

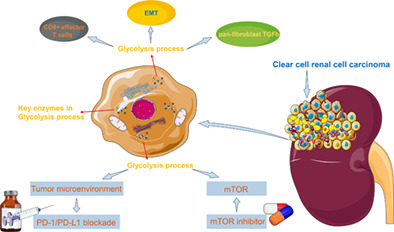
Abstract
In clear cell renal cell carcinoma (ccRCC), glycolysis is enhanced mainly because of the increased expression of key enzymes in glycolysis. Hence, the discovery of new molecular biomarkers for glycolysis may help guide and establish a precise system of diagnosis and treatment for ccRCC. Expression profiles of 1079 tumor samples of ccRCC patients (including 311 patients treated with everolimus or nivolumab) were downloaded from public databases. Proteomic profiles of 232 ccRCC samples were obtained from Fudan University Shanghai Cancer Center (FUSCC). Biological changes, tumor microenvironment and prognostic differences were explored between samples with various glycolysis characteristics. There were significant differences in CD8+ effector T cells, epithelial‐to‐mesenchymal transition and pan‐fibroblast TGFb between the Low and High glyScore groups. The tumor mutation burden of the Low glyScore group was lower than that of the High glyScore group. And higher glyScore was significantly associated with worse overall survival (OS) in 768 ccRCC patients (P < .0001). External validation in FUSCC cohort also indicated that glyScore was of strong ability for predicting OS (P < .05). GlyScore may serve as a biomarker for predicting everolimus response in ccRCC patients due to its significant associations with progression‐free survival (PFS). And glyScore may also predict overall survival in patients treated with nivolumab. We calculated the glyScore in ccRCC and the defined glyScore was of strong ability for predicting OS. In addition, glyScore may also serve as a biomarker for predicting PFS in patients treated with everolimus and could predict OS in patients treated with nivolumab.
Keywords: biomarker, clear cell renal cell carcinoma, everolimus, glycolysis, nivolumab, tumor microenvironment
What's new?
Numerous enzymes and biomarkers involved in the glycolytic process are dysregulated in clear cell renal cell carcinoma. Here, using public and hospital gene/protein expression profiles, the authors analysed differences in biological changes, tumour microenvironment, and prognosis between clear cell renal cell carcinoma samples with various glycolysis characteristics. They identified a glycolysis signature with a strong ability to predict overall survival. In addition, the calculated glyScore showed potential as a biomarker for predicting progression‐free survival in patients treated with everolimus and overall survival in patients treated with nivolumab.
Abbreviations
- AUC
area under the curve
- ccRCC
clear cell renal cell carcinoma
- CNV
copy number variations
- DEG
differentially expressed genes
- EMT
epithelial‐mesenchymal transformation
- GSVA
gene set variation analysis
- mTOR
mammalian target of rapamycin
- OS
overall survival
- PFS
progression‐free survival
- RCC
renal cell carcinoma
- ROC
receiver operating characteristic
- ssGSEA
single‐sample gene set enrichment analysis
- TCGA
the cancer genome atlas
- TGFb
transforming growth factor‐β
- TME
tumor microenvironment
- VHL
von Hippel‐Lindau
1. INTRODUCTION
Renal cell carcinoma (RCC) is a heterogeneous disease encompassing different histological subtypes. 1 Among the histologic types, clear cell RCC (ccRCC) derived from proximal tubular epithelium accounts for 70% to 80% of RCCs. Mortality is high for aggressive ccRCC and up to 30% of patients have been diagnosed preliminarily with metastatic ccRCC. 2 , 3 , 4 Localized ccRCC usually can be treated by traditional surgical resection or ablation to obtain satisfactory results. 5 However, some patients with aggressive ccRCC are not suitable for surgery, and chemotherapy or radiotherapy cannot be used because of the tumor heterogeneity. 6 Thus, the carcinogenic mechanisms of ccRCC, including metabolism or molecular processes, are important considerations.
Research on tumors has a long tradition, covering their occurrence, development, aggression and metastasis. About 100 years ago, Otto Warburg found that cancer cells tended to generate their energy by fermentation even in aerobic conditions while working on in vitro biopsies of human tumor samples. The explanation for this observation is the high glycolysis rate followed by lactic acid fermentation, even in the presence of abundant oxygen, that cancer cells use to produce their energy. This was later called the Warburg effect, which has led to several breakthroughs for various tumor types 7 and is the basis for the diagnostic 18‐fluoro‐2‐deoxyglucose‐positron emission tomography (FDG‐PET) scan. An inversely proportional relationship between FDG uptake and differentiation of cancers manifested in a disorder of the transition from glycolysis to oxidative phosphorylation was detected in cancer stem cells. 8 The Warburg effect can lead to various mutations in cancer cells, consequently, tumors with different proliferation or metastases characteristics appear, which is one of the primary reasons why the treatment of even the same kind of cancer can be difficult. 9 , 10 Glycolysis plays a major role in the genesis and development of cancers, and the intrinsic regulatory mechanisms of genes and other molecules involved in glycolysis have been the focus of many recent studies.
The best‐known genetic feature of ccRCC is the von Hippel‐Lindau (VHL)/hypoxia‐inducible factors (HIF) oxygen‐sensing pathway, in which deletion or mutation of the VHL tumor suppressor gene leads to the increase and accumulation of hypoxia‐inducible factors (HIFs), thereby activating processes, such as cell proliferation, infiltration, neovascularization and metastases, ultimately leading to the development of ccRCC. 11 Many enzymes play important roles in balancing oxidative phosphorylation and glycolysis. In the tricarboxylic acid (TCA) cycle, fumarate hydratase and succinate dehydrogenase are necessary for mitochondrial respiration, and mutations in either of the two genes encoding these enzymes can disrupt the oxidative process, which means that affected cells will rely more on glycolysis. 12 Besides, mutations in genes encoding AMP‐activated protein kinase, pyruvate dehydrogenase kinase 1, lactate dehydrogenase A and pyruvate kinase M2 also alter the metabolism in ccRCC. 13 , 14 Variation of the gluconeogenesis enzyme fructose‐1,6‐bisphosphatase was found to suppress gluconeogenesis and raise the glycolytic flux in RCC cells. 15 Furthermore, polybromo 1 (PBRM1), SET domain containing 2, histone lysine methyltransferase (SETD2), BRCA1‐associated protein (BAP1) and phosphatase and tensin homolog (PTEN) can determine the remodeling of chromatin structure, accelerating the growth of ccRCC. 16 Thus, understanding the genetic basis of ccRCC, and the development of a comprehensive system to assess or score ccRCCs are crucial for the diagnosis, treatment and prognosis of ccRCC.
Bioinformatics provides a huge and convenient platform for studying the mechanisms of multiple diseases, including enormous databases, strong analysis tools and assessment methods. 17 In this study, we obtained gene/protein expression profiles of 1311 samples of ccRCC and made an integrated analysis to explore glycolysis characteristics of ccRCC and identify a potential glycolysis signature to predict prognosis and drug response.
2. MATERIALS AND METHODS
2.1. Data downloading and normalization
A total of 1311 samples of ccRCC were well enrolled for analysis. Expression data of 768 ccRCC samples from the Cancer Genome Atlas (TCGA), European Molecular Biology Laboratory (EMBL), International Cancer Genome Consortium (ICGC), Clinical Proteomic Tumor Analysis Consortium (CPTAC) were collected. Proteomic profiles of 232 ccRCC samples were obtained from Fudan University Shanghai Cancer Center (FUSCC). Transcriptomic (RNA‐seq) and clinical data of 311 ccRCC samples from patients treated with Everolism or nivolumab were obtained from Table S4 appended to the published paper. 18 The expressed values were converted and standardized for data merging, and the batch effect was removed for subsequent analysis. Somatic mutation data, transcriptome data, copy number variation (CNV) data and sample phenotype data were downloaded from the cancer genome atlas‐kidney renal clear cell carcinoma (TCGA‐KIRC) database (https://xenabrowser.net/datapages/). The somatic mutation data covered 336 samples. There were two sets of CNV data: one set contained the CNV results sorted by the Xena Functional Genomics Explorer, which is specific to whether a CNV is present in a gene; and the other set was the sample DNAcopy file downloaded from the Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov/). Set 1 was used to facilitate grouping later and Set 2 was used to implement the GISTIC2 analysis. The transcriptome data consisted of one set of RNA sequencing (RNA‐seq) data, labeled TCGA‐KIRC, with 607 samples. Expression data were obtained for 602 of the samples, including 531 tumor samples and 71 normal samples, after excluding samples without survival information. E‐MTAB‐3267 expression microarray data, including 53 tumor samples and six normal samples, were downloaded from EMBL's ArrayExpress database (https://www.ebi.ac.uk/arrayexpress/). The RECA‐EU transcriptome data were downloaded from the ICGC (https://dcc.icgc.org/) database, and 91 tumor samples and 45 normal samples were obtained after excluding samples without survival information. Transcriptome data for ccRCC samples were obtained from the CPTAC database (https://cptac-data-portal.georgetown.edu/study-summary/S050), including 93 tumor samples and 75 normal samples. The expression levels of the RNA‐seq samples were converted from FPKM (fragments per kilobase of transcript per million mapped reads) to TPM (transcripts per million), and log2(TPM + 1) was taken. The expression levels in the microarray data were already given as log2, and therefore no processing was required. These four sets of expression data were combined and the intersection of gene expression levels was used for further analyses. The sva package in R was used to remove the batch effect. We first merge the dataset by taking the intersection, then, elimination of batch effect was carried out based on ComBat function in SVA package and known batch information. Sample IDs of all publicly available datasets were listed in Table S6.
2.2. Extraction of glycolysis‐related genes and unsupervised clustering
The REACTOME_GLYCOLYSIS set of 72 genes was downloaded from the Molecular Signatures Database (MSigDB; (https://www.gsea-msigdb.org/gsea/msigdb) together with 186 gene sets (Canonical Pathways gene sets derived from the KEGG pathway database). Gene sets of 23 infiltrating cells were downloaded from the previous study. 19 Gene sets of biological processes such as angiogenesis, CD8+ effector T cells, epithelial‐to‐mesenchymal transition stages (EMT1, EMT2, EMT3) and pan‐fibroblast TGFb were downloaded from the previous study. 20 The expression values of 72 glycolytic genes were extracted using unscreened TCGA‐KIRC expression data, and a boxplot of these genes was drawn using the ggpubr package in R to show the differences in gene expression between tumor and normal tissues. The CNV data for the glycolytic genes were extracted, the frequencies of amplification and deletion were counted, and a dot plot was drawn. TCGA‐KIRC samples in MAF format were imported using the maftools package in R to display the somatic mutation spectrum. A Circos map of the glycolytic genes was drawn using the RCircos package in R to visualize the positions of the glycolytic genes on the human reference genome hg38. A principal component analysis (PCA, Table S5) of the expression matrix of 72 glycolytic genes was performed using the pca3d package in R, and a 3D PCA map was drawn. The expression matrix of 72 glycolytic genes was extracted from the combined data using the ConsensusClusterPlus package in R and saved as the input file for unsupervised clustering. The parameter settings were: maximum classification number maxK = 6; repeated sampling reps = 1000; proportion of selected samples pItem = 0.8; proportion of features pFeature = 1; clustering algorithm clusterAlg = “PAM”; and distance = “Pearson.” The output results were integrated, K values were screened and the classification of each sample was obtained. Clinical data of the samples were integrated. Survival analysis was performed using the survival package in R, and a Kaplan‐Meier survival curve of glycolysis.cluster was plotted using the survminer package in R. Log‐rank was used as the statistical test, with P‐value <.05 set as the significant survival difference between groups.
2.3. Gene set variation analysis
For the different glycolysis.clusters, every two clusters formed a group. In every intra‐group, the expression matrices of all the genes in the two clusters were extracted and combined with the reference gene set c2.cp.kegg.v7.1.symbols as the input file for the gene set variation analysis (GSVA) package in R. The enrichment score of each gene set in each sample was obtained. Then, the limma package in R was used for gene set difference analysis, and the screening threshold was set as Benjamini‐Hochberg corrected P‐value <.05. The top 20 differential gene sets were extracted, and the ComplexHeatmap package in R was used to draw the enrichment score heat map of the differential gene sets, with grouping and data source labels.
2.4. Assessing the proportion and differences of 23 infiltrating immune cells
The expression matrices of all the genes in the combined data and the gene sets of 23 infiltrating cells were used as the input file for the GSVA package to carry out single‐sample gene set enrichment analysis (ssGSEA); the enrichment score was taken as the content of each cell. A boxplot was drawn using the ggpubr package, and a log‐rank test was conducted to indicate the differences among the 23 infiltrating cells in different glycolysis.cluster groups. Differentiated cells were selected, and the hazard ratio and P‐values were obtained by univariate Cox risk regression analysis using the survival package. Forest plots were obtained using the forestplot package in R to visualize the prognostic effect of the differentiated cells. Differences in the enrichment scores for the angiogenesis, CD8+ effector T cells, EMT1, EMT2, EMT3 and pan‐fibroblast TGFb processes among the glycolysis.cluster groups also were demonstrated using the methods described above. Gene sets were replaced and ssGSEA was used to obtain enrichment scores for each of the six biological processes in each sample. A boxplot using the ggpubr package, and a log‐rank test were conducted.
2.5. Construction of glycolysis gene signatures
For different glycolysis clusters, every two clusters formed a group. The limma package was used to analyze the differential genes, and the threshold of differential gene screening was log2 (fold change) = log2 (1.25) and corrected P‐value <.05. Finally, several groups of differentially expressed genes were obtained, and the intersection was taken as the final differential gene. The expression matrix of the above differentially expressed genes was extracted as the input file, and the ConsensusClusterPlus package was used to identify glycolysis gene clusters. Clinical data of the samples were integrated, and survival analysis of the glycolysis gene clusters was performed using Kaplan‐Meier methods. Univariate Cox risk regression analysis was performed using the survival package for the detected differential genes with P‐values <.05. For the selected genes, the random forest model was constructed using the randomForestSRC package in R, then the important characteristic variables were chosen as the glycolysis gene signatures. The glycolysis score (glyScore) was calculated by PCA as: , where i is the expression value of the glycolysis‐related gene.
2.6. Identifying biological changes behind the glyScore and exploring its potential use in predicting immunotherapy response
The REACTOME_GLYCOLYSIS gene set and six groups of the top 10 differential gene sets obtained by GSVA were integrated, and ssGSEA was performed. The enrichment score of the gene sets combined with the glyScore was used to calculate a Pearson correlation coefficient matrix and P‐value. The correlation plot of the upper triangle was plotted using the corrplot package in R; P‐values <.01 were considered significant. Colored dots indicate the corresponding correlation coefficients. Based on the Tumor Immune Dysfunction and Exclus (TIDE) (http://tide.dfci.harvard.edu/) immunotherapy outcomes prediction, a 5‐year ROC (receiver operating characteristic) curve was plotted using the survivalROC package in R.
2.7. A proteomic cohort from FUSCC
We performed genomic and proteomic profiling of 232 paired Chinese ccRCC samples with the median follow‐up time for 85 months (range, 3‐138 months) from FUSCC. Firmiana and the human National Center for Biotechnology Information (NCBI) RefSeq protein database (updated on April 7, 2013, 32 015 entries) were used in processing the Raw files. Mass tolerances were 20 ppm for precursor and 50 mmu for product ions. Up to two missed cleavages were allowed. Precursor ion score were limited to +2, +3 and +4. A decoy database was also utilized to search the data so that protein identifications were accepted at FDR of 1%. A label‐free intensity‐based absolute quantification (iBAQ) approach was used for calculating the protein quantifications. A dynamic regression function was built based on common peptides in tumor/adjacent samples. Based on the correlation value R2, Firmiana chooses a linear or quadratic function for regression to calculate the retention time 935 (RT) of corresponding hidden peptides and checks the existence of the extracting ion current (XIC) based on the m/z and calculated RT. The peak area values of existing XICs were determined by the program and were calculated as parts of corresponding proteins. Proteins with at least one unique peptide with a 1% FDR at the peptide level were selected for further analysis. The fraction of total (FOT) was used to represent the normalized abundance of a particular protein across samples and it was defined as a protein's iBAQ divided by the total iBAQ of all proteins identified in each sample. FOT values were multiplied by 105 for ease of presentation and missing values were assigned 10‐5. A total of 16 915 proteins and 11 678 somatic variants were obtained. Proteome raw datasets are publicly available at the iProX data portal: https://www.iprox.cn/page/PSV023.html;?url=1633189560349iEVs, with a password Ygzk. We used median value to define High and Low glyScore groups. To explore the potential differences in TME between High and Low glyScore groups, we used immunohistochemistry to estimate the expression level of CD70 (67749‐1‐Ig, Proteintech), CD80 (66406‐1‐Ig, Proteintech), CD86 (91882S; Cell Signaling Technology) and PD‐L1 (ab205921; Abcam) according to procedures as previously described. 21
2.8. Genomic and clinical data with immune checkpoint blockade and mTOR inhibitor therapy for ccRCC
One immunotherapeutic cohort—advanced ccRCC treated with PD‐1 blockade and mammalian target of rapamycin (mTOR) inhibition—was included. We obtained the genomic, transcriptomic and clinical data from Table S4 appended to the published paper. Then, we tested the glycolysis signature in the cohort and assessed the associations between the glyScore and drug response. 18
2.9. Statistical analysis
A comparison between the two groups in the boxplots was conducted using the Wilcox test, and the Kruskal‐Wallis rank‐sum test was used to compare the multi groups. To visualize the molecular types of the samples, Sankey diagrams of the glycolysis cluster, glycolysis gene cluster, glyScore grouping and survival status of 768 tumor samples were plotted using the ggalluvial package in R. The maftools package in R was used to visualize the somatic mutation map and CNV peak figure. To visualize the CNV data, the results of the GISTIC2 analysis of the sample files were necessary, but because the GISTIC2 results in TCGA database were not complete, the GISTIC2 analysis was implemented using the DNAcopy file. The methods and parameter settings were according to those detailed in the CNV Analysis Pipeline (https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/CNV_Pipeline/). The ComplexHeatmap package was used to draw the enrichment score heat map of the differential gene sets. The Pearson correlation coefficient of glycolytic genes was calculated, with the P‐value cutoff set as <.00001 and the absolute value of the correlation coefficient set as >.45. Genes were clustered by consistent clustering, and the prognostic effects of genes were determined by univariate Cox risk regression. The results were compiled into a table and imported into Cytoscape (3.7.2) to generate a gene interaction network map (Table 1).
TABLE 1.
The basic information of datasets used in our study
| Databases | Data label | Data platform | Tumor samples | Normal samples | Total samples |
|---|---|---|---|---|---|
| TCGA | TCGA‐KIRC | RNAseq | 531 | 71 | 602 |
| EMBL | E‐MTAB‐3267 | Affymetrix GeneChip Human Gene 1.0 ST array | 53 | 6 | 59 |
| ICGC | RECA‐EU | RNAseq | 91 | 45 | 136 |
| CPTAC | ccRCC | RNAseq | 93 | 75 | 168 |
| CM‐025 (CheckMate‐025) | CM‐025 | RNAseq | 311 | 0 | 311 |
| FUSCC | FUSCC | Proteome | 232 | 232 | 464 |
Abbreviations: ccRCC, clear cell renal cell carcinoma; CPTAC, Clinical Proteomic Tumor Analysis Consortium; EMBL, European Molecular Biology Laboratory; FUSCC, Fudan University Shanghai Cancer Center; ICGC, International Cancer Genome Consortium; TCGA, the cancer genome atlas; TCGA‐KIRC, the cancer genome atlas‐kidney renal clear cell carcinoma.
3. RESULTS
3.1. Glycolysis pattern of ccRCC (TCGA cohort)
We found that 81% of the glycolytic genes in the TCGA cohort were differentially expressed between normal and cancer tissues, as shown in Figure 1A. The CNV frequencies of the glycolytic genes showed that PFKFB4, SEC13, NUP210, ENO1 and NUP43 had high frequencies of deletion (14.3%, 13.8%, 13.6%, 5.1% and 3.9%, respectively), whereas HC3, GNPDA1 and PPP2CA had high frequencies of amplification (22.8%, 20.9% and 19.7%, respectively), as shown in Figure 1B. The mutation frequencies were highest for RANBP2 and NUP205 (3% and 2%, respectively) (Figure 1C ). The overall mutation frequency of the glycolytic genes was 22.92%, which may indicate broad transcriptional changes of glycolysis in ccRCC. The positions of the 72 glycolytic genes in the reference genome hg38 are shown in Figure 1D.
FIGURE 1.
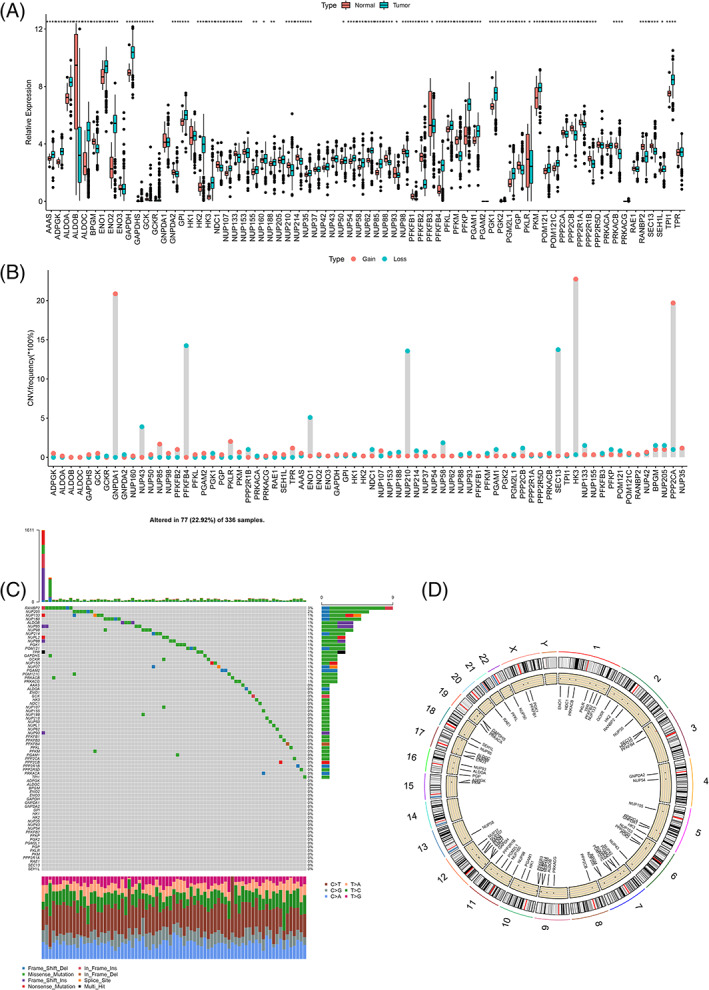
The overall display of glycolytic genes using the cancer genome atlas (TCGA) data. (A) Differential expression of 72 glycolytic genes in unscreened the cancer genome atlas‐kidney renal clear cell carcinoma (TCGA‐KIRC) tumor tissues and normal tissues. (B) Copy number variations (CNV) variation frequency of 536 samples of 72 glycolytic genes. (C) The spectrum of TCGA‐KIRC somatic mutation. (D) The genomic locations of 72 glycolytic genes [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Identifying four glycolysis patterns of ccRCC
Correlation analyses indicated close relationships between glycolytic genes and the glycolytic genes could be divided into three clusters based on consensus clustering (Figure 2A). Among the glycolytic genes, 36 genes were positively associated with prognosis and 11 genes were negatively associated with prognosis in 768 ccRCC patients (Table S1). Based on consensus clustering, 768 ccRCC samples were divided into four subgroups (glycolysis cluster A, B, C, D) and there were significant survival differences between the four subgroups (Figure 2B,C). The glycolysis cluster C was significantly associated with better overall survival. The functional enrichment analyses of the results of the GSVA for glycolysis clusters A, B and C identified six groups, as shown in Figure 2D‐F. These results showed that glycolysis cluster C genes involved in the fatty acid metabolism, beta‐alanine metabolism and limonene and pinene degradation pathways were significantly highly expressed. Conversely, glycolysis cluster B genes in these pathways were significantly lowly expressed, and glycolysis cluster A genes in these pathways were moderately expressed. The enrichment results for glycolysis cluster D genes are shown in Figure S1. The CIBERSORT results (Figure 2G) indicated that, except for eosinophil, monocyte and Type.17.T.Helper cell in different glycolysis.clusters, there were very significant differences in the contents of the other 20 infiltrating cells, and most of the infiltrating cells in glycolysis cluster B were higher than those of the other three clusters. For the 20 types of infiltrating cells, the prognostic effect was evaluated. As shown in Figure 2H, immature dendritic cells, mast cells and neutrophils were favorable prognostic factors of ccRCC, whereas Activated CD4 T cells, activated dendritic cells, macrophage, MDSC and Natural killer T cells were risk prognostic factors.
FIGURE 2.
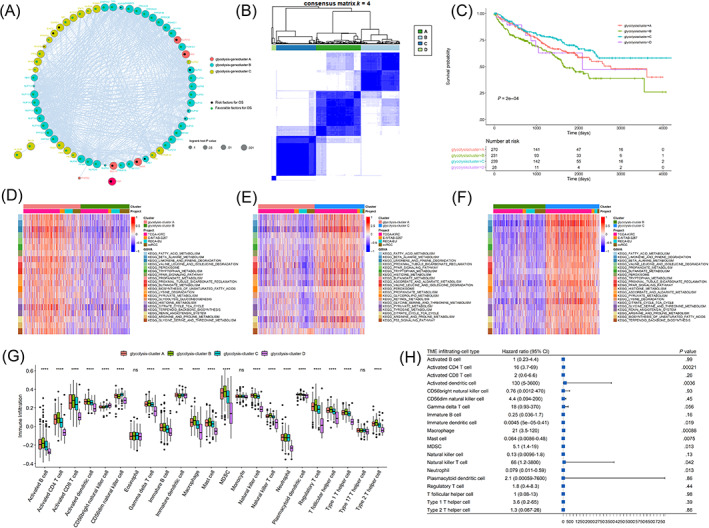
Unsupervised clustering of glycolytic genes (glycolysis cluster). (A) Glycolytic gene interaction network map, different colors represented different gene classifications, the size of gene nodes corresponded to the log‐rank test P‐value of Cox risk regression analysis. The smaller P‐value was, the more significant the prognostic effect was, and the larger the node was. Green dots in the nodes indicated favorable prognostic factors and black dots indicated risk factors. (B) The consistent clustering diagram. (C) Kaplan‐Meier survival curves of glycolysis cluster. D‐F represented top20 differential gene sets of glycolysis‐cluster A vs glycolysis‐cluster B, glycolysis‐cluster A vs glycolysis‐cluster C, glycolysis‐cluster B vs glycolysis‐cluster C, respectively (based on BH‐corrected P‐value sequencing). ssGSEA assessed the proportions and differences of 23 types of cells in different glycolysis clusters. (G) The boxplot showed the difference in the proportions of 23 infiltrating cells in different glycolysis clusters. (H) In the prognostic forest plot of differential infiltrating cells, each row represented one type of infiltrating cell. The third column showed the distribution of hazard ratio (HR) at 95% confidence intervals graphically, where the value of the horizontal axis corresponding to the blue box represented HR [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Four glycolysis clusters could be further simplified into three glycolysis gene clusters
Most of the glycolytic genes in glycolysis Clusters B and D were lowly expressed, whereas most of the glycolytic genes in glycolysis Clusters A and C were relatively highly expressed. Glycolytic gene expression patterns in glycolysis Cluster D were extremely different from those in the other three groups. The difference analysis between pairs of different glycolysis clusters identified a total of 223 overlapping differentially expressed genes (DEGs) (Figure 3A, Table S2). Based on the expression pattern of the DEGs, the four glycolysis clusters could be further simplified into three glycolysis gene clusters (Figure 3B). Consistent clustering of the DEGs identified three glycolysis gene clusters named gene cluster A, gene cluster B and gene cluster C, which contained 320, 185 and 263 ccRCC samples, respectively (Table S4). The enrichment scores of Angiogenesis, CD8+ effector T cells, EMT1, EMT2, EMT3 and pan‐fibroblast TGFb were significantly different in different glycolysis gene clusters (Figure 3C). Survival analysis indicated a very significant survival difference among three of the clusters, and that gene cluster C had a survival advantage (P‐value < .0001), as shown in Figure 3D. The gene ontology (GO) enrichment results showed that the DEGs were involved mainly inorganic anion transport and small molecule catabolic process under the biological process category, apical part of the cell and apical plasma membrane under the cellular component category, and symporter activity and solute sodium symporter activity under the molecular function category (Figure 3E, Table S3). Fifty‐three glycolytic genes were significantly differentially expressed among the glycolysis gene clusters, whereas the other 19 genes, GAPDHS, NDC1, NUP107, NUP155, NUP205, NUP37, NUP42, NUP62, NUP85, NUP98, PFKFB2, PFKFB3, PGAM2, PGK2, PKM, PRKACA, PRKACG, TPI1 and TPR, were not differentially expressed (Figure 3F).
FIGURE 3.
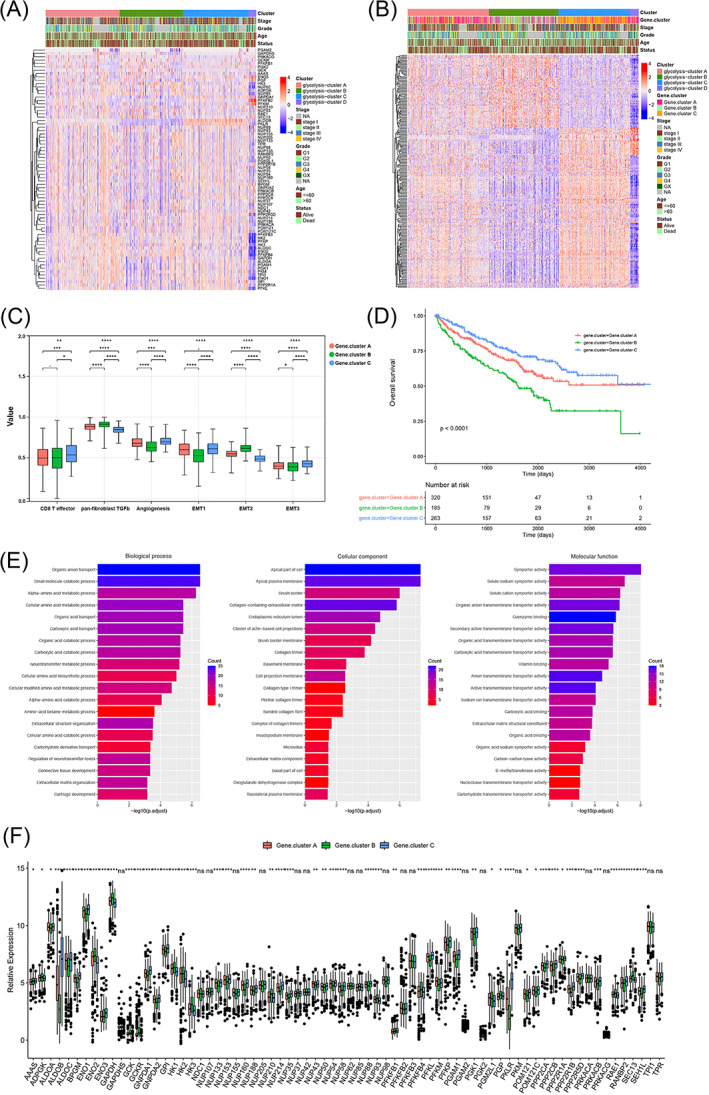
The relevance between different clusters and clinical features. Gray in the column indicated missing comment information of samples (A). Heat map (B) of 223 differential genes expression. Gray in the column indicated missing comment information of samples. (C) The difference of the enrichment scores of tumor‐related biological processes in different glycolysis gene clusters. Survival analysis of glycolysis gene cluster and expression of glycolytic genes in different glycolysis gene clusters (D). Enrichment bar chart. The color difference represented the number of enriched genes, the value on the horizontal axis represented the significant degree of enrichment, the higher, the more significant (E). Boxplot showing the expression of 72 glycolytic genes in different glycolysis gene clusters (F) [Color figure can be viewed at wileyonlinelibrary.com]
3.4. Calculating glycolysis score and evaluating its potential clinical and biological significance
The glycolysis scores (glyScore) of each sample were calculated (Figure 4A, Table S4). The Kaplan‐Meier curves of glyScore showed that there was a significant survival difference between the High and Low glyScore groups, and the Low glyScore group had a significant survival advantage (Figure 4B). Glyscore was significantly higher in both glycolysis cluster B/glycolysis gene cluster B (Figure 4C,D). And there is a trend that a higher glyScore may be associated with a higher tumor grade and tumor stage (Figure 4E,F). And enrichment scores of CD8+ effector T cells, EMT1, EMT2 and pan‐fibroblast TGFb were significantly different among the glyScore groups (Figure 4G). We evaluated the expression level of immune checkpoint proteins and found that most of the immune checkpoint proteins were upregulated significantly in High glyScore group, which indicated that High glyScore may be associated with a suppressive tumor microenvironment (Figure 4H). GSVA results also indicated that glyScore was significantly associated with fatty acid metabolism, beta‐alanine metabolism, peroxisome, etc. (Figure 4I). These results indicated that glyScore may be associated with various clinical characteristics and biological processes.
FIGURE 4.
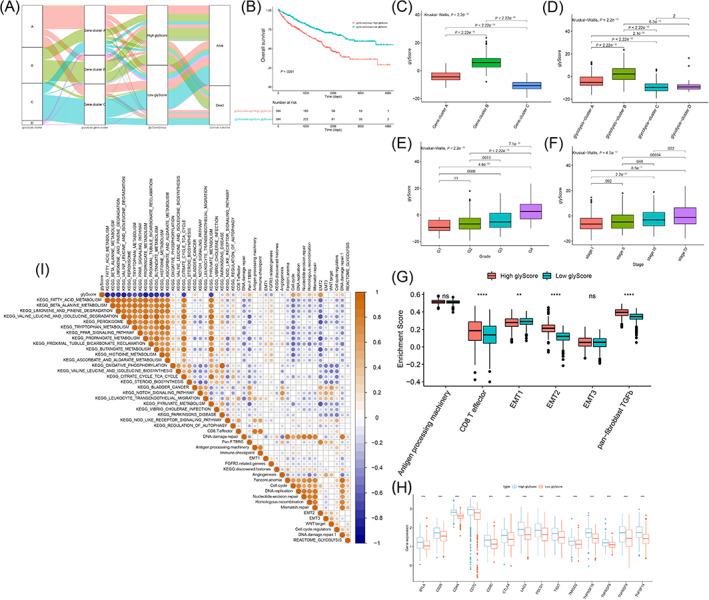
Calculating the glyScore. (A) The Sankey diagram showed the grouping and molecular typing of 768 samples. Kaplan‐Meier survival curves of glycolysis‐score (B). (C) The difference of glyScore among glycolysis clusters. (D) The difference of glyScore among glycolysis gene clusters. (E) The difference of glyScore among different stages of clear cell renal cell carcinoma (ccRCC). (F) The difference of glyScore among different grades of ccRCC. (G) The difference of the related biological processes enrichment scores between the glyScore groups. (H) Expression level of immune checkpoint proteins. (I) Correlation map of glycolysis‐score, glycolytic genes (REACTOME_GLYCOLYSIS gene set) and differential enrichment pathway. The shade of dots represented correlation degree, yellow was a positive correlation, blue was a negative correlation, and white was irrelevant. The size of the dots indicated the absolute value of the correlation. The larger the absolute value of the correlation, the larger the dots. Statistical test P‐value < .01, and the correlation coefficient that did not pass the statistical test was not shown [Color figure can be viewed at wileyonlinelibrary.com]
3.5. The landscape of High and Low glyScore groups and potential predictive capabilities of immune checkpoint blockade response of glyScore
Tumor mutation burden of the Low glyScore group was lower than that of the High glyScore group, and the G‐score of 5q35 of the Low glyScore group was higher than that of the High glyScore group (Figure 5A‐D). The TIDE prediction score was significantly higher in the High glyScore group, which implies that patients in the High glyScore group may not benefit from immune checkpoint blockade (ICB) therapy (Figure 5E). The area under the curve (AUC) of the 5‐year ROC curve for the glyScore was 0.649 (Figure 5F). To find out if the glyScore could behave better when it is included in a multi cox regression model, we enrolled samples with recorded tumor stage and grade for further analysis. In the revised version of our manuscript, we integrated the glyScore with tumor stage, grade to construct a glyScore‐associated nomogram as depicted in Figure 5G. The nomogram‐predicted OS is relatively similar with observed OS (Figure 5H) and the ROC‐AUC (prediction time = 5 years) value of nomogram is highest (Figure 5I, AUC = 0.760). These results indicated that glyScore could serve as a significant tool in predicting patients' overall survival.nomogram.
FIGURE 5.
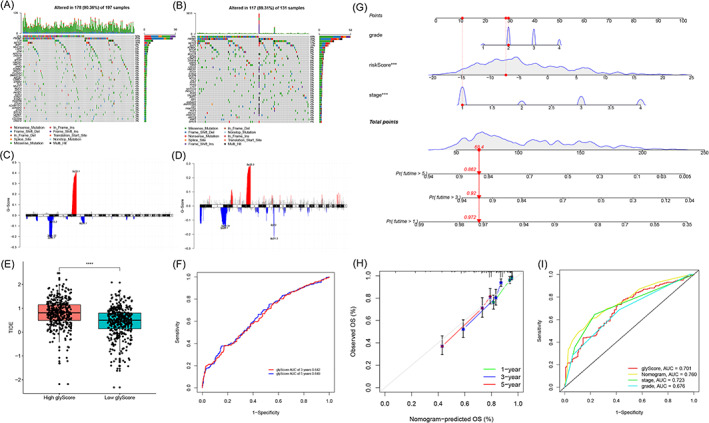
Landscape of High and Low glycolysis‐score groups and predictive ability of immune checkpoint blockade (ICB) response of the glyScore. (A) The spectrum of somatic mutations in the Low glyScore group. (B) The spectrum of somatic mutations in the High glyScore group. (C) Copy number variations (CNV) peak in the Low glyScore group. (D) CNV peak in the High glyScore group. (E) The difference of TIDE prediction scores between the glyScore groups. (F) The receiver operating characteristic (ROC) curve for glyScore's 5‐year survival. (G) Glycolysis‐related nomogram. (H) Calibration of the nomogram. (I) ROC curves of nomogram, glyScore, stage and grade (prediction time = 5 years) [Color figure can be viewed at wileyonlinelibrary.com]
3.6. GlyScore could predict patients' overall survival in FUSCC cohort and may be associated with drug response
In FUSCC cohort, higher glyScore was still significantly associated with a worse prognosis (Figure 6A) and we further validate the glyScore‐associated nomogram in FUSCC cohort, the ROC‐AUC (prediction time = 5 years) value is 0.934 (Figure 6B), which also indicated the stableness of the nomogram. Thus, external validation indicated the stableness of the glyScore in predicting prognosis. To explore more potential clinical significance, we tested the glyScore in two cohorts containing patients treated with everolimus or nivolumab. Although no significant correlations between glyScore and overall survival were observed (Figure 6C), higher glyScore was significantly associated with worse progression‐free survival (PFS) in patients treated with everolimus (Figure 6D, P < .01). Although no significant correlations between glyScore and PFS were observed (Figure 6F), higher glyScore was also significantly associated with worse overall survival in patients treated with nivolumab (Figure 6E, P < .05). As depicted in Figure 6G, we further validated the expression level of immune checkpoint proteins in FUSCC cohort and we found that CD70, CD80, CD86 and PDL1 were all elevated in High glyScore group. Our findings proved that higher glyScore may be associated with suppressive tumor microenvironment.
FIGURE 6.
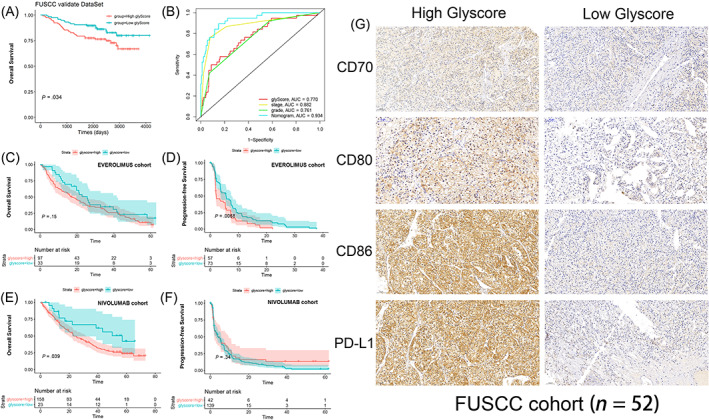
External validation of the glyScore. (A, B) Survival and receiver operating characteristic (ROC) curves based on glyScore and nomogram in Fudan University Shanghai Cancer Center (FUSCC) cohort. (C‐F) Overall survival and progression‐free survival curves based on glyScore in patients treated with everolimus or nivolumab. (G) Representative immunohistochemistry (IHC) images of immune checkpoint proteins [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
Metabolic deregulation is an important factor in many cancers. 22 ccRCC has been proved to be a complex disease resulted from multiple reasons, the role of metabolism should be taken into account critically, the components of TME, the energy production, even the immune surveillance of cancers can be altered by the reprogramming of metabolic pathways. 23 Thus, besides the genetic and epigenetic reasons, metabolism especially glycolysis in ccRCC should be paid more attention, after all the heterogeneity of ccRCC has make troubles for clinicians.
The Warburg effect has been a vital hallmark of various kinds of tumors, importantly, aerobic glycolysis supports adequate ATP for cancer cell. 24 Numerous enzymes and biomarkers involved in the glycolytic process would be dysregulated in ccRCC. 25 , 26 Thus, representative signatures or effective classification of glycolytic genes would be valuable for the diagnosis and prognosis of ccRCC.
For years, kinds of classification methods of ccRCC are conducted. Clinicopathologic types of ccRCC are regarded as convenient and practical, SSIGN (stage, size, grade and necrosis) scoring system has been once extensively adopted with ccRCC patients that accepted radical nephrectomy, this prediction model serves for clinicians to evaluate the possible prognosis according to the tumor characteristics. 27 Although this prediction model appears useful for partial nephrectomy as well, 28 it seems not comprehensive merely thinking about clinical factors. A more precise tool namely ClearCode34 is based on gene expression signatures, which supplies risk stratification (good/poor risk, ccA/ccB) for nonmetastatic and metastatic ccRCC, 29 , 30 it is more beneficial for assessing the risk of recurrence and death. Compared to above 2 models, Büttner et al 23 exploited a novel risk score called S3‐score, meaning the S3 regions of the proximal tubules in ccRCC tumors, S3‐score reflected diverse carcinogenic pathways relatively to ccA/ccB signature, while combining above 3 scoring systems, S3‐score will provide more value of prognosis, even clinical treatment choice. 32
For the glycolysis‐related genes (GRGs), Xing et al 33 established a novel model depended on 10 genes, their constructed model is validated by multiple cohorts and can predict the OS of ccRCC. After analyzing plentiful datasets, Zhang et al 34 focused on the overall biological pathway, and filtered 13 GRGs for a risk model, of all the genes can represent relatively different pathways and outcomes for ccRCC patients. Similarly, Lv et al 35 built a 7‐mRNA signature for indicating the prognosis, even the reaction of TKI and immune‐therapy. Also, Xu et al 36 constructed another glycolysis‐related risk signature from the perspective of tumor microenvironment, they suggested some crucial genes, CD44, PLOD1, PLOD2, to be the essential glycolytic risk genes, they put forward an important view that these glycolysis‐related genes were strongly linked to immune microenvironment of ccRCC.
As we know, tumor immune microenvironment suggests the communication between tumor cells and surrounding materials, indicating the occurrence and development of cancer. Based on the tumor microenvironment, Xu et al 37 set immunophenotyping clusters and validated some immune checkpoint molecules for ccRCC, they found that “hot” or “cold” clusters showed the gene mutations and clinical characteristics, so predicted the outcomes of ccRCC patients. Different from this kind of classification, we provided a more detailed method for classifying glycolysis genes; furthermore, we set up glyScore for precisely correlating between biological process and patients' clinical features and OS of ccRCC. Also, our system can indicate the effect of ICB therapy. In response to the advanced RCC, ICB would be in the first‐line therapy, 38 from our scoring system, patients in different glyScore groups can be defined as high or low response of ICB therapy, so as to apply for more suitable treatment.
Our scoring system and prediction model developed an innovative approach to classify the ccRCC in the light of glycolytic genes. Other than regular clustering analysis, we first took a route to screen differential genes between tumor and normal tissues, clustering these genes presented different state of expression in different clusters, the highlight of our system was the clustering of overlap differential genes between primary gene.clusters, this further clustering method make the classification of genes easier to identify, meanwhile, the survival analysis, screening glycolysis‐gene signatures and subsequent calculation of glycolysis‐score were all based on the gene.clusters, this novel clustering idea exhibited a well‐rounded system that can be more clinically relevant.
In summary, metabolic features in ccRCC perform remarkably, cells, gene identity, TME and immune response are influenced by various metabolic transformations. 39 Aiming to metabolism appears to indeed effective, whether from the perspective of diagnosis, treatment, or risk prediction.
AUTHOR CONTRIBUTIONS
The work reported in the paper has been performed by the authors, unless clearly specified in the text. Dingwei Ye, Hailiang Zhang and Yuanyuan Qu defined the theme of the study and discussed analysis, interpretation and presentation. Xi Tian, Yue Wang, Wenhao Xu, Haidan Tang and Shuxuan Zhu drafted the manuscript, analyzed the data, developed the algorithm, and explained the results. Aihetaimujiang Anwaier, Wangrui Liu, Wenfeng Wang, Wenkai Zhu and Jiaqi Su participated in the collection of relevant data and helped draft the manuscript.
FUNDING INFORMATION
This work is supported by Grants from the National Key Research and Development Program of China (No. 2019YFC1316005), National Natural Science Foundation of China (Nos. 81772706, 81802525 and 81902568), Shanghai Science and Technology Committee (Nos. 20ZR1413100 and 18511108000) and Shanghai Sailing Program (No.19YF1409700).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
The ethics approval and consent to participate in the current study were approved and consented to by the ethics committee of Fudan University Shanghai Cancer Center.
Supporting information
Appendix S1 Supporting Information
Table S1 Results of consistent clustering and prognostic analysis
Table S2 223 overlapped differentially expressed genes
Table S3 GO enrichment results of 223 overlapped differentially expressed genes
Table S4 Clinical information and clustering of 768 tumor samples
Table S5 Principal component analysis
Table S6 Sample IDs of all publicly available datasets
ACKNOWLEDGEMENT
We thank the TCGA, EMBL, ICGC and CPTAC database for providing NGS data and clinical information of ccRCC.
Tian X, Wang Y, Xu W, et al. Special issue “The advance of solid tumor research in China”: Multi‐omics analysis based on 1311 clear cell renal cell carcinoma samples identifies a glycolysis signature associated with prognosis and treatment response. Int J Cancer. 2023;152(1):66‐78. doi: 10.1002/ijc.34121
Xi Tian, Yue Wang, Wenhao Xu, Haidan Tang, Shuxuan Zhu are contributed equally to the study.
Funding information National Key Research and Development Program of China, Grant/Award Number: 2019YFC1316005; National Natural Science Foundation of China, Grant/Award Numbers: 81772706, 81802525, 81902568; Shanghai Sailing Program, Grant/Award Number: 19YF1409700; Shanghai Science and Technology Committee, Grant/Award Numbers: 18511108000, 20ZR1413100
Contributor Information
Yuanyuan Qu, Email: 19211230017@fudan.edu.cn.
Hailiang Zhang, Email: zhanghl918@alu.fudan.edu.cn.
Dingwei Ye, Email: 20111230051@fudan.edu.cn.
DATA AVAILABILITY STATEMENT
DNA copy file and RNA‐seq data, labeled TCGA‐KIRC, with 607 samples were downloaded from (https://portal.gdc.cancer.gov/). The data of the E‐MTAB‐3267 expression microarray was downloaded from EMBL database (https://www.ebi.ac.uk/arrayexpress/). The transcriptome data of RECA‐EU were downloaded from ICGC (https://dcc.icgc.org/) database. Transcriptome data for ccRCC samples in the CPTAC database were downloaded from https://cptac-data-portal.georgetown.edu/study-summary/S050. The sample IDs of all publicly available datasets used in this study are listed in Table S4. The proteomic profiles of the 232 ccRCC from FUSCC are publicly available at the iProX data portal: https://www.iprox.cn/page/PSV023.html;?url=1633189560349iEVs, with a password Ygzk. Further details and other data that support the findings of this study are available from the corresponding author upon request.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Teng R, Liu Z, Tang H, et al. HSP60 silencing promotes Warburg‐like phenotypes and switches the mitochondrial function from ATP production to biosynthesis in ccRCC cells. Redox Biol. 2019;24:101218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sanchez DJ, Simon MC. Genetic and metabolic hallmarks of clear cell renal cell carcinoma. Biochim Biophys Acta Rev Cancer. 2018;1870(1):23‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Turajlic S, Xu H, Litchfield K, et al. Deterministic evolutionary trajectories influence primary tumor growth: TRACERx renal. Cell. 2018;173(3):595‐610.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hsieh JJ, Purdue MP, Signoretti S, et al. Renal cell carcinoma. Nat Rev Dis Primers. 2017;3:17009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hu J, Chen Z, Bao L, et al. Single‐cell transcriptome analysis reveals Intratumoral heterogeneity in ccRCC, which results in different clinical outcomes. Mol Ther. 2020;28(7):1658‐1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crooks DR, Linehan WM. The Warburg effect in hominis: isotope‐resolved metabolism in ccRCC. Nat Rev Urol. 2018;15(12):731‐732. [DOI] [PubMed] [Google Scholar]
- 8. Riester M, Xu Q, Moreira A, Zheng J, Michor F, Downey RJ. The Warburg effect: persistence of stem‐cell metabolism in cancers as a failure of differentiation. Ann Oncol. 2018;29(1):264‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu M, Quek LE, Sultani G, Turner N. Epithelial‐mesenchymal transition induction is associated with augmented glucose uptake and lactate production in pancreatic ductal adenocarcinoma. Can Metab. 2016;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Icard P, Shulman S, Farhat D, Steyaert JM, Alifano M, Lincet H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist Updat. 2018;38:1‐11. [DOI] [PubMed] [Google Scholar]
- 11. Courtney KD, Bezwada D, Mashimo T, et al. Isotope tracing of human clear cell renal cell carcinomas demonstrates suppressed glucose oxidation in vivo. Cell Metab. 2018;28(5):793‐800.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sourbier C, Ricketts CJ, Matsumoto S, et al. Targeting ABL1‐mediated oxidative stress adaptation in fumarate hydratase‐deficient cancer. Cancer Cell. 2014;26(6):840‐850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chappell JC, Payne LB, Rathmell WK. Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers. J Clin Invest. 2019;129(2):442‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Linehan WM, Rouault TA. Molecular pathways: fumarate hydratase‐deficient kidney cancer—targeting the Warburg effect in cancer. Clin Cancer Res. 2013;19(13):3345‐3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grasmann G, Smolle E, Olschewski H, Leithner K. Gluconeogenesis in cancer cells—repurposing of a starvation‐induced metabolic pathway? Biochim Biophys Acta Rev Cancer. 2019;1872(1):24‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ran X, Xiao J, Zhang Y, et al. Low intratumor heterogeneity correlates with increased response to PD‐1 blockade in renal cell carcinoma. Ther Adv Med Oncol. 2020;12:1758835920977117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Braun DA, Hou Y, Bakouny Z, et al. Interplay of somatic alterations and immune infiltration modulates response to PD‐1 blockade in advanced clear cell renal cell carcinoma. Nat Med. 2020;26(6):909‐918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang B, Wu Q, Li B, Wang D, Wang L, Zhou YL. m(6)A regulator‐mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol Cancer. 2020;19(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD‐L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu W, Anwaier A, Ma C, et al. Multi‐omics reveals novel prognostic implication of SRC protein expression in bladder cancer and its correlation with immunotherapy response. Ann Med. 2021;53(1):596‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miranda‐Gonçalves V, Lameirinhas A, Henrique R, Baltazar F, Jerónimo C. The metabolic landscape of urological cancers: new therapeutic perspectives. Cancer Lett. 2020;477:76‐87. [DOI] [PubMed] [Google Scholar]
- 23. Büttner F, Winter S, Rausch S, et al. Survival prediction of clear cell renal cell carcinoma based on gene expression similarity to the proximal tubule of the nephron. Eur Urol. 2015;68(6):1016‐1020. [DOI] [PubMed] [Google Scholar]
- 24. Wang K‐J, Meng X‐Y, Chen J‐F, et al. Emodin induced necroptosis and inhibited glycolysis in the renal cancer cells by enhancing ROS. Oxid Med Cell Longev. 2021;2021:8840590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang J, Zhao X, Li X, Peng J, Yang W, Mi S. HMGCR inhibition stabilizes the glycolytic enzyme PKM2 to support the growth of renal cell carcinoma. PLoS Biol. 2021;19(4):e3001197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. He Y, Wang X, Lu W, et al. PGK1 contributes to tumorigenesis and sorafenib resistance of renal clear cell carcinoma via activating CXCR4/ERK signaling pathway and accelerating glycolysis. Cell Death Dis. 2022;13(2):118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Donini M, Buti S, Lazzarelli S, et al. Dose‐finding/phase II trial: bevacizumab, immunotherapy, and chemotherapy (BIC) in metastatic renal cell cancer (mRCC). Antitumor effects and variations of circulating T regulatory cells (Treg). Target Oncol. 2015;10(2):277‐286. [DOI] [PubMed] [Google Scholar]
- 28. Nuñez S, Saez JJ, Fernandez D, et al. T helper type 17 cells contribute to anti‐tumour immunity and promote the recruitment of T helper type 1 cells to the tumour. Immunology. 2013;139(1):61‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frank I, Blute ML, Cheville JC, Lohse CM, Weaver AL, Zincke H. An outcome prediction model for patients with clear cell renal cell carcinoma treated with radical nephrectomy based on tumor stage, size, grade and necrosis: the SSIGN score. J Urol. 2002;168(6):2395‐2400. [DOI] [PubMed] [Google Scholar]
- 30. Parker WP, Cheville JC, Frank I, et al. Application of the stage, size, grade, and necrosis (SSIGN) score for clear cell renal cell carcinoma in contemporary patients. Eur Urol. 2017;71(4):665‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Serie DJ, Joseph RW, Cheville JC, et al. Clear cell type a and B molecular subtypes in metastatic clear cell renal cell carcinoma: tumor heterogeneity and aggressiveness. Eur Urol. 2017;71(6):979‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brooks SA, Brannon AR, Parker JS, et al. ClearCode34: A prognostic risk predictor for localized clear cell renal cell carcinoma. Eur Urol. 2014;66(1):77‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xing Q, Zeng T, Liu S, Cheng H, Ma L, Wang Y. A novel 10 glycolysis‐related genes signature could predict overall survival for clear cell renal cell carcinoma. BMC Cancer. 2021;21(1):381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y, Chen M, Liu M, Xu Y, Wu G. Glycolysis‐related genes serve as potential prognostic biomarkers in clear cell renal cell carcinoma. Oxid Med Cell Longev. 2021;2021:6699808‐6699820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lv Z, Qi L, Hu X, Mo M, Jiang H, Li Y. Identification of a novel glycolysis‐related gene signature correlates with the prognosis and therapeutic responses in patients with clear cell renal cell carcinoma. Front Oncol. 2021;11:633950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu F, Guan Y, Xue L, et al. The effect of a novel glycolysis‐related gene signature on progression, prognosis and immune microenvironment of renal cell carcinoma. BMC Cancer. 2020;20(1):1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu W, Anwaier A, Ma C, et al. Prognostic Immunophenotyping clusters of clear cell renal cell carcinoma defined by the unique tumor immune microenvironment. Front Cell Dev Biol. 2021;9:785410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Klümper N, Ralser DJ, Zarbl R, et al. Promoter hypomethylation is a negative prognostic biomarker at initial diagnosis but predicts response and favorable outcome to anti‐PD‐1 based immunotherapy in clear cell renal cell carcinoma. J Immunother Cancer. 2021;9(8):e002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Büttner F, Winter S, Rausch S, et al. Clinical utility of the S3‐score for molecular prediction of outcome in non‐metastatic and metastatic clear cell renal cell carcinoma. BMC Med. 2018;16(1):108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information
Table S1 Results of consistent clustering and prognostic analysis
Table S2 223 overlapped differentially expressed genes
Table S3 GO enrichment results of 223 overlapped differentially expressed genes
Table S4 Clinical information and clustering of 768 tumor samples
Table S5 Principal component analysis
Table S6 Sample IDs of all publicly available datasets
Data Availability Statement
DNA copy file and RNA‐seq data, labeled TCGA‐KIRC, with 607 samples were downloaded from (https://portal.gdc.cancer.gov/). The data of the E‐MTAB‐3267 expression microarray was downloaded from EMBL database (https://www.ebi.ac.uk/arrayexpress/). The transcriptome data of RECA‐EU were downloaded from ICGC (https://dcc.icgc.org/) database. Transcriptome data for ccRCC samples in the CPTAC database were downloaded from https://cptac-data-portal.georgetown.edu/study-summary/S050. The sample IDs of all publicly available datasets used in this study are listed in Table S4. The proteomic profiles of the 232 ccRCC from FUSCC are publicly available at the iProX data portal: https://www.iprox.cn/page/PSV023.html;?url=1633189560349iEVs, with a password Ygzk. Further details and other data that support the findings of this study are available from the corresponding author upon request.