

Abstract
Proteins are naturally formed by domains edging their functional and structural properties. A domain out of the context of an entire protein can retain its structure and to some extent also function on its own. These properties rationalize construction of artificial fusion multidomain proteins with unique combination of various functions. Information on the specific functional and structural characteristics of individual domains in the context of new artificial fusion proteins is inevitably encoded in sequential order of composing domains defining their mutual spatial positions. So the challenges in designing new proteins with new domain combinations lie dominantly in structure/function prediction and its context dependency. Despite the enormous body of publications on artificial fusion proteins, the task of their structure/function prediction is complex and nontrivial. The degree of spatial freedom facilitated by a linker between domains and their mutual orientation driven by noncovalent interactions is beyond a simple and straightforward methodology to predict their structure with reasonable accuracy. In the presented manuscript, we tested methodology using available modeling tools and computational methods. We show that the process and methodology of such prediction are not straightforward and must be done with care even when recently introduced AlphaFold II is used. We also addressed a question of benchmarking standards for prediction of multidomain protein structures—x‐ray or Nuclear Magnetic Resonance experiments. On the study of six two‐domain protein chimeras as well as their composing domains and their x‐ray structures selected from PDB, we conclude that the major obstacle for justified prediction is inappropriate sampling of the conformational space by the explored methods. On the other hands, we can still address particular steps of the methodology and improve the process of chimera proteins prediction.
Keywords: 3D structure prediction, fusion proteins, molecular simulations, x‐ray crystallography
1. INTRODUCTION
Protein domains (most commonly described as structurally and functionally independent protein units) are the basic building blocks by which nature can create an almost unlimited space of combinations. However, nature exploits only a small portion of such portfolio. 1 In addition, only a smaller portion of natural proteins is structurally characterized. The narrow coverage of the three‐dimensional (3D) structure space of proteins is further limited by the fact that almost two‐third of experimentally determined structures are only one‐domain segments of larger existing proteins. This is in sharp contrast to the fact that more than 65% of eukaryotic proteins have multidomain character.
Structural domains are key elements in understanding complex protein 3D structure, protein function, and its origin. 2 Some proteins consist of only one domain, whereas some proteins contain up to dozens of domains. The same domain usually occurs in several different proteins in a specific arrangement. In other words, when comparing a particular domain occurring in a group of other domains, one can follow linear N‐ to C‐ termini domain order in proteins where this group of domains is observed together. However, there is also evidence of perturbation of the preferred order for several domains. 3 , 4 Such unusual organization of domains in a protein can provide novel functions different from the representative group even if particular domain composition is the same.
Strategic usage of protein engineering approaches such as directed evolution, de novo design, and computational approaches has opened improvement of proteins properties and more importantly allow us to engineer novel artificial fusion proteins composed of two and more functional protein domains with improved functional features. The fusion of protein domains in different orders and compositions offers to build a unique novel artificial protein with unique properties. Nowadays, artificial proteins are routinely used in the protein‐engineering field covering many disciplines from basic to applied science, medicine, and industry. 5 Fusion proteins are a vast subset of proteins, represented by, for example: affinity or soluble tags linked to proteins to allow easier purification or crystallization procedures (6xHis‐tag, maltose‐binding protein [MBP] or albumin‐binding protein [ABP]; thioredoxin [TRX] and B1 domain of protein G [called GB1 domain]). 6 , 7 Green fluorescent protein, widely used in cell localization and in vivo assays, is another representative of fusion proteins and their utilization. 8 Other strategies of protein engineering cover metabolic engineering where a fusion of several enzymes from a single metabolic pathway can considerably speed up and cheapen multienzyme reaction steps. 9 , 10 Additional significant fusion protein applications lie in the design of protein therapeutics such as functional antibodies derivates (bispecific, single chain, multivalent antibodies) in cancer immunotherapy 11 , 12 , 13 or protein vaccines design as COVID‐19 pandemic showed recently. 14 In other cases, protein chimeras can provide several specific functions for high‐risk goals, such as therapeutics direct localization, protective transfer or protection against external environment causing, for example, fast degradation of a therapeutic in the human body, and so on. 15 , 16 , 17 The fusion protein approach covers plenty of promising strategies to targeted therapies that avoid side effects during treatment of patients and increase therapeutics effects. 18 , 19
Therefore, novel protein design and engineering approaches call for an essential understanding of principles that govern the mutual orientation of domains in chimeric proteins and a careful design of their composition and order. However, there is one major obstacle in designing artificial protein chimeras and their structural properties in silico as such chimeras have no natural analogs, and therefore a single template (commonly used by structure predictors based on homology modeling) is not sufficient for preliminary structure predictions. 20 This remains an issue even if advanced methodology such as the one applied in domain enhance modeling (DEMO) 21 or AlphaFold 22 tools are in place. The DEMO uses distance profiles from templates detected via domain‐level structure comparison. Even though individual domains usually have well‐characterized structures, their interaction interfaces in chimera proteins made up of completely unrelated domains may not have a direct parallel counterpart in naturally occurring proteins. The recently introduced AlhaFold is in principle able to predict any structure from a sequence, including multidomain proteins but detailed analysis of accuracy of a huge amount of data is still missing. 23 In addition, we have to answer the principal question in chimera protein design—how strong the interactions between fused domains are or whether the domains interact at all. Preliminary theoretical studies can help answer this question prior to the experimental part of protein chimera preparation, so the major experimental difficulties can be avoided. Another important parameter for fusion proteins design is a linker—usually flexible connections between composing domains. This represents important constraint for conformational portfolio of final compositions and is expected to shift enthalpy/entropy contribution in the final structure.
It is generally accepted that native proteins form domain–domain interfaces (DDIs) in a nonrandom way, which is analogous to interactions establishing protein–protein interfaces (PPIs). 24 Based on this assumption, we can utilize these principles in the structure prediction of chimeric proteins. 21 Similar difficulties with predicting a wide variety of PPIs are already being challenged by revelations made in the mechanism of protein crystallization. The currently popular “sticky patch” model of crystallization is based on cohesive interaction of specific surfaces. 25 There are also online services that aim to predict whether the studied interface is most likely to be biologically functional or just a result of the crystallization process. 26 , 27 Such analysis is unfortunately only indicative since there is no sharp boundary between these two types of interfaces revealed yet.
Due to packing forces, crystal structures may not be always relevant to the actual form of a protein in solution. 28 , 29 , 30 The problem of predicting the structure 31 of multidomain proteins is thus essentially twofold. First, there is no universal method for predicting such multidomain proteins, and second, there is not necessarily a suitable dataset on which to build and validate such a method. Therefore, it would be appropriate to have some approach of predicting the structure of multidomain proteins, which would be more “ab initio” with respect to the possible positions of domains relative to each other.
In the proposed study, we focus on the development of a simple and illustrative methodology of tertiary structure predictions for protein chimeras based on comparison with already experimentally determined structures of individual domains and their fusions. The ultimate goal of the study was to compare results of in silico predictions with respective protein structures determined by x‐ray crystallography and discuss the appropriateness of such benchmark. Molecular dynamics (MD) simulations were used to test the conformational variability of the designed structures and their isolated domain components, as it was shown to provide valuable information unavailable from static structures. 32 We did not pay specific attention in the selected cases to the role of interdomain linkers representing conformational and spatial constraints for mutual domain arrangement and did not focus on the role of linker as a specific parameter for local concentration increase. On the other hand, we are aware of the fact that the length and composition of the linker can play very important role in the character and structure of the designed chimeras as documented in the literature. 33 , 34 To summarize, we provide a survey on the minimalistic design approach of chimera fusion proteins by means of currently used molecular modeling methods to determine the most important parameters of the approach and its practical applicability.
2. MATERIALS AND METHODS
2.1. Dataset preparation
The dataset of chimeric proteins was built based on the Protein Data Bank 35 entries. The artificial origin of potential candidates was checked carefully to avoid a naturally occurring interface (or its analog) between composing domains. The final selection took into consideration several additional criteria: protein structure was determined by x‐ray crystallography; the overall sequence length of chimera was shorter than 500 residues; resolution better than 1.5 A, the fusion construct consisted of a “functional/leading domain” (the domain of the main interest) and the additional “tag domain” (TRX, MBP, GFP, etc.) and only nonliganded structures were preferentially taken into account. It is necessary to say that the process of the selection was not straightforward due to the fact that PDB does not contain statistically large ensemble of distinct two‐domain structures composed of heterogeneous and at some time artificial domains. The final dataset consisted of the following structures with the respective PDB ID in parentheses: GFP‐ubiquitin (3ai5), MBP‐YS1 monobody (3csg), TRX‐UHM domain (3dxb), MBP‐CARD domain (4ifp), SMT3‐isomerase (5v8t) and endolysin‐GRAM (5yqr). Additionally, each domain of a particular chimera had at least two other representatives in PDB, the only exception is the GRAM domain in 5yqr with no available structure of the single domain.
2.2. Domain assignment and initial structure preparation
Chimeric protein structures were decomposed into the individual domains and linkers based on the comparison with other PDB entries containing the domain in question either as a single domain or in a macromolecular complex. The sequence alignment of chimeric proteins and the native domain sequences was performed using Clustal Omega tool. 36 Specific domain boundaries were selected based on a two‐step procedure. At first, UniProt sequence domain annotations were considered. 37 However, final boundaries were adjusted after thorough visual inspection and comparison with homologous structures using PyMOL software 38 (version 1.7.2.1). All missing residues in the x‐ray structures were modeled with ModLoop web server 39 if necessary.
2.3. Tertiary structure prediction
Two approaches of protein chimera structure prediction were adopted using the Ab Initio Domain Assembly Server (AIDA) 20 and ClusPro web servers. 40 The aim of the dual approach was to compare already designed tool for structure prediction of multidomain proteins and the approach where identification of potential domain–domain interface serves as the criteria for model construction.
AIDA is a tool developed for prediction of the structural assemblies of multidomain proteins. Based on the provided input, AIDA builds an initial model that is afterward optimized by a “coarse‐grained” potential. If the 3D structures of individual domains are submitted, AIDA treats them as rigid during the modeling process, but the rest of the polypeptide chain (linkers) is kept flexible and optimized as well as the respective orientation of the rigid (domain) parts. In the end, the lowest energy structure is obtained and proposed as the plausible model. For prediction of the studied chimeric protein structures, the coordinates of individual domains from the resolved crystal structures of protein chimeras were used but random displacement and reorientation of both domains were initially applied. The submissions were repeated 10 times to assess the reproducibility of the final predictions.
ClusPro web server v. 2.0 is primarily intended for rigid body protein docking, optionally constrained by experimental evidence such as small‐angle x‐ray scattering (SAXS) profile and distance constraints. ClusPro performs an exhaustive docking of poses using Fast‐Fourier‐Transformation (FFT)–based algorithm. Finally, up to 30 optimized poses with the best scoring are reported. However, ClusPro provides several scoring functions, and the calculations are conducted with four different potentials (“balanced,” “electrostatics‐favored,” “hydrophobic‐favored,” and “van der Waals + electrostatics”), which weigh differently internal energetic contributions. The individual potentials are suitable for different kinds of protein–protein interfaces and the user is expected to decide on the best scoring potential based on the expertise of the studied system. In our study, we preferred the “balanced” potential because it does not bias toward electrostatics or hydrophobic interfaces. Additionally, the distance restraints between the termini of both domains were applied to limit the docking procedure. The restraints were introduced individually for each chimeric protein to mimic the effect of the interdomain linker. Specifically, they were applied between Cα atoms of the last and the first residues of two linked domain, and the length was set to be at most (n + 1) ×3.8 Å, where n stands for the number of residues in the linker. Finally, the 3D structure of the linker in a docked domain complex was modeled by ModLoop web server. 39
2.4. Analysis of interfaces between composing domains
The size and the structural properties of interfaces between fused domains were characterized for the whole data set of experimental and predicted structures. In case of experimental crystallographic structures, analysis of interaction interfaces was extended by investigation of additional crystallographic interfaces between the domains and their symmetry‐related counterparts. All interface analyses were performed by Protein Interfaces Surfaces and Assemblies (PISA) and PROtein binDIng enerGY prediction (PRODIGY) web tools. 27 , 41
PISA estimates the stability in solution of all plausible macromolecular assemblies, which can be generated by crystallographic symmetries. The thermodynamic model of PISA takes particular interactions at the interfaces into account (hydrogen bonds, salt bridges, and disulfide bonds), size of the interacting area, and the entropic penalties due to a symmetry of the complex.
PRODIGY predicts free energy of binding by means of a simple regression model based on the number of contacts between charged, polar, and apolar residues, and the fraction of noninteracting surface. A similar predictor, PRODIGY‐CRYSTAL, was used to classify the interfaces as biological (functional) or crystallographic (artifacts of crystalline environment).
2.5. MD simulation details and analysis
All individual domains, predicted models and crystallographic structures from PDB were processed and simulated using the same protocol. All these steps were conducted using the GROMACS 5.1.2 molecular dynamics package. 42 The initial coordinates for MD simulations were adopted from the three sources: (1) results of the crystallographic experiments; (2) selected AIDA predictions; and (3) selected ClusPro docking‐based models. Proteins were described by AMBER14SB force field and water molecules by explicit TIP3P model. 43 , 44 The simulated protein was embedded in a periodic dodecahedron box with the minimum distance of 1.5 nm between the protein and the walls of the box. Ionic strength was set to 150 mM using the appropriate amount of sodium and chloride ions.
Selection of initial configurations was followed by energy minimization by the steepest descent algorithm to remove close atomic contacts. The initial velocities were generated randomly to match Maxwell‐Boltzmann distribution at 300 K. The solvent molecules were equilibrated for 200 ps while the structure of the protein was constrained by harmonic restraints. The production runs were conducted for 500 ns in three independent replicates for each simulated structure. The control simulations of individual domains were run for 100 ns.
The leap‐frog variant of Verlet algorithm was used to integrate the equation of motion. The linear constraint solver (LINCS) algorithm 45 maintained the bond lengths constrained at their equilibrium values. Particle Mesh Ewald method was used for treatment of electrostatic interactions. 46 The Lennard‐Jones potential was cut off at 10 Å. The temperature (300 K) and the pressure (1 bar) were controlled by the v‐rescale thermostat and Parrinello–Rahman barostat, respectively. 47 , 48 The trajectories were recorded in 1 ps intervals.
Dynamics and structural deviations in course of simulations were quantified by root mean square deviation of atomic positions of Cα atoms in the secondary structure elements (Cα‐RMSD) upon their optimal superposition on a reference structure. These analyses were performed by dedicated GROMACS tools.
Principal component analysis (PCA) of the trajectories was conducted on Cα‐RMSD from the selected reference structures as the features. The set of reference structures includes all initial crystallographic and predicted models and the final frames of all conducted simulations. Hence, each frame of a simulation was typically annotated by 16 Cα‐RMSD values. This representation was reduced to two‐dimensional representation by standard PCA procedure without prior normalization of input data. The PCA was performed in the NumPy numeric library 49 for Python language. Plots and graphs were elaborated using the matplotlib library for Python. Molecular visualizations were done by the PyMol molecular visualization system. 38
3. RESULTS
This study follows the workflow sketched schematically in Figure 1. First, the dataset of chimeric proteins from Protein Data Bank (PDB) was collected. Afterward, the boundaries of the individual domains and linker regions were identified. In addition to the experimental crystallographic structures, the 3D structure prediction of the chimeric constructs was performed. Both experimental and predicted structures prior to MD simulations were compared and analyzed. The first static approach, focused on structural descriptors characterizing the mutual interaction of the linked domains was applied. Then, dynamic approach focused on stability and dynamics of the chimeric constructs by means of molecular dynamics simulations was investigated.
FIGURE 1.
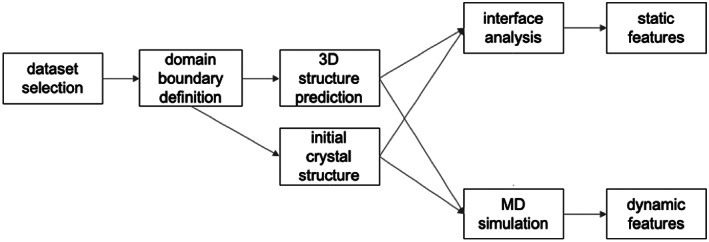
The workflow adopted in the study of chimeric multidomain proteins prediction and assessment
3.1. Selection of protein chimeras and their characteristics
As already mentioned, six artificial two‐domain protein chimeras from PDB 19 were selected (see Section 2 for details). Their basic characteristics are listed in Table S1. This dataset of two‐domain proteins manifests a variety of interdomain interface sizes (ranging from 0 to 755 Å2). The linkers connecting domains in chimeric constructs vary in length from 2 to 8 amino acid residues (Table S1 in the Supporting Information).
3.2. Tertiary structure prediction
The structure of all six chimeric proteins was predicted de novo by AIDA—assembler of multi‐domain proteins and ClusPro—protein–protein docking tool. Linkers for structures obtained by ClusPro were modeled separately.
Structures predicted by the AIDA were found strongly dependent on the initial orientation of the submitted domains. Due to the fact that AIDA reports only a single final model, we performed 10 predictions with randomized orientations of domains. The resulting structures typically fell into several structural clusters with a strong preference for one or two. Only the prediction of 3CSG chimera always finished in one cluster. Nevertheless, no significant hits among the predicted models in terms of Cα‐RMSD to crystallographic structure were found as demonstrated in Table S2. Moreover, AIDA did not even preserve the structure of crystallographic models if domains were provided in the corresponding experimental orientation (see models AIDA #11 in Table S2). The reason could be that the x‐ray structures are not stable minima in a coarse‐grained potential used in AIDA for guiding the optimization procedure. On the other hand, there are other distinctive minima determined by the used potential manifested by distinct structural clusters gathering the optimized structures. For the following MD simulations, we selected representatives of the two most populated clusters (except for 3CSG).
For each pair of domains, ClusPro provided a variable number of models (from 10 to 30) for four differently weighted potential functions. Generally, no significant match between any model and the reference x‐ray crystal structures from PDB was detected by Cα‐RMSD (see Table SI3) value at the best ranked positions. Nevertheless, ClusPro predicted the closest match for the 4IFP construct. As a quick assessment of the influence of different ClusPro potentials we compared the top predictions for the “balanced,” “electrostatic‐favored” and “vdW + Elec “potential and the results deviated in Cα‐RMSD between 3 and 4.5Å. The best match (~1 Å) between predicted and reference PDB structure was reached for 4IFP, which was practically identical to the x‐ray reference. Interestingly, the model was produced using “electrostatic‐favored” potential and it was ranked at seventh position between proposed identified “best” models. The five other predicted chimeric constructs deviated significantly more from the reference structures and any closer matches were found. Interestingly, the structures with relatively low Cα‐RMSD of ~4 Å were identified for 3DXB and 5V8T, but they occurred at 19th‐ and 17th‐ranked positions in the proposed models. No potential (“balanced”, “electrostatics‐favored”, “hydrophobic‐favored” and “vdW + Elec”) performed substantially better than the others so we decided to use the “balanced” potential for all obtained models. For the following MD simulations, we therefore selected the top ranked model obtained by “balanced” potential as a rational choice in a situation without any prior knowledge of the experimentally determined structures.
3.3. Interface analysis
Interfaces between domains in the chimeric proteins were analyzed in all x‐ray structures and all models prior to the MD simulations. Terminal extensions of domains (residues not visible in the crystal structures) were excluded from the analysis due to their inherent flexibility as well as the linker regions. Excluding the linkers, the domain–domain interactions could have been approximated by analogous protein–protein interactions of independent (unchained) domains.
The PISA and PRODIGY tools enabled calculation of the binding free energies and classification of the interface (stable/unstable, biological/crystal). The results of the interface analysis are summarized in Table S4. The interface areas varied broadly among the x‐ray structures and the predicted models. Nonetheless, three general situations were observed as follows:
the interface of the experimental structure is larger than the predicted (4ifp, 5v8t),
the interface of the experimental structure is smaller than any predicted interface (3ai5, 5yqr), and
the interface of the experimental structure falls in the range of predicted interface sizes (3csg, 3dxb).
The interfaces with larger area than predicted (Group 1) manifested also the highest absolute size, and they were ranked at the first position among other intermolecular interfaces in crystals detected by PISA. Analogously, the small experimental interfaces (group 2 and 3dxb structure) were ranked lower in size among other intermolecular interfaces, which can be interpreted as the fact that they play a more important role in stability of the crystal. The 3csg structure seems to fall in the category of the intermediate case. The area of the interdomain interface was relatively high, but there was still a larger interface between adjacent molecules in the crystalline lattice.
Additionally, a clear trend in interface areas of predicted models was noticed. ClusPro always produced a model with a higher interface size than AIDA. It is not a surprise that ClusPro also predicted interfaces involving more hydrogen bonds and salt bridges than AIDA. The number of these interactions was often even higher than for the interfaces found in the reference crystal structures (see Table S4 in Supporting Information). This finding may reflect different scoring functions of both methods, or alternatively, better capability of ClusPro to find complementary surfaces on the domains than AIDA.
The predicted free energy of binding (association) obtained by PISA and PRODIGY differed noticeably (see Table S4). PRODIGY consistently assigned lower ΔG than PISA, and only mild correlation in ΔG (Pearson's r = .648) was found between these methods. Interestingly, the domain interface from the x‐ray structures never manifested lower ΔG than in any predicted model. Models constructed by ClusPro scored exceptionally well and almost always provided the lowest estimated ΔG (except 5v8t).
Classification of interfaces by PRODIGY, which predicts the interface as biologically relevant (stable in solution) or as crystallization artifact, never confirmed strongly the biological character of interdomain interfaces (see Table S4). The classifier either predicted an interface as a crystallization artifact (values below 50%) or undecided (values around 50%). Neither the crystal structures were classified as stable in solution by PISA (not shown) and PRODIGY (biological). However, these analyses were calibrated on interactions of unchained proteins and therefore may be misleading in case of linked domains. The linked domains could form stable and metastable interfaces between surfaces of lower respective affinity due to the local concentration effect and the topological constraints.
3.4. MD simulation of chimeras
Figure 2 shows the structure of chimeric proteins chosen for MD simulations—the experimental x‐ray structures and models obtained by AIDA and ClusPro. All models of individual protein were simulated at the same conditions to eliminate the effect of various box sizes, the number of water and ion molecules (see Table S5 in Supporting Information for details).
FIGURE 2.
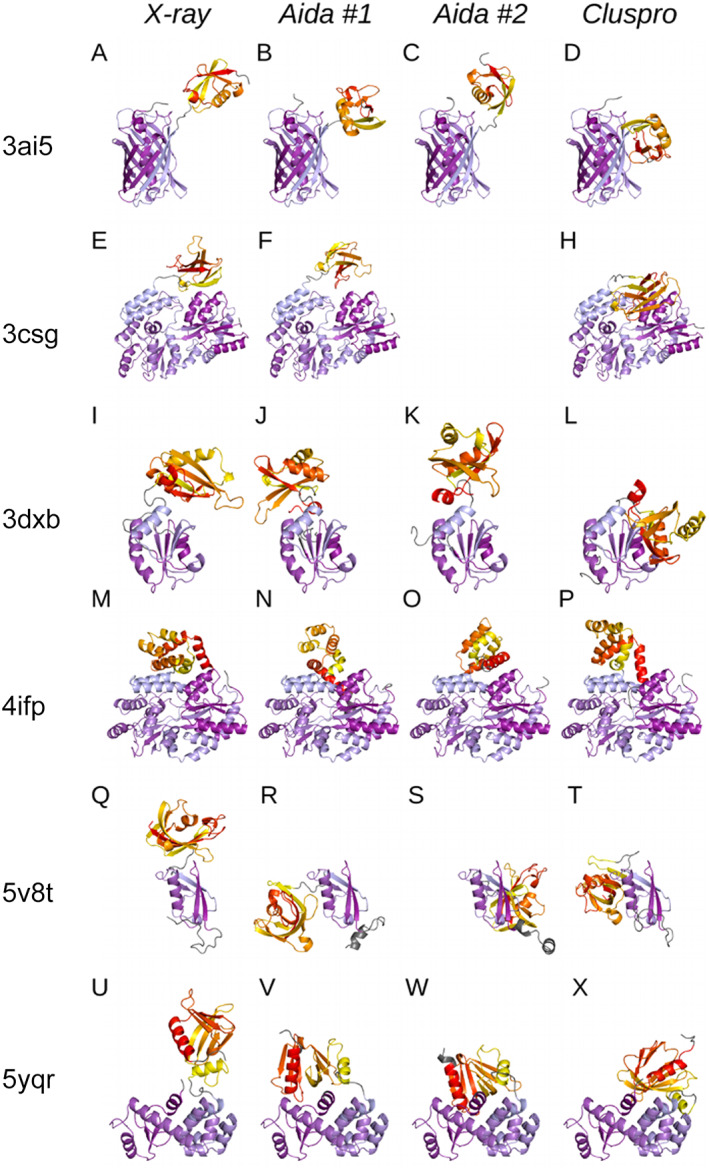
Overview of studied crystal structures and their models. Crystal structures selected for the study (the first column), two representative structure predictions from AIDA tool (the second and third columns) and structure prediction from ClusPro tool (the last column); one row corresponds to a single chimeric protein. Crystal structure and its corresponding models are aligned to the first N‐terminal domain (colored in shades of blue) for simpler comparison. C‐terminal domains are colored in yellow‐red gradient, linkers and terminal extensions (“nondomain” parts of the proteins) are colored in gray. (A–D) GFP‐ubiquitin (3AI5), (E–H): MBP‐YS1 mono‐body (3CSG), (I–L): TRX‐UHM domain (3DXB), (M–P): MBP‐CARD domain (4IFP), (Q–T): SMT3‐isomerase (5V8T) and (U–X): endolysin‐GRAM (5YQR)
MD simulations were intended to
verify the stability of interdomain interfaces of x‐ray and predicted structures in the aqueous environment as the interactions with the solvent are known to be the important factor in macromolecular complex formation and stability, 26
investigate the dynamic behavior and the fluctuations of the domains and the interfaces between them, and
refine the predicted structures and observe, whether they converge toward their experimentally determined counterparts or whether all of the structures (both predicted and x‐ray) move toward a comparable conformation.
3.5. MD data analysis
The stability of individual domains during simulations was firstly verified by their RMSD with the initial structure. The distribution of RMSD values in the course of simulations is shown in Figure S6. Stable conformation of domains was observed in the vast majority of performed simulations except for a few simulations with MBP (4ifp) and T4 lysozyme (5ybq). We observed that MBP exhibited transitions between closed and open conformation (corresponding to the state with and without maltose, respectively). A comparison of both states is provided in Figure S7. Although 4ifp structure was resolved with the maltose in the binding pocket, no maltose was present in our simulations. Hence, all simulations derived from 4ifp structure started in the closed conformation, which is not the preferred state for the apo‐MBP domain. 50 , 51 However, in the simulations initiated from the x‐ray structure (4ifp), the MBP did not manifest any significant deviation from the closed conformation at the simulated time scale (500 ns) as demonstrated in Figure S6. We speculate that the transitions observed in some simulation of AIDA and ClusPro models could be triggered by spatial rearrangement of the MBP and the NLRP1 domain in the chimeric construct. On the other hand, no conformational transitions of the MBP domain were observed in simulations of the 3csg construct, which were initialized with MBP in the open conformation.
The analogous conformational transitions were observed for the T4 lysozyme domain in the 5yqr chimera. The structure of T4 lysozyme consists of two subdomains, which may vary their respective orientation by hinge‐bending motions. 52 The interconversion between the open and close state takes place on the tens of microseconds time scale in solution through multiple intermediated states, which are known to be trapped in various x‐ray structures. 53 The 5yqr x‐ray structure involves the T4 lysozyme in the closed form adopted usually by this domain in the crystalline environment (see Figure S8 in).
The dynamics of the chimeric constructs in the course of simulations were assessed by Cα‐RMSD of the secondary structure elements. The distributions of Cα‐RMSD are plotted in Figure 3 for all simulations conducted in this study. Generally, most of the performed simulations manifested dynamic behavior and sampled conformations distant from the initial structure in terms of Cα‐RMSD.
FIGURE 3.
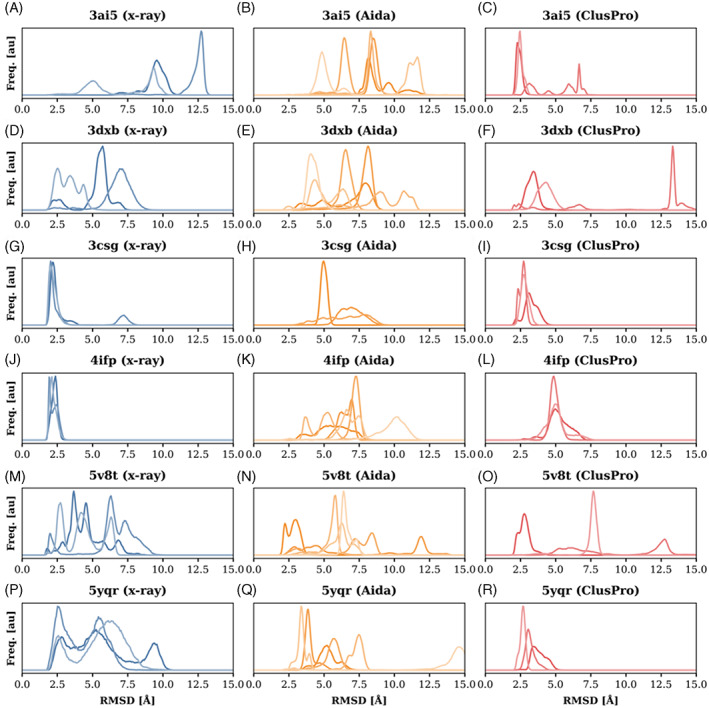
Distribution of RMSD from the initial structure as collected in course of the simulations. The three (six) curves shown for each chimeric protein in each plot (A–R) represent histograms obtained from three independent simulations of the same initial structure. In the case of AIDA (plots B, E, K, N, and Q), the plots involve both AIDA models resulting in six plotted curves.
This finding holds also for the crystallographic structures (Figure 3A,D,G,J,M,P). Only two chimeric proteins, 4ifp and 3csg (Figure 3G,J) maintained consistently the original orientation of the domains during simulations starting from their crystallographic structures for a significant portion of the simulation. The typical Cα‐RMSD from the initial coordinates for these proteins remained below 2.5 Å. Other chimeric proteins (3ai5, 3dxb, 5v8t, and 5yqr) populated the initial crystal conformation only transiently.
The simulations starting from the AIDA and ClusPro models diverged significantly from the initial conformations in all studied cases, see Figure 3. Nonetheless, some ClusPro structures manifested metastable behavior. For example, two of three simulations of the ClusPro 3ai5 model did not deviate from the initial model, but the last diverged quickly (Figure 3C). A similar metastability was observed for other ClusPro models, namely 3csg, 5v8t, and 5yqr (Figure 3I,O,R). Models generated by AIDA deviated to a larger extent than the ClusPro models and almost no simulation manifested metastability of the initial structure (except 5v8t model, Figure 3N).
Simulations of de novo predicted structures were further examined for similarity with their reference structures determined by x‐ray crystallography. Statistics on the corresponding Cα‐RMSD are shown in Table S9 in Supporting Information. Almost all the models significantly differed from their reference x‐ray structure even after the 500 ns simulation where certain type of convergence to the reference structure was expected. Importantly, we even did not detect any consistent tendency to evolve toward the reference x‐ray structures. The only exception represents a model of Thioredoxin and UHM domain chimera obtained by the AIDA prediction (reference PDB ID: 3dxb), where Cα‐RMSD values dropped to the minimal value of 2.7 Å in two independent simulation of AIDA #1 model. Nevertheless, this structure was not preferentially populated and the simulations evolved further toward higher Cα‐RMSD. The same conclusions were drawn for other simulation accidentally reaching low Cα‐RMSD, for example, a single simulation of 3ai5 AIDA #1 and #2 model and ClusPro model of 4ifp (see Table S9).
MD simulations provided useful, yet insufficient insight into available conformational space of the chimeric proteins. Figure 4 presents a two‐dimensional sketch of conformational landscape as obtained by projection of trajectories on the two most significant principal components (see Section 2 for details). This analysis revealed the fragmented picture delivered by the MD simulations. Figure 4 indicates that MD simulations mostly generated nonoverlapping trajectories if they started from different initial conformations (e.g., Figure 4C,F). Moreover, often a triplet of simulations initiated from the same model diverged in the sampled conformational space. This lack of recurrency might be caused both by the limited temporal sampling of the simulations (500 ns) and the character of the underlying free energy landscape. On the other hand, this analysis confirmed the stability of x‐ray structures 4ifp and 3csg (Figure 4C,D), whose trajectories populated restricted regions of conformation space. Other x‐ray structures manifested higher variability in sampling and their trajectories crossed unique regions as well as regions populated by other simulations.
FIGURE 4.
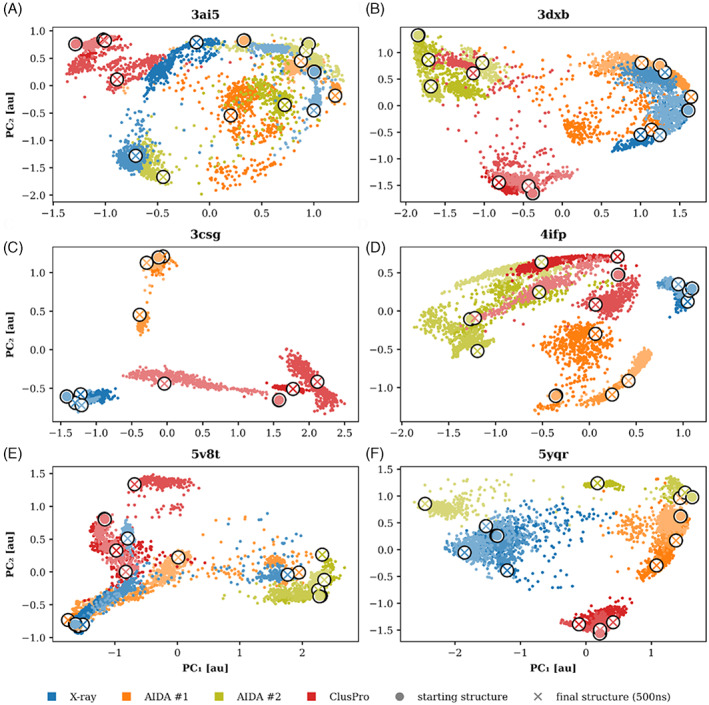
Projection of trajectories on two major principal components obtained by PCA. The features for PC analysis were RMSD from the starting and final structure of all simulations. The two AIDA models, ClusPro model and x‐ray are distinguished by colors (orange, olive, red, and blue, respectively). The three independent simulations are indicated by different saturation of the colors.
Additionally, the geometric descriptors of the protein molecule, such as radius of gyration (Rg) and solvent accessible surface area (SASA) were monitored in the course of simulations. Figure S10 shows Rg/SASA plots for each chimeric construct. These plots complement the findings about nonconvergent sampling during MD simulations provided in Figure 4. Although there is generally more overlap in Rg/SASA plot between individual simulations, this effect is rather caused by lower structural resolution of these features. On the other hand, in many cases (see, e.g., panels C, D, F in Figure S10), the simulations clearly sampled different areas of the Rg/SASA plot. The plots show no general and shared trends present in behavior of the simulated structures such as compaction and loosening. It might indicate that the used force field is not excessively biased towards compact structures and describes the behavior of the domains realistically.
The Rg/SASA plot revealed that the initial structures, both crystallographic and predicted, represent in some cases the extreme values of Rg or SASA. For example, the x‐ray structures 3csg and 5v8t (plots C and E in Figure S10) had the lowest SASA among the initial models and the value of SASA quickly increased during the simulations. Analogously, the initial ClusPro models manifested the lowest SASA for 3ai5, 3dxb, 4ifp, and 5yqr (plots A, B, D, and F in Figure S10). In these cases, the SASA also increased rapidly in the course of the simulation. A similar behavior can be observed less frequently for Rg, which can be seen, for example, for the x‐ray structure of 3csg (plot C in Figure S10) or the ClusPro model of 3ai5 (plot A in Figure S10).
Finally, we examined the conformational flexibility of the linkers connecting individual domains. The conformation of individual amino acid residues in the linker region was monitored by the backbone ψ and φ torsions in course of the simulations starting from the crystallographic structures. The corresponding Ramachandran plots are shown in Figures S11–S16 for 3ai5, 3csg, 3dxb, 4ifp, 5v8t, and 5yqr constructs, respectively. Except for 4ifp (Figure S14), all linkers contains at least one Gly residue, which sampled all accessible conformational states. In addition, also non‐Gly residues often occupied more than a single conformational basin. These observations suggest the flexible character of most of these linkers, implying that the linkers do not significantly dictate the respective orientation of the domains. The linker in 4ifp is only two residues long and therefore it deviates from the others. Its conformation is mostly extended, but the Leu‐372 residue is also forced to sample the energetically disadvantageous region in the upper right quadrant of the Ramachandran plot (see Figure S14).
4. DISCUSSION
Although the protein structure prediction has developed significantly in the last decades, most of the effort was put into structural predictions of single domains. There is a plethora of methods, approaches and tools, whose performance is assessed regularly at critical assesment of protein structure prediction (CASP) 54 competition. Many of them are publicly available to the users from the structural biologist community as web services. On the other hand, methods and programs for prediction of 3D structure of multidomain proteins and specifically protein chimeras composed of unrelated domains, represent only a small fraction of available tools. With no exceptions, the structure of a multidomain protein is produced as an assembly of domains. It follows the general belief that the composing domains are stable themselves and they do not manifest a structural variability in the isolated state and as parts of various multidomain proteins. Under this assumption, the only degrees of freedom are the respective position and orientation of the domains, and the structure of linker regions, which connect individual domains. Nonetheless, this problem still remains generally unsolved despite the remarkable effort resulting in some tools (e.g., DOME, SAXSDom, SynLinker, MDA, Rosetta) 55 , 56 , 57 , 58 , 59 , 60 which make the predictions possible to some extent. The effort is tightly connected as well with reliable prediction of protein–protein interactions and structural prediction of quaternary complexes of biomolecules within CAPRI 61 initiative.
The most successful approaches in protein structure predictions until introduction of AlphaFold 22 utilize homology modeling. Similar protein sequences usually share the same 3D structure, the latter being even more conserved. However, a significant portion of known proteins lack a relative with a solved 3D structure, which can be used as a template for homology modeling. The lack of 3D structures in PDB also hinders the utility of the approach on predicting protein–protein interactions and structure of multidomain proteins, which can be also reason attributed to relatively modest performance of AlphaFold due to relatively poor training set for machine learning approach in these cases. Although the individual domains can be modeled according to corresponding templates, their assembly is usually beyond the scope of homology modeling toolkits.
Prediction of structures of artificial multidomain proteins by construction misses any natural templates. Therefore, predictions of their structure must rely on physical approaches and exhaustive sampling strategies. At the same time, artificial multidomain proteins represent a challenging strategy for engineering of novel protein function and subject of gain‐of‐function studies. Therefore, it is of utter importance to understand and reliably predict their structure and relate it to their function.
In our study, we embraced a hypothesis, that spatial arrangement of domains in multidomain proteins is maintained dominantly by favorable/unfavorable domain–domain interactions at their interfaces. Our approach thus reflects the selection of tools employed in the study. Both AIDA but at more large extent the ClusPro targets on identification of interfaces between domains, which favor energetically their noncovalent interactions.
MD simulations of predicted models did not converge to the reference structure, although there were several attempts directed toward the benchmark crystal structure. Slightly higher stability of predicted models obtained from ClusPro tool compared to the AIDA tool can be easily explained by the difference in the level of model resolution. ClusPro tool considers all‐atom structures of both domains, whereas the AIDA procedure works only with structures represented by main‐chain atoms and side‐chain centers. Thus, further optimization of AIDA models by MD simulation was expected to provide more representative predictions.
In both methods, the role of the linker is not fully acknowledged. It is initially treated as fully flexible in AIDA, or nonexistent in ClusPro approach. Therefore, the effect of the linker is captured as a simple distance constraint between adjacent termini of both domains by the ClusPro predictions. Naturally, the short linker must inevitably restrict the pool of available interdomain orientations. Some linkers are considered as flexible, whereas others offer only a limited number of conformations. These rigid conformations can further limit the respective position and orientation of the domains. In addition to a simple geometric conception, linkers are known to influence the properties and behavior of multidomain proteins in a complex way. 33 , 34 Our results from molecular dynamics simulations suggest that the linkers in chosen constructs allow flexibility in positioning of the domains. This suggests that the conformation of linkers studied in this work is rather determined by the interaction of the domains than the other way. However, the exact effects of linkers remain to be elucidated. We could not address this question in this work due to the limited representability of the dataset.
Hydration plays an important role in stability of the interfaces. The hydration effects are considered by both docking methods, either implicitly as part of the effective interaction potential (AIDA) or empirically approximated based on the contact potential (ClusPro). Additionally, the stability of the predicted interfaces can be assessed by independent tools, such as a PISA and PRODIGY, which are trained on x‐ray structures. Nonetheless, prediction of stability of an interface is a challenging task, regardless if an estimate of Gibbs binding energy is required or binary classification (stable/unstable) suffices.
Although it was not original intention to compare proposed methodology with the most powerful structure prediction method nowadays—the AlphaFold II we were tempted to compare its prediction performance on the set of selected proteins (all contained in PDB). To illustrate the differences of our approach and the current state‐of‐the‐art in protein structure prediction, we applied AlphaFold II on sequences of all studied targets. The Table 1 below shows RMSD values obtained after superimposition of the solved x‐ray structures to best models made by AlphaFold 2.
TABLE 1.
RMSD of the x‐ray structures superimposed to best models by AlphaFold 2
| PDB code | RMSD(x‐ray vs. AF2) (Å) |
|---|---|
| 3AI5 | 0.373 |
| 3CSG | 6.572 |
| 3DXB | 12.268 |
| 4IFP | 0.430 |
| 5V8T | 5.062 |
| 5YQR | 1.171 |
In order to better illustrate the problem of prediction of the interdomain position, RMSD values were plotted as a function of residue number (see Figure S17). It can be clearly seen that the RMSD values by residue increase significantly for the second (nonaligned to the reference) domain in all structures since the different interdomain positioning relative to the experimental structure automatically incurs large gains in RMSD values. This can be seen visually (Figure S18), where one of the domains always corresponds better to the reference structure than the other domain. To summarize the comparison between AF II predicted structures and their x‐ray targets, we can report significant deviation between the model and the target for three of six proteins. The same number of structures were predicted with relatively low RMSD so the accuracy of the AFII was about 50% for the studied proteins.
One of the problems in assessment of the tested approach is related to the selection of target/structure benchmarks. Because all of the studied chimeras were compared with their structures obtained by x‐ray we had to take into accounts additional limits and conditions. Primarily—how much the computational methodology has to reflect the crystal structure environment or at which extent the crystal environment constraints conformational space of the produced chimeras. It is necessary to say that this problem was only partially addressed by PRODIGY and PISA analyses. Regarding the obtained results, we can conclude that the proposed approach is more suitable for prediction of structures in solution due to the fact that MD simulations are relatively easy to be run in solvent environment and no effect of crystal environment is necessary to take into account. The presented study clearly shows that there is no simple approach and straightforward methodology for prediction of fusion proteins primarily due to the fact that the performed sampling is insufficient because multiple minima of the chimeras can be comparable at energy level but substantially different at conformational level. Therefore, we cannot conclude that x‐ray structures are not reasonable benchmarks neither that they are due to the fact that crystal structure environment is impossible to apply for new predicted chimeras. In this study, we show that proposed methodology does not work unless we will define conditions under which these benchmarks could be reliably used. Results of the presented study and studies focused on comparison x‐ray and NMR structures 28 , 29 , 30 , 31 clearly show that we are in a “twilight zone” and no simple and accepted conditions for benchmarking is easy to set and apply.
5. CONCLUSION
The major conclusion of this study is the finding that structure prediction of novel chimera proteins needs a solid methodology justification and analyses which would lead to a reliable prediction tool. The tested approaches using publicly available tools and simulation programs did not lead to the full agreement with experimentally determined structure by x‐ray crystallography (even the AF II program succeeded in 50% of cases). Advanced procedure using extensive MD simulation runs of crystal structures further highlighted conformationally unstable character of two‐domain protein outside the crystal environment when most of the chimeras exceeded maximal RMSD of 5 Å during the 500 ns simulation run (except for MBP constructs ‐ figure F3, plot J and both of its domains ‐ figure S6, x‐ray ‐ domain #1 and #2). It is necessary to add that the simulation performed mimicked in solution srather than crystal structure environment.
To sum up, predicted models by AIDA and CLUSPRO did not correspond to the original crystal structures and MD simulation of models in general did not lead to the better overlap with original crystal structure. MD simulations of crystal structures in some cases revealed relatively high flexibility of mutual domain orientation apparently caused by characters of domains and linker composition and length. All these results led us to the conclusion that it is primarily insufficient sampling of conformational space in the proposed method and that we cannot state that crystallographic structures of two‐domain protein chimeras are or are not suitable to be good benchmarks for in solution predicted structures.
Nonetheless, the inability of the prediction methods to reproduce the crystal structures cannot be considered as their complete failure only due to apparently insufficient sampling. Our results showed that it could be problematic to relate the crystal structure of a fusion protein to its behavior in solution. At some extent, the crystal structures of fusion proteins could be even potentially misleading, since their conformation might be driven by alternative interfaces available in the crystal lattice between other protein molecules in (between) the unit cell.
In conclusion, there are aspects that should be considered thoroughly when preparing novel chimeric protein constructs, such as linker length or the overall stability of individual domains used for fusion. However, the interface naturally established between the fused domains and its size was proved to be the main determining factor for general structural flexibility of a single molecule (taken from the crystal environment) in the aqueous environment. Therefore, it is vital to carefully design and characterize interaction interfaces within the chimeric construct in order to create multidomain protein with rigid domain organization.
AUTHOR CONTRIBUTIONS
Jiri Vondrasek: Conceptualization; data curation; formal analysis; funding acquisition; investigation; resources; supervision; validation; writing the draft; writing‐review and editing. Jiří Vymětal: Conceptualization; data curation; formal analysis; investigation; supervision; validation; writing‐original draft; writing‐review and editing. Kateřina Mertová: MD simulations, data curation, analysis, writing. Kristýna Boušová: Methodology; writing‐review and editing. Konstantinos Tripsianes: Data curation; funding acquisition; supervision. Josef Šulc: Formal analysis, simulations, writing‐review, editing manuscript.
CONFLICT OF INTERESTS
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author declares that there are no potential conflicts of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/prot.26398.
Supporting information
Appendix S1 Supporting Information
ACKNOWLEDGMENTS
Computational resources were supplied by the project “e‐Infrastruktura CZ” and ELIXIR CZ (e‐INFRA CZ LM2018140, ELIXIR CZ LM2018131) supported by the Ministry of Education, Youth and Sports of the Czech Republic. The authors thank members of the Bioinformatics group for stimulating discussion. European Regional Development Fund; OP RDE, Grant/Award Number: CZ.02.1.01/0.0/0.0/16_019/0000729; Grantova Agentura České Republiky, Grant/Award Number: GA19‐03488S; Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Grant/Award Number: RVO: 61388963; MEYS CR and National Program for Sustainability II, Grant/Award Number: CEITEC 2020 (LQ1601).
Vymětal J, Mertová K, Boušová K, Šulc J, Tripsianes K, Vondrasek J. Fusion of two unrelated protein domains in a chimera protein and its 3D prediction: Justification of the x‐ray reference structures as a prediction benchmark. Proteins. 2022;90(12):2067‐2079. doi: 10.1002/prot.26398
Funding information MEYS CR and National Program for Sustainability II, Grant/Award Number: CEITEC 2020 (LQ1601); Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Grant/Award Number: RVO: 61388963; Grantova Agentura České Republiky, Grant/Award Number: GA19‐03488S; European Regional Development Fund, Grant/Award Number: CZ.02.1.01/0.0/0.0/16_019/0000729; the Ministry of Education, Youth and Sports, Grant/Award Numbers: LM2018131, LM2018140
DATA AVAILABILITY STATEMENT
All relevant data are available on request to the corresponding author. The primary data used in the paper are downloadable from the Protein Data Bank (http://www.rcsb.org).
REFERENCES
- 1. Naveenkumar N, Kumar G, Sowdhamini R, Srinivasan N, Vishwanath S. Fold combinations in multi‐domain proteins. Bioinformation. 2019;15:342‐350. doi: 10.6026/97320630015342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dawson NL, Lewis TE, Das S, et al. CATH: an expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 2017;45:D289‐D295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dunn HA, Ferguson SS. PDZ protein regulation of G protein‐coupled receptor trafficking and signaling pathways. Mol Pharmacol. 2015;88:624‐639. doi: 10.1124/mol.115.098509 [DOI] [PubMed] [Google Scholar]
- 4. Kummerfeld SK, Teichmann SA. Protein domain organisation: adding order. BMC Bioinformatics. 2009;10:39. doi: 10.1186/1471-2105-10-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yu K, Liu C, Kim BG, Lee DY. Synthetic fusion protein design and applications. Biotechnol Adv. 2015;33:155‐164. doi: 10.1016/j.biotechadv.2014.11.005 [DOI] [PubMed] [Google Scholar]
- 6. Young CL, Britton ZT, Robinson AS. Recombinant protein expression and purification: a comprehensive review of affinity tags and microbial applications. Biotechnol J. 2012;7:620‐634. doi: 10.1002/biot.201100155 [DOI] [PubMed] [Google Scholar]
- 7. Waugh DS. Crystal structures of MBP fusion proteins. Protein Sci. 2016;25:559‐571. doi: 10.1002/pro.2863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li S, Chen LX, Peng XH, et al. Overview of the reporter genes and reporter mouse models. Anim Model Exp Med. 2018;1:29‐35. doi: 10.1002/ame2.12008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Navale GR, Sharma P, Said MS, et al. Enhancing epi‐cedrol production in Escherichia coli by fusion expression of farnesyl pyrophosphate synthase and epi‐cedrol synthase. Eng Life Sci. 2019;19:606‐616. doi: 10.1002/elsc.201900103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fan L, Wang Y, Tuyishime P, et al. Engineering artificial fusion proteins for enhanced methanol bioconversion. Chembiochem. 2018;19:2465‐2471. doi: 10.1002/cbic.201800424 [DOI] [PubMed] [Google Scholar]
- 11. Teillaud JL. Engineering of monoclonal antibodies and antibody‐based fusion proteins: successes and challenges. Expert Opin Biol Ther. 2005;5:S15‐S27. doi: 10.1517/14712598.5.1.S15 [DOI] [PubMed] [Google Scholar]
- 12. Beck A, Reichert JM. Therapeutic fc‐fusion proteins and peptides as successful alternatives to antibodies. MAbs. 2011;3:415‐416. doi: 10.4161/mabs.3.5.17334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hutmacher C, Neri D. Antibody‐cytokine fusion proteins: biopharmaceuticals with immunomodulatory properties for cancer therapy. Adv Drug Deliv Rev. 2019;141:67‐91. doi: 10.1016/j.addr.2018.09.002 [DOI] [PubMed] [Google Scholar]
- 14. Wang LL, Xu JY, Kong Y, et al. Engineering a novel antibody‐peptide bispecific fusion protein against MERS‐CoV. Antibodies. 2019;8. doi: 10.3390/antib8040053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Caravella JA, Wang DP, Glaser SM, Lugovskoy A. Structure‐guided design of antibodies. Curr Comput Aided Drug des. 2010;6:128‐138. [PubMed] [Google Scholar]
- 16. Iyengar ARS, Gupta S, Jawalekar S, Pande AH. Protein chimerization: a new frontier for engineering protein therapeutics with improved pharmacokinetics. J Pharmacol Exp Ther. 2019;370:703‐714. doi: 10.1124/jpet.119.257063 [DOI] [PubMed] [Google Scholar]
- 17. Kintzing JR, Interrante MVF, Cochrane JR. Emerging strategies for developing next‐generation protein therapeutics for cancer treatment. Trends Pharmacol Sci. 2016;37:993‐1008. doi: 10.1016/j.tips.2016.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trang VH, Zhang XQ, Yumul RC, et al. A coiled‐coil masking domain for selective activation of therapeutic antibodies. Nat Biotechnol. 2019;37:761. doi: 10.1038/s41587-019-0135-x [DOI] [PubMed] [Google Scholar]
- 19. Yang Y, Aloysius H, Inoyama D, Chen Y, Hu L. Enzyme‐mediated hydrolytic activation of prodrugs. Acta Pharm Sin B. 2011;1:143‐159. doi: 10.1016/j.apsb.2011.08.001 [DOI] [Google Scholar]
- 20. Xu D, Jaroszewski L, Li Z, Godzik A. AIDA: ab initio domain assembly for automated multi‐domain protein structure prediction and domain‐domain interaction prediction. Bioinformatics. 2015;31:2098‐2105. doi: 10.1093/bioinformatics/btv092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou XG, Hu J, Zhang CX, Zhang GJ, Zhang Y. Assembling multidomain protein structures through analogous global structural alignments. Proc Natl Acad Sci U S A. 2019;116:15930‐15938. doi: 10.1073/pnas.1905068116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583‐589. doi: 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Varadi M, Anyango S, Deshpande M, et al. AlphaFold protein structure database: massively expanding the structural coverage of protein‐sequence space with high‐accuracy models. Nucleic Acids Res. 2022;50:D439‐D444. doi: 10.1093/nar/gkab1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Verma R, Pandit SB. Unraveling the structural landscape of intra‐chain domain interfaces: implication in the evolution of domain‐domain interactions. PLoS One. 2019;14:e0220336. doi: 10.1371/journal.pone.0220336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nanev CN. Application of mean‐separation‐works method to protein crystal nucleation. Cryst Res Technol. 2008;43:229‐233. doi: 10.1002/crat.200711085 [DOI] [Google Scholar]
- 26. Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774‐797. doi: 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 27. Jimenez‐Garcia B, Elez K, Koukos PI, Bonvin AM, Vangone A. PRODIGY‐crystal: a web‐tool for classification of biological interfaces in protein complexes. Bioinformatics. 2019;35:4821‐4823. doi: 10.1093/bioinformatics/btz437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carlon A, Ravera E, Parigi G, Murshudov GN, Luchinat C. Joint X‐ray/NMR structure refinement of multidomain/multisubunit systems. J Biomol NMR. 2019;73:265‐278. doi: 10.1007/s10858-018-0212-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Garbuzynskiy SO, Melnik BS, Lobanov MY, Finkelstein AV, Galzitskaya OV. Comparison of X‐ray and NMR structures: is there a systematic difference in residue contacts between X‐ray and NMR‐resolved protein structures? Proteins. 2005;60:139‐147. doi: 10.1002/prot.20491 [DOI] [PubMed] [Google Scholar]
- 30. Schneider M, Fu XR, Keating AE. X‐ray vs. NMR structures as templates for computational protein design. Proteins. 2009;77:97‐110. doi: 10.1002/prot.22421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Qian B, Raman S, Das R, et al. High‐resolution structure prediction and the crystallographic phase problem. Nature. 2007;450:259‐264. doi: 10.1038/nature06249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Childers MC, Daggett V. Insights from molecular dynamics simulations for computational protein design. Mol Syst Des Eng. 2017;2:9‐33. doi: 10.1039/C6ME00083E [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang JH, Yun J, Shang ZG, Zhang XH, Pan BR. Design and optimization of a linker for fusion protein construction. Prog Nat Sci Int. 2009;19:1197‐1200. doi: 10.1016/j.pnsc.2008.12.007 [DOI] [Google Scholar]
- 34. Chen X, Zaro JL, Shen WC. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65:1357‐1369. doi: 10.1016/j.addr.2012.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Madeira F, Park YM, Lee J, et al. The EMBL‐EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47:W636‐W641. doi: 10.1093/nar/gkz268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bateman A. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506‐D515. doi: 10.1093/nar/gky1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. DeLano WL, Lam JW. PyMOL: a communications tool for computational models. Abstr Pap Am Chem Soc. 2005;230:U1371‐U1372. [Google Scholar]
- 39. Fiser A, Sali A. ModLoop: automated modeling of loops in protein structures. Bioinformatics. 2003;19:2500‐2501. doi: 10.1093/bioinformatics/btg362 [DOI] [PubMed] [Google Scholar]
- 40. Kozakov D, Hall DR, Xia B, et al. The ClusPro web server for protein‐protein docking. Nat Protoc. 2017;12:255‐278. doi: 10.1038/nprot.2016.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Krissinel E. Advances in PISA software for macromolecular assembly predictions from CCP4. Acta Cryst A. 2015;71:S40. doi: 10.1107/S2053273315099362 [DOI] [Google Scholar]
- 42. Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model. 2001;7:306‐317. doi: 10.1007/s008940100045 [DOI] [Google Scholar]
- 43. Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696‐3713. doi: 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926‐935. doi: 10.1063/1.445869 [DOI] [Google Scholar]
- 45. Hess B. P‐LINCS: a parallel linear constraint solver for molecular simulation. J Chem Theory Comput. 2008;4:116‐122. doi: 10.1021/ct700200b [DOI] [PubMed] [Google Scholar]
- 46. Cheatham TE, Miller JL, Fox T, Darden TA, Kollman PA. Molecular‐dynamics simulations on solvated biomolecular systems ‐ the particle mesh Ewald method leads to stable trajectories of DNA, RNA, and proteins. J Am Chem Soc. 1995;117:4193‐4194. doi: 10.1021/ja00119a045 [DOI] [Google Scholar]
- 47. Martonak R, Laio A, Parrinello M. Predicting crystal structures: the Parrinello‐Rahman method revisited. Phys Rev Lett. 2003;90:75503. doi: 10.1103/PhysRevLett.90.075503 [DOI] [PubMed] [Google Scholar]
- 48. Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. Molecular‐dynamics with coupling to an external Bath. J Chem Phys. 1984;81:3684‐3690. doi: 10.1063/1.448118 [DOI] [Google Scholar]
- 49. Ranjani J, Sheela A, Meena KP. Combination of NumPy, SciPy and Matplotlib/Pylab ‐ a good alternative methodology to MATLAB ‐A Comparative Analysis. IEEE; 2019. [Google Scholar]
- 50. Wang Y, Tang C, Wang EK, Wang J. Exploration of multi‐state conformational dynamics and underlying global functional landscape of maltose binding protein. PLoS Comput Biol. 2012;8:e1002471. doi: 10.1371/journal.pcbi.1002471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tang C, Schwieters CD, Clore GM. Open‐to‐closed transition in apo maltose‐binding protein observed by paramagnetic NMR. Nature. 2007;449:1078‐U12. doi: 10.1038/nature06232 [DOI] [PubMed] [Google Scholar]
- 52. Mchaourab HS, Oh KJ, Fang CJ, Hubbell WL. Conformation of T4 lysozyme in solution hinge‐bending motion and the substrate‐induced conformational transition studied by site‐directed spin labeling. Biochemistry. 1997;36:307‐316. doi: 10.1021/bi962114m [DOI] [PubMed] [Google Scholar]
- 53. Yirdaw RB, Mchaourab HS. Direct observation of T4 lysozyme hinge‐bending motion by fluorescence correlation spectroscopy. Biophys J. 2012;103:1525‐1536. doi: 10.1016/j.bpj.2012.07.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. Critical assessment of methods of protein structure prediction (CASP)‐round XIII. Proteins. 2019;87:1011‐1020. doi: 10.1002/prot.25823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Matsuno S, Ohue M, Akiyama Y. Multidomain protein structure prediction using information about residues interacting on multimeric protein interfaces. Biophys Physicobiol. 2020;17:2‐13. doi: 10.2142/biophysico.BSJ-2019050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu CC, Chin JX, Lee DY. SynLinker: an integrated system for designing linkers and synthetic fusion proteins. Bioinformatics. 2015;31:3700‐3702. doi: 10.1093/bioinformatics/btv447 [DOI] [PubMed] [Google Scholar]
- 57. Hou J, Adhikari B, Tanner JJ, Cheng JL. SAXSDom: modeling multidomain protein structures using small‐angle X‐ray scattering data. Proteins. 2020;88:775‐787. doi: 10.1002/prot.25865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hertig S, Goddard TD, Johnson GT, Ferrin TE. Multidomain assembler (MDA) generates models of large multidomain proteins. Biophys J. 2015;108:2097‐2102. doi: 10.1016/j.bpj.2015.03.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dukka BKC. Recent advances in sequence‐based protein structure prediction. Brief Bioinform. 2017;18:1021‐1032. doi: 10.1093/bib/bbw070 [DOI] [PubMed] [Google Scholar]
- 60. Wollacott AM, Zanghellini A, Murphy P, Baker D. Prediction of structures of multidomain proteins from structures of the individual domains. Protein Sci. 2007;16:165‐175. doi: 10.1110/ps.062270707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wodak SJ, Velankar S, Sternberg MJE. Modeling protein interactions and complexes in CAPRI: seventh CAPRI evaluation meeting, April 3‐5 EMBL‐EBI, Hinxton, UK. Proteins. 2020;88:913‐915. doi: 10.1002/prot.25883 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information
Data Availability Statement
All relevant data are available on request to the corresponding author. The primary data used in the paper are downloadable from the Protein Data Bank (http://www.rcsb.org).