which inhibits p27KIP1 transcription and unleashes FOXO-dependent Cyclins D1 and D2 expression [276]. This signaling axis has been shown to be a phylogenetically conserved cell growth control mechanism [278] supporting a tight functional link between insulin/IGF signals and FOXO in the control of cell growth and other metabolic actions in vivo [275]. Nonetheless, gene ablation studies in drosophila have shown that, while dFOXO overexpression causes organ size reduction by targeting 4EBP, its gene knockout does not cause body or organs overgrowth despite an increase in cell number [279]. This suggests that a specific compensatory or size regulatory checkpoints are present downstream to FOXO transcription factors functionally independent from this TF family effects on cell cycle.
Jun/Fos (AP-1)
The Jun/Fos heterodimer known as AP1 is a Pol-II-associated transcription co-activator regulated by serum, IGFs and other growth factors through direct phosphorylation of c-FOS allowing its dimer partner c-Jun to enhance the function of CyclinD1 and other genes required for G1 progression [280]. Specifically, mitogens inducing sustained ERK activation [281] along with its downstream signal target p90RSK phosphorylate c-Fos C-terminus domain, enhancing c-Fos protein stability and potentiating its activity towards the induction of target genes involved in the G1/S checkpoint. The same ERK signaling, via phosphorylation and transcriptional upregulation of the AP-1 transcriptional complex induces downregulation of antiproliferative genes that inhibit G1 progression such as sox6, jun-d, gadd45a, and tob1, among others [250]. Recently, a positive feedback cross talk at the transcriptional and post-transcriptional level between AP-1 and the YAP/TAZ dimer has been demonstrated for the reciprocally targeted genes supporting the tight interaction between the mitogenic signals involved in immediate early transcription and the Hippo pathway contributing to a coordinated cell proliferation and organ growth [282].
Myc/Max
Like c-Fos, also Myc is an early transcription factor phosphorylated by ERK (p90RSK?) and CK2 [283] enhancing its stability and consequent ability to promote G1/S cell cycle transition [280]. A specific mechanism triggered by insulin and serum (containing IGFs) involves p90RSK phosphorylation of MAD leading to its ubiquitin degradation. Since MAD competes with Myc/Max dimer assembly in GF-deprived conditions, its p90RSK-induced degradation favors Myc/Max dimer formation and downstream activation of its G1/S phase promoting genes [284]. Myc regulates this critical checkpoint by several consensual mechanisms. These include (a) the antagonization of the activity and/or expression of cell cycle inhibitors p15, ARF, p21 and p27. Specifically, on p27 (b) Myc induces its proteasomal degradation through the upregulation of the SCFSKP2 complex which regulates Cul1, Cks1, and Skp2 (reviewed in [285]; (c) Myc directly stabilizes the CyclinE/CDK2 complex by dimerizing with Max [286] and mediating the mTORC2-dependent up-regulation of E2F1 [287] which is an established transcriptional activator of Cyclin E [288]. Finally, (d) Myc represses the CDKN1B promoter by direct interaction and inhibition of FOXO3a, transcription factor known to upregulate CDKN1B expression [289]. Interestingly, Myc regulates mitochondrial functions such as oxygen consumption, pyruvate/lactate production and ATP generation which have been found to be an essential requirement to enable rapid cell cycle entry [290]. Another mechanism used by Myc to promote cellular growth beyond its Polymerase-II dependent transcriptional activity targeting G1 phase genes, relates to its Pol-II independent ability to stimulate Polymerase-I-mediated transcription of the ribosomal DNA gene which is a key mechanism for ribosomal biogenesis and the housekeeping gene products synthesis occurring in S phase. Myc exert such direct stimulatory effect by binding and activating Pol-I core TF SL1 at the promoter level [291].
E2F
E2F regulates the expression of genes relevant for cell proliferation most of which involved with the G1/S cell cycle checkpoint and has a specific role in determining the correct timing for G2 cell cycle transition [292]. E2F exerts its function as an heterodimer combination of two groups of conserved gene products (E2F1-5 and DP1-3), mainly via conditional and sequential dimer interaction and complex formation with the RB (“pocket”) proteins (also known as p107 and p130) where the unbound E2F dimer form has activating functions while the E2F dimer-RB complexed form exerts repressing transcriptional functions (reviewed in [293]. E2F has been found regulated by mTORC2 through MYC which establishes a direct exploitable axis linking the IGF signal to its transcriptional functions [287]. E2F activity is further regulated by physical interaction with p53 (reviewed in [294] Sp1 [295], Cyclin A [292] and Cyclin F [296].
Egr-1
The interest between this early response gene and the IGF system stands on the positive feedback between its induction by mitogenic growth factors and its mediated transcriptional activation of the Igf2 and Igf1r genes [297,298]. Egr-1 has been found to regulate directly multiple tumor suppressor genes including TGFβ1, p53, Fibronectin and PTEN [299]. In particular, for the PTEN regulation, Egr-1 has been found to be part of an in vivo AKT-Egr1-ARF-PTEN as part of a feedback inhibition on the PI3K signal [300]. Interestingly, the involvement of Egr-1 implies its Sumoylation which requires post-transcriptional priming provided by direct phosphorylation from AKT [300].
Insulin/IGFs-induced ribosomal biogenesis by RNA Polymerase I&III and growth/cell-cycle-regulated transcription by RNA Polymerase II
The process of ribosomal biogenesis refers to the sum of the events leading to the synthesis of mature ribosomal subunits in the cell. To grasp the relevance and potential of ribosomal biogenesis for both cellular growth and proliferation maybe the most compelling case can be made by the results of the Telomere-to-Telomere Consortium project for the full sequencing of the human genome in which the number of ribosomal genes for the haploid genome has been clearly established at 209 copies (409-/+9 per diploid cell) [302]. This level of intrinsically designed redundancy allows a tremendous burst in protein translation along with the enzymatic activities required to synthesize all other non-proteic components of the cell. It further implies that sustained and coordinated control signal is required by hormones and growth factors of the insulin and IGF family towards ensuring the required amount of ribosomal content and activity in the growing/proliferating cell. The ribosome structure contains dedicated constitutive RNAs and proteins and their synthetic process is regulated by all three human polymerases, each responsible for the synthesis of a dedicated component. Specifically, Polymerase I and III for its structural RNA components, and Polymerase II for the genes coding for the ribosomal proteins (reviewed by [303]. Furthermore, this process is regulated during cell-cycle [304,305] and stimulated by Insulin and IGFs [306–308]. In particular, a number of evidences have shown a rapid activation of ribosomal gene transcription by Polymerase I to be activated by Insulin and IGFs within 10 minutes and to reach maximal activation by 30 minutes [309–311]. As expected, given the macromolecular and the above mentioned heterogeneous composition of the ribosome, a large amount of mechanisms need to participate to the control of its synthesis and function. Only at the level of the ribosomal gene transcription by Polymerase I, insulin-, IGFs- and cell-cycle-regulated kinases such as ERK, PI3K110α and CK2, have been shown to increase (via direct phosphorylative events) Polymerase-I transcription factors RRN3-TIFIA and UBF [108,312–314]. Similarly, insulin and IGF-regulated factors have been involved in the regulation of Polymerase-III [307] which gene products are key structural requirements for both ribosomal structure and the overall translational function. Since animal gene ablation studies have demonstrated the role of the IRS1-PI3K-PDK1-AKT-mTORC1-S6K1 axis in growth and cell cycle control (as summarized in Table 2), and given the dependence of growing cells from the structural genes transcribed by Pol-I and Pol-III activities, it is reasonable to consider them as rate limiting mechanisms for cell growth. Although both Pol-I and Pol-III activities are cell cycle regulated and Pol-I activity is specifically silenced at mitosis through p53-dependent and independent mechanisms [304,315], they do not seem to have a direct role in supporting the mitotic process. While the key biological contribute of Polymerase I and III genes relates to the synthetic apparatus of the cell mainly conveyed in activities occurring in the homonymous (“S”: Synthetic) cell cycle phase, Polymerase II-transcribed gene products and underlying transcriptional activity is required for both cellular growth and division. Specifically, Polymerase II involvement in both cell growth and cell-cycle has been shown based upon the established evidences proving its direct involvement (1) in ribosomal biogenesis through the transcription of all the ribosomal proteins which constitute the ribosome macromolecular complex (reviewed by [303]) and, (2) through the Cell-Cycle specific role of its core transcription factor component TAF250 also referred as TAF1, part of its TAFIID complex, which has been shown to correspond to the gene product CCG1, a transcription factor required for G1/S transition [316]. Since TAF1 is a phylogenetically required gene for the G1/S boundary, this makes Polymerase II activity a key transducer of proliferative signals being directly involved in all the cell cycle boundary junctions (G1/S and G2/M). Nonetheless, TAF1, has also been found to stimulate Pol-I activity outside the TAFIID complex via UBF carboxy terminal tail interaction [317] although the physiologic and pathologic relevance of this mechanism remains to be determined. Indeed, several studies have shown that cells spanning from prokaryotic to human types bear an intrinsic mechanism for the coordination of cell growth versus cell division [318]. Although we have a better picture about the molecular transducers involved in the process triggering either cell growth (affecting cell size) and proliferation (by number of mitoses) as well as having gained a broader understanding of the Insulin and IGFs signal mediated by its receptors, the exact mechanism for the coordination of these two co-dependent and partially and intrinsically overlapping processes, remain to be elucidated. The few hypotheses that are supported by several authors and the current review are represented in figures 3 and 4 where particular emphasis has been given to the relative and/or predominant activation of rate-limiting transducers of the IGF signal considering as ultimate growth and mitotic gatekeepers all three RNA polymerases, namely Pol-I/III as exquisitely favoring S phase and cellular size, and Polymerase II, covering all cell-cycle phases but specifically bearing an intrinsic critical role for both G1/S and G2/M checkpoints. In this scenario, considering the double and parallel role of growth factor signal and nutrients availability (either in defect and in excess) to regulate growth and/or mitosis, the hypotheses fitting our current knowledge have been revisited and graphically conveyed in Figures 3 and 4 considering that they are an exemplification of a more integrated but widely established growth and cell division signal network discussed herein and further summarized in Figure 2 and Tables 2, 3 and 4.
Figure 4.
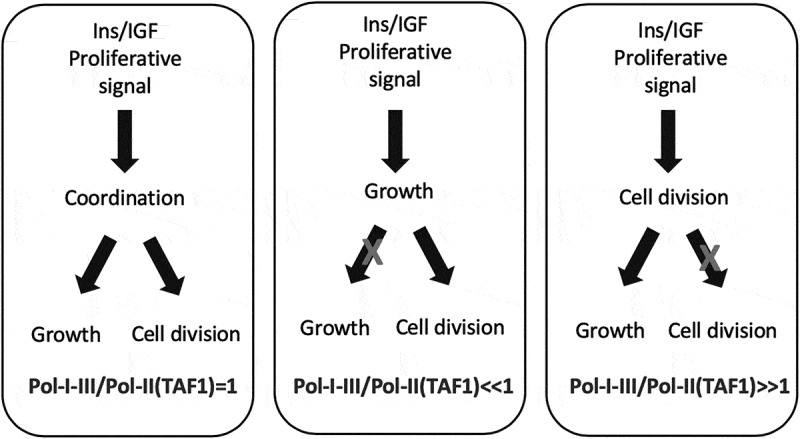
Proposed relationship between cellular growth, cell division and relative Polymerases activities in response to the Insulin/IGF proliferative signal. Refer to text for further explanation. [337](renumber to 336).
Considerations and perspectives
Our present critical analysis of retrospective and current research on the role of the IGF system and cell cycle related signal in mediating growth and proliferation can be reflected, respectively, in: (a) the revisited role of the IGF ligands in physiology and abnormal growth/proliferative conditions such as cancer, (b) the revisited role of their cell signaling transducing receptors, and (c) the functional distinction of the oncogenic- versus tumor-suppressing intracellular kinase network towards regulation of the cellular, machinery underlying the growth/mitotic effects. As far as the actual role of the IGF ligands for the regulation of cellular, tissues and body growth in mammalians, we find that, beyond the large amount of in vitro evidence produced in regards to the affinity between each insulin/IGF ligand and their highly conserved RTKs (namely, IGF1R, InsR-A/B, and their heterodimeric hybrids), an additional tier has been missing or underscored towards proper evaluation of the growth/proliferative effects of IGF-I, IGF-II and insulin in vivo. This missing frame relates to the IGF ligands and receptors contextual mode of action depending on the place and timing of their expression and protein stability determining actual physical interaction and signal duration. We believe that this very simple concept established by both clinical and experimental work has been diluted and is often missing in the experimental design leading to reductionistic conclusions with the result of flattening critical biological differences that could be otherwise properly exploited towards sound actionable interventions. Specifically, insulin production, secretion and rapid clearance under physiologic conditions, let alone its harmful hypoglycemic effect when sustained levels of insulin are produced in a normal human body, per se allows to rule out insulin from sustaining pathological proliferative tissue growth in vivo. This with the only exception of pancreatic insulinoma as a benign condition linked to its restricted paracrine/autocrine effect. This consideration leaves IGF-I and IGF-II as the main IGF factors functionally affecting cell growth and division under physiologic and pathologic conditions. This, we believe, is where retrospective and current findings allow to draw novelty that can guide field scientists to establish new findings and working hypotheses. In fact, although to date the amount of experimental work on the IGF ligands has been focusing on IGF-I in vitro, giving the wrong perception of IGF-II as a complementary factor for the activation of the IGF1R, specific and strong lines of evidence place indeed IGF-II at the very heart of the IGF system-mediated control of the growth and mitogenic cellular program. We here stress out that contextual expression holds the interpretation key for understanding the growth effects mediated by IGF-I compared to those mediated by IGF-II. Namely, IGF-I exerts its physiological functions mostly via endocrine and paracrine action mode. Specifically, IGF-I endocrine effect is linked to GH stimuli, while its paracrine effect is linked to its secretion by stromal tissue which provided a physiological IGF trophic growth stimuli for the maintenance of cell and tissue proteic structures in the whole body linked to its anabolic (insulin-like) promoting cellular effect. On the contrary, most of the IGF-II production and growth/proliferative-promoting effects are tightly linked to its autocrine loop, which is mostly noticeable during fetal growth and in malignancy. We wont reiterate herein the revisited role of the IGF ligands/receptors and IGFBP network which has been discussed elsewhere [29,319]. Instead, upon critical review of the work related to the IGF binding proteins we find that: (1) there are no consistent and reproducible evidences in the literature supporting a key role from any of the previously classified IGF Binding Proteins (IGFBPs) towards exerting their physiological role as IGF-inhibiting factors by purely or mostly interfering with the IGF-growth/proliferative signal in vivo; this suggests that other IGF-independent functions are predominant in the whole organism; (2) the only demonstrated inhibitor of the IGF growth/proliferative effects with a validated effect in vivo is the IGF-II scavenging transmembrane protein traditionally known as Igf2r/mannose-6-phosphate “receptor” for which we propose the physiologically and functionally adherent new acronym of SpI2-6 for “Scavenger TM-protein for Igf2 and mannose-6-phosphate”. Although cumulative evidence has established that the binding of IGF-II to SpI2-6 does not actively trigger an active intracellular response, and it would have been sufficient to revise such misleading definition much earlier, we believe it is still important to adopt the proposed new acronym so to be able to restrict the wording “receptor” to any other gene product with measurable high affinity for a given ligand which triggers a pro-active biological effect. This typically involves an increase of the receptor enzymatic activity and the consequent activation of a second messenger system (where the first messenger is provided by the ligand). On the contrary, SpI2-6 has consistently proven its IGF-II scavenging role towards buffering the stimulatory autocrine effect of both circulating and secreted IGF-II under both physiological and pathological conditions. This function of SpI2-6 per se supports its established tumor-suppressing capability as widely demonstrated by gene knockout studies. As far as the analysis of the IGFs- regulated intracellular transducing network in terms of oncogenic versus tumor-suppressing activities towards the control of cellular, tissue and whole body growth, the present review confirmed that, constitutive gene ablation/silencing of the oncogenic components of the IGF signaling clearly associates with a noticeable cell size/number defect and an impaired/small phenotype. On the contrary, the constitutive ablation of tumor-suppressing components of the IGF growth/proliferative signal (namely, GSK3β, 4EBP1 and FOXO1A) do not lead to an expected overgrowth/hyper-sized phenotype (eg as observed in the SpI2-6/igf2r KO mouse). In spite of this, the reviewed work supports that a minimum level of redundancy in IGF growth/proliferation signals must be in place to allow cellular and whole organism viability. The general perception is that the oncogenic components of the cellular pathway regulated by this GF/RTK family carries higher redundant potential compared to its tumor-suppressing counterpart towards control of the same growth/proliferation effects. In other words, while the constitutive gene knockout of an oncogenic-acting component of the IGF signal preserves a functional cross talk between the cell-cycle machinery and the supporting pre-mitotic metabolic growth program (active during S phase), the constitutive block of a tumor-suppressing component of the IGF pathway appears, somehow, to cause loss of coordination between the molecular events supporting cell growth versus the events solely dedicated to cell division. This view is consistent with the results of the gene KO studies for major IGF-regulated tumor-suppressing transducers, namely, 4EBP1, GSK3β, and FOXOA1. Accordingly, in all these cases, the constitutive gene block does not result in an overgrowth phenotype suggesting functional disconnection between cellular growth and cell cycling. A condition, which is minimally compliant with a viable organism. This hypothesis is further epitomized by a study in GSK3−/− mice where, although single gene ablation is not compatible with life, this growth-suppressing gene deletion in the pancreatic tissue is still able to rescue the major proliferative and metabolic defect caused by the parallel block of the insulin receptor towards rescuing normal pancreatic cells and organ growth [215]. Although an exponential amount of novel findings is expected to fill-in the blanks on the mechanistic details by which the IGF family of ligands and TK receptors regulates growth and proliferation going forward, we believe that several pieces of a previously challenging puzzle have finally come into place. This provides the long-awaited evidence-based landscape for these factors’ mechanism of action which is mature for biologically sound molecular interventions.
Conclusions
The IGF family of ligands and receptors are major drivers of cellular growth and division. Under physiological circumstances they contribute to cell growth through two broad biological strategies. The first strategy involves the activation of the cell synthetic program. Although under diversified biological circumstances, these IGFs/IGF receptors effects are achieved via upregulation of most anabolic signaling pathways. Specifically, the IGFs signal enhances the rate-limiting activities of a specific set of enzymes along with the RNA-Polymerasic machinery regulating cell growth and proliferation acting at transcriptional and post-transcriptional checkpoints. Among these regulatory crossroads are protein translation, ribosomal biogenesis, lipid synthesis associated to pre-mitotic cell membranes expansion, and nucleic acids pool synthesis and specific gene expression. Furthermore, IGFs and their receptors enhance both directly and indirectly the molecular events responsible for G1/S and G2/M cell-cycle transitions. These effects are exerted in vivo as a result of the integrated effect of IGF-I and IGF-II via respective activation of the IGF1R and the Insulin receptor fetal variant (IR-A), along with their heterologous (hybrid) receptors counterparts. While paracrine IGF-I plays a growth/division promoting role under physiologic and in part under pathologic conditions, autocrine IGF-II plays a major function in angiogenic/invasive cancer cells via activation of both the IGF1R and the IR-A. These ligand/receptors interactions determines a series of acute and delayed signaling events depending upon the length of the ligand availability as well as on the contextual/ectopic expression of their intracellular signaling components (isoforms, paralogs and mutated fusion variants). The balance between ligand/receptor protein expression and interaction duration along with the co-expression of downstream signaling components and relative protein variants affects the net outcome of such signal towards cellular growth and division. The acquired structural/functional knowledge of the involved IGF signal components reviewed herein shed light on the positive and negative feedbacks generated at core intermediate signal checkpoints leading to the distinctive and coordinated regulation of cell size and number. Overall, strategies meant to block the growth and proliferative effects exerted by the IGF system in the personalized medicine age will reach a desirable predictive/preventive/interventional level upon adoption of proteo-transcriptomic profiling of the patient cancer cell along with the translation of the knowledge discussed herein into smart IT tools.
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Fujita-Yamaguchi Y, Choi S, Sakamoto Y, et al. Purification of insulin receptor with full binding activity. J Biol Chem. 1983. Apr 25;258(8):5045–5049. [PubMed] [Google Scholar]
- 2.Kasuga M, Fujita-Yamaguchi Y, Blithe DL, et al. Tyrosine-specific protein kinase activity is associated with the purified insulin receptor. Proc Natl Acad Sci U S A. 1983. Apr;80(8):2137–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ebina Y, Ellis L, Jarnagin K, et al. The human insulin receptor cDNA: the structural basis for hormone-activated transmembrane signalling. Cell. 1985. Apr;40(4):747–758. [DOI] [PubMed] [Google Scholar]
- 4.Ullrich A, Bell JR, Chen EY, et al. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature. 1985. Feb-Mar 28-06;313(6005):756–761. [DOI] [PubMed] [Google Scholar]
- 5.LeBon TR, Jacobs S, Cuatrecasas P, et al. Purification of insulin-like growth factor I receptor from human placental membranes. J Biol Chem. 1986. Jun 15;261(17):7685–7689. [PubMed] [Google Scholar]
- 6.Ullrich A, Gray A, Tam AW, et al. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986. Oct;5(10):2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hunter T. The proteins of oncogenes. Sci Am. 1984. Aug;251(2):70–79. [DOI] [PubMed] [Google Scholar]
- 8.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999. Jun 15;13(12):1501–1512. [DOI] [PubMed] [Google Scholar]
- 9.Uhlen M, Fagerberg L, Hallstrom BM, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015. Jan 23;347(6220):1260419. [DOI] [PubMed] [Google Scholar]
- 10.Scalia P, Giordano A, Martini C, et al. Isoform- and Paralog-Switching in IR-Signaling: When Diabetes Opens the Gates to Cancer. Biomolecules. 2020. Nov 30;10(12):1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grandjean V, Smith J, Schofield PN, et al. Increased IGF-II protein affects p57kip2 expression in vivo and in vitro: implications for Beckwith-Wiedemann syndrome. Proc Natl Acad Sci U S A. 2000. May 9;97(10):5279–5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morison IM, Becroft DM, Taniguchi T, et al. Somatic overgrowth associated with overexpression of insulin-like growth factor II. Nat Med. 1996. Mar;2(3):311–316. [DOI] [PubMed] [Google Scholar]
- 13.Kim SY, Lee SM, Kwon GE, et al. Maternal dyslipidemia and altered cholesterol metabolism in early pregnancy as a risk factor for small for gestational age neonates. Sci Rep. 2021. Oct 26;11(1):21066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooke NE, et al. Normal and aberrant growth in children. Melmed S, Koenig R, Rosen C, et al., editors. Williams Textbook of Endocrinology. 13th. Vol. 25, Philadelphia PA: Elsevier; 2016:964–1073. [Google Scholar]
- 15.Menon RK, Sperling MA. Insulin as a growth factor. Endocrinol Metab Clin North Am. 1996. Sep;25(3):633–647. [DOI] [PubMed] [Google Scholar]
- 16.Chao W, D’Amore PA. IGF2: epigenetic regulation and role in development and disease. Cytokine Growth Factor Rev. 2008. Apr;19(2):111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holly JMP, Biernacka K, Perks CM. The Neglected Insulin: IGF-II, a Metabolic Regulator with Implications for Diabetes, Obesity, and Cancer. Cells. 2019. Oct 6;8(10):1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halje M, Nordin M, Bergman D, et al. Review: The effect of insulin-like growth factor II in the regulation of tumour cell growth in vitro and tumourigenesis in vivo. Vivo. 2012. Jul-Aug;26(4):519–526. [PubMed] [Google Scholar]
- 19.Ziegler AN, Schneider JS, Qin M, et al. IGF-II promotes stemness of neural restricted precursors. Stem Cells. 2012. Jun;30(6):1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimura G, Kasuya J, Giannini S, et al. Insulin-like growth factor (IGF) system components in human prostatic cancer cell-lines: LNCaP, DU145, and PC-3 cells. Int j urol. 1996. Jan;3(1):39–46. [DOI] [PubMed] [Google Scholar]
- 21.Sciacca L, Costantino A, Pandini G, et al. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene. 1999. Apr 15;18(15):2471–2479. [DOI] [PubMed] [Google Scholar]
- 22.Sciacca L, Mineo R, Pandini G, et al. In IGF-I receptor-deficient leiomyosarcoma cells autocrine IGF-II induces cell invasion and protection from apoptosis via the insulin receptor isoform A. Oncogene. 2002. Nov 28;21(54):8240–8250. [DOI] [PubMed] [Google Scholar]
- 23.Pandini G, Frasca F, Mineo R, et al. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem. 2002. Oct 18;277(42):39684–39695. [DOI] [PubMed] [Google Scholar]
- 24.Yang L, Li J, Ran L, et al. Phosphorylated insulin-like growth factor 1 receptor is implicated in resistance to the cytostatic effect of gefitinib in colorectal cancer cells. J Gastrointest Surg. 2011. Jun;15(6):942–957. [DOI] [PubMed] [Google Scholar]
- 25.Scalia P, Pandini G, Carnevale V, et al. Identification of a novel EphB4 phosphodegron regulated by the autocrine IGFII/IRA axis in malignant mesothelioma. Oncogene. 2019. Aug 01;38(31):5987–6001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quinn KA, Treston AM, Unsworth EJ, et al. Insulin-like growth factor expression in human cancer cell lines. J Biol Chem. 1996. May 10;271(19):11477–11483. [DOI] [PubMed] [Google Scholar]
- 27.Christofori G, Naik P, Hanahan D. A second signal supplied by insulin-like growth factor II in oncogene-induced tumorigenesis. Nature. 1994. Jun 2;369(6479):414–418. [DOI] [PubMed] [Google Scholar]
- 28.Ulanet DB, Ludwig DL, Kahn CR, et al. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc Natl Acad Sci U S A. 2010. Jun 15;107(24):10791–10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scalia P, Giordano A, Williams SJ. The IGF-II-Insulin Receptor Isoform-A; Autocrine Signal in Cancer: Actionable Perspectives. Cancers (Basel). 2020. Feb 5;12(2):366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaneda A, Wang CJ, Cheong R, et al. Enhanced sensitivity to IGF-II signaling links loss of imprinting of IGF2 to increased cell proliferation and tumor risk. Proc Natl Acad Sci U S A. 2007;104(52):20926–20931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humbel RE. Insulin-like growth factors I and II. Eur J Biochem. 1990. Jul 5;190(3):445–462. [DOI] [PubMed] [Google Scholar]
- 32.Ballard FJ, Read LC, Francis GL, et al. Binding properties and biological potencies of insulin-like growth factors in L6 myoblasts. Biochem J. 1986. Jan 1;233(1):223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daughaday WH, Hall K, Raben MS, et al. Somatomedin: proposed designation for sulphation factor. Nature. 1972. Jan 14;235(5333):107. [DOI] [PubMed] [Google Scholar]
- 34.Morgan DO, Edman JC, Standring DN, et al. Insulin-like growth factor II receptor as a multifunctional binding protein. Nature. 1987. Sep 24-30;329(6137):301–307. [DOI] [PubMed] [Google Scholar]
- 35.MacDonald RG, Pfeffer Suzanne R, Coussens L, et al. A Single Receptor Binds Both Insulin-Like Growth Factor II and Mannose-6-Phosphate. Science. 1988. Mar 04;239(4844):1134–1137. [DOI] [PubMed] [Google Scholar]
- 36.Mottola C, MacDonald RG, Brackett JL, et al. Purification and amino-terminal sequence of an insulin-like growth factor-binding protein secreted by rat liver BRL-3A cells. J Biol Chem. 1986. Aug 25;261(24):11180–11188. [PubMed] [Google Scholar]
- 37.Roth RA. Structure of the Receptor for Insulin-Like Growth Factor II: The Puzzle Amplified. Science. 1988. Mar 11;239(4845):1269–1271. [DOI] [PubMed] [Google Scholar]
- 38.Oates AJ, Schumaker LM, Jenkins SB, et al. The mannose 6-phosphate/insulin-like growth factor 2 receptor (M6P/IGF2R), a putative breast tumor suppressor gene. Breast Cancer Res Treat. 1998. Feb;47(3):269–281. [DOI] [PubMed] [Google Scholar]
- 39.O’Gorman DB, Costello M, Weiss J, et al. Decreased insulin-like growth factor-II/mannose 6-phosphate receptor expression enhances tumorigenicity in JEG-3 cells. Cancer Res. 1999. Nov 15;59(22):5692–5694. [PubMed] [Google Scholar]
- 40.Chen Z, Ge Y, Landman N, et al. Decreased expression of the mannose 6-phosphate/insulin-like growth factor-II receptor promotes growth of human breast cancer cells. BMC cancer. 2002. Jul 30;2(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hughes J, Surakhy M, Can S, et al. Maternal transmission of an Igf2r domain 11: IGF2 binding mutant allele (Igf2r(I1565A)) results in partial lethality, overgrowth and intestinal adenoma progression. Sci Rep. 2019;9(1):11388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hankins GR, De Souza AT, Bentley RC, et al. M6P/IGF2 receptor: a candidate breast tumor suppressor gene. Oncogene. 1996. May 2;12(9):2003–2009. [PubMed] [Google Scholar]
- 43.Casella SJ, Han VK, D’Ercole AJ, et al. Insulin-like growth factor II binding to the type I somatomedin receptor. Evidence for two high affinity binding sites. J Biol Chem. 1986. Jul 15;261(20):9268–9273. [PubMed] [Google Scholar]
- 44.Papa V, Pezzino V, Costantino A, et al. Elevated insulin receptor content in human breast cancer. J Clin Invest. 1990. Nov;86(5):1503–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Milazzo G, Giorgino F, Damante G, et al. Insulin receptor expression and function in human breast cancer cell lines. Cancer Res. 1992. Jul 15;52(14):3924–3930. [PubMed] [Google Scholar]
- 46.Frittitta L, Vigneri R, Stampfer MR, et al. Insulin receptor overexpression in 184B5 human mammary epithelial cells induces a ligand-dependent transformed phenotype. J Cell Biochem. 1995. Apr;57(4):666–669. [DOI] [PubMed] [Google Scholar]
- 47.Milazzo G, Yip CC, Maddux BA, et al. High-affinity insulin binding to an atypical insulin-like growth factor-I receptor in human breast cancer cells. J Clin Invest. 1992. Mar;89(3):899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pezzino V, Papa V, Milazzo G, et al. Insulin-like growth factor-I (IGF-I) receptors in breast cancer. Ann N Y Acad Sci. 1996. Apr 30;784(1 Challenges an):189–201. [DOI] [PubMed] [Google Scholar]
- 49.Massague J, Blinderman LA, Czech MP. The high affinity insulin receptor mediates growth stimulation in rat hepatoma cells. J Biol Chem. 1982. Dec 10;257(23):13958–13963. [PubMed] [Google Scholar]
- 50.McFarland DC. Nutritional and developmental roles of insulin-like growth factors between species: a brief history and introduction. J Nutr. 1998. Feb;128(2 Suppl):300S–301S. [DOI] [PubMed] [Google Scholar]
- 51.Bailyes EM, Nave BT, Soos MA, et al. Insulin receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues: quantification of individual receptor species by selective immunoprecipitation and immunoblotting. Biochem J. 1997. Oct 1;327(Pt 1):209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Louvi A, Accili D, Efstratiadis A. Growth-promoting interaction of IGF-II with the insulin receptor during mouse embryonic development. Dev Biol. 1997. Sep 1;189(1):33–48. [DOI] [PubMed] [Google Scholar]
- 53.Frasca F, Pandini G, Scalia P, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol. 1999. May;19(5):3278–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dynkevich Y, Rother KI, Whitford I, et al. Tumors, IGF-2, and hypoglycemia: insights from the clinic, the laboratory, and the historical archive. Endocr Rev. 2013. Dec;34(6):798–826. [DOI] [PubMed] [Google Scholar]
- 55.Morcavallo A, Gaspari M, Pandini G, et al. Research resource: New and diverse substrates for the insulin receptor isoform A revealed by quantitative proteomics after stimulation with IGF-II or insulin. Mol Endocrinol. 2011. Aug;25(8):1456–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pandini G, Medico E, Conte E, et al. Differential gene expression induced by insulin and insulin-like growth factor-II through the insulin receptor isoform A. J Biol Chem. 2003. Oct 24;278(43):42178–42189. [DOI] [PubMed] [Google Scholar]
- 57.Nagao H, Cai W, Wewer Albrechtsen NJ, et al. Distinct signaling by insulin and IGF-1 receptors and their extra- and intracellular domains. Proc Natl Acad Sci U S A. 2021. Apr 27;118(17). DOI: 10.1073/pnas.2019474118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rotwein P. The complex genetics of human insulin-like growth factor 2 are not reflected in public databases. J Biol Chem. 2018. Mar 23;293(12):4324–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baserga R. The decline and fall of the IGF-I receptor. J Cell Physiol. 2013. Apr;228(4):675–679. [DOI] [PubMed] [Google Scholar]
- 60.Allard JB, Duan C. IGF-Binding Proteins: Why Do They Exist and Why Are There So Many? Front Endocrinol (Lausanne). 2018. Apr 09;9:9. DOI: 10.3389/fendo.2018.00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Forbes B, McCarthy P, Norton R. Insulin-Like Growth Factor Binding Proteins: A Structural Perspective. Front Endocrinol (Lausanne). 2012. Mar 02;3:3. DOI: 10.3389/fendo.2012.00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gupta MB. The role and regulation of IGFBP-1 phosphorylation in fetal growth restriction. J Cell Commun Signal. 2015;9(2):111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin YW, Weng XF, Huang BL, et al. IGFBP-1 in cancer: expression, molecular mechanisms, and potential clinical implications. Am J Transl Res. 2021;13(3):813–832. [PMC free article] [PubMed] [Google Scholar]
- 64.Gray A, Aronson WJ, Barnard RJ, et al. Global Igfbp1 deletion does not affect prostate cancer development in a c-Myc transgenic mouse model. J Endocrinol. 2011. Dec;211(3):297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duan C, Ding J, Li Q, et al. Insulin-like growth factor binding protein 2 is a growth inhibitory protein conserved in zebrafish. Proc Natl Acad Sci U S A. 1999. Dec 21;96(26):15274–15279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wood TL, Rogler LE, Czick ME, et al. Selective alterations in organ sizes in mice with a targeted disruption of the insulin-like growth factor binding protein-2 gene. Mol Endocrinol. 2000. Sep;14(9):1472–1482. [DOI] [PubMed] [Google Scholar]
- 67.Pickard A, McCance DJ. IGF-Binding Protein 2 - Oncogene or Tumor Suppressor? Front Endocrinol (Lausanne). 2015;6:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mehta HH, Gao Q, Galet C, et al. IGFBP-3 is a metastasis suppression gene in prostate cancer. Cancer Res. 2011;71(15):5154–5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blouin M-J, Bazile M, Birman E, et al. Germ line knockout of IGFBP-3 reveals influences of the gene on mammary gland neoplasia. Breast Cancer Res Treat. 2015. Feb 01;149(3):577–585. [DOI] [PubMed] [Google Scholar]
- 70.Lau MM, Stewart CE, Liu Z, et al. Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes Dev. 1994. Dec 15;8(24):2953–2963. [DOI] [PubMed] [Google Scholar]
- 71.Morrione A, Valentinis B, Xu SQ, et al. Insulin-like growth factor II stimulates cell proliferation through the insulin receptor. Proc Natl Acad Sci U S A. 1997. Apr 15;94(8):3777–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ludwig T, Eggenschwiler J, Fisher P, et al. Mouse Mutants Lacking the Type 2 IGF Receptor (IGF2R) Are Rescued from Perinatal Lethality inIgf2andIgf1rNull Backgrounds. Dev Biol. 1996. Aug 01;177(2):517–535. [DOI] [PubMed] [Google Scholar]
- 73.Papa V, Russo P, Gliozzo B, et al. An intact and functional soluble form of the insulin receptor is secreted by cultured cells. Endocrinology. 1993;133(3):1369–1376. [DOI] [PubMed] [Google Scholar]
- 74.Sun XJ, Rothenberg P, Kahn CR, et al. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature. 1991. Jul 4;352(6330):73–77. [DOI] [PubMed] [Google Scholar]
- 75.Araki E, Sun XJ, Haag BL 3rd, et al. Human skeletal muscle insulin receptor substrate-1. Characterization of the cDNA, gene, and chromosomal localization. Diabetes. 1993. Jul;42(7):1041–1054. [DOI] [PubMed] [Google Scholar]
- 76.White MF. The insulin signalling system and the IRS proteins. Diabetologia. 1997. Jul;40(Suppl 2):S2–17. [DOI] [PubMed] [Google Scholar]
- 77.Valentinis B, Navarro M, Zanocco-Marani T, et al. Insulin receptor substrate-1, p70S6K, and cell size in transformation and differentiation of hemopoietic cells. J Biol Chem. 2000. Aug 18;275(33):25451–25459. [DOI] [PubMed] [Google Scholar]
- 78.Kaburagi Y, Yamashita R, Ito Y, et al. Insulin-induced cell cycle progression is impaired in chinese hamster ovary cells overexpressing insulin receptor substrate-3. Endocrinology. 2004. Dec;145(12):5862–5874. [DOI] [PubMed] [Google Scholar]
- 79.Ikink GJ, Hilkens J. Insulin receptor substrate 4 (IRS4) is a constitutive active oncogenic driver collaborating with HER2 and causing therapeutic resistance. Mol Cell Oncol. 2017;4(2):e1279722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shoelson SE, Chatterjee S, Chaudhuri M, et al. YMXM motifs of IRS-1 define substrate specificity of the insulin receptor kinase. Proc Natl Acad Sci U S A. 1992. Mar 15;89(6):2027–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Skolnik EY, Lee CH, Batzer A, et al. The SH2/SH3 domain-containing protein GRB2 interacts with tyrosine-phosphorylated IRS1 and Shc: implications for insulin control of ras signalling. EMBO J. 1993. May;12(5):1929–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sawka-Verhelle D, Tartare-Deckert S, White MF, et al. Insulin receptor substrate-2 binds to the insulin receptor through its phosphotyrosine-binding domain and through a newly identified domain comprising amino acids 591-786. J Biol Chem. 1996. Mar 15;271(11):5980–5983. [DOI] [PubMed] [Google Scholar]
- 83.Gustafson TA, He W, Craparo A, et al. Phosphotyrosine-dependent interaction of SHC and insulin receptor substrate 1 with the NPEY motif of the insulin receptor via a novel non-SH2 domain. Mol Cell Biol. 1995. May;15(5):2500–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ruderman NB, Kapeller R, White MF, et al. Activation of phosphatidylinositol 3-kinase by insulin. Proc Natl Acad Sci U S A. 1990. Feb;87(4):1411–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alessi DR, Andjelkovic M, Caudwell B, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996. Dec 2;15(23):6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 86.Alessi DR, James SR, Downes CP, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997. Apr 1;7(4):261–269. [DOI] [PubMed] [Google Scholar]
- 87.Liu AX, Testa JR, Hamilton TC, et al. AKT2, a member of the protein kinase B family, is activated by growth factors, v-Ha-ras, and v-src through phosphatidylinositol 3-kinase in human ovarian epithelial cancer cells. Cancer Res. 1998. Jul 15;58(14):2973–2977. [PubMed] [Google Scholar]
- 88.Harrington LS, Findlay GM, Gray A, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004. Jul 19;166(2):213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang HH, Lipovsky AI, Dibble CC, et al. S6K1 regulates GSK3 under conditions of mTOR-dependent feedback inhibition of Akt. Mol Cell. 2006. Oct 20;24(2):185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang J, Gao Z, Yin J, et al. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J Biol Chem. 2008;283(51):35375–35382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shin S, Wolgamott L, Yu Y, et al. Glycogen synthase kinase (GSK)-3 promotes p70 ribosomal protein S6 kinase (p70S6K) activity and cell proliferation. Proc Natl Acad Sci U S A. 2011. Nov 22;108(47):E1204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia. 2012. Oct;55(10):2565–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Scalia P, Heart E, Comai L, et al. Regulation of the Akt/Glycogen synthase kinase-3 axis by insulin-like growth factor-II via activation of the human insulin receptor isoform-A. J Cell Biochem. 2001;82(4):610–618. [DOI] [PubMed] [Google Scholar]
- 94.Medema RH, de Vries-Smits Am, van der Zon Gc, et al. Ras activation by insulin and epidermal growth factor through enhanced exchange of guanine nucleotides on p21ras. Mol Cell Biol. 1993. Jan;13(1):155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yin Y, Hua H, Li M, et al. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Res. 2016. Jan;26(1):46–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yoneyama Y, Inamitsu T, Chida K, et al. Serine Phosphorylation by mTORC1 Promotes IRS-1 Degradation through SCFbeta-TRCP E3 Ubiquitin Ligase. iScience. 2018. Jul 27;5:1–18. DOI: 10.1016/j.isci.2018.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wu S, Zhou B, Xu L, et al. IRS-2, but not IRS-1, can sustain proliferation and rescue UBF stabilization in InR or InR defective signaling of 32D myeloid cells. Cell Cycle. 2009. Oct 1;8(19):3218–3226. [DOI] [PubMed] [Google Scholar]
- 98.Xuan S, Szabolcs M, Cinti F, et al. Genetic analysis of type-1 insulin-like growth factor receptor signaling through insulin receptor substrate-1 and −2 in pancreatic beta cells. J Biol Chem. 2010;285(52):41044–41050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Araki E, Lipes MA, Patti ME, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994. Nov 10;372(6502):186–190. [DOI] [PubMed] [Google Scholar]
- 100.Tamemoto H, Kadowaki T, Tobe K, et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature. 1994. Nov 10;372(6502):182–186. [DOI] [PubMed] [Google Scholar]
- 101.Bruning JC, Winnay J, Cheatham B, et al. Differential signaling by insulin receptor substrate 1 (IRS-1) and IRS-2 in IRS-1-deficient cells. Mol Cell Biol. 1997. Mar;17(3):1513–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rabiee A, Kruger M, Ardenkjaer-Larsen J, et al. Distinct signalling properties of insulin receptor substrate (IRS)-1 and IRS-2 in mediating insulin/IGF-1 action. Cell Signal. 2018. Jul;47:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tartare-Deckert S, Sawka-Verhelle D, Murdaca J, et al. Evidence for a differential interaction of SHC and the insulin receptor substrate-1 (IRS-1) with the insulin-like growth factor-I (IGF-I) receptor in the yeast two-hybrid system. J Biol Chem. 1995. Oct 6;270(40):23456–23460. [DOI] [PubMed] [Google Scholar]
- 104.Guo S, Dunn SL, White MF. The reciprocal stability of FOXO1 and IRS2 creates a regulatory circuit that controls insulin signaling. Mol Endocrinol. 2006. Dec;20(12):3389–3399. [DOI] [PubMed] [Google Scholar]
- 105.Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004. Sep 21;14(18):1650–1656. [DOI] [PubMed] [Google Scholar]
- 106.Tzatsos A. Raptor binds the SAIN (Shc and IRS-1 NPXY binding) domain of insulin receptor substrate-1 (IRS-1) and regulates the phosphorylation of IRS-1 at Ser-636/639 by mTOR. J Biol Chem. 2009. Aug 21;284(34):22525–22534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kim SJ, DeStefano MA, Oh WJ, et al. mTOR complex 2 regulates proper turnover of insulin receptor substrate-1 via the ubiquitin ligase subunit Fbw8. Mol Cell. 2012. Dec 28;48(6):875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Drakas R, Tu X, Baserga R. Control of cell size through phosphorylation of upstream binding factor 1 by nuclear phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2004. Jun 22;101(25):9272–9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ozoe A, Sone M, Fukushima T, et al. Insulin receptor substrate-1 associates with small nucleolar RNA which contributes to ribosome biogenesis. Front Endocrinol (Lausanne). 2014;5:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Piper AJ, Clark JL, Mercado-Matos J, et al. Insulin Receptor Substrate-1 (IRS-1) and IRS-2 expression levels are associated with prognosis in non-small cell lung cancer (NSCLC). PloS one. 2019;14(8):e0220567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Luo X, Fan S, Huang W, et al. Downregulation of IRS-1 promotes metastasis of head and neck squamous cell carcinoma. Oncol Rep. 2012. Aug 01;28(2):659–667. [DOI] [PubMed] [Google Scholar]
- 112.Manohar S, Yu Q, Gygi SP, et al. The Insulin Receptor Adaptor IRS2 is an APC/C Substrate That Promotes Cell Cycle Protein Expression and a Robust Spindle Assembly Checkpoint. Mol Cell Proteomics. 2020. Sep;19(9):1450–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lowenstein EJ, Daly RJ, Batzer AG, et al. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell. 1992. Aug 7;70(3):431–442. [DOI] [PubMed] [Google Scholar]
- 114.Li W, Nishimura R, Kashishian A, et al. A new function for a phosphotyrosine phosphatase: linking GRB2-Sos to a receptor tyrosine kinase. Mol Cell Biol. 1994. Jan;14(1):509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wick KR, Werner ED, Langlais P, et al. Grb10 inhibits insulin-stimulated insulin receptor substrate (IRS)-phosphatidylinositol 3-kinase/Akt signaling pathway by disrupting the association of IRS-1/IRS-2 with the insulin receptor. J Biol Chem. 2003. Mar 7;278(10):8460–8467. [DOI] [PubMed] [Google Scholar]
- 116.Ramos FJ, Langlais PR, Hu D, et al. Grb10 mediates insulin-stimulated degradation of the insulin receptor: a mechanism of negative regulation. Am J Physiol Endocrinol Metab. 2006. Jun;290(6):E1262–6. [DOI] [PubMed] [Google Scholar]
- 117.Yu Y, Yoon SO, Poulogiannis G, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011. Jun 10;332(6035):1322–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kasus-Jacobi A, Perdereau D, Auzan C, et al. Identification of the rat adapter Grb14 as an inhibitor of insulin actions. J Biol Chem. 1998. Oct 2;273(40):26026–26035. [DOI] [PubMed] [Google Scholar]
- 119.Goenaga D, Hampe C, Carré N, et al. Molecular determinants of Grb14-mediated inhibition of insulin signaling. Mol endocrinol (Baltimore, Md). 2009;23(7):1043–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Perdereau D, Cailliau K, Browaeys-Poly E, et al. Insulin-induced cell division is controlled by the adaptor Grb14 in a Chfr-dependent manner. Cell Signal. 2015. Apr;27(4):798–806. [DOI] [PubMed] [Google Scholar]
- 121.Pronk GJ, McGlade J, Pelicci G, et al. Insulin-induced phosphorylation of the 46- and 52-kDa Shc proteins. J Biol Chem. 1993. Mar 15;268(8):5748–5753. [PubMed] [Google Scholar]
- 122.Harmer SL, DeFranco AL. Shc contains two Grb2 binding sites needed for efficient formation of complexes with SOS in B lymphocytes. Mol Cell Biol. 1997;17(7):4087–4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Collins LR, Ricketts WA, Yeh L, et al. Bifurcation of cell migratory and proliferative signaling by the adaptor protein Shc. J Cell Biol. 1999. Dec 27;147(7):1561–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xu Y, Guo DF, Davidson M, et al. Interaction of the adaptor protein Shc and the adhesion molecule cadherin. J Biol Chem. 1997. May 23;272(21):13463–13466. [DOI] [PubMed] [Google Scholar]
- 125.Bandaru P, Kondo Y, Kuriyan J. The Interdependent Activation of Son-of-Sevenless and Ras. Cold Spring Harb Perspect Med. 2019. Feb 1;9(2):a031534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Saha M, Carriere A, Cheerathodi M, et al. RSK phosphorylates SOS1 creating 14-3-3-docking sites and negatively regulating MAPK activation. Biochem J. 2012. Oct 1;447(1):159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Corbalan-Garcia S, Yang SS, Degenhardt KR, et al. Identification of the mitogen-activated protein kinase phosphorylation sites on human Sos1 that regulate interaction with Grb2. Mol Cell Biol. 1996. Oct;16(10):5674–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Denley A, Kang S, Karst U, et al. Oncogenic signaling of class I PI3K isoforms. Oncogene. 2008. Apr 17;27(18):2561–2574. [DOI] [PubMed] [Google Scholar]
- 129.Zhao JJ, Cheng H, Jia S, et al. The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci U S A. 2006. Oct 31;103(44):16296–16300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fox M, Mott HR, Owen D. Class IA PI3K regulatory subunits: p110-independent roles and structures. Biochem Soc Trans. 2020;48(4):1397–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rodriguez-Viciana P, Warne PH, Dhand R, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994. Aug 18;370(6490):527–532. [DOI] [PubMed] [Google Scholar]
- 132.Madsen RR, Vanhaesebroeck B, Semple RK. Cancer-Associated PIK3CA Mutations in Overgrowth Disorders. Trends Mol Med. 2018. Oct;24(10):856–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lawlor MA, Mora A, Ashby PR, et al. Essential role of PDK1 in regulating cell size and development in mice. EMBO J. 2002;21(14):3728–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pullen N, Dennis PB, Andjelkovic M, et al. Phosphorylation and activation of p70s6k by PDK1. Science. 1998. Jan 30;279(5351):707–710. [DOI] [PubMed] [Google Scholar]
- 135.Dutil EM, Toker A, Newton AC. Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1). Curr Biol. 1998. Dec 17-31;8(25):1366–1375. [DOI] [PubMed] [Google Scholar]
- 136.Castel P, Ellis H, Bago R, et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kalpha Inhibition. Cancer cell. 2016. Aug 8;30(2):229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Scortegagna M, Lau E, Zhang T, et al. PDK1 and SGK3 Contribute to the Growth of BRAF-Mutant Melanomas and Are Potential Therapeutic Targets. Cancer Res. 2015;75(7):1399–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell. 2017. Apr 20;169(3):381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sarbassov DD, Guertin DA, Ali SM, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005. Feb 18;307(5712):1098–1101. [DOI] [PubMed] [Google Scholar]
- 140.Moon Z, Wang Y, Aryan N, et al. Serine 396 of PDK1 is required for maximal PKB activation. Cell Signal. 2008. Nov;20(11):2038–2049. [DOI] [PubMed] [Google Scholar]
- 141.Bellacosa A, Chan TO, Ahmed NN, et al. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998. Jul 23;17(3):313–325. [DOI] [PubMed] [Google Scholar]
- 142.Frias MA, Thoreen CC, Jaffe JD, et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006. Sep 19;16(18):1865–1870. [DOI] [PubMed] [Google Scholar]
- 143.Fan Q, Wang Q, Cai R, et al. The ubiquitin system: orchestrating cellular signals in non-small-cell lung cancer. Cell Mol Biol Lett. 2020;25(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gonzalez E, McGraw TE. Insulin-modulated Akt subcellular localization determines Akt isoform-specific signaling. Proc Natl Acad Sci U S A. 2009. Apr 28;106(17):7004–7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nguyen le XT, Mitchell BS. Akt activation enhances ribosomal RNA synthesis through casein kinase II and TIF-IA. Proc Natl Acad Sci U S A. 2013. Dec 17;110(51):20681–20686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen K, Jiao X, Di Rocco A, et al. Endogenous Cyclin D1 Promotes the Rate of Onset and Magnitude of Mitogenic Signaling via Akt1 Ser473 Phosphorylation. Cell Rep. 2020. Sep 15;32(11):108151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cristofano AD, Pesce B, Cordon-Cardo C, et al. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998. Aug 01;19(4):348–355. [DOI] [PubMed] [Google Scholar]
- 148.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012. May 01;13(5):283–296. [DOI] [PubMed] [Google Scholar]
- 149.Nguyen KT, Tajmir P, Lin CH, et al. Essential role of Pten in body size determination and pancreatic beta-cell homeostasis in vivo. Mol Cell Biol. 2006. Jun;26(12):4511–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Myers MP, Pass I, Batty IH, et al. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A. 1998. Nov 10;95(23):13513–13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998. May 29;273(22):13375–13378. [DOI] [PubMed] [Google Scholar]
- 152.Al-Khouri AM, Ma Y, Togo SH, et al. Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J Biol Chem. 2005. Oct 21;280(42):35195–35202. [DOI] [PubMed] [Google Scholar]
- 153.Lee YR, Chen M, Lee JD, et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science. 2019. May 17;364(6441). DOI: 10.1126/science.aau0159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Papa A, Wan L, Bonora M, et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 2014;157(3):595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017. Mar 9;168(6):960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cai SL, Tee AR, Short JD, et al. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J Cell Biol. 2006. Apr 24;173(2):279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Garami A, Zwartkruis FJ, Nobukuni T, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003. Jun;11(6):1457–1466. [DOI] [PubMed] [Google Scholar]
- 158.Manning BD, Tee AR, Logsdon MN, et al. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002. Jul;10(1):151–162. [DOI] [PubMed] [Google Scholar]
- 159.Long X, Lin Y, Ortiz-Vega S, et al. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005. Apr 26;15(8):702–713. [DOI] [PubMed] [Google Scholar]
- 160.Hanrahan J, Blenis J. Rheb activation of mTOR and S6K1 signaling. Methods Enzymol. 2006;407:542–555. [DOI] [PubMed] [Google Scholar]
- 161.Gao X, Pan D. TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev. 2001. Jun 1;15(11):1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Roux PP, Ballif BA, Anjum R, et al. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A. 2004. Sep 14;101(37):13489–13494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ma L, Chen Z, Erdjument-Bromage H, et al. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005. Apr 22;121(2):179–193. [DOI] [PubMed] [Google Scholar]
- 164.Rosner M, Hofer K, Kubista M, et al. Cell size regulation by the human TSC tumor suppressor proteins depends on PI3K and FKBP38. Oncogene. 2003. Jul 31;22(31):4786–4798. [DOI] [PubMed] [Google Scholar]
- 165.Scott PH, Brunn GJ, Kohn AD, et al. Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1998. Jun 23;95(13):7772–7777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Guertin DA, Stevens DM, Thoreen CC, et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006. Dec;11(6):859–871. [DOI] [PubMed] [Google Scholar]
- 167.Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002. Jul 26;110(2):163–175. [DOI] [PubMed] [Google Scholar]
- 168.Jacinto E, Facchinetti V, Liu D, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006. Oct 6;127(1):125–137. [DOI] [PubMed] [Google Scholar]
- 169.Avruch J, Hara K, Lin Y, et al. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene. 2006. Oct 16;25(48):6361–6372. [DOI] [PubMed] [Google Scholar]
- 170.Holz MK, Blenis J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J Biol Chem. 2005. Jul 15;280(28):26089–26093. [DOI] [PubMed] [Google Scholar]
- 171.Holz MK, Ballif BA, Gygi SP, et al. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005. Nov 18;123(4):569–580. [DOI] [PubMed] [Google Scholar]
- 172.Sabatini DM, Erdjument-Bromage H, Lui M, et al. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994. July 15;78(1):35–43. [DOI] [PubMed] [Google Scholar]
- 173.Yang H, Jiang X, Li B, et al. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature. 2017;552(7685):368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004. Nov;6(11):1122–1128. [DOI] [PubMed] [Google Scholar]
- 175.Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004. Jul 27;14(14):1296–1302. [DOI] [PubMed] [Google Scholar]
- 176.Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006. Apr 21;22(2):159–168. [DOI] [PubMed] [Google Scholar]
- 177.Chou PC, Rajput S, Zhao X, et al. mTORC2 Is Involved in the Induction of RSK Phosphorylation by Serum or Nutrient Starvation. Cells. 2020. Jun 27;9(7):1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Li X, Gao T. mTORC2 phosphorylates protein kinase Cζ to regulate its stability and activity. EMBO Rep. 2014. Feb;15(2):191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Baffi TR, Lordén G, Wozniak JM, et al. mTORC2 controls the activity of PKC and Akt by phosphorylating a conserved TOR interaction motif. Sci Signal. 2021;14(678):eabe4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Briaud I, Dickson LM, Lingohr MK, et al. Insulin receptor substrate-2 proteasomal degradation mediated by a mammalian target of rapamycin (mTOR)-induced negative feedback down-regulates protein kinase B-mediated signaling pathway in beta-cells. J Biol Chem. 2005. Jan 21;280(3):2282–2293. [DOI] [PubMed] [Google Scholar]
- 181.Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011. Aug;1(3):248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008. Sep;118(9):3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kinkade CW, Castillo-Martin M, Puzio-Kuter A, et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008. Sep;118(9):3051–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sridharan S, Basu A. Distinct Roles of mTOR Targets S6K1 and S6K2 in Breast Cancer. Int J Mol Sci. 2020;21(4):1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Shima H, Pende M, Chen Y, et al. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 1998. Nov 16;17(22):6649–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gentilella A, Kozma SC, Thomas G. A liaison between mTOR signaling, ribosome biogenesis and cancer. Biochim Biophys Acta. 2015. Jul;1849(7):812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020. Apr 01;21(4):183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ben-Hur V, Denichenko P, Siegfried Z, et al. S6K1 alternative splicing modulates its oncogenic activity and regulates mTORC1. Cell Rep. 2013. Jan 31;3(1):103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ben-Sahra I, Manning BD. mTORC1 signaling and the metabolic control of cell growth. Curr Opin Cell Biol. 2017. Apr;45:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Szwed A, Kim E, Jacinto E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol Rev. 2021. Jul 1;101(3):1371–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 2009. Nov;29(21):5657–5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Liu P, Gan W, Inuzuka H, et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat Cell Biol. 2013. Nov;15(11):1340–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Julien L-A, Carriere A, Moreau J, et al. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30(4):908–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Chauvin C, Koka V, Nouschi A, et al. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene. 2014. Jan 23;33(4):474–483. [DOI] [PubMed] [Google Scholar]
- 195.Rosner M, Hengstschlager M. Nucleocytoplasmic localization of p70 S6K1, but not of its isoforms p85 and p31, is regulated by TSC2/mTOR. Oncogene. 2011. Nov 3;30(44):4509–4522. [DOI] [PubMed] [Google Scholar]
- 196.Martineau Y, Azar R, Bousquet C, et al. Anti-oncogenic potential of the eIF4E-binding proteins. Oncogene. 2013. Feb 7;32(6):671–677. [DOI] [PubMed] [Google Scholar]
- 197.Muller D, Lasfargues C, El Khawand S, et al. 4E-BP restrains eIF4E phosphorylation. Translation (Austin). 2013;1(2):e25819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Mendez R, Myers MG Jr., White MF, et al. Stimulation of protein synthesis, eukaryotic translation initiation factor 4E phosphorylation, and PHAS-I phosphorylation by insulin requires insulin receptor substrate 1 and phosphatidylinositol 3-kinase. Mol Cell Biol. 1996. Jun;16(6):2857–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiology (Bethesda). 2006. Oct;21:362–369. [DOI] [PubMed] [Google Scholar]
- 200.Sukarieh R, Sonenberg N, Pelletier J. The eIF4E-binding proteins are modifiers of cytoplasmic eIF4E relocalization during the heat shock response. Am J Physiol Cell Physiol. 2009. May;296(5):C1207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Culjkovic-Kraljacic B, Skrabanek L, Revuelta MV, et al. The eukaryotic translation initiation factor eIF4E elevates steady-state m(7)G capping of coding and noncoding transcripts. Proc Natl Acad Sci U S A. 2020. Oct 27;117(43):26773–26783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Llanos S, Garcia-Pedrero JM, Morgado-Palacin L, et al. Stabilization of p21 by mTORC1/4E-BP1 predicts clinical outcome of head and neck cancers. Nat Commun. 2016. Feb 2;7(1):10438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Jiang H, Coleman J, Miskimins R, et al. Expression of constitutively active 4EBP-1 enhances p27Kip1 expression and inhibits proliferation of MCF7 breast cancer cells. Cancer Cell Int. 2003. Feb 18;3(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tsukiyama-Kohara K, Poulin F, Kohara M, et al. Adipose tissue reduction in mice lacking the translational inhibitor 4E-BP1. Nat Med. 2001. Oct 01;7(10):1128–1132. [DOI] [PubMed] [Google Scholar]
- 205.Teleman AA, Chen Y-W, Cohen SM. 4E-BP functions as a metabolic brake used under stress conditions but not during normal growth. Genes Dev. 2005;19(16):1844–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Maurer U, Preiss F, Brauns-Schubert P, et al. GSK-3 – at the crossroads of cell death and survival. J Cell Sci. 2014;127(7):1369–1378. [DOI] [PubMed] [Google Scholar]
- 207.Mora A, Sakamoto K, McManus EJ, et al. Role of the PDK1-PKB-GSK3 pathway in regulating glycogen synthase and glucose uptake in the heart. FEBS Lett. 2005. Jul 4;579(17):3632–3638. [DOI] [PubMed] [Google Scholar]
- 208.Shaw M, Cohen P, Alessi DR. Further evidence that the inhibition of glycogen synthase kinase-3β by IGF-1 is mediated by PDK1/PKB-induced phosphorylation of Ser-9 and not by dephosphorylation of Tyr-216. FEBS Lett. 1997;416(3):307–311. [DOI] [PubMed] [Google Scholar]
- 209.Welsh GI, Proud CG. Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem J. 1993;294(Pt 3):625–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sung CK, Choi WS, Scalia P. Insulin-stimulated glycogen synthesis in cultured hepatoma cells: differential effects of inhibitors of insulin signaling molecules. J Recept Signal Transduct Res. 1998. Jul-Nov;18(4–6):243–263. [DOI] [PubMed] [Google Scholar]
- 211.Welcker M, Orian A, Jin J, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101(24):9085–9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Diehl JA, Cheng M, Roussel MF, et al. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998. Nov 15;12(22):3499–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Goode N, Hughes K, Woodgett JR, et al. Differential regulation of glycogen synthase kinase-3 beta by protein kinase C isotypes. J Biol Chem. 1992. Aug 25;267(24):16878–16882. [PubMed] [Google Scholar]
- 214.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004. Feb;29(2):95–102. [DOI] [PubMed] [Google Scholar]
- 215.Tanabe K, Liu Z, Patel S, et al. Genetic deficiency of glycogen synthase kinase-3beta corrects diabetes in mouse models of insulin resistance. PLoS Biol. 2008. Feb;6(2):e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Fernández-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer. 2011;2(3):344–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Prior IA, Hood FE, Hartley JL. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020;80(14):2969–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kasid A, Lippman ME. Estrogen and oncogene mediated growth regulation of human breast cancer cells. J Steroid Biochem. 1987;27(1–3):465–470. [DOI] [PubMed] [Google Scholar]
- 219.Sell C, Dumenil G, Deveaud C, et al. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol Cell Biol. 1994. Jun;14(6):3604–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Harvey JJ. An Unidentified Virus Which Causes the Rapid Production of Tumours in Mice. Nature. 1964. Dec 12;204(4963):1104–1105. [DOI] [PubMed] [Google Scholar]
- 221.Hancock JF, Magee AI, Childs JE, et al. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989. Jun 30;57(7):1167–1177. [DOI] [PubMed] [Google Scholar]
- 222.Hancock JF, Cadwallader K, Paterson H, et al. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 1991. Dec;10(13):4033–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Hancock JF, Cadwallader K, Marshall CJ. Methylation and proteolysis are essential for efficient membrane binding of prenylated p21K-ras(B). EMBO J. 1991. Mar;10(3):641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007. May 14;26(22):3113–3121. [DOI] [PubMed] [Google Scholar]
- 225.Arvidsson AK, Rupp E, Nånberg E, et al. Tyr-716 in the platelet-derived growth factor beta-receptor kinase insert is involved in GRB2 binding and Ras activation. Mol Cell Biol. 1994;14(10):6715–6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kovalski JR, Bhaduri A, Zehnder AM, et al. The Functional Proximal Proteome of Oncogenic Ras Includes mTORC2. Mol Cell. 2019. Feb 21;73(4):830–844 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Gillies TE, Pargett M, Silva JM, et al. Oncogenic mutant RAS signaling activity is rescaled by the ERK/MAPK pathway. Mol Syst Biol. 2020. Oct;16(10):e9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Nakamura K, Ichise H, Nakao K, et al. Partial functional overlap of the three ras genes in mouse embryonic development. Oncogene. 2008. May 8;27(21):2961–2968. [DOI] [PubMed] [Google Scholar]
- 229.Johnson L, Greenbaum D, Cichowski K, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997. Oct 1;11(19):2468–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Niault TS, Baccarini M. Targets of Raf in tumorigenesis. Carcinogenesis. 2010. Jul;31(7):1165–1174. [DOI] [PubMed] [Google Scholar]
- 231.Tran TH, Chan AH, Young LC, et al. KRAS interaction with RAF1 RAS-binding domain and cysteine-rich domain provides insights into RAS-mediated RAF activation. Nat Commun. 2021. Feb 19;12(1):1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Hmitou I, Druillennec S, Valluet A, et al. Differential regulation of B-raf isoforms by phosphorylation and autoinhibitory mechanisms. Mol Cell Biol. 2007. Jan;27(1):31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Galabova-Kovacs G, Kolbus A, Matzen D, et al. ERK and beyond: insights from B-Raf and Raf-1 conditional knockouts. Cell Cycle. 2006. Jul;5(14):1514–1518. [DOI] [PubMed] [Google Scholar]
- 234.Martinez Fiesco JA, Durrant DE, Morrison DK, et al. Structural insights into the BRAF monomer-to-dimer transition mediated by RAS binding. Nat Commun. 2022. Jan 25;13(1):486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Wojnowski L, Stancato LF, Zimmer AM, et al. Craf-1 protein kinase is essential for mouse development. Mech Dev. 1998. Aug;76(1–2):141–149. [DOI] [PubMed] [Google Scholar]
- 236.Hüser M, Luckett J, Chiloeches A, et al. MEK kinase activity is not necessary for Raf-1 function. EMBO J. 2001;20(8):1940–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Mercer K, Giblett S, Oakden A, et al. A-Raf and Raf-1 work together to influence transient ERK phosphorylation and Gl/S cell cycle progression. Oncogene. 2005. Aug 4;24(33):5207–5217. [DOI] [PubMed] [Google Scholar]
- 238.Venkatanarayan A, Liang J, Yen I, et al. CRAF dimerization with ARAF regulates KRAS-driven tumor growth. Cell Rep. 2022. Feb 08;38(6):110351. [DOI] [PubMed] [Google Scholar]
- 239.Eblen ST, Slack-Davis JK, Tarcsafalvi A, et al. Mitogen-activated protein kinase feedback phosphorylation regulates MEK1 complex formation and activation during cellular adhesion. Mol Cell Biol. 2004. Mar;24(6):2308–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Brady SC, Coleman ML, Munro J, et al. Sprouty2 association with B-Raf is regulated by phosphorylation and kinase conformation. Cancer Res. 2009. Sep 1;69(17):6773–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Pratilas CA, Taylor BS, Ye Q, et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A. 2009. Mar 17;106(11):4519–4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Scalia P, Williams SJ, Ventura E, et al. The Onco-genomic Landscape of Malignant Melanoma: The Tumor Microenvironment comes of Age. J Cancer Res Clin Oncol. 2020;3(1):1–8. [Google Scholar]
- 243.Alessi DR, Saito Y, Campbell DG, et al. Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J. 1994;13(7):1610–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Schaeffer HJ, Catling AD, Eblen ST, et al. MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science. 1998. Sep 11;281(5383):1668–1671. [DOI] [PubMed] [Google Scholar]
- 245.Teis D, Wunderlich W, Huber LA. Localization of the MP1-MAPK scaffold complex to endosomes is mediated by p14 and required for signal transduction. Dev Cell. 2002. Dec;3(6):803–814. [DOI] [PubMed] [Google Scholar]
- 246.Procaccia S, Ordan M, Cohen I, et al. Direct binding of MEK1 and MEK2 to AKT induces Foxo1 phosphorylation, cellular migration and metastasis. Sci Rep. 2017. Feb 22;7(1):43078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Giroux S, Tremblay M, Bernard D, et al. Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr Biol. 1999. Apr 8;9(7):369–372. [DOI] [PubMed] [Google Scholar]
- 248.Bélanger L-F, Roy S, Tremblay M, et al. Mek2 is dispensable for mouse growth and development. Mol Cell Biol. 2003;23(14):4778–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Sturgill TW, Ray LB, Erikson E, et al. Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature. 1988. Aug 25;334(6184):715–718. [DOI] [PubMed] [Google Scholar]
- 250.Yamamoto T, Ebisuya M, Ashida F, et al. Continuous ERK activation downregulates antiproliferative genes throughout G1 phase to allow cell-cycle progression. Curr Biol. 2006. Jun 20;16(12):1171–1182. [DOI] [PubMed] [Google Scholar]
- 251.Eblen ST. Extracellular-Regulated Kinases: Signaling From Ras to ERK Substrates to Control Biological Outcomes. Adv Cancer Res. 2018;138:99–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Schmidt M, Fernandez de Mattos S, van der Horst A, et al. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol. 2002. Nov;22(22):7842–7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Dijkers PF, Medema RH, Pals C, et al. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol Cell Biol. 2000. Dec;20(24):9138–9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Yang JY, Zong CS, Xia W, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008. Feb;10(2):138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Brunet A, Pages G, Pouyssegur J. Constitutively active mutants of MAP kinase kinase (MEK1) induce growth factor-relaxation and oncogenicity when expressed in fibroblasts. Oncogene. 1994. Nov;9(11):3379–3387. [PubMed] [Google Scholar]
- 256.Mansour SJ, Matten WT, Hermann AS, et al. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994. Aug 12;265(5174):966–970. [DOI] [PubMed] [Google Scholar]
- 257.Brummer T, Naegele H, Reth M, et al. Identification of novel ERK-mediated feedback phosphorylation sites at the C-terminus of B-Raf. Oncogene. 2003. Dec 4;22(55):8823–8834. [DOI] [PubMed] [Google Scholar]
- 258.Ritt DA, Monson DM, Specht SI, et al. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol Cell Biol. 2010. Feb;30(3):806–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Rozakis-Adcock M, van der Geer P, Mbamalu G, et al. MAP kinase phosphorylation of mSos1 promotes dissociation of mSos1-Shc and mSos1-EGF receptor complexes. Oncogene. 1995. Oct 5;11(7):1417–1426. [PubMed] [Google Scholar]
- 260.Fritsche L, Neukamm SS, Lehmann R, et al. Insulin-induced serine phosphorylation of IRS-2 via ERK1/2 and mTOR: studies on the function of Ser675 and Ser907. Am J Physiol Endocrinol Metab. 2011. May;300(5):E824–36. [DOI] [PubMed] [Google Scholar]
- 261.Furuta H, Yoshihara H, Fukushima T, et al. IRS-2 deubiquitination by USP9X maintains anchorage-independent cell growth via Erk1/2 activation in prostate carcinoma cell line. Oncotarget. 2018. Sep 21;9(74):33871–33883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Saba-El-Leil MK, Vella FDJ, Vernay B, et al. An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep. 2003;4(10):964–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Pages G, Guerin S, Grall D, et al. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science. 1999. Nov 12;286(5443):1374–1377. [DOI] [PubMed] [Google Scholar]
- 264.Bruning JC, Gillette JA, Zhao Y, et al. Ribosomal subunit kinase-2 is required for growth factor-stimulated transcription of the c-Fos gene. Proc Natl Acad Sci U S A. 2000. Mar 14;97(6):2462–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Erikson RL. Regulation of cell proliferation by oncogene products and growth factors. Somat Cell Mol Genet. 1987. Jul;13(4):459–461. [DOI] [PubMed] [Google Scholar]
- 266.Lara R, Seckl MJ, Pardo OE. The p90 RSK family members: common functions and isoform specificity. Cancer Res. 2013. Sep 1;73(17):5301–5308. [DOI] [PubMed] [Google Scholar]
- 267.Fujita N, Sato S, Tsuruo T. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2003. Dec 5;278(49):49254–49260. [DOI] [PubMed] [Google Scholar]
- 268.Chen RH, Abate C, Blenis J. Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc Natl Acad Sci U S A. 1993. Dec 1;90(23):10952–10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Torres MA, Eldar-Finkelman H, Krebs EG, et al. Regulation of ribosomal S6 protein kinase-p90(rsk), glycogen synthase kinase 3, and beta-catenin in early Xenopus development. Mol Cell Biol. 1999. Feb;19(2):1427–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Douville E, Downward J. EGF induced SOS phosphorylation in PC12 cells involves P90 RSK-2. Oncogene. 1997. Jul 24;15(4):373–383. [DOI] [PubMed] [Google Scholar]
- 271.De S, Campbell C, Venkitaraman AR, et al. Pulsatile MAPK Signaling Modulates p53 Activity to Control Cell Fate Decisions at the G2 Checkpoint for DNA Damage. Cell Rep. 2020;30(7):2083–2093.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Albeck JG, Mills GB, Brugge JS. Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol Cell. 2013. Jan 24;49(2):249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Dufresne SD, Bjorbaek C, El-Haschimi K, et al. Altered extracellular signal-regulated kinase signaling and glycogen metabolism in skeletal muscle from p90 ribosomal S6 kinase 2 knockout mice. Mol Cell Biol. 2001. Jan;21(1):81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Kops GJ, Medema RH, Glassford J, et al. Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol Cell Biol. 2002. Apr;22(7):2025–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Puig O, Tjian R. Transcriptional feedback control of insulin receptor by dFOXO/FOXO1. Genes Dev. 2005. Oct 15;19(20):2435–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Essaghir A, Dif N, Marbehant CY, et al. The transcription of FOXO genes is stimulated by FOXO3 and repressed by growth factors. J Biol Chem. 2009;284(16):10334–10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Sang T, Cao Q, Wang Y, et al. Overexpression or silencing of FOXO3a affects proliferation of endothelial progenitor cells and expression of cell cycle regulatory proteins. PloS one. 2014;9(8):e101703–e101703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Demontis F, Perrimon N. Integration of Insulin receptor/Foxo signaling and dMyc activity during muscle growth regulates body size in Drosophila. Development. 2009. Mar;136(6):983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Junger MA, Rintelen F, Stocker H, et al. The Drosophila forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J Biol. 2003;2(3):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Murphy LO, MacKeigan JP, Blenis J. A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol Cell Biol. 2004. Jan;24(1):144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995. Jul 14;270(28):16483–16486. [DOI] [PubMed] [Google Scholar]
- 282.Koo JH, Plouffe SW, Meng Z, et al. Induction of AP-1 by YAP/TAZ contributes to cell proliferation and organ growth. Genes Dev. 2020. Jan 1;34(1–2):72–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Berberich SJ, Cole MD. Casein kinase II inhibits the DNA-binding activity of Max homodimers but not Myc/Max heterodimers. Genes Dev. 1992. Feb;6(2):166–176. [DOI] [PubMed] [Google Scholar]
- 284.Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci U S A. 2008. May 6;105(18):6584–6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Garcia-Gutierrez L, Delgado MD, Leon J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes (Basel). 2019. Mar 22;10(3):244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Vlach J, Hennecke S, Alevizopoulos K, et al. Growth arrest by the cyclin-dependent kinase inhibitor p27Kip1 is abrogated by c-Myc. EMBO J. 1996;15(23):6595–6604. [PMC free article] [PubMed] [Google Scholar]
- 287.Zou Z, Chen J, Liu A, et al. mTORC2 promotes cell survival through c-Myc-dependent up-regulation of E2F1. J Cell Biol. 2015. Oct 12;211(1):105–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci U S A. 1995. Dec 19;92(26):12146–12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Chandramohan V, Mineva ND, Burke B, et al. c-Myc represses FOXO3a-mediated transcription of the gene encoding the p27(Kip1) cyclin dependent kinase inhibitor. J Cell Biochem. 2008. Aug 15;104(6):2091–2106. [DOI] [PubMed] [Google Scholar]
- 290.Morrish F, Neretti N, Sedivy JM, et al. The oncogene c-Myc coordinates regulation of metabolic networks to enable rapid cell cycle entry. Cell Cycle. 2008. Apr 15;7(8):1054–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Grandori C, Gomez-Roman N, Felton-Edkins ZA, et al. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol. 2005. Mar;7(3):311–318. [DOI] [PubMed] [Google Scholar]
- 292.Krek W, Xu G, Livingston DM. Cyclin A-kinase regulation of E2F-1 DNA binding function underlies suppression of an S phase checkpoint. Cell. 1995. Dec 29;83(7):1149–1158. [DOI] [PubMed] [Google Scholar]
- 293.Johnson DG, Schneider-Broussard R. Role of E2F in cell cycle control and cancer. Front Biosci. 1998. Apr 27;3(4):d447–8. [DOI] [PubMed] [Google Scholar]
- 294.Polager S, Ginsberg D. p53 and E2f: partners in life and death. Nat Rev Cancer. 2009. Oct;9(10):738–748. [DOI] [PubMed] [Google Scholar]
- 295.Lin SY, Black AR, Kostic D, et al. Cell cycle-regulated association of E2F1 and Sp1 is related to their functional interaction. Mol Cell Biol. 1996. Apr;16(4):1668–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Rosner HI, Sorensen CS. E2F transcription regulation: an orphan cyclin enters the stage. EMBO J. 2019. Oct 15;38(20):e103421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Bae SK, Bae MH, Ahn MY, et al. Egr-1 mediates transcriptional activation of IGF-II gene in response to hypoxia. Cancer Res. 1999. Dec 1;59(23):5989–5994. [PubMed] [Google Scholar]
- 298.Ma Y, Cheng Q, Ren Z, et al. Induction of IGF-1R expression by EGR-1 facilitates the growth of prostate cancer cells. Cancer Lett. 2012. Apr 28;317(2):150–156. [DOI] [PubMed] [Google Scholar]
- 299.Baron V, Adamson ED, Calogero A, et al. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFbeta1, PTEN, p53, and fibronectin. Cancer Gene Ther. 2006. Feb;13(2):115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Yu J, Zhang SS, Saito K, et al. PTEN regulation by Akt-EGR1-ARF-PTEN axis. EMBO J. 2009. Jan 7;28(1):21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Hanke S, Mann M. The phosphotyrosine interactome of the insulin receptor family and its substrates IRS-1 and IRS-2. Mol Cell Proteomics. 2009. Mar;8(3):519–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Nurk S, Koren S, Rhie A, et al. The complete sequence of a human genome. Science. 2022. Apr;376(6588):44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Pelletier J, Thomas G, Volarevic S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat Rev Cancer. 2018. Jan;18(1):51–63. [DOI] [PubMed] [Google Scholar]
- 304.Klein J, Grummt I. Cell cycle-dependent regulation of RNA polymerase I transcription: the nucleolar transcription factor UBF is inactive in mitosis and early G1. Proc Natl Acad Sci U S A. 1999. May 25;96(11):6096–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.White RJ, Gottlieb TM, Downes CS, et al. Cell cycle regulation of RNA polymerase III transcription. Mol Cell Biol. 1995. Dec;15(12):6653–6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Surmacz E, Kaczmarek L, Ronning O, et al. Activation of the ribosomal DNA promoter in cells exposed to insulinlike growth factor I. Mol Cell Biol. 1987. Feb;7(2):657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Michels AA, Robitaille AM, Buczynski-Ruchonnet D, et al. mTORC1 directly phosphorylates and regulates human MAF1. Mol Cell Biol. 2010. Aug;30(15):3749–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Rideout EJ, Marshall L, Grewal SS. Drosophila RNA polymerase III repressor Maf1 controls body size and developmental timing by modulating tRNAiMet synthesis and systemic insulin signaling. Proc Natl Acad Sci U S A. 2012. Jan 24;109(4):1139–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.DePhilip RM, Rudert WA, Lieberman I. Preferential stimulation of ribosomal protein synthesis by insulin and in the absence of ribosomal and messenger ribonucleic acid formation. Biochemistry. 1980. Apr 15;19(8):1662–1669. [DOI] [PubMed] [Google Scholar]
- 310.Hammond ML, Bowman LH. Insulin stimulates the translation of ribosomal proteins and the transcription of rDNA in mouse myoblasts. J Biol Chem. 1988. Nov 25;263(33):17785–17791. [PubMed] [Google Scholar]
- 311.Scalia, P , Comai, L. Regulation of Polymerase-I Transcription by Insulin-Like Growth Factor-II. Third International Symposium on Polymerase I & III. Craig SP ed. (Gene Expression (Cognizant Comm)). June 5-9. Asilomar Conference Grounds, Pacific Grove, Monterrey CA. 2002; Vol 10. 263–270. [Google Scholar]
- 312.Lin CY, Navarro S, Reddy S, et al. CK2-mediated stimulation of Pol I transcription by stabilization of UBF-SL1 interaction. Nucleic Acids Res. 2006;34(17):4752–4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Bierhoff H, Dundr M, Michels AA, et al. Phosphorylation by casein kinase 2 facilitates rRNA gene transcription by promoting dissociation of TIF-IA from elongating RNA polymerase I. Mol Cell Biol. 2008. Aug;28(16):4988–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Zhao J, Yuan X, Frodin M, et al. ERK-dependent phosphorylation of the transcription initiation factor TIF-IA is required for RNA polymerase I transcription and cell growth. Mol Cell. 2003. Feb;11(2):405–413. [DOI] [PubMed] [Google Scholar]
- 315.Zhai W, Comai L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol Cell Biol. 2000. Aug;20(16):5930–5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Hisatake K, Hasegawa S, Takada R, et al. The p250 subunit of native TATA box-binding factor TFIID is the cell-cycle regulatory protein CCG1. Nature. 1993. Mar 01;362(6416):179–181. [DOI] [PubMed] [Google Scholar]
- 317.Lin CY, Tuan J, Scalia P, et al. The cell cycle regulatory factor TAF1 stimulates ribosomal DNA transcription by binding to the activator UBF. Curr Biol. 2002. Dec 23;12(24):2142–2146. [DOI] [PubMed] [Google Scholar]
- 318.Neufeld TP, Edgar BA. Connections between growth and the cell cycle. Curr Opin Cell Biol. 1998. Dec;10(6):784–790. [DOI] [PubMed] [Google Scholar]
- 319.Belfiore A, Malaguarnera R, Vella V, et al. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr Rev. 2017. Oct 1;38(5):379–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Baker J, Liu JP, Robertson EJ, et al. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993. Oct 8;75(1):73–82. [PubMed] [Google Scholar]
- 321.Zhang H, Liu J, Li CR, et al. Deletion of Drosophila insulin-like peptides causes growth defects and metabolic abnormalities. Proc Natl Acad Sci U S A. 2009. Nov 17;106(46):19617–19622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Oldham S, Stocker H, Laffargue M, et al. The Drosophila insulin/IGF receptor controls growth and size by modulating PtdInsP3 levels. Development. 2002;129(17):4103–4109. [DOI] [PubMed] [Google Scholar]
- 323.Accili D, Drago J, Lee EJ, et al. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat Genet. 1996. Jan;12(1):106–109. [DOI] [PubMed] [Google Scholar]
- 324.Brogiolo W, Stocker H, Ikeya T, et al. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr Biol. 2001. Feb 20;11(4):213–221. [DOI] [PubMed] [Google Scholar]
- 325.Sun XJ, Wang L-M, Zhang Y, et al. Role of IRS-2 in insulin and cytokine signalling. Nature. 1995. Sep 01;377(6545):173–177. [DOI] [PubMed] [Google Scholar]
- 326.Bohni R, Riesgo-Escovar J, Oldham S, et al. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell. 1999. Jun 25;97(7):865–875. [DOI] [PubMed] [Google Scholar]
- 327.Leevers SJ, Weinkove D, MacDougall LK, et al. The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J. 1996;15(23):6584–6594. [PMC free article] [PubMed] [Google Scholar]
- 328.Dummler B, Tschopp O, Hynx D, et al. Life with a single isoform of Akt: mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol Cell Biol. 2006;26(21):8042–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Chen WS, Xu PZ, Gottlob K, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15(17):2203–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Verdu J, Buratovich MA, Wilder EL, et al. Cell-autonomous regulation of cell and organ growth in Drosophila by Akt/PKB. Nat Cell Biol. 1999. Dec 01;1(8):500–506. [DOI] [PubMed] [Google Scholar]
- 331.Goberdhan DC, Paricio N, Goodman EC, et al. Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3-kinase signaling pathway. Genes Dev. 1999;13(24):3244–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Potter CJ, Huang H, Xu T. Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell. 2001. May 4;105(3):357–368. [DOI] [PubMed] [Google Scholar]
- 333.Zhang H, Stallock JP, Ng JC, et al. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 2000;14(21):2712–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Doble BW, Patel S, Wood GA, et al. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell. 2007. Jun;12(6):957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Hoeflich KP, Luo J, Rubie EA, et al. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature. 2000. Jul 01;406(6791):86–90. [DOI] [PubMed] [Google Scholar]
- 336.Montagne J, Stewart MJ, Stocker H, et al. Drosophila S6 Kinase: A Regulator of Cell Size. Science. 1999;285(5436):2126–2129. [DOI] [PubMed] [Google Scholar]
- 337.Hosaka T, Biggs WH 3rd, Tieu D, et al. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci U S A. 2004;101(9):2975–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.