

Summary
Other than genetically engineered mice, few reliable platforms are available for the study of hematopoietic stem cell (HSC) quiescence. Here we present a platform to analyze HSC cell cycle quiescence by combining culture conditions that maintain quiescence with a CRISPR-Cas9 genome editing system optimized for HSCs. We demonstrate that preculture of HSCs enhances editing efficiency by facilitating nuclear transport of ribonucleoprotein complexes. For post-editing culture, mouse and human HSCs edited based on non-homologous end joining and cultured under low-cytokine, low-oxygen, and high-albumin conditions retain their phenotypes and quiescence better than those cultured under the proliferative conditions. Using this approach, HSCs regain quiescence even after editing by homology-directed repair. Our results show that low-cytokine culture conditions for gene-edited HSCs are a useful approach for investigating HSC quiescence ex vivo.
Keywords: hematopoietic stem cell, genome editing, quiescence, stem cell culture, CRISPR-Cas9
Graphical abstract
Highlights
-
•
Preculture of HSPCs influences RNP nuclear transport and gene editing efficiency
-
•
We report culture conditions optimized for post-editing return to quiescence
-
•
Our results establish an ex vivo platform for quiescence studies of edited HSCs
Motivation
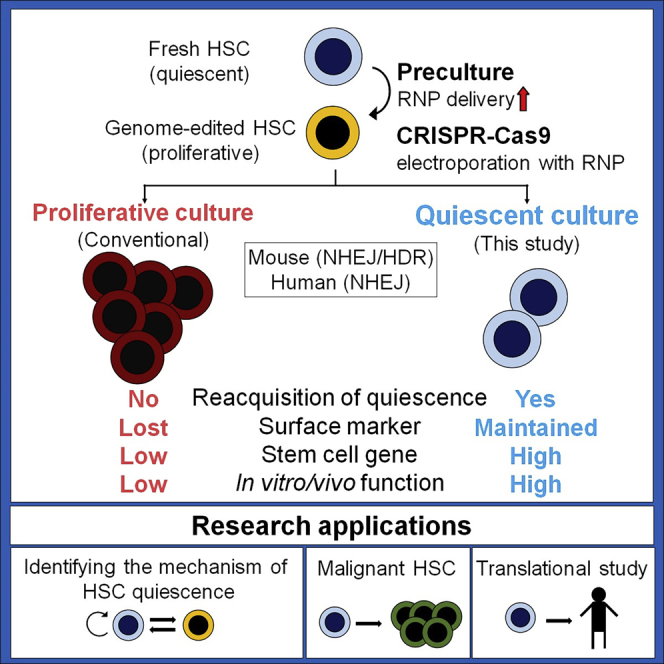
Although most hematopoietic stem cells (HSCs) are quiescent, maintaining and investigating HSC quiescence ex vivo remains challenging. Genetically engineered mice are a reliable approach for quiescent study, but it is time consuming and has limited translation to human HSCs. To establish an accessible platform for the study of the biology of quiescent HSCs, we have adapted and optimized a low-cytokine, low-oxygen, and high-albumin culture system that restores and maintains HSC quiescence following genome editing. This method can revert genome-edited HSCs generated by both non-homologous end joining (NHEJ) and homology-directed repair (HDR) to quiescence and is a useful tool for investigating HSC quiescence.
Shiroshita et al. establish an accessible platform to study the biology of quiescent HSCs by adapting and optimizing a low-cytokine, low-oxygen, and high-albumin culture system that restores and maintains HSC quiescence following genome editing. Using this approach, HSCs regain quiescence after editing by non-homologous end-joining or homology-directed repair.
Introduction
Hematopoietic stem cells (HSCs) maintain lifelong hematopoiesis in the bone marrow (BM). The majority of HSCs are quiescent at steady state.1 HSC function is closely related to quiescence,2 and label-retaining assays,3,4 cell cycle indicators,5,6 and genetic ablation of cyclin-dependent kinase inhibitors7,8,9 confirmed that repopulation capacity is highest in quiescent HSCs. HSC quiescence is precisely regulated by metabolic status, reactive oxygen species (ROS), mitochondrial activity, autophagy, lysosomal activity, and CDK6 levels.10,11,12,13,14 HSCs generate committed progenitors in response to various stressors and signals.15 Several groups have reported that, once activated, quiescent HSCs undergo functional changes in response to stimulation by factors such as interferon (IFN), lipopolysaccharide (LPS), and 5-fluorouracil (5-FU).16,17,18,19,20,21,22 Although these studies advanced the field of HSC biology, HSC quiescence remains incompletely understood, and many aspects of this process are unknown. For example, although the ability of activated HSCs to revert to quiescence in vivo is well known, the regulatory mechanisms for this phenomenon remain elusive. The use of genetically engineered mice is a promising approach to further investigate HSC quiescence, but this approach is time consuming, and the number of target genes is limited. Findings in mouse models are often not translatable to human HSCs. Therefore, new tools to study the biology of human HSC quiescence in physiological and pathological states are required.
CRISPR-Cas9 genome editing has revolutionized the study of HSCs23,24,25 and their therapeutic application.26 Genome editing using the conventional CRISPR-Cas9 system induces non-homologous end joining (NHEJ), resulting in introduction of insertions or deletions (indels) into the genome.27,28,29 Although genome editing via NHEJ is reportedly less affected by the cell cycle status than genome editing via homology-directed repair (HDR),30,31,32,33,34 it is not fully understood whether there are differences in the genome editing efficiency within hematopoietic stem/progenitor cell (HSPC) subpopulations. Various methods reportedly improve the genome editing efficiency,35,36,37,38,39 genome editing accuracy,40 and repopulation capacity32,41 of HSCs. Preclinical and clinical trials of HSC genome editing are ongoing to treat various genetic diseases, including thalassemia,42,43 sickle cell disease,43,44,45,46,47 and congenital immunodeficiency.48,49,50
Genome editing of a mixture of HSCs and progenitors has been reported.51,52 Although these reports are valuable, HSC-optimized genome editing protocols are necessary, considering the unique biological properties of quiescent HSCs. It is unclear why preculture of HSCs is necessary not only for HDR, which requires entry into S phase, but also for NHEJ, which can occur in quiescent cells.35 This prompted the hypothesis that HSCs have specific gene editing mechanisms related to quiescence. Proliferative culture conditions have been widely used before and after genome editing. HSCs gradually lose their repopulation capacity upon prolonged exposure to proliferative conditions,53 highlighting the necessity of proliferative culture conditions that maintain HSC function. Recently, ex vivo culture methods for mouse and human HSPCs have been established.54,55 These methods overcome the functional decline after ex vivo proliferative culture and are useful for obtaining sufficient HSC numbers for application in translational studies. As another approach to maintaining HSC function, our group and others have focused on retaining HSC quiescence in vitro.56,57,58,59 These two culture methods, proliferative and quiescent culture, are conceptually different, especially pertaining to the cell cycle. Therefore, to study the biology of quiescent HSCs, further development and improvement of quiescent culture-based methods is necessary. Based on this concept, controlling the cell cycle following genome editing is worth considering. Attempts have been made to revert edited cells to quiescence using chemical compounds.31
In this study, we adapted and optimized a low-cytokine, low-oxygen, and high-albumin culture condition that maintains quiescence of HSCs (quiescence-maintaining condition)57 following genome editing and demonstrated that this condition can revert genome-edited HSCs generated by NHEJ and HDR to quiescence. This strategy preserves quiescent HSCs that retain the same surface marker phenotypes as HSCs in BM. In addition, the transcriptional profile and repopulation capacity of genome-edited HSCs cultured under the quiescence-maintaining condition are better maintained than those of genome-edited HSCs cultured under the proliferative condition. We adapted this method to the editing of human HSPCs and confirmed its usefulness in this context. Collectively, our method preserves quiescent HSCs after genome editing and enables investigation of the biology and genetics of HSC quiescence under near-physiological conditions.
Results
Efficient editing of quiescent HSCs by NHEJ requires preculture
To analyze factors that affect the editing efficiency of HSPCs, we used single guide RNAs (sgRNAs) targeting CD45 and GFP (CD45-sgRNA and GFP-sgRNA, respectively). sgRNA targeting the Rosa26 locus served as a negative control. HSPCs were electroporated to transfect a ribonucleoprotein (RNP) complex containing sgRNA and Cas9 (Figure S1A).35,60 Homozygous or heterozygous mutation using CD45-sgRNA and GFP-sgRNA was evaluated by picking single-cell-derived colonies (Figure S1B). sgRNA produced by in vitro transcription (IVT) reportedly induces IFN signaling, and this phenomenon is prevented by using chemically modified sgRNA.61,62,63 We performed quantitative PCR (qPCR) to assess the gene expression and colony-forming capacity of HSPCs treated with IVT-produced or chemically modified sgRNAs in our system. The expression levels of IFN signaling-related genes (Ifnb1, Oas2, Ddx58/RIG-I, and Isg15) tended to increase upon treatment with IVT-produced sgRNA, as previously reported, but these increases were not significant (Figure S1C). We next analyzed the effects of the differentiation status and culture before genome editing. Freshly isolated HSPCs or HSPCs cultured for various durations were electroporated with the RNP complex, and the editing efficiency was analyzed on day 3 after electroporation (Figure 1A). The editing efficiency differed among freshly isolated HSPC fractions; it was >40% for granulocyte/monocyte progenitors (GMPs), <20% for HSCs, and variable (10%–30%) for multipotent progenitors (MPPs) and common myeloid progenitors (CMPs) (Figure 1B). The genome editing efficiency of all the HSPC fractions precultured overnight was higher than that of freshly isolated cells (blue bars in Figure 1B), consistent with the previous finding that preculture is required for genome editing.35,64 Notably, preculture almost abrogated the differences in editing efficiency among HSPC fractions (red bars in Figure 1B).
Figure 1.

The cell cycle status determines the genome editing efficiency of HSPC fractions
(A) Experimental design.
(B) Frequencies of CD45− edited cells among HSCs, MPP1 cells, MPP2 cells, MPP3 cells, CMP cells, and GMP cells precultured with 50 ng/mL SCF and 50 ng/mL TPO (mean ± SD, n = 6–12 per group from two independent experiments).
(C) EdU incorporation assay using freshly isolated (fresh) and precultured (preculture) HSCs and GMPs (mean ± SD, n = 3 biological replicates).
(D) Correlation of the frequency of cells in S/G2/M phases and the genome editing efficiency among HSPC fractions.
Given that HSCs are more quiescent than differentiated progenitors,2 we speculated that the differences in genome editing efficiency of HSPCs and the effects of preculture are related to the cell cycle status. The frequency of cycling cells determined by 5-ethynyl-2′-deoxyuridine (EdU) labeling was significantly lower among freshly isolated HSCs than among freshly isolated GMPs, whereas the frequencies of EdU+ cells in these populations increased and were comparable following preculture with cytokines overnight (Figure 1C). Within the freshly isolated HSPC fraction, the genome editing efficiency and cell cycle status determined by Ki67 and Hoechst 33342 staining (Figure 1D) or in vivo EdU incorporation (Figure S1D) were positively correlated. These data suggest that genome editing occurs efficiently in cycling cells. Thus, even in the case of genome editing with NHEJ, which can occur in cells not in S/G2 phases, including quiescent cells, a sufficient preculture period to activate the cell cycle of HSCs is needed.
The preculture condition, but not the post-culture condition, defines the genome editing efficiency
To further determine the optimal genome editing condition for HSCs, we tested several variables, including the preculture duration, cytokine combination, number of cells subjected to editing per experiment, and post-electroporation culture (post-culture) conditions (Figure 2A). The frequency of CD45− edited cells increased in proportion to the preculture duration, consistent with previous reports (Figure 2B).31,35,64 Regarding cytokine combinations, the frequency of CD45− edited cells was highest using 50 ng/mL stem cell factor (SCF) and 50 ng/mL thrombopoietin (TPO), whereas preculture with low concentrations of cytokines or a single cytokine (SCF or TPO) decreased genome editing efficiency (Figure 2C). In addition, this preculture condition resulted in a genome editing efficiency comparable with preculture in polyvinyl alcohol (PVA)-based medium, and the preculture period was shorter than in previous reports (Figure S2A).65 Furthermore, the number of cells loaded on the electrode tip did not affect the genome editing efficiency, making it possible to edit as few as 250 HSCs (Figure S2B). Notably, although high cytokine concentrations were required for preculture, changing the post-culture medium to maintain quiescence of HSCs (1.5 ng/mL SCF, 1 ng/mL TPO, 4% bovine serum albumin [BSA] supplemented with fatty acids and cholesterol, and 1% O2; STAR Methods)57 did not compromise the genome editing efficiency (Figure 2D). Our results indicate that the preculture condition, not the post-culture condition, defines genome editing efficiency.
Figure 2.

The preculture condition defines the genome editing efficiency and increases the nucleofection efficiency of HSPCs
(A) Experimental design.
(B) Optimization of the preculture duration for genome editing of HSCs (mean ± SD, n = 2–3 per group from three independent experiments).
(C) Optimization of the cytokines included during preculture for efficient genome editing of HSCs (mean ± SD, n = 6–9 per group from three independent experiments).
(D) Genome editing efficiency of CD45 in HSCs post-cultured under the indicated cytokine conditions following electroporation (mean ± SD, n = 6 per group from two independent experiments).
(E) Annexin V analysis of LSK cells at 2 days after electroporation. Mock cells and cells electroporated with RNP with/without preculture were compared (mean ± SD, n = 3 per group from independent cell cultures).
(F) Cell number of mock- or RNP-electroporated LSK cells with/without preculture (mean ± SD, n = 4 per group from four independent cell cultures).
(G) Nucleofection efficiency (ATTO550+) of edited LSK cells with/without preculture (mean ± SD, n = 3 per group from three independent electroporations).
(H) Time course analysis of the nucleofection efficiency (mean ± SD, n = 3 per group from three independent electroporations).
(I) Confocal imaging of fresh and precultured LSK cells electroporated with ATTO550-labeled RNP. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). The negative control (NTC) used non-labeled RNP. Representative images are shown.
(J) Frequency of nuclei containing ATTO550-labeled RNP (n = 10 cells per group).
(K) FPKM of Kpna2 and Kpnb1 in fresh and precultured HSCs determined from RNA-seq analysis (n = 3 per group from three biological replicates).
Preculture improves the nuclear localization of RNPs and upregulates nuclear import-related genes
Based on the results described above, fresh HSCs are poorly edited by NHEJ compared with hematopoietic progenitor cells (Figures 1B, 2B, and 2C). Quiescent HSCs mainly use the NHEJ-mediated DNA repair mechanism66; therefore, preculture may improve editing efficiency by improving viability or transfection efficiency after electroporation. 2 days after electroporation, the frequency of apoptotic cells was unaffected by preculture (Figure 2E). As demonstrated in a prior study,35 the number of freshly isolated HSPCs (Lin−Sca-1+c-Kit+ [LSK] cells) after RNP electroporation was similar to that of mock-electroporated cells until 72 h post-electroporation. Contrastingly, the number of LSK cells electroporated with RNP after preculture decreased about to 80% compared with mock-electroporated cells (Figure 2F). This suggested that preculture did not allow LSK cells to electroporation. Next, we tested whether HSPC preculture changes the transfection efficiency of primitive HSPCs. Fresh or precultured LSK cells were electroporated with ATTO550-labeled or non-labeled gRNA, and the transfection efficiency (the frequency of ATTO550+ cells) was analyzed by flow cytometry. A higher proportion of ATTO550+ cells than ATTO550− cells was CD45−, suggesting that the transfection efficiency can be monitored using ATTO550-labeled gRNA (Figure S2C). As shown in previous experiments (Figures 1B and 2B), CD45− cells were less frequent among fresh LSK cells than among precultured LSK cells. The nucleofection efficiency of precultured LSK cells was significantly higher than that of fresh LSK cells 24 h after electroporation (92.7% ± 1.1% vs. 67.0% ± 5.7%) (Figures 2G and S2E). To determine when these differences occurred, we performed a time-course analysis of transfection efficiency. The transfection efficiency significantly differed between freshly isolated and precultured LSK cells immediately after electroporation, and this difference persisted for at least 24 h (Figure 2H).
Next, we investigated why fresh HSPCs had relatively high transfection efficiency (about 70% for LSK cells, as shown in Figure 2G) but were inefficiently edited (less than 20% of CD45− cells among HSCs, as shown in Figures 1B and 2B). The flow cytometry histograms of cells electroporated with ATTO550 were biphasic, and ATTO550high and ATTO550low populations could be detected. ATTO550high cells were dominant among precultured LSK cells (Figures S2D and S2E). Considering that preculture improved the efficiency of gene editing, we hypothesized that the ATTO550high population contained cells with nuclear localization of RNP. Indeed, confocal imaging confirmed that the co-localization frequency of RNP and nuclei was significantly increased in precultured LSK cells compared with fresh LSK cells (84.5% ± 8.3% vs. 47.5% ± 8.3%) (Figures 2I and 2J). The ATTO550high population of fresh LSK cells also exhibited RNP localized to the nucleus (Figure S2F), which was compatible with the low editing efficiency of fresh LSK cells (Figure S2C). We next examined the efficiency of RNP nuclear transfer by nucleofection, an improved transfection method for nucleic acid and proteins into the cytoplasm and nucleus. Nuclear localization of RNP after nucleofection (64.3% ± 5.8%) was higher than that of fresh LSK cells (47.5% ± 8.3%) (Figures 2I, 2J, S2G, and S2H). However, RNP nuclear localization after nucleofection of precultured LSK cells significantly increased (84.6% ± 2.6%) relative to fresh LSK cells and was similar to that of electroporated precultured LSK cells (84.5% ± 8.3%) (Figures 2I, 2J, S2G, and S2H). Thus, in freshly isolated LSK cells, nucleofection introduces RNPs to the nucleus more efficiently than conventional electroporation, but even conventional electroporation of precultured LSK cells introduced more RNPs to the nucleus than did nucleofection of freshly isolated LSK cells. In support of these observations, RNA sequencing (RNA-seq) revealed that gene expression of Kpna2 (Importin α2) and nuclear import-related genes (Rcc1, Ran, Ranbp1, and Kpnb1) was significantly increased after preculture (Figures 2K and S2I). On the other hand, transmission electron microscopy showed no increase in the number of nuclear pores (Figures S2J and S2K).
Our data suggest that preculture improves the nucleofection efficiency of HSPCs by upregulating nuclear import machinery and increasing the nuclear localization of RNPs.
HSCs re-enter quiescence after genome editing under the quiescence-maintaining condition
Although preculture improves the efficiency of editing, cell cycle changes and proliferation induced by preculture perturb the function of HSCs.1,67,68 Several studies induced this reversion in retrovirally transduced primate and human HSPCs.31,69 We thus sought to optimize post-editing culture conditions to restore quiescence to edited HSCs using physiological factors. We investigated if the quiescence-maintaining culture condition that we reported previously positively affects HSCs following genome editing at the early phase (day 2 after editing) and late phase (day 7 after editing) (Figure 3A).57 After culture under the quiescence-maintaining condition, 60% of LSK cells in the early and late phases retained the surface marker phenotype of HSCs, which was higher than the corresponding percentage under the conventional proliferative culture or PVA-based condition (Figures S3A and S3B). In the early phase, the total cell number was significantly lower, but the frequency of HSCs was significantly higher, under the quiescence-maintaining condition than under the proliferative and PVA-based conditions (Figures 3B–3D). These differences were also observed in the late phase (Figures 3E–3G). More than 60% of cells did not express CD45 in the late phase (Figures 3G and S3B). These results suggest that the quiescence-maintaining condition maintains phenotypic HSCs (pHSCs) even after genome editing.
Figure 3.

Post-culture induces reversion of edited HSCs to quiescence
(A) Experimental design for genome editing.
(B) Total cell counts of HSCs edited using CD45-sgRNA in the early phase after editing (mean ± SD, n = 3 per group from independent cell cultures).
(C) HSC counts among HSCs edited using CD45-sgRNA in the early phase after editing (mean ± SD, n = 3 per group from independent cell cultures).
(D) Frequency of pHSCs among HSCs edited using CD45-sgRNA in the early phase after editing (mean ± SD, n = 3 per group from independent cell cultures).
(E) Total cell counts of HSCs edited using CD45-sgRNA in the late phase after editing (mean ± SD, n = 14 per group from independent cell cultures).
(F) Frequency of pHSCs among HSCs edited using CD45-sgRNA in the late phase after editing (mean ± SD, n = 14 per group from independent cell cultures).
(G) Frequency of CD45− cells in the late phase after editing (mean ± SD, n = 6 per group from independent cell cultures).
(H) EdU incorporation assay of genome-edited HSCs in the early phase after editing (mean ± SD, n = 6 per group from two independent experiments).
(I) EdU incorporation assay of genome-edited HSCs in the late phase after editing (mean ± SD, n = 3–6 per group from independent cell cultures).
(J) Frequency of Evi1+ cells in the CD48− LSK fraction of CD45− (edited) or CD45+ (non-edited) cells in the late phase after editing (mean ± SD, n = 16 per group from independent cell cultures).
Next, we investigated whether genome-edited HSCs could revert to a quiescent state and, if so, their reversion kinetics. To test this, EdU incorporation was examined at several time points. The EdU+ fraction of HSCs was increased after preculture (Figure 1C). In the early phase after editing, the percentage of EdU+ LSK cells was significantly lower under the quiescence-maintaining condition than under the PVA-based condition (Figure 3H), and the percentage of CD48− LSK cells under the quiescence-maintaining condition was significantly higher than under other conditions (Figure S3C). About 25% of LSK cells under the quiescence-maintaining condition were EdU+, which suggested that reversion to quiescence was not fully completed in the early phase. In the late phase, CD48− LSK cells incorporated less EdU than CD48+ LSK cells under all culture conditions tested (Figure 3I). Importantly, about 10% of cells in the CD48− LSK fraction were EdU+ under the quiescence-maintaining condition (Figure 3I). This was similar to the frequency for HSCs cultured under the quiescence-maintaining condition without electroporation and lower than the frequencies under the conventional proliferative and PVA-based conditions (Figures 3I and S3D). These results indicate that the cell cycle begins to slow down in genome-edited HSCs cultured under the quiescence-maintaining condition 2 days after editing and these cells revert to a more dormant state at 7 days after editing.
The Evi1 expression level predicts HSC capacity; therefore, we monitored Evi1 expression following genome editing of HSCs from Evi1-GFP mice.70,71 The frequency of Evi1-GFP+ cells was inversely correlated with the cytokine concentration in culture without electroporation and was highest upon culture in the presence of 0.5 ng/mL SCF and 1 ng/mL TPO (Figure S3E). Consistently, the frequency of Evi1-GFP+ cells within the CD48− LSK fraction (Figure 3J) was lower under the proliferative condition than under the quiescence-maintaining condition. Of note, the frequency of Evi1-GFP+ cells was identical among CD45+ and CD45− cells regardless of the post-culture condition (Figure 3J).
These results suggest that the quiescence-maintaining condition gradually reverts edited HSCs activated by preculture to a dormant state, which maintains pHSCs without compromising the genome editing efficiency.
HSCs reverted to quiescence after genome editing transcriptionally resemble freshly isolated HSCs
To determine whether HSCs cultured under the quiescence-maintaining condition retain HSC characteristics, we performed RNA-seq of the following nine samples: (1) freshly isolated endothelial protein C receptor (EPCR)+ HSCs (fresh), (2) EPCR+ HSCs precultured for 1 day in the presence of 50 ng/mL SCF and 50 ng/mL TPO (preculture), (3) EPCR+ HSCs cultured under the quiescence-maintaining condition at 2 days after electroporation without RNP (day 2-mock), (4) EPCR+ HSCs cultured under the quiescence-maintaining condition at 7 days (day 7-mock), (5) genome-edited EPCR+ HSCs with quiescence-maintaining post-culture at 2 days (day 2-CRISPR-quiescent), (6) genome-edited EPCR+ HSCs with quiescence-maintaining post-culture at 7 days (day 7-CRISPR-quiescent), (7) genome-edited EPCR+ HSCs with proliferative post-culture at 2 days (day 2-CRISPR-proliferative), (8) genome-edited EPCR+ HSCs with proliferative post-culture at 7 days (day 7-CRISPR-proliferative), and (9) genome-edited EPCR+ HSCs with PVA-based post-culture at 2 days (day 2-CRISPR-PVA) (Figure 4A). The gating strategy for cell sorting is shown in Figure S4A.
Figure 4.

RNA-seq
(A) Experimental design for RNA-seq.
(B) PCA of RNA-seq data.
(C) Volcano plots comparing the expression levels of various genes between day 2-CRISPR-quiescent and day 2-CRISPR-proliferative.
(D) Hierarchical clustering of single-sample GSEA of HSPC-related gene sets by gene set variation analysis (GSVA).
(E) GSEA of upregulated gene sets, comparing HSCs and MPPs between day 7-CRISPR-quiescent and day 7-CRISPR-proliferative.
(F) GSEA of downregulated gene sets, comparing HSCs and MPPs between day 7-CRISPR-quiescent and day 7-CRISPR-proliferative.
(G) GSEA of upregulated gene sets comparing HSCs and MPPs between preculture and day 7-CRISPR-quiescent.
Principal-component analysis (PCA) of RNA-seq profiles showed several transcriptional differences in short-term (2 days after electroporation) and long-term (7 days after electroporation) samples. First, the coordinates of precultured HSCs were vastly different from those of fresh HSCs (Figure 4B). Second, focusing on the short-term samples, the coordinates of the quiescence-maintaining culture samples (day 2-mock and day 2-CRISPR-quiescent HSCs) were close to those of preculture HSCs but formed a different cluster than day 2-CRIPSR-proliferative and day 2-CRISPR-PVA HSCs (Figure 4B). The PC1 loading was characterized by expression of quiescent HSC-related genes, such as Vwf, Procr, Pdzk1ip1, Trim47, and Sult1a1 (Table S1). Day 2-CRISPR-quiescent HSCs more highly expressed Vwf, Pdzk1ip1, Trim47, Sult1a1, Robo4, Cdkn1c, Ctsf, and Sox18 than day 2-CRISPR-PVA and day 2-CRISPR-proliferative HSCs (Figure 4C). Third, the coordinates of day 7-mock HSCs, which were cultured under the quiescence-maintaining condition but not precultured, were closer to those of fresh HSCs than those of day 2-mock HSCs. The coordinates of day 7-CRISPR-quiescent HSCs were different from those of day 7-mock and closer to those of day 2-mock and day 2-CRISPR-quiescent HSCs in the PC1 axis (Figure 4B). This suggested that culture under the quiescence-maintaining condition could incompletely revert transcriptional changes induced by preculture 2 days after electroporation and had the potential to maintain a gene expression profile similar to that of fresh HSCs unless preculture was performed. The coordinates of day 7-CRISPR-proliferative HSCs were located opposite those of fresh HSCs and formed a different cluster from day 7-CRISPR-quiescent HSCs (Figure 4B). Day 7-CRISPR-quiescent HSCs maintained expression of HSC-related genes such as Vwf, Esam, Robo4, Ramp2, and Cdkn1c better than day 7-CRISPR-proliferative HSCs (Figure S4B), whereas other HSC-related genes, such as Mllt3 and Procr, were downregulated in day 7-CRISPR-quiescent HSCs compared with fresh HSCs (Figure S4C). These results suggest that the quiescence-maintaining condition can alleviate transcriptional changes in genome-edited HSCs compared with the proliferative condition.
Gene set enrichment analysis (GSEA) revealed that fresh, day 2-mock, day 7-mock, day 2-CRISPR-quiescent, and day 7-CRISPR-quiescent HSCs had similar enrichment profiles of hallmark gene sets (Figure S4D). E2F target genes were downregulated in day 7-CRISPR-quiescent HSCs activated by preculture (Figure S4E) compared with CRISPR-proliferative HSCs (Figure S4F) in addition to other cell cycle-related genes, including Myc target genes (Figure S4G), consistent with the EdU incorporation assay (Figures 1C and S3D). The enrichment profile of HSPC-related gene sets showed that day 2-mock, day 7-mock, and day 2-CRISPR-quiescent HSCs were more similar to fresh HSCs than day 2-and day 7-CRISPR-proliferative HSCs and CRISPR-PVA HSCs (Figure 4D). Day 7-CRISPR-quiescent HSCs had an enrichment profile that was more similar to that of fresh HSCs than day 7-CRISPR-proliferative HSCs (Figure 4D). For instance, genes highly expressed in HSCs compared with MPPs were more enriched in day 7-CRISPR-quiescent HSCs than in day 7-CRISPR-proliferative HSCs and vice versa (Figures 4E and 4F), whereas HSC genes were downregulated compared with preculture HSCs (Figure 4G).
These results suggest that HSCs cultured under the quiescence-maintaining condition reverted to quiescence soon after genome editing and transcriptionally retained HSC phenotypes more than HSCs cultured under the proliferative and PVA-based conditions.
Edited HSCs maintain their stem cell potential in vitro and in vivo
pHSCs do not necessarily retain HSC functions72; therefore, we next tested whether edited HSCs subjected to quiescence-maintaining post-culture retain their repopulation potential in vitro and in vivo. Freshly isolated or genome-edited HSCs subjected to quiescence-maintaining or proliferative post-culture for 7 days were clonally sorted and subjected to a single-cell colony formation assay (Figure S5A). The colony-forming capacity of genome-edited HSCs subjected to quiescence-maintaining post-culture was highest (216 of 243 wells; Figure S5B) and was even superior to that of fresh HSCs (208 of 384 wells; Figure S5C). The colony-forming capacity of genome-edited HSCs subjected to proliferative post-culture was lower (Figures S5D and S5E). Thus, HSCs reverted to quiescence after genome editing retain their colony formation capacity.
Next we evaluated whether the in vivo repopulating capacity of HSCs was affected by editing or post-culture. To this end, we prepared HSCs (CD45.2+) derived from Ubc-GFP mice and edited them at the GFP locus. Edited HSCs were cultured under the quiescence-maintaining condition (quiescent group), proliferative condition (proliferative group), and PVA-based condition (PVA). Freshly isolated GFP+ HSCs (fresh) and GFP+ HSCs not subjected to editing procedures and cultured under the quiescence-maintaining condition (culture group) served as controls. Cultured cells were transplanted into lethally irradiated recipient mice (CD45.1+) with competitor cells (CD45.1+) 2 days (Figure S5F) or 10 days after electroporation (Figure 5A). GFP-sgRNA was validated (Figures S1A and S1B). To test the repopulation capacity of edited HSCs, we compared the chimerism of donor-derived total (CD45.2+) cells in the control groups (fresh and precultured groups) with that of edited (CD45.2+GFP−) cells in different post-culture conditions (quiescent, proliferative, and PVA based). First, to determine how post-culture conditions affect HSC function in vivo on a short-term basis, we competitively transplanted edited HSCs 2 days after electroporation (day 3). Donor-derived chimerism in peripheral blood was comparable between fresh HSCs and the post-culture groups (Figure S5G). Peripheral blood (PB differentiation was similar between groups 4 months after transplantation (Figure S5H). Also, donor-derived chimerism of BM cells and HSCs in each edited group was similar to that of fresh HSCs (Figures S5I and S5J). This suggests that, after short-term culture, edited HSCs are functionally similar to fresh HSCs regardless of post-culture conditions.
Figure 5.

Edited HSCs under the quiescence-maintaining condition retain their stem cell potential in vivo
(A) Experimental design of the transplantation assay.
(B) Peripheral blood chimerism of donor-derived cells following primary transplantation of day 11 (mean ± SD, n = 5–6).
(C) Peripheral blood chimerism of donor-derived myeloid cells, B cells, and T cells following primary transplantation of day 11 (mean ± SD, n = 5–6).
(D) BM chimerism of donor-derived cells following primary transplantation of day 11 (mean ± SD, n = 5–6).
(E) Donor-derived HSC chimerism following primary transplantation of day 11 (mean ± SD, n = 5–6).
(F) Peripheral blood chimerism of donor-derived cells following secondary transplantation of day 11 (mean ± SD, n = 5–6).
(G) Peripheral blood chimerism of donor-derived myeloid cells, B cells, and T cells 4 months after secondary transplantation of day 11 (mean ± SD, n = 5–6).
(H) BM chimerism of donor-derived cells following secondary transplantation of day 11 (mean ± SD, n = 5–6).
(I) Donor-derived HSC chimerism following secondary transplantation of day 11 (mean ± SD, n = 5–6).
Next, to test the functional effect of editing and long-term culture on HSCs, we transplanted HSCs 10 days after editing or HSCs cultured for 10 days without editing (day 11). Following the primary transplantation, donor-derived chimerism of the quiescent group in peripheral blood was generally superior to that of the proliferative group but lower than that of the culture group (Figure 5B). Chimerism of myeloid and B cells 4 months after transplantation did not significantly differ between the quiescent and culture groups, but that of T cells was lower in the quiescent group than in the culture group (Figure 5C). Chimerism of donor-derived BM cells and HSCs in the quiescent group was significantly higher than that in the proliferative group and equivalent to that in the culture group (Figures 5D and 5E). To confirm the serial transplantation capacity, we performed secondary transplantation. Following secondary transplantation, donor-derived chimerism in peripheral blood did not differ between the quiescent and culture groups (Figure 5F). 4 months after secondary transplantation, chimerism of myeloid and B cells was equivalent between the quiescent and culture groups, but that of T cells was lower in the quiescent group than in the culture group (Figure 5G). Chimerism of donor-derived BM cells and HSCs was equivalent between the quiescent and culture groups (Figures 5H and 5I). The proliferative group showed no donor-derived chimerism at 1 month following secondary transplantation, and all recipient mice thereafter died, probably because competitor cells alone were insufficient to repopulate the BM (Figure 5F).
These data demonstrate that all short-term culture conditions tested maintained a similar functionality of edited HSCs compared with fresh HSCs. In long-term culture, edited HSCs maintained sufficient function only under quiescence-maintaining culture conditions, although editing modestly decreased HSC function in this context.
Quiescent culture is beneficial for HSCs edited by HDR
In addition to editing by NHEJ, analyzed above, we tested the effects of the quiescence-maintaining condition on HSCs edited by HDR. To deliver the HDR template, we used validated sgRNAs for the Rosa26 locus and recombinant adenovirus-associated virus serotype 6 (AAV6) expressing EGFP under the control of the CAG promoter (Figure S6A). LSK cells were electroporated with RNP following AAV6 transduction. To enhance AAV6 transduction, precultured and electroporated LSK cells were cultured under a high-cytokine condition for an additional 24 h. Then they were switched to the quiescence-maintaining or proliferative condition (Figure 6A). The efficiency of HDR-based editing was about 15% (Figure 6B). Insertion of the GFP sequence into the Rosa26 locus was confirmed by Sanger sequencing of GFPhigh cells (Figure S6B). Using CD150+CD34− LSK cells edited by HDR, we tested the effect of the quiescence-maintaining condition upon short-term culture. On day 2 after transduction, the total cell number was significantly lower under the quiescence-maintaining condition than under the PVA-based and proliferative conditions (Figure 6C). The frequency of CD150+CD34− LSK cells was significantly higher under the PVA-based condition than under the quiescence-maintaining condition, which was superior to the proliferative condition (Figures 6D, S6C, and S6D). However, the frequency and number of CD150+CD34−CD48− LSK cells were better maintained under the quiescence-maintaining condition than under the PVA-based and proliferative conditions (Figures 6E,6F, and S6C–S6D). The frequency of GFPhigh cells was higher under the quiescence-maintaining and proliferative conditions than under the PVA-based condition (Figure S6F). The frequencies of GFPhigh CD150+CD34− LSK cells and CD150+CD48− LSK cells were significantly higher under the quiescence-maintaining condition than under the proliferative and PVA-based conditions (Figures 6G and S6G). To determine how HDR using AAV6 affects the phenotype of edited HSCs we compared the expression of SLAM markers between GFPhigh and GFP−/low LSK cells (Figure 6H). The frequency of CD150+CD48− LSK cells was lower in the GFPhigh LSK fraction than within the GFP−/low fraction under all three culture conditions (Figure 6H). However, this effect was alleviated by the quiescence-maintaining condition, and CD150+CD48− LSK cells were better retained (Figure 6H).
Figure 6.

Quiescent culture is beneficial for HSCs edited by HDR
(A) Experimental design for HDR-based editing.
(B) Representative flow cytometry histogram of GFP expression.
(C) Total cell numbers 2 days after AAV6 transduction (mean ± SD, n = 3 from independent cell cultures).
(D) Frequency of CD150+CD34− LSK cells 2 days after AAV6 transduction (mean ± SD, n = 3 from independent cell cultures).
(E) Frequency of CD150+CD34−CD48− LSK cells 2 days after AAV6 transduction (mean ± SD, n = 3 from independent cell cultures).
(F) Number of CD150+CD34−CD48− LSK cells 2 days after AAV6 transduction (mean ± SD, n = 3 from independent cell cultures).
(G) Frequency of GFPhigh CD150+CD34− LSK cells 2 days after AAV6 transduction (mean ± SD, n = 3 from independent cell cultures).
(H) SLAM marker expression within the LSK fraction 2 days after AAV6 transduction in GFP−/low cells (left) and GFPhigh cells (right) (mean ± SD, n = 3 from independent cell cultures).
(I) Frequencies of GFPhigh and GFP−/low cells in G0 phase 2 days after AAV6 transduction (mean ± SD, n = 3 from independent cell cultures).
(J) Number of colonies derived from GFPhigh LSK cells cultured under the quiescence-maintaining, PVA-based, and proliferative conditions 2 days after editing (mean ± SD, n = 3 per group).
Next we examined the cell cycle status after HDR-based editing. The frequency of cells in G0 phase was significantly higher under the quiescence-maintaining condition than under the PVA-based and proliferative conditions (Figure S6H). Higher frequencies of GFPhigh and GFP−/low cells were in G0 phase under the quiescence-maintaining condition (Figure 6I). These results suggest that HSCs cultured under the quiescence-maintaining condition can revert to quiescence following HDR-based editing.
Finally, we performed a colony formation assay using HSCs edited by HDR to evaluate their progenitor function. The number of colonies derived from GFPhigh cells was similar among the three culture conditions, and these colonies expressed GFP, as confirmed by genomic PCR (Figures 6J, S6I, and S6J).
These results show that phenotypic and quiescent HSCs were better maintained under the quiescence-maintaining condition than under the proliferative condition after HDR-based editing.
Quiescent culture maintains primitive human HSPCs and reverts these cells to quiescence upon NHEJ-based editing
We next assessed the effect of the quiescence-maintaining condition for post-culture of human HSPCs after NHEJ-based editing of the CD45 locus.
First, we investigated the numbers and surface marker expression of human HSPCs in the short term after gene editing (2 days). We used cord blood-derived CD34+CD38−CD45RA− or CD34+CD38−CD45RA−CD90+ cells and compared four culture conditions: the quiescence-maintaining condition, the UM171-based condition, and two proliferative conditions (proliferative condition 1 and proliferative condition 2) (Figure 7A). The editing efficiency was evaluated according to human CD45 expression. The total cell number was significantly lower under the quiescence-maintaining condition than under the other conditions (Figure 7B). The frequency of CD34+EPCR+CD90+ cells, which include cells with engraftment potential after ex vivo culture,73 was 40% under the UM171-based condition, which was the highest frequency among the four culture conditions (Figures 7C and S7A). However, the frequency of CD34+EPCR+CD90+ cells was significantly higher under the quiescence-maintaining condition than under proliferative conditions 1 and 2 (Figure 7C). hCD45 was efficiently knocked out under all culture conditions (Figure S7B). Cell cycle analysis revealed that the frequency of cells in G0 phase was significantly higher under the quiescence-maintaining condition than under the UM171-based condition, proliferative condition 1, and proliferative condition 2 (Figures 7D, S7C, and S7D). Thus, phenotypic and quiescent human HSPCs were better maintained under the quiescence-maintaining condition than under the proliferative condition, although the frequency of CD34+EPCR+CD90+ cells was lower under the quiescence-maintaining condition than under the UM171-based condition.
Figure 7.

Quiescent culture reverts human HSPCs to quiescence and maintains their primitive state after NHEJ-based editing
(A) Experimental design for editing human HSPCs.
(B) Total number of genome-edited CD34+CD38−CD45RA− cells 2 days after electroporation (mean ± SD, n = 3–6 from independent cell cultures).
(C) Frequency of CD34+EPCR+CD90+ cells among genome-edited CD34+CD38−CD45RA− cells 2 days after electroporation (mean ± SD, n = 3–6 from independent cell cultures).
(D) Frequency of cells in G0 phase derived from genome-edited CD34+CD38−CD45RA− cells 2 days after electroporation (mean ± SD, n = 3 from independent cell cultures).
(E) Number of colonies formed by genome-edited CD34+CD38−CD45RA−CD90+ cells (mean ± SD, n = 3 per group).
(F) Total number of genome-edited CD34+CD38−CD45RA−CD90+ cells 7 days after electroporation (mean ± SD, n = 3 from independent cell cultures).
(G) Frequency of CD34+CD45− cells among genome-edited CD34+CD38−CD45RA−CD90+ cells 7 days after electroporation (mean ± SD, n = 3 from independent cell cultures).
Next we performed a colony assay using CD34+CD38−CD45RA−CD90+ cells. Colony numbers were lower under the quiescence-maintaining and UM171-based conditions than under proliferative condition 1 and 2 (Figure 7E). However, the number of colony-forming unit (CFU)-GEMMs was higher under the quiescence-maintaining and UM171-based conditions than under proliferative condition 2 (Figure 7E). This result was confirmed using another cord blood sample (Figure S7E). These results suggest that the quiescence-maintaining and UM171-based conditions sustain more primitive cells than proliferative condition 2.
Finally, we tested the effect of the quiescence-maintaining condition in the longer term (day 7 after editing). The number of cultured cells was significantly lower under the quiescence-maintaining condition than under proliferative condition 1 (Figure 7F). The frequency of CD34+ cells was significantly higher under the quiescence-maintaining condition (Figure S7F). hCD45 expression was efficiently decreased in both culture conditions (Figure S7G). Accordingly, the frequency of CD34+CD45− cells was significantly higher under the quiescence-maintaining condition than under proliferative condition 1 (Figure 7G).
These results show that phenotypic and quiescent human HSPCs are maintained better under the quiescence-maintaining condition than under the proliferative condition in the short and long term after NHEJ-based gene editing.
Discussion
Through an analysis of quiescence in genome-edited HSCs, we present approaches for ex vivo study of quiescence in HSCs that do not require knockout mice. In this study, we demonstrated that (1) the genome editing efficiency of HSCs is primarily dependent on preculture but not post-culture conditions because of improvement of RNP transport to the nucleus, (2) quiescence-maintaining post-culture maintains the undifferentiated state of HSCs edited by NHEJ or HDR and reverts these cells to a quiescent state, and (3) this method can be applied to human HSPCs.
HSCs are quiescent compared with progenitors.1,2 Recent advances in culture methods revealed that maintenance of HSCs in quiescence in vitro helps retain their function.56,57,58 Furthermore, Shin et al.31 used genome editing in combination with mTOR and GSK3 inhibitors and showed that pharmacological control of the cell cycle effectively increases the HDR/NHEJ ratio in human mobilized peripheral blood CD34+ HSPCs.
These reports and our study provide an alternative approach to that used in another study, which performed highly efficient genome editing of HSCs under proliferative conditions using PVA-based medium.65 One advantage of our approach is that HSCs are maintained in a more physiological condition with less proliferation in the absence of chemical compounds, and the frequency (>30%) of pHSCs among total cells is higher, which enables the functions of genes specifically relevant for quiescence of HSCs to be studied. Koide et al.74 reported that the surface marker phenotype of HSCs (CD48− LSK cells) cultured under the PVA-based condition reflects their in vivo repopulation capacity.74 This suggests that retaining the HSC phenotype after genome editing is an alternative approach to maintaining functional HSCs. Notably, re-entry into quiescence requires adjustment of the concentrations of only two essential cytokines for HSCs (SCF and TPO). RNA-seq data indicated that reversion to quiescence after genome editing occurred in the early phase, and this was beneficial for maintaining HSC function compared with the proliferative condition. Considering the transcriptional alteration after genome editing, additional factors relevant to HSC quiescence, such as small-molecule compounds and transforming growth factor β,75,76 may further improve the properties of edited HSCs so that they are more similar to fresh quiescent HSCs.
Only limited studies have performed HSC-specialized genome editing,31,51,65,77 and there is room to improve the genome editing conditions for HSCs. Although HDR occurs exclusively during S/G2/M phases, our data suggest that genome editing via NHEJ also positively correlates with the cell cycle. It has been reported that transfected plasmid DNA is incorporated into the nucleus during telophase.78 Preculture is generally used for lentiviral transduction of HSCs. These observations suggest that nuclear delivery of exogenous DNA may be related to the cell cycle or cell division. In the setting of genome editing, the transfection efficiency of RNP determines the editing outcome in human CD34+ HSPCs.79 Interestingly, preculture dramatically improved delivery of RNP to the nucleus, and the transfection efficiency also increased, but this effect was moderate. This is attributed to the activation of cytosol-nucleus transport by preculture and improved delivery of RNP to the nucleus.80
Edited HSCs subjected to quiescence-maintaining post-culture showed relatively low donor-derived chimerism early after transplantation of day 11 and alteration of the transcriptome following genome editing. These differences from fresh HSCs indicate that further improvements in quiescence-maintaining culture as well as preculture or electroporation conditions are needed. Preculture might negatively affect HSC function based on the finding that day 7-mock HSCs formed a different cluster from day 7-CRISPR-quiescent HSCs in transcriptome analysis. To prevent proliferation and differentiation of HSCs, several compounds, such as PVA,65 UM171,81 and an mTOR inhibitor,31 have been introduced for genome editing to maintain and/or expand primitive HSPCs. Alternatively, Seki and Rutz82 reported that IL-7 improves the transfection efficiency and gene knockout efficiency of resting mouse and human T cells without increasing expression of differentiation markers during preculture. This suggests that the original phenotype and quiescence might be maintained by optimization of the preculture conditions. In future studies, it may be valuable to test similar approaches for quiescent HSCs, such as a new preculture condition that avoids loss of quiescence or electroporation of nuclear transport proteins together with RNP.
The present study established and optimized a platform to reintroduce HSCs that have been cultured in proliferation conditions for genome editing back to a quiescent state using physiological factors composed of the BM environment, such as fatty acids, low cytokines, and hypoxia. This method facilitates study of the biology and gene function in quiescent HSCs.
Limitations of the study
Our study has several limitations that should be considered in its interpretation. First, the methods reported here did not improve editing efficiency over that of a previous report,35 but we demonstrated how preculture improved the gene editing efficiency. As shown in Figures 2B and 2C, HSC editing efficiency is defined by preculture conditions (Figures 2B and 2C), suggesting that further improvements in preculture conditions could further increase editing efficiency. Second, we did not precisely determine when HSCs regain quiescence after gene editing. Based on cell cycle analysis (Figures S3D and S4D), the cell cycle begins to slow 2 days post-editing, and the edited HSCs return to a more dormant state over the following 5 days. Defining the cell cycle kinetics of HSCs ex vivo will be important in determining when HSCs regain quiescence after editing. This will be investigated in future studies using G0 reporters.6 Third, engraftment capacity was not improved over that of proliferative conventional/PVA-based conditions55 under short-term (day 2 post-electroporation) or long-term (day 10 post-electroporation) culture in quiescence-maintaining conditions after gene editing (Figures 5 and S5). Importantly, the chimerism of genome-edited HSCs in quiescence-maintaining conditions was comparable with that of fresh HSCs (day 3 transplantation) or culture-only HSCs (day 11 transplantation) and superior to that of proliferative conditions in long-term culture. This suggests that our method has similar engraftment capacity and poses further benefits for longer periods of time. Further optimization of culture conditions could improve engraftment capacity after gene editing. Fourth, the in vitro functions of HDR-edited mouse HSCs and HDR-edited human HSCs were successfully restored to the quiescent state (Figures 6I and 7D), but their transplantation capacity was not assessed. This will be evaluated in imminent future studies, as the application of our approach to clinical settings is our long-term objective. Finally, in HDR experiments, the frequency of CD150+CD48−LSK cells was lower in the GFPhigh fraction than in the GFP−/low fraction (Figure 6H). Undifferentiated cells may have low HDR efficiency, or GFPhigh HSCs (edited by HDR) could be more prone to differentiation than GFP−/low HSCs. We have developed a platform that should enable the study of mouse and human HSC quiescence after gene editing by NHEJ. To clarify how the quiescence-maintaining post-culture condition affects the in vivo function of HDR-edited mouse and human HSCs, BM transplantation using HDR-edited HSCs should be evaluated in future studies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD4-PerCP-Cy5.5 (clone: RM4-5) | TONBO biosciences | Cat# 65-0042-U100; RRID: AB_2621876 |
| Anti-mouse CD8a-PerCP-Cy5.5 (clone: 53-6.7) | TONBO biosciences | Cat# 65-0081-U100; RRID: AB_2621882 |
| Anti-mouse B220-PerCP-Cy5.5 (clone: RA3-6B2) | TONBO biosciences | Cat# 65-0452-U100; RRID: AB_2621892 |
| Anti-mouse B220-APC (clone: RA3-6B2) | BioLegend | Cat# 103212; RRID: AB_312997 |
| Anti-mouse Ter-119-PerCP-Cy5.5 (clone: TER-119) | TONBO biosciences | Cat# 65-5921-U100 |
| Anti-mouse Gr1 (Ly-6G/6C)-PerCP-Cy5.5 (clone: RB6-8C5) | BioLegend | Cat# 108428; RRID: AB_893558 |
| Anti-mouse Gr1-PE-Cy7 (clone: RB6-8C5) | TONBO biosciences | Cat# 60-5931-U100; RRID: AB_2621870 |
| Anti-mouse Mac1 (CD11b)-PerCP-Cy5.5 (clone: M1/70) | TONBO biosciences | Cat# 65-0112-U100; RRID: AB_2621885 |
| Anti-mouse Mac1-PE-Cy7 (clone: M1/70) | TONBO biosciences | Cat# 60-0112-U100; RRID: AB_2621836 |
| Anti-mouse CD45.1-PE (clone: A20) | BD biosciences | Cat# 553776; RRID: AB_395044 |
| Anti-mouse CD45.2-BV421 (clone: 104) | BD biosciences | Cat# 562895; RRID: AB_2737873 |
| Anti-mouse CD45.2-Alexa Four 700 (clone: 104) | BioLegend | Cat# 109822; RRID: AB_493731 |
| Anti-mouse Sca-1 (Ly-6A/E)-PE-Cy7 (clone: E13-161.7) | BioLegend | Cat# 122514; RRID: AB_756199 |
| Anti-mouse Sca-1-Alexa488 (clone: E13-161.7) | BioLegend | Cat# 122516; RRID: AB_756201 |
| Anti-mouse Sca-1-BV785 (clone: D7) | BioLegend | Cat# 108139; RRID AB_2565957 |
| Anti-mouse c-Kit (CD117)-APC-Cy7 (clone: 2B8) | BioLegend | Cat# 105826; RRID: AB_1626278 |
| CD117 MicroBeads Mouse | Miltenyi Biotec | Cat# 130-091-224 |
| Anti-mouse CD150-PE (clone: TC15-12F12.2) | BioLegend | Cat# 115904; RRID: AB_313683 |
| Anti-mouse CD150-BV421 (clone: TC15-12F12.2) | BioLegend | Cat# 115926; RRID: AB_2562190 |
| Anti-mouse CD150-BV785 (clone: TC15-12F12.2) | BioLegend | Cat# 115937; RRID: AB_2565962 |
| Anti-mouse CD48-FITC (clone: HM48-1) | BioLegend | Cat# 103404; RRID: AB_313019 |
| Anti-mouse CD48-PE (clone: HM48-1) | BioLegend | Cat# 103406; RRID: AB_313021 |
| Anti-mouse CD48-APC (clone: HM48-1) | BioLegend | Cat# 103412; RRID: AB_571997 |
| Anti-mouse CD48-BV510 (clone: HM48-1) | BD biosciences | Cat# 563536; RRID: AB_2738266 |
| Anti-mouse CD48-Biotin (clone: HM48-1) | BioLegend | Cat# 103410; RRID: AB_528827 |
| Fc-block (anti-mouse CD16/32) (clone: 2.4-G2) | BD biosciences | Cat# 553142; RRID: AB_394657 |
| Anti-mouse CD16/32-BV510 (clone: 2.4G2) | BD biosciences | Cat# 740111; RRID: AB_2739869 |
| Anti-mouse CD16/32- PE (clone: 93) | BioLegend | Cat# 101307; RRID: AB_312807 |
| Anti-mouse CD16/32- Alexa700 (clone: A93) | Thermo Fisher Scientific | Cat# 56-4321-80; RRID: AB_493994 |
| Anti-mouse CD34-FITC (clone: RAM34) | BD biosciences | Cat# 562608; RRID: AB_11154576 |
| Anti-mouse CD34-BV421 (clone: MEC14.7) | BioLegend | Cat# 119321; RRID: AB_10900980 |
| Anti-mouse Flt3 (CD135)-APC (clone: A2F10) | BioLegend | Cat# 135310; RRID: AB_2107050 |
| Anti-mouse CD201 (EPCR)-PE (clone: RCR-16) | BioLegend | Cat# 141504; RRID: AB_10895909 |
| Anti-mouse CD45-FITC (clone: 30-F11) | BD biosciences | Cat# 553080; RRID: AB_394609 |
| Anti-mouse CD45-PE (clone: 30-F11) | BioLegend | Cat# 103134; RRID: AB_2562559 |
| Anti-mouse CD45-BV421 (clone: 30-F11) | BioLegend | Cat# 103134; RRID: AB_2562559 |
| Anti-mouse CD45-BV786 (clone: 30-F11) | BD biosciences | Cat# 564225; RRID: AB_2716861 |
| Anti-mouse CD45-Biotin (clone: 30-F11) | BioLegend | Cat# 103104; RRID: AB_312969 |
| Anti-Streptavidin-Alexa Fluor 555 (clone: TER-119) | Thermo Fisher Scientific | Cat# S21381; RRID: AB_2307336 |
| CD34 MicroBeads Human | Miltenyi Biotec | Cat# 130-046-702 |
| Anti-human CD34-FITC (clone: 581) | BD Bioscience | Cat# 560942; RRID: AB_396150 |
| Anti-human CD34-APC (clone: 561) | BioLegend | Cat# 343607; RRID: AB_2074356 |
| Anti-human CD38-PerCP-Cy5.5 (clone: HIT2) | BD Bioscience | Cat# 551400; RRID: AB_394184 |
| Anti-human CD45RA-PE (clone: HI100) | BD Bioscience | Cat# 555489; RRID: AB_395880 |
| Anti-human CD90-PE-Cy7 (clone: 5E10) | BioLengend | Cat# 328123; RRID: AB_2561692 |
| Anti-human CD201 (EPCR)-BV421 (clone: RCR-252) | BD Bioscience | Cat# 743552; RRID: AB_2741576 |
| Anti-human CD201 (EPCR)-PE (clone: RCR-401) | BioLegend | Cat# 351903; RRID: AB_10897801 |
| Anti-human CD45-PE (clone: 2D1) | BioLegend | Cat # 368510; RRID: AB_2566370 |
| Anti-mouse Ki67-eFlour660 (clone: SolA15) | eBioscience | Cat# 50-5698-82; RRID: AB_2574235 |
| Anti-mouse/human Ki67-FITC (clone: 11F6) | BioLegend | Cat# 151212; RRID: AB_2814055 |
| Annexin V-APC | BioLegend | Cat# 640920; RRID: AB_2561515 |
| Chemicals, peptides, and recombinant proteins | ||
| PBS | Nacalai Tesque | Cat# 14249-24 |
| SF-O3 | SEKISUI MEDICAL | SS1303 |
| DMEM/Ham’s-F12 medium | Nacalai Tesque | Cat# 11581-15 |
| RPMI1640 | Nacalai Tesque | Cat# 30263-95 |
| StemSpan SFEM | STEMCELL Technologies | Cat# 09650 |
| HemEX-Type9A | Cell Science & Technology Institute | Cat# A5P00P01C |
| MethoCult GF M3434 | STEMCELL Technologies | Cat# 03444 |
| MethoCult H4434 | STEMCELL Technologies | Cat# 04434 |
| Insulin-Transferrin-Selenium-Ethanolamine (ISTX) 1000x | Thermo Fisher Scientific | Cat# 51500-056 |
| 2-mercapto ethanol (2-ME) 1000x | Thermo Fisher Scientific | Cat# 21985-023 |
| Penicillin | Meiji Seika | PGLD755 |
| Streptomycin sulfate | Meiji Seika | SSDN1013 |
| Fetal bovine serum | Thermo Fisher Scientific | Cat# 10270-106 |
| Bovine serum albumin | Sigma Aldrich | Cat# A4503-100G |
| Palmitic acid | Wako Pure Chemical Corporation | Cat# 165-00102 |
| Oleic acid | Sigma Aldrich | Cat# O1383-1G |
| Cholesterol | Sigma Aldrich | Cat# C3045-5G |
| Sodium Hydroxide | Wako Pure Chemical Corporation | Cat# 191-01665 |
| Recombinant Murine SCF | PeproTech | Cat# 250-03 |
| Recombinant Human TPO | PeproTech | Cat# 300-18 |
| Recombinant Murine G-CSF | PeproTech | Cat# 250-05 |
| Recombinant Human SCF | PeproTech | Cat# 250-31L |
| Recombinant Human FLT3L | PeproTech | Cat# 300-19 |
| Recombinant Human IL-3 | PeproTech | Cat# 200-03 |
| Recombinant Human IL-6 | PeproTech | Cat# 200-06 |
| DMSO | Sigma Aldrich | Cat# D8418 |
| UM171 | Selleck | Cat# 7608 |
| Lymphoprep | STEMCELL Technologies | Cat# 07811 |
| BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit | Thermo Fisher Scientific | Cat# BDB554714 |
| 5-ethynyl-2′-deoxyuridine (EdU) | Tokyo Chemical industry | Cat# E1057 |
| Tris-hydroxypropyltriazolymethylamine (THPTA) | Click Chemistry Tools | Cat# 1010 |
| AFDye 647 Picolyl Azide | Click Chemistry Tools | Cat# 1300-1 |
| Copper (Brenet et al.)75 Sulfate Pentahydrate | Tokyo Chemical Industry Co., Ltd. | Cat# 7758-99-8 |
| Ascorbic acid | Tokyo Chemical Industry Co., Ltd. | Cat# 03428-72 |
| Click-iT Plus EdU Alexa Flour 647 Flow Cytometry Assay Kit | Thermo Fisher Scientific | Cat# C10634 |
| Propidium iodide | Lifetechnologies | Cat# P3566 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat# H3570 |
| TrueCut Cas9 Protein v2 | Thermo Fisher Scientific | Cat# A36299 |
| Flow-Check Fluorospheres | Beckman Coulter | Cat# 7547053 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat# 34094 |
| Critical commercial assays | ||
| Neon Transfection System 10μL Kit | Thermo Fisher Scientific | Cat# MPK1096 |
| CUGA7 sgRNA Synthesis Kit | Nippon Gene | Cat# 314-08691 |
| TaKaRa Ex Taq | Takara Bio Inc | Cat# RR001A |
| Q5 High-Fidelity 2x Master Mix | New England BioLabs | Cat# M0492S |
| NucleoSpin Tissue XS | Macherey Nagel | Cat# 740901.50 |
| Wizard SV Gel and PCR Clean-Up System | Promega | Cat# A9282 |
| Annexin V-PE Apoptosis Detection Kit | BD Biosciences | Cat# 559763 |
| RNeasy Mini Kit | QIAGEN | Cat# 74136 |
| SuperScript VILO cDNA Synthesis Kit | Thermo Fisher Scientific | Cat# 11754050 |
| TB Green Premix Ex Taq II | Takara Bio | Cat# RR820D |
| AAVpro Purification Kit | Takara Bio | Cat# 6666 |
| AAV Helper Free Expression System | Cell Biolabs., Inc | Cat# VPK-402 |
| Deposited data | ||
| RNA-seq | This paper | DNA Data Bank of Japan (DDBJ): Accession #DRA014998 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6JJmsSlc | Japan SLC, Inc. | http://www.jslc.co.jp/english/index2.htm |
| Mouse: C57BL/6J-Ly5.1 | CLEA Japan, Inc | N/A |
| Mouse: C57BL/6-Tg(Ubc-GFP)30Scha/J | The Jackson Laboratory | JAX stock #004353 |
| Mouse: Evi1-IRES-GFP mice | Kataoka et al.70 | N/A |
| Oligonucleotides | ||
| sgRNA Common primer: AAAAGCACCGACTCGGTGCC | This paper | N/A |
| sgRNA Reversed primer: AAAAGCACCGACTCGGTGCCA CTTTTTCAAGTTGATAACGGAC TAGCCTTATTTTAACTTGCT ATT TCTAGCTCTAAAAC |
This paper | N/A |
| Rosa sgRNA target Forward primer: ttaatacgactcactataggACTCCAGTCT TTCTAGAAGAgttttagagctagaaatagc |
Chu et al.60 | N/A |
| Mouse CD45 sgRNA target Forward primer: ttaatacgactcactataGGGTTTGTGGCTCAAA CTTCgttttagagctagaaatagc |
This paper | N/A |
| GFP sgRNA target Forward primer: ttaatacgactcactataGGGCGAGGAGCTGTT CACCGgttttagagctagaaatagc |
Gundry et al.35 | |
| Rosa forward primer: CCAAAGTCGCTCTGAGTTGTTATCAGT | This paper | N/A |
| Rosa reverse primer: GGAGCGGGAGAAATGGATATGAAG |
This paper | N/A |
| Rosa sequence primer: ACATAGTCTAACTCGCGACAC |
This paper | N/A |
| Mouse CD45 forward primer: AGAAGCCATTGCACTGACTTTG |
This paper | N/A |
| Mouse CD45 reverse primer: GTGTGATCTTTCCCCGAAACAT |
This paper | N/A |
| Mouse CD45 sequence primer: CTGCAAAGAGGACCCTTTACAGT |
This paper | N/A |
| Ubc-GFP forward primer: GTTCACCTTGATGCCGTTCT |
This paper | N/A |
| Ubc-GFP reverse and sequence primer: CACCCGTTCTGTTGGCTTAT | This paper | N/A |
| Rosa sgRNA: ACUCCAGUCUUUCUAG AAGA |
Synthego Chu et al.60 |
N/A |
| Mouse CD45 sgRNA: GGGUUUGUGGCU CAAACUUC |
Synthego | N/A |
| AAVS1 sgRNA: GGGGCCACUAGGGA CAGGAU |
Synthego Bak et al.37 |
N/A |
| Human CD45 sgRNA: GGUGCUGGUGUUGGGCGCAC | Synthego Gundry et al.35 |
N/A |
| GFP sgRNA: GGGCGAGGAGCU GUUCACCG |
Synthego Gundry et al.35 |
N/A |
| Alt-R CRISPR-Cas9 crRNA XT for CD45: GGGTTTGTGGCTCAAACTTC |
IDT | N/A |
| Alt-R CRISPR-Cas9 tracRNA | IDT | N/A |
| Alt-R CRISPR-Cas9 tracRNA ATTO | IDT | N/A |
| AAV LHA in-out forward primer (Rosa genome): CTCTGGGGGAGTCGTTTTACC | This paper | N/A |
| AAV LHA in-out reverse primer (CAG promoter): CCAGGCGGGCCATTTACC | This paper | N/A |
| AAV RHA in-out forward primer (EGFP): AACGA GAAGCGCGATCACAT |
This paper | N/A |
| AAV RHA in-out reverse primer (Rosa genome): ACAGCCTCGATTTGTGGTGT | This paper | N/A |
| AAV titration qPCR forward primer: GGAACCCCTAGTGATGGAGTT | Aurnhammer et al.83 | N/A |
| AAV titration qPCR reverse primer: CGGCCTCAGTGAGCGA | Aurnhammer et al.83 | N/A |
| Infb1 qPCR forward primer: CCTGGAGCAGCTGAATGGAA | Takara Bio | N/A |
| Infb1 qPCR reverse primer: TGGATGGCAAAGGCAGTGAA | Takara Bio | N/A |
| Oas2 qPCR forward primer: GGCCTGGTACAGCCTTGGAA | Takara Bio | N/A |
| Oas2 qPCR reverse primer: AGTCTTTGCCAGATCACTCCAGAA | Takara Bio | N/A |
| Ddx58 (RIG-I) qPCR forward primer: AGCCAAGGATGTCTCCGAGGAA | Liu et al.84 | N/A |
| Ddx58 (RIG-I) qPCR reverse primer: ACACTGAGCACGCTTTGTGGAC | Liu et al.84 | N/A |
| Isg15 qPCR forward primer: CATCCTGGTGAGGAACGAAAGG | Liu et al.84 | N/A |
| Isg15 qPCR reverse primer: CTCAGCCAGAACTGGTCTTCGT | Liu et al.84 | N/A |
| β-actin qPCR forward primer: CATCCGTAAAGACCTCTATGCCAAC | Takara Bio | N/A |
| β-actin qPCR reverse primer: ATGGAGCCACCGATCCACA | Takara Bio | N/A |
| Software and algorithms | ||
| FlowJo version 10.7.2 | Tree Star | https://www.flowjo.com/solutions/flowjo |
| SnapGene | GLS Biotech | https://www.snapgene.com/ |
| Molecular Biology tool | Benchling | https://www.benchling.com |
| TIDE | Brinkman et al.85 | https://tide.nki.nl |
| Prism v7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Imaris 9.8.2 | Oxford Instruments | https://imaris.oxinst.com/support/imaris-release-notes/9-8-0 |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| R v.4.1.0 | R Development Core Team (2008) | http://www.r-project.org |
| DESeq2 | Love et al.86 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| fGSEA | Korotkevich et al.87 | http://bioconductor.org/packages/release/bioc/html/fgsea.html |
| GSVA | Hanzelmann et al.88 | http://bioconductor.org/packages/release/bioc/html/GSVA.html |
| Others | ||
| Neon Transfection System | Thermo Fisher Scientific | Cat# MPK5000 |
| Neon Transfection System 10μL Kit | Thermo Fisher Scientific | Cat# MPK1096 |
| Nucleofector 2b | Lonza | AAB-1001 |
| Human CD34+ Cell Nucleofector Kit | Lonza | PVA-1003 |
| NanoDrop Onec Microvolume UV-Vis Spectrophotometer | Thermo Fisher Scientific | Cat# ND-ONEC-W |
| Hiseq1500 | Illumina | |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Keiyo Takubo (keiyot@gmail.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
The RNA-seq data reported in this paper have been deposited at DNA Data Bank of Japan (DDBJ) (accession number: DRA014998) and is available at the time of publication.
-
•
This paper did not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Mice
C57BL/6J mice (8–19 weeks old, purchased from SLC Japan or CLEA Japan) were used in all experiments, unless otherwise stated. C57BL/6-Ly5.1 congenic mice purchased from CLEA Japan were used for competitive repopulation assays. Ubc-GFP reporter mice (C57BL/6-Tg(Ubc-GFP)30Scha/J)89 were purchased from The Jackson Laboratory. All mice were bred in the animal facility at the National Center for Global Health and Medicine under specific pathogen-free conditions and fed ad libitum. Mice were euthanized by cervical dislocation. Animal experiments were approved by the National Center for Global Health and Medicine. Both male and female mice were used in experiments.
Preparation of human CD34+ cord blood cells
Cord blood was collected with written informed consent from the mother before delivery at the Japanese Red Cross Cord Blood Bank. Cord blood experiments were approved by the ethical committees of the Japanese Red Cross Cord Blood Bank and National Center for Global Health and Medicine. The sample was processed using Lymphoprep (GE Healthcare) density gradient medium to isolate mononuclear cells. CD34 enrichment was performed by magnetic cell separation using a MACS CD34 Microbead Kit (Miltenyi Biotec). CD34+ enriched cells were frozen until use. Frozen cells were thawed in vials in a 37°C water bath and transferred to a 15 mL tube. RPMI-1640 medium containing 10% fetal calf serum (FCS) was slowly added to the suspension while gently swirling to fill the tube. The suspension was centrifuged at 200 × g for 15 min at room temperature. Supernatants were aspirated and the wash step was repeated. An antibody cocktail (50 μL of phosphate-buffered saline (PBS) containing 2% FCS plus 10 μL of anti-CD34-FITC, 2 μL of anti-CD38-PerCP-Cy5.5, 5 μL of anti-CD90-PE-Cy7, and 10 μL of anti-CD45RA-PE) was added to the suspension and kept on ice for 30 min. Cells were washed once with PBS containing 2% FCS and resuspended in PBS containing 2% FCS supplemented with 0.1% propidium iodide (PI). Cells were sorted into StemSpan SFEM-I using a FACSAria IIIu instrument.
Method details
Cell preparation
Mouse BM cells were isolated from both femurs, tibiae, and the pelvis. Femurs, tibiae, and the pelvis were flushed with PBS containing 2% FCS using a 21-gauge needle (Terumo) and a 10 mL syringe (Terumo) to collect the BM plug. The plug was dispersed by repeatedly passing it through the needle. The suspension was centrifuged at 680 × g for 5 min at 4°C. Cells were then lysed with lysis buffer (0.17 M NH4Cl, 1 mM EDTA, and 10 mM NaHCO3) on ice for 1 min, washed with two volumes of PBS containing 2% FCS, and centrifuged at 680 × g for 5 min at 4°C. Cells were resuspended in PBS containing 2% FCS, filtered through a 40 μm nylon mesh (BD Biosciences), centrifuged at 680 × g for 5 min at 4°C, and treated with an anti-CD16/32 antibody to block the Fc receptor (2 μL/mouse) for 5 min at 4°C. Thereafter, anti-c-Kit magnetic beads (Miltenyi) were added at a vol/vol ratio of 1/5 to 1/10, and samples were incubated for 15 min at 4°C. After two washes with PBS containing 2% FCS to remove the antibody, c-Kit+ cells were isolated using Auto-MACS Pro (Miltenyi) and the Possel-S program. Isolated cells were centrifuged at 340 × g for 5 min and stained with antibodies for flow cytometry.
Cell culture
For mouse HSPCs, SF-O3 medium (Sekisui Medical), StemSpan SFEM-I (STEMCELL Technologies), custom medium (GMEP), and HemaEx-Type9A medium (Cell Science and Technology) were used for cell sorting and editing experiments, and all media were supplemented with 1% penicillin/streptomycin. SF-O3, StemSpan SFEM-I, and custom medium was used for culture under proliferative conditions. HemaEx-Type9A medium was used for culture under PVA-based conditions after supplementation with cytokines, 1% recombinant human insulin, recombinant human transferrin, sodium selenite, and ethanolamine (ITSE) (Thermo Fisher Scientific). Custom medium was used for culture under quiescence-maintaining condition as previously reported with slight modifications.57,90 Briefly, custom medium was DMEM/F-12 containing 4% w/v BSA (Sigma-Aldrich), 100 μg/mL palmitic acid (Wako Pure Chemical Corporation), 100 μg/mL oleic acid (Sigma-Aldrich), and 20 μg/mL cholesterol (Sigma-Aldrich) adjusted to pH 7.6 ± 0.1 using NaOH (Wako Pure Chemical Corporation). Sodium pyruvate was excluded and 1 mM sodium lactate was added. Cytokines, 55 μM 2-mercaptoethanol (Thermo Fisher Scientific), and 0.1% ITSE were added to custom medium before culture. The cytokine conditions were 0.5–1.5 ng/mL SCF and 1 ng/mL TPO for the quiescence-maintaining condition, 50–100 ng/mL SCF and 50–100 ng/mL TPO for the proliferative condition, and 10 ng/mL SCF and 100 ng/mL TPO in a fibronectin-coated plate for the PVA-based condition. For culture of GMPs under the proliferative condition, 100 ng/mL SCF and 5 ng/mL G-CSF were used. Custom medium lacking 4% BSA was added to dilute cells with/without electroporation and attained the desired number of cells per well in 10 μL, which was dispensed into 200 μL of culture medium. Culture conditions were 1% O2 and 5% CO2 or 20% O2 and 5% CO2 at 37°C in appropriate humidified incubators. For 7 days cultures, the medium was not changed. For longer periods of culture (10 days), 150–170 μL of medium was changed every day.
For human HSPCs, cells were cultured in SFEM-I containing 100 ng/mL human SCF (hSCF), 100 ng/mL TPO, 100 ng/mL human FLT3L (hFLT3L), and 35 nM UM171 in 1% O2 for 48 h before electroporation. The post-culture conditions were medium containing 4% BSA supplemented with 3.0 ng/mL hSCF and 3.0 ng/mL TPO in 1% O2 for the quiescence-maintaining condition, SFEM-I containing 100 ng/mL hSCF, 50 ng/mL TPO, 100 ng/mL hFLT3L, and 35 nM UM171 in 20% O2 for the UM171-based condition, and SFEM-I or SF-O3 containing 100 ng/mL hSCF, 100 ng/mL TPO, 100 ng/mL hFLT3L, 20 ng/mL human IL-3, and 20 ng/mL IL-6 in 20% O2 for the proliferative condition.
Flow cytometry and cell sorting
After selecting c-Kit+ cells using magnetic beads, the murine HSPC fraction was labeled as follows. For staining of C57BL/6J mice, lineage marker (CD4, CD8a, Gr-1, Mac-1, Ter-119, B220)-PerCP-Cy5.5, c-Kit-APC-Cy7, Sca-1-PE-Cy7, Sca-1-BV785, CD150-PE, CD150-BV421, CD48-FITC, EPCR-PE, Flt3-APC, FcγRII/III-Alexa Fluor 700, and CD34-FITC were used. For staining of Ubc-GFP and Evi1-IRES-GFP mice, lineage marker (CD4, CD8a, Gr-1, Mac-1, Ter-119, B220)-PerCP-Cy5.5, c-Kit-APC-Cy7, Sca-1-PE-Cy7, CD150-BV421, CD48-PE, and Flt3-APC were used. A total of 0.5 μL of each antibody was used per mouse. Cells were resuspended in 0.5–2 mL of PBS containing 2% FCS and 0.1% PI, and sorted using an FACSAria IIIu instrument into SF-O3 medium or StemSpan SFEM-I. Murine HSCs were defined as CD150+CD48-Flt3−LSK91 or CD150+CD34−LSK.55 MPPs were sub-fractionated into MPP1 (CD150−CD48-Flt3−LSK), MPP2 (CD150+CD48+Flt3−LSK), MPP3 (CD150−CD48+Flt3- LSK), and MPP4 (Flt3+LSK). Myeloid and lymphoid progenitors were defined as CMPs (Lin−CD34+FcγRII/III−), GMPs (Lin−CD34+FcγRII/III+), MEPs (Lin−CD34−FcγRII/III−), and CLPs (Lin−Sca-1lowc-KitlowIL-7R+). Data were analyzed using FlowJo™ software.
In vitro sgRNA synthesis
The sgRNA target sequences were determined based on previous reports for Rosa26-1,60 and GFP.35 The target sequences of CD45 were determined by reference to benchling (https://www.benchling.com). Each sgRNA was synthesized in vitro using a CUGA7 gRNA Synthesis Kit (Nippon Gene) following the manufacturer’s instructions. Briefly, to generate template DNA, PCR was performed using ExTaq (TaKaRa Bio Inc.) and three primers, namely, a sgRNA common primer, a sgRNA reverse primer, and each forward primer including the T7 promoter and target sequence. To achieve optimal transcription by the T7 RNA promoter in the CUGA 7 enzyme reaction, guanine was added to the beginning of the target sequence if it did not already start with guanine. The PCR conditions were 98°C for 3 min, followed by 35 cycles of 98°C for 30 s, 60°C for 30 s, and 72°C for 30 s, and then 72°C for 3 min for final extension. To synthesize sgRNA, template DNA was incubated with CUGA7 Enzyme Solution at 37°C for 2 h, and then at 37°C for 15 min after addition of 2 μL of DNase I to remove template DNA (final volume 22 μL). To avoid decreasing the sgRNA yield, 578 μL of gRNA Binding Buffer was added and thoroughly mixed. Six hundred microliters of the mixture was applied to a spin column and centrifuged at 16,000 g for 1 min at 4°C. The column was washed by adding 750 μL of gRNA Wash Buffer and the centrifugation was repeated. To elute sgRNA, the spin column was centrifuged after reaction with 20 μL of RNase-free water at room temperature for 3 min. The sgRNA concentration was determined using a NanoDrop Onec instrument (Thermo Fisher Scientific). sgRNA was diluted to a concentration of 1.5 μg/μL and cryopreserved at −80°C until use.
Chemically modified sgRNAs
Chemically modified sgRNAs with three terminal nucleotides at both the 5′ and 3′ ends containing 2′ O-methyl 3′ phosphonothioate were purchased from Synthego.
CRISPR-Cas9
Cells sorted using a FACSAria IIIu instrument were cultured in SF-O3 medium or SFEM-I supplemented with 50 ng/mL SCF and 50 ng/mL TPO (preculture medium) in 20% O2 and 5% CO2 for 16–24 h. For most experiments, 1.0–3.0×104 HSCs were sorted and precultured in a 96-well plate or 12-well dish at a density lower than 1.0×105/mL. RNP complex preparation and the electroporation conditions were previously described.35 In brief, 3 μg of Cas9 protein (Thermo Fisher Scientific, TrueCut Cas9 Protein v2) and 3 μg of sgRNA were incubated for 15 min at room temperature in a final volume of 6 μL and kept at 4°C until use. Cultured cells were resuspended in 30 μL of Buffer T and added to the RNP to yield a total volume of 36 μL. Cells were electroporated using the Neon Transfection System (Thermo Fisher Scientific) and the following conditions: 1700 V, 20 ms, and one pulse for mouse HSPCs and 1600 V, 10 ms, and three pulses for human HSPCs. Alternatively, a Lonza Nucleofector 2b (program: U-008) and a CD34+ Cell Nucleofector Kit (PVA-1003, Lonza) were used for nucleofection. Cell suspension was transferred to post-culture medium.
Evaluation of gene editing efficiency
The knockout efficiencies of CD45 and GFP were compared with that of Rosa using FACSAria IIIu and MACSQuant instruments. Mock cells were also analyzed. For staining of C57BL/6J mice, lineage marker (CD4, CD8a, Gr-1, Mac-1, Ter-119, B220)-PerCP-Cy5.5, c-Kit-APC-Cy7, Sca-1-PE-Cy7, CD150-PE, CD48-FITC, CD34-BV421, CD45-FITC, and CD45-BV421 were used. For staining of Ubc-GFP and Evi1-IRES-GFP mice, lineage marker (CD4, CD8a, Gr-1, Mac-1, Ter-119, B220)-PerCP-Cy5.5, c-Kit-APC-Cy7, Sca-1-PE-Cy7, CD150-PE, CD48-APC, and CD45-BV421 were used. To evaluate the gene editing efficiency, genomic DNA was extracted from cells using NucleoSpin Tissue XS (Macherey Nagel) or QuickExtract DNA Extraction Solution (Epicentre) at 2–3 days after electroporation. To evaluate homozygous or heterozygous mutations, single cell-derived colonies was picked at 7 days after culture in MethoCult and DNA was extracted. PCR was performed using Q5 High-Fidelity DNA Polymerase (New England BioLabs) and the following conditions: 98°C for 30 s, followed by 35 cycles of 98°C for 10 s, 67°C for 30 s, and 72°C for 30 s, and then 72°C for 2 min for final extension. PCR products were purified using Wizard SV Gels and the PCR Clean-Up System (Promega) following the manufacturer’s instructions. The TIDE assay85 was performed for sequencing data of each PCR product obtained by Sanger sequencing. The sgRNA with the best editing efficiency was used in subsequent experiments.
Evaluation of the transfection efficiency of RNP
ATT550-labeled or non-labeled tracrRNA and crRNA were purchased from IDT. tracRNA and crRNA were duplexed according to the manufacturer’s recommendation. For staining of LSK cells, lineage marker (CD4, CD8a, Gr-1, Mac-1, Ter-119, B220)-PerCP-Cy5.5, c-Kit-APC-Cy7, and Sca-1-BV785 were used. Fresh or precultured LSK cells were electroporated with ATT550-labeled or non-labeled RNP. Mock-electroporated cells were prepared. Cells were cultured under the proliferative condition and stained with DAPI before analysis. The frequency of ATTO550+ cells was determined by comparison with samples treated with non-labeled RNP as a negative control.
AAV6 production and AAV infection
The AAV Hepler Free System (Takara Bio Inc) was used to produce AAV6. In brief, AAV6 vector plasmids were cloned into the pAAV-CMV plasmid containing ITRs from AAV serotype 2. The vector contained 400 bp homology arms flanking the CRISRP-Cas9 cut site and a reporter gene (EGFP) under the control of the CAG promoter followed by polyA. AAV6 particles were produced in AAV293T cells transfected with the pRC6 vector containing the Cap gene and the pHelper vector containing helper genes using the standard PEI transfection protocol. AAV293T cells were harvested after 72 h AAV6 was purified using a AAVpro Purification Kit (Takara Bio Inc.) according to the manufacturer’s instructions and stored at −80°C until use. Alternative method was also used as previously described.92 AAV6 virus was titered by measuring the number of vector genomes using qPCR as described previously.83 For HDR-based editing experiments, electroporated cells without RNP (mock) were immediately added to 100 μL of StemSpan SFEM-I containing 100 ng/mL SCF and 100 ng/mL TPO at 37°C. The AAV volume was kept lower than 20% of the total culture volume as previously reported.93 Fresh medium was added after overnight culture, and the medium was changed to the post-culture condition at 24 h after transduction. A mock-electroporated control was included in most experiments and a MOI (vector genomes/cell) of 10,000–50,000 was used. Proper integration of the AAV donor DNA into the genomic Rosa locus was confirmed by PCR with a forward primer that recognized the AAV construct (within EGFP) and a reverse primer that recognized the genome (in Rosa).
EdU incorporation assay
EdU (Tokyo Chemical Industry) was diluted to a concentration of 5 mg/mL (in vivo master stock) or 10 mM (in vitro master stock) using PBS and preserved at −30°C. For the in vivo EdU assay, 1 mg of EdU (200 μL from the master stock) was intra-orbitally injected per mouse. After labeling for 2 h, mice were sacrificed, BMMNCs were isolated, and Fc block was performed as described in the cell preparation section. For the in vitro EdU assay, the 10 mM stock of EdU was added to the culture medium at a final concentration of 10 μM and incubated at 37°C for 2 h. Anti-lineage (CD4, CD8a, Gr-1, Mac-1, Ter-119, B220)-PerCP-Cy5.5, anti-c-Kit-APC-Cy7, anti-Sca-1-Alexa Fluor 488, anti-CD150-BV785, anti-CD48-BV510, anti-CD48-Biotin, anti-Streptavidin-Alexa Fluor 555, anti-CD45-BV421, and anti-CD45-BV786 were used to detect surface antigens. After staining, cells were fixed at 4°C for 20 min and permeabilized at room temperature for 15 min using BD Cytofix/Cytoperm buffer (Thermo Fisher Scientific). To collect all cultured cells, cells were centrifuged at 5000 rpm for 5 min after fixation. For the click reaction, cells were incubated with AFDye 647 picolyl azide (Click Chemistry Tools), 2 mM THPTA (Click Chemistry Tools), 2 mM CuSO4 (Tokyo Chemical Industry), and 10 mM ascorbic acid (Tokyo Chemical Industry) (final volume 250 μL/sample) at 37°C for 30 min in the dark. Ascorbic acid was freshly prepared for each experiment. Cells were washed with Cytoperm buffer, filtered, and analyzed with a BD FACSAria IIIu instrument. A fluorescence minus one (FMO) sample of AFDye647 was prepared for each experiment. Alternatively, a Click-iT EdU cell Proliferation Kit (ThermoFisher Scientific) was used. For surface marker staining, anti-lineage (CD4, CD8a, Gr-1, Mac-1, Ter-119, B220)-PerCP-Cy5.5, anti-c-Kit-APC-Cy7, anti-Sca-1-BV785, anti-CD150-BV421, anti-CD48-FITC, anti-CD45-Biotin, and anti-Streptavidin-Alexa Fluor 555 were used.
Cell cycle analysis
Hoechst 33,342 and Ki-67-eFlour660 (for mouse HSPCs) or Ki-67-FITC (for human HSPCs) were used to analyze the cell cycle. BMMNCs were treated with Fc block (2 μL/sample) for 10 min at 4°C. Anti-lineage (CD4, CD8a, Gr-1, Mac-1, Ter-119, B220)-PerCP-Cy5.5, anti-c-Kit-APC-Cy7, anti-Sca-1-PE-Cy7, anti-CD150-PE, anti-FcγR-PE, anti-CD48-FITC, and anti-CD34-FITC were used to detect surface antigens. Stained samples were washed and centrifuged at 340 g at 4°C for 5 min. Cells were fixed and permeabilized using BD Cytofix/Cytoperm, according to the manufacturer’s instructions. Samples were washed and suspended in 250 μL of PBS containing 2% FCS together with 10 μg/mL Hoechst 33342, filtered, and analyzed with a BD FACSAria IIIu instrument.
Apoptosis analysis
Cultured HSCs were stained with DAPI (Invitrogen) and Annexin V-APC (BioLegend), according to the manufacturers’ instructions. Cells were resuspended in 250 μL of PBS containing 2% FCS and 0.1% PI. Apoptotic (Annexin V+) cells were detected using a BD FACSAria IIIu instrument.
Colony-forming assay
For mouse HSPCs, total cells derived from 500 edited LSK cells were added to 3 mL of MethoCult M3434 (STEMCELL Technologies) and plated in triplicate. For human HSPCs, total cells derived from 500 edited CD34+CD38−CD45RA−CD90+ cells were added to 4 mL of MethoCult H4434 (STEMCELL Technologies) and plated in triplicate. Plates were incubated at 37°C in 5% CO2. The number of colonies in each sample was scored at 7 days (for mouse HSPCs) or 14 days (for human HSPCs).
Single-cell colony formation assay
HSCs were precultured and CRISPR-Cas9 was performed using CD45-sgRNA. Edited cells were cultured under the proliferative condition (50 ng/mL SCF and 50 ng/mL TPO) in 20% O2 or quiescence-maintaining condition (0.5–1.5 ng/mL SCF and 1 ng/mL TPO) in 1% O2. CD45− HSCs were sorted into single cells at 7 days after electroporation and their colony forming capacity was compared with that of freshly isolated HSCs upon culture in the presence of 20 ng/mL SCF and 20 ng/mL TPO in 20% O2 for 10 days. Colony size was defined by the total number of live cells within each well, which was calculated from the raw cell count using a MACSQuant instrument.
cDNA synthesis and RT-qPCR
Edited LSK cells were sorted at 6 h after electroporation. Total RNA was extracted using an RNeasy Mini Kit (Qiagen). Complementary DNA was reverse-transcribed using Superscript VILO (Invitrogen). qPCR was performed using SYBR Premix ExTaq IIa (TaKaRa Bio) according to the manufacturer’s instructions. Gene expression levels were measured using the ABI 7500 Fast Real-Time PCR System with TaqMan Gene Expression Assay Mixes (Applied Biosystems) under the following conditions: 95°C for 10 s followed by 40 cycles of 95°C for 5 s and 60°C for 34 s. Expression levels were determined as 2∧ (Ct value −mean Ct value of β-actin) and normalized against those in mock-electroporated samples as a control, unless otherwise stated.
BM transplantation
Genome-edited HSCs (CD150+CD48-Flt3- LSK cells) from Ubc-GFP mice were used as donor cells. For BM transplantation of ex vivo cultured HSCs, cells were transplanted at day 10 (medium change every day) after electroporation. Donor cells were retro-orbitally transplanted together with 0.5 × 106 BMMNCs from untreated C57BL/6-Ly5.1 mice. Donor cells were transplanted into C57BL/6-Ly5.1 congenic recipient mice that had been lethally irradiated (9.0 Gy using an MBR-1520R instrument from Hitachi Power Solutions, 125 kV, 10 mA, 0.5 mm Al filter, and 0.2 mm Cu filter) with sevoflurane anesthesia. At 1, 2, 3, and 4 months after BM transplantation, peripheral blood was collected, and the percentage of donor-derived cells and their differentiation status were determined using a MACSQuant instrument. Forty to eighty microliters of peripheral blood was sampled from the retro-orbital plexus using heparinized glass capillary tubes (Drummond Scientific) and suspended in 1 mL of PBS containing heparin. To analyze white blood cells, the blood suspension was centrifuged at 340 × g for 3 min. The supernatant was discarded and the pellet was resuspended in 1 mL of PBS containing 1.2% w/v dextran (200 kDa, Nacalai Tesque) for 45–60 min at room temperature. The sample was centrifuged at 340 × g for 3 min, and the pellet was resuspended in 0.17 M NH4Cl solution to lyse residual red blood cells for 5–10 min until the suspension became clear. Cells were resuspended in 50 μL of PBS containing 2% FCS and 0.3 μL of Fc block. Surface antigens were stained using the following antibody panel: Gr1-PE-Cy7, Mac-1-PE-Cy7, B220-APC, CD4-PerCP-Cy5.5, CD8a-PerCP-Cy5.5, CD45.1-PE, and CD45.2-BV421 for Ubc-GFP mice. A total of 0.3 μL of each antibody was added per sample. The frequency of donor-derived cells was calculated using the following equation:
100 x donor-derived (Ly5.2+Ly5.1-) cells (%)/(donor-derived cells (%) + competitor- or recipient-derived (Ly5.2-Ly5.1+) cells (%)).
Myeloid cells, B cells, T cells, and red blood cells were Gr-1+ or Mac-1+, B220+, CD4+ or CD8+, and Ter119+, respectively. Total cell chimerism represents the frequency of donor-derived Ly5.2+Ly5.1- cells (for fresh and mock-edited cells) or GFP−Ly5.2+Ly5.1- cells (for RNP-edited cells) over the frequency of Ly5.2-Ly5.1+ cells relative to total mononuclear or non-lysed cells. At 4 months after BM transplantation, the frequency of donor-derived cells in BM was determined using one femur and tibia per recipient. After counting BM cells using a TC10 automated counter (Bio-Rad), equal volumes of cell suspensions (20–30% of total volume) from each recipient were pooled and 2 × 106 cells were resuspended in PBS containing 2% FCS. The cell suspension (200 μL/recipient) was retro-orbitally injected into lethally (9.0 Gy) irradiated Ly5.1+ recipients using a 1 mL syringe and 27-gauge needle. Remaining cells were stained to assess BM chimerism. For BM analysis, anti-lineage-PerCP-Cy5.5, anti-c-Kit-APC-Cy7, anti-Sca-1-PE-Cy7, anti-CD150-BV421, anti-CD48-APC, anti-Ly5.1-PE, and anti-Ly5.2-Alexa Fluor 700 were used to detect surface antigens. A total of 1 μL of each antibody was added per sample. To determine the genome editing efficiency after transplantation, donor-derived cells were sorted and evaluated by TIDE (see evaluation of gene editing efficiency).
Immunocytochemistry
LSK cells electroporated with ATTO550-labeled or non-labeled gRNA were cytospin and fixed in 4% paraformaldehyde. Nuclei were identified by DAPI. Confocal immunofluorescence microscopy was performed with a confocal laser scanning microscope (LSM880; Zeiss). Images were analyzed using Imaris 9.8.2 software (Oxford Instruments) according to the manufacturer’s instructions. For visualization, confocal images were rendered into 3D volumes and represented as a maximum intensity projection, unless indicated otherwise. To quantify delivery of RNP to the nucleus, confocal images were rendered into 2D with Ortho Slicer mode. Then, the co-localization area of DAPI (nucleus) and ATTO550 (RNP) was calculated using the following equation:
100 x area of ATTO550 that colocalized with DAPI (%)/total area of ATTO550 (%)
Non-labeled gRNA was used as a negative control.
Transmission electron microscopy
Transmission Electron Microscopy was conducted as previously described with some modifications.94 To image small number of FACS sorted cells, cells were pre-stained with Evans blue for better detection of cell pellet.95 Cell pellets were processed and embedded in Araldite resin and sectioned at 80nm using ultramicrotome Leica Ultracut UCT microtome. Sections were placed on a 200 mesh copper grid and imaged by JEOL JEM-1400Plus. The number of nuclear pores was counted using ImageJ.
RNA-seq and data analysis
Libraries for RNA-seq were prepared using 3000 freshly isolated EPCR+ HSCs and the LSK fraction of cultured cells. Cells were lysed in 350 μL of RLT buffer from a RNeasy kit plus 7 μL of 2-mercaptoethanol and stored at −80°C until use. Total RNA was extracted using an RNeasy Plus Micro Kit (Qiagen) and cDNA was synthesized using an SMART-Seq HT Kit for Sequencing (Clontech). Double-stranded cDNA was fragmented using an S220 Focused-ultrasonicator (Covaris) and cDNA libraries were generated using a NEBNext Ultra DNA Library Prep Kit (New England BioLabs). Sequencing was performed using a HiSeq1500 system (Illumina) with a single-read sequencing length of 60 bp. Sequences were aligned to the mouse genome (mm10) with the annotation data from iGenomes (Illumina) using Hisat2. Mapped reads were counted using StringTie. Downstream analyses were performed using R (v4.1.0) programs.96 Count data were normalized and analyzed using DESeq2 (v1.30.1)86 to detect differentially expressed genes. PCA was performed using the prcomp function for 500 genes with the highest variation among samples after transforming the raw count data using the vst function from the DESeq2 package. GSEA was performed using the fGSEA (v1.16.0) package97 with the rank statistics according to the log fold changes of the DESeq2 results. GSVA were performed using the GSVA package.88 Gene sets subjected to fGSEA or GSVA were downloaded from MsigDB (https://www.gsea-msigdborg/gsea/msigdb). Custom gene sets were generated to investigate HSPC-related gene set enrichment (Table S2).
Quantification and statistical analysis
Statistical analysis
Data are presented as means ± SD, unless otherwise stated. For multiple comparisons, statistical significance was determined by the Tukey-Kramer multiple comparisons test. An unpaired two-tailed Student’s t test was used for experiments with two groups. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. n.s., not significant.
Acknowledgments
We thank all members of the Takubo laboratory for their indispensable support, K. Brownhill for helping to prepare the manuscript, Dr. Takehiko Yamamoto of Keio University for advice regarding Nucleofector, Drs. Ravindra Majeti and Yusuke Nakauchi of Stanford University for support regarding human HSPC KI experiments, and Dr. Satoshi Yamazaki of Tsukuba University for advice regarding PVA-based medium. K.S. was supported in part by a KAKENHI grant from the Ministry of Education, Culture, Sports, Science and Technology (MEXT)/Japan Society for the Promotion of Science (JSPS) (21J11016). H.K. was supported in part by a KAKENHI grant from MEXT/JSPS (19K17847 and 21K08431) and a grant from the National Center for Global Health and Medicine. D.K. was supported in part by a KAKENHI grant from MEXT/JSPS (19K17877, 21J01690, and 22K08493), JB research grants, and the Takeda Science Foundation. K.T. was supported in part by KAKENHI grants from MEXT/JSPS (20K21621, 21H02957, and 22K19550), grants from the National Center for Global Health and Medicine, a grant from the Japan Health Research Promotion Bureau, Japan Agency for Medical Research and Development grants (JP18ck0106444, JP18ae0201014, JP20bm0704042, and JP20gm1210011), a grant from the Takeda Science Foundation, and the MEXT Joint Usage/Research Center Program at the Advanced Medical Research Center, Yokohama City University.
Author contributions
K.S., H.K., Y.S., S.F., S.T., M.H., N.I., K.A., S.K., and K. Kobayashi performed the study and analyzed the data. S.W., D.K., Y.M., A.N.-I., M.K., A.I., S.O., and K. Kataoka provided scientific advice and materials. K.S., H.K., and K.T. wrote the manuscript. K.T. conceived the project and supervised the research.
Declarations of interests
The authors declare no competing interests.
Published: December 5, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2022.100354.
Supplemental information
References
- 1.Trumpp A., Essers M., Wilson A. Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 2010;10:201–209. doi: 10.1038/nri2726. [DOI] [PubMed] [Google Scholar]
- 2.Passegué E., Wagers A.J., Giuriato S., Anderson W.C., Weissman I.L. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson A., Laurenti E., Oser G., van der Wath R.C., Blanco-Bose W., Jaworski M., Offner S., Dunant C.F., Eshkind L., Bockamp E., et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 4.Foudi A., Hochedlinger K., Van Buren D., Schindler J.W., Jaenisch R., Carey V., Hock H. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat. Biotechnol. 2009;27:84–90. doi: 10.1038/nbt.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang S., Ma Y., Wang L., Li X., Dong Y., Wang J., Cheng T., Dong F., Ema H. Expansion of quiescent hematopoietic stem cells under stress and nonstress conditions in mice. Stem Cell Rev. Rep. 2022;18:2388–2402. doi: 10.1007/s12015-022-10380-6. [DOI] [PubMed] [Google Scholar]
- 6.Fukushima T., Tanaka Y., Hamey F.K., Chang C.H., Oki T., Asada S., Hayashi Y., Fujino T., Yonezawa T., Takeda R., et al. Discrimination of dormant and active hematopoietic stem cells by G(0) marker reveals dormancy regulation by cytoplasmic calcium. Cell Rep. 2019;29:4144–4158.e7. doi: 10.1016/j.celrep.2019.11.061. [DOI] [PubMed] [Google Scholar]
- 7.Cheng T., Rodrigues N., Shen H., Yang Y., Dombkowski D., Sykes M., Scadden D.T. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 8.Zou P., Yoshihara H., Hosokawa K., Tai I., Shinmyozu K., Tsukahara F., Maru Y., Nakayama K., Nakayama K.I., Suda T. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell. 2011;9:247–261. doi: 10.1016/j.stem.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Matsumoto A., Takeishi S., Kanie T., Susaki E., Onoyama I., Tateishi Y., Nakayama K., Nakayama K.I. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2011;9:262–271. doi: 10.1016/j.stem.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 10.García-Prat L., Kaufmann K.B., Schneiter F., Voisin V., Murison A., Chen J., Chan-Seng-Yue M., Gan O.I., McLeod J.L., Smith S.A., et al. TFEB-mediated endolysosomal activity controls human hematopoietic stem cell fate. Cell Stem Cell. 2021;28:1838–1850.e10. doi: 10.1016/j.stem.2021.07.003. [DOI] [PubMed] [Google Scholar]
- 11.Liang R., Arif T., Kalmykova S., Kasianov A., Lin M., Menon V., Qiu J., Bernitz J.M., Moore K., Lin F., et al. Restraining lysosomal activity preserves hematopoietic stem cell quiescence and potency. Cell Stem Cell. 2020;26:359–376.e7. doi: 10.1016/j.stem.2020.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ito K., Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014;15:243–256. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filippi M.D., Ghaffari S. Mitochondria in the maintenance of hematopoietic stem cells: new perspectives and opportunities. Blood. 2019;133:1943–1952. doi: 10.1182/blood-2018-10-808873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laurenti E., Frelin C., Xie S., Ferrari R., Dunant C.F., Zandi S., Neumann A., Plumb I., Doulatov S., Chen J., et al. CDK6 levels regulate quiescence exit in human hematopoietic stem cells. Cell Stem Cell. 2015;16:302–313. doi: 10.1016/j.stem.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laurenti E., Göttgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature. 2018;553:418–426. doi: 10.1038/nature25022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venezia T.A., Merchant A.A., Ramos C.A., Whitehouse N.L., Young A.S., Shaw C.A., Goodell M.A. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2004;2:e301. doi: 10.1371/journal.pbio.0020301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takizawa H., Fritsch K., Kovtonyuk L.V., Saito Y., Yakkala C., Jacobs K., Ahuja A.K., Lopes M., Hausmann A., Hardt W.D., et al. Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell. 2017;21:225–240.e5. doi: 10.1016/j.stem.2017.06.013. [DOI] [PubMed] [Google Scholar]
- 18.Pietras E.M., Lakshminarasimhan R., Techner J.M., Fong S., Flach J., Binnewies M., Passegué E. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J. Exp. Med. 2014;211:245–262. doi: 10.1084/jem.20131043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Busch K., Klapproth K., Barile M., Flossdorf M., Holland-Letz T., Schlenner S.M., Reth M., Höfer T., Rodewald H.R. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015;518:542–546. doi: 10.1038/nature14242. [DOI] [PubMed] [Google Scholar]
- 20.Baldridge M.T., King K.Y., Boles N.C., Weksberg D.C., Goodell M.A. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez-Fraticelli A.E., Wolock S.L., Weinreb C.S., Panero R., Patel S.H., Jankovic M., Sun J., Calogero R.A., Klein A.M., Camargo F.D. Clonal analysis of lineage fate in native haematopoiesis. Nature. 2018;553:212–216. doi: 10.1038/nature25168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Essers M.A.G., Offner S., Blanco-Bose W.E., Waibler Z., Kalinke U., Duchosal M.A., Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez-Fraticelli A.E., Weinreb C., Wang S.W., Migueles R.P., Jankovic M., Usart M., Klein A.M., Lowell S., Camargo F.D. Single-cell lineage tracing unveils a role for TCF15 in haematopoiesis. Nature. 2020;583:585–589. doi: 10.1038/s41586-020-2503-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowling S., Sritharan D., Osorio F.G., Nguyen M., Cheung P., Rodriguez-Fraticelli A., Patel S., Yuan W.C., Fujiwara Y., Li B.E., et al. An engineered CRISPR-cas9 mouse line for simultaneous readout of lineage histories and gene expression profiles in single cells. Cell. 2020;181:1410–1422.e27. doi: 10.1016/j.cell.2020.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tothova Z., Krill-Burger J.M., Popova K.D., Landers C.C., Sievers Q.L., Yudovich D., Belizaire R., Aster J.C., Morgan E.A., Tsherniak A., Ebert B.L. Multiplex CRISPR/Cas9-Based genome editing in human hematopoietic stem cells models clonal hematopoiesis and myeloid neoplasia. Cell Stem Cell. 2017;21:547–555.e8. doi: 10.1016/j.stem.2017.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naldini L. Genetic engineering of hematopoiesis: current stage of clinical translation and future perspectives. EMBO Mol. Med. 2019;11:e9958. doi: 10.15252/emmm.201809958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bothmer A., Phadke T., Barrera L.A., Margulies C.M., Lee C.S., Buquicchio F., Moss S., Abdulkerim H.S., Selleck W., Jayaram H., et al. Characterization of the interplay between DNA repair and CRISPR/Cas9-induced DNA lesions at an endogenous locus. Nat. Commun. 2017;8:13905. doi: 10.1038/ncomms13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brinkman E.K., Chen T., de Haas M., Holland H.A., Akhtar W., van Steensel B. Kinetics and fidelity of the repair of Cas9-induced double-strand DNA breaks. Mol. Cell. 2018;70:801–813.e6. doi: 10.1016/j.molcel.2018.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Overbeek M., Capurso D., Carter M.M., Thompson M.S., Frias E., Russ C., Reece-Hoyes J.S., Nye C., Gradia S., Vidal B., et al. DNA repair profiling reveals nonrandom outcomes at Cas9-mediated breaks. Mol. Cell. 2016;63:633–646. doi: 10.1016/j.molcel.2016.06.037. [DOI] [PubMed] [Google Scholar]
- 30.Janssen J.M., Chen X., Liu J., Gonçalves M.A.F.V. The chromatin structure of CRISPR-Cas9 target DNA controls the balance between mutagenic and homology-directed gene-editing events. Mol. Ther. Nucleic Acids. 2019;16:141–154. doi: 10.1016/j.omtn.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin J.J., Schröder M.S., Caiado F., Wyman S.K., Bray N.L., Bordi M., Dewitt M.A., Vu J.T., Kim W.T., Hockemeyer D., et al. Controlled cycling and quiescence enables efficient HDR in engraftment-enriched adult hematopoietic stem and progenitor cells. Cell Rep. 2020;32:108093. doi: 10.1016/j.celrep.2020.108093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Genovese P., Schiroli G., Escobar G., Tomaso T.D., Firrito C., Calabria A., Moi D., Mazzieri R., Bonini C., Holmes M.C., et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uchida N., Li L., Nassehi T., Drysdale C.M., Yapundich M., Gamer J., Haro-Mora J.J., Demirci S., Leonard A., Bonifacino A.C., et al. Preclinical evaluation for engraftment of CD34(+) cells gene-edited at the sickle cell disease locus in xenograft mouse and non-human primate models. Cell Rep. Med. 2021;2 doi: 10.1016/j.xcrm.2021.100247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Humbert O., Radtke S., Samuelson C., Carrillo R.R., Perez A.M., Reddy S.S., Lux C., Pattabhi S., Schefter L.E., Negre O., et al. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci. Transl. Med. 2019;11:eaaw3768. doi: 10.1126/scitranslmed.aaw3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gundry M.C., Brunetti L., Lin A., Mayle A.E., Kitano A., Wagner D., Hsu J.I., Hoegenauer K.A., Rooney C.M., Goodell M.A., Nakada D. Highly efficient genome editing of murine and human hematopoietic progenitor cells by CRISPR/Cas9. Cell Rep. 2016;17:1453–1461. doi: 10.1016/j.celrep.2016.09.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lattanzi A., Meneghini V., Pavani G., Amor F., Ramadier S., Felix T., Antoniani C., Masson C., Alibeu O., Lee C., et al. Optimization of CRISPR/Cas9 delivery to human hematopoietic stem and progenitor cells for therapeutic genomic rearrangements. Mol. Ther. 2019;27:137–150. doi: 10.1016/j.ymthe.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bak R.O., Dever D.P., Reinisch A., Cruz Hernandez D., Majeti R., Porteus M.H. Multiplexed genetic engineering of human hematopoietic stem and progenitor cells using CRISPR/Cas9 and AAV6. Elife. 2017;6:e27873. doi: 10.7554/eLife.27873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hendel A., Bak R.O., Clark J.T., Kennedy A.B., Ryan D.E., Roy S., Steinfeld I., Lunstad B.D., Kaiser R.J., Wilkens A.B., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tran N.T., Sommermann T., Graf R., Trombke J., Pempe J., Petsch K., Kühn R., Rajewsky K., Chu V.T. Efficient CRISPR/Cas9-Mediated gene knockin in mouse hematopoietic stem and progenitor cells. Cell Rep. 2019;28:3510–3522.e5. doi: 10.1016/j.celrep.2019.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schiroli G., Conti A., Ferrari S., Della Volpe L., Jacob A., Albano L., Beretta S., Calabria A., Vavassori V., Gasparini P., et al. Precise gene editing preserves hematopoietic stem cell function following transient p53-mediated DNA damage response. Cell Stem Cell. 2019;24:551–565.e8. doi: 10.1016/j.stem.2019.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zonari E., Desantis G., Petrillo C., Boccalatte F.E., Lidonnici M.R., Kajaste-Rudnitski A., Aiuti A., Ferrari G., Naldini L., Gentner B. Efficient ex vivo engineering and expansion of highly purified human hematopoietic stem and progenitor cell populations for gene therapy. Stem Cell Rep. 2017;8:977–990. doi: 10.1016/j.stemcr.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S., et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 44.Wu Y., Zeng J., Roscoe B.P., Liu P., Yao Q., Lazzarotto C.R., Clement K., Cole M.A., Luk K., Baricordi C., et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019;25:776–783. doi: 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeWitt M.A., Magis W., Bray N.L., Wang T., Berman J.R., Urbinati F., Heo S.J., Mitros T., Muñoz D.P., Boffelli D., et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016;8:360ra134. doi: 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park S.H., Lee C.M., Dever D.P., Davis T.H., Camarena J., Srifa W., Zhang Y., Paikari A., Chang A.K., Porteus M.H., et al. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019;47:7955–7972. doi: 10.1093/nar/gkz475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lattanzi A., Camarena J., Lahiri P., Segal H., Srifa W., Vakulskas C.A., Frock R.L., Kenrick J., Lee C., Talbott N., et al. Development of β-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci. Transl. Med. 2021;13:eabf2444. doi: 10.1126/scitranslmed.abf2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pavel-Dinu M., Wiebking V., Dejene B.T., Srifa W., Mantri S., Nicolas C.E., Lee C., Bao G., Kildebeck E.J., Punjya N., et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat. Commun. 2019;10:1634. doi: 10.1038/s41467-019-09614-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schiroli G., Ferrari S., Conway A., Jacob A., Capo V., Albano L., Plati T., Castiello M.C., Sanvito F., Gennery A.R., et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci. Transl. Med. 2017;9:eaan0820. doi: 10.1126/scitranslmed.aan0820. [DOI] [PubMed] [Google Scholar]
- 50.Chang C.W., Lai Y.S., Westin E., Khodadadi-Jamayran A., Pawlik K.M., Lamb L.S., Jr., Goldman F.D., Townes T.M. Modeling human severe combined immunodeficiency and correction by CRISPR/Cas9-Enhanced gene targeting. Cell Rep. 2015;12:1668–1677. doi: 10.1016/j.celrep.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 51.Wagenblast E., Azkanaz M., Smith S.A., Shakib L., McLeod J.L., Krivdova G., Araújo J., Shultz L.D., Gan O.I., Dick J.E., Lechman E.R. Functional profiling of single CRISPR/Cas9-edited human long-term hematopoietic stem cells. Nat. Commun. 2019;10:4730. doi: 10.1038/s41467-019-12726-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pavani G., Laurent M., Fabiano A., Cantelli E., Sakkal A., Corre G., Lenting P.J., Concordet J.P., Toueille M., Miccio A., Amendola M. Ex vivo editing of human hematopoietic stem cells for erythroid expression of therapeutic proteins. Nat. Commun. 2020;11:3778. doi: 10.1038/s41467-020-17552-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morrison S.J., Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–1074. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 54.Fares I., Chagraoui J., Gareau Y., Gingras S., Ruel R., Mayotte N., Csaszar E., Knapp D.J.H.F., Miller P., Ngom M., et al. Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science. 2014;345:1509–1512. doi: 10.1126/science.1256337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilkinson A.C., Ishida R., Kikuchi M., Sudo K., Morita M., Crisostomo R.V., Yamamoto R., Loh K.M., Nakamura Y., Watanabe M., et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature. 2019;571:117–121. doi: 10.1038/s41586-019-1244-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cabezas-Wallscheid N., Buettner F., Sommerkamp P., Klimmeck D., Ladel L., Thalheimer F.B., Pastor-Flores D., Roma L.P., Renders S., Zeisberger P., et al. Vitamin A-retinoic acid signaling regulates hematopoietic stem cell dormancy. Cell. 2017;169:807–823.e19. doi: 10.1016/j.cell.2017.04.018. [DOI] [PubMed] [Google Scholar]
- 57.Kobayashi H., Morikawa T., Okinaga A., Hamano F., Hashidate-Yoshida T., Watanuki S., Hishikawa D., Shindou H., Arai F., Kabe Y., et al. Environmental optimization enables maintenance of quiescent hematopoietic stem cells ex vivo. Cell Rep. 2019;28:145–158.e9. doi: 10.1016/j.celrep.2019.06.008. [DOI] [PubMed] [Google Scholar]
- 58.Oedekoven C.A., Belmonte M., Bode D., Hamey F.K., Shepherd M.S., Che J.L.C., Boyd G., McDonald C., Belluschi S., Diamanti E., et al. Hematopoietic stem cells retain functional potential and molecular identity in hibernation cultures. Stem Cell Rep. 2021;16:1614–1628. doi: 10.1016/j.stemcr.2021.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iriuchishima H., Takubo K., Matsuoka S., Onoyama I., Nakayama K.I., Nojima Y., Suda T. Ex vivo maintenance of hematopoietic stem cells by quiescence induction through Fbxw7α overexpression. Blood. 2011;117:2373–2377. doi: 10.1182/blood-2010-07-294801. [DOI] [PubMed] [Google Scholar]
- 60.Chu V.T., Weber T., Graf R., Sommermann T., Petsch K., Sack U., Volchkov P., Rajewsky K., Kühn R. Efficient generation of Rosa26 knock-in mice using CRISPR/Cas9 in C57BL/6 zygotes. BMC Biotechnol. 2016;16:4. doi: 10.1186/s12896-016-0234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim S., Koo T., Jee H.G., Cho H.Y., Lee G., Lim D.G., Shin H.S., Kim J.S. CRISPR RNAs trigger innate immune responses in human cells. Genome Res. 2018;28:367–373. doi: 10.1101/gr.231936.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schubert M.S., Cedrone E., Neun B., Behlke M.A., Dobrovolskaia M.A. Chemical modification of CRISPR gRNAs eliminate type I interferon responses in human peripheral blood mononuclear cells. J. Cytokine Biol. 2018;3:121. doi: 10.4172/2576-3881.1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wienert B., Shin J., Zelin E., Pestal K., Corn J.E. In vitro-transcribed guide RNAs trigger an innate immune response via the RIG-I pathway. PLoS Biol. 2018;16 doi: 10.1371/journal.pbio.2005840. e2005840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shi X., Kitano A., Jiang Y., Luu V., Hoegenauer K.A., Nakada D. Clonal expansion and myeloid leukemia progression modeled by multiplex gene editing of murine hematopoietic progenitor cells. Exp. Hematol. 2018;64:33–44.e5. doi: 10.1016/j.exphem.2018.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilkinson A.C., Dever D.P., Baik R., Camarena J., Hsu I., Charlesworth C.T., Morita C., Nakauchi H., Porteus M.H. Cas9-AAV6 gene correction of beta-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nat. Commun. 2021;12:686. doi: 10.1038/s41467-021-20909-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mohrin M., Bourke E., Alexander D., Warr M.R., Barry-Holson K., Le Beau M.M., Morrison C.G., Passegué E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7:174–185. doi: 10.1016/j.stem.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gothot A., van der Loo J.C., Clapp D.W., Srour E.F. Cell cycle-related changes in repopulating capacity of human mobilized peripheral blood CD34(+) cells in non-obese diabetic/severe combined immune-deficient mice. Blood. 1998;92:2641–2649. [PubMed] [Google Scholar]
- 68.Habibian H.K., Peters S.O., Hsieh C.C., Wuu J., Vergilis K., Grimaldi C.I., Reilly J., Carlson J.E., Frimberger A.E., Stewart F.M., Quesenberry P.J. The fluctuating phenotype of the lymphohematopoietic stem cell with cell cycle transit. J. Exp. Med. 1998;188:393–398. doi: 10.1084/jem.188.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takatoku M., Sellers S., Agricola B.A., Metzger M.E., Kato I., Donahue R.E., Dunbar C.E. Avoidance of stimulation improves engraftment of cultured and retrovirally transduced hematopoietic cells in primates. J. Clin. Invest. 2001;108:447–455. doi: 10.1172/jci12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kataoka K., Sato T., Yoshimi A., Goyama S., Tsuruta T., Kobayashi H., Shimabe M., Arai S., Nakagawa M., Imai Y., et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J. Exp. Med. 2011;208:2403–2416. doi: 10.1084/jem.20110447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arai F., Stumpf P.S., Ikushima Y.M., Hosokawa K., Roch A., Lutolf M.P., Suda T., MacArthur B.D. Machine learning of hematopoietic stem cell divisions from paired daughter cell expression profiles reveals effects of aging on self-renewal. Cell Syst. 2020;11:640–652.e5. doi: 10.1016/j.cels.2020.11.004. [DOI] [PubMed] [Google Scholar]
- 72.Chen J.Y., Miyanishi M., Wang S.K., Yamazaki S., Sinha R., Kao K.S., Seita J., Sahoo D., Nakauchi H., Weissman I.L. Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature. 2016;530:223–227. doi: 10.1038/nature16943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fares I., Chagraoui J., Lehnertz B., MacRae T., Mayotte N., Tomellini E., Aubert L., Roux P.P., Sauvageau G. EPCR expression marks UM171-expanded CD34(+) cord blood stem cells. Blood. 2017;129:3344–3351. doi: 10.1182/blood-2016-11-750729. [DOI] [PubMed] [Google Scholar]
- 74.Koide S., Sigurdsson V., Radulovic V., Saito K., Zheng Z., Lang S., Soneji S., Iwama A., Miharada K. CD244 expression represents functional decline of murine hematopoietic stem cells after in vitro culture. iScience. 2022;25:103603. doi: 10.1016/j.isci.2021.103603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brenet F., Kermani P., Spektor R., Rafii S., Scandura J.M. TGFβ restores hematopoietic homeostasis after myelosuppressive chemotherapy. J. Exp. Med. 2013;210:623–639. doi: 10.1084/jem.20121610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yamazaki S., Iwama A., Takayanagi S.i., Eto K., Ema H., Nakauchi H. TGF-beta as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood. 2009;113:1250–1256. doi: 10.1182/blood-2008-04-146480. [DOI] [PubMed] [Google Scholar]
- 77.Dong Y., Bai H., Dong F., Zhang X.B., Ema H. Gene knockout in highly purified mouse hematopoietic stem cells by CRISPR/Cas9 technology. J. Immunol. Methods. 2021;495:113070. doi: 10.1016/j.jim.2021.113070. [DOI] [PubMed] [Google Scholar]
- 78.Haraguchi T., Koujin T., Shindo T., Bilir Ş., Osakada H., Nishimura K., Hirano Y., Asakawa H., Mori C., Kobayashi S., et al. Transfected plasmid DNA is incorporated into the nucleus via nuclear envelope reformation at telophase. Commun. Biol. 2022;5:78. doi: 10.1038/s42003-022-03021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nasri M., Mir P., Dannenmann B., Amend D., Skroblyn T., Xu Y., Schulze-Osthoff K., Klimiankou M., Welte K., Skokowa J. Fluorescent labeling of CRISPR/Cas9 RNP for gene knockout in HSPCs and iPSCs reveals an essential role for GADD45b in stress response. Blood Adv. 2019;3:63–71. doi: 10.1182/bloodadvances.2017015511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maggio I., Zittersteijn H.A., Wang Q., Liu J., Janssen J.M., Ojeda I.T., van der Maarel S.M., Lankester A.C., Hoeben R.C., Gonçalves M.A.F.V. Integrating gene delivery and gene-editing technologies by adenoviral vector transfer of optimized CRISPR-Cas9 components. Gene Ther. 2020;27:209–225. doi: 10.1038/s41434-019-0119-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Haltalli M.L.R., Wilkinson A.C., Rodriguez-Fraticelli A., Porteus M. Hematopoietic stem cell gene editing and expansion: state-of-the-art technologies and recent applications. Exp. Hematol. 2022;107:9–13. doi: 10.1016/j.exphem.2021.12.399. [DOI] [PubMed] [Google Scholar]
- 82.Seki A., Rutz S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J. Exp. Med. 2018;215:985–997. doi: 10.1084/jem.20171626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aurnhammer C., Haase M., Muether N., Hausl M., Rauschhuber C., Huber I., Nitschko H., Busch U., Sing A., Ehrhardt A., Baiker A. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum. Gene Ther. Methods. 2012;23:18–28. doi: 10.1089/hgtb.2011.034. [DOI] [PubMed] [Google Scholar]
- 84.Liu H., Cui Y., Bai Y., Fang Y., Gao T., Wang G., Zhu L., Dong Q., Zhang S., Yao Y., et al. The tyrosine kinase c-Abl potentiates interferon-mediated antiviral immunity by STAT1 phosphorylation. iScience. 2021;24:102078. doi: 10.1016/j.isci.2021.102078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. Preprint at bioRxiv. 2021 doi: 10.1101/060012. [DOI] [Google Scholar]
- 88.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schaefer B.C., Schaefer M.L., Kappler J.W., Marrack P., Kedl R.M. Observation of antigen-dependent CD8+ T-cell/dendritic cell interactions in vivo. Cell. Immunol. 2001;214:110–122. doi: 10.1006/cimm.2001.1895. [DOI] [PubMed] [Google Scholar]
- 90.Kobayashi H., Takubo K. Protocol for the maintenance of quiescent murine hematopoietic stem cells. STAR Protoc. 2020;1:100078. doi: 10.1016/j.xpro.2020.100078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kiel M.J., Yilmaz O.H., Iwashita T., Yilmaz O.H., Terhorst C., Morrison S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 92.Sano H., Kobayashi K., Yoshioka N., Takebayashi H., Nambu A. Retrograde gene transfer into neural pathways mediated by adeno-associated virus (AAV)-AAV receptor interaction. J. Neurosci. Methods. 2020;345:108887. doi: 10.1016/j.jneumeth.2020.108887. [DOI] [PubMed] [Google Scholar]
- 93.Bak R.O., Dever D.P., Porteus M.H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 2018;13:358–376. doi: 10.1038/nprot.2017.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nakamura-Ishizu A., Matsumura T., Stumpf P.S., Umemoto T., Takizawa H., Takihara Y., O'Neil A., Majeed A.B.B.A., MacArthur B.D., Suda T. Thrombopoietin metabolically primes hematopoietic stem cells to megakaryocyte-lineage differentiation. Cell Rep. 2018;25:1772–1785.e6. doi: 10.1016/j.celrep.2018.10.059. [DOI] [PubMed] [Google Scholar]
- 95.Kumar S., Ciraolo G., Hinge A., Filippi M.D. An efficient and reproducible process for transmission electron microscopy (TEM) of rare cell populations. J. Immunol. Methods. 2014;404:87–90. doi: 10.1016/j.jim.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.R Core Team . R Foundation for Stastical Computing; 2021. R: A Language and Environment for Stastical Computing. [Google Scholar]
- 97.Sergushichev A.A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. Preprint at bioRxiv. 2016 doi: 10.1101/060012. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The RNA-seq data reported in this paper have been deposited at DNA Data Bank of Japan (DDBJ) (accession number: DRA014998) and is available at the time of publication.
-
•
This paper did not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.