

SUMMARY
Mammalian genomes are organized into three-dimensional DNA structures called A/B compartments that are associated with transcriptional activity/inactivity. However, whether these structures are simply correlated with gene expression or are permissive/impermissible to transcription has remained largely unknown because we lack methods to measure DNA organization and transcription simultaneously. Recently, we developed RNA & DNA (RD)-SPRITE, which enables genome-wide measurements of the spatial organization of RNA and DNA. Here we show that RD-SPRITE measures genomic structure surrounding nascent pre-mRNAs and maps their spatial contacts. We find that transcription occurs within B compartments—with multiple active genes simultaneously colocalizing within the same B compartment—and at genes proximal to nucleoli. These results suggest that localization near or within nuclear structures thought to be inactive does not preclude transcription and that active transcription can occur throughout the nucleus. In general, we anticipate RD-SPRITE will be a powerful tool for exploring relationships between genome structure and transcription.
In brief
Using RD-SPRITE to simultaneously map 3D genome structure and nascent RNA transcription genome-wide, Goronzy et al. find that RNA polymerase II transcription can occur within multiple compartments in the nucleus, including regions previously associated with inactive transcription, such as the B compartment and close to the nucleolus.
Graphical Abstract
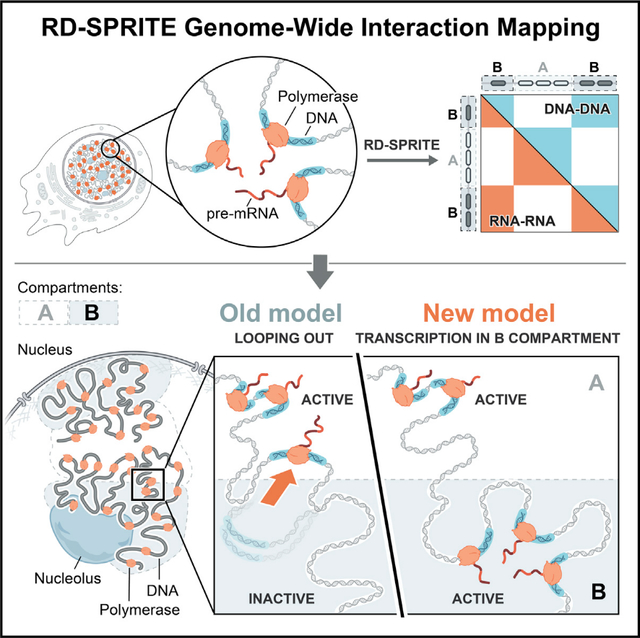
INTRODUCTION
The three-dimensional (3D) arrangement of DNA in the nucleus is thought to be important for regulating critical nuclear processes such as DNA replication and transcription.1–3 Accordingly, there have been significant efforts to map DNA structure across different cell types using proximity ligation methods like 3C,4 Hi-C,5–7 and related variants.2,8 These methods have identified several structural features including chromosome territories, A/B compartments, topologically associating domains (TADs), loops, and enhancer-promoter interactions. However, which of these are critical for gene regulation and other cellular functions remains unclear.
The main reason that structure-function relationships within the nucleus are poorly understood is that current methods cannot simultaneously measure transcriptional states and 3D genome organization.9,10 Instead, analysis of the functional consequences of nuclear structure relies on correlations between distinct measurements of DNA organization and gene expression profiles generated from a combination of experimental methods (e.g., Hi-C and RNA-seq) in different populations of cells. These measurements capture an ensemble of many individual cells, each of which may contain heterogeneous functional states and structures, making the direct comparison between 3D structure and transcription challenging.5,11
To highlight this limitation, consider A and B compartments, which refer to alternating sets of DNA regions that broadly partition chromosomes; DNA regions within one compartment preferentially interact with each other (e.g., A-A) rather than with neighboring regions of the other (e.g., A-B). Early studies found that A compartments are enriched for genomic DNA regions containing actively transcribed RNA polymerase II (Pol II) genes, whereas B compartments are depleted for active Pol II genes and enriched for repressive chromatin marks.7,12 As such, these compartments are generally thought to represent spatial organization of transcriptionally active (A) and inactive (B) Pol II genes within distinct regions of the nucleus.3,10,13–15
In contrast to this general observation, there are specific genes located within B compartments that are actively transcribed.12 This is predominantly explained by a model where actively transcribed DNA loci “loop out” of the inactive (B) compartment to localize within an active (A) compartment.1,16–19 In this model, Pol II transcription does not occur within B compartments; actively transcribed genes may appear to be within them simply because of the ensemble nature of compartment (e.g., Hi-C, SPRITE) and gene expression measurements (e.g., RNA-seq). In support of this “looping out” model, single-cell microscopy measurements have shown that individual active genes can be located away from the remainder of the chromosome from which they are transcribed,16,18–20 that the promoter regions of active genes in B compartments can form local associations with the A compartment,21 and that transcribed genomic loci (measured by interactions between pre-mRNAs) do not form A/B compartments.16
Yet, there are other observations to suggest that transcription may occur within both A and B compartments: many A/B compartment boundaries remain the same between distinct cell states despite major changes in gene expression programs,12 and direct recruitment of various gene loci to the nuclear lamina (a compartment associated with transcriptional silencing and located within B compartments) does not always lead to transcriptional repression for all genes.22–25 Accordingly, whether localization of genes within B compartments or other nuclear structures that have been associated with inactive Pol II transcription and repressive heterochromatin (such as the nucleolus and nuclear lamina)22,26,27 precludes Pol II transcription or is simply correlated with inactive transcription remains unclear.
Recently, we developed RNA & DNA SPRITE (RD-SPRITE), which enables simultaneous multiway measurements of DNA and RNA organization in the nucleus.28 In our previous study, we focused on the spatial localization of ncRNAs and their roles in seeding nuclear organization. However, RD-SPRITE also measures localization of mRNAs, including individual nascent pre-mRNAs at their transcriptional loci. Because RNA represents the functional output of transcription, this approach allows us to directly measure both 3D genome organization and transcription at the same location within the nucleus. Here, we show that RD-SPRITE can be used to assess the relationship between structural organization and transcriptional activity within different structural compartments.
RESULTS
RD-SPRITE measures nascent and mature mRNAs at precise locations in the cell
RD-SPRITE uses split-and-pool barcoding to measure the spatial organization of individual RNA and DNA molecules within the cell. The fundamental measurement unit of RD-SPRITE is the SPRITE cluster, which contains multiple RNA and DNA molecules that are in close proximity within a single cell.28,29 Using these clusters, we can measure multiway RNA and DNA contacts, including RNA-RNA, RNA-DNA, and DNA-DNA contacts, within higher-order structures in the cell (Figure 1A). We previously showed that RD-SPRITE can accurately measure the 3D spatial organization of DNA and RNA in the nucleus, including DNA structures such as chromosome territories, A/B compartments, TADs, and loops as well as DNA and RNA within nuclear bodies such as the nucleolus, nuclear speckles, and histone locus body.28,30
Figure 1. RD-SPRITE measures nascent and mature mRNAs at precise locations in the cell.
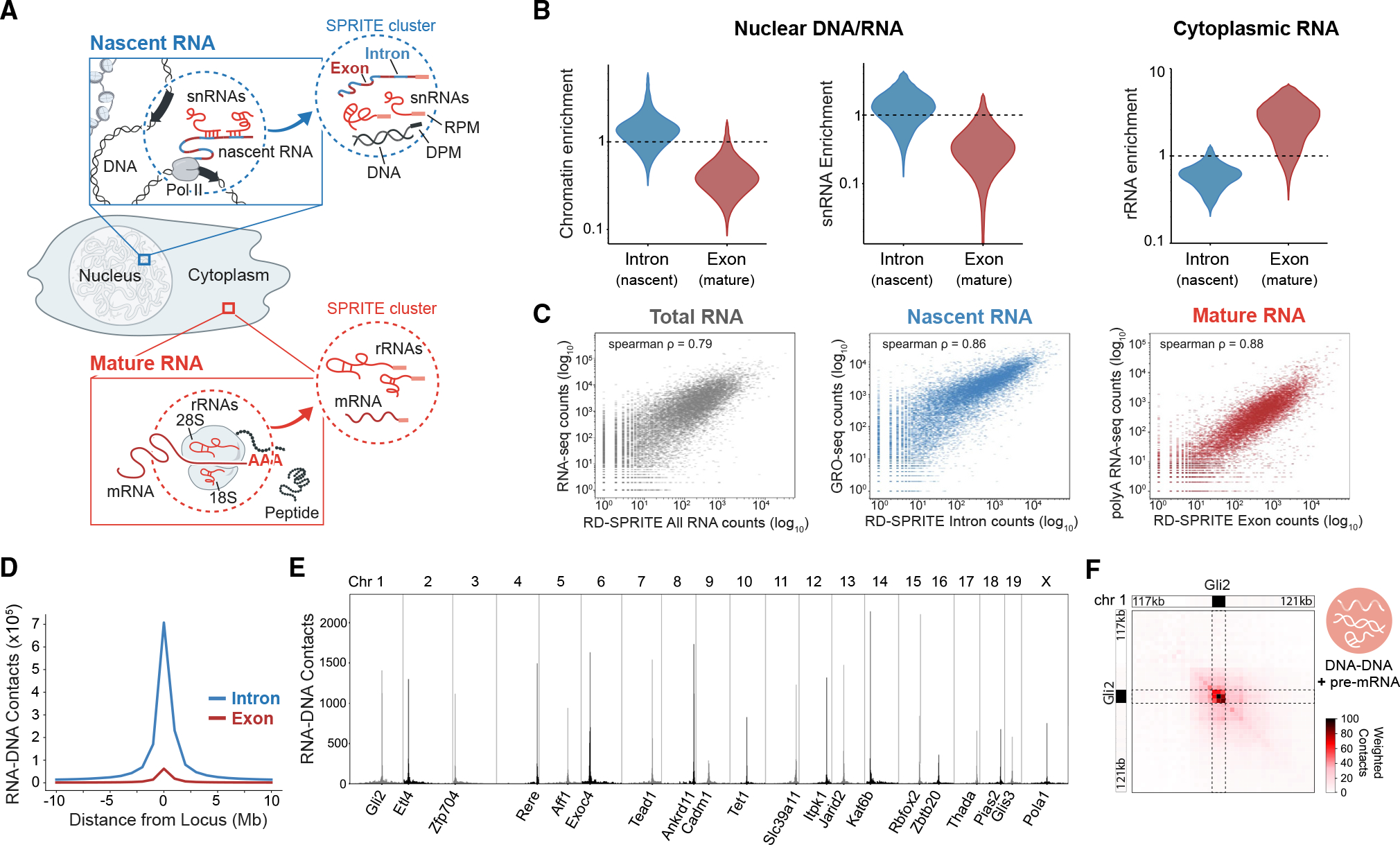
(A) Schematic of nascent pre-mRNAs (blue), mature mRNAs (red), and their respective molecular interactions mapped using RNA & DNA SPRITE. Zoom-ins show nascent pre-mRNA contacts in the nucleus (top) and mature mRNA contacts in the cytoplasm (bottom). The specific RNA (RPM) or DNA (DPM) molecules measured within RD-SPRITE clusters are shown in the dotted circles.
(B) Contact frequency enrichment scores of introns (blue) or exons (red) with chromatin (left), snRNAs (middle), or rRNAs (right) measured using RNA-DNA or RNA-RNA interactions.
(C) Correlations between RD-SPRITE RNA abundance and total RNA-seq31 (left), RD-SPRITE introns and GRO-seq32 (middle), and RD-SPRITE exons and polyA-selected RNA-seq (right).
(D) Aggregated total RNA-DNA contacts of introns or exons with DNA regions surrounding their genomic loci. Shown is the total weighted contact frequency of all RNAs within these populations contacting 1 megabase (Mb) genomic DNA windows from 10 Mbs up- and downstream of the transcriptional start site.
(E) Examples of weighted RNA-DNA interactions for selected pre-mRNAs (1-Mb resolution). The genomic locus for each pre-mRNA is annotated on the x axis.
(F) Weighted DNA-DNA interactions for transcriptionally active loci at the Gli2 gene locus on chromosome 1 (100-kb resolution). Interactions of transcriptionally active loci are defined as the DNA contacts occurring within multiway SPRITE clusters containing both nascent Gli2 pre-mRNA transcripts and multiple DNA reads.
Here, we sought to explore whether RD-SPRITE can measure the 3D organization of distinct populations of mRNAs—including nascent and mature mRNAs—and their quantitative levels at various locations in the cell. To do this, we examined the RNA-RNA and RNA-DNA contacts in our RD-SPRITE dataset collected from mouse embryonic stem cells. Specifically, we focused on intronic reads as a surrogate for nascent pre-mRNAs and exonic reads as a surrogate for mature mRNAs. We reasoned that newly transcribed (nascent) pre-mRNAs should be preferentially located on chromatin in proximity to their genomic DNA locus, while fully spliced (mature) mRNAs should be associated with ribosomal RNAs (rRNAs) in the cytoplasm. Consistent with this, we find that intronic reads in RD-SPRITE represent nascent pre-mRNAs in that they are (1) enriched on chromatin, (2) enriched for contacts with various small nuclear RNAs (snRNAs) such as U1 and U2, which are involved in pre-mRNA splicing in the nucleus, and (3) depleted for contacts with cytoplasmic RNAs, such as rRNAs (Figure 1B). In contrast, exonic reads show properties consistent with mature mRNAs in that they are (1) depleted on chromatin, (2) depleted for contacts with snRNAs, and (3) enriched for contacts with rRNAs (Figure 1B). Together, these data demonstrate that RD-SPRITE can detect both classes of mRNAs located in different parts of the cell and distinguish between their localization patterns.
We next tested whether RD-SPRITE can quantitatively measure the relative abundance of these distinct mRNA populations. First, we measured whether the overall RNA levels measured by RD-SPRITE correlate with total RNA-seq measurements and observed a strong correlation between the levels of RNAs measured by each approach (Spearman p = 0.79) (Figure 1C).31 Next, we focused specifically on nascent pre-mRNA levels by comparing transcription levels estimated from intronic reads in RD-SPRITE and found them to be highly correlated with those estimated from global run-on and sequencing (GRO-seq) assays,32 which measure transcription levels of mRNAs (Spearman p = 0.86). Finally, we observed a strong correlation between exonic reads measured by RD-SPRITE and mature mRNA levels measured by polyA-selected RNA-seq (Spearman p = 0.88).
To confirm the localization of nascent RNAs at their genomic loci, we measured the DNA contacts of pre-mRNAs (RNA-DNA contacts) and found them to be enriched for contacts with their genomic loci (Figures 1D and 1E). Next, we explored whether RD-SPRITE can detect the 3D structure at these actively transcribing DNA loci. To do this, we mapped the DNA-DNA contacts of SPRITE clusters containing a specific nascent pre-mRNA and found that the DNA contacts are highly enriched surrounding the locus from which the pre-mRNA is transcribed (Figure 1F).
Taken together, these results demonstrate that RD-SPRITE accurately distinguishes distinct populations of mRNAs within the cell, enables quantitative measurement of their transcription levels, and detects the genomic contacts and 3D structure around individual pre-mRNAs.
Genomic DNA located within B compartments can be actively transcribed
Because RD-SPRITE accurately measures both nascent RNA transcripts and higher-order DNA organization genome-wide, we used it to explore the global structure of genomic DNA regions undergoing Pol II transcription. Specifically, we generated a genome-wide DNA-DNA contact matrix using SPRITE clusters containing nascent pre-mRNAs. We reasoned that if most genes within B compartments loop out and reposition into A compartments when actively transcribed (the “looping out” model), then we would see a single active compartment in the DNA-DNA contact matrix of actively transcribed regions. Conversely, if genes are transcribed within B compartments, then we would observe both A and B compartments within this DNA-DNA contact matrix (Figure 2A). In fact, the genomic DNA structures generated from only actively transcribed clusters show clear chromosome territories and intra-compartment structures comparable to those observed when measuring DNA-DNA contacts across all SPRITE clusters (Figure 2B). The A/B compartment structure seen in transcribed clusters closely corresponds to A/B compartments defined using principal eigenvector analysis on the DNA contacts measured from all SPRITE clusters (Figure S1, STAR Methods). This suggests that genes in the B compartment do not “loop out” as they are transcribed but instead remain in the B compartment.
Figure 2. Genomic DNA located within B compartments can be actively transcribed.
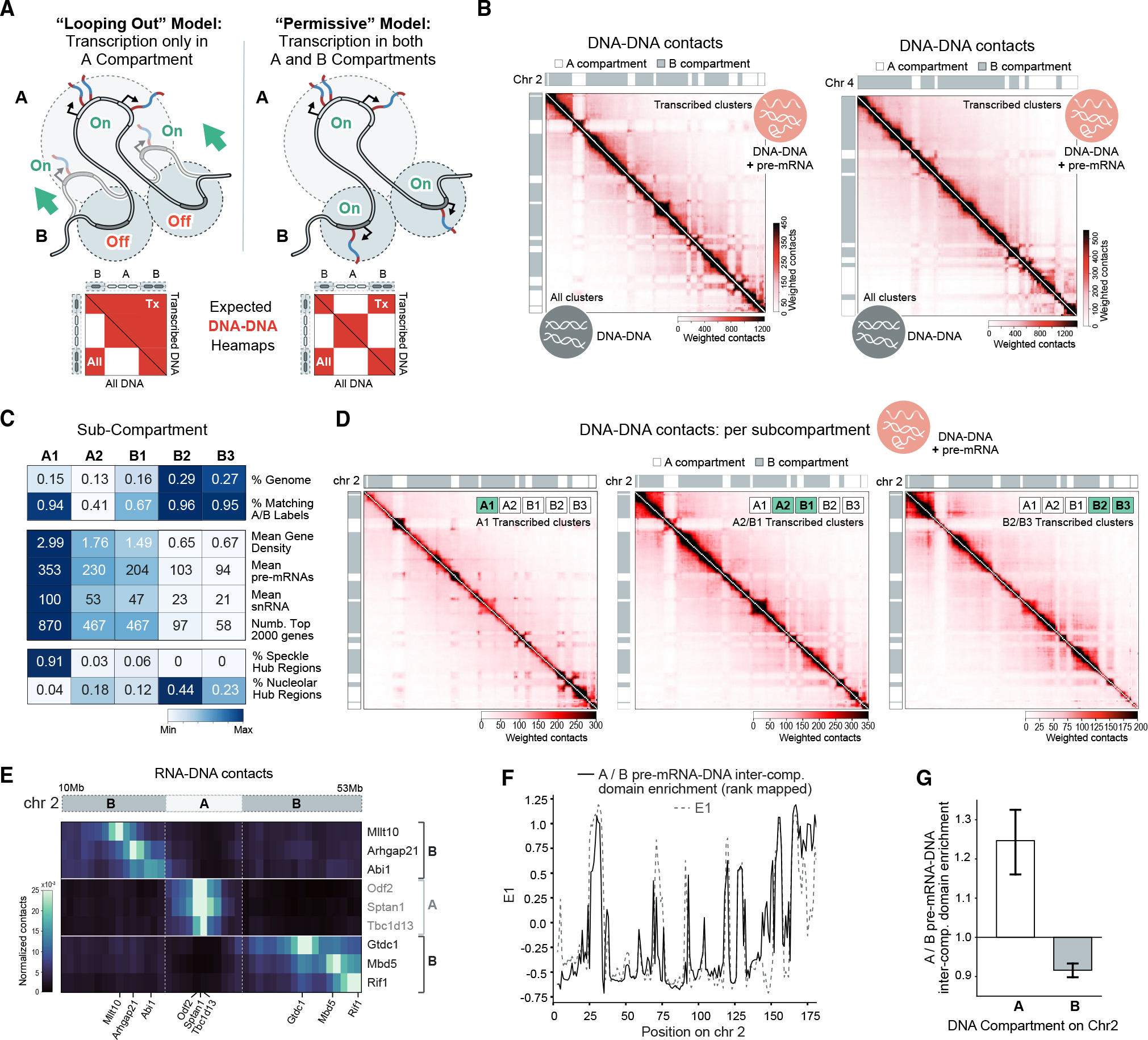
(A) Two models of RNA Pol II gene transcription within A or B compartments and the expected DNA-DNA interaction matrices for actively transcribed loci. The “looping out” model requires B compartment genes to loop into the A compartment to be transcribed, and the corresponding DNA-DNA matrix generated from transcribed DNA regions (left heatmap, upper diagonal) would not have compartment structure, while the heatmap generated from all DNA regions would have compartment structure (lower diagonal). In the “permissive” model, transcription of B compartment genes occurs without a change in genomic structure, and the corresponding DNA-DNA matrix from transcribed DNA regions (right heatmap, upper diagonal) would have A/B compartment structure.
(B) Weighted DNA-DNA interaction heatmaps for SPRITE clusters containing nascent pre-mRNAs (upper diagonal) versus all SPRITE clusters (lower diagonal). Chromosomes 2 and 4 are shown as examples.
(C) Feature profiles of A/B sub-compartments at 100-kilobase (kb) resolution. Characteristics include percentage (%) of genome assigned to each sub-compartment (top), % of 100-kb regions for each sub-compartment matching the corresponding “super-compartment” labels (i.e., A1-A, B1-B) calculated by principal eigenvector analysis of RD-SPRITE (second), mean number of protein-coding genes per 100-kb DNA region (third), mean weighted RNA-DNA contacts of pre-mRNAs per 100-kb DNA region (fourth), mean weighted RNA-DNA contacts of small nuclear RNAs (snRNAs) per 100-kb DNA region (fifth), number of top 2,000 genes within each sub-compartment (sixth), and percentage of speckle or nucleolar hub regions within each sub-compartment (seventh and eighth).
(D) Weighted DNA-DNA interaction heatmaps for actively transcribing SPRITE clusters containing nascent pre-mRNAs of genes in various sub-compartments.
(E) Unweighted RNA-DNA interactions of nascent pre-mRNAs at the B-A-B compartment boundaries near the front end of chromosome 2 (10–53 Mb).
(F) Inter-compartment pre-mRNA-DNA contact enrichment score for A versus B compartment genes (black solid line) and the first eigenvector (E1) (gray dotted line) along chromosome 2. Enrichment scores were rank-remapped to E1 for direct comparison (STAR Methods).
(G) Mean inter-compartment pre-mRNA-DNA enrichment scores for A versus B compartment genes on A (left) or B (right) compartment genomic regions of chromosome 2. Error bars show 95% bootstrapped confidence intervals.
While it is commonly described as a single compartment, the B compartment is in fact heterogeneous. Compartment structures can also be defined using five sub-compartments, three of which (B1, B2, B3) are considered B-like but differ in repressive chromatin modifications, gene density, and nuclear location33,34 (Figure 2C); B2 and B3 are highly enriched for chromatin features associated with transcriptional repression, while B1 has chromatin features more closely resembling the A2 sub-compartment. Because of this, we considered the possibility that our observations of transcription within the B compartment might be restricted to B1. To explore this, we focused on a set of highly expressed nascent pre-mRNAs in RD-SPRITE and found these genes to be located within all three B sub-compartments (Figure 2C, STAR Methods). Focusing specifically on the sub-compartments associated with repressive features (B2 or B3), we measured the DNA organization (DNA-DNA contacts) when pre-mRNAs are actively transcribed. We selected individual SPRITE clusters that contain reads for nascent pre-mRNAs located within B2 or B3 and generated a DNA-DNA heatmap (Figure 2D). We found that actively transcribed genomic regions within these sub-compartments maintain DNA-DNA contacts with other B2 and B3 regions and do not contact neighboring A1 sub-compartment genomic regions. Conversely, when we used clusters containing pre-mRNAs from genes within the A1 sub-compartment to generate a DNA-DNA heatmap, we observed preferential contacts with other A1 regions but not contacts with neighboring B compartment DNA regions. Together, these results demonstrate that active transcription can occur within all B sub-compartments.
To further validate that B compartment structures are observed when B compartment genes are actively transcribed, we explored the DNA contacts of nascent pre-mRNAs. RD-SPRITE detects long-distance RNA-DNA interactions between nascent pre-mRNAs and genomic DNA sites beyond their transcriptional loci (Figure 1E). To investigate the underlying genomic DNA structure during active transcription of B compartment genes, we looked for A/B compartment structures in these long-range RNA-DNA contacts. Indeed, beyond RNA-DNA contacts between pre-mRNAs and their own loci, B compartment pre-mRNAs are enriched for contacts with DNA regions located in neighboring B compartments and depleted for contacts with DNA regions located within A compartments (Figures 2E–2G). Because these nascent RNA transcripts are located near their gene locus, this confirms that genes contained within B compartments do not “loop out” when transcribed.
Together, these results indicate that localization of genes within B compartments does not preclude transcription.
Nascent pre-mRNAs organize within genome-wide structures resembling A/B compartments
We next wondered whether multiple, simultaneously transcribed genes organize together within the B compartment. To explore this, we generated an RNA-RNA contact matrix to measure the genome-wide spatial organization of nascent pre-mRNAs (Figures 3A and 3B). Because the number of observed RNA contacts is dependent on expression level, we focused on the 2,000 most highly expressed genes to ensure high-confidence measurements of individual pre-mRNA contacts (Table S1). These highly expressed genes include those located within both A and B compartments (1,216 A genes and 784 B genes) and display comparable expression levels (Figures S1C and S2A). We sorted these pre-mRNAs by the genomic position of their gene locus and observed clear structural patterns, including the following: (1) preferential contacts between pre-mRNAs that are transcribed from the same chromosome, reminiscent of chromosome territories (Figure 3A), and (2) alternating blocks of highly interacting pre-mRNAs within individual chromosomes, reminiscent of A/B compartments (Figure 3B). In contrast, contact matrices generated between mature mRNAs (exons) do not display preferential contact frequencies based on their genomic positions, consistent with their localization in the cytoplasm (Figures S3A and S3B).
Figure 3. Nascent pre-mRNAs organize within genome-wide structures resembling A/B compartments.
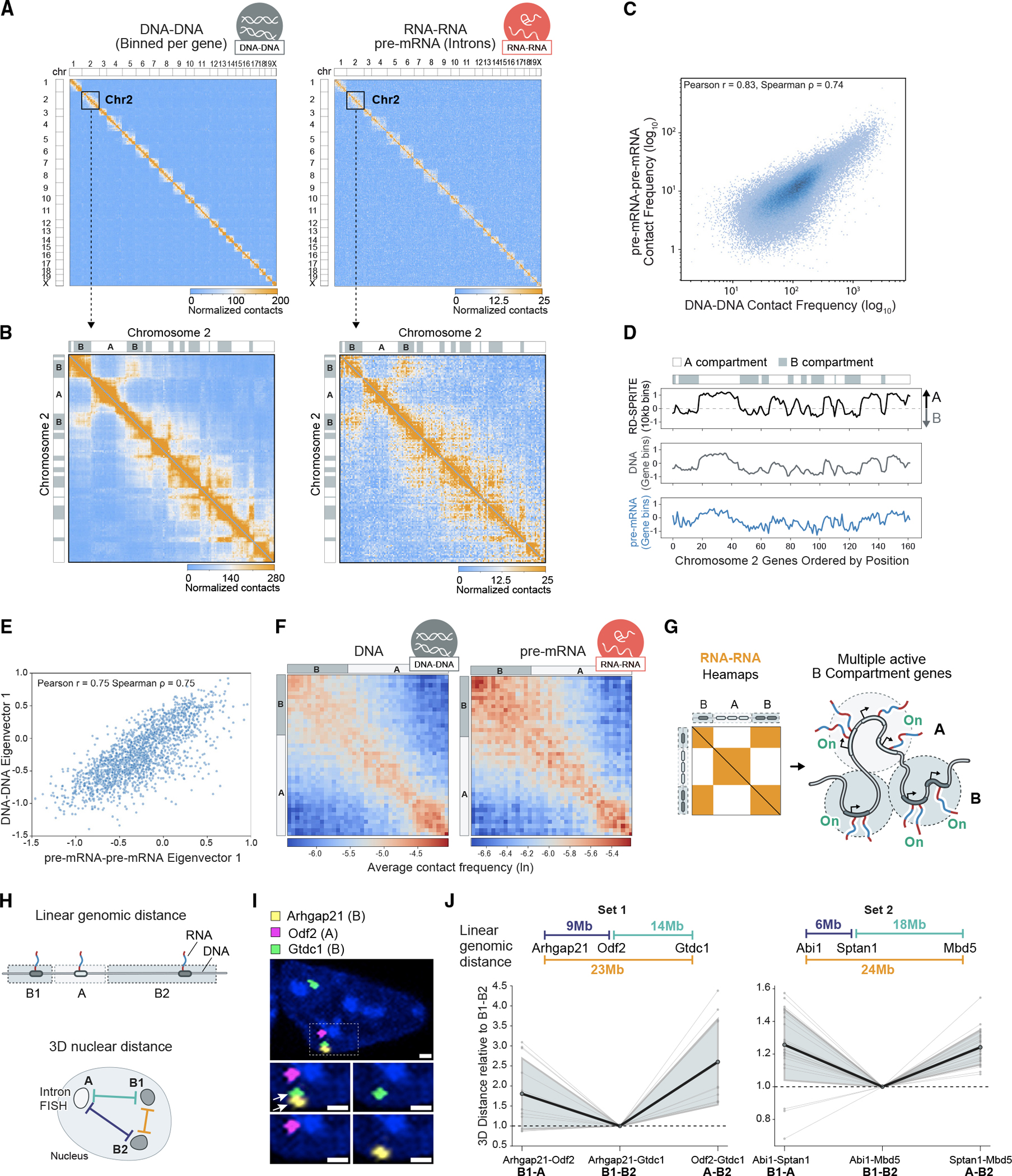
(A) Gene-level DNA-DNA and nascent pre-mRNA RNA-RNA contact matrices. Unweighted RNA-RNA contacts between the top 2,000 expressed pre-mRNAs are shown (STAR Methods). DNA-DNA matrices are binned by the genomic loci of genes used in the RNA-RNA matrix. Genes (and pre-mRNAs) are sorted based on their genomic position.
(B) Zoom-in of gene-level DNA-DNA and nascent pre-mRNA RNA-RNA contact matrices for chromosome 2.
(C) Correlation of genome-wide, intrachromosomal contact frequencies for gene-level DNA-DNA (x axis) and nascent pre-mRNA RNA-RNA (y axis) contact matrices.
(D) Comparison of the first eigenvector (E1) calculated from a genome-wide 10-kb binned DNA-DNA contact matrix (top), gene-level binned DNA-DNA contact matrix (middle), and nascent pre-mRNA RNA-RNA contact matrix (bottom) along chromosome 2. A/B indicator bar along the top shows compartment assignments based on the value of the 10-kb binned E1.
(E) Correlation of E1 calculated from gene-level DNA-DNA (y axis) and nascent pre-mRNA RNA-RNA (x axis) contact matrices.
(F) Saddle plots generated from the gene-level DNA-DNA and nascent pre-mRNA RNA-RNA contact matrices. Plots show the average interactions between groups of genes ordered by their compartment signals calculated from a 10-kb binned DNA-DNA matrix. A/B indicator bars along the axes indicate the compartments of the genes.
(G) Model of RNA Pol II transcription of multiple genes within B compartments and the expected RNA-RNA interaction matrix.
(H) Schematic of intron RNA-FISH design. Nascent transcripts from two B compartment genes located on opposite sides of an A compartment gene in linear genomic space (left) were probed, and the 3D distance between pairs (right) was measured.
(I) Representative microscopy image of intron RNA-FISH for Arhgap21 (B compartment gene), Odf2 (A compartment gene), and Gtdc1 (B compartment gene). Both alleles of Gtdc1 are expressed. Transcripts from a single chromosome are boxed. Arrows highlight the B compartment genes coming together in 3D space. Scale bar represents 1 μm.
(J) Parallel coordinates plot of pairwise 3D distances measured by intron RNA-FISH (B1-A and A-B2). Distances were normalized to the B1-B2 pair distance for each cell to account for differences in cell size. Each gray line indicates a measurement from a single cell, bolded black lines indicate the mean, and shaded gray regions indicate the standard deviation. Probed genes and the linear genomic distances between DNA loci are listed on top of each plot. Measurements are from n = 12 cells containing Arhgap21, Gtdc1, and Odf2 triplets and n = 27 cells containing Abi1, Mbd5, and Sptan1 triplets.
To determine whether these intrachromosomal structural patterns correspond to A/B compartments, we compared them to the 3D structure of their corresponding genomic DNA loci. We generated a DNA-DNA contact matrix for these highly expressed genes (gene-level heatmap) and observed highly similar intrachromosomal patterns in the DNA-DNA and the pre-mRNA RNA-RNA contact maps (Pearson r = 0.83), but not between gene-level DNA and mature mRNA contact maps (Pearson r = 0.04) (Figures 3C and S3C). Next, we defined A/B compartments using nascent RNA-RNA contacts and asked whether their quantitative (eigenvector) values matched those defined using DNA-DNA contacts. First, we ensured that A/B compartment scores based on the gene-level DNA-DNA contacts were similar to those measured across the genome (Pearson r = 0.87, STAR Methods) to confirm that this gene-level analysis is comparable to genome-wide analysis (Figure 3D). Second, we compared the gene-level DNA-DNA eigenvectors to those calculated from the nascent RNA-RNA contact matrix and found a strong correlation (Pearson r = 0.75) (Figure 3E). Finally, we grouped RNA-RNA contacts based on A/B compartment definitions from genomic DNA and found that pre-mRNAs transcribed from B compartments display a high contact frequency with other pre-mRNAs transcribed from B compartments, but not with pre-mRNAs transcribed from loci contained within A compartments, and vice versa (Figure 3F). In contrast, mature mRNAs do not display any preferential interactions between A/B regions (Figures S3D–S3F).
To ensure that the observed compartmentalization of nascent RNAs is not a unique feature of highly expressed genes, we explored compartmentalization properties across mRNAs that span a broad range of expression levels (i.e., the top 10,000 most abundant pre-mRNAs) (Figure S4A) and observed preferential A-A and B-B contacts and depletion of neighboring A-B contacts (Figure S4B). Indeed, zooming in on chromosome 2, we detected clear B-A-B compartment structures, comparable to those measured for the most abundant 2,000 pre-mRNAs, within the RNA-RNA contacts of lower expression genes (Figure S4C). This indicates that the organization of pre-mRNAs within A/B compartments and transcription within the B compartment is observed across a range of expression levels and all classes of transcribed Pol II genes.
These results are consistent with our observations that actively transcribed genes are spatially organized into A/B compartments and that multiple genes are simultaneously transcribed within B compartments (Figure 3G). If transcription only occurred in a single active compartment, we would expect nascent RNAs to globally interact with each other (Figure S3G). Instead, our RNA-RNA heatmaps clearly demonstrate that compartmentalization occurs among nascent transcripts; we observe distinct groups of pre-mRNAs interacting with each other while excluding other nearby transcripts. Importantly, this pre-mRNA compartmentalization, which closely matches the corresponding A/B compartment definitions of DNA, is observed in the RNA-RNA contacts a priori, independent of any compartment calls from DNA-DNA contacts.
To confirm these observations using an orthogonal assay, we performed RNA-fluorescence in situ hybridization (FISH) and measured whether B compartment pre-mRNAs interact more closely with each other than with pre-mRNAs in neighboring A compartments. Specifically, we generated probes against introns of six pre-mRNAs within chromosome 2; each set of three probes corresponded to two mRNAs from distinct B compartments and one from an intervening A compartment (Figure 3H). Consistent with our RD-SPRITE measurements, we find that the pre-mRNAs transcribed from the two B compartments are closer in 3D space than they are to the pre-mRNA transcribed from the A compartment. This occurs even though the two genes in the B compartments (B-B pairs) are farther apart in linear space than the A-B pairs (Figures 3I and 3J).
Together, these results indicate that nascent pre-mRNA transcripts from genes located in both the A and B compartments organize into structures such as chromosomal territories and A/B compartment structures. This highlights the power of RD-SPRITE and its ability to measure long-distance interactions of nascent pre-mRNAs genome-wide to uncover the spatial organization of RNA in the nucleus.
Transcription of RNA Pol II genes can occur in proximity to the nucleolus
We next explored whether Pol II transcription occurs near the nucleolus, a nuclear body that is organized around active transcription and processing of RNA Polymerase I (Pol I) transcribed pre-ribosomal RNAs.35,36 Previous studies have shown that genomic DNA regions positioned near the nucleolus are associated with inactive Pol II transcription and heterochromatin marks.26,27,37,38 However, whether proximity to the nucleolus is simply correlated with inactive transcription or whether organization around the nucleolus precludes Pol II transcription remains unknown.
To explore this, we utilized the ability of RD-SPRITE to measure long-range RNA and DNA organization around the nucleolus (Figure 4A).28,30 We reasoned that if proximity to the nucleolus precludes transcription, DNA regions would loop away when they are transcribed (Figure 4B), and this would result in SPRITE clusters containing nascent pre-mRNAs depleted for nucleolar contacts. In contrast, if transcription can occur near the nucleolus, we would detect preferential contacts between nascent pre-mRNAs of nucleolar-proximal genes and nucleolar RNAs.
Figure 4. Transcription of RNA polymerase II genes can occur in proximity to the nucleolus.
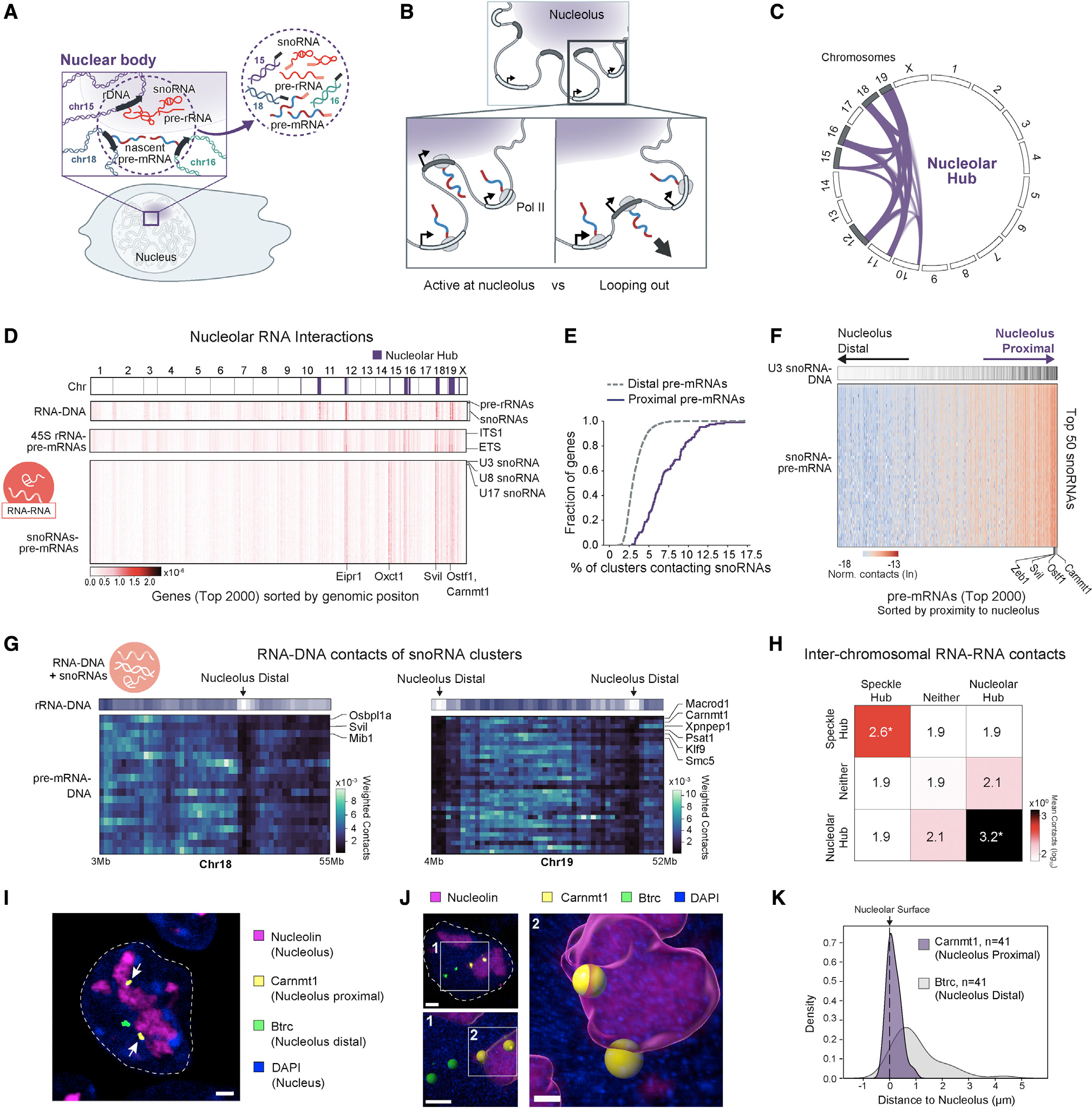
(A) Schematic of the molecular interactions occurring near the nucleolus and their corresponding RNA and DNA interactions measured within an RD-SPRITE cluster (circle). Because RD-SPRITE clusters can capture long-distance interactions, a single cluster can measure multiple interacting RNA (pre-mRNAs) and DNA molecules (genomic loci) around RNA bodies (containing 45S rRNA and snoRNAs).
(B) Two models of RNA Pol II gene transcription for nucleolar-proximal genes.
(C) Diagram of inter-chromosomal DNA contacts between nucleolar hub regions. Chromosomes in gray contain ribosomal DNA genes.
(D) RNA-DNA interactions (top box) corresponding to RNAs known to reside in the nucleolus (y axis; snoRNAs and 45S pre-rRNAs) with genomic loci (x axis; binned per gene) are shown along the top. RNA-RNA interactions (bottom two boxes) between the top 2,000 expressed pre-mRNAs (x axis; STAR Methods) and nucleolar RNAs (y axis; 45S pre-rRNA and snoRNAs) are shown. Genes are ordered along the x axis based on genomic position. Three components of 45S pre-rRNA spacers (ITS1, ITS2, 3′ETS) and the top 50 snoRNAs in descending order by contact frequency are along the y axis. DNA loci within the nucleolar hub are annotated in purple.
(E) Cumulative density of pre-mRNA contacts with snoRNAs for nucleolar proximal (purple) and nucleolar distal (gray) genes.
(F) Nascent pre-mRNA-snoRNA contact matrix for the top 2,000 genes. Genes are ordered based on their distance to the nucleolus, defined by contact frequency of the genomic locus to nucleolar hub regions in RD-SPRITE. Heatmap of U3 snoRNA-DNA density at the genomic bin corresponding to each gene is shown.
(G) Unweighted pre-mRNA-DNA contacts occurring in SPRITE clusters containing snoRNAs for nucleolar genes of chromosome 18 (left) and 19 (right). 45S pre-rRNA density (RNA-DNA contact frequency) is shown as a heatmap, indicating nucleolar close (purple) and far (white) regions.
(H) Average unweighted inter-chromosomal RNA-RNA contacts of the top 2,000 nascent pre-mRNAs grouped by hub (speckle hub, nucleolar hub, neither). *p < 0.01. p values were calculated relative to an expected distribution generated by randomizing RNAs reads across SPRITE clusters and calculating the resulting inter-chromosomal RNA-RNA contact frequency (STAR Methods).
(I) Immunofluorescence (IF) combined with intron RNA-FISH for two genes on chromosome 19: Carnmt1 (yellow), a nucleolar-proximal gene, and Btrc (green), a nucleolar-distal gene. Both alleles of Carnmt1 are transcribed while located adjacent to the nucleolus (Nucleolin; purple). Nucleus is demarcated with DAPI. Arrows highlight the nucleolar-proximal pre-mRNAs located adjacent to nucleoli. Scale bar represents 2 μm.
(J) 3D surface representation of intron RNA-FISH for Carnmt1 (yellow) and Btrc (green) and IF for Nucleolin (purple). Zoom-out (upper left, scale bar represents 2 μm) shows original FISH and IF signals in the entire cell. Zoom-ins show spheres corresponding to FISH signal and nucleolar surface (lower left, scale bar represents 2 μm) and the two transcribed Carnmt1 alleles intersecting the nucleolar surface (right, scale bar represents 0.7 μm).
(K) Distribution of 3D distances between the nucleolar surface and Carnmt1 (purple) or Btrc (gray) nascent transcripts quantified from intron RNA-FISH and IF images (n = 22 cells).
First, we defined the genomic DNA regions proximal to the nucleolus (the “nucleolar hub”) based on DNA contact frequency with nucleolar RNAs, such as 45S pre-ribosomal RNAs (rRNAs) and small nucleolar RNAs (snoRNAs), and inter-chromosomal DNA-DNA contacts (Table S2). We previously showed that these genomic DNA regions are proximal to the nucleolus30 (STAR Methods, Figures 4C and S5A). Next, we analyzed whether nascent pre-mRNAs transcribed from these nucleolar-proximal DNA loci co-occur in SPRITE clusters with snoRNAs, suggesting that they are transcribed when they are physically close to the nucleolus and do not “loop out” during transcription. Indeed, pre-mRNAs from nucleolar-proximal genes display strong enrichment for snoRNA contacts, whereas nascent pre-mRNAs transcribed from nucleolar-distal genes exhibit few snoRNA contacts (Figures 4D and 4E). In fact, the frequency of snoRNA to pre-mRNA contacts was positively correlated with the nucleolar proximity of the pre-mRNA’s genomic locus (Pearson r = 0.75; Figures 4F and S5B), while the transcriptional levels of the pre-mRNAs were not (Pearson r = −0.02; Figure S2B). This suggests that RNA Pol II transcription can occur close to the nucleolus.
To explore the underlying genomic DNA structure of nucleolar-proximal genes when they are actively transcribed, we measured the RNA-DNA contacts for these pre-mRNAs (Figure 4G). We reasoned that if these DNA loci loop away from the nucleolus when they are transcribed, nucleolar proximal pre-mRNAs would exhibit reduced interactions with nucleolar hub DNA regions and increased interactions with neighboring non-nucleolar hub regions. Instead, we observed that nascent pre-mRNAs of these genes frequently contact other nucleolar-proximal DNA regions and are depleted at neighboring non-nucleolar hub regions.
Because our results suggest that genes are transcribed when they are near the nucleolus, we wondered whether multiple actively transcribed genes organize together around the nucleolus. Specifically, we explored if nascent pre-mRNAs of nucleolar-associated genes display preferential inter-chromosomal contacts. To do this, we took the genome-wide inter-chromosomal contact matrix between all nascent pre-mRNAs (Figure 3A) and aggregated the mRNAs into three groups: speckle hub genes, nucleolar hub genes, or neither (Figure 4H, Tables S2 and S3). We observed enrichment of inter-chromosomal contacts between these nucleolar hub nascent pre-mRNAs (p value < 0.01, STAR Methods and Figure S5C), suggesting that the nucleolar hub DNA regions from multiple chromosomes remain organized together in space during transcription and that genes from multiple chromosomes are simultaneously transcribed at the nucleolus.
To confirm this observation, we performed intron RNA-FISH for two genes located on chromosome 19, one within the nucleolar hub (Carnmt1) and one far from it (Btrc, Figure 4I, Videos S1 and S2). Carnmt1 is actively transcribed while positioned adjacent to the nucleolus (Figure 4I). We measured the distance to the nucleolus for 41 alleles of each gene across 22 cells and observed that ~50% (21/41) of Carnmt1 alleles are transcribed within 0.1 μm of the nucleolar surface. Indeed, even when the allele is directly contacting the nucleolus (distance ≤ 0 μm), we observe transcription in ~1/3 of measured Carnmt1 alleles (Figures 4J and 4K). In contrast, we rarely observe Btrc transcribed near the nucleolus, even though it is located on the same chromosome as Carnmt1 (Figure 4K).
Together, these results demonstrate that proximity to the nucleolus does not preclude transcription of Pol II genes.
DISCUSSION
Here, we showed that RD-SPRITE enables simultaneous measurement of 3D DNA structure and nascent RNA transcription to map the DNA contacts of pre-mRNAs, the 3D structure of actively transcribed genomic loci, and the global organization of nascent pre-RNAs. Previously, the question of whether certain nuclear structures are impermissible to transcription was unresolved because we lacked the ability to map DNA and RNA contacts simultaneously at high resolution across the genome. Existing methods are unable to do this because they either focus exclusively on DNA structure (e.g., Hi-C) or map RNA and DNA via proximity ligation (e.g., GRID-seq), which is limited to pairwise interactions and therefore cannot simultaneously measure 3D DNA structure and nascent RNA localization or RNA-RNA interactions between pre-mRNAs. Using RD-SPRITE, we can simultaneously measure RNA-RNA, RNA-DNA, and DNA-DNA contacts genome-wide and therefore generate global profiles of the 3D structures associated with nascent transcripts.
We leveraged these features of RD-SPRITE to show that transcription of genomic DNA occurs within both A and B compartments as well as at genomic DNA regions that are proximal to the nucleolus. Our results demonstrate that DNA does not need to reposition into an “active” compartment in the nucleus to be transcribed and that gene localization within B compartments or near the nucleolus does not preclude Pol II transcription. Furthermore, we found that nascent pre-mRNAs—including those within A compartments, B compartments, and near the nucleolus—localize with other transcripts from their respective nuclear structures; this is reminiscent of DNA organization such as chromosomal territories, A/B compartments, and inter-chromosomal interactions around the nucleolus. While we focused on exploration of inactive compartments, we note that this approach can also be used to explore other structural features and transcription, including enhancer-promoter contacts.
Our findings argue against the “looping out” model whereby active genes need to move out of inactive compartments to contact active compartments when transcribed. Previous evidence for this model came primarily from imaging studies that observed that individual DNA loci can loop away from their chromosome territories when transcriptionally active.18,39 Additional studies using nascent RNA-FISH did not detect chromosome territories or compartment-like structures, suggesting that pre-mRNAs organized within a single active compartment.16 It was therefore postulated that DNA structure detected by Hi-C and similar approaches may capture ensemble DNA organization across a population of cells rather than the organization of DNA loci that are actively transcribed. However, these imaging approaches were not able to simultaneously measure both RNA and DNA with high sensitivity on a genome-wide scale; therefore, their inability to measure these structures likely reflects limited resolution rather than support for this model. Using RD-SPRITE, we can detect the long-range RNA-RNA interactions of lower abundance RNAs, such as nascent pre-mRNAs, which enables us to generate high-depth, genome-wide RNA-RNA contact maps for thousands of RNAs.
Altogether, our results demonstrate that transcription can occur throughout the nucleus, including within regions that have typically been viewed as inactive. Thus, the simple idea of “active” and “inactive” compartments—distinct structural domains within the nucleus that are globally permissive or impermissible for transcription—is likely inaccurate. Consistent with this, previous studies have shown that transcription can occur at genes proximal to the nuclear lamina22,25,40,41 and that Pol II can freely access the inactive X chromosome heterochromatin domain during X chromosome inactivation.42
Because spatial organization does not appear to dictate transcriptional state, arrangement of DNA into A/B compartments likely reflects other features of these genomic regions. Indeed, our study and others9 suggest that transcription is unlikely to be the sole factor driving compartmentalization of the genome. Genomic DNA regions within transcriptionally inert sperm cells9,43,44 as well as cells treated with various transcriptional inhibitors45,46 are still partitioned into A/B compartments. Additionally, A/B compartments can be invariant across cell states, even when undergoing large-scale changes in gene expression.12 One possibility is that these compartments reflect differential gene density: DNA regions contained within A compartments are generally gene dense, whereas those in B compartments are generally gene poor. This would explain why A/B compartments are correlated with transcriptional activity but may not regulate transcription state, because gene-dense regions are more likely to be transcriptionally active. Other possible contributors to compartmentalization include A/T sequence content of the genome, the prevalence of SINE and LINE elements,47 and the replication timing of DNA.48,49 In fact, multiple studies have found that early and late replicating domains correspond to A and B compartments, respectively.48,50 Yet another possibility is that these compartments reflect patterns of histone modifications. While the precise features that drive compartment organization are unknown, our results suggest that these compartments do not define transcriptional state, and additional work is needed to understand what role, if any, spatial compartments play in gene regulation.
Limitations of the study
Because RD-SPRITE does not quantify absolute physical 3D distances, our results do not directly measure how close actively transcribed genes are to each other or from structures such as the nucleolus. Instead, we assess the relative proximity between molecules by calculating contact frequency. While our results suggest that transcription can occur near the nucleolus, the measurements cannot determine whether transcription is occurring directly within—or at a defined distance from—the nucleolus. However, we have validated our observations for representative loci using FISH and find strong concordance between distances measured by microscopy and SPRITE data (here and in Quinodoz et al., 201830).
While our data suggest that large-scale, global repositioning of B compartment genomic regions into an A compartment is not required for transcription, we cannot exclude the possibility that small scale, local structural reorganization may occur (e.g., promoter regions loop out and contact each other21) or that individual genes may relocate upon transcription. For instance, certain transcribed nucleolar genes may be located further from the nucleolus than their inactive counterparts but remain within the B compartment and in proximity to the nucleolus. Alternatively, multiple B compartment genes may undergo local structural changes to organize together when transcribed, while remaining distinct from other active A compartment genes. While these questions remain to be addressed, our data clearly indicate that genes remain compartmentalized when transcribed and do not reorganize into a single active compartment.
Finally, our study focused on mouse ES cells, and therefore we cannot exclude the possibility that other cell types might display distinct properties. Furthermore, it remains possible that other spatial compartments in the nucleus that have not yet been studied might preclude Pol II transcription. Future work will be needed to comprehensively map transcriptional states and 3D genome structure in other cell types and extend these observations to additional cell types and nuclear compartments.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for further information, resources, and reagents should be directed to and will be fulfilled by the lead contact, Mitchell Guttman (mguttman@caltech.edu).
Materials availability
This study did not generate new unique materials.
Data and code availability
This paper analyzes existing, publicly available RD-SPRITE data (GEO: GSE151515). PolyA RNA-seq data generated in this work has been deposited at GEO: GSE211287 and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit polyclonal anti-Nucleolin | Abcam | Cat# ab22758; RRID:AB_776878 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Doxycycline | Sigma | D9891 |
|
| ||
| Deposited data | ||
|
| ||
| SPRITE data | Quinodoz et al.28 | GEO:GSE151515 |
| Ribo-Depleted RNA RNA-seq data in mESC | Sigova et al.31 | GEO:GSM903663 |
| PolyA RNA-seq data in mESC | This Study | GEO: GSE211287 |
| GRO-seq data in mES cells | Jonkers et al.32 | GEO:GSE48895 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse: pSM44 ES cell line | This Study | pSM44 (dox-inducible Xist) |
|
| ||
| Software and algorithms | ||
|
| ||
| SPRITE pipeline 2.0 (v0.2) | This Study | https://github.com/GuttmanLab/sprite2.0-pipeline |
| Bowtie2 (v2.3.5) | Langmead and Salzberg51 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Hisat (v2.1.0) | Kim et al.52 | http://www.ccb.jhu.edu/software/hisat/index.shtml |
| Samtools (v1.4) | Li et al.53 | http://samtools.sourceforge.net/ |
| Bedtools (v2.30.0) | Quinlan and Hall54 | https://bedtools.readthedocs.io/en/latest/ |
| Trim Galore! (v0.6.2) | Felix Krueger (The Babraham Institute) | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Subread (v2.0.3) | Liao et al.55,56 | http://subread.sourceforge.net/ |
| Cooler (v0.8.5) | Abdennur and Mirny57 | https://github.com/open2c/cooler |
| Cooltools (v0.4.1) | 10.5281/zenodo.5214125 | https://github.com/open2c/cooltools |
| Scipy (v1.7.1) | Virtanen et al.58 | https://scipy.org/ |
The original code for the SPRITE analysis pipeline is available on Github at https://github.com/GuttmanLab/sprite2.0-pipeline. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines used in this study
We used the following cell line in this study: Female ES cells (pSM44 ES cell line) derived from a 129 × castaneous F1 mouse cross. These cells express Xist from the endogenous locus under control of a tetracycline-inducible promoter. The dox-inducible Xist gene is present on the 129 allele, enabling allele-specific analysis of Xist induction and X chromosome silencing.
Cell culture conditions
All mouse ES cells were grown at 37°C under 7% CO2 on plates coated with 0.2% gelatin (Sigma, G1393-100ML) and 1.75 μg/mL laminin (Life Technologies Corporation, #23017015) in serum-free 2i/LIF media composed as follows: 1:1 mix of DMEM/F-12 (GIBCO) and Neurobasal (GIBCO) supplemented with 1x N2 (GIBCO), 0.5x B-27 (GIBCO 17504-044), 2 mg/mL bovine insulin (Sigma), 1.37 μg/mL progesterone (Sigma), 5 mg/mL BSA Fraction V (GIBCO), 0.1 mM 2-mercaptoethanol (Sigma), 5 ng/mL murine LIF (GlobalStem), 0.125 μM PD0325901 (SelleckChem) and 0.375 μM CHIR99021 (SelleckChem). 2i inhibitors were added fresh with each medium change. Medium was replaced every 2448 h depending on culture density, and cells were passaged every 72 h using 0.025% Trypsin (Life Technologies) supplemented with 1mM EDTA and chicken serum (1/100 diluted; Sigma), rinsing dissociated cells from the plates with DMEM/F12 containing 0.038% BSA Fraction V.
METHODS DETAILS
RD-SPRITE dataset and computational pipeline
The RNA-DNA SPRITE dataset was previously generated in female PSM44 mouse embryonic stem cells treated for 24hr with doxycycline for induction of Xist expression and can be found under the GEO accession number GSE151515.28 RD-SPRITE data processing pipeline details were described in https://github.com/GuttmanLab/sprite2.0-pipeline. The final pipeline output is a cluster file containing reads grouped into SPRITE clusters based on shared SPRITE barcode sequences. Each SPRITE cluster may contain repeat-masked DNA reads aligned to mm10, RNA reads annotated with Gencode vM25 gene annotations and repeatmasker annotations and/or repeat RNA reads aligned to a custom genome of repeat RNAs.
Gene identification was improved to avoid mis-annotations of genes. Intronic RNA reads were defined as RPM-containing reads that aligned to the genome and were uniquely annotated as the intron of a protein-coding gene. Similarly, exonic RNA reads were defined as RPM-containing reads that aligned to the genome and were uniquely annotated as the exon of a protein-coding gene. For example, mRNA reads overlapping with snoRNAs or other repetitive sequences were excluded. Unless stated otherwise, all analyses were based on SPRITE clusters of size 2–1000 reads. These cluster sizes were chosen to be consistent with the analysis in our previous papers, where we showed that many known structures such as TADs, compartments, RNA-DNA and RNA-RNA interactions, etc., occur within SPRITE clusters containing 2–1000 reads.28,30 We also previously showed that the A/B compartments identified in these cluster sizes are highly correlated with those observed by Hi-C.
RD-SPRITE data analysis
Exon and intron enrichment scores
A “contact enrichment score” was devised to compare the contact profiles of intronic and exonic RNA reads. Specifically, the frequency of an intronic or exonic read co-occurring in the same SPRITE cluster as another molecular species - such as chromatin (DNA reads, defined by DPM tags), small nuclear RNAs (U1, U2) or ribosomal RNAs (18S, 28S) was calculated. Contact scores were generated by sorting all clusters with a gene of interest based on whether they also contained the other molecule of interest. The contact and no-contact frequency scores were computed by taking the sum of 1/(cluster size) for all clusters with or without the other molecule, respectively. Summing 1/(cluster size) accounts for contact distance - larger clusters indicate further contact distances and are proportionally down-weighted. A final contact score for a gene is the ratio of contact to no-contact frequency scores. This procedure was performed for intronic and exonic reads. Enrichment was computed by normalizing contact scores to the median contact score of all intronic and exonic contact scores. Outlier genes with very few annotated exonic or intronic reads had extreme scores that were not necessarily representative of intronic or exonic reads as a class. These were removed by setting a minimum contact and no contact threshold of two clusters in each category for a given gene. The median 90% of intronic and exonic contact enrichment scores were plotted as violin plots using the python plotting package seaborn.
Locus enrichment scores
To map the genome-wide localization profiles of intronic or exonic RNA reads relative to their genomic locus, the contact frequency between RNA transcripts (intronic or exonic reads) and each region of the genome (binned at 1Mb resolution) was calculated. 1Mb resolution was selected for this analysis such that most gene loci were located within only a single genomic bin (genomic locus bin). The raw contact frequency was defined as the number of SPRITE clusters in which a specific RNA transcript read (intronic or exonic) and a given genomic bin co-occur. The normalized contact frequency was calculated by weighting each SPRITE cluster by a scaling factor proportional to its size (2/n, where n is the total number of reads in the cluster). The normalized contact frequency profiles for intronic and exonic reads of each gene were summed over all genes, with the contact profiles centered on the genomic locus bin +/− 10Mb. The contact profiles for antisense (- strand genes) were reversed before summing to account for gene orientation.
Expression correlations
Expression levels for each gene were calculated by counting the number of annotated intronic RNA reads and/or annotated exonic RNA reads in SPRITE clusters. The intronic expression levels were compared to GRO-Seq expression levels, from NCBI GEO (GSE48895 accession).32 The exonic expression levels were compared to a polyA-selected RNA-seq library generated from PSM44 after 24hr dox induction (the identical cell line/conditions used for RD-SPRITE). The total expression levels were compared to Ribo-depleted RNA-seq expression levels from GEO Accession: GSM903663.31
Selecting the top 2000 introns
The top 2000 pre-mRNAs with highest coverage in the RD-SPRITE dataset were determined by counting the number of RNA reads in SPRITE clusters of size 2–1000 that were annotated as an intron of each protein-coding gene. To remove potential mis-annotations or non-representative genes, genes with transcript lengths greater than 1 Mb were filtered out. Additionally, to remove redundant or overlapping annotations, genes with intersecting annotations or annotations within 1000 bp of each other were filtered, keeping only the most abundant. This second filter allowed for any DNA locus to be uniquely assigned to only a single gene. From the final filtered list, the top 2000 pre-mRNA genes were selected.
DNA-DNA contact matrices
DNA-DNA contact matrices were generated from all SPRITE clusters of total size 2–1000 reads. Raw contact frequency was calculated at 1Mb resolution by counting the number of SPRITE clusters in which pairs of genomic bins co-occur. 1Mb resolution was selected for DNA-DNA heatmaps to enable visualization of genome-wide patterns (ie. chromosomal territories, A/B compartments). Normalized contact frequency was calculated by dividing each genomic contact by a scaling factor proportional to SPRITE cluster size (specifically, n/2 where n is the total number of reads in the SPRITE cluster).29 Normalized contact frequency maps were corrected using ICE normalization, a matrix balancing algorithm commonly used for correcting Hi-C contact maps using CoolTools.59
To analyze the 3D structure associated with a single active gene locus (Figure 1F), DNA-DNA contact maps were generated from a subset of SPRITE clusters that contained an intronic RNA transcript of the gene of interest. Cluster size normalized contact maps were generated at 100kb resolution by mapping the interactions between all pairs of DNA within these clusters, as described above. 100kb resolution was selected to allow for visualization of contact enrichment at the gene-locus level. These contact frequency maps were corrected using a modified ICE normalization strategy. Specifically, because cluster subsets may be enriched or depleted for certain genomic regions or contacts, the assumptions for typical ICE normalization of a matrix do not apply. To correct these matrices for any genome coverage bias present in the entire SPRITE dataset, ICE bias factors from DNA-DNA contact matrices generated with all clusters were applied to the matrices generated from cluster subsets.
To analyze the 3D-structure associated with nascent transcription for all active regions or sets of genes (e.g. A1, A2, B1, B2, etc.), DNA-DNA contact maps were generated from a subset of SPRITE clusters that contained a specific set of RNA transcripts. In the case of mapping all active regions, SPRITE clusters that had DNA reads and at least one RNA read annotated as the intron of a protein coding gene were selected. Cluster size normalized contact maps were generated at 1Mb resolution from this subset of clusters, as described above. 1Mb resolution was selected to enable visualization of genome-wide structural patterns (ie. chromosomal territories and A/B compartments). These contact maps were ICE normalized using the modified ICE normalization strategy for cluster subsets, as described above.
Genome-wide eigenvector calculations for A/B compartment identification
Genome-wide eigenvectors were calculated from SPRITE DNA-DNA contact maps to define reference A/B compartments. First, SPRITE clusters of sizes 2–1000 DNA reads were converted to a cooler format, a standard format for HiC interaction data, using the ‘cloud-pairs” function of cooler.57 The pairs of contacts within SPRITE clusters were individually written out and weighted by the n/2 scaling factor, where n is the number of DNA reads in the cluster. Next, cooler files were generated at various resolutions (10kb, 100kb, 1Mb) using the cooler function “coursen” and matrix balancing weights were calculated using the cooler function “balance”. Finally, eigenvectors were calculated at these resolutions using the HiC analysis software cooltools. 1Mb resolution eigenvectors were used to define A/B compartment domains for genome-wide or chromosome-wide analysis; 100kb resolution eigenvectors were used to match sub compartment resolution (see below); 10kb resolution eigenvectors were used to define compartments on a gene-resolution level and assign individual genes to either compartment.
RNA-DNA contact maps
Genome-wide localization profiles were generated for individual pre-mRNAs by calculating the contact frequency of intronic RNA reads for that gene and genomic DNA binned at various resolutions (10Mb, 1Mb, 100kb). A range of DNA binning resolutions was used to measure contacts occurring at different size scales – i.e., gene-locus specific contacts (100kb), intra-compartment contacts (1Mb) and long-range, chromosome-wide contacts (10Mb). Raw contact frequency was calculated by counting the number of SPRITE clusters in which an intronic RNA read and a DNA read, mapped to its corresponding genomic bin, co-occurred. Weighted contact frequency was calculated by scaling raw contacts with a scaling factor proportional to cluster size, as described for DNA-DNA contact matrices. To account for differences in gene expression when comparing RNA-DNA localization profiles across genes, the genome-wide RNA-DNA contacts for each gene were normalized to one.
Aggregate inter-compartment domain RNA-DNA contact frequencies were computed using the unweighted RNA-DNA contact profiles of the top 2000 genes at 1Mb genomic bin resolution. 1Mb resolution was selected to define A/B compartment domains and resolve compartment boundaries. First, for each gene, the A/B compartment domain containing the gene locus was masked. Then, the inter-compartment domain contacts for all A compartment genes and all B compartment genes were summed separately. To account for the difference in number of A and B compartment genes, the aggregated contact frequencies were normalized to their respective medians on a per-chromosome basis. Finally, the ratio of A-to-B frequency was used to generate an enrichment score. When the ratio is > 1, the inter-compartment domain contact frequency with A genes is higher; when the ratio is < 1, the inter-compartment domain contact frequency with B genes is higher. This enrichment score was further aggregated across all A compartment regions and all B compartment regions on chromosome 2. To compare the magnitude of the enrichment score to the magnitude of eigenvector 1 (E1), the enrichment score along chromosome 2 was plotted after rank re-mapping to E1; all enrichment score values and all eigenvector values along chromosome 2 were ordered each from greatest to least. The top enrichment score was assigned the value of the top eigenvector, the second highest enrichment score was assigned the value of the second highest eigenvector and so forth.
RNA-RNA contact matrices
RNA-RNA contact matrices were generated by computing the contact frequency between RNA-RNA pairs. Contact frequency was defined as the number of SPRITE clusters containing both transcripts. RNA-RNA contacts were not weighted by cluster size.
For all of the top 2000 genes, their intronic or exonic RNA transcripts were used to generate RNA-RNA contact matrices between nascent pre-mRNAs or mature mRNAs, respectively. Matrices were ordered by the genomic position of these genes and normalized using ICE normalization (also used for matrix balancing HiC data) to account for differences in RNA expression.
DNA-DNA contact matrices by gene
DNA-DNA contact matrices were generated on a gene-level to directly compare to the corresponding RNA-RNA matrices of the same genes. Specifically, instead of calculating the contact frequency between genomic bins of DNA, raw frequencies were calculated by annotating each DNA fragment with its respective gene locus (similar to RNA annotations but ignoring strand) and counting the number of SPRITE clusters containing pairs of interacting gene loci. These matrices were normalized using ICE normalization.
Eigenvector calculations for gene-binned contact maps
Eigenvectors were calculated from RNA-RNA contact matrices or gene-resolution DNA-DNA contact matrices using these same HiC analysis software packages to define A/B compartments (see above). Pre-computed, raw contact matrices were converted into a cooler format by assigning the contacts of each gene to the 10kb genomic bin located in the center of the gene annotation. Coolers were balanced using the cooler function “balance”. Finally, eigenvectors were calculated using the HiC analysis software cooltools. Signs of eigenvectors for individual chromosomes were matched to the eigenvectors calculated from the entire genome.
Saddle plots
Saddle plots were generated from the RNA-RNA and gene-resolution DNA-DNA contact matrices using cooltools. To enable direct comparisons, the ordering of genes was the same for all saddle plots. The genes were sorted and grouped into 40 bins based on eigenvector 1 (E1) of their genomic positions from the genome-wide eigenvector calculation using RD-SPRITE. The RNA-RNA and gene-resolution DNA-DNA contact matrices were then aggregated based on these groups. The total interaction sum and count were used to calculate an average contact frequency per group.
RNA-RNA contacts for gene expression levels
Genes were grouped into five expression levels of 2000 genes each based on pre-mRNA abundance in RD-SPRITE clusters. Specifically, genes were ordered by number of associated intron-containing RNA reads and grouped into 0–1999, 2000–3999, 4000–5999, 6000–7999 and 8000–9999, with the 0–1999 group containing the most abundant pre-mRNA genes. For each group, RNA-RNA contacts were mapped between pre-mRNAs as previously described. Individual gene-based contacts were collapsed into A and B compartment contacts for each chromosome. A and B compartments for each gene were assigned based on E1 calculated at 10kb resolution from RD-SPRITE data, as described above. The genome-wide, by-chromosome A/B compartment contact matrix was normalized using ICE normalization. Finally, the total intrachromosomal A-A, A-B and B-B contacts were calculated and displayed as a 2-by-2 matrix. For the top three expression levels (0–1999, 2000–3999, 4000–5999), the gene-based RNA-RNA contacts were additionally collapsed into contiguous domains of A and B along each chromosome (instead of one A and one B group per chromosome). The resulting contact matrix was normalized using ICE normalization. A similar domain level analysis was not performed for the lowest expression levels because of sparsity in pre-mRNA reads.
Sub-compartment analysis
Annotations for the A/B sub-compartments in mouse embryonic stem cells were kindly provided by the Jian Ma laboratory at Carnegie Mellon University. These subcompartment annotations were only used for the analyses shown in Figures 2C and 2D. Sub-compartment annotations were based on a 5 state Gaussian Hidden Markov Model and were at 100 kb resolution. The features of the sub-compartments were profiled using the RD-SPRITE dataset. Specifically, A/B compartment labels, assigned based on the principal eigenvector calculated at 100 kb resolution using RD-SPRITE, were compared to the A1/A2/B1/B2/B3 and the number of mismatched compartments (e.g. A compartment with B1/B2/B3 sub-compartment) was calculated. Next, the weighted RNA-DNA contacts of nascent pre-mRNAs or of small nuclear RNAs (U1, U2) across all regions of a single sub-compartment annotation were averaged. To determine whether genes in all sub-compartments were expressed, the top 2000 pre-mRNA genes were assigned to their respective sub-compartments. If a gene annotation intersected multiple sub-compartments, it was assigned to the sub-compartment with maximum representation (ie. most covered base pairs); each gene could only be counted for one sub-compartment. Chromosome X genes were excluded from this analysis because of the lack of sub-compartment assignments.
Weighted DNA-DNA contact matrices for transcribing regions containing pre-mRNAs from each sub-compartment were generated as described above. Specifically, a subset of SPRITE clusters containing intronic RNA transcripts of genes located in a given sub-compartment were selected. Then, the DNA-DNA contact frequency from this cluster subset was calculated. Any of the top 2000 genes were assigned to a given sub-compartment if a portion of the gene annotation intersected the given sub-compartment. Thus, genes could be included in more than one sub-compartment for this analysis.
Nucleolar and speckle hub definition
Nucleolar and Speckle Hubs were previously defined using inter-chromosomal contacts of DNA SPRITE at 1Mb resolution in mES cells.30 Briefly, it was found that two, mutually-exclusive sets of DNA regions showed enriched inter-chromosomal contacts within a set but not with DNA regions of the other set of interacting loci.
To improve these annotations in this manuscript, the hubs were recalculated using the RD-SPRITE dataset and including RNA enrichment information. Instead of all SPRITE clusters being included to generate a DNA-DNA heatmap and measure inter-chromosomal contact enrichment, we only used SPRITE clusters containing known RNAs functionally associated with the respective nuclear body (nucleolus/speckles) being mapped. For the nucleolar hub, clusters were selected using small nucleolar RNAs (snoRNAs) or pre-rRNAs (45s rRNA); for the speckle hub, clusters were selected using small nuclear RNAs (e.g. U1, U2, or other snRNA ‘biotype’ genes). DNA-DNA contact frequency was calculated at 1Mb resolution (the same resolution as used for the original hub definition) from these cluster subsets and was not weighted by cluster size, in order to maximize the information from larger clusters which we have found are enriched for interactions around nuclear bodies.30 The resulting raw heatmaps were balanced using the ICE bias factors of the DNA-DNA heatmap calculated using all clusters, as described above. Inter-chromosomal contacts were hierarchically clustered using the python package g.cluster.hierarchy.58 Hierarchical clustering was converted into flat clusters using the fcluster function. Upon clustering, a single cluster of genomic bins nearly matching the previously annotated “inactive” hub (for the snoRNA/pre-rRNA workup) or “active” hub (for the snRNA workup) was apparent. These genomic bins within these clusters were redefined as the “Nucleolar” Hub and “Speckle” Hub.
Distance to nucleolus using SPRITE contacts
Genes located within the nucleolar hub annotations are defined as “nucleolus proximal” while genes located outside are defined as “nucleolus distal”.
Additionally, a continuous metric for distance to nucleolus was generated using DNA-DNA contact frequencies. For each 1Mb bin of the genome, the total inter-chromosomal contact frequency with nucleolar hub DNA region was calculated and genes were assigned the distance of the 1Mb genomic bin in which they are located.
snoRNA-RNA and pre-rRNA to pre-mRNA contacts
For each of the top 2000 genes, the contact frequency between nascent pre-mRNAs and 2 sets of nucleolar hub RNAs, defined as individual snoRNAs or the three components of 45S pre-rRNA (ITS1, ITS2, 3’ETS), was calculated by counting the number of SPRITE clusters containing both an intronic read and a snoRNA/pre-rRNA read. A heatmap of snoRNA-RNA contacts was generated using only the top 50 snoRNAs with the highest contact frequency to the set of top 2000 genes. To account for differences in gene expression, the contacts for each gene were normalized to the total number of intronic reads for that gene (independent of contact with snoRNAs). To account for differences in snoRNA abundance, the total contacts of each snoRNA with the set of top 2000 genes was normalized to 1. The contact matrix between pre-rRNA and pre-mRNAs was similarly normalized.
Contact matrices were ordered in two ways: by genomic position and by distance to nucleolus. In both cases, snoRNAs/pre-rRNAs are ordered along the y axis with pre-rRNAs on top, followed by snoRNAs in the order from most frequently to least frequently contacting the set of genes.
snoRNA-DNA and pre-rRNA to DNA contacts at Gene level
For comparison to the snoRNA-RNA and pre-rRNA-RNA contact matrices, the snoRNA-DNA and pre-rRNA-DNA contacts per gene were generated. Raw contact frequencies were calculated by counting the number of clusters in which a specific snoRNA or pre-rRNA and a DNA read overlapping a gene annotation co-occur. To account for biases in DNA coverage, the raw frequencies per gene were divided by the total DNA coverage of that gene locus. To account for differences in snoRNA or pre-RNA abundance, the total contact of each individual RNA was normalized to 1.
RNA-DNA contacts for snoRNA containing clusters
DNA localization profiles for nascent pre-mRNAs of nucleolar genes were calculated using a subset of clusters containing snoRNAs. Specifically, we selected clusters that contained the top 100 snoRNAs and mapped the RNA-DNA contacts within these clusters. We calculated the contact frequency between intronic RNA reads of nucleolar proximal genes and genomic DNA binned at 1Mb. 1Mb resolution was selected because we previously defined nucleolar hub regions at this resolution.30 Raw contact frequencies and normalized contact profiles were generated as described in the RNA-DNA contact maps section above.
RNA-seq experiments and data processing
PolyA-selected RNA-seq of mES cells
RNA-seq libraries of dox-induced PSM44 mouse embryonic stem cells (mES cells) were prepared using a double poly-A selection step prior to RNA library preparation (described in the Guttman Lab CLAP protocol; https://guttmanlab.caltech.edu/files/2021/08/CLAPprotocol_combined_word.pdf). Specifically, NEBNext Magnetic Oligo d(T)25 Beads (NEB, S1419S) were prepared by washing twice with RNA binding buffer (50 mM HEPES pH 7.5, 1000 mM LiCl, 2.5 mM EDTA, 0.1% Triton X-100). Total RNA was diluted in HEPES buffer, heated to 65°C for 5 min and then cooled to 4°C to denature RNA. Prepared Oligo dT(25) beads were mixed with denatured RNA and incubated at room temperature for 10 min to allow for RNA binding. These beads were then washed twice with RNA Wash Buffer (50 mM HEPES pH 7.5, 300 mM LiCl, 2.5 mM EDTA, 0.1% Triton X-100). Polyadenylated RNA was eluted from beads by heating to 80°C for 2 min in HEPES Elution Buffer (5mM HEPES pH 7.5, 1.0 mM EDTA), followed by a hold at 25°C. This capture step (bind, wash and elute) was repeated, for two total capture steps, using the same Oligo dT beads. Specifically, to re-capture the polyA-selected RNA, RNA binding buffer was added to the mixture of beads and eluted RNA and the mixture was incubated at RT for 10 min. These beads were then washed twice with RNA Wash Buffer. The final selected polyA transcripts were eluted in HEPES Elution buffer by heating to 80°C for 2 min and holding at 25°C. The eluted beads were immediately placed on a magnet until the solution cleared and the cleared solution was transferred to a new tube. cDNA generation and library prep were performed as described in the Guttman Lab CLAP protocol after this.
Data processing and read annotation
Libraries were sequenced on a HiSeq 2500 (90 cycle × 125 cycle). Adapters were trimmed from raw paired-end fastq files using Trimmomatic v0.38. Trimmed reads were then aligned to GRCm38.p6 with the Ensembl GRCm38 v95 gene model annotation using Hisat2 v2.1.01 with a high penalty for soft-clipping (–sp 1000,1000) and excluding mixed or discordant alignments (–no-mixed –no-discordant). Unmapped reads and reads with a low MapQ score (samtools view -bq 20) were filtered out. Mapped reads were annotated with the featureCounts tool from the subread package v1.6.455,56 using the Gencode release M25 annotations for GRCm38.p6 and a subset of the Repeat and Transposable element annotation from the Hammel lab, identical to the annotation strategy for genome-aligned RNA reads of RD-SPRITE. Reads that received a single annotation for a protein coding gene were counted and correlated with intronic read counts from RD-SPRITE.
Microscopy experiments
Intron RNA fluorescence in situ (RNA-FISH)
RNA-FISH experiments were performed with ViewRNA ISH Cell Assay (ThermoFisher, QVC0001) protocol following manufacturer instructions with minor modifications.28,60 First, pSM44 mES cells were fixed on coverslips with 4% formaldehyde in PBS for 15 min at room temperature followed by permeabilization 0.5% Triton X-100 in 1x PBS (RNAse-free) for 10 min at room temperature. Then, coverslips with cells were washed twice with 1x PBS (RNAse-free) and either dehydrated with 70% ethanol and stored for up to one week at −20C or used directly for the next step. Next, coverslips were washed one more time with 1x PBS and incubated with the desired combination of RNA FISH probes (custom probe design from Affymetrix) in Probe Set Diluent at 40°C for at least three hours. Coverslips were then rinsed once with 1x PBS, twice with Wash Buffer for 10 min, and rinsed once more with PBS before incubating in PreAmplifier Mix Solution at 40°C for 45 min. This step was repeated for the Amplifier Mix Solution and Label Probe Solution. After all three steps of amplification were performed followed by washes, coverslips were incubated with 1x DAPI in PBS at room temperature for 15 min and subsequently mounted onto glass slides using ProLong Gold with DAPI (Invitrogen, P36935).
RNA-FISH & immunofluorescence
For IF combined with in situ RNA visualization, the ViewRNA Cell Plus (Thermo Fisher Scientific, 88-19000-99) kit was used following the RNA-FISH part of protocol from above with minor modifications. First, pSM44 mES cells were fixed on coverslips with 4% formaldehyde in PBS for 15 min at room temperature followed by permeabilization with 0.5% Triton X-100 in 1x PBS for 10 min at room temperature. Next, immunostaining was performed starting with two washes of coverslips with 1x PBS (RNAse-free) and blocking with blocking buffer (kit) with addition of RNAse inhibitor (kit) for 30 min. Then, coverslips were incubated with primary antibody for 3 h at room temperature in a blocking buffer with RNAse inhibitor (anti-Nucleolin Abcam Cat# ab22758, RRID:AB 776878, 1:500). After incubation, cells were washed 3 times in 1x PBS (RNAse-free) and incubated for 1 h at room temperature with secondary antibody labeled with Alexa fluorophores (Invitrogen, Alexa 555) diluted in 1x PBS (1:500). Next, coverslips were washed three times in 1x PBS (RNAse-free) and RNA-FISH protocol was performed starting with probe incubation step (described above). After the final wash, coverslips were rinsed in ddH2O, mounted with ProLong Gold with DAPI (Invitrogen, P36935), and stored at 4°C until acquisition.
Image quantification and analysis
RNA-FISH only images were acquired with Zeiss LSM 800 with the 63× oil objective and collected every 0.3 μm for 16 Z-stacks, IF/RNA-FISH images were acquired with Zeiss LSM 980 with the 63× oil objective and collected every 0.3 μm for 16 Z-stacks.
Image analysis was performed using an Icy (v2.3) software followed by custom written python script for x, y, z euclidean distance measurements. Briefly, a region of interest corresponding to each nucleus was determined using DAPI staining. Next, in each nucleus, intron spots were indentified based on a local intensity threshold. Only nuclei with at least one spot for each target probed and a maximum of two spots per individual target (corresponding to the individual alleles) were kept for further analysis; nuclei that did not meet criteria were discarded. Then, the x, y, z position of each intron spot was determined and used to calculate euclidean distance between all possible pairs of gene alleles. Using this matrix of interactions, nuclei were selected for further analysis only if they contained one or two full sets of triplet alleles (B-A-B) and each pair of alleles within the triplet was in a proximity of less than 20 units. This allows us to focus on triplets of genes that come from the same allele.
Imaris software v8 from Bitplane (Oxford Instruments Company) was used to visualize Nucleolin and intron-RNA-FISH localization. Distances were measured from the middle of the 3D spot constructed from allele intensity to the 3D surface constructed from the nucleolin signal.
QUANTIFICATION AND STATISTICAL ANALYSIS
Details of statistical analyses performed in this paper including analyses packages can be found in the figure legends, main text, and STAR Methods. Spearman correlation coefficients and Pearson correlation coefficients were calculated using the stats module of the scipy python package.58 Mann-Whitney U test was performed using the stats module of the scipy python package.58 Precision measures such as mean, median, quartiles, standard deviation, bootstrapped confidence intervals are described in the corresponding figure legends.
Significance of inter-chromosomal RNA-RNA contacts
To compute the significance of inter-chromosomal RNA-RNA contacts between speckle hub or nucleolar hub genes, the RNA-RNA contacts between the top 2000 pre-mRNAs were randomly permuted to generate an expected distribution for contact frequency. Specifically, the pre-mRNA reads associated with these genes were randomized across the clusters containing them. The RNA-RNA contacts for the permuted clusters were calculated, the gene-based RNA-RNA contact map were normalized using ICE, and the inter-chromosomal contacts were collapsed into speckle hub genes, nucleolar hub genes, or neither. This procedure was repeated 100 times to generate an expected distribution of mean inter-chromosomal contacts. The observed value was compared to the expected distribution to generate a p value.
Supplementary Material
Highlights.
RD-SPRITE enables simultaneous mapping of nascent transcription and DNA structure
Pol II transcription occurs within both A and B compartments and proximal to nucleoli
Nascent pre-mRNAs organize within chromosome territories and A/B compartments
Our findings argue against structural domains that preclude RNA Pol II transcription
ACKNOWLEDGMENTS
We thank Kyle Xiong and Jian Ma for sub-compartment annotations for mESCs, Mario Blanco for comments on the manuscript, Peter Chovanec for help with SPRITE experiments and analyses, Shawna Hiley for editing, and Inna-Marie Strazhnik for illustrations. This work was funded by NIH 4DN (U01 DA040612 and U01 HL130007); NIH Directors’ Transformative Research Award (R01 DA053178); the Chan Zuckerberg Initiative; an HHMI Gilliam and Hanna H. Gray Fellowship (S.A.Q.); NIH UCLA-Caltech Medical Scientist Training Program (T32GM008042) (I.G.); American Cancer Society Fellowship (N.O.); Caltech BBE fellowship (J.W.J.); and NIH T32 GM 7616-40, NIH NRSA CA247447, Josephine De Karman Fellowship Trust, and the UCLA-Caltech Medical Scientist Training Program (P.B.). Imaging was performed in the Biological Imaging Facility with the support of the Caltech Beckman Institute. Sequencing was performed in the Millard and Muriel Jacobs Genetics and Genomics facility with support from Igor Antoshechkin.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
S.A.Q. and M.G. are inventors on a patent covering the SPRITE method.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111730.
REFERENCES
- 1.Pombo A, and Dillon N (2015). Three-dimensional genome architecture: players and mechanisms. Nat. Rev. Mol. Cell Biol. 16, 245–257. 10.1038/nrm3965. [DOI] [PubMed] [Google Scholar]
- 2.Dekker J, Belmont AS, Guttman M, Leshyk VO, Lis JT, Lomvardas S, Mirny LA, O’Shea CC, Park PJ, Ren B, et al. (2017). The 4D nucleome project. Nature 549, 219–226. 10.1038/nature23884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Misteli T (2020). The self-organizing genome: principles of genome architecture and function. Cell 183, 28–45. 10.1016/j.cell.2020.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dekker J, Rippe K, Dekker M, and Kleckner N (2002). Capturing chromosome conformation. Science 295, 1306–1311. 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 5.Dekker J (2016). Mapping the 3D genome: aiming for consilience. Nat. Rev. Mol. Cell Biol. 17, 741–742. 10.1038/nrm.2016.151. [DOI] [PubMed] [Google Scholar]
- 6.Gibcus JH, and Dekker J (2013). The hierarchy of the 3D genome. Mol. Cell 49, 773–782. 10.1016/j.molcel.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lieberman-aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions revelas folding principles of the human genome. Science 326, 289–293. 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kempfer R, and Pombo A (2019). Methods for mapping 3D chromosome architecture. Nat. Rev. Genet. 21, 207–226. 10.1038/s41576-019-0195-2. [DOI] [PubMed] [Google Scholar]
- 9.van Steensel B, and Furlong EEM (2019). The role of transcription in shaping the spatial organization of the genome. Nat. Rev. Mol. Cell Biol. 20, 327–337. 10.1038/s41580-019-0114-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oudelaar AM, and Higgs DR (2021). The relationship between genome structure and function. Nat. Rev. Genet. 22, 154–168. 10.1038/s41576-020-00303-x. [DOI] [PubMed] [Google Scholar]
- 11.Finn EH, Pegoraro G, Brandã0 HB, Valton A-L, Oomen ME, Dekker J, Mirny L, and Misteli T (2017). Heterogeneity and intrinsic variation in spatial genome organziation. Preprint at bioRxiv. 10.1101/171801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rowley MJ, and Corces VG (2018). Organizational principles of 3D genome architecture. Nat. Rev. Genet. 19, 789–800. 10.1038/s41576-018-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falk M, Feodorova Y, Naumova N, Imakaev M, Lajoie BR, Leonhardt H, Joffe B, Dekker J, Fudenberg G, Solovei I, and Mirny LA (2019). Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 570, 395–399. 10.1038/s41586-019-1275-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dekker J, and Mirny L (2016). The 3D genome as moderator of chromosomal communication. Cell 164, 1110–1121. 10.1016/j.cell.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah S, Takei Y, Zhou W, Lubeck E, Yun J, Eng CHL, Koulena N, Cronin C, Karp C, Liaw EJ, et al. (2018). Dynamics and spatial genomics of the nascent transcriptome by intron seqFISH. Cell 174, 363–376.e16. 10.1016/j.cell.2018.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferrai C, de Castro IJ, Lavitas L, Chotalia M, and Pombo A (2010). Gene positioning. Cold Spring Harb. Perspect. Biol. 2, a000588. 10.1101/cshperspect.a000588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahy NL, Perry PE, Gilchrist S, Baldock RA, and Bickmore WA (2002). Spatial organization of active and inactive genes and noncoding DNA within chromosome territories. J. Cell Biol. 157, 579–589. 10.1083/jcb.200111071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahy NL, Perry PE, and Bickmore WA (2002). Gene density and transcription influence the localization of chromatin outside of chromosome territories detectable by FISH. J. Cell Biol. 159, 753–763. 10.1083/jcb.200207115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williamson I, Berlivet S, Eskeland R, Boyle S, Illingworth RS, Paquette D, Dostie J, and Bickmore WA (2014). Spatial genome organization: contrasting views from chromosome conformation capture and fluorescence in situ hybridization. Genes Dev. 28, 2778–2791. 10.1101/gad.251694.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu H, Harris H, Olshansky M, Eliaz Y, Krishna A, Kalluchi A, et al. (2021). Fine-mapping of nuclear compartments using ultra-deep Hi-C shows that active promoter and enhancer elements localize in the active A compartment even when adjacent sequences do not. bioRxiv. 10.1101/2021.10.03.462599. [DOI] [Google Scholar]
- 22.van Steensel B, and Belmont AS (2017). Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169, 780–791. 10.1016/j.cell.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reddy KL, Zullo JM, Bertolino E, and Singh H (2008). Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 452, 243–247. 10.1038/nature06727. [DOI] [PubMed] [Google Scholar]
- 24.Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, and Bickmore WA (2008). Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 4, e1000039. 10.1371/journal.pgen.1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumaran RI, and Spector DL (2008). A genetic locus targeted to the nuclear periphery in living cells maintains its transcriptional competence. J. Cell Biol. 180, 51–65. 10.1083/jcb.200706060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Zhang Y, Zhang R, van Schaik T, Zhang L, Sasaki T, Peric-Hupkes D, Chen Y, Gilbert DM, van Steensel B, et al. (2021). SPIN reveals genome-wide landscape of nuclear compartmentalization. Genome Biol. 22, 36. 10.1186/s13059-020-02253-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Padeken J, and Heun P (2014). Nucleolus and nuclear periphery: velcro for heterochromatin. Curr. Opin. Cell Biol. 28, 54–60. 10.1016/j.ceb.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 28.Quinodoz SA, Jachowicz JW, Bhat P, Ollikainen N, Banerjee AK, Goronzy IN, Blanco MR, Chovanec P, Chow A, Markaki Y, et al. (2021). RNA promotes the formation of spatial compartments in the nucleus. Cell 184, 5775–5790.e30. 10.1016/j.cell.2021.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quinodoz SA, Bhat P, Chovanec P, Jachowicz JW, Ollikainen N, Detmar E, Soehalim E, and Guttman M (2022). SPRITE: a genome-wide method for mapping higher-order 3D interactions in the nucleus using combinatorial split-and-pool barcoding. Nat. Protoc. 17, 36–75. 10.1038/s41596-021-00633-y. [DOI] [PubMed] [Google Scholar]
- 30.Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, et al. (2018). Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174, 744–757.e24. 10.1016/j.cell.2018.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sigova AA, Mullen AC, Molinie B, Gupta S, Orlando DA, Guenther MG, Almada AE, Lin C, Sharp PA, Giallourakis CC, and Young RA (2013). Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc. Natl. Acad. Sci. USA 110, 2876–2881. 10.1073/pnas.1221904110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jonkers I, Kwak H, and Lis JT (2014). Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife. 10.7554/eLife.02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiong K, and Ma J (2019). Revealing Hi-C subcompartments by imputing inter-chromosomal chromatin interactions. Nat. Commun. 10, 5069. 10.1038/s41467-019-12954-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pederson T (2011). The nucleolus. Cold Spring Harb. Perspect. Biol. 3, 0006388–a715. 10.1101/cshperspect.a000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mélèse T, and Xue Z (1995). The nucleolus: an organelle formed by the act of building a ribosome. Curr. Opin. Cell Biol. 7, 319–324. 10.1016/0955-0674(95)80085-9. [DOI] [PubMed] [Google Scholar]
- 37.Németh A, and Längst G (2011). Genome organization in and around the nucleolus. Trends Genet. 27, 149–156. 10.1016/j.tig.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Kresoja-Rakic J, and Santoro R (2019). Nucleolus and rRNA gene chromatin in early embryo development. Trends Genet. 35, 868–879. 10.1016/j.tig.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boyle S, Rodesch MJ, Halvensleben HA, Jeddeloh JA, and Bickmore WA (2011). Fluorescence in situ hybridization with high-complexity repeat-free oligonucleotide probes generated by massively parallel synthesis. Chromosome Res. 19, 901–909. 10.1007/s10577-011-9245-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, and van Steensel B (2008). Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453, 948–951. 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- 41.Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SWM, Solovei I, Brugman W, Gräf S, Flicek P, Kerkhoven RM, van Lohuizen M, et al. (2010). Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell 38, 603–613. 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collombet S, Rall I, Dugast-Darzacq C, Heckert A, Halavatyi A, le Saux A, Dailey G, Darzacq X, and Heard E (2021). RNA polymerase II depletion from the inactive X chromosome territory is not mediated by physical compartmentalization. Preprint at bioRxiv. 10.1101/2021.03.26.437188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jung YH, Sauria MEG, Lyu X, Cheema MS, Ausio J, Taylor J, and Corces VG (2017). Chromatin states in mouse sperm correlate with embryonic and adult regulatory landscapes. Cell Rep. 18, 1366–1382. 10.1016/j.celrep.2017.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Battulin N, Fishman VS, Mazur AM, Pomaznoy M, Khabarova AA, Afonnikov DA, Prokhortchouk EB, and Serov OL (2015). Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome Biol. 16, 77. 10.1186/s13059-015-0642-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang Y, Huang J, Lun K, Li B, Zheng H, Li Y, Zhou R, Duan W, Wang C, Feng Y, et al. (2020). Genome-wide analyses of chromatin interactions after the loss of Pol I, Pol II, and Pol III. Genome Biol. 21, 158. 10.1186/s13059-020-02067-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barutcu AR, Blencowe BJ, and Rinn JL (2019). Differential contribution of steady-state RNA and active transcription in chromatin organization. EMBO Rep. 20, e48068. 10.15252/embr.201948068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu JY, Chang L, Li T, Wang T, Yin Y, Zhan G, Han X, Zhang K, Tao Y, Percharde M, et al. (2021). Homotypic clustering of L1 and B1/Alu repeats compartmentalizes the 3D genome. Cell Res. 31, 613–630. 10.1038/s41422-020-00466-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sima J, Chakraborty A, Dileep V, Michalski M, Klein KN, Holcomb NP, Turner JL, Paulsen MT, Rivera-Mulia JC, Trevilla-Garcia C, et al. (2019). Identifying cis elements for spatiotemporal control of mammalian DNA replication. Cell 176, 816–830.e18. 10.1016/j.cell.2018.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu J, and Gilbert DM (2007). Proliferation-dependent and cell cycle-regulated transcription of mouse pericentric heterochromatin. J. Cell Biol. 179, 411–421. 10.1083/jcb.200706176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marchal C, Sima J, and Gilbert DM (2019). Control of DNA replication timing in the 3D genome. Nat. Rev. Mol. Cell Biol. 20, 721–737. 10.1038/s41580-019-0162-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360. 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 56.Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108. 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abdennur N, and Mirny LA (2020). Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36, 311–316. 10.1093/bioinformatics/btz540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, et al. (2020). SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272. 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Venev S, Abdennur N, Goloborodko A, Flyamer I, Fudenberg G, Nuebler J, Galitsyna A, Akgol B, Abraham S, Kerpedjiev P, et al. (2021). open2c/cooltools v0.4.1.
- 60.Jachowicz JW, Strehle M, Banerjee AK, Blanco MR, Thai J, and Guttman M (2022). Xist spatially amplifies SHARP/SPEN recruitment to balance chromosome-wide silencing and specificity to the X chromosome. Nat. Struct. Mol. Biol. 29, 239–249. 10.1038/s41594-022-00739-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available RD-SPRITE data (GEO: GSE151515). PolyA RNA-seq data generated in this work has been deposited at GEO: GSE211287 and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit polyclonal anti-Nucleolin | Abcam | Cat# ab22758; RRID:AB_776878 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Doxycycline | Sigma | D9891 |
|
| ||
| Deposited data | ||
|
| ||
| SPRITE data | Quinodoz et al.28 | GEO:GSE151515 |
| Ribo-Depleted RNA RNA-seq data in mESC | Sigova et al.31 | GEO:GSM903663 |
| PolyA RNA-seq data in mESC | This Study | GEO: GSE211287 |
| GRO-seq data in mES cells | Jonkers et al.32 | GEO:GSE48895 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse: pSM44 ES cell line | This Study | pSM44 (dox-inducible Xist) |
|
| ||
| Software and algorithms | ||
|
| ||
| SPRITE pipeline 2.0 (v0.2) | This Study | https://github.com/GuttmanLab/sprite2.0-pipeline |
| Bowtie2 (v2.3.5) | Langmead and Salzberg51 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Hisat (v2.1.0) | Kim et al.52 | http://www.ccb.jhu.edu/software/hisat/index.shtml |
| Samtools (v1.4) | Li et al.53 | http://samtools.sourceforge.net/ |
| Bedtools (v2.30.0) | Quinlan and Hall54 | https://bedtools.readthedocs.io/en/latest/ |
| Trim Galore! (v0.6.2) | Felix Krueger (The Babraham Institute) | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Subread (v2.0.3) | Liao et al.55,56 | http://subread.sourceforge.net/ |
| Cooler (v0.8.5) | Abdennur and Mirny57 | https://github.com/open2c/cooler |
| Cooltools (v0.4.1) | 10.5281/zenodo.5214125 | https://github.com/open2c/cooltools |
| Scipy (v1.7.1) | Virtanen et al.58 | https://scipy.org/ |
The original code for the SPRITE analysis pipeline is available on Github at https://github.com/GuttmanLab/sprite2.0-pipeline. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.