

Abstract
Type 2 diabetes mellitus is a recognized risk factor for HCC in patients with liver disease, independent from the etiology of their liver disease. Hence, prevention and treatment of type 2 diabetes mellitus and its underlying cause, insulin resistance, should be considered a treatment target for patients with liver disease. The drug armamentarium for diabetes is wide and consists of agents with insulin‐sensitizing activity, agents that stimulate insulin secretion, insulin itself, and agents that reduce gastrointestinal and urinary glucose absorption. From an endocrinology perspective, the main goal of treatment is the achievement of euglycemia; however, in patients at risk of, or with known underlying liver disease, the choice of diabetic medication as it relates to potential hepatic carcinogenesis remains complex and should be carefully considered. In the last decade, increasing evidence has suggested that metformin may reduce the risk of HCC, whereas evidence for other classes of diabetic medications, particularly some of the newer agents including the sodium glucose cotransporter‐2 inhibitors and glucagon‐like peptide‐1 receptor agonists, is fewer and often inconsistent. In this review, we aim to summarize the current evidence on the potential effects of the most widely used diabetic agents on liver cancer tumorigenesis.
Abbreviations
- AMPK
activated protein kinase
- aOR
adjusted OR
- DPP‐4
dipeptidyl peptidase‐4
- GLP1
glucagon‐like peptide‐1
- GLP‐1RA
GLP‐1 receptor agonist
- ICI
immune check‐point inhibitor
- IGF‐1
insulin‐like growth factor 1
- IR
insulin resistance
- mTOR
mammalian target of rapamycin
- OS
overall survival
- PFS
progression‐free survival
- PPARγ
peroxisome proliferator‐activated receptor gamma
- ROS
reactive oxygen species
- RR
relative risk
- SGLT2
sodium glucose cotransporter 2
- T2DM
type 2 diabetes mellitus
- TZD
thiazolidinedione
INTRODUCTION
Type 2 diabetes mellitus (T2DM) is a well‐established, independent risk factor for both NAFLD and HCC.[ 1 , 2 ] It has been shown that the risk of developing HCC is 2.5–4‐fold higher in patients with T2DM, independently of the presence of cirrhosis or of the etiology of the underlying liver disease.[ 3 , 4 , 5 , 6 , 7 , 8 ] Additionally, the risk of HCC appears to be higher in patients with long‐standing and poorly controlled diabetes.[ 9 , 10 ] Given these clear associations, it is of utmost importance that hepatologists and primary care providers both screen for and ensure appropriate management of diabetes to prevent the development of liver disease and mitigate risk factors for HCC development and/or recurrence.
The precise pathophysiologic mechanisms behind the association between T2DM and HCC are not completely understood, and in‐depth discussion of the putative mechanisms at the basis of this association goes beyond the aims of this manuscript, although some hypotheses postulated to explain this association are reported here in order to provide an introduction to the potential role of T2DM medications on this relationship. Briefly, insulin resistance (IR) and activation of the insulin receptor and insulin‐like growth factor 1 (IGF‐1) signaling pathways are among the main determinants in the initiation and progression of hepatocarcinogenesis.[ 11 , 12 , 13 ] In fact, it has been shown that hepatoma cells overexpress IGF‐1 and insulin receptor substrate‐1, suggesting its importance in HCC development.[ 14 ] To this point, data from the Human Protein Atlas demonstrate that high expression of the IGF‐1 receptor predicts poorer survival in patients with liver cancer.[ 15 ] IR has a variety of metabolic and molecular effects that cause inflammation, oxidative stress with resultant DNA damage, and stimulation of cellular pathways that play a role in cellular growth and proliferation, all leading to potential HCC development. Additionally, IR leads to a systemic redistribution of substrate that is predicted to be permissive for tumor growth: Insulin‐resistant skeletal muscle and liver fail to take up glucose in response to insulin, but there is little evidence that insulin resistance develops in tumor cells. Therefore, insulin resistance redistributes glucose toward the tumor, where it has been known for a century to accelerate pathology.[ 16 ] Finally, IR also leads to changes in visceral adipose tissue, including increased fatty acid oxidation and liberation along with changes in the inflammatory and adipokine secretory profile, resulting in increased levels of TNF‐alpha, IL‐6 and leptin, which further result in states of hepatic inflammation and fibrosis, perpetuate insulin resistance, and result in tumorigenesis.[ 17 ]
In the hepatocytes, IR also results in steatosis through alterations in lipoprotein metabolism. In one study by Bugianesi et al., patients with NAFLD and IR had increased de novo hepatic lipogenesis of up to 25% compared with 5% in control subjects.[ 18 ] The resultant hepatic steatosis results in the activation of the mammalian target of rapamycin (mTOR)/NF‐kB pathway, which inhibits the expression of the tumor suppressor phosphatase and tensin homologue in hepatocytes.[ 19 , 20 ] Moreover, hepatic IR alters glucose metabolism by enhancing hepatic gluconeogenesis and glycogenolysis, which lead to hyperglycemia, glucose intolerance, and, when glucolipotoxicity damages the pancreatic beta‐cell sufficiently to render beta‐cell insulin secretion unable to match increasing demand for insulin resulting from IR, results in diabetes.[ 21 , 22 ] These alterations in lipid and glucose metabolism stimulate microsomal peroxidases and cause mitochondrial damage and excess production of reactive oxygen species (ROS). The consequences of such oxidative stress include increased inflammatory cytokine release, direct DNA damage, mitochondrial dysfunction, mutations in the p53 onco‐suppressor gene and the activation and suppression of genes involved in antioxidant pathways, cellular proliferation, and metabolism.[ 2 , 23 , 24 ] Additionally, as insulin is an important mitogen, the state of hyperinsulinemia resulting from IR results in the up‐regulation and/or cross‐activation of multiple growth factor receptors (e.g., IGF‐1), which play a key role in cellular proliferation and inhibition of apoptosis. Indeed, IGF‐1 and its receptor (IGF‐1R) signaling are known to regulate stem cell pluripotency and differentiation to trigger cell proliferation, organ development, and tissue regeneration during embryonic development. Thus, an unbalanced IGF/IGF‐1R signaling can promote cancer‐cell proliferation and activate cancer reprogramming in tumor tissues, especially in the liver.[ 25 ] Insulin receptor substrate‐1 (IRS‐1) activity is also increased as a result of insulin receptor and IGF‐1 signaling, and this results in the activation of multiple cytokine pathways including phosphoinositide‐3‐kinase/AKT/mTOR and mitogen‐activated protein kinases, which modify cell cycle and thus cellular proliferation. Additionally, IRS‐1 may play a role in preventing TGF‐β–mediated apoptosis.[ 26 , 27 , 28 ] Finally, insulin also increases hepatic growth hormone receptor levels and down‐regulates IGF‐1 binding protein, resulting in increased bioavailability of IGF‐1 and further potentiating its tumorigenic effects.[ 29 ]
In summary, through the intricate and synergistic effects of insulin resistance and resultant hepatic steatosis, there is increased production of ROS leading to subsequent DNA damage and alterations in cytokine and adipokine production, which affect various cellular pathways involved in cell proliferation, anti‐apoptosis and inflammation, all leading to a pro‐carcinogenic environment.
T2DM medications and HCC
Given the increased risk of HCC in patients with T2DM and the potential hepatoprotective features of some anti‐diabetic agents (Fig. 1), recent research has focused on the effects of anti‐diabetic therapy on the incidence and outcome of HCC.
FIGURE 1.
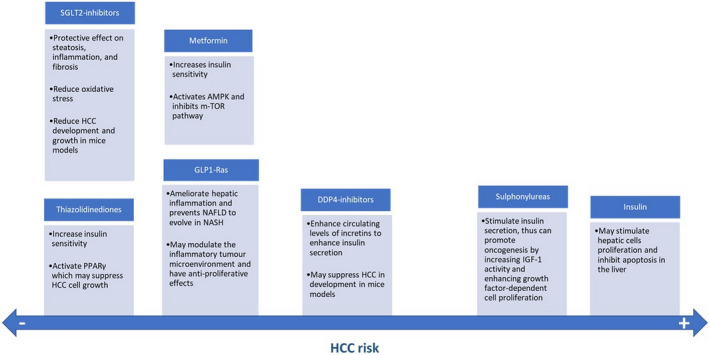
Postulated mechanisms by which the mostly used anti‐diabetic medications may favor or prevent hepatocarcinogenesis. Metformin, TZDs, SGLT2 inhibitors, GLP1‐Ras, and DDP4‐inhibitors may protect from HCC development and growth, whereas sulphonylureas and insulin favor HCC development
Metformin
Metformin was first identified as a potential hepatoprotective medication in 2005 when it was shown to reduce the risk of cancer in patients with diabetes.[ 30 ] Since that time, several studies have demonstrated its potential preventative role against HCC development.[ 31 , 32 , 33 , 34 ] As an example, a metanalysis by Zhang et al. including three cohort studies and four case‐control studies with more than 16,000 diabetic subjects, showed that metformin treatment was associated with a 76% reduced risk of HCC (relative risk [RR] 0.24, 95% CI 0.13–0.46, p < 0.001).[ 32 ] Importantly, the protective effect of metformin remained significant after adjusting for other risk factors such as the etiology of liver disease.[ 32 ] In the same year, another metanalysis by Singh et al. showed similar results, with an overall 50% reduced risk of incident HCC among metformin‐treated patients (adjusted OR [aOR] 0.50, 95% CI 0.34–0.73, p < 0.05).[ 31 ] Consistent with the results by Zhang et al., when restricting the analysis to studies that adjusted for the effect of other anti‐diabetic medications, the chemo‐preventive effect of metformin was still significant and relevant (aOR 0.78, 95% CI 0.75–0.83, p < 0.05). Likewise, in a subgroup analysis of studies that adjusted for duration and/or severity of T2DM, the chemo‐preventive effect of metformin still persisted.[ 31 ] These findings were recently confirmed in a study showing that metformin use was associated with a significantly decreased risk of HCC in T2DM (OR 0.59, 95% CI 0.51–0.68, p < 0.001).[ 34 ] Additionally, this study indicated that metformin use was associated with a decreased all‐cause mortality in diabetic patients with HCC (HR 0.74, 95% CI 0.66–0.83, p = 0.037).[ 34 ] This finding is in accordance with those from a previous report showing a reduced overall mortality risk among metformin users (HR 0.68, 95% CI 0.61–0.75).[ 35 ]
Metformin has multiple potential mechanisms by which it may inhibit HCC development and growth. First, metformin reduces hepatic glucose production, thus indirectly reducing circulating insulin levels and mitigating effects of hyperinsulinemia on cell cycle and inflammation.[ 36 ] Additionally, the anti‐neoplastic effect of metformin may be mediated by activation of adenosine monophosphate–activated protein kinase (AMPK), which has various downstream effects including inhibition of mTOR pathway, which plays a key role in proliferation through control of the cell cycle, activation of the immune system, alterations in cancer stem cell activation, along with anti‐angiogenic effects.[ 36 ] Moreover, compared with exogenous insulin and insulin secretagogues, which raise plasma insulin levels and promote body weight gain, metformin has the benefit of stimulating moderate weight loss through the activation of AMPK.[ 36 ] This latter effect is of particular importance in patients with concomitant obesity and NAFLD, as weight loss is the key treatment target for the achievement of metabolic control, regression of steatosis and steatohepatitis, and, secondarily, for the reduction of HCC development.[ 37 , 38 ] Finally, metformin use has further immune‐biologic effects, as it can lead to positive changes in the gut microbiome that can affect glucose metabolism—a finding that appears to be mediated by the effect(s) of the drug on short‐chain fatty acid–producing bacteria, especially butyrate‐producing taxa, and by the increase in Akkermansia muciniphila, which improves antitumor immune CD8+ T‐cell infiltration and activity.[ 39 ] Moreover, metformin use is associated with an improvement in liver biochemistry and histology in patients with NAFLD, a finding that may explain its favorable influence on the prognosis of patients with advanced liver disease and T2DM.[ 40 , 41 , 42 , 43 , 44 ]
Statins are frequently prescribed in patients with T2DM, as dyslipidemia is a common comorbidity and known element of the metabolic syndrome. Statin co‐utilization has been hypothesized to confound the prior evidence for metformin as a chemo‐preventive agent for HCC in population‐level studies. For example, in the aforementioned retrospective cohort study by Kaplan et al., no demonstrable impact of metformin on HCC or hepatic decompensation was shown after adjusting for concomitant statin use.[ 35 ] We could argue that dual treatment with metformin and statins may play synergistically in the HCC risk reduction, as shown by a study by Tseng et al., which enrolled 21,900 ever‐users and a matched‐pair cohort of 21,900 never‐users of metformin.[ 45 ] The results confirmed the chemo‐preventive role of metformin, and the subanalysis assessing concomitant treatment with statins showed a potential protective synergistic effect. Indeed, metformin had a protective effect independently from the concomitant treatment with statins, but the group under the dual treatment showed the lowest risk of HCC (HR 0.46, 95% CI 0.39–0.54, p < 0.0001 vs. metformin‐only treatment: HR 0.66, 95% CI 0.59–0.74, p < 0.0001).[ 45 ] Additionally, another recent study did demonstrate an anti‐neoplastic effect of metformin independent from statin use.[ 46 ]
Metformin has not only been associated with a reduced risk of HCC and mortality, but it has also been shown to prolong overall survival (OS) among patients with cirrhosis.[ 44 , 47 , 48 ] A retrospective study conducted at the Mayo Clinic of 250 patients with diabetes and cirrhosis taking metformin at the initial visit showed that patients who continued metformin during the observation period had a significantly longer median survival than those who discontinued metformin (11.8 vs. 5.6 years in the overall cohort, p < 0.0001; 11.8 vs. 6.0 years for Child‐Pugh class A patients, p = 0.006; and 7.7 vs. 3.5 years for class B/C patients, p = 0.04), even after adjusting for other variables (HR 0.43, 95% CI 0.24–0.78, p = 0.005).[ 44 ] Notably, the administration of metformin was safe, as no adverse reactions were reported and none of the patients developed metformin‐associated lactic acidosis during follow‐up. These results were confirmed in a nested case‐control study by Bosetti et al. and in a recently published Thai study of 1061 patients with cirrhosis followed up for at least 5 years.[ 47 , 48 ] In this latter study, the outcomes of 719 metformin users were compared with those of 342 metformin never‐users: Consistent with previous results, metformin‐exposed patients had a significantly lower risk of developing HCC (HR 0.48, 95% CI 0.36–0.61, p < 0.001), and the median survival of metformin users was almost twice that of metformin never‐users (6.9 and 3.88 years, respectively; p = 0.003).[ 47 ] The small numbers for the subgroup analysis between each Barcelona Clinic Liver Cancer (BCLC) stage of disease did not allow us to investigate whether metformin could give a survival advantage in each HCC stage group.[ 47 ]
Evidence regarding the potential effects of metformin in patients with HCC has been shown by a prior meta‐analysis, which revealed that metformin significantly prolonged the survival of diabetic patients with HCC who were treated with curative intent, whereas it had no effect in patients with more advanced stages of cancer.[ 49 ] Consistent with this finding, Seo et al. found a significantly lower risk for HCC‐specific mortality (HR 0.38, 95% CI 0.30–0.49) and for retreatment events (HR 0.41, 95% CI 0.33–0.52) among metformin‐treated patients.[ 50 ] Finally, a recent multicenter study reported that diabetic patients with HCC on metformin treatment had a significantly longer median OS than those who were not treated with metformin (22 vs. 15 months, p = 0.019), and that this effect was more relevant among patients with potentially curative stages of HCC.[ 51 ] A unique finding is the potential negative impact on survival that metformin may have when used in combination with sorafenib, a well‐known tyrosine kinase inhibitor used in the treatment of HCC. Schulte et al. demonstrated that co‐treatment with metformin tended to have a negative impact on survival in patients treated with sorafenib (7 vs. 11 months, p = 0.088).[ 51 ] This finding may be explained by the potential of metformin to confer resistance to sorafenib, as was previously shown by Casadei Gardini et al. in their study of 279 patients, in which 19% were on metformin therapy and 12% were on insulin therapy; for patients with HCC on metformin, the use of sorafenib was associated with poor progression‐free survival (PFS) and OS: 1.9 and 6.6 months, respectively, compared with 3.7 and 10.8 months, respectively, for patients without T2DM, and 8.4 and 16.6 months, respectively, for patients on insulin (p < 0.0001).[ 52 ] The analysis from the biopsy samples suggested that tumors that developed during chronic treatment with metformin had intrinsic mechanisms of resistance to metformin, which may have led to resistance to sorafenib.[ 52 ] Conversely, Chung et al. found there was no difference in PFS and OS among metformin‐treated and non‐metformin‐users receiving sorafenib for HCC recurrence after hepatic resection, although the study included a low number of patients—a fact that could have prevented the results from showing a true effect of metformin.[ 53 ] Additionally, there is also evidence for a positive effect of metformin in patients undergoing sorafenib treatment[ 54 ]; thus, given the inconsistencies in available studies, it remains unclear as to the true effects of metformin in sorafenib users.
With regard to the potential effects of metformin on the outcomes of patients treated with immune check‐point inhibitor (ICI)–based therapies, which currently represent the first‐line systemic treatment option for patients with advanced HCC, data on primary liver cancer are still scanty, but the evidence emerging from studies on other cancer types are intriguing. Indeed, recent literature has shown that some concomitant medications, such as proton‐pump inhibitors, antibiotics and corticosteroids, may negatively impact the response to these drugs—not because of pharmacodynamics and pharmacokinetic interactions, but through immune‐modulatory effects, not only systemically and within the tumor microenvironment, but also through the modulation of the gut microbiome.[ 55 , 56 , 57 , 58 ] Interestingly, changes in gut microbiome driven by medications appear to play a pivotal role in the response to ICIs. Specifically, a decreased concentration of Akkermansia muciniphila may reduce the effectiveness of immunotherapy. Metformin increases Akkermansia muciniphila concentration in the gut, which improves antitumor immune CD8+ T‐cell infiltration and activity; this finding might explain the recent evidence of a prolonged OS, PFR, and response rate among patients with non–small‐cell lung cancer treated with metformin.[ 59 ] However, evidence of an absence of association between metformin use and response to ICIs has been presented as well[ 60 ]; hence, until further evidence regarding the eventual role of metformin in response to immunotherapy is available, definite conclusions cannot be drawn.
Overall, although evidence suggests that metformin could decrease the risk of HCC in diabetic patients, the magnitude and severity of liver disease, concomitant treatment with statins, use of sorafenib, and duration of diabetes all remain potentially confounding factors that have limited consistent findings in various studies. Moreover, even if recent studies suggest that metformin could worsen the prognosis of patients treated with sorafenib, more consistent evidence from large populations are needed to draw definite conclusions and further elaborate the underlying cellular mechanisms responsible for these findings. Notably, given the changing paradigm in systemic therapies, further studies assessing the effects of metformin on patients undergoing treatment with immunotherapy and/or other tyrosine kinase inhibitors are still needed.[ 61 ]
Thiazolidinediones
Thiazolidinediones (TZDs) improve glycemic control by activating peroxisome proliferator–activated receptor gamma (PPARγ), leading to insulin sensitization and enhanced glucose metabolism. Expression of PPARγ has been shown to suppress HCC cell growth in mice and in vitro models.[ 62 ] TZDs may be advantageous for patients at risk for HCC, as they not only increase insulin sensitivity, but they have demonstrated anti‐tumorigenic effects via cell cycle arrest, induction of apoptosis, inhibition of cell invasion, along with stimulation of anti‐angiogenic and pro‐differentiation pathways.[ 63 ]
The potential effects of TZDs on HCC incidence have been investigated by several retrospective and metanalytical studies with contrasting results. A nationwide case‐control study that was run in Taiwan included a total 606,583 patients with T2DM, without a history of cancer.[ 64 ] Patients with incident cancer of liver, colon, lung, and urinary bladder were included, as cases and up to four age‐matched and sex‐matched controls were selected by risk‐set sampling. A significantly lower risk of liver cancer incidence was found for any use of rosiglitazone (OR 0.73, 95% CI 0.65–0.81) or pioglitazone (OR 0.83, 95% CI 0.72–0.95), and the protective effects were stronger for higher cumulative dosage and longer duration.[ 52 ] These results are consistent with those found by other retrospective studies, mostly conducted in Asia, which showed a reduced risk for HCC among TZD‐treated diabetics as compared with non‐TZD users.[ 65 , 66 , 67 , 68 , 69 ] Interestingly, in a recent study the use of pioglitazone was also found to reduce the risk of HCC recurrence in a cohort of overweight patients treated for HCV infection. This study was important, as it shed light on some of the mechanisms in which pioglitazone may reduce the risk of HCC recurrence, with overweight status being an important factor. Excess adiposity is a risk factor for IR, and as previously described, increased levels of insulin is associated with increased HCC risk, given its growth‐promoting effects. The authors of this study hypothesized that pioglitazone may suppress HCC recurrence through improvement of IR, as supported by the finding of a reduced homeostasis model assessment–IR score, as well as reduced serum insulin levels, in patients treated on the drug—a finding that has also been confirmed by other reports.[ 67 , 68 , 70 , 71 ] With regard to the meta‐analytic evidence, in 2013 two studies reported TZD use to be associated with a significantly decreased incidence of HCC.[ 72 , 73 ] However, another metanalysis in the same year did not identify a significant protective association, although notably there was significant heterogeneity across studies.[ 31 ] More recently, a systematic review/meta‐analysis including seven studies, 280,567 participants, and 19,242 HCC cases found that TZDs use was associated with an overall reduced HCC risk (aOR 0.92, 95% CI 0.86–0.97, I 2 = 43%), which was confirmed only among Asian subjects (aOR0.90, 95% CI 0.83–0.97), but not in Western subjects (aOR 0.95, 95% CI 0.87–1.04).[ 73 ] Potential theories as to the lack of protection in Western subjects was hypothesized to be due to genetic polymorphisms, which may affect TZD alterations on insulin sensitivity and also on drug metabolism.
Conversely, other studies have demonstrated that TZDs may not be associated with a reduced risk of HCC and may in fact be associated with increased cardiovascular adverse events in patients with cirrhosis.[ 74 ] For example, a study compared the risks for all‐cause mortality, major adverse cardiovascular events, and hepatic outcomes in 1705 patients with T2DM and cirrhosis based on TZD use. For the patients on TZD, the aHRs (95% CIs) for stroke, ischemic heart disease, and heart failure were 1.81 (1.28–2.55), 1.59 (1.03–2.44), and 2.09 (1.22–3.60), respectively. Conversely, all‐cause mortality, HCC, decompensated cirrhosis, and hepatic failure risks did not differ significantly between TZD users and nonusers. Therefore, given the lack of hepatic benefit with TZD use, the authors expressed caution in prescribing TZDs to patients with T2DM and cirrhosis, given the higher risk of major adverse cardiovascular complications.[ 74 ] It is worth noting, however, that because TZDs are at best second‐line for T2DM, there is a risk of bias in observational studies, because patients treated with TZDs will be the ones who have not achieved adequate glycemic control with first‐line therapy. In addition, socioeconomic factors may play a role, as TZD are far cheaper than sodium glucose cotransporter 2 (SGLT2), glucagon‐like peptide‐1 (GLP1), and dipeptidyl peptidase‐4 (DPP‐4) modulating drugs.
Overall, there is insufficient evidence to recommend against or in favor of the use of TZDs in patients with comorbid T2DM and cirrhosis, as the potential benefit on liver‐related outcomes has yet to be consistently demonstrated. Moreover, because there is evidence of increased cardiovascular complications in patients with cirrhosis treated with TZDs, these drugs should be prescribed with caution in subjects who have significant risk factors for, or history of, cardiovascular disease. Note, however, that the American Association for the Study of Liver Disease (AASLD) currently recommends the use of TZDs to treat patients with biopsy‐proven NASH, which is a common complication in patients with T2DM.[ 75 ]
Sulfonylureas
Sulfonylureas, which are insulin secretagogues, have been associated with an increased risk of HCC. Sulfonylureas stimulate insulin secretion by closing ATP‐sensitive potassium channels in pancreatic β cells. By increasing insulin secretion, and exogenous insulin itself, this drug class can promote oncogenesis either directly or indirectly by increasing IGF‐1 activity, resulting in abnormal stimulation of multiple cellular signaling cascades, enhancing growth factor–dependent cell proliferation, and affecting cell metabolism.[ 76 ]
The available evidence suggests that sulfonylureas significantly increase the risk of HCC.[ 48 , 69 , 77 , 78 , 79 ] Lee et al. documented an almost 2‐fold increased risk of HCC for sulfonylurea users, with the risk being higher for those taking glimepiride as opposed to gliclazide.[ 77 , 78 ] These results are in accordance with those from Bosetti et al., who found an increased HCC risk (OR 1.39, 95% CI 0.98–1.99) in patients treated with this drug class,[ 48 ] but slightly in contrast to those from the Kawaguchi et al. nested case‐control study that showed an even higher risk for HCC development in patients treated with second‐generation sulfonylureas (gliclazide, glibenclamide), as the OR was 6.831, with a 95% CI between 1.954 and 23.881 (p < 0.0026).[ 79 ] At odds with the finding by Lee et al., this association was not observed with the third‐generation sulfonylurea glimepiride. However, the baseline characteristics of the included subjects differ between the two studies: Indeed, Lee et al. included and monitored T2DM patients of whom only a minority had comorbid liver diseases, whereas the cohort of Kawaguchi et al. was made up of patients with T2DM and concomitant HCV infection, and a high prevalence of cirrhosis. Therefore, we feel that these results are not directly comparable. Interestingly, in the stratified analyses of the cohort by Kawaguchi et al., the impact of sulfonylureas was more evident in patients without cirrhosis than in those with cirrhosis. Severity of liver fibrosis was the strongest risk factor for hepatocarcinogenesis; therefore, in patients with cirrhosis, the authors suggested the potential carcinogenic activity of exogenous insulin, and a second‐generation sulfonylureas was less evident in comparison to the carcinogenic mechanisms seen in cirrhosis. The carcinogenic risk of different classes of sulfonylureas demonstrated inconsistent results in studies, but it is worth nothing that glimepiride may lead to reduced ability to stimulate insulin secretion, and gliclazide may act as a potential free‐radical scavenger and aid in DNA repair.[ 80 , 81 ] The clear effect of sulfonylureas to increase HCC risk further confirms the role of insulin as a carcinogenic factor in those at risk for HCC.
Insulin
As described previously, insulin is a potent mitogen associated with up‐regulation of various growth factors that lead to stimulation of cytokine pathways related to cell proliferation and inhibition of apoptosis.[ 82 ] In the past decade, increasing evidence has shown a higher risk of HCC incidence in diabetic patients treated with insulin. Indeed, high fasting serum insulin levels (> 6.10 µU/ml) have been associated with a 2.36‐fold risk of HCC when compared with low fasting insulin levels (< 2.75 µU/ml).[ 83 ]
In 2013, Schlesinger et al. conducted a prospective analysis among 363,426 EPIC‐cohort study participants with self‐reported diabetes.[ 84 ] During 8.5 years of follow‐up, 176 HCC cases were identified. Independent from other factors including body mass index and waist‐to‐height ratio, diabetes was associated with a higher risk of HCC (RR 2.17, 95% CI 1.36–3.47), and treatment with insulin conferred the highest risk of HCC (RR 5.25, 95% CI 2.93–9.44), whereas no association was observed for participants without insulin treatment.[ 84 ] Unfortunately, important factors including other medications used other than insulin and status of cirrhosis were not available, which could have confounded the results. Similar results were found by Bosetti et al., who analyzed the role of various antidiabetic drugs on HCC risk in a large population‐based study from Italy.[ 48 ] Increased risks of HCC were found with the use of insulin (OR 3.73, 95% CI 2.52–5.51), with a higher risk associated with longer treatment duration (OR 2.52 for < 1 year, 5.41 for 1–2 years, and 6.01 for ≥ 2 years; p < 0.001), and sulfonylureas (OR 1.39, 95% CI 0.98–1.99).[ 48 ] Similarly, a recent study showed a 2‐fold increased risk of HCC for patients treated with insulin (aOR 1.9, 95% CI: 0.8–4.6).[ 69 ] These results are in accordance with those from the metanalysis by Singh et al., who found an increased risk of HCC for insulin users (OR 2.61, 95% CI 1.46–4.65).[ 31 ]
Interestingly, an association has also been found between survival of patients with advanced HCC who are concurrently treated with insulin and sorafenib. Among 279 patients consecutively treated with sorafenib, the use of sorafenib in patients undergoing chronic treatment with metformin was associated with a median PFS of 1.9 months (95% CI 1.8–2.3) compared with 8.5 months (95% CI 5.3–11.4) for those on insulin (p < 0.0001).[ 52 ] Metformin plus sorafenib–treated patients showed a median OS of 6.6 months (95% CI 4.6–8.7) compared to 16.6 months (95% CI 14.5–25.5) for the insulin group (p < 0.0001). The effect of metformin on clinical outcomes was also investigated in relation to the objective response rate. Patients treated chronically with metformin and sorafenib showed a higher percentage of progression at the first CT scan re‐evaluation than those treated with insulin or the non‐diabetic group (75.8% vs. 14.7% vs. 38.8%, respectively). The risk of progression was higher in those taking metformin+ sorafenib than in the insulin plus sorafenib group (HR 2.91, 95% CI 1.84–4.6).[ 52 ] Similar results were observed for survival (HR 2.74, 95% CI 1.69–4.43, p < 0.0001). However, these results may not reflect a potential advantage for insulin users, but may rather be explained by the hypothetical resistance to sorafenib in metformin‐treated patients, as the survival of patients treated with metformin was similar to the survival of BCLC C untreated patients and to the control arm of the sorafenib registration study.[ 52 , 85 , 86 ]
DPP‐4 inhibitors
DPP‐4 inhibitors work by increasing circulating levels of incretins, such as GLP‐1 and glucose‐dependent insulinotropic polypeptide (GIP), to enhance insulin secretion and inhibit glucagon secretion. The potential role of these drugs in HCC development has not been investigated so far. To the best of our knowledge, only one clinical study has been published considering the hepatic outcomes of DDP‐4 inhibitors.[ 87 ] This study evaluated the risks of mortality, cardiovascular events, and hepatic outcomes between DPP‐4 inhibitor users and nonusers with comorbid diabetes and compensated cirrhosis. The incidence rate of decompensated cirrhosis during follow‐up was 2.20 and 1.53 per 100 patient‐years (aHR 1.35, 95% CI 1.03–1.77, p < 0.05) for DPP‐4 inhibitor users and nonusers, respectively. The aHRs (95%CI) of variceal bleeding and hepatic failure were 1.67 (1.11–2.52) and 1.35 (1.02–1.79), respectively, for DPP‐4 inhibitor users over nonusers (p < 0.05).[ 87 ] The risk of all‐cause mortality, HCC, and major cardiovascular events between DPP‐4 inhibitor users and nonusers were not statistically different. Possible explanations for the higher risk of decompensating events rely on the findings from animal studies showing that DPP‐4 inhibitors can elevate GLP‐1 and GIP levels in splanchnic and portal circulation, which can promote nitric‐oxide production and accelerate portal vein inflow.[ 88 ] One pharmacovigilance study found that DPP‐4 inhibitors were associated with a higher risk of gastrointestinal venous thromboembolism,[ 89 ] which may then increase portal pressure and result in complications such as variceal bleeding. However, a meta‐analysis of DPP‐4 inhibitors did not indicate a risk of hepatic injury,[ 90 ] and a prospective study investigating the efficacy and safety of sitagliptin in 122 diabetic patients with chronic liver disease (including 19 patients with cirrhosis) suggested that sitagliptin can be safely administered.[ 91 ]
In vivo and in vitro studies have suggested that DDP‐4 inhibition may prevent HCC development in mice both indirectly in a glucose metabolism–independent manner by suppressing NASH development and directly by suppressing HCC development through the activated infiltration of lymphocytes into the tumor, although evidence is still scarce and further data are needed before understanding the potential carcinogenic effects of this medication in humans.[ 92 , 93 ]
SGLT2 inhibitors
SGLT2 inhibitors are a new class of oral hypoglycemic agents that work by decreasing glucose reabsorption in the renal proximal tubules, leading to increased urinary glucose excretion and reduced blood glucose levels as a result of the decreased glucose area under the curve (i.e., decreased glucose load presented over the course of the day).
Various animal models of NAFLD treated with SGLT2 inhibitors have demonstrated a protective effect on steatosis, inflammation, and fibrosis.[ 94 , 95 , 96 , 97 , 98 , 99 ] To date, there are no clinical data on whether these drugs might protect from—or favor—carcinogenesis in patients with cirrhosis. However, in an experimental mouse model of NASH, canaglifozin altered glutathione metabolism and reduced oxidative stress in adipose tissue; moreover, it inhibited the development of hepatic fibrosis and HCC.[ 100 ] Another study showed that in a mouse model of diabetes and NASH‐related HCC, canagliflozin had anti‐steatotic and anti‐inflammatory effects that attenuated the development of NASH and prevented the development of HCC, partly due to the induction of cell cycle arrest and/or apoptosis, as well as the reduction of tumor growth through the direct inhibition of SGLT2 in tumor cells.[ 101 ] Using multi‐omic analysis of metabolomics, canaglifozin has been found to alter mitochondrial oxidative phosphorylation metabolism, fatty acid metabolism, and purine and pyrimidine metabolism, resulting in metabolic reprogramming of HCC cell lines and suppression of HCC cell proliferation.[ 102 ] Additionally, in vitro studies have demonstrated that canaglifozin suppresses human HCC cell growth by inducing cell cycle arrest and apoptosis.[ 103 ] Finally, canaglifozin may reduce the human HCC xenograft tumor burden, independent from glycemic status (in vivo), and attenuate the proangiogenic activities of human HCC cells[ 103 ]—an effect that is potentiated with the combination of canaglifozin and tenegliptin, a DDP‐4 inhibitor.[ 104 ] Again, due to lack of clinical data, recommendations on this class of medication for use in patients with T2DM with liver disease cannot yet be made.
GLP‐1RAs
GLP‐1RAs constitute an anti‐diabetic class that acts as insulin secretagogues and reduce postprandial glucose levels in patients with T2DM. Additionally, GLP‐1, which is an intestinal peptide made by gut neuroendocrine cells, has been shown to stimulate pancreatic beta cell proliferation, inhibit glucagon secretion, increase satiety through alterations in the hypothalamic signaling pathways, and delay gastric emptying.[ 105 ] Recently, several animal and human studies have emphasized the role of GLP‐1RAs in ameliorating liver fat accumulation, alleviating the inflammatory environment and preventing NAFLD progression to NASH.[ 106 ] Intuitively, the amelioration of hepatic inflammation and the prevention of NASH should also help indirectly to reduce the risk of HCC development, given our knowledge that NASH portends a higher risk of HCC compared with NAFLD,[ 107 ] but to date, there is no clinical evidence on whether GLP1‐RAs might prevent HCC occurrence. In vitro and in vivo studies have shown that liraglutide and exenatide, despite being insulin secretagogues, may possess antitumor efficacy by modulating the inflammatory microenvironment of HCC and inducing autophagy and senescence, although evidence of an effect in patients with liver disease at risk of HCC is still lacking.[ 108 , 109 ]
CONCLUSIONS
The bidirectional relationship between diabetes and chronic liver diseases is well established. Indeed, advanced liver disease may favor the onset of T2DM, while T2DM is a recognized risk factor for cirrhosis and its complications, including HCC.[ 6 , 8 , 9 , 110 ] Several preclinical and clinical studies have shown that diabetic medications may influence the risk of HCC development, as summarized in Table 1. Most available clinical data have evaluated the effects of metformin, TZD, sulfonylureas and insulin, whereas data on the more novel glucose‐lowering agents (i.e., DDP‐4 inhibitors, SGLT‐2‐inhibitors, and GLP1‐RAs) are preliminary and lack large clinical studies. Notably, to date there are still no large multicenter randomized clinical trials for any class of diabetic medications. Metformin is the first‐line treatment option for T2DM.[ 111 ] Considering that metformin has consistently been reported to reduce the risk of HCC in diabetic patients while also potentially improving the prognosis of patients with an established HCC diagnosis at early stages, this drug can be considered the first‐line option for patients with advanced liver disease.[ 31 , 32 , 34 , 35 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 112 , 113 ] Moreover, the available evidence suggests that metformin can be safely administered in well‐selected patients with T2DM and cirrhosis, with limited risk of lactic acidosis.[ 114 , 115 , 116 ] However, due to the reported cases of this complication, and to the frequent incidence of renal impairment in patients with chronic liver disease, it seems reasonable to implement caution while prescribing metformin in patients with decompensated liver disease, particularly considering the recent addition of numerous effective therapeutic options to the anti‐diabetes arsenal. Additionally, given the potential for metformin to induce resistance to sorafenib and portend a worse survival in patients co‐treated with these medications, caution again must be implemented when considering use in patients with advanced HCC under consideration of sorafenib treatment.[ 51 , 52 , 53 , 54 , 117 ] Evidence regarding the effects of metformin when used in combination with other systemic therapies, including other tyrosine‐kinase inhibitors or ICIs, is still needed. Although data obtained in other oncological settings do not appear to support a positive effect of concomitant metformin and ICI treatment, there is still no solid evidence in patients with HCC.[ 61 ]
TABLE 1.
Clinical studies on the impact of anti‐diabetic medications on the risk of HCC
| Reference | Study design | Anti‐diabetic medication | Study population | No. of patients (HCC incidence: n; %) | Patients with cirrhosis (%) | Results |
|---|---|---|---|---|---|---|
| Singh, 2013[ 31 ] | Metanalysis of RCTs and observational studies | Metformin | Patients with T2DM | 334,307 (22,650; 6.8%) | NR | Metformin reduced by 50% HCC incidence (OR = 0.50, 95% CI 0.34–0.73); sulfonylurea increased HCC risk by 62% (OR = 1.62, 95% CI 1.16–2.24); insulin use increased the risk by 161% (OR = 2.61, 95% CI 1.46–4.65); TZDs did not modify the risk of HCC (OR = 0.54, 95% CI 0.28–1.02) |
| SUs | ||||||
| TZDs | ||||||
| Insulin | ||||||
| Zhang, 2013[ 32 ] | Metanalysis of observational studies | Metformin | Patients with T2DM | 16,739 (752; 3.4%) | NR | Metformin significantly reduced the risk of HCC (RR = 0.24, 95% CI 0.13–0.46, p < 0.001) |
| Li, 2021[ 34 ] | Metanalysis of observational studies | Metformin | Patients with T2DM | 1,452,265 (NR) | NR | Metformin decreases the HCC risk with a random effects model (OR/RR = 0.59, 95% CI 0.51–0.68, p < 0.001) and decreases all‐cause mortality of HCC in T2DM with a random effects model (HR = 0.74, 95% CI 0.66–0.83, I 2 = 49.6%, p = 0.037) |
| Donadon, 2010[ 112 ] | Retrospective case‐control study | Metformin | HCC cases compared to matched patients with cirrhosis and controls (general population) | 610 HCC cases | — | Metformin, compared with SUs or insulin therapy, was associated with a significant reduction of the risk for HCC vs. controls and vs. liver cirrhosis cases (OR = 0.15, 95% CI 0.04–0.50, p = 0.005; and OR = 0.16, 95% CI 0.06–0.46, p = 0.0006, respectively) |
| 618 matched patients with cirrhosis | ||||||
| 1696 controls | ||||||
| Kaplan, 2021[ 35 ] | Retrospective cohort study | Metformin | Subjects with liver cirrhosis (58.2% with comorbid T2DM: 53.8% before the diagnosis of cirrhosis and 4.8% following the diagnosis of cirrhosis) | 74,984 (NR) | 100% | Metformin was not associated with reduced HCC after adjustment for concomitant statin exposure; for patients with diabetes before diagnosis of cirrhosis but no before metformin exposure, metformin was associated with reduced mortality (HR = 0.72 95% CI 0.35–0.97) but not with reduced HCC or hepatic decompensation (p < 0.05) |
| Bosetti, 2015[ 48 ] | Retrospective case‐control study | Metformin | Subjects with T2DM with an HCC diagnosis compared to controls with T2DM only | 190 T2DM subjects with HCC; 3772 controls | NR | Increased risks of HCC were found for use of insulin (OR = 3.73, 95% CI 2.52–5.51), SUs (OR = 1.39, 95% CI 0.98–1.99), and repaglinide (OR = 2.12, 95% CI 1.38–3.26), whereas a reduced risk was found for use of metformin (OR = 0.57, 95% CI 0.41–0.79) |
| SUs | ||||||
| TZDs | ||||||
| Insulin | ||||||
| Alpha‐glucosidase inhibitors | ||||||
| Incretins | ||||||
| Repaglinide | ||||||
| Tseng, 2018[ 45 ] | Retrospective cohort study | Metformin | Patients with T2DM treated with metformin and matched metformin never‐users | Among 21,900 metformin ever‐users: 462 (2.11%); among 21,900 metformin never users: 619 (2.83%) | Among metformin ever‐users: 3.3%; among metformin never‐users: 5.1% | Decreased risk for HCC in metformin ever‐users: HR = 0.45 (95% CI 0.41–0.49), p < 0.001; the risk remained significantly decreased also in the subanalysis considering only subjects with cirrhosis (HR = 0.51, 95% CI 0.43–0.60) |
| Tangjarusritaratorn, 2021[ 47 ] | Retrospective cohort study | Metformin | 1061 patients with T2DM with cirrhosis | Among 719 metformin users: 125 (17.4%); among 342 metformin never‐users: 128 (37.4%) | 100% | Metformin exposure was associated with a decreased incidence of HCC, with a HR of 0.48 (95% CI 0.36–0.62, p < 0.001) |
| Vilar‐Gomez, 2019[ 113 ] | Retrospective cohort study | Metformin | 191 patients with T2DM with NASH and advanced liver fibrosis | Among 110 metformin ever‐users: 7 (6%); among 81 metformin never‐users: 21 (26%) | Among metformin ever‐users: 85%; among metformin never‐users: 88% | The 10‐year cumulative incidence of HCC was significantly lower in metformin users (12%, 95% CI 7–18) as compared with nonusers (40%, 95% CI 35–48), which corresponds with a subhazard ratio = 0.25 (95% CI 0.11–0.58; (p < 0.01) |
| Vilar‐Gomez, 2021[ 46 ] | Retrospective cohort study | Metformin | Patients with biopsy‐proven NASH with Child–Pugh A cirrhosis | 299 (39; 13%) | 100% | Metformin use was associated with a significant reduction in risk of HCC (aHR = 0.78; 95% CI 0.69–0.96); metformin significantly reduced the risk of hepatic decompensation and HCC only in subjects with HbA1c levels > 7.0% (aHR = 0.97, 95% CI 0.95–0.99; and aHR = 0.67, 95% CI 0.43–0.94, respectively) |
| Chang, 2012[ 64 ] | Retrospective case‐control study | TZDs | Patients with T2DM | 606,583 (10,741; 1.7%) | NR | A significantly decreased risk of HCC was found for any use of rosiglitazone (OR 0.73, 95% CI 0.65–0.81) and pioglitazone (OR 0.83, 95% CI 0.72–0.95) and for metformin (OR = 0.77, 95% CI 0.69–0.85), in contrast to the aORs = 2.35 (95% CI 2.21–2.49) for short‐acting insulin and 1.05 (95% CI 0.93–1.18) for sulfonylurea |
| SUs | ||||||
| Metformin | ||||||
| Huang, 2017[ 65 ] | Retrospective population‐based cohort study | TZDs | 76,349 patients with T2DM (TDZ users and TDZ ever‐users) | Among 3026 TZD users: 46 (1.5%); among 12,104 TZD never‐users: 200 (1.6%) | Among TZD users: 2973 (98.25%); among TZD never‐users: 11,784 (97.36%) | Patients in the TZD cohort had a 0.53‐fold lower risk of cancer compared with the risk for patients in the non‐TZD cohort (aHR = 0.53, 95% CI 0.38–0.77) |
| Lai, 2020[ 66 ] | Retrospective population‐based case‐control study | TZDs | 23,580 subjects with T2DM with newly diagnosed HCC and 23,580 matched controls | — | NR | A negative association was found between HCC and every 1‐year increase of cumulative duration of TZDs use (aOR = 0.94, 95% CI 0.92–0.97) |
| Yip, 2020[ 67 ] | Retrospective cohort study | TZDs | Diabetic patients with chronic hepatitis B | 28,999 (2655; 9.2%) | 5261 (18.1%) | At univariate analysis, TZDs protected from HCC incidence (HR = 0.38, 95% CI 0.20–0.71, p < 0.002) but not in the multivariate analysis (HR = 0.57, 95% CI 0.28–1.14, p = 0.111) |
| Hassan, 2010[ 69 ] | Retrospective case‐control study | Metformin | 420 patients with HCC and T2DM | — | NR | The aORs (95%CI) were 0.3 (0.2–0.6), 0.3 (0.1–0.7), 7.1 (2.9–16.9), 1.9 (0.8–4.6), and 7.8 (1.5–40.0) for those treated with biguanides, TZDs, SUs, insulin, and dietary control, respectively (p < 0.05 for all except insulin [p = 0.1]) |
| SUs | ||||||
| TZDs | ||||||
| Insulin | ||||||
| Yen, 2021[ 74 ] | Retrospective cohort study | TZDs | 1705 TZD users and 1705 matched TZD‐nonusers, with cirrhosis | Among TZD users: 147 (8.6%); among TZD nonusers: 170 (9.9%) | 100% | The HCC risk did not differ significantly between TZD users and non‐users (aHR = 0.85, 95% CI 0.68–1.06, p = 0.15) |
| Wang, 2013[ 72 ] | Metanalysis including 1 RCT, 2 case control, and 2 cohort studies | TZDs | 900,522 patients with T2DM | — | NR | TZDs were associated with a significantly lower risk HCC (HR = 0.73; 95% CI 0.63–0.85, p < 0.005) |
| Arvind, 2021[ 73 ] | Metanalysis of observational studies | TZDs | 562,360 patients with T2DM | — | — | TZDs were associated with reduced HCC risk (aOR = 0.92, 95% CI 0.86–0.97, I 2 = 43%); alpha‐glucosidase inhibitors were associated with increased HCC incidence (aOR = 1.08; 95% CI 1.02–1.14, I 2 = 21%); SUs were associated with increased HCC risk (aOR = 1.06, 95%CI 1.02 –1.11, I 2 = 75%); meglitinide use was not associated with HCC incidence (aOR = 1.19; 95% CI 0.89–1.60, I 2 = 72%) |
| Meglitinides | ||||||
| SUs | ||||||
| Alpha‐glucosidase inhibitors | ||||||
| Lee, 2019[ 77 ] | Nationwide nested case‐control study | SUs | 241 HCC cases and 1205 matched controls | — | Cases: 96 (39.8%); controls: 19 (1.6%) | SUs and glinide use were significantly higher in the HCC group (OR = 1.84 [95% CI 1.19–2.86] and OR = 2.00 [95% CI 1.29–3.08], respectively) than in the control group, whereas metformin use was lower in the HCC group than in the control group (OR = 0.59, 95% CI 0.43–0.81) |
| Metformin | ||||||
| Insulin | ||||||
| Glinide | ||||||
| TZDs | ||||||
| DDP4 inhibitors | ||||||
| Kawaguchi, 2010[ 79 ] | Nested case‐control study | SUs | 138 HCC cases and 102 non‐HCC controls; all patients had T2DM and HCV | — | Cases: 101 (73.2%); controls: 34 (33%) | Insulin and a second‐generation SUs were significantly associated with an incidence of HCC (OR = 2.969, 95% CI 1.293–6.819, p < 0.0103; and OR = 6.831, 95% CI 1.954–23.881, p < 0.0026, respectively); in stratified analyses, the impact of these antidiabetic agents was more evident in patients without cirrhosis than in those with cirrhosis; there were no significant differences in the use of a third‐generation SU, a‐glucosidase inhibitor, glinide, metformin or TZDs between HCC and non‐HCC groups |
| Insulin | ||||||
| Alpha‐glucosidase inhibitors | ||||||
| Glinide | ||||||
| Metformin | ||||||
| TZDs | ||||||
| Schlesinger, 2013[ 84 ] | Observational prospective study | Insulin | Patients with T2DM | 363,426 (176; 0.04%) | — | The risk for HCC was particularly higher in participants treated with insulin (RR = 6.19, 95% CI 3.50–10.98, p < 0.05) |
| Yen, 2021[ 87 ] | Observational case control study | DDP4‐inhibitors | 2828 propensity score–matched DPP‐4 inhibitor users and nonusers from a cohort of T2DM with compensated liver cirrhosis | 100% | The risk of HCC was not statistically different between DDP4 inhibitors users and non‐users; DDP4‐inhibitor users had higher risks of decompensated cirrhosis (aHR = 1.35, 95% CI 1.03–1.77) and hepatic failure (aHR = 1.35, 95% CI 1.02–1.79) and of variceal bleeding (aHR = 1.67, 95% CI 1.11–2.52) |
Abbreviations: NR, not reported; RCT, randomized controlled trial; RR, relative risk; SU, sulphonylureas.
With regard to TZDs, most studies suggest this class of medication appears to protect from HCC development, although we acknowledge the inconsistency of these results. Hence, in light of the paucity of data regarding its potential chemo‐preventive role and advantage on liver‐related events, along with the concern for increased cardiovascular events in patients with cirrhosis, it appears premature to recommend the preferential treatment with these drugs in patients with comorbid cirrhosis and diabetes.[ 31 , 48 , 64 , 65 , 66 , 67 , 68 , 69 , 72 , 73 , 74 ] Again, for patients with biopsy‐proven NASH, which is a common complication in patients with diabetes, the use of the TZD pioglitazone is recommended by the AASLD, given the evidence that suggests it induces regression of fibrosis in this class of patients.[ 75 , 118 , 119 , 120 , 121 , 122 , 123 ]
Unlike metformin and TZDs, sulfonylureas have consistently been reported to significantly increase the risk of HCC, especially for patients treated with second‐generation sulfonylureas.[ 48 , 69 , 77 , 78 , 79 ] The effects of sulfonylureas on HCC were more evident in patients without cirrhosis; this finding was similar to those on TZDs and may find an explanation in the increased intrinsic tumorigenic mechanisms found in cirrhosis. Due to the potential tumorigenic effect of this drug class, together with a high risk of inducing hypoglycemia, one could argue that sulfonylureas might not be the best treatment option for patients with advanced liver disease. As suggested by Chung et al., who proposed a treatment algorithm for patients with comorbid T2DM and chronic liver disease, GLP‐1 receptor agonists, DPP‐4 inhibitors, and SGLT‐2 inhibitors may be preferred, even in patients with mild‐to‐moderate hepatic impairment, given their low risk of hypoglycemia and the potential protective effect on HCC development.[ 92 , 93 , 100 , 101 , 103 , 108 , 109 , 124 ] However, because safety data on subgroups of patients with chronic liver disease are scarce, caution and monitoring of liver function tests should be taken, especially in patients treated with DDP4 inhibitors, given the potential risk of hepatic decompensation.[ 89 , 90 ] Data on the potential effects of alpha‐glucosidase inhibitors are poor and controversial[ 48 , 73 , 123 ]; therefore, their potential role as preventive treatments for HCC needs further investigation. With regard to insulin, clinical evidence has consistently shown an increased risk of HCC among insulin‐treated patients as compared with cohorts treated with other anti‐diabetic medications.[ 31 , 48 , 69 , 84 ] The risk seems to rise for longer treatment duration; however, whether this is reflective of a more severe course of diabetes with a secondarily inherent higher carcinogenic effect, or of a true independent higher pro‐neoplastic activity of the medication itself, remains unclear.
It is worth emphasizing that most of the studies assessing the effects of various diabetic medications on HCC incidence did not adjust for the same confounders (i.e., cirrhosis, diabetes duration and severity, etiology of liver disease, and nature of comparator group) and for the degree of metabolic control of diabetes. As the presence of diabetes may predict more severe liver dysfunction, treatment of diabetes (and treatment with some drugs) may just represent a proxy of more advanced liver disease, and most studies have not truly adjusted for the competing mortality related to liver failure. Moreover, most diabetic patients were on multiple glucose‐lowering agents simultaneously; hence, the nature of the control group for each drug consisted of patients treated with multiple diabetic medications, each one with an inherent cancer‐modifying effect. Diabetes duration and severity may increase the risk of HCC, and the prescription of multiple antidiabetic agents and insulin is reflective of more severe glycemic impairment. Finally, although some studies tried to adjust their analyses for co‐prescription of other drugs with a putative effect on HCC development such as statins, it is difficult to truly rule out the potential effect attributable to the use of diabetic medications alone. Thus, notwithstanding the increasing evidence in this field, it is difficult to interpret whether the risk modification for any one agent is real or confounded by diabetes severity, exposure to different glucose‐lowering medications, or various of other medications commonly used in patients affected by the metabolic syndrome. However, based on the current evidence, we could argue that metformin may be considered the first‐line treatment option for patients with advanced compensated chronic liver disease, and that in case of unsatisfactory glycemic control, it would be prudent to avoid sulfonylureas and insulin in favor of newer glucose‐lowering drugs such as TZDs, SGLT‐2 inhibitors, DDP4‐inhibitors and GLP1‐RAs, particularly in light of the data supporting their potential protective effect against HCC development. However, in the case of the selection of SGLT‐2 inhibitors, special attention should be paid to the potential occurrence of urinary tract infections, which are a common complication of these drugs due to their intrinsic mechanism of action that causes glycosuria, and that actually represent one the most frequent infections in patients with cirrhosis.[ 125 ] GLP1‐RAs may be preferred in patients needing to lose weight, as these drugs favor weight loss, whereas TZDs should be avoided in patients at high risk of cardiovascular events. Conversely, in the case of patients with decompensated liver disease, insulin remains the safest option, as consistent evidence has shown its efficacy and safety in this subpopulation, and these patients are often candidates to liver transplantation independently of the presence of HCC, thus decreasing the potential HCC risks associated with long‐term treatment. With regard to non‐insulin agents, until specific guidelines are created, use of these classes of drugs may be considered in compensated and mildly decompensated cirrhosis (i.e., Child‐Pugh class A or B), with the recommendation of caution and strict monitoring of liver and renal function tests, whereas all of them should be avoided in cases of severely decompensated cirrhosis (i.e., Child‐Pugh class C or score > 9), in which insulin is the sole safe treatment option.[ 126 ]
In a context of an increasing prevalence of diabetes and of metabolic‐associated liver disease, further well‐designed, large randomized controlled trials are needed to understand how to optimize the diabetes treatment in patients with cirrhosis, to prevent cardiovascular, hepatologic, and malignant complications.
FUTURE PERSPECTIVES
Due to the progressive, dramatic decrease in patients affected by chronic viral hepatitis as the etiologic agent of liver disease and HCC, at least in the Western world, the burden of patients with NAFLD and HCC is expected to proportionally rise in the near future.[ 127 ] Surveillance for HCC in patients with NAFLD may be hampered by the high prevalence of the disease in the general population, thus making the surveillance not cost‐effective.[ 37 ] However, the presence of diabetes has consistently been associated with a clinically relevant increase in the risk of HCC in patients without cirrhosis with NAFLD, thus helping narrowing down the population who may need to undergo surveillance.[ 128 ] How use of (certain) diabetes medications might affect this risk and further help restrict surveillance to limited subgroups of patients is yet unknown, although it is likely that that the evaluation of large prescription databases might help to provide indications on this issue in the future.
Currently, HCC represents the most common indication for liver transplantation in the United States, and NASH represents the fastest cause for advanced liver disease and HCC.[ 129 , 130 ] Between 10% and 15% of patients undergoing liver transplantation are affected by T2DM, and the 5‐year cumulative incidence of posttransplant diabetes may be as high as 35%.[ 131 ] Given that metformin is considered the anti‐diabetic treatment of choice following liver transplantation,[ 132 ] analysis of a large transplant database may help to identify whether, and how, anti‐diabetic treatment might modulate HCC recurrence, as hypothesized in a study performed in patients with viral etiology of disease.[ 133 ]
Finally, given the rapidly emerging landscape of HCC treatment with ICIs and the recent hypothesis that patients with NAFLD and HCC may be inherently “resistant” to these highly effective drugs due to aberrant T‐cell activation,[ 134 ] it may be of interest to assess whether the use of diabetes medications might counteract, or modulate, this negative effect, as the use of metformin might promote intratumoral T‐cell infiltration, and there is experimental evidence that metformin may reverse the reduced intrahepatic T‐cell density determined by a high‐fat diet in a NAFLD/NASH‐HCC zebrafish model.[ 135 , 136 ]
All in all, we feel that—due to the rapid changing landscape of chronic liver disease etiology and to the high prevalence of T2DM in these patients—it is likely that in the future we will witness an exponential increase in the knowledge regarding not only the modulating effect(s) potentially exerted by diabetes medicine on the risk of HCC, but also on how use of anti‐diabetic drugs might help focus interventions, and how their use might possibly help enhance the effects of anti‐tumoral therapies.
CONFLICT OF INTEREST
Dr. Strazzabosco consults for Engitix.
AUTHOR CONTRIBUTIONS
Maria Corina Plaz Torres: conception and design, drafting the article, preparation, creation of the published work, specifically writing the initial draft, final approval of the version to be published. Ariel Jaffe: conception and design, drafting the article, revising the article for intellectual content, final approval of the version to be published. Rachel Perry: drafting the article, revising the article for intellectual content, final approval of the version to be published. Elisa Marabotto: drafting the article, revising the article for intellectual content, final approval of the version to be published. Mario Strazzabosco: conception and design, revising the article for intellectual content, supervision, final approval of the version to be published. Edoardo G. Giannini: conception and design, revising the article for intellectual content, supervision, final approval of the version to be published.
ACKNOWLEDGMENTS
Supported by the Yale Liver Center National Institutes of Health (P30 DK034989), Clinical and Translational Core, and the National Institutes of Health (R37 CA258261‐01A1). Open Access Funding provided by Universita degli Studi di Genova within the CRUI‐CARE Agreement.
Plaz Torres MC, Jaffe A, Perry R, Marabotto E, Strazzabosco M, Giannini EG. Diabetes medications and risk of HCC. Hepatology. 2022;76:1880–1897. 10.1002/hep.32439
Mario Strazzabosco and Edoardo G. Giannini contributed equally to this work.
Contributor Information
Mario Strazzabosco, Email: mario.strazzabosco@yale.edu.
Edoardo G. Giannini, Email: edoardo.giannini@unige.it.
REFERENCES
- 1. Allaire M, Nault JC. Type 2 diabetes–associated hepatocellular carcinoma: a molecular profile. Clin Liv Dis. 2016;8:53–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mantovani A, Targher G. Type 2 diabetes mellitus and risk of hepatocellular carcinoma: spotlight on nonalcoholic fatty liver disease. Ann Transl Med. 2017;5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adami H‐O, Chow W‐H, Nyren O, Berne C, Linet MS, Ekbom A, et al. Excess risk of primary liver cancer in patients with diabetes mellitus. J Natl Cancer Inst. 1996;88:1472–7. [DOI] [PubMed] [Google Scholar]
- 4. El‐Serag HB, Richardson PA, Everhart JE. The role of diabetes in hepatocellular carcinoma: a case‐control study among United States Veterans. Am J Gastroenterol. 2001;96:2462–7. [DOI] [PubMed] [Google Scholar]
- 5. Davila JA, Morgan RO, Shaib Y, McGlynn KA, El‐Serag HB. Diabetes increases the risk of hepatocellular carcinoma in the United States: a population based case control study. Gut. 2005;54:533–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. El‐Serag HB, Hampel H, Javadi F. The association between diabetes and hepatocellular carcinoma: a systematic review of epidemiologic evidence. Clin Gastroenterol Hepatol. 2006;4:369–80. [DOI] [PubMed] [Google Scholar]
- 7. Wang C, Wang X, Gong G, Ben Q, Qiu W, Chen YI, et al. Increased risk of hepatocellular carcinoma in patients with diabetes mellitus: a systematic review and meta‐analysis of cohort studies. Int J Cancer. 2012;130:1639–48. [DOI] [PubMed] [Google Scholar]
- 8. Yang WS, Va P, Bray F, Gao S, Gao J, Li HL, et al. The role of pre‐existing diabetes mellitus on hepatocellular carcinoma occurrence and prognosis: a meta‐analysis of prospective cohort studies. PLoS One. 2011;6:e27326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. El‐Serag HB, Tran T, Everhart JE. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology. 2004;126:460–8. [DOI] [PubMed] [Google Scholar]
- 10. Yoo JJ, Cho EJ, Han K, Heo SS, Kim BY, Shin DW, et al. Glucose variability and risk of hepatocellular carcinoma in patients with diabetes: a nationwide population‐based study. Cancer Epidemiol Biomarkers Prev. 2021;30:974–81. [DOI] [PubMed] [Google Scholar]
- 11. Kanda T, Goto T, Hirotsu Y, Masuzaki R, Moriyama M, Omata M. Molecular mechanisms: connections between nonalcoholic fatty liver disease, steatohepatitis and hepatocellular carcinoma. Int J Mol Sci. 2020;21:1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smedile A, Bugianesi E. Steatosis and hepatocellular carcinoma risk. Eur Rev Med Pharmacol Sci. 2005;9:291–3. [PubMed] [Google Scholar]
- 13. Hung CH, Wang JH, Hu TH, Chen CH, Chang KC, Yen YH, et al. Insulin resistance is associated with hepatocellular carcinoma in chronic hepatitis C infection. World J Gastroenterol. 2010;16:2265–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang P, Kang D, Cao W, Wang Y, Liu Z. Diabetes mellitus and risk of hepatocellular carcinoma: a systematic review and meta‐analysis. Diabetes Metab Res Rev. 2012;28:109–22. [DOI] [PubMed] [Google Scholar]
- 15. Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357:eaan2507. [DOI] [PubMed] [Google Scholar]
- 16. Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. [DOI] [PubMed] [Google Scholar]
- 17. Jaffe A, Strazzabosco M. Metabolic syndrome and liver cancer. In: Doria C, Rogart JN, editors. Hepato‐Pancreato‐Biliary Malignancies: Diagnosis and Treatment in the 21st Century. Cham, Switzerland: Springer International Publishing; 2021. p. 1–19. [Google Scholar]
- 18. Bugianesi E, McCullough AJ, Marchesini G. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology. 2005;42:987–1000. [DOI] [PubMed] [Google Scholar]
- 19. Vinciguerra M, Carrozzino F, Peyrou M, Carlone S, Montesano R, Benelli R, et al. Unsaturated fatty acids promote hepatoma proliferation and progression through downregulation of the tumor suppressor PTEN. J Hepatol. 2009;50:1132–41. [DOI] [PubMed] [Google Scholar]
- 20. Laurent‐Puig P, Zucman‐Rossi J. Genetics of hepatocellular tumors. Oncogene. 2006;25:3778–86. [DOI] [PubMed] [Google Scholar]
- 21. Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6:87–97. [PubMed] [Google Scholar]
- 22. Bica C, Sandu C, Suceveanu A, Sarbu E, Stoica R, Gherghiceanu F, et al. Non‐alcoholic fatty liver disease: a major challenge in type 2 diabetes mellitus (Review). Exp Ther Med. 2020;20:2387–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bugianesi E. Non‐alcoholic steatohepatitis and cancer. Clin Liver Dis. 2007;11:191–207, x–xi. [DOI] [PubMed] [Google Scholar]
- 24. Hu W, Feng Z, Eveleigh J, Iyer G, Pan J, Amin S, et al. The major lipid peroxidation product, trans‐4‐hydroxy‐2‐nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis. 2002;23:1781–9. [DOI] [PubMed] [Google Scholar]
- 25. Ngo M‐H, Jeng H‐Y, Kuo Y‐C, Diony Nanda J, Brahmadhi A, Ling T‐Y, et al. The role of IGF/IGF‐1R signaling in hepatocellular carcinomas: stemness‐related properties and drug resistance. Int J Mol Sci. 2021;22:1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leclercq IA, Da Silva MA, Schroyen B, Van Hul N, Geerts A. Insulin resistance in hepatocytes and sinusoidal liver cells: mechanisms and consequences. J Hepatol. 2007;47:142–56. [DOI] [PubMed] [Google Scholar]
- 27. Hashimoto E, Tokushige K. Hepatocellular carcinoma in non‐alcoholic steatohepatitis: growing evidence of an epidemic? Hepatol Res. 2012;42:1–14. [DOI] [PubMed] [Google Scholar]
- 28. Tanaka S, Mohr L, Schmidt EV, Sugimachi K, Wands JR. Biological effects of human insulin receptor substrate‐1 overexpression in hepatocytes. Hepatology. 1997;26:598–604. [DOI] [PubMed] [Google Scholar]
- 29. Pollak MN, Schernhammer ES, Hankinson SE. Insulin‐like growth factors and neoplasia. Nat Rev Cancer. 2004;4:505–18. [DOI] [PubMed] [Google Scholar]
- 30. Evans JM, Donnelly LA, Emslie‐Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Anti‐diabetic medications and the risk of hepatocellular cancer: a systematic review and meta‐analysis. Am J Gastroenterol. 2013;108:881–91; quiz 892. [DOI] [PubMed] [Google Scholar]
- 32. Zhang H, Gao C, Fang L, Zhao H‐C, Yao S‐K. Metformin and reduced risk of hepatocellular carcinoma in diabetic patients: a meta‐analysis. Scand J Gastroenterol. 2013;48:78–87. [DOI] [PubMed] [Google Scholar]
- 33. Cunha V, Cotrim HP, Rocha R, Carvalho K, Lins‐Kusterer L. Metformin in the prevention of hepatocellular carcinoma in diabetic patients: a systematic review. Ann Hepatol. 2020;19:232–7. [DOI] [PubMed] [Google Scholar]
- 34. Li Q, Xu H, Sui C, Zhang H. Impact of metformin use on risk and mortality of hepatocellular carcinoma in diabetes mellitus. Clin Res Hepatol Gastroenterol. 2022;46:101781. [DOI] [PubMed] [Google Scholar]
- 35. Kaplan DE, Serper M, John BV, Tessiatore KM, Lerer R, Mehta R, et al. Effects of metformin exposure on survival in a large national cohort of patients with diabetes and cirrhosis. Clin Gastroenterol Hepatol. 2021;19:2148–60.e2114. [DOI] [PubMed] [Google Scholar]
- 36. Gallagher EJ, LeRoith D. Diabetes, cancer, and metformin: connections of metabolism and cell proliferation. Ann N Y Acad Sci. 2011;1243:54–68. [DOI] [PubMed] [Google Scholar]
- 37. Plaz Torres MC, Bodini G, Furnari M, Marabotto E, Zentilin P, Strazzabosco M, et al. Surveillance for hepatocellular carcinoma in patients with non‐alcoholic fatty liver disease: universal or selective? Cancers (Basel). 2020;12:1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. European Association for the Study of the Liver . EASL Clinical Practice Guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69:182–236. [DOI] [PubMed] [Google Scholar]
- 39. Verdura S, Cuyas E, Martin‐Castillo B, Menendez JA. Metformin as an archetype immuno‐metabolic adjuvant for cancer immunotherapy. Oncoimmunology. 2019;8:e1633235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015;528:262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, et al. Corrigendum: disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2017;545:116. [DOI] [PubMed] [Google Scholar]
- 42. Bugianesi E, Gentilcore E, Manini R, Natale S, Vanni E, Villanova N, et al. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am J Gastroenterol. 2005;100:1082–90. [DOI] [PubMed] [Google Scholar]
- 43. Lee H, Ko G. Effect of metformin on metabolic improvement and gut microbiota. Appl Environ Microbiol. 2014;80:5935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang X, Harmsen WS, Mettler TA, Kim WR, Roberts RO, Therneau TM, et al. Continuation of metformin use after a diagnosis of cirrhosis significantly improves survival of patients with diabetes. Hepatology (Baltimore, MD). 2014;60:2008–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tseng CH. Metformin and risk of hepatocellular carcinoma in patients with type 2 diabetes. Liver Int. 2018;38:2018–27. [DOI] [PubMed] [Google Scholar]
- 46. Vilar‐Gomez E, Calzadilla‐Bertot L, Wong VW, Castellanos M, Aller‐de la Fuente R, Eslam M, et al. Type 2 diabetes and metformin use associate with outcomes of patients with nonalcoholic steatohepatitis‐related, Child‐Pugh a cirrhosis. Clin Gastroenterol Hepatol. 2021;19:136–45.e136. [DOI] [PubMed] [Google Scholar]
- 47. Tangjarusritaratorn T, Tangjittipokin W, Kunavisarut T. Incidence and survival of hepatocellular carcinoma in type 2 diabetes patients with cirrhosis who were treated with and without metformin. Diabetes Metab Syndr Obes. 2021;14:1563–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bosetti C, Franchi M, Nicotra F, Asciutto R, Merlino L, La Vecchia C, et al. Insulin and other antidiabetic drugs and hepatocellular carcinoma risk: a nested case‐control study based on Italian healthcare utilization databases. Pharmacoepidemiol Drug Saf. 2015;24:771–8. [DOI] [PubMed] [Google Scholar]
- 49. Zhou J, Ke Y, Lei X, Wu T, Li Y, Bao T, et al. Meta‐analysis: the efficacy of metformin and other anti‐hyperglycemic agents in prolonging the survival of hepatocellular carcinoma patients with type 2 diabetes. Ann Hepatol. 2020;19:320–8. [DOI] [PubMed] [Google Scholar]
- 50. Seo Y‐S, Kim Y‐J, Kim M‐S, Suh K‐S, Kim SB, Han CJ, et al. Association of metformin use with cancer‐specific mortality in hepatocellular carcinoma after curative resection: a nationwide population‐based study. Medicine. 2016;95:e3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schulte L, Scheiner B, Voigtländer T, Koch S, Schweitzer N, Marhenke S, et al. Treatment with metformin is associated with a prolonged survival in patients with hepatocellular carcinoma. Liver Int. 2019;39:714–26. [DOI] [PubMed] [Google Scholar]
- 52. Casadei Gardini A, Faloppi L, De Matteis S, Foschi FG, Silvestris N, Tovoli F, et al. Metformin and insulin impact on clinical outcome in patients with advanced hepatocellular carcinoma receiving sorafenib: validation study and biological rationale. Eur J Cancer. 2017;86:106–14. [DOI] [PubMed] [Google Scholar]
- 53. Chung Y‐K, Hwang S, Song G‐W, Lee Y‐J, Kim K‐H, Ahn C‐S, et al. Absence of antitumor effects of metformin in sorafenib‐treated patients with hepatocellular carcinoma recurrence after hepatic resection and liver transplantation. Ann Hepatobiliary Pancreat Surg. 2018;22:297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cho YY, Yu SJ, Lee HW, Kim DY, Kang W, Paik Y‐H, et al. Clinical characteristics of long‐term survivors after sorafenib treatment for unresectable hepatocellular carcinoma: a Korean National Multicenter Retrospective Cohort study. J Hepatocell Carcinoma. 2021;8:613–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hussain N, Naeem M, Pinato DJ. Concomitant medications and immune checkpoint inhibitor therapy for cancer: causation or association? Hum Vaccin Immunother. 2021;17:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Buti S, Bersanelli M, Perrone F, Tiseo M, Tucci M, Adamo V, et al. Effect of concomitant medications with immune‐modulatory properties on the outcomes of patients with advanced cancer treated with immune checkpoint inhibitors: development and validation of a novel prognostic index. Eur J Cancer. 2021;142:18–28. [DOI] [PubMed] [Google Scholar]
- 57. Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell. 2018;33:570–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Malmberg K‐J. Effective immunotherapy against cancer. Cancer Immunol Immunother. 2004;53:879–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Afzal MZ, Dragnev K, Sarwar T, Shirai K. Clinical outcomes in non‐small‐cell lung cancer patients receiving concurrent metformin and immune checkpoint inhibitors. Lung Cancer Manag. 2019;8:LMT11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Svaton M, Zemanova M, Zemanova P, Kultan J, Fischer O, Skrickova J, et al. Impact of concomitant medication administered at the time of initiation of nivolumab therapy on outcome in non‐small cell lung cancer. Anticancer Res. 2020;40:2209–17. [DOI] [PubMed] [Google Scholar]
- 61. Plaz Torres MC, Lai Q, Piscaglia F, Caturelli E, Cabibbo G, Biasini E, et al. Treatment of hepatocellular carcinoma with immune checkpoint inhibitors and applicability of first‐line atezolizumab/bevacizumab in a real‐life setting. J Clin Med. 2021;10:3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shen GS, Zhou HB, Zhang H, Chen B, Liu ZP, Yuan Y, et al. The GDF11‐FTO‐PPARgamma axis controls the shift of osteoporotic MSC fate to adipocyte and inhibits bone formation during osteoporosis. Biochim Biophys Acta Mol Basis Dis. 2018;1864:3644–54. [DOI] [PubMed] [Google Scholar]
- 63. Okumura T. Mechanisms by which thiazolidinediones induce anti‐cancer effects in cancers in digestive organs. J Gastroenterol. 2010;45:1097–102. [DOI] [PubMed] [Google Scholar]
- 64. Chang CH, Lin JW, Wu LC, Lai MS, Chuang LM, Chan KA. Association of thiazolidinediones with liver cancer and colorectal cancer in type 2 diabetes mellitus. Hepatology. 2012;55:1462–72. [DOI] [PubMed] [Google Scholar]
- 65. Huang MY, Chung CH, Chang WK, Lin CS, Chen KW, Hsieh TY, et al. The role of thiazolidinediones in hepatocellular carcinoma risk reduction: a population‐based cohort study in Taiwan. Am J Cancer Res. 2017;7:1606–16. [PMC free article] [PubMed] [Google Scholar]
- 66. Lai S‐W, Lin C‐L, Liao K‐F. Association of hepatocellular carcinoma with thiazolidinediones use: a population‐based case‐control study. Medicine. 2020;99:e19833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yip T‐F, Wong V‐S, Chan H‐Y, Tse Y‐K, Hui V‐K, Liang LY, et al. Thiazolidinediones reduce the risk of hepatocellular carcinoma and hepatic events in diabetic patients with chronic hepatitis B. J Viral Hepat. 2020;27:904–14. [DOI] [PubMed] [Google Scholar]
- 68. Sumie S, Kawaguchi T, Kawaguchi A, Kuromatsu R, Nakano M, Satani M, et al. Effect of pioglitazone on outcome following curative treatment for hepatocellular carcinoma in patients with hepatitis C virus infection: a prospective study. Mol Clin Oncol. 2015;3:115–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hassan MM, Curley SA, Li D, Kaseb A, Davila M, Abdalla EK, et al. Association of diabetes duration and diabetes treatment with the risk of hepatocellular carcinoma. Cancer. 2010;116:1938–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bajaj M, Suraamornkul S, Pratipanawatr T, Hardies LJ, Pratipanawatr W, Glass L, et al. Pioglitazone reduces hepatic fat content and augments splanchnic glucose uptake in patients with type 2 diabetes. Diabetes. 2003;52:1364–70. [DOI] [PubMed] [Google Scholar]
- 71. Tiikkainen M, Häkkinen AM, Korsheninnikova E, Nyman T, Mäkimattila S, Yki‐Järvinen H. Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes. 2004;53:2169–76. [DOI] [PubMed] [Google Scholar]
- 72. Wang F, Zhao SZ, Zhang MY, Ma YL, Zhang P, Qin HL. Decreased risk of liver cancer with thiazolidinediones therapy in patients with type 2 diabetes: results from a meta‐analysis. Hepatology. 2013;58:835–6. [DOI] [PubMed] [Google Scholar]
- 73. Arvind A, Memel ZN, Philpotts LL, Zheng H, Corey KE, Simon TG. Thiazolidinediones, alpha‐glucosidase inhibitors, meglitinides, sulfonylureas, and hepatocellular carcinoma risk: a meta‐analysis. Metabolism. 2021;120:154780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yen F‐S, Wei JC‐C, Chiu L‐T, Hsu C‐C, Hou M‐C, Hwu C‐M. Thiazolidinediones were associated with higher risk of cardiovascular events in patients with type 2 diabetes and cirrhosis. Liver Int. 2021;41:110–22. [DOI] [PubMed] [Google Scholar]
- 75. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67:328–57. [DOI] [PubMed] [Google Scholar]
- 76. Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer‐related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29:254–8. [DOI] [PubMed] [Google Scholar]
- 77. Lee JY, Jang SY, Nam CM, Kang ES. Incident hepatocellular carcinoma risk in patients treated with a sulfonylurea: a nationwide, nested, case‐control study. Sci Rep. 2019;9:8532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lee JY, Kim G, Lee YH, Lee BW, Cha BS, Nam CM, et al. Comparison of hepatocellular carcinoma risk between patients treated with glimepiride and gliclazide. Diabetes Metab. 2019;45:83–5. [DOI] [PubMed] [Google Scholar]
- 79. Kawaguchi T, Taniguchi E, Morita Y, Shirachi M, Tateishi I, Nagata E, et al. Association of exogenous insulin or sulphonylurea treatment with an increased incidence of hepatoma in patients with hepatitis C virus infection. Liver Int. 2010;30:479–86. [DOI] [PubMed] [Google Scholar]
- 80. Jennings PE, Belch JJ. Free radical scavenging activity of sulfonylureas: a clinical assessment of the effect of gliclazide. Metabolism. 2000;49:23–6. [DOI] [PubMed] [Google Scholar]
- 81. Sliwinska A, Blasiak J, Kasznicki J, Drzewoski J. In vitro effect of gliclazide on DNA damage and repair in patients with type 2 diabetes mellitus (T2DM). Chem Biol Interact. 2008;173:159–65. [DOI] [PubMed] [Google Scholar]
- 82. Wu J, Zhu AX. Targeting insulin‐like growth factor axis in hepatocellular carcinoma. J Hematol Oncol. 2011;4:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gupta SP, Mittal A, Sathian B, Jha DK. Elevated serum insulin is an independent risk factor for hepatocellular carcinoma: a case control study from Nepal. Asian Pac J Cancer Prev. 2013;14:7331–3. [DOI] [PubMed] [Google Scholar]
- 84. Schlesinger S, Aleksandrova K, Pischon T, Jenab M, Fedirko V, Trepo E, et al. Diabetes mellitus, insulin treatment, diabetes duration, and risk of biliary tract cancer and hepatocellular carcinoma in a European cohort. Ann Oncol. 2013;24:2449–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Giannini EG, Farinati F, Ciccarese F, Pecorelli A, Rapaccini GL, Di Marco M, et al. Prognosis of untreated hepatocellular carcinoma. Hepatology. 2015;61:184–90. [DOI] [PubMed] [Google Scholar]
- 86. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J‐F, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90. [DOI] [PubMed] [Google Scholar]
- 87. Yen FS, Wei JC, Yip HT, Hwu CM, Hou MC, Hsu CC. Dipeptidyl peptidase‐4 inhibitors may accelerate cirrhosis decompensation in patients with diabetes and liver cirrhosis: a nationwide population‐based cohort study in Taiwan. Hepatol Int. 2021;15:179–90. [DOI] [PubMed] [Google Scholar]
- 88. Ussher JR, Drucker DJ. Cardiovascular biology of the incretin system. Endocr Rev. 2012;33:187–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gouverneur A, Lair A, Arnaud M, Bégaud B, Raschi E, Pariente A, et al. DPP‐4 inhibitors and venous thromboembolism: an analysis of the WHO spontaneous reporting database. Lancet Diabetes Endocrinol. 2020;8:365–7. [DOI] [PubMed] [Google Scholar]
- 90. Ligueros‐Saylan M, Foley JE, Schweizer A, Couturier A, Kothny W. An assessment of adverse effects of vildagliptin versus comparators on the liver, the pancreas, the immune system, the skin and in patients with impaired renal function from a large pooled database of Phase II and III clinical trials. Diabetes Obes Metab. 2010;12:495–509. [DOI] [PubMed] [Google Scholar]
- 91. Asakawa M, Mitsui H, Akihisa M, Sekine T, Niitsu Y, Kobayashi A, et al. Efficacy and safety of sitagliptin for the treatment of diabetes mellitus complicated by chronic liver injury. Springerplus. 2015;4:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Nishina S, Yamauchi A, Kawaguchi T, Kaku K, Goto M, Sasaki K, et al. Dipeptidyl peptidase 4 inhibitors reduce hepatocellular carcinoma by activating lymphocyte chemotaxis in mice. Cell Mol Gastroenterol Hepatol. 2019;7:115–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kawakubo M, Tanaka M, Ochi K, Watanabe A, Saka‐Tanaka M, Kanamori Y, et al. Dipeptidyl peptidase‐4 inhibition prevents nonalcoholic steatohepatitis‐associated liver fibrosis and tumor development in mice independently of its anti‐diabetic effects. Sci Rep. 2020;10:983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Honda Y, Imajo K, Kato T, Kessoku T, Ogawa Y, Tomeno W, et al. The selective SGLT2 inhibitor ipragliflozin has a therapeutic effect on nonalcoholic steatohepatitis in mice. PLoS One. 2016;11:e0146337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Obata A, Kubota N, Kubota T, Iwamoto M, Sato H, Sakurai Y, et al. Tofogliflozin improves insulin resistance in skeletal muscle and accelerates lipolysis in adipose tissue in male mice. Endocrinology. 2016;157:1029–42. [DOI] [PubMed] [Google Scholar]
- 96. Nakano S, Katsuno K, Isaji M, Nagasawa T, Buehrer B, Walker S, et al. Remogliflozin etabonate improves fatty liver disease in diet‐induced obese male mice. J Clin Exp Hepatol. 2015;5:190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Qiang S, Nakatsu Y, Seno Y, Fujishiro M, Sakoda H, Kushiyama A, et al. Treatment with the SGLT2 inhibitor luseogliflozin improves nonalcoholic steatohepatitis in a rodent model with diabetes mellitus. Diabetol Metab Syndr. 2015;7:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Watanabe Y, Nakayama K, Taniuchi N, Horai Y, Kuriyama C, Ueta K, et al. Beneficial effects of canagliflozin in combination with pioglitazone on insulin sensitivity in rodent models of obese type 2 diabetes. PLoS One. 2015;10:e0116851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tahara A, Kurosaki E, Yokono M, Yamajuku D, Kihara R, Hayashizaki Y, et al. Effects of SGLT2 selective inhibitor ipragliflozin on hyperglycemia, hyperlipidemia, hepatic steatosis, oxidative stress, inflammation, and obesity in type 2 diabetic mice. Eur J Pharmacol. 2013;715:246–55. [DOI] [PubMed] [Google Scholar]
- 100. Shiba K, Tsuchiya K, Komiya C, Miyachi Y, Mori K, Shimazu N, et al. Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Sci Rep. 2018;8:2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Jojima T, Wakamatsu S, Kase M, Iijima T, Maejima Y, Shimomura K, et al. The SGLT2 inhibitor canagliflozin prevents carcinogenesis in a mouse model of diabetes and non‐alcoholic steatohepatitis‐related hepatocarcinogenesis: association with SGLT2 expression in hepatocellular carcinoma. Int J Mol Sci. 2019;20:5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Nakano D, Kawaguchi T, Iwamoto H, Hayakawa M, Koga H, Torimura T. Effects of canagliflozin on growth and metabolic reprograming in hepatocellular carcinoma cells: multi‐omics analysis of metabolomics and absolute quantification proteomics (iMPAQT). PLoS One. 2020;15:e0232283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kaji K, Nishimura N, Seki K, Sato S, Saikawa S, Nakanishi K, et al. Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. Int J Cancer. 2018;142:1712–22. [DOI] [PubMed] [Google Scholar]
- 104. Ozutsumi T, Namisaki T, Shimozato N, Kaji K, Tsuji Y, Kaya D, et al. Combined treatment with sodium‐glucose cotransporter‐2 inhibitor (Canagliflozin) and dipeptidyl peptidase‐4 inhibitor (teneligliptin) alleviates NASH progression in a non‐diabetic rat model of steatohepatitis. Int J Mol Sci. 2020;21:2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Dhir G, Cusi K. Glucagon like peptide‐1 receptor agonists for the management of obesity and non‐alcoholic fatty liver disease: a novel therapeutic option. J Investig Med. 2018;66:7–10. [DOI] [PubMed] [Google Scholar]
- 106. Sofogianni A, Filippidis A, Chrysavgis L, Tziomalos K, Cholongitas E. Glucagon‐like peptide‐1 receptor agonists in non‐alcoholic fatty liver disease: an update. World J Hepatol. 2020;12:493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Cholankeril G, Patel R, Khurana S, Satapathy SK. Hepatocellular carcinoma in non‐alcoholic steatohepatitis: current knowledge and implications for management. World J Hepatol. 2017;9:533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Krause GC, Lima KG, Levorse V, Haute GV, Gassen RB, Garcia MC, et al. Exenatide induces autophagy and prevents the cell regrowth in HepG2 cells. EXCLI J. 2019;18:540–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Krause GC, Lima KG, Dias HB, da Silva EFG, Haute GV, Basso BS, et al. Liraglutide, a glucagon‐like peptide‐1 analog, induce autophagy and senescence in HepG2 cells. Eur J Pharmacol. 2017;809:32–41. [DOI] [PubMed] [Google Scholar]
- 110. Orsi E, Grancini V, Menini S, Aghemo A, Pugliese G. Hepatogenous diabetes: is it time to separate it from type 2 diabetes? Liver Int. 2017;37:950–62. [DOI] [PubMed] [Google Scholar]
- 111. Standards of medical care in diabetes—2017: summary of revisions. Diabetes Care. 2016;40:S4–5. [DOI] [PubMed] [Google Scholar]
- 112. Donadon V, Balbi M, Mas MD, Casarin P, Zanette G. Metformin and reduced risk of hepatocellular carcinoma in diabetic patients with chronic liver disease. Liver Int. 2010;30:750–8. [DOI] [PubMed] [Google Scholar]
- 113. Vilar‐Gomez E, Vuppalanchi R, Desai AP, Gawrieh S, Ghabril M, Saxena R, et al. Long‐term metformin use may improve clinical outcomes in diabetic patients with non‐alcoholic steatohepatitis and bridging fibrosis or compensated cirrhosis. Aliment Pharmacol Ther. 2019;50:317–28. [DOI] [PubMed] [Google Scholar]
- 114. Chung W, Promrat K, Wands J. Clinical implications, diagnosis, and management of diabetes in patients with chronic liver diseases. World J Hepatol. 2020;12:533–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Brackett CC. Clarifying metformin’s role and risks in liver dysfunction. J Am Pharm Assoc. 2010;50:407–10. [DOI] [PubMed] [Google Scholar]
- 116. Richy FF, Sabidó‐Espin M, Guedes S, Corvino FA, Gottwald‐Hostalek U. Incidence of lactic acidosis in patients with type 2 diabetes with and without renal impairment treated with metformin: a retrospective cohort study. Diabetes Care. 2014;37:2291–5. [DOI] [PubMed] [Google Scholar]
- 117. Bhat M, Chaiteerakij R, Harmsen WS, Schleck CD, Yang JD, Giama NH, et al. Metformin does not improve survival in patients with hepatocellular carcinoma. World J Gastroenterol. 2014;20:15750–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Promrat K, Lutchman G, Uwaifo GI, Freedman RJ, Soza A, Heller T, et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology. 2004;39:188–96. [DOI] [PubMed] [Google Scholar]
- 119. Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, et al. A placebo‐controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–307. [DOI] [PubMed] [Google Scholar]
- 120. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Aithal GP, Thomas JA, Kaye PV, Lawson A, Ryder SD, Spendlove I, et al. Randomized, placebo‐controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135:1176–84. [DOI] [PubMed] [Google Scholar]
- 122. Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz‐Lopez C, et al. Long‐term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus. Ann Intern Med. 2016;165:305–15. [DOI] [PubMed] [Google Scholar]
- 123. Zhao Y, Wang Y, Lou H, Shan L. Alpha‐glucosidase inhibitors and risk of cancer in patients with diabetes mellitus: a systematic review and meta‐analysis. Oncotarget. 2017;8:81027–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Lu X, Xu C, Dong J, Zuo S, Zhang H, Jiang C, et al. Liraglutide activates nature killer cell‐mediated antitumor responses by inhibiting IL‐6/STAT3 signaling in hepatocellular carcinoma. Transl Oncol. 2021;14:100872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Dionigi E, Garcovich M, Borzio M, Leandro G, Majumdar A, Tsami A, et al. Bacterial infections change natural history of cirrhosis irrespective of liver disease severity. Am J Gastroenterol. 2017;112:588–96. [DOI] [PubMed] [Google Scholar]
- 126. Tacelli M, Celsa C, Magro B, Giannetti A, Pennisi G, Spatola F, et al. Antidiabetic drugs in NAFLD: the accomplishment of two goals at once? Pharmaceuticals (Basel). 2018;11:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. 2018;67:123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Simon TG, Roelstraete B, Sharma R, Khalili H, Hagstrom H, Ludvigsson JF. Cancer risk in patients with biopsy‐confirmed nonalcoholic fatty liver disease: a population‐based cohort study. Hepatology. 2021;74:2410–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Yang JD, Larson JJ, Watt KD, Allen AM, Wiesner RH, Gores GJ, et al. Hepatocellular carcinoma is the most common indication for liver transplantation and placement on the waitlist in the United States. Clin Gastroenterol Hepatol. 2017;15:767–75.e763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Younossi ZM, Stepanova M, Ong J, Trimble G, AlQahtani S, Younossi I, et al. Nonalcoholic steatohepatitis is the most rapidly increasing indication for liver transplantation in the United States. Clin Gastroenterol Hepatol. 2021;19:580–9.e585. [DOI] [PubMed] [Google Scholar]
- 131. Azhie A, Sheth P, Hammad A, Woo M, Bhat M. Metabolic complications in liver transplantation recipients: how we can optimize long‐term survival. Liver Transpl. 2021;27:1468–78. [DOI] [PubMed] [Google Scholar]
- 132. Sharif A. Should metformin be our antiglycemic agent of choice post‐transplantation? Am J Transplant. 2011;11:1376–81. [DOI] [PubMed] [Google Scholar]
- 133. Shen C, Peng C, Shen B, Zhu Z, Xu N, Li T, et al. Sirolimus and metformin synergistically inhibit hepatocellular carcinoma cell proliferation and improve long‐term survival in patients with HCC related to hepatitis B virus induced cirrhosis after liver transplantation. Oncotarget. 2016;7:62647–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Pfister D, Núñez NG, Pinyol R, Govaere O, Pinter M, Szydlowska M, et al. NASH limits anti‐tumour surveillance in immunotherapy‐treated HCC. Nature. 2021;592:450–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Xia W, Qi X, Li M, Wu YU, Sun L, Fan X, et al. Metformin promotes anticancer activity of NK cells in a p38 MAPK dependent manner. Oncoimmunology. 2021;10:1995999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. de Oliveira S, Houseright RA, Graves AL, Golenberg N, Korte BG, Miskolci V, et al. Metformin modulates innate immune‐mediated inflammation and early progression of NAFLD‐associated hepatocellular carcinoma in zebrafish. J Hepatol. 2019;70:710–21. [DOI] [PMC free article] [PubMed] [Google Scholar]