

Abstract
Traditionally, the monolayer (two-dimensional) cell cultures are used for initial evaluation of the effectiveness of anticancer drugs. In particular, these experiments provide the IC50 curves that determine drug concentration that can inhibit growth of a tumor colony by half when compared to the cells grown with no exposure to the drug. Low IC50 value means that the drug is effective at low concentrations, and thus will show lower systemic toxicity when administered to the patient. However, in these experiments cells are grown in a monolayer, all well exposed to the drug, while in vivo tumors expand as three-dimensional multicellular masses, where inner cells have a limited access to the drug. Therefore, we performed computational studies to compare the IC50 curves for cells grown as a two-dimensional monolayer and a cross section through a three-dimensional spheroid. Our results identified conditions (drug diffusivity, drug action mechanisms and cell proliferation capabilities) under which these IC50 curves differ significantly. This will help experimentalists to better determine drug dosage for future in vivo experiments and clinical trials.
Keywords: Drugs efficacy, IC50 values, Monolayer versus spheroid cell cultures
Mathematics Subject Classification: 92-08, 92B05, 92C50
1. Introduction
In general, drug–dose response curves are used to measure and analyze the relationship between a drug’s inhibitory capabilities associated with its respective concentrations. Inhibitory concentration curves denoted ICx are dose response curves that allow for determining the drug concentration required to reduce a population of viable cells by x%, when compared to the cells grown with no exposure to the drug (Chou 2006). This change in cell population size could be a result of increased cell death or suppressed cell proliferation. Drug discovery and pharmacology studies use the IC50 values to determine drug effectiveness (potency). Low IC50 value means that the drug is potent at low concentrations, and thus will show lower systemic toxicity when administered to the patient. The drug–dose response curves are also used to identify synergistic combination therapies and drug interactions mechanisms (Yu et al. 2015; Tallarida 2011; Chevereau and Bollenbach 2015).
However, the typical experiments to determine the IC50 curves are performed in two-dimensional (2D) monolayer cell cultures in Petri dishes (Fig. 1a). The cells are covered with a medium of uniform drug concentration and grown for 72 h which is a timeframe long enough for cells to divide 1–2 times and to observe drug effects without cells reaching confluence (Turner and Charlton 2005; Hafner et al. 2016). In contrast, in vivo tumors develop as three-dimensional (3D) masses of tightly packed tumor cells, and thus, their response to therapeutic interventions may be different than in 2D experiments. To test drug potency in vitro in a way to preserve geometry of typical in vivo tumors, the 3D cultures of multicellular spheroids were developed (Pampaloni et al. 2007; Laurent et al. 2013; Weiswald et al. 2015). In these experiments, the 3D spheroids are first formed either by proliferation from single seeded cells or by aggregation of individual cells seeded together (Weiswald et al. 2015; Rodrigues et al. 2018). The spheroids are then covered with a medium with a uniformly dissolved drug (Fig. 1b); similar like it is done in the 2D experiments. However, a significantly different culture geometry results in a limited access to the drug inside the spheroid, and thus, in a different overall response when compared to results from 2D cell cultures. While several studies addressed drug efficacy in 2D versus 3D experiments (Eichler et al. 2015; Imamura et al. 2015; Fontoura et al. 2020), each used a different seeding protocol and assessment method making result cross-examination difficult. Therefore, we designed computational models as in silico analogues of the 2D monolayer and a cross section through the 3D multicellular spheroid cell cultures (Fig. 1) to compare side-by-side how identical cells respond to identical drugs in these two settings starting with the same number of cells. To our knowledge, this is the first comprehensive computational comparison between drug efficacy simulated using analogues of 2D and 3D cell cultures.
Fig. 1.
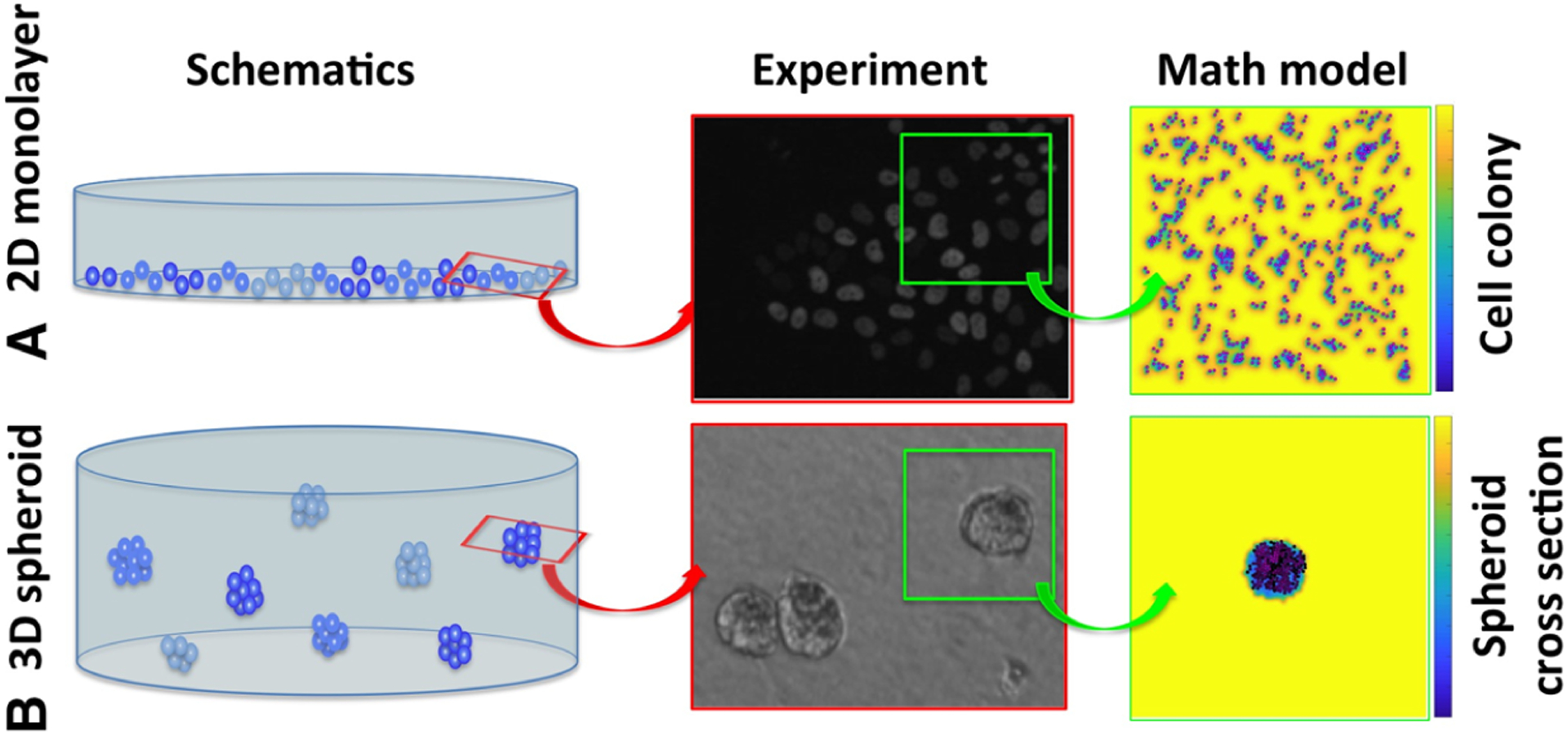
Schematics of 2D and 3D cell cultures and their mathematical analogues. a A schematics of the 2D monolayer cell culture, a corresponding fluorescent microscopy image of a part of the Petri dish, and a snapshot from our in silico analogue model of the 2D cell culture. b A schematics of the 3D multicellular spheroid culture, a corresponding bright field microscopy image of a part of the well plate, and a snapshot from our in silico analogue model of the cross section through the 3D spheroid. The background color represents drug concentration to illustrate local drug gradients from high concentration (yellow) to low concentration (blue). Experimental images courtesy of the Moffitt Analytic Microscopy Core
In this paper, we first describe a mathematical framework used to model the 2D monolayer cell culture and the cross section through a 3D multicellular spheroid culture (Sect. 2). Numerical implementation of these models is described in “Appendix A”. These two models are then used to test conditions (drug diffusivity, drug action mechanisms and cell proliferation capabilities) under which the IC50 values are either similar or significantly different between 2D and 3D cultures. The method of fitting the IC50 curves to simulated data is presented in Sect. 3. The analysis of results for cytotoxic drugs is presented in Sect. 4.1, and for anti-mitotic drugs in Sect. 4.2. We summarize our results with the discussion in Sect. 5.
2. The Mathematical Model
From a mathematical modeling perspective of tumor cell populations, the models can be classified into two types: continuous or discrete. Continuous models treat the population of tumor cells as a continuous density distribution, which usually is described by a system of ordinary or partial differential equations. On the other hand, in discrete models each cell in the population is represented by a discrete object (an agent) that follows a set of prescribed rules. Thus, these models are often referred as agent-based models (ABMs). Depending on the research question, the one or the other mathematical modeling approach is considered though both have their strengths and weaknesses. For a review on the comparison of the two types of models, see Schaller and Meyer-Hermann (2006). A continuous model is relatively easier to analyze analytically and computationally, but it fails to capture the individual cell-to-cell interactions and cellular heterogeneity. On the other hand, the ABM models focus on interactions between the individual cells, but are not easy to analyze analytically, if not impossible, and the computation time increases significantly with the number of cells involved. Recently, researchers have been focusing on developing hybrid discrete-continuous models which combine both approaches with the goal of maximizing their advantages and minimizing their drawbacks. More information on the hybrid discrete-continuous models can be found in Chamseddine and Rejniak (2020).
Here, we use a hybrid discrete-continuous model in which tumor cells interact physically with one another and react to a drug dissolved in a surrounding medium. The cells are modeled as individual off-lattice agents, and drug concentration is described by the continuous partial differential equation. We previously used a similar mathematical framework to model in vivo tumors and the emergence of drug-induced resistance (Gevertz et al. 2015; Perez-Velazquez et al. 2016; Shah et al. 2016; Karolak et al. 2019a; Perez-Velazquez and Rejniak 2020). Here, we adjusted this framework to model in vitro cell cultures, both the 2D monolayer and the cross section through the 3D spheroid. In addition to different culture geometry, we also extended the previous work by considering different mechanisms of drug action, i.e., the cytotoxic and anti-mitotic drugs. Below, we provide all model equations and spatial setups of all performed computational experiments.
2.1. Discrete Description of Tumor Cells
Each cell in our model is treated as a separate entity characterized by several individually regulated properties. The position of kth cell is denoted by Xk(t), current cell age by , cell maturation age, which is the age when the cell is ready to divide, by , the number of nearby neighboring cells, , and a level of the drug accumulated inside the cell by . For simplicity, we assume that all cells have the same diameter RD. The state of the kth cell at time t is denoted by . The initial state of the kth cell is .
Note that the cell maturation age, , is the only cell feature that does not depend on time t, and it is assigned explicitly upon cell birth. However, to avoid cell synchronized divisions, we impose up to 15% of differences in the duration of the cell cycle (Tyson et al. 2012). Thus, we set
| (1) |
where Amat is the average maturation age for the whole cell population, and ω is randomly chosen from a uniform distribution from [0, 2.5] h.
The cell Ci is considered to be a neighbor of cell Ck at time t, if it is located within the neighborhood radius Rneigh from cell Ck, that is:
| (2) |
We consider here the neighborhood radius equal to two cell diameters (Rneigh = 2RD) which accounts for two layers around the host cell (as in Perez-Velazquez et al. 2016; Karolak et al. 2019b). The initial number of neighboring cells, , depends on the initial cell configuration (see Sect. 2.3). As the cells divide, die or move around, the number of neighboring cells, , varies with time.
The cell spatial dynamics is modeled by Newton’s second law of motion:
| (3) |
where fk, i is the interaction force between two neighboring cells Ck and Ci, denotes the force against viscosity of the surrounding medium, and mk is the mass of the kth cell. Newton’s second law of motion has been used to address the dynamics of cell-to-cell interactions, see the review in Murray et al. (2009) and the references therein.
The cell interaction forces arise when two neighboring cells, Ck and Ci, come into too close contact. If the distance between the cells’ centers is smaller than the cell diameter RD, the repulsive Hooke’s forces fk, i and fi, k = − fk, i are exerted to preserve cells’ volumes:
| (4) |
where ℱ is the constant spring stiffness, and the spring resting length is equal to cell diameter RD. Since the cell can be exposed to interactions with multiple neighbors, the total force acting on the kth cell is the sum of all repulsive forces fk, 1+ fk, 2+···+ fk, M between the kth cell and its M neighbors.
Cell dynamics is thus governed by the equations of motion where the connecting springs are overdamped and system returns to equilibrium without oscillations. Hence,
| (5) |
The viscous force, , of the kth cell is modeled as proportional to its velocity. That is,
| (6) |
where ν denotes the media viscosity coefficient. Then, substituting (5) and (6) in (3), we obtain that the cell motion is determined by
| (7) |
Clearly, cell’s age progresses at the same rate as time progresses; hence,
| (8) |
This equation has an exact solution , where is the time at which the cell was born. However, we will keep the differential equation form for consistency with other equations in numerical implementation described in “Appendix A”.
When the kth cell reaches its maturation age, , it will divide unless it is over-crowded. The overcrowding means that the number of neighboring cells, , located within the neighborhood radius, Rneigh, exceeds the prescribed threshold 𝒩max. If the cell is overcrowded, its proliferation is suppressed until the space becomes available. Here, the overcrowding threshold was determined computationally to be equal to 10 cells.
Upon division of the kth cell, two daughter cells and are created instantaneously. They are placed symmetrically near the mother cell in the random direction θ chosen from a uniform discrete distribution [0,2π]. The locations of the daughter cells are defined by
| (9) |
Since they are placed at the distance smaller than cell diameter, the repulsive forces between daughter cells are activated. Furthermore, this may also result in daughter cell placement near other cells. Therefore, multiple repulsive forces will be applied to resolve potential cell overlap, and the cells will be pushed away until the whole cell cluster reaches an equilibrium configuration.
The age of each newly born daughter cell is initialized to zero,
| (10) |
The cell maturation age is inherited from its mother cell; however, a small noise term is added to avoid synchronization of the cell cycles,
| (11) |
where ωi is randomly chosen from a uniform distribution [0,2.5] h.
The level of drug accumulated by the mother cell is divided equally between the two daughter cells
| (12) |
The overall rate of change in drug accumulated by the kth cell is modeled in the most simple way as absorption at a constant rate ργ
| (13) |
This equations has an exact solution: , provided that drug concentrations at the locations visited by the cell are not lower that cell’s demand. Thus, special precautions will be taken in the numerical implementation described in “Appendix A” to account for these situations.
We model here two different mechanisms of drug action. In the case of a cytotoxic drug, the cell dies immediately after accumulation of a lethal dose of the drug γ max. In the case of an anti-mitotic drug, the cell dies when it attempts to divide (in the mitotic phase of the cell cycle) after accumulated drug exceeds the lethal dose γ max. The dead cells are removed from the system.
2.2. Continuous Description of Drug Kinetics
The change in drug concentration γ(x, t) at location x = (x, y) within the domain Ω, depends on drug diffusivity and on the uptake by the tumor cells located nearby. The partial differential equation (PDE) describing the spatiotemporal drug dynamics is given by:
| (14) |
where 𝒟γ is the drug diffusion coefficient, ργ is the cellular uptake rate, N is the total number of cells located in the neighborhood of radius Rγ defined by the indicator function χ:
| (15) |
We assume that there is no loss or gain of the drug along the domain boundaries ∂Ω. Hence, we impose Neumann-type boundary conditions
where x ∈ ∂Ω, where n is the inward pointing normal.
The drug is supplied only once, at the beginning of the simulation and its concentration in the extracellular space, that is in the space outside the cells (Ω \ ⋃ Ck), at time t = 0 is uniform,
2.3. Initial Cellular Configurations
Since our goal is to compare the IC50 values achieved in the computational analogues of the 2D and 3D cell cultures when the same cells are exposed to the same drug, we consider two different initial configurations that correspond to these laboratory experiments. In both cases, we start with the same initial number of cells (315) that was determined computationally to ensure that the cells will not grow to confluence during the simulated 72 h. This is consistent with laboratory experiments. We reproduced the cell monolayer culture (Fig. 2a) and the cross section though the cell spheroid culture (Fig. 2b), respectively.
Fig. 2.
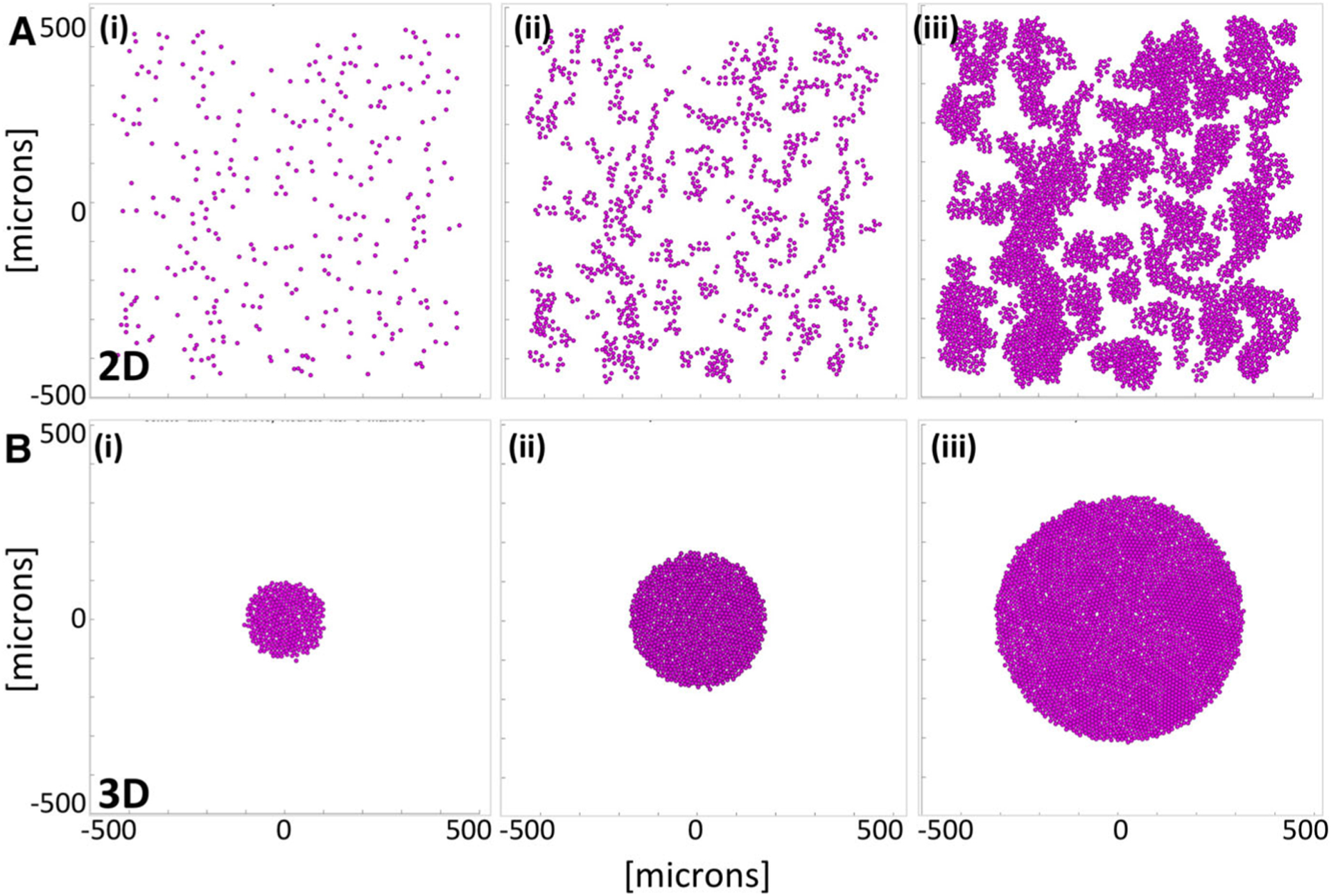
Tumor growth in 2D and 3D cell cultures with no drug (the control case). a A computational model of the monolayer cell culture with sparsely seeded initial 315 cells (i), the daughter cells divide and spread throughout the domain (ii) for 72 h of the simulated time reaching 4516 cells (iii). b A computational model of the cross section though the spheroid cell culture with initial cluster of 315 cells (i), the non-overcrowded cells divide and expand (ii) for 72 h of the simulated time reaching 3183 cells (iii)
Modeling the monolayer cell culture
The two-dimensional computational model is designed to reproduce the evolution of cells seeded sparsely in the domain filled with a dissolved drug. This is an analogue of a 2D cell monolayer culture in the Petri dish where all cells are exposed to the drug dissolved in the surrounding medium (Chou 2006; Yu et al. 2015). Each simulation starts with 315 cells that are randomly distributed within a domain. The cells are monitored for 72 h, and the final number of viable cells is recorded. During this computational experiment, the cells are allowed to divide, absorb the drug and die. Upon division, the daughter cells are placed randomly nearby the mother cell and the repulsive forces are applied between overlapping cells until the new stable configuration is achieved.
The cells could also become growth-arrested due to contact inhibition if their configuration reached confluence. Upon death, the cells are removed from the system which could create free space and initiate new cell divisions. Figure 2a(i)–(iii) shows three snapshots from a monolayer simulation with no drug.
Modeling a cross section of the multicellular spheroid culture
The two-dimensional computational model is designed to reproduce the central cross section of 3D multicellular spheroid culture. In these laboratory experiments, the cells are first grown in the dish until they form a packed sphere and then are embedded into the medium mixed with a drug (Adcock et al. 2015; Friedrich et al. 2009). We reproduce this experimental design by starting each simulation with 315 cells that formed a circular cell cluster. The cells are then monitored for 72 h and are allowed to proliferate, absorb the drug or die, as in the monolayer case. However, the difference in initial configurations between these two computational models results in distinct population dynamics. In the spheroid model, the cells inside the cluster are overcrowded and thus stop proliferating due to cellular contact inhibition. Figure 2b(i)–(iii) shows three snapshots from a multicellular spheroid simulation with no drug.
Drug initial distribution in the monolayer and spheroid models
The initial drug concentration γ(x, t0) = γ0 is uniform throughout the extracellular space outside the cells, that is in Ω \ ⋃Ck. Since in the monolayer cell culture model the cells are located sparsely inside the domain, drug concentration γ0 is imposed uniformly in the whole domain (and on each grid point in the numerical implementation). In contrast, in the spheroid experiments, the drug is initially present outside the 3D culture only. To mimic the lack of the drug inside the spheroid in numerical implementation, the grid points inside the spheroid are left with no drug and drug diffusion occurs at the border of the cell cluster in the medium. However, in both computational models, drug absorption by the cells leads to local depletion of the drug (see the last column in Fig. 1). All physical and computational model parameters are summarized in Table 1.
Table 1.
Physical and computational model parameters
| Cellular and microenvironmental parameters | ||
| Cell diameter | RD = 10 μm, cell radius R = 5μm | Shashni et al. (2018), NCI-60 Human Tumor Cell Lines Screen [Internet] (2015) |
| Spring stiffness | ℱ = 50 μg/μm · s2 | Baumgartner and Drenckhahn (2002), Rejniak and Dillon (2007) |
| Mass viscosity | v = 250 μg/μm · s | Kane et al. (2018) |
| Neighborhood radius | Rneigh = 2RD = 20 μm | Perez-Velazquez et al. (2016), Karolak et al. (2019b) |
| Overcrowding cell number | 𝒩max = 10 cells | estimated |
| Average maturation age | 𝒜mat = 18, 30, 50 h | Hafner et al. (2016), Mehrara et al. (2007) |
| Drug parameters | ||
| Diffusion coefficient | 𝒟γ = 1, 10−2, 10−4 μm2/s | Schmidt and Wittrup (2009) |
| Drug uptake radius | Rγ = 5 μm | Cell radius |
| Baseline drug concentration | = 1 mM | |
| Initial drug concentration | γ0 vary from 0 to 103 mM | Fallahi-Sichani et al. (2013) |
| Cellular uptake rate | ργ = 5 × 10−8 ng/μm3s | 0.1/s |
| Cellular death threshold | γmax = 2 × 10−6 ng/μm3 | 2 |
| Numerical parameters | ||
| Domain size | [−500, 500] μm × [−500, 500] μm | |
| Mesh width | Δx = Δy = 5 μm | Cell radius |
| Time step | Δt = 5 s | FTCS stability |
3. Fitting the Inhibitory Concentration (IC50) Curves
The inhibitory concentration curve (called also the drug response curve or the IC50 curve) is described by the following Hill equation (Gardner 2000; Levasseur et al. 1998):
| (16) |
where f0 is the control effect (the plateau observed for low drug concentrations), f∞ is the background effect (the plateau observed for large drug concentrations), β is the curve slope, and IC50 value is the curve inflection point at which the drug maximal effect (f0 − f∞) decreases by 50%. The curve slope β is a measure of variability in drug response—the steeper the slope is, the more homogeneous the drug response. The value of f∞ describes drug efficacy—the lower the f∞, the higher the beneficial effect (often denoted by Emax, maximal effect). The IC50 value is correlated with drug potency, i.e., the amount of drug necessary to produce the effect—the lower the IC50 value, the more potent the drug (Meyer et al. 2019). The relationship between drug potency, drug efficacy and the IC50 curve shape is shown in Fig. 3.
Fig. 3.
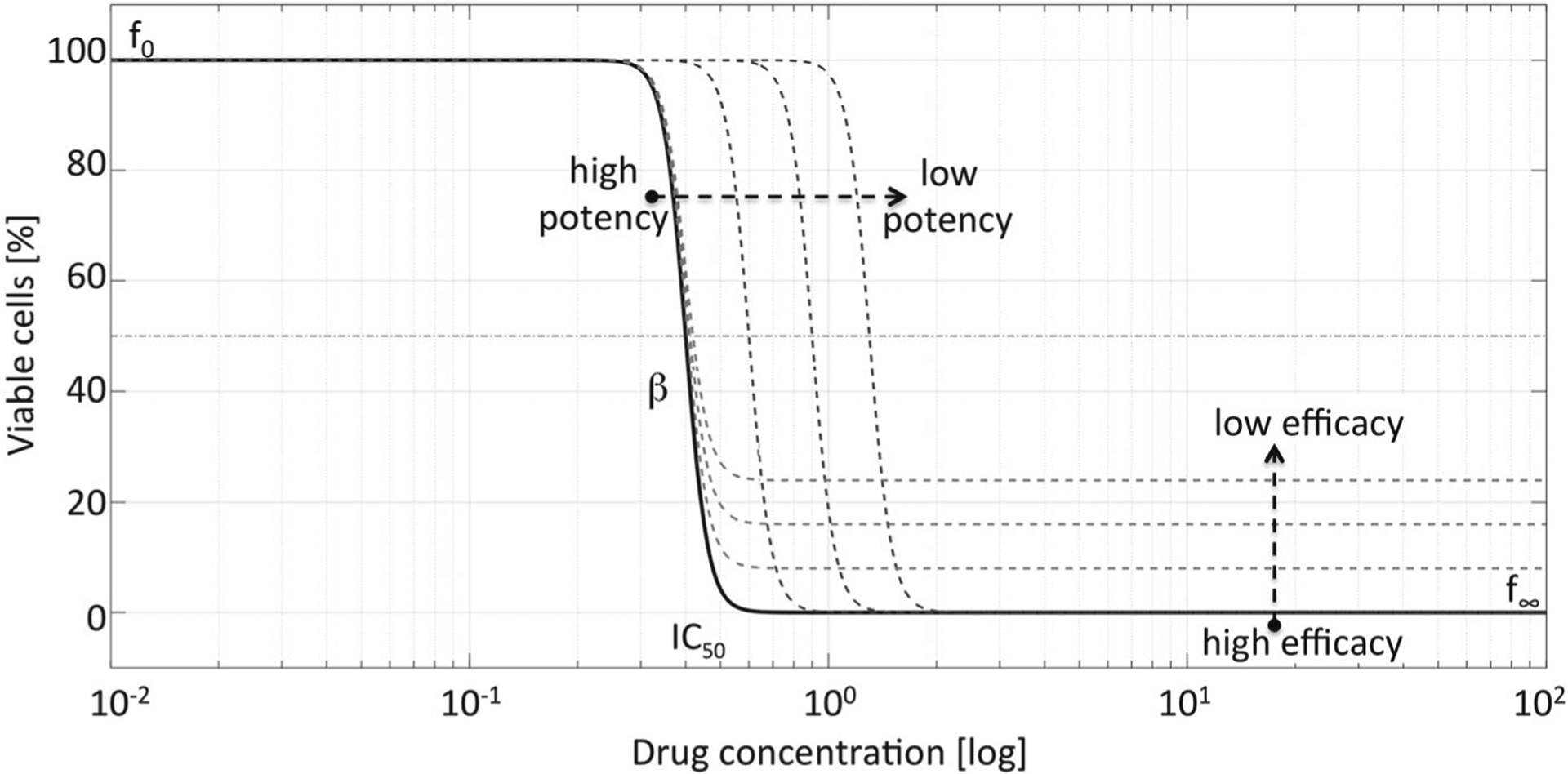
Drug potency and efficacy reflected in IC50 curves. The shape of the IC50 curve depends on the control effect (f0), variability of cell population response (slope β), drug potency (IC50 value), and drug efficacy (background effect f∞)
To draw the inhibitory concentration curves and determine the half-inhibitory concentration value IC50, we perform computational experiments for the monolayer and spheroid cultures with initial drug concentrations varied from γ0=0 to 103 mM. Each simulation starts with 315 cells arranged either in the monolayer or spheroid configuration. The total number of cells that remained after 72 h of the simulated time is recorded. For each drug concentration, we repeat the simulation 3 times to determine the average number of viable cells. These numbers were normalized using the average count of cells grown with no drug. With this process, we obtained the normalized cell counts f (γ0) for each tested drug concentration γ0. For simplicity, we focus on drugs that have high efficacy, f∞ = 0 and assume that 100% of viable cells remained at end of control experiment, i.e., f0 = 100. Thus, we consider the following simplified Hill function:
| (17) |
where γ0 is the drug concentration and f (γ0) is the normalized cell count for drug concentration γ0. The Hill coefficient β and the half-inhibitory concentration value IC50 were determined by fitting the Hill function Eq. (17) to the simulated data using the MATLAB built-in function fit that minimizes
where are the simulated normalized cell count data at the tested drug concentrations γ0, i and are the normalized cell counts given by the Hill function Eq. (17).
4. Results
Our main goal is to examine under which conditions the IC50 values calculated from 2D monolayer and 3D spheroid culture data are similar, and under which conditions they are distinct. This will allow us to determine, whether the typical 2D cell culture experiments are sufficient in assessing efficacy of the anti-cancer drugs, or if the experiments should be carried using the 3D spheroid cultures. In particular, we are interested how the IC50 values depend on drug mechanism of action, on drug diffusivity and on vital properties of the tumor cells. We consider here three cell lines that differ in their doubling times and span division ages consistent with reported experimental data (Hafner et al. 2016; Leander et al. 2014). We perform simulations for three distinct drug diffusivity values that correspond to therapeutic compounds used in the clinic, such as small molecule drugs, nanoparticles or antibodies, as reported in the literature (Schmidt and Wittrup 2009; Karolak et al. 2018). We also take into consideration two distinct killing mechanisms characteristic for clinically applicable drugs (Lipp and Hartmann 2008; Kustermann et al. 2013): the cytotoxic drugs (Sect. 4.1) and the anti-mitotic drugs (Sect. 4.2). In order to cross-examine the results for both drugs, we assume that each drug is absorbed by the cells at the same constant rate and the same drug concentration is lethal for the cells.
4.1. Cytotoxic Anti-Cancer Drugs
In the case of cytotoxic drugs, cell survival is regulated only by the level of the absorbed drug, and the cell dies if the amount of the accumulated drug exceeds the predefined threshold. We first examined whether the IC50 values for the 2D and 3D cultures depend on how fast the drug is diffusing within each cell culture. Next, we examine whether the drug-induced cell death depends on how fast the cells are dividing.
The impact of drug diffusivity on IC50 values in 2D and 3D cell cultures
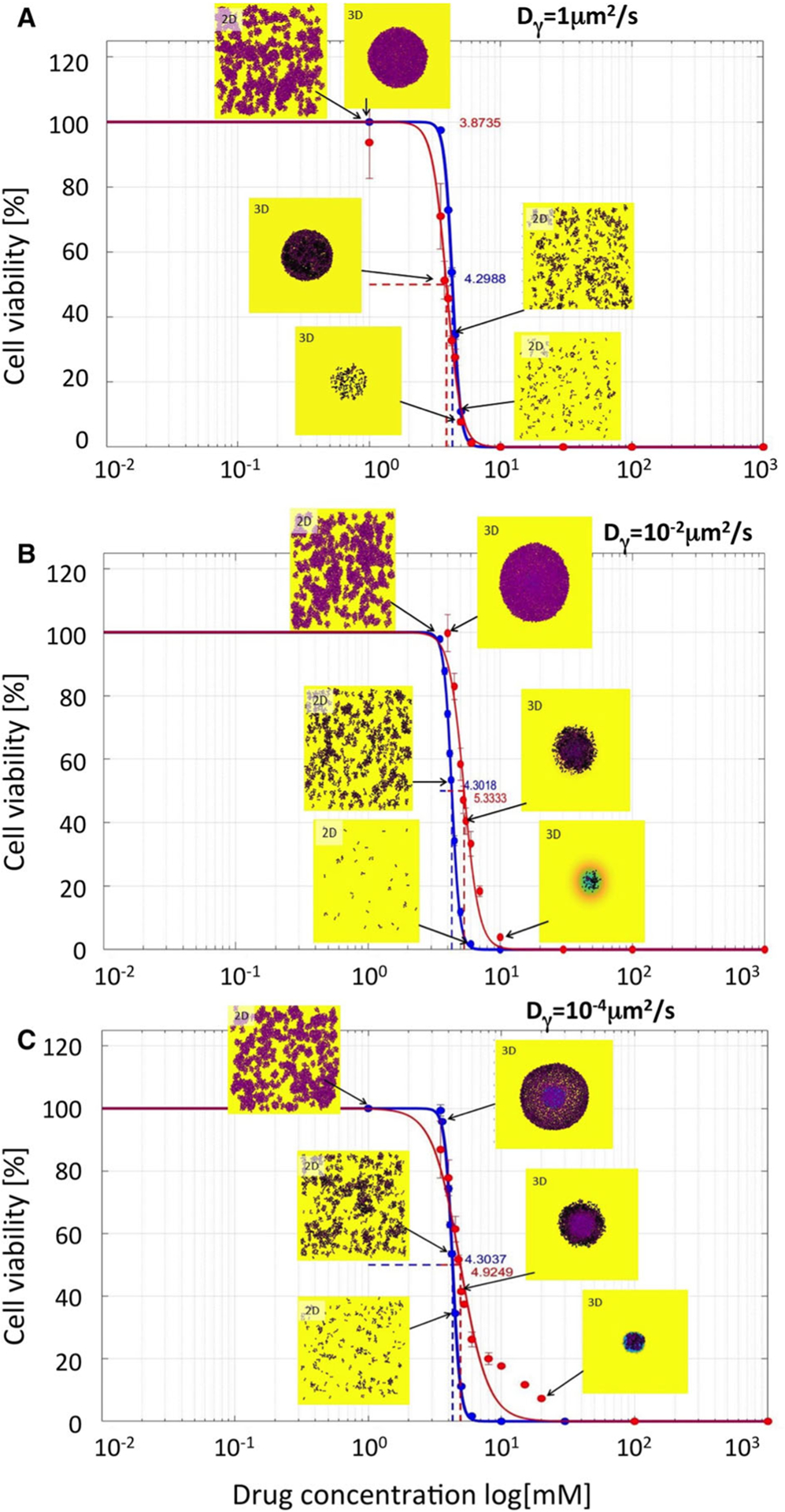
We considered three distinct diffusion coefficients: 𝒟γ = 1, 10−2, 10−4 μm2/s. In each case, we perform several computational simulations using initial drug concentrations γ0 between 0 and 103 mM for both a monolayer and a spheroid cultures. These results are summarized in Fig. 4a–c.
Fig. 4.
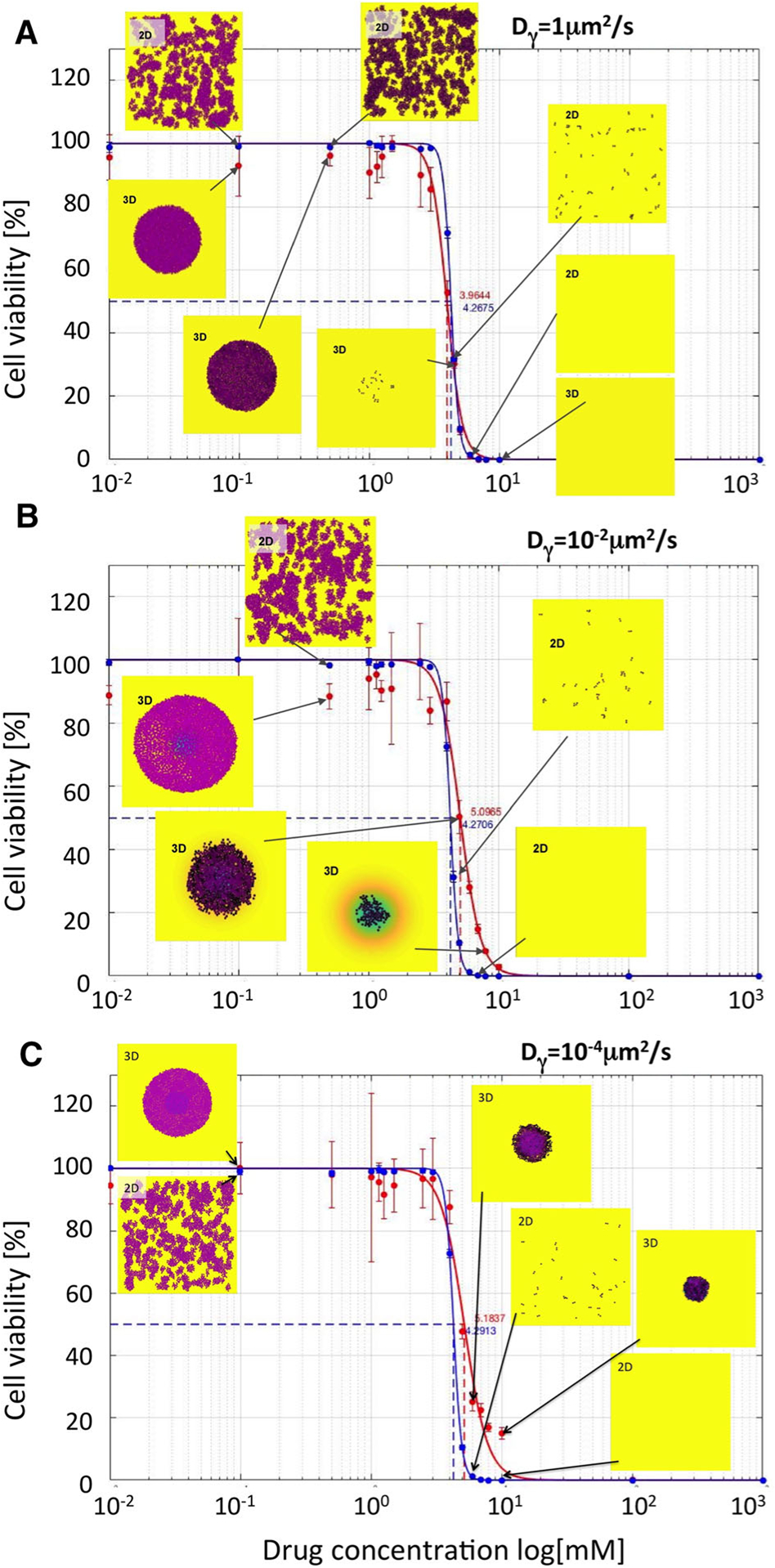
Comparison of IC50 curves and values for cytotoxic drugs. IC50 curves for monolayer cultures (blue curves) and cross sections though spheroid cultures (red curves) and their corresponding IC50 values are shown for the following diffusivity values: a Dγ = 1μm2/s; IC50=4.2675 for 2D and 3.9644 for 3D cultures; b Dγ = 10−2μm2/s; IC50=4.2706 for 2D and 5.0965 for 3D cultures; c Dγ = 10−4μm2/s; IC50=4.2913 for 2D and 5.1837 for 3D cultures. The vertical lines show standard deviation values. Insets show final cell configurations for the selected drug concentrations. Cell colors indicate the level of absorbed drug (low-pink, high-black). Background colors indicate the level of the remaining drug (high-yellow, medium-green, low-blue). The drug was supplied uniformly only once, at the beginning of each simulation. The cell maturation time for these simulations was 18 h
For each drug diffusivity, the simulations are repeated 3 times, and the average number of viable cells is used to calculate the IC50 curves, separately for the 2D monolayer cultures (blue curves) and 3D multicellular spheroid cultures (red curves). All IC50 curves have quite similar shapes, and the corresponding IC50 values are of the same order of magnitude with less than 20% of difference. It can be observed that with the diminishing diffusion coefficients, the IC50 values for the spheroid cultures are slightly higher than for the 2D cultures, that is, consistent with the fact that slowly diffusing drugs are not able to effectively penetrate the tightly packed cell clusters. Therefore, the cells in the 3D spheroids could survive better the drug insult. This is also confirmed by the final cellular configurations shown in the insets in Fig. 4. For higher concentrations of slowly diffusing drugs, some cells in the 3D spheroids have survived after 72 h of exposure to the drug, while no cells remained in the 2D cell cultures exposed to the same drug concentrations. For example, when cells were exposed to the initial drug concentration of γ0 = 10 mM, there were no cell remaining in both 2D and 3D experiments for the highest diffusion case of 1 μm2/s (Fig. 4a), while 105 and 315 cells remained in the 3D spheroid model for diffusion of 10−2μm2/s (Fig. 4b) and 10−4μm2/s (Fig. 4c).
In summary, slower drug diffusion resulted in more pronounced changes in the IC50 values for the spheroid model in comparison with the IC50 values for the monolayer model, which were not significantly different.
The impact of cell maturation ages on IC50 values in 2D and 3D cell cultures
Since the amount of the absorbed drug depends on how long the cell is exposed to it, we examined the relationship between the IC50 curves as the average cell maturation age Amat increases from 18, to 30, to 50 h, while all other parameters were kept fixed. These results are shown in Fig. 5a–c.
Fig. 5.
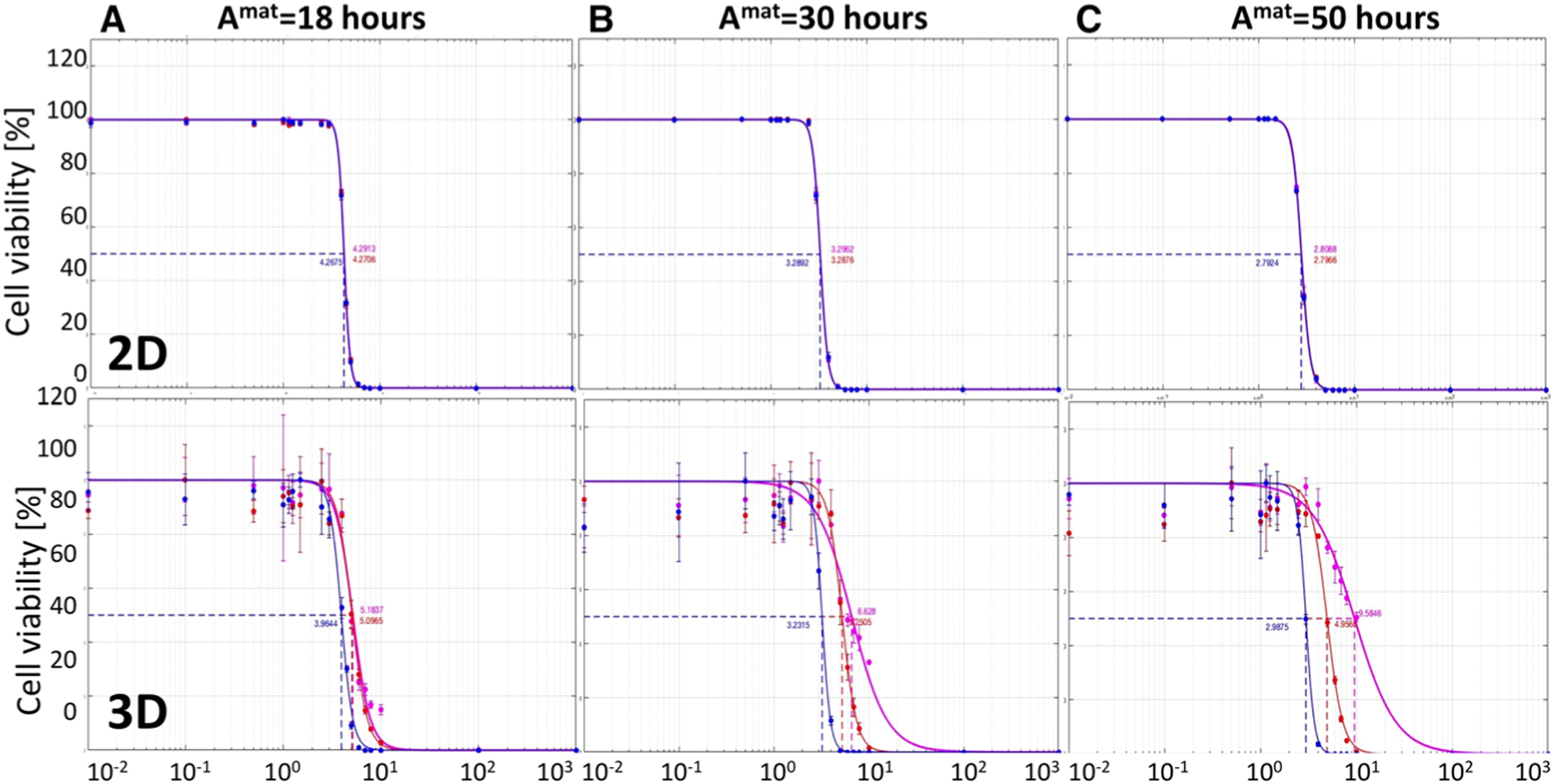
Comparison of IC50 curves for three cell lines of different maturation ages. IC50 curves and values for drugs with diffusion coefficients of Dγ = 1μm2/s (blue), 10−2μm2/s (red), and 10−4μm2/s (magenta) in the monolayer culture model (top) and the cross section though the spheroid culture model (bottom) for the maturation times of a Amat = 18 h, b Amat = 30 h, and c Amat = 50 h. All data are presented after 72 h of the simulated time
For each maturation time, we perform simulations for both the monolayer (top row in Fig. 5) and spheroid (bottom row) cultures. We consider three diffusion coefficients: 𝒟γ = 1μm2/s (blue curves), 𝒟γ = 10−2μm2/s (red curves) and 𝒟γ = 10−4μm2/s (magenta curves). The corresponding IC50 values for all simulated cases are summarized in Table 2. These results indicate that in the monolayer model cell maturation time and drug diffusivity do not effect the IC50 curves, and thus have insignificant role in inhibiting growth of the whole cell population. On the other hand, in the spheroid model, the changes in the IC50 values are more pronounced. For a fixed cell maturation age, the IC50 values differ at lease twofold between the fastest and the slowest diffusing drug. Moreover, in the case of slowly proliferating cells, this difference is almost fivefold. For faster diffusing drugs, the IC50 values are similar despite the differences in cell maturation ages, but in the cases of slower spreading drugs, the corresponding IC50 values increase with the increased maturation.
Table 2.
IC50 values for cytotoxic drug diffusion coefficients Dγ and cell maturation ages Amat, for both monolayer and spheroid in silico cultures
| Dγ μm2/s | Amat = 18 h | Amat = 30 h | Amat = 50 h |
|---|---|---|---|
| Summary of IC50 values in the monolayer model | |||
| 1 | 4.2675 | 3.2892 | 2.7924 |
| 10−2 | 4.2706 | 3.2876 | 2.7966 |
| 10−4 | 4.2913 | 3.2962 | 2.8088 |
| Summary of IC50 values in the cross section spheroid model | |||
| 1 | 3.9644 | 3.2315 | 2.9875 |
| 10−2 | 5.0965 | 5.2505 | 4.9565 |
| 10−4 | 5.1837 | 6.6280 | 9.5846 |
In summary, the 2D monolayer model is not able to differentiate between the inhibitory effects of cytotoxic drugs of different diffusivity for a range of tumor cells with diverse proliferation dynamics. This is because all cells in the monolayer culture are equally exposed to the drug. On the other hand, in the 3D spheroid culture model, both factors: drug diffusivity and frequency of cell proliferation affect the IC50 values noticeably, with values increasing for slower diffusing drugs and for slower proliferating cells. This is an effect of a more realistic tumor geometry in which tightly packed cells are more difficult to penetrate by the drug dissolved in the surrounding external medium.
4.2. Anti-mitotic Anti-cancer Drug
The anti-mitotic drugs are designed to interfere with microtubule functions which are essential in the cell division process. Cells exposed to these drugs cannot progress to the mitotic phase of the cell cycle and die. Therefore, cells’ survival depends on both the level of the accumulated drug and the current cell cycle phase. In particular, if the absorbed drug exceeds the predefined threshold, the cell remains viable until it attempts to divide. For the anti-mitotic drugs, we first examined whether the IC50 values for the 2D and 3D cultures depend on drug diffusivity within the 2D and 3D cell cultures. Next, we tested whether the drug-induced cell death depends on cell maturation age, that is on how fast the cells are dividing.
The impact of drug diffusivity on IC50 values in 2D and 3D cell cultures
As in the previous case, we considered three different drug diffusion rates: 𝒟γ = 1, 10−2, 10−4 μm2/s and performed several computational simulations using initial drug concentrations γ0 between 0 and 103 mM. Each simulation is seeded with 315 cells arranged either sparse in the domain (a monolayer culture) in a circular configuration (a cross section through a spheroid culture). For each combination of parameters, we perform 3 simulations and the average data are used to generate the IC50 curves for the 2D monolayer cultures (blue curves) and 3D multicellular spheroid cultures (red curves). These results are presented in Fig. 6a–c.
Fig. 6.
Comparison of IC50 curves and values for anti-mitotic drugs. IC50 curves for monolayer cultures (blue curves) and cross sections though spheroid cultures (red curves) and their corresponding IC50 values are shown for the following diffusivity values: a Dγ = 1μm2/s; IC50=4.2988 for 2D and 3.8735 for 3D cultures; b Dγ = 10−2μm2/s; IC50=4.3018 for 2D and 5.3333 for 3D cultures; c Dγ = 10−4μm2/s; IC50=4.3037 for 2D and 4.8457 for 3D cultures. The vertical lines show standard deviation values. Insets show final cell configurations for the selected drug concentrations. Cell colors indicate the level of absorbed drug (low-pink, high-black). Background colors indicate the level of the remaining drug (high-yellow, medium-green, low-blue). The drug was supplied uniformly only once, at the beginning of each simulation. The cell maturation time for these simulations was 18 h
Similarly, as in the cytotoxic drug case, the IC50 values for the 2D cell cultures do not differ significantly despite distinct drug diffusivity. Moreover, for fast proliferating cells, the IC50 curves for both 2D and 3D cultures are overlapping (Fig. 6a), while for the slowly proliferating cells, the IC50 curves for the spheroid cultures attain higher half-inhibitory values (Fig. 6b, c). In contrast to the cytotoxic case, the 3D cell colonies of cells exposed to higher concentrations of anti-mitotic drugs are less compact. This is an effect of the delayed death of the cells located in the middle of the spheroid. These cells have already absorbed a lethal level of the drug, but remain in the dormant state due to overcrowding. Once the space for their division becomes available, these cells make an attempt to divide and die due to the drug action. This, in turn, creates space for division of other cells that might instead die if they accumulated a lethal dose of the drug. This may create a domino effect that results in more cell death and less compact spheroid structure. This effect may also take place in 2D cell cultures, but because these cells are seeded more sparsely, it is not so apparent as in the 3D spherical cultures.
The impact of cell maturation ages on IC50 values in 2D and 3D cell cultures
Next, we focused on the relationship between the IC50 values for the anti-mitotic drugs and cell maturation age. We considered again three cell lines characterized by distinct maturation ages: Amat = 18, 30 or 50 h, respectively. These results are summarized in Fig. 7a–c where the IC50 curves for 2D monolayer culture are shown in red and for the 3D spheroid culture in blue. The corresponding IC50 values are summarized in Table 3.
Fig. 7.
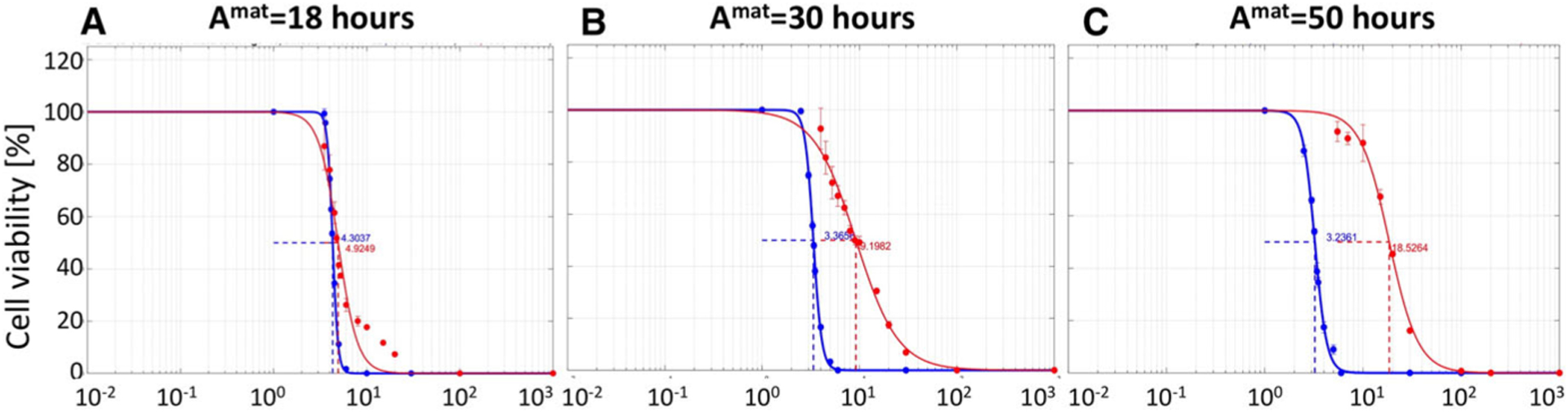
Comparison of IC50 curves for three cell lines of different maturation ages. IC50 curves for drug diffusion coefficient of Dγ = 10−4μm2/s and cell maturation ages of a Amat=18 h, b Amat=30 h, and c Amat=50 h, in 2D cell monolayer culture (blue) and 3D cell spheroid culture (red). All data are presented after 72 h of the simulated time
Table 3.
IC50 values for anti-mitotic drug diffusion coefficients Dγ and cell maturation ages Amat, for both monolayer and spheroid in silico cultures
| Dγ μm2/s | Amat = 18 h | Amat = 30 h | Amat = 50 h |
|---|---|---|---|
| Summary of IC50 values in the 2D model | |||
| 1 | 4.2988 | 3.3495 | 3.2190 |
| 10−2 | 4.3018 | 3.3641 | 3.1989 |
| 10−4 | 4.3037 | 3.3656 | 3.2361 |
| Summary of IC50 values in the 3D model | |||
| 1 | 3.8735 | 3.3896 | 3.4942 |
| 10−2 | 5.3333 | 5.7667 | 7.4052 |
| 10−4 | 4.8457 | 9.0081 | 18.148 |
We again observe the trend that IC50 values increase when drug diffusivity decreases and cell maturation age increases for the 3D cell cultures; however, these differences between IC50 values and curves are even more pronounced for anti-mitotic drugs in comparison with cytotoxic drugs. In fact, for very slowly proliferating cells (Amat = 50 h), there is an order of magnitude difference between IC50 values for fast diffusing (Dγ = 1μm2/s) and slowly diffusing (Dγ = 10−4μm2/s) anti-mitotic drugs (red curves in Fig. 7a, c). This IC50 value is also an order of magnitude larger than the corresponding value for the cytotoxic drugs. In case of the 2D cell monolayer cultures, there is no significant difference in the IC50 values despite differences in drug diffusivity and cell proliferation speed. This is similar to the case of the cytotoxic drugs. For the anti-mitotic drug, we observe a non-monotone relationship between drug diffusivity and IC50 values when the average maturation age of the cells is 18 h (see Table 3, when Amat = 18 h). To confirm this non-monotone relationship, we repeated the experiment for the anti-mitotic drug with drug diffusivity Dγ set to 1, 10−2 and 10−4 μm2/s when Amat = 18 h. Results, not shown here, confirm our initial observations.
In summary, the 2D monolayer model is again not able to distinguish between the inhibitory effects of anti-mitotic drugs of different diffusivity for a range of tumor cells with diverse proliferation dynamics. Again, this is attributed to the fact that all cells in the 2D monolayer culture are equally exposed to the drug. In contrast, in the 3D spheroid culture model, both drug diffusivity and cell proliferation time have noticeable effect on the IC50 values which increase for slowly diffusing drugs and for slowly proliferating cells. While this trend is similar for cytotoxic and anti-mitotic cells, the overall increase in IC50 values is larger for the anti-mitotic cells. Actually, it is an order of magnitude larger for the extreme case (Amat = 50 h and Dγ = 10−4μm2/s), showing the crucial role that the drug mechanism of cell killing plays in determining the drug inhibitory effect.
5. Discussion
In this paper, we addressed an issue of comparing the potency of anti-cancer drugs using 2D monolayer and 3D spheroid cell cultures. Traditionally, the monolayer cultures are used for evaluation of how a given drug effects growth dynamics of a given cell line or malignant cells derived from a patient’s tumor. These experiments provide information about whether the drug exerts the expected effect and what is the minimal drug concentration needed to observe this effect. The drugs can be then compared to one another using the IC50 values which indicate drug concentration that inhibits growth of tumor cell colony by half. By comparing IC50 values for different drugs, one can assess which of them is effective at lower concentrations.
However, it has been shown experimentally that the 2D cell cultures do not recreate tumor features observed in vivo, such as tumor morphology, cell phenotypes and cell-cell interactions, tumor heterogeneity, and the composition of tumor microenvironment (Pampaloni et al. 2007; Hoarau-Vechot et al. 2018; Belgodere et al. 2018). Therefore, tumor cells’ response to anti-cancer drugs and drug penetration through the tumor tissue might not be faithfully captured in the monolayer cultures. To test this hypothesis, we performed computational studies to compare the IC50 curves generated from the analogues of the two-dimensional cell monolayer culture and the three-dimensional multicellular spheroid culture, when the same cells are exposed to the same drugs for the same period of time. We considered hypothetical drugs of various sizes (and thus different diffusivity) and different cell killing mechanisms (cytotoxic and anti-mitotic). Our results indicated that in simulations of 2D cell cultures the IC50 values were similar, indicating the same drug potency despite different drug characteristics and cell properties. However, in the simulations of 3D multicellular spheroid cultures, both classes of drugs showed significant differences in the the IC50 values for different drug diffusivity and cells of different cell cycles. This strongly suggest that 2D cell cultures are not able to differentiate the half-inhibitory effects for distinct drugs, and the 3D multicellular spheroid cultures should be rather used to assess effective drug concentrations in laboratory experiments.
While in this manuscript, we consider hypothetical drugs of biologically relevant properties, our ultimate goal for our future work is to apply these analyses to specific drugs and specific cell lines/tumors for which we can provide experimental validation. Certain assumptions of our model can also be refined in the future. For example, some cytotoxic drugs can induce cell necrosis, while others result in cell lysis. In the former case, cells will remain in the system, though they would stop absorbing the drug. In the latter case, dying cells may release toxins that have a direct impact on neighboring cells’ survival. The process of dead cell clearance may also be modeled in a more realistic way by using additional time lag, in contrast to instantaneous cell removal, as it is modeled in the current version. Moreover, we observed in our simulations that anti-mitotic drugs may result in less compact spheroid structure due to sudden death of inner cells. This took place when some inner dying cells created space for their immediate neighbors to divide, but the cells died instead when they entered into mitotic phase of their cell cycle. It is observed that some in vivo tumors, such as melanomas, are less dense and contain more interstitial fluid that would suggest more intratumoral death. However, further studies are needed to confirm if the mechanism suggested by our simulations faithfully explains empirical observations.
In summary, to our knowledge, this work is the first comprehensive computational study comparing drug efficacy in analogues of the 2D and 3D cell cultures. Our simulations indicate that monolayer cell cultures may provide misleading results, since the produced IC50 values were almost identical for several cases for which spheroid cultures resulted in significantly distinct IC50 values. Thus, our main message from this study is to advocate for using the 3D cell cultures as a standard for testing drug efficacy, which is not currently practiced by the experimental laboratories and pharmacology industry.
Acknowledgements
This project was initiated as a part of the Preparation for Industrial Careers in Mathematical Sciences (PIC-Math) Award (to NT) from Mathematical Association of America (MAA) and Society of Industrial and Applied Mathematics (SIAM). Support was provided by the National Science Foundation, NSF Grant DMS 1345499 (to NT as PIC-Math Faculty). Author NT would like to acknowledge the support of NSF Grant DMS 1515442. This work was supported by the National Institutes of Health National Cancer Institute Grant NIH/NCI U01-CA202229 (to KAR). This work was supported in part by the Shared Resources at the H. Lee Moffitt Cancer Center and Research Institute, an NCI designated Comprehensive Cancer Center through the National Institutes of Health Grant P30-CA076292.
Appendix A: Numerical Implementation of Model Equations
Our computational model combines the off-lattice individual cells with the continuous PDE for the drug concentration. The drug concentration equation Eq. (14) is numerically solved on the regular Cartesian grid x = (x, y), while cells’ positions are defined off-lattice . The exchange of information between these two computational structures takes advantage of the indicator function Eq. (15).
A finite difference scheme is used to approximate the solution of the differential equations modeling cell relocation and drug kinetics. Let Δt denote the time step, and let cell velocity be discretized by the first-order difference equation, then the cell motion Eq. (7) is discretized as
| (18) |
Thus, the location of the kth cell at the next time step (t + Δt) can be determined by
| (19) |
Using the same finite difference approximation for Eq. (8), the age of the kth cell at the next time step is given by
| (20) |
Similarly, let the rate of change of the drug accumulated in the kth cell be discretized by the first-order finite difference scheme, then Eq. (13) becomes,
| (21) |
However, if there is not enough drug in cell vicinity (within the cell radius RD) to be absorbed by the kth cell, then the cell would uptake all the drug available nearby. Hence, the drug accumulated by the kth cell at time (t + Δt) will be computed by
| (22) |
The domain Ω is discretized into a square grid with equal spacing between grid points Δx = Δy. Solution of the reaction-diffusion equation Eq. (14) modeling drug concentration in the domain is then approximated using a forward finite-difference approximation in time and centered finite-difference approximation in space. Thus, an approximation of the solution of Eq. (14) is obtained by the following numerical method:
| (23) |
Numerical stability of this forward in time centered in space (FTCS) method is ensured by satisfying the following condition:
which is satisfied for the maximum diffusion value considered in this study (see Table 1 for parameter values), and the chosen time step and grid size. Thus, the numerical method FTCS for all parameter values is stable.
Other numerical methods could be used to approximate Eq. (14), such as an unconditionally stable fully implicit method or an IMEX method that implicit–explicit combines an implicit scheme for diffusion with an explicit scheme for the reaction terms (Ascher et al. 1995; Madzvamuse 2006). This, however, would impose higher computation costs.
In the case, when cell’s demand for drug absorption is higher than the drug available in the medium, all available drug will be depleted; thus, the cellular uptake component is equal to the minimum between the drug demanded by the cells and the drug available:
| (24) |
References
- Adcock AF, Trivedi G, Edmondson R, Spearman C, Yang L (2015) Three-dimensional (3D) cell cultures in cell-based assays for in-vitro evaluation of anticancer drugs. J Anal Bioanal Tech 6:247 [Google Scholar]
- Ascher UM, Ruuth SJ, Wetton BTR (1995) Implicit-explicit methods for time-dependent PDEs, SIAM. J Numer Anal 32(3):797–823 [Google Scholar]
- Baumgartner W, Drenckhahn D (2002) Transmembrane cooperative linkage in cellular adhesion. Eur J Cell Biol 81:161–168 [DOI] [PubMed] [Google Scholar]
- Belgodere JA, King CT, Bursavich JB, Burow ME, Martin EC, Jung JP (2018) Engineering breast cancer microenvironments and 3D bioprinting. Front Bioeng Biotechnol 6:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamseddine I, Rejniak KA (2020) Hybrid modeling frameworks of tumor development and treatment. WIREs Syst Biol Med 12(1):e1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevereau G, Bollenbach T (2015) Systematic discovery of drug interaction mechanisms. Mol Syst Biol 11(4):807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T-C (2006) Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58(3):621–81 [DOI] [PubMed] [Google Scholar]
- Eichler M, Jahnke H-G, Krinke D, Müller A, Schmidt S, Azendorf R, Robitzki AA (2015) A novel 96-well multielectrode array based impedimetric monitoring platform for comparative drug efficacy analysis on 2D and 3D brain tumor cultures. Biosens Bioelectron 67:582–589 [DOI] [PubMed] [Google Scholar]
- Fallahi-Sichani M, Honarnejad S, Heiser LM, Gray JW, Sorger PK (2013) Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat Chem Biol 9(11):708–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontoura JC, Viezzer C, dos Santos FB, Ligabue RA, Weinlich R, Puga RD, Antonow D, Severino P, Bonorino C (2020) Comparison of 2D and 3D cell culture models for cell growth, gene T expression and drug resistance. Mater Sci Eng, C 107:110264. [DOI] [PubMed] [Google Scholar]
- Friedrich J, Seidel C, Ebner R, Kunz-Schughart LA (2009) Spheroid-based drug screen: considerations and practical approach. Nat Protoc 4(3):309–24 [DOI] [PubMed] [Google Scholar]
- Gardner SN (2000) A mechanistic, predictive model of dose-response curves for cell cycle phase-specific and -nonspecific drugs. Cancer Res 60(5):1417–25 [PubMed] [Google Scholar]
- Gevertz JL, Aminzare Z, Norton K-A, Perez-Velazquez J, Volkening A, Rejniak KA (2015) Emergence of anti-cancer drug resistance: exploring the importance of the microenvironmental niche via a spatial model. In: Jackson T, Radunskaya A (eds) Applications of dynamical systems in biology and medicine, vol 158. The IMA volumes in mathematics and its applications. Springer, Berlin, pp 1–34 [Google Scholar]
- Hafner M, Niepel M, Lee S, Chung M, Sorger PK (2016) Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat Methods 13(6):521–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoarau-Vechot J, Rafii A, Touboul C, Pasquier J (2018) Halfway between 2D and animal models: are 3D cultures the ideal tool to study cancer-microenvironment interactions? Int J Mol Sci 19:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura Y, Mukohara T, Shimoto Y, Funakoshi Y, Chayahara N, Toyoda M, Kiyota N, Takao S, Kono S, Nakatsura T, Minami H (2015) Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol Rep 33:1837–1843 [DOI] [PubMed] [Google Scholar]
- Kane KIW, Lucumi-Moreno E, Lehr CM, Hachi S, Dannert R, Sanctuary R, Wagner C, Fleming RMT, Baller J (2018) Determination of the rheological properties of Matrigel for optimum seeding conditions in microfluidic cell cultures. AIP Adv 8:125332 [Google Scholar]
- Karolak A, Estrella VC, Huynh AS, Chen T, Vagner J, Morse DL, Rejniak KA (2018) Targeting ligand specificity linked to tumor tissue topological heterogeneity via single-cell micro-pharmacological modeling. Sci Rep 8:3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolak A, Agrawal S, Lee S, Rejniak KA (2019a) Single-cell-based in silico models: a tool for dissecting tumor heterogeneity. In: Narayan S (ed) Encyclopedia of biomedical engineering, vol 3. Elsevier, Amsterdam, p 130 [Google Scholar]
- Karolak A, Poonja S, Rejniak KA (2019b) Morphophenotypic classification of tumor organoids as an indicator of drug exposure and penetration potential. PLoS Comput Biol 15(7):e1007214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustermann S, Boess F, Buness A, Schmitz M, Watzele M, Weiser T, Singer T, Suter L, Roth A (2013) A label-free, impedance-based real time assay to identify drug-induced toxicities and differentiate cytostatic from cytotoxic effects. Toxicol In Vitro 27:1589–1595 [DOI] [PubMed] [Google Scholar]
- Laurent J, Frongia C, Cazales M, Mondesert O, Ducommun B, Lobjois V (2013) Multicellular tumor spheroid models to explore cell cycle checkpoints in 3D. BMC Cancer 13:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leander R, Allen EJ, Garbett SP, Tyson DR, Quaranta V (2014) Derivation and experimental comparison of cell-division probability densities. J Theor Biol 359:129–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levasseur LM, Slocum HK, Rustum YM, Greco WR (1998) Modeling of the time-dependency of in vitro drug cytotoxicity and resistance. Cancer Res 58:5749–5761 [PubMed] [Google Scholar]
- Lipp H-P, Hartmann JT (2008) Cytostatic and cytotoxic drugs. In: Aronson JK (ed) Side effects of drugs. Elsevier, Amsterdam [Google Scholar]
- Madzvamuse A (2006) Time-stepping schemes for moving grid finite elements applied to reaction-diffusion systems on fixed and growing domains. J Comput Phys 214:239–263 [Google Scholar]
- Mehrara E, Forssell-Aronsson E, Ahlman H, Bernhardt P (2007) Specific growth rate versus doubling time for quantitative characterization of tumor growth rate. Cancer Res 67(8):3970–5 [DOI] [PubMed] [Google Scholar]
- Meyer CT, Wooten DJ, Paudel BB, Bauer J, Hardeman KN, Westover D, Lovly CM, Harris LA, Tyson DR, Quaranta V (2019) Quantifying drug combination synergy along potency and efficacy axes. Cell Syst 8(2):97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, Edwards CM, Tindall MJ, Maini PK (2009) From a discrete to a continuum model of cell dynamics in one dimension. Phys Rev E 80:031912. [DOI] [PubMed] [Google Scholar]
- NCI-60 Human Tumor Cell Lines Screen [Internet] (2015). https://dtp.cancer.gov/discovery_development/nci-60/
- Pampaloni F, Reynaud EG, Stelzer EH (2007) The third dimension bridges the gap between cell culture and live tissue. Nat Rev Mol Cell Biol 8(10):839–45 [DOI] [PubMed] [Google Scholar]
- Perez-Velazquez J, Rejniak KA (2020) Drug-induced resistance in micrometastases: analysis of spatiotemporal cell lineages. Front Physiol 11:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Velazquez J, Gevertz JL, Karolak A, Rejniak KA (2016) Microenvironmental niches and sanctuaries: a route to acquired resistance. Adv Exp Med Biol 936:149–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rejniak KA, Dillon RH (2007) A single cell-based model of the ductal tumour microarchitecture. Comput Math Methods Med 8(1):51–69 [Google Scholar]
- Rodrigues T, Kundu B, Silva-Correia J, Kundu SC, Oliveira JM, Reis RL, Correlo VM (2018) Emerging tumor spheroids technologies for 3D in vitro cancer modeling. Pharmacol Ther 184:201–211 [DOI] [PubMed] [Google Scholar]
- Schaller G, Meyer-Hermann M (2006) Continuum versus discrete model: a comparison for multicellular tumour spheroids. Philos Trans R Soc A Math Phys Eng Sci 364:1443–1464 [DOI] [PubMed] [Google Scholar]
- Schmidt MM, Wittrup KD (2009) A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol Cancer Ther 8:2861–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah AB, Rejniak KA, Gevertz JL (2016) Limiting the development of anti-cancer drug resistance in a spatial model of micrometastases. Math Biosci Eng 13(6):1185–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashni B, Ariyasu S, Takeda R, Suzuki T, Shiina S, Akimoto K, Maeda T, Aikawa N, Abe R, Osaki T, Itoh N, Aoki S (2018) Size-based differentiation of cancer and normal cells by a particle size analyzer assisted by a cell-recognition PC software. Biol Pharm Bull 41(4):487–503 [DOI] [PubMed] [Google Scholar]
- Tallarida RJ (2011) Quantitative methods for assessing drug synergism. Genes Cancer 2(11):1003–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner R, Charlton JS (2005) Assessing the minimum number of data points required for accurate IC50 determination. Assay Drug Dev Technol 3:525–531 [DOI] [PubMed] [Google Scholar]
- Tyson DR, Garbett SP, Frick PL, Quaranta V (2012) Fractional proliferation: a method to deconvolve cell population dynamics from single-cell data. Nat Methods 9(9):923–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiswald LB, Bellet D, Dangles-Marie V (2015) Spherical cancer models in tumor biology. Neoplasia 17(1):1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Kahen E, Cubitt CL, McGuire J, Kreahling J, Lee J, Altiok S, Lynch CC, Sullivan DM, Reed DR (2015) Identification of synergistic, clinically achievable, combination therapies for osteosarcoma. Sci Rep 5:16991. [DOI] [PMC free article] [PubMed] [Google Scholar]