

Abstract
Background
The developmental toxicity potential (dTP) concentration from the devTOX quickPredict (devTOX qP ) assay, a metabolomics‐based human induced pluripotent stem cell assay, predicts a chemical's developmental toxicity potency. Here, in vitro to in vivo extrapolation (IVIVE) approaches were applied to address whether the devTOX qP assay could quantitatively predict in vivo developmental toxicity lowest effect levels (LELs) for the prototypical teratogen valproic acid (VPA) and a group of structural analogues.
Methods
VPA and a series of structural analogues were tested with the devTOX qP assay to determine dTP concentration and we estimated the equivalent administered doses (EADs) that would lead to plasma concentrations equivalent to the in vitro dTP concentrations. The EADs were compared to the LELs in rat developmental toxicity studies, human clinical doses, and EADs reported using other in vitro assays. To evaluate the impact of different pharmacokinetic (PK) models on IVIVE outcomes, we compared EADs predicted using various open‐source and commercially available PK and physiologically based PK (PBPK) models. To evaluate the effect of in vitro kinetics, an equilibrium distribution model was applied to translate dTP concentrations to free medium concentrations before subsequent IVIVE analyses.
Results
The EAD estimates for the VPA analogues based on different PK/PBPK models were quantitatively similar to in vivo data from both rats and humans, where available, and the derived rank order of the chemicals was consistent with observed in vivo developmental toxicity. Different models were identified that provided accurate predictions for rat prenatal LELs and conservative estimates of human safe exposure. The impact of in vitro kinetics on EAD estimates is chemical‐dependent. EADs from this study were within range of predicted doses from other in vitro and model organism data.
Conclusions
This study highlights the importance of pharmacokinetic considerations when using in vitro assays and demonstrates the utility of the devTOX qP human stem cell‐based platform to quantitatively assess a chemical's developmental toxicity potency.
Keywords: developmental toxicity potency, in vitro to in vivo extrapolation, pharmacokinetics, valproic acid (VPA) analogues
Abbreviations
- 4‐ene‐VPA
2‐propyl‐4‐pentenoic acid or 2‐propylpent‐4‐enoic acid (IUPAC name)
- AC50
half‐maximal activity concentration
- CASRN
chemical abstracts service registry number
- Clint
in vitro intrinsic clearance
- C max
max plasma concentration
- C ss
steady state plasma concentration
- devTOX qP
devTOX quickPredict assay
- DMPA
2,2‐dimethylvaleric acid or 2,2‐dimethylpentanoic acid (IUPAC name)
- dTP
in vitro developmental toxicity potential
- EAD
equivalent administered dose
- EBA
2‐ethylbutyric acid or 2‐ethylbutanoic acid (IUPAC name)
- EC10
effect concentration at which 10% effect is observed compared to the control
- EHA
2‐ethylhexanoic acid
- EPA
U.S. Environmental Protection Agency
- fu
fraction of chemical unbound to plasma protein
- GP
GastroPlus
- HA
hexanoic acid
- HTS
high‐throughput screening
- httk
R package for high throughput toxicokinetics
- iPSC
induced pluripotent stem cell
- IVIVE
in vivo to in vitro extrapolation
- LEL
lowest effect level
- MHA
2‐methylhexanoic acid
- MPA
2‐methylpentanoic acid
- NMRI
Naval Medical Research Institute
- o/c ratio
ornithine to cystine ratio
- OPERA
OPEn Structure‐activity/property Relationship App
- PA
4‐pentenoic acid or pent‐4‐enoic acid (IUPAC name)
- PBPK
physiologically based pharmacokinetic
- PHA
2‐propylheptanoic acid
- PK/TK
Pharmacokinetics/toxicokinetics
- PPK
population‐based pharmacokinetic
- QSAR
quantitative structure‐activity relationship
- TP
in vitro cytotoxicity potential
- VPA
valproic acid
1. INTRODUCTION
Birth defects affect approximately 3% of all babies born in the United States each year (CDC, 2020). Many of these defects may be caused by in utero exposure to various pharmaceutical and environmental chemicals, including metals, pesticides, and industrial solvents (Weinhold, 2009). To identify chemicals that may pose a risk to the developing fetus, developmental toxicity tests utilizing in vivo animal models are frequently used. However, these in vivo tests are expensive and time‐consuming and require a large number of animals, making it impractical to test the more than 80,000 chemicals registered for commercial use in the United States (Luz & Tokar, 2018).
Compared to animal tests, in vitro assays can provide more mechanistic insights. Use of human cell lines can also improve human pathway relevance, helping to address the difficulties inherent to interspecies extrapolation that is necessary when animal models are used. In vitro assays can often be converted to a high‐throughput platform, which greatly accelerates testing and reduces overall costs. Over the past decade, significant advances have been made to overcome the challenges associated with implementation of high‐throughput screening (HTS) assays (Richard et al., 2021; Shukla, Huang, Austin, & Xia, 2010). Screening 10,000 chemicals in a few weeks has become routine in some facilities. Efforts in both the public and private sectors are exploring how HTS data can accelerate the evaluation of potentially toxic chemicals while providing insights into mechanisms of human toxicity. For example, a European Union research program focused on human cell‐ and mechanism‐based toxicological assessment (EU‐ToxRisk, 2020) has developed several case studies to address alternative models in regulatory decision‐making. One case study investigated the teratogenic potency of valproic acid (VPA) analogues using alternative methods.
VPA is a short‐chain aliphatic acid used as an anticonvulsant and antiepileptic drug that is known to be teratogenic in humans and animals. VPA exposure during pregnancy causes spina bifida and minor facial malformations in children (Lammer, Sever, & Oakley, 1987; Ornoy, 2009). VPA is also teratogenic in most animal species tested, although animals are considered to be less susceptible to its teratogenic effects. In rats, which is a commonly used animal model for identifying potential teratogens, prenatal VPA exposure caused a significant reduction in fetal weight starting at a daily dose of 200 mg/kg. At higher doses, VPA also induces abnormal vertebrae, ribs and craniofacial dysmorphia, cardiovascular defects and hydronephrosis (Binkerd, Rowland, Nau, & Hendrickx, 1988).
VPA and its analogues were tested with the devTOX quickPredict™ assay (devTOX qP ), an in vitro human induced pluripotent stem cell (iPSC)‐based assay that predicts a chemical's developmental toxicity potential (dTP) based on changes in cell metabolism. Stem cells are ideal for examining developmental toxicity due to their capacity to differentiate into nearly any cell type. Earlier studies demonstrated that the teratogenic potency ranking based on the devTOX qP assay was consistent with in vivo developmental toxicity potency for sets of environmental chemicals (Kleinstreuer et al., 2011), pharmaceutical compounds (Palmer et al., 2013), and retinoic acid and its analogues (Palmer et al., 2017). The assay was also used to screen 1,065 chemicals from the U.S. EPA's ToxCast program and achieved ~80% accuracy when compared to results from in vivo animal developmental toxicity studies (Zurlinden et al., 2020). However, whether concentrations that induce a response in the devTOX qP assay can be used to quantitatively predict in vivo exposure causing developmental toxicity has not been evaluated.
In vitro to in vivo extrapolation (IVIVE) of dosimetry, also known as reverse dosimetry, translates bioactive chemical concentrations to external exposures that would be predicted to result in plasma concentrations equal to the in vitro bioactive concentrations. Translating in vitro measurements into estimates of in vivo outcomes is a complex process requiring understanding of the pharmacokinetics and pharmacodynamics of both in vitro and in vivo systems. The IVIVE outcome relies on both in vitro and in vivo kinetics. Due to a lack of in vitro kinetics data, the nominal concentration was often used to derive the bioactive concentration for IVIVE. However, Efforts started recently to characterize chemical kinetics in in vitro assay systems. For example, Armitage et al. demonstrated a mass‐balance model that considers essential assay components (e.g., media volume, cell number, head space, percent of serum) and physicochemical properties to calculate chemical distribution in vitro (Armitage, Wania, & Arnot, 2014).
In addition, a pharmacokinetic (PK) model describing in vivo kinetics is needed for reverse dosimetry (Figure 1). PK models used for IVIVE can vary in complexity. At one extreme is a simple one‐compartment model that relies on assumptions of linear dose‐response and steady‐state kinetics. In contrast, complex multi‐compartment physiologically based PK (PBPK), or toxicokinetic (PBTK), models use tissue‐specific partition coefficients to calculate dynamic chemical concentrations in plasma and tissues (Andersen, 2003). To evaluate developmental effects, a pregnancy‐specific PBPK model simulating in utero exposure is preferred. A pregnancy‐specific model is inherently more complex than conventional PBPK model due to growth of and interaction between maternal and embryo/fetal tissues. Many of the parameters such as compartmental volumes and cardiac output, which are treated as constants in conventional PBPK models, must be formulated as time‐varying quantities for pregnancy PBPK models due to the significant physiological and anatomical changes occurring during the course of gestation (Young et al., 1997). Both open‐source and commercial platforms are available for generic and pregnancy‐specific PBPK modeling (Pearce, Strope, Setzer, Sipes, & Wambaugh, 2017; Simulations Plus Inc, 2020).
FIGURE 1.
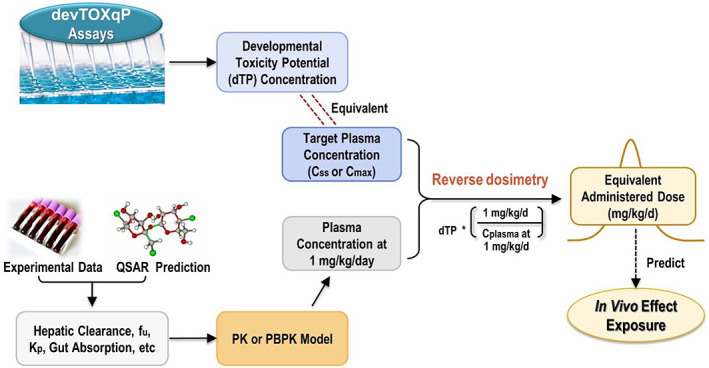
Predicting equivalent administered dose using an in vitro bioactivity concentration in the devTOX quickPredict (devTOX qP ) assay. The developmental toxicity potential (dTP) concentration provided by the devTOX qP assay is used as the target plasma concentration, expressed as either a steady‐state (C ss) or maximum (C max) concentration. Experimental or predicted values for input parameters are used to populate a pharmacokinetic or physiologically based pharmacokinetic model, which estimates plasma concentration at a given dose (mg/kg/day). Reverse dosimetry is carried out to estimate an equivalent administered dose that results in the plasma concentration equal to the in vitro dTP concentration.
In the current study, we performed IVIVE to predict in vivo developmental toxicity dose levels by estimating equivalent administered doses (EADs) that would result in maternal and/or fetal blood concentrations equivalent to the dTP concentrations from the devTOX qP assay. The in vitro‐derived EADs were compared to lowest effect levels (LELs) in rat developmental toxicity studies or human therapeutic doses, and to EADs from a recent OECD case study based on different sets of in vitro data (OECD, 2020). We evaluated the impact of in vitro kinetics and different modeling approaches on EAD estimates and identified the type of PK/PBPK models that best predicted oral doses that would induce developmental toxicity in vivo. This work relates in vitro developmental toxicity potency to in vivo measurements for VPA analogues, promoting confidence in use of these approaches in chemical prioritization and hazard assessment.
2. METHODS
2.1. In vitro assay data
VPA and nine structural analogues were tested with the devTOX qP assay using human iPS cells (HYR0103, ATCC®ACS‐1007™, Manassas, VA) to determine dTP concentrations as previously described (Palmer et al., 2017). The devTOX qP assay (Stemina Biomarker Discovery, Inc., Madison, Wisconsin) is a biomarker‐based human pluripotent stem cell assay for developmental toxicity screening across a dose‐response range (Palmer et al., 2013, 2017). The devTOX qP assay has been developed and validated using both human embryonic stem (hES) cells and iPS cells; the current work was carried out using iPS cells. The assay measures changes in the abundance (metabolism) of two metabolite biomarkers, ornithine and cystine, from which the ornithine to cystine (o/c) ratio is calculated at different chemical concentration. Both ornithine and cystine were identified as indicators of developmental toxicity. Perturbation of biochemical pathways that include these biomarkers has been shown to be associated with teratogenic mechanisms in vivo (Palmer et al., 2013). Using a set of chemicals with known information on developmental toxicity, the o/c ratios were shown to be predictive of developmental toxicity potency (Palmer et al., 2013). From the o/c ratio response curve (Figure S1), the dTP concentrations can be derived and used for determining in vitro developmental toxicity potency (Palmer et al., 2013, 2017). The dTP concentration is the interpolated concentration at which the o/c ratio response curve drops below the predefined developmental toxicity threshold, which was developed based on a set of pharmaceuticals with known human teratogenic potential. Conversely, the toxicity potential (TP) concentration is defined as the concentration at which cell viability drops below the same threshold. The TP concentration is used for assessing overall toxicity of a chemical (Figure S1). The dTP concentration was used for subsequent IVIVE analysis as it measures bioactivity relevant to teratogenic mechanisms.
2.2. Armitage model adjustment to in vitro concentrations
To calculate the free medium concentration corresponding to nominal concentration, the Armitage mass balance model contained in the high throughput toxicokinetic (httk) R package was used (Pearce et al., 2017). A set of assay‐specific and chemical‐specific parameters are required as inputs for executing the Armitage model. The values for assay‐specific parameters (e.g., system temperature, number of wells on plate, total volume of wells) were provided by Stemina Biomarker Discovery, Inc. The values for physiochemical parameters (e.g., logP, Henry's law constant, water solubility and melting point) were obtained using the Open Structure‐activity/property Relationship App (OPERA) (Mansouri, Grulke, Judson, & Williams, 2018).
2.3. In vivo developmental toxicity study data
The rat LELs from in vivo developmental toxicity studies for VPA and its analogues were obtained from the scientific literature. Information extracted include strains, dosing range, exposure routes, dosing period, and LELs for maternal and fetal toxicities. LELs for both maternal and fetal toxicities were combined to derive the range of developmental toxicity LELs.
2.4. Pharmacokinetic parameters
Values required as inputs for the PK and PBPK models included fraction of chemical unbound to plasma protein (fu) and intrinsic clearance rate (Clint) in primary hepatocytes. Experimental fu and Clint values in primary hepatocytes were obtained from literature sources (Mansouri et al., 2018; OECD, 2020) or provided by Dr. Ciaran Fisher (Certara). For Clint values that fall below the limit of quantitation for the assay (e.g., PHA), a value two‐fold below the limit of quantitation was used. When experimental values are not available, predicted values from OPERA (v2.7) were used (Mansouri et al., 2018). All other model input parameters, for example, uptake rates from gut, tissue‐to‐plasma partition coefficients, were obtained from the U.S. Environmental Protection Agency's (EPA) high‐throughput toxicokinetics (httk) R package (Pearce et al., 2017) or commercial software GastroPlus® v9.7 (Simulations Plus Inc, 2020). Structural similarity between VPA and the nine other analogues was calculated based on Tanimoto similarity score and extended connectivity fingerprints (ECFP4) (Table S3). ECFP x are circular topological fingerprints that measure the features x radius from the center of the chemical, where x is 0, 2, 4, 6 (O'Boyle & Sayle, 2016).
2.5. PK models and EAD estimates
Four different PK and PBPK models of different complexity were used to estimate the daily EAD (expressed as mg/kg body weight/day) from the oral route of exposure (Figure 2). The simplest model (Figure 2a) was a one‐compartment population‐based PK (PPK) model (Wetmore et al., 2012) that assumes 100% absorption and calculates total clearance as the sum of hepatic clearance and renal clearance calculated based on passive glomerular filtration rate. For a given dose of test substance, this model estimates the upper 95th percentile steady‐state plasma concentration (C ss) for a Monte Carlo simulated population of 10,000 samples, which accounts for variability due to individual parameters such as liver size, hepatic clearance rate. Using this model, EADs were estimated that would result in the total C ss equal to the dTP concentration from the devTOX qP assay.
FIGURE 2.
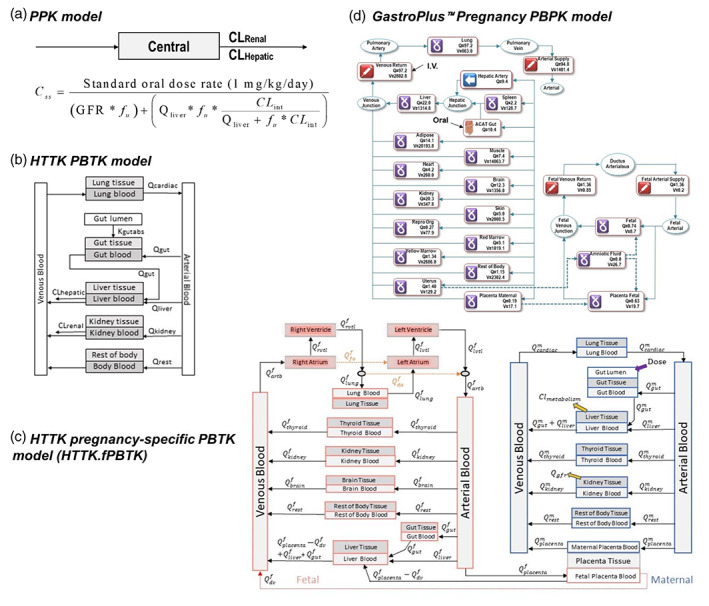
Structures of pharmacokinetic (PK) models used for in vivo to in vitro extrapolation analyses. (a) The population‐based pharmacokinetic model is a one‐compartment, open‐source population‐based PK model. (b) The httk.PBTK model is a generalized PBTK model provided in the Environmental Protection Agency (EPA) httk R package. (c) The httk.fPBTK model is a human pregnancy specific PBTK model provided in the EPA httk R package. Adapted from Sfeir et al. (2020). (d) A human pregnancy physiologically based pharmacokinetic model included in GastroPlus software. Expressions such as “Qliver”, “Qkidney”, represent the blood flow rate in liters/hr (L/h) into and through that tissue. CLHepatic, hepatic clearance (L/h); CLint, intrinsic hepatic clearance (L/h); CLRenal, renal clearance (L/h); GFR, glomerular filtration rate (L/h); PBTK, physiologically based toxicokinetic; Qf, fetal tissue blood flow; Qm, maternal tissue blood flow; V, tissue volume.
The EAD derived from the total chemical concentration was calculated as:
| (1) |
The httk.PBTK model (Figure 2b) is a generalized open‐source physiologically based toxicokinetic (PBTK) model obtained from the httk R package (v1.8.1) (Pearce et al., 2017). The model includes compartments for artery, vein, lung, gut, liver, kidney, and rest‐of‐body, with each tissue compartment described by perfusion rate‐limited kinetics, in which the blood flow to the tissue becomes the rate‐limiting process (Jones & Rowland‐Yeo, 2013). For this model, tissue: plasma partition coefficients were calculated using the modified Schmitt method provided in the httk R package (Schmitt, 2008). The model assumes that elimination of chemical is mainly through hepatic and renal clearance. The model generates simulations using default body weights of 70 kg for human and 0.25 kg for rat. For IVIVE, the model was applied to predict C max following an oral dose of 1 mg/kg/day. As linear relationship between dose and plasma concentration is assumed in httk.PBTK model, the EAD corresponding to the dTP concentration was calculated as:
| (2) |
The httk.fPBTK model (Figure 2c) is a human pregnancy‐specific PBTK model obtained from the httk R package (v1.8.1) (Kapraun, Wambaugh, Woodrow Setzer, & Judson, 2019). The httk.fPBTK model includes fetal and maternal instances of each of the tissue compartments included in the generalized model. In addition, the model includes four fetal cardiac compartments, a fetal brain compartment, maternal and fetal thyroid compartments, and maternal and fetal placenta blood compartments. For IVIVE, the model is used to simulate a 30‐year‐old pregnant female with body weight of 61 kg and a two‐week oral chemical exposure of 1 mg/kg/day starting at 12 weeks of gestation. Based on these inputs, the model predicts both maternal and fetal plasma C max. Same as httk.PBTK model, linear relationship between dose and plasma concentration is assumed in httk.fPBTK model. Therefore, the EAD that would result in maternal or fetal plasma C max equal to the dTP concentration is calculated as:
| (3) |
A pregnancy PBPK model from the commercial GastroPlus software (v9.7, Figure 2d) was also used in our analysis. The GastroPlus (GP) pregnancy model is a human 12‐week gestation model built to simulate drug exposure via maternal ingestion of a delayed‐release formulation (enteric coated tablet or capsule). Compartments in this model include maternal lung, liver, spleen, gut, adipose tissue, muscle, heart, brain, kidney, skin, reproductive tract, red marrow, yellow marrow, uterus, placenta, and rest‐of‐body, as well as fetus, amniotic fluid, and fetal placenta. The model simulates a 30‐year‐old American female with pre‐pregnancy body weight of 61 kg and a two‐week oral chemical exposure of 1 mg/kg/day starting at 12 weeks of gestation. To calculate the EAD that leads to a maternal or fetal plasma C max equal to dTP concentrations, we first used Equation (3) to have a rough estimation on EADs, then we adjusted EAD values by comparing simulated maternal or fetal C max at the EADs to dTP concentrations. For chemicals with C max plateau reached, the dose at the starting point of plateau is used as EAD.
Additionally, we obtained plasma C max values for VPA at given clinical doses from PharmaPendium (Elsevier, Amsterdam, Netherlands, 2021), based on which we applied linear extrapolation to estimate EAD corresponding to dTP concentration.
| (4) |
3. RESULTS
3.1. Comparison of in vitro dTP and TP concentrations
As noted above, the devTOX qP assay produces two statistics for each chemical tested. The dTP concentration describes developmental toxicity potency, while the TP describes the concentration where a chemical is cytotoxic in human pluripotent stem cells (Palmer et al., 2013; Palmer et al., 2017). Since the dTP concentration is used for predicting the developmental toxicity potency of a chemical, it was used as the in vitro activity concentration input for the IVIVE analysis. The dTP concentration for all of the VPA analogues was lower than or within two‐fold of the TP concentration (Table 1). Two analogues, EBA and MPA, did not decrease iPS cell viability at the concentrations tested in this study (Table 1).
TABLE 1.
In vitro activity concentrations derived from the devTOX qP assay
| CASRN | Chemical name a | Abbreviation | Structure similarity | dTP (μM) | TP (μM) | dTPanalogue/dTPVPA | In vivo potency b | References | |
|---|---|---|---|---|---|---|---|---|---|
| 99‐66‐1 | Valproic acid | VPA | 1 | 236 | 318 | 1.0 |
|
Eikel, Lampen, and Nau (2006), Nau, Hauck, and Ehlers (1991), Nau and Löscher (1986) | |
| 149‐57‐5 | 2‐Ethylhexanoic acid | EHA | 0.77 | 399 | 390 | 1.7 |
|
Hauck, Wegner, Blumtritt, Fuhrhop, and Nau (1990) | |
| 31080‐39‐4 | 2‐Propylheptanoic acid | PHA | 0.58 | 546 | 425 | 2.3 |
|
Eikel et al. (2006) | |
| 1575‐72‐0 | 2‐Propyl‐4‐pentenoic acid | 4‐ene‐VPA | 0.64 | 611 | 636 | 2.6 |
|
Eikel et al. (2006), Nau et al. (1991), Nau and Löscher (1986) | |
| 1185‐39‐3 | 2,2‐Dimethylpentanoic acid c | DMPA | 0.35 | 784 | 1,745 | 3.3 |
|
Nau et al. (1991), Nau and Löscher (1986) | |
| 142‐62‐1 | Hexanoic acid | HA | 0.36 | 838 | 1,022 | 3.6 | ND | ||
| 591‐80‐0 | 4‐Pentenoic acid | PA | 0.48 | 913 | 719 | 3.9 |
|
Nau et al. (1991), Nau and Löscher (1986) | |
| 4536‐23‐6 | 2‐Methylhexanoic acid | MHA | 0.21 | 976 | 1,631 | 4.1 |
|
||
| 88‐09‐5 | 2‐Ethylbutyric acid | EBA | 0.6 | 1,071 | NE | 4.5 |
|
Nau et al. (1991), Nau and Löscher (1986) | |
| 97‐61‐0 | 2‐Methylpentanoic acid | MPA | 0.59 | 1,248 | NE | 5.3 | ND |
Abbreviations: NE, no effect detected within the exposure range tested; ND, not determined.
Note: The “+” and “−” signs indicate positive and negative results in the in vivo studies, respectively: +, slightly positive; ++, positive; +++, very positive.
The chemicals are sorted from the lowest to highest dTP values.
Potency relative to VPA based on results in the NMRI exencephaly‐mouse model using decision criteria in Eikel et al. (2006).
2,2‐Dimethylpentanoic acid and 2,2‐Dimethylvaleric acid are synonyms.
VPA has the lowest dTP concentration (Table 1, Figure 3), indicating that it is the most potent developmental toxicant of all the chemicals tested in this study. For VPA, the dTP and TP concentrations from a similar assay but using hES cells have also been reported (Palmer et al., 2013), and were found to be slightly different (dTP of 91 μM and TP of 1,114 μM), but within the expected range for variability (3‐fold), especially considering that different cell lines were used. Assay specific information and results comparison can be found in Table S1. Interestingly, the o/c ratio dose‐response curve for PA was biphasic and differed from VPA and the other analogues (Figure 3). This effect was the result of increased cystine uptake prior to cell death, which may be due to an adaptive response to oxidative stress.
FIGURE 3.
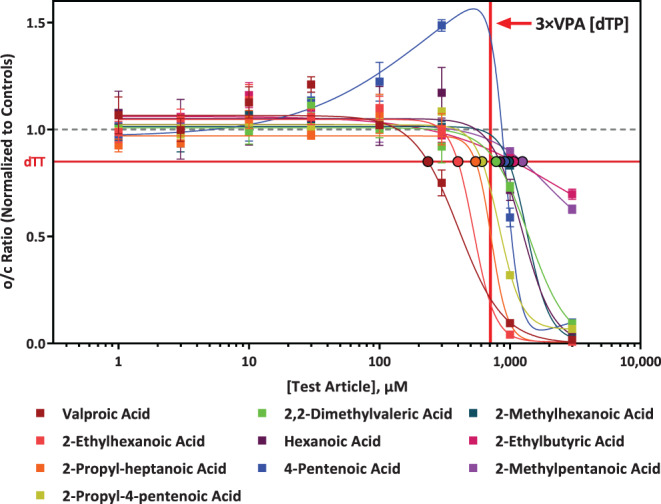
Comparison of valproic acid (VPA) analogue response in the devTOX quickPredict assay. The x‐axis is the analogue concentration (μM) of the test article and the y‐axis is the vehicle‐treatment normalized (fold change) values for the o/c ratio of each analogue. The horizontal red line represents the developmental toxicity threshold (0.85), the dashed horizontal gray line represents 1.0. The vertical red line indicated the concentration that is three times more than the developmental toxicity potential (dTP) concentration of VPA. The solid squares are mean values and error bars are the standard error of the mean. If not shown, error bars are smaller than the size of the symbol. The black outlined circles of various colors indicate the dTP concentrations for the corresponding analogue.
In addition, chemical structure similarity between VPA and the rest of chemicals were calculated based on ECFP4 fingerprints and the Simplified Molecular Input Line Entry Specification (SMILES), which is a widely used chemical notation system designed for chemical information (Weininger, 1988) (Table 1). Based on dTP concentration, EHA is the second most potent analogue and also has the highest structure similarity score. PHA, 4‐ene‐VPA, EBA, MPA have relatively higher structure similarity score (0.58–0.64) (Table 1). However, based on dTP concentration, 4‐ene VPA and PHA are considered to have high potency, while EBA and MPA show lowest potency.
3.2. Assay‐specific and chemical‐specific parameters
The parameters describing the devTOX qP assay system are listed in Table S2. Chemical specific parameters (e.g., physiochemical parameter, fu and Clint) used in IVIVE analyses are listed in Table S3. The fu values for ten VPA analogues are ≥0.15, indicating these substances are highly unbound to plasma protein.
Experimental Clint values in primary hepatocytes for five chemicals (EHA, MHA, EBA, MPA, VPA) were obtained from a recent OECD report (OECD, 2020). Experimental Clint values for two additional chemicals, HA and PHA, were obtained through communication with one co‐author of the OECD report. For Clint values that fall below the limit of quantitation for the assay (e.g., PHA), a default value for Clint that was two‐fold below the limit of quantitation was used (Table S3). The range of experimental values for Clint is 0.07–33.6 (μl/min/106), suggesting a large variation in clearance among these chemicals.
The same fu and Clint values for each chemical were used regardless of PK/PBPK model type. However, for other parameters required for populating PK/PBPK models (e.g., uptake rate of chemical from the gut, tissue to plasma partition coefficients) default values provided by httk package or GastroPlus v9.7 were used respectively. The use of these default values is to evaluate the impact of different modeling approaches on EAD estimates under routine applications.
3.3. In vivo developmental toxicity study data
We performed a literature search to obtain rat in vivo developmental toxicity data for VPA and nine VPA analogues (Table 2). Search terms included the CASRN or chemical name for each chemical in combination with “developmental toxicology,” “teratogen,” “teratogenicity,” or “developmental toxicity.” From the articles returned, we selected articles with relevant information in the titles and abstracts for data extraction. The LELs inducing toxicities in both dams and fetuses were collected and combined to define a range of developmental toxicity LELs, which we used to evaluate the performance of the IVIVE analyses. To be inclusive, general adverse effects such as maternal body weight changes are also included (Narotsky et al., 1994).
TABLE 2.
Rat LELs obtained from in vivo assays
| Chemical | Dose range (mg/kg/day) | Strains | Route | Gestational dosing period | LEL: Maternal toxicity (mg/kg/day) | LEL: Fetal toxicity (mg/kg/day) | References |
|---|---|---|---|---|---|---|---|
| VPA | 0–800 | Sprague–Dawley | NA | GD 8–17 | NA | 200 | (Binkerd et al., 1988) |
| 0–400 | Sprague–Dawley | Corn oil gavage | GD 6–15 | 400 | 200 | (Narotsky, Francis, & Kavlock, 1994) | |
| EHA | 0–600 | Wistar | Drinking water | GD 6–19 | 600 | 100 | Pennanen, Tuovinen, Huuskonen, and Komulainen (1992) |
| 0–1,000 | Fischer 344 | NA | GD 6–15 | 500 | 250 | Hendrickx et al. (1993) | |
| PA | 0–100 | Sprague–Dawley | Corn oil gavage | GD 6–15 | 75 | 50 | (Narotsky et al., 1994) |
| EBA | 0–200 | Sprague–Dawley | Corn oil gavage | GD 6–15 | 150 | NA | (Narotsky et al., 1994) |
| MPA | 0–250 | Sprague–Dawley | Corn oil gavage | GD 6–15 | 188 | NA | (Narotsky et al., 1994) |
Abbreviations: EBA, 2‐ethylbutyric acid or 2‐ethylbutanoic acid (IUPAC name); EHA, 2‐ethylhexanoic acid; GD, gestation days; LEL, lowest effect level; MPA, 2‐methylpentanoic acid; NA, not available; PA, 4‐pentenoic acid or pent‐4‐enoic acid (IUPAC name); VPA, valproic acid.
Rat development toxicity study data are only available for five chemicals (EHA, PA, VPA, EBA, and MPA). For EBA and MPA, LELs only for maternal adverse effects are available. For those chemicals with LELs for both maternal and fetal toxicities, LELs for fetal toxicity are generally lower than those for maternal toxicity, suggesting that fetal toxic endpoints are more sensitive to chemical exposure.
Since LEL data was only available for a subset of the analogues, we compared the relative potency of the analogues in vitro to the relative potency in vivo as determined in the Naval Medical Research Institute (NMRI) exencephaly‐mouse model (Eikel et al., 2006), which had results for 8 of the 10 analogues (Table 1). The mouse model is described in detail in an early publication (Nau, Zierer, Spielmann, Neubert, & Gansau, 1981). Using the dTP concentration of VPA as a baseline, we calculated the relative developmental toxicity potency of each chemical by determining ratio of each analogue's dTP concentration relative to VPA's dTP concentration (Table 1). A higher ratio indicates a low relative potency (less likely to be developmentally toxic), while a lower ratio indicates a relatively high potency (more likely to be developmentally toxic). Using this ratio, the relative potency of the analogues can be categorized into two groups. Analogues with high potency in the NMRI exencephaly‐mouse model elicited a response in human iPS cells <3‐fold of the VPA dTP concentration, whereas the dTP concentrations for analogues with little to no effect this model were >3‐fold higher than VPA's dTP.
3.4. EAD estimates using rat nonpregnancy models and comparison to rat LELs
Table 3 compares EAD estimates from the two rat nonpregnancy models (PPK and httk.PBTK models) to the rat developmental toxicity LELs. Except for one chemical, EADs produced using PPK model are 1.2–6.4‐fold higher than those estimated using httk.PBTK model. The EAD difference between the two models for all chemicals are <10‐fold, ranging from 1.9 to 6.5‐fold, which is expected considering the differences between the two models regarding model structure and types of plasma concentrations used for IVIVE (Table 3). Figure 4 compared EADs to oral rat LELs for the five chemicals with rat developmental toxicity study data. Both PPK and httk.PBTK models accurately predict the lowest LEL (within 1.5‐fold) for EHA. Both models overpredicted the rat LELs for PA, EBA, and MPA, and underpredicted the rat LEL for VPA. Of the two models, the httk.PBTK model provided more conservative, and accurate, estimates of rat LELs. The EADs produced from the httk.PBTK model were within 3.5‐fold of rat LEL range for all chemicals except for VPA.
TABLE 3.
Rat EAD estimates (mg/kg/day) compared to rat developmental toxicity LELs
| Chemical | EAD using PPK model a | EAD using Httk.PBTK model a | Developmental toxicity LEL (mg/kg/day)b | Ratio (max EAD vs min EAD) | ||
|---|---|---|---|---|---|---|
| Using nominal dTP Conc. | Using free medium Conc. | Using nominal dTP Conc. | Using free medium Conc. | |||
| VPA | 33.3 | 14.3 | 13.5 | 5.8 | 200–400 | 5.7 |
| EHA | 95.3 | 45.5 | 77.3 | 36.9 | 100–600 | 2.6 |
| PHA | 27.5 | 6.3 | 39.5 | 9.1 | NA | 6.3 |
| 4‐ene‐VPA | 119.6 | 95.7 | 77.1 | 61.7 | NA | 1.9 |
| DMPA | 936.6 | 505.8 | 388.3 | 209.7 | NA | 4.5 |
| HA | 2,921.8 | 2,308.2 | 652.7 | 515.6 | NA | 5.7 |
| PA | 947.1 | 931.1 | 147.7 | 145.2 | 50–75 | 6.5 |
| MHA | 924.3 | 573.1 | 331.4 | 205.5 | NA | 4.5 |
| EBA | 2,708.3 | 2,260.4 | 517.4 | 431.8 | 150 | 6.3 |
| MPA | 2,957.1 | 2,407.2 | 574.2 | 467.4 | 188 | 6.3 |
Abbreviations: Conc., concentration; DMPA, 2,2‐Dimethylvaleric acid or 2,2‐dimethylpentanoic acid (IUPAC name); EAD, equivalent administered dose; EBA, 2‐ethylbutyric acid or 2‐ethylbutanoic acid (IUPAC name); EHA, 2‐ethylhexanoic acid; HA, hexanoic acid; LEL, lowest effect level; MHA, 2‐methylhexanoic acid; MPA, 2‐methylpentanoic acid; NA, not available; PA, 4‐pentenoic acid or pent‐4‐enoic acid (IUPAC name); PHA, 2‐propylheptanoic acid; VPA, valproic acid.
EAD values in boldface indicate values were within 3.5‐fold of the lowest or highest rat LELs.
Data were extracted from rat studies with oral, repeat dosing unless indicated otherwise. Data sources and experimental details are listed in Table 2.
FIGURE 4.
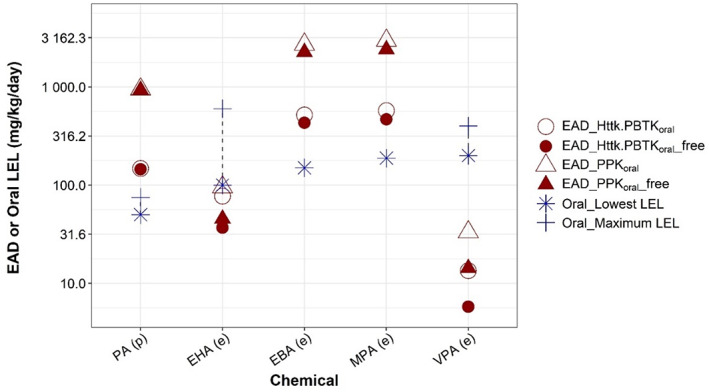
Comparison of equivalent administered doses (EADs) to oral rat lowest effect levels (LELs) for selected valproic acid analogues. The figure represents data provided in Table 3. EAD values predicted from devTOX quickPredict assay nominal developmental toxicity potential concentrations (open symbols) and free medium concentration (filled symbols) using one‐compartment population‐based pharmacokinetic (triangles) or httk.PBTK model (circles) are compared to the lowest and highest rat LELs (star and cross, respectively). Dashed lines highlight the range from lowest to highest in vivo LELs. The chemicals are ordered based on their lowest LELs, from low (left) to high (right). Whether predicted (p) or experimental (e) Clint values were used to generate EAD is indicated parenthetically on x‐axis labels.
Both nominal dTP concentration and corresponding free medium concentration (predicted using the Armitage mass balance model) were used in IVIVE. To evaluate the impact of the two types of in vitro concentration on IVIVE outcomes, ratios of free medium versus nominal concentration for each chemical were calculated (Table S4). In general, EAD estimates using free concentration are lower than those obtained from the nominal concentration for all chemicals, with a ratio spanning from 0.23 to 0.98 for all chemicals (Table S4). The predicted free concentration is about 80% of corresponding nominal concentration for 4 chemicals (EBA, MPA, HA, and 4‐ene‐VPA), 40–60% of nominal concentration for the other four chemicals (EHA, VPA, DMPA, and MHA), almost the same (98%) as nominal concentration for PA, and the smallest ratio is 23% for PHA.
3.5. EAD estimates using human regular and pregnancy‐specific PK/PBPK models
The human EADs predicted using nonpregnancy models (i.e., PPK and httk.PBTK models) and pregnancy‐specific PBPK models (i.e., httk.fPBTK and GP pregnancy models) are shown in Figure 5. Compared to regular PK models, pregnancy PBPK models have separate fetus compartments, so they can simulate dynamic change of both maternal and fetal plasma concentrations. To determine which of the two plasma concentrations gives more conservative estimates of in vivo LELs, EADs corresponding to both maternal and fetal plasma C max were calculated for pregnancy models.
FIGURE 5.
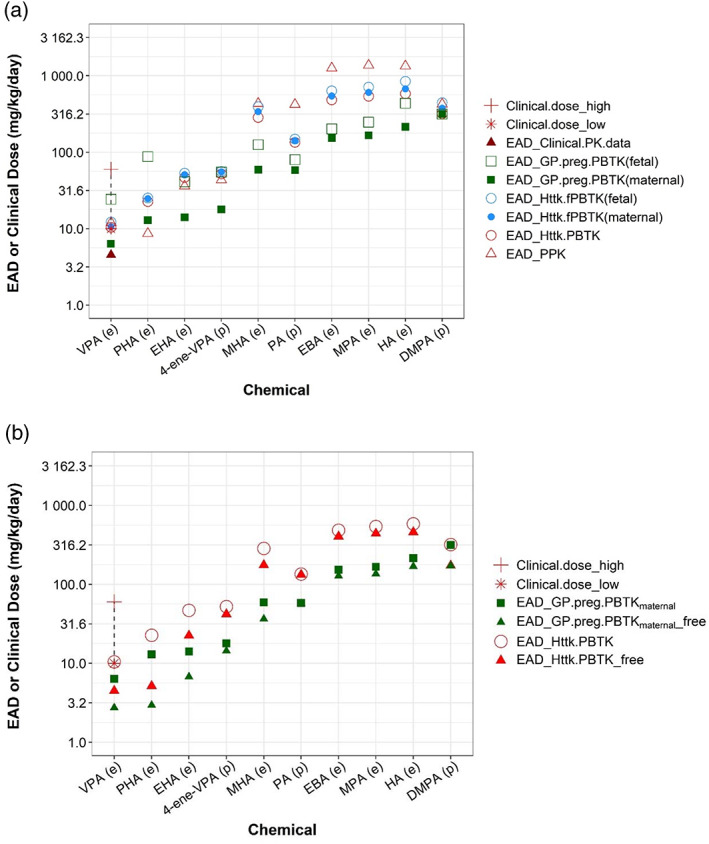
Human equivalent administered doses (EADs) estimated from various pharmacokinetic (PK)/physiologically based PK (PBPK) models. (a) Human EAD values estimated from nominal developmental toxicity potential (dTP) concentrations using various PK models (circles and squares) and clinical PK data were presented with different symbols. (b) Human EAD estimates between using free and nominal dTP concentration were compared for selected PK models. The chemicals are ordered from left to right based on lowest EAD values from low to high. Letters “(e)” or “(p)” on x‐axis labels indicate, respectively, whether experimental or quantitative structure‐activity relationship‐predicted Clint values were used to parameterize a PK model. For valproic acid, the dashed line highlights the range from lowest (“*”) to highest (“+”) clinical dose. Data for both figures are provided in Table S5.
EADs estimated using the httk pregnancy model (httk.fPBTK, Figure 5 blue circles) are similar (< 1.5‐fold) to those using the httk nonpregnancy model (httk.PBTK, Figure 5, red open circle). Using the same PK/PBPK models, EAD values for humans are in general lower than those for rat (Table 3, Table S5), suggesting that at similar external doses, humans would achieve higher plasma concentrations as compared to rat. For both pregnancy models (i.e., httk.fPBTK and Gp.preg.PBTK) EAD estimates with maternal C max as target concentration are less than those when fetal plasma C max was chosen as target concentration. This suggests that compared to maternal C max, a higher exposure level is needed for fetal C max to reach the same target concentration. This observation is expected considering the existence of blood placenta barrier between mother and fetus. EAD calculated from different PK/PBPK models varies. The variation in these EADs is ~1.4‐10‐fold across PK/PBPK models and chemicals (Table S5, Column of “Ratio_max EAD vs min EAD”). The GastroPlus pregnancy model with maternal C max as target plasma concentration (Figure 5a, solid square) provided the lowest, thus most conservative, EAD estimates for all chemicals except for PHA. For VPA, it also produces an EAD value (6.4, solid square) that is closest to the EAD value (4.6, red solid triangle) calculated by linear extrapolation of human clinical dose‐C max data reported in the PharmaPendium (Figure 5a).
For VPA, we were able to get information on its clinical usage. Our results showed that all models produce EADs that approximate the minimum clinical dose of VPA. The lowest EAD was provided by the GP pregnancy model, which was 1.6‐fold less than the lowest clinical dose of VPA (Figure 5a). This result suggests that the human EADs derived from the devTOX qP assay dTP provide a quantitative estimate of the VPA clinical dose, and indicates that the VPA clinical dose may exert developmental toxicities. This observation is consistent with the established warning that VPA is not safe in pregnancy (Diav‐Citrin et al., 2008; Jentink et al., 2010).
Figure 5b showed EAD comparison between two PBPK models using free and nominal dTP concentrations. Same as shown for rat, EAD estimates using free concentration are lower than those obtained from the nominal concentration for all chemicals, with a ratio spanning from 0.23 to 0.98 for all chemicals (Figure 5b, Table S4).
3.6. Comparison of EAD estimates to literature reported values
A recent OECD case study explored the possibility of using read‐across information from a subset of the VPA structural analogues tested here to predict the developmental toxicity of the target compound, MHA (OECD, 2020). In the study, several structural related aliphatic carboxylic acids of MHA were selected and tested using a battery of in vitro testing models that are relevant to developmental toxicity. These models include Zebrafish Embryo Test (ZET), ZET ceratohyal angle assay, mouse Embryonic Stem cell Test (mEST), iPSC‐based neurodevelopmental model (UKN1), and Chemically Activated LUciferase eXpression (CALUX) reporter gene assay. The methods and suitability of these in vitro models are described in detail in the OECD report (OECD, 2020).
In the report, in vitro effect concentrations (e.g., EC10) for each endpoint measured with these models and corresponding mouse or human oral equivalent doses (OEDs) were presented (OECD, 2020). We extracted all human OEDs (Table S6) from the report and calculated the OED range across all assays (OEDall) for five VPA analogues (row 4, Table 4). The OEDall ranges are quite large with fold differences between minimum and maximum OEDs spanning from 24‐ to ~1,200‐fold, indicating a large variation in endpoint sensitivity among these assays. To narrow this down and be conservative, we selected only OED values (OEDnarrow) derived from the most sensitive endpoint in each in vitro model and calculated another OED range (row 5, Table 4).
TABLE 4.
Comparison of human EADs predicted from different assays and PBPK models
| EBA | MPA | MHA | EHA | VPA | |
|---|---|---|---|---|---|
| EAD_Httk.PBTK a | 484.7 (404.5) | 541.2 (440.6) | 285.5 (176.0) | 47.0 (22.4) | 10.4 (4.5) |
| EAD_Gp.preg.PBTK a maternal | 153.2 (127.8) | 166.9 (135.8) | 59.12 (36.5) | 14.1 (6.7) | 6.4 (2.7) |
| Range of all human OEDs reported (OECD, 2020) b | 34.4–826.6 | 35.2–1,353.3 | 2.3–928.4 | 0.74–901.3 | 0.11–51.9 |
| Range of human OEDs estimated from most sensitive endpoints | 35.8–357.8 | 37.3–261.1 | 2.3–286.3 | 2.6–10 | 0.11–3.2 |
| OEDs from the most sensitive endpoints in each in vitro model category (OECD, 2020) | |||||
| ZET assay: Pericardial and/or yolk edema, Total embryo EC10 | 53.3 | 84.5 | 55.1 | 4.8 | 2.8 |
| ZET CHA reporter assay: Scoliosis/lordosis, Total embryo EC10 | 124.9 | 44.5 | NA | 0.74 | 0.1 |
| mEST assay: HDAC inhibition, day10, medium‐unbound EC10 | 357.8 | 261.1 | 286.3 | 10 | 2 |
| UKN1 assay: HDAC inhibition, medium‐unbound EC10 | 161.4 | 186.6 | 254.8 | 6 | 3 |
| CALUX reporter assay: Minimum cell‐total EC10 | 35.8 | 37.3 | 2.3 | 2.6 | 3.2 |
Abbreviations: CALUX, Chemically Activated LUciferase eXpression reporter gene assays; CHA, ceratohyal angle assay that assesses the morphological appearance of the ceratohyal; D3, ES cells; EAD, equivalent administered dose; EBA, 2‐ethylbutyric acid or 2‐ethylbutanoic acid (IUPAC name); EC10, effect concentration at which 10% effect is observed compared to the control; EHA, 2‐ethylhexanoic acid; HDAC, histone deacetylase; mEST, mouse embryonic stem cell test; MHA, 2‐methylhexanoic acid; MPA, 2‐methylpentanoic acid; OED, oral equivalent dose; PBPK, physiologically based pharmacokinetic; PBTK, physiologically based toxicokinetic; UKN1, iPSC‐based neurodevelopmental model; VPA, valproic acid; ZET, zebrafish embryo test.
EADs corresponding to nominal (outside of parenthesis) or free medium (inside of parenthesis) dTP concentrations. EAD values in boldface indicate those values that were within the range of human OEDs corresponding to the most sensitive endpoints in each in vitro model category (OECD, 2020).
The individual human oral equivalent dose extracted from literature are contained in Table S6.
Next, we compared EADs estimated using the devTOX qP assay and httk.PBTK or Gp.preg.PBTK models to these two OED ranges. Regardless the type of PBPK we used, all EADs we estimated fell within the “all‐inclusive” OED ranges, which is not surprising as OEDall range is quite large. When compared to the OEDnarrow range that covers only the most sensitive endpoints in each in vitro model category, EADs estimated using Gp.preg.PBTK model simulating maternal plasma C max were within range for most chemicals (row 3, Table 4), whereas EADs estimated using httk.PBTK model were outside of the range for four chemicals (row 2, Table 4), albeit within an order of magnitude. The difference between EADs predicted from nominal dTP concentrations and the high end of the OEDnarrow range is ≤2‐fold for EBA and MPA, 3.25‐fold for VPA, and 4.7‐fold for EHA, and the difference is even smaller for EADs predicted from free medium concentrations. In general, our EAD values are comparable to literature values, providing an additional layer of validation for the approach combining the devTOX qP assay and IVIVE.
4. DISCUSSION
Reliable, predictive, human cell‐based in vitro developmental toxicity assays are needed for rapidly screening chemicals for potential developmental toxicity. Despite the challenging nature of this endpoint, much progress has been made in using iPSCs for such assays. Before widespread use of iPSC‐based assays in toxicity assessment, validation is needed to ensure the assay's outcomes correlate well to in vivo assay results (Luz & Tokar, 2018). IVIVE is a useful tool to evaluate the correlation between an in vivo toxic effect and activity measured in an in vitro assay that is presumed to be toxicologically relevant based on the represented biology. For chemicals lacking in vivo toxicity data, IVIVE can be used to predict potentially toxic in vivo doses from in vitro assay measurements, expediting the safety assessment process.
In this study, we used in vitro dTP concentrations derived from the devTOX qP assay and applied various PK/PBPK models to estimate in vivo doses in rats and humans that would exert potential developmental toxicity. We evaluated the effects of using different modeling approaches (e.g., different PK model structures and platform, free medium versus nominal concentration) on IVIVE outcomes. LELs from rat in vivo developmental toxicity studies, human clinical doses, and oral equivalent doses reported in a recent OECD case study publication were used to validate our model predictions. The close agreement between EADs produced by the PK/PBPK models and in vivo data suggests that the devTOX qP assay in combination with IVIVE can be used to quantitatively predict in vivo developmental toxicity levels.
4.1. Nontargeted IVIVE versus targeted IVIVE
Several important factors need to be considered when conducting IVIVE: biological relevance of the in vitro assay to in vivo endpoints, assay‐ and chemical‐specific data to inform in vitro and in vivo kinetics, and suitability of the PK/PBPK models for reverse dosimetry. Whether an in vitro assay measures interaction with a molecular target that can be linked to in vivo toxicity through an adverse outcome pathway (AOP) or demonstrated biological plausibility impacts IVIVE applicability. “Nontargeted” IVIVE uses in vitro assays with nonspecific molecular targets for an in vivo outcome, which is useful for prioritizing chemicals for further testing. One good example of such application is to use half‐maximal activity concentration (AC50) values from all in vitro ToxCast assays (>500) measuring diverse endpoints to predict EAD distributions, and choose the lowest EAD to be compared to actual human exposure to determine testing priority for chemicals (Wetmore et al., 2012). A chemical with EAD estimates far above the actual human exposure has low testing priorities, while those with EAD estimates close to or below human exposures will have high testing priorities.
However, to quantitively predict in vivo doses exerting any specific toxic outcome, in vitro assays that measure an endpoint targeting to the same AOP or informing the same mechanism of action is preferred for IVIVE, referred to here as “targeted IVIVE.” As a good example of this “targeted IVIVE,” Casey et al. used a set of in vitro assays measuring key aspects of estrogen receptor pathway activation to accurately predict LELs of rodent uterotrophic bioassays (Casey et al., 2018). Applying a similar rationale, here we used an iPSC‐based assay that provides information on dTP to predict rat developmentally toxic LELs and human clinical doses for VPA and nine closely related structural analogues and achieved good agreement between EADs and in vivo data. In fact, for VPA, the range of EADs (2.7–24.4 mg/kg/day) derived from the in vitro dTP concentration predicted to result in human developmental toxicity was substantially closer to the reported teratogenic clinical doses (10–60 mg/kg/day) than the LEL derived from the rat developmental toxicity study (200 mg/kg/day), in addition to being more protective.
4.2. Effect of model type on EAD prediction
Our analysis evaluated the performance of several types of PK/PBPK models with different structures and applicability for IVIVE. We used both open‐source and proprietary PK models to generate EAD estimates that would produce maternal or fetal plasma concentrations equal to dTP concentrations in the devTOX qP assay. However, no pregnancy‐specific rat PBPK models were available from either open‐source and proprietary platforms. Due to significant physiology differences in pregnancy between humans and rats, extrapolation from humans to rat via allometric modeling is not feasible. Thus, only nonpregnancy models were used for rat IVIVE, while both nonpregnant and pregnant models were used for human IVIVE.
The PK/PBPK models used in this analysis had vastly different complexity (Figure 2). Therefore, some variations were expected in the predictive performance between these models. Our results showed ~1.4‐10‐fold variation in human EAD estimates across models and chemicals (Table S5). Surprisingly, we did not see significant differences in EAD estimates produced by the httk.PBTK (nonpregnancy) and httk.fPBTK (pregnancy) models. The closeness between the predictions produced by these two models is likely due to the simulated early gestation age (12 weeks). This finding suggests that, for early pregnancy predictions of EAD, the httk.PBTK model can approximate httk.fPBTK models in simulating maternal and fetal plasma concentrations, justifying our approach of using httk.PBTK model for predicting rat developmental toxicity LELs. Of all human models, the GP pregnancy model that simulated maternal plasma concentration provided the lowest EAD estimates for majority of tested chemicals, suggesting that this is the most conservative approach for risk evaluation. All PK/PBPK models produce EADs that approximated the minimum clinical dose of VPA, which is in agreement with observations that VPA clinical doses are not safe for pregnant women (Diav‐Citrin et al., 2008; Jentink et al., 2010).
Both httk.PBTK and httk.fPBTK models assume constant absorption rate and clearance, thus plasma C max is linearly related to external dose in those approaches. On the other hand, the GastroPlus pregnancy model incorporates nonlinear components in chemical absorption, protein binding and clearance mechanism, leading to potential nonlinear relationships between external dose and plasma C max. Nonlinearities in absorption and bioavailability can cause increases in chemical concentrations that are disproportionately high or low relative to the change in external dose (Ludden, 1991). Nonlinear pharmacokinetics also occur when the amount of chemical exceeds the capacity of the enzymes to metabolize it (Stein & Peletier, 2018). To determine possible existence of nonlinear PK, and to validate the EADs predicted under linearity assumption, we simulated maternal and fetal C max at the predicted EADs using GastroPlus pregnancy model and compared these C max values to the corresponding dTP concentrations. We found that for the range of EAD estimates, the linear assumption between external dose and C max largely holds true for most VPA analogues except for 2,2‐dimethylpentanoic acid (DMPA). On a dose‐plasma C max curve for DMPA, a C max plateau is reached at ~20,000 mg/day, suggesting a possible absorption saturation (Figure S2). Further increases in dosing amount did not significantly increase C max. Thus, it is difficult to estimate an EAD that would yield plasma C max equal to the dTP concentration for DMPA. Instead, the dose at the starting point of saturation plateau is used as EAD for DMPA, providing a more conservative estimate.
The httk models do not incorporate nonlinear PK, limiting their use for simulating nonlinear kinetics. However, the linearity assumption between external dose and C max holds true for the majority of chemicals at the dose ranges tested. Considering the attractive features of being open‐source and high throughput, the httk models demonstrate great potential in IVIVE applications and can be readily accessed via the National Toxicology Program Integrated Chemical Environment (https://ice.ntp.niehs.nih.gov/) (Abedini et al., 2021).
4.3. Effect of pharmacokinetic parameters on EAD prediction
The lack of accurate experimental data for key pharmacokinetic parameters (e.g., Clint) is often the limiting factor in developing IVIVE approaches for large chemical sets. Fortunately, experimental data for Clint are available for several VPA analogues (OECD, 2020). The Clint range for these VPA analogues is 0.07–33.6 μl/min/106, covering a relatively large range from low to high. In general, chemicals with low clearance tend to have a lower EAD estimate, as low clearance rate will cause chemical accumulation in the body, thus less exposure is needed for C max to reach the target concentration.
When experimental values are not available, open‐source QSAR models provided in OPERA (v2.7) (Mansouri et al., 2018) can be used to predict the fu and Clint of a chemical. Using in silico models to adequately estimate input parameter values for PK and PBPK modeling greatly expands the utility of IVIVE for risk assessment. However, the reliability of QSAR predictions depends on the number and structural diversity of the chemicals included in the model's training set (Cherkasov et al., 2014). The training set for the OPERA (v2.7) Clint model includes experiment values for several VPA analogues, which ensures that VPA‐like chemicals are included within the model's applicability domain.
4.4. Adjusting for in vitro nominal concentration
IVIVE relies on in vitro measurements of bioactivity to predict EADs in exposed subjects. As a result of lacking experimental in vitro kinetics data, the nominal concentration has routinely been used to derive the effect concentration for IVIVE. However, theoretically, only the chemical portion unbound to plasma protein or culture vessel should be considered available for uptake into cells and exert toxicity (Groothuis et al., 2015). Therefore, the free medium concentration would be a better representative for the true effective concentration.
In our study, both nominal and free medium dTP concentration were used for IVIVE. The ratio of free versus nominal concentration in devTOX qP assay system was calculated using a published mass‐balance model that considers essential assay components and physicochemical characteristics (Armitage et al., 2014). The free:nominal concentration ratio was 0.23–0.98 across chemicals (Table S4). EAD adjustment with these ratios led to lower EADs across all chemicals, suggesting a more conservative EAD estimate when free medium concentration is used. These biologically more relevant values represent relative intrinsic potencies of the chemicals to induce effects in these various in vitro systems. The fu parameter is particularly important in affecting free medium concentration. For example, the chemical that had the lowest ratio of free versus nominal concentration (0.23, for PHA) also has the lowest fu value (0.057), whereas the chemical with the highest ratio (0.98, for PA) also has the second highest fu value (0.66) (Table S4).
4.5. Chemical structure similarity analysis and comparison to OECD studies
Chemical structure similarity between VPA and the rest of chemicals were also calculated (Table 1) and used to help understand the potency differences between VPA analogues. EHA is the second most potent analogues based on devTOX qP dTP concentration. Interestingly, it also has the highest structure similarity score to VPA. However, four other chemicals (PHA, 4‐ene‐VPA, EBA, MPA) had relatively high structure similarity scores (0.58–0.64), but their potencies (based on dTP concentration) were different (Table 1). For example, PHA and EBA had almost the same structure similarity score relative to VPA (0.58 vs. 0.6), but there is approximately a two‐fold difference in their dTP concentrations (546 μM vs. 1,071 μM). This discrepancy could be explained by data shown in the OECD report (OECD, 2020), which suggested that side‐chain length is related to the teratogenic potency of these chemicals, and PHA has the longest side chains while EBA has the smallest side chain. Our result suggests that chemical structure information may help explain differences in toxicity potency of this series of aliphatic carboxylic acids, but the exact functional group or moiety that is related to potency needs further investigation.
In the recent OECD case study, multiple in vitro testing models with various endpoints relevant to developmental toxicity, for example, pericardial and yolk edema, cranio‐facial deformation for zebrafish embryo test, HDAC inhibition assay using mouse embryonic stem cell, were used for IVIVE of the VPA analogues. In the OECD report, the nominal EC10 values were also converted into biologically more relevant EC10 values by assay‐specific in vitro biokinetics models that consider factors of evaporation, plastics binding, and binding to serum lipids and protein, etc (OECD, 2020). Two ranges were calculated to summarize OED data from the OECD report, one includes data from all in vitro assays (OEDall), the other only includes the most sensitive endpoint of each in vitro assay (OEDnarrow). The OEDall range is quite large with fold differences between the lowest and highest OEDs spanning from 24‐ to ~1,200‐ fold across the tested chemicals, suggesting a large variation in in vitro assay/endpoint sensitivity. The CALUX reporter assay and ZET CHA reporter assay seem more sensitive than the rest of assays in detecting developmental toxicity, which is not surprising as reporter gene assays were generally observed to possess greater endpoint sensitivity than other assay types (Dreier, Connors, & Brooks, 2015). In the OECD case study, there was another in vitro assay using human stem cells, that is, human neural embryonic cells (UKN1 assay), which was also used for IVIVE for the VPA analogues. The UKN1 assay measures changes in gene expression and differentiation using human neural embryonic cells and provides valuable information on a chemical's neurodTP (OECD, 2020). However, the assay is not adaptable to high throughput platform because the differentiation needs to be done in 6 or 12 well plates. In comparison, devTOX qP assay can be adapted to a medium/HTS platform, has a relatively short testing period (3‐day), and high balanced accuracy in predicting in vivo developmental toxicity for a wide variety of chemicals (Zurlinden et al., 2020), making it suitable in screening chemicals for dTP in a high throughput manner.
Using devTOX qP assay and PBPK models, all EADs estimated from dTP concentration fell within the OEDall range. Depending on the type of PBPK models used, the EADs were either within the OEDnarrow range or ≤ 5‐fold different when compared to the high end of the OEDnarrow range. In all, our EAD values are comparable to the literature reported values that used a diverse set of in vitro assays, further validating our IVIVE approach. However, considering the large variance in assay/endpoint sensitivity, how to consolidate results from multiple assays with various molecular initiation and signaling events remains a challenging question to answer.
When examining this type of approach for pairing in vitro testing with IVIVE projections, a detailed understanding on mode of action or AOP for toxicity endpoints (e.g., developmental toxicity) and the relevance of an in vitro assay endpoint to specific molecular and biological process leading to an adverse outcome (e.g., dysmorphic effects) will facilitate establishing confidence and supporting widespread application. The predictive accuracy of all in silico models (e.g., QSAR and PBPK models) should be evaluated, and efforts are also needed to characterize applicability domain and model uncertainty. Work is underway to account for population variability in the PBPK models (Cohen Hubal et al., 2019), and promote use of guidance documents for model reporting and validation to help users determine the suitability of a model for a specific IVIVE application (Bell et al., 2018; OECD, 2021).
5. CONCLUSIONS
In summary, this study demonstrated the application of the devTOX qP assay and IVIVE in predicting rat oral developmental toxicity LELs and human clinical doses for VPA and its analogues. We evaluated the impact of using various PK models with different complexities and in vitro kinetics on IVIVE outcomes. The EAD variations observed when using different PK models are within expected ranges. Minimal differences in model performance between open‐source nonpregnancy (httk.PBTK), pregnancy‐specific (httk.fPBRK), and commercial (GP) models in human EAD predictions were observed. The httk.PBTK model provided the most accurate predictions of rat LELs for the majority of the VPA analogues. The EAD derived from the human iPS cell‐based devTOX qP assay was a better predictor of the human teratogenic clinical dose for VPA than the LELs from the rat developmental toxicity study. This work highlights the utility of IVIVE to support assessment of developmental toxicity potency based on in vitro assays.
CONFLICT OF INTERESTS
Jessica Palmer and Elizabeth Donley work for Stemina, which developed the devTOX qP assay. The rest authors declare no competing financial interests. This manuscript and the views expressed herein are those of the authors and do not necessarily reflect the views or policies of the U.S. Food and Drug Administration, the U.S. Environmental Protection Agency, NIEHS, or any other U.S. government entity.
Supporting information
Appendix S1 Supplementary Information
Table S6
ACKNOWLEDGMENTS
The authors thank Dr. Ciaran Fisher (Certara) and Ms. Lara Ward (Cyprotex) for their input on experimental intrinsic clearance data measured using primary hepatocytes for VPA and its analogues. The authors thank Dr. David Hines for review of R script for applying httk models. The authors also thank technical reviewers, Dr. Pei‐Li Yao (NIEHS), and Dr. Kristen Ryan (NIEHS) for constructive feedback. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
Chang, X. , Palmer, J. , Lumen, A. , Lee, U. J. , Ceger, P. , Mansouri, K. , Sprankle, C. , Donley, E. , Bell, S. , Knudsen, T. B. , Wambaugh, J. , Cook, B. , Allen, D. , & Kleinstreuer, N. (2022). Quantitative in vitro to in vivo extrapolation for developmental toxicity potency of valproic acid analogues. Birth Defects Research, 114(16), 1037–1055. 10.1002/bdr2.2019
Funding information The Intramural Research Program of the National Institute of Environmental Health Sciences (NIEHS) supported this work. Technical support was provided by Inotiv‐RTP, under NIEHS contract HHSN273201500010C.
Contributor Information
Xiaoqing Chang, Email: xiaoqing.chang@inotivco.com.
Jessica Palmer, Email: JPalmer@stemina.com.
Annie Lumen, Email: alumen@amgen.com.
Un Jung Lee, Email: unjung.lee@einsteinmed.org.
Patricia Ceger, Email: patricia.ceger@inotivco.com.
Kamel Mansouri, Email: kamel.mansouri@nih.gov.
Catherine Sprankle, Email: Catherine.Sprankle@inotivco.com.
Elizabeth Donley, Email: BDonley@stemina.com.
Shannon Bell, Email: Shannon.Bell@inotivco.com.
Thomas B. Knudsen, Email: Knudsen.Thomas@epa.gov
John Wambaugh, Email: Wambaugh.John@epa.gov.
Bethany Cook, Email: Bethany.Cook@inotivco.com.
David Allen, Email: Dave.Allen@inotivco.com.
Nicole Kleinstreuer, Email: nicole.kleinstreuer@nih.gov.
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article
REFERENCES
- Abedini, J. , Cook, B. , Bell, S. , Chang, X. , Choksi, N. , Daniel, A. B. , … Kleinstreuer, N. (2021). Application of new approach methodologies: ICE tools to support chemical evaluations. Computational Toxicology, 20(November), 100184. 10.1016/j.comtox.2021.100184 [DOI] [Google Scholar]
- Andersen, M. E. (2003). Toxicokinetic modeling and its applications in chemical risk assessment. Toxicology Letters, 138(1–2), 9–27. 10.1016/s0378-4274(02)00375-2 [DOI] [PubMed] [Google Scholar]
- Armitage, J. M. , Wania, F. , & Arnot, J. A. (2014). Application of mass balance models and the chemical activity concept to facilitate the use of in vitro toxicity data for risk assessment. Environmental Science & Technology, 48(16), 9770–9779. 10.1021/es501955g [DOI] [PubMed] [Google Scholar]
- Bell, S. M. , Chang, X. , Wambaugh, J. F. , Allen, D. G. , Bartels, M. , Brouwer, K. L. R. , … Kleinstreuer, N. C. (2018). In vitro to in vivo extrapolation for high throughput prioritization and decision making. Toxicology in Vitro, 47(March), 213–227. 10.1016/j.tiv.2017.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binkerd, P. E. , Rowland, J. M. , Nau, H. , & Hendrickx, A. G. (1988). Evaluation of valproic acid (VPA) developmental toxicity and pharmacokinetics in Sprague‐Dawley rats. Fundamental and Applied Toxicology, 11, 485–493. 10.1016/0272-0590(88)90112-1 [DOI] [PubMed] [Google Scholar]
- Casey, W. M. , Chang, X. , Allen, D. G. , Ceger, P. C. , Choksi, N. Y. , Hsieh, J.‐H. , … Kleinstreuer, N. C. (2018). Evaluation and optimization of pharmacokinetic models for in vitro to in vivo extrapolation of estrogenic activity for environmental chemicals. Environmental Health Perspectives, 126(9), 97001. 10.1289/EHP1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC . 2020. Data & statistics on birth defects | CDC. Centers for Disease Control and Prevention. January 23, 2020. https://www.cdc.gov/ncbddd/birthdefects/data.html
- Cherkasov, A. , Muratov, E. N. , Fourches, D. , Varnek, A., II , Baskin, I. I. , Cronin, M. , … Tropsha, A. (2014). QSAR modeling: Where have you been? Where are you going to? Journal of Medicinal Chemistry, 57(12), 4977–5010. 10.1021/jm4004285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diav‐Citrin, O. , Shechtman, S. , Weinbaum, D. , Wajnberg, R. , Avgil, M. , Di Gianantonio, E. , … Ornoy, A. (2008). Paroxetine and fluoxetine in pregnancy: A prospective, multicentre, controlled, observational study. British Journal of Clinical Pharmacology, 66(5), 695–705. 10.1111/j.1365-2125.2008.03261.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier, D. A. , Connors, K. A. , & Brooks, B. W. (2015). Comparative endpoint sensitivity of in vitro estrogen agonist assays. Regulatory Toxicology and Pharmacology: RTP, 72(2), 185–193. 10.1016/j.yrtph.2015.04.009 [DOI] [PubMed] [Google Scholar]
- Eikel, D. , Lampen, A. , & Nau, H. (2006). Teratogenic effects mediated by inhibition of histone deacetylases: Evidence from quantitative structure activity relationships of 20 valproic acid derivatives. Chemical Research in Toxicology, 19(2), 272–278. 10.1021/tx0502241 [DOI] [PubMed] [Google Scholar]
- Elsevier , Amsterdam , Netherlands . 2021. PharmaPendium. 2021. https://www.pharmapendium.com/
- EU‐ToxRisk . 2020. EU‐ToxRisk. 2020. https://www.eu-toxrisk.eu/
- Groothuis, F. A. , Heringa, M. B. , Nicol, B. , Hermens, J. L. M. , Blaauboer, B. J. , & Kramer, N. I. (2015). Dose metric considerations in in vitro assays to improve quantitative in vitro‐in vivo dose extrapolations. Toxicology, 332(June), 30–40. 10.1016/j.tox.2013.08.012 [DOI] [PubMed] [Google Scholar]
- Hauck, R. S. , Wegner, C. , Blumtritt, P. , Fuhrhop, J. H. , & Nau, H. (1990). Asymmetric synthesis and teratogenic activity of (R)‐ and (S)‐2‐ethylhexanoic acid, a metabolite of the plasticizer Di‐(2‐Ethylhexyl)phthalate. Life Sciences, 46(7), 513–518. 10.1016/0024-3205(90)90007-e [DOI] [PubMed] [Google Scholar]
- Hendrickx, A. G. , Peterson, P. E. , Tyl, R. W. , Fisher, L. C. , Fosnight, L. J. , Kubena, M. F. , … Katz, G. V. (1993). Assessment of the developmental toxicity of 2‐ethylhexanoic acid in rats and rabbits. Fundamental and Applied Toxicology, 20(2), 199–209. 10.1093/toxsci/20.2.199 [DOI] [PubMed] [Google Scholar]
- Hubal, C. , Elaine, A. , Wetmore, B. A. , Wambaugh, J. F. , El‐Masri, H. , Sobus, J. R. , & Bahadori, T. (2019). Advancing internal exposure and physiologically‐based toxicokinetic modeling for 21st‐century risk assessments. Journal of Exposure Science & Environmental Epidemiology, 29(1), 11–20. 10.1038/s41370-018-0046-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentink, J. , Bakker, M. K. , Nijenhuis, C. M. , Wilffert, B. , Lolkje, T. W. d. , & den Berg, J.‐v. (2010). Does folic acid use decrease the risk for spina bifida after in utero exposure to valproic acid? Pharmacoepidemiology and Drug Safety, 19(8), 803–807. 10.1002/pds.1975 [DOI] [PubMed] [Google Scholar]
- Jones, H. M. , & Rowland‐Yeo, K. (2013). Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT: Pharmacometrics & Systems Pharmacology, 2(8), e63. 10.1038/psp.2013.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapraun, D. F. , Wambaugh, J. F. , Woodrow Setzer, R. , & Judson, R. S. (2019). Empirical models for anatomical and physiological changes in a human mother and fetus during pregnancy and gestation. PLoS One, 14(5), e0215906. 10.1371/journal.pone.0215906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstreuer, N. C. , Smith, A. M. , West, P. R. , Conard, K. R. , Fontaine, B. R. , Weir‐Hauptman, A. M. , … Cezar, G. G. (2011). Identifying developmental toxicity pathways for a subset of ToxCast chemicals using human embryonic stem cells and metabolomics. Toxicology and Applied Pharmacology, 257(1), 111–121. 10.1016/j.taap.2011.08.025 [DOI] [PubMed] [Google Scholar]
- Lammer, E. J. , Sever, L. E. , & Oakley, G. P. (1987). Teratogen update: Valproic acid. Teratology, 35(3), 465–473. 10.1002/tera.1420350319 [DOI] [PubMed] [Google Scholar]
- Ludden, T. M. (1991). Nonlinear pharmacokinetics: Clinical implications. Clinical Pharmacokinetics, 20(6), 429–446. 10.2165/00003088-199120060-00001 [DOI] [PubMed] [Google Scholar]
- Luz, A. L. , & Tokar, E. J. (2018). Pluripotent stem cells in developmental toxicity testing: A review of methodological advances. Toxicological Sciences, 165(1), 31–39. 10.1093/toxsci/kfy174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri, K. , Grulke, C. M. , Judson, R. S. , & Williams, A. J. (2018). OPERA models for predicting physicochemical properties and environmental fate endpoints. Journal of Cheminformatics, 10(1), 10. 10.1186/s13321-018-0263-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narotsky, M. G. , Francis, E. Z. , & Kavlock, R. J. (1994). Developmental toxicity and structure‐activity relationships of aliphatic acids, including dose‐response assessment of valproic acid in mice and rats. Fundamental and Applied Toxicology, 22(2), 251–265. [DOI] [PubMed] [Google Scholar]
- Nau, H. , Hauck, R. S. , & Ehlers, K. (1991). Valproic acid‐induced neural tube defects in mouse and human: Aspects of chirality, alternative drug development, pharmacokinetics and possible mechanisms. Pharmacology & Toxicology, 69(5), 310–321. 10.1111/j.1600-0773.1991.tb01303.x [DOI] [PubMed] [Google Scholar]
- Nau, H. , & Löscher, W. (1986). Pharmacologic evaluation of various metabolites and analogs of Valproic acid: Teratogenic potencies in mice. Fundamental and Applied Toxicology, 6(4), 669–676. 10.1016/0272-0590(86)90180-6 [DOI] [PubMed] [Google Scholar]
- Nau, H. , Zierer, R. , Spielmann, H. , Neubert, D. , & Gansau, C. (1981). A new model for embryotoxicity testing: Teratogenicity and pharmacokinetics of valproic acid following constant‐rate administration in the mouse using human therapeutic drug and metabolite concentrations. Life Sciences, 29(26), 2803–2813. 10.1016/0024-3205(81)90541-5 [DOI] [PubMed] [Google Scholar]
- O'Boyle, N. M. , & Sayle, R. A. (2016). Comparing structural fingerprints using a literature‐based similarity benchmark. Journal of Cheminformatics, 8(July), 36. 10.1186/s13321-016-0148-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- OECD . 2020. ENVIRONMENT DIRECTORATE JOINT MEETING OF THE CHEMICALS COMMITTEE AND THE WORKING PARTY ON CHEMICALS, PESTICIDES AND BIOTECHNOLOGY Case Study on the Use of Integrated Approaches to Testing and Assessment for READ‐ACROSS BASED FILLING OF DEVELOPMENTAL TOXICITY DATA GAP FOR METHYL HEXANOIC ACID . Series on Testing and Assessment, No. 325. https://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV%2fJM%2fMONO(2020)21&docLanguage=en&msclkid=cce48a99a62111ecb8a11d0da24f83ae
- OECD . 2021. Guidance Document on the Characterisation, Validation and Reporting of Physiologically Based Kinetic (PBK) Models for Regulatory Purposes . Series on Testing and Assessment, No. 331. https://www.oecd.org/chemicalsafety/risk-assessment/guidance-document-on-the-characterisation-validation-and-reporting-of-physiologically-based-kinetic-models-for-regulatory-purposes.pdf
- Ornoy, A. (2009). Valproic acid in pregnancy: How much are we endangering the embryo and fetus? Reproductive Toxicology, 28(1), 1–10. 10.1016/j.reprotox.2009.02.014 [DOI] [PubMed] [Google Scholar]
- Palmer, J. A. , Smith, A. M. , Egnash, L. A. , Colwell, M. R. , Donley, E. L. R. , Kirchner, F. R. , & Burrier, R. E. (2017). A human induced pluripotent stem cell‐based in vitro assay predicts developmental toxicity through a retinoic acid receptor‐mediated pathway for a series of related retinoid analogues. Reproductive Toxicology (Elmsford, N.Y.), 73, 350–361. 10.1016/j.reprotox.2017.07.011 [DOI] [PubMed] [Google Scholar]
- Palmer, J. A. , Smith, A. M. , Egnash, L. A. , Conard, K. R. , West, P. R. , Burrier, R. E. , … Kirchner, F. R. (2013). Establishment and assessment of a new human embryonic stem cell‐based biomarker assay for developmental toxicity screening. Birth Defects Research Part B: Developmental and Reproductive Toxicology, 98(4), 343–363. 10.1002/bdrb.21078 [DOI] [PubMed] [Google Scholar]
- Pearce, R. , Strope, C. , Setzer, W. , Sipes, N. , & Wambaugh, J. (2017). Httk: R package for high‐throughput toxicokinetics. Journal of Statistical Software, 79(4), 1–26. 10.18637/jss.v079.i04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennanen, S. , Tuovinen, K. , Huuskonen, H. , & Komulainen, H. (1992). The developmental toxicity of 2‐ethylhexanoic acid in Wistar rats. Fundamental and Applied Toxicology, 19(4), 505–511. 10.1016/0272-0590(92)90088-Y [DOI] [PubMed] [Google Scholar]
- Richard, A. M. , Huang, R. , Waidyanatha, S. , Shinn, P. , Collins, B. J. , Thillainadarajah, I. , … Tice, R. R. (2021). The Tox21 10K compound library: Collaborative chemistry advancing toxicology. Chemical Research in Toxicology, 34(2), 189–216. 10.1021/acs.chemrestox.0c00264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt, W. (2008). General approach for the calculation of tissue to plasma partition coefficients. Toxicology in Vitro, 22(2), 457–467. 10.1016/j.tiv.2007.09.010 [DOI] [PubMed] [Google Scholar]
- Sfeir, M. (2020). Evaluation of a rapid, multi‐chemical human gestational physiologically based toxicokinetic model (conference abstract). Conference abstract presented at the Society of Toxicology 59th Annual Meeting, 2939, March 14. https://eventpilotadmin.com/web/page.php?page=IntHtml&project=SOT20&id=688
- Shukla, S. J. , Huang, R. , Austin, C. P. , & Xia, M. (2010). The future of toxicity testing: A focus on in vitro methods using a quantitative high‐throughput screening platform. Drug Discovery Today, 15(23–24), 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simulations Plus Inc . 2020. GastroPlus Software. http://www.simulations-plus.com
- Stein, A. M. , & Peletier, L. A. (2018). Predicting the onset of nonlinear pharmacokinetics. CPT: Pharmacometrics & Systems Pharmacology, 7(10), 670–677. 10.1002/psp4.12316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhold, B. (2009). Environmental factors in birth defects: What we need to know. Environmental Health Perspectives, 117(10), A440–A447. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2897222/ [Google Scholar]
- Weininger, D. (1988). SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. Journal of Chemical Information and Computer Sciences, 28(1), 31–36. 10.1021/ci00057a005 [DOI] [Google Scholar]
- Wetmore, B. A. , Wambaugh, J. F. , Ferguson, S. S. , Sochaski, M. A. , Rotroff, D. M. , Freeman, K. , … Thomas, R. S. (2012). Integration of dosimetry, exposure, and high‐throughput screening data in chemical toxicity assessment. Toxicological Sciences, 125(1), 157–174. [DOI] [PubMed] [Google Scholar]
- Young, J. F. , Branham, W. S. , Sheehan, D. M. , Baker, M. E. , Wosilait, W. D. , & Luecke, R. H. (1997). Physiological ‘constants’ for PBPK models for pregnancy. Journal of Toxicology and Environmental Health, 52(5), 385–401. 10.1080/00984109708984072 [DOI] [PubMed] [Google Scholar]
- Zurlinden, T. J. , Saili, K. S. , Rush, N. , Kothiya, P. , Judson, R. S. , Houck, K. A. , … Knudsen, T. B. (2020). Profiling the ToxCast library with a pluripotent human (H9) stem cell line‐based biomarker assay for developmental toxicity. Toxicological Sciences, 174(2), 189–209. 10.1093/toxsci/kfaa014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supplementary Information
Table S6
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article