

Abstract
Mitochondria were described as early as 1890 as ubiquitous intracellular structures by Ernster and Schatz (1981) [1]. Since then, the accretion of knowledge in the past century has revealed much of the molecular details of mitochondria, ranging from mitochondrial origin, structure, metabolism, genetics, and signaling, and their implications in health and disease. We now know that mitochondria are remarkably multifunctional and deeply intertwined with many vital cellular processes. They are quasi-self organelles that still possess remnants of its bacterial ancestry, including an independent genome. The mitochondrial free radical theory of aging (MFRTA), which postulated that aging is a product of oxidative damage to mitochondrial DNA, provided a conceptual framework that put mitochondria on the map of aging research. However, several studies have more recently challenged the general validity of the theory, favoring novel ideas based on emerging evidence to understand how mitochondria contribute to aging and age-related diseases. One prominent topic of investigation lies on the fact that mitochondria are not only production sites for bioenergetics and macromolecules, but also regulatory hubs that communicate and coordinate many vital physiological processes at the cellular and organismal level. The bi-directional communication and coordination between the co-evolved mitochondrial and nuclear genomes is especially interesting in terms of cellular regulation. Mitochondria are dynamic and adaptive, rendering their function sensitive to cellular context. Tissues with high energy demands, such as the brain, seem to be uniquely affected by age-dependent mitochondrial dysfunction, providing a foundation for the development of novel mitochondrial-based therapeutics and diagnostics.
Keywords: Mitochondria, Genomic instability, Aging, Longevity, Mitonuclear, Communication, Mitochondrial-derived peptides, Oxidative stress, Immunity, Inflammation
1. Introduction
Dietary interventions, nutrient sensing pathways, and metabolic homeostasis have profound effects on lifespan and/or healthspan in a broad range of model organisms. Upon the discovery that reducing caloric intake, known as caloric restriction, could extend lifespan in rats, considerable advances on the effect of dietary components and feeding patterns on aging have been made in the past century. For instance, diets that are low in proteins or specific amino acids, ketogenic diets, intermittent fasting, fasting-mimicking diets, and time-restricted feeding are known to promote healthy aging. Genetic studies in C. elegans, D. melanogaster, and mice have also paved the way to the current understanding that nutrient sensing pathways also play a pivotal role in regulating aging. First discovered in C. elegans, single gene manipulations were shown to effectively extend lifespan [2,3]. Soon after, several other genes were also identified to regulate lifespan, many of which turned out to be interconnected in the context of insulin and insulin-like signaling pathways. Parallel pathways in D. melanogaster were discovered, providing a strong genetic foundation to the aging process. Meanwhile, in mice, deficiencies in the growth hormone and its downstream insulin-like growth factor 1 (IGF-1) axis were revealed as major regulators of aging in mammals [4]. These pathways reflect a conserved metabolic network with profound effects on lifespan and/or healthspan.
Mitochondria are supreme metabolic entities that evidently originated from bacteria some 2 billion years ago. The remnants of their bacterial ancestry are evident even today, including an independent genome with polycistronic genes, usage of a distinct genetic code, asexual mode of division (i.e. fission). They are multifunctional organelles that not only produce the great majority of cellular ATP, but also function as major regulatory hubs that coordinate essential cellular processes, including programmed cell death, immune response, macro-molecular synthesis (e.g. steroids, heme), calcium regulation, and intracellular and endocrine signaling. The multifunctional and adaptive nature of mitochondria makes their role in aging a complex moving target. Recent advances in mitochondrial research have moved the field of aging forward in various avenues, including geroscience (Fig. 1). Yet, a coherent molecular map that integrates the various layers of mitochondrial functions during aging is far from complete. Technological advances, including mitochondrial genome editing [5–7], imaging [8,9], bioinformatics [10–12], and emerging vertebrate model organisms [13–15] hold much promise in revealing deeper and comprehensive molecular details of mitochondrial function during aging and in age-related diseases such as Alzheimer’s disease (AD). In this review, we discuss some of the recent advances made on the role of mitochondria in aging and age-related diseases, with special emphasis on the brain.
Fig. 1.
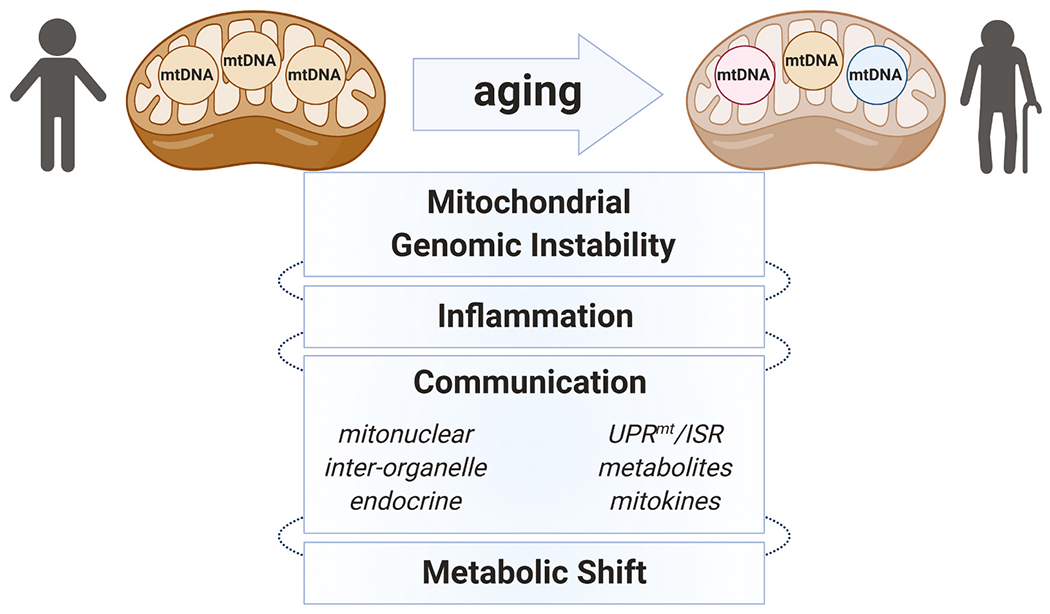
Mitochondria are multifunctional organelles that are extensively integrated with many cellular activities. The broad actions of mitochondria that are adaptive to cellular contexts render their role in aging highly complex and a moving target. mtDNA mutations and heteroplasmy increase with age, which are strongly implicated in aging phenotypes and age-related diseases. Mitochondria are also heavily involved in immune responses, including mtDNA-induced stimulation of inflammatory pathways. Further, mitochondria are regulatory hubs that communicate and coordinate many vital physiological processes at the cellular (i.e. other subcellular compartments (inter-organelle), including the nucleus(mitonuclear)) and organismal level (i.e. endocrine). Several methods of communication are employed, including proteostasis signaling (i.e. UPRmt and ISR), mitochondrial metabolites, and mitokines (e.g. mitochondrial-derived peptides (MDPs)). Multiple age-dependent mitochondrial dysfunctions are thought to ultimately cause maladaptive metabolic shifts and reduce organismal fitness, contributing to aging phenotypes and age-related disabilities/diseases.
2. Mitochondrial genomic instability
Mitochondria generate the great majority of cellular ATP by transferring electrons harnessed from nutrients through the electron transfer chain (ETC) to oxygen during oxidative phosphorylation (OXPHOS). However, during this process, electrons can react with oxygen and generate reactive oxygen species (ROS), largely from complex I and III of the ETC [16,17]. Mitochondria are the major source of intracellular ROS production. Subsequently, mitochondrial ROS, particularly hydroxyl radicals, can react with and damage macromolecules, including proteins, nucleic acids, phospholipids, thereby impairing their function. Whereas proteins and lipids are turned over without permanent damage, unrepaired ROS-inflicted DNA damage can persist and accumulate over time. mtDNA is thought to be more vulnerable to ROS-mediated mutagenesis, largely due to its proximity to the production site. Denham Harman, in his mitochondrial free radical theory of aging (MFRTA), postulated that aging and degenerative diseases are attributed to the progressive accumulation of ROS-mediated deleterious mtDNA mutations [18–25]. Consistently, oxidative damage to macromolecules was observed during aging in multiple organisms [26,27] and long-lived model organisms were reported to exhibit higher expression of antioxidant enzymes [28]. Multiple studies in various model organisms provide inconsistent results, indicating a complex role of antioxidants in regulating lifespan that is largely unclear [29–38]. Further, the presumably inferior DNA repair capacity of mitochondria, compared to the nucleus [39] lent weight to the MFRTA. However, more recent studies show that mitochondria are capable of repairing oxidative mtDNA lesions [40]. In addition, protection from ROS can be conferred by the nucleoid complex that binds to mtDNA [39], distancing from the respiratory chain (i.e. site of ROS production) [41–43], mitochondrial dynamics [44,45], and mitophagy [46]. While mtDNA mutations accumulate with age [47,48], oxidative damage in aged tissues of model organisms and humans is considerably lower than expected and rather modest [49–51]. Instead, prominent age-dependent mtDNA mutations are attributed to replication errors introduced by mitochondrial DNA polymerase γ (POLG). Indeed, mice expressing mutant POLG that are defective in proofreading during mtDNA replication have exhibit supraphysiological mtDNA mutation loads (~2500-fold in the homozygous Polgmut/mut and ~500-fold higher in the Polg+/mut) and exhibited premature aging phenotypes [49,52]. Yet, despite the fact that both homo- and heterozygous mutants harbored mtDNA mutations far exceeding that observed during aging, only the homozygous mice (Polgmut/mut) experienced shortened lifespan, indicating that mtDNA mutation load alone does not determine lifespan [53–55] and that a more complex manifestation of mitochondrial genomic instability is likely involved [56]. Although the MFRTA provided a valuable conceptual foundation for aging research its validity has been challenged [57]. Alternative roles for ROS, such as mitonuclear redox signaling [58–61], may provide further insight into their role in aging.
3. Mitochondria and inflammation
Aging is accompanied by a chronic state of low-grade inflammation referred to as ‘inflammaging’, which is interconnected with other major mechanisms of aging and age-related diseases [62]. Thought to largely result from chronic stimulation of the innate immune system, inflammaging leads to immune dysfunction characterized by impaired response to infection [63] and stimulation (vaccination) [64,65] and aberrant inflammatory signaling [66]. Mitochondria are critical mediators of innate immune response to viral infections [67,68], mitochondrial stress [69], and can signal through inflammasomes [70], toll-like receptor (TLR) signaling [71–73], and interferons [74]. The innate immune system recognizes intruding foreign organisms by pathogen-associated molecular patterns via pattern recognition receptors (PRRs), such as TLRs. Meanwhile, sterile inflammation can occur upon recognition of cellular damage by damage-associated molecular patterns via PRRs, such as mitochondrial DNA (mtDNA) and formyl peptides that are bacterial-like due to the prokaryotic origin of mitochondria and released during cellular stress [74,75]. Notably, the release of mtDNA from mitochondria is a regulated process that communicates the damage to other subcellular compartments or to distal cells. Deficiencies in mitochondrial transcription factor A cause considerable mitochondrial stress and triggers the ejection of mtDNA [74,76]. Oxidative stress also causes mtDNA to be released into the cytoplasm through pores formed by VDAC (voltage-dependent anion channel) oligomers in the mitochondrial outer membrane [77]. Further, apoptosis leads to mtDNA release following mitochondrial outer membrane permeabilization (MOMP) driven by the activation pro-apoptotic BCL-2 proteins (e.g. BAX and BAK) [78]. The mitochondrial inner membrane extrudes into the cytosol and becomes permeabilized following the widening of BAX/BAK-mediated MOMP, allowing mitochondrial export of mtDNA [79]. Another mechanism for mtDNA transport across the mitochondrial inner membrane is through the mitochondrial permeability transition pore (mPTP), which spans the mitochondrial inner membrane in response to mitochondrial calcium concentration and cellular stress [80–82]. mtDNA that is released into the cytosol can then bind to cGAS (cyclic guanosine monophosphate–adenosine monophosphate synthase) and relay an immune response via the activation of STING (stimulator of interferon genes), such as type I interferons (IFNs) and IFN-stimulated genes [71,74,83,84]. Notably, the cGAS/STING pathway is involved in cellular senescence and is strongly linked to the pro-inflammatory senescence-associated secretory phenotype [85–87], indicating an immunological role of mitochondria in cellular senescence. In addition to cGAS-STING activation, stress-induced damage and release of mtDNA also promotes nuclear DNA repair, indicating a role for mtDNA as a sensor and communicator of genotoxic stress that guards the nuclear genome [69]. mtDNA is also exported out of the cell and detected as circulating cell-free mtDNA (ccf-mtDNA) in extracellular fluid [88,89] and cerebrospinal fluid (CSF) [90,91]. ccf-mtDNA provides an novel mechanism for mitochondrial communication between distal tissues [92] and are implicated in neurological disorders [93–95] and systemic inflammatory conditions [96]. Further, psychological stress [97–101] and age [102] increase ccf-mtDNA levels, suggesting the possibility that mtDNA may connect the aging mind and inflammation. Mitochondrial cytokines (mitokines), including growth differentiation factor 15, fibroblast growth factor 21, and mitochondrial-encoded humanin, have been suggested to mediate age-dependent adaptive mitochondrial anti-inflammatory responses [103].
4. Mitochondrial communication
Mitochondria communicate in a variety of ways to coordinate cellular processes, including metabolism, stress response, and adaptive nuclear gene expression. The mode and scope of mitochondrial communication is continuously being unveiled and has shown to be involved in key intra- and inter-cellular processes. To maintain cellular homeostasis under a continuously changing cellular context, mitochondria communicate to the nucleus to relay proteotoxic and metabolic stress and inflammatory signals. Several mediators of mitochondrial communication have been identified, including nuclear-encoded proteins, mitochondrial-encoded peptides, metabolites, inorganic molecules, and mtDNA itself. Here, we discuss some aspects of mitochondrial communication, particularly to the nucleus.
4.1. Mitochondrial signaling and proteostasis
The observation that mitochondrial stress triggers an adaptive transcriptional response in the nucleus was first reported in mammalian cells; the loss of mtDNA [104] or the accumulation of unfolded proteins in the mitochondrial matrix [105] triggered the expression of nuclear-encoded mitochondrial-targeted heat shock chaperones. Such mitochondria-to-nucleus proteotoxic signaling that induces a nuclear transcriptional program is largely known as the mitochondrial unfolded protein response (UPRmt). Considerable advances in understanding the molecular mechanisms of UPRmt were made using the model organism C. elegans, including the transcription factors ATFS-1 and DVE-1 [106–112]. UPRmt induces adaptive nuclear gene expression, in part, through chromatin remodeling via histone H3K9 methylation by MET-2 and LIN-65 [113] and H3K27 demethylation by histone demethylases (jmjd-1.2 and jmjd-3.1) [111]. Notably, UPRmt in C. elegans is not confined to intracellular signaling but can also act on distal cells; neuronal OXPHOS perturbation can activate UPRmt in the intestine and extend lifespan in C. elegans [114].
In mammals, a more general adaptive stress response program exists. The integrated stress response (ISR) is activated by various insults including proteotoxicity, nutrient deprivation, oxidative stress, and viral infection [115]. ISR is induced during aging and is implicated in age-related functional decline and diseases of the brain [116]. Age-dependent activation of ISR is, in part, driven by increased protein misfolding and reduced protein synthesis, which impedes brain functions (e.g. long-term memory) and contributes to pathology (e.g. AD [117]). ISR is mediated by 4 sensor kinases (PERK, GCN2, PKR, and HRI) that relay stress signals by converging on a single phosphorylation site (Ser51) of the eukaryotic translation initiation factor 2 (eIF2) protein [115]. Phospho-eIF2 ultimately leads to a global reduction of protein synthesis, while selectively promoting the expression of specific stress-responsive proteins that contain upstream ORFs (uORFs), such as ATF4 [118]. Notably, ISR is necessary for UPRmt in metazoans but not in C. elegans, suggesting conserved and distinct mitochondrial protein stress signaling between the two organisms. The accretion of recent evidence indicates the involvement of other nuclear-encoded mitochondrial stress sensors that signal to the nucleus, which may feed into mitochondrial ISR. GPS2 (G-Protein Pathway Suppressor 2) is a nuclear-encoded mitochondrial-resident protein that translocates to the nucleus upon mitochondrial depolarization to activate multiple transcriptional programs that overlaps with those induced by UPRmt [119,120]. GPS2 is involved in chromatin remodeling via the H3K9me2/3 histone demethylase JMJD2A/KDM4A [119]. Further, multiple nuclear-encoded proteins that reside in mitochondria translocate to the nucleus upon stimulation [111,121], indicating a regulatory network that maintains mitochondrial homeostasis.
4.2. Mitochondrial metabolites and nuclear gene regulation
Mitochondria communicate to the nucleus using metabolites to provide a metabolic context for nuclear gene regulation. The relative abundance of specific metabolites directly reflects nutrient availability, metabolic demand, cellular redox state and/or mitochondrial function, which can be modulated by diet and exercise. The metabolites serve as substrates and cofactors for various enzymatic modifications that regulate chromatin remodeling and consequent transcriptional regulation of multiple genes [122]. Many of the metabolites are intermediates from the tricarboxylic acid (TCA) cycle, including ATP, acetyl-CoA, α-ketoglutarate (α-KG), fumarate, succinate, and 2-hydroxyglutarate (2-HG), which regulate enzymes that write, read, and erase the post-translational modifications on histones or modify DNA and RNA. Interestingly, mitochondrial metabolites relay actionable information regarding the state of mtDNA heteroplasmy, which increases with age and is linked with aging phenotypes [123–125], to the nucleus [126]. High levels of heteroplasmy lead to reduced, mitochondrial-derived acetyl-CoA levels and subsequent reduction in histone H4 acetylation [126]. mtDNA heteroplasmy also alters mitochondrial nicotinamide adenine dinucleotide (NAD+/NADH) ratio, which likely affects histone acetylation. Midlevel heteroplasmy causes an increase in α-ketoglutarate concentration that may negatively regulate histone H3 methylation. Here, we discuss the mechanisms by which metabolites mediate mitonuclear communication via epigenetic modification.
Histone methylation and acetylation are key post-translational modifications that regulate epigenetics based on the canonical “histone code” [127]. The methylation of histone tails, which occurs at lysine and arginine residues [128], assists as a docking site for chromodomain epigenetic readers on histone [122]. Histone methylation is associated with both repression and activation of transcription depending on the genomic context. Histone methyltransferases (HMTs) and demethylases (HDMs) allow for the enzymatic transfer or removal of a methyl group to and from lysine and arginine residues. HMT utilizes S-adenosylmethionine (SAM) as a cofactor, which is derived from methionine and ATP through the one-carbon folate cycle that shuttles between the cytoplasm and mitochondria [129,130]. Conversely, S-adenosyl-homocysteine (SAH), a byproduct of HMT activity, inhibits HMT activity [131]. HDM utilizes mitochondrial-derived metabolites as cofactors [122,132,133]. Lysine-specific histone demethylase (LSD) and Jumonji C domain-containing protein (JMJD) families are two major conserved families of HDM [111,134]. LSD utilizes flavin adenine dinucleotide (FAD) as a cofactor, which is generated de novo in mitochondria and cytoplasm from the essential vitamin riboflavin (vitamin B2) [135]. JMJD proteins use iron (Fe2+) and the TCA cycle intermediate α-KG as cofactors. This type of HDM can be inhibited by other TCA intermediates such as fumarate, succinate, and the onco-metabolite 2-hydroxyglutarate (2-HG) [136,137].
DNA methylation also modifies chromatin structure and inhibits gene expression by single nucleotide structural modification. DNA methyltransferases (DNMTs) also use SAM as a methyl donor to convert naked cytosine to 5-methylcytosine (5mC) [138]. DNA methylation can be passively reversed by replication-dependent dilution if not maintained or actively reversed through stepwise oxidation of 5mC by the ten-eleven translocation (TET) enzymes, coupled with or thymine DNA glycosylase (TDG)-mediated excision and based excision repair (BER) of the oxidized 5mC forms [139,140]. TET enzymes use Fe2+ and α-KG as cofactors and are inhibited by fumarate, succinate, and 2-HG, similar to the HDM JMJD [136].
Histone acetylation is associated with chromatin opening (i.e. euchromatin) and is therefore associated with active transcription [141,142]. Histone acetyltransferases (HATs) and deacetylases (HDACs) are enzymes that add or remove an acetyl group, respectively. Acetyl-CoA provides the acetyl group necessary for HAT activity. Acetyl-CoA exists both in and out of mitochondria. Mitochondrial-derived acetyl-CoA typically results from complex metabolic activities, including ATP generation through OXPHOS. Cytoplasmic acetyl-CoA serves as building blocks for lipids, steroids, amino acids, and cofactor for HATs. Besides acetyl-CoA, acyl-CoA derivatives such as propionyl-CoA, butyryl-CoA, crotonyl-CoA, and succinyl-CoA also contribute to the activity of HATs [143]. Enzymes that produce acyl-CoAs are linked to fatty acid β-oxidation in mitochondria [144]. There are two different families of HDACs, of which one is Zn2+-dependent histone deacetylases (HDACs) and the other one being NAD+-dependent sirtuin deacetylases (SIRTs). The sirtuin family of HDACs use NAD+ as a cofactor, which provides reducing equivalents (i.e. NADH) for OXPHOS, thus reflects the metabolic state of a cell [145]. In addition to metabolites, metabolic enzymes can reside in the nucleus for direct on-site generation of substrates and co-factors (e.g. acetyl-CoA) for epigenetic regulation [146]. Notably, RNA is also subject to modification, which is relatively recently identified compared to histone and DNA modification [147] and known to undergo ~150 different alterations [148]. RNA-modifying enzymes are reported to travel between mitochondria and nucleus, thereby offering a direct link of nucleus and mitochondrial communication [149]. For example, nuclear-encoded tRNA-modifying enzyme, pseudouridine synthase 1 (PUS1), modify both mitochondrial and cytoplasmic RNAs.
The ability of a cell to sense its metabolic state is key to maintaining homeostasis under an ever-changing environment. This is consistent with the fact that, as mentioned above, nutrient sensing pathways can regulate the aging process. Here, we highlight the fact that the nucleus is directly under the influence of mitochondrial metabolites that fluctuate depending on the rate of metabolic processes and cause chemical modifications to key regulatory components of gene expression. In other words, the nucleus fine-tunes gene regulation based on the cellular fuel gauge (i.e. metabolites) to effectively mount adaptive responses and promote cellular fitness.
4.3. Mitochondrial-Derived Peptides (MDPs)
The mtDNA has been long considered to encode 13 proteins that are key components of the electron transport chain complexes [1]. However, the recent identification of short genes in the mtDNA and nDNA reveals a more complex proteome of mitochondrial basis. Peptides derived from short open reading frames (sORFs) that are encoded in the mtDNA are collectively referred to as mitochondrial-derived peptides (MDPs). The selection pressure that drove the retention of two independent genomes, while the entire mtDNA can be found scattered across the nuclear genome [150], indicates that genomic consolidation was technically possible. Thus, a surpassing benefit(s) of choosing a dual-genomic system, over a single consolidated genomic system, likely increased cellular and/or organismal fitness. MDPs may reflect an ancient communication method that was retained based on the benefit of possessing inherent mitochondrial messengers. A lingering question lies regarding the role mtDNA, which divides asexually and is prone to higher mutation frequency, in influencing the genetics of aging. Currently, there are eight published MDPs, including humanin [151–153], six small humanin-like peptides (SHLP1-6) [154], and mitochondrial ORF of the 12S ribosomal RNA type-c (MOTS-c) [155]. Notably, MDPs are detected in cells/tissues and in circulation, indicating dual intracellular and endocrine roles.
Humanin is the first MDP that was identified, which was initially discovered as a neuro-protective factor from unaffected brain fractions of an autopsy-diagnosed AD patient [151–153]. Indeed, humanin has been shown to protect neurons from multiple cytotoxic insults, including those related to AD (e.g. familial AD genes and β-amyloid) [151–153,156–159]. Humanin is encoded within the mitochondrial 16S rRNA gene as a 75 bp sORF. Later, six additional MDPs were also identified within the 16S rRNA and named SHLP1–6. Humanin and SHLP expression is dynamic and alters with age, which reflects its role in adaptive stress response. In mice, humanin levels in tissues and in circulation have been shown to decline with age (2 vs 13 mo.) [160]. In humans, circulating humanin levels during aging have been reported to decline [(45–65 vs 65–80 vs 81–110 yrs.) [160]; (39 vs 60 yrs.) [161]] and increase (21–113 yrs.) [162]. The underlying reason for the varying reports on humanin levels is likely complex, but considering that humanin is a stress-responsive factor that adapts to physiological contexts, further studies in healthy subjects that control for biological and technical variables (e.g. genetic background, sex, physical status, sample collection time/methods) are needed to better interpret the significant of circulating peptide levels.
Central administration of humanin or SHLP2 improves insulin sensitivity in mice [160], indicating a role for regulating neuronal and/or glial function. As discussed below, MOTS-c also acts in the brain [163]. Notably, circulating humanin levels were decreased in AD and MELAS (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes) patients, suggesting that further studies on peptide levels from patients with age-related diseases will likely provide additional insight [164]. Further, circulating humanin levels were considerably higher in centenarian offsprings compared to their age-matched counterparts [164].
Considerable expression of transcripts from the mitochondrial rRNA loci (i.e. 12S and 16S rRNA) have been reported previously in mononuclear cells under interferon-inducing conditions, but the originating genes were not identified nor annotated [165]. Whereas humanin and SHLPs are derived from the 16S rRNA, MOTS-c is encoded within the 12S rRNA [155]. MOTS-c is detected in various tissues (e.g. brain, liver, skeletal and cardiac muscle) and in circulation [155], thus has been dubbed a mitochondrial hormone [166] or a mitochondrial cytokine (mitokine) [167,168]. MOTS-c acts as a regulator of metabolic homeostasis, in part, by targeting the skeletal muscle and enhancing insulin sensitivity. In fact, MOTS-c prevented diet-induced and reversed age-dependent insulin resistance in mice. Notably, MOTS-c translocates to the nucleus in response to metabolic stress and directly regulates adaptive nuclear gene expression to promote homeostasis [169]. This indicates that the co-evolved mitochondrial and nuclear genomes cross-regulate each other and coordinate cellular responses [170,171].
MOTS-c was predicted to enhance physical performance and mediate some of the physiological benefits of exercise, in part, based on its effect on skeletal muscle, glucose metabolism, and AMPK signaling [155,172–175]. Indeed, exercise increases MOTS-c levels in skeletal muscle and in circulation in humans [176] and mice [177]. Further, MOTS-c injections can significantly extend the running capacity of mice on a treadmill, regardless of their age [176]. RNA-seq analyses revealed that MOTS-c promotes proteostasis in skeletal muscle in a HSF-1-dependent manner [176]. Further, exercise induces MOTS-c actions in the brain. Regular exercise and intracerebroventricular (ICV) treatment with the exerkine IL-6 can both induce the expression of MOTS-c in hypothalamic POMC neurons in an ROS-dependent manner [163], consistent with exercise-induced increase in skeletal muscle and in circulation [176,177]. MOTS-c mediates mitohormetic responses in the brain that connect with peripheral tissues, such as fat. Mild mitochondrial ribosomal stress specifically in the hypothalamic POMC neurons, induced by heterozygous deletion of the nuclear-encoded mitochondrial ribosome component CRIF1 (Pomc-Cre; Crif1f/+), increases the expression of MOTS-c and β-endorphin [163]. In these mice, mild mitoribosomal stress in the hypothalamic POMC neurons triggers UPRmt in distal adipose tissue, promotes browning of white adipose tissue (WAT), increases metabolic turnover, and protects from obesity. Consistently, central MOTS-c administration (ICV injections) recapitulates the adipose phenotype of Pomc-Cre; Crif1f/+ mice, including the distal induction of UPRmt and WAT browning [163]. Systemic MOTS-c treatment (intraperitoneal (IP) injections) has been shown to induce similar physiological effects [155,176–178], but whether such responses are mediated by direct neuronal regulation is yet to be determined. Further, although MOTS-c is detected in whole brain samples, it is unclear whether they are synthesized by neurons and glia and/or represent circulating peptides that crossed the blood brain barrier [155]. In neurons, MOTS-c entered the nucleus [163] and regulated POMC expression in coordination with STAT3 [163]. Finally, exercise and ICV treatment with the exerkine IL-6 both induced the expression of MOTS-c in hypothalamic POMC neurons in an ROS-dependent manner [163], consistent with exercise-induced increase in skeletal muscle and in circulation [176,177].
sORF that encode for bioactive peptides are continuously being identified from both mitonuclear genomes, and more sORFs are expected to be annotated followed by functional analyses [179–182]. The fact that MOTS-c can directly regulate nuclear gene expression and that nuclear-encoded factors are known to regulate mitochondrial function indicates our mitonuclear genomes have co-evolved to coordinate gene expression to establish a dynamic yet unified cellular network. The advent of mtDNA engineering technologies, it may not be far before targeted mutagenesis could provide powerful genetic tools for basic research and clinical correction of age-dependent and hereditary mtDNA alterations.
5. Mitochondria in the aging brain
The brain is a major consumer of energy thus is heavily reliant on mitochondria for survival and function. Mitochondria have key roles in neurons, including the biosynthesis of neurotransmitters and their regulation through ATP production and calcium handling [183–187]. The number and morphological status of mitochondria at the synapse affect brain function by changing synaptic strength with age as demonstrated using 3D electron microscopy reconstructions of the brain area linked with working memory in monkeys [188]. The variability of presynaptic strength is also affected by axonal mitochondrial motility, which regulates the release of neurotransmitters [189]. Although neuronal mitochondria exist abundantly in axons, its turnover and biogenesis largely occur in the soma, which requires active mitochondrial transport [190], which is coordinated with axonal growth [191]. Mitochondria can dock or pause in areas with high metabolic demands (e.g. dendritic spines) and resume movement depending on cellular cues [190]. Aging is associated with reduced mitochondrial transport with respect to total movement duration, distance, and duty cycle (portion of time spent in transit) in old mice (23–25 mo.) [192]. Further, mitochondria-free regions increase in axons and lengths of transported mitochondria along the axon decrease with aging in rats [192]. Mitochondria are transported via motor proteins (i.e. kinesins and dyneins) across microtubules that can be destabilized by tau hyperphosphorylation. Such microtubule disruption would negatively impact mitochondrial mobility and provide a connection between mitochondrial dysfunction and Alzheimer’s disease (AD) [193,194]. Further, mitochondria may be involved in the aberrant processing of amyloid precure protein (APP), providing an amyloid connection to the pathogenesis of AD [195,196]. Also, mitochondria may influence the sex dimorphic nature of AD [197] as estradiol, upon binding to estrogen receptors (ER), localizes to mitochondria and modulates mtDNA gene expression, respiratory capacity, mitochondrial antioxidant defenses, and calcium buffering capacity, which in turn can affect neuronal plasticity [198–201].
Considering that a single neuron can have upwards of thousands of mitochondria [202], maintaining mitochondrial fitness would require well-tuned orchestration of multiple processes, including mobility, energetics, dynamics (i.e. fission and fusion), mitophagy, and biogenesis. Thus, as described below, the role of mitochondrial communication/signaling for successful coordination of complex processes is likely a key factor. In addition, mitochondrial quality control at the genetic level is important as mtDNA heteroplasmy, which increases with age in the brain and other tissues, impairs memory retention [203].
6. Conclusion
Mitochondria are the chief metabolic organelle that not only serves as production sites for bioenergetic units and a myriad of macromolecules, but also as prominent regulatory entities that have a stake in a wide range of physiological processes from inflammation to nuclear gene regulation. Considering that eukaryotic existence can be largely attributed to the establishment of mitochondria at all stages of evolution [204], their broad involvement in cellular functions is not surprising. The early endosymbiotic relationship can be seen as an infection, which, together with the fact the immunity and metabolism co-evolved [62], conceptually supports the fact that metabolic pathways are key regulators of aging [205,206] and immunity [207–211] and the intricate involvement of mitochondria. Further, the mitochondrial and nuclear genomes have co-evolved for the past ~2 billion years as the ancestral cells became functionally more complex [212,213]. It is likely that the cellular network is indeed synthesized by factors from both genomes that co-regulate each other to coordinate adaptive gene expression to maximize cellular fitness. Indeed, currently unknown force of selection likely opted a dual genomic cellular system over a unified singular genome, which evidently was fully possible as the entire mtDNA sequence, although degenerate, is scattered across the nuclear genome [214,215]. “Mitochondrial function” undoubtedly encompasses a broad range of cellular processes that have key roles in aging. However, the multiple functions appear to be mechanistic moving targets that change with age, in part, due to their dynamically adaptive and extensively interconnected nature; what occurs in youth may not necessarily be applicable in old age. Thus, a holistic and evolutionary perspective would benefit investigations into how mitochondria contribute to aging and age-related diseases.
Acknowledgments
We are grateful to members of the Lee laboratory for discussions and feedback. This work was funded by NIH R01AG052258 and R01GM136837 to CL and the American Federation for Aging Research (AFAR) and the Larry L. Hillblom Foundation (LLHF) fellowship to JMS.
Footnotes
Competing interests
C.L. is a consultant and shareholder of CohBar, Inc. All other authors declare no competing interests.
References
- [1].Ernster L, Schatz G, Mitochondria: a historical review, J. Cell Biol 91 (1981) 227s–255s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kenyon C, The first long-lived mutants: discovery of the insulin/IGF-1 pathway for ageing, Philos. Trans. R. Soc. B Biol. Sci 366 (2011) 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Finch CE, Ruvkun G, The genetics of aging, Annu. Rev. Genom. Hum. Genet 2 (2001) 435–462. [DOI] [PubMed] [Google Scholar]
- [4].Bartke A, Sun LY, Longo V, Somatotropic signaling: trade-offs between growth, reproductive development, and longevity, Physiol. Rev 93 (2013) 571–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gammage PA, Moraes CT, Minczuk M, Mitochondrial genome engineering: the revolution may not be CRISPR-Ized, Trends Genet. 34 (2018) 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gammage PA, Viscomi C, Simard M-L, Costa ASH, Gaude E, Powell CA, Van Haute L, McCann BJ, Rebelo-Guiomar P, Cerutti R, Zhang L, Rebar EJ, Zeviani M, Frezza C, Stewart JB, Minczuk M, Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo, Nat. Med 24 (2018) 1691–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mok BY, De Moraes MH, Zeng J, Bosch DE, Kotrys AV, Raguram A, Hsu F, Radey MC, Peterson SB, Mootha VK, Mougous JD, Liu DR, A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing, Nature 583 (2020) 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang C, Taki M, Sato Y, Tamura Y, Yaginuma H, Okada Y, Yamaguchi S, A photostable fluorescent marker for the superresolution live imaging of the dynamic structure of the mitochondrial cristae, Proc. Natl. Acad. Sci. USA 116 (2019) 15817–15822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Parlakgül G, Arruda AP, Cagampan E, Pang S, Güney E, Lee Y, Hess HF, Xu CS, Hotamışlıgil GS, High Resolution 3D Imaging of Liver Reveals a Central Role for Subcellular Architectural Organization in Metabolism, Cold Spring Harbor Laboratory, 2020. [Google Scholar]
- [10].Da Silveira WA, Fazelinia H, Rosenthal SB, Laiakis EC, Kim MS, Meydan C, Kidane Y, Rathi KS, Smith SM, Stear B, Ying Y, Zhang Y, Foox J, Zanello S, Crucian B, Wang D, Nugent A, Costa HA, Zwart SR, Schrepfer S, Elworth RAL, Sapoval N, Treangen T, Mackay M, Gokhale NS, Horner SM, Singh LN, Wallace DC, Willey JS, Schisler JC, Meller R, McDonald JT, Fisch KM, Hardiman G, Taylor D, Mason CE, Costes SV, Beheshti A, Comprehensive multi-omics analysis reveals mitochondrial stress as a central biological hub for spaceflight impact, Cell 183 (1185–1201) (2020), e1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Khan S, Ince-Dunn G, Suomalainen A, Elo LL, Integrative omics approaches provide biological and clinical insights: examples from mitochondrial diseases, J. Clin. Investig 130 (2020) 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, Goodman RP, Grabarek Z, Haas ME, Hung WHW, Joshi PR, Jourdain AA, Kim SH, Kotrys AV, Lam SS, McCoy JG, Meisel JD, Miranda M, Panda A, Patgiri A, Rogers R, Sadre S, Shah H, Skinner OS, To T-L, Melissa Wang H., Ward PS, Wengrod J, Yuan C-C, Calvo SE, Mootha VK, MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations, Nucleic Acids Res. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Baumgart M, Priebe S, Groth M, Hartmann N, Menzel U, Pandolfini L, Koch P, Felder M, Ristow M, Englert C, Guthke R, Platzer M, Cellerino A, Longitudinal RNA-seq analysis of vertebrate aging identifies mitochondrial complex I as a small-molecule-sensitive modifier of lifespan, Cell Syst. 2 (2016) 122–132. [DOI] [PubMed] [Google Scholar]
- [14].Willemsen D, Cui R, Reichard M, Valenzano DR, Intra-species differences in population size shape life history and genome evolution, eLife 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dario, érénice B, Param, Zhang E, Paul, Hu C-K, Clément-Ziza M, Willemsen D, Cui R, Harel I, Ben, Yee M-C, Sabrina, Carlos, Beyer A, Eric, Brunet A, The African Turquoise Killifish Genome Provides Insights into Evolution and Genetic Architecture of Lifespan. 163: 1539–1554, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cadenas E, Davies KJA, Mitochondrial free radical generation, oxidative stress, and aging11This article is dedicated to the memory of our dear friend, colleague, and mentor Lars Ernster (1920–1998), in gratitude for all he gave to us, Free Radic. Biol. Med 29 (2000) 222–230. [DOI] [PubMed] [Google Scholar]
- [17].St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD, Topology of superoxide production from different sites in the mitochondrial electron transport chain, J. Biol. Chem 277 (2002) 44784–44790. [DOI] [PubMed] [Google Scholar]
- [18].Harman D, The biologic clock: the mitochondria? J. Am. Geriatr. Soc 20 (1972) 145–147. [DOI] [PubMed] [Google Scholar]
- [19].Harman D, Alzheimer’s disease pathogenesis: role of aging, Ann. N. Y. Acad. Sci 1067 (2006) 454–460. [DOI] [PubMed] [Google Scholar]
- [20].Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA, Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging, Science 309 (2005) 481–484. [DOI] [PubMed] [Google Scholar]
- [21].Linnane A, Ozawa T, Marzuki S, Tanaka M, Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases, Lancet 333 (1989) 642–645. [DOI] [PubMed] [Google Scholar]
- [22].Miquel J, An update on the oxygen stress–mitochondrial mutation theory of aging: genetic and evolutionary implications, Exp. Gerontol 33 (1998) 113–126. [DOI] [PubMed] [Google Scholar]
- [23].Bandy B, Davison AJ, Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging? Free Radic. Biol. Med 8 (1990) 523–539. [DOI] [PubMed] [Google Scholar]
- [24].Harman D, Aging: a theory based on free radical and radiation chemistry, J. Gerontol 11 (1956) 298–300. [DOI] [PubMed] [Google Scholar]
- [25].Viña J, The free radical theory of frailty: mechanisms and opportunities for interventions to promote successful aging, Free Radic. Biol. Med 134 (2019) 690–694. [DOI] [PubMed] [Google Scholar]
- [26].Halliwell B, Biochemistry of Oxidative Stress, Portland Press Ltd, 2007. [DOI] [PubMed] [Google Scholar]
- [27].Cadenas E, Davies KJ, Mitochondrial free radical generation, oxidative stress, and aging, Free Radic. Biol. Med 29 (2000) 222–230. [DOI] [PubMed] [Google Scholar]
- [28].Lei XG, Zhu J-H, Cheng W-H, Bao Y, Ho Y-S, Reddi AR, Holmgren A, Arnér ESJ, Paradoxical roles of antioxidant enzymes: basic mechanisms and health implications, Physiol. Rev 96 (2016) 307–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pomatto LCD, Davies KJA, Adaptive homeostasis and the free radical theory of ageing, Free Radic. Biol. Med 124 (2018) 420–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Longo VD, Gralla EB, Valentine JS, Superoxide dismutase activity is essential for stationary phase survival in Saccharomyces cerevisiae. Mitochondrial production of toxic oxygen species in vivo, J. Biol. Chem 271 (1996) 12275–12280. [DOI] [PubMed] [Google Scholar]
- [31].Unlu ES, Koc A, Effects of deleting mitochondrial antioxidant genes on life span, Ann. N. Y Acad. Sci 1100 (2007) 505–509. [DOI] [PubMed] [Google Scholar]
- [32].Doonan R, McElwee JJ, Matthijssens F, Walker GA, Houthoofd K, Back P, Matscheski A, Vanfleteren JR, Gems D, Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans, Genes Dev. 22 (2008) 3236–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Martin I, Jones MA, Rhodenizer D, Zheng J, Warrick JM, Seroude L, Grotewiel M, Sod2 knockdown in the musculature has whole-organism consequences in Drosophila, Free Radic. Biol. Med 47 (2009) 803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wicks S, Bain N, Duttaroy A, Hilliker AJ, Phillips JP, Hypoxia rescues early mortality conferred by superoxide dismutase deficiency, Free Radic. Biol. Med 46 (2009) 176–181. [DOI] [PubMed] [Google Scholar]
- [35].Zhang Y, Unnikrishnan A, Deepa SS, Liu Y, Li Y, Ikeno Y, Sosnowska D, Van Remmen H, Richardson A, A new role for oxidative stress in aging: the accelerated aging phenotype in Sod1(−/)(−) mice is correlated to increased cellular senescence, Redox Biol. 11 (2017) 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fabrizio P, Liou LL, Moy VN, Diaspro A, Valentine JS, Gralla EB, Longo VD, SOD2 functions downstream of Sch9 to extend longevity in yeast, Genetics 163 (2003) 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Melov S, Ravenscroft J, Malik S, Gill MS, Walker DW, Clayton PE, Wallace DC, Malfroy B, Doctrow SR, Lithgow GJ, Extension of life-span with superoxide dismutase/catalase mimetics, Science 289 (2000) 1567–1569. [DOI] [PubMed] [Google Scholar]
- [38].Zhang Y, Liu Y, Walsh M, Bokov A, Ikeno Y, Jang YC, Perez VI, Van Remmen H, Richardson A, Liver specific expression of Cu/ZnSOD extends the lifespan of Sod1 null mice, Mech. Ageing Dev 154 (2016) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lee SR, Han J, Mitochondrial nucleoid: shield and switch of the mitochondrial genome, Oxid. Med. Cell Longev (2017), 8060949, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bohr VA, Stevnsner T, De Souza-Pinto NC, Mitochondrial DNA repair of oxidative damage in mammalian cells, Gene 286 (2002) 127–134. [DOI] [PubMed] [Google Scholar]
- [41].Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency, Cell 155 (2013) 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cogliati S, Enriquez JA, Scorrano L, Mitochondrial cristae: where beauty meets functionality, Trends Biochem. Sci 41 (2016) 261–273. [DOI] [PubMed] [Google Scholar]
- [43].Kopek BG, Shtengel G, Xu CS, Clayton DA, Hess HF, Correlative 3D superresolution fluorescence and electron microscopy reveal the relationship of mitochondrial nucleoids to membranes, Proc. Natl. Acad. Sci. USA 109 (2012) 6136–6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC, Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations, Cell 141 (2010) 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Prevost CT, Peris N, Seger C, Pedeville DR, Wershing K, Sia EA, Sia RAL, The influence of mitochondrial dynamics on mitochondrial genome stability, Curr. Genet 64 (2018) 199–214. [DOI] [PubMed] [Google Scholar]
- [46].Pickles S, Vigie P, Youle RJ, Mitophagy and quality control mechanisms in mitochondrial maintenance, Curr. Biol 28 (2018) R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cortopassi GA, Arnheim N, Detection of a specific mitochondrial DNA deletion in tissues of older humans, Nucleic Acids Res. 18 (1990) 6927–6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Larsson NG, Somatic mitochondrial DNA mutations in mammalian aging, Annu. Rev. Biochem 79 (2010) 683–706. [DOI] [PubMed] [Google Scholar]
- [49].Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA, Loeb LA, Mitochondrial point mutations do not limit the natural lifespan of mice, Nat. Genet 39 (2007) 540–543. [DOI] [PubMed] [Google Scholar]
- [50].Kennedy SR, Salk JJ, Schmitt MW, Loeb LA, Ultra sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage, PLoS Genet. 9 (2013), e1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ameur A, Stewart JB, Freyer C, Hagstrom E, Ingman M, Larsson NG, Gyllensten U, Ultra-deep sequencing of mouse mitochondrial DNA: mutational patterns and their origins, PLoS Genet. 7 (2011), e1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG, Premature ageing in mice expressing defective mitochondrial DNA polymerase, Nature 429 (2004) 417–423. [DOI] [PubMed] [Google Scholar]
- [53].Szczepanowska K, Trifunovic A, Origins of mtDNA mutations in ageing, Essays Biochem. 61 (2017) 325–337. [DOI] [PubMed] [Google Scholar]
- [54].Szczepanowska K, Trifunovic A, Mitochondrial DNA mutations and aging. The Human Mitochondrial Genome, Elsevier, 2020, pp. 221–242. [Google Scholar]
- [55].Ng LF, Ng LT, van Breugel M, Halliwell B, Gruber J, Mitochondrial DNA damage does not determine C. elegans lifespan, Front. Genet 10 (2019) 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hämäläinen RH, Landoni JC, Ahlqvist KJ, Goffart S, Ryytty S, Rahman MO, Brilhante V, Icay K, Hautaniemi S, Wang L, Laiho M, Suomalainen A, Defects in mtDNA replication challenge nuclear genome stability through nucleotide depletion and provide a unifying mechanism for mouse progerias, Nat. Metab 1 (2019) 958–965. [DOI] [PubMed] [Google Scholar]
- [57].Pomatto LCD, Davies KJA, Adaptive homeostasis and the free radical theory of ageing, Free Radic. Biol. Med 124 (2018) 420–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Schieber M, Chandel Navdeep S., ROS function in redox signaling and oxidative stress, Curr. Biol 24 (2014) R453–R462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sies H, Jones DP, Reactive oxygen species (ROS) as pleiotropic physiological signalling agents, Nat. Rev. Mol. Cell Biol 21 (2020) 363–383. [DOI] [PubMed] [Google Scholar]
- [60].Ristow M, Schmeisser S, Extending life span by increasing oxidative stress, Free Radic. Biol. Med 51 (2011) 327–336. [DOI] [PubMed] [Google Scholar]
- [61].Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M, Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress, Cell Metab. 6 (2007) 280–293. [DOI] [PubMed] [Google Scholar]
- [62].Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A, Inflammaging: a new immune–metabolic viewpoint for age-related diseases, Nat. Rev. Endocrinol 14 (2018) 576–590. [DOI] [PubMed] [Google Scholar]
- [63].Oishi Y, Manabe I, Macrophages in age-related chronic inflammatory diseases, npj Aging Mech. Dis 2 (2016) 16018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ciabattini A, Nardini C, Santoro F, Garagnani P, Franceschi C, Medaglini D, Vaccination in the elderly: the challenge of immune changes with aging, Semin. Immunol 40 (2018) 83–94. [DOI] [PubMed] [Google Scholar]
- [65].Shaw AC, Panda A, Joshi SR, Qian F, Allore HG, Montgomery RR, Dysregulation of human Toll-like receptor function in aging. 10: 346–353, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Qian F, Wang X, Zhang L, Chen S, Piecychna M, Allore H, Bockenstedt L, Malawista S, Bucala R, Shaw AC, Fikrig E, Montgomery RR, Age-associated elevation in TLR5 leads to increased inflammatory responses in the elderly, Aging Cell 11 (2012) 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS, Mitochondrial DNA stress primes the antiviral innate immune response, Nature 520 (2015) 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ren Z, Ding T, Zuo Z, Xu Z, Deng J, Wei Z, Regulation of MAVS expression and signaling function in the antiviral innate immune response, Front. Immunol 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wu Z, Oeck S, West AP, Mangalhara KC, Sainz AG, Newman LE, Zhang X-O, Wu L, Yan Q, Bosenberg M, Liu Y, Sulkowski PL, Tripple V, Kaech SM, Glazer PM, Shadel GS, Mitochondrial DNA stress signalling protects the nuclear genome, Nat. Metab 1 (2019) 1209–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Nakahira K, Haspel JA, Rathinam VAK, Lee S-J, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AMK, Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome, Nat. Immunol 12 (2011) 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ, Circulating mitochondrial DAMPs cause inflammatory responses to injury, Nature 464 (2010) 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zhang J-Z, Liu Z, Liu J, Ren J-X, Sun T-S, Mitochondrial DNA induces inflammation and increases TLR9/NF-κB expression in lung tissue, Int. J. Mol. Med 33 (2014) 817–824. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [73].Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, Shlomchik MJ, Coffman RL, Candia A, Mehal WZ, Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9, J. Clin. Investig 126 (2016) 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].West AP, Shadel GS, Mitochondrial DNA in innate immune responses and inflammatory pathology, Nat. Rev. Immunol 17 (2017) 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zindel J, Kubes P, DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation, Annu. Rev. Pathol. Mech. Dis 15 (2020) 493–518. [DOI] [PubMed] [Google Scholar]
- [76].West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, Macduff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS, Mitochondrial DNA stress primes the antiviral innate immune response, Nature 520 (2015) 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, Zhu J, Yoon HE, Wang X, Kerkhofs M, Kang H, Brown AL, Park S-J, Xu X, Zandee Van Rilland E, Kim MK, Cohen JI, Kaplan MJ, Shoshan-Barmatz V, Chung JH, VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease, Science 366 (2019) 1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bock FJ, Tait SWG, Mitochondria as multifaceted regulators of cell death, Nat. Rev. Mol. Cell Biol 21 (2020) 85–100. [DOI] [PubMed] [Google Scholar]
- [79].Riley JS, Quarato G, Cloix C, Lopez J, O’Prey J, Pearson M, Chapman J, Sesaki H, Carlin LM, Passos JF, Wheeler AP, Oberst A, Ryan KM, Tait SW, Mitochondrial inner membrane permeabilisation enables mt DNA release during apoptosis, EMBO J. 37 (2018), e99238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Patrushev M, Kasymov V, Patrusheva V, Ushakova T, Gogvadze V, Gaziev AI, Release of mitochondrial DNA fragments from brain mitochondria of irradiated mice, Mitochondrion 6 (2006) 43–47. [DOI] [PubMed] [Google Scholar]
- [81].García N, García JJ, Correa F, Chävez E, The permeability transition pore as a pathway for the release of mitochondrial DNA. 76: 2873–2880, 2005. [DOI] [PubMed] [Google Scholar]
- [82].García N, Chävez E, Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size, Life Sci. 81 (2007) 1160–1166. [DOI] [PubMed] [Google Scholar]
- [83].Ablasser A, Chen ZJ, cGAS in action: expanding roles in immunity and inflammation, Science 363 (2019) eaat8657. [DOI] [PubMed] [Google Scholar]
- [84].Riley JS, Tait SW, Mitochondrial DNA in inflammation and immunity, EMBO Rep. 21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Yang H, Wang H, Ren J, Chen Q, Chen ZJ, cGAS is essential for cellular senescence, Proc. Natl. Acad. Sci. USA 114 (2017) E4612–E4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Glück S, Guey B, Gulen MF, Wolter K, Kang T-W, Niklas, Bridgeman A, Rehwinkel J, Zender L, Ablasser A, Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence, Nat. Cell Biol 19 (2017) 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ogrodnik M, Salmonowicz H, Jurk D, Passos JF, Expansion and cell-cycle arrest: common denominators of cellular senescence, Trends Biochem. Sci 44 (2019) 996–1008. [DOI] [PubMed] [Google Scholar]
- [88].Trumpff C, Marsland AL, Sloan RP, Kaufman BA, Picard M, Predictors of ccf-mtDNA reactivity to acute psychological stress identified using machine learning classifiers: a proof-of-concept, Psychoneuroendocrinology 107 (2019) 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Picard M, Wallace DC, Burelle Y, The rise of mitochondria in medicine, Mitochondrion 30 (2016) 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Peng Y, Zheng D, Zhang X, Pan S, Ji T, Zhang J, Shen H-Y, Wang H-H, Cell-free mitochondrial DNA in the CSF: a potential prognostic biomarker of anti-NMDAR encephalitis, Front. Immunol 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Podlesniy P, Trullas R, Biomarkers in Cerebrospinal Fluid: Analysis of Cell-Free Circulating Mitochondrial DNA by Digital PCR, Springer, New York, 2018, pp. 111–126. [DOI] [PubMed] [Google Scholar]
- [92].Thurairajah K, Briggs GD, Balogh ZJ, The source of cell-free mitochondrial DNA in trauma and potential therapeutic strategies, Eur. J. Trauma Emerg. Surg 44 (2018) 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Gambardella S, Limanaqi F, Ferese R, Biagioni F, Campopiano R, Centonze D, Fornai F, ccf-mtDNA as a potential link between the brain and immune system in neuro-immunological disorders, Front. Immunol 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Lowes H, Pyle A, Duddy M, Hudson G, Cell-free mitochondrial DNA in progressive multiple sclerosis, Mitochondrion 46 (2019) 307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Lowes H, Pyle A, Santibanez-Koref M, Hudson G, Circulating cell-free mitochondrial DNA levels in Parkinson’s disease are influenced by treatment, Mol. Neurodegener 15 (2020) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Boyapati RK, Tamborska A, Dorward DA, Ho G-T, Advances in the understanding of mitochondrial DNA as a pathogenic factor in inflammatory diseases. 6: 169, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Lindqvist D, Wolkowitz OM, Picard M, Ohlsson L, Bersani FS, Fernström J, Westrin Å, Hough CM, Lin J, Reus VI, Epel ES, Mellon SH, Circulating cell-free mitochondrial DNA, but not leukocyte mitochondrial DNA copy number, is elevated in major depressive disorder, Neuropsychopharmacology 43 (2018) 1557–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Lindqvist D, Fernström J, Grudet C, Ljunggren L, Träskman-Bendz L, Ohlsson L, Westrin Å, Increased plasma levels of circulating cell-free mitochondrial DNA in suicide attempters: associations with HPA-axis hyperactivity, Transl. Psychiatry 6 (2016), e971 e971–e971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Hummel EM, Hessas E, Müller S, Beiter T, Fisch M, Eibl A, Wolf OT, Giebel B, Platen P, Kumsta R, Moser DA, Cell-free DNA release under psychosocial and physical stress conditions, Transl. Psychiatry 8 (2018) 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Picard M, Trumpff C, Burelle Y, Mitochondrial psychobiology: foundations and applications, Curr. Opin. Behav. Sci 28 (2019) 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Trumpff C, Marsland AL, Basualto-Alarcón C, Martin JL, Carroll JE, Sturm G, Vincent AE, Mosharov EV, Gu Z, Kaufman BA, Picard M, Acute psychological stress increases serum circulating cell-free mitochondrial DNA, Psychoneuroendocrinology 106 (2019) 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Pinti M, Cevenini E, Nasi M, De Biasi S, Salvioli S, Monti D, Benatti S, Gibellini L, Cotichini R, Stazi MA, Trenti T, Franceschi C, Cossarizza A, Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”, Eur. J. Immunol 44 (2014) 1552–1562. [DOI] [PubMed] [Google Scholar]
- [103].Conte M, Martucci M, Chiariello A, Franceschi C, Salvioli S, Mitochondria, immunosenescence and inflammaging: a role for mitokines? Semin. Immunopathol 42 (2020) 607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Martinus RD, Garth GP, Webster TL, Cartwright P, Naylor DJ, Høj PB, Hoogenraad NJ, Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome, Eur. J. Biochem 240 (1996) 98–103. [DOI] [PubMed] [Google Scholar]
- [105].Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ, A mitochondrial specific stress response in mammalian cells, EMBO J. 21 (2002) 4411–4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D, ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans, Dev. Cell 13 (2007) 467–480. [DOI] [PubMed] [Google Scholar]
- [107].Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D, The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans, Mol. Cell 37 (2010) 529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Lin Y-F, Schulz AM, Pellegrino MW, Lu Y, Shaham S, Haynes CM, Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response, Nature 533 (2016) 416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D’Amico D, Moullan N, Potenza F, Schmid AW, Rietsch S, Counts SE, Auwerx J, Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity, Nature 552 (2017) 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Tian Y, Garcia G, Bian Q, Kristan, Joe L, Wolff S, Barbara, Dillin A, Mitochondrial stress induces chromatin reorganization to promote longevity and UPR mt, Cell 165 (2016) 1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Merkwirth C, Jovaisaite V, Durieux J, Matilainen O, Sabine Pedro, Kristan Evan, Mouchiroud L, Sarah Murillo V., Suzanne Reuben, Auwerx J, Dillin A, Two conserved histone demethylases regulate mitochondrial stress-induced longevity, Cell 165 (2016) 1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM, Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation, Science 337 (2012) 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Tian Y, Garcia G, Bian Q, Steffen KK, Joe L, Wolff S, Meyer BJ, Dillin A, Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt), Cell 165 (2016) 1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Durieux J, Wolff S, Dillin A, The cell-non-autonomous nature of electron transport chain-mediated longevity, Cell 144 (2011) 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Costa-Mattioli M, Walter P, The integrated stress response: From mechanism to disease, Science 368 (2020) eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Krukowski K, Nolan A, Frias ES, Boone M, Ureta G, Grue K, Paladini M-S, Elizarraras E, Delgado L, Bernales S, Walter P, Rosi S, Small molecule cognitive enhancer reverses age-related memory decline in mice, eLife 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Chang RC, Wong AK, Ng HK, Hugon J, Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease, Neuroreport 13 (2002) 2429–2432. [DOI] [PubMed] [Google Scholar]
- [118].Anderson NS, Haynes CM, Folding the mitochondrial UPR into the integrated stress response, Trends Cell Biol. 30 (2020) 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Cardamone MD, Tanasa B, Cederquist CT, Huang J, Mahdaviani K, Li W, Rosenfeld MG, Liesa M, Perissi V, Mitochondrial retrograde signaling in mammals is mediated by the transcriptional cofactor GPS2 via direct mitochondria-to-nucleus translocation, Mol. Cell 69 (757–772) (2018) 757–772.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Quiros PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, Gygi SP, Auwerx J, Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals, J. Cell Biol 216 (2017) 2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Monaghan RM, Whitmarsh AJ, Mitochondrial proteins moonlighting in the nucleus, Trends Biochem. Sci 40 (2015) 728–735. [DOI] [PubMed] [Google Scholar]
- [122].Wiese M, Bannister AJ, Two genomes, one cell: mitochondrial-nuclear coordination via epigenetic pathways, Mol. Metab 38 (2020), 100942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Burgstaller JP, Kolbe T, Havlicek V, Hembach S, Poulton J, Piálek J, Steinborn R, Rülicke T, Brem G, Jones NS, Johnston IG, Large-scale genetic analysis reveals mammalian mtDNA heteroplasmy dynamics and variance increase through lifetimes and generations, Nat. Commun 9 (2018) 2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Tranah GJ, Katzman SM, Lauterjung K, Yaffe K, Manini TM, Kritchevsky S, Newman AB, Harris TB, Cummings SR, Mitochondrial DNA m.3243A > G heteroplasmy affects multiple aging phenotypes and risk of mortality, Sci. Rep 8 (2018) 11887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Kann LM, Rosenblum EB, Rand DM, Aging, mating, and the evolution of mtDNA heteroplasmy in Drosophila melanogaster. 95: 2372–2377, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Kopinski PK, Janssen KA, Schaefer PM, Trefely S, Perry CE, Potluri P, Tintos-Hernandez JA, Singh LN, Karch KR, Campbell SL, Doan MT, Jiang H, Nissim I, Nakamaru-Ogiso E, Wellen KE, Snyder NW, Garcia BA, Wallace DC, Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy, Proc. Natl. Acad. Sci. USA 116 (2019) 16028–16035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Carlberg C, Molnár F, The Histone Code, Springer, Singapore, 2018, pp. 75–88. [Google Scholar]
- [128].Bannister AJ, Kouzarides T, Regulation of chromatin by histone modifications, Cell Res. 21 (2011) 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Jambhekar A, Dhall A, Shi Y, Roles and regulation of histone methylation in animal development, Nat. Rev. Mol. Cell Biol 20 (2019) 625–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Michalak EM, Burr ML, Bannister AJ, Dawson MA, The roles of DNA, RNA and histone methylation in ageing and cancer, Nat. Rev. Mol. Cell Biol 20 (2019) 573–589. [DOI] [PubMed] [Google Scholar]
- [131].Deguchi T, Barchas J, Inhibition of transmethylations of biogenic amines by s-adenosylhomocysteine enhancement of transmethylation by adenosylhomocysteinase, J. Biol. Chem 246 (1971) 3175–3181. [PubMed] [Google Scholar]
- [132].Rabhi N, Hannou SA, Froguel P, Annicotte J-S, Cofactors as metabolic sensors driving cell adaptation in physiology and disease, Front. Endocrinol 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Martínez-Reyes I, Chandel NS, Mitochondrial TCA cyde metabolites control physiology and disease, Nat. Commun 11 (2020) 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Shi YG, Tsukada YI, The discovery of histone demethylases, Cold Spring Harb. Perspect. Biol 5 (2013), a017947 a017947–a017947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Janssen JJE, Grefte S, Keijer J, De Boer VCJ, Mito-nuclear communication by mitochondrial metabolites and its regulation by b-vitamins, Front. Physiol 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, Guan KL, Inhibition of -KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors, Genes Dev. 26 (2012) 1326–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Ye D, Guan K-L, Xiong Y, Metabolism, activity, and targeting of D- and L-2-hydroxyglutarates, Trends Cancer 4 (2018) 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Lyko F, The DNA methyltransferase family: a versatile toolkit for epigenetic regulation, Nat. Rev. Genet 19 (2018) 81–92. [DOI] [PubMed] [Google Scholar]
- [139].Breiling A, Lyko F, Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond, Epigenet. Chromatin 8 (2015) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Wu X, Zhang Y, TET-mediated active DNA demethylation: mechanism, function and beyond, Nat. Rev. Genet 18 (2017) 517–534. [DOI] [PubMed] [Google Scholar]
- [141].Eberharter A, Becker PB, Histone acetylation: a switch between repressive and permissive chromatin, EMBO Rep. 3 (2002) 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Martire S, Banaszynski LA, The roles of histone variants in fine-tuning chromatin organization and function, Nat. Rev. Mol. Cell Biol 21 (2020) 522–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Sabari BR, Zhang D, Allis CD, Zhao Y, Metabolic regulation of gene expression through histone acylations, Nat. Rev. Mol. Cell Biol 18 (2017) 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Houten SM, Violante S, Ventura FV, Wanders RJA, The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders, Annu. Rev. Physiol 78 (2016) 23–44. [DOI] [PubMed] [Google Scholar]
- [145].Cantó C, Keir, Auwerx J, NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus, Cell Metab. 22 (2015) 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Boukouris AE, Zervopoulos SD, Michelakis ED, Metabolic enzymes moonlighting in the nucleus: metabolic regulation of gene transcription, Trends Biochem. Sci 41 (2016) 712–730. [DOI] [PubMed] [Google Scholar]
- [147].Shi H, Wei J, He C, Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers, Mol. Cell 74 (2019) 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Boccaletto P, Machnicka MA, Purta E, Piątkowski P, Bagiński B, Wirecki TK, Valérie, Ross R, Limbach PA, Kotter A, Helm M, Bujnicki JM, MODOMICS: a database of RNA modification pathways. 2017 update, Nucleic Acids Res. 46 (2018) D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Bohnsack MT, Sloan KE, The mitochondrial epitranscriptome: the roles of RNA modifications in mitochondrial translation and human disease, Cell. Mol. Life Sci 75 (2018) 241–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Ricchetti M, Tekaia F, Dujon B, Continued colonization of the human genome by mitochondrial DNA, PLoS Biol. 2 (2004), e273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Hashimoto Y, Ito Y, Niikura T, Shao Z, Hata M, Oyama F, Nishimoto I, Mechanisms of neuroprotection by a novel rescue factor humanin from Swedish mutant amyloid precursor protein, Biochem. Biophys. Res. Commun 283 (2001) 460–468. [DOI] [PubMed] [Google Scholar]
- [152].Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, Sobue G, Koide T, Tsuji S, Lang J, Kurokawa K, Nishimoto I, A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Abeta, Proc. Natl. Acad. Sci. USA 98 (2001) 6336–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Tajima H, Niikura T, Hashimoto Y, Ito Y, Kita Y, Terashita K, Yamazaki K, Koto A, Aiso S, Nishimoto I, Evidence for in vivo production of Humanin peptide, a neuroprotective factor against Alzheimer’s disease-related insults, Neurosci. Lett 324 (2002) 227–231. [DOI] [PubMed] [Google Scholar]
- [154].Cobb LJ, Lee C, Xiao J, Yen K, Wong RG, Nakamura HK, Mehta HH, Gao Q, Ashur C, Huffman DM, Wan J, Muzumdar R, Barzilai N, Cohen P, Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers, Aging 8 (2016) 796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, Kim SJ, Mehta H, Hevener AL, de Cabo R, Cohen P, The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance, Cell Metab. 21 (2015) 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Ikonen M, Liu B, Hashimoto Y, Ma L, Lee KW, Niikura T, Nishimoto I, Cohen P, Interaction between the Alzheimer’s survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis, Proc. Natl. Acad. Sci. USA 100 (2003) 13042–13047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, Reed JC, Humanin peptide suppresses apoptosis by interfering with Bax activation, Nature 423 (2003) 456–461. [DOI] [PubMed] [Google Scholar]
- [158].Jung SS, Van Nostrand WE, Humanin rescues human cerebrovascular smooth muscle cells from Abeta-induced toxicity, J. Neurochem 84 (2003) 266–272. [DOI] [PubMed] [Google Scholar]
- [159].Xu X, Chua CC, Gao J, Chua KW, Wang H, Hamdy RC, Chua BH, Neuroprotective effect of humanin on cerebral ischemia/reperfusion injury is mediated by a PI3K/Akt pathway, Brain Res. 1227 (2008) 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Muzumdar RH, Huffman DM, Atzmon G, Buettner C, Cobb LJ, Fishman S, Budagov T, Cui L, Einstein FH, Poduval A, Hwang D, Barzilai N, Cohen P, Humanin: a novel central regulator of peripheral insulin action, PLoS One 4 (2009), e6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Bachar AR, Scheffer L, Schroeder AS, Nakamura HK, Cobb LJ, Oh YK, Lerman LO, Pagano RE, Cohen P, Lerman A, Humanin is expressed in human vascular walls and has a cytoproteetive effect against oxidized LDL-induced oxidative stress, Cardiovasc. Res 88 (2010) 360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Conte M, Ostan R, Fabbri C, Santoro A, Guidarelli G, Vitale G, Mari D, Sevini F, Capri M, Sandri M, Monti D, Franceschi C, Salvioli S, Human aging and longevity are characterized by high levels of mitokines, J. Gerontol. A Biol. Sci. Med. Sci 74 (2019) 600–607. [DOI] [PubMed] [Google Scholar]
- [163].Kang GM, Min SH, Lee CH, Km JY, Lim HS, Choi MJ, Jung SB, Park JW, Kim S, Park CB, Dugu H, Choi JH, Jang WH, Park SE, Cho YM, Kim JG, Kim KG, Choi CS, Kim YB, Lee C, Shong M, Kim MS, Mitohormesis in hypothalamic POMC neurons mediates regular exercise-induced high-turnover metabolism, Cell Metab. 33 (334–349) (2021), e336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Yen K, Mehta HH, Kim S-J, Lue Y, Hoang J, Guerrero N, Port J, Bi Q, Navarrete G, Brandhorst S, Lewis KN, Wan J, Swerdloff R, Mattison JA, Buffenstein R, Breton CV, Wang C, Longo V, Atzmon G, Wallace D, Barzilai N, Cohen P, The mitochondrial derived peptide humanin is a regulator of lifespan and healthspan, Aging 12 (2020) 11185–11199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Tsuzuki T, Nomiyama H, Setoyama C, Maeda S, Shimada K, Pestka S, The majority of cDNA clones with strong positive signals for the interferon-induction-specific sequences resemble mitochondrial ribosomal RNA genes, Biochem. Biophys. Res. Commun 114 (1983) 670–676. [DOI] [PubMed] [Google Scholar]
- [166].Zarse K, Ristow M, A mitochondrially encoded hormone ameliorates obesity and insulin resistance, Cell Metab. 21 (2015) 355–356. [DOI] [PubMed] [Google Scholar]
- [167].Mottis A, Herzig S, Auwerx J, Mitocellular communication: shaping health and disease, Science 366 (2019) 827–832. [DOI] [PubMed] [Google Scholar]
- [168].Tan JX, Finkel T, Mitochondria as intracellular signaling platforms in health and disease, J. Cell Biol 219 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Kim KH, Son JM, Benayoun BA, Lee C, The mitochondrial-encoded peptide MOTS-c translocates to the nucleus to regulate nuclear gene expression in response to metabolic stress, Cell Metab. 28 (516–524) (2018) 516–524.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Benayoun BA, Lee C, MOTS-c: a mitochondrial-encoded regulator of the nucleus, BioEssays 41 (2019), 1900046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Sloan DB, Warren JM, Williams AM, Wu Z, Abdel-Ghany SE, Chicco AJ, Havird JC, Cytonuclear integration and co-evolution, Nat. Rev. Genet 19 (2018) 635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Hunter P, Exercise in a bottle, EMBO Rep. 17 (2016) 136–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Li S, Laher I, Exercise pills: at the starting line, Trends Pharmacol. Sci 36 (2015) 906–917. [DOI] [PubMed] [Google Scholar]
- [174].Thevis M, Schanzer W, Emerging drugs affecting skeletal muscle function and mitochondrial biogenesis - potential implications for sports drug testing programs, Rapid Commun. Mass Spectrom 30 (2016) 635–651. [DOI] [PubMed] [Google Scholar]
- [175].Knoop A, Thomas A, Thevis M, Development of a mass spectrometry based detection method for the mitochondrion-derived peptide MOTS-c in plasma samples for doping control purposes, Rapid Commun. Mass Spectrom 33 (2019) 371–380. [DOI] [PubMed] [Google Scholar]
- [176].Reynolds JC, Lai RW, Woodhead JST, Joly JH, Mitchell CJ, Cameron-Smith D, Lu R, Cohen P, Graham NA, Benayoun BA, Merry TL, Lee C, MOTS-c is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis, Nat. Commun 12 (2021) 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Guo Q, Chang B, Yu Q-L, Xu S-T, Yi X-J, Cao S-C, Adiponectin treatment improves insulin resistance in mice by regulating the expression of the mitochondrial-derived peptide MOTS-c and its response to exercise via APPL1–SIRT1–PGC-1α, Diabetologia 63 (2020) 2675–2688. [DOI] [PubMed] [Google Scholar]
- [178].Lu Tang, Xue Liu, Wang Zhang, Luo Chen, Mitochondrial-derived peptide MOTS-c increases adipose thermogenic activation to promote cold adaptation, Int. J. Mol. Sci 20 (2019) 2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Chen J, Brunner A-D, Cogan JZ, Nuñez JK, Fields AP, Adamson B, Itzhak DN, Li JY, Mann M, Leonetti MD, Weissman JS, Pervasive functional translation of noncanonical human open reading frames, Science 367 (2020) 1140–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Couso J-P, Patraquim P, Classification and function of small open reading frames, Nat. Rev. Mol. Cell Biol 18 (2017) 575–589. [DOI] [PubMed] [Google Scholar]
- [181].Saghatelian A, Couso JP, Discovery and characterization of smORF-encoded bioactive polypeptides, Nat. Chem. Biol 11 (2015) 909–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Makarewich CA, Olson EN, Mining for micropeptides, Trends Cell Biol. 27 (2017) 685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Mattson MP, Gleichmann M, Cheng A, Mitochondria in neuroplasticity and neurological disorders, Neuron 60 (2008) 748–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Hou Y, Ouyang X, Wan R, Cheng H, Mattson MP, Cheng A, Mitochondrial superoxide production negatively regulates neural progenitor proliferation and cerebral cortical, Development 30 (2012) 2535–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Yun J, Finkel T Mitohormesis, Mitohormesis, Cell Metab. 19 (2014) 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Raefsky SM, Mattson MP, Adaptive responses of neuronal mitochondria to bioenergetic challenges: roles in neuroplasticity and disease resistance, Free Radic. Biol. Med 102 (2017) 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Rizzuto R, De Stefani D, Raffaello A, Mammucari C, Mitochondria as sensors and regulators of calcium signalling, Nat. Rev. Mol. Cell Biol 13 (2012) 566–578. [DOI] [PubMed] [Google Scholar]
- [188].Hara Y, Yuk F, Puri R, Janssen WGM, Rapp PR, Morrison JH, Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment, Proc. Natl. Acad. Sci. USa 111 (2014) 486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Sun T, Qiao H, Pan P-Y, Chen Y, Sheng Z-H, Motile axonal mitochondria contribute to the variability of presynaptic strength, Cell Rep. 4 (2013) 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Sheng Z-H, Cai Q, Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration, Nat. Rev. Neurosci 13 (2012) 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Morris RL, Hollenbeck PJ, The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth, J. Cell Sci 104 (1993) 917–927. [DOI] [PubMed] [Google Scholar]
- [192].Takihara Y, Inatani M, Eto K, Inoue T, Kreymerman A, Miyake S, Ueno S, Nagaya M, Nakanishi A, Iwao K, Takamura Y, Sakamoto H, Satoh K, Kondo M, Sakamoto T, Goldberg JL, Nabekura J, Tanihara H, In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS, Proc. Natl. Acad. Sci. USA 112 (2015) 10515–10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Pérez MJ, Jara C, Quintanilla RA, Contribution of Tau pathology to mitochondrial impairment in neurodegeneration, Front. Neurosci 12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Cheng Y, Bai F, The association of Tau with mitochondrial dysfunction in Alzheimer’s disease, Front. Neurosci 12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Lin MT, Beal MF, Alzheimer’s APP mangles mitochondria. 12: 1241–1243, 2006. [DOI] [PubMed] [Google Scholar]
- [196].Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, De. Groof AJC, Madra M, Ikenouchi J, Umeda M, Bird TD, Sturley SL, Schon EA, Upregulated function of mitochondria-associated ER membranes in Alzheimer disease, EMBO J. 31 (2012) 4106–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Pike CJ, Sex and the development of Alzheimer’s disease, J. Neurosci. Res 95 (2017) 671–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Henderson VW, Brinton RD, Menopause and Mitochondria, Elsevier, 2010, pp. 77–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Yao J, Brinton RD, Estrogen Regulation of Mitochondrial Bioenergetics, Elsevier, 2012, pp. 327–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Nilsen J, Brinton RD, Mechanism of estrogen-mediated neuroprotection: Regulation of mitochondrial calcium and Bcl-2 expression, Proc. Natl. Acad. Sci. USA 100 (2003) 2842–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Rettberg JR, Yao J, Brinton RD, Estrogen: a master regulator of bioenergetic systems in the brain and body, Front. Neuroendocrinol 35 (2014) 8–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Misgeld T, Schwarz TL, Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture, Neuron 96 (2017) 651–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Mark Marciniak C., Eckel-Mahan K, McManus M, Crimi M, Waymire K, Chun, Masubuchi S, Friend N, Koike M, Chalkia D, Macgregor G, Sassone-Corsi P, Douglas, Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition, Cell 151 (2012) 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Lane N, Martin WF, Eukaryotes really are special, and mitochondria are why. 112: E4823–E4823, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Lopez-Otin C, Galluzzi L, Freije JM, Madeo F, Kroemer G, Metabolic control of longevity, Cell 166 (2016) 802–821. [DOI] [PubMed] [Google Scholar]
- [206].Finkel T, The metabolic regulation of aging, Nat. Med 21 (2015) 1416–1423. [DOI] [PubMed] [Google Scholar]
- [207].Loftus RM, Finlay DK, Immunometabolism: cellular metabolism turns immune regulator, J. Biol. Chem 291 (2016) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].O’Neill LAJ, Kishton RJ, Rathmell J, A guide to immunometabolism for immunologists, Nat. Rev. Immunol 16 (2016) 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Hotamisligil GS, Foundations of immunometabolism and implications for metabolic health and disease, Immunity 47 (2017) 406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Pence BD, Yarbro JR, Aging impairs mitochondrial respiratory capacity in classical monocytes, Exp. Gerontol 108 (2018) 112–117. [DOI] [PubMed] [Google Scholar]
- [211].Assmann N, Finlay DK, Metabolic regulation of immune responses: therapeutic opportunities, J. Clin. Investig 126 (2016) 2031–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Sunnucks P, Morales HE, Lamb AM, Pavlova A, Greening C, Integrative approaches for studying mitochondrial and nuclear genome co-evolution in oxidative phosphorylation, Front. Genet 8 (2017) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Quiros PM, Mottis A, Auwerx J, Mitonuclear communication in homeostasis and stress, Nat. Rev. Mol. Cell Biol 17 (2016) 213–226. [DOI] [PubMed] [Google Scholar]
- [214].Lopez JV, Yuhki N, Masuda R, Modi W, O’Brien SJ, Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat, J. Mol. Evol 39 (1994) 174–190. [DOI] [PubMed] [Google Scholar]
- [215].Timmis JN, Ayliffe MA, Huang CY, Martin W, Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes, Nat. Rev. Genet 5 (2004) 123–135. [DOI] [PubMed] [Google Scholar]