

Abstract
Purpose
The aim of this study was to evaluate changes in body weight, liver weight, blood glucose, liver injury markers, pro-inflammatory factors and oxidative stress marker levels in obese mice with HFD induced NAFLD after semaglutide use.
Patients and methods
The 24 C57BL6J mice were randomly divided into three groups (NCD, HFD and Sema) for the assessment of metabolic status, inflammatory factor and oxidative stress marker levels, liver histopathology in mice. Liver metabolomics was determined by liquid chromatography/mass spectrometry (LC-MS) method.
Results
The mice body weight, liver weight, blood glucose, TG, TCHO, LDL and pro-inflammatory factors were significantly reduced after semaglutide. Meanwhile, semaglutide increased the SOD level. Semaglutide treatment significantly improved the pathological changes such as hepatocyte steatosis, balloon degeneration and lymphoid foci by HE. It also significantly reduced lipid droplet by Oil Red O. The mitochondria were swollen, the volume increased, the cristae were partially broken and reduced, the intramembrane matrix was partially dissolved, and the mitophagy structure was visible in the visual field. There were 6 metabolites down-regulated and 2 metabolites significantly up-regulated after semaglutide treatment.
Conclusions
Semaglutide can reduce blood glucose level and liver fat accumulation and play an anti-inflammatory role in advanced NAFLD that due to the effects of HFD.
Keywords: semaglutide, hepatic, NAFLD, hepatic fibrosis, metabolomics
Introduction
Nonalcoholic fatty liver disease (NAFLD) is becoming an increasingly prevalent form of chronic liver disease in the Western world. NAFLD is a multifactorial clinicopathological syndrome characterized by hepatocyte lipid accumulation and hepatocyte steatosis, including simple fatty liver and nonalcoholic steatohepatitis (NASH). NAFLD can progress from simple steatosis (i.e., nonalcoholic fatty liver [NAFL]) to NASH, cirrhosis, and cancer. In addition to steatosis, NASH is characterized by inflammation, ballooning degeneration, and fibrosis, which ultimately leads to liver fibrosis and end-stage cirrhosis (1). Hepatocellular carcinoma in the setting of non-cirrhotic nonalcoholic fatty liver disease and the metabolic syndrome: The global incidence of NAFLD is 6-35% (median 20%) (2, 3). Interestingly, NAFLD with hepatic fibrosis is strongly associated with hepatic and cardiovascular mortality (4, 5). It is well known that diet control combined with physical exercise can reduce the occurrence of fatty liver, especially diet control. However, dietary changes do not treat established NAFLD. GLP-1 receptor agonists have been shown to be effective in the treatment of NAFLD patients with diabetes (6), but there is lack of evidence for its efficacy and mechanism of action in non-diabetic NAFLD patients (7). As a GLP-1 receptor agonist, semaglutide has good clinical efficacy in the treatment of NAFLD with diabetes (8). The purpose was to investigate whether semaglutide has anti-inflammatory and anti-fibrotic effects on non-diabetic NAFLD while improving hepatic steatosis. Therefore, we constructed an obesity model with advanced NAFLD without diabetes assessed by liver histology. To further explore the potential mechanism of semaglutide intervention in NAFLD. Metabolomics mainly studies the metabolic pathways of small molecules and their metabolites. At present, metabolomics technology has been widely used in the research of NAFLD drug treatment, such as: drug effect evaluation, drug screening, and drug action mechanism research (9). This study was the first to investigate the effect of semaglutide on liver metabolomics. The hepatic metabolomic profile was determined by liquid chromatography/mass spectrometry (LC-MS) method to explore the regulatory targets and underlying mechanisms of semaglutide on non-diabetic NAFLD.
Materials and methods
Animal model
Male C57BL/6J mice (6 weeks old) were purchased from Hebei Ivivo Biotechnology Co., Ltd. (Shijiazhuang, China). All mice fed in specific-pathogen free (SPF)-room under the 12h-alternation cycle of day/night, temperature of (24 ± 2) °C, and relative humidity of (60 ± 10) %. Following one week of acclimation, mice were randomly divided into normal diet group (4% fat, 20% carbohydrate, and 20% protein, NCD group, n=9) and high-fat diet (60% fat, 20% carbohydrate, and 20% protein, HFD group, n=18). After 12 weeks of feeding, mice in the HFD group were subdivided into high-fat diet control group (HFD, n=9) and semaglutide intervention group (HFD+sema, n=9). Specifically, mice in the HFD+sema group were intraperitoneally injected with 30 μmol/kg semaglutide (Novo Nordisk, Copenhagen, Denmark) once daily for another 12 weeks. The highest dose of semaglub was selected on the basis of previously published studies in mice (10). The mice in the NFD and HFD groups received an equal of phosphate-buffered saline (PBS). Mice were anesthetized by intraperitoneal injection of pentobarbital sodium (40mg/kg) and sacrificed on the 24th week. After that, blood samples and liver tissues were collected and stored for further analyses.
Intraperitoneal glucose tolerance test
Intraperitoneal glucose tolerance test (IPGTT) was conducted referring to previous study (11). Specifically, mice were fasted overnight, followed by intraperitoneal injection of glucose (2 g/kg). Blood samples were collected at 0, 15, 30, 60, 90, and 120 min and measured by glucometer (German Roche Rokan total glucose meter).
Biochemical analysis
Blood was collected and then centrifuged to obtain the serum. Serum levels of alanine transaminase (ALT), aspartate transaminase (AST), triglyceride (TG), total cholesterol (TC), low-density lipoprotein (LDL) and high-density lipoprotein (HDL), and were measured using corresponding kits (Nanjing Jiancheng, Nanjing, China).
Measurement of inflammatory cytokines and oxidative stress marker
The concentrations of inflammatory cytokines including tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and IL-1β, and oxidative stress marker including malonaldehyde (MDA) and superoxide dismutase (SOD) were detected by ELISA kits (Lianke, Hangzhou, China).
HE staining
Liver tissues were fixed with 4% paraformaldehyde, embedded in paraffin, and sliced into 5 µm sections. Afterwards, the sections were dewaxed, rehydrated, and stained with hematoxylin-eosin (HE). Images were captured by microscope (ECLIPSE Ci-L, Nikon, Tokyo, Japan).
Oil red O staining
Liver tissues were frozen followed by cutting into 10 μm thick. According to manufacture instruction, the sections were stained by Oil red O for lipid deposition evaluation. Finally, the area of lipid deposition was quantified with Image-pro plus 6.0 software (Media Cybermetrics, Rockville, MD, USA), thereby calculating the percentage of area of lipid deposition.
Masson staining
To observe the extent of liver fibrosis, MASSON staining kit (G1006-100ml, CR2202127, Servicebio) was employed in the study. Briefly, 4 μm slices were dehydrated with a series of ethanol concentration, and then dyed with Wiegert’s iron haematoxylin solution. After staining with Lichun red acidic magenta solution, slices were differentiated with phosphomolybdic acid, dyed with aniline blue dye solution, and rinsed with glacial acetic acid. Images were captured by microscopy (ECLIPSE Ci-L, Nikon, Tokyo, Japan).
Transmission electron microscopy
Liver tissues were dissected from mice and then immediately fixed in 2.5% glutaraldehyde at 4°C. After washing, tissues were post-fixed in 2% osmium tetroxide, dehydrated in ethanol/propylene oxide and embedded in epoxy resin. Then, liver tissues were cut at 70 nm thickness using ultramicrotome (Leica UC7, Leica), and next stained with uranyl acetate and lead solution. Until the sections were dry, images were captured by transmission electron microscope (HT7700, Hitachi, Tokyo, Japan), and the number of autophagosomes and autophagolysosomes was mounted by Image-proplus 6.0 (Media Cybernetics, Rockville, MD, USA).
Liver metabolic profiling
Liver samples containing internal standard were prepared, ground, and extracted with ice bath ultrasound, followed by centrifugation. The extracts were put into liquid chromatography-mass spectrometry (LC-MS) vial, dried under nitrogen, and re-dissolved in the combination of methanol and water. Afterwards, samples were silenced at -20 °C for 2 h and centrifuged to obtain the supernatant. Supernatant was filtrated and collected for analysis using LC-MS platform. Metabolomics data were preprocessed using the Progenesis QI v2.3 (Waters Corp., Milford, MA).
Analysis of metabolic data
Multivariate statistical analysis, such as principal component analysis (PCA), partial least squares discriminant analysis (PLS-DA) and orthogonal partial least squares discriminant analysis (OPLS-DA), was carried out after normalization of the preprocessed data matrix. Univariate analysis mainly focused on univariate description and statistical inference, and Student’s test and Fold change (FC) analysis was used to compare metabolites between two groups. The screening criteria for differential metabolites were variable important in projection (VIP) of OPLS-DA greater than 1, and p-value of Student’s test less than 0.05 or FC greater than 1, and P-value less than 0.05.
Metabolic pathway analysis
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was performed for differential metabolites. If the influence value of the pathway was higher than 0.05 and the P-value was less than 0.05, the pathway was considered to be significantly correlated. Bubble mapping were employed to display the metabolic enrichment pathway.
Statistical analysis
Data were expressed as mean ± standard deviation (SD) and analyzed using the SPSS 23.0. The differences amongst different groups were compared with one-way ANOVA followed by LSD test. Statistically significance was accepted at P<0.05 level (*),P<0.01 level (**), P<0.001 level (***), P>0.05 level (ns).
Ethic approval statement
The animal use protocol for this study has been reviewed and approved by the Animal Ethics Committee of Hebei General Hospital. The experiment was implemented in accordance with the experimental protocol, strictly regulated the operating procedures, and did not violate the ethical requirements.
Results
Semaglutide ameliorated HFD-induced obesity and blood glucose
To examine the function of semaglutide on NAFLD, C57BL/6J mice were fed with HFD to establish the NAFLD model in vivo. Results displayed that body weight in the HFD group was increased rapidly and significantly changed after the first week of feeding, compared with NCD group (P<0.05). However, the body weight was significantly reduced in the first week after administration of semaglutide (P<0.05). Additionally, the body weight of HFD+Sema group was close to that of the NCD group at the end of the 12 th week of treatment ( Figure 1A ). Correspondingly, liver weight in the HFD group were outstandingly elevated, in contrast to NCD group (P<0.05), but were declined in the HFD+Sema group (P<0.05) ( Figure 1B ). IFGTT test indicated that blood glucose was notably reduced at 15 minutes following intervention with semaglutide (P<0.05). At 60 minutes, the blood glucose level of the HFD+sema group was close to the blood glucose level of the NCD group ( Figure 1C ). Collectively, aforementioned findings clarified that semaglutide can effectively reduce the blood glucose and body weight in HFD-induced NAFLD model.
Figure 1.
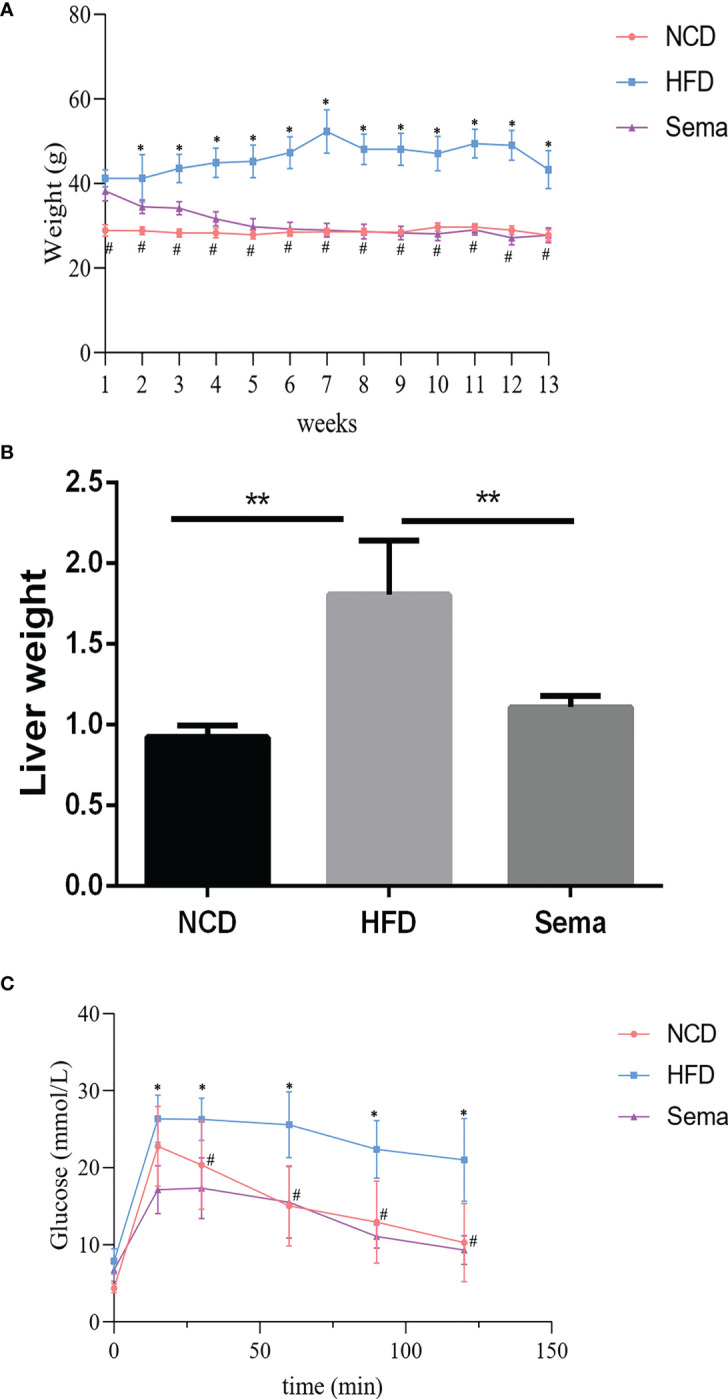
Semaglutide ameliorated HFD-induced obesity and blood glucose. (A) The body weight of NFD, HFD, HFD+sema group at the end of the 12th week of treatment was shown; (B) The liver weight of NCD, HFD, HFD+sema group at the end of the 12th week of treatment was shown; (C) Glucose tolerance was determined by IFGTT test; NCD, normal diet; HFD, high-fat diet; sema, semaglutide; IPGTT, Intraperitoneal glucose tolerance test. # means HFD vs Sema; * means NCD vs HFD.
Semaglutide alleviated the blood lipid profile and liver damage in HFD-induced NAFLD model
At the end of 24 weeks of feeding, the levels of TG, TC and LDL in HFD group were notably increased in contrast to NCD group (P<0.05). After administration of semaglutide, the contents of TG, TC and LDL were markedly down-regulated (P<0.05). However, there was no significant difference in HDL level among the different groups (P>0.05) ( Figures 2A–D ). Besides, semaglutide relieved the liver damage, as evidenced by the reduction of ALT and AST level in serum (P<0.05) ( Figures 2E, F ). Thus, we believed that semaglutide can relieve dyslipidemia and liver injury to a certain degree.
Figure 2.

Semaglutide alleviated the blood lipid profile and liver damage in HFD-induced NAFLD model. The levels of TG (A), TC (B), LDL (C), and HDL (D) were shown; The contents of ALT (E) and AST (F) in serum were shown; TG, triglyceride; TC, total cholesterol; LDL, low-density lipoprotein; HDL, high-density lipoprotein; ALT, alanine transaminase; AST, aspartate transaminase; HFD, high-fat diet; NAFLD, non-alcoholic fatty liver disease.
Semaglutide reduced inflammation and oxidative stress response in HFD-induced NAFLD model
The contents of TNF-α ( Figure 3A ), IL-6 ( Figure 3B ), IL-1β ( Figure 3C )and insulin ( Figure 3F )in the HFD group were higher than those of NCD group (P<0.05). By comparison with HFD group, these levels in the HFD+sema were remarkably decreased (P<0.05). Additionally, we also observed that HFD accelerated the oxidative stress response, as demonstrated by the reduction of MDA expression and elevation of SOD expression (P<0.05) ( Figures 3D, E ). Those data indicated that semaglutide may improve NAFLD through mechanisms of anti-inflammatory and anti-oxidative stress.
Figure 3.

Semaglutide reduced inflammation and oxidative stress response in HFD-induced NAFLD model. The contents of TNF-α (A), IL-6 (B), IL-1β (C) and insulin (F) were determined by ELISA assay; The levels of MDA (D) and SOD (E) were shown; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; IL-1β, interleukin-1β; MDA, malonaldehyde; SOD, superoxide dismutase; HFD, high-fat diet; NAFLD, non-alcoholic fatty liver disease, INS, insulin.
Effects of semaglutide on liver histology changes in NAFLD model
HE staining showed that liver sections in NCD group had normal structures. Liver cells were plump and regularly arranged without obvious pathological changes in NCD group. In the liver tissue of HFD group, a large area of diffuse hepatocyte steatosis were seen, with a large number of round vacuoles (black arrows) of different sizes around the nucleus. More common hepatocyte watery degeneration, swollen cells, cytoplasmic loose (blue arrows), pale staining and focal lymphocytic infiltration were observed. After administration of semaglutide, the lesions including hepatocyte steatosis, ballooning degeneration and lymphoid foci (yellow arrows) were significantly improved ( Figure 4A ). Additionally, no abnormality in hepatocyte morphology and no lipid droplet were formatted in NCD group. Oil red O staining of liver tissue stated that the lipid deposition in the HFD group was aggravated, whereas semaglutide treatment remarkably reduced the percentage of lipid droplet (yellow circle) area (P<0.05) ( Figure 4B ). Masson staining was utilized to reflect the degree of hepatic fibrosis, and this study indicated that the poor percentage of collagen fibrin (black circle) in HFD group. And it was decreased after semaglutide treatment ( Figure 4C ). Because the time of modeling was too short, fibrosis in the liver was not obvious, so fibrosis was not obvious after the administration of semaglutide. Furthermore, TEM presented that the mitochondria in HFD group were slightly swollen, the mitochondrial cristae were partially broken and reduced, the intramembrane matrix was partially dissolved, and the mitochondrial microautophagy structure and ribosomes was visible. Consistent with the preservation of hepatic structures, semaglutide ameliorated the mitochondrial structure with less swollen and mitochondrial cristae with relatively more organized compared to NAFLD mice ( Figure 4D ). Taken together, those results supported the protecting role of semaglutide against NAFLD via reducing hepatocyte steatosis, lipid deposition, and mitochondrial damage.
Figure 4.

Effects of semaglutide on liver histology changes in NAFLD model. (A) Representative images of liver tissues stained with HE staining; (B) Representative images of lipid deposition stained with Oil Red O; (C) Representative images of liver fibrosis stained with Masson; (D) TEM images showed the mitochondria, mitochondrial microautophagy structure and ribosomes; NAFLD, non-alcoholic fatty liver disease.
Effects of semaglutide on liver metabolomics in HFD-induced NAFLD model
To understand the action of semaglutide on the metabolic profile alteration, we further conducted the LC-MS of liver tissues from mice. First, The PCA score plot indicated that there was a clear trend of separation between groups, indicating significant metabolic and relatively stable system ( Figure 5A ). OPLS-DA score plot showed all groups were distinguished clearly. Additionally, the triangle representing the HFD+sema group moved to the dot representing the NCD group, indicating that semaglutide significantly modulated metabolic status of HFD group. OPLS-DA score plot displayed that manifest separation between NCD and HFD group or HFD and HFD+sema group ( Figures 5B, C ). All these results showed that the LC-MS system was with good reproducibility and stability.
Figure 5.
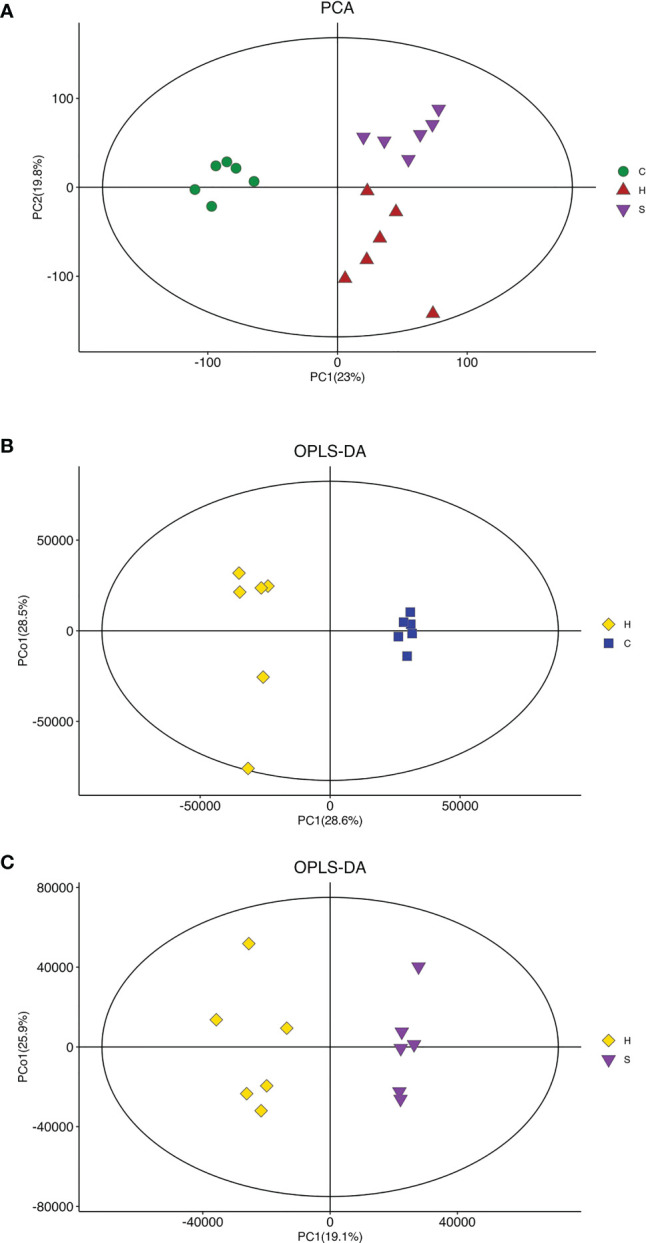
Effects of semaglutide on liver metabolomics in HFD-induced NAFLD model. (A) PCA was utilized to analyze the metabolites from NFD and HFD-fed rats with and without semaglutide treatment for 24 weeks; (B, C) OPLS-DA was utilized to analyze the metabolites between NCD and HFD group or HFD and HFD+sema group; PLS-DA, partial least squares discriminant analysis; OPLS-DA, orthogonal partial least squares discriminant analysis; NAFLD, non-alcoholic fatty liver disease.
Based on the screening criteria with VIP greater than 1 and P-value less than 0.05, we found that differential metabolites mainly included amino acids, fatty acids, carbohydrates, nitrogenous compounds, and cholines, as shown in Heatmap analysis ( Figure 6A ). Heatmap Pearson’s correlation analysis was conducted to evaluate the correlation of differential metabolites ( Figure 6B ). The red and blue colors in the plot represented the up-regulated and down-regulated correlation intensity. According to the correlation coefficient, cluster analysis suggested the interaction among some metabolites.
Figure 6.

(A) Heatmap analysis of differential metabolites with VIP of OPLS-DA greater than 1, and P-value of Student’s test less than 0.05; VIP, variable important in projection; OPLS-DA, orthogonal partial least squares discriminant analysis. (B) Heatmap Pearson’s correlation analysis was conducted to evaluate the correlation of differential metabolites. Some metabolites were negatively related (blue), and some were positively related (red).
Additionally, we also adopted the criteria with FC greater than 1 and P-value less than 0.05, and it turned out that 19 differential metabolites were up-regulated and 13 differential metabolites were down-regulated in the HFD group compared with NFD group ( Figure 7A ; Table 1 ). However, semaglutide treatment down-regulated 10 metabolites and upregulated 4 metabolites ( Figure 7B ; Table 2 ). The results showed that 6 up-regulated metabolites in the HFD group were altered by semaglutide treatment including L-Histidinol, Arachidonic acid, Glutathione, LysoPC(16:0), TG(16:0/16:1(9Z)/18:1(9Z) and 4,5-LTA4. On the other hand, 2 down-regulated metabolites in HFD group were also restored by semaglutide treatment including L-Isoleucine and Methacholine ( Table 3 ).
Figure 7.

Differential metabolites with FC greater than 1 and P-value less than 0.05 were shown. The volcano plots (A, B) of differential metabolites in the mice of NCD vs HFD and HFD vs HFD+Sema groups; FC, fold change; NCD, normal diet; HFD, high-fat diet; Sema, semaglutide.
Table 1.
The DEPs between HFD/NCD (FC value>1, upregulated proteins, FC value<1, downregulated proteins).
| KEGG | Metabolites | Annotation | FC value | P value |
|---|---|---|---|---|
| C00860 | L-Histidinol | Histidine metabolism | 1.18894 | 0.034 |
| C00103 | Glucose 1-phosphate | Galactose metabolism | 1.68831 | 0.01 |
| C00243 | Alpha-Lactose | Galactose metabolism | 10.3004 | 0.002 |
| C00984 | D-Galactose | Galactose metabolism | 3.17289 | 0.0002 |
| C01613 | Stachyose | Galactose metabolism | 66.0569 | 0.0002 |
| C00219 | Arachidonic acid | Arachidonic acid metabolism | 1.80676 | 0.005 |
| C00208 | D-Maltose | Carbohydrate digestion and absorption | 20.8401 | 0.004 |
| C01355 | Levan | Carbohydrate digestion and absorption | 20.6935 | 0.0002 |
| C00051 | Glutathione | ABC transporters | 5.50334 | 0.0005 |
| C00669 | gamma-Glutamylcysteine | Glutathione metabolism | 7.08569 | 0.0003 |
| C00670 | Glycerophosphocholine | Glycerophospholipid metabolism | 3.57497 | 0.00015 |
| C04230 | LysoPC(16:0) | Glycerophospholipid metabolism | 1.75061 | 0.0157 |
| C04230 | LysoPC(20:4(8Z,11Z,14Z,17Z) | Glycerophospholipid metabolism | 1.87344 | 0.01289 |
| C00064 | L-Glutamine | D-Glutamine and D-glutamate metabolism |
9.0535 | 0.0277 |
| C00819 | D-Glutamine | D-Glutamine and D-glutamate metabolism | 13.8482 | 0.02468 |
| C00721 | Dextrin | Starch and sucrose metabolism | 17.2959 | 0.00171 |
| C00422 | TG(16:0/16:1(9Z)/18:1(9Z) | Insulin resistance | 1.6681 | 0.02452 |
| C00043 | Uridine diphosphate-N-acetylglucosamine | Insulin resistance | 3.31479 | 0.04066 |
| C02645 | 4,5-LTA4 | 4.88241 | 0.0392 | |
| C00439 | Formimino-L-glutamic acid | Histidine metabolism | 0.03011 | 0.00682 |
| C02741 | Phosphoribosyl-AMP | Histidine metabolism | 0.02699 | 0.00181 |
| C04896 | PhosphoribosylformiminoAICAR-phosphate | Histidine metabolism | 0.00694 | 0.00331 |
| C04916 | Phosphoribulosylformimino-AICAR-P | Histidine metabolism | 0.02819 | 0.00011 |
| C05570 | Ergothioneine | Histidine metabolism | 0.00216 | 0.00012 |
| C05575 | L-Histidine trimethylbetaine | Histidine metabolism | 0.01523 | 0.00054 |
| C05960 | 15-keto-PGF2alpha | Arachidonic acid metabolism | 0.0399 | 0.00021 |
| C05964 | 11-dehydro-TXB2 | Arachidonic acid metabolism | 0.39868 | 0.00192 |
| C14748 | 20-HETE | Arachidonic acid metabolism| | 0.3891 | 0.00035 |
| C14749 | 19-HETE | Arachidonic acid metabolism| | 0.1698 | 0.00013 |
| C00004 | NADH | Aldosterone synthesis and secretion | 0.12352 | 0.00952 |
| C00624 | N-Acetyl-L-glutamic acid | Arginine biosynthesis | 0.45795 | 0.01823 |
| C01227 | DHA | Steroid hormone biosynthesis | 0.22734 | 0.00024 |
| C07471 | Methacholine | 0.13864 | 0.00295 |
Table 2.
The DEPs between HFD/Semaglutide(FC value>1, upregulated proteins, FC value<1, downregulated proteins).
| KEGG | Metabolites | Annotation | FC value | P value |
|---|---|---|---|---|
| C00422 | TG(16:0/18:1(9Z)/18:2(9Z,12Z) | Regulation of lipolysis in adipocytes | 1.40119 | 0.02682 |
| C00696 | Prostaglandin D2 | Neuroactive ligand-receptor interaction | 3.9372 | 0.04731 |
| C00245 | Taurine | Neuroactive ligand-receptor interaction | 2.253 | 0.00295 |
| C00079 | L-Phenylalanine | Aminoacyl-tRNA biosynthesis | 0.532 | 0.00072 |
| C00082 | L-Tyrosine | Aminoacyl-tRNA biosynthesis | 0.52658 | 0.00017 |
| C00148 | L-Proline | Aminoacyl-tRNA biosynthesis | 0.74646 | 0.0108 |
| C00183 | L-Valine | Aminoacyl-tRNA biosynthesis | 0.45524 | 0.00679 |
| C00407 | L-Isoleucine | Aminoacyl-tRNA biosynthesis | 0.70402 | 0.00783 |
| C00020 | Adenosine monophosphate | Regulation of lipolysis in adipocytes | 0.55494 | 0.00972 |
| C01772 | 2-Hydroxycinnamic acid | Phenylalanine metabolism | 0.62917 | 0.00091 |
| C00077 | L-Ornithine | Arginine biosynthesis | 0.73271 | 0.0135 |
| C00624 | N-Acetyl-L-glutamic acid | Arginine biosynthesis | 0.58908 | 0.00803 |
| C07471 | Methacholine | 0.3277 | 0.00881 |
Table 3.
The up-regulated and down-regulated metabolites in the HFD group were altered by semaglutide.
| KEGG | Metabolites | Average(H) | Average(C) | Average(S) |
|---|---|---|---|---|
| C00860 | L-Histidinol↑ | 18153811 | 15268907.04 | 15139503 |
| C00219 | Arachidonic acid↑ | 998326.112 | 552549.249 | 57032.5321 |
| C00051 | Glutathione↑ | 16980724.2 | 3085533.34 | 603878.8711 |
| C04230 | LysoPC(16:0) ↑ | 84352861.5 | 48184770.88 | 55474026.6 |
| C00422 | TG(16:0/16:1(9Z)/18:1(9Z) ↑ | 2217434.67 | 1329316.561 | 1381633.49 |
| C02645 | 4,5-LTA4↑ | 474530.935 | 97192.03688 | 57098.8542 |
| C00407 | L-Isoleucine↓ | 13394228.1 | 15857001.7 | 19025355.7 |
| C07471 | Methacholine↓ | 370441.79 | 2671875.99 | 1130433.9 |
KEGG enrichment analysis stated that the principal differentially metabolites following semaglutide treatment were involved in metabolic pathways of liver. The top ten hepatic pathways included the histidine metabolism, regulation of lipolysis in adipocytes, arachidonic acid metabolism, aminoacyl-tRNA biosynthesis, ABC transporters, aldosterone synthesis and secretion, galactose metabolism, carbohydrate digestion and absorption, choline metabolism in cancerhe, and insulin resistance ( Figure 8 ).
Figure 8.

KEGG enrichment analysis stated the top ten hepatic pathways via bubble mapping. (A) A histogram (B) bubble mapping, KEGG, Kyoto Encyclopedia of Genes and Genomes.
Discussion
The HFD mouse model was prone to liver steatosis and can replicate most of the metabolic characteristics of human NAFLD. Thus, this model can be used to investigate NAFLD and related metabolic syndromes (12). In the present study, body weight, and liver weight were significantly higher in the HFD group than in the control group after 12 weeks of HFD feeding. There were studies using the same or similar dietary composition and the same duration to successfully induce steatosis, inflammation and no fibrosis (12, 13). In addition, abnormal serum transaminases, serum and hepatic lipid levels. And NAFLD histology features steatosis, inflammation, and ballooning. These characteristics indicated that the rat NAFLD model has been successfully constructed.
There was a study reported that NAFLD exhibits elevated levels of SOD and oxidative stress, which lead to changes in mitochondrial function (14). Consistent with the published research results, we detected the increase of SOD, a marker of oxidative stress in the blood. Under the electron microscope, mild mitochondrial swelling, increased volume, local fragmentation and reduction of cristae, and local dissolution of the intramembrane matrix, and mitochondrial micro-autonomy can be seen in the visual field. Large areas of diffuse hepatocyte steatosis with numerous round vacuoles of various sizes surrounding the nucleus were seen in HFD liver tissue. Watery degeneration of hepatocytes and focal infiltration of lymphocytes are more common. Semaglutide can significantly improve the pathological changes of hepatocyte steatosis, ballooning degeneration and lymphoid foci. Semaglutide relieves lipid accumulation in the liver. The HFD group had significant collagen deposition, and semaglutide exhibited significant anti-fibrotic activity, greatly reducing the percentage of collagen area. All pathological results confirmed that semaglutide alleviated hepatic lipid accumulation, inflammation, and fibrosis morphologically, and led to changes in mitochondrial structure.
Wang et al. showed that FA and its metabolites are detrimental factors in the development of NAFLD (1). The major pathophysiological mechanisms of NAFLD involve increased FA accumulation and decreased mitochondrial FA oxidation. It leads to liver cell lipid metabolism dysfunction and lipid accumulation as well as hepatocyte damage caused by activation of tumor necrosis factor-α and stimulation of reactive oxygen species (ROS) production (1). Under normal physiological conditions, the synthesis and degradation of free fatty acids, triglycerides and total cholesterol in the body are in a dynamic equilibrium (15). Hepatic steatosis is caused by an imbalance in hepatic lipid accumulation. Due to the breakdown of visceral adipose tissue fat, the synthesis of new fat in the liver, the increase in the consumption of a high-calorie high-fat diet, and the reduction of hepatic mitochondrial β-oxidation. Ultimately leads to increased production and accumulation of plasma free fatty acids, low-density lipoproteins, and triglycerides (16).
L-Histidinol is an enzyme that regulates histidine biosynthesis. Studies have reported that excessive intake of histidine can cause hepatic steatosis (17). May be due to an imbalance between lipid transport in the liver and free fatty acid uptake into hepatocytes. LysoPC(16:0) is a phospholipid whose elevated levels increase the risk of liver injury and oxidative stress, and phospholipid metabolism is closely related to the pathogenesis of NAFLD. The concentration of LysoPC(16:0) metabolite was higher in patients with different degrees of hepatic fat accumulation (18). L-Isoleucine is a Branched-chain amino acids (BCAA). Previous studies have reported that BCAAs can promote fatty acid β-oxidation and ketone body production (19). β-oxidation is thought to be the main mechanism of hepatic lipid reduction and is involved in the upregulation of peroxisome and mitochondrial genes (20). Methacholine is an acetylcholine receptor agonist. Studies have found that patients with long-term parenteral nutrition lack of choline, after choline supplementation, there will be a reversal of hepatic steatosis and a decrease in serum transaminases. Pathological changes reappeared after choline supplementation was discontinued (21, 22). L-Isoleucine and Methacholine were significantly down-regulated in HFD group and NFD group, but were significantly up-regulated after semaglutide intervention. Semaglutide reduces lipid droplet formation, inhibits adipose tissue lipolysis and regulates phospholipid homeostasis by downregulating L-Histidinol, LysoPC(16:0) and TG. Consistent with our results in liver histology, by increasing L-Isoleucine and Methacholine levels to promote fatty acid β-oxidation and ketone body production, changes in metabolites alleviated hepatic lipid accumulation.
The pro-inflammatory cytokines (IL-6 and TNF-α) released during the pathophysiology of NAFLD are mainly derived from macrophages (23). IL-6 is both a pro- and an anti-inflammatory factor in many biological processes. TNF-α is a central link in the inflammatory cascade and can promote the release of other pro-inflammatory substances, thereby accelerating the inflammatory process (24). IL-6 and TNF-α can directly reflect the degree of inflammatory response in the body (25). Consistent with our experimental results, semaglutide inhibited the up-regulation of pro-inflammatory factors (TNF-α, IL-6, IL-1β, MDA). Semaglutide reduced but did not fully normalize hepatic inflammation. This may be related to the dose and timing of semaglutide.
Arachidonic acid, a polyunsaturated fatty acid metabolized by cyclooxygenase and lipoxygenase pathways, can mediate inflammation and participate in the development of NAFLD. The study of Hall (26) et al. showed that phospholipid membrane is remodeled in late NAFLD stage, and arachidonic acid is released from phospholipid membrane, thereby aggravating cell damage and inflammatory response. Arachidonic acid can be converted into LTA4 (5,6-epoxide of arachidonic acid), and 4,5-LTA4 is a precursor of leukotrienes. Leukotrienes are a class of classical pro-inflammatory lipid metabolism intermediates that trigger and amplify inflammatory responses (27). Inflammation is an important factor leading to liver damage. This study is consistent with previous studies: arachidonic acid and 4,5- LTA 4 are significantly up-regulated in HFD group, while the metabolites prostaglandin D2 and 4,5-LTA4 of arachidonic acid are significantly down regulated after semaglutide treatment. Therefore, semaglutide may reduce the inflammatory response of NAFLD and liver cell damage in mice by down regulating the expression of inflammatory factors arachidonic acid and 4,5- LTA 4. A limitation of this study is the lack of further confirmation by in vitro experiments, which should be further evaluated in future studies.
Conclusion
Semaglutide can reduce blood glucose level and liver fat accumulation and play an anti-inflammatory role in advanced NAFLD that due to the effects of HFD.
Data availability statement
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by The Animal Ethics Committee of Hebei General Hospital.
Author contributions
SN and SC conceived and designed the study. SN, QR, LY, and XinC provided materials and samples. LY, QR, SN, XP, XiaC, HZ and ZL collected and assembled the data. SN and SC analyzed and interpreted the data. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Hebei Provincial Central leading Local Science and Technology Development funds Project (206Z7702G).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- 1. Berlanga A, Guiu-Jurado E, Porras JA, Auguet T. Molecular pathways in non-alcoholic fatty liver disease. Clin Exp Gastroenterol (2014) 7:221–39. doi: 10.2147/CEG.S62831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Powell EE, Wong VW, Rinella M. Non-alcoholic fatty liver disease. Lancet (London England) (2021) 397:2212–24. doi: 10.1016/S0140-6736(20)32511-3 [DOI] [PubMed] [Google Scholar]
- 3. Djordjevic DB, Zdravkovic M, Nagorni A, Manolis A, Tsioufis C, Lovic D. A critical approach of guideline therapeutic recommendations for NAFLD. Curr Vasc Pharmacol (2018) 16:228–38. doi: 10.2174/1570161115666170621080228 [DOI] [PubMed] [Google Scholar]
- 4. Polyzos SA, Kang ES, Tsochatzis EA, Kechagias S, Ekstedt M, Xanthakos S, et al. Commentary: Nonalcoholic or metabolic dysfunction-associated fatty liver disease? the epidemic of the 21st century in search of the most appropriate name. Metabolism: Clin Exp (2020) 113:154413. doi: 10.1016/j.metabol.2020.154413 [DOI] [PubMed] [Google Scholar]
- 5. Polyzos SA, Kountouras J, Mantzoros CS. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism: Clin Exp (2019) 92:82–97. doi: 10.1016/j.metabol.2018.11.014 [DOI] [PubMed] [Google Scholar]
- 6. Bifari F, Manfrini R, Dei Cas M, Berra C, Siano M, Zuin M, et al. Multiple target tissue effects of GLP-1 analogues on non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH). Pharmacol Res (2018) 137:219–29. doi: 10.1016/j.phrs.2018.09.025 [DOI] [PubMed] [Google Scholar]
- 7. Sumida Y, Yoneda M. Current and future pharmacological therapies for NAFLD/NASH. J Gastroenterol (2018) 53:362–76. doi: 10.1007/s00535-017-1415-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. New Engl J Med (2021) 384:1113–24. doi: 10.1056/NEJMoa2028395 [DOI] [PubMed] [Google Scholar]
- 9. Safaei A, Arefi Oskouie A, Mohebbi SR, Rezaei-Tavirani M, Mahboubi M, Peyvandi M, et al. Metabolomic analysis of human cirrhosis, hepatocellular carcinoma, non-alcoholic fatty liver disease and non-alcoholic steatohepatitis diseases. Gastroenterol Hepatol bed to bench (2016) 9:158–73. [PMC free article] [PubMed] [Google Scholar]
- 10. Dalbøge LS, Christensen M, Madsen MR, Secher T, Endlich N, Drenic V, et al. Nephroprotective effects of semaglutide as mono- and combination treatment with lisinopril in a mouse model of hypertension-accelerated diabetic kidney disease. Biomedicines (2022) 10. doi: 10.3390/biomedicines10071661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mori N, Kurata M, Yamazaki H, Hosokawa H, Nadamoto T, Inoue K, et al. Intragastric administration of allyl isothiocyanate reduces hyperglycemia in intraperitoneal glucose tolerance test (IPGTT) by enhancing blood glucose consumption in mice. J Nutr Sci vitaminol (2013) 59:56–63. doi: 10.3177/jnsv.59.56 [DOI] [PubMed] [Google Scholar]
- 12. Xu ZJ, Fan JG, Ding XD, Qiao L, Wang GL. Characterization of high-fat, diet-induced, non-alcoholic steatohepatitis with fibrosis in rats. Digestive Dis Sci (2010) 55:931–40. doi: 10.1007/s10620-009-0815-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fan JG, Ding XD, Wang GL, Xu ZJ, Tian LY, Zheng XY. [Expression of uncoupling protein 2 and its relationship to the content of adenosine triphosphate in the nonalcoholic fatty livers of rats fed a high-fat diet]. Zhonghua gan zang bing za zhi = Zhonghua ganzangbing zazhi = Chin J Hepatol (2005) 13:374–7. doi: 10.3760/j.issn:1007-3418.2005.05.017 [DOI] [PubMed] [Google Scholar]
- 14. Shen B, Zhao C, Wang Y, Peng Y, Cheng J, Li Z, et al. Aucubin inhibited lipid accumulation and oxidative stress via Nrf2/HO-1 and AMPK signalling pathways. J Cell Mol Med (2019) 23:4063–75. doi: 10.1111/jcmm.14293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Byrne CD, Targher G. NAFLD: A multisystem disease. J Hepatol (2015) 62:S47–64. doi: 10.1016/j.jhep.2014.12.012 [DOI] [PubMed] [Google Scholar]
- 17. Aoyama Y, Takagi M, Yoshida A. Lipid alterations in the liver and serum of rats in histidine-excess and copper deficiency. J Nutr Sci vitaminol (1999) 45:773–83. doi: 10.3177/jnsv.45.773 [DOI] [PubMed] [Google Scholar]
- 18. Xu Q, Zhang Y, Zhang X, Liu L, Zhou B, Mo R, et al. Medium-chain triglycerides improved cognition and lipid metabolomics in mild to moderate alzheimer's disease patients with APOE4(-/-): A double-blind, randomized, placebo-controlled crossover trial. Clin Nutr (Edinburgh Scotland) (2020) 39:2092–105. doi: 10.1016/j.clnu.2019.10.017 [DOI] [PubMed] [Google Scholar]
- 19. Honda T, Ishigami M, Luo F, Lingyun M, Ishizu Y, Kuzuya T, et al. Branched-chain amino acids alleviate hepatic steatosis and liver injury in choline-deficient high-fat diet induced NASH mice. Metabolism: Clin Exp (2017) 69:177–87. doi: 10.1016/j.metabol.2016.12.013 [DOI] [PubMed] [Google Scholar]
- 20. Perakakis N, Joshi A, Peradze N, Stefanakis K, Li G, Feigh M, et al. The selective peroxisome proliferator-activated receptor gamma modulator CHS-131 improves liver histopathology and metabolism in a mouse model of obesity and nonalcoholic steatohepatitis. Hepatol Commun (2020) 4:1302–15. doi: 10.1002/hep4.1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Buchman AL, Dubin MD, Moukarzel AA, Jenden DJ, Roch M, Rice KM, et al. Choline deficiency: A cause of hepatic steatosis during parenteral nutrition that can be reversed with intravenous choline supplementation. Hepatol (Baltimore Md.) (1995) 22:1399–403. doi: 10.1002/hep.1840220510 [DOI] [PubMed] [Google Scholar]
- 22. Buchman AL, Ament ME, Sohel M, Dubin M, Jenden DJ, Roch M, et al. Choline deficiency causes reversible hepatic abnormalities in patients receiving parenteral nutrition: proof of a human choline requirement: a placebo-controlled trial. JPEN. J parenteral enteral Nutr (2001) 25:260–8. doi: 10.1177/0148607101025005260 [DOI] [PubMed] [Google Scholar]
- 23. Cobbina E, Akhlaghi F. Non-alcoholic fatty liver disease (NAFLD) - pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab Rev (2017) 49:197–211. doi: 10.1080/03602532.2017.1293683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ceccarelli S, Panera N, Mina M, Gnani D, De Stefanis C, Crudele A, et al. LPS-induced TNF-α factor mediates pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty liver disease. Oncotarget (2015) 6:41434–52. doi: 10.18632/oncotarget.5163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yagi S, Hirata M, Miyachi Y, Uemoto S. Liver regeneration after hepatectomy and partial liver transplantation. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21218414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hall Z, Bond NJ, Ashmore T, Sanders F, Ament Z, Wang X, et al. Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatol (Baltimore Md.) (2017) 65:1165–80. doi: 10.1002/hep.28953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Di Gennaro A, Haeggström JZ. The leukotrienes: immune-modulating lipid mediators of disease. Adv Immunol (2012) 116:51–92. doi: 10.1016/B978-0-12-394300-2.00002-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author.