

Abstract
Objective
Esophageal adenocarcinoma (EAC) arises in the setting of Barrett’s esophagus, an intestinal metaplastic precursor lesion that can develop in patients with chronic gastroesophageal reflux disease (GERD). Here, we investigated the role of acidic bile salts, the mimicry of reflux, in activation of NOTCH signaling in EAC.
Design
This study utilized public databases, EAC cell line models, L2-IL1β transgenic mouse model and human EAC tissue samples to identify mechanisms of NOTCH activation under reflux conditions.
Results
Analysis of public databases demonstrated significant upregulation of NOTCH signaling components in EAC. In vitro studies demonstrated nuclear accumulation of active NOTCH1 cleaved fragment (NICD) and upregulation of NOTCH targets in EAC cells in response to reflux conditions. Additional investigations identified DLL1 as the predominant ligand contributing to NOTCH1 activation under reflux conditions. We discovered a novel crosstalk between APE1 redox function, reflux-induced inflammation, and DLL1 up-regulation where NF-κB can directly bind to and induce the expression of DLL1. The APE1 redox function was crucial for activation of the APE1-NF-kB-NOTCH axis and promoting cancer cell stem-like properties in response to reflux conditions. Overexpression of APE1 and DLL1 were detected in gastroesophageal junctions of the L2-IL1ß transgenic mouse model and human EAC tissue microarrays. DLL1 high levels were associated with poor overall survival in patients with EAC.
Conclusion
These findings underscore a unique mechanism that links redox balance, inflammation, and embryonic development (NOTCH) into a common pro-tumorigenic pathway that is intrinsic to EAC cells.
Introduction
Esophageal cancer is the sixth leading cause of cancer death worldwide[1]. Esophageal adenocarcinoma (EAC), the predominant histopathological type of esophageal cancer in the U.S. and Western countries, has been increasing rapidly over the past 40 years[2]. EAC patients have a 5-year survival rate below 20%[3, 4]. Hence, there is an urgent need to identify novel molecular mechanisms and find new therapeutic targets for EAC. Chronic gastroesophageal reflux disease (GERD) is characterized by abnormal exposure of the lower esophagus to a mixture of acidic gastric juice and bile salts. GERD is the main risk factor for metaplastic Barrett’s esophagus (BE) and its progression to EAC [5] [6]. GERD conditions in patients with BE and EAC induce DNA damage along with aberrant activation of pro-inflammatory and pro-tumorigenic signaling pathways such as NF-κB and STAT3 pathways[7, 8].
Apurinic/apyrimidinic endonuclease (APE1), also known as APEX or redox factor 1 (REF1), is a dual-functional protein that plays important roles in both DNA base excision repair and reduction-oxidation (redox) transcriptional regulation[9]. APE1 interacts with a number of redox-dependent transcription factors to induce transcription of target genes [10, 11]. APE1 maintains the reduced status of cysteine residues on these transcription factors to enhance their DNA-binding affinity on target genes [10, 12]. Aberrant overexpression of APE1 has been described in multiple cancers, including EAC[13, 14, 15]. Dysregulation of APE1 is closely related to cell proliferation, cell survival, angiogenesis, metastasis and drug resistance[16, 17]. Exposure to acidic bile salts (ABS), the mimic of the reflux condition under GERD, induces oxidative stress and genotoxic DNA damage, with subsequent upregulation of APE1 to promote cell survival in EAC cells[15, 18].
The NOTCH pathway is a highly conserved cell signaling system, playing both oncogenic and tumor suppressive functions, depending on the cancer type and cellular context[19, 20]. NOTCH signaling promotes cell differentiation, apoptosis, invasion, cancer stem-like properties, therapeutic resistance and tumor immune escape[21, 22, 23]. Mammals possess four different NOTCH receptors (NOTCH 1 – 4) and five Delta/Serrate/Lag2 (DSL) ligands, referred to as JAG1, JAG2, DLL1, DLL3 and DLL4[21]. Once binding to a ligand from neighboring cells, the NOTCH receptor undergoes consecutive proteolytic cleavages by ADAM17 and γ-secretase[21], releasing the NOTCH intracellular domain (NICD) into the nucleus. NICD, the active form of receptor that forms a ternary complex with CBF1Su(H)–LAG1 (CSL) and protein mastermind-like 1 (MAML1) to transcriptionally regulate downstream targets such as HES and HEY gene families[21, 24].
Although NOTCH activation has been reported in BE and EAC[25, 26], the mechanisms underlying its activation in EAC and under reflux conditions remain largely unknown. This study demonstrates upregulation of NOTCH signaling in response to exposure to acidic bile salts, mimicking reflux conditions. Activation of NOTCH signaling occurred in an APE1-redox-dependent manner. Our findings uncover DLL1 as a critical ligand, activated by the APE1-NF-kB axis, to induce activation of NOTCH and promote stem-like properties in EAC tumorigenesis.
Materials and Methods
For detailed resources, please refer to online supplemental materials and methods and online supplemental tables 1–3.
Results
NOTCH signaling is activated in EAC patients and ABS-provoked cell lines.
Gene Set Enrichment Analysis (GSEA) of the TCGA and GEO datasets of EAC patient samples predicted a significant alteration of NOTCH signaling (Supplemental Figure 1, A–C). Using the TCGA-EAC dataset, we detected significant upregulation of mRNA levels of two receptors (NOTCH1 and NOTCH3), four ligands (DLL1, DLL4, JAG1 and JAG2) and several downstream targets (HES1, HEY1 and HEYL) (Figure 1A). Analysis of the GSE13898 dataset (75 primary EAC and 15 BE) demonstrated similar results (Figure 1B). Because chronic GERD is the primary risk factor for BE-derived EAC, we decided to validate the expression levels of NOTCH signaling components under reflux conditions (ABS exposure) using EAC cell lines. The results demonstrated consistent ABS-induced expression of several genes involved in the NOTCH signaling cascade, including NOTCH1, DLL1, DLL4, HES1 and HEY1 (Figure 1C and Supplemental Figure 1D). In addition, western blot analysis demonstrated a remarkable increase in the total DLL1 protein level, including the full length and cleaved c-terminal proteolytic fragment of DLL1 (30kDa), following exposure to ABS (Figure 1D and Supplemental Figure 1E). Consistent with activation of NOTCH, we observed an increase in the NOTCH1 intracellular domain (NICD) (120kDa), the active form of NOTCH1 (Figure 1D and Supplemental Figure 1E). These results suggest up-regulation and activation of NOTCH signaling components in response to reflux conditions (ABS).
Figure 1.
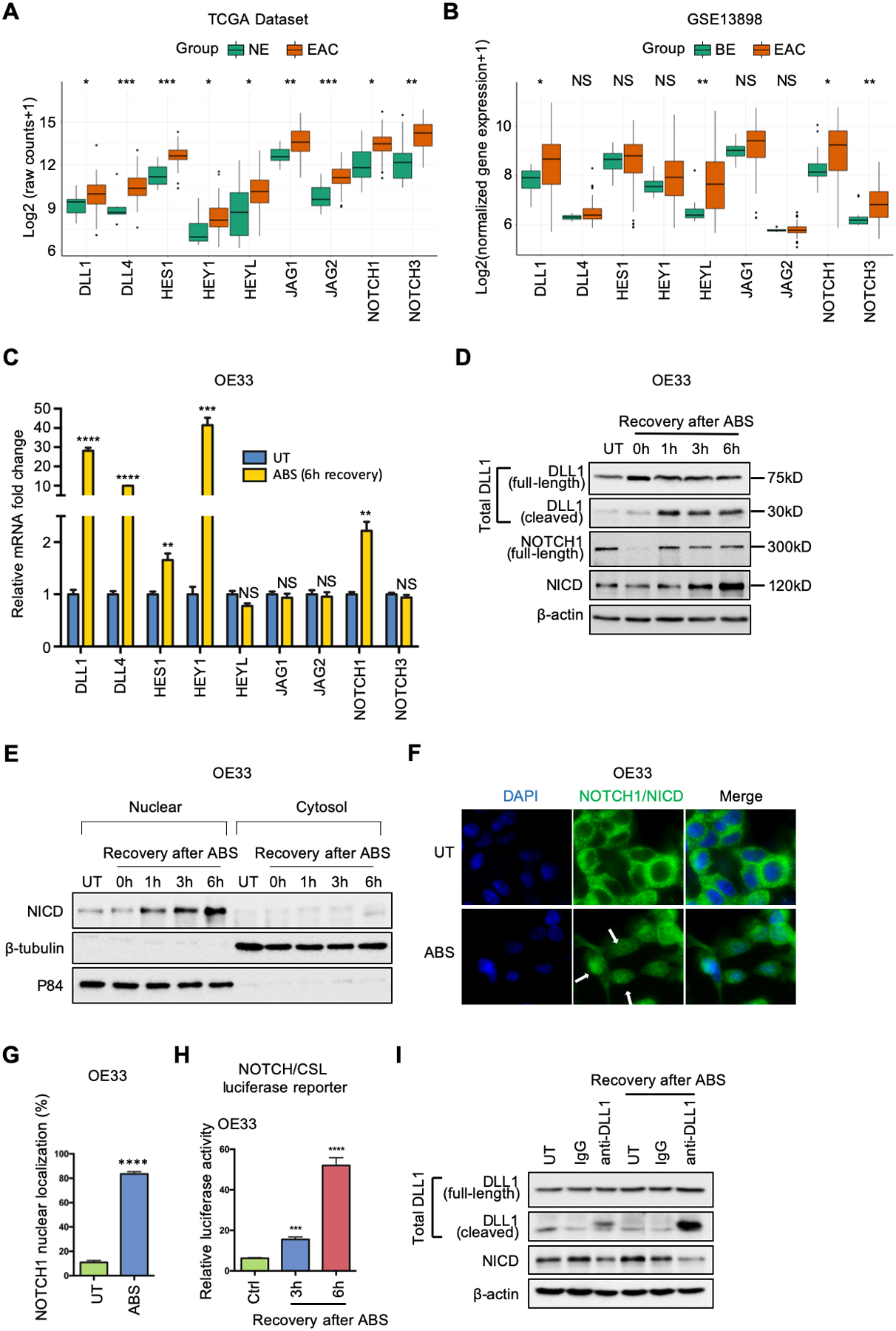
NOTCH signaling is activated in EAC patients and ABS-provoked cell lines. (A) and (B) Grouped boxplot showing gene expression of core NOTCH signaling components in EAC/BE and normal esophagus tissues in TCGA-EAC database (A) and GSE13898 dataset (B), respectively. (C) qRT-PCR showing mRNA expression levels of key components of NOTCH pathway in OE33 cells after 6h-recovery from ABS exposure. (D) Western blots of total DLL1 (including full-length DLL1 at 75kDa and cleaved DLL1 at 30kDa), full-length NOTCH1 (300kDa), active NOTCH1 intracellular domain (NICD, 120kDa) and β-actin in OE33 cells during indicated recovery time courses after ABS exposure. (E) OE33 cells were harvested for cytosol/nuclear fractionation during 6h-recovery after ABS exposure. Induction of NICD was examined by Western blots. β -tubulin and p84 were used as loading control for cytosol fraction and nuclear fractions, respectively. (F) Representative immunofluorescent staining images of NOTCH1/NICD in OE33 cells after ABS exposure. DAPI was used for nuclear staining. (G) The percentage of NOTCH1 nuclear localization was quantified using three independent fields. (H) NOTCH/CSL luciferase assays were performed in OE33 cells. (I) Anti-DLL1 antibody neutralization blocked NOTCH1 cleavage and activation in OE33 cells. The cells were incubated with DLL1 antibody (10μg/ml) or IgG control (10μg/ml) in full medium before and during recovery of ABS exposure. All quantification analyses were shown as mean ± SEM. Statistical significance was calculated using the Wilcoxon test in the public datasets for two group comparisons. t test was performed to analyze the experimental data for two group comparisons. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, NS no significance.
The hallmark of canonical activation of NOTCH signaling is nuclear localization of NICD, which triggers transcription of downstream target genes. To confirm the nuclear translocation of NICD in response to ABS, we performed cytosol/nuclear fractionation in OE19 and OE33 cells. The results showed an accumulation of NICD in the nucleus in response to ABS exposure (Figure 1E and Supplemental Figure 1F). Immunofluorescence staining of NOTCH1/NICD demonstrated membranous immunostaining of NOTCH in the control (untreated) cells whereas cells exposed to ABS showed nuclear immunostaining indicative of cleaved NOTCH (NICD) (Figure 1, F–G and Supplemental Figure 1, G–H). To confirm the transcriptional activity of ABS-induced nuclear NICD, we performed NOTCH/CSL luciferase reporter assays as a measure of NOTCH transcription activity. There was a significant increase in luciferase activity (P<.001) in response to ABS treatment compared with the control group (Ctrl) (Figure 1H and Supplemental Figure 1I).
As shown in Figure 1D and Supplemental Figure 1E, we detected ABS-induced increase and cleavage of DLL1, an important NOTCH ligand. To determine if DLL1 was indeed a crucial ligand for activation of NOTCH under ABS conditions, we performed a DLL1 antibody neutralization experiment. DLL1 neutralization not only reduced NICD at a base level (Figure 1I, lane 3 vs lane 2) but also abrogated ABS-induced NOTCH1 cleavage compared with the IgG control (Figure 1I, lane 6 vs lane 5). These results indicate that DLL1 is a critical ligand for ABS-induced NOTCH1 cleavage and activation.
NF-κB is an upstream transcription factor for DLL1 that directly binds to the DLL1 promoter
As we detected changes in DLL1 at the mRNA level in response to ABS, our next step was to identify the upstream transcription factors (TFs) that are induced with ABS and play a role in induction of DLL1 mRNA expression. First, we identified the differentially expressed genes (DEGs) defined by distinct DLL1 mRNA expression levels (DLL1-high versus DLL1-low) in the TCGA-EAC database. Based on the DEGs, we performed GSEA (http://software.broadinstitute.org/gsea/msigdb/collections.jsp#C3) and utilized a web-based tool (TRRUST, https://www.grnpedia.org/trrust/Network_search_form.php) for TF target prediction. GSEA predicted 100 potential TFs critical for DLL1 transcription (Supplemental Table 4, p.adjust < 6e-34), while 29 TFs were identified as candidates by TRRUST (Supplemental Table 5). Overlapping the 100 enriched TFs of GESA and the TRRUST candidates highlighted 6 TFs, which included NF-κB (Figure 2A). GSEA of the TCGA-EAC database confirmed enrichment of the NF-κB signature in DLL1-high compared with DLL1-low EAC samples (Figure 2B). We further performed RNA-seq using OE33 cells, following exposure to ABS, compared to untreated control. RNA-seq analysis of OE33 cells demonstrated an increase in the expression levels of DLL1 and several NOTCH receptors (NOTCH1, NOTCH2 and NOTCH3), following exposure to ABS, as compared to untreated control (Supplemental Figure 2A). GSEA analysis showed significant enrichment of the NF-κB signature (Figure 2C and Supplemental Figure 2B). Using Western blot analysis, we confirmed the increase in NF-kB phosphorylation (S536) (Figure 2D) and transcription activity (Figure 2E) following exposure to ABS. Consistent with these data, immunofluorescence staining demonstrated an increase in nuclear NF-kB immunostaining following exposure to ABS (Figure 2, F–G). Of note, ectopic overexpression of p65 led to induction of DLL1 mRNA expression, suggesting a possible direct role of NF-kB in transcription regulation of DLL1 (Figure 2, H–I).
Figure 2.
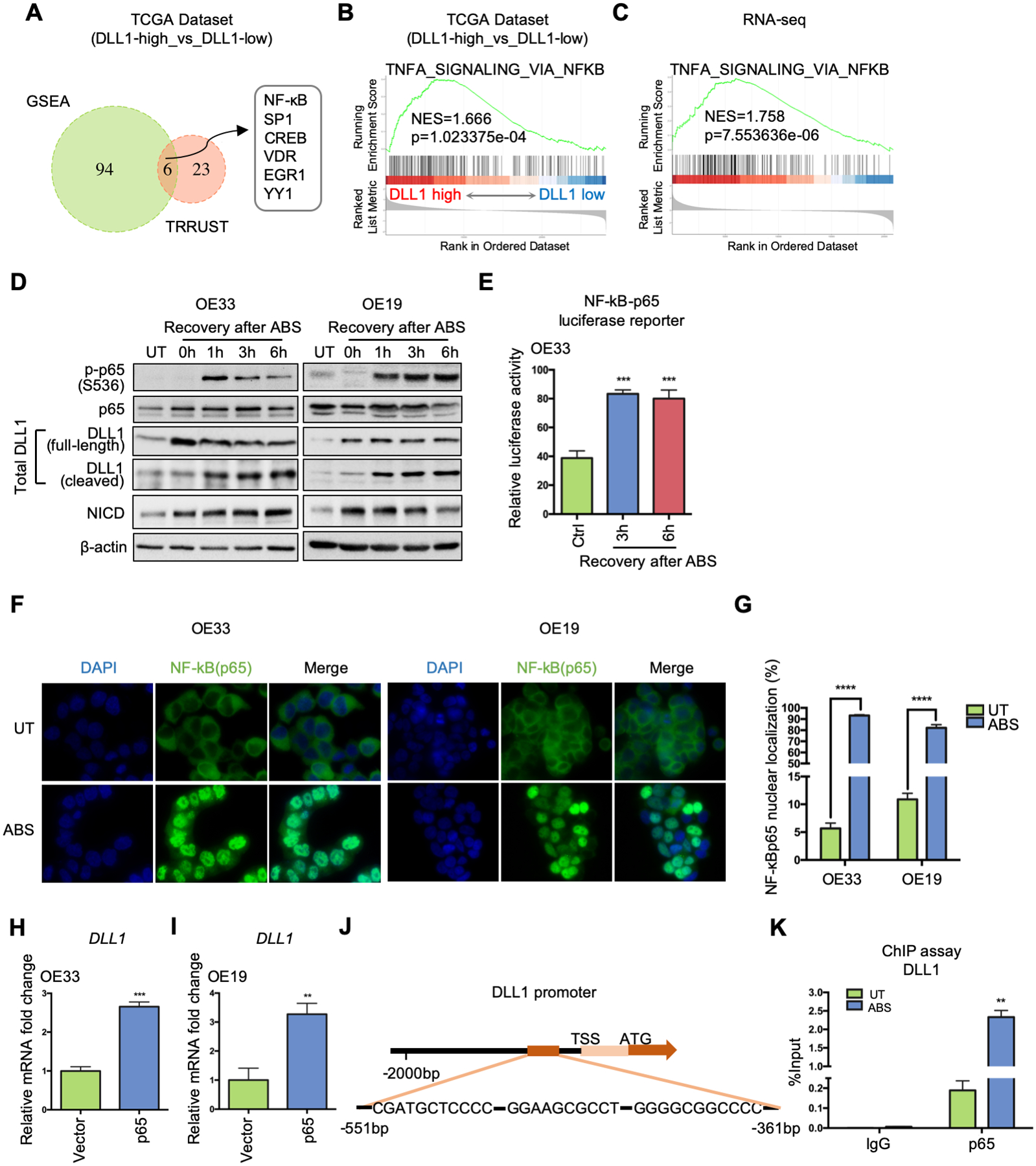
NF-κB is critical for ABS-induced DLL1 transcription by directly binding to DLL1 promoter region. (A) DLL1-expression-based transcription factor enrichment analysis by GSEA and TRRUST website indicates NF-κB as one of the key transcriptional regulators in DLL1-high tumor samples in the TCGA-EAC database. (B) GSEA of NF-κB signaling in the TCGA-EAC database, comparing DLL1-high EAC with DLL1-low EAC. (C) GSEA of NF-κB pathway in the RNA-seq datasets of OE33 cells, comparing control cells with ABS-treated cells (shCtrl vs shCtrl+ABS). (D) Western blots show protein level change of phosphor-p65, total p65, total DLL1 (including full-length ligand and its cleaved fragment), NICD and β-actin during 6h-recovery after ABS exposure in OE33 (left) and OE19 (right) cells. (E) NF-κB-p65 luciferase assay shows NF-κB transcriptional activity were elevated after ABS exposure in OE33 cells. Untreated cells worked as control (Ctrl). (F) Representative immunofluorescent staining images of NF-κB (p65) in OE33 (left) and OE19 cells (right) showing nuclear translocation after ABS exposure. DAPI was used for nuclear staining. (G) Percentage of NF-κB (p65) nuclear localization was quantified using three independent fields. (H) and (I) DLL1 mRNA expression was detected after OE33 (H) and OE19 (I) cells were transfected with a p65 overexpression vector or control (Vector). (J) The ChIP primers amplified the DNA fragment containing three adjacent potential NF-κB binding sites in DLL1 promoter, −551bp to −361bp from transcription start site (TSS). (K) ChIP-qPCR was performed in OE33 cells by using NF-κB antibody to pull down chromatin after ABS exposure, compared to the untreated control. IgG works as control. Statistical data are shown as mean ± SEM. **p<0.01, ***p<0.001 and ****p<0.0001 as calculated by t test for two group comparisons.
Based on the above results, we analyzed the DLL1 promoter to identify potential NF-κB binding sites using three online tools: JASPAR 2020 (http://jaspar.genereg.net/), Promo (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) and TRANSFAC-Gene Regulation (http://gene-regulation.com/pub/databases.html. This analysis identified eight potential binding sites of NF-κB on the DLL1 promoter (Supplemental Figure 2C). To confirm direct binding, we performed a ChIP assay using a phospho-NF-κB-p65 antibody, followed by qPCR using six primers’ pairs (Supplemental Table 3) that cover all potential binding sites. ChIP-qPCR results indicated that only one primer set covering three adjacent predicted binding sites (−551bp to – 361bp) (Figure 2J) was amplified in the ChIP samples. This primer set showed a 12-fold increase of NF-κB binding on the DLL1 promoter after ABS exposure compared with the untreated group (UT) (Figure 2K). There was no significant amplification detected in the IgG pull-down control sample (Figure 2K). These novel findings confirmed the direct binding of NF-κB on the DLL1 promoter.
Exposure to ABS promotes NOTCH activation in an APE1-redox-dependent manner
Exposure of esophageal cells to reflux conditions generates high levels of reactive oxygen species and oxidative stress necessitating activation of cellular redox mechanisms for the protection of transcription networks and maintaining cellular homeostasis. Our previous studies identified APE1 as an important signaling event in conditions of ABS exposure [8, 13, 15]. APE1 redox function is critical for protecting cellular signaling and activation of redox-dependent TFs such as STAT3 under reflux conditions [8]. We, therefore, hypothesized that APE1 played a critical role in promoting NF-κB transcription activity with subsequent transcriptional induction of DLL1 and NOTCH activation.
Indeed, ectopic expression of APE1 increased the levels of the three proteins (Figure 3A). The NF-κB luciferase reporter assay consistently supported the role of APE1, showing induction of the luciferase activity with ectopic expression of APE1 (Figure 3B). Using qRT-PCR, we found that APE1 overexpression upregulated DLL1 mRNA expression compared with the control (Vector) (Figure 3C and Supplemental Figure 3A). The NOTCH/CSL luciferase reporter assay confirmed that ectopic expression of APE1 significantly increased NOTCH luciferase activity, a measure of NOTCH transcription activity (Figure 3D and Supplemental Figure 3B). Based on these results, we next determined if changes in NF-κB-p65 phosphorylation, DLL1 induction and NOTCH activation are APE1-dependent under reflux conditions (ABS). Western blot analysis demonstrated an increase in APE1, phospho-p65, total DLL1, and active NICD (120kDa), following ABS treatment (Figure 3E). APE1 silencing using siRNA abolished the ABS-induced increase in phospho-p65, DLL1 and NICD protein levels (Figure 3F). We also used OE33 cells with stable APE1 knockdown (shAPE1) or control (shCtrl) for the NF-κB luciferase reporter assays. The results indicated that APE1 suppression abrogated ABS-enhanced NF-κB transcription activity (Figure 3G). Transient knockdown of APE1 (siAPE1) abolished ABS-induced DLL1 transcripts (Figure 3H and Supplemental Figure 3C) and NOTCH/CSL luciferase activity (Figure 3I and Supplemental Figure 3D). Interestingly, DLL4 was not reduced upon APE1 knockdown (Supplemental Figure 3, E–F), further confirming that DLL1 is the crucial NOTCH ligand in EAC. Collectively, these results indicate that changes in DLL1 and NOTCH activity are APE1-dependent under reflux conditions (ABS).
Figure 3.
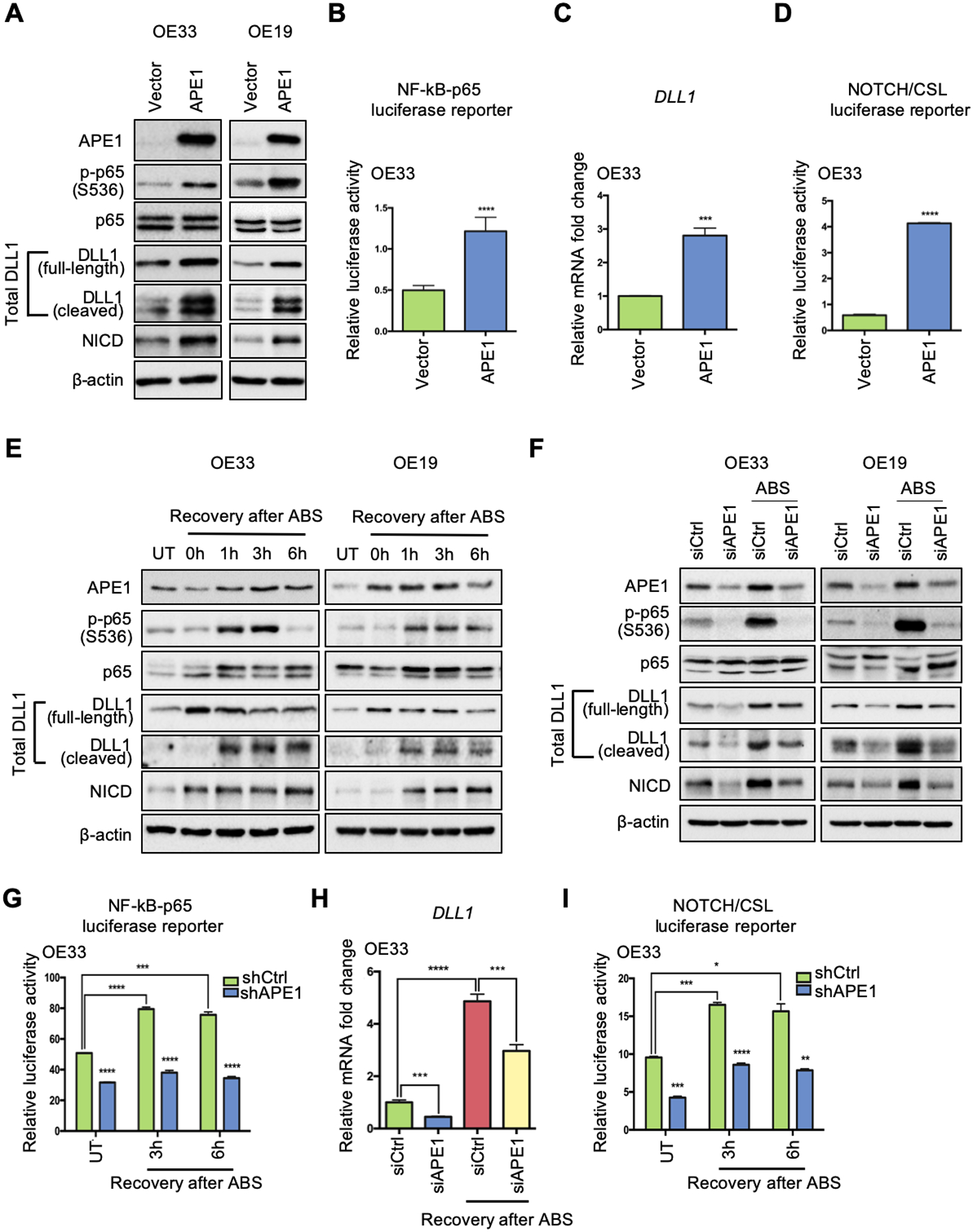
APE1 plays an essential role in ABS-induced NF-κB-DLL1-NOTCH activation. (A) Western blots were used to detect protein levels of interested genes after ectopic APE1 overexpression in OE33 (left) and OE19 (right) cells. (B) NF-κB-p65 luciferase activity was examined after APE1 overexpression. (C) qRT-PCR for DLL1 mRNA expression was performed in the condition of APE1 overexpression. (D) NOTCH/CSL luciferase reporter assay was performed to confirm NOTCH transcriptional activity after APE1 overexpression. (E) Western blots were used for detecting protein expression of APE1, phosphor-p65, total p65, total DLL1, NICD and β-actin at different time points of recovery from ABS exposure in OE33 (left) and OE19 (right) cells. (F) OE33 (left) and OE19 (right) cells were transfected with APE1 siRNA (siAPE1) or control siRNA (siCtrl) before ABS exposure; protein levels of interested genes were examined by Western blot analysis. (G) OE33 cells with stable APE1 knockdown (shAPE1) or control (shCtrl) were exposed to ABS, NF-κB-p65 activity luciferase reporter assay was performed at 3h and 6h recovery time points after ABS exposure. (H) APE1 knockdown (siAPE1) blocked ABS-induced DLL1 upregulation in OE33 as comparing to control cells (siCtrl). (I) NOTCH/CSL luciferase activity was checked in APE1-knockdown (shAPE1) and scramble shRNA control (shCtrl) OE33 cells with or without ABS exposure. Statistical data are shown as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 as calculated by t test for two group comparisons.
To determine the role of APE1 redox function in the ABS-induced NF-κB-DLL1-NOTCH activation, we utilized a stable shAPE1 cell line for reconstitution with wild-type APE1 (WT) or APE1-redox-deficient-mutant (C65A) under ABS conditions. The results indicated that the wild-type APE1, not the redox-deficient-mutant, restored protein levels of phospho-p65 (Ser536), DLL1 and NICD in response to ABS (Figure 4A). In addition, the increase in phosphor-p65, DLL1 and NICD by ABS was abrogated by E3330, a pharmacologic small molecule inhibitor of APE1 redox activity (Figure 4B). The use of E3330 also abrogated the ABS-induced NF-κB luciferase reporter activity (Figure 4C), DLL1 expression (Figure 4, D–E), and NOTCH/CSL luciferase reporter activity (Figure 4, F–G). These results indicate that APE1 redox function was required for NF-κB activation, induction, and cleavage of DLL1, and NOTCH1 cleavage and transcription activation under reflux conditions. To further validate the connection between APE1 and DLL1, we performed a Pearson correlation analysis of normalized gene expression in the GEO dataset of NE, BE and EAC (GSE13898). The results demonstrated a correlation between APE1 and NOTCH signaling components (Figure 5A). Of note, DLL1 was the only ligand that had a strong positive correlation with APE1 in EAC patients and also significantly induced in our ABS-treated EAC cell models (Figure 5B, R = 0.66, p = 4.6e-16). Immunofluorescence analysis demonstrated an increase in nuclear APE1 immunostaining levels together with up-regulation of intracytoplasmic DLL1 after ABS treatment, as compared with untreated controls (UT) (Figure 5, C–D). Similar results were detected using 3D organotypic culture model of OE33 cells (Figure 5, E–F). Consistent with these findings, we detected an increase in the cleaved extracellular N-terminal fragment (NTF) of DLL1 in the conditioned medium after 3h- and 6h-recovery from ABS exposure compared to UT controls (Supplemental Figure 4). These results demonstrate an important role of APE1 redox function in the activation of NOTCH signaling via NF-kB-dependent transcription regulation of DLL1.
Figure 4.
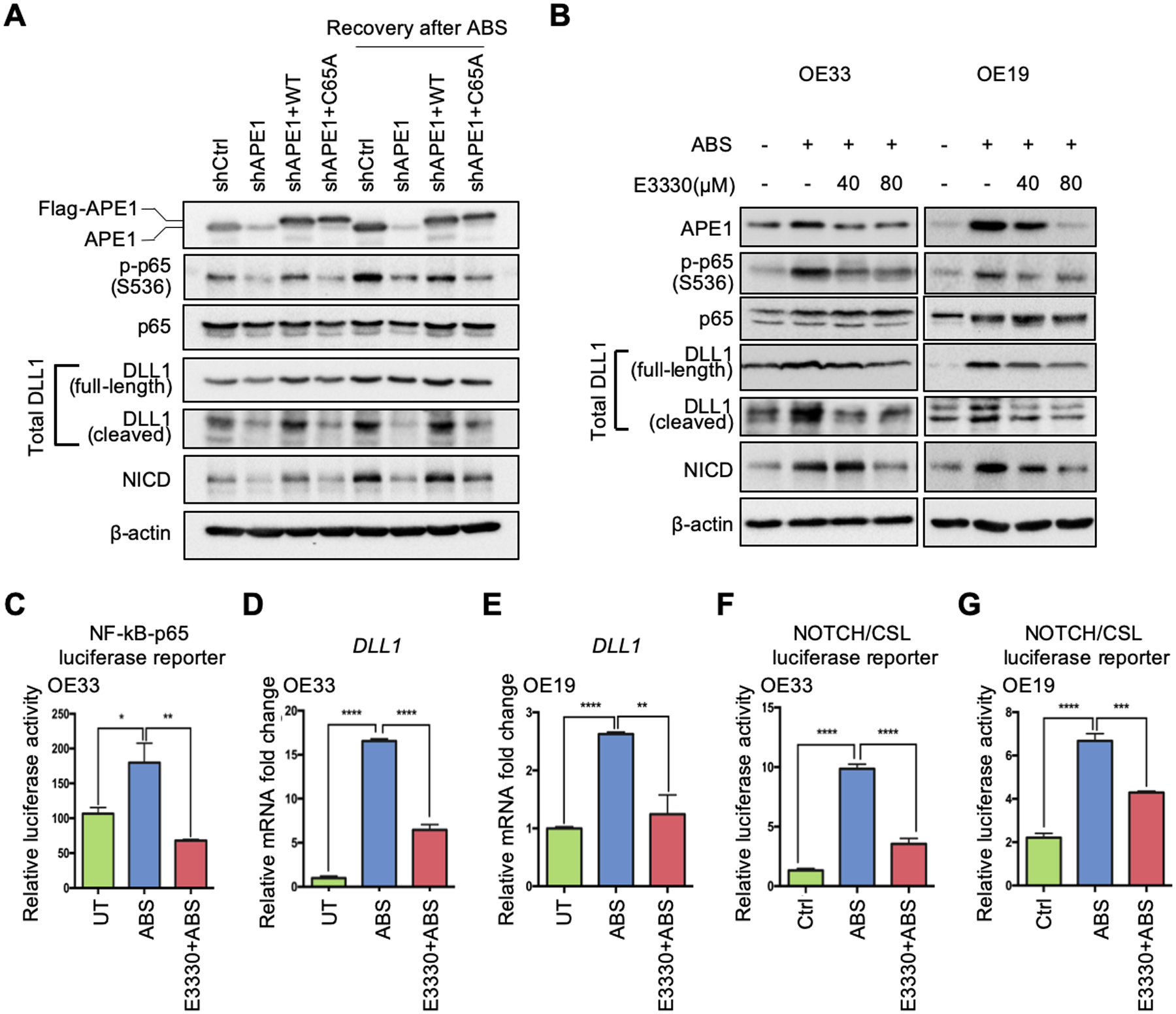
APE1 regulates NF-κB-DLL1-NOTCH axis through its redox function in response to ABS. (A) The APE1 stable silencing OE33 cells (shAPE1) were transfected with APE1-wild-type (WT) overexpression plasmid or APE1-redox-deficient-mutant (C65A) plasmid in the absence or presence of ABS exposure for Western blots. (B) Western blots were used to detect protein levels of interested genes in the condition of ABS treatment with or without APE1 redox inhibitor, E3330, in OE33 (left) and OE19 (right) cells. (C) OE33 cells were maintained with 80 μM E3330 before and after ABS exposure, and then NF-κB-p65 activity luciferase reporter assay was performed. (D) and (E) qRT-PCR was used to detect DLL1 mRNA change in the condition of ABS exposure with or without E3330 application (80 μM) in OE33 (D) and OE19 (E) cells. (F) and (G) Luciferase reporter assays were performed to examine NOTCH transcriptional activity in the condition of ABS treatment with or without E3330 in OE33 (F) and OE19 (G) cells. Statistical data are shown as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 as calculated by t test for two group comparisons.
Figure 5.
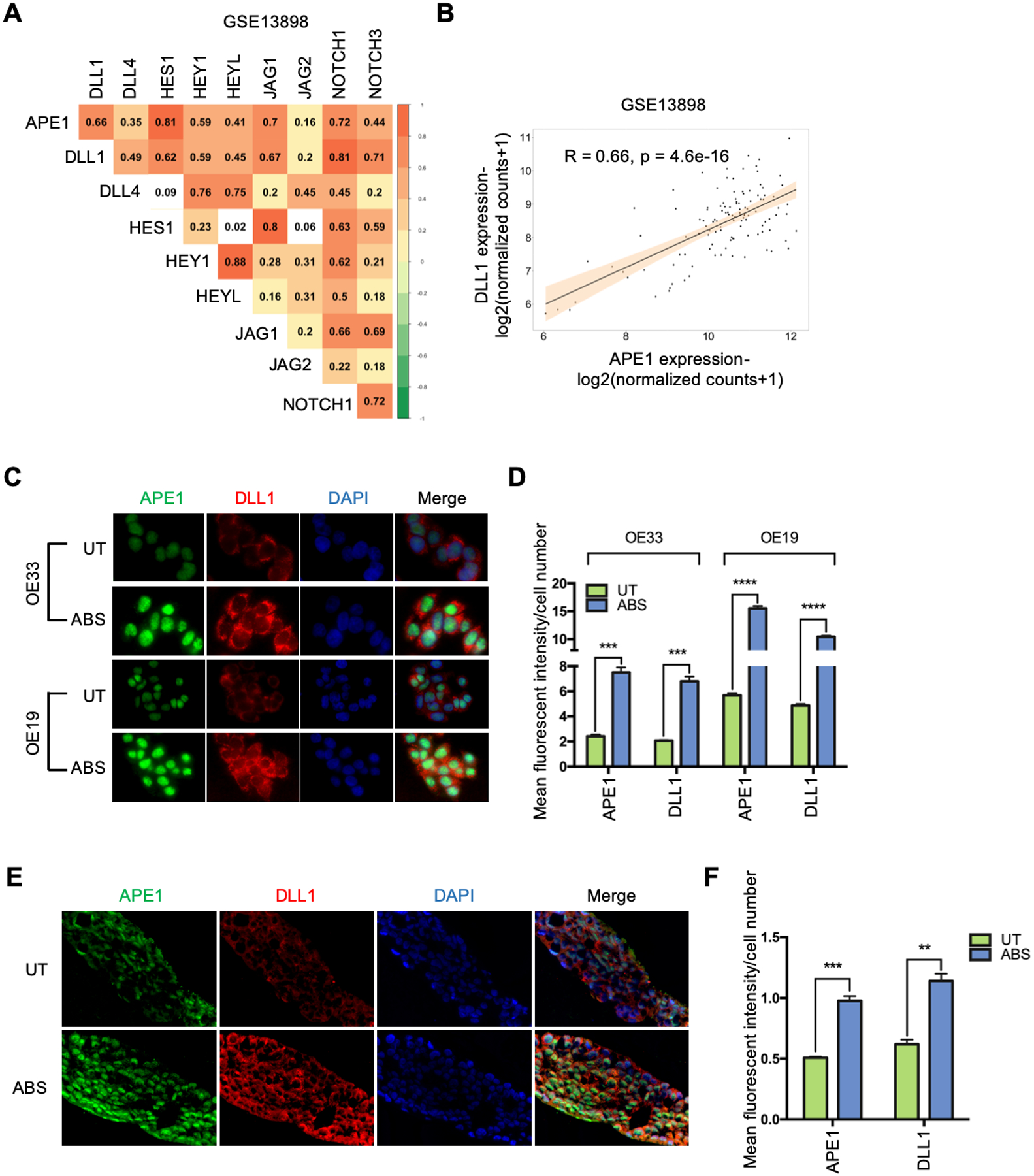
APE1 is correlated and co-overexpressed with DLL1 under ABS exposure. (A) Pearson correlation analysis of APE1 and NOTCH signaling components in a GEO dataset of NE, BE and EAC (GSE13898). (B) APE1 has significant positive correlation with DLL1 at transcription level (R = 0.66, p = 4.6e-16) in the GSE13898 dataset. (C) Representative immunofluorescent staining images of APE1 (green) and DLL1 (red) in OE33 (upper) and OE19 (lower) cells with or without ABS treatment. (D) Mean fluorescent intensity of APE1 and DLL1 was quantified using three independent fields in OE33 (left) and OE19 (right) cells. (E) Representative immunofluorescent staining images of APE1 (green) and DLL1 (red) in OE33-derived 3D organotypic culture with or without ABS treatment. (F) Mean fluorescent intensity of APE1 and DLL1 was quantified using three independent fields. Statistical data are shown as mean ± SEM. **p<0.01, ***p<0.001 and ****p<0.0001 as calculated by t test for two group comparisons.
EAC tumorosphere formation is enhanced in response to acidic bile salts
NOTCH signaling plays a critical role in driving and maintaining stem cell properties [25, 27, 28]. We, therefore, investigated if the APE1-NOTCH axis promotes stem-like features in response to reflux conditions. To mimic the clinical conditions of GERD, we exposed OE33 cells to repeated episodes of ABS cocktail (200uM, pH=5.5) 20 min per day for 14 days, followed by a 3D tumorosphere formation assay [29]. OE33 cells with repeated ABS exposure displayed significant augmentation of tumorosphere number and growth compared with UT controls (Figure 6, A–C). Immunofluorescence staining demonstrated concomitant overexpression APE1 and DLL1 (Supplemental Figure 5, A–B). Western blots showed that cells exposed to repeated ABS treatment had higher levels of APE1, phospho-p65, DLL1 and NICD (Figure 6D), confirming activation of this signaling cascade in response to reflux conditions. To determine if these phenotypic and molecular changes were dependent on APE-redox function, we maintained OE33-derived tumorospheres with APE1-redox-specific inhibitor, E3330, for 3 consecutive days before imaging. E3330 (50μM and 100μM) gradually reduced the number and size of tumorospheres (Figure 6, E–G) compared with UT controls. The changes of immunofluorescence intensity (Supplemental Figure 5, C–D) and molecular signaling (Figure 6H) were also reversed. We also used a recent derivative of E3330, APX2009, which is a potent inhibitor of APE1 redox function [30, 31]. APX2009 decreased protein levels of phospho-p65, total DLL1 (both full-length and cleaved ligands) and NICD in the tumorospheres compared to the untreated group (Supplemental Figure 5E).
Figure 6.
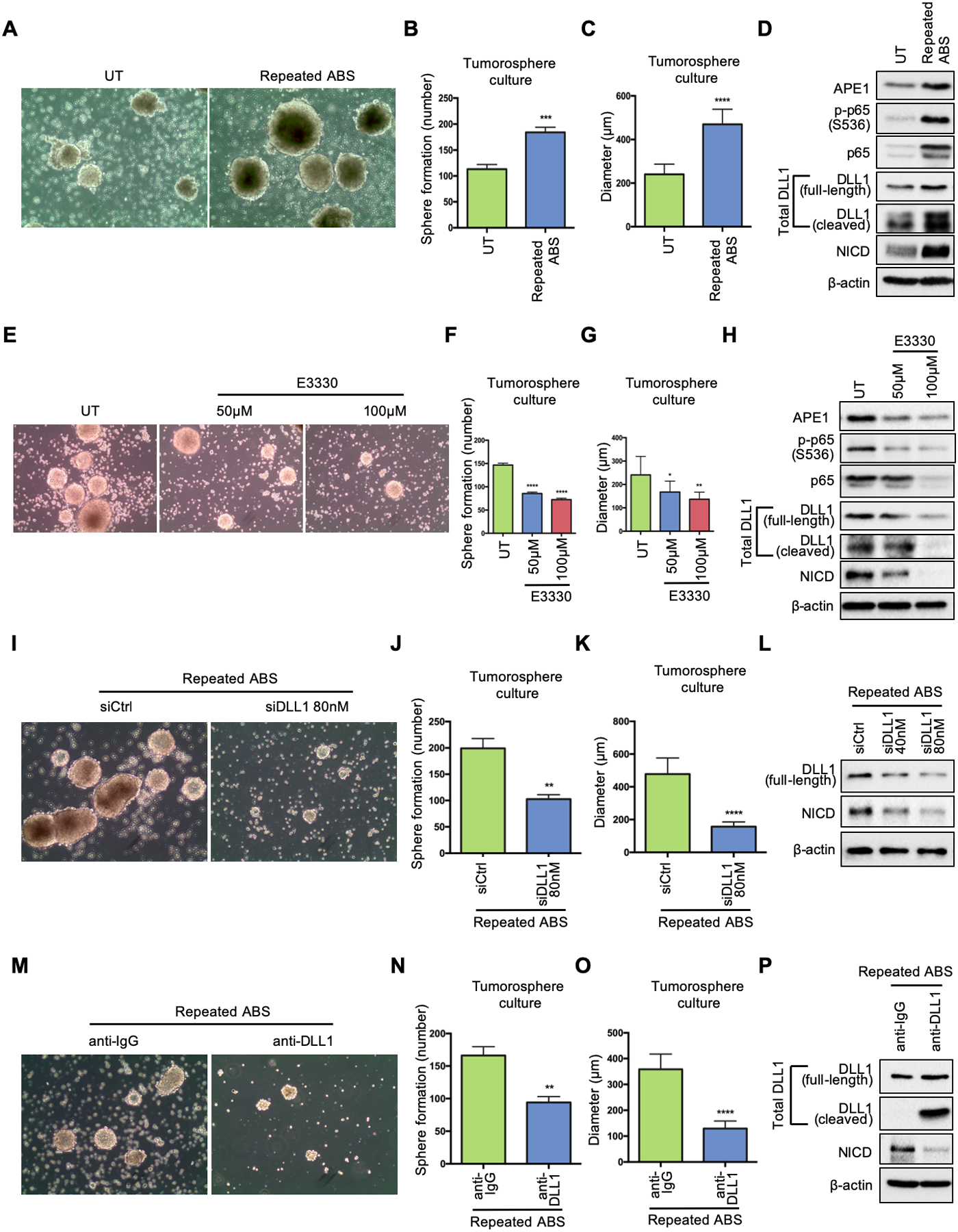
EAC tumorosphere formation is enhanced in response to acidic bile salts. (A) OE33 cells were exposed to ABS (200uM, pH5.5) 20min per day for 14 days. Then the cells were seeded for 3D tumorosphere culture. Untreated OE33 cells (UT) were used as a control. Representative images of the OE33-derived tumorospheres under white light. (B) and (C) Quantification of number (B) and size (C) of the tumorospheres in A. (D) Western blots of APE1, phosphor-p65, total p65, total DLL1(full-length and cleaved form), NICD and β-actin in the tumorospheres with and without repeated ABS exposure. (E) Tumorospheres derived from untreated OE33 cells were incubated with or without indicated doses of E3330 during culture; representative images of the OE33-derived tumorospheres with E3330 under white light. (F) and (G) Quantification of number (F) and size (G) of the tumorospheres in E. (H) Western blots of APE1, phosphor-p65, total p65, total DLL1(full-length and cleaved form), NICD and β-actin in the tumorospheres with E3330. (I) Tumorospheres derived from repeated-ABS-treated OE33 cells were transfected with DLL1 siRNA (siDLL1) or scrambled control (siCtrl); representative images of the tumorospheres with or without transient knockdown of DLL1 under white light. (J) and (K) Quantification of number (J) and size (K) of the tumorospheres in I. (L) Western blots of full-length DLL1, NICD and β-actin in the tumorospheres with and without indicated concentration of DLL1 siRNA. (M) Tumorospheres derived from repeated-ABS-treated OE33 cells were incubated with DLL1 neutralizing antibody or anti-IgG; representative images of the tumorospheres with DLL1 neutralization under white light. (N) and (O) Quantification of number (N) and size (O) of the tumorospheres in M. (P) Western blots of total DLL1 (full-length and cleaved form), NICD and β-actin in the tumorospheres with DLL1 neutralizing antibody or anti-IgG. Statistical data are shown as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 as calculated by t test for two group comparisons.
To further confirm the vital role of DLL1 in NOTCH-mediated cancer cells’ expansion in response to reflux, we transfected the tumorospheres derived from repeated-ABS-treated OE33 cells with DLL1 siRNA. Transient knockdown of DLL1 led to a remarkable decrease in tumorospheres’ number and size (Figure 6, I–K) compared with control siRNA (siCtrl). In addition, western blots verified the DLL1 knockdown efficiency and demonstrated a reduced protein level of active NICD (Figure 6L). Similarly, the use of DLL1 neutralizing antibody abrogated tumorospheres’ growth (Figure 6, M–O) and showed a decrease in NICD protein level (Figure 6P) compared with the IgG control. These results demonstrate an important role of APE1-NOTCH signaling axis in promoting stem-like properties in EAC cells in response to reflux.
DLL1 is upregulated in neoplastic mouse and human esophageal tissues and correlates with poor prognostic outcomes in human EAC
We validated our findings in vivo using L2-IL-1β transgenic mouse model of BE/EAC [32]. These mice develop neoplastic lesions at the gastro-esophageal junctions that include high-grade dysplasia (HGD) or EAC lesion following 6–8 months of exposure to deoxycholic acid (DCA) in drinking water (Figure 7A). Immunofluorescence staining demonstrated high levels of APE1 and DLL1 in the neoplastic glandular areas at the gastro-esophageal junctions. APE1 immunostaining was predominantly nuclear, whereas DLL1 was detected on the cell surface (Figure 7A). We also examined the protein expression levels of APE1 and DLL1 in human EAC tissues. We detected a co-overexpression pattern of APE1 and DLL1 by immunofluorescence (Figure 7B) and IHC staining (Figure 7C). To evaluate the correlation of APE1 and DLL1 in human EAC and normal esophageal tissues, protein levels of APE1 and DLL1 were examined by IHC staining on tissue microarrays (Figure 7D). We observed a co-expression pattern of APE1 and DLL1 in EAC tissues; IHC scores are summarized in Supplemental Table 6. Statistical analysis of IHC index scores showed that protein levels of APE1 and DLL1 were significantly upregulated in EAC tissues compared to the normal esophagus (Figure 7, E–F). Pearson correlation analysis revealed strong positive association between APE1 and DLL1 protein levels (Figure 7G, R = 0.73, p = 1.7e-12). Survival analysis based on the TCGA database indicated poor prognostic outcome with a lower overall survival rate for EAC patients with high levels of DLL1 (Figure 7H) or HES5, a NOTCH downstream transcription target (Figure 7I). A cartoon summarizing our findings is shown in Figure 8.
Figure 7.
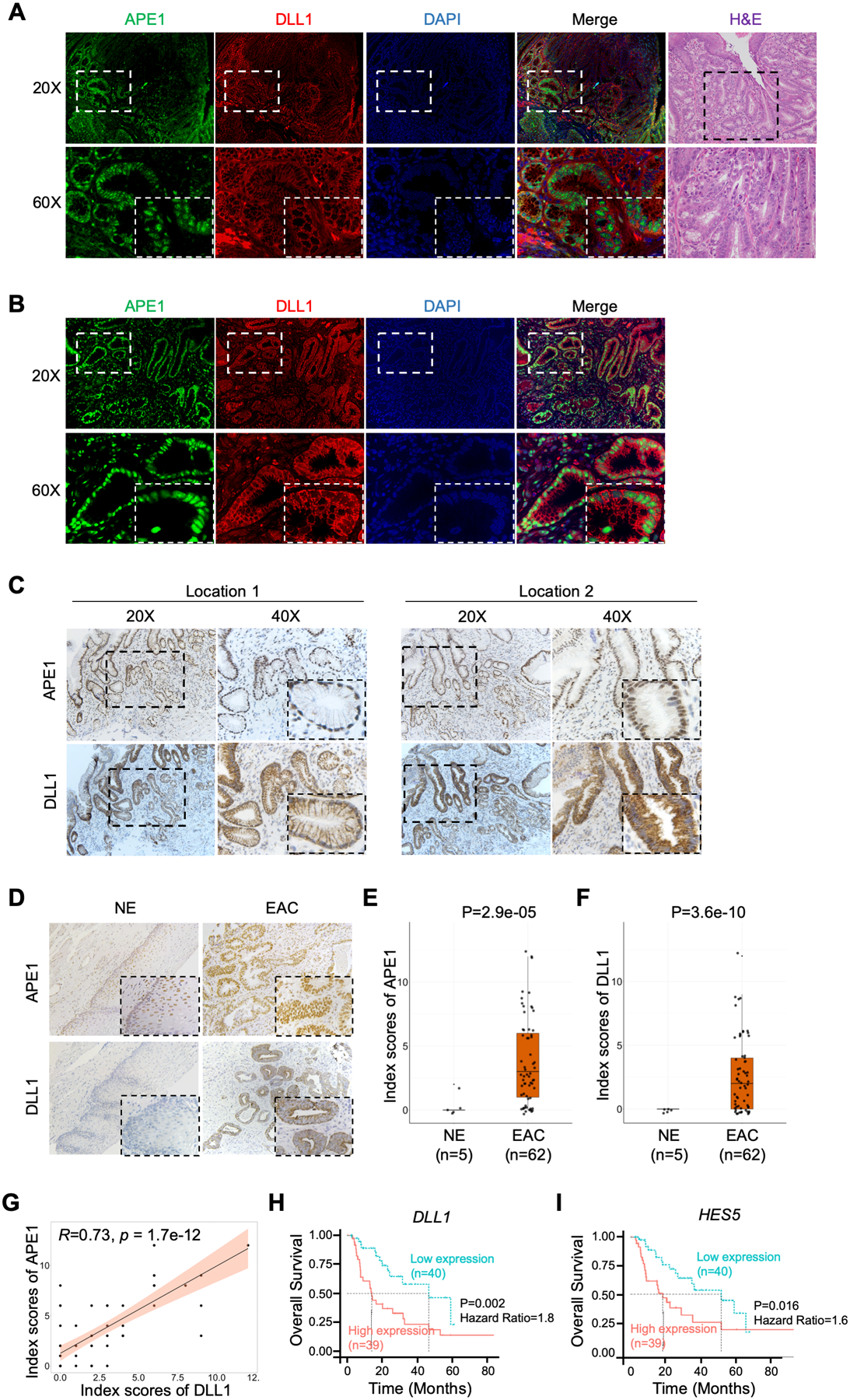
DLL1 is upregulated in neoplastic mouse and human esophageal tissues and correlates with poor prognostic outcomes in human EAC. (A) and (B) Representative immunofluorescent staining images of APE1 (green) and DLL1 (red) in EAC tissue samples from pL2-IL1ß transgenic mouse (A) and human EAC tissue (B). DAPI was used for nuclear staining. (C) and (D) Representative IHC staining images of APE1 and DLL1 using the slides of the same human EAC tissue as B (C) and human EAC tissue microarrays (TMA) (D). (E) and (F) Comparisons of IHC index scores of APE1 (E) and DLL1 (F) between normal esophageal epithelium (n=5) and EACs (n=62) in human EAC TMA were shown. (G) Pearson correlation analysis of the IHC index scores of APE1 and DLL1 protein from human EAC TMA. (H) and (I) Kaplan-Meier plots were used for the survival analysis in the TCGA-EAC database, comparing DLL1-high patients with DLL1-low patients (H), or comparing HES5-high patients with HES5-low patients (I).
Figure 8.
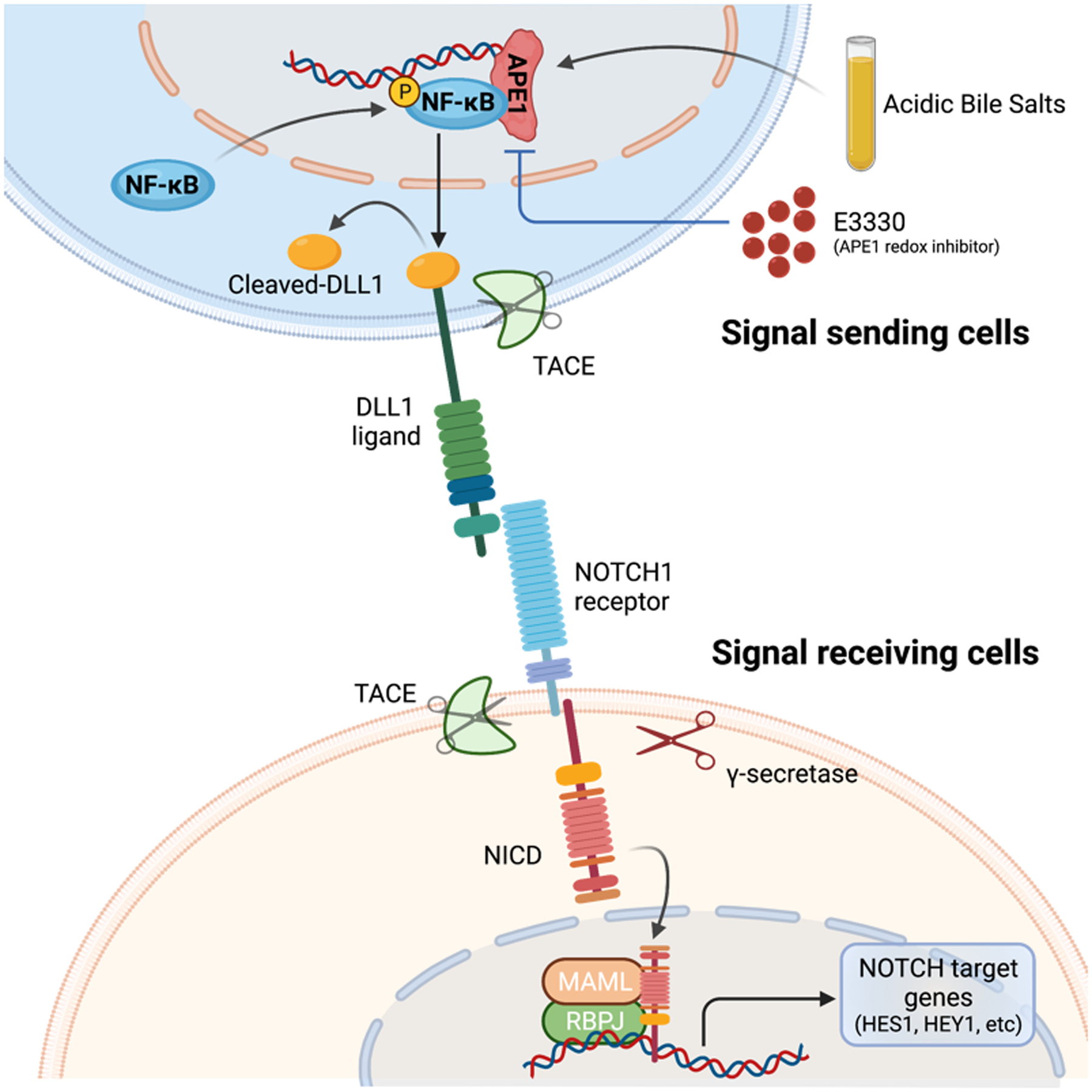
Schematic summary of APE1-redox-mediated NF-κB-DLL1-NOTCH activation in response to ABS in EAC. Exposure to ABS, the in vitro mimic of GERD, increases APE1 protein level and activates APE1-redox-dependent transcription factor, NF-κB, which consequently upregulates DLL1 transcription by directly binding to DLL1 promoter region in the signal sending cells. Accumulated DLL1 protein on cell surface facilitates NOTCH1 cleavage in the signal receiving cells, releasing the active form of NOTCH1 receptor, NICD, which is translocated into cell nucleus, forms transcriptional complex with RBPJ and MAML, and activates transcription of downstream targets like HES1 and HEY1. APE1 redox-specific inhibitor, E3330, effectively blocks this ABS-activated NF-κB/DLL1/NICD signaling axis.
Discussion
The incidence of BE and EAC has increased at alarming rates over the past four decades [2, 33]. GERD, the main risk factor for EAC, is a clinical condition present throughout stages of BE-associated esophageal tumorigenesis [34]. Exposure of esophageal cells to reflux generates high levels of oxidative stress forcing cells to develop adaptive signaling mechanisms to maintain redox and cellular homeostasis. Although earlier studies have shown overexpression and activation of NOTCH signaling in EAC tumorigenesis [25, 35], the molecular mechanisms governing activation of NOTCH in EAC under reflux conditions remain unknown. In this study, our results uncover the underlying mechanism of NOTCH activation in EAC. We report a novel signaling axis linking intracellular oxidative stress sensor (APE1), GERD-induced chronic inflammation (NF-kB), and NOTCH activation in EAC.
While an active scientific debate continues about origin of BE [36, 37, 38, 39], several recent studies support that BE originates from Lgr5+ progenitor cells[39] or Dclk1+ tuft cells[40] of gastric cardia. Aberrant NOTCH activation resulted in accelerated development of metaplastic BE-like lesions in the L2-IL1β mice [32]. Our analysis of public datasets demonstrated activation of NOTCH molecular signature in EAC. NOTCH is an evolutionary highly conserved cell-fate-control signaling pathway, which evolved with the initial appearance of multicellular organisms and conserved in all living organisms[21]. NOTCH signaling is essential for generating and maintaining cancer stem cell (CSC) characteristics in EAC cells [25]. NOTCH activation can also induce transcriptomic alterations of multiple oncogenic pathways, such as c-MYC, KRAS, and Wnt/β-catenin [40], creating a pro-tumorigenic environment for BE progression.
Although a recent study suggested that NOTCH signaling mediates differentiation in BE-like lesions with progression to adenocarcinoma in the L2-IL-1ß mouse model[39], this study did not address the mechanisms of NOTCH activation. Using in vitro and in vivo models, we detected significant up-regulation of NOTCH signaling components in response to reflux conditions. DLL1 was the most consistently up-regulated NOTCH ligand in the public datasets and in response to reflux conditions in our models. DLL1 is one of the three mammalian NOTCH ligands that belongs to Delta family. DLL1 overexpression promoted NOTCH signaling and cancer stem-like features in several tumor types[41, 42]. We detected an increase in the full-length and cleaved intracellular form of DLL1. Interestingly, we found that ABS-induced transcriptional upregulation of DLL1 with cleavage. DLL1 binds to NOTCH receptors to mediate its proteolytic cleavages that, in turn, release the Notch-intracellular domain (NICD) that enters the nucleus to bind to other transcription co-activators to induce expression of NOTCH targets [21]. We detected an increase in NICD with induction of expression of several NOTCH targets, such as HES1 and HEY1, following exposure to ABS. Of note, the use of neutralizing DLL1 antibody abrogated ABS-mediated NOTCH activation, confirming its critical role in reflux conditions. These data indicate that reflux conditions play an important role in activation of NOTCH through DLL1.
Exposure to acidic bile salts during reflux conditions generates high levels of reactive oxygen species and oxidative stress with activation of pro-inflammatory signaling in EAC [43, 44, 45]. Through bioinformatics analysis of public datasets, NF-kB signature was prominent in EAC where GSEA demonstrated enrichment for NF-kB signature in tumors expressing high levels of DLL1. We identified and confirmed novel active NF-kB binding sites on the DLL1 promoter. Although the mechanisms by which NF-kB activation is induced and maintained under oxidative reflux conditions are not entirely understood, our data imply that APE1 redox function plays an important role in the process. APE1 has a unique redox function that protects and promotes transcription factors’ activities and their binding affinity to target genes’ promoters [46]. High levels of APE1, also known as REF1, have been described in EAC [8, 13, 15]. We found a positive correlation between APE1 and DLL1 in human EAC. These novel results uncover a novel link between inflammation and activation of NOTCH, showing crosstalk between APE1-redox, NF-kB, and DLL1/NOTCH1 signaling in EAC. It is worth mentioning that the APE1 redox function can promote activation of the STAT3 transcription factor in response to ABS in EAC[14]. We propose APE1 as an important redox protein that promotes pro-inflammatory oncogenic transcription factors in response to reflux conditions in EAC tumorigenesis.
Using repeated short exposures to ABS over 14 days to mimic chronic reflux conditions, we found that ABS not only activated APE1//NF-κB/DLL1/NOTCH signaling but also significantly enhanced tumorospheres formation capacity. APE1 small molecule redox inhibitors suppressed tumorospheres formation. We detected co-overexpression of APE1 and DLL1 in neoplastic lesions at the gastro-esophageal junction in the L2-IL1β mice. A similar expression pattern of APE1 and DLL1 was present in EAC tissue samples, where EAC patients with DLL1-high expression levels had poorer clinical outcomes and shorter overall survival than those with DLL1-low levels.
In summary, our findings link inflammation (NF-kB), redox (APE1), and NOTCH signaling in EAC tumorigenesis under reflux conditions, providing a new perspective on the complex crosstalk of signaling networks. The results call for the development of novel therapeutic strategies that target APE1-redox or NOTCH signaling in EAC patients.
Supplementary Material
What is already known on this topic?
Apurinic/apyrimidinic endonuclease (APE1) is aberrantly overexpressed in multiple cancer types, including esophageal adenocarcinoma (EAC). Exposure to chronic gastroesophageal reflux, the main risk factor for Barrett’s esophagus (BE)-originated esophageal tumorigenesis, induces APE1 dysregulation, activates intricate networks of redox-dependent transcription factors and promotes cancer cell survival.
Although activation of NOTCH signaling was reported in BE and EAC, the mechanisms underlying its activation in reflux conditions remain largely unknown.
What this study adds?
Using 2D and 3D in vitro models as well as mouse and human data, we report a novel signaling axis where APE1 promotes activation of NOTCH through redox-dependent NF-κB activation and DLL1 induction. High levels of DLL1 are associated with poor overall survival in patients with EAC.
How might this study affect research, practice or policy?
EAC is one of the leading causes of cancer death in the U.S. and Western countries, where standard chemotherapy has limited efficacy. Our findings provide a new perspective for the crosstalk between signaling networks, suggesting that targeting NOTCH or APE1-redox function can be a promising therapeutic strategy in EAC patients.
Acknowledgments
The research reported in this publication was supported by grants from the U.S. National Institutes of Health (NIH) (El-Rifai, W: R01CA206563, R01CA224366 and P01CA268991). LC received a scholarship from the China Scholarship Council. The Sylvester Comprehensive Cancer Center (P30CA240139) shared resources were used in this study. The contents of this work are solely the authors’ responsibility and do not necessarily represent the official views of the National Institutes of Health or the University of Miami.
Funding
This study was funded by National Institutes of Health (Grant number: R01CA206563, R01CA224366, and P01CA268991).
Footnotes
Competing interests None declared.
Ethics approval
The Institutional Research Ethics Committee approved the study design of de-identified human tissues or data. Written informed consents were received from all patients prior to participation. All animal studies were carried out following the protocols approved by the Institutional Animal Care and Use Committee of the University of Miami (UM-20-110).
Data availability statement
Data are available in a public, open access repository. The data of RNA expression profiles and clinical information of EACs were downloaded from The Cancer Genome Atlas (TCGA) official website (https://portal.gdc.cancer.gov/repository). The Gene Expression Omnibus (GEO) datasets, including GSE13898, GSE92396 and GSE37203, were retrieved from National Center for Biotechnology Information (NCBI) GEO database (https://www.ncbi.nlm.nih.gov/). The transcription factor target prediction gene sets can be accessed from the Molecular Signatures Database (MSigDB) (http://software.broadinstitute.org/gsea/msigdb/collections.jsp#C3).
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA: a cancer journal for clinicians 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Edgren G, Adami H-O, Weiderpass E, Nyrén O. A global assessment of the oesophageal adenocarcinoma epidemic. Gut 2013;62:1406–14. [DOI] [PubMed] [Google Scholar]
- 3.Gavin A, Francisci S, Foschi R, Donnelly D, Lemmens V, Brenner H, et al. Oesophageal cancer survival in Europe: a EUROCARE-4 study. Cancer epidemiology 2012;36:505–12. [DOI] [PubMed] [Google Scholar]
- 4.Njei B, McCarty TR, Birk JW. Trends in esophageal cancer survival in United States adults from 1973 to 2009: a SEER database analysis. Journal of gastroenterology and hepatology 2016;31:1141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pera M, Manterola C, Vidal O, Grande L. Epidemiology of esophageal adenocarcinoma. Journal of surgical oncology 2005;92:151–9. [DOI] [PubMed] [Google Scholar]
- 6.Lagergren J. Adenocarcinoma of oesophagus: what exactly is the size of the problem and who is at risk? Gut 2005;54:i1–i5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng DF, Hu TL, Soutto M, Belkhiri A, El-Rifai W. Loss of glutathione peroxidase 7 promotes TNF-alpha-induced NF-kappaB activation in Barrett’s carcinogenesis. Carcinogenesis 2014;35:1620–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhat AA, Lu H, Soutto M, Capobianco A, Rai P, Zaika A, et al. Exposure of Barrett’s and esophageal adenocarcinoma cells to bile acids activates EGFR–STAT3 signaling axis via induction of APE1. Oncogene 2018;37:6011–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vascotto C, Cesaratto L, Zeef LA, Deganuto M, D’Ambrosio C, Scaloni A, et al. Genome - wide analysis and proteomic studies reveal APE1/Ref - 1 multifunctional role in mammalian cells. Proteomics 2009;9:1058–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shah F, Logsdon D, Messmann RA, Fehrenbacher JC, Fishel ML, Kelley MR. Exploiting the Ref-1-APE1 node in cancer signaling and other diseases: from bench to clinic. NPJ precision oncology 2017;1:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cardoso AA, Jiang Y, Luo M, Reed AM, Shahda S, He Y, et al. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1–STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PloS one 2012;7:e47462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Luo M, Marasco D, Logsdon D, LaFavers KA, Chen Q, et al. Inhibition of apurinic/apyrimidinic endonuclease I’s redox activity revisited. Biochemistry 2013;52:2955–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu H, Bhat AA, Peng D, Chen Z, Zhu S, Hong J, et al. APE1 upregulates MMP-14 via redox-sensitive ARF6-mediated recycling to promote cell invasion of esophageal adenocarcinoma. Cancer research 2019;79:4426–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhat AA, Lu H, Soutto M, Capobianco A, Rai P, Zaika A, et al. Exposure of Barrett’s and esophageal adenocarcinoma cells to bile acids activates EGFR-STAT3 signaling axis via induction of APE1. Oncogene 2018;37:6011–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong J, Chen Z, Peng D, Zaika A, Revetta F, Washington MK, et al. APE1-mediated DNA damage repair provides survival advantage for esophageal adenocarcinoma cells in response to acidic bile salts. Oncotarget 2016;7:16688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Acharya A, Das I, Chandhok D, Saha T. Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxidative medicine and cellular longevity 2010;3:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jorgenson TC, Zhong W, Oberley TD. Redox imbalance and biochemical changes in cancer. Cancer research 2013;73:6118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sriramajayam K, Peng D, Lu H, Zhou S, Bhat N, McDonald OG, et al. Activation of NRF2 by APE1/REF1 is redox-dependent in Barrett’s related esophageal adenocarcinoma cells. Redox Biology 2021;43:101970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aster JC, Pear WS, Blacklow SC. The varied roles of notch in cancer. Annual Review of Pathology: Mechanisms of Disease 2017;12:245–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ntziachristos P, Lim JS, Sage J, Aifantis I. From fly wings to targeted cancer therapies: a centennial for notch signaling. Cancer cell 2014;25:318–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nature Reviews Cancer 2011; 11:338–51. [DOI] [PubMed] [Google Scholar]
- 22.Jin Y, Wang M, Hu H, Huang Q, Chen Y, Wang G. Overcoming stemness and chemoresistance in colorectal cancer through miR-195–5p-modulated inhibition of notch signaling. Int J Biol Macromol 2018;117:445–53. [DOI] [PubMed] [Google Scholar]
- 23.Radtke F, Fasnacht N, Macdonald HR. Notch signaling in the immune system. Immunity 2010;32:14–27. [DOI] [PubMed] [Google Scholar]
- 24.Hori K, Sen A, Artavanis-Tsakonas S. Notch signaling at a glance. Journal of cell science 2013;126:2135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, Da Silva TG, Jin K, Han X, Ranganathan P, Zhu X, et al. Notch signaling drives stemness and tumorigenicity of esophageal adenocarcinoma. Cancer research 2014;74:6364–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song S, Maru DM, Ajani JA, Chan C-H, Honjo S, Lin H-K, et al. Loss of TGF-β adaptor β2SP activates notch signaling and SOX9 expression in esophageal adenocarcinoma. Cancer research 2013;73:2159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hovinga KE, Shimizu F, Wang R, Panagiotakos G, Van Der Heijden M, Moayedpardazi H, et al. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem cells 2010;28:1019–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu J, Ye X, Fan F, Xia L, Bhattacharya R, Bellister S, et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer cell 2013;23:171–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson S, Chen H, Lo P-K. In vitro tumorsphere formation assays. Bio-protocol 2013;3:e325–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pasha SPBS, Sishtla K, Sulaiman RS, Park B, Shetty T, Shah, et al. Ref-1/APE1 inhibition with novel small molecules blocks ocular neovascularization. Journal of Pharmacology and Experimental Therapeutics 2018;367:108–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Logsdon DP, Shah F, Carta F, Supuran CT, Kamocka M, Jacobsen MH, et al. Blocking HIF signaling via novel inhibitors of CA9 and APE1/Ref-1 dramatically affects pancreatic cancer cell survival. Scientific reports 2018;8:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quante M, Bhagat G, Abrams JA, Marache F, Good P, Lee MD, et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer cell 2012;21:36–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lagergren J, Lagergren P. Recent developments in esophageal adenocarcinoma. CA: a cancer journal for clinicians 2013;63:232–48. [DOI] [PubMed] [Google Scholar]
- 34.Coleman HG, Xie S-H, Lagergren J. The epidemiology of esophageal adenocarcinoma. Gastroenterology 2018;154:390–405. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z, Chen J, Capobianco AJ. The Notch signaling pathway in esophageal adenocarcinoma. Cellular and Molecular Biology 2015;61:24–32. [PubMed] [Google Scholar]
- 36.Nicholson AM, Graham TA, Simpson A, Humphries A, Burch N, Rodriguez-Justo M, et al. Barrett’s metaplasia glands are clonal, contain multiple stem cells and share a common squamous progenitor. Gut 2012;61:1380–9. [DOI] [PubMed] [Google Scholar]
- 37.Jiang M, Li H, Zhang Y, Yang Y, Lu R, Liu K, et al. Transitional basal cells at the squamous–columnar junction generate Barrett’s oesophagus. Nature 2017;550:529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leedham SJ, Preston SL, McDonald SA, Elia G, Bhandari P, Poller D, et al. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut 2008;57:1041–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kunze B, Wein F, Fang H-Y, Anand A, Baumeister T, Strangmann J, et al. Notch signaling mediates differentiation in Barrett’s esophagus and promotes progression to adenocarcinoma. Gastroenterology 2020;159:575–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kunze B, Middelhoff M, Maurer H, Agibalova T, Anand A, Bührer A-M, et al. Notch signaling drives development of Barrett’s metaplasia from Dclk1-positive epithelial tuft cells in the murine gastric mucosa. Scientific reports 2021;11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar S, Srivastav RK, Wilkes DW, Ross T, Kim S, Kowalski J, et al. Estrogen-dependent DLL1-mediated Notch signaling promotes luminal breast cancer. Oncogene 2019;38:2092–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang C, Hai L, Zhu M, Yu S, Li T, Lin Y, et al. Actin cytoskeleton regulator Arp2/3 complex is required for DLL1 activating Notch1 signaling to maintain the stem cell phenotype of glioma initiating cells. Oncotarget 2017;8:33353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dvorak K, Payne CM, Chavarria M, Ramsey L, Dvorakova B, Bernstein H, et al. Bile acids in combination with low pH induce oxidative stress and oxidative DNA damage: relevance to the pathogenesis of Barrett’s oesophagus. Gut 2007;56:763–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang HY, Hormi-Carver K, Zhang X, Spechler SJ, Souza RF. In benign Barrett’s epithelial cells, acid exposure generates reactive oxygen species that cause DNA double-strand breaks. Cancer research 2009;69:9083–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng D-F, Hu T-L, Soutto M, Belkhiri A, El-Rifai W. Glutathione peroxidase 7 suppresses bile salt-induced expression of pro-inflammatory cytokines in Barrett’s carcinogenesis. Journal of Cancer 2014;5:510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxidants & redox signaling 2009;11:601–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available in a public, open access repository. The data of RNA expression profiles and clinical information of EACs were downloaded from The Cancer Genome Atlas (TCGA) official website (https://portal.gdc.cancer.gov/repository). The Gene Expression Omnibus (GEO) datasets, including GSE13898, GSE92396 and GSE37203, were retrieved from National Center for Biotechnology Information (NCBI) GEO database (https://www.ncbi.nlm.nih.gov/). The transcription factor target prediction gene sets can be accessed from the Molecular Signatures Database (MSigDB) (http://software.broadinstitute.org/gsea/msigdb/collections.jsp#C3).