

Abstract
Vibrio cholerae can switch from a smooth to a wrinkled or rugose colony phenotype characterized by the secretion of a polysaccharide that enables the bacteria to survive harsh environmental conditions. In order to understand the genetic basis of rugosity, we isolated TnphoA-induced stable, smooth mutants of two O1 El Tor rugose strains and mapped the insertion sites in several of the mutants using a modified Y-adapter PCR technique. One of the TnphoA insertions was mapped to the first gene of the vps region that was previously shown to encode the rugose polysaccharide biosynthesis cluster. Three insertions were mapped to a previously unknown hlyA-like gene, also in the vps region. Five other insertions were found in loci unlinked to the vps region: (i) in the epsD gene (encodes the “secretin” of the extracellular protein secretion apparatus), (ii) in a hydG-like gene (encodes a ς54-dependent transcriptional activator similar to HydG involved in labile hydrogenase production in Escherichia coli, (iii) in a gene encoding malic acid transport protein upstream of a gene similar to yeiE of E. coli (encodes a protein with similarities to LysR-type transcriptional activators), (iv) in dxr (encodes 1-deoxy-d-xylulose 5-phosphate reductoisomerase), and (v) in the intergenic region of lpd and odp (encode enzymes involved in the pyruvate dehydrogenase complex formation). These data suggest the involvement of a complex regulatory network in rugose polysaccharide production and highlight the general utility of the Y-adapter PCR technique described here for rapid mapping of transposon insertion sites.
Vibrio cholerae, the etiologic agent of cholera, is a gram-negative bacterium and an environmental species that occupies a variety of aquatic niches (5). V. cholerae exists both as planktonic or free swimming forms and surface-attached biofilm communities (9). An epidemic cycle in humans is initiated with the ingestion of the bacteria through contaminated food or water, followed by colonization of the human intestinal tract and multiplication. Production of cholera toxin leads to the diarrheal symptoms and the eventual release of more bacteria into the environment.
Cholera exists in both endemic and epidemic forms. In countries where cholera is endemic, such as Bangladesh and India, cholera epidemics occur in seasonal peaks, and the bacteria seem to disappear during interepidemic periods and reappear simultaneously at multiple focal points during the next wave of the epidemic (11). Survival of the organism during interepidemic periods has long been a major question. It has been suggested that zoo- and phytoplanktons serve as reservoirs of the bacteria and that the bacteria enter into a viable but nonculturable state and yet retain their virulence (4, 7, 28, 41).
V. cholerae has also been shown to have another survival form, termed “rugose,” in which it produces an exopolysaccharide in response to different stress conditions (39, 40). The rugose polysaccharide has been shown to confer resistance to a variety of agents and conditions (chlorine, UV, starvation stress, and an acidic environment in the human intestine) and also to provide a gelatinous matrix in which the organism survives these extreme stress conditions (19, 27, 34, 35; E. W. Rice, C. J. Johnson, R. M. Clark, K. R. Fox, D. J. Reasoner, M. E. Dunnigan, P. Panigrahi, J. A. Johnson, and J. G. Morris, Jr., Letter, Lancet 340:740, 1992). Although the physiological significance of rugosity can be surmised from the many adaptive features conferred by rugose polysaccharide, the environmental signals that trigger its production are still poorly understood.
Strains belonging to many serogroups have been shown to switch from smooth to rugose with the exception of O1 classical biotype strains (unpublished data and reference 42). The genetic basis of this difference and that of the production and regulation of rugose polysaccharide are not fully understood at the present time. A region on the V. cholerae chromosome, termed vps, that encodes the rugose polysaccharide (termed EPSEl Tor) biosynthesis genes has been identified, although the individual genes and the organization of this region were not reported (42).
Spontaneous reversion of smooth to rugose and vice versa can occur at a low but detectable frequency which might be due to a poorly understood switching or phase variation mechanism (27). Conditions that induce rugose production have been identified: growth in alkaline peptone water (APW) (27) and minimal salts at low temperature (34). The normal medium used to select for V. cholerae (TCBS agar) precludes the isolation of the rugose form of V. cholerae (27).
We were interested in identifying the genetic regions involved in the rugose phenotype. Transposons have been used extensively for mutagenesis and deciphering the function of genes. We have previously reported the isolation of TnphoA-induced smooth mutants that lost the ability to produce rugose polysaccharide. Sixteen of the smooth mutants were characterized further. None of these reverted to the rugose phenotype even under rugose-inducing conditions such as growth in APW. Seven of the 16 mutants had more than one TnphoA insertion, and 14 of the 16 mutants had cointegration of the transposon delivery vector (1). As reported earlier by us, one of the mutants was mapped to the epsD gene by conventional cloning of the TnphoA junction and sequencing. Involvement of the extracellular protein secretion (EPS) pathway in rugose production was further demonstrated by construction of targeted epsD and epsE mutants and complementation of these mutants by the respective wild-type plasmid clones (1).
In our attempts to map the insertion sites in other mutants, we faced two problems. The first was that conventional mapping involved cloning of the TnphoA insertion utilizing the antibiotic marker (Kanr) present on the transposon (1, 3). This was time-consuming, and we were unsuccessful in cloning TnphoA insertions from strains that had cointegrates of the transposon delivery vector. The second problem we faced was that many of the mutants had multiple insertions and the transposon junctions appeared to have undergone genetic rearrangements, which rendered it even more difficult to clone and interpret the results. Various PCR-based approaches have been devised for the identification of transposon-flanking sequences, including inverse PCR (21), single-specific-primer PCR (31), and targeted gene-walking PCR using random primers (23). We devised an alternative PCR-based method, modified Y-adapter PCR (15, 24), for specific amplification of transposon junction sequences. This technique prevents nonspecific amplification and requires the sequence information of only transposon specific sequences. Additionally, this method can simultaneously amplify multiple insertions present within a mutant.
MATERIALS AND METHODS
The bacterial strains and culture conditions used here have been described earlier (1).
Isolation of TnphoA mutants and mapping of the TnphoA insertions.
TnphoA, on suicide vector pRT733 (33), was introduced by conjugation, into rugose isolates of strains C6706 and N16961. Transconjugants were plated onto Luria-Bertani agar containing kanamycin and polymyxin B to select for cells that acquired the transposon, TnphoA, and then screened for smooth colonies. These colonies were grown in APW (22) to check whether the shift from rugose to smooth is not due to phase variation but due to the inactivation of a gene essential for the rugose phenotype.
To determine the number and location of the TnphoA insertions in each mutant, pulsed-field gel electrophoresis (PFGE) (2) of SfiI-digested chromosomal DNAs of wild-type and mutant strains was carried out on a CHEF-DRII mapper (Bio-Rad Laboratories). The DNA was transferred onto nylon membrane (MSI, Westboro, Mass.) by capillary transfer and hybridized with a 32P-labeled 2.6-kb BglII TnphoA fragment under stringent conditions (16). Since SfiI does not cut within TnphoA, each hybridized band corresponded to a single insertion.
Y-adapter PCR.
The method involves four steps (Fig. 1): (i) restriction digestion of the chromosomal DNA of the transposon containing strain with a four-base-cutting restriction enzyme in order to obtain fairly short fragments; (ii) formation of a Y-adapter; (iii) ligation of the Y-adapter to the digested DNA; and (iv) PCR amplification with a Y-adapter and transposon specific primers, so that only the transposon junction fragments are amplified. In our experiments, 25 to 30 ng of chromosomal DNA was digested with Sau3A1 to completion at 37°C for 2 h (Fig. 1, step 1). The adapter was prepared by the following procedure. The sequences of the oligonucleotides were as follows: oligonucleotides A1, 5′-TAGCGTCCGGCGCAGCGACGGCCAG-3′ (the noncomplementary Y region is italicized and the Y-adapter primer sequence is underlined), and A2, 5′-GATCCTGGCCGTCGGCTGTCTGTCGGCGC-3′. A total of 1 μg of oligonucleotide A2 was phosphorylated at the 5′ end using T4 polynucleotide kinase (PNK). After phosphorylation, PNK was denatured and 1 μg of oligonucleotide A1 was added along with 10× annealing buffer (1 M NaCl; 100 mM Tris-HCl, pH 8.0; 10 mM EDTA, pH 8.0) in a final volume of 20 μl. This mixture was then heated at 65°C for 10 min, followed by slow cooling to room temperature for 30 min, resulting in the formation of Y-adapter at a final concentration of 100 ng/μl (Fig. 1, step 2). The Y-adapter has a noncomplementary region on one end and a Sau3A1 sticky end (italicized and underlined in oligonucleotide A2) on the other end. A total of 5 ng of this DNA was ligated to 100 ng of the Y-adapter in a 5-μl reaction volume at 16°C overnight. After ligation, the reaction mixture was diluted with water to a final volume of 80 μl and heated at 65°C for 10 min to inactivate T4 DNA ligase (Fig. 1, step 3). Then, 2-μl aliquots were used as template for PCR. The PCR primers were as follows: adapter primer, 5′-TAGCGTCCGGCGCAGCGAC-3′ (underlined part of oligonucleotide A1); and Tn primer, 5′-GAAAGGTTCCGTTCAGGA-3′. The adapter primer sequence is of the same strand as the noncomplementary 5′ Y region and therefore cannot anneal to the adapter itself. The fragments that do not have the TnphoA end sequence will not be amplified in the PCR. However, the Tn primer anneals to the fragments containing transposon end sequences during the first PCR cycle and extends DNA synthesis into the Y region of the ligated Y adapter (Fig. 1, step 4a); thus, the adapter primer can now anneal and synthesize the second strand (Fig. 1, step 4b). In subsequent cycles (Fig. 1, step 4c) the two primers can selectively amplify the fragments containing TnphoA junction sequences under stringent PCR conditions. Since the transposon ends are inverted repeats [the DNA sequences are the same except for a single mismatch at the transposon left end: 5′-GAAAGGTTCCGT(T/C)CAGGA-3′], the primer can anneal to two Sau3A1 fragments (i.e., the transposon left and right end fragments). As a result, for each TnphoA insertion two PCR products are obtained. The PCR mixture (20 μl) consisted of 2 μl of the template, 2 μl of the 10× PCR buffer (Boehringer Mannheim Corp.), 1.5 mM MgCl2, 50 μM concentrations of the deoxynucleoside triphosphates, a 200 nM concentration of each primer, and 1 U of AmpliTaq. The PCR consisted of 28 cycles of 94°C for 30 s, 56°C for 2 min, and 72°C for 30 s. The PCR products were cloned into pCR2.1 vector (Invitrogen Corp.) and sequenced using T7 and M13 reverse primers.
FIG. 1.

Schematic of the Y-adapter PCR. In step 1, chromosomal DNAs of the TnphoA insertion containing strains are digested with restriction enzyme Sau3A1. In step 2, a Y-adapter linker (the Y region is noncomplementary) is prepared from oligonucleotides A1 and A2. In step 3, the Y-adapter is ligated to the restriction fragments. In step 4, the adapter-ligated DNAs are used as templates for PCRs with a Y-adapter (indicated as the 5′ PCR primer [P]) and a transposon-specific primer. Since the 5′ primer sequence is from the same strand as the Y region, it requires a first round of PCR with the Tn primer (step 4a). The resulting single-stranded DNA serves as the template for annealing of the “P primer” in the second round of PCR (step 4b). The resulting PCR products from subsequent cycles are cloned and sequenced to identify the transposon junction.
Construction of a hydG::Kanr mutant.
A 2.6-kb fragment containing the hydG gene and
the gene upstream of it (lysyl tRNA synthetase) was PCR amplified
(primers
5′-TCC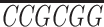 GTCGGTGGTTTTGATCGTGT-3′
and
5′-CGGGATCCCGCTAAGTCAGAGTTTTTATCGC-3′),
and the PCR product was digested with SacII and
BamHI and cloned into similarly digested pBluescript vector.
The resulting plasmid pAA2311 was digested with SphI and
MluNI and blunt ended with T4 DNA polymerase, which created
a small 26-bp deletion in the hydG open reading frame (ORF);
in its place a Kanr cassette (SmaI fragment)
from pUC18k3 (18) was introduced. The resulting plasmid
pAA2348 was digested with SacII-BamHI, and the
3.4-kb fragment containing hydG::Kanr
was blunt ended and ligated to SalI-digested and blunt-ended
pCVD442 (6). The resulting plasmid, pAA263, was used to
introduce the hydG::Kanr knockout into
the rugose strain (N16961-R) as described earlier (1).
GTCGGTGGTTTTGATCGTGT-3′
and
5′-CGGGATCCCGCTAAGTCAGAGTTTTTATCGC-3′),
and the PCR product was digested with SacII and
BamHI and cloned into similarly digested pBluescript vector.
The resulting plasmid pAA2311 was digested with SphI and
MluNI and blunt ended with T4 DNA polymerase, which created
a small 26-bp deletion in the hydG open reading frame (ORF);
in its place a Kanr cassette (SmaI fragment)
from pUC18k3 (18) was introduced. The resulting plasmid
pAA2348 was digested with SacII-BamHI, and the
3.4-kb fragment containing hydG::Kanr
was blunt ended and ligated to SalI-digested and blunt-ended
pCVD442 (6). The resulting plasmid, pAA263, was used to
introduce the hydG::Kanr knockout into
the rugose strain (N16961-R) as described earlier (1).
RESULTS
Y-adapter PCR mapping of TnphoA insertion sites.
We mapped the TnphoA insertion junctions in 16 mutants using the Y-adapter PCR method. Figure 2 shows the results of the PCR from seven of the TnphoA insertion mutants. Since the transposon-specific primer can anneal to both ends of the transposon, we obtained two fragments in four of the reactions (NS1, NS25, S11, and S16). In three other reactions (S1, S13, and S20) only one product was obtained, and it is possible that one of the TnphoA junctions is missing due to genetic rearrangements, although doublets (fragments of the same size) resulting from amplification of both the TnphoA ends cannot be ruled out. We have determined the identity of the PCR products by subcloning and sequencing the PCR products. In the case of NS1, as reported by us earlier by conventional cloning of the TnphoA junction, the PCR fragment yielded the expected sequence of the epsD gene (1). The other genes identified from the sequences of the PCR fragments and their products based on similarities to other proteins in the database are presented in Table 1.
FIG. 2.

Agarose gel electrophoresis of the Y-adapter PCR products. Size marker, 1-kb ladder. In four PCRs, the two PCR products resulting from amplification of the left and right TnphoA junctions can be seen. In three reactions, only one product is seen (S1, S13, and S20). The asterisk indicates the position of the primers.
TABLE 1.
Mapping of TnphoA insertion sites by Y-adapter PCR
| Mutant | No. of TnphoA insertions | Amplicon size (bp) | Gene | Similar protein/organism | Putative function | Reference or accession no. |
|---|---|---|---|---|---|---|
| NS1 | 1 | 234 | epsD 3′ | EpsD/Vibrio cholerae | Type II secretion | 1 |
| 410 | epsD 5′ | EpsD/Vibrio cholerae | Type II secretion | |||
| NS25 | 1 | 300 | hydG 3′ | HydG/Escherichia coli | Response regulator of hydrogenase 3 | P14375 |
| 400 | hydG 5′ | HydG/Escherichia coli | Response regulator of hydrogenase 3 | |||
| S1 | 1 | 700 | hypothetical | Y576/Methanococcus jannaschii | Malic acid transporter protein | Q57996 |
| S2 | 1 | 700 | dxr | Dxr/Escherichia coli | DXP reductoisomerase | P45568 |
| S7 | 1 | 700 | tetR | Tet protein | Tetracycline resistance | AAA77664 |
| S8 | 1 | 700 | tetR | Tet protein | Tetracycline resistance | AAA77664 |
| 400 | hly | Hemolysin/Vibrio cholerae | Hemolysin-cytolysin precursor | CAA36035 | ||
| S10 | 1 | 500 | lpd | DldH/Escherichia coli | Dihydrolipoamide dehydrogenase | P00391 |
| 300 | odp | Odp2/Escherichia coli | Dihydrolipoamide acetyltransferase | P06959 | ||
| S11 | 2 | 580 | Not done | |||
| 700 | tetR | Tet protein | Tetracycline resistance | AAA77664 | ||
| S13 | 2 | 700 | hly | Hemolysin/Vibrio cholerae | Hemolysin-cytolysin precursor | CAA36035 |
| S16 | 2 | 600 | RNA-dependent RNAP | AAC96311 | ||
| 700 | vps | EPSEl Tor biosynthesis | AAF04237 | |||
| S17 | 1 | 700 | Vector (?) | |||
| 380 | hlyA | Hemolysin/Vibrio cholerae | Hemolysin-cytolysin precursor | CAA36035 | ||
| S18 | 2 | None | Primer dimer | |||
| S20 | 1 | 700 | tetR | Tet protein | Tetracycline resistance | AAA77664 |
Mutations in S3 and S4 (two insertions), as well as S14 (three insertions), could not be mapped since there was no PCR amplification in these reactions. Insertions in mutants S7, S8, S11, S17, and S20 amplified a Tet fragment or a vector sequence, which is part of the transposon delivery vector. In S18, the PCR-amplified fragment upon sequencing turned out to be the primer dimer. In some of the mutants PCR amplification did not yield the two or four expected fragments (S1, S2, S7, and S20 [expected two fragments] and S11, S13, S16, and S18 [four fragments] which is probably due to rearrangements at the TnphoA insertion site on one or both junctions. However, 9 of the 16 mutants could be mapped, and the inactivated genes are described below.
vps region.
We expected that many of our TnphoA insertion sites would map within the vps region recently reported by Yildiz and Schoolnik (42). These authors reported the mapping of multiple mini-Tn5 km2-induced smooth mutants to a single region on the V. cholerae chromosome. However, the organization of the vps region was not reported. Using the recently released whole genome sequence of V. cholerae (8) (http://www.tigr.org/tdb/mdb/mdb.html), we analyzed the vps region. The entire region is 26,458 bp located adjacent to acrD encoding acriflavin resistance at the left junction. The gene immediately downstream of the vps region at the right junction shows very weak similarity (24%) to hypothetical proteins in Bacillus subtilis (yitA and yuxA), followed by glyA that encodes serine hydroxymethyltransferase. The ORFs within the vps region and the predicted functions of the proteins are shown in Fig. 3. The vps region has two clusters (ORFs VC0916 to VC0927 and ORFs VC0934 to VC0937) with similarities to known polysaccharide biosynthesis genes interrupted by a cluster (ORFs VC0928 to VC0933) encoding seemingly unrelated functions. The genes downstream of ORF VC0937 do not appear to be involved in polysaccharide biosynthesis. Upstream of the vps cluster between acrD (VC0914) and ORF VC0916 is a 426-bp intergenic region that contains putative ς70 promoter elements.
FIG. 3.

Genetic organization of the VPS region. The DNA sequence of the V. cholerae VPS region was obtained from the TIGR microbial genome database. The entire region in the map is 33,024 bp, and the vps region is 26,458 bp. Sequence analysis was done by the NCI BLAST server program, and the recently published annotation of the vps region (8) has been used to define the products of the ORFs. acrD (VC0914) is at the left junction of the vps cluster, and yitA (VC0940) and glyA (VC0941) are at the right junction. There is a 426-bp intergenic region containing the vps promoter. There are 22 ORFs arranged in three clusters: ORFs VC0916 to VC0927 and ORFs VC0934 to VC0937, separated by the cluster of ORFs from VC0927 to VC0933. The middle cluster genes apparently encode functions unrelated to polysaccharide biosynthesis. The arrowheads indicate the positions of the TnphoA insertions in the vps region. The three hly::TnphoA insertions were in close proximity to each other.
Location of TnphoA insertions within the vps region.
The TnphoA insertion in mutant S16 was found to be in the first ORF of the vps region (42). The mini-Tn5 insertion in the vps3 mutant of Yildiz and Schoolnik (42) was found in this ORF as well. A second fragment from this mutant mapped to a gene with a weak similarity to RNA-dependent RNA polymerase, and the significance of this is currently unknown. Sequencing of the TnphoA junctions of the insertions in S8, S13, and S17 indicated similarities to an hlyA gene, indicating that these are insertions in ORF VC0930. The TnphoA insertions in these hly mutants (hlyA::TnphoA) probably affect rugose expression by a polar effect on the polysaccharide biosynthesis genes (ORFs VC0934 to VC0937) downstream of it.
Mutations that mapped outside of the vps region.
Five TnphoA insertions mapped outside of the vps region. We demonstrated earlier the involvement of one of the regions, eps (VC2723 to VC2734), in rugose production (1). Here we report the identification of the other genes.
hydG.
The insertion in one of the TnphoA mutants (NS25) was mapped to a gene (ORF VC0665) whose product has similarities to HydG, which is involved in labile hydrogenase expression in Escherichia coli (32). HydG belongs to the two-component signal transduction system of the NtrBC family (13, 20). A fragment of a gene (designated s54act5) similar to hydG in V. cholerae was identified in a search for ς54-dependent transcriptional activators by degenerate primer PCR, and it was further shown that a mutation in this gene did not affect colonization in an infant mouse model (12). Our data (described below) suggests that hydG (s54act5) is involved in rugose production.
Construction of a hydG::Kanr mutant and complementation of the mutant.
Earlier it has been shown that inactivation of hydG (s54act5 ORF) does not affect colonization of V. cholerae in an infant mouse model and therefore suggested a different role for this transcriptional activator (12). The TnphoA insertion in the NS25 smooth mutant suggested that HydG might be involved in rugose production. In order to test this idea and further validate the reliability of the Y-adapter technique (that it is not a nonspecific amplification of a random fragment), we constructed a targeted hydG mutant and verified the effect of the mutation on the rugose phenotype. We obtained the complete hydG gene sequence of V. cholerae from the microbial genome database of the Institute for Genomic Research (TIGR) and constructed a targeted deletion insertion mutant of this gene as described in Materials and Methods. The suicide plasmid carrying the hydG::Kanr gene (pAA263) was introduced into the rugose variant of an El Tor strain, N16961. Selection for the chromosomal knockout was carried out as described earlier (1). The resulting hydG::Kanr mutant was smooth, indicating that hydG positively regulates rugose expression. Unlike the epsD::Kanr mutant, which exhibited a partial secretion phenotype (the colonies are opaque), the hydG::Kanr mutant was completely smooth and translucent (Fig. 4). This defect could be complemented by the hydG+ plasmid pAA2311, as was the original TnphoA mutant, NS25. Similarly, the epsD::Kanr mutant AA10 could also be complemented by an epsD+ plasmid (pDSK-2) (Fig. 4).
FIG. 4.

Colony morphology of wild-type smooth and rugose variants of the N16961 strain. The left panel shows the streak plates, and the right panels show the magnified individual colonies on the respective plates. (Top panel) N16961 R, wild-type rugose; N16961 S, wild-type smooth. (Middle panel) NS25 and AA54, hydG::TnphoA and hydG::Kanr mutants, respectively; AA10, epsD::Kanr. Only AA54 and AA10 colonies are shown in the middle right panel. The NS25 colonies were similar to the AA54 colonies. (Bottom panel) AA10 complemented by the epsD+ plasmid pDSK-2 and NS25 and AA54 complemented by hydG+ plasmid. pAA435 is a pWSK29 derivative containing the hydG+ gene (SacII-BamHI) from pAA2311 (see the text).
Other genes.
Three TnphoA insertions were found in genes whose effect on inhibiting rugose production is not quite obvious by the predicted function of the product of the gene inactivated. This could be explained by polar effects on genes downstream of the insertion site or by an as-yet-unidentified role for that gene in rugose expression. These insertions are described below, and their role in rugose production is only speculative at the present time.
yeiE.
The TnphoA insertion site in mutant S1 was found to be in a gene encoding a malic acid transport protein. However, closer examination of the V. cholerae DNA sequence in the vicinity of the TnphoA insertion (obtained from TIGR microbial genome database [8]) indicated that, immediately downstream of the TnphoA insertion site, there is an ORF VC2324 (yeiE) that encodes a protein with similarities to LysR-type transcriptional activators (30). We hypothesize that the S1 TnphoA insertion affects the expression of yeiE, which in turn affects the expression of rugose biosynthesis genes.
lpd and odp.
The TnphoA insertion in mutant S10 was found to be in the intergenic region of the lpd and odp genes (ORFs VC2412 and VC2413), which encode the dihydrolipoamide dehydrogenase and acetyltransferase enzymes, respectively. In E. coli, these two enzymes, along with pyruvate dehydrogenase, form the pyruvate dehydrogenase complex, which catalyzes the NAD-linked oxidative decarboxylation of pyruvate to acetyl coenzyme A and CO2 (25). Downstream of the lpd and odp genes, several genes resembling polysaccharide biosynthesis genes (VC2407 to VC2420) are present (8). The genetic organization of the lpd region (yadF-hpt-opaR-lpd) in V. parahaemolyticus is very interesting. In this organism, upstream of the lpd gene is a luxR homolog, opaR, which has been implicated in opaque-to-translucent switching (17). The expression of OpaR is regulated by genetic rearrangements (small deletion) in the promoter region of the opaR gene (17). The opaR homolog in V. cholerae, hapR, has the organization yadF-hpt-hapR (ORFs VC0586 to VC0584), and the genes downstream of hapR are different (10); we found from analyzing the V. cholerae genome sequence (obtained from the TIGR genome database) that the lpd gene is located in another part of the chromosome.
dxr.
The TnphoA insertion in mutant S2 mapped to the dxr gene (ORF VC2254) that encodes the enzyme 1-deoxy-d-xylulose 5-phosphate reductoisomerase. This enzyme simultaneously catalyzes the intramolecular rearrangement and reduction of 1-deoxy-d-xylulose 5-phosphate (DXP) to form 2-C-methyl-d-erythritol 4-phosphate (MEP). It constitutes a key enzyme of an alternative mevalonate-independent pathway for isopentenyl diphosphate biosynthesis (14). Examination of the genes in the vicinity of dxr revealed genes resembling polysaccharide biosynthesis genes (VC2247 to VC2250), raising the possibility of a polar effect of this TnphoA insertion.
DISCUSSION
Rapid mapping of transposon insertion sites.
The Y-adapter PCR technique described in this report is a very useful method for rapid mapping of any transposon insertion junction, especially where there are multiple insertions within the same mutant. We have identified insertions in genes known to be involved in rugose production such as vps and several new genes such as an hlyA-like gene, eps, and hydG. Insertion mutants of genes whose products are not so obviously involved in rugose production, such as lpd-odp, yeiE, and dxr, will be reconstructed by targeted nonpolar Kanr cassette mutagenesis, and the phenotypes will be verified before drawing any conclusion. Also, strains with multiple insertions need to be linked to phenotypes. Hence, at this time the involvement of these three genes in rugose production is only speculative.
Of the 10 mutants that hybridized to a 291-kb fragment in PFGE and Southern analysis (1), only 1 (NS1) had a TnphoA insertion at the epsD locus (1). The other nine mutants appear to have insertions in other loci since they did not amplify epsD in the present study with Y-adapter PCR. It is possible that these nine insertions might be in loci unlinked to the eps operon in the same fragment. Also, SfiI digestion of the V. cholerae chromosome results in multiple fragments in the 291-kb range. Some of the nine insertions might be in the other fragments in the 291-kb range.
Some of the mutants that hybridized to a 291-kb fragment had more than one insertion (S3, S4, S11, S13, S14, and S16). Yet these mutants did not yield the expected number of PCR fragments (i.e., two for each insertion). We speculate that this is due to the loss of transposon ends at the insertion site. It is possible that the 291-kb insertion in the eps operon had lost the transposon ends and hence did not amplify the eps insertion junction.
Finally, one cautionary note on the use of Y-adapter PCR for mapping multiple insertions in the same mutant: the Y-adapter PCR products from mutants with multiple insertions might yield fragments of the same or similar sizes that may not be resolved on an agarose gel (Fig. 2). One may have to sequence multiple clones of the PCR products to rule out this possibility. Alternatively, one may have to use different enzymes (Sau3A1 was used in the present study) to digest the genomic DNA in the first step of the Y-adapter PCR protocol which might result in different-sized junction fragments for each insertion.
Working model for vps gene cluster expression and regulation.
Based on all the available data, we propose the following working model for the regulation of synthesis and secretion of rugose polysaccharide. V. cholerae possesses a distinct set of biosynthesis genes (vps operon) for rugose polysaccharide. The expression of the vps operon is probably governed by multiple regulatory circuits (environmental signals such as nutrient limitation and/or starvation, UV light, chemicals, and other stresses and also by cell density-dependent signals). Watnik and Kolter proposed that the formation of a biofilm is initiated when bacteria swim to an abiotic surface using the polar flagella and loosely attach to the surface with type IV pili (MSHA), resulting in the formation of microcolonies (36–38). We hypothesize that when the cell density increases in the microcolony, cell density-dependent signaling occurs. We speculate that the quorum-sensing signal molecules (homoserine lactones) activate a membrane-bound sensor protein, most likely by phosphorylation. This signal in turn is transmitted to a relay protein, perhaps HydG, that transmits the signal to HapR. HapR is a LuxR-type regulatory protein involved in positive regulation of hemagglutinin-protease gene hap (10). Since HapR appears to be a negative regulator of the vps operon (hapR mutants express rugose polysaccharide constitutively) (10), we hypothesize that HapR in a nonphosphorylated state acts as the repressor of the vps operon. When the phosphorylation signal is transferred from HydG to HapR (phosphorylated state), the vps operon is derepressed.
A second level of regulation is exerted by the type II protein secretion system, since eps mutants are defective in rugose production. We hypothesize that the eps mutations affect rugose production by affecting the membrane localization of the sensor protein or another unidentified secreted protein. Eps mutants are known to have an altered outer membrane protein profile (29). This can profoundly alter the signal transduction process. Our present data, however, do not rule out a direct role for the EPS system in the secretion of rugose polysaccharide. In other words, rugose polysaccharide may utilize the type II protein secretion channel for its secretion.
The LysR-type transcriptional activator, YeiE, may exert another level of control. LysR-type transcriptional activators act on divergently transcribed operons (26). The vps operon does not contain any divergently transcribed polysaccharide biosynthesis genes, although there are two divergent promoters (between VC0928 and VC0929 and between VC0930 and VC0931) in the middle cluster (Fig. 3). The significance of this genetic arrangement remains to be determined. It is also possible that the YeiE target is not the vps operon and that its effect on rugose production is due to yet another regulator.
In addition to cell density-dependent signaling, there is a switching mechanism that acts either directly at the vps promoter or indirectly through an intermediate that is regulated by switching. One of the ways that this can be effected is by an “on-off” expression of the repressor HapR itself. In V. parahaemolyticus, the expression of OpaR (LuxR homolog) is controlled by genetic rearrangements in the opaR promoter region. OpaR in turn affects the shift from the opaque to the translucent phenotype (17). There may be other environmental signaling pathways, such as nutrient limitations, stringent response, and SOS regulation, that control rugose production. Our future studies will be aimed at investigating these various pathways.
ACKNOWLEDGMENTS
We thank Lisa Sadzewicz (University of Maryland Biopolymer Laboratory) for DNA sequencing, Jim Kaper for useful comments on the manuscript, and Shafaq Presswala for excellent technical assistance.
This work was supported by a VA/DOD grant on emerging infectious diseases to J.G.M., a Department of Veterans Affairs grant to J.A.J., PHS grant AI 35729 to J.A.J., and a University of Maryland intramural grant to S.S.
REFERENCES
- 1.Ali A, Johnson J A, Franco A A, Metzger D J, Connell T D, Morris J G, Jr, Sozhamannan S. Mutations in the extracellular protein secretion pathway genes (eps) interfere with rugose polysaccharide production in and motility of Vibrio cholerae. Infect Immun. 2000;68:1967–1974. doi: 10.1128/iai.68.4.1967-1974.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cameron D N, Khambaty F M, Wachsmuth I K, Tauxe R V, Barrett T J. Molecular characterization of Vibrio choleraeO1 strains by pulsed-field gel electrophoresis. J Clin Microbiol. 1994;32:1685–1690. doi: 10.1128/jcm.32.7.1685-1690.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiang S L, Mekalanos J J. Use of signature tagged transposon mutagenesis to identify Vibrio choleraegenes critical for colonization. Mol Microbiol. 1998;27:797–805. doi: 10.1046/j.1365-2958.1998.00726.x. [DOI] [PubMed] [Google Scholar]
- 4.Colwell R R, Brayton P R, Grimes D J, Roszak D B, Huq A, Palmer L M. Viable but non-culturable Vibrio choleraeand related pathogens in the environment: implications for release of genetically engineered microorganisms. Bio/Technology. 1985;3:817–820. [Google Scholar]
- 5.Colwell R R, Huq A. Vibrios in the environment: viable but nonculturable Vibrio cholerae. In: Wachsmuth I K, Blake P A, Olsvik O, editors. Vibrio cholerae and cholera: molecular to global perspectives. Washington, D.C.: ASM Press; 1994. pp. 117–133. [Google Scholar]
- 6.Donnenberg M S, Kaper J B. Construction of an eae deletion mutant of enteropathogenic Escherichia coliby using a positive selection suicide vector. Infect Immun. 1991;59:4310–4317. doi: 10.1128/iai.59.12.4310-4317.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Felsenfeld O. The survival of cholera vibrios. In: Barua D, Burrows W, editors. Cholera. Philadelphia, Pa: The W.B. Saunders Co.; 1974. pp. 359–366. [Google Scholar]
- 8.Heidelberg J F, Eisen J A, Nelson W C, Clayton R A, Gwinn M L, Dodson R J, Haft D H, Hickey E K, Peterson J D, Umayam L, Gill S R, Nelson K E, Read T D, Tettelin H, Richardson D, Ermolaeva M D, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann R D, Nierman W C, White O. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature. 2000;406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Islam M S, Drasar B S, Sack R B. The aquatic flora and fauna as reservoirs of Vibrio cholerae: a review. J Diarrhoeal Dis Res. 1994;12:87–96. [PubMed] [Google Scholar]
- 10.Jobling M G, Holmes R K. Characterization of hapR, a positive regulator of the Vibrio cholerae HA/protease gene hap, and its identification as a functional homologue of the Vibrio harveyi luxRgene. Mol Microbiol. 1997;26:1023–1034. doi: 10.1046/j.1365-2958.1997.6402011.x. [DOI] [PubMed] [Google Scholar]
- 11.Kaper J B, Morris J G, Jr, Levine M M. Cholera. Clin Microbiol Rev. 1995;8:48–86. doi: 10.1128/cmr.8.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klose K E, Novik V, Mekalanos J J. Identification of multiple ς54-dependent transcriptional activators in Vibrio cholerae. J Bacteriol. 1998;180:5256–5259. doi: 10.1128/jb.180.19.5256-5259.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kustu S, North A K, Weiss D S. Prokaryotic transcriptional enhancers and enhancer-binding proteins. Trends Biochem Sci. 1991;16:397–402. doi: 10.1016/0968-0004(91)90163-p. [DOI] [PubMed] [Google Scholar]
- 14.Kuzuyama T, Takahashi S, Takagi M, Seto H. Characterization of 1-deoxy-d-xylulose 5-phosphate reductoisomerase, an enzyme involved in isopentenyl diphosphate biosynthesis, and identification of its catalytic amino acid residues. J Biol Chem. 2000;275:19928–19932. doi: 10.1074/jbc.M001820200. [DOI] [PubMed] [Google Scholar]
- 15.Kwon Y M, Ricke S C. Efficient amplification of multiple transposon flanking sequences. J Microbiol Methods. 2000;41:195–199. doi: 10.1016/s0167-7012(00)00159-7. [DOI] [PubMed] [Google Scholar]
- 16.Maniatis T, Fritsch E F, Sambrook J. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1982. [Google Scholar]
- 17.McCarter L L. OpaR, a homolog of Vibrio harveyi LuxR, controls opacity of Vibrio parahaemolyticus. J Bacteriol. 1998;180:3166–3173. doi: 10.1128/jb.180.12.3166-3173.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Menard R, Sansonetti P J, Parsot C. Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexnerientry into epithelial cells. J Bacteriol. 1993;175:5899–5906. doi: 10.1128/jb.175.18.5899-5906.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizunoe Y, Wai S N, Takede A, Yoshida S-I. Isolation and characterization of rugose form of Vibrio choleraeO139 strain MO10. Infect Immun. 1999;67:958–963. doi: 10.1128/iai.67.2.958-963.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morett E, Segovia L. The ς54bacterial enhancer-binding protein family: mechanism of action and phylogenetic relationship of their functional domains. J Bacteriol. 1993;175:6067–6074. doi: 10.1128/jb.175.19.6067-6074.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ochman H, Gerber A S, Hartl D L. Genetic applications of an inverse polymerase chain reaction. Genetics. 1988;120:621–623. doi: 10.1093/genetics/120.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pal S C. Laboratory diagnosis. In: Barua D, Greenough III W B, editors. Cholera. New York, N.Y: Plenum Medical Book Co.; 1992. pp. 229–251. [Google Scholar]
- 23.Parker J D, Rabinovitch P S, Burmer G C. Targeted gene walking polymerase chain reaction. Nucleic Acids Res. 1991;19:3055–3060. doi: 10.1093/nar/19.11.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prashar Y, Weissman S M. Analysis of differential gene expression by display of 3′ end restriction fragments of cDNAs. Proc Natl Acad Sci USA. 1996;93:659–663. doi: 10.1073/pnas.93.2.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quail M A, Haydon D J, Guest J R. The pdhR-aceEF-lpd operon of Escherichia coliexpresses the pyruvate dehydrogenase complex. Mol Microbiol. 1994;12:95–104. doi: 10.1111/j.1365-2958.1994.tb00998.x. [DOI] [PubMed] [Google Scholar]
- 26.Rhee K Y, Opel M, Ito E, Hung S-P, Arfin S M, Hatfield G W. Transcriptional coupling between the divergent promoters of a prototypic LysR-type regulatory system, the ilvYC operon of Escherichia coli. Proc Natl Acad Sci USA. 1999;96:14294–14299. doi: 10.1073/pnas.96.25.14294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rice E W, Johnson C H, Clark R M, Fox K R, Reasoner D J, Dunnigan M E, Panigrahi P, Johnson J A, Morris J G., Jr Vibrio choleraeO1 can assume a “rugose” survival form that resists killing by chlorine, yet retains virulence. Int J Environ Health Res. 1993;3:89–98. [Google Scholar]
- 28.Roszak D B, Colwell R R. Survival strategies of bacteria in the natural environment. Microbiol Rev. 1987;51:365–379. doi: 10.1128/mr.51.3.365-379.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sandkvist M, Michel L O, Hough L P, Morales V M, Bagdasarian M, Koomey M, DeRita V J, Bagdasarian M. General secretion pathway (eps) genes required for toxin secretion and outer membrane biogenesis in V. cholerae. J Bacteriol. 1997;179:6994–7003. doi: 10.1128/jb.179.22.6994-7003.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schell M A. Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol. 1993;47:597–626. doi: 10.1146/annurev.mi.47.100193.003121. [DOI] [PubMed] [Google Scholar]
- 31.Shyamala V, Ames G F. Genome walking by single-specific-primer polymerase chain reaction. Gene. 1989;84:1–8. doi: 10.1016/0378-1119(89)90132-7. [DOI] [PubMed] [Google Scholar]
- 32.Stoker K, Reijnders W N, Oltmann L F, Stouthamer A H. Initial cloning and sequencing of hydHG, an operon homologous to ntrBC and regulating the labile hydrogenase activity in Escherichia coliK-12. J Bacteriol. 1989;171:4448–4456. doi: 10.1128/jb.171.8.4448-4456.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor R K, Manoil C, Mekalanos J J. Broad-host-range vectors for delivery of TnphoA: use in genetic analysis of secreted virulence determinants of Vibrio cholerae. J Bacteriol. 1989;171:1870–1878. doi: 10.1128/jb.171.4.1870-1878.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wai S N, Mizunoe Y, Takade A, Kawabata S-I, Yoshida S-I. Vibrio choleraeO1 strain TSI-4 produces the exopolysaccharide materials that determine colony morphology, stress resistance, and biofilm formation. Appl Environ Microbiol. 1998;64:3648–3655. doi: 10.1128/aem.64.10.3648-3655.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wai S N, Mizunoe Y, Yoshida S-I. How Vibrio choleraesurvive during starvation. FEMS Microbiol Lett. 1999;180:123–131. doi: 10.1111/j.1574-6968.1999.tb08786.x. [DOI] [PubMed] [Google Scholar]
- 36.Watnick P I, Fuller K J, Kolter R. A role for the mannose sensitive hemagglutinin in biofilm formation by Vibrio choleraeEl Tor. J Bacteriol. 1999;181:3606–3609. doi: 10.1128/jb.181.11.3606-3609.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watnick P I, Kolter R. Steps in the development of a Vibrio choleraeEl Tor biofilm. Mol Microbiol. 1999;34:586–595. doi: 10.1046/j.1365-2958.1999.01624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watnick P I, Kolter R. Biofilm, city of microbes. J Bacteriol. 2000;182:2675–2679. doi: 10.1128/jb.182.10.2675-2679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.White P B. The rugose variant of vibrios. J Pathol Bacteriol. 1938;46:1–6. [Google Scholar]
- 40.White P B. The characteristic hapten and antigen of rugose races of cholera and El Tor vibrios. J Pathol Bacteriol. 1940;50:160–164. [Google Scholar]
- 41.Xu H-S, Roberts N, Singleton F L, Attwell R W, Grimes D J, Colwell R R. Survival and viability of non-culturable Escherichia coli and Vibrio choleraein the estuarine and marine environment. Microb Ecol. 1982;8:313–323. doi: 10.1007/BF02010671. [DOI] [PubMed] [Google Scholar]
- 42.Yildiz F H, Schoolnik G K. Vibrio choleraeO1 El Tor: identification of a gene cluster required for the rugose colony type, exopolysaccharide production, chlorine resistance, and biofilm formation. Proc Natl Acad Sci USA. 1999;96:4028–4033. doi: 10.1073/pnas.96.7.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]