

Summary
Graph neural networks (GNNs) have received increasing attention because of their expressive power on topological data, but they are still criticized for their lack of interpretability. To interpret GNN models, explainable artificial intelligence (XAI) methods have been developed. However, these methods are limited to qualitative analyses without quantitative assessments from the real-world datasets due to a lack of ground truths. In this study, we have established five XAI-specific molecular property benchmarks, including two synthetic and three experimental datasets. Through the datasets, we quantitatively assessed six XAI methods on four GNN models and made comparisons with seven medicinal chemists of different experience levels. The results demonstrated that XAI methods could deliver reliable and informative answers for medicinal chemists in identifying the key substructures. Moreover, the identified substructures were shown to complement existing classical fingerprints to improve molecular property predictions, and the improvements increased with the growth of training data.
Keywords: explainable artificial intelligence, graph neural networks, quantitative assessments, molecular property benchmarks, XAI applications
Graphical abstract
Highlights
-
•
Establishing XAI benchmarks for quantitative evaluation of XAI methods
-
•
Evaluating the explainability of XAI methods combined with GNN models
-
•
Comparing with human experts to identify key substructures of molecular toxicity
-
•
Utilizing the interpretations for promoting molecular property predictions
The bigger picture
Graph neural networks (GNNs) have received increasing attention because of their expressive power on topological data, but they are still criticized for their lack of interpretability. Explainable artificial intelligence (XAI) methods have been developed, but they are limited to qualitative analyses without quantitative assessments. Benefiting from extensive labor in drug discovery, molecular property datasets have been well studied with high-quality ground truths of substructures, but they have not been well accumulated in the assessments of XAI methods. Furthermore, the learned interpretations have neither been quantitatively compared with human experts nor proven to provide informative answers. For this purpose, we have established five molecular XAI benchmarks to quantitatively assess XAI methods on GNN models and made comparisons with human experts. The quantitative assessments of XAI methods would fasten the development of novel approaches and extend their applications.
Quantitative assessment of the interpretability is a challenge for the development of XAI methods due to the lack of prior knowledge. We established five molecular XAI benchmarks to quantitatively evaluate XAI methods applied on GNN models and made comparisons with human experts. Experimental results demonstrated that current XAI methods could deliver reliable and informative explanations for chemists in identifying the key substructures.
Introduction
Graph neural networks (GNNs) have received increasing attention across various areas because of their expressive power on topological data,1 including social networks,2,3 recommendation systems,4,5 and natural science.6,7 Despite their promise, GNN models are still criticized for their lack of interpretability, and these models are often considered “black box.”8 It is necessary to interpret the models through explainable methods.
To interpret GNN models, many explainable artificial intelligence (XAI) methods have been developed. For example, GNNExplainer9 explains GNNs through mutual information between the predictions and explanations. PGExplainer10 learns a parameterized model to predict whether an edge is important. Unfortunately, these methods are only qualitatively assessed on real-world datasets such as social network (Reddit-Binary)9 and sentiment graph (Graph-SST2),8 mainly because these datasets are difficult to summarize their interpretable substructural "ground truth" for quantitative evaluations. In contrast, prior knowledge of explainability has already existed in scientific datasets, especially in chemistry and drug-discovery fields. For example, the interpretable substructural patterns for several molecular properties (e.g., hepatotoxicity and mutagenicity) have been well explored and carefully reviewed by previous studies.11,12
Benefiting from the accumulated data in drug discovery, several XAI studies have been devoted to interpreting molecular substructures. For example, Rodríguez-Pérez et al.13 interpreted relevant features and substructures for compound activity prediction through the Shapley additive explanations. Pope et al.14 identified key substructures in adverse-effect prediction by the attribution method,15 which was originally developed for CNN models. Jiménez-Luna et al.16 highlighted molecular features and structural elements on GNN models through the integrated gradient (IG) attribution. Since these methods focused on nodes or edges individually according to their attributions, they did not consider substructures such as functional groups. For this reason, Jin et al.17 employed the Monte Carlo tree search (MCTS) to extract molecular substructures that are likely responsible for the property. Unfortunately, all these studies relied on qualitative judgments in case studies due to a lack of ground truths. In addition, these XAI studies have not been rigorously compared with human experts on explainability tasks to demonstrate that their learned interpretations are reliable. The comparison with human experts is widespread in scientific experiments, especially in chemistry,18,19 for example, asking medicinal chemists to predict molecular solubility20 and compound prioritization21 during drug discovery.
In parallel, Sanchez-Lengeling et al.22 attempted to quantitively assess XAI methods through synthetic molecular benchmarks, where subgraph logic rules were defined by whether molecules contained particular substructures such as benzene, fluoride, and carbonyl groups. Though such definitions have built relationships between graphs and labels, the simple chemical rules are not applicable to realistic scenarios. For instance, in toxicity prediction, the key substructures (also known as structural alerts) often involve dozens of fragments that are strongly associated with increased occurrences of toxicity. These fragments need to be determined from literature reports11 and statistical analyses.23,24 Jose Jimenez-Luna et al.25 have established the activity cliffs benchmark to evaluate the feature attribution methods and showed that machine-learning models with accurate predictions could bring meaningful inspiration to chemists, but their analyses are limited by their benchmarking and predictive performance. In the case of property cliffs,26 as molecular counterfactuals,27,28 it is even more complicated because slight modifications of molecular structures will cause huge changes in molecular properties, sometimes from strong binding to non-binding with the target molecules. The collections of these realistic and complex scenario datasets will certainly bring big challenges but also opportunities to the development of XAI methods.
On the other hand, though XAI methods are widely considered helpful to improve model prediction performance,29 the current XAI methods, despite affording interpretable predictions, have not really led to improvements in prediction performance.30 For example, Yu et al.31 recognized the compressed subgraph through the graph information bottleneck and used it to predict the molecular properties. However, their prediction performances have not been improved, especially on the MUTAG dataset. Hao et al.32 generated subgraph patterns through reinforcement learning and applied them to predict the graph labels, but they only showed their interpreting models with reasonable performance without comparative assessments. Therefore, it is desirable to propose a scheme to advance the model performance through the learned interpretations.
In this study, we have established five XAI benchmark datasets, including two synthetic and three experimental datasets, to quantitatively assess the interpretability of XAI methods. Through these benchmarks, we comprehensively evaluated six commonly used XAI methods combined with four types of GNN models and made a direct comparison with seven medicinal chemists of different experience levels. The results demonstrated that current XAI methods could deliver reliable and informative answers for medicinal chemists in identifying the key substructures. Based on the learned interpretations, we developed a data-driven fingerprint that could complement the classical fingerprints in molecular property predictions, and the improvement continued to increase with the growth of training data. To our best knowledge, this is the first time that a comparison of XAI methods with human experts on explainability tasks has been made. In addition, it is also important to indicate the ability to promote model performance through learned interpretations.
In summary, our study offers three main contributions:
-
1.
We established five benchmark datasets, including two synthetic and three experimental datasets, to enable quantitative assessments of XAI methods.
-
2.
We comprehensively evaluated six commonly used XAI methods combined with four types of GNN models and made a direct comparison with medicinal chemists.
-
3.
We developed a data-driven fingerprint based on the learned interpretations that could complement the classical fingerprints in molecular property predictions.
Results
Framework overview
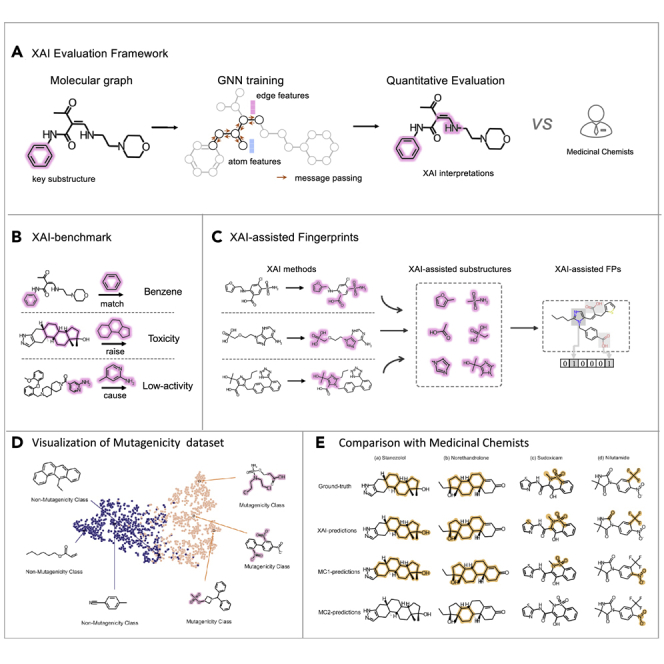
As shown in Figure 1, we established five XAI benchmark datasets, including two synthetic and three experimental datasets. Two synthetic datasets determined the ground-truth substructures through particular subgraphs of three-membered rings (3MRs) and benzene, respectively. Two out of three experimental datasets comprised multiple hand-crafted substructures causing hepatotoxicity and Ames mutagenicity, respectively. Another experimental dataset automatically computed the substructures by comparing one pair of similar compounds with similar structures but significantly different CYP3A4 inhibitions (i.e., property cliff). Based on the datasets, GNN models were trained through four state-of-the-art GNN architectures, which were then, respectively, interpreted through six commonly used XAI methods. These combinations were quantitively evaluated and compared with seven medicinal chemists of different levels. The results demonstrated that current XAI methods could deliver reliable and informative answers for medicinal chemists in identifying key substructures. Based on the learned interpretations, we encoded the learned structural descriptors into a binary bit string as the data-driven fingerprint, which was employed to supplement the classic fingerprint (extended-connectivity fingerprints [ECFP]) in five commonly used molecular property prediction benchmarks.
Figure 1.

Schematic view of the XAI study
(A) XAI evaluation framework: different prediction models are first trained using state-of-the-art GNN models, which are then interpreted through all XAI methods. The interpretations are quantitatively assessed and compared with experienced medicinal chemists.
(B) XAI benchmarks including two particular subgraphs (two synthetic benchmarks), the conjunction of multiple substructures, or a local transformation between two molecular graphs (i.e., property cliff).
(C) XAI-assisted fingerprints: the high-frequency key substructures predicted by XAI methods are encoded as fingerprints to input machine-learning models for predicting properties.
Benchmarking XAI methods with GNN models for recognizing key substructures
To quantitatively assess the interpretability of XAI methods over GNN models, we evaluated the explanation performance on five benchmark datasets with combinations of four GNN and six XAI methods, leading to 5∗4∗6 attribution-area under the receiver operating characteristic curve (AUROC) and accuracy values. The prediction performance of the four state-of-the-art GNN models is shown in Table 1. The superiority of these GNN models has been demonstrated from the performance comparisons with various deep-learning models, as shown in Tables S1–S3. When averaging over all six XAI methods and five datasets for GNN models (Figure 2A), the communicative message-passing neural network (CMPNN) achieved overall the highest average attribution-AUC of 0.671, while GraphSAGE achieved the lowest value of 0.523. GraphNET and graph attention network (GAT) ranked 2nd and 3rd, respectively. This agreed with a previous study22 that vanilla GCN (GraphSAGE) and GAT are often inferior to delicate graph convolutional network (GCN) models such as GraphNET and CMPNN. This difference was consistent in the synthetic and realistic benchmarks, where CMPNN still performed the best with attribution-AUROC values of 0.827 and 0.566, respectively.
Table 1.
Prediction performance on the XAI benchmarks
| Model | Dataset |
Synthetic benchmarks |
Experimental benchmarks |
|||
|---|---|---|---|---|---|---|
| 3MR |
Benzene |
Mutagenicity |
Hepatotoxicity |
CYP450 |
||
| Metric | AUROC | AUROC | AUROC | ACC | AUROC | |
| ML | RF | 0.995 | 0.997 | 0.871 | 0.517 | 0.818 |
| XGBoost | 0.972 | 0.980 | 0.873 | 0.525 | 0.868 | |
| GNN | GraphSAGE | 0.996 | 0.999 | 0.872 | 0.584 | 0.864 |
| GAT | 0.996 | 0.999 | 0.877 | 0.602 | 0.865 | |
| GraphNET | 0.999 | 1.000∗ | 0.973 | 0.619 | 0.976 | |
| CMPNN | 1.000∗ | 1.000∗ | 0.981∗ | 0.681∗ | 0.979∗ | |
The best results are marked with asterisks
Figure 2.

Quantitative assessments
(A and B) The average attribution-AUROC values for (A) four GNN models and (B) six XAI methods.
(C) Detailed attribution accuracies for combinations of all XAI methods and GNN models.
(D) The predicted substructures by representative combinations of CMPNN or IG for the CAS 33301-41-6 molecule.
(E) The embeddings learned by CMPNN + IG over the mutagenicity dataset shown by t-SNE, together with representatively 3 mutagenic and 3 non-mutagenic molecules.
When averaging for XAI methods (Figure 2B), integrated gradients (IG) led to the highest AUROC of 0.675, followed by the GradInput and GradCAM with values of 0.629 and 0.559, respectively. SmoothGrad and class activation map (CAM) achieved AUC values of 0.467 and 0.520, which are essentially the same as the 0.513 achieved randomly. This is likely because SmoothGrad and CAM are unstable across different GNN model architectures. They were originally developed for CNN models so that the average AUROC values would be dragged down by the poor performance in combinations with different GNN architectures. In terms of accuracy, the trends are similar for both GNN models and XAI methods (Figure S1).
Instead of the overall accuracies over datasets, we also computed the accuracy for each molecule in the datasets. As shown in Figure 2C, CMPNN + IG has overall the best performance on all five datasets. Here, we did not show AUROC values since they cannot be calculated for negative molecules without key substructures.
As an example, we selected the CAS 33301-41-6 molecule from the mutagenicity dataset interpreted by IG over four GNN models and the other four XAI methods over CMPNN models (Figure 2D; see also Table S4). When using the IG method, CMPNN truly identified the connected substructure, while GraphNET falsely identified another separate fragment. GAT and GraphSAGE methods partly identified the actual substructure but falsely focused on other substructures such as benzene and nitrogen atoms. The correct identification by CMPNN should attribute to its robust inclusion of both node and edge interactions during the communicative message passing. When comparing different XAI methods, IG could correctly recognize the substructures, while GradInput and GradCAM identified redundant substructures. This is likely because IG eliminates the errors by interpolating gradients from zero embeddings to the input vector. We further projected the embeddings learned by IG over CMPNN, and the learned embeddings from t-stochastic neighbor embedding (t-SNE)33 were able to clearly separate two classes of molecules and correctly identify their key substructures for the mutagenicity (Figure 2E) and benzene (Figure S2) datasets.
Comparison with medicinal chemists to identify key substructures of molecular hepatotoxicity
To assess the actual ability of the interpretation methods, we made a comparison with 7 medicinal chemists (MCs) of different levels (9 months to 30 years). We randomly selected 50 unlabeled molecules from the test set of the hepatotoxicity dataset and predicted the hepatotoxicity and key substructures. In real-world scenarios, it is a challenging task for MCs to determine the human hepatotoxicity accurately even with their background knowledge and experience.23,34 As shown in Figure 3A, the CMPNN + IG model achieved an accuracy of 0.920, much higher than all MCs (detailed in Table S5). MCs achieved accuracies between 0.500 and 0.680, with the best one achieved by the most experienced MC1 (30 years). The prediction accuracies of MCs had a positive but weak correlation (Pearson correlation coefficient [PCC] = 0.530, Wilcoxon paired signed-rank test, p < 0.01) with the working years. Though MC1 is above the precision-recall curve by CMPNN + IG (Figure 3B), MC1 over-estimated the hepatotoxicity with a high recall but relatively low precision.
Figure 3.

Comparison of the XAI (CMPNN + IG) method with medicinal chemists
The accuracy on the hepatotoxicity dataset along the predicted confidence score by (A) XAI or the working years of medicinal chemists (MCs), (B) precision-recall curve, (C) attribution accuracy for identified substructures, (D) attribution precision-recall curves, and (E) the identified substructures by XAI and MCs.
In terms of the substructure identification, the CMPNN + IG model achieved an attribution accuracy of 0.839, slightly lower than 0.852 achieved by the 2nd experienced MC2 (21 years) but higher than other MCs (Figure 3C). According to the attribution precision-recall curve by CMPNN + IG, MC2 was slightly above our ROC curve because of his over-definition of the toxic substructures, while other MCs were below the curve (Figure 3D).
The differences were further shown by four representative molecules (Figure 3E). The CMPNN + IG model correctly identified the key substructures for stanozolol and norethandrolone molecules. It also predicted most of the substructure atoms for the sudoxicam and nilutamide molecules but missed separate substructures. By comparison, MC1 was conservative to predict smaller substructures, while MC2 tended to predict a larger substructure containing the key substructures. For example, MC1 missed the sulfonamide moiety that is known to be associated with hepatotoxicity35 for the Sudoxicam molecule. For the nilutamide molecule, both MC1 and MC2 are disturbed by the NO2− group, causing them to miss the actual halogen atoms. Note that the substructures misidentified by MCs may also be the new potential structural alerts that have not yet been confirmed, but it is a fair comparison for XAI methods and human experts to quantitatively evaluate with consistent ground truths.
XAI-assisted FPs for molecular property prediction
Since the XAI method is comparable with MCs in the previous experiments, it is interesting to know how the identified substructures could promote the performance of machine-learning models. Herein, we used the interpreted substructures identified by XAI methods as an XAI-assisted or data-driven FP. The FP was combined with Morgan-FP (ECFP)36 and input to the random forest technique for molecular property predictions on five popular benchmarks, including three classification datasets (BBBP, BACE, and HIV from the OGB benchmark37) and two regression datasets (IGC50 and LD50 collected by AGBT38). We did not use XAI-FP individually because it only reflects the substructures relevant to properties without containing other molecular structural information. The random forest technique was utilized because of its balance of performance and interpretability.39
As shown in Figures 4A and 4B, the inclusion of our FP achieved AUCs of 0.832, 0.855, and 0.724 over three classification tasks and R2s of 0.638 and 0.648 over two regression tasks. These are higher than those from only Morgan-FP by 2.59%, 0.35%, and 4.32% on classification tasks and 8.36% and 5.63% on regression tasks, respectively. The improvements are not trivial because Morgan-FP development reached a bottleneck, and the addition of other FPs brought little improvement.40,41 As a result, the combination of MACCS, another popular FP, is on average 2.302% lower than our FP. Figure S3 detailed all AUROCs and the comparison of predicted and experimental values.
Figure 4.

The performances of Morgan-FP, Morgan-FP + MACCS-FP, and Morgan-FP + XAI-FP by random forest
(A) The AUROC of three classification tasks.
(B) The on two regression tasks (IGC50 and LD50).
(C) The AUROC values on the OGB-HIV dataset by Morgan-FP and XAI-assisted fingerprints trained with different sample sizes.
Since the interpretations were learned from the training data, we would like to know whether the growth of training data could further promote the improvements. By tested on the largest dataset (OGB-HIV; Figure 4C), the AUROC increased with the size of the training data. The learned FP hurt the Morgan-FP performance when using only 100 training samples but outperformed Morgan-FP at ∼1,000 samples. The FP reached the level of MACCS FP with ∼5,000 samples and of CMPNN with ∼9,000 samples. The performance had did not converge until 3 × 104 training samples. Therefore, the addition of training samples was expected to improve the performance, indicating the advantages of such data-driven methods.
Discussion
In this study, we have established five XAI benchmarks, including synthetic and experimental benchmarks, in the context of drug discovery. Through these benchmarks, we quantitatively evaluated six commonly used XAI methods combined with four types of GNN models and made a direct comparison with different levels of MCs. The results demonstrated that current XAI methods could deliver reliable and informative answers for MCs in identifying both molecular toxicity and key substructures. Based on the interpretations, our XAI-assisted FP was shown to promote the predictions of molecular property.
While many XAI methods have been developed to interpret GNN models, they have not been quantitatively evaluated due to a lack of high-quality data or ground truths. Benefiting from the accumulated knowledge and laboratory data in drug discovery, we have established five molecular datasets with well-defined ground truths, enabling quantitative assessments of state-of-the-art XAI methods combined with GNN models. To our best knowledge, this is the first time a comparison of XAI methods with human experts on the explainability tasks has been made. The comparison with human experts indicated that current XAI methods could deliver reliable and informative answers for MCs in identifying the key substructures.
Another challenge in XAI development is to go beyond the model interpretations. Though several studies31,32 attempted to integrate the interpretations back into the GNN models, the results were frustrated with no effect or even negative impacts. This is partly caused by the noise in their datasets and a lack of ground truths. Here, we encode the learned interpretation into the data-driven FP and input it into the random forest together with classical fingerprints. We prove that the learned interpretations are complementary to the classical FPs and that the strategy can improve the performance in molecular property predictions. More importantly, the improvements brought by the data-driven FP continue incrementing with the growth of training data. This represents a potential direction of XAI applications not only in drug discovery but also XAI methods in general fields.
Despite the merits of this study, we focus on post-processing XAI methods that do not fully combine the advantages of GNN and XAI techniques. It might be desired to develop a highly accurate and mechanically interpretable GNN model. We hope that further development of new XAI methods in GNNs would greatly benefit from our meaningful benchmarking and rigorous framework. Secondly, current methods are purely model based, which strongly depends on high-quality scientific datasets. In the future, it might be necessary to include prior knowledge through a knowledge graph42 so as to process other noisy data like multi-omics or social networks data.
Experimental procedures
Resource availability
Lead contact
The lead contact is Yuedong Yang (yangyd25@mail.sysu.edu.cn).
Materials availability
There are no newly generated materials.
Materials and methods
XAI benchmarks
To evaluate the interpretability of XAI methods over GNN models, we established different levels of benchmarks from easy to hard: two synthetic datasets containing particular subgraphs (benzene and 3MRs), two datasets of handcrafted substructures causing toxicity (hepatotoxicity and Ames mutagenicity), and one dataset of substructures automatically computed according to pairs of molecules with similar structures but different molecular activities. The statistics of XAI benchmarks are shown in Table S6.
Synthetic datasets. Following the strategy of Sanchez-Lengeling et al.,22 we established two synthetic datasets to identify whether a molecular graph contains particular substructures. By substructure matching through RDKit program, we identified 6,000 and 1,406 positive compounds containing benzene and 3MRs, respectively, from the lead-like subset (50,000 compounds) in ZINC15.43 For comparison, we randomly selected 6,000 and 1,746 compounds’ unmatched molecules as negative samples, respectively.
Experimental datasets. In real-world scenarios, molecular properties usually result from dozens of substructures, which are much more difficult to identify than the particular substructures. Here, we compiled two toxicity datasets containing hand-crafted substructures (hepatotoxicity and Ames mutagenicity). The hepatotoxicity data were collected from Liu et al.,23 including 174 hepatotoxic, 230 possible hepatotoxic, and 183 non-hepatotoxic compounds. Among these, 12 molecular substructures have been identified as key substructures for human liver injuries. The Ames mutagenicity data were collected from Hansen et al.,44 including 6,506 compounds and corresponding Ames mutagenicity values. From the data, 46 toxic substructures were summarized by Sushko et al.11 These handcrafted substructures would be used as ground truths for assessing XAI methods.
In addition, we compiled another more challenging dataset to automatically determine substructures without handcrafted rules. For this purpose, we employed the property cliffs in the Cytochrome P450 3A4 (CYP3A4) inhibitions experimentally measured by Veith et al.,45 which includes 3,626 active inhibitors/substrates and 5,496 inactive compounds. The active compounds were compared with inactive ones through MMPA46 in the RDKit package. This led to 106 molecular pairs and corresponding substructures involving 46 active and 51 inactive compounds. During the GNN model training, all these involved compounds involved in found pairs will be put into the test set.
Formulating molecular explanation task for GNNs
In molecular GNNs, a molecule is represented using a graph with node representation for atoms and edge representation for chemical bonds, where and are the number of atoms and bonds and and are the lengths of their representations, respectively. A trained GNN model over the graph is interpreted by input into an explanation method (XAI) to fit the prediction by generating , where is the predicted importance of atoms or key subgraphs by the XAI method and is the predicted importance of edges.
GNN
To benchmark XAI methods, we employed four popular GNN architectures (GraphSAGE,47 GAT,48 GraphNET,49 and CMPNN50) that mostly differed in their message-passing strategies. GraphSAGE is the first method to generate node embeddings by sampling and aggregating features from its local neighborhood. It relies on a neighborhood sampling scheme to improve the effectiveness of message passing. Instead, the GAT incorporates the attention mechanism into the message-passing step. It computes the relative weights between two connected nodes following a self-attention strategy, which shows an impressive improvement over GraphSAGE on the classification tasks. Differently, GraphNET and CMPNN take the edge features into consideration during message passing. While GraphNET updates the node representation by aggregating its neighbor information with edge features for message passing, the CMPNN improves the molecular embedding by strengthening the message interactions between nodes and edges through a communicative kernel.
XAI methods
We focus on the XAI methods that explain GNN models as an external explainer in the testing phase without changing the models. The XAI methods include attribution-based (CAM,51 SmoothGrad,52 GradInput,53 GradCAM,15 IGs54) and subgraph recognition methods (MCTS17), totaling 6 methods as detailed below.
CAM attributes on GNN models by multiplying the final convolutional layer’s feature map activations act with the output weights of the last message-passing layer over the node and edge features as
| (Equation 1) |
GradInput obtains the attributions based on the partial derivatives of the input node and edge features as
| (Equation 2) |
Therefore, GradInput is defined as the element-wise product of the partial derivative with the input node and edge features:
| (Equation 3) |
GradCAM extended GradInput by utilizing the element-wise product of the activations in the intermediate GNN convolution layer and the gradients of the node and edge features in the intermediate layer:
| (Equation 4) |
SmoothGrad was a strategy used to combine the GradInput attribution method by averaging the attributions obtained from noise-perturbed inputs:
| (Equation 5) |
IG integrates the element-wise product of the interpolated input graph and the gradient of property with respect to interpolated graphs. The interpolated graphs are interpolated between the actual input G and a counterfactual input :
| (Equation 6) |
MCTS extracted the candidate rationales from molecules with the help of a property predictor. The root of the search tree is the molecular graph G, and each state in the search tree is a subgraph derived from a sequence of bond deletions.
As a control, we also included random attributions drawn from a uniform distribution.
Evaluation metrics for XAI
Following the evaluation strategy of previous studies,8,9 we assessed the explanation performance of XAI methods by quantifying how well the interpretations match the ground-truth key substructures. If the GNN model is “right for the right reasons,” we expect the XAI methods to highlight the correct ground-truth subgraphs in the input graph. Therefore, we used attribution-accuracy (ACC) and attribution-AUROC metrics to evaluate the XAI methods.
Attribution-ACC. The attribution-ACC is defined as the node-level accuracy for those important nodes/edges in explanation predictions compared with those in the ground truths. Formally, the metric attribution-ACC is computed as
| (Equation 7) |
where is the number of nodes in a molecular graph and denotes the ground-truth label for node . Here, is the prediction that the node is an important node, and the indicator function returns 1 if and are equal and returns 0 otherwise.
Attribution-AUROC. The attribution-AUROC is defined as the area under the receiver operating characteristic curve from the atom attribution scores. Formally, the metric attribution-AUROC is computed as
| (Equation 8) |
where is the descending rank value of atom in the substructures among all predicted attribution scores and and n represent the number of atoms in the substructures and not in the substructures, respectively. Particularly, the subgraph recognition methods such as MCTS could not be evaluated by attribution-AUROC since their outputs are directly subgraphs rather than attribution scores.
There are still many other metrics for explainability. For example, stability often measures how interpretations change when the input graph is perturbed, but it is not suitable for molecule-related tasks because the molecules with structural changes/perturbations are quite different from the original molecules.
GNN for property prediction
Since XAI methods depend on GNN models, we first introduce how to train four GNN models, which are implemented by Pytorch and run on Ubuntu Linux 18 with NVIDIA 3090 GPUs. In practice, the node and edge features used in GNN models are listed in Table S7 and are computed by open-source package RDKit. In our experiments, the XAI benchmarks were randomly split into training, validation, and test sets using a proportion of 8:1:1. We have applied a hyper-parameter searching module to explore the best performance for each GNN model on the validation set. The hyper-parameters of the four types of GNN models are detailed in Table S8. With the well-trained GNN models, we evaluated their prediction performances on the test set and further applied them to the XAI studies.
Assessment of XAI methods
To quantitatively assess the interpretability of XAI methods on GNN models, the explanation experiments were also evaluated on the test sets. These test set are consistent with the prediction experiments of GNNs. These XAI methods do not require retraining of the GNN models, so we applied the XAI methods to the well-trained models and interpret the predictions of GNN. Our goal is to judge whether the GNN models have learned the implicit relationship between a molecular graph and its corresponding properties. Therefore, the GNN model does not learn the labels of the key substructures of molecules. For the interpretations, the XAI methods calculated the importance of atoms and edges or recognized the key substructures that are associated with the property, and then the interpretations by XAI are quantitatively assessed by comparisons with predefined key substructures. Particularly, in the property cliff dataset, we assessed the interpretations in terms of molecular pairs.
Comparison with MCs
Human experts are frequently used in surveys about the future of research areas in science. In our experiments, we invited 7 MCs from Galixir Technologies (Shenzhen, Guangdong, China) and asked them to predict the molecular hepatotoxicity and their corresponding key substructures. The selection of the MCs was carefully planned to keep diversity. We selected MCs with different levels of working experience, including experts (MC1: 30 working years, and MC2: 20 working years) that are experienced in both academic and industry. The other 5 MCs have working experience ranging from 9 months to 10 years. All these MCs have been involved in experimental projects on hepatotoxicity and have background knowledge on this particular topic.
Before the experiment began, it was suggested that they study the relevant literature (except the benchmark literature), but they were not allowed to access additional information during the experiments. We also have made the test set of 50 compounds and results marked by experts available at https://github.com/biomed-AI/MolRep.
XAI-assisted FP generation
The model interpretations would be able to be used as additional features to enhance the predictive performance of the machine-learning (ML)/AI models. Firstly, we counted and selected the substructures that XAI method considered to be the most critical for a certain molecular property. In our experiments, we selected top-K substructures (K = 50 or 100) with the highest frequency. Particularly, the substructures only consisting of one or two carbon atoms will be removed in the selection process. And then, we utilized these high-frequency key substructures to construct structural molecular FPs as the XAI-assisted FP. The XAI-assisted FP is encoded into a binary bit string, and each bit corresponds to whether the selected substructures have matched the molecule.
XAI-assisted FP experiments
To demonstrate that the effectiveness of the XAI-assisted FP, we conducted the XAI-assisted FP experiments with random forest (RF) models on five public benchmarks including three classification datasets (BBBP, BACE, and HIV) and two regression datasets (IGC50 and LD50). The statistics of the datasets are shown in Table S9. We also conducted the experiments on the XAI benchmarks (Table S10). For each dataset, the XAI-assisted FP was generated from the training set following our generation strategy. In our experiments, we compared the performance of Morgan-FP, the combination of Morgan-FP and MACCS-FP, and the combination of Morgan-FP and XAI-assisted FP. And, we have performed a hyper-parameter searching on these RF models to explore the best performance, and the hyper-parameters are detailed in Table S11. Therefore, we can demonstrate the effectiveness of the XAI-assisted FPs in promoting the performance of molecular property prediction. In addition, we conducted experiments on the HIV dataset to explore the impact of XAI-FP with the increase of training data. The HIV dataset was down-sampled to 100, 1,000, 5,000, 10,000, and 30,000 molecules, and each subsampled data were used to construct XAI-assisted FP.
Acknowledgments
The authors thank MCs of Galixir Technologies, Ltd., who participated in the human benchmarking study. The authors are also grateful to Chengtao Li, Jixian Zhang, and the three anonymous reviewers, whose constructive comments were helpful for strengthening this article. This study has been supported by the National Key R&D Program of China (2020YFB0204803), National Natural Science Foundation of China (61772566 and 62041209), Guangdong Key Field R&D Plan (2019B020228001 and 2018B010109006), Introducing Innovative and Entrepreneurial Teams (2016ZT06D211), and Guangzhou S&T Research Plan (202007030010).
Author contributions
J.R., S.Z., and Y.Y. devised the project and the main conceptual ideas. J.R. and S.Z. developed the methodology and performed the experiments. J.R. and S.Z. wrote the manuscript in consultation with Y.L. and Y.Y. All authors discussed the results and contributed to the final manuscript.
Declaration of interests
This work was done when J.R. worked as an intern at Galixir Technologies, Ltd., and S.Z. currently works for Galixir Technologies, Ltd.
Published: November 10, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.patter.2022.100628.
Supplemental information
Data and code availability
All data and code used in the experiments are available at https://github.com/biomed-AI/MolRep. The codes are also publicly available on Zenodo (https://doi.org/10.5281/zenodo.7176794)
References
- 1.Zhou J., Cui G., Hu S., Zhang Z., Yang C., Liu Z., Wang L., Li C., Sun M. Graph neural networks: a review of methods and applications. AI Open. 2020;1:57–81. doi: 10.1016/j.aiopen.2021.01.001. [DOI] [Google Scholar]
- 2.Fan W., Ma Y., Li Q., He Y., Zhao E., Tang J., et al. The World Wide Web Conference. Association for Computing Machinery; New York: 2019. Graph neural networks for social recommendation; pp. 417–426. [Google Scholar]
- 3.Guo Z., Wang H. A deep graph neural network-based mechanism for social recommendations. IEEE Trans. Industr. Inform. 2021;17:2776–2783. doi: 10.1109/TII.2020.2986316. [DOI] [Google Scholar]
- 4.Berg R. van den, Kipf T.N., Welling M. Graph convolutional matrix completion. Preprint at arXiv. 2017 doi: 10.48550/arXiv.1706.02263. [DOI] [Google Scholar]
- 5.Ying R., He R., Chen K., Eksombatchai P., Hamilton W.L., Leskovec J. Graph convolutional neural networks for web-scale recommender systems. Proc. ACM SIGKDD Int. Conf. Knowl. Discov. Data Min. 2018:974–983. doi: 10.1145/3219819.3219890. [DOI] [Google Scholar]
- 6.Sanchez-Gonzalez A., Heess N., Springenberg J.T., Merel J., Riedmiller M., Hadsell R., Battaglia P. 35th International Conference on Machine Learning, ICML 2018. PMLR; 2018. Graph networks as learnable physics engines for inference and control; pp. 7097–7117. [Google Scholar]
- 7.Gilmer J., Schoenholz S.S., Riley P.F., Vinyals O., Dahl G.E. 34th International Conference on Machine Learning, ICML 2017. PMLR; 2017. Neural message passing for quantum chemistry; pp. 2053–2070. [Google Scholar]
- 8.Yuan H., Yu H., Gui S., Ji S. arXiv; 2020. Explainability in Graph Neural Networks: A Taxonomic Survey. [DOI] [PubMed] [Google Scholar]
- 9.Ying R., Bourgeois D., You J., Zitnik M., Leskovec J. In: Advances in Neural Information Processing Systems. Wallach H.M., Larochelle H., Beygelzimer A., d’Alché-Buc F., Fox E.A., Garnett R., editors. NIH Public Access; 2019. GNNExplainer: generating explanations for graph neural networks; p. 9240. [PMC free article] [PubMed] [Google Scholar]
- 10.Luo D., Cheng W., Xu D., Yu W., Zong B., Chen H., Zhang X. Parameterized explainer for graph neural network. Adv. Neural Inf. Process. Syst. 2020:19620–19631. [Google Scholar]
- 11.Sushko I., Salmina E., Potemkin V.A., Poda G., Tetko I.V. ToxAlerts: a web server of structural alerts for toxic chemicals and compounds with potential adverse reactions. J. Chem. Inf. Model. 2012;52:2310–2316. doi: 10.1021/ci300245q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baell J.B., Holloway G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 13.Rodríguez-Pérez R., Bajorath J. Interpretation of compound activity predictions from complex machine learning models using local approximations and shapley values. J. Med. Chem. 2020;63:8761–8777. doi: 10.1021/acs.jmedchem.9b01101. [DOI] [PubMed] [Google Scholar]
- 14.Pope P.E., Kolouri S., Rostami M., Martin C.E., Hoffmann H. Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition. IEEE; 2019. Explainability methods for graph convolutional neural networks; pp. 10764–10773. [DOI] [Google Scholar]
- 15.Selvaraju R.R., Cogswell M., Das A., Vedantam R., Parikh D., Batra D. Grad-CAM: visual explanations from deep networks via gradient-based localization. Int. J. Comput. Vis. 2020;128:336–359. doi: 10.1007/s11263-019-01228-7. [DOI] [Google Scholar]
- 16.Jiménez-Luna J., Skalic M., Weskamp N., Schneider G. Coloring molecules with explainable artificial intelligence for Preclinical relevance assessment. J. Chem. Inf. Model. 2021;61:1083–1094. doi: 10.1021/ACS.JCIM.0C01344/SUPPL_FILE/CI0C01344_SI_002.PDF. [DOI] [PubMed] [Google Scholar]
- 17.Jin W., Barzilay R., Jaakkola T. 37th International Conference on Machine Learning, ICML 2020. PMLR; 2020. Multi-objective molecule generation using interpretable substructures; pp. 4799–4809. [Google Scholar]
- 18.Fischer A., Smieško M., Sellner M., Lill M.A. Decision making in structure-based drug discovery: visual inspection of docking results. J. Med. Chem. 2021;64:2489–2500. doi: 10.1021/acs.jmedchem.0c02227. [DOI] [PubMed] [Google Scholar]
- 19.Lajiness M.S., Maggiora G.M., Shanmugasundaram V. Assessment of the consistency of medicinal chemists in reviewing sets of compounds. J. Med. Chem. 2004;47:4891–4896. doi: 10.1021/jm049740z. [DOI] [PubMed] [Google Scholar]
- 20.Boobier S., Osbourn A., Mitchell J.B.O. Can human experts predict solubility better than computers? J. Cheminform. 2017;9:63. doi: 10.1186/s13321-017-0250-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kutchukian P.S., Vasilyeva N.Y., Xu J., Lindvall M.K., Dillon M.P., Glick M., Coley J.D., Brooijmans N. Inside the mind of a medicinal chemist: the role of human bias in compound prioritization during drug discovery. PLoS One. 2012;7:e48476. doi: 10.1371/journal.pone.0048476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez-Lengeling B., Wei J., Lee B., Reif E., Wang P.Y., Qian W.W., et al. In: Advances in Neural Information Processing Systems. Larochelle H., Ranzato M., Hadsell R., Balcan M.-F., Lin H.-T., editors. Thirty-fourth Conference on Neural Information Processing Systems; 2020. Evaluating attribution for graph neural networks; pp. 5898–5910. [Google Scholar]
- 23.Liu R., Yu X., Wallqvist A. Data-driven identification of structural alerts for mitigating the risk of drug-induced human liver injuries. J. Cheminform. 2015;7:4–8. doi: 10.1186/s13321-015-0053-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hewitt M., Enoch S.J., Madden J.C., Przybylak K.R., Cronin M.T.D. Hepatotoxicity: a scheme for generating chemical categories for read-across, structural alerts and insights into mechanism(s) of action. Crit. Rev. Toxicol. 2013;43:537–558. doi: 10.3109/10408444.2013.811215. [DOI] [PubMed] [Google Scholar]
- 25.Jiménez-Luna J., Skalic M., Weskamp N. Benchmarking molecular feature attribution methods with activity cliffs. J. Chem. Inf. Model. 2022;62:274–283. doi: 10.1021/acs.jcim.1c01163. [DOI] [PubMed] [Google Scholar]
- 26.Stumpfe D., Hu Y., Dimova D., Bajorath J. Recent progress in Understanding activity cliffs and their utility in medicinal chemistry. J. Med. Chem. 2013;57:18–28. doi: 10.1021/JM401120G. [DOI] [PubMed] [Google Scholar]
- 27.Numeroso D., Bacciu D. Proceedings of International Joint Conference on Neural Networks. Institute of Electrical and Electronics Engineers (IEEE); 2021. MEG: generating molecular counterfactual explanations for deep graph networks; pp. 1–8. [Google Scholar]
- 28.Wellawatte G.P., Seshadri A., White A.D. Model agnostic generation of counterfactual explanations for molecules. Chem. Sci. 2022;13:3697–3705. doi: 10.1039/d1sc05259d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiménez-Luna J., Grisoni F., Schneider G. Drug discovery with explainable artificial intelligence. Nat. Mach. Intell. 2020;2:573–584. doi: 10.1038/s42256-020-00236-4. [DOI] [Google Scholar]
- 30.Linardatos P., Papastefanopoulos V., Kotsiantis S. Explainable ai: a review of machine learning interpretability methods. Entropy. 2021;23:E18–E45. doi: 10.3390/e23010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu J., Xu T., Rong Y., Bian Y., Huang J., He R. Graph information bottleneck for subgraph recognition. Int. Conf. Learn. Represent. 2020 [Google Scholar]
- 32.Yuan H., Tang J., Hu X., Ji S. Towards model-level explanations of graph neural networks. Virtual Event. 2020 doi: 10.1145/3394486.3403085. [DOI] [Google Scholar]
- 33.Van Der Maaten L., Hinton G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008;9:2579–2625. [Google Scholar]
- 34.O’Brien P.J., Irwin W., Diaz D., Howard-Cofield E., Krejsa C.M., Slaughter M.R., Gao B., Kaludercic N., Angeline A., Bernardi P., et al. High concordance of drug-induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell-based model using high content screening. Arch. Toxicol. 2006;80:580–604. doi: 10.1007/s00204-006-0091-3. [DOI] [PubMed] [Google Scholar]
- 35.Khalili H., Soudbakhsh A., Talasaz A.H. Severe hepatotoxicity and probable hepatorenal syndrome associated with sulfadiazine. Am. J. Health Syst. Pharm. 2011;68:888–892. doi: 10.2146/ajhp100516. [DOI] [PubMed] [Google Scholar]
- 36.Rogers D., Hahn M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010;50:742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 37.Hu W., Fey M., Zitnik M., Dong Y., Ren H., Liu B., et al. In: Advances in Neural Information Processing Systems. Larochelle H., Ranzato M., Hadsell R., Balcan M.-F., Lin H.-T., editors. Thirty-fourth Conference on Neural Information Processing Systems; 2020. Open graph benchmark: datasets for machine learning on graphs; pp. 22118–22133. [Google Scholar]
- 38.Chen D., Gao K., Nguyen D.D., Chen X., Jiang Y., Wei G.W., Pan F. Algebraic graph-assisted bidirectional transformers for molecular property prediction. Nat. Commun. 2021;12:3521. doi: 10.1038/s41467-021-23720-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Louppe G. Understanding random forests: from theory to practice. arXiv. 2014 doi: 10.48550/arXiv.1407.7502. [DOI] [Google Scholar]
- 40.Tseng Y.J., Hopfinger A.J., Esposito E.X. The great descriptor melting pot: Mixing descriptors for the common good of QSAR models. J. Comput. Aided Mol. Des. 2012;26:39–43. doi: 10.1007/s10822-011-9511-4. [DOI] [PubMed] [Google Scholar]
- 41.Xie L., Xu L., Kong R., Chang S., Xu X. Improvement of prediction performance with Conjoint molecular fingerprint in deep learning. Front. Pharmacol. 2020;11:606668. doi: 10.3389/fphar.2020.606668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng S., Rao J., Song Y., Zhang J., Xiao X., Fang E.F., Yang Y., Niu Z. PharmKG: a dedicated knowledge graph benchmark for bomedical data mining. Brief. Bioinform. 2021;22:bbaa344. doi: 10.1093/bib/bbaa344. [DOI] [PubMed] [Google Scholar]
- 43.Sterling T., Irwin J.J. Zinc 15 - Ligand discovery for Everyone. J. Chem. Inf. Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hansen K., Mika S., Schroeter T., Sutter A., ter Laak A., Steger-Hartmann T., Heinrich N., Müller K.R. Benchmark data set for in silico prediction of Ames mutagenicity. J. Chem. Inf. Model. 2009;49:2077–2081. doi: 10.1021/ci900161g. [DOI] [PubMed] [Google Scholar]
- 45.Veith H., Southall N., Huang R., James T., Fayne D., Artemenko N., Shen M., Inglese J., Austin C.P., Lloyd D.G., Auld D.S. Comprehensive characterization of cytochrome P450 isozyme selectivity across chemical libraries. Nat. Biotechnol. 2009;27:1050–1055. doi: 10.1038/nbt.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hussain J., Rea C. Computationally efficient algorithm to identify matched molecular pairs (MMPs) in large data sets. J. Chem. Inf. Model. 2010;50:339–348. doi: 10.1021/ci900450m. [DOI] [PubMed] [Google Scholar]
- 47.Hamilton W.L., Ying R., Leskovec J. Inductive representation learning on large graphs. Adv. Neural Inf. Process. Syst. 2017;30 [Google Scholar]
- 48.Veličković P., Casanova A., Liò P., Cucurull G., Romero A., Bengio Y. 6th International Conference on Learning Representations, ICLR 2018 - Conference Track Proceedings. International Conference on Learning Representations (ICLR); 2018. Graph attention networks. [Google Scholar]
- 49.Battaglia P.W., Hamrick J.B., Bapst V., Sanchez-Gonzalez A., Zambaldi V., Malinowski M., et al. arXiv; 2018. Relational Inductive Biases, Deep Learning, and Graph Networks. [DOI] [Google Scholar]
- 50.Song Y., Zheng S., Niu Z., Fu Z.H., Lu Y., Yang Y. International Joint Conferences on Artificial Intelligence (IJCAI); 2020. Communicative representation learning on attributed molecular graphs; pp. 2831–2838. [Google Scholar]
- 51.Zhou B., Khosla A., Lapedriza A., Oliva A., Torralba A. Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition. Institute of Electrical and Electronics Engineers (IEEE); 2016. Learning deep features for Discriminative Localization; pp. 2921–2929. [Google Scholar]
- 52.Smilkov D., Thorat N., Kim B., Viégas F., Wattenberg M. arXiv; 2017. SmoothGrad: Removing Noise by Adding Noise. [DOI] [Google Scholar]
- 53.Shrikumar A., Greenside P., Kundaje A. 34th International Conference on Machine Learning, ICML 2017. PMLR; 2017. Learning important features through propagating activation differences; pp. 4844–4866. [Google Scholar]
- 54.Sundararajan M., Taly A., Yan Q. 34th International Conference on Machine Learning. ICML 2017; 2017. Axiomatic attribution for deep networks; pp. 5109–5118. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and code used in the experiments are available at https://github.com/biomed-AI/MolRep. The codes are also publicly available on Zenodo (https://doi.org/10.5281/zenodo.7176794)